

Abstract
Viral diseases remain a major cause of death worldwide. Despite advances in vaccine and antiviral drug technology, each year over three million people die from a range of viral infections. Predominant viruses include human immunodeficiency virus, hepatitis viruses, and gastrointestinal and respiratory viruses. Now more than ever, robust, easily mobilised and cost-effective antiviral strategies are needed to combat both known and emerging disease threats. RNA interference and small interfering (si)RNAs were initially hailed as a “magic bullet”, due to their ability to inhibit the synthesis of any protein via the degradation of its complementary messenger RNA sequence. Of particular interest was the potential for attenuating viral mRNAs contributing to the pathogenesis of disease that were not able to be targeted by vaccines or antiviral drugs. However, it was soon discovered that delivery of active siRNA molecules to the infection site in vivo was considerably more difficult than anticipated, due to a number of physiological barriers in the body. This spurred a new wave of investigation into nucleic acid delivery vehicles which could facilitate safe, targeted and effective administration of the siRNA as therapy. Amongst these, cationic polymer delivery vehicles have emerged as a promising candidate as they are low-cost and easy to produce at an industrial scale, and bind to the siRNA by non-specific electrostatic interactions. These nanoparticles (NPs) can be functionally designed to target the infection site, improve uptake in infected cells, release the siRNA inside the endosome and facilitate delivery into the cell cytoplasm. They may also have the added benefit of acting as adjuvants. This chapter provides a background around problems associated with the translation of siRNA as antiviral treatments, reviews the progress made in nucleic acid therapeutics and discusses current methods and progress in overcoming these challenges. It also addresses the importance of combining physicochemical characterisation of the NPs with in vitro and in vivo data.
Keywords: Polymer, RNA interference, Antiviral therapeutics, Nucleic acid therapeutics, Quantitative nanomechanical atomic force microscopy, Physicochemical characterisation
The Challenge of Managing Ongoing and Emerging Infectious Diseases
The Global Burden of Viral Disease
For centuries, infectious diseases were the major cause of human morbidity and mortality. Today, cardiovascular disease and cancer are the highest-rated killers. However, viral diseases such as lower respiratory tract infections, HIV/AIDS, measles, hepatitis viruses and diarrhoeal diseases still remain among the leading causes of death and disability, particularly in developing countries (http://www.who.int/topics/mortality/en/; Fig. 1). The combined mortality rates associated with viral infection total over three million deaths per year.
Fig. 1.
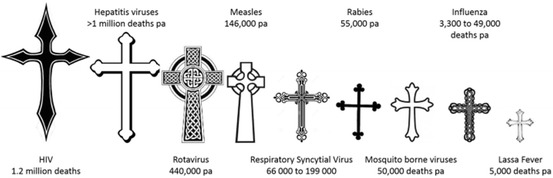
Current annual deaths due to major viral infections
Against a backdrop of established and persistent viral infections is the challenge of managing the threat of new epidemics from old and new infectious diseases (Fig. 2) [1]. Examples include recent outbreaks of new strains of the Ebola virus, which swept through Africa in 2014 and 2015, killing at least 11,000 people [2]; the swine flu epidemic of 2009, which spread around the world between April and December, resulting in an estimated 151,700 to 575,400 deaths [3]; and the emergence of a new, highly pathogenic strain of the Zika virus in South America, causing microcephaly in newborns, which has already spread to 67 countries, including the USA [4].
Fig. 2.
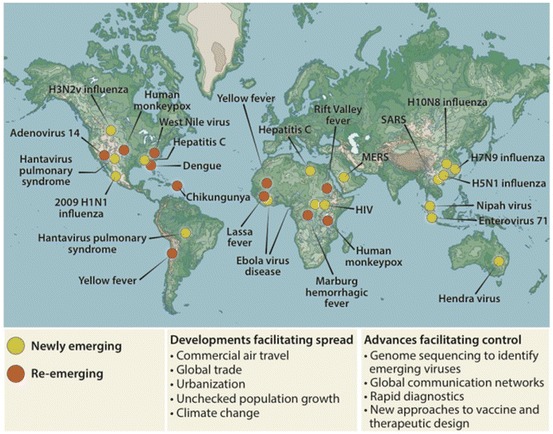
Mapping emerging viral diseases. Examples of sites where diseases caused by new and re-emerging viruses have appeared
Reproduced with permission from Marston et al., Science Translational Medicine, 2014 [1]
Over the past century, we have also observed the emergence of numerous new zoonotic infections which have had significant impact on the global burden of disease (Fig. 2). In particular, highly pathogenic avian influenza strains originating in poultry have contributed to a total of 856 laboratory-confirmed cases and 452 human deaths since 2003 [5]. Other examples of zoonotic spillover events include the emergence of new viruses originating in bats, such as the henipaviruses, and severe acute respiratory syndrome coronavirus (SARS-CoV) [6], all of which have caused outbreaks with high mortality rates and continue to threaten communities globally.
Viral infections in the agricultural industry are also of major concern, as infection of livestock can act as a reservoir for potential zoonotic pandemics. In addition, infectious outbreaks in crops and farming animals poses a significant threat to our economic and food security [7]. As we move into the future, an increasing socioeconomic burden of diseases is untenable. Thus there is a global need for intervention strategies to conserve and maintain human, plant and animal health and to secure our economic welfare [7, 8].
An Urgent Need for New Antiviral Strategies
The development of therapeutics against nine of the highest-impact human diseases has progressed rapidly over the past 50 years, leading to the commercial translation of over 90 antiviral drugs (Fig. 3). A range of different targeting approaches have been utilised to block the infection cycle of several viruses including HIV, hepatitis B virus (HBV), hepatitis C virus (HCV), herpesvirus, human cytomegalovirus, varicella-zoster virus, respiratory syncytial virus, influenza virus and external anogenital warts caused by human papillomavirus [9]. To date, combinations of these treatment and prevention strategies have saved millions of lives, in addition to generating revenue predicted to reach $36.44 billion by 2019 for the global antiviral market.
Fig. 3.
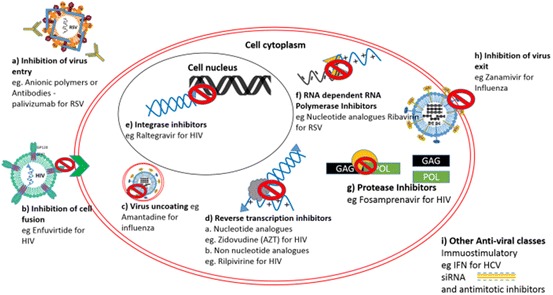
Current antiviral mechanisms. Nine classes of antiviral drugs have been developed, including inhibitors of a viral entry, b cell fusion, c viral uncoating, d viral integrases, e reverse transcriptases, f RNA-dependent RNA polymerases, g proteases, h viral exit, and i immunostimulatory or nucleic acid therapies [9]
Despite fervent efforts in the field of antiviral research and development, many persistent or newly emerging viral diseases in humans and livestock animals still do not have low-cost, safe or readily mobilised treatments or vaccines. Global health will be left in an increasingly vulnerable position as a result of a combination of factors that include an increased number of emerging disease outbreaks, limited availability of effective drug and vaccine candidates, development of resistant strains of pathogens, disease vectors carrying viruses into previously uninfected areas, the ease of movement of infected hosts via air travel, and declining rates of vaccination (Fig. 2) [1]. Thus it is essential that we move swiftly towards establishing improved detection, vaccination and treatment strategies.
Nucleic Acid Therapeutics
Nucleic acid therapy is being investigated as an alternative treatment solution for a wide range of infectious diseases, many of which are becoming resistant to standard drug interventions or which no longer respond reliably to traditional clinical management strategies. Three main approaches for nucleic acid therapies are currently under investigation.
Gene addition The host genome is supplemented with exogenous genetic material, resulting in the overexpression of naturally occurring immune factors to mount an improved immune defence against the pathogen [10].
Antisense oligonucleotides (ASO) ASOs are short (13–25) single-stranded DNA molecules that hybridise to a unique target sequence within viral messenger (m)RNA expressed in the host cell, thus modulating gene expression [11].
RNA interference (RNAi) This method is similar to ASO regulation; however, here gene expression is altered by small interfering (si)RNAs [12]. siRNAs are a class of double-stranded RNA, 20–25 base pairs in length, which use the microRNA pathway. By binding to complementary nucleotide sequences, they are able to direct the degradation of complementary mRNA, hence regulating the expression of the protein product.
The first gene addition therapy treatment was licensed in 2003 in China to treat head and neck squamous cell carcinoma. This treatment was an adenovirus-based medicine that delivered a functional copy of the p53 gene via recombinant adenovirus-p53 (Gendicine; Shenzhen SiBiono Gene Technologies) [13]. Since then, a further three gene addition therapeutics have been approved for market.
Glybera was approved in the European Union in 2012 [14] for the treatment of lipoprotein lipase deficiency, with an adeno-associated viral vector engineered to express lipoprotein lipase in the muscle [15]. In 2015, a joint advisory committee of the US Food and Drug Administration (FDA) voted to approve talimogene laherparepvec (T-Vec) with final approval granted by the FDA [16]. This attenuated, replication-competent oncolytic herpes simplex 1 virus was engineered to induce the expression of an immunostimulatory cytokine, granulocyte–macrophage colony-stimulating factor, for the treatment of melanoma [17]. Finally, GlaxoSmithKline are anticipating the imminent release of Strimvelis, a gamma retrovirus-mediated ex vivo gene therapy, in late 2016. Strimvelis is designed to treat the rare but severe combined immune deficiency disorder which arises from an adenosine deaminase deficiency [18]. Whilst the main focus of gene therapy to date has been on host genetic disorders, the benefits of this type of therapeutic strategy are not limited to mutations in human genes. As the field expands, this type of gene therapy may also be used to attenuate or inhibit the spread of the infectious pathogens, including viruses.
The FDA has also approved three ASO drugs. Fomivirsen, a phosphorothioate-modified oligodeoxynucleotide targeting the major immediate-early region proteins of human cytomegalovirus, was licensed by the FDA in August 1998 [11]. Intraocular administration of fomivirsen is used in the treatment of cytomegalovirus retinitis in immunocompromised patients, including those with AIDS. In 2013, FDA approval was granted for an ASO treatment, mipomersen, for the treatment of homozygous familial hypercholesterolaemia. This is a rare genetic disorder that leads to excessive levels of apolipoprotein B (apoB) and low-density lipoprotein cholesterol and results in premature cardiovascular disease [19]. However, because of the side effects of this treatment, administration is reserved for severe cases of the disorder. Finally, Alicaforsen [20] is used in the treatment of inflammatory bowel disease caused by ulcerative colitis. The FDA has designated the therapy as a fast-track candidate for unmet medical need in the orphan drug category.
Despite the translational success of gene addition and ASO therapies, there are no siRNA-based therapeutics currently on the market, due to their limited success in clinical trials [21]. Factors contributing to the failure of these therapeutics include the instability of siRNA and rapid clearance from the body, inability to efficiently enter target cells and an inability to reach the cell cytoplasm. To overcome these delivery challenges, particular effort is being focussed on the development of suitable nanoparticle (NP) delivery vehicles, which protect and facilitate siRNA delivery to its target site of action. The history of the discovery and development of siRNA-based therapies and the success and obstacles associated with this strategy are reviewed in greater detail below.
RNA Interference
The Discovery of RNA Interference
RNA interference (RNAi) is believed to have emerged as an antiviral defence system in eukaryotes, whereby the invading viral RNA genome is recognised and degraded in order to inhibit virus replication [22, 23]. The process of RNAi was first observed with the inhibition of RNA transcripts expressed in virus-infected transgenic plants [24]. A model proposed by Waterhouse et al. [24] hypothesised that the observed post-transcriptional gene silencing was mediated by an RNA-dependent mechanism present in the cell cytoplasm, and required a double-stranded (ds)RNA template for sequence-specific recognition and degradation of the foreign mRNA transcript.
Around the same time, other research groups were noting a similar mechanism in alternate eukaryotic systems (Fig. 4). In 1998, Fire and Mello [12, 25] published analogous findings in the nematode Caenorhabditis elegans. Injection of the nematode with double-stranded RNA (dsRNA) targeting the unc22 gene eliminated expression of an endogenous mRNA transcript. The cause of transcript suppression was proven to be due to the hybridisation between the injected dsRNA and the complementary endogenous mRNA. The hybridisation event ultimately resulted in mRNA cleavage by a dsRNA-dependent RNA polymerase and inhibition of protein production. This work confirmed the molecular mechanism underpinning the phenotypic observations made by Waterhouse et al., and thus the term “RNA interference” was coined.
Fig. 4.
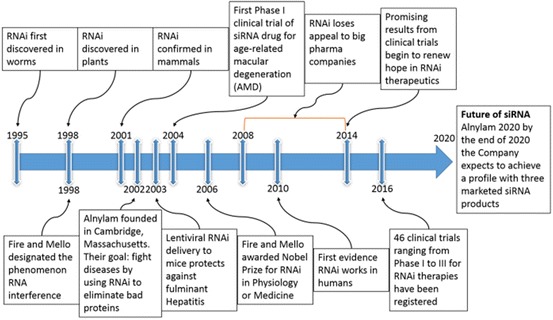
RNAi discovery timeline. 1995: First indication that sense RNA was as effective as antisense RNA for suppressing gene expression in C. elegans [26]. 1998: Waterhouse describes the process of RNAi in plants, whilst Fire and Mello coin the term “RNA interference” [12, 24]. 2001: First evidence that the RNAi mechanism is present in mammals [27]. 2002: Alnylam Pharmaceuticals founded by Phillip D. Zamore, Thomas Tuschl and Phillip Sharp. 2003: First evidence that RNAi is effective in inhibiting fulminant hepatitis virus in a mouse model [28]. 2004: Acuity Pharmaceuticals performs first phase I clinical trial of siRNA. 2006: Andrew Fire and Craig Mello awarded the Nobel Prize in Physiology or Medicine for RNA interference–gene silencing by double-stranded RNA. 2008–2014: Due to lack of success in clinical trials, many of the large pharmaceutical companies stop investing in siRNA technology. 2010: Davies describes first evidence of siRNA-mediated inhibition in humans [29]. 2014: RNAi begins to show promising clinical progress [30]. 2016: A total of 46 registered siRNA clinical trials for RNAi therapeutics from phase I to phase III trials, most involving siRNA NP formulations (http://clinicaltrials.gov). Future: By the end of 2020, Alnylam Pharmaceuticals, still the largest RNAi-dedicated company, expects to achieve a company profile with three marketed products and ten RNAi therapeutic clinical programs—including four in late stages of development (http://www.alnylam.com)
As a result of the rapid advances in gene technology that followed from this groundbreaking research, Fire and Mello were awarded the Nobel Prize in Physiology or Medicine in 2006 (the shortest period ever from discovery to award). RNAi seized the world’s attention because it offered a way to attenuate the production of any protein within a cell, including those associated with disease states. Since this revolutionary discovery, a staggering amount of research has been undertaken to harness this molecular pathway as a therapeutic strategy for the treatment of cancer, genetic disorders and viral infections.
Endogenous RNAi Pathway
We now know that RNA interference is a process that has been adapted over evolutionary time to contribute to the regulation of eukaryotic protein synthesis. The RNAi pathway has been identified in all eukaryotes [23]. Similarly, all eukaryotes encode a variety of short RNA strands, typically referred to as microRNA (miRNA), that are not destined for translation but as a secondary level of transcriptional control [31, 32].
miRNAs can be transcribed in the nucleus either as primary (pri-)miRNAs containing ∼70 nucleotide long stem–loop structures or encoded within mRNA introns [33] (Fig. 5). Two proteins, the RNase III enzyme Drosha [34] and the dsRNA binding protein DGCR8, make up the microprocessor complex that cleaves the pri-miRNA to the precursor (pre-)miRNA [35]. Pre-miRNAs are then transported into the cytoplasm by a nucleocytoplasmic transport factor exportin-5 [36]. Mature miRNA production requires a final cleavage step to remove the loop structure and to produce the short dsRNA. This is performed by another RNase III enzyme, Dicer. The miRNA then associates with the multiprotein RNA-induced silencing complex (RISC), which comprises Argonaute 2, Dicer, and the human immunodeficiency virus trans-activation response RNA-binding protein. The miRNA is unwound and the guide miRNA strand is integrated into the RISC complex. The remaining strand, the passenger strand, is degraded as a RISC complex substrate. The RISC-bound miRNA can then recognise and bind complementary mRNA, such as viral RNA, in the host cell cytoplasm. RISC then cleaves the target mRNA at the position facing nucleotides 10 and 11 of the siRNA, leading to degradation of the mRNA and inhibiting any subsequent protein production. For a comprehensive review of this process, see [37].
Fig. 5.
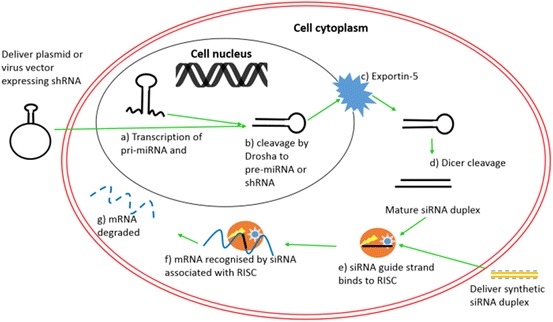
RNAi pathway. a Transcription of pri-miRNA or shRNA; b Drosha cleavage produces pre-miRNA; c miRNA exported into cytoplasm; d Dicer cleavage produces mature miRNA; e association with RISC complex; f recognition and cleavage of target mRNA; g mRNA degraded, no protein production
siRNA as an Antiviral Therapeutic
Following the discovery of RNAi pathway in C. elegans [12], researchers found that the introduction of exogenous, synthetically synthesised siRNA molecules into cells could also enable specific and targeted mRNA transcript silencing [27]. In mammalian cells, siRNA is typically composed of synthetic dsRNA molecules 20–25 base pairs in length with 3′ overhangs on each strand (Fig. 6a; Table 1) [27]. Plasmid or virus vector-based delivery of siRNA has also been explored (Fig. 6b–d; Table 1), whereby the siRNA is produced as short hairpin (sh)RNA [38], shRNAmirs that mimic natural miRNA structures [39, 40] or multiple hairpins in the nucleus [41, 42]. These then enter the pathway using exportin-5 to exit the nucleus (Fig. 5). The ability of synthetic siRNAs to selectively target and degrade mRNA paves the way for a novel class of antiviral therapeutics capable of inhibiting protein viral production and suppressing viral infection.
Fig. 6.
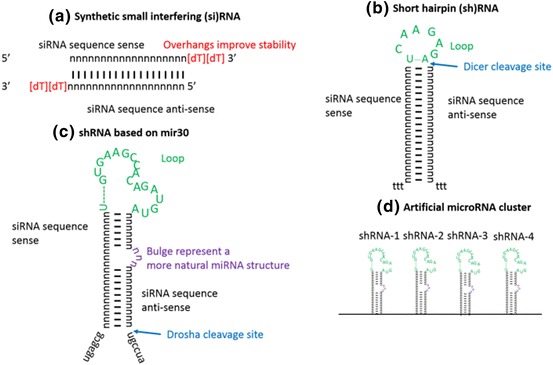
RNAi strategies. a Synthetically designed small interfering RNAs; b plasmid-based short hairpin RNAs; c sRNAs based on the design of natural mir-30 believed to enhance recognition by the RNAi pathway; d delivery of multiple shRNAmir clusters for improved silencing and inhibition of escape mutants
Table 1.
Advantages and disadvantages of the different RNAi-based silencing systems
| RNAi molecule | Activity | Advantage | Disadvantage |
|---|---|---|---|
|
MicroRNAs [32] |
Naturally occurring small RNA molecule responsible for regulation of mRNA translation |
Naturally occurring, some are known to be involved in cancer and virus regulation [43–45] Active in the cytoplasm Enters RISC directly |
Highly unstable in physiological conditions May have multiple mRNA targets, resulting in off-target side effects |
|
Synthetic small interfering RNA (siRNA) [27] |
Synthetic double-stranded RNA molecule with complementary sequence to target gene |
Specifically designed to target the gene of interest, minimising off-target effects Highly effective in laboratory environments Active in the cytoplasm Enters RISC directly |
Highly unstable in physiological conditions May activate an immune response |
|
Short hairpin RNA (shRNA) [21] |
Artificial RNA molecule with an artificially designed tight hairpin loop. One stem sequence is complementary to the target gene |
Can be delivered as a plasmid or virus vector with a poll III promoter for more stable and longer-lasting treatment Enters the RNAi pathway at an earlier step than siRNA, which may be advantageous for activity |
Needs to be delivered to the nucleus, requiring crossing of the nuclear membrane May activate an immune response May result in excess RNAi molecule expression, leading to saturation of the natural miRNA pathways and resulting in severe toxicities [46] |
|
Mirised short hairpin RNA (shRNAmir) |
Artificial RNA molecule with a hairpin loop and bulges derived from a native microRNA. One stem sequence is complementary to the target gene |
More stable as delivered via a plasmid or virus vector Mimics the native miRNAs more closely, therefore recognised by the RNAi machinery more accurately Enters the RNAi pathway at an earlier step than siRNA |
Needs to be delivered to the nucleus, requiring crossing of the nuclear membrane May activate an immune response May result in excess RNAi molecule expression, leading to saturation of the natural miRNA pathways and resulting in severe toxicities [46] |
|
Artificial MicroRNA cluster [42] |
Artificial RNA molecule with multiple stem loops derived from a native microRNA cluster, i.e. mir-17-92, stem sequences have been replaced to be complementary to the target gene/s |
As above, with the additional benefit of delivering multiple RNAi molecules for targeting of several sites of one gene or different genes Avoidance of escape mutants |
As above First RNAi molecule is expressed at higher levels than remaining molecules [47] |
Multiple studies have shown that siRNAs can inhibit a wide range of viruses (Table 2). All viruses have an mRNA step in their replication cycle for production of viral proteins; therefore, even viruses with DNA-based genomes can be targeted (Fig. 7; Table 2). The attraction of siRNA as a unique antiviral therapeutic is because synthetic siRNAs can be rapidly developed against any virus, provided that the sequence of the viral RNA is known. Furthermore, conserved target sequences have been identified for several virus families, thus offering promise for the development of pan-family therapeutic strategies, which could provide protection against multiple viruses in one therapeutic [48, 49].
Table 2.
Antiviral siRNAs or shRNAs designed and validated
| Virus family | Genome | Genes targeted | |
|---|---|---|---|
|
Herpes simplex virus |
Herpesviridae | Linear double-stranded DNA genome | UL29.2 (a viral DNA binding protein affecting virus replication) UL54, UL29 and UL27 |
|
Human immunodeficiency virus |
Retroviridae | Single-stranded positive-sense RNA virus with a DNA intermediate | HIV-1 core proteins Gag, Rev, Tat and the 3′-LTR |
|
Hepatitis C virus [63] |
Flaviviridae | Positive-sense single-stranded RNA virus | Viral internal ribosome entry sequence pseudoknot |
|
Human parainfluenza virus [64] |
Paramyxoviridae | Negative-sense single-stranded RNA viruses | Essential viral RNA-dependent RNA polymerase subunits, L and P |
|
Respiratory syncytial virus |
Paramyxoviridae | Negative-sense single-stranded RNA viruses | Nucleocapsid and essential viral RNA-dependent RNA polymerase subunits, L and P, |
|
Hepatitis B virus |
Hepadnaviridae | Circular DNA, but not fully double-stranded | 3.5-kb pre-genomic RNA encoding the core protein/HBeAg and polymerase-reverse transcriptase |
|
Human papillomavirus 18 [67] |
Papillomaviridae | Circular, double-stranded viral genome | Early proteins 6 and 7 mRNAs |
|
Measles virus [68] |
Paramyxoviridae | Negative-sense-single-stranded RNA viruses | Nucleocapsid (N), phosphoprotein (P), and polymerase (L) mRNAs |
|
Rotavirus [69] |
Reoviridae | 11 segments of dsRNA | Virus spike protein VP4 |
|
Ebola virus [57] |
Filoviridae | Negative-sense single-stranded RNA linear genome | Viral protein 35, RNA polymerase |
|
Hendra virus [70] |
Paramyxoviridae | Negative-sense single-stranded RNA genome | N, P and M mRNA |
|
Influenza |
Orthomyxoviridae | Negative-sense segmented single-stranded RNA genome | Nucleoprotein, RNA Polymerase subunit B |
|
SARS coronavirus [74] |
Coronaviridae | Positive-sense single-stranded RNA genome | Spike protein-coding and ORF1b (NSP12) regions |
|
Vesicular stomatitis virus [64] |
Rhabdoviridae | Negative-sense single-stranded RNA genome | Essential viral RNA-dependent RNA polymerase subunits L and P |
|
Foot and mouth disease virus [75] |
Picornaviridae | Positive-sense single-stranded RNA genome | 3D protein and 3′ untranslated region |
|
Bovine viral diarrhoea virus [76] |
Flaviviridae | Positive-sense single-stranded RNA genome | 5′ non-translated (NTR) region and the regions encoding the C, NS4B and NS5A proteins |
|
Chicken anaemia virus [77] |
Circoviridae | Single-stranded circular DNA genome | Viral protein (VP) 1, or overlapping sections of VP2/3 and VP1/2 |
|
Marek’s disease virus [78] |
Herpesviridae | Linear double-stranded DNA genome (10) which contains ~80 genes | gB glycoprotein and ICP4 transcriptional regulatory mRNAs |
Fig. 7.
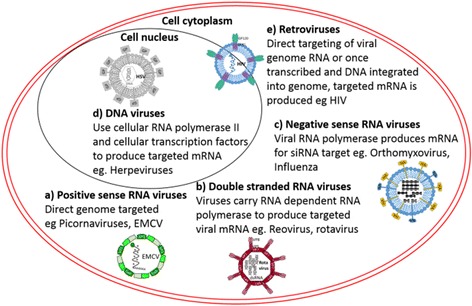
siRNA targeting of DNA and RNA viruses—role of RNA in the viral life cycle. a Positive-sense RNA viruses, where the viral genome is directly targeted; b double-stranded RNA viruses, where the genome is transcribed by a viral protein to produce the targeted mRNA; c negative-sense RNA viruses, where the genome is transcribed by a viral protein to produce the targeted mRNA; d DNA viruses, which use the host cell transcription factors to produce the targeted mRNA; e retroviruses, similar to positive-sense RNA genomes, where the genome can be directly targeted, or once integrated into the cell DNA, viral mRNA produced can also be targeted
Advances in high-throughput sequencing technology allow the rapid adaptation of the targeting siRNA therapy to emerging or new viral strains and viruses conferring drug resistance [50–54]. This is of the utmost importance in outbreak response and management efforts, as the speed at which a therapeutic can be developed and produced in large quantities for treatment is often a major bottleneck to management strategies. Industry has also recognised this need and have now made the fast translation of large quantities of therapeutic product feasible [55, 56]. For instance, in the case of the recent Ebola outbreak, a newly designed siRNA targeting the emerged Ebola virus Makona strain was produced and incorporated into a lipid NP system within 8 weeks [57], and was deployed for compassionate use during the outbreak. This is remarkable compared to an average of 20 months that it takes to produce a new vaccine.
However, despite the promise that siRNA-based therapy holds for the treatment of viral disease, limited success in clinical trials has restricted the production and marketing of siRNA therapeutics. Understanding and overcoming the limitations and barriers to siRNA delivery, therefore, is essential for maximising the potential of this novel therapeutic approach in modern medicine.
Delivery of Antiviral siRNAs
Issues
Multiple factors contribute to the main obstacles to efficient siRNA delivery (Fig. 8). The key obstacles are listed below.
siRNA is highly unstable in bodily fluids due to RNAses present in serum and mucosal layers, which rapidly degrade the siRNA phosphate backbone. Thus, efficient protection from RNAses must be sought [79].
Due to its small size (less than 5 nm), siRNA is rapidly cleared via renal filtration. Up to 40 times higher concentrations of siRNA have been observed in the kidneys and in excreted urine compared to other tissues 1 h following intravenous injection [80]. In another study, 10% of the siRNA dose was found in the kidneys after only 30 min [81].
During viral infection, there is rarely an enhanced permeability retention (EPR) effect in the infected tissue. This is in contrast to delivery models of cancer therapeutics, where a combination of poor lymphatic drainage and leaky vasculature allows accumulation of the therapeutic at the target site. Thus it is important to find ways to increase the circulation times of serum siRNA for successful delivery strategies.
As with all nucleic acids, the phosphate backbone of the siRNA results in an overall negatively charged particle (Fig. 9). The outer leaflet of the cell membranes also carries a slight negative charge, therefore repelling the naked siRNA from binding and efficiently entering the cell. In order to overcome this problem, the negative charge of the siRNA should be shielded to allow for efficient transport across the cell membrane.
Typically, siRNA therapeutics cross the cell membrane via the endocytic pathway and thus must escape the endosome before they can interact with the RISC complex in the cytoplasm. An effective delivery of siRNA must therefore be able to efficiently disrupt the endosome in order to deliver the siRNA to the cytoplasm prior to being transported to and degraded in a lysosome [82].
Fig. 8.
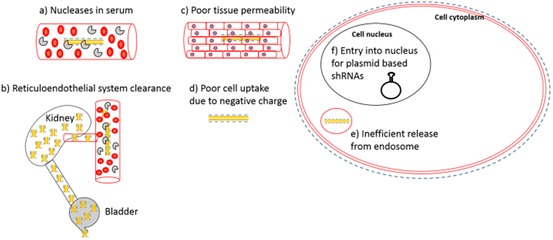
Obstacles to the uptake of naked siRNA. a Degradation of siRNA by nucleases in the serum; b clearance through the liver and kidney by the reticuloendothelial system; c poor tissue permeability; d poor cell uptake due to negative charge on cell membrane and siRNA; e inefficient release from the endosome
Fig. 9.
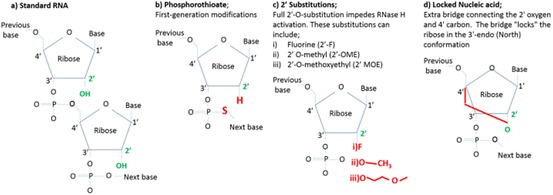
Oligonucleotide modifications. a The chemical structure of unmodified RNA bases. The 2′ OH group in green is the distinguishing feature and target of modifications. b Phosphorothioate modification, where the non-bridging oxygen on the phosphate linkage is replaced with a sulfur atom (red); c additional 2′-O- substitutions; d locked nucleic acids, where the ribose moiety is modified with an extra bridge connecting the 2′ oxygen and 4′ carbon
Therefore, unless delivered directly to the site of treatment, naked siRNA is ineffective as a therapeutic.
Current Methods for Improving Uptake
Significant work has been undertaken towards the development of carriers for RNAi. Table 3 summarises some of the advantages and disadvantages of the different methods (for a comprehensive review see [83]). Some of the more advanced methods currently in clinical trials are then described.
Table 3.
Advantages and disadvantages of commonly used NPs for siRNA delivery
| NP material | Advantages | Disadvantages |
|---|---|---|
|
Cationic block polymers |
Relatively simple synthesis Inexpensive High throughput Large choice of functionalities (e.g. biodegradable units) Stimulus-responsive polymers for “smart” delivery |
High polydispersity Variable silencing efficiency Balance between high toxicity (at high MW) and efficient cell transport (also at high MW) Potential for protein corona formation resulting in rapid clearance |
|
Cationic dendrimers [86] |
Low polydispersity Large choice of functionalities Molecular architecture and biodistribution can be accurately tuned |
Complex synthetic routes Balance required between high toxicity (at high MW) and efficient cell transport (also at high MW) Potential for protein corona formation and rapid clearance |
|
Cationic lipid formulations |
Ease of formulation Limited synthesis required (self-assembly) Rapid and well-characterised cell internalisation Commercially available materials |
Low siRNA loading efficiency High production cost More limited choice of functionalities (cf. polymers) Leakage or degradation of loaded materials Tendency to aggregate Some toxicity |
|
Metallic NPs (Ag/Au) [90] |
High control of NP shape and morphology Ease of preparation Ease of surface modification Biocompatibility (Au) |
siRNA bound to surface of particle, so limited protection offered Particle aggregation Toxicity (Ag) Non-biodegradable |
|
Quantum dots (QDs) [91] |
Inherent fluorescence of QDs enables dual imaging and therapeutic (theranostic) systems |
Metal core toxicity More complex surface functionalisation chemistry required (cf. Ag/Au NPs) Particle aggregation |
|
Viral delivery [92] |
High silencing efficiency Highly monodisperse |
Safety concerns Expensive May trigger immune response, neutralising efficacy |
Modified siRNAs
To overcome the issue of serum degradation, chemical modification of siRNA nucleotides has been investigated [93–95]. The 2′-OH of the ribonucleotide distinguishes RNA from DNA and is the catalytic target of RNases (Fig. 9a). Modification of the nucleotide at the 2′-O-position of the sugar has been shown to increase siRNA stability against RNAses. Such adjustments include a phosphorothioate modification, where the non-bridging oxygen on the phosphate linkage is replaced with a sulfur atom; 2′-O-methoxyethyl (2′-O-MOE) and 2′-deoxy-2′-fluoro (2′-F) substitutions that impede RNAse activation; and locked nucleic acids (LNA) that lock the 2′ group to the carbon 4′ group to inhibit RNAse degradation (Fig. 9b–d) [79, 94, 96].
The most advanced antiviral siRNA clinical trials using modified siRNAs were conducted by Alnylam Pharmaceuticals to treat respiratory syncytial virus (RSV) using 2′-O-MOE-modified anti-RSV siRNA (Fig. 10) [97, 98]. The first trial was a safety and pharmacokinetic study analysing single doses up to 150 mg or multiple doses over 5 days. None of the 101 adults in the cohort showed any adverse reactions [97]. Alnylam followed on with human trials [99], where 85 healthy human subjects were infected and nasal washes were assayed for antiviral effects and clinical efficacy. The ALN-RSV01-treated groups saw a 38.1% reduction in the rate of infection, with a trend toward lower viral titres, demonstrating for the first time that a respiratory-delivered siRNA could significantly reduce RSV infection [99].
Fig. 10.

Alnylam’s RSV-modified siRNA. a siRNA sense strand 5′–3′ 2′OME modified bases indicated in red; deoxynucleotide overhangs indicated in green; b siRNA antisense strand 5′–3′ modified bases indicated in red
Two further trials, phase IIa and b, assessed the ability of ALN-RSV01 to inhibit bronchiolitis obliterans syndrome following RSV infection in lung transplant recipients [98, 100]. Both trials resulted in a significant reduction in the presentation of bronchiolitis obliterans syndrome from RSV infection in lung transplant patients. These results have led Alnylam to discuss strategies for moving the drug forward with the FDA for registration. Alnylam has also conducted clinical trials with siRNAs directly conjugated with the asialoglycoprotein receptor ligand N-acetylgalactosamine (GalNAc). A GalNac-conjugated transthyretin (TTR) siRNA has entered phase III trials, with >90 TTR knockdown in serum for treatment of TTR-mediated amyloidosis [119]. The second candidate is a treatment for haemophilia and related blood disorders, targeting anti-thrombin 3 (AT3). A recent press release from Alnylam indicated that >60 AT3 knockdown in serum was achieved in the phase I trial (http://www.alnylam.com/capella).
Lipids
Commercially available lipid-based transfection reagents such as Lipofectamine™ and FuGENE® 6 have been used in in vitro studies on several occasions. These reagents have demonstrated successful delivery of siRNAs targeting influenza and protection from lethal challenge in animal models [72]. However, due to their toxicity, the translation of these agents into viable siRNA carriers for applications in humans has not been explored [88].
Nonetheless, a significant body of literature is available to demonstrate that other lipid-based systems may be suitable for therapeutic siRNA delivery in humans (Fig. 11). These studies have been comprehensively reviewed [21, 87, 89, 101]. During the 2014–2015 Ebola outbreak, for example, clinical trials were conducted for an anti-Ebola siRNA-lipid-bound small interfering RNA (siRNA) known as TKM-130803 [102]. Administration of TKM-130803 at a dose of 0.3 mg/kg per day by intravenous infusion for 7 days to adult patients with severe Ebola virus disease did not improve survival when compared to historic controls. This was despite promising results in experimentally infected non-human primate models [57], and was attributed to the fact that the delivery occurred during an advanced disease stage, by which time the viral load and existing organ damage was overwhelming [102]. In addition to obstacles pertaining the timing of therapeutic administration, lipid nanocarriers often have a tendency to aggregate, and the active ingredient can be degraded during storage. This makes large-scale manufacturing and cold chain storage difficult, particularly in remote and arid settings.
Fig. 11.
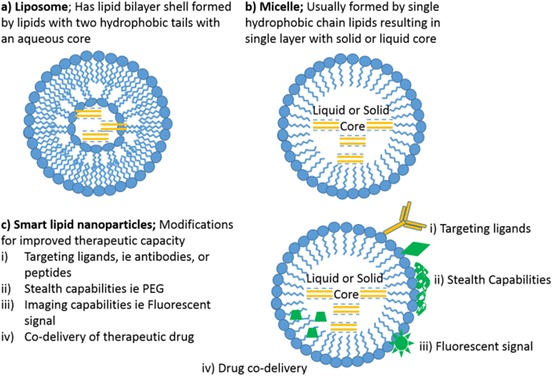
Lipid NP structures. a Liposome; b micelle; c smart lipid NPs
Viral Vectors
The most commonly used viral vectors include adenovirus, lentivirus and retroviruses (Fig. 12). Retroviruses have the ability to integrate into the host genome, allowing continuous expression of the RNAi treatment, whilst adenoviruses do not. Viral vectors have given significant hope to the long-lasting treatment of HIV and hepatitis B and C [62, 89, 92]. Clinical trials of siRNA delivery using viral vectors have been conducted in HIV patients, in which participants underwent transplantation of lentiviral-transformed haematopoietic progenitor cells designed to express three anti-HIV siRNAs. This would theoretically reinforce the individuals’ immune repertoire and control HIV viraemia [92, 103]. Clinical trials demonstrated that this approach was safe and transiently reduced viral load and increased CD4 immune cell count; however, this was not sufficient to produce a clinical benefit. Significantly more work is required to improve the transduction and expansion of the haematopoietic progenitor cells.
Fig. 12.
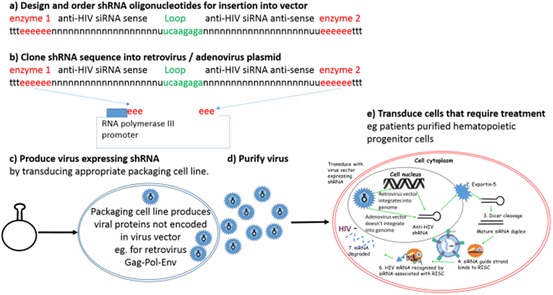
Development of viral vectors. a Design the shRNA sequences required using online algorithms and oligonucleotide companies; b clone shRNA oligonucleotides into the commercially available viral vector plasmid that suits the expression profile required; c transfect plasmids into specialised commercially available packaging cell lines that produce the replication factors required for the specific virus vector; d purify the virus that is produced, and e transduce the cells that require treatment
Polymers
Since the 1970s, the use of polymeric NP formulations has rapidly expanded [85]. Polymers offer greater stability, more control over structure and drug loading, increased opportunities for high-throughput synthesis compared to lipid-based systems and better quality control. Polymers have also been adapted for specific functions, to provide stimuli-responsive properties including responses to pH, redox, enzyme and light.
Cationic polymers show the greatest promise for the delivery of siRNA, as they can electrostatically bind and condense siRNA into NPs and improve cellular uptake [104]. However, with the use of cationic polymers there is a fine balance between membrane disruption that results in toxicity and membrane disruption that results in transfection. Both synthetic and natural polymers have been explored as candidates, and they must be non-toxic, non-immunogenic and non-carcinogenic, and should ultimately be degraded or cleared from the body via excretion. Multiple polymer compositions and architectures have been investigated for their suitability for siRNA delivery. Some examples include block co-polymers (di-blocks [105] and tri-blocks [71, 106]), polymeric micelles [107], dendrimers [108] and star polymers [109–111] (Fig. 13). Subtle differences in architectures and the physicochemical properties of the NP complexes (i.e. size, stiffness, surface potential, morphology, hydrophobicity) [112, 113] can have significant effects on the in vitro and in vivo delivery efficiency [85]. Thus polymer composition should be carefully considered when designing such delivery vehicles.
Fig. 13.
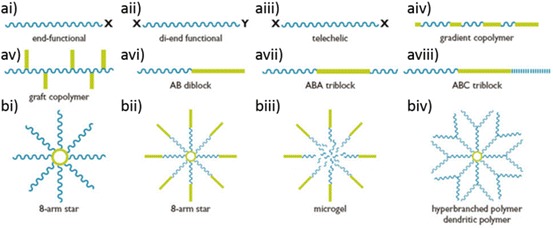
Possible polymer structures. a Linear polymers: (ai–iii) structures from a single monomer, with different end functionalities for modifications; (aiv–viii) multiple monomer derivations, either (aiv) gradient polymers, with two monomers interspersed in the polymer; (av) grafted co-polymer, a linear polymer with second monomer arms; (avi) AB di-block, two different monomer blocks; (avii) ABA tri-block, with one monomer block flanked by a different monomer block; or (aviii) three monomer blocks. b Star polymers: (bi) 8-armed star with homologous or heterologous (mikto) arms; (bii) 8-armed star with AB di-block arms; (biii) microgel centre of polymer is cross-linked (biv) dendrimer or hyperbranched polymer. Image courtesy of Dr. San H. Thang CSIRO
Only 7 of the 60 clinical trials using siRNAs have involved a polymeric carrier system. The first targeted delivery of siRNA was accomplished in humans in 2008 using a complex cyclodextrin-based polymer with transferrin for targeting and polyethylene glycol (PEG) for stabilisation. Known as CALAA-01 from Calando Pharmaceuticals, this targeted NP was administered intravenously for the treatment of solid tumours [29]. However, the phase I trial had to be terminated, as 21% of the patients discontinued the study due to adverse side effects. Mild, dose-dependent elevations in some serum cytokines were also measured; all elevations were transient in nature, peaking at 4–6 h post-infusion and returning to baseline by 24 h [21, 114].
Another phase I clinical trial was conducted for siG12D-Local Drug EluteR (LODER), a biodegradable polymeric implant surgically placed in the tumour and designed for slow release of an siRNA therapeutic. The trial was for treatment of locally advanced pancreatic cancer in combination with chemotherapy in order to target the mutated KRAS gene, which is the hallmark of pancreatic cancer [115, 116]. Patients in the study had median survival of 16 months, compared to a median survival of 10–13 months when treated with just chemotherapy. Successful completion of this trial has paved the way for a phase IIb trial scheduled to begin in February 2017 (ClinicalTrials.gov; NCT01676259).
Polymers such as poly(lactic acid) (PLA), poly(glycolic acid) (PGA) and polycaprolactone (PCL), and their co-polymers such as poly(lactide)-co-(glycolide) (PLGA), have been approved by the FDA. To date, however, no polymeric NP gene delivery system has been approved, given the unknown long-term toxicity and the low gene transfection efficiency of nanomaterials in vivo [117]. Only one antiviral polymeric carrier, a Dynamic PolyConjugate, has reached clinical trials [118]. Nevertheless, several in vitro and in vivo models have demonstrated inhibition in viral challenge models. Some of the most promising candidates are outlined in Table 4.
Table 4.
Clinical trials of polymer-based RNAi NPs
| Polymer-based NP | RNAi molecule | Target | Trial phase | Results | Company |
|---|---|---|---|---|---|
|
Cyclodextrin-based NP CALAA-01 [29] |
M2 subunit of ribonucleotide reductase (RRM2) siRNA | Solid tumours | Phase I | Terminated due to toxic side effects | Calando Pharmaceuticals |
|
Biodegradable polymer matrix implant Local Drug EluteR [116] |
Mutated KRAS gene siRNA | Pancreatic cancer | Phase I | Increased survival rates | Silenseed Ltd. |
|
Biodegradable polymer matrix implant Local Drug EluteR [116] |
Mutated KRAS gene siRNA | Pancreatic cancer | Phase II recruiting | N/A | Silenseed Ltd. |
|
Dynamic PolyConjugates ARC-520 [118] |
Anti-HBV siRNA | Hepatitis B virus | Phase I | Mostly well tolerated in healthy volunteers, a few side effects noted | Arrowhead Research |
|
Dynamic PolyConjugates ARC-520 [118] |
Anti-HBV siRNA | Hepatitis B virus | Phase I | ~50% decrease in viral load | Arrowhead Research |
|
Dynamic PolyConjugates ARC-520 |
Anti-HBV siRNA | Hepatitis B virus | Phase II | Terminated December 2016 | Arrowhead Research |
|
Dynamic PolyConjugates ARC-AAT |
Alpha-1 antitrypsin (AAT) siRNA | Alpha-1 antitrypsin deficiency (AATD) | Phase I | Withdrawn by company | Arrowhead Research |
|
Jet PEI (SNS01-T) |
Anti-eIF5A siRNA | Relapsed or refractory B cell malignancies | Phase I | Tolerated at 0.2 mg/kg | Sevion Therapeutics |
|
Jet PEI (SNS01-T) |
Anti-eIF5A siRNA | Relapsed or refractory B cell malignancies | Phase II | Withdrawn by company | Sevion Therapeutics |
Polymer-Based Antiviral siRNA Delivery
Hepatitis B
The most clinically advanced example of a polymeric delivery vehicle used to treat viral infection comes from Arrowhead Pharmaceuticals. Arrowhead have produced a series of Dynamic PolyConjugates (DPCs) [119] consisting of an amphipathic polymer shielded by PEG with a reversibly attached targeting ligand. One DPC (ARC-520) for combatting chronic HBV has reached phase II clinical trials. ARC-520 consists of two synthetic modified cholesterol-conjugated siRNAs (Fig. 14a) and is co-administered with a hepatocyte targeting peptide N-acetylglucosamine (NAG) that is conjugated to an amphipathic membrane active polymer, melittin-like peptide (NAG-MLP) (Fig. 14b). Both components are endocytosed by their respective receptors on hepatocytes (Fig. 14c). Once in the endosome the NAG component is released allowing MLP to interact with the endosomal membrane and facilitate siRNA release. At concentrations of 4.0 mg/kg administered to healthy volunteers by IV (or 3.0 mg/kg to patients with chronic HBV), ARC-520 appears to be well tolerated and also shows a statistically significant reduction in HBV antigen levels [118]. A November 2016 press release by Arrowhead Pharmaceuticals announced that trials for ARC-520 would be discontinued in favor of their new proprietary subcutaneous and extra-hepatic delivery system (DPCsq™). This was due to a decision by the regulatory committees to halt trials until primate deaths in non-clinical toxicology study were further investigated.
Fig. 14.
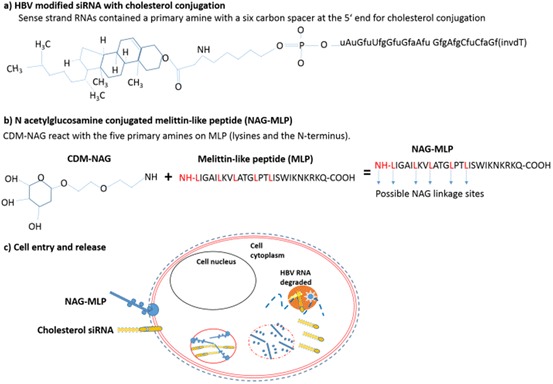
ARC520. a A co-administered modified cholesterol-conjugated siRNA and b NAG-MLP polymer. c Both components are endocytosed by the respective receptors. In the acidic endosome, NAG is released and MLP is able to associate and disrupt the endosome membrane. This allows siRNA to be released into the cytoplasm, where it can associate with RISC, leading to HBV RNA degradation
Herpes Simplex Virus
Herpes simplex virus (HSV) represents a significant problem for world health, as it can facilitate co-infections such as HIV, chlamydia and syphilis. Polymer NPs have been shown to be promising delivery agents for the in vivo inhibition of HSV. For example, Steinbach et al. developed PLGA-based polymer–siRNA NPs formed using a double-emulsion solvent evaporation technique (Fig. 15). In the first step, siRNAs targeting UL28 (a protein from HSV required for the cleavage and packaging of viral DNA) and nectin-1 (which acts as an HSV receptor) were bound to cationic polyamines (spermidine) to form polyplexes [59], before mixing with poly(lactic-co-glycolic acid) (PLGA). The initial emulsion was then added to 5% polyvinyl alcohol (PVA) and sonicated to create the second emulsion. Dynamic light scattering (DLS) and scanning electron microscopy (SEM) analysis revealed spherical particles approximately 190 nm in diameter. A dose response was observed in in vivo studies, whereby mice inoculated with a lethal dose of HSV-2 and topically treated with PLGA NPs showed increased survival from ~9 days (in untreated mice) to >28 days (in PLGA NP-treated mice). This study provided promising proof-of-concept validation for the development of topical treatment strategies with siRNA NP formulations.
Fig. 15.
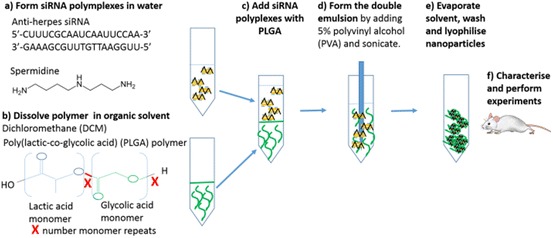
Genital herpes PLGA siRNA NPs formed by a double-emulsion solvent evaporation technique: a form amine/siRNA polyplexes; b add to PLGA; c subject to sonication to form emulsion; d evaporate and wash NPs; e characterise and perform biological assays
Human Immunodeficiency Virus
A number of research groups have also begun to investigate the application of polymeric nanocarriers as delivery vehicles for the treatment or prevention of human immunodeficiency virus (HIV). Whilst exploring novel application strategies for HIV therapeutics in the female genital tract, Gu and co-workers developed a biodegradable film loaded with siRNA-complexed NPs (Fig. 16) [120]. The NP complexes were formed similarly to the HSV PLGA particles using double-emulsion solvent evaporation. Initially, polyplexes of polyethylenimine (PEI) and anti-SNAP-23 siRNA (a SNARE protein required for HIV particle production) were formed (Fig. 16a). A protective layer of PLGA–polyethylene glycol (PEG) was added (Fig. 16b) and the surface of the NPs was further functionalised with anti-HLA-DR antibodies that target the membrane of HLA-DR+ immune cells in the vaginal mucosa (Fig. 16c). These were then embedded in the PVA, λ-carrageenan, glycerine and PEG film (Fig. 16d). Upon application to an in vitro mucosal co-culture model, the film degraded, freeing the NPs to cross the mucosal layer and epithelial cells. Delivery of the siRNA-complexed polymers to the HLA-DR+ cells was successful and demonstrated significant knockdown of SNAP-23 expression (Fig. 16e). Systems such as this offer promising platforms for administration of siRNA therapeutics to mucosa-lined organs such as the female genital tract.
Fig. 16.
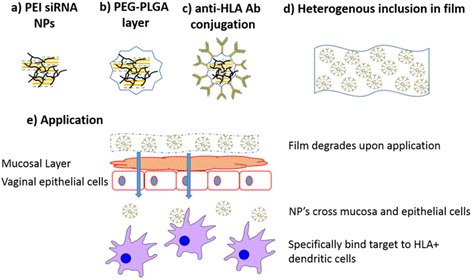
The design of the HIV novel biodegradable film loaded with siRNA-complexed NPs: a form PEI/siRNA polyplexes; b layer with PLGA; c conjugate with targeting antibodies; d include in film; e apply to vagina, as the film degrades, releasing NPs
In addition, Eszterhas et al. harnessed commercially available cationic polymeric transfection agents (INTERFERin®) to deliver siRNA to human endometrial and cervical tissue explants [121]. In ex vivo tissue samples, attenuated expression of the cell receptors CD4 and CCR5 was observed, alongside protection from an HIV challenge 48 h after NP incubation. In in vivo trials, the delivery of murine CD4 siRNA reduced CD4 transcripts over several days. This demonstrates that attenuation of host cell proteins—in this case receptors involved in viral entry—can also aid in attenuating viral infectivity.
Sulfated polysaccharides such as chitosan have shown antiviral properties against a range of viruses, including HIV, by blocking viral entry and virus–cell fusion [122]. Chitosan has also been used for the delivery of nucleic acids [123, 124]. Boyapalle et al. developed multifunctional chitosan–lipid nanocomplexes by combining the antiviral activity of chitosan with delivery of a plasmid expressing four anti-HIV siRNAs in a lipid NP [61]. When applied as a cream vaginally to rhesus monkeys on days −6, −3 and 0 before infection, a significant decrease in viral load was detected up to day 42 of infection. However, in the control monkeys, the viral load peaked and plateaued from day 34. This formulation effectively delivered the plasmids encoding siRNA to provide significant protection against HIV in both in vitro and in vivo models, without adverse effects.
Influenza
In 2004, Ge et al. reported the inhibition of influenza virus in a mouse model after intravenous delivery of PEI/siRNA polyplexes at a nitrogen: phosphate ratio of 5:1 [73]. Delivery of 120 µg of siRNA to mice 3 h prior to infection resulted in >1000-fold reduction in lung virus titres in some mice. Despite this encouraging result, a limited number of delivery applications using PEI have emerged, as it is known to be toxic and is not suitable for therapeutic delivery [125].
Work performed by Hinton et al. [71, 106] used cationic tri-block polymers based on 2-(dimethylamino)ethyl methacrylate (DMAEMA) and poly(ethylene glycol) (PEG) (synthesised via reversible addition-fragmentation chain transfer [RAFT] polymerisation), to deliver anti-influenza siRNA (siAnti-Flu) to inhibit virus replication in chicken embryos [71] (Fig. 17). Within 6 h, polymer had been clearly internalised by cells in the chorioallantoic membrane (CAM), the site of influenza replication. At higher magnification, it became evident that polymer was present in intracellular vesicles, whilst no toxicity was observed in the CAM. When the siAnti-Flu polymer complex was delivered, an average 2.5 and 1.5 log decrease in virus titre was observed compared to PBS- and control siRNA-treated embryos.
Fig. 17.
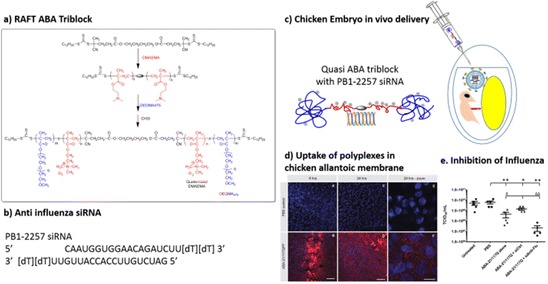
Influenza siRNA delivery via ABA tri-blocks using reversible addition fragmentation chain transfer (RAFT)-mediated free radical polymerisation. Adapted from [71]. a construction of ABA tri-block; b siAnti-Flu used; c schematic of delivery of polyplex and influenza to chicken allantoic fluid; d uptake of polyplex in the chorioallantoic membrane, the main site of influenza replication; e inhibition of influenza measured by tissue culture infective dose 50%
Of interest, the polymer used in this study was also shown to induce an interferon response in the CAM cells when injected without siRNA into the allantoic fluid of eggs. In comparison, a lower level of IFN response was observed with polymer carrying the siRNAs. This effect was hypothesised to be a result of the cells’ response to the charge of the polymer. Whilst this phenomenon is still under exploration, an induction of INF response in host cells based on polymer charge may be beneficially harnessed in the treatment of virus infection.
Safety and Immune System Activation and Potential Adjuvant Effects of Nanocarriers
The characteristics of siRNA-loaded cationic polymer NPs must be carefully tuned to allow binding, cell entry and endosome escape whilst minimising toxicity. Considerations of immune stimulation are also important. As demonstrated in the CALAA-01 phase I clinical trial [21, 114], high doses of polymer NP treatment may result in transient increases of serum cytokines, potentially inducing a T helper (Th)2-mediated immune response [114]. Previous work with gold NPs and other engineered NP systems (ENPS) have also shown that these particles can activate the NLRP3 (NOD-like receptor family pyrin domain-containing 3) inflammasome pathways, a danger-associated molecular pattern (DAMP) non-infectious immune response [126] (Fig. 18).
Fig. 18.
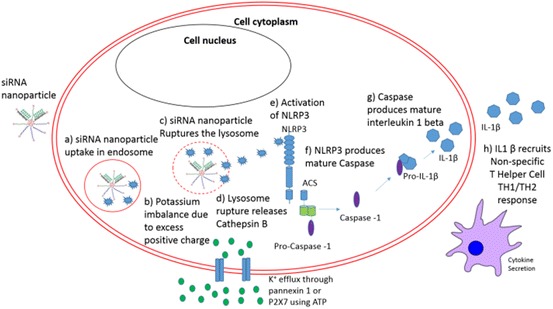
DAMP response. a Uptake of NPs by cell; inflammasome activation by endocytosed NPs can include; b uptake of highly charged particles, opens ion channels resulting K+ efflux to compensate for the charge imbalance activating cathepsin B [129]; c cationic NPs could induce a proton sponge effect which ruptures the lysosomal compartment; d Lysosome rupture releases lysosomal content, e.g. cathepsin B [130, 131]; e Cathepsin B activates NLRP3; f NLRP3 produces mature caspase-1; g caspase-1 converts pro-interleukins such as (IL)1β into mature IL1β; h active IL1β and other cytokines are secreted, which attracts and stimulates a T helper-2 (TH2) response [132]
Activation of NLRP3 stimulates a Th2 response, resulting in interferon (IFN) α and γ expression, and is hypothesised to be dependent on the physicochemical properties of nanomaterials including NP size, shape and coating. The T helper response is important in antiviral immunity, as it secretes IFNs and other cytokines to attract lymphocytes and direct the immune response. A T helper response is also important for B cell activation, for a high level of antibody production, and for inducing cellular memory conferring long-term protective immunity [127, 128]. Immune cells including macrophages and dendritic cells are largely responsible for this activation; however, other cells such as fibroblast and epithelial cells can also induce this response.
Interestingly, common adjuvants including alum or aluminium salts appear to work through the same NLRP3 inflammasome pathway following disruption of the lysosomes after endocytosis [128, 132, 133]. Other lipid-based adjuvants such as lipopolysaccharides can induce the NLRP3 inflammasome by activation of pathogen-associated molecular patterns, which recognises foreign antigens through Toll-like receptor 4 residing on the cell membrane. Therefore, polymers that are able to induce a DAMP response similar to an adjuvant whilst delivering antiviral siRNAs may be beneficial in acute antiviral treatment [134, 135]. This approach is currently being investigated for polymer–peptide-based vaccines and mRNA and pDNA vaccines against a range of viruses [136–143].
However, we must be mindful of these types of effects during the administration of long-term treatment strategies. Inducing a DAMP response could be problematic if frequently or extensively stimulated. High levels of antibodies produced against delivery vehicles could also lead to amplification of the immune response, which may lead to dangerous and off-target side effects. Over time, these responses may also contribute to reduced effectiveness of the therapeutic. For example, rapid clearance of liposomes from the bloodstream, known as the accelerated blood clearance phenomenon, has been reported following repeated treatments [144, 145]. Therefore, a fine balance must be maintained between factors such as dose, composition of the delivery agent and frequency of administration in order to achieve maximum effectiveness of the siRNA therapeutic.
In Vitro Characterisation of Physicochemical Properties of Polymer NPs
As mentioned above, the chemical and physical parameters of the polymer and the siRNA-bound NP complex can have a substantial influence on tissue targeting, cell uptake, toxicity and gene silencing ability of the therapeutic agent. Thus, it is essential to identify important correlations between physicochemical properties of the siRNA/polymer complex and the efficacy of viral inhibition in vitro and in vivo. Comprehensive chemical characterisation can usually be achieved using a combination of nuclear magnetic resonance spectroscopy and chromatographic techniques [146–148]. Tools for the accurate quantification of NP physical parameters (e.g. size, morphology, stiffness and surface charge) are less well defined, and the choice of available techniques can be confusing. This section will discuss the strengths and weaknesses of some of the complementary techniques available for quantification of physical parameters.
Measurement of Surface Potential
Zeta Potential
Accurate quantification of the charge density of polymer NPs after siRNA complexation is very important. If too high, the complexes may be cytotoxic [149, 150], and if too low, they may have insufficient positive charge to bind to the cell membrane, hindering endocytosis [151, 152]. Zeta potential (ZP) is commonly used to estimate the surface charge of NPs. Dispersions of charged particles in an electrolyte solution form an electrical double layer (EDL) of charged ions around the particle (Fig. 18). When placed in an electric field, the charged particles move towards an oppositely charged electrode (electrophoresis). Light scattering is typically used to determine the velocity of the particles undergoing electrophoresis, which in turn can be related to the electrical potential at a plane within the EDL (the ZP) [153].
Particle Sizing Techniques to Determine the Hydrodynamic Radius
NP size can affect the ability of the particle to undergo cell uptake [154, 155] and the pathway by which cell uptake occurs [156, 157]. The difference between the hydrodynamic radius and the NP core size can also provide insight into delivery efficiency. Below is a brief overview of some of the most common methods for determining the size of polymeric NPs.
Dynamic Light Scattering
Dynamic light scattering (DLS) is a technique whereby a laser is passed through a suspension of NPs and the intensity of scattered light is measured. Brownian motion of the particles causes fluctuations in the detected intensities that can be used to estimate hydrodynamic radii (R h) of particles via the Stokes–Einstein equation (Fig. 19a) [158].
Fig. 19.
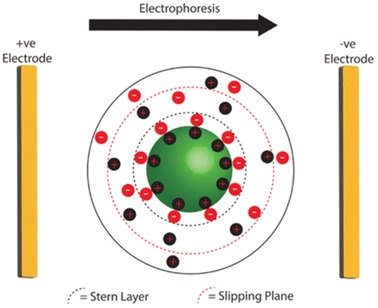
Schematic of the electrical double layer formed by a charged particle in an electrolyte solution. During electrophoresis, the charged particle will migrate towards an electrode, and a slipping plane between mobile and static ions (red dashed line) is established. The electrical potential at this plane is known as the zeta potential
The popularity of DLS as a particle sizing tool stems from the fact that quantitative results from large populations of NPs in solution can be obtained quickly and easily, and due to the short timescales required for data collection (1–10 min), time-resolved studies can be performed. Furthermore, combined DLS and ZP instruments allow a large proportion of physical NP characterisation to be performed on the same instrument.
DLS has a number of limitations that should be carefully considered when performing particle size measurements. The intensity of the scattered light (I) is sensitive to the presence of adsorbing groups, fluorophores and the radius (r) of particles in solution (I ∝ r 6). Thus coloured or fluorescent samples should be avoided, and the presence of large particles such as dust or other contaminants will cause R h to be overestimated. Samples must be very pure, homogenous and non-aggregated; thus time-consuming and potentially damaging preparation protocols (e.g. extensive sonication) are often required [153]. To prevent underestimation of particle size, it is important that each incident photon undergoes no more than one scattering event [159]. Thus, for polymer NPs targeting virus-challenged cells, maximum working concentrations for DLS are often lower than clinically relevant concentrations, and important inter-particle forces that may affect efficacy can be missed [160].
Nanoparticle Tracking Analysis (NTA)
Nanoparticle tracking analysis (NTA) combines laser scattering microscopy and NP tracking to determine their Rh (Fig. 19b). NTA does not rely on ensemble measurements or show increased sensitivity to larger particles, and is better suited for measuring polydisperse samples, can estimate particle concentration and has higher resolution than DLS [158]. This is particularly useful in the case of polymer NPs, which often show considerable variation in particle size due to molecular weight distribution.
Whilst NTA provides a more accurate measurement of R h compared to DLS, it also has its own limitations. The NTA instrument is more difficult to operate and has less automated parameter optimisation, making NTA measurements time-consuming and labor-intensive [158]. The concentration range of NTA, like that of DLS, is limited [158]. However, because the majority of polymeric siRNA complexes are polydisperse, containing multiple populations of NPs with similar Rh, NTA should provide greater confidence in particle size measurements.
Particle Morphology, True Particle Size and More by Electron Microscopy and Atomic Force Microscopy
Deviations from a spherical morphology seen in many polymer NPs has been shown to affect cell uptake [154, 161]. Microscopy techniques such as SEM, transmission electron microscopy and atomic force microscopy (AFM) offer sub-nanometer resolution that can distinguish the shape and morphology of individual polymer complexes and provide a true (non-hydrodynamic) particle size (Fig. 20a, b) [162].
Fig. 20.
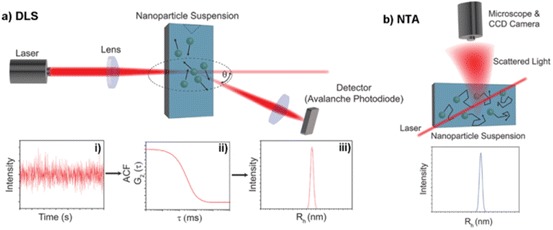
Dynamic light scattering (DLS) and nanoparticle tracking analysis (NTA) as tools for determining NP hydrodynamic radius (R h). a (i) DLS estimates R h through ensemble measurements of scattering intensity at angle θ over time; (ii) autocorrelation functions (ACF) are generated from which a diffusion constant (D) is estimated; (iii) R h is calculated from the estimated D via the Stokes–Einstein equation, and analysis of many ACF is combined to generate a histogram of R h vs. intensity. b NTA tracks the movement of individual NPs; R h is calculated for each particle, giving NTA greater resolution and no intensity bias to larger NPs
Electron Microscopy
Like all nanoscale analytical techniques, there are limitations, the most obvious being that for SEM, TEM and AFM, particles must be adsorbed to a solid substrate before imaging, which may affect the morphology. Additional coatings such as metallic thin films or heavy metal stains such as uranyl acetate are often required to enhance contrast in EM [163–165], which may alter sample morphology and introduce imaging artefacts. Furthermore, electron microscopes typically operate in a vacuum, requiring dehydration of the NPs before imaging. Dehydration can be avoided using cryoSEM or cryoTEM; however, these techniques are expensive and complex, requiring considerable user expertise [165]. Environmental scanning electron microscopy (ESEM) operates under a reduced vacuum, where water remains in a liquid phase, allowing the investigation of NPs in a more physiological environment, although the resolution of ESEM is typically lower than that of SEM [166].
Atomic Force Microscopy
Tapping mode AFM is well suited to polymer NP morphology characterisation (Fig. 20a, b), as it can operate with little sample preparation and in a fluid environment with relative ease. The scan speeds associated with typical AFM experiments (0.5–1 Hz) make this technique slow. However, the development of high-speed AFM units that operate at scan speeds of 30 Hz or higher may mean that the slow scan rates associated with AFM will soon be a thing of the past [167].
NP stiffness has been shown both theoretically [113] and experimentally [168, 169] to have a large influence on cell uptake and potentially endosome escape [111]. Techniques that monitor the force exerted on a statically oscillating AFM tip by a substrate have been used for many years to determine the stiffness of materials, but their lack of spatial resolution makes it difficult to quantify individual NPs. Recent developments including quantitative nanomechanical atomic force microscopy (QNM-AFM) have enabled the mapping of the mechanical properties of a surface with nanoscale resolution, allowing the stiffness of individual NPs to be mapped [111]. This has the potential to revolutionise the physical characterisation of polymer NPs. The mode of operation differs slightly among manufacturers, but advances in software and hardware generally allow the measurement of the force on the AFM tip to be recorded for every pixel as the tip is rastered across a substrate. Stiffness moduli can be extracted from force vs. distance curves using numerical algorithms (e.g. Sneddon or Derjaguin–Müller–Toporov [DMT] analysis) to provide nanoscale maps of stiffness (Fig. 20c–e) [170]. The ability to routinely measure the mechanical properties of individual NPs using QNM-AFM will enable a greater understanding of how stiffness (and other mechanical properties) affect cell uptake.
Ideally, analysis of the physicochemical properties of targeted nanocarriers should harness a combination of the above techniques. Correlation of physical parameters such as particle size, stiffness or surface charge with biological phenomena such as cell uptake or gene silencing can then be used as a basis for the rational design of future generations of polymeric delivery vehicles (Fig. 21).
Fig. 21.
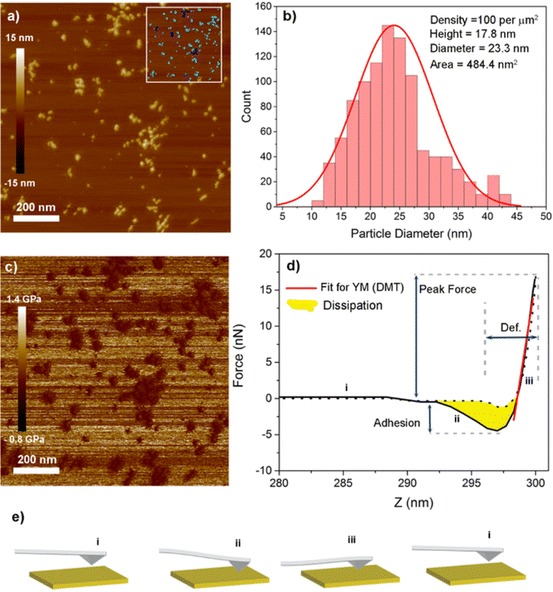
AFM as a tool for NP characterisation. a Topographical image of cationic star polymer siRNA complexes. Inset shows the results of a particle analysis tool (Bruker NanoScope Analysis software): individual particles (turquoise) contribute to statistics whilst aggregated particles are eliminated (dark blue). b A histogram showing distribution of particle diameters; text on the top right shows mean values of parameters calculated by the analysis tool. c QNM-AFM image recorded simultaneously using PeakForce Tapping mode (Bruker). d A representative force–distance curve generated for every pixel in the QNM-AFM image. The curve is fitted in the repulsive regime (iii) in order to calculate Young’s modulus (YM) (red line). e Graphical representation of the deflection of the AFM tip as it oscillated through one cycle
Future for Polymer/siRNA Antiviral Therapeutics
Despite the initial interest in siRNA as an antiviral therapeutic, pharmaceutical companies have become increasingly hesitant to invest in new research in this space, given the limited success demonstrated in clinical trials. Despite these limitations, however, research around new polymer delivery vehicles and devices is a highly competitive field, with continual improvement and innovation. Furthermore, reports from recent clinical trials have suggested that siRNA therapies may also be effective when delivered as a prophylactic rather than in a post-exposure therapeutic regimen [102]. Along with the adjuvant-like effect of some NP carriers, new avenues of application are emerging for siRNA therapeutics. Whilst current research around siRNA delivery is directed towards cancer treatment regimes, it is highly likely that a successful clinical trial will rekindle interest in siRNA as an antiviral therapeutic. In such a dynamic and fast-paced global economy, siRNA delivery strategies still hold promise for combatting many of the world’s persistent and emerging viral diseases.
Abbreviations
- AFM
Atomic force microscopy
- AIDS
Acquired immune deficiency syndrome
- ALN-RSV01
Alnylam anti-RSV therapeutic
- apoB
Apolipoprotein B
- ASO
Antisense oligonucleotide
- CAM
Chorioallantoic membrane
- DAMP
Danger-associated molecular pattern non-infectious immune response
- dsRNA
Double-stranded RNA
- DLS
Dynamic light scattering
- DMAEMA
2-(Dimethylamino)ethyl methacrylate
- EPR
Enhanced permeability retention effect
- FDA
US Food and Drug Administration
- HBV
Hepatitis B virus
- HCV
Hepatitis C virus
- HIV
Human immunodeficiency virus
- HSV
Herpes simplex virus
- ICAM-1
Intercellular adhesion molecule 1
- IBD
Inflammatory bowel disease
- IFN
Interferon
- LODER
Local Drug EluteR
- mRNA
Messenger RNA
- miRNA
MicroRNA
- NAG-MLP
N-Acetylglucosamine-melittin-like peptide
- NLRP3
NOD-like receptor family pyrin domain-containing protein complex
- NP
Nanoparticle
- NPs
Nanoparticles
- NTA
Nanoparticle tracking analysis
- PEG
Polyethylene glycol
- PEI
Polyethylenimine
- PLGA
Poly(lactic-co-glycolic acid)
- PVA
Polyvinyl alcohol
- QN-AFM
Quantitative nanomechanical atomic force microscopy
- RISC
RNA induced silencing complex
- RAFT
Reversible addition-fragmentation chain transfer
- RNA
Ribonucleic acid
- RNAi
RNA interference
- RSV
Respiratory syncytial virus
- SEM
Scanning electron microscopy
- shRNA
Short hairpin RNA
- shRNAmir
Short hairpin RNA designed based on native microRNA
- siAnti-Flu
anti-influenza siRNA
- siRNA
Small interfering RNA
- Th
T helper
- T-Vec
Talimogene laherparepvec
- UC
Ulcerative colitis
Footnotes
This article is part of the Topical Collection “Polymeric Gene Delivery Systems”: edited by Yiyun Cheng.
References
- 1.Marston HD, Folkers GK, Morens DM, Fauci AS. Emerging viral diseases: confronting threats with new technologies. Sci Transl Med. 2014 doi: 10.1126/scitranslmed.3009872. [DOI] [PubMed] [Google Scholar]
- 2.The Centers for Disease Control and Prevention (2016) 2014-2016 Ebola Outbreak in West Africa. Available at: https://www.cdc.gov/vhf/ebola/outbreaks/2014-west-africa/. Accessed 15 Oct 2016
- 3.Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng P-Y, Bandaranayake D, Breiman RF, Brooks WA, Buchy P, Feikin DR, Fowler KB, Gordon A, Hien NT, Horby P, Huang QS, Katz MA, Krishnan A, Lal R, Montgomery JM, Mølbak K, Pebody R, Presanis AM, Razuri H, Steens A, Tinoco YO, Wallinga J, Yu H, Vong S, Bresee J, Widdowson M-A. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis. 2009;12(9):687–695. doi: 10.1016/S1473-3099(12)70121-4. [DOI] [PubMed] [Google Scholar]
- 4.The World Health Organisation (2016) Emergencies Zika virus and complications. Available at: http://www.who.int/emergencies/zika-virus/en/. Accessed 15 Oct 2016
- 5.The World Health Organisation (2016) Cumulative number of confirmed human cases of avian influenza A(H5N1) reported to WHO. Available at: http://www.who.int/influenza/human_animal_interface/H5N1_cumulative_table_archives/en/. Accessed 15 Oct 2016
- 6.Plowright RK, Eby P, Hudson PJ, Smith IL, Westcott D, Bryden WL, Middleton D, Reid PA, McFarlane RA, Martin G, Tabor GM, Skerratt LF, Anderson DL, Crameri G, Quammen D, Jordan D, Freeman P, Wang L-F, Epstein JH, Marsh GA, Kung NY, McCallum H. Ecological dynamics of emerging bat virus spillover. Proc R Soc B Biol Sci. 2015;282(1798):20142124. doi: 10.1098/rspb.2014.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arruda PHE, Stevenson GW, Killian ML, Burrough ER, Gauger PC, Harmon KM, Magstadt DR, Yoon K-J, Zhang J, Madson DM, Piñeyro P, Derscheid RJ, Schwartz KJ, Cooper VL, Halbur PG, Main RG, Sato Y, Arruda BL. Outbreak of H5N2 highly pathogenic avian Influenza A virus infection in two commercial layer facilities: lesions and viral antigen distribution. J Vet Diagn Invest. 2016 doi: 10.1177/1040638716658929. [DOI] [PubMed] [Google Scholar]
- 8.Group WB (2014) The economic impact of the 2014 Ebola epidemic: short and medium term estimates for West Africa. World Bank Group
- 9.De Clercq E, Li G. Approved antiviral drugs over the Past 50 Years. Clin Microbiol Rev. 2016;29(3):695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang D, Gao G. State-of-the-art human gene therapy: part II. Gene therapy strategies and applications. Discov Med. 2014;18(98):151–161. [PMC free article] [PubMed] [Google Scholar]
- 11.Geary RS, Henry SP, Grillone LR. Fomivirsen. Clin Pharmacokinet. 2002;41(4):255–260. doi: 10.2165/00003088-200241040-00002. [DOI] [PubMed] [Google Scholar]
- 12.Fire A, Xu S, Montgomery M, Kostas S, Driver S, Mello C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 13.G-x Chen, Zhang S, X-h He, S-y Liu, Ma C, Zou X-P. Clinical utility of recombinant adenoviral human p53 gene therapy: current perspectives. OncoTargets Ther. 2014;7:1901–1909. doi: 10.2147/OTT.S50483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yla-Herttuala S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European Union. Mol Ther. 2012;20(10):1831–1832. doi: 10.1038/mt.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaudet D, de Wal J, Tremblay K, Déry S, van Deventer S, Freidig A, Brisson D, Méthot J. Review of the clinical development of alipogene tiparvovec gene therapy for lipoprotein lipase deficiency. Atheroscler Suppl. 2010;11(1):55–60. doi: 10.1016/j.atherosclerosissup.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Fong Y. Oncolytic treatment for cancer recommended for approval. Mol Ther. 2015;23(7):1131. doi: 10.1038/mt.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andtbacka RHI, Ross M, Puzanov I, Milhem M, Collichio F, Delman KA, Amatruda T, Zager JS, Cranmer L, Hsueh E, Chen L, Shilkrut M, Kaufman HL. Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM Phase III Clinical Trial. Ann Surg Oncol. 2016 doi: 10.1245/s10434-016-5286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yla-Herttuala S. ADA-SCID gene therapy endorsed by european medicines agency for marketing authorization. Mol Ther. 2016;24(6):1013–1014. doi: 10.1038/mt.2016.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rader DJ, Kastelein JJP (2014) Lomitapide and mipomersen. Two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial. Hypercholesterolemia 129(9):1022–1032. doi:10.1161/circulationaha.113.001292 [DOI] [PubMed]
- 20.Marafini I, Di Fusco D, Calabrese E, Sedda S, Pallone F, Monteleone G. Antisense approach to inflammatory bowel disease: prospects and challenges. Drugs. 2015;75(7):723–730. doi: 10.1007/s40265-015-0391-0. [DOI] [PubMed] [Google Scholar]
- 21.Zuckerman JE, Davis ME. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat Rev Drug Discov. 2015;14(12):843–856. doi: 10.1038/nrd4685. [DOI] [PubMed] [Google Scholar]
- 22.Obbard DJ, Gordon KHJ, Buck AH, Jiggins FM. The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc B Biol Sci. 2009;364(1513):99–115. doi: 10.1098/rstb.2008.0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerutti H, Casas-Mollano JA. On the origin and functions of RNA-mediated silencing: from protists to man. Curr Genet. 2006;50(2):81–99. doi: 10.1007/s00294-006-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waterhouse PM, Graham MW, Wang M-B. Virus resistance and gene silencing in plants can be induced by simultaneous expression of sense and antisense RNA. Proc Natl Acad Sci. 1998;95(23):13959–13964. doi: 10.1073/pnas.95.23.13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fire A, Albertson D, Harrison SW, Moerman DG. Production of antisense RNA leads to effective and specific inhibition of gene expression in C. elegans muscle. Development. 1991;113(2):503–514. doi: 10.1242/dev.113.2.503. [DOI] [PubMed] [Google Scholar]
- 26.Guo S, Kemphues KJ. Par-1, a gene required for establishing polarity in C. elegans embryos, encodes a putative Ser/Thr kinase that is asymmetrically distributed. Cell. 1995;81(4):611–620. doi: 10.1016/0092-8674(95)90082-9. [DOI] [PubMed] [Google Scholar]
- 27.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 28.Song E, Lee S-K, Wang J, Ince N, Ouyang N, Min J, Chen J, Shankar P, Lieberman J. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003;9(3):347–351. doi: 10.1038/nm828. [DOI] [PubMed] [Google Scholar]
- 29.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6(3):659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 30.Bender E (2014) The second coming of RNAi. The scientist, vol. 28. LabX Media Group
- 31.Matranga C, Zamore PD. Small silencing RNAs. Curr Biol. 2007;17(18):R789–R793. doi: 10.1016/j.cub.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 32.H-o Iwakawa, Tomari Y. The functions of MicroRNAs: mRNA decay and translational repression. Trends Cell Biol. 2015;25(11):651–665. doi: 10.1016/j.tcb.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14(10a):1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425(6956):415–419. doi:http://www.nature.com/nature/journal/v425/n6956/suppinfo/nature01957_S1.html [DOI] [PubMed]
- 35.Landthaler M, Yalcin A, Tuschl T. The human digeorge syndrome critical region gene 8 and its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol. 2004;14(23):2162–2167. doi: 10.1016/j.cub.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17(24):3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson RC, Doudna JA. Molecular mechanisms of RNA interference. Annu Rev Biophys. 2013;42(1):217–239. doi: 10.1146/annurev-biophys-083012-130404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296(5567):550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 39.Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ, Siolas D, Hu G, Paddison PJ, Schlabach MR, Sheth N, Bradshaw J, Burchard J, Kulkarni A, Cavet G, Sachidanandam R, McCombie WR, Cleary MA, Elledge SJ, Hannon GJ (2005) Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet 37 (11):1281–1288. doi:http://www.nature.com/ng/journal/v37/n11/suppinfo/ng1650_S1.html [DOI] [PubMed]
- 40.Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai Dan Y, Barbosa Inês AM, Kwon Jung S, Guan Y, Sinha N, Zuber J. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5(6):1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 41.ter Brake O, Konstantinova P, Ceylan M, Berkhout B (2006) Silencing of HIV-1 with RNA interference: a multiple shRNA approach. Mol Ther 14(6):883–892. doi:http://www.nature.com/mt/journal/v14/n6/suppinfo/mt20061372s1.html [DOI] [PubMed]
- 42.Liu YP, Haasnoot J, ter Brake O, Berkhout B, Konstantinova P. Inhibition of HIV-1 by multiple siRNAs expressed from a single microRNA polycistron. Nucleic Acids Res. 2008;36(9):2811–2824. doi: 10.1093/nar/gkn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammond SM. RNAi, microRNAs, and human disease. Cancer Chemother Pharmacol. 2006;58(1):63–68. doi: 10.1007/s00280-006-0318-2. [DOI] [PubMed] [Google Scholar]
- 44.Dickey LL, Hanley TM, Huffaker TB, Ramstead AG, O’Connell RM (2017) Lane TE MicroRNA 155 and viral-induced neuroinflammation. J Neuroimmunol. doi: 10.1016/j.jneuroim.2017.01.016 [DOI] [PMC free article] [PubMed]
- 45.Trobaugh DW, Klimstra WB. MicroRNA regulation of RNA virus replication and pathogenesis. Trends Mol Med. 2017;23(1):80–93. doi: 10.1016/j.molmed.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA (2006) Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441(7092):537–541. doi:http://www.nature.com/nature/journal/v441/n7092/suppinfo/nature04791_S1.html [DOI] [PubMed]
- 47.Hinton TM, Doran TJ. Inhibition of chicken anaemia virus replication using multiple short-hairpin RNAs. Antiviral Res. 2008;80(2):143–149. doi: 10.1016/j.antiviral.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 48.Alvarez R, Elbashir S, Borland T, Toudjarska I, Hadwiger P, John M, Roehl I, Morskaya SS, Martinello R, Kahn J, Van Ranst M, Tripp RA, DeVincenzo JP, Pandey R, Maier M, Nechev L, Manoharan M, Kotelianski V, Meyers R. RNA interference-mediated silencing of the respiratory syncytial virus nucleocapsid defines a potent antiviral strategy. Antimicrob Agents Chemother. 2009;53(9):3952–3962. doi: 10.1128/AAC.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barik S (2009) Treating respiratory viral diseases with chemically modified, second generation intranasal siRNAs. In: Sioud M (ed) siRNA and miRNA gene silencing: from bench to bedside. Humana Press, Totowa, NJ, pp 1–11. doi:10.1007/978-1-60327-547-7_16 [DOI] [PMC free article] [PubMed]
- 50.Hoenen T, Groseth A, Rosenke K, Fischer RJ, Hoenen A, Judson SD, Martellaro C, Falzarano D, Marzi A, Squires RB, Wollenberg KR, de Wit E, Prescott J, Safronetz D, van Doremalen N, Bushmaker T, Feldmann F, McNally K, Bolay FK, Fields B, Sealy T, Rayfield M, Nichol ST, Zoon KC, Massaquoi M, Munster VJ, Feldmann H. Nanopore sequencing as a rapidly deployable ebola outbreak tool. Emerg Infect Dis. 2016;22(2):331–334. doi: 10.3201/eid2202.151796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jeffrey RK, Michael RW, Suzanne M, Jason TL, Brett B, Lawrence F, Fahn T, Karla P, Joseph WD, Timothy M, Randal JS, Kurt ES, James P, Stacey B, Joseph F, Jens HK, Lisa H, Daniel JP, Pardis CS, Mariano S-L, Fatorma KB, Gustavo P. Monitoring of Ebola virus makona evolution through establishment of advanced genomic capability in liberia. Emerg Infect Dis J. 2015;21(7):1135. doi: 10.3201/eid2107.150522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greninger AL, Naccache SN, Federman S, Yu G, Mbala P, Bres V, Stryke D, Bouquet J, Somasekar S, Linnen JM, Dodd R, Mulembakani P, Schneider BS, Muyembe-Tamfum J-J, Stramer SL, Chiu CY. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015;7:99. doi: 10.1186/s13073-015-0220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. siRNA selection server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 2004;32(Web Server issue):W-130–W-134. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu ZJ, Mathews DH. OligoWalk: an online siRNA design tool utilizing hybridization thermodynamics. Nucleic Acids Research. 2008;36(Web Server issue):W-104–W-108. doi: 10.1093/nar/gkn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tedebark U, Scozzari A, Werbitzky O, Capaldi D, Holmberg L (2011) Industrial-scale manufacturing of a possible oligonucleotide cargo CPP-based drug. In: Langel Ü (ed) Cell-penetrating peptides: methods and protocols. Humana Press, Totowa, pp 505–524. doi:10.1007/978-1-60761-919-2_36 [DOI] [PubMed]
- 56.Aalto AP, Sarin LP, van Dijk AA, Saarma M, Poranen MM, Arumäe U, Bamford DH. Large-scale production of dsRNA and siRNA pools for RNA interference utilizing bacteriophage ϕ6 RNA-dependent RNA polymerase. RNA. 2007;13(3):422–429. doi: 10.1261/rna.348307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thi EP, Mire CE, Lee ACH, Geisbert JB, Zhou JZ, Agans KN, Snead NM, Deer DJ, Barnard TR, Fenton KA, MacLachlan I, Geisbert TW. Lipid nanoparticle siRNA treatment of Ebola-virus-Makona-infected nonhuman primates. Nature. 2015;521(7552):362–365. doi: 10.1038/nature14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paavilainen H, Lehtinen J, Romanovskaya A, Nygårdas M, Bamford DH, Poranen MM, Hukkanen V. Inhibition of clinical pathogenic herpes simplex virus 1 strains with enzymatically created siRNA pools. J Med Virol. 2016 doi: 10.1002/jmv.24578. [DOI] [PubMed] [Google Scholar]
- 59.Steinbach JM, Weller CE, Booth CJ, Saltzman WM. Polymer nanoparticles encapsulating siRNA for treatment of HSV-2 genital infection. J Controlled Release. 2012;162(1):102–110. doi: 10.1016/j.jconrel.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Capodici J, Karikó K, Weissman D. Inhibition of HIV-1 infection by small interfering RNA-mediated RNA interference. J Immunol. 2002;169(9):5196–5201. doi: 10.4049/jimmunol.169.9.5196. [DOI] [PubMed] [Google Scholar]
- 61.Boyapalle S, Xu W, Raulji P, Mohapatra S, Mohapatra SS. A multiple siRNA-based anti-HIV/SHIV microbicide shows protection in both In Vitro and In Vivo models. PLoS One. 2015;10(9):135288. doi: 10.1371/journal.pone.0135288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spanevello F, Calistri A, Del Vecchio C, Mantelli B, Frasson C, Basso G, Palù G, Cavazzana M, Parolin C. Development of lentiviral vectors simultaneously expressing multiple siRNAs against CCR5, vif and tat/rev genes for an HIV-1 gene therapy approach. Mol Ther Nucleic Acids. 2016;5(4):e312. doi: 10.1038/mtna.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moon J-S, Lee S-H, Kim E-J, Cho H, Lee W, Kim G-W, Park H-J, Cho S-W, Lee C, Oh J-W. Inhibition of hepatitis C virus in mice by a small interfering RNA targeting a highly conserved sequence in viral IRES Pseudoknot. PLoS One. 2016;11(1):e0146710. doi: 10.1371/journal.pone.0146710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barik S. Control of nonsegmented negative-strand RNA virus replication by siRNA. Virus Res. 2004;102(1):27–35. doi: 10.1016/j.virusres.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 65.Hamasaki K, Nakao K, Matsumoto K, Ichikawa T, Ishikawa H, Eguchi K. Short interfering RNA-directed inhibition of hepatitis B virus replication. FEBS Lett. 2003;543(1–3):51–54. doi: 10.1016/S0014-5793(03)00400-9. [DOI] [PubMed] [Google Scholar]
- 66.Wang J, Feng S-S, Wang S, Z-y Chen. Evaluation of cationic nanoparticles of biodegradable copolymers as siRNA delivery system for hepatitis B treatment. Int J Pharm. 2010;400(1–2):194–200. doi: 10.1016/j.ijpharm.2010.08.026. [DOI] [PubMed] [Google Scholar]
- 67.Cho JS, Lee S-W, Kim Y-M, Kim D, Kim D-Y, Kim Y-T. Long (27-nucleotides) small inhibitory RNAs targeting E6 protein eradicate effectively the cervical cancer cells harboring human papilloma virus. Obstet Gynecol Sci. 2015;58(3):210–216. doi: 10.5468/ogs.2015.58.3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reuter T, Weissbrich B, Schneider-Schaulies S, Schneider-Schaulies J. RNA interference with measles virus N, P, and L mRNAs efficiently prevents and with matrix protein mRNA enhances viral transcription. J Virol. 2006;80(12):5951–5957. doi: 10.1128/JVI.02453-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Déctor MA, Romero P, López S, Arias CF. Rotavirus gene silencing by small interfering RNAs. EMBO Rep. 2002;3(12):1175. doi: 10.1093/embo-reports/kvf234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McCaskill JL, Marsh GA, Monaghan P, Wang L-F, Doran T, McMillan NAJ. Potent inhibition of Hendra virus infection via RNA Interference and poly I: C immune activation. PLoS One. 2013;8(5):e64360. doi: 10.1371/journal.pone.0064360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hinton TM, Challagulla A, Stewart CR, Guerrero-Sanchez C, Grusche FA, Shi SN, Bean AG, Monaghan P, Gunatillake PA, Thang SH, Tizard ML. Inhibition of influenza virus in vivo by siRNA delivered using ABA triblock copolymer synthesized by reversible addition-fragmentation chain-transfer polymerization. Nanomedicine. 2014;9(8):1141–1154. doi: 10.2217/nnm.13.119. [DOI] [PubMed] [Google Scholar]
- 72.Tompkins SM, Lo C-Y, Tumpey TM, Epstein SL. Protection against lethal influenza virus challenge by RNA interference in vivo. Proc Natl Acad Sci USA. 2004;101(23):8682–8686. doi: 10.1073/pnas.0402630101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ge Q, Filip L, Bai A, Nguyen T, Eisen HN, Chen J. Inhibition of influenza virus production in virus-infected mice by RNA interference. Proc Natl Acad Sci USA. 2004;101(23):8676–8681. doi: 10.1073/pnas.0402486101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li B-j, Tang Q, Cheng D, Qin C, Xie FY, Wei Q, Xu J, Liu Y, Zheng B-j, Woodle MC, Zhong N, Lu PY (2005) Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat Med 11(9):944–951. doi:http://www.nature.com/nm/journal/v11/n9/suppinfo/nm1280_S1.html [DOI] [PMC free article] [PubMed]
- 75.Gismondi MI, Ortiz XP, Currá AP, Asurmendi S, Taboga O. Artificial microRNAs as antiviral strategy to FMDV: structural implications of target selection. J Virol Methods. 2014;199:1–10. doi: 10.1016/j.jviromet.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 76.Lambeth LS, Moore RJ, Muralitharan MS, Doran TJ. Suppression of bovine viral diarrhea virus replication by small interfering RNA and short hairpin RNA-mediated RNA interference. Vet Microbiol. 2007;119(2–4):132–143. doi: 10.1016/j.vetmic.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 77.Hinton TM, Doran TJ. Inhibition of chicken anaemia virus replication using multiple short-hairpin RNAs. Antiviral Res. 2008;80(2):143–149. doi: 10.1016/j.antiviral.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 78.Chen M, Payne WS, Dunn JR, Chang S, Zhang HM, Hunt HD, Dodgson JB. Retroviral delivery of RNA interference against Marek’s disease virus in vivo. Poult Sci. 2009;88(7):1373–1380. doi: 10.3382/ps.2009-00070. [DOI] [PubMed] [Google Scholar]
- 79.Braasch DA, Jensen S, Liu Y, Kaur K, Arar K, White MA, Corey DR. RNA interference in mammalian cells by chemically-modified RNA. Biochemistry. 2003;42(26):7967–7975. doi: 10.1021/bi0343774. [DOI] [PubMed] [Google Scholar]
- 80.van de Water FM, Boerman OC, Wouterse AC, Peters JGP, Russel FGM, Masereeuw R Intravenously administered short interfering RNA accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab Dispos. 2006;34(8):1393–1397. doi: 10.1124/dmd.106.009555. [DOI] [PubMed] [Google Scholar]
- 81.Kang L, Wang RF, Yan P, Liu M, Zhang CL, Yu MM, Cui YG, Xu XJ. Noninvasive visualization of RNA delivery with 99mTc-radiolabeled small-interference RNA in tumor xenografts. J Nucl Med. 2010;51(6):978–986. doi: 10.2967/jnumed.109.069906. [DOI] [PubMed] [Google Scholar]
- 82.Shorter SA, Gollings AS, Gorringe-Pattrick MAM, Coakley JE, Dyer PDR, Richardson SCW. The potential of toxin-based drug delivery systems for enhanced nucleic acid therapeutic delivery. Expert Opin Drug Deliv. 2016 doi: 10.1080/17425247.2016.1227781. [DOI] [PubMed] [Google Scholar]
- 83.Mishra DK, Balekar N, Mishra PK. Nanoengineered strategies for siRNA delivery: from target assessment to cancer therapeutic efficacy. Drug Deliv Transl Res. 2017 doi: 10.1007/s13346-016-0352-5. [DOI] [PubMed] [Google Scholar]
- 84.Mokhtarzadeh A, Alibakhshi A, Hashemi M, Hejazi M, Hosseini V, de la Guardia M, Ramezani M. Biodegradable nano-polymers as delivery vehicles for therapeutic small non-coding ribonucleic acids. J Controll Release. 2017;245:116–126. doi: 10.1016/j.jconrel.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 85.Lakkireddy HR, Bazile D. Building the design, translation and development principles of polymeric nanomedicines using the case of clinically advanced poly(lactide(glycolide))-poly(ethylene glycol) nanotechnology as a model: an industrial viewpoint. Adv Drug Deliv Rev. 2016 doi: 10.1016/j.addr.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 86.Chen C, Posocco P, Liu X, Cheng Q, Laurini E, Zhou J, Liu C, Wang Y, Tang J, Col VD, Yu T, Giorgio S, Fermeglia M, Qu F, Liang Z, Rossi JJ, Liu M, Rocchi P, Pricl S, Peng L. Mastering dendrimer self-assembly for efficient siRNA delivery: from conceptual design to In vivo efficient gene silencing. Small. 2016;12(27):3667–3676. doi: 10.1002/smll.201503866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leung AKK, Tam YYC, Cullis PR (2014) Chapter four—lipid nanoparticles for short interfering RNA delivery. In: Leaf Huang DL, Ernst W (eds) Advances in genetics, vol. 88, Academic Press, pp 71–110. doi: 10.1016/B978-0-12-800148-6.00004-3 [DOI] [PMC free article] [PubMed]
- 88.Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Controlled Release. 2006;114(1):100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 89.Zatsepin TS, Kotelevtsev YV, Koteliansky V. Lipid nanoparticles for targeted siRNA delivery—going from bench to bedside. Int J Nanomed. 2016;11:3077–3086. doi: 10.2147/IJN.S106625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peng X-H, Qian X, Mao H, Wang AY, Chen Z, Nie S, Shin DM. Targeted magnetic iron oxide nanoparticles for tumor imaging and therapy. Int J Nanomed. 2008;3(3):311–321. doi: 10.2147/ijn.s2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Derfus AM, Chen AA, Min D-H, Ruoslahti E, Bhatia SN. Targeted quantum dot conjugates for siRNA delivery. Bioconj Chem. 2007;18(5):1391–1396. doi: 10.1021/bc060367e. [DOI] [PubMed] [Google Scholar]
- 92.DiGiusto DL, Stan R, Krishnan A, Li H, Rossi JJ, Zaia JA. Development of hematopoietic stem cell based gene therapy for HIV-1 infection: considerations for proof of concept studies and translation to standard medical practice. Viruses. 2013;5(11):2898–2919. doi: 10.3390/v5112898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Allerson CR, Sioufi N, Jarres R, Prakash TP, Naik N, Berdeja A, Wanders L, Griffey RH, Swayze EE, Bhat B. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J Med Chem. 2005;48(4):901–904. doi: 10.1021/jm049167j. [DOI] [PubMed] [Google Scholar]
- 94.Chiu Y-L, Rana TM. siRNA function in RNAi: a chemical modification analysis. RNA-a Publ RNA Soc. 2003;9(9):1034–1048. doi: 10.1261/rna.5103703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Amarzguioui M, Holen T, Babaie E, Prydz H. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003;31(2):589–595. doi: 10.1093/nar/gkg147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Egli M, Minasov G, Tereshko V, Pallan PS, Teplova M, Inamati GB, Lesnik EA, Owens SR, Ross BS, Prakash TP, Manoharan M. Probing the influence of stereoelectronic effects on the biophysical properties of oligonucleotides: comprehensive analysis of the RNA affinity, nuclease resistance, and crystal structure of ten 2′-O-ribonucleic acid modifications. Biochemistry. 2005;44(25):9045–9057. doi: 10.1021/bi050574m. [DOI] [PubMed] [Google Scholar]
- 97.DeVincenzo J, Cehelsky JE, Alvarez R, Elbashir S, Harborth J, Toudjarska I, Nechev L, Murugaiah V, Vliet AV, Vaishnaw AK, Meyers R. Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV) Antiviral Res. 2008;77(3):225–231. doi: 10.1016/j.antiviral.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 98.Gottlieb J, Zamora MR, Hodges T, Musk AW, Sommerwerk U, Dilling D, Arcasoy S, DeVincenzo J, Karsten V, Shah S, Bettencourt BR, Cehelsky J, Nochur S, Gollob J, Vaishnaw A, Simon AR, Glanville AR. ALN-RSV01 for prevention of bronchiolitis obliterans syndrome after respiratory syncytial virus infection in lung transplant recipients. J Heart Lung Transpl. 2016;35(2):213–221. doi: 10.1016/j.healun.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 99.DeVincenzo J, Lambkin-Williams R, Wilkinson T, Cehelsky J, Nochur S, Walsh E, Meyers R, Gollob J, Vaishnaw A. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc Natl Acad Sci USA. 2010;107(19):8800–8805. doi: 10.1073/pnas.0912186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zamora MR, Budev M, Rolfe M, Gottlieb J, Humar A, DeVincenzo J, Vaishnaw A, Cehelsky J, Albert G, Nochur S, Gollob JA, Glanville AR. RNA interference therapy in lung transplant patients infected with respiratory syncytial virus. Am J Respir Crit Care Med. 2011;183(4):531–538. doi: 10.1164/rccm.201003-0422OC. [DOI] [PubMed] [Google Scholar]
- 101.Lin Q, Chen J, Zhang Z, Zheng G. Lipid-based nanoparticles in the systemic delivery of siRNA. Nanomedicine. 2013;9(1):105–120. doi: 10.2217/nnm.13.192. [DOI] [PubMed] [Google Scholar]
- 102.Dunning J, Sahr F, Rojek A, Gannon F, Carson G, Idriss B, Massaquoi T, Gandi R, Joseph S, Osman HK, Brooks TJG, Simpson AJH, Goodfellow I, Thorne L, Arias A, Merson L, Castle L, Howell-Jones R, Pardinaz-Solis R, Hope-Gill B, Ferri M, Grove J, Kowalski M, Stepniewska K, Lang T, Whitehead J, Olliaro P, Samai M, Horby PW, for the R-TKMtt Experimental treatment of ebola virus disease with TKM-130803: a single-arm phase 2 clinical trial. PLoS Med. 2016;13(4):e1001997. doi: 10.1371/journal.pmed.1001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mitsuyasu RT, Zack JA, Macpherson JL, Symonds GP. Phase I/II clinical trials using gene-modified adult hematopoietic stem cells for HIV: lessons learnt. Stem Cells Int. 2011 doi: 10.4061/2011/393698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Smith D, Holley AC, McCormick CL. RAFT-synthesized copolymers and conjugates designed for therapeutic delivery of iRNA. Polym Chem. 2011;2(7):1428–1441. doi: 10.1039/c1py00038a. [DOI] [Google Scholar]
- 105.Scales CW, Huang FQ, Li N, Vasilieva YA, Ray J, Convertine AJ, McCormick CL. Corona-stabilized interpolyelectrolyte complexes of SiRNA with nonimmunogenic, hydrophilic/cationic block copolymers prepared by aqueous RAFT polymerization. Macromolecules. 2006;39(20):6871–6881. doi: 10.1021/ma061453c. [DOI] [Google Scholar]
- 106.Hinton TM, Guerrero-Sanchez C, Graham J, Le TPT, Muir BW, Shi S, Tizard ML, Gunatillake P, McLean K, Thang SH. The effect of RAFT-derived cationic block copolymer structure on gene silencing efficiency. Biomaterials. 2012;33:7631. doi: 10.1016/j.biomaterials.2012.06.090. [DOI] [PubMed] [Google Scholar]
- 107.Flores JD, Xu XW, Treat NJ, McCormick CL. Reversible “self-locked” micelles from a zwitterion-containing triblock copolymer. Macromolecules. 2009;42(14):4941–4945. doi: 10.1021/ma900517w. [DOI] [Google Scholar]
- 108.Cloninger MJ. Biological applications of dendrimers. Curr Opin Chem Biol. 2002;6(6):742–748. doi: 10.1016/S1367-5931(02)00400-3. [DOI] [PubMed] [Google Scholar]
- 109.Rosselgong J, Williams EGL, Le TP, Grusche F, Hinton TM, Tizard M, Gunatillake P, Thang SH. Core degradable star RAFT polymers: synthesis, polymerization, and degradation studies. Macromolecules. 2013;46(23):9181–9188. doi: 10.1021/ma402122z. [DOI] [Google Scholar]
- 110.Teo J, McCarroll JA, Boyer C, Youkhana J, Sagnella SM, Duong HTT, Liu J, Sharbeen G, Goldstein D, Davis TP, Kavallaris M, Phillips PA. A rationally optimized nanoparticle system for the delivery of RNA interference therapeutics into pancreatic tumors in vivo. Biomacromolecules. 2016;17(7):2337–2351. doi: 10.1021/acs.biomac.6b00185. [DOI] [PubMed] [Google Scholar]
- 111.Dearnley M, Reynolds NP, Cass P, Wei X, Shi S, Mohammed AA, Le TP, Gunatillake P, Tizard M, Thang SH, Hinton TM. Comparing gene silencing and physiochemical properties in siRNA bound cationic star-polymer complexes. Biomacromolecules. 2016;17(11):3532–3546. doi: 10.1021/acs.biomac.6b01029. [DOI] [PubMed] [Google Scholar]
- 112.Scomparin A, Polyak D, Krivitsky A, Satchi-Fainaro R. Achieving successful delivery of oligonucleotides—from physico-chemical characterization to in vivo evaluation. Biotechnol Adv. 2015;33(6):1294–1309. doi: 10.1016/j.biotechadv.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 113.Yi X, Shi X, Gao H. Cellular uptake of elastic nanoparticles. Phys Rev Lett. 2011;107(9):098101. doi: 10.1103/PhysRevLett.107.098101. [DOI] [PubMed] [Google Scholar]
- 114.Zuckerman JE, Gritli I, Tolcher A, Heidel JD, Lim D, Morgan R, Chmielowski B, Ribas A, Davis ME, Yen Y. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc Natl Acad Sci. 2014;111(31):11449–11454. doi: 10.1073/pnas.1411393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ramot Y, Rotkopf S, Gabai RM, Zorde Khvalevsky E, Muravnik S, Marzoli GA, Domb AJ, Shemi A, Nyska A. Preclinical safety evaluation in rats of a polymeric matrix containing an siRNA drug used as a local and prolonged delivery system for pancreatic cancer therapy. Toxicol Pathol. 2016;44(6):856–865. doi: 10.1177/0192623316645860. [DOI] [PubMed] [Google Scholar]
- 116.Golan T, Khvalevsky EZ, Hubert A, Gabai RM, Hen N, Segal A, Domb A, Harari G, David EB, Raskin S, Goldes Y, Goldin E, Eliakim R, Lahav M, Kopleman Y, Dancour A, Shemi A, Galun E. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget. 2015;6(27):24560–24570. doi: 10.18632/oncotarget.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Keles E, Song Y, Du D, Dong W-J, Lin Y. Recent progress in nanomaterials for gene delivery applications. Biomater Sci. 2016;4(9):1291–1309. doi: 10.1039/C6BM00441E. [DOI] [PubMed] [Google Scholar]
- 118.Gish RG, Yuen M-F, Chan HLY, Given BD, Lai C-L, Locarnini SA, Lau JYN, Wooddell CI, Schluep T, Lewis DL. Synthetic RNAi triggers and their use in chronic hepatitis B therapies with curative intent. Antiviral Res. 2015;121:97–108. doi: 10.1016/j.antiviral.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 119.Rozema DB, Lewis DL, Wakefield DH, Wong SC, Klein JJ, Roesch PL, Bertin SL, Reppen TW, Chu Q, Blokhin AV, Hagstrom JE, Wolff JA. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci USA. 2007;104(32):12982–12987. doi: 10.1073/pnas.0703778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gu J, Yang S, Ho EA. Biodegradable film for the targeted delivery of siRNA-loaded nanoparticles to vaginal immune cells. Mol Pharm. 2015;12(8):2889–2903. doi: 10.1021/acs.molpharmaceut.5b00073. [DOI] [PubMed] [Google Scholar]
- 121.Eszterhas SK, Ilonzo NO, Crozier JE, Celaj S, Howell AL. Nanoparticles containing siRNA to silence CD4 and CCR5 reduce expression of these receptors and inhibit HIV-1 infection in human female reproductive tract tissue explants. Infect Dis Rep. 2011;3:11. doi: 10.4081/idr.2011.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Artan M, Karadeniz F, Karagozlu MZ, Kim M-M, Kim S-K. Anti-HIV-1 activity of low molecular weight sulfated chitooligosaccharides. Carbohydr Res. 2010;345(5):656–662. doi: 10.1016/j.carres.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 123.Park S, Jeong EJ, Lee J, Rhim T, Lee SK, Lee KY. Preparation and characterization of nonaarginine-modified chitosan nanoparticles for siRNA delivery. Carbohydr Polym. 2013;92(1):57–62. doi: 10.1016/j.carbpol.2012.08.116. [DOI] [PubMed] [Google Scholar]
- 124.Nielsen EJB, Nielsen JM, Becker D, Karlas A, Prakash H, Glud SZ, Merrison J, Besenbacher F, Meyer TF, Kjems J, Howard KA. Pulmonary gene silencing in transgenic EGFP mice using aerosolised chitosan/siRNA nanoparticles. Pharm Res. 2010;27(12):2520–2527. doi: 10.1007/s11095-010-0255-y. [DOI] [PubMed] [Google Scholar]
- 125.Xue HY, Liu S, Wong HL. Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomedicine (London, England) 2014;9(2):295–312. doi: 10.2217/nnm.13.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sun B, Wang X, Ji Z, Li R, Xia T. NLRP3 inflammasome activation induced by engineered nanomaterials. Small (Weinheim an der Bergstrasse, Germany) 2013;9:1595–1607. doi: 10.1002/smll.201201962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gregory AE, Williamson D, Titball R. Vaccine delivery using nanoparticles. Front Cell Infect Microbiol. 2013 doi: 10.3389/fcimb.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Marrack P, McKee AS, Munks MW. Towards an understanding of the adjuvant action of aluminium. Nat Rev Immunol. 2009;9(4):287–293. doi: 10.1038/nri2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14(9):1583–1589. doi:http://www.nature.com/cdd/journal/v14/n9/suppinfo/4402195s1.html [DOI] [PubMed]
- 130.Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol. 2012;9:20. doi: 10.1186/1743-8977-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Thomas TP, Majoros I, Kotlyar A, Mullen D, Banaszak Holl MM, Baker JR. Cationic Poly(amidoamine) dendrimer induces lysosomal apoptotic pathway at therapeutically relevant concentrations. Biomacromolecules. 2009;10(12):3207–3214. doi: 10.1021/bm900683r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Eisenbarth SC, Colegio OR, O/’Connor W, Sutterwala FS, Flavell RA (2008) Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453(7198):1122–1126. doi:http://www.nature.com/nature/journal/v453/n7198/suppinfo/nature06939_S1.html [DOI] [PMC free article] [PubMed]
- 133.Sokolovska A, Hem SL, HogenEsch H. Activation of dendritic cells and induction of CD4+ T cell differentiation by aluminum-containing adjuvants. Vaccine. 2007;25(23):4575–4585. doi: 10.1016/j.vaccine.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 134.Akagi T, Baba M, Akashi M. Development of vaccine adjuvants using polymeric nanoparticles and their potential applications for anti-HIV vaccine. Yakugaku Zasshi. 2007;127(2):307–317. doi: 10.1248/yakushi.127.307. [DOI] [PubMed] [Google Scholar]
- 135.Whitehead KA, Dahlman JE, Langer RS, Anderson DG. Silencing or stimulation? siRNA delivery and the immune system. Annu Rev Chem Biomol Eng. 2011;2(1):77–96. doi: 10.1146/annurev-chembioeng-061010-114133. [DOI] [PubMed] [Google Scholar]
- 136.Chahal JS, Khan OF, Cooper CL, McPartlan JS, Tsosie JK, Tilley LD, Sidik SM, Lourido S, Langer R, Bavari S, Ploegh HL, Anderson DG. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc Natl Acad Sci. 2016;113(29):E4133–E4142. doi: 10.1073/pnas.1600299113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Francica JR, Lynn GM, Laga R, Joyce MG, Ruckwardt TJ, Morabito KM, Chen M, Chaudhuri R, Zhang B, Sastry M, Druz A, Ko K, Choe M, Pechar M, Georgiev IS, Kueltzo LA, Seymour LW, Mascola JR, Kwong PD, Graham BS, Seder RA. Thermoresponsive polymer nanoparticles co-deliver RSV F trimers with a TLR-7/8 adjuvant. Bioconj Chem. 2016;27(10):2372–2385. doi: 10.1021/acs.bioconjchem.6b00370. [DOI] [PubMed] [Google Scholar]
- 138.Miyata K, Mohri K, Egawa T, Endo R, Morimoto N, Ochiai K, K-i Hiwatari, Tsubaki K, Tobita E, Uto T, Baba M, Sakuma S. Demonstration of d-octaarginine-linked polymers as promising adjuvants for mucosal vaccination through influenza virus challenge. Bioconj Chem. 2016;27(8):1865–1871. doi: 10.1021/acs.bioconjchem.6b00283. [DOI] [PubMed] [Google Scholar]
- 139.Hiremath J, K-i Kang, Xia M, Elaish M, Binjawadagi B, Ouyang K, Dhakal S, Arcos J, Torrelles JB, Jiang X, Lee CW, Renukaradhya GJ. Entrapment of H1N1 influenza virus derived conserved peptides in PLGA nanoparticles enhances T cell response and vaccine efficacy in pigs. PLoS One. 2016;11(4):e0151922. doi: 10.1371/journal.pone.0151922. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 140.Sarkar I, Garg R, van Drunen Littel-van den Hurk S. Formulation of the respiratory syncytial virus fusion protein with a polymer-based combination adjuvant promotes transient and local innate immune responses and leads to improved adaptive immunity. Vaccine. 2016;34(42):5114–5124. doi: 10.1016/j.vaccine.2016.08.053. [DOI] [PubMed] [Google Scholar]
- 141.Bivas-Benita M, Bar L, Gillard GO, Kaufman DR, Simmons NL, Hovav A-H, Letvin NL. Efficient generation of mucosal and systemic antigen-specific CD8(+) T-cell responses following pulmonary DNA immunization. J Virol. 2010;84(11):5764–5774. doi: 10.1128/JVI.02202-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bivas-Benita M, Gillard GO, Bar L, White KA, Webby RJ, Hovav AH, Letvin NL. Airway CD8(+) T cells induced by pulmonary DNA immunization mediate protective anti-viral immunity. Mucosal Immunol. 2013;6(1):156–166. doi: 10.1038/mi.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bolhassani A, Javanzad S, Saleh T, Hashemi M, Aghasadeghi MR, Sadat SM. Polymeric nanoparticles: potent vectors for vaccine delivery targeting cancer and infectious diseases. Human Vaccin Immunother. 2014;10(2):321–332. doi: 10.4161/hv.26796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shiraishi K, Kawano K, Maitani Y, Aoshi T, Ishii KJ, Sanada Y, Mochizuki S, Sakurai K, Yokoyama M. Exploring the relationship between anti-PEG IgM behaviors and PEGylated nanoparticles and its significance for accelerated blood clearance. J Controlled Release. 2016;234:59–67. doi: 10.1016/j.jconrel.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 145.Ishihara T, Takeda M, Sakamoto H, Kimoto A, Kobayashi C, Takasaki N, Yuki K, K-i Tanaka, Takenaga M, Igarashi R, Maeda T, Yamakawa N, Okamoto Y, Otsuka M, Ishida T, Kiwada H, Mizushima Y, Mizushima T. Accelerated blood clearance phenomenon upon repeated injection of PEG-modified PLA-nanoparticles. Pharm Res. 2009;26(10):2270–2279. doi: 10.1007/s11095-009-9943-x. [DOI] [PubMed] [Google Scholar]
- 146.Jacob S, Kennedy JP. Synthesis, characterization and properties of octa-arm polyisobutylene-based star polymers. Polym Synth Polym Polym Compl. 1999;146:1–38. doi: 10.1007/3-540-49424-3_1. [DOI] [Google Scholar]
- 147.Charleux B, Faust R. Synthesis of branched polymers by cationic polymerization. Branched Polym I. 1999;142:1–69. doi: 10.1007/3-540-68310-0_1. [DOI] [Google Scholar]
- 148.Pridgen EM, Alexis F, Langer RS, Farohkzad OC (2009) Biodegradable, targeted nanoparticle drug delivery formulation for cancer therapy. Methods in bioengineering: nanoscale bioengineering and nanomedicine, pp 197–222
- 149.Naha PC, Davoren M, Lyng FM, Byrne HJ. Reactive oxygen species (ROS) induced cytokine production and cytotoxicity of PAMAM dendrimers in J774A.1 cells. Toxicol Appl Pharmacol. 2010;246(1–2):91–99. doi: 10.1016/j.taap.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 150.McNerny DQ, Leroueil PR, Baker JR. Understanding specific and nonspecific toxicities: a requirement for the development of dendrimer-based pharmaceuticals. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2(3):249–259. doi: 10.1002/wnan.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chung TH, Wu SH, Yao M, Lu CW, Lin YS, Hung Y, Mou CY, Chen YC, Huang DM. The effect of surface charge on the uptake and biological function of mesoporous silica nanoparticles 3T3-L1 cells and human mesenchymal stem cells. Biomaterials. 2007;28(19):2959–2966. doi: 10.1016/j.biomaterials.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 152.Cho EC, Xie JW, Wurm PA, Xia YN. Understanding the role of surface charges in cellular adsorption versus internalization by selectively removing gold nanoparticles on the cell surface with a I-2/KI etchant. Nano Lett. 2009;9(3):1080–1084. doi: 10.1021/nl803487r. [DOI] [PubMed] [Google Scholar]
- 153.Bhattacharjee S. DLS and zeta potential—what they are and what they are not? J Controlled Release. 2016;235:337–351. doi: 10.1016/j.jconrel.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 154.Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci USA. 2008;105(33):11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ferrari R, Lupi M, Colombo C, Morbidelli M, D’Incalci M, Moscatelli D. Investigation of size, surface charge, PEGylation degree and concentration on the cellular uptake of polymer nanoparticles. Colloid Surf B-Biointerfaces. 2014;123:639–647. doi: 10.1016/j.colsurfb.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 156.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin-and caveolae-mediated endocytosis. Biochem J. 2004;377:159–169. doi: 10.1042/bj20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lai SK, Hida K, Man ST, Chen C, Machamer C, Schroer TA, Hanes J. Privileged delivery of polymer nanoparticles to the perinuclear region of live cells via a non-clathrin, non-degradative pathway. Biomaterials. 2007;28(18):2876–2884. doi: 10.1016/j.biomaterials.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 158.Filipe V, Hawe A, Jiskoot W. Critical evaluation of nanoparticle tracking analysis (NTA) by nanosight for the measurement of nanoparticles and protein aggregates. Pharm Res. 2010;27(5):796–810. doi: 10.1007/s11095-010-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Panchal J, Kotarek J, Marszal E, Topp EM. Analyzing subvisible particles in protein drug products: a comparison of dynamic light scattering (DLS) and resonant mass measurement (RMM) Aaps J. 2014;16(3):440–451. doi: 10.1208/s12248-014-9579-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhou C, Qi W, Lewis EN, Carpenter JF. Concomitant Raman spectroscopy and dynamic light scattering for characterization of therapeutic proteins at high concentrations. Anal Biochem. 2015;472:7–20. doi: 10.1016/j.ab.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 161.Florez L, Herrmann C, Cramer JM, Hauser CP, Koynov K, Landfester K, Crespy D, Mailander V. How shape influences uptake: interactions of anisotropic polymer nanoparticles and human mesenchymal stem cells. Small. 2012;8(14):2222–2230. doi: 10.1002/smll.201102002. [DOI] [PubMed] [Google Scholar]
- 162.Troiber C, Kasper JC, Milani S, Scheible M, Martin I, Schaubhut F, Küchler S, Rädler J, Simmel FC, Friess W, Wagner E. Comparison of four different particle sizing methods for siRNA polyplex characterization. Eur J Pharm Biopharm. 2013;84(2):255–264. doi: 10.1016/j.ejpb.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 163.Winey M, Meehl JB, O’Toole ET, Giddings TH. Conventional transmission electron microscopy. Mol Biol Cell. 2014;25(3):319–323. doi: 10.1091/mbc.E12-12-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hacker C, Asadi J, Pliotas C, Ferguson S, Sherry L, Marius P, Tello J, Jackson D, Naismith J, Lucocq JM. Nanoparticle suspensions enclosed in methylcellulose: a new approach for quantifying nanoparticles in transmission electron microscopy. Sci Rep. 2016 doi: 10.1038/srep25275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kratosova G, Dedkova K, Vavra I, Ciampor F (2014) Intracellular delivery II: fundamentals and applications. In: Prokop A, Iwasaki Y, Harada A (eds) Intracellular Delivery Ii: Fundamentals and applications, vol. 7. Fundamental Biomedical Technologies. Springer, Dordrecht, pp 1–479. doi:10.1007/978-94-017-8896-0
- 166.Donald AM. The use of environmental scanning electron microscopy for imaging wet and insulating materials. Nat Mater. 2003;2(8):511–516. doi: 10.1038/nmat898. [DOI] [PubMed] [Google Scholar]
- 167.Ando T. High-speed atomic force microscopy coming of age. Nanotechnology. 2012;23(6):27. doi: 10.1088/0957-4484/23/6/062001. [DOI] [PubMed] [Google Scholar]
- 168.Sun J, Zhang L, Wang J, Feng Q, Liu D, Yin Q, Xu D, Wei Y, Ding B, Shi X, Jiang X. Tunable Rigidity of (Polymeric Core)-(Lipid Shell) Nanoparticles for Regulated Cellular Uptake. Adv Mater. 2015;27(8):1402. doi: 10.1002/adma.201404788. [DOI] [PubMed] [Google Scholar]
- 169.Anselmo AC, Zhang M, Kumar S, Vogus DR, Menegatti S, Helgeson ME, Mitragotri S. Elasticity of nanopartides influences their blood circulation, phagocytosis, endocytosis, and targeting. ACS Nano. 2015;9(3):3169–3177. doi: 10.1021/acsnano.5b00147. [DOI] [PubMed] [Google Scholar]
- 170.Dokukin ME, Sokolov I. Quantitative mapping of the elastic modulus of soft materials with harmonix and peakforce QNM AFM modes. Langmuir. 2012;28(46):16060–16071. doi: 10.1021/la302706b. [DOI] [PubMed] [Google Scholar]