

Abstract
Introduction
Since acute respiratory tract infections inflict a high burden of disease in children worldwide, a multiplex reverse transcription polymerase chain reaction combined with a microwell hybridization assay (m-RT-PCR–ELISA) to detect 19 different respiratory pathogens was developed and validated.
Methods
A total of 430 respiratory specimens were retrospectively tested in parallel by both the advanced 19-valent m-RT-PCR–ELISA as well as by culture or individual RT-PCR assays used in clinical routine.
Results
The mean (median) sensitivity of the m-RT-PCR–ELISA in the retrospective test was 93.3% (95.1%; range 83.3–100 %), and the mean (median) specificity was 99.8 and 100 % (range 98.6–100 %), respectively. The mean positive predictive value was 99.3 % (range 93.4–100 %) and the mean negative predictive value was 95.3 % (range 98.4–100 %). Feasibility and clinical value of the 19-valent method was prospectively shown on 16,231 incoming clinical specimens from patients between 0 and 16 years of age with acute respiratory tract infections admitted to pediatric hospitals or private practices from October 2003 to June 2010 in three regions in Germany (Kiel, Mainz, Freiburg; Freiburg to June 2007 only). At least one microorganism was detected in 10,765 of 16,231 (66.3 %) clinical specimens: 5,044 RV, 1,999 RSV, 1,286 AV, 944 EV, 737 seasonal IVA, 173 pandemic IVA H1N1-2009, 899 MPV, 518 CV, 383 PIV3, 268 PIV1, 259 Mpn, 205 IVB, 164 PIV2, 144 PIV4, 103 Bp, 29 Cpn and 29 Bpp, while reovirus and Lpn were not present in these specimens from a pediatric population. More than one organism could be detected in 13.4 % of the specimens.
Conclusions
The m-RT-PCR–ELISA evaluated here improves the spectrum for diagnosing respiratory infections and is a feasible instrument for individual diagnostic and epidemiological studies.
Keywords: m-RT-PCR-ELISA, Respiratory tract infections, Validation, Sensitivity, Diagnosis, Enterovirus (EV), Influenza virus type A (IVA), Influenza virus type B (IVB), Respiratory syncytial virus (RSV), Parainfluenza virus type 1 (PIV1), Parainfluenza virus type 2 (PIV2), Parainfluenza virus type 3 (PIV3), Parainfluenza virus type 4 (PIV4), Adenovirus (AV), Rhinovirus (RV), Human metapneumovirus (MPV), Coronavirus (CV), Reovirus (ReoV), Mycoplasma pneumoniae (Mpn), Chlamydophila pneumoniae (Cpn), Bordetella pertussis (Bp), Bordetella parapertussis (Bpp), Legionella pneumophila (Lpn)
Introduction
Acute respiratory tract infections inflict a high burden of disease in children worldwide [1]. While some important causes like Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis are commensals of the upper respiratory tract, other viral and bacterial agents are considered to be regular pathogens and their detection from a clinical specimen is considered as proof of infection. These include enterovirus (EV), influenza virus type A (IVA) and type B (IVB), respiratory syncytial virus (RSV), parainfluenza virus type 1 (PIV1), type 2 (PIV2), type 3 (PIV3), and type 4 (PIV4), adenovirus (AV), rhinovirus (RV), human metapneumovirus (MPV), coronavirus (CV), reovirus (RV), Mycoplasma pneumoniae (Mpn), Chlamydophila pneumoniae (Cpn), Bordetella pertussis (Bp), Bordetella parapertussis (Bpp), and Legionella pneumophila (Lpn). For these, molecular amplification techniques are advantageous as compared to culture techniques, because the results are available earlier, lower cost, and higher sensitivity. Moreover, the diagnostic laboratory handling of viable pathogens is not required.
Our group has developed and used multiplex polymerase chain reaction (PCR) testing for acute respiratory infection (ARI) pathogens since 1996 [2, 3] to allow for a comprehensive laboratory-based surveillance. In the meantime, additional methods have been described [4–10].
We previously reported on a multiplex reverse transcription polymerase chain reaction combined with a microwell hybridization assay (m-RT-PCR-ELISA) [2] that allows the simultaneous detection of nine important respiratory pathogens (RNA viruses EV, IVA, IVB, PIV1, PIV3, RSV, and a DNA virus AV, as well as the bacteria Cpn and Mpn). The feasibility of the m-RT-PCR-ELISA on clinical specimens was tested [2] and it was validated for the diagnosis of respiratory tract infections on clinical specimens and on culture supernatants in comparison to the referring gold standard [3], and descriptive epidemiological studies based on the results obtained with this method have been reported [11–26].
The present study describes an advanced 19-valent m-RT-PCR-ELISA to detect RNA viruses EV, RV, IVA, IVB, PIV1, PIV2, PIV3, PIV4, RSV, MPV, CVOC43, CV229E, ReoV, and a DNA virus (AV), as well as the bacteria Cpn, Mpn, Bp, Bpp, and Lpn.
Methods
Specimens
For validation of the m-RT-PCR, four different approaches were used:
-
(i)
Supernatants from cell culture and negative controls (all blinded) as used for the validation of the previously described 9-valent m-RT-PCR were received from different laboratories [3].
-
(ii)
Analytical sensitivity of the multiplex approach in comparison to the corresponding single primer testing was determined on culture supernatants of the organisms [3].
-
(iii)
Clinical specimens from frozen stocks (−80 °C) which had been collected between 1995 and 2004 at the Institute of Virology, Rotterdam, the Netherlands, were tested by m-RT-PCR techniques versus culture for sensitivity and specificity (here called the “retrospective test”).
-
(iv)
The feasibility and clinical usefulness of the method was evaluated in the pediatric University Hospital of Kiel between October 2003 and June 2010 and from 2007 additionally in the laboratory in Mainz on incoming specimens (n = 16,316), predominantly nasopharyngeal aspirates in NaCl (0.9 %) from hospitalized children in Kiel, Mainz, and Freiburg (specimens from Freiburg until June 2007 only).
Clinical specimens from Kiel were not stored in virus medium, and, thus, their use in virus culture was known to result in a reduced sensitivity [3]. To avoid this bias, in the retrospective testing, only clinical specimens from the Department of Virology, Rotterdam, stored in virus medium (calf serum) at −80 °C were used. The specimens used for this part of the evaluation (n = 430) were nasopharyngeal aspirates, swabs, or bronchoalveolar lavage (BAL). The gold standard was defined as the result of culture, a secondary RT-PCR in an external laboratory, or from sequence information derived on organisms detected with one of the methods within a specimen, especially for differentiation within the picornavirus group (EV and RV).
A total of 18 specimens (17 sputum and one BAL) from 15 patients historically characterized as Legionella pneumophila cases and three negative controls (all sputum) were obtained from the Dr. Ijzerman (Haarlem, the Netherlands). Legionella cases were defined if one of the diagnostic tests (urinary antigen, culture, or PCR) historically performed in Haarlem was positive. The specimens were retested in Kiel by the 19-valent m-RT-PCR-ELISA. Since the retesting of specimens by culture or individual PCR in Haarlem may become “negative” due to the long storage of specimens, the historically determined “positive cases” were used as the standard for sensitivity calculations.
A total of 112 clinical specimens (92 Amies Swab Transport System, 20 nasopharyngeal aspirates) were tested by the culture method on Bordetella-selective media (Oxoid, Wesel, Germany) and differentiated by the agglutination test using sera for B. pertussis and B. parapertussis (Genzyme Virotech GmbH, Rüsselsheim, Germany) and by an individual Bordetella PCR for the presence of B. pertussis and B. parapertussis [27]. In parallel, m-RT-PCR-ELISA was performed. Furthermore, 177 specimens from patients with typical clinical symptoms were tested by an individual Bordetella PCR protocol [27] and in comparison by the m-RT-PCR-ELISA.
For the retrospective study (iii), clinical specimens were thawed and divided. One aliquot was sent on dry ice to the laboratory in Kiel. After arrival, the specimens were immediately stored at −80 °C without thawing until they were used for retesting by cell culture (Rotterdam) and m-RT-PCR-ELISA (Kiel). All samples were blinded, and the real contents of the samples were not disclosed until the results of testing were revealed to the cooperating laboratory.
For Mpn and Cpn, no clinical specimens were available for evaluation from external laboratories; therefore, 60 Mpn and seven Cpn blinded culture supernatants were tested [3].
Furthermore, culture supernatants of adenovirus type 1 to type 40 and type 42 to type 51 were tested with the multiplex approach to test the sensitivity/specificity of the method for this pathogen.
No comparison tests could be done on reovirus, because no clinical or blinded culture supernatants were available. Only culture supernatants of the reovirus types 1, 2, and 3 could be tested as positive controls. For influenza A (2009) H1N1, a plasmid kindly provided by Dr. Schweiger, RKI Berlin, Germany, was used as a positive control, as previously described [28].
Nucleic acid extraction
After the isolation of total nucleic acids, the purified nucleic acids were eluted in a total of 50 µl elution buffer, and 4.5 µl of this was used as a template for the RT-PCR-ELISA procedures, as previously described [3].
For the determination of the analytical sensitivity, the culture supernatant was diluted by tenfold logarithmic steps in NaCl (0.9 %) up to a dilution factor of 10−10. The nucleic acids were prepared from this dilution series as described above and were tested according to the protocol of the m-RT-PCR-ELISA and by a single primer RT-PCR-ELISA (see “Individual RT-PCR procedures”) with a primer pair specific for one organism and classified as described below (see “Analysis of PCR products and classification of results”).
Multiplex RT-PCR
Target sequences or regions for amplification were predominantly selected from the literature, and, if necessary, adapted for the multiplex PCR protocol (Tables 1, 2, and 3). The procedure of m-RT-PCR was as follows: 4.5 µl of the nucleic acid preparations from specimens were included in the reverse transcription (RT) reaction in a final volume of 15 µl (10.5 µl RT buffer + 4.5 µl template). The setup of buffer for 15 reactions (nine specimens plus controls) was as follows: 51 µl first strand buffer [5× conc.] (Invitrogen), 77 µl dNTP [10 mM] (Amersham), 25.5 µl DTT [0.1 M] (Invitrogen), 12.8 µl hexanucleotide mixture [10× conc.] (Roche Diagnostika), 6.4 µl RNAsin [40 U/µl] (Promega), and 6.4 µl SuperScript II [200 U/µl] (Invitrogen). The RT was performed using Superscript II reverse transcriptase from Invitrogen for 10 min at 25 °C, followed by 50 min at 42 °C and 5 min at 90 °C, and then finally chilled to 4 °C. The RT and PCR were performed on a BioRad iCycler (BioRad Laboratories GmbH, Munich, Germany) or a Perkin-Elmer GeneAmp PCR System 9600 Thermocycler (Perkin-Elmer, Branchburg, NJ, USA). For the PCR, the AccuPrime™ Taq DNA Polymerase System (Invitrogen GmbH, Karlsruhe, Germany) was used. The PCR was performed in parallel in two tubes containing Primer Mix A and Mix B, respectively. Primers and concentrations used in the mix are given in Table 1 and the sequences are shown in Table 2. The setup of PCR mixtures for 15 reactions was performed on ice as follows. Mix A: 394 µl nuclease-free water (Promega GmbH, Mannheim, Germany), 68 µl PCR buffer I (AccuPrime System, Invitrogen), 27 µl Primer Mix A, 13.6 µl AccuPrime Taq DNA Polymerase (Invitrogen), and 6.8 µl of digoxigenin-11-dUTP (Roche Diagnostics, Germany), and for Mix B 2/3 of the volumes: 263 µl nuclease-free water, 45 µl PCR buffer I, 18 µl Primer Mix B, 9.1 µl AccuPrime Taq DNA Polymerase (Invitrogen), and 4.5 µl of digoxigenin-11-dUTP. 20 µl of this Mix B were transferred to 14 new PCR tubes each and 6 µl of the c-DNA from the 15-µl RT reaction were added as the PCR template. 30 µl of Mix A were added to the remaining (9 µl) c-DNA from each RT tube. The protocol for the PCR was: 2 min activation at 94 °C and, afterwards, 35 cycles at 94 °C for 30 s, 55 °C for 20 s, 72 °C for 40 s, followed by one final extension for 10 min at 72 °C and a cooling step to 4 °C. The ramping times were adjusted as follows: 36 s for heating from 55 to 94 °C, 25 s for cooling from 94 to 55 °C, and 31 s for heating from 55 to 72 °C. The performance of this m-RT-PCR is optimal using the AccuPrime System (Invitrogen) and other enzymes or a different thermocycler may need adaptation. Especially, the ramping times of thermocyclers may be different and should be controlled.
Table 1.
Primers used in primer Mix A or B and corresponding probes for ELISA
| Organism | Primer pair | Fragment size (bp) | Probe | Target region | [pmol/µl mix] used for each primer in Mix A/B | Concentration of probe in hybridization buffer for ELISA (pmol/ml) | References |
|---|---|---|---|---|---|---|---|
| AV | ADV1/ADV2 | 134 | ADV3 | Hexon gene | 5.5 pmol/Mix A | 10 | [42] |
| Bpp | Bp1/Bp3 | 121 | BP5 | Porin gene | 16.5 pmol/Mix A | 10 | [27] |
| Bp | Bp-FP2/BpRP2 | 194 | Bp-S2 | IS481 | 5.5 pmol/Mix A | 10 | [43, 44] |
| Cpn | Cpn-A/CpnB-1 | 468 | Cpn-C | 16s-rRNA | 5.5 pmol/Mix A | 10 | [45] |
| CV229E | Cor229E-FP5/Cor229E-RP5 | 240 | Cor229E-S5 | RdRp/PolGen | 5.5 pmol/Mix B | 10 | [46] and Dr. J. Ziebuhr, personal communication |
| CVOC43 | CorOC43-FP4/CorOC43RP4 | 240 | CorOC43-S4 | M-Gen | 11 pmol/Mix B | 10 | [47] |
| CVOC43 2nd | CorOC43-FP5/CorOC43RP5 | 240 | CorOC43-S5 | RdRp/PolGen | 5.5 pmol/Mix B | 10 | [46] and Dr. J. Ziebuhr, personal communication |
| EV | EV1/EV2 | 154 | EV3 | 5′ UTR | 5.5 pmol/Mix A | 10 | [48] |
| IVA | INFA1/INFA2 | 190 | INFA3 | NSP | 5.5 pmol/Mix A | 10 | [49] |
| IVB | INFB1/INFB2 | 241 | INFB3a | NSP | 5.5 pmol/Mix A | 10 | [49] |
| Lpn | Lpn-FP1/Lpn-RP1 | 156 | Lpn-S1 | 5s to 23s rRNA | 5.5 pmol/Mix A | 10 | [50] |
| Mpn | MP1/MP2 | 277 | MP3 | 16s-rRNA | 5.5 pmol/Mix A | 10 | [51] |
| MPV | HMPV-FP3/hMPV-RP3 | 151 | hMPV-S3 | N-gene | 5.5 pmol/Mix B | 10 | [52] and Dr. Fouchier, personal communication |
| PIV1 | PIV1-1/Piv1-2 | 180 | PIV1C2 | HN-gene | 5.5 pmol/Mix A | 10 | [53] |
| PIV2 | PIV2-FP4/PIV2-RP4 | 205 | PIV2-S4 | NP-gen | 5.5 pmol/Mix A | 10 | [54] |
| PIV3 | Pip3+/Pis3− | 225 | Pis3+ −S1 | HN-gene | 5.5 pmol/Mix A | 10 | [35] |
| PIV4 | Piv4-FP1/PIV4-RP1 | 443 | Piv4-S1 | PP-gene | 11 pmol/Mix A | 10 | [55] |
| Picorna | hRV1-c/hRV2-b | 395 | hRV-S1/RV-S1 | 5′ UTR | 5.5 pmol/Mix B | 10 each | [56, 57] |
| Reo | Reo-FP4/Reo-RP2 | 365 | Reo-S3 and Reo-S4 | L1 gene | 5.5 pmol/Mix B | 10 each | [58] |
| RSV | RSV1-b/RSV2-b | 245 | RSV3/RSV3b and RSV6/RSV6b | FPF1-gen | 5.5 pmol/Mix A | 5 each | [59] |
| Pan. H1N1 (2009) | H1SWS/H1SWAs1 | 81 | H1SWP | HA-gen | 5.5 pmol/Mix B | 10 | [28] |
Table 2.
Sequences of primers
| Organism | Primer name | 5′–3′ sequence | Organism | Primer name | 5′–3′ sequence |
|---|---|---|---|---|---|
| AV | ADV1 | GCCGAGAAGGGCGTGCGCAGGTA | Lpn | Lpn-FP1 | GAAACGTATCGTGTAAACTCTG |
| AV | ADV2 | ATGACTTTTGAGGTGGATCCCATGGA | Lpn | Lpn-RP1 | TATCATTGGCGCGGAAATGTTT |
| Bpp | Bp1 | TGCAACATCCTGTCCCCTTAATCC | Mpn | MP1 | AAGGACCTGCAAGGGTTCGT |
| Bpp | Bp3 | CGTCCACCAGGGGTGGTAGGAGAT | Mpn | MP2 | CTCTAGCCATTACCTGCTAA |
| Bp | Bp-FP2 | GGTGTGAAGATTCAATAGGTTGT | MPV | hMPV-FP3 | CATGCTATATTAAAAGAGTCTC |
| Bp | Bp-RP2 | GCCGCTTCAGGCACACAAAC | MPV | hMPV-RP3 | TCTGCAGCATATTTGTAATCAG |
| Cpn | Cpn-A | TGACAACTGTAGAAATACAGC | PIV1 | PIV1-1 | CACATCCTTGAGTGATTAAGTTTGATGA |
| Cpn | CpnB-1 | GGGCGCCTCTCTCCTATAAAT | PIV1 | PIV1-2 | ATTTCTGGAGATGTCCCGTAGGAGAAC |
| PIV2 | PIV2-FP4 | CCTGATACCCTTAATCACCA | |||
| PIV2 | PIV2-RP4 | CATTGATTCTCCCTTGTTGT | |||
| CV229E | Cor229E-FP5 | TCTTAAATACGCCATATCTGG | PIV3 | Pip3+ | CTGTAAACTCAGACTTGGTA |
| CV229E | Cor229E-RP5 | TCACACTTAGGATAGTCCCA | PIV3 | Pis3− | TAAATCTTGTTGTTGAGATTG |
| CVOC43 | CorOC43-FP4 | GGAGTTTCAACCCAGAAACAA | PIV4 | Piv4-FP1 | CTGAACGGTTGCATTCAGGT |
| CVOC43 | CorOC43-FP5 | TTTGAAATATGCTATTAGTGC | PIV4 | Piv4-RP1 | TTGCATCAAGAATGAGTCCT |
| CVOC43 | CorOC43-RP4 | CGCTTATCCTGTCAAGAAAACC | Picorna | hRV1-c | CAAGCACTTCTGTTTCCCCG |
| CVOC43 | CorOC43-RP5 | TCACACTTAGGATAATCCCA | Picorna | hRV2-b | GAAACACGGACACCCAAAGTAGT |
| EV | EV1 | ATTGTCACCATAAGCAGCCA | Reo 1-3 | Reo-FP4 | CCATTTATGGGGGTTCCTGC |
| EV | EV2 | TCCTCCGGCCCCTGAATGCG | Reo 1-3 | Reo-RP2 | ATCATTAATCCATCATCACCTT |
| IVA | INFA1 | AAGGGCTTTCACCGAAGAGG | RSV | RSV1-b | CATTTGTTATAGGCATATCATTG |
| IVA | INFA2 | CCCATTCTCATTACTGCTTC | RSV | RSV2-b | CTTAACCAGCAAAGTGTTAGAC |
| IVB | INFB1 | ATGGCCATCGGATCCTCAAC | Pan. H1N1 (2009) | H1SWS | CATTTGAAAGGTTTGAGATATTCCC |
| IVB | INFB2 | TGTCAGCTATTATGGAGCTG | Pan. H1N1 (2009) | H1SWAs1 | GGACATGCTGCCGTTACACC |
Table 3.
Sequences of probes for ELISA
| Organism | Name | 5′–3′ sequence |
|---|---|---|
| AV | ADV3 | CTCGATGACGCCGCGGTGC |
| Bpp | Bp5-(Sonde) | AGGAGCTTGTTGCATTGCGAT |
| Bp | Bp-S2 | AGCCCGGCCGGATGAACACCC |
| Cpn | Cpn-C | TCTTGCTACCTTCTGTACTAA |
| CV229E | Cor229E-S5 | AAGTTTTATGGCGGGTGGGA |
| CVOC43 | CorOC43-S4 | GGCTATTCTTGGGCAGATTTG |
| CVOC43 | CorOC43-S5 | AAATTTTATGGTGGCTGGGA |
| EV | EV3 | GAAACACGGACACCCAAAGTA |
| IVA | INFA3 | GTCCTCATCGGAGGACTTGAATGGAATGAT |
| IVB | INFB3a | CCAATTTGGTCAAGAGCACCGATTATCACC |
| Lpn | Lpn-S1 | CGACTATAGCGATTTGGAACC |
| Mpn | MP3 | ACTCCTACGGGAGGCAGCAGTA |
| MPV | hMPV-S3 | TGCAATGATGAGGGTGTCACTGC |
| PIV1 | PIV1C2 | TACCTTCATTATCAATTGGTGATGCAATATATG |
| PIV2 | PIV2-S4 | TGTATGACTGCTCCTGATCA |
| PIV3 | Pis3+-S1 | ACTCCCAAAGTTGATGAAAGAT |
| PIV4 | Piv4-S1 | AAAGAATTAGGTGCAACCAGTC |
| Picorna | hRV-S1 | GCATTCAGGGGCCGGAG |
| Reo 1-3 | Reo-S3 | GGCCGATATCGGGAATGCAGAA |
| Reo 1-3 | Reo-S4 | GGCCTATATCTGGAATGCAGAA |
| RV | RV-S1 | TCTAGCCTGCGTGGCTGC |
| RV | RV-S1-c | TCTAGCCTGCGTGGCGGC |
| RSV | RSV3 | CCTGCATTAACACTAAATTC |
| RSV | RSV3-b | CCTGCATTAACACTGAATTC |
| RSV | RSV6 | CCTGCATTGACACTAAATTC |
| RSV | RSV6-b | CCTGCATTGACACTGAATTC |
| Pan. H1N1 (2009) | H1SWP | ACAAGTTCATGGCCCAATCATGACTCG |
As negative controls (2), blank reagent that contained H2O was used instead of nucleic acid. As positive controls (2) for each m-RT-PCR run, total cellular nucleic acid extracted from virus and/or bacterial stocks were used as the template.
Pandemic (H1N1) 2009 influenza A virus was later on included in the m-RT-PCR-ELISA. Although rapid tests can be a very useful tool in pandemic (H1N1) 2009 influenza diagnosis, experts still recommend RT-PCR tests [28]. The primers used were those recommended by the German National Reference Center for Influenza [29] and integrated in Mix B of the m-RT-PCR-ELISA instead of the reovirus primers and probes. The performance of the PCR as single primer PCR and in the multiplex approach was tested first on positive control material derived from the German National Reference Center for Influenza. Later, the m-RT-PCR-ELISA was compared to the real-time format of this RT-PCR on positive clinical specimens.
Individual RT-PCR procedures
Single primer RT-PCR-ELISA specific for one organism was performed as previously described [3]. Individual PCR procedures in comparison to the m-RT-PCR-ELISA were performed at the Institute of Virology, Erasmus MC, Rotterdam, by Dr. Niesters. Comparison of the PCRs for B. pertussis and B. parapertussis performed in Kiel has been described previously [27].
Analysis of PCR products and classification of results
Electrophoretic separation of PCR products and microwell hybridization analysis was performed as previously described [2], and the sequences and concentrations used as capture probes are given in Tables 1 and 3. For the detection of RSV, four detection probes were used [12]. RSV3 and RSV6 were combined in one hybridization buffer and RSV3b and RSV6b in a second hybridization buffer. Likewise, the two capture probes for reovirus and coronavirus OC43 were each combined in one hybridization mix. The optical density was measured at OD405, with a reference at OD490. The run was considered to be valid if all negative control values were <0.2 OD units at OD405. The classification of results was performed as previously described [3]. The complete m-RT-PCR-ELISA procedure was guided by Excel-based software, which calculates the volumes for RT-PCR and gives a pipetting pattern for the ELISA microwell plates, as well as classification of the ELISA results as described above.
To prevent carry-over contamination within the laboratory, precautions were taken as previously described [3].
Rapid shell vial culture assay
Virus isolation was performed on tertiary monkey kidney (tMK), human embryo lung cells, A549 cells, and MDCK cells [30, 31]. After 48 h of culture, one of the glass slides from each specimen was tested by indirect immunofluorescence assay (IDFA) and cultures were followed for a maximum of 14 days, as summarized in [3].
Gold standard
The broad spectrum of organisms detectable with the m-RT-PCR-ELISA makes it difficult to define one method as the gold standard for all ARI-causing pathogens. Rhinoviruses, for example, are more difficult to culture, and differentiation between enteroviruses is even more problematic, even by RT-PCR, because, in most cases, the 5′ untranslated region is used as the target sequence and differentiation is accomplished by hybridization. Therefore, the standard for these organisms was first to establish the diagnosis as “organisms from the picornavirus group”. In case of differences in the classification, the nucleic acid sequences of the questionable organisms was determined and this information was used to define the gold standard. Sequence information of specimens containing MPV, PIV2, and PIV4 were also the determining factor for the gold standard, if differences between two methods were detected. Sequencing of PCR fragments was performed by the sequencing service of Qiagen (Hilden, Germany). Culture results (cytopathic effect and/or IDFA) from historical and/or retesting were used as the gold standard for Cpn, Mpn, IVA, IVB, RSV, MPV, PIV1, 3, and AV. For Bp and Bpp, the culture result and/or Bordetella-specific individual PCR was defined as the gold standard. For Lpn, the “case” specified as a positive result of one diagnostic test (urinary antigen test, culture, or PCR) was defined as the gold standard.
Due to the fact that it is difficult or even impossible to detect more than one pathogen within one specimen by culture methods or RT-PCR techniques using a single primer, these multiple detections of pathogens in one specimen by m-RT-PCR-ELISA were classified as follows:
If a single organism detected by culture was detected by m-RT-PCR ELISA, the result was concordant also if additional organisms were detected by the multiplex approach.
If the additional organisms were proven by an individual single primer PCR and/or by sequencing of the PCR fragment, the secondary and tertiary organisms in a specimen were defined to be correct as well.
Administration of data
The results from the m-RT-PCR-ELISA on the blinded specimens were sent to the cooperating laboratories from which the specimens were received, and, subsequently, the contents of the samples were disclosed. The total number of samples and the number of samples for a referring pathogen were used to the calculate sensitivity, specificity, and positive and negative predictive values. They were retested in parallel by culture and m-RT-PCR-ELISA. According to the central limit theorem of biostatistics, the mean or mean percentage becomes normally distributed at a sample size between 20 and 30. Therefore, the aim was to test for at least 25 samples for each pathogen to obtain robust estimates of the referring parameters [32].
Results
Figure 1 shows a 2 % agarose gel of the RT-PCR on positive controls (upper part divided into Mix B and A) and on clinical specimens (lower part divided into Mix B and A). The fragment sizes and the pathogen to Mix allocation are listed in Table 1. The correlation of fragment sizes and ELISA signals based on hybridization with the pathogen-specific probe was clearly visible for all pathogens, as also tested by individual RT-PCR-ELISA. No cross-reactivity could be detected between the pathogens tested. For a result to be determined as “positive”, both gel and ELISA had to be “positive”.
Fig. 1.
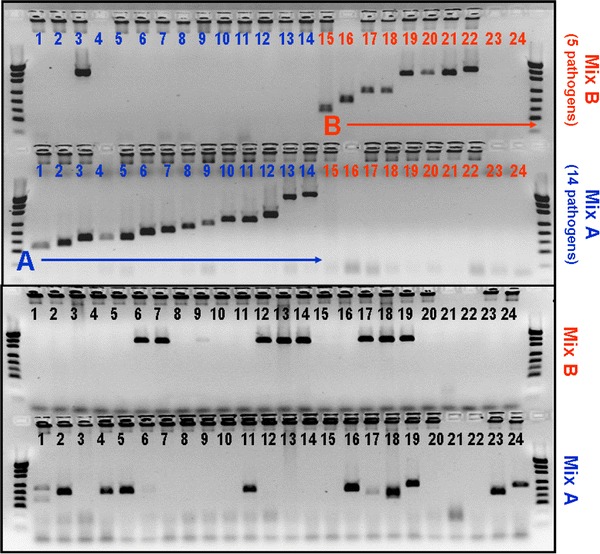
Detection of pathogens in positive controls and clinical specimens (gel electrophoresis, agarose gel 2 % stained with ethidium bromide). Upper part 10 µl RT-PCR product amplified with primer Mix A (bottom) on positive controls in lanes: 1 Bpp, 2 AV, 3 EV, 4 Lpn, 5 PIV1, 6 IVA, 7 Bp, 8 PIV2, 9 PIV3, 10 RSV, 11 IVB, 12 Mpn, 13 PIV4, 14 Cpn, and 10 µl RT-PCR product amplified with primer Mix B (top) on positive controls in lanes: 15 MPV, 16 SARS coronavirus (not included in regular m-RT-PCR), 17 CV229E, 18 CVOC43, 19 Reo1, 20 Reo2, 21 Reo3, 22 RV, and negative control for RT-PCR (lane 23) and PCR (lane 24). For fragment sizes, see Table 1. Lower part lanes 1 to 9 and 11 to 19 are 10 µl RT-PCR product amplified with primer Mix A (bottom) and Mix B (top) on clinical specimens from a routine RT-PCR run. Negative preparation controls (lanes 10 and 20), negative control for RT-PCR (lane 21) and PCR (lane 22), and positive controls for preparation Bp (lane 23), PIV2 (lane 24) (954 m)
Identification of organisms in cell culture supernatants
Cell culture supernatants as previously described [3] and used for the validation of the previously described 9-valent m-RT-PCR were tested with the 19-valent m-RT-PCR-ELISA. All 36 specimens (5 AV, 13 EV, 7 IVA, and 6 IVB) tested the respective pathogens correctly, as described before [3]. Additionally, the 3 PIV2 and one more IVA, which was not detected by the 9-valent method, were detected by the 19-valent m-RT-PCR-ELISA. As for the 9-valent method, one specimen positive for IVB was also identified as “positive” for IVA by the 19-valent m-RT-PCR-ELISA. All 30 of the 60 specimens with Mpn were identified correctly as “positives” by the 19-valent m-RT-PCR-ELISA, so the concordance of the results of m-RT-PCR-ELISA to culture was 100 %. Within ten specimens positive for C. pneumoniae (n = 5), C. trachomatis (n = 2), C. psittaci (n = 1) or negative (n = 2), the five C. pneumoniae were identified to be “positive” by the highly specific primer pair and probe for Cpn. The C. trachomatis or C. psittaci strains were not detected by this primer and probe. All 49 different adenovirus types (culture supernatants) could be detected with the 19-valent m-RT-PCR-ELISA.
The new pandemic influenza A virus (H1N1) 2009 and avian H5N1 could not be detected by the primers for seasonal influenza originally used in this m-RT-PCR. Due to the importance of the (H1N1) 2009 virus, the m-RT-PCR-ELISA was extended for the detection of this pathogen in October 2009 and it was possible to detect positive controls and positive samples with the specific primers if integrated in Mix B (see also “Methods”).
Analytical sensitivity
The analytical sensitivity of the m-RT-PCR-ELISA (Table 4) and of the corresponding single primer RT-PCR-ELISA was tested on nucleic acid preparations from serial tenfold dilutions of the culture supernatants. The amount of template present in each RT-PCR reaction corresponds to the amount of template present in 12 µl of culture supernatant, and TCID50 values were determined on 1 ml of culture (or dilution). These experiments revealed that the m-RT-PCR-ELISA in comparison to culture was less sensitive for PIV1, as sensitive for Cpn, EV, MPV, PIV3, and RSV, and more sensitive for AV, IVA, and IVB. Comparison of the single primer test with the multiplex approach showed a higher sensitivity for the detection of Cpn and PIV1 in the single primer test and the same sensitivity for the other pathogens. By using a 40-cycle PCR protocol, the sensitivity could be increased (see Table 4: comparison of single 40/single 35 and single 40/multi 35). To improve sensitivity, the single primer test was performed on all questionable results with a 40-cycle PCR protocol (see “Individual RT-PCR procedures”).
Table 4.
Comparison of multiplex, single primer PCR, and culture
| Detection limit at dilution step | Comparison | ||||||
|---|---|---|---|---|---|---|---|
| Single | Multi | Culture | Single/multi 35 | Single 40/single 35 | Single 40/multi 35 | Multi 35/culture | |
| AV | 10−6 | 10−6 | 10−5 | Comparable | More sensitive | More sensitive | More sensitive |
| Bp | 10−5 | 10−5 | Comparable | ||||
| Bpp | 10−3 | 10−3 | Comparable | ||||
| Cpn A4 | 10−5 | 10−4 | 10−4 | More sensitive | Comparable | More sensitive | Comparable |
| CV229E | 10−2 | 10−2 | Comparable | Comparable | Comparable | ||
| CVOC43 | 10−2 | 10−2 | Comparable | ||||
| EV (CoxB1) | 10−7 | 10−7 | 10−7 | Comparable | Comparable | Comparable | Comparable |
| IVA | 10−6 | 10−6 | 10−5 | Comparable | More sensitive | More sensitive | More sensitive |
| IVB | 10−6 | 10−6 | 10−3 | Comparable | More sensitive | More sensitive | More sensitive |
| Lpn | 10−6 | 10−6 | Comparable | ||||
| Mpn | 10−6 | 10−6 | Comparable | More sensitive | More sensitive | ||
| MPV | 10−4 | 10−4 | 10−4 | Comparable | Comparable | ||
| PIV1 | 10−4 | 10−3 | 10−6 | More sensitive | More sensitive | More sensitive | Less sensitive |
| PIV2 | 10−5 | 10−5 | Comparable | ||||
| PIV3 | 10−3 | 10−3 | 10−3 | Comparable | More sensitive | More sensitive | Comparable |
| PIV4 | 10−2 | 10−2 | Comparable | ||||
| Reo | 10−4 | 10−4 | Comparable | ||||
| RSV | 10−5 | 10−5 | 10−5 | Comparable | More sensitive | More sensitive | Comparable |
| RV | 10−4 | 10−4 | Comparable | ||||
Identification of organisms in clinical specimens of unknown contents
A total of 430 clinical specimens (nasopharyngeal aspirates, swabs, or BAL) were taken from a pool of retained specimens used already for the 9-valent method [3] and “newer” specimens, both obtained from the Department of Virology, Rotterdam, and stored in virus medium (calf serum) at −80 °C. Specimens for comparison to the 19-valent m-RT-PCR-ELISA were tested in parallel with culture and partially (CV, RV, EV, AV) with additional PCR at the Institute of Virology in Rotterdam. The specimens included: 41 RSV, 38 PIV1, 20 PIV2, 47 PIV3, 9 PIV4, 18 MPV, 33 IVA, 27 IVB, 21 EV, 71 RV, 12 CV, and 50 AV. A total of 289 specimens with 65 Bp and 21 Bpp were tested in comparison to an individual single PCR (n = 177) and additionally by culture (n = 112). A set of 18 specimens containing 15 specimens classified as Lpn cases were tested also in parallel to culture and PCR. Cpn and Mpn could be tested only by positive culture supernatants as described above. For ReoV types 1, 2, and 3, only culture supernatants were available and tested positive with the m-RT-PCR-ELISA.
The comparison of m-RT-PCR-ELISA with the gold standard on clinical specimens for the single pathogens is shown in Table 5. The sensitivity on clinical specimens from Rotterdam was between 83.3 % for CV and MPV up to 100 % for the picornaviruses (EV/RV), IVA, and PIV4. The specificity of the m-RT-PCR-ELISA was over 99 % for most of the organisms. The five RV which were only positive in the m-RT-PCR-ELISA were not tested by sequencing and two of them were from the first set of specimens from Rotterdam which were not tested for RV by culture. Thus, it is possible that these specimens were not detected correctly by culture. IVB, PIV1, CV, and MPV were not detected correctly in 11.1, 15.8, 16.7, and 16.7 % respectively. The sensitivity for B. pertussis and B. parapertussis as tested in comparison to culture and/or an individual single PCR was 98.5 and 95.2 %, respectively. The mean sensitivity for the method, calculated on the results for this group of pathogens (without Cpn, Mpn, and Lpn) is 93.3 % (95.1 % median), with a range from 83.3 to 100 %. No specimens were available for reoviruses and no testing was performed.
Table 5.
Comparison of m-RT-PCR-ELISA to the gold standard
| Pathogen | Number of pathogens in the “golden standard” | Number of specimens tested for this organism | Detected correctly | Detected incorrectly | Sensitivity (%) | Specificity (%) | Positive predictive value (%) | Negative predictive value (%) | Efficacy (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Positive | Negative | Positive | Negative | ||||||||
| RSV | 41 | 289 | 37 | 247 | 1 | 4 | 90.2 | 99.6 | 97.4 | 98.4 | 98.3 |
| PIV1 | 38 | 429 | 32 | 391 | 0 | 6 | 84.2 | 100.0 | 100.0 | 98.5 | 98.6 |
| PIV2 | 20 | 181 | 19 | 161 | 0 | 1 | 95.0 | 100.0 | 100.0 | 99.4 | 99.4 |
| PIV3 | 47 | 430 | 46 | 382 | 1 | 1 | 97.9 | 99.7 | 97.9 | 99.7 | 99.5 |
| PIV4 | 9 | 289 | 9 | 280 | 0 | 0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| MPV | 18 | 292 | 15 | 274 | 0 | 3 | 83.3 | 100.0 | 100.0 | 98.9 | 99.0 |
| IVA | 33 | 292 | 33 | 258 | 1 | 0 | 100.0 | 99.6 | 97.1 | 100.0 | 99.7 |
| IVB | 27 | 292 | 24 | 265 | 0 | 3 | 88.9 | 100.0 | 100.0 | 98.9 | 99.0 |
| EV | 21 | 400 | 21 | 379 | 0 | 0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| RV | 71 | 430 | 71 | 354 | 5 | 0 | 100.0 | 98.6 | 93.4 | 100.0 | 98.8 |
| CV | 12 | 178 | 10 | 166 | 0 | 2 | 83.3 | 100.0 | 100.0 | 98.8 | 98.9 |
| AV | 50 | 428 | 45 | 377 | 1 | 5 | 90.0 | 99.7 | 97.8 | 98.7 | 98.6 |
| Lpn a | 15 | 18 | 8 | 3 | 0 | 7 | 53.3 | 100.0 | 100.0 | 30.0 | 61.1 |
| Bp b | 65 | 289 | 64 | 223 | 1 | 1 | 98.5 | 99.6 | 98.5 | 99.6 | 99.3 |
| Bpp b | 21 | 289 | 20 | 268 | 0 | 1 | 95.2 | 100.0 | 100.0 | 99.6 | 99.7 |
| Cpn c | 5 | 10 | 5 | 5 | 0 | 0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| Mpn c | 30 | 60 | 30 | 30 | 0 | 0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
aThe results for Lpn are based on so-called “Lpn cases” defined from clinical symptoms and/or on UAG test and/or historical PCR results
bFor Bp and Bpp, the results were taken from parallel testing in the lab in Kiel with different methods, as described in “Methods”
cFor Cpn and Mpn, the results were taken from a blinded test on culture supernatants. No specimens were available for ReoV
For Lpn, the sensitivity was only 53.3 %, but one has to bear in mind that the gold standard for the detection of Lpn was based on the results of the historically performed tests (culture or PCR or urinary antigen test). It is noteworthy that the parallel retesting of the specimens in Haarlem generally failed; all specimens were negative on retesting. No specimen could be identified as positive by culture and only two specimens were identified as positive by the Lpn-specific individual single PCR. In contrast, the m-RT-PCR-ELISA correctly detected Lpn in eight specimens.
For Cpn and Mpn, no clinical specimens were available and, thus, the results presented in Table 5 were taken from a blinded test on culture supernatants. Taking into account all the results available (including Cpn, Mpn, and Lpn), the overall sensitivity and specificity (mean) was 91.8 and 99.8 %, respectively.
Identification of organisms in clinical specimens for epidemiological surveillance
In order to evaluate the feasibility of the m-RT-PCR-ELISA for surveillance, 16,231 clinical specimens were tested prospectively. These were predominantly nasopharyngeal aspirates or nasopharyngeal swabs and BAL in NaCl (0.9 %) from hospitalized children below 16 years of age sampled between July 2003 and June 2010 in three areas of Germany (Kiel, Mainz, Freiburg; Freiburg until June 2007). The number of samples tested over time and the proportion of samples with positive PCR results can be seen in Fig. 2. The number of specimens peaked periodically during all “cold seasons” from November to April, and this correlated with an increased number of samples with positive m-RT-PCR results. A total of 10,765 (66.3 %) of the 16,231 clinical specimens were tested “positive”. Of the isolates, 5,044 (31.1 %) were RV, 1,999 (12.3 %) were RSV, 1,286 (7.9 %) were AV, 944 (5.8 %) were EV, 737 (4.5 %) were seasonal IVA plus 173 (7.6 % of the season 2009/2010) were new pandemic IVA (H1N1) 2009, 899 (5.5 %) were MPV, 518 (3.2 %) were CV, 383 (2.4 %) were PIV3, 268 (1.7 %) were PIV1, 259 (1.6 %) were Mpn, 205 (1.3 %) were IVB, 164 (1.0 %) were PIV2, 144 (0.9 %) were PIV4, 103 (0.6 %) were Bp, 29 (0.2 %) were Cpn, and 29 (0.2 %) were Bpp on the basis of all specimens tested. Lpn and ReoV were not detected in any specimen. The new pandemic IVA (H1N1) 2009 was first tested in the season 2009/2010 (since October 2009, and from this time on, no further testing for ReoV has been performed. The epidemiological pattern is shown in Fig. 2.
Fig. 2.
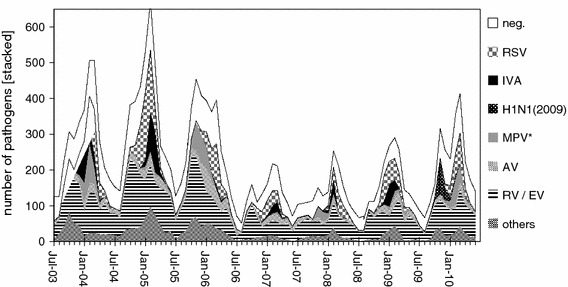
Pathogens detected in clinical specimens. Seasonality of pathogens detected with the m-RT-PCR-ELISA between July 2003 and June 2010 in 16,316 clinical specimens, predominantly nasopharyngeal aspirates or nasopharyngeal swabs and BAL in NaCl (0.9 %). The numbers of pathogens detected are stacked in the graph. Shown are the pathogens with more than 5 % of detection within the group of positive specimens. These are RV, EV, AV, IVA, RSV, MPV, and the pandemic influenza H1N1 2009 shown for the season 2009/2010. Together with negative specimens, the upper line of negatives (neg.) illustrates the total amount of specimens tested. The fraction of others in the bottom of the graph represents the total detection of CV, IVB, PIV1, PIV2, PIV3, PIV4, Mpn, Bp, Bpp, and Cpn
The sensitivity for pandemic influenza virus (H1N1) 2009 in Mix B was tested on clinical specimens identified as “positive” for the pandemic IVA (H1N1) 2009 in comparison to the real-time PCR method, as previously published [29]. The detection limit on clinical specimens was approximately at cycle threshold (ct) values between 32 and 34. Specimens identified by real-time assay with ct values above 35 could not be detected by the multiplex approach, which was performed with “only” 35 cycles.
Simultaneous detection of two or more organisms
The m-RT-PCR revealed evidence of simultaneous infection with more than one organism: while an ARI pathogen was detected in 10,765 of 16,231 (66.43 %) specimens, 8,534 (52.5 %) specimens were “positive” for a single pathogen only. More than one pathogen was detected in 2,182 (13.4 %) specimens, of which 1,937 (88.7 %) tested “positive” for two pathogens, 211 (9.7 %) for three, 30 (1.4 %) for four, and 0.2 % specimens for five, six, and seven pathogens. AV, EV, CV, Bp, and Bpp were among the most common microorganisms observed in specimens testing positive for multiple pathogens.
Conclusions
In order to provide a technique for the rapid detection of a wide array of non-colonizing organisms, the former m-RT-PCR-ELISA method [2, 3] was extended to include ten additional pathogens. With this test, it is now possible to detect most of the non-colonizing organisms of the upper respiratory tract with one m-RT-PCR protocol.
To assess the integrity of the primer pairs and probes in this extended m-RT-PCR assay, RNA and DNA from viral and bacterial stocks were assayed in the presence of all nonhomologous primer pairs, and no cross-reactions were detected, documenting the high degree of specificity of this assay. Positive controls of all pathogens could clearly be detected by the method.
Unfortunately, there was no single local laboratory with routine use of conventional techniques for all pathogens to validate the method directly on incoming specimens. Therefore, a multistep approach involving several institutions, bacteriologists, and virologists was used. The most crude test on cell culture supernatants (i) and tests for analytical sensitivity (ii) were performed to verify the test in general and to evaluate the sensitivity of the chosen primers as used in single and multiplex formats. The retrospective studies on clinical specimens in comparison to culture (iii) including individual mono RT-PCRs were conducted to determine the clinical sensitivity, and the feasibility of the method (iv) for epidemiological approaches was tested prospectively on 16,231 clinical specimens.
All pathogens from culture supernatants were detected as described before [3]. With the extension of the spectrum of pathogens, the 19-valent method also detected PIV2 viruses plus one more IVA, possibly due to a slightly higher sensitivity. The spectrum of enteroviruses included Coxsackie types B1 to B6, type A16, echo virus types 6, 7, 11, 24, 30, and 12 different influenza viruses (types A and B). Furthermore, the tests on different types of adenoviruses revealed that all 49 tested types could be detected. Sequence alignments with the primers and probes used for, e.g., enterovirus, influenza viruses, and others, revealed that these were capable of detecting many more types of human pathogens within these groups. The primers used for influenza as described in Table 2 are not able to detect the avian H5N1 virus. For this, a separate primer pair and probe were used. In case of a human H5N1 infection, it is important to test for this pathogen directly by single primer PCR, prompted by the history of exposure.
In addition, with the m-RT-PCR assay, it was possible to integrate primers and probes for the detection of SARS coronaviruses. A distinct amplification product of 190 bp with a positive ELISA signal could be detected on inactivated human serum containing SARS coronavirus, as shown in Fig. 1 lane 16 Mix B. The SARS primers are as described by Drosten et al. [33, 34] and the human serum and RNA transcript for control experiments were kindly provided by Prof. Drosten (Institute of Virology, University of Bonn Medical Centre, Bonn, Germany). However, the labs in Kiel and Mainz were not equipped to handle materials potentially containing SARS and, thus, the diagnostic for SARS with the m-RT-PCR-ELISA was not offered as a routine diagnostic.
The analytical sensitivity was tested on serial tenfold dilutions of culture supernatants. The results for TCID50 values determined on culture were available only for nine pathogens. The m-RT-PCR in most cases was comparable or more sensitive as compared to culture. Only for PIV1 did the m-RT-PCR-ELISA show a lower sensitivity versus culture. Since the single primer RT-PCR ELISA showed a tenfold higher sensitivity for PIV1 as compared to the multiplex approach, one must assume that the reduced sensitivity is based on the use in the multiplex approach rather than on the design of primers itself. As discussed earlier [3], the PIV3 F-gene primers originally used in the 9-valent m-RT-PCR were changed to the hemagglutinin–neuraminidase gene primers as described by Echevarría et al. [35], because the F-gene primers failed to detect all PIV3 in clinical specimens. The extended m-RT-PCR uses the hemagglutinin–neuraminidase gene as target sequences for PIV1 and PIV3, and the latter gave comparable results in analytical sensitivity to culture. For PIV1 and also for Cpn, the single primer detection was more sensitive by a factor of ten.
For IVA, IVB, and AV, the m-RT-PCR-ELISA was more sensitive compared to culture, as it was in the 9-valent approach [3]. For all of the comparisons to TCID50 culture supernatants, the samples had been frozen for more than 2 years before the determination of the analytical sensitivity by m-RT-PCR-ELISA. Therefore, the analytical sensitivity might even be underestimated.
In summary, based on these results, the multiplex approach was only less sensitive by a factor of 10 for 2 of the 19 pathogens in comparison to single primer RT-PCR. This appears to be quite acceptable for use of the method in clinical routine. The sensitivity could be enhanced by increasing the cycle number from 35 to 40. However, this may result in some weak unspecific fragments on agarose gel in the multiplex approach, especially on viscous and/or mucous specimens. Therefore, multiplex method was performed with 35 cycles and to confirm specimens with questionable results in ELISA or weak fragments on agarose gel by single primer RT-PCR with 40 cycles, which was more sensitive in 80 % of the 35 cycle/40 cycle comparison tested.
The pathogen most commonly identified was RV. Why this is consistent with other studies in children with acute respiratory tract infections [36, 37], it raises the question as to what extent the presence of RV represents carriage, incubation, or convalescence is unknown [36]. Even the detection of a virus from the lower respiratory tract is no proof for its disease-causing abilities. Nevertheless, the high frequency at which RV was identified from nasopharyngeal specimens of infants and young children admitted to our hospitals with ARI suggests a causative correlation of the agent and the observed ARI symptoms. More definite support for considering RV as a “true ARI pathogen” comes from the fact that in situ hybridization has demonstrated that RV may trigger inflammatory processes in infected cells and tissues [37]. Future studies will have to show if specific microorganisms which, to date, are considered to be “true pathogens”, may, in fact, colonize certain populations under specific circumstances.
Evidence of the simultaneous detection of nucleic acids from two or more pathogens within one specimen from children with ARI was observed by m-RT-PCR in 13.4 % of all specimens, and the simultaneous detections were most frequently with CV, AV, EV, and Bp. Simultaneous detections have also been observed by others using molecular techniques in a multiplex format [4, 7, 38–41]. However, the results of simultaneous detections have to be judged with care, because m-RT-PCR, like other “non-quantitative” PCR-based techniques, only detect fragments of nucleic acids. Thus, a positive PCR result only allows the conclusion that a specific part of the genome of a pathogen was present. It is not possible to draw conclusions about the viability of the respective pathogens. Quantitative PCR assays have the advantage of calculating the amount of pathogens present in specimens (only roughly for respiratory specimens) and may give additional guidance to judge whether the pathogen is, in fact, responsible for the infection. In specimens, e.g., from the lower respiratory tract, which may contain encapsulated material, it is possible that non-viable pathogens from earlier infections are present. Also, some pathogens, such as adenoviruses, may persist in the airways for a long time without causing disease.
The epidemiological pattern of the ARI pathogens detected by 19-valent m-RT-PCR can be taken from Fig. 2, which shows “time-limited yearly epidemics” for RSV, MPV, and IVA, as well as the year-round endemic presence of RV, EV, and AV. Further detailed analysis of the seasonality and epidemiology of the pathogens detected with this method is published elsewhere [18, 20, 21].
The m-RT-PCR-ELISA evaluated here improves the diagnostic yield in terms of the overall sensitivity, as well as the spectrum of coverage for diagnosing respiratory infections. It can be performed with little requirement for equipment and at low costs (approximately 50 € including all materials like tips, gloves, and also the retesting of specimens). Given the lack of specificity of clinical symptoms associated with ARI caused by an organism, multiplexed molecular testing has become more commonplace [4–10, 41].
Further development of the m-RT-PCR-ELISA should focus on the inclusion of newer respiratory pathogens like coronavirus NL63 and HUK1 or bocaviruses, although the clinical relevance of the latter is still under evaluation. Also, the integration of primers for the detection of “new” viruses like the pandemic influenza strain (H1N1) 2009 was possible and enables the differentiation of such pandemic infectious agents from others.
Acknowledgments
We appreciate the excellent technical assistance of B. Reckewitz, N. Esly, and E. Budo-Guetaifi, and we thank I. Chaloupka, Institute for Medical Microbiology, Department of Virology, TU Munich, Germany; E. Jacobs and R. Dumke, Institute of Medical Microbiology, University Hospital, Dresden, Germany; R.P. Verkooyen, Erasmus University, Rotterdam, the Netherlands; M. Maaß, Institute for Medical Microbiology, University of Lübeck, Germany; E. Staube, Institute of Clinical Microbiology, University Hospital, Jena, Germany; Dr. B. Schweiger, RKI, Berlin, Germany, for providing several specimens and strains used in this study, C. Drosten, Institute of Virology, University of Bonn Medical Centre, Bonn, Germany; R. Fouchier, Erasmus Medical Center, Rotterdam, the Netherlands; J. Ziebuhr, University of Giessen, Germany and Dr. Ijzerman, Haarlem, the Netherlands, for providing and retesting of the Legionella pneumophila strains. This work was supported by the Bundesministerium für Bildung und Forschung (BMBF) via the Deutsches Zentrum für Luft- und Raumfahrt e.V. by the research grant (01KI9910/1) for “PID-ARI.net”, a pediatric infectious diseases network on acute respiratory tract infections.
Conflict of interest
There is no actual or potential conflict of interest in relation to this article.
References
- 1.Leowski J. Mortality from acute respiratory infections in children under 5 years of age: global estimates. World Health Stat Q. 1986;39:138–144. [PubMed] [Google Scholar]
- 2.Gröndahl B, Puppe W, Hoppe A, Kühne I, Weigl JA, Schmitt HJ. Rapid identification of nine microorganisms causing acute respiratory tract infections by single-tube multiplex reverse transcription-PCR: feasibility study. J Clin Microbiol. 1999;37:1–7. doi: 10.1128/jcm.37.1.1-7.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puppe W, Weigl JA, Aron G, Gröndahl B, Schmitt HJ, Niesters HG, Groen J. Evaluation of a multiplex reverse transcriptase PCR ELISA for the detection of nine respiratory tract pathogens. J Clin Virol. 2004;30:165–174. doi: 10.1016/j.jcv.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Brunstein JD, Cline CL, McKinney S, Thomas E. Evidence from multiplex molecular assays for complex multipathogen interactions in acute respiratory infections. J Clin Microbiol. 2008;46:97–102. doi: 10.1128/JCM.01117-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gruteke P, Glas AS, Dierdorp M, Vreede WB, Pilon JW, Bruisten SM. Practical implementation of a multiplex PCR for acute respiratory tract infections in children. J Clin Microbiol. 2004;42:5596–5603. doi: 10.1128/JCM.42.12.5596-5603.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kehl SC, Henrickson KJ, Hua W, Fan J. Evaluation of the Hexaplex assay for detection of respiratory viruses in children. J Clin Microbiol. 2001;39:1696–1701. doi: 10.1128/JCM.39.5.1696-1701.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Létant SE, Ortiz JI, Bentley Tammero LF, Birch JM, Derlet RW, Cohen S, Manning D, McBride MT. Multiplexed reverse transcriptase PCR assay for identification of viral respiratory pathogens at the point of care. J Clin Microbiol. 2007;45:3498–3505. doi: 10.1128/JCM.01712-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nolte FS, Marshall DJ, Rasberry C, Schievelbein S, Banks GG, Storch GA, Arens MQ, Buller RS, Prudent JR. MultiCode-PLx system for multiplexed detection of seventeen respiratory viruses. J Clin Microbiol. 2007;45:2779–2786. doi: 10.1128/JCM.00669-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Syrmis MW, Whiley DM, Thomas M, Mackay IM, Williamson J, Siebert DJ, Nissen MD, Sloots TP. A sensitive, specific, and cost-effective multiplex reverse transcriptase-PCR assay for the detection of seven common respiratory viruses in respiratory samples. J Mol Diagn. 2004;6:125–131. doi: 10.1016/S1525-1578(10)60500-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Templeton KE, Scheltinga SA, Beersma MF, Kroes AC, Claas EC. Rapid and sensitive method using multiplex real-time PCR for diagnosis of infections by influenza A and influenza B viruses, respiratory syncytial virus, and parainfluenza viruses 1, 2, 3, and 4. J Clin Microbiol. 2004;42:1564–1569. doi: 10.1128/JCM.42.4.1564-1569.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wasem S, Weichert S, Walther S, Weigl JA, Puppe W, Ihorst G, Schmitt HJ, Forster J. Lower respiratory tract disease in children: constant pathogens—constant management?! Klin Padiatr. 2008;220:291–295. doi: 10.1055/s-2007-990301. [DOI] [PubMed] [Google Scholar]
- 12.Weigl JA, Puppe W, Schmitt HJ. Incidence of respiratory syncytial virus-positive hospitalizations in Germany. Eur J Clin Microbiol Infect Dis. 2001;20:452–459. doi: 10.1007/s100960100527. [DOI] [PubMed] [Google Scholar]
- 13.Weigl JA, Puppe W, Schmitt HJ. The incidence of influenza-associated hospitalizations in children in Germany. Epidemiol Infect. 2002;129:525–533. doi: 10.1017/S0950268802007707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weigl JA, Puppe W, Schmitt HJ. Can respiratory syncytial virus etiology be diagnosed clinically? A hospital-based case–control study in children under two years of age. Eur J Epidemiol. 2003;18:431–439. doi: 10.1023/A:1024213400297. [DOI] [PubMed] [Google Scholar]
- 15.Weigl JA, Puppe W, Schmitt HJ. Variables explaining the duration of hospitalization in children under two years of age admitted with acute airway infections: does respiratory syncytial virus have a direct impact? Klin Padiatr. 2004;216:7–15. doi: 10.1055/s-2004-817688. [DOI] [PubMed] [Google Scholar]
- 16.Weigl JA, Puppe W, Gröndahl B, Schmitt HJ. Epidemiological investigation of nine respiratory pathogens in hospitalized children in Germany using multiplex reverse-transcriptase polymerase chain reaction. Eur J Clin Microbiol Infect Dis. 2000;19:336–343. doi: 10.1007/s100960050490. [DOI] [PubMed] [Google Scholar]
- 17.Weigl JA, Puppe W, Rockahr S, Schmitt HJ. Burden of disease in hospitalized RSV-positive children in Germany. Klin Padiatr. 2002;214:334–342. doi: 10.1055/s-2002-35365. [DOI] [PubMed] [Google Scholar]
- 18.Weigl JA, Puppe W, Belke O, Neusüss J, Bagci F, Schmitt HJ. The descriptive epidemiology of severe lower respiratory tract infections in children in Kiel, Germany. Klin Padiatr. 2005;217:259–267. doi: 10.1055/s-2004-820352. [DOI] [PubMed] [Google Scholar]
- 19.Weigl JA, Puppe W, Belke O, Neusüss J, Bagci F, Schmitt HJ. Population-based incidence of severe pneumonia in children in Kiel, Germany. Klin Padiatr. 2005;217:211–219. doi: 10.1055/s-2004-822699. [DOI] [PubMed] [Google Scholar]
- 20.Weigl JA, Puppe W, Meyer CU, Berner R, Forster J, Schmitt HJ, Zepp F. Ten years’ experience with year-round active surveillance of up to 19 respiratory pathogens in children. Eur J Pediatr. 2007;166:957–966. doi: 10.1007/s00431-007-0496-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weigl JA, Puppe W, Meyer CU, Berner R, Forster J, Schmitt HJ, Zepp F. PID-ARI.net—a pediatric infectious diseases network on acute respiratory infections and the added value of a multilevel research network. Klin Padiatr. 2008;220:281–286. doi: 10.1055/s-2007-985815. [DOI] [PubMed] [Google Scholar]
- 22.du Prel JB, Puppe W, Gröndahl B, Knuf M, Weigl JA, Schaaff F, Schmitt HJ. Are meteorological parameters associated with acute respiratory tract infections? Clin Infect Dis. 2009;49:861–868. doi: 10.1086/605435. [DOI] [PubMed] [Google Scholar]
- 23.Gröndahl B, Puppe W, Weigl J, Schmitt HJ. Comparison of the BD Directigen Flu A+B Kit and the Abbott TestPack RSV with a multiplex RT-PCR ELISA for rapid detection of influenza viruses and respiratory syncytial virus. Clin Microbiol Infect. 2005;11:848–850. doi: 10.1111/j.1469-0691.2005.01223.x. [DOI] [PubMed] [Google Scholar]
- 24.Huck B, Scharf G, Neumann-Häfelin D, Puppe W, Weigl J, Falcone V. Novel human metapneumovirus sublineage. Emerg Infect Dis. 2006;12:147–150. doi: 10.3201/eid1201.050772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schützle H, Weigl J, Puppe W, Forster J, Berner R. Diagnostic performance of a rapid antigen test for RSV in comparison with a 19-valent multiplex RT-PCR ELISA in children with acute respiratory tract infections. Eur J Pediatr. 2008;167:745–749. doi: 10.1007/s00431-007-0581-1. [DOI] [PubMed] [Google Scholar]
- 26.Toschke AM, Arenz S, von Kries R, Puppe W, Weigl JA, Höhle M, Heininger U. No temporal association between influenza outbreaks and invasive pneumococcal infections. Arch Dis Child. 2008;93:218–220. doi: 10.1136/adc.2006.098996. [DOI] [PubMed] [Google Scholar]
- 27.Li Z, Jansen DL, Finn TM, Halperin SA, Kasina A, O’Connor SP, Aoyama T, Manclark CR, Brennan MJ. Identification of Bordetella pertussis infection by shared-primer PCR. J Clin Microbiol. 1994;32:783–789. doi: 10.1128/jcm.32.3.783-789.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pongthanapisith V, Sukasem C, Premchaiporn K, Srichantaratsamee C, Chantratita W. Clinical performance of three rapid diagnostic tests for influenza virus in nasopharyngeal specimens to detect novel swine-origin influenza viruses. Infection. 2011;39:105–111. doi: 10.1007/s15010-011-0092-x. [DOI] [PubMed] [Google Scholar]
- 29.Panning M, Eickmann M, Landt O, Monazahian M, Olschläger S, Baumgarte S, Reischl U, Wenzel JJ, Niller HH, Günther S, Hollmann B, Huzly D, Drexler JF, Helmer A, Becker S, Matz B, Eis-Hübinger A, Drosten C. Detection of influenza A(H1N1)v virus by real-time RT-PCR. Euro Surveill. 2009;14:pii=19329. [PubMed]
- 30.Engler HD, Selepak ST. Effect of centrifuging shell vials at 3,500 × g on detection of viruses in clinical specimens. J Clin Microbiol. 1994;32:1580–1582. doi: 10.1128/jcm.32.6.1580-1582.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rothbarth PH, Groen J, Bohnen AM, de Groot R, Osterhaus AD. Influenza virus serology—a comparative study. J Virol Methods. 1999;78:163–169. doi: 10.1016/S0166-0934(98)00174-8. [DOI] [PubMed] [Google Scholar]
- 32.Dawson B, Trapp RG. Probability and related topics for making inferences about data. In: Dawson B, Trapp RG, editors. Basic and clinical biostatistics. 3. New York: Lange Medical Books/McGraw-Hill; 2001. pp. 82–84. [Google Scholar]
- 33.Drosten C, Chiu LL, Panning M, Leong HN, Preiser W, Tam JS, Günther S, Kramme S, Emmerich P, Ng WL, Schmitz H, Koay ES. Evaluation of advanced reverse transcription-PCR assays and an alternative PCR target region for detection of severe acute respiratory syndrome-associated coronavirus. J Clin Microbiol. 2004;42:2043–2047. doi: 10.1128/JCM.42.5.2043-2047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drosten C, Günther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguière AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Müller S, Rickerts V, Stürmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 35.Echevarría JE, Erdman DD, Swierkosz EM, Holloway BP, Anderson LJ. Simultaneous detection and identification of human parainfluenza viruses 1, 2, and 3 from clinical samples by multiplex PCR. J Clin Microbiol. 1998;36:1388–1391. doi: 10.1128/jcm.36.5.1388-1391.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arden KE, McErlean P, Nissen MD, Sloots TP, Mackay IM. Frequent detection of human rhinoviruses, paramyxoviruses, coronaviruses, and bocavirus during acute respiratory tract infections. J Med Virol. 2006;78:1232–1240. doi: 10.1002/jmv.20689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renwick N, Schweiger B, Kapoor V, Liu Z, Villari J, Bullmann R, Miething R, Briese T, Lipkin WI. A recently identified rhinovirus genotype is associated with severe respiratory-tract infection in children in Germany. J Infect Dis. 2007;196:1754–1760. doi: 10.1086/524312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benson R, Tondella ML, Bhatnagar J, Carvalho Mda G, Sampson JS, Talkington DF, Whitney AM, Mothershed E, McGee L, Carlone G, McClee V, Guarner J, Zaki S, Dejsiri S, Cronin K, Han J, Fields BS. Development and evaluation of a novel multiplex PCR technology for molecular differential detection of bacterial respiratory disease pathogens. J Clin Microbiol. 2008;46:2074–2077. doi: 10.1128/JCM.01858-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lam WY, Yeung AC, Tang JW, Ip M, Chan EW, Hui M, Chan PK. Rapid multiplex nested PCR for detection of respiratory viruses. J Clin Microbiol. 2007;45:3631–3640. doi: 10.1128/JCM.00280-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marshall DJ, Reisdorf E, Harms G, Beaty E, Moser MJ, Lee WM, Gern JE, Nolte FS, Shult P, Prudent JR. Evaluation of a multiplexed PCR assay for detection of respiratory viral pathogens in a public health laboratory setting. J Clin Microbiol. 2007;45:3875–3882. doi: 10.1128/JCM.00838-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reijans M, Dingemans G, Klaassen CH, Meis JF, Keijdener J, Mulders B, Eadie K, van Leeuwen W, van Belkum A, Horrevorts AM, Simons G. RespiFinder: a new multiparameter test to differentially identify fifteen respiratory viruses. J Clin Microbiol. 2008;46:1232–1240. doi: 10.1128/JCM.02294-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hierholzer JC, Halonen PE, Dahlen PO, Bingham PG, McDonough MM. Detection of adenovirus in clinical specimens by polymerase chain reaction and liquid-phase hybridization quantitated by time-resolved fluorometry. J Clin Microbiol. 1993;31:1886–1891. doi: 10.1128/jcm.31.7.1886-1891.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kösters K, Reischl U, Schmetz J, Riffelmann M, Wirsing von König CH. Real-time LightCycler PCR for detection and discrimination of Bordetella pertussis and Bordetella parapertussis. J Clin Microbiol. 2002;40:1719–1722. doi: 10.1128/JCM.40.5.1719-1722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reischl U, Lehn N, Sanden GN, Loeffelholz MJ. Real-time PCR assay targeting IS481 of Bordetella pertussis and molecular basis for detecting Bordetella holmesii. J Clin Microbiol. 2001;39:1963–1966. doi: 10.1128/JCM.39.5.1963-1966.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaydos CA, Quinn TC, Eiden JJ. Identification of Chlamydia pneumoniae by DNA amplification of the 16S rRNA gene. J Clin Microbiol. 1992;30:796–800. doi: 10.1128/jcm.30.4.796-800.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stephensen CB, Casebolt DB, Gangopadhyay NN. Phylogenetic analysis of a highly conserved region of the polymerase gene from 11 coronaviruses and development of a consensus polymerase chain reaction assay. Virus Res. 1999;60:181–189. doi: 10.1016/S0168-1702(99)00017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vabret A, Mouthon F, Mourez T, Gouarin S, Petitjean J, Freymuth F. Direct diagnosis of human respiratory coronaviruses 229E and OC43 by the polymerase chain reaction. J Virol Methods. 2001;97:59–66. doi: 10.1016/S0166-0934(01)00343-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rotbart HA, Sawyer MH, Fast S, Lewinski C, Murphy N, Keyser EF, Spadoro J, Kao SY, Loeffelholz M. Diagnosis of enteroviral meningitis by using PCR with a colorimetric microwell detection assay. J Clin Microbiol. 1994;32:2590–2592. doi: 10.1128/jcm.32.10.2590-2592.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Claas EC, Sprenger MJ, Kleter GE, van Beek R, Quint WG, Masurel N. Type-specific identification of influenza viruses A, B and C by the polymerase chain reaction. J Virol Methods. 1992;39:1–13. doi: 10.1016/0166-0934(92)90120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alexiou-Daniel S, Stylianakis A, Papoutsi A, Zorbas I, Papa A, Lambropoulos AF, Antoniadis A. Application of polymerase chain reaction for detection of Legionella pneumophila in serum samples. Clin Microbiol Infect. 1998;4:144–148. doi: 10.1111/j.1469-0691.1998.tb00377.x. [DOI] [PubMed] [Google Scholar]
- 51.van Kuppeveld FJ, van der Logt JT, Angulo AF, van Zoest MJ, Quint WG, Niesters HG, Galama JM, Melchers WJ. Genus- and species-specific identification of mycoplasmas by 16S rRNA amplification. Appl Environ Microbiol. 1992;58:2606–2615. doi: 10.1128/aem.58.8.2606-2615.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maertzdorf J, Wang CK, Brown JB, Quinto JD, Chu M, de Graaf M, van den Hoogen BG, Spaete R, Osterhaus AD, Fouchier RA. Real-time reverse transcriptase PCR assay for detection of human metapneumoviruses from all known genetic lineages. J Clin Microbiol. 2004;42:981–986. doi: 10.1128/JCM.42.3.981-986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fan J, Henrickson KJ. Rapid diagnosis of human parainfluenza virus type 1 infection by quantitative reverse transcription-PCR-enzyme hybridization assay. J Clin Microbiol. 1996;34:1914–1917. doi: 10.1128/jcm.34.8.1914-1917.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yuasa T, Bando H, Kawano M, Tsurudome M, Nishio M, Kondo K, Komada H, Ito Y. Sequence analyses of the 3′ genome end and NP gene of human parainfluenza type 2 virus: sequence variation of the gene-starting signal and the conserved 3′ end. Virology. 1990;179:777–784. doi: 10.1016/0042-6822(90)90145-H. [DOI] [PubMed] [Google Scholar]
- 55.Aguilar JC, Pérez-Breña MP, García ML, Cruz N, Erdman DD, Echevarría JE. Detection and identification of human parainfluenza viruses 1, 2, 3, and 4 in clinical samples of pediatric patients by multiplex reverse transcription-PCR. J Clin Microbiol. 2000;38:1191–1195. doi: 10.1128/jcm.38.3.1191-1195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andeweg AC, Bestebroer TM, Huybreghs M, Kimman TG, de Jong JC. Improved detection of rhinoviruses in clinical samples by using a newly developed nested reverse transcription-PCR assay. J Clin Microbiol. 1999;37:524–530. doi: 10.1128/jcm.37.3.524-530.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pitkäranta A, Arruda E, Malmberg H, Hayden FG. Detection of rhinovirus in sinus brushings of patients with acute community-acquired sinusitis by reverse transcription-PCR. J Clin Microbiol. 1997;35:1791–1793. doi: 10.1128/jcm.35.7.1791-1793.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leary TP, Erker JC, Chalmers ML, Wetzel JD, Desai SM, Mushahwar IK, Dermody TS. Detection of reovirus by reverse transcription-polymerase chain reaction using primers corresponding to conserved regions of the viral L1 genome segment. J Virol Methods. 2002;104:161–165. doi: 10.1016/S0166-0934(02)00058-7. [DOI] [PubMed] [Google Scholar]
- 59.Paton AW, Paton JC, Lawrence AJ, Goldwater PN, Harris RJ. Rapid detection of respiratory syncytial virus in nasopharyngeal aspirates by reverse transcription and polymerase chain reaction amplification. J Clin Microbiol. 1992;30:901–904. doi: 10.1128/jcm.30.4.901-904.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]