

Abstract
Genome regulation requires control of chromosomal organization by SMC–kleisin complexes. The cohesin complex contains the Smc1 and Smc3 subunits which associate with the kleisin Scc1 to form a ring-shaped complex that can topologically engage chromatin to regulate chromatin structure. Release from chromatin involves opening of the ring at the Smc3–Scc1 interface in a reaction that is controlled by acetylation and engagement of the Smc ATPase head domains. To understand the underlying molecular mechanisms, we have determined the 3.2 Å cryo-EM structure of the ATPγS-bound, hetero-trimeric cohesin ATPase head module, and the 2.1 Å crystal structure of a nucleotide-free Smc1–Scc1 subcomplex from Saccharomyces cerevisiae and Chaetomium thermophilium. We found that ATP-binding and Smc1–Smc3 heterodimerization promote conformational changes within the ATPase which are transmitted to the Smc coiled-coil domains. Remodeling of the coiled-coil domain of Smc3 abrogates the binding surface for Scc1 thus leading to ring opening at the Smc3–Scc1 interface.
Keywords: Cohesin, Chromatin folding, SMC, cryoEM, genome regulation
Introduction
The cohesin complex is a member of the SMC–kleisin protein family, which facilitates genome organization, transcription, repair, and segregation across all domains of life1. A fundamental and conserved property of SMC–kleisin enzymes is their capacity to topologically engage DNA2. Cohesin is thought to regulate genome architecture through coordinated cycles of intra-chromatid DNA capture, loop extrusion, and release2–5. Co-entrapment of sister chromatids by cohesin allows the complex to establish chromosome cohesion, a prerequisite for the equal distribution of genetic information between daughter cells during eukaryotic cell division6.
The core cohesin trimer comprises Smc1–3 and the α-kleisin Scc1. The N- and C- termini of Smc1 and 3 form composite ABC ATPase domains intersected by an extended, ~45nm length of antiparallel coiled-coil which folds in its central region to produce a ‘hinge’ domain through which Smc1–3 heterodimerise. The α-kleisin subunit bridges the SMC ATPase lobes, thus sealing the tripartite ring7,8. The topological entrapment and release of chromatin substrate requires controlled opening and closure of the cohesin ring, however the underlying molecular mechanisms have remained elusive.
Initial entrapment of chromatin by cohesin is achieved via an accessory loading complex, Scc2–4, which stimulates the cohesin ATPase during G1 to facilitate DNA capture predominantly at the centromere9–12. The release of DNA can occur either through cleavage of Scc1 by the protease separase, an event that triggers the metaphase-to-anaphase transition, or through opening of the Smc3–NScc1 interface or ‘gate’8,13,14.
The second, dynamic mode of release is catalyzed by the disassociation factor, Wapl, which collaborates with the HEAT proteins Pds5 and Scc3 to disengage the Smc3–NScc1 gate, and is counteracted by acetylation of the Smc3 nucleotide binding domain (bound) in S phase13,15,16. Hence, dynamic DNA entry and exit require an independent set of co-factors which utilize the same cohesin ATPase domain to achieve functionally distinct outcomes. Furthermore, loading involves the SMC hinge, and is not impaired by artificial closure of either of the two SMC–kleisin interfaces17,18. Conversely, fusion of the Smc3–NScc1 gate is sufficient to abolish Wapl-mediated release entirely13,19,20.
ABC-type ATPases, such as cohesin, share a conserved mechanism of action in which ATP-driven conformational changes propagate into adjacent domains to fulfill various functions21,22. In cohesin, the integrity of both ATPases is important for efficient chromatin loading, whereas the impairment of the Smc3 ATP-binding site specifically abolishes disengagement of the Smc3–NScc1 interface and cohesin release16,23–25. Thus, ATP-mediated SMC head engagement by cohesin presumably remodels the complex in a manner which results in ring opening, whether to promote DNA capture or release. Comprehensive understanding of cohesin function will therefore require a full structural and biochemical understanding of its ATPase cycle and the regulation thereof.
Here we report a cryo-EM structure of the heterotrimeric cohesin Smc3–Scc1–Smc1 ATPase module in complex with the nucleotide analogue ATPγS, and the X-ray crystal structure of the Smc1–CScc1 subcomplex in the absence of bound nucleotide. Our structures show that dimerization and nucleotide engagement of the cohesin ATPase are coupled to opening of the Smc3–NScc1 gate, and that re-closure of the ring is likely contingent on hydrolysis or release of bound nucleotide.
Results
Cryo-EM structure of the cohesin ATPase head module
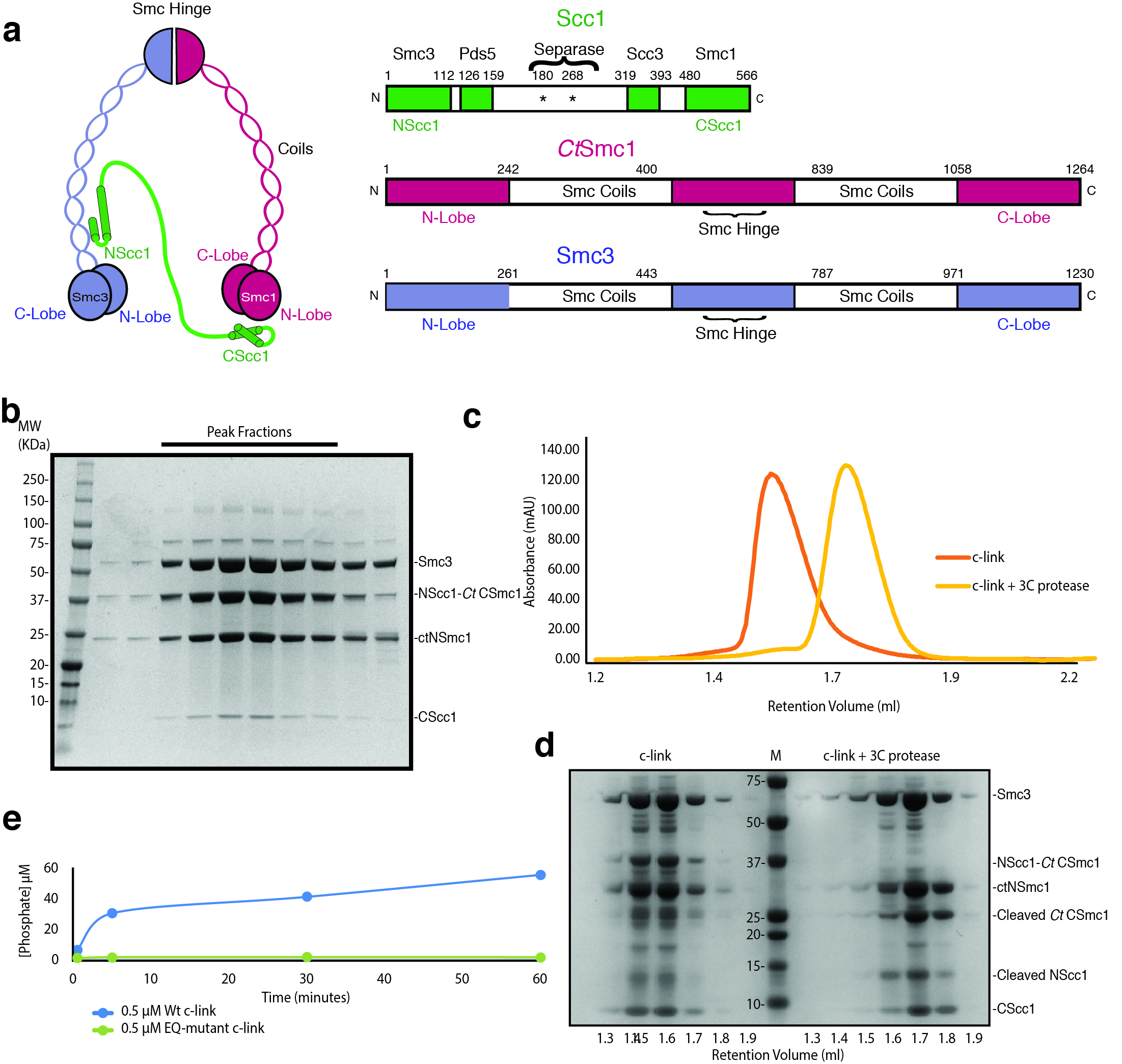
To obtain a heterotrimeric Smc3–Scc1–Smc1 ATPase head module, we fused Saccharomyces cerevisiae NScc1 (ScNScc1) to the C-lobe of the Smc1 ATPase from Chaetomium thermophilum (CtSmc1), as ScSmc1 proved insoluble in Escherichia coli, and co-expressed this construct in E. coli with the remaining head module constituents: the ScSmc3 ATPase, the N-lobe CtSmc1 and its binding partner, ScCScc1 (Fig. 1a). Co-purification of the Smc3–Scc1–CtSmc1 head module, termed c-link (‘linked’ cohesin), showed that a direct physical interaction of NScc1 with the coiled coil of Smc38 is maintained in this construct (Fig. 1a, Extended Data Fig. 1a, b). An ATPase assay demonstrates that c-link, but not a variant containing Walker B site mutations (CtSmc1 E1198Q–Smc3 E1155Q) which prevent ATP hydrolysis26, is an active ATPase, thus showing that our procedure allows purification of a functional cohesin head module (Extended Data Fig. 1e).
Figure 1. CryoEM structure of the cohesin ATPase head module.

a, Domain organisation and cartoon depiction of c-link. All construct used are from Saccharomyces cerevisiae unless indicated Chaetomium thermophilium (Ct). b, SDS-PAGE analysis of disulphide crosslinked c-link (’x-linked’). c, Low-pass filtered density map of the cohesin head module (blue: Smc3; Green: CScc1; red: CtSmc1). d, Cryo-EM densities at 2.5 RMSD (gray mesh) are superimposed on the atomic model of the ATPγS–Mg2+ molecules. Conserved elements of the ABC ATPase modules are labelled for orientation. e, Cryo-EM density at 2.5 RMSD of the Signature motif helix from CtSmc1. f, Cryo-EM density at 1.5 RMSD of the coiled coil from Smc3. There is no apparent density for NScc1. g, Ribbon diagram of the model of the cohesin head module with each colored according to each subunit.
To stabilize ATPase head engagement, we introduced two pairs of cysteine residues at the predicted SMC heterodimerization interface and induced disulphide bond formation by addition of copper phenanthroline, in the presence of ATP. The cross-linking procedure resulted in the essentially complete conversion of Smc1–Smc3 monomers into a single species on SDS-PAGE (Fig. 1b; Extended Data Fig. 1b). We collected a cryo-EM dataset from cross-linked c-link, vitrified in the presence of ATPγS–Mg2+, that yielded a map with an overall resolution of 3.2Å, using the gold standard Fourier shell correlation (FSC) = 0.143 criteria (Fig. 1c, Extended Data Fig. 2d).
Density corresponding to two ATPγS–Mg2+ molecules was observed between the respective Walker A and Walker B motifs of each subunit, and the cognate signature motif of its partner (Fig. 1d). Most side chains in the core of the ATPase were readily distinguishable, as are those of the conserved Walker A, B, and Signature motifs (Fig. 1e, Extended Data Fig. 2f-g). Whereas the base of the flexible coiled-coil segments was well-resolved (Extended Data Fig. 2f-g), density toward the periphery of these domains was mostly limited to main chain atoms and bulky side-chains (Fig. 1f). We failed to detect density for NScc1 bound to the coiled coil of Smc3 (Fig. 1f, Extended Data Fig. 2g) even though this domain was present in the expressed protein and purification of the c-link complex relied on initial binding of NScc1 to the Smc3 (Extended Data Fig. 1d). Our interpretation of the absence of density for NScc1 is that engagement of the Smc ATPase heads releases the kleisin from Smc3.
We determined the 2.1Å X-ray crystal structure of CtSmc1 bound to ScCScc1 domain by molecular replacement, using the Saccharomyces cerevisiae ortholog7 and found, as expected, high structural conservation (Extended Data Fig. 3a and Table 1). As the CtSmc1–CScc1 structure was obtained in the absence of bound nucleotide, it provides high-resolution details concerning the unliganded conformation of this subcomplex (Extended Data Fig. 3c). To obtain a final model of the cohesin head complex, we docked the CtSmc1–CScc1 and the Smc3 structures8 into the cryo-EM map and rebuilt and refined the atomic models (Fig. 1g and Table 2).
Table 1. Xray data collection, phasing and refinement statistics.
|
CtSmc1–Scc1 (PDB: 6QPQ) |
|
|---|---|
| Data collection | ESRF MASSIF ID30A-1 |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 80.7, 111.1, 166.1 |
| Resolution (Å) | 45.70–2.09 |
| No. reflections | 82453 (12513) |
| Rsym | 6.2 (121)* |
| I / σI | 13.4 (1.1)* |
| CC 1/2 | 0.99 (0.49) |
| Completeness (%) | 98.8 (94.2)* |
| Redundancy | 4.5 (4.4)* |
| Refinement | |
| Resolution (Å) | 45.70–2.09 |
| Rwork / Rfree | 0.21 / 0.24 |
| No. atoms | 15801 |
| Smc1# | 6431 |
| Scc1# | 1302 |
| B-factors (mean; Å2) | |
| Smc1# | 63.7 |
| Scc1# | 69.9 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.002 |
| Bond angles (°) | 0.46 |
Values in parentheses are for highest-resolution shell
Averages are shown for the two copies in the asymmetric unit
Table 2. Cryo-EM data collection, refinement and validation statistics.
| Smc3–Scc1–CtSmc1 complex (EMD-4614, PDB 6QPW) |
|
|---|---|
| Data collection and processing | |
| Magnification | 165,000 |
| Voltage (kV) | 300 |
| Electron exposure (e–/Å2) | 42.08 |
| Defocus range (μm) | 1.25-2.5 |
| Pixel size (Å) | 0.81 |
| Symmetry imposed | C1 |
| Initial particle images (no.) | 290,821 |
| Final particle images (no.) | 178,162 |
| Map resolution (Å) | 3.2 |
| FSC threshold | 0.143 |
| Map resolution range (Å) | 3.2-10 |
| Refinement | |
| Initial model used (PDB code) | 4UX3, 6QPQ |
| Model resolution (Å) | 3.2 |
| FSC threshold | 0.143 |
| Model resolution range (Å) | 3.2-10 |
| Map sharpening B factor (Å2) | -124 |
| Model composition | |
| Nonhydrogen atoms | 6784 |
| Protein residues | 839 |
| Ligands | 4 |
| B factors (Å2) | |
| Protein | 85.99 |
| Ligand | 53.76 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.987 |
| Validation | |
| MolProbity score | 1.66 |
| Clashscore | 5.83 |
| Poor rotamers (%) | 0.14 |
| Ramachandran plot | |
| Favored (%) | 95.02 |
| Allowed (%) | 4.92 |
| Disallowed (%) | 0.00 |
Remodeling of the Smc3–NScc1 interface
To understand the conformational changes associated with ATP-mediated Smc3–Smc1 head engagement, we superposed the ATPase N-lobes of the unliganded CtSmc1–CScc1 structure and of the previously determined Smc3–NScc1 structure (Extended Data Fig. 3c, d)8 with our cryo-EM model. This showed that the Smc1 and Smc3 coiled coils adopt variable conformations in the complex (Fig. 2a). This conformational change is due to flexibility in the region at the base of the coiled-coil and drives the coiled-coils toward each other by ~ 10° for Smc1 and ~ 16° for Smc3 (Fig. 2a). The underlying structural basis is a concerted movement of the Smc signature-coupling helices and Q-loops, in which the eponymous glutamine residues engage the Mg2+ and γ phosphate of ATPγS (Extended Data Fig. 3c, d). The associated displacement of α3 is propagated by the signature coupling helices, α4, to the N-terminus of α5, the base of the coiled coil (Fig. 2b, c and Supplementary Video 1). In Smc3, the piston-like movement of α4 results in ~7.3Å displacement of the N-terminus of α5 (Fig. 2d and Supplementary Video 1). The cysteine crosslinks are positioned in flexible loops between secondary structural elements (Extended Data Fig. 3e). Structural analysis shows that the cross-linked residues are conformationally mobile and the surrounding regions undistorted (Extended Data Fig. 3f). Thus, we conclude that the cross-linking reaction merely serves to prevent dissociation of the catalytic complex, and does not otherwise influence the conformation of the cohesin ATPase.
Figure 2. Conformational changes in the engaged cohesin head module lead to remodeling of the NScc1–Smc3 interface.

a, Conformational changes in the coiled coil of the ATPγS and Mg2+ engaged cohesin head module as compared to homodimeric Smc3 (PDB 4UX3) and apo CtSmc1 (both in grey) are indicated with curved arrows. The binding site of NScc1 (green) is indicated. Inset: zoomed view of the ATPase active site. The directions of movements are indicated with arrows in α3, the signature-coupling helix α4 and the N-terminal end of the coiled coil α5 in the C-lobe of CtSmc1 (red) and Smc3 (blue). b, Details of the displacement of α3, the signature-coupling helix α4 and the coiled coil α5 in CtSmc1 (bound and apo forms are depicted in red and grey respectively). c, and d, Details of the displacement of Smc3 coiled-coil and signature-coupling helices upon cohesin head engagement and DNA exit gate release (hetero- and homodimeric forms of Smc3 are depicted in blue and grey respectively). For clarity, NScc1 is omitted from the homodimeric form. e, Surface representation of the Smc3 coiled coil (blue) from the ‘closed’ Smc3–NScc1 gate structure. PDB:4UX3. Amino acid residues from NScc1 (green; left) engage Smc3 through a set of surface pockets lined by conserved hydrophobic amino acid residues of Smc3 (right). f, Surface representation of the rearranged Smc3 coiled coil (blue). The NScc1 binding site is occluded by remodeling of the cognate Smc3 interface. g, C-link complexes, harboring a 3C protease in NScc1, were cross-linked and incubated with 3C prior to immobilization on Ni2+-conjugated beads. Quantification of NScc1 retention by c-link complexes in the presence or absence of nucleotide is depicted in a scatter plot. Representative silver-stained SDS-PAGE bands are boxed, and the corresponding proteins indicated. Data from two independent experiments are plotted. Data for the graph and uncropped gel images are available as source data.
The conformational rearrangement of the coiled coil of Smc3 likely explains why NScc1 is displaced upon Smc3–Smc1 head engagement. NScc1contains an extended α-helix, α3, which together with α2 forms a four helical bundle with Smc3’s coiled coil (Fig. 2e)8. A set of conserved hydrophobic residues of NScc1 including L75, I78, Y82, L89, and L97 bind into pockets on the surface of the Smc3 coiled coil (Fig. 2e). The rearrangement of the coiled coil upon SMC head engagement results in complete reconfiguration of this NScc1–binding surface (Fig. 2f). Due to the displacement of the N-terminus of α5, critical binding pockets such as those provided by F175 and L179 of Smc3 are remodeled, resulting in abolishment of the NScc1 binding site. A morph between the crystallographically-defined states and the cryo-EM structure illustrates how these conformational transitions lead to remodeling of the binding surface, and NScc1 displacement (Supplementary Video 1). Overall, SMC head engagement results in concerted movements in α3 and the signature coupling helices α4 which are propagated to the base of the coiled coil thus resulting in dissociation of the Smc3–NScc1 gate (Supplementary Video 1). Previously, it has been demonstrated that non-hydrolysable ATP analogues, and Walker B mutant cohesin, which binds but does not hydrolyze ATP, are capable of executing the release reaction in vitro25 and in vivo16, respectively. Our structure thus corroborates the model that the conformational changes induced by nucleotide-binding and ATPase heterodimerization are sufficient to disengage NScc1 from Smc3.
Hence, to assess whether SMC head engagement and nucleotide-binding are sufficient to release NScc1 from the isolated cohesin head module, we produced disulphide cross-linked c-link, proteolytically cleaved the linker connecting NScc1 to the C-terminus of CtSmc1, and monitored retention of NScc1 on Ni-NTA beads by c-link in the presence and absence of nucleotide. By quantifying relative NScc1 intensity, we determined that incubation of the cohesin head module with ATP and ATPγS results in the release of ~60% and ~80% of bound NScc1, respectively, relative to a nucleotide-free control (Fig. 2g). Therefore, nucleotide-mediated engagement of the SMC ATPase heads, and the ensuing conformational changes described by our cryo-EM structure, are a central mechanistic event underlying NScc1 release and cohesin ring opening. As such nucleotide-dependent NScc1 release is observed with cross-linked c-link, we conclude that the cross-linking procedure does not interfere with the structure of the cohesin ATPase, nor its ability to engage nucleotides. Additional ATPase assays would be required to assess the impact of cross-linking on ATP hydrolysis.
The Smc3 and Smc1 ATPase sites are structurally distinct
The acetylation of the Smc3 NBD at K112 and K113 by Eco1 is an essential determinant of cohesion establishment, apparently by antagonizing disengagement of the Smc3–NScc1 gate13,15,19,20,25,27–29. The structure of the Smc3–NScc1 complex revealed, however, that these residues are quite distant from the interface and may not modulate the interaction directly8. In the core cohesin trimer, suppressor mutants which artificially restore viability in the absence of cohesin acetylation congregate in the vicinity of the Smc3 ATPase site (AS1) but not in the Smc1 ATPase site (AS2)13,15,16,23,24,30. Acetyl-mimicking or acetyl-lysine proximate residues, and those which destabilize the composite Smc3 ATP-binding site all result in increased stability of the NScc1–Smc3 interface16,23,24. Modelling release suppressing residues onto our structure enables the classification of these mutants according to their predicted and previously observed impact on different facets of cohesin release (Fig. 3b). One such group of mutations clusters within or proximal to the acetyl-lysine pair and probably uncouple AS1 from allosteric regulation without influencing basal rates of ATP hydrolysis13,15,25,31.
Figure 3. The Smc3 and Smc1 ATPase sites are structurally distinct.

a, Amino acid residue interactions in the ATPγS-engaged Smc3–CtSmc1 heterodimer and comparison with the hypothetical apo Smc heterodimer interface. Interactions (i.e. hydrogen bonds and salt bridges) are indicated by yellow dashed lines. b, Mutations which suppress cohesin release (green) cluster at the Smc3 ATPase site. Residues in close proximity to the Smc3 acetyl-lysines and which may influence allosteric regulation of the ATPase are depicted in the top inset. Suppressors within the Smc3 ATPase directly influence hydrolysis (bottom inset). c, Model of the cohesin ATPase cycle (Clockwise from left-hand side). DNA capture is mediated by Scc2–4 and depends on ATP. DNA release is catalyzed by Wapl–Pds5. Heterodimerization and ATP-binding causes conformational changes within the Smc3 coiled-coil domain, stabilized by a ‘latch’-like function of the Q-loop (inset) which lead to Scc1 displacement and cohesin release. ATP hydrolysis permits the cycle to resume. Cohesin is liberated from this cycle by acetylation of Smc3, which counteracts release and establishes cohesion (right-hand side).
A further group, however, occurs within the Smc3 ATPase interface AS1 and impair hydrolysis directly. Despite the fact that mutations within the Smc1 and Smc3 signature motifs, in particular, impair ATP hydrolysis by cohesin to a similar extent, only the former suppress its release from DNA16,23,24,32. Hence, AS1 and AS2 have been proposed to contribute differentially to cohesin release. To understand the molecular basis of this reported functional asymmetry, we analyzed the SMC heterodimerization interfaces in greater detail. Engagement of the N-lobe of the Smc3 ATPase domain with the C-lobe of Smc1 to form AS1 results in burial of ~550Å2 (Fig. 3a). The equivalent buried surface in the Smc1 ATPase domain, AS2, is considerably more extensive ~1200Å2 (Fig. 3a). SMC head engagement results in remodeling of this interface: E1124Smc3 hydrogen bonds to N61Smc1 while Q1125Smc3 directly engages D64Smc1. Additional hydrogen bonds are made by K1116Smc3, N1118Smc3 and E1119Smc3 with Q1248Smc1 and N1251Smc1. Upon Smc1 engagement, the Smc3 signature motif, connecting the signature helix α11 and β7, moves by ~4Å towards the signature-coupling helix α4, thus apparently contributing to its displacement (Supplementary Video 2).
Overall, as there is a direct relationship between buried surface area and the strength of the interaction, it is possible that mutations in AS1 have a relatively greater effect while the equivalent Smc3 mutations are more permissive for SMC head engagement and NScc1 release, given the more extensive AS2 interface16,23. A more parsimonious explanation, arising from our structure, for these mutants is that conformational changes in Smc3 leading to kleisin release are inhibited by destabilization of AS1. Thus, the apparent paucity of protein-protein contacts at AS1 likely facilitates allosteric control of both ATP hydrolysis and the Smc3–NScc1 gate, and explain this regulatory focus at the Smc3 ATPase23,24.
Discussion
Recent data show that ATP-dependent Smc2–Smc4 head dimerization induces dissociation of the N-kleisin from the Smc2 coiled coil to open the condensin ring 33. A comparison of the ATPγS-bound CtSmc1 structure to Smc2 and Smc4 as well as the nucleotide free and ATPγS-bound form of bacterial SMC ATPases and Rad50 showed that the conformational changes associated with ATP-mediated head engagement are conserved between SMCs (Extended Data Fig. 4). SMC coiled-coil arms can adopt a conformation in which they are intimately aligned in a manner refractory to ATPase engagement34–40,41,42. Thus, it is possible that controlled remodeling of the SMC arms to promote or preclude ATP-sandwich formation is a central paradigm for the regulation of SMC–kleisin function. Our results suggest that the cohesin release factors are not an absolute requirement for conformational changes within the isolated Smc1–Smc3–Scc1module per se. Hence, Pds5 and Wapl may function in release by promoting or stabilizing head engagement, and that acetylation potentially establishes cohesion by blocking this activity. Correspondingly, a recent study reported that acetylation of Smc3 almost completely eliminates engagement of the cohesin ATPase 39.
In summary, our structures demonstrate that head engagement can promote a structural intermediate in the cohesin ATPase cycle which is incompatible with NScc1 binding, providing direct structural insight into the mechanism by which cohesin is released from chromosomes (Fig. 3c). As ATP hydrolysis and SMC head engagement are also required for DNA loading and translocation, it will be essential to determine how NScc1 release is regulated by Pds5–Wapl and Scc210,11,25. Future studies will be required to extend these insights to the cohesin holocomplex. Given the exceptional architectural conservation of this interface amongst related complexes, we anticipate that the mechanism described here is likely a universal feature of SMC–kleisin gate opening, the regulation of which may be a fundamental factor governing their functional diversification across evolution.
Methods
Protein purification
All protein complexes were purified according to essentially identical procedures. Following overexpression in E.Coli, according to a published auto-induction procedure43, cells were lysed using a microfluidizer (Microfluidics) in 0.5 M NaCl, 50 mM Tris-HCl pH 7.5, 20 mM Imidazole, and 0.5 mM TCEP. Resultant lysate was clarified by centrifugation and applied to IMAC sepharose resin (GE Healthcare) coupled to Co2+. Bound proteins were washed with lysis buffer, and eluted in lysis buffer supplemented with 300 mM Imidazole. Protein complexes were subsequently further purified by anion exchange via a linear gradient of buffer QA (50 mM Tris-HCl pH 7.5, 0.5 mM TCEP) against QB (1 M NaCl, 50 mM Tris-HCl pH 7.5, 0.5 mM TCEP) (MonoQ, GE Healthcare), and gel filtration chromatography (Superdex 200 16/60, GE Healthcare) in SEC buffer (300 mM NaCl, 40 mM HEPES pH 7.5). For complexes intended for disulphide crosslinking, it was essential to maintain 0.5 mM TCEP in all buffers preceding the final gel filtration step, during which reducing agent was omitted, in order to prevent spontaneous disulphide formation.
To determine the influence of the NScc1–CtSmc1 linker on co-migration of the individual Smc3–NScc1 and CtSmc1–CScc1 complexes, c-link was incubated at 4°C for 30 mins in SEC buffer in the presence and absence of 0.5 μg 3C protease, at a concentration of 5 μM. Samples were then applied to a Superose 6 3.2/300 column (GE Healthcare) equilibrated in SEC buffer and coupled to a Microäkta device (GE Healthcare). Resultant protein fractions were analyzed by SDS-PAGE, followed by Coomassie Blue staining.
Crystallization and structure determination
Crystals of CtSmc1–CScc1 were grown by hanging-drop vapor diffusion at 4 °C by mixing equal volumes of protein at 15 mg ml-1 and crystallization solution containing 0.1 M Tris pH 8.5, 30% PEG300. The crystals were flash frozen in liquid nitrogen, using well solution supplemented with 15% glycerol as a cryoprotectant. Diffraction data were recorded with a Pilatus3 2M detector at the European Synchrotron Radiation facility on beamline ID30A-1/MASSIF-144 using an X-ray wave-length 0.966 Å at 100 K. Location and optimal centering of crystals were determined automatically as described previously45. The beam diameter was selected automatically to match the crystal volume of highest homogeneous quality46. Data were processed with XDS47 and imported into CCP4 format using AIMLESS48.
The structure was determined by molecular replacement using Phaser49 using the ySmc1–CScc1 complex7 as a search model. Iterative manual model building and refinement in Coot and PHENIX, respectively, were pursued to derive a final model. The final CtSmc1–Scc1 model was refined to 2.09 Å resolution with an Rwork and an Rfree of 21% and 24%, respectively (Table S1). Analysis of the refined structure by MolProbity showed that there are no residues in disallowed regions of the Ramachandran plot. The MolProbity all atom clash score was 2.5.
Site directed mutagenesis
All mutagenesis was performed using the QuikChange Lightning kit (Agilent) according to the manufacturer’s instructions. To facilitate disulphide crosslinking of c-link, CtSmc1 residues L1160 and A1201, and Smc3 A1159 and N1204 were mutated to cysteine. To prevent adventitious crosslinking, we introduced the mutation C56S into Scc1.
ATP hydrolysis assays
ATPase activity of c-link and the indicated mutant was quantified using a Malachite green assay (BioAssay Systems, Hayward, CA, USA). 0.5 μM protein was preincubated for 10 minutes at room temperature in ATPase buffer containing (20 mM HEPES pH 7.5, 200 mM NaCl, 4 mM MgCl2). Reactions were initiated by the addition of 2 mM ATP and aliquots of 10 μl removed at the time intervals indicated, and quenched with ATPase buffer supplemented with 100 mM EDTA. To quantify phosphate concentration, 80 μl of 0.2x Malachite green reagent was added and allowed to develop at room temperature for 60 minutes. Thereafter, the absorbance at 622 nm was measured immediately with a CLARIOstar plate reader (BMG Labtech, Germany).
Disulphide crosslinking
Following a 5-minute incubation at room temperature with 2 mM ATP (pH 7.0; Sigma) in a buffer containing 300 mM NaCl, 40 mM HEPES pH 7.5, 4 mM MgSO4, at a protein concentration of 10 μM, crosslinking was initiated by addition of copper phenanthroline (Santa Cruz) to a final concentration of 25 μM and allowed to proceed for 30 minutes, at which point the reaction was quenched with an equal volume of 300 mM NaCl, 40 mM HEPES pH 7.5, 100 mM EDTA. All crosslinking reactions were immediately further purified via size-exclusion chromatography.
NScc1 retention assay
To cleave the NScc1–CtSmc1 linker, disulphide crosslinked c-link complexes were incubated with 0.5 μg 3C protease for 30 minutes at 4 °C. Following cleavage, 22.5 pmol of c-link was immobilized on 60 μl Dynabeads His-Tag (Invitrogen) in wash buffer (150 mM NaCl, 40 mM HEPES pH 7.5, 5% Glycerol, 5 mM MgCl2, 0.01% IGEPAL-CA630, 20 mM Imidazole), washed three times to exclude unbound protein and divided equally between three 1.5 ml Eppendorf tubes. Release of NScc1 was then assayed by 3 consecutive washes in wash buffer supplemented with 0.1 mM nucleotide where indicated. Each wash step was performed at room temperature for a duration of 10 minutes. Beads were then resuspended in SDS loading buffer and bound proteins analyzed by SDS-PAGE followed by silver staining. Intensity of NScc1 retained was quantified using ImageJ (NIH), and normalized according to relative intensity of c-link bands in the nucleotide negative and positive reactions. Source gels are provided as supplementary data.
EM Grid Preparation
Quantifoil R1.2/1.3 300 mesh copper holey-carbon grids (Quantifoil Micro Tools GmbH) were glow discharged (EasyGlow, Pelco) for 30s at 30mA under a pressure of 0.25mbar. Immediately thereafter, 3 μl of sample (at a concentration of 1 μM, supplemented with 2 mM ATPγS, and 4 mM MgSO4) was applied to the carbon side, blotted (blot force and duration of 5 and 1s, respectively) and flash frozen in liquid ethane using a Vitrobot Mark IV (ThermoFisher) maintained at a temperature of 270K and a humidity of 100%.
EM Data Processing
Micrograph movies were recorded using a Titan Krios (FEI, ThermoFisher) operating at 300keV, equipped with a K2-Summit (Gatan, ThermoFisher) direct electron detector configured in counting mode. Dose, magnification, and effective pixel size are detailed in Table S2. Micrograph movie alignment, dose-weighting and summing was performed using MOTIONCORR250. Contrast-transfer function estimation was performed using GCTF51. Initial particle sets were picked with Gautomatch (Kai Zhang, MRC-LMB), extracted, and subjected to 2D classification in Relion 2.152. Resulting classes corresponding to protein complexes were subsequently employed as references for auto-picking in Relion 2.1. Following several iterations of 2D classification, classes exhibiting clear secondary structure features were selected for 3D classification, using a simulated density map generated from a hypothetical model of the Smc3–Smc1 heterodimer low-pass filtered to a resolution of 40 Å as a reference. After iterative rounds of 3D classification, well-defined classes were selected and global refinement was first performed unmasked, and the resulting maps employed to produce soft masks for a final masked refinement.
Cryo-EM Model Refinement
All models were refined by an initial round of rigid body modelling in Chimera53 followed by several iterations of real-space refinement (minimization, Atomic Displacement Factors and local refinement), using the ySmc3–yNScc1 and CtSmc1–yCScc1 crystal structures as references for geometry restraint generation, and manual correction in Phenix54 and Coot55 respectively. The Smc3 coiled-coil domain was manually corrected against a map filtered to 4.0 Å using Phenix Auto-Sharpen, which provided a more reliable trace for main-chain residues (Extended Data Fig. 2g)
Extended Data
Extended Data Fig. 1. Cohesin domain organization and purification of c-link complexes.

a, Domain organization and cartoon depiction of the cohesin complex. b, SDS-PAGE analysis of a representative c-link purification. Bands corresponding to each subunit are indicated. c, Size-exclusion chromatography analysis of c-link prior to (orange) and following 3C-mediated cleavage (yellow) of the NScc1–CtCSmc1 linker. Elution volume of molecular weight standards are shown. d, SDS-PAGE analysis of indicated fractions corresponding c, (c-link left of marker; 3C-treated c-link to the right). e, ATPase assays of wild-type (Wt) and Walker B (EQ) mutant c-link. A single experiment was performed at the indicated protein concentration. Data for the graphs in c and e are available as source data.
Extended Data Fig. 2. Cryo-EM data processing and validation.

a, Following initial 2D classification, several iterations of 3D classification were conducted. Classes are presented as 3D volumes. Percentages of particles sorted into each class are displayed below. The selected final 3D class is boxed, and was proceeded by a further round of 2D classification prior to masked global consensus refinement. b, A representative micrograph is shown. c, Angular distribution plot of final 3D consensus refinement. d, Fourier shell correlation plot. Final overall resolution is 3.2 Å (when FSC=0.143). e, Local resolution of the EM density map (Å) as computed by ResMap. f, EM density and modelled residues corresponding to catalytic motifs of the CtSmc1 ATPase domain, and the coiled-coils (RMSD 2.5). g, EM density and modelled residues corresponding to catalytic motifs of the Smc3 ATPase domain, and the coiled-coils (RMSD 2.5).
Extended Data Fig. 3. Comparative structural analysis of the cohesin ATPase.

a, Structural alignment-based superposition of the RecA N-lobes of apo CtSmc1–CScc1 (red) and ATPγS-bound ySmc1–CScc1 complex (grey; PDB code 1W1W). Cα root-mean-square deviation [RMSD] = 0.98 Å. b, The Smc3–NScc1 ATPγS complex (PDB code 4UX3). c, Relative motions of α-helices within the ctSmc1 ATPase upon ATPγS binding and head heterodimerization. d, Relative motions of α-helices within the ySmc3 ATPase upon ATPγS binding and head heterodimerization. e, The cross-links are positioned in loops between secondary structural elements. f, Structural details around the cross-linked disulfides. Smc3 N1204 and CtSmc1 L1160 are closely apposed in the modeled heterodimer (grey). Replacement of these residues by cysteine allows cross-linking without major distortions in the Smc heterodimer. The cystine disulfide bonds are indicated in yellow.
Extended Data Fig. 4. Nucleotide-induced conformational changes in SMC ATPases.

a, Structural alignment based on ATPγS-bound CtSMC1, the nucleotide-free form of Bacillus subtilis (Bs) SMC and the ATPγS-bound form of Geobacillus stearothermophilus (Gs) SMC. b, Nucleotide free (grey) and bound (green) Pyrococccus furiosis (Pf) Rad50 conformations. Nucleotide binding induces an ~35° C-lobe rotation. c, Nucleotide free (teal) form of Chaetomium thermophilium (Ct) Smc2 and ATPγS-bound form of CtSmc1 (red). d, Nucleotide free (blue) form of CtSmc4 and ATPγS-bound form of CtSmc1 (red). All structural superpositions were done using the SMC N-lobe.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank M. Hons and M. Karuppasamy (EMBL) for training K.W.M. in cryo-EM, M.W. Bowler (EMBL) for X-ray data collection, S. Cusack (EMBL) and D. Barford (MRC-LMB) for their support, and P. V. Sauer (UC Berkeley) for critical reading of the manuscript.
Funding: This work was funded by EMBL.
Footnotes
Reporting Summary statement. Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability. Cryo-EM reconstructions are deposited in the Electron Microscopy Data Bank (EMDB accession number: EMD-4614). The Smc3–Scc1–Smc1, and CtSmc1–CScc1structures are deposited in the Protein Databank (PDB accession numbers 6QPW and 6QPQ, respectively). Source Data for Fig. 2g and Extended Data Fig. 1c, e are available with the paper online.
Author contributions: K.W.M., L.Y. and D.P. conceived the study. K.W.M. and L.Y. designed c-link. K.W.M. and D.P. devised the crosslinking strategy. Y.L. crystallized CtSmc1–CScc1. K.W.M. and F.W. collected cryo-EM data. K.W.M. processed cryo-EM data. Molecular models were built and refined by K.W.M. and D.P. K.W.M. and D.P. wrote the manuscript with input from L.Y and F.W.
Competing interests: The authors declare no competing interests.
References
- 1.Yatskevich S, Rhodes J, Nasmyth K. Organization of Chromosomal DNA by SMC Complexes. Annu Rev Genet. 2019 doi: 10.1146/annurev-genet-112618-043633. [DOI] [PubMed] [Google Scholar]
- 2.Hassler M, Shaltiel IA, Haering CH. Towards a Unified Model of SMC Complex Function. Curr Biol. 2018;28:R1266–R1281. doi: 10.1016/j.cub.2018.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirano T. Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell. 2016;164:847–857. doi: 10.1016/j.cell.2016.01.033. [DOI] [PubMed] [Google Scholar]
- 4.Kim Y, Shi Z, Zhang H, Finkelstein IJ, Yu H. Human cohesin compacts DNA by loop extrusion. Science. 2019 doi: 10.1126/science.aaz4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davidson IF, et al. DNA loop extrusion by human cohesin. Science. 2019 doi: 10.1126/science.aaz3418. [DOI] [PubMed] [Google Scholar]
- 6.Nasmyth K. Cohesin: a catenase with separate entry and exit gates? Nat Cell Biol. 2011;13:1170–1177. doi: 10.1038/ncb2349. [DOI] [PubMed] [Google Scholar]
- 7.Haering CH, et al. Structure and stability of cohesin's Smc1-kleisin interaction. Mol Cell. 2004;15:951–964. doi: 10.1016/j.molcel.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 8.Gligoris TG, et al. Closing the cohesin ring: structure and function of its Smc3-kleisin interface. Science. 2014;346:963–967. doi: 10.1126/science.1256917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinshaw SM, Makrantoni V, Harrison SC, Marston AL. The Kinetochore Receptor for the Cohesin Loading Complex. Cell. 2017;171:72–84 e13. doi: 10.1016/j.cell.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petela NJ, et al. Scc2 Is a Potent Activator of Cohesin's ATPase that Promotes Loading by Binding Scc1 without Pds5. Mol Cell. 2018;70 doi: 10.1016/j.molcel.2018.05.022. 1134-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murayama Y, Uhlmann F. Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature. 2014;505:367–371. doi: 10.1038/nature12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munoz S, Minamino M, Casas-Delucchi CS, Patel H, Uhlmann F. A Role for Chromatin Remodeling in Cohesin Loading onto Chromosomes. Mol Cell. 2019;74:664–673 e665. doi: 10.1016/j.molcel.2019.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan KL, et al. Cohesin's DNA exit gate is distinct from its entrance gate and is regulated by acetylation. Cell. 2012;150:961–974. doi: 10.1016/j.cell.2012.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uhlmann F, Wernic D, Poupart MA, Koonin EV, Nasmyth K. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell. 2000;103:375–386. doi: 10.1016/s0092-8674(00)00130-6. [DOI] [PubMed] [Google Scholar]
- 15.Rowland BD, et al. Building sister chromatid cohesion: smc3 acetylation counteracts an antiestablishment activity. Mol Cell. 2009;33:763–774. doi: 10.1016/j.molcel.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 16.Beckouet F, et al. Releasing Activity Disengages Cohesin's Smc3/Scc1 Interface in a Process Blocked by Acetylation. Mol Cell. 2016;61:563–574. doi: 10.1016/j.molcel.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gruber S, et al. Evidence that Loading of Cohesin Onto Chromosomes Involves Opening of Its SMC Hinge. Cell. 2006;127:523–537. doi: 10.1016/j.cell.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 18.Srinivasan M, et al. The Cohesin Ring Uses Its Hinge to Organize DNA Using Non-topological as well as Topological Mechanisms. Cell. 2018;173 doi: 10.1016/j.cell.2018.04.015. 1508-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buheitel J, Stemmann O. Prophase pathway-dependent removal of cohesin from human chromosomes requires opening of the Smc3–Scc1 gate. The EMBO Journal. 2013;32:666–676. doi: 10.1038/emboj.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eichinger CS, Kurze A, Oliveira RA, Nasmyth K. Disengaging the Smc3/kleisin interface releases cohesin from Drosophila chromosomes during interphase and mitosis. EMBO J. 2013;32:656–665. doi: 10.1038/emboj.2012.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hopfner KP, Tainer JA. Rad50/SMC proteins and ABC transporters: unifying concepts from high-resolution structures. Curr Opin Struct Biol. 2003;13:249–255. doi: 10.1016/s0959-440x(03)00037-x. [DOI] [PubMed] [Google Scholar]
- 22.Locher KP. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat Struct Mol Biol. 2016;23:487–493. doi: 10.1038/nsmb.3216. [DOI] [PubMed] [Google Scholar]
- 23.Elbatsh AMO, et al. Cohesin Releases DNA through Asymmetric ATPase-Driven Ring Opening. Mol Cell. 2016;61:575–588. doi: 10.1016/j.molcel.2016.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber RG, et al. Impairing Cohesin Smc1/3 Head Engagement Compensates for the Lack of Eco1 Function. Structure. 2016;24:1991–1999. doi: 10.1016/j.str.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Murayama Y, Uhlmann F. DNA Entry into and Exit out of the Cohesin Ring by an Interlocking Gate Mechanism. Cell. 2015;163:1628–1640. doi: 10.1016/j.cell.2015.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu B, et al. ATP hydrolysis is required for relocating cohesin from sites occupied by its Scc2/4 loading complex. Curr Biol. 2011;21:12–24. doi: 10.1016/j.cub.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ben-Shahar T, et al. Eco1-Dependent Cohesin Acetylation During Establishment of Sister Chromatid Cohesion. Science. 2008;321:563–566. doi: 10.1126/science.1157774. [DOI] [PubMed] [Google Scholar]
- 28.Unal E, et al. A molecular determinant for the establishment of sister chromatid cohesion. Science. 2008;321:566–569. doi: 10.1126/science.1157880. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, et al. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell. 2008;31:143–151. doi: 10.1016/j.molcel.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Guacci V, et al. A novel mechanism for the establishment of sister chromatid cohesion by the ECO1 acetyltransferase. Mol Biol Cell. 2015;26:117–133. doi: 10.1091/mbc.E14-08-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ladurner R, et al. Cohesin's ATPase activity couples cohesin loading onto DNA with Smc3 acetylation. Curr Biol. 2014;24:2228–2237. doi: 10.1016/j.cub.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camdere G, Guacci V, Stricklin J, Koshland D. The ATPases of cohesin interface with regulators to modulate cohesin-mediated DNA tethering. eLife. 2015;4 doi: 10.7554/eLife.11315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hassler M, et al. Structural Basis of an Asymmetric Condensin ATPase Cycle. Mol Cell. 2019;74:1175–1188 e1179. doi: 10.1016/j.molcel.2019.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huis in 't Veld PJ, et al. Characterization of a DNA exit gate in the human cohesin ring. Science. 2014;346:968–972. doi: 10.1126/science.1256904. [DOI] [PubMed] [Google Scholar]
- 35.Soh YM, et al. Molecular Basis for SMC Rod Formation and Its Dissolution upon DNA Binding. Mol Cell. 2015;57:290–303. doi: 10.1016/j.molcel.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kulemzina I, et al. A Reversible Association between Smc Coiled Coils Is Regulated by Lysine Acetylation and Is Required for Cohesin Association with the DNA. Mol Cell. 2016;63:1044–1054. doi: 10.1016/j.molcel.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Chao WCH, et al. Structure of the cohesin loader Scc2. Nature communications. 2017;8 doi: 10.1038/ncomms13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diebold-Durand ML, et al. Structure of Full-Length SMC and Rearrangements Required for Chromosome Organization. Mol Cell. 2017 doi: 10.1016/j.molcel.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chapard C, Jones R, van Oepen T, Scheinost JC, Nasmyth K. Sister DNA Entrapment between Juxtaposed Smc Heads and Kleisin of the Cohesin Complex. Mol Cell. 2019;75:224–237 e225. doi: 10.1016/j.molcel.2019.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vazquez Nunez R, Ruiz Avila LB, Gruber S. Separate Compartments for Chromosome Entrapment and DNA Binding during SMC translocation. bioRxiv. 2018 [Google Scholar]
- 41.Burmann F, et al. A folded conformation of MukBEF and cohesin. Nat Struct Mol Biol. 2019;26:227–236. doi: 10.1038/s41594-019-0196-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams GJ, et al. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat Struct Mol Biol. 2011;18:423–431. doi: 10.1038/nsmb.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 44.Bowler MW, et al. MASSIF-1: a beamline dedicated to the fully automatic characterization and data collection from crystals of biological macromolecules. Journal of synchrotron radiation. 2015;22:1540–1547. doi: 10.1107/S1600577515016604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Svensson O, Malbet-Monaco S, Popov A, Nurizzo D, Bowler MW. Fully automatic characterization and data collection from crystals of biological macromolecules. Acta crystallographica Section D, Biological crystallography. 2015;71:1757–1767. doi: 10.1107/S1399004715011918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svensson O, Gilski M, Nurizzo D, Bowler MW. Multi-position data collection and dynamic beam sizing: recent improvements to the automatic data-collection algorithms on MASSIF-1. Acta crystallographica Section D, Structural biology. 2018;74:433–440. doi: 10.1107/S2059798318003728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta crystallographica Section D, Biological crystallography. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta crystallographica Section D, Biological crystallography. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCoy AJ, et al. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang K. Gctf: Real-time CTF determination and correction. J Struct Biol. 2016;193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheres SH. Processing of Structurally Heterogeneous Cryo-EM Data in RELION. Methods Enzymol. 2016;579:125–157. doi: 10.1016/bs.mie.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 53.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 54.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica. Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 56.Schrodinger L. The PyMOL Molecular Graphics System, Version 2.0. 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.