

Abstract
Super-resolution fluorescence microscopy has become an important catalyst for discovery in the life sciences. In STimulated Emission Depletion (STED) microscopy, a pattern of light drives fluorophores from a signal-emitting on-state to a non-signalling off-state. Only emitters residing in a sub-diffraction volume around an intensity minimum are allowed to fluoresce, rendering them distinguishable from the nearby, but dark fluorophores. STED routinely achieves resolution in the few tens of nanometers range in biological samples and is suitable for live imaging. Here, we review the working principle of STED and provide general guidelines for successful STED imaging. The strive for ever higher resolution comes at the cost of increased light burden. We discuss techniques to reduce light exposure and mitigate its detrimental effects on the specimen. These include specialized illumination strategies as well as protecting fluorophores from photobleaching mediated by high-intensity STED light. This opens up the prospect of volumetric imaging in cells and tissues with diffraction-unlimited resolution in all three spatial dimensions in living samples.
Keywords: Fluorescence Microscopy; Optical Imaging; Super-Resolution Microscopy; Nanoscopy; Stimulated Emission Depletion Microscopy; STED, Protected STED; Live Imaging; Photodamage
1. Introduction
Light microscopy is an extremely valuable tool for the life sciences. Its broad usefulness is underscored by the following features: (1) Sample preparation is often straightforward. (2) Light microscopy is uniquely suited to attain molecular contrast and, in multicolour measurements, to visualize mutual spatial arrangements. (3) Fluorescence microscopy boasts exquisitely high signal-to-background ratio with detection efficiency ultimately down to the single-molecule level. (4) Optical sectioning facilitates high-contrast volumetric imaging. (5) Light microscopy allows analysing living systems. These features set optical microscopy apart from other valuable biological imaging approaches, such as electron microscopy (EM), which has exquisite spatial resolution but is incompatible with imaging of living specimens and offers more limited access to molecular information.
1.1. The diffraction limit
Conventional light microscopes are fundamentally limited in their spatial resolution by diffraction of light waves to about half the wavelength of light or ~200 nm1–4. Accordingly, features residing closer to each other than this diffraction resolution limit cannot be discerned, thus preventing the analysis of fine structural details. Many biologically relevant entities, such as cellular organelles or (macro)molecular machineries are assembled on much finer spatial scales. The diffraction limit only applies to “lens-based” microscopy, i.e. methods that operate in the optical “far field”, where the components of the imaging system are positioned at distances from the structure of interest that are large compared to the wavelength of light. In contrast to “near-field” methods5,6, far-field imaging permits analysing structures inside cells or tissues non-invasively.
1.2. Super-resolution microscopy
Several powerful methods have emerged that break the diffraction resolution limit. Diffraction-unlimited optical “nanoscopy” or “super-resolution” fluorescence microscopy approaches offer spatial resolution that is no longer conceptually limited7–14. These methods routinely achieve resolution of tens of nanometers in fixed and living samples and now reach into the single-digit nanometer domain15,16. They can be broadly categorized into “coordinate-targeted” methods, such as STimulated Emission Depletion (STED) microscopy, which is the focus of this review, and single-molecule based “coordinate-stochastic” methods. Both rely on preparing fluorophores within a diffraction-limited zone in distinguishable molecular states and reading them out sequentially7,17. Any appropriately controllable state transition that turns fluorophores “on” and “off” or, more generally speaking, that renders them distinguishable is suited.
In the single-molecule based methods8, a sparse subset of fluorophores is activated in each imaging frame and the location of these spatially well separated single molecule emission events is pinpointed with high precision. Thus, a super-resolved image builds up cumulatively from many consecutive imaging frames. These methods include STochastic Optical Reconstruction Microscopy (STORM)18, (Fluoroescence) PhotoActivation Localization Microscopy (PALM,19; fPALM,20), and a series of other variants which differ in the precise mechanism of fluorophore activation, e. g. Direct-(d)STORM and Ground State Depletion microscopy followed by Individual Molecule return (GSIDM)21,22. Also distinguishing between transiently bound and diffusing molecules in (u)PAINT ((universal) Point Accumulation for Imaging in Nanoscale Topography23–25) and DNA-PAINT26 generates the required sparse highlighting of target molecules.
In coordinate-targeted nanoscopy, a pattern of light determines the regions where fluorescence is allowed to originate from, i.e. where fluorophores reside in a signal-emitting “on”-state, on spatial scales finer than the diffraction limit (Fig. 1). The prototypical example is STED microscopy27–29. Here, a light pattern transiently silences fluorophores by stimulated emission except for those residing in the immediate (sub-diffraction vicinity of an intensity minimum, ideally an intensity “zero”. The STED light pattern is spatially overlapped with the excitation light.
Figure 1.

STED nanoscopy. a Schematic of molecular states for nanoscopy. b Top: Energy diagram. S0, singlet ground state; S1, first excited state; τfluo, fluorescence lifetime. Curved arrows: photons. Bottom: Excitation and emission spectra of typical fluorophore with wavelengths for excitation and STED (vertical lines) and detection window. c Left: In conventional laser-scanning microscopy, all fluorophores within a diffraction-limited laser focus are excited simultaneously (orange stars) and are hence indistinguishable. Middle and right: In STED nanoscopy, a light pattern superimposed with the excitation focus transiently silences fluorophores (black stars) except for those located at an intensity zero, thus making fluorophores distinguishable on sub-diffraction length scales. IS, saturation intensity; Imax, maximum STED intensity. d Timing in STED nanoscopy. The scale is zoomed into an individual excitation (cyan) and STED (red) pulse pair. Fluorescence signal time course (orange) is indicated for the location of the STED light intensity zero. Signal integration time is typically (tens of) μs at each scan position. Colour code applies to all figures.
In addition, several techniques have pushed the limit of resolution while still remaining conceptually limited by diffraction These do not require on/off switching of fluorophores and hence often offer fast and gentle imaging conditions where only moderate resolution increase over conventional microscopy is required. For example, 4Pi-30 and I5M-31 microscopy increase the resolution along the optical (z-)axis by exploiting interferometric effects. Structured illumination microscopy in its linear variant yields a resolution enhancement by a factor of two over conventional widefield imaging32,33. However, in its nonlinear variant, conceptually unlimited resolution increase is achieved via saturation of fluorophores34,35.
An interesting alternative super-resolution approach has recently emerged. “Expansion microscopy” increases effective spatial resolution by embedding in and linking the sample to a swellable hydrogel that is then isotropically expanded36. Here, rather than increasing optical instrument resolution, the physical separation between fluorophores in the sample is increased. Effective resolution is thus increased, presently down to ~25 nm on conventional light microscopes37–39. By design, this method is limited to fixed samples.
In this review, we focus on strategies to improve STED microscopy. We will first briefly recall some of the basic features of the interaction of light with biological matter in fluorescence light microscopy. These considerations apply to diffraction-limited as well as super-resolution microscopy but merit special attention in nanoscale optical imaging because they are more likely to become limiting factors here.
1.3. Interaction of light with biological samples and fluorophores
In fluorescence microscopy, absorption of an excitation light photon by a fluorophore followed by emission of a fluorescence photon is the desired mode of interaction between light and fluorophores (Fig. 1b)40. Here, fluorophores are excited from the ground state (typically a singlet state S0) to the first excited state S1. The excited state lifetime of fluorescent labels is typically a few nanoseconds (~1-4 ns). The emitted fluorescence light is of longer wavelength because some energy is dissipated via vibrational transitions, leading to a “Stokes shift” between the maxima of excitation and emission spectra.
Direct absorption of light by biological specimens
Many cellular constituents absorb light, which is usually undesired in fluorescence microscopy. Often, absorption cross sections rise for shorter wavelengths. For example, absorption by proteins drastically increases in the blue and UV range due to the presence of aromatic amino acid side chains. Similarly, nucleic acids absorb in the UV. Many further cell and tissue resident chromophores, such as e. g. hemoglobin or cytochrome c, display distinct absorption spectra. All of these lump together to a wavelength-dependent tissue absorption coefficient41,42 that decreases from the UV/blue to the red/near infrared range, such that long wavelengths prove advantageous for imaging.
Autofluorescence
Along the same lines, some of the cellular constituents not only absorb light but also act as endogenous fluorophores. Although they typically exhibit low quantum yield (i.e. low probability of emitting a fluorescence photon upon excitation) they may collectively contribute a significant undesired background signal, reducing signal-to-background ratio in fluorescence imaging. Such autofluorescence is often excited more efficiently with blue than with red light, thus again rendering the red to near infrared spectral region an attractive choice for imaging40. Endogenous fluorophores typically exhibit short fluorescence lifetime, such that time-resolved fluorescence detection can be useful for suppressing this unwanted background.
Scattering and aberrations
Under ideal conditions, imaging light would traverse the biological specimen in an unperturbed manner as in a perfectly transparent and homogeneous sample of the appropriate refractive index, thus producing an ideal focus in the sample or on the detector. However, scattering of light at refractive index inhomogeneities in biological samples can be a major factor limiting imaging performance in diffraction-limited microscopy but even more so in super-resolution microscopy, especially when aspiring to image deep in tissue samples. While the precise dependence on wavelength varies with the size of the scatterers, there is overall a steep decline of scattering cross section with increasing wavelength42–44.
Similarly, a mismatch between sample refractive index and the optical design of the objective lens including immersion, as well as spatial variation of the sample’s refractive index may induce optical errors (aberrations), deteriorating imaging performance.
Spectral window for imaging
Absorption by most cellular constituents decreases with wavelength, just as scattering does. The absorption cross section of water rises in the near infrared region, thus defining in between a spectral “window” in the red to near infrared region that is often considered desirable for optical imaging45.
Mechanisms of sample damage
Absorption of light by molecular constituents of cells and tissues or fluorescent labels may lead to physiological or structural alterations of the sample which are particularly of concern when imaging living specimens. The precise mechanisms of sample damage depend on the characteristics of the light exposure. They include direct absorption with covalent bond breakage and radical formation, especially for short wavelength light and deposition of excessive energy leading to thermal damage.
Photobleaching
Fluorophores mediate an important and often dominant part of photodamage. Mechanisms of photobleaching include excitation of fluorophores to higher lying molecular states Sn. From there, they are likely to transition to triplet states Tm (“inter-system crossing”) or even dissociate in case they are excited to a non-binding state46 (Fig. 3a). As fluorophores bleach, radicals are generated that may lead to a reaction cascade involving reactive oxygen species, such as singlet oxygen. These compounds constitute a redox burden and may react with cellular constituents in the vicinity, damaging molecules important to the cell’s function. Ultimately this may overwhelm cellular redox buffering and repair mechanisms and lead to irreversible damage and cell death.
Figure 3.

Strategies to reduce light exposure and photobleaching in STED nanoscopy. a Dark/triplet state relaxation (T-Rex). Energy schematic for the relevant molecular states in STED microscopy. Fluorophores normally cycle between the ground state S0 and the first excited state S1 (same colour code for transitions as in Fig. 1). Excited state absorption drives fluorophores to higher lying molecular states Sn. Fluorophores undergoing intersystem crossing (ISC) to the triplet manifold may also absorb excitation and/or STED light and are particularly prone to chemical reactions, including radical generation. Residence times in triplet states are long (μs) compared to lifetimes of singlet excited states (ns). Dark (or triplet) state relaxation allows fluorophores to relax back to the singlet manifold. b RESCue STED. Left: Sample structure (shaded areas) with colour code for light exposure at each scan position (pixel). Right: Three exemplary photon counts as a function of time at different pixels. Blue: lower threshold is not reached at the decision time. Excitation and STED beams are turned off. Yellow: enough photons are collected to extrapolate from the readout time to the full pixel dwell time with the desired signal-to-noise ratio. Lasers are turned off. Pink: the full pixel dwell time is used for collecting the signal. c DyMIN STED. Three exemplary light exposure sequences (top to bottom) at individual pixels. Position 1: No signal is collected in diffraction-limited mode. Lasers are turned off. Position 2: Signal is present in diffraction-limited mode. STED is turned on at the first power level. Since no more signal is present, a further increase in STED power would not yield additional information. Position 3: STED power is successively stepped up to the maximum value to separate fluorophores. Stars: fluorophores. Dark orange: normalized on-state probability defining resolution; light orange: resolution in previous intensity step; r, scan position; I, intensity. d In MINFIELD STED, the field of view is restricted to a region smaller than the extent of the doughnut beam, such that the region of interest is spared from exposure to the STED-light intensity maxima.
Compared to diffraction-limited microscopy, super-resolution imaging with its on/off switching cycles and increased light doses and intensities puts an additional burden on the sample and a higher demand on fluorophores. Clearly, if perturbation of the sample is overwhelming, no informative imaging data can be collected. But also in less severe situations, bleaching reduces the number of fluorophores available for the measurement and thus decreases signal-to-noise ratio (SNR) and hence the meaningful resolution that can be dialled into a STED measurement47,48. Moreover, in order to decode the spatial distribution of target molecules, they must be decorated with fluorescent labels reporting on their positions. The fraction of target molecules associated with a functional fluorophore determines whether they are adequately sampled. Taking into account the sampling theorem, this labelling density is thus a direct determinant for the achievable biologically meaningful resolution, which is again jeopardized by photobleaching.
While there is general consensus that reduction of light exposure and photobleaching is highly desirable49, systematic studies on the effects of light exposure on (living) biological specimens in super-resolution imaging are relatively sparse. For example, the Sauer group has analysed light exposure effects in single-molecule based super-resolution imaging50. For STED microscopy, the Bewersdorf group has recently obtained encouraging results, indicating that a parameter regime can be found where high resolution data can be obtained from living cells such that a substantial fraction of cells remain viable for prolonged periods of time after imaging51. These authors also give valuable advice for reducing phototoxicity in live-cell STED nanoscopy. Promising developments are under way that reduce light exposure and/or photobleaching in STED microscopy. For example, a combination of reduced photobleaching and increased on/off state contrast in an approach called “protected STED”52 enabled decoding the structure of a neuron in a tissue volume of a living brain slice with 3D diffraction-unlimited resolution (Fig. 2). Excitingly, in single-molecule based nanoscopy, efficiency of photon usage previously deemed impossible has been achieved with the MINFLUX concept15,53.
Figure 2.
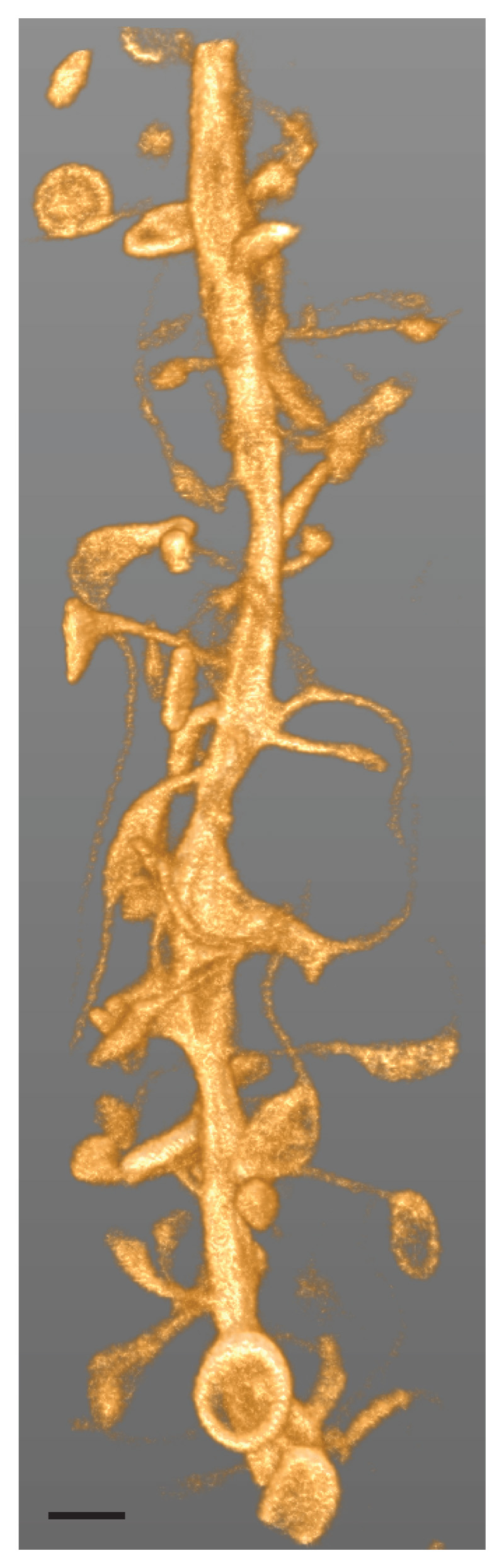
Diffraction-unlimited imaging of a portion of a neuronal dendrite in living brain tissue with resolution increase in xy- and z-directions. Volume rendering of a 3D-image stack comprising 56 z-slices recorded with protected STED. Actin is labelled by an intracellularly expressed nanobody fused to a reversibly photoswitchable fluorescent protein (rsEGFP2) after viral delivery in an organotypic hippocampal brain slice. A multitude of dendritic spines, the postsynaptic compartments of excitatory synapses, emanate from the dendritic shaft. Spine necks exhibit sub-diffraction widths. In some spine heads, peculiar ring-like actin structures can be discerned. Scale bar: 1 μm. Reproduced with permission from Ref. Danzl et al.52.
1.4. Scope of this review: maximizing performance in STED nanoscopy
Super-resolution imaging requires special attention to several factors in order to generate a faithful representation of the specimen. These include sample preparation and labelling approaches, instrument performance, and the specifics of the measurement process itself. There are a range of excellent reviews on STED microscopy that cover various important aspects, e.g. references:7,11,54–59.
In this review, we first give some basic considerations for optimizing STED measurements. We then put an emphasis on strategies to reduce light exposure as well as photobleaching. Whether perturbation of the sample can be kept at acceptably low levels decisively influences to what extent meaningful information can be obtained from a measurement and the resolution that can be dialled into a STED experiment, especially for obtaining volumetric datasets of biological specimens with super-resolution in all three spatial dimensions. We further discuss correction of optical errors and parallelized, non-point scanning implementations. The latter promise increased acquisition speed of large sample areas and volumes.
2. Principle of STED nanoscopy
In STED nanoscopy, the excitation light distribution is spatially overlapped with an additional STED light pattern that drives fluorophores from the excited state S1 to the ground state S0 via stimulated emission, such that spontaneous emission of a fluorescence photon cannot take place27,28 (Fig. 1). The STED light thus drives fluorophores to a non-signalling off-state (Fig. 1a)7. The photon generated by stimulated emission is of the same wavelength as the STED laser and blocked from detection by optical filters. This additional S1 → S0 pathway effectively outcompetes the spontaneous transition to the ground state60 if the STED light intensity is sufficiently high. The intensity where a certain fraction of fluorophores (typically defined as 50 %) are turned off is called saturation intensity IS. It is specific for each combination of fluorophore and STED light characteristics, including STED wavelength.
The STED light distribution features maxima and, crucially, minima that ideally have zero intensity. The shape of the STED light pattern is itself limited by diffraction. However, by increasing the overall power in the STED beam, the saturation intensity is reached near the intensity zero and the STED light pattern is thus able to define on-state and off-state regions on a spatial scale that is finer than the diffraction limit. It sets the coordinates where fluorescence is allowed to originate from directly in the sample.
In most practical realizations, the excitation light takes on the shape of a diffraction-limited laser focus and the STED light distribution features a single intensity minimum with a “doughnut”-shaped light distribution in the focal region (Fig. 1c), resulting in a single-point scanning implementation. Such a doughnut-shaped focus can be created by a 2π-helical phase modulation of the incoming wave front in the back focal plane of the objective lens61. Whereas the doughnut beam only increases resolution in the focal plane (xy-direction), an additional z-STED light pattern (“z-doughnut” or “bottle beam”, created by a π-phase retardation of the central portion of the beam)29 may be employed to also increase resolution along the optical axis (z-direction). Most practical implementations of single-point scanning STED microscopes are based on a confocal design for facile optical sectioning, but other configurations, such as two-photon (2P) excitation combined with STED, have also been implemented62–66. However, a particular geometry is not part of the STED concept. Resolution increase in the z-direction can e. g. also be reached with a more elaborate, interferometric 4pi-STED approach67, which has yielded isotropic 3D STED resolution68. The STED light pattern may also be implemented with other light distributions, as long as they feature appropriate intensity zeros, including e. g. highly parallelized or light-sheet based approaches, as discussed in sections 9 and 10.
Resolution in STED nanoscopy
The smallest distance d0 that can be resolved in a conventional light microscope is approximately where NA is the numerical aperture of the objective lens and λ the wavelength of light. In STED mode, one may approximate the STED light intensity profile around the minimum as a parabola and assume exponential decay of fluorescence ability as a function of STED light intensity. This directly yields a smallest resolvable distance with Imax being the applied peak intensity47,61,69,70. Note that this expression for resolution is no longer fundamentally limited but approaches zero for large values of Imax with the lower bound effectively being set by the size of individual fluorophores (~ 1 nm). Resolution only scales with the square root of Imax, such that increasing resolution by applying more STED power becomes progressively harder. This sets practical limits to the achievable resolution in view of photobleaching and finite quality of the STED intensity zero. In fact, the highest resolution STED measurements have been achieved with extremely photostable nitrogen vacancy centers71.
Beyond this simple consideration, the spatially varying on/off state contrast generated by the STED light pattern directly translates to the effective point spread function (PSF) of the STED microscope (taking also the excitation and detection point spread functions into account)47,48,52. The effective PSF corresponds to the instrument response to a point-like emitter in the sample. For example, a diffraction-limited pedestal is created in the effective PSF if fluorophores in designated off-regions have a finite probability for residing in the on-state, e. g. due to spontaneous emission before full action of the STED light or erroneous excitation by STED light from S0 to S1 in a process called “Anti-Stokes” excitation. Convolution of such an imperfect effective PSF with the sample structure during image formation may lead to severe degradation of (super-resolution) signal-to-noise ratio and complete loss of super-resolution information in densely packed areas52. Therefore, the achieved on/off state contrast is a crucial determinant for performance of STED and other coordinate-targeted super-resolution modalities and is distinct from the common notion of “image contrast” in microscopy.
Timing considerations
The spatially varying on/off state contrast is “written” into the molecular states of the fluorescent labels within a time period shorter than the excited state lifetime of the fluorphores (Fig. 1d). This (sub-) nanosecond timescale is fast compared to the processes we commonly address in biological measurements. STED can hence be used to read out extremely fast biological processes and imaging rates of 125 frames per second have been achieved, limited only by the rate of photon emission from the sample72. Even diffusion of a small molecule in solution through a volume corresponding to the diffraction-limited focus generated by a high-NA objective takes place on the scale of (tens of) microseconds. Therefore, movement of freely diffusing fluorophores does not lead to appreciable blurring of the locally and “instantaneously” created on/off state contrast. Accordingly, STED is suited to assess the distribution of diffusing dye molecules, both intracellularly73 and extracellularly74. Similarly, in STED-Fluorescence Correlation Spectroscopy (STED-FCS), the STED process is used to confine the observation volume for diffusion measurements to sub-diffraction spatial scales75. Signal integration time at each scan position (pixel dwell time) is typically in the (tens of) μs range.
Most STED implementations rely on pulsed lasers for excitation and STED. Excitation pulses are chosen considerably shorter than the excited state lifetime and typically have durations of up to ~100 ps. For 2P-excitation STED microscopy, typically femtosecond excitation pulses are employed, just as in conventional 2P-microscopy76. Optimization over the years have shown that STED pulses with a duration of hundreds of ps to ~1 ns are advantageous77, especially if combined with “time gating” of fluorescence detection to maintain high state contrast, as discussed in the next section. This regime mitigates high peak photon fluxes that may lead to unwanted photophysical effects and excessive photobleaching. In fact, STED pulses were originally obtained from mode-locked femtosecond lasers and performance increased substantially when pulse stretching via high refractive index glass rods and long optical fibers (~100 m) was introduced. Nowadays, pulsed fiber lasers with the appropriate pulse length are a popular choice.
The excitation pulse sets the time when fluorophores are transferred to S1. The STED pulse must arrive at the same time as the excitation pulse at the sample in order to drive the transition S1 → S0. Therefore, excitation and STED pulses must be carefully synchronized.
In continuous wave (CW) STED microscopy, STED is implemented with a CW-laser but the excitation laser typically operates as a pulsed laser78,79. The CW STED laser obviates the need for pulse synchronization. However, it also entails that in the gaps between excitation pulses the sample is exposed to STED light that does not contribute to image formation. The pulsed excitation again serves to set the time when fluorophores are transferred to the excited state, as required for time gating of fluorescence detection.
Time gating of fluorescence detection
With a precisely defined time point for transfer to S1, the opportunity arises to synchronize fluorescence detection with excitation, detecting only photons emitted in a selected time window of several nanoseconds duration (Fig. 1d)80–82. This is attractive because with long (~1 ns) STED pulses or CW STED, in the initial phase of the action of the STED light, there is a finite probability for spontaneous emission also in designated off-regions. This decreases on/off state contrast and hence signal-to-background ratio and resolution. With time-gated detection, fluorescence photons are collected starting only after full action of the STED light, e. g. ~1 ns after each excitation pulse. Accordingly, photons that were spontaneously emitted in designated off regions before full action of the STED light are preferentially rejected, thus increasing on/off state contrast. The detection window is closed after most of the fluorescence emission has taken place (e. g. after ~10 ns, corresponding to a few fluorescence lifetimes). Time gating can also be helpful for rejecting autofluorescence and, especially in low repetition rate implementations, noise from technical sources or stray light.
Multi-colour and co-localization measurements
Super-resolution optical imaging is uniquely positioned to investigate spatial relationships between different molecules. In STED microscopy, combinations of two77,83, three84,85, or four86 fluorophores are available that can be addressed with the same STED beam but distinguished in their emission and/or excitation properties. In this configuration, one and the same STED light intensity minimum sets the fluorescence emission coordinates for all fluorophores simultaneously, thereby abrogating all alignment and magnification errors between colour channels. This facilitates high-confidence co-localization studies with minimized systematic errors.
Generalization of the concept
The STED approach has been generalized to any appropriately light-addressable molecular transition making fluorophores distinguishable in the framework of the “REversible Saturable Optical Fluorescence Transitions” (RESOLFT) concept69,87–89. Specifically, switching between the metastable on- and off-states of dedicated reversibly photoswitchable fluorophores has made coordinate-targeted nanoscopy at low light intensities with long observation times possible90. Compared to STED, the long timescales associated with photoswitching (commonly hundreds of microseconds photoswitching time at each scan position) bear the advantage that low light intensities are sufficient to drive the off-transition but come with the drawback of reduced imaging speed. The ~100 μs- to ms-timescale for generating state contrast means that this approach at present is not suitable for imaging with rapidly diffusing molecules as labels. Reversibly photoswitchable fluorescent proteins (RSFPs)91–93 and fluorescent dyes94–97 are undergoing intense development with intriguing prospects for the future.
3. Basic considerations for optimizing performance in STED nanoscopy
STED nanoscopy is straightforward in terms of data acquisition and analysis as it directly generates super-resolved images without the need for sophisticated data processing. However, for satisfactory performance it needs care in instrument and experiment design. While imaging of a single or a few planes with resolution increase only laterally is typically easy to accomplish, requirements in terms of repeated imaging capability and reduction of sample light exposure are much more stringent if one aspires to reconstruct an entire volume of a cell or tissue with resolution enhancement in all three spatial directions.
In particular, the following basic considerations help avoid loss of signal and excessive fluorophore bleaching.
-
(1)
The major factor determining whether fluorophores residing in a designated on-region generate signal is the quality of the intensity zero. Any non-zero STED light intensity Imin at the minimum effectively turns off fluorophores. For example, if Imax/IS ≈ 100 for ~10-fold resolution increase, already 1% of peak intensity leaking into the intensity minimum would mean Imin = IS, reducing fluorescence signal by half. Any loss of signal requires additional excitation intensity or integration time to recover the signal-to-noise ratio, increasing light burden on the sample potentially up to a parameter regime where excessive photobleaching effectively precludes recording of high resolution data. The quality of the intensity minimum of an xy-STED doughnut is critically influenced by (i) the polarization of the STED light, which must be perfectly circular and match the helicity of the phase ramp used to generate the doughnut-shaped light distribution; (ii) sample-induced scattering of STED light into the intensity minimum, limiting tissue penetration; (iii) optical aberrations of the STED beam induced by the optical system or the sample; (iv) and a spectral width of the STED laser beyond ~10 nm may become problematic as different wavelengths may form their intensity minima at slightly offset spatial positions, eliminating an overall intensity zero. For other STED light distributions, different considerations apply. The quality of the intensity zero of a z-STED pattern (“z-doughnut”) is exquisitely sensitive to spherical aberration, an optical error that frequently arises due to refractive index mismatch between the sample and the immersion medium/objective lens design.
-
(2)
State cycling takes place in regions where both excitation light and STED light are present. In a perfectly aligned STED microscope, the intensity minimum of the STED light pattern and the intensity maximum of the excitation focus coincide. Any slight misalignment causes the excitation maximum to coincide with regions of higher STED intensity, leading to loss of signal, excessive state cycling, and photobleaching.
-
(3)
The STED process is exquisitely sensitive to timing of excitation and STED pulses on the sub-ns timescale. Incorrect timing or timing jitter leads to reduced action of the STED light, thus sacrificing state contrast and resolution for a given STED light exposure. Equally, correct setting of detection time gating maximizes state contrast and hence information for a given light dose.
-
(4)
The STED wavelength is typically chosen in the long wavelength tail of the emission spectrum of the fluorophore. The choice of STED wavelength is governed by two opposing factors: wavelengths close to the emission maximum increase the cross section for stimulated emission98 and reduce the intensity required to achieve a given resolution. However, with decreasing STED wavelength, the probability for directly exciting fluorophores (S0 → S1) via Anti-Stokes excitation increases, thus deteriorating on/off-state contrast.
-
(5)
STED light may drive additional photophysical processes depending sensitively on the fluorophore’s energy structure, which is, however, typically not well characterized spectroscopically99. STED light may excite a fluorophore from the ground state in a single- or two-photon process or drive excited state absorption from S1 to higher lying molecular states Sn. Fluorophores may transition to triplet states via inter-system crossing where they may also absorb STED light (triplet state absorption) and may populate higher lying triplet states Tm. Therefore, resistance to photobleaching and overall imaging performance may differ considerably between STED vs. diffraction-limited mode and among fluorophores. The choice of fluorophore requires testing under STED conditions, for which further systematic studies would be desirable. Long STED pulses (~1 ns) lead to lower peak pulse intensities, mitigating unwanted processes that depend non-linearly on intensity, such as two-photon transitions driven by the STED laser.
-
(6)
Last but not least, sample light exposure, photobleaching and thus overall imaging performance can be optimized most easily by a prudent choice of imaging parameters. In STED microscopy, resolution scales with the square root of the applied intensity. Hence, it gets quadratically harder to “squeeze out” additional resolution and marginal benefit of higher STED power decreases. Following the sampling theorem, increased resolution also requires smaller step size for scanning (smaller pixel size). Light exposure scales quadratically with the inverse pixel size for acquiring a single plane. Similarly, excessive oversampling with smaller than necessary pixel size should be avoided.
4. Labeling is a decisive factor for STED microscopy performance
4.1. Sample preparation and choice of fluorophores
The quality of sample preparation profoundly influences the imaging outcome. Evidently, any shortcomings of fixation or labeling procedures, such as nanoscale distortions of the sample structure from chemical fixation, are much more evident and more limiting in super-resolution imaging than in diffraction-limited microscopy and often warrant optimization and specialized protocols. Sufficiently high labelling density is central to resolving a structure of interest. In addition, the labelling strategy may entail a “linkage error”, i.e. a spatial displacement between target molecules and fluorophores. Specifically, the size of primary and secondary antibodies (each ~ 10 nm) in immuno-stainings leads to errors that are substantial on the scale of the resolution currently achieved in super-resolution imaging. Smaller probes, like e. g. nanobodies, promise more accurate reporting on target molecule positions100. Specificity of labeling has a major influence on signal-to-background ratio, which is a key determinant of STED performance.
Fluorescent proteins, like green fluorescent protein (GFP) have been employed in STED101. However, broadly speaking, high performance synthetic fluorescent dyes offer higher photostability and photon budgets, i.e. number of emitted photons before photobleaching, than fluorescent proteins. Therefore, it is advisable to use fluorescent dyes as labels for STED if possible. Also for live imaging, an increasing palette of synthetic dyes is becoming available102–106. These can be targeted to specific molecules of interest via a range of methods, such as SNAP tag107, Halo tag108, click chemistry109, or small peptide tags110. Fluorescent proteins can also be highlighted with dye-coupled specific antibodies or nanobodies111.
Imaging buffers that scavenge reactive oxygen species may further improve performance in STED microscopy51,112. An intriguing development are “self-healing” dyes with protective groups113–118.
4.2. Replenishment of fluorophores facilitates repeated imaging
A simple strategy for mitigating the effect of photobleaching on the imaging outcome is to replenish bleached fluorophores during the imaging process. Dissociation of affinity binders with moderate affinity frees the binding site after photobleaching for another binder carrying a new label, thus constantly turning over flurophores. A good example is the actin binding peptide Lifeact119, which has been used to reconcile molecular specificity with replenishment of fluorophores in STED imaging of neuronal structures120. Similarly, replenishing membrane stains profit from fluorophore turnover121. Fluorophore-conjugated DNA-oligonucleotides that bind to complementary barcoding sequences introduced into the sample during labelling can also be used for exchanging fluorophores122–125.
Fluorophore replenishment is particularly easy to implement if fine morphological features, rather than molecular arrangements, are of interest. A prototypical example is STED-based analysis of the elaborate shapes of dendritic spines, the postsynaptic specializations of many excitatory synapses in the central nervous system, in living neurons expressing a fluorescent protein in the cytosol73. Similarly, single neurons or other cells can be accessed in whole cell mode with a patch clamp pipette filled with a dye containing solution.
A similar approach can also be applied for staining the extracellular rather than intracellular spaces. Here, cellular structures are demarcated as “shadows” in the bath of extracellularly applied dye, hence the term “SUper-resolution SHadow Imaging” (SUSHI)74. This has yielded fascinating images of the architecture of neuronal tissue. Also here, bleached dye molecules are simply substituted with fresh ones by diffusion.
It should be noted that replenishment of fluorophores does not reduce photobleaching, but rather the effect of photobleaching on image quality. If fluorophores bleach, they create the same radicals, metabolic burden and photochemical damage as with other labelling strategies, albeit with different implications for intra- and extracellularly generated toxic products.
In these approaches, sample light exposure remains unaltered in first approximation. Therefore, direct absorption of the imaging light by the tissue is still equally of concern. However, in some cases, such as bath application of fluorophores, the local concentration of fluorophores can be set very high, such that low excitation power and/or short pixel dwell times are sufficient to collect enough signal.
5. Dark/triplet state relaxation increases the photon budget
During repeated excitation and (stimulated) emission cycles, fluorophores may transition to the triplet manifold by inter-system crossing (Fig. 3a). Fluorophores residing in triplet states are “dark” and do not contribute to signal generation. However, they are typically not inert. They may still absorb excitation or STED light, causing excitation to higher lying triplet states, and undergo chemical reactions46. A typical outcome is the formation of radicals that react with cellular targets and damage them permanently.
Evidently, it is desirable to keep fluorophore population in triplet/dark states low. Triplet states are typically long-lived and the fraction of fluorophores residing in dark states depends on the rates at which they are pumped there and at which they relax back to the singlet manifold, which differ between fluorophores for given light exposure.
There is a simple and general strategy to minimize dark/triplet state population: Allowing time in the imaging procedure for fluorophores to relax back to the singlet manifold, with relaxation timescales on the order of ~1 μs. This is applicable both to diffraction-limited126 and super-resolution STED imaging127 and leads to substantial signal increase and reduced bleaching.
Strategies to increase dark state relaxation include (i) low repetition rate excitation, (ii) fast scanning with accumulation of the signal over individual image lines or frames that are repeatedly queried, and (iii) dedicated short breaks during image acquisition without light exposure.
Low repetition rate gives fluorophores time to relax between individual excitation events. The original implementation demonstrated a significant improvement at 250 kHz over 80 MHz pulse repetition rate127, albeit at correspondingly increased acquisition times. However, also higher, more user-friendly pulse repetition rates in the low to ~20 MHz range seem to offer an improvement over e. g. the 80 MHz typically dictated by mode-locked titanium:sapphire lasers.
Fast scanning has been implemented with a range of technologies: In STED, resonant mechanical scanners have been employed early on79,128, achieving video-rate nanoscopy. Here, lines are queried repeatedly and signal is accumulated. Scanning without mechanically moving parts further increases scan speed. Acousto-optical scanning129, where scan speed is limited by travelling times of acoustic waves, enabled triplet state relaxation in conventional 2P-microscopy. Even faster scanning is possible via electro-optical scanners, such that signal can be accumulated frame-wise with maximized triplet state relaxation until the desired signal-to-noise ratio is reached72.
Even with a conventional scanner based on galvanometric mirrors, dividing up the total acquisition time for each scan position by accumulating several line scans of shorter duration or brief laser blanking at each scan position may yield an improvement of the imaging outcome.
6. Adaptive illumination strategies reduce overall light exposure
Light exposure can be significantly reduced if excitation and STED illumination are only applied as necessary to obtain the desired information or resolution. This simple thought forms the basis for several related techniques.
In RESCue (REduction of State transition Cycles) STED, the fluorescence photon counts are compared to user-set thresholds during acquisition130 (Fig. 3b). If the photon count at a scan position does not exceed a lower threshold after a predefined fraction of the acquisition time, it is set to zero assuming that no emitter is present. For bright signals surpassing an upper threshold at any time during acquisition, the photon count can be extrapolated to the remaining dwell time while maintaining sufficient SNR. In both cases, all lasers are turned off for the remaining dwell time to spare the sample from unnecessary light exposure; only if neither case applies, acquisition continues for the full dwell time. Reliably deciding between the three scenarios requires a certain number of photons. Still, depending on sample structure, sizeable reductions in light dose and photobleaching may be achieved, with largest benefits for bright and sparse samples (Fig. 4). The RESCue approach is adapted from previous developments in confocal microscopy131 and has also been applied to RSFP-based RESOLFT132.
Figure 4.

RESCue STED attenuates photobleaching compared to conventional STED. Two regions of the same nucleus of a fixed U-2 OS cell immunostained for the nuclear pore protein Nucleoporin 153 with Abberior Star Red were each imaged 30 times using conventional STED (upper sequence) or RESCue STED (lower sequence). In an initial confocal probing step of 5 μs duration, a decision level of 30 photon counts was used to determine whether STED mode should be used at the respective pixel. For RESCue STED, four decision times with corresponding lower thresholds (lTh) were used for early laser shutdown: 1 count after 14% of the pixel dwell time, 3 counts after 24%, 6 counts after 37% and 16 counts after 79%. Upper threshold (uTh) that triggered premature laser shutoff and signal extrapolation was 50 counts. Pixel size was set to 15 nm with a dwell time of 40 μs, laser power at the sample was 1.7 μW for 640 nm excitation and 103 mW for 775 nm STED in both conditions. Scale bars: 500 nm. Note that the colour lookup table is different for frame #1 vs. the later frames.
In DyMIN (DYnamic intensity MINimum) STED133, full STED power is only applied where high resolution is required. Only if fluorescence is detected in the diffraction-limited probe step, the STED laser is turned on to silence fluorophores at the periphery of the excitation volume. If signal is still emitted from the smaller volume, STED power (PSTED) is consecutively stepped up to narrow the effective PSF until the desired resolution is reached. If, on the other hand, no signal is collected, a further power increase would not yield additional information. Probe steps could e. g. be PSTED = 0 for diffraction-limited resolution, PSTED = Pmax/4 for ~ half the final resolution, and the highest STED power Pmax for maximum resolution. Significant reductions in STED light exposure can be achieved, potentially sparing sample regions that do not contain structures of interest from intense STED exposure.
In “multilevel STED”, the STED illumination intensity for multicolour imaging is adjusted according to the requirements for each (spectrally distinct) fluorophore to achieve the same resolution, given differing cross sections for stimulated emission at a fixed STED wavelength85.
For sub-micrometer-sized regions of interest, the scan range can be narrowed down such that the high-intensity crest of the STED doughnut remains outside and never scans across the nanoscale structures of interest (MINFIELD STED, Fig. 3d, Göttfert et al. 134). Increasing the total power of the STED beam steepens the gradient around the intensity minimum and improves resolution.
7. Multiple off-state transitions for protecting fluorophores
The realization that fluorophores and other labels are not just passive “colouring agents” but offer a molecular state structure that can be manipulated and therefore harnessed to improve imaging was the key to breaking the diffraction limit12. STED is the paradigmatic example for fluorophores being deterministically transferred between a signalling on-state and a non-signalling off-state. Similarly, switching between an off- and an on-state lies at the heart of the single-molecule based super-resolution techniques.
While controlling a single off-transition is sufficient for nanoscopy, controlling multiple state transitions opens up refined manipulation of molecular states and synergistic action for diffraction-unlimited image generation. In multiple off-state transitions (MOST) nanoscopy, two (or more) off-state transitions are exploited to generate the on/off state contrast required for coordinate-targeted nanoscopy52. In its prototypical implementation as “protected STED” (Fig. 5), the two off-state transitions were realized by stimulated emission, as in conventional STED microscopy, and reversible photoswitching in RSFPs. This scheme has two important implications:
It introduces a second off-state OFF2. If appropriately chosen, transfer to this state protects fluorophores from photobleaching associated with high STED light intensities, thus increasing repeated imaging capability.
When two independent off-transitions act to define on- and off-regions, the achievable on/off state contrast is higher due to synergistic action of the two off-transitions.
Figure 5.

Multiple off-state transitions for nanoscopy: protected STED. a Molecular state diagram for super-resolution imaging with multiple off-state transitions (MOST): In STED microscopy, a single off-transition is utilized to create the state contrast (grey box). In MOST nanoscopy, an additional off-state OFF2 protects fluorophores and enhances state contrast. Wavelengths are exemplary for protected STED with rsEGFP variants. b In conventional STED, only fluorophores near intensity minima, where I < IS, are allowed to fluoresce (green stars). In order to confine this region tightly, molecules away from the minimum (black stars) are exposed to high STED light intensities. c In protected STED mode, molecules are transferred to OFF2 prior to subjecting them to STED (grey circles). is the intensity required to transfer molecules to OFF2. d Sequence of light pulses and fluorescence detection at each scan position for protected STED imaging with corresponding light patterns in the focal plane. e Protected STED increases repeated imaging capability. Image series of living cells expressing keratin-rsEGFP2, imaged with conventional STED (top) and protected STED (bottom) with the same resolution and brightness in the first frame. Scale bar: 1 μm. Adapted from Danzl et al.52.
In protected STED, fluorophores are pre-emptively transferred to OFF2 in regions of high STED light intensity before applying the combined excitation and STED pulses. Fluorophores in these regions are still exposed to the high STED light intensities but as they reside in OFF2 they essentially do not interact with the STED (and excitation) light (Fig. 5c). They are hence spared from repeated excitation/stimulated emission cycles and unwanted photophysical processes. This translates into reduced photobleaching and increased repeated imaging capability (Fig. 5e). A further effect contributes to reduced photobleaching: In first approximation, both off-transitions take equivalent roles for increasing resolution and their relative contribution can be tuned at will. Therefore, STED power can be reduced while still taking advantage of the improved state contrast over nanoscopy using reversible photoswitching as a single off-transition.
Improved state contrast is easily rationalized when modelling the effective PSF52. It is instructive to first briefly recall the situation with a single off-transition. Any off-transition is expected to suffer from some imperfection in creating on/off state contrast, corresponding to a finite population of fluorophores in the signalling state also in designated off-regions. In STED nanoscopy, this might arise from spontaneous emission before the STED light can take full action or from Anti-Stokes excitation, wheras in RESOLFT nanoscopy with photoswitchable fluorophores, “photoswitching background” arises from incomplete transfer to or spontaneous return from the deactivated configuration. Imperfect state contrast directly leads to a diffraction-limited pedestal in the effective PSF in addition to the desired sharp peak. During image generation in the microscope, the sample structure is convolved with the effective PSF, where, especially in dense regions of the sample, summation over these diffraction-limited components severely reduces (super-resolved) signal to (diffraction-limited) background ratio and hence the achievable resolution. In contrast, if also a second off-transition is operative, the situation changes. The effective PSF is just the product of the individual spatially varying probability distributions for assuming the on-state associated with each of the individual on- and off-state transitions and also the detection PSF. This product implies that a diffraction-limited pedestal left from one off-state transition is counteracted by multiplication with the probability distribution of sub-diffraction width stemming from the other off-transition. The combination of repeated imaging capability and enhanced state contrast enabled 3D super-resolved imaging of volumes of living brain tissue where individual neurons were highlighted and reconstructed in 3D (Figure 2).
Several prerequisites have to be fulfilled for protected STED. Trivially, the fluorophore must be a sufficiently good STED label to begin with. Similarly, it must sustain a large number of on/off photoswitching cycles, i.e. low “switching fatigue” for the second off-transition to be protective against photobleaching. STED light should not be absorbed by the putatively protective off-state OFF2. In addition, the state transitions must be sufficiently decoupled from each other. For example, the intense STED laser could in principle drive a two-photon transition from the deactivated to the activated configuration, jeopardizing the protective effect. However, the ns-laser pulses of a STED laser are expected to drive two-photon transitions much less efficiently than the femtosecond pulses typically used for 2P-excitation or 2P-uncaging. For the reversibly photoswitchable GFP (rsEGFP) variants90,91,135, 2P-activation by STED light was negligible in typical imaging parameter regimes. In the initial proof-of concept, protected STED has only been realized with rsEGFPs. However, the protective effect per se is also present in other reversibly photoswitchable fluorophores52. An intriguing prospect for the future are improved reversibly photoswitchable synthetic fluorophores94–97, which promise increased photostability and photon budgets over fluorescent proteins also for protected STED.
One drawback of protected STED over conventional STED is the requirement for additional light pulses for fluorophore activation and deactivation. While the deactivation step can be chosen shorter than in RSFP-based RESOLFT nanoscopy, it still introduces a prolonged pixel dwell time as compared to conventional STED. An obvious route to increase imaging speed is through parallelization (see section 9).
8. Adaptive optics for correcting optical errors
Aberrations (optical errors) degrade image quality and can be introduced both by imperfections in the microscope’s optics (“system aberrations”) or by refractive index mismatch and inhomogeneity in the sample. In diffraction-limited microscopy, aberrations prevent the formation of a tight focus, spreading the signal across a larger volume and ultimately degrading signal-to-noise ratio and image contrast. STED microscopy is considerably more susceptible to aberrations. They deform the shape of the light distributions and both aberrations and scattering divert laser intensity into STED light minima.
Aberrations adversely affect STED microscopy in several unique ways: (1) Achievable state contrast and resolution depend critically on the quality of the intensity minimum. A non-zero intensity minimum will decrease signal, elicit unnecessary state cycling, and increase photobleaching. Increasing laser power or acquisition time to counteract low signal-to-noise ratio will worsen detrimental effects further. (2) Spreading of the excitation and STED light patterns may increase their overlap, resulting in increased state cycling and photobleaching. (3) Deformations of excitation and/or STED light patterns may result in regions of insufficient fluorescence suppression, leading to sidelobes and artefacts in the final image.
The effects of aberrations on both the STED light distribution136 and the effective PSFs137 have been studied computationally. Notably, coma aberrations in a 3D STED system lead to a translation of the intensity minima for the xy-doughnut and z-STED pattern in opposing directions, resulting in non-overlapping minima and signal loss in an otherwise perfectly-aligned system138.
Spherical aberrations in STED microscopy can often simply be minimized by using an objective lens and immersion medium matched to the sample refractive index, e. g. with glycerol120,139 or water objectives140 normally also featuring a correction collar.
Adaptive optics approaches promise to restore image quality in aberrating samples and are an area of active development in various fields of microscopy141. Here, an active optical element (e. g. deformable mirror, DM or spatial light modulator, SLM) is used to pre-compensate system- and sample-induced aberrations. The same devices can potentially be used to additionally imprint phase patterns onto the wavefront to emulate discrete optical elements, including the phase masks for forming xy- and z-STED patterns. At present, many STED implementations use an SLM instead of conventional (polymer) phase masks to shape the STED light patterns, such that the same SLM can be used for correction of system or sample-induced aberrations without any additional optical elements or intensity losses. The STED patterns are the main determinant for image contrast and resolution, such that here aberrations are more problematic than for the regularly focused excitation foci. Already with a single SLM, both the STED xy-doughnut and the z-STED light pattern can be generated and corrected for system aberrations142. The same hardware can also be used to execute an auto-alignment routine overlaying the excitation and depletion beams143.
Excitation beams can be corrected either by adding an additional SLM to the excitation path144, by passing both excitation and STED beams over a common DM145 or by a double-pass geometry where the same SLM is used to correct both excitation and STED beams146.
Clearly a major challenge in adaptive optics is to determine the correction pattern to dial in. Two principal approaches exist: (1) measuring the wavefront directly or (2) iteratively optimizing an image quality metric while different corrections are applied. The first requires a high signal-to-noise measurement of sample-induced aberrations from the dim fluorescence signal; the second necessitates multiple measurements on the sample during the optimization process. Therefore, either approach subjects the sample to additional light exposure, potentially prematurely bleaching it.
9. Parallelization of image acquisition
Single-point scanning is the most wide-spread implementation of STED microscopy, usually combined with confocal detection. Here, the necessary STED light intensities are reached with readily available laser sources and optical sectioning is achieved with a comparatively simple optical setup. With its action on the (sub-)ns timescale, the STED process itself is extremely fast. However, point scanning limits high-speed recordings to small fields of view.
Parallelization by reading out from many STED light intensity minima simultaneously is an attractive route towards high-speed super-resolution imaging of large fields of view. In an initial implementation, both the illumination and STED beams, as well as detection, were multiplexed four-fold147. Considerably higher degrees of parallelization have been achieved using standing wave patterns. They can be created in the focal plane by interfering a pair of laser beams focused off-axis through the objective. Incoherently superimposing two perpendicular standing waves results in an optical lattice featuring a periodic pattern of thousands of intensity minima and maxima as used for STED148,149) or over a hundred-thousand for RSFP-based RESOLFT135). Conveniently, the optical lattice setup can be designed such that it is achromatic, facilitating colour multiplexing150. For parallelized readout, a high-speed camera is used and the optical lattice is scanned across one unit cell (spanning one period of the lattice) to build a super-resolved image. In highly parallelized STED148,149, the available laser power is spread across large areas, thus limiting the achievable degree of parallelization for a given target resolution. Reversible photoswitching in RESOLFT135 offers an advantage because it requires only low light intensities. Similarly, protected STED is expected to facilitate large scale parallelization. Here, improved on/off state contrast and resolution are achieved already at low STED light intensities52 due to the synergistic contribution of both off-transitions. Accordingly, while the availability of high-power lasers is a limiting factor for parallelization in STED, this requirement would be substantially reduced in parallelized protected STED.
In addition to increased imaging speed, the use of standing waves has two important implications: (1) They provide an up to 15-fold steeper slope around intensity minima and hence require lower peak intensity Imax than the prevalent doughnut implementation149. (2) Each maximum borders several intensity minima and thus contributes to confining fluorescence in more than a single location.
In parallelized approaches, excess state cycling can be decreased if also the excitation light is patterned148,150–152, such that the STED light intensity maxima coincide with the minima of the excitation pattern.
Yet, crosstalk between the closely spaced intensity minima makes it necessary to restrict signal collection by the camera to the central pixels corresponding to the intensity minima, resulting in tight “digital” pinholes (e. g. ~ 1/5 of an Airy disk135). The pinhole diameter can be increased to transmit more signal if a sparsely spaced (excitation or) photoactivation pattern, matching the diffraction limited resolution, is used, e. g. activating only at every fourth minimum of the off-switching pattern in RSFP-based RESOLFT153, albeit at the expense of slower acquisition because the patterns have to be scanned across the larger unit cell, now determined by the photoactivation pattern periodicity.
Despite offering massive parallelization in the image plane, existing implementations based on optical lattices achieve only diffraction-limited resolution along the optical axis. Interference with a fifth, on-axis beam has been suggested for resolution enhancement in 3D154, but experimental demonstration is still lacking.
10. Light-sheet implementations of STED and RESOLFT
In light sheet fluorescence microscopy, a plane of the sample is selectively illuminated with a thin sheet of light, providing optical sectioning and sparing the remainder of the sample from unnecessary light exposure. Light sheet microscopy thus ensures fast, gentle, and high-contrast fluorescence imaging. Light sheets are created either by focusing with a cylindrical lens (“static” light sheet,155), by scanning a laser beam rapidly through the sample, resulting in a “virtual” sheet156, or more elaborate schemes, such as designing the light pattern in the back focal plane of the objective lens to achieve a “lattice” light sheet157. Resolution perpendicular to the light sheet, i.e. axially with respect to the detection objective, is governed by the product of the PSF of the detection objective and the light sheet.
To achieve axial super-resolution, light sheet illumination has been combined with STED and RSFP-based RESOLFT: A “hollow” light sheet silences fluorophores above and below the central plane, reducing the effective sheet thickness. A STED implementation demonstrated up to 2.5 fold increased axial resolution158,159, whereas 5–12 fold improved axial resolution was reported using RSFP-based RESOLFT160. Lateral resolution was unaltered and determined by the PSF of the detection objective.
Due to the diffractive nature of Gaussian beams, light sheet thickness and size of the field of view are interlinked. The use of (theoretically) non-diffracting Bessel beams for light sheet generation promises not only larger uniformly-illuminated field of views, but, equally importantly, also less susceptibility to scattering due to the self-reconstructing nature of Bessel beams161,162. Several approaches to suppress the Bessel beam’s sidelobes while maintaining its self-reconstructing properties have been demonstrated163,164. Simulations have shown that the Bessel beam’s sidelobes can also be efficiently suppressed using stimulated emission165 while retaining the self-reconstructing ability in scattering samples166. An initial experimental demonstration has achieved a twofold resolution enhancement167.
11. Outlook
STED microscopy has come a long way in the last decades: The concept was first proposed 25 years ago and experimentally demonstrated soon thereafter. Since then, STED has undergone a dramatic evolution in terms of performance, ease of use, and accessibility for non-expert labs. It has been serving as a valuable tool for biological research and will do so even more in the future. In this endeavour, each advancement in imaging technology will help to address a new class of biological problems.
Enabling progress is expected in various aspects of STED nanoscopy and other super-resolution techniques. As optical resolution will further increase, ever finer macromolecular arrangements inside cells will become accessible. It will be exciting to visualize these directly in the native spatial context, with cells residing in their complex natural 3D tissue environments. For specific questions, this promises more trustworthy biological data than obtainable from cells artificially grown on a coverslip and incentivizes further super-resolution developments towards 3D-tissue imaging and increased depth penetration. It will be equally exciting to see continued progress in nanoscale imaging of living specimens to follow the dynamic evolution of the biological system. For this, further development of strategies to reduce light exposure, photobleaching, and phototoxicity in live-cell nanoscopy are in high demand. This in turn will boost 3D-diffraction-unlimited analysis of entire tissue volumes. Here, for live measurements, as well as for sufficiently high throughput in fixed tissue analysis, further development of parallelized approaches promises to deliver the required increase in acquisition speed. In parallel to advancing new imaging concepts and developments on the instrumentation side, substantial gain can be expected from new developments in labelling technology. Here, augmented brightness, photostability, and physicochemical properties of the labels will be important catalysts for progress, both for fixed and living specimens. New approaches to multicolour super-resolution imaging will provide high content of molecular information for decoding the three-dimensional nanoarchitecture of cells and tissues. These examples serve to illustrate that a range of exciting developments are on the horizon and we can expect that they will drive fascinating biological discoveries.
Acknowledgements
J.G.D. and P.V. gratefully acknowledge funding by the Austrian Science Fund (FWF; I 3600-B27). W.J. gratefully acknowledges funding by a Human Frontier Science Program long term fellowship (HFSP LT000557/2018).
References
- [1].Abbe E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Archiv für Mikroskopische Anatomie. 1873;9:413–468. doi: 10.1007/bf02956173. ISSN 0176-7348. [DOI] [Google Scholar]
- [2].Verdet E. Leçons d’optique physique. Vol. 1 Victor Masson et fils; Paris: 1869. [Google Scholar]
- [3].Rayleigh JW. On the theory of optical images, with special reference to the microscope. Philosophical Magazine. 1896;5(42):167–195. doi: 10.1080/14786449608620902. [DOI] [Google Scholar]
- [4].von Helmholtz H. Die theoretische Grenze für die Leistungsfähigkeit der Mikroskope. Ann Phys Chem. 1874:557–584. [Google Scholar]
- [5].Ash EA, Nicholls G. Super-resolution aperture scanning microscope. Nature. 1972;237:510–512. doi: 10.1038/237510a0. ISSN 0028-0836. [DOI] [PubMed] [Google Scholar]
- [6].Betzig E, Trautman JK, Harris TD, Weiner JS, Kostelak RL. Breaking the diffraction barrier – optical microscopy on a nanometric scale. Science. 1991;251:1468–1470. doi: 10.1126/science.251.5000.1468. ISSN 0036-8075. [DOI] [PubMed] [Google Scholar]
- [7].Hell SW. Microscopy and its focal switch. Nature Methods. 2009;6(1):24–32. doi: 10.1038/nmeth.1291. [DOI] [PubMed] [Google Scholar]
- [8].Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell. 2010;143:1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Toomre Derek, Bewersdorf Joerg. A new wave of cellular imaging. Annual Review of Cell and Developmental Biology. 2010;26(1):285–314. doi: 10.1146/annurev-cellbio-100109-104048. [DOI] [PubMed] [Google Scholar]
- [10].Liu Zhe, Lavis Luke D, Betzig Eric. Imaging live-cell dynamics and structure at the single-molecule level. Molecular Cell. 2015;58(4):644–659. doi: 10.1016/j.molcel.2015.02.033. ISSN 1097-2765. [DOI] [PubMed] [Google Scholar]
- [11].Sahl Steffen J, Hell Stefan W, Jakobs Stefan. Fluorescence nanoscopy in cell biology. Nature Reviews Molecular Cell Biology. 2017;18(11):685. doi: 10.1038/nrm.2017.71. [DOI] [PubMed] [Google Scholar]
- [12].Hell SW. Nanoscopy with focused light (Nobel Lecture) Angewandte Chemie International Edition. 2015;54(28):8054–8066. doi: 10.1002/anie.201504181. [DOI] [PubMed] [Google Scholar]
- [13].Betzig Eric. Single molecules, cells, and super-resolution optics (Nobel Lecture) Angewandte Chemie International Edition. 2015;54(28):8034–8053. doi: 10.1002/anie.201501003. ISSN 1521-3773. [DOI] [PubMed] [Google Scholar]
- [14].Moerner WE. Single-molecule spectroscopy, imaging, and photocontrol: foundations for super-resolution microscopy (Nobel Lecture) Angewandte Chemie International Edition. 2015;54(28):8067–8093. doi: 10.1002/anie.201501949. [DOI] [PubMed] [Google Scholar]
- [15].Balzarotti Francisco, Eilers Yvan, Gwosch Klaus C, Gynnå Arvid H, Westphal Volker, Stefani Fernando D, Elf Johan, Hell Stefan W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science. 2016;355(6325):606–612. doi: 10.1126/science.aak9913. [DOI] [PubMed] [Google Scholar]
- [16].Dai Mingjie, Jungmann Ralf, Yin Peng. Optical imaging of individual biomolecules in densely packed clusters. Nature Nanotechnology. 2016;11:798–807. doi: 10.1038/nnano.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hell Stefan W. Far-field optical nanoscopy. Science. 2007;316(5828):1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- [18].Rust Michael J, Bates Mark, Zhuang Xiaowei. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nature Methods. 2006;3(10):793–796. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313(5793):1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- [20].Hess Samuel T, Girirajan Thanu PK, Mason Michael D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophysical Journal. 2006;91(11):4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angewandte Chemie International Edition. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- [22].Fölling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, Hell SW. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nature Methods. 2008;5:943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- [23].Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proceedings of the National Academy of Sciences. 2006;103(50):18911–18916. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Giannone Gregory, Hosy Eric, Levet Florian, Constals Audrey, Schulze Katrin, Sobolevsky Alexander I, Rosconi Michael P, Gouaux Eric, Tampé Robert, Choquet Daniel, Cognet Laurent. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophysical Journal. 2010;99(4):1303–1310. doi: 10.1016/j.bpj.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lew MD, Lee SF, Ptacin JL, Lee MK, Twieg RJ, Shapiro L, Moerner WE. Three-dimensional superresolution colocalization of intracellular protein superstructures and the cell surface in live Caulobacter crescentus. Proceedings of the National Academy of Sciences. 2011;108(46):E1102–E1110. doi: 10.1073/pnas.1114444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jungmann Ralf, Steinhauer Christian, Scheible Max, Kuzyk Anton, Tinnefeld Philip, Simmel Friedrich C. Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Letters. 2010;10(11):4756–4761. doi: 10.1021/nl103427w. [DOI] [PubMed] [Google Scholar]
- [27].Hell Stefan W, Wichmann Jan. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Optics Letters. 1994;19(11):780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- [28].Klar Thomas A, Hell Stefan W. Subdiffraction resolution in far-field fluorescence microscopy. Optics Letters. 1999;24(14):954–956. doi: 10.1364/ol.24.000954. [DOI] [PubMed] [Google Scholar]
- [29].Klar Thomas A, Jakobs Stefan, Dyba Marcus, Egner Alexander, Hell Stefan W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proceedings of the National Academy of Sciences. 2000;97(15):8206–8210. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hell SW, Stelzer EHK. Properties of a 4Pi confocal fluorescence microscope. Optical Society of America. Journal A: Optics, Image Science, and Vision. 1992;9:2159–2166. doi: 10.1364/josaa.9.002159. [DOI] [Google Scholar]
- [31].Gustafsson MGL, Agard DA, Sedat JW. I5M: 3D widefield light microscopy with better than 100 nm axial resolution. Journal of Microscopy. 1999;195:10–16. doi: 10.1046/j.1365-2818.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- [32].Gustafsson MGL. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. Journal of Microscopy. 2000;198(2):82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- [33].Gustafsson Mats GL, Shao Lin, Carlton Peter M, Rachel Wang CJ, Golubovskaya Inna N, Zacheus Cande W, Agard David A, Sedat John W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophysical Journal. 2008;94(12):4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gustafsson MGL. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proceedings of the National Academy of Sciences. 2005;102(37):13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Heintzmann R, Jovin TM, Cremer C. Saturated patterned excitation microscopy – a concept for optical resolution improvement. Journal of the Optical Society of America. A, Optics, image science, and vision. 2002;19(8):1599–1609. doi: 10.1364/josaa.19.001599. [DOI] [PubMed] [Google Scholar]
- [36].Chen F, Tillberg PW, Boyden ES. Expansion microscopy. Science. 2015;347(6221):543–548. doi: 10.1126/science.1260088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chang Jae-Byum, Chen Fei, Yoon Young-Gyu, Jung Erica E, Babcock Hazen, Jeong Seuk Kang, Asano Shoh, Suk Ho-Jun, Pak Nikita, Tillber Paul W, Wassie Asmamaw T, et al. Iterative expansion microscopy. Nature Methods. 2017;14:593–599. doi: 10.1038/nmeth.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Truckenbrodt Sven, Maidorn Manuel, Crzan Dagmar, Wildhagen Hanna, Kabatas Selda, Rizzoli Silvio O. X10 expansion microscopy enables 25-nm resolution on conventional microscopes. EMBO Reports. 2018;19(9):e45836. doi: 10.15252/embr.201845836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Truckenbrodt Sven, Sommer Christoph, Rizzoli Silvio O, Danzl Johann G. A practical guide to optimization in X10 expansion microscopy. Nature Protocols. 2019 doi: 10.1038/s41596-018-0117-3. [DOI] [PubMed] [Google Scholar]
- [40].Lakowicz Joseph R. Principles of Fluorescence Spectroscopy. 3 edition. Springer Science + Business Media; 2006. [Google Scholar]
- [41].Sandell Julia L, Zhu Timothy C. A review of in-vivo optical properties of human tissues and its impact on PDT. Journal of Biophotonics. 2011;4(11–12):773–787. doi: 10.1002/jbio.201100062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jacques Steven L. Optical properties of biological tissues: a review. Physics in Medicine and Biology. 2013;58:37–61. doi: 10.1088/0031-9155/58/11/R37. [DOI] [PubMed] [Google Scholar]
- [43].Ntziachristos Vasilis. Going deeper than microscopy: The optical imaging frontier in biology. Nature Methods. 2010;7(8):603–614. doi: 10.1038/nmeth.1483. [DOI] [PubMed] [Google Scholar]
- [44].Sharpe James. Optical Projection Tomography. 2004:209–228. doi: 10.1146/annurev.bioeng.6.040803.140210. [DOI] [PubMed] [Google Scholar]
- [45].Hong Guosong, Antaris Alexander L, Dai Hongjie. Near-infrared fluorophores for biomedical imaging. Nature Biomedical Engineering. 2017;1(1):0010. doi: 10.1038/s41551-016-0010. [DOI] [Google Scholar]
- [46].Eggeling C, Widengren J, Rigler R, Seidel CAM. Applied Fluorescence in Chemistry, Biology and Medicine. Springer Berlin Heidelberg; 1999. Photostability of fluorescent dyes for single-molecule spectroscopy: Mechanisms and experimental methods for estimating photobleaching in aqueous solution; pp. 193–240. [DOI] [Google Scholar]
- [47].Leutenegger Marcel, Eggeling Christian, Hell Stefan W. Analytical description of STED microscopy performance. Optics Express. 2010;18(25):26417. doi: 10.1364/oe.18.026417. [DOI] [PubMed] [Google Scholar]
- [48].Tortarolo Giorgio, Castello Marco, Diaspro Alberto, Koho Sami, Vicidomini Giuseppe. Evaluating image resolution in stimulated emission depletion microscopy. Optica. 2018;5(1):32–35. doi: 10.1364/OPTICA.5.000032. [DOI] [Google Scholar]
- [49].Laissue P Philippe, Alghamdi Rana A, Tomancak Pavel, Reynaud Emmanuel G, Shroff Hari. Assessing phototoxicity in live fluorescence imaging. Nature Methods. 2017;14(7):657–661. doi: 10.1038/nmeth.4344. [DOI] [PubMed] [Google Scholar]
- [50].Wäldchen Sina, Lehmann Julian, Klein Teresa, van de Linde Sebastian, Sauer Markus. Light-induced cell damage in live-cell super-resolution microscopy. Scientific Reports. 2015;5(1) doi: 10.1038/srep15348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kilian Nicole, Goryaynov Alexander, Lessard Mark D, Hooker Giles, Toomre Derek, Rothman James E, Bewersdorf Joerg. Assessing photodamage in live-cell STED microscopy. Nature Methods. 2018;15(10):755–756. doi: 10.1038/s41592-018-0145-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Danzl Johann G, Sidenstein Sven C, Gregor Carola, Urban Nicolai T, Ilgen Peter, Jakobs Stefan, Hell Stefan W. Coordinate-targeted fluorescence nanoscopy with multiple off states. Nature Photonics. 2016;10(2):122. doi: 10.1038/nphoton.2015.266. [DOI] [Google Scholar]
- [53].Eilers Yvan, Ta Haisen, Gwosch Klaus C, Balzarotti Francisco, Hell Stefan W. MINFLUX monitors rapid molecular jumps with superior spatiotemporal resolution. Proceedings of the National Academy of Sciences. 2018;115(24):6117–6122. doi: 10.1073/pnas.1801672115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gould Travis J, Pellett Patrina A, Bewersdorf Joerg. STED Microscopy. Wiley-VCH; 2013. [DOI] [Google Scholar]
- [55].Eggeling C, Willig KI, Sahl SJ, Hell SW. Lens-based fluorescence nanoscopy. Quarterly Reviews of Biophysics. 2015;48(2):178–243. doi: 10.1017/S0033583514000146. [DOI] [PubMed] [Google Scholar]
- [56].Hell Stefan W, Sahl Steffen J, Bates Mark, Zhuang Xiaowei, Heintzmann Rainer, Booth Martin J, Bewersdorf Joerg, Shtengel Gleb, Hess Harald, Tinnefeld Philip, Honigmann Alf, et al. The 2015 super-resolution microscopy roadmap. Journal of Physics D: Applied Physics. 2015;48(44) doi: 10.1088/0022-3727/48/44/443001. 443001. [DOI] [Google Scholar]
- [57].Blom Hans, Widengren Jerker. Stimulated emission depletion microscopy. Chemical Reviews. 2017;117(11):7377–7427. doi: 10.1021/acs.chemrev.6b00653. [DOI] [PubMed] [Google Scholar]
- [58].Vicidomini Giuseppe, Bianchini Paolo, Diaspro Alberto. STED super-resolved microscopy. Nature Methods. 2018;15(3):173–182. doi: 10.1038/nmeth.4593. [DOI] [PubMed] [Google Scholar]
- [59].Birk Udo J. Super-Resolution Microscopy. Wiley VCH Verlag GmbH; 2017. ISBN 3527341331. [DOI] [Google Scholar]
- [60].Rittweger E, Rankin BR, Westphal V, Hell SW. Fluorescence depletion mechanisms in super-resolving STED microscopy. Chemical Physics Letters. 2007;442(4-6):483–487. doi: 10.1016/j.cplett.2007.06.017. [DOI] [Google Scholar]
- [61].Keller Jan, Schönle Andreas, Hell Stefan W. Efficient fluorescence inhibition patterns for RESOLFT microscopy. Optics Express. 2007;15(6):3361. doi: 10.1364/oe.15.003361. [DOI] [PubMed] [Google Scholar]
- [62].Moneron Gael, Hell Stefan W. Two-photon excitation STED microscopy. Optics Express. 2009;17(17):14567–14573. doi: 10.1364/oe.17.014567. [DOI] [PubMed] [Google Scholar]
- [63].Ding Jun B, Takasaki Kevin T, Sabatini Bernardo L. Supraresolution imaging in brain slices using stimulated-emission depletion two-photon laser scanning microscopy. Neuron. 2009;63:429–437. doi: 10.1016/j.neuron.2009.07.011. ISSN 0896-6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Li Q, Wu SSH, Chou KC. Subdiffraction-limit two-photon fluorescence microscopy for GFP-tagged cell imaging. Biophysical Journal. 2009;97(12):3224–3228. doi: 10.1016/j.bpj.2009.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bianchini P, Harke B, Galiani S, Vicidomini G, Diaspro A. Single-wavelength two-photon excitation-stimulated emission depletion (SW2PE-STED) superresolution imaging. Proceedings of the National Academy of Sciences. 2012;109(17):6390–6393. doi: 10.1073/pnas.1119129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bethge Philipp, Chéreau Ronan, Avignone Elena, Marsicano Giovanni, Nägerl Valentin U. Two-photon excitation STED microscopy in two colors in acute brain slices. Biophysical Journal. 2013;104(4):778–785. doi: 10.1016/j.bpj.2012.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Dyba M, Hell SW. Focal spots of size λ/23 open up far-field florescence microscopy at 33 nm axial resolution. Physical Review Letters. 2002;88(16):163901. doi: 10.1103/PhysRevLett.88.163901. [DOI] [PubMed] [Google Scholar]
- [68].Schmidt Roman, Wurm Christian A, Jakobs Stefan, Engelhardt Johann, Egner Alexander, Hell Stefan W. Spherical nanosized focal spot unravels the interior of cells. Nature Methods. 2008;5(6):539–544. doi: 10.1038/nmeth.1214. [DOI] [PubMed] [Google Scholar]
- [69].Hell Stefan W. Toward fluorescence nanoscopy. Nature Biotechnology. 2003;21(11):1347. doi: 10.1038/nbt895. [DOI] [PubMed] [Google Scholar]
- [70].Harke Benjamin, Keller Jan, Ullal Chaitanya K, Westphal Volker, Schönle Andreas, Hell Stefan W. Resolution scaling in STED microscopy. Optics Express. 2008;16(6):4154–4162. doi: 10.1364/oe.16.004154. [DOI] [PubMed] [Google Scholar]
- [71].Rittweger Eva, Han Kyu Young, Irvine Scott E, Eggeling Christian, Hell Stefan W. STED microscopy reveals crystal colour centres with nanometric resolution. Nature Photonics. 2009;3(3):144. doi: 10.1038/nphoton.2009.2. [DOI] [Google Scholar]
- [72].Schneider Jale, Zahn Jasmin, Maglione Marta, Sigrist Stephan J, Marquard Jonas, Chojnacki Jakub, Kräusslich Hans-Georg, Sahl Steffen J, Engelhardt Johann, Hell Stefan W. Ultrafast, temporally stochastic STED nanoscopy of millisecond dynamics. Nature Methods. 2015;12(9):827–830. doi: 10.1038/nmeth.3481. [DOI] [PubMed] [Google Scholar]
- [73].Nägerl U Valentin, Willig Katrin I, Hein Birka, Hell Stefan W, Bonhoeffer Tobias. Live-cell imaging of dendritic spines by STED microscopy. Proceedings of the National Academy of Sciences. 2008;105(48) doi: 10.1073/pnas.0810028105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Tønnesen Jan, Inavalli VVG Krishna, Nägerl U Valentin. Super-resolution imaging of the extracellular space in living brain tissue. Cell. 2018;172(5):1108–1121.e15. doi: 10.1016/j.cell.2018.02.007. [DOI] [PubMed] [Google Scholar]
- [75].Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, Polyakova S, Belov VN, Hein B, von Middendorff C, Schönle A, Hell SW. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2009;457:1159–1162. doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- [76].Denk Winfried, Strickler James H, Webb Watt W. Two-photon laser scanning fluorescence microscopy. Science. 1990;248(4951):73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- [77].Göttfert Fabian, Wurm Christian A, Mueller Veronika, Berning Sebastian, Cordes Volker C, Honigmann Alf, Hell Stefan W. Coaligned dual-channel STED nanoscopy and molecular diffusion analysis at 20 nm resolution. Biophysical Journal. 2013;105(1):L01–L03. doi: 10.1016/j.bpj.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Willig KI, Harke B, Medda R, Hell SW. STED microscopy with continuous wave beams. Nature Methods. 2007;4(11):915–918. doi: 10.1038/nmeth1108. [DOI] [PubMed] [Google Scholar]
- [79].Moneron Gael, Medda Rebecca, Hein Birka, Giske Arnold, Westphal Volker, Hell Stefan W. Fast STED microscopy with continuous wave fiber lasers. Optics Express. 2010;18(2):1302–1309. doi: 10.1364/oe.18.001302. [DOI] [PubMed] [Google Scholar]
- [80].Vicidomini Giuseppe, Moneron Gael, Han Kyu Y, Westphal Volker, Ta Haisen, Reuss Matthias, Engelhardt Johann, Eggeling Christian, Hell Stefan W. Sharper low-power STED nanoscopy by time gating. Nature Methods. 2011;8(7):571. doi: 10.1038/nmeth.1624. [DOI] [PubMed] [Google Scholar]
- [81].Moffitt Jeffrey R, Osseforth Christian, Michaelis Jens. Time-gating improves the spatial resolution of STED microscopy. Optics Express. 2011;19(5):4242–4254. doi: 10.1364/oe.19.004242. [DOI] [PubMed] [Google Scholar]
- [82].Vicidomini Giuseppe, Schönle Andreas, Ta Haisen, Han Kyu Young, Moneron Gael, Eggeling Christian, Hell Stefan W. STED nanoscopy with time-gated detection: Theoretical and experimental aspects. PLoS ONE. 2013;8(1):e54421. doi: 10.1371/journal.pone.0054421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Tønnesen Jan, Nadrigny Fabien, Willig Katrin I, Wedlich-Söldner Roland, Nägerl U Valentin. Two-color STED microscopy of living synapses using a single laser-beam pair. Biophysical Journal. 2011;101(10):2545–2552. doi: 10.1016/j.bpj.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Görlitz Frederik, Hoyer Patrick, Falk Henning, Kastrup Lars, Engelhardt Johann, Hell Stefan W. A STED microscope designed for routine biomedical applications. Progress In Electromagnetics Research. 2014;147:57–68. doi: 10.2528/pier14042708. [DOI] [Google Scholar]
- [85].Sidenstein Sven C, DEste Elisa, Böhm Marvin J, Danzl Johann G, Belov Vladimir N, Hell Stefan W. Multicolour multilevel sted nanoscopy of actin/spectrin organization at synapses. Scientific Reports. 2016;6 doi: 10.1038/srep26725. 26725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Winter Franziska R, Loidolt Maria, Westphal Volker, Butkevich Alexey N, Gregor Carola, Sahl Steffen J, Hell Stefan W. Multicolour nanoscopy of fixed and living cells with a single STED beam and hyperspectral detection. Scientific Reports. 2017 doi: 10.1038/srep46492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Hell SW, Jakobs S, Kastrup L. Imaging and writing at the nanoscale with focused visible light through saturable optical transitions. Applied Physics A. 2003;77(7):859–860. doi: 10.1007/s00339-003-2292-4. [DOI] [Google Scholar]
- [88].Hofmann Michael, Eggeling Christian, Jakobs Stefan, Hell Stefan W. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proceedings of the National Academy of Sciences. 2005;102(49):17565–17569. doi: 10.1073/pnas.0506010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Andresen M, Wahl MC, Stiel AC, Gräter F, Schäfer LV, Trowitzsch S, Weber G, Eggeling C, Grubmüller H, Hell SW, Jakobs S. Structure and mechanism of the reversible photoswitch of a fluorescent protein. Proceedings of the National Academy of Sciences. 2005;102:13070–13074. doi: 10.1073/pnas.0502772102. ISSN 0027-8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Grotjohann T, Testa I, Leutenegger M, Bock H, Urban NT, Lavoie-Cardinal F, Willig KI, Eggeling C, Jakobs S, Hell SW. Diffraction-unlimited all-optical imaging and writing with a photochromic GFP. Nature. 2011;478(7368):204–208. doi: 10.1038/nature10497. [DOI] [PubMed] [Google Scholar]
- [91].Grotjohann Tim, Testa Ilaria, Reuss Matthias, Brakemann Tanja, Eggeling Christian, Hell Stefan W, Jakobs Stefan. rsEGFP2 enables fast RESOLFT nanoscopy of living cells. eLife. 2012;1 doi: 10.7554/elife.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Brakemann T, Stiel AC, Weber G, Andresen M, Testa I, Grotjohann T, Leutenegger M, Plessmann U, Urlaub H, Eggeling C, Wahl MC, et al. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nature Biotechnology. 2011;29(10):942–947. doi: 10.1038/nbt.1952. [DOI] [PubMed] [Google Scholar]
- [93].Pennacchietti Francesca, Serebrovskaya Ekaterina O, Faro Aline R, Shemyakina Irina I, Bozhanova Nina G, Kotlobay Alexey A, Gurskaya Nadya G, Bodén Andreas, Dreier Jes, Chudakov Dmitry M, Lukyanov Konstantin A, et al. Fast reversibly photoswitching red fluorescent proteins for live-cell RESOLFT nanoscopy. Nature Methods. 2018;15(8):601–604. doi: 10.1038/s41592-018-0052-9. [DOI] [PubMed] [Google Scholar]
- [94].Kwon Jiwoong, Hwang Jihee, Park Jaewan, Han Gi Rim, Han Kyu Young, Kim Seong Keun. RESOLFT nanoscopy with photoswitchable organic fluorophores. Scientific Reports. 2015;5(1) doi: 10.1038/srep17804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Roubinet Benoît, Bossi Mariano L, Alt Philipp, Leutenegger Marcel, Shojaei Heydar, Schnorrenberg Sebastian, Nizamov Shamil, Irie Masahiro, Belov Vladimir N, Hell Stefan W. Carboxylated photoswitchable diarylethenes for biolabeling and super-resolution RESOLFT microscopy. Angewandte Chemie International Edition. 2016;55(49):15429–15433. doi: 10.1002/anie.201607940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Shao Baihao, Baroncini Massimo, Qian Hai, Bussotti Laura, Di Donato Mariangela, Credi Alberto, Aprahamian Ivan. Solution and solid-state emission toggling of a photochromic hydrazone. Journal of the American Chemical Society. 2018;140(39):12323–12327. doi: 10.1021/jacs.8b07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Xiong Yaoyao, Jentzsch Andreas Vargas, Osterrieth Johannes WM, Sezgin Erdinc, Sazanovich Igor V, Reglinski Katharina, Galiani Silvia, Parker Anthony W, Eggeling Christian, Anderson Harry L. Spironaphthoxazine switchable dyes for biological imaging. Chemical Science. 2018;9(11):3029–3040. doi: 10.1039/c8sc00130h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Vicidomini Giuseppe, Moneron Gael, Eggeling Christian, Rittweger Eva, Hell Stefan W. STED with wavelengths closer to the emission maximum. Optics Express. 2012;20(5):5225. doi: 10.1364/oe.20.005225. [DOI] [PubMed] [Google Scholar]
- [99].Hotta Jun-ichi, Fron Eduard, Dedecker Peter, Janssen Kris PF, Li Chen, Müller Klaus, Harke Benjamin, Bückers Johanna, Hell Stefan W, Hofkens Johan. Spectroscopic rationale for efficient stimulated-emission depletion microscopy fluorophores. JACS. 2010;132:5021–5023. doi: 10.1021/ja100079w. ISSN 0002-7863. [DOI] [PubMed] [Google Scholar]
- [100].Muyldermans Serge. Nanobodies: Natural single-domain antibodies. Annual Review of Biochemistry. 2013;82(1):775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- [101].Willig Katrin I, Kellner Robert R, Medda Rebecca, Hein Birka, Jakobs Stefan, Hell Stefan W. Nanoscale resolution in GFP-based microscopy. Nature Methods. 2006;3(9):721–723. doi: 10.1038/nmeth922. [DOI] [PubMed] [Google Scholar]
- [102].Lukinavičius G, Umezawa K, Olivier N, Honigmann A, Yang G, Plass T, Mueller V, Reymond L, Corrêa IR, Jr, Luo ZG, Schultz C, et al. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nature Chemistry. 2013;5(2):132–139. doi: 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- [103].Grimm Jonathan B, English Brian P, Chen Jiji, Slaughter Joel P, Zhang Zhengjian, Revyakin Andrey, Patel Ronak, Macklin John J, Normanno Davide, Singer Robert H, Lionnet Timothée, et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nature Methods. 2015;12(3):244–250. doi: 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Bottanelli Francesca, Kromann Emil B, Allgeyer Edward S, Erdmann Roman S, Baguley Stephanie Wood, Sirinakis George, Schepartz Alanna, Baddeley David, Toomre Derek K, Rothman James E, Bewersdorf Joerg. Two-colour live-cell nanoscale imaging of intracellular targets. Nature Communications. 2016;7(1) doi: 10.1038/ncomms10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Butkevich Alexey N, Mitronova Gyuzel Yu, Sidenstein Sven C, Klocke Jessica L, Kamin Dirk, Meineke Dirk NH, D’Este Elisa, Kraemer Philip-Tobias, Danzl Johann G, Belov Vladimir N, Hell Stefan W. Fluorescent rhodamines and fluorogenic carbopyronines for super-resolution STED microscopy in living cells. Angewandte Chemie International Edition. 2016;55(10):3290–3294. doi: 10.1002/anie.201511018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Grimm Jonathan B, Muthusamy Anand K, Liang Yajie, Brown Timothy A, Lemon William C, Patel Ronak, Lu Rongwen, Macklin John J, Keller Philipp J, Ji Na, Lavis Luke D. A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nature Methods. 2017;14(10):987–994. doi: 10.1038/nmeth.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Mollwitz Birgit, Brunk Elizabeth, Schmitt Simone, Pojer Florence, Bannwarth Michael, Schiltz Marc, Rothlisberger Ursula, Johnsson Kai. Directed evolution of the suicide protein o6-alkylguanine-DNA alkyltransferase for increased reactivity results in an alkylated protein with exceptional stability. Biochemistry. 2012;51(5):986–994. doi: 10.1021/bi2016537. [DOI] [PubMed] [Google Scholar]
- [108].Los Georgyi V, Encell Lance P, McDougall Mark G, Hartzell Danette D, Karassina Natasha, Zimprich Chad, Wood Monika G, Learish Randy, Ohana Rachel Friedman, Urh Marjeta, Simpson Dan, et al. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chemical Biology. 2008;3(6):373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- [109].Rostovtsev Vsevolod V, Green Luke G, Fokin Valery V, Sharpless K Barry. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angewandte Chemie International Edition. 2002;41(14):2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::aidanie2596>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- [110].Crivat Georgeta, Taraska Justin W. Imaging proteins inside cells with fluorescent tags. Trends in Biotechnology. 2012;30(1):8–16. doi: 10.1016/j.tibtech.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Ries Jonas, Kaplan Charlotte, Platonova Evgenia, Eghlidi Hadi, Ewers Helge. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nature Methods. 2012;9(6):582–584. doi: 10.1038/nmeth.1991. [DOI] [PubMed] [Google Scholar]
- [112].Kasper Robert, Harke Benjamin, Forthmann Carsten, Tinnefeld Philip, Hell Stefan W, Sauer Markus. Single-molecule STED microscopy with photostable organic fluorophores. Small. 2010;6(13):1379–1384. doi: 10.1002/smll.201000203. [DOI] [PubMed] [Google Scholar]
- [113].Liphardt Bodo, Liphardt Bernd, Lüttke W. Laser dyes with intramolecular triplet quenching. Optics Communications. 1981;38:207–210. doi: 10.1016/0030-4018(81)90325-4. ISSN 0030-4018. [DOI] [Google Scholar]
- [114].Altman Roger B, Terry Daniel S, Zhou Zhou, Zheng Qinsi, Geggier Peter, Kolster Rachel A, Zhao Yongfang, Javitch Jonathan A, Warren J David, Blanchard Scott C. Cyanine fluorophore derivatives with enhanced photostability. Nature Methods. 2011;9(1):68–71. doi: 10.1038/nmeth.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].van der Velde Jasper HM, Ploetz Evelyn, Hiermaier Matthias, Oelerich Jens, de Vries Jan Willem, Roelfes Gerard, Cordes Thorben. Mechanism of intramolecular photostabilization in self-healing cyanine fluorophores. ChemPhysChem. 2013;14(18):4084–4093. doi: 10.1002/cphc.201300785. [DOI] [PubMed] [Google Scholar]
- [116].Zheng Qinsi, Jockusch Steffen, Rodríguez-Calero Gabriel G, Zhou Zhou, Zhao Hong, Altman Roger B, Abruña Héctor D, Blanchard Scott C. Intramolecular triplet energy transfer is a general approach to improve organic fluorophore photostability. Photochemical & Photobiological Sciences. 2016;15(2):196–203. doi: 10.1039/c5pp00400d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].van der Velde Jasper HM, Oelerich Jens, Huang Jingyi, Smit Jochem H, Jazi Atieh Aminian, Galiani Silvia, Kolmakov Kirill, Gouridis Giorgos, Eggeling Christian, Herrmann Andreas, Roelfes Gerard, Cordes Thorben. A simple and versatile design concept for fluorophore derivatives with intramolecular photostabilization. Nature Communications. 2016;7(1) doi: 10.1038/ncomms10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].van der Velde Jasper HM, Smit Jochem H, Hebisch Elke, Punter Michiel, Cordes Thorben. Self-healing dyes for super-resolution fluorescence microscopy. Journal of Physics D: Applied Physics. 2018;52(3):034001. doi: 10.1088/1361-6463/aae752. [DOI] [Google Scholar]
- [119].Riedl Julia, Crevenna Alvaro H, Kessenbrock Kai, Yu Jerry Haochen, Neukirchen Dorothee, Bista Michal, Bradke Frank, Jenne Dieter, Holak Tad A, Werb Zena, Sixt Michael, Wedlich-Soldner Roland. Lifeact: a versatile marker to visualize F-actin. Nature Methods. 2008;5(7):605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Urban Nicolai T, Willig Katrin I, Hell Stefan W, Nägerl U Valentin. STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophysical Journal. 2011;101(5):1277–1284. doi: 10.1016/j.bpj.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Spahn Christoph, Grimm Jonathan B, Lavis Luke D, Lampe Marko, Heilemann Mike. Whole-cell, 3D, and multicolor STED imaging with exchangeable fluorophores. Nano Letters. 2018;19(1):500–505. doi: 10.1021/acs.nanolett.8b04385. [DOI] [PubMed] [Google Scholar]
- [122].Jungmann Ralf, Avendaño Maier S, Woehrstein Johannes B, Dai Mingjie, Shih William M, Yin Peng. Multiplexed 3d cellular super-resolution imaging with DNA-PAINT and exchange-PAINT. Nature Methods. 2014;11(3):313–318. doi: 10.1038/nmeth.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Beater Susanne, Holzmeister Phil, Lalkens Birka, Tinnefeld Philip. Simple and aberration-free 4color-STED - multiplexing by transient binding. Optics Express. 2015;23(7):8630. doi: 10.1364/oe.23.008630. [DOI] [PubMed] [Google Scholar]
- [124].Schueder Florian, Strauss Maximilian T, Hoerl David, Schnitzbauer Joerg, Schlichthaerle Thomas, Strauss Sebastian, Yin Peng, Harz Hartmann, Leonhardt Heinrich, Jungmann Ralf. Universal super-resolution multiplexing by DNA exchange. Angewandte Chemie International Edition. 2017;56(14):4052–4055. doi: 10.1002/anie.201611729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Wang Yu, Woehrstein Johannes B, Donoghue Noah, Dai Mingjie, Avendaño Maier S, Schackmann Ron CJ, Zoeller Jason J, Wang Shan Shan H, Tillberg Paul W, Park Demian, Lapan Sylvain W, et al. Rapid sequential in situ multiplexing with DNA exchange imaging in neuronal cells and tissues. Nano Letters. 2017;17(10):6131–6139. doi: 10.1021/acs.nanolett.7b02716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Donnert Gerald, Eggeling Christian, Hell Stefan W. Major signal increase in fluorescence microscopy through dark-state relaxation. Nature Methods. 2007;4(1):81. doi: 10.1038/nmeth986. [DOI] [PubMed] [Google Scholar]
- [127].Donnert Gerald, Keller Jan, Medda Rebecca, Alexandra Andrei M, Rizzoli Silvio O, Lührmann Reinhard, Jahn Reinhard, Eggeling Christian, Hell Stefan W. Macromolecular-scale resolution in biological fluorescence microscopy. Proceedings of the National Academy of Sciences. 2006;103(31):11440–11445. doi: 10.1073/pnas.0604965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Westphal Volker, Lauterbach Marcel A, Di Nicola Angelo, Hell Stefan W. Dynamic far-field fluorescence nanoscopy. New Journal of Physics. 2007;9(12):435. doi: 10.1088/1367-2630/9/12/435. [DOI] [Google Scholar]
- [129].Chen Xiaowei, Leischner Ulrich, Varga Zsuzsanna, Jia Hongbo, Deca Diana, Rochefort Nathalie L, Konnerth Arthur. LOTOS-based two-photon calcium imaging of dendritic spines in vivo. Nature Protocols. 2012;7(10):1818–1829. doi: 10.1038/nprot.2012.106. [DOI] [PubMed] [Google Scholar]
- [130].Staudt Thorsten, Engler Andreas, Rittweger Eva, Harke Benjamin, Engelhardt Johann, Hell Stefan W. Far-field optical nanoscopy with reduced number of state transition cycles. Optics Express. 2011;19(6):5644–5657. doi: 10.1364/oe.19.005644. [DOI] [PubMed] [Google Scholar]
- [131].Hoebe RA, Van Oven CH, Gadella TWJ, Dhonukshe PB, Van Noorden CJF, Manders EMM. Controlled light-exposure microscopy reduces photobleaching and phototoxicity in fluorescence live-cell imaging. Nature Biotechnology. 2007;25(2):249–253. doi: 10.1038/nbt1278. [DOI] [PubMed] [Google Scholar]
- [132].Dreier Jes, Castello Marco, Coceano Giovanna, Cáceres Rodrigo, Plastino Julie, Vicidomini Giuseppe, Testa Ilaria. Smart scanning for low-illumination and fast RESOLFT nanoscopy in vivo. Nature Communications. 2019;10(1) doi: 10.1038/s41467-019-08442-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Heine Jörn, Reuss Matthias, Harke Benjamin, D’Este Elisa, Sahl Steffen J, Hell Stefan W. Adaptive-illumination STED nanoscopy. Proceedings of the National Academy of Sciences. 2017;114(37):9797–9802. doi: 10.1073/pnas.1708304114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Göttfert Fabian, Pleiner Tino, Heine Jörn, Westphal Volker, Görlich Dirk, Sahl Steffen J, Hell Stefan W. Strong signal increase in STED fluorescence microscopy by imaging regions of subdiffraction extent. Proceedings of the National Academy of Sciences. 2017;114(9):2125–2130. doi: 10.1073/pnas.1621495114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Chmyrov Andriy, Keller Jan, Grotjohann Tim, Ratz Michael, d’Este Elisa, Jakobs Stefan, Eggeling Christian, Hell Stefan W. Nanoscopy with more than 100,000 ‘doughnuts’. Nature Methods. 2013;10(8):737–740. doi: 10.1038/nmeth.2556. [DOI] [PubMed] [Google Scholar]
- [136].Antonello Jacopo, Burke Daniel, Booth Martin J. Aberrations in stimulated emission depletion (STED) microscopy. Optics Communications. 2017;404:203–209. doi: 10.1016/j.optcom.2017.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Li Yanghui, Zhou Hui, Liu Xiaoyu, Li Yuxue, Wang Le. Effects of aberrations on effective point spread function in STED microscopy. Applied Optics. 2018;57(15):4164. doi: 10.1364/ao.57.004164. [DOI] [PubMed] [Google Scholar]
- [138].Antonello Jacopo, Kromann Emil B, Burke Daniel, Bewersdorf Joerg, Booth Martin J. Coma aberrations in combined two- and three-dimensional STED nanoscopy. Optics Letters. 2016;41(15):3631. doi: 10.1364/ol.41.003631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Berning S, Willig KI, Steffens H, Dibaj P, Hell SW. Nanoscopy in a living mouse brain. Science. 2012;335(6068):551–551. doi: 10.1126/science.1215369. [DOI] [PubMed] [Google Scholar]
- [140].Chéreau Ronan, Tønnesen Jan, Nägerl U Valentin. STED microscopy for nanoscale imaging in living brain slices. Methods. 2015;88:57–66. doi: 10.1016/j.ymeth.2015.06.006. [DOI] [PubMed] [Google Scholar]
- [141].Booth Martin J. Adaptive optical microscopy: the ongoing quest for a perfect image. Light: Science & Applications. 2014;3(4):e165. doi: 10.1038/lsa.2014.46. [DOI] [Google Scholar]
- [142].Lenz Martin O, Sinclair Hugo G, Savell Alexander, Clegg James H, Brown Alice CN, Davis Daniel M, Dunsby Chris, Neil Mark AA, French Paul MW. 3-D stimulated emission depletion microscopy with programmable aberration correction. Journal of Biophotonics. 2013;7(1–2):29–36. doi: 10.1002/jbio.201300041. [DOI] [PubMed] [Google Scholar]
- [143].Gould Travis J, Kromann Emil B, Burke Daniel, Booth Martin J, Bewersdorf Joerg. Auto-aligning stimulated emission depletion microscope using adaptive optics. Optics Letters. 2013;38(11):1860–1862. doi: 10.1364/ol.38.001860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Gould Travis J, Burke Daniel, Bewersdorf Joerg, Booth Martin J. Adaptive optics enables 3D STED microscopy in aberrating specimens. Optics Express. 2012;20(19):20998–21009. doi: 10.1364/oe.20.020998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Patton Brian R, Burke Daniel, Owald David, Gould Travis J, Bewersdorf Joerg, Booth Martin J. Three-dimensional STED microscopy of aberrating tissue using dual adaptive optics. Optics Express. 2016;24(8):8862. doi: 10.1364/oe.24.008862. [DOI] [PubMed] [Google Scholar]
- [146].Görlitz Frederik, Guldbrand Stina, Runcorn Timothy H, Murray Robert T, Jaso-Tamame Angel L, Sinclair Hugo G, Martinez-Perez Enrique, Taylor James R, Neil Mark AA, Dunsby Christopher, French Paul MW. easySLM-STED: Stimulated emission depletion microscopy with aberration correction, extended field of view and multiple beam scanning. Journal of Biophotonics. 2018;11(11):e201800087. doi: 10.1002/jbio.201800087. [DOI] [PubMed] [Google Scholar]
- [147].Bingen Pit, Reuss Matthias, Engelhardt Johann, Hell Stefan W. Parallelized STED fluorescence nanoscopy. Optics Express. 2011;19(24):23716–23726. doi: 10.1364/oe.19.023716. [DOI] [PubMed] [Google Scholar]
- [148].Yang Bin, Przybilla Frédéric, Mestre Michael, Trebbia Jean-Baptiste, Lounis Brahim. Large parallelization of STED nanoscopy using optical lattices. Optics Express. 2014;22(5):5581–5589. doi: 10.1364/oe.22.005581. [DOI] [PubMed] [Google Scholar]
- [149].Bergermann Fabian, Alber Lucas, Sahl Steffen J, Engelhardt Johann, Hell Stefan W. 2000-fold parallelized dual-color STED fluorescence nanoscopy. Optics Express. 2015;23(1):211. doi: 10.1364/oe.23.000211. [DOI] [PubMed] [Google Scholar]
- [150].Chmyrov Andriy, Leutenegger Marcel, Grotjohann Tim, Schönle Andreas, Keller-Findeisen Jan, Kastrup Lars, Jakobs Stefan, Donnert Gerald, Sahl Steffen J, Hell Stefan W. Achromatic light patterning and improved image reconstruction for parallelized RESOLFT nanoscopy. Scientific Reports. 2017;7:44619. doi: 10.1038/srep44619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Yang Bin, Przybilla Frédéric, Mestre Michael, Trebbia Jean-Baptiste, Lounis Brahim. Using optical lattice for STED parallelization. In: Verma Prabhat, Egner Alexander., editors. Nanoimaging and Nanospectroscopy II. SPIE; 2014. [DOI] [Google Scholar]
- [152].Yang B, Fang C-Y, Chang H-C, Treussart F, Trebbia J-B, Lounis B. Polarization effects in lattice-STED microscopy. Faraday Discuss. 2015;184:37–49. doi: 10.1039/c5fd00092k. [DOI] [PubMed] [Google Scholar]
- [153].Masullo Luciano A, Bodén Andreas, Pennacchietti Francesca, Coceano Giovanna, Ratz Michael, Testa Ilaria. Enhanced photon collection enables four dimensional fluorescence nanoscopy of living systems. Nature Communications. 2018;9(1) doi: 10.1038/s41467-018-05799-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Xue Yi, So Peter TC. Three-dimensional super-resolution high-throughput imaging by structured illumination STED microscopy. Optics Express. 2018;26(16):20920. doi: 10.1364/oe.26.020920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Huisken Jan, Swoger Jim, Del Bene Filippo, Wittbrodt Joachim, Stelzer Ernst HK. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305:1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- [156].Keller Philipp J, Schmidt Annette D, Wittbrodt Joachim, Stelzer Ernst HK. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 2008;322:1065–1069. doi: 10.1126/science.1162493. [DOI] [PubMed] [Google Scholar]
- [157].Chen Bi-Chang, Legant Wesley R, Wang Kai, Shao Lin, Milkie Daniel E, Davidson Michael W, Janetopoulos Chris, Wu Xufeng S, Hammer John A, Liu Zhe, English Brian P, et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science. 2014;346(6208):1257998. doi: 10.1126/science.1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Friedrich Mike, Gan Qiang, Ermolayev Vladimir, Harms Gregory S. STED-SPIM: Stimulated emission depletion improves sheet illumination microscopy resolution. Biophysical Journal. 2011;100:43–45. doi: 10.1016/j.bpj.2010.12.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Friedrich Mike, Harms Gregory S. Axial resolution beyond the diffraction limit of a sheet illumination microscope with stimulated emission depletion. Journal of Biomedical Optics. 2015;20(10):106006. doi: 10.1117/1.jbo.20.10.106006. [DOI] [PubMed] [Google Scholar]
- [160].Hoyer Patrick, Gustavo de Medeiros, Balázs Bálint, Norlin Nils, Besir Christina, Hanne Janina, Kräusslich Hans-Georg, Engelhardt Johann, Sahl Steffen J, Hell Lars. Breaking the diffraction limit of light-sheet fluorescence microscopy by RESOLFT. Proceedings of the National Academy of Sciences. 2016;113(13):3442–3446. doi: 10.1073/pnas.1522292113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Fahrbach Florian O, Rohrbach Alexander. A line scanned light-sheet microscope with phase shaped self-reconstructing beams. Optics Express. 2010;18(23):24229–24244. doi: 10.1364/oe.18.024229. [DOI] [PubMed] [Google Scholar]
- [162].Planchon Thomas A, Gao Liang, Milkie Daniel E, Davidson Michael W, Galbraith James A, Galbraith Catherine G, Betzig Eric. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nature Methods. 2011;8(5):417–423. doi: 10.1038/nmeth.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Fahrbach Florian O, Gurchenkov Vasily, Alessandri Kevin, Nassoy Pierre, Rohrbach Alexander. Self-reconstructing sectioned Bessel beams offer submicron optical sectioning for large fields of view in light-sheet microscopy. Optics Express. 2013;21(9):11425–11440. doi: 10.1364/oe.21.011425. [DOI] [PubMed] [Google Scholar]
- [164].Fahrbach Florian O, Gurchenkov Vasily, Alessandri Kevin, Nassoy Pierre, Rohrbach Alexander. Light-sheet microscopy in thick media using scanned Bessel beams and two-photon fluorescence excitation. Optics Express. 2013;21(11):13824–13839. doi: 10.1364/oe.21.013824. [DOI] [PubMed] [Google Scholar]
- [165].Zhang P, Goodwin PM, Werner JH. Fast, super resolution imaging via Bessel-beam stimulated emission depletion microscopy. Optics Express. 2014;22(10):12398. doi: 10.1364/oe.22.012398. [DOI] [PubMed] [Google Scholar]
- [166].Gohn-Kreuz Cristian, Rohrbach Alexander. Light-sheet generation in inhomogeneous media using self-reconstructing beams and the STED-principle. Optics Express. 2016;24(6):5855. doi: 10.1364/oe.24.005855. [DOI] [PubMed] [Google Scholar]
- [167].Gohn-Kreuz Cristian, Rohrbach Alexander. Light needles in scattering media using self-reconstructing beams and the STED principle. Optica. 2017;4(9):1134. doi: 10.1364/optica.4.001134. [DOI] [PubMed] [Google Scholar]