

Introduction
Ischemic stroke, the most common type of stroke, causes high rate of long-term disability and significant economic burden.1 Current therapeutic options for ischemic stroke include thrombolysis and thrombectomy, which only help less than 10% of patients due to the relatively narrow therapeutic windows.2 Additionally, these treatments are associated with severe complications, such as secondary hemorrhage.2 One important pathology of ischemic stroke is blood-brain barrier (BBB) breakdown.
The BBB is a physical barrier that separates the central nervous system (CNS) and systemic circulation. By preventing the entry of blood components and toxic macromolecules into the brain, the BBB maintains the homeostasis of the CNS. Accumulating evidence suggests that BBB disruption is a common pathology in a variety of neurological disorders,3–5 including stroke. More importantly, BBB breakdown also plays a causal role in some diseases. For example, it has been shown that BBB disruption induces secondary brain injury and actively contributes to the pathogenesis of ischemic stroke.5–7 Over the past century, we have made substantial progress on the cellular constituents of the BBB, including endothelial cells (ECs), pericytes, and astrocytes. The non-cellular component of the BBB–the basement membrane (BM), on the other hand, is understudied and its importance in BBB function has just started to be elucidated.
Here, we first introduce the structure and function of the BM in detail. Next, we summarize how the BM and its individual components change in ischemic stroke. Furthermore, we discuss potential therapeutic targets at the BBB and critical questions that need future research with a focus on the BM. Our goals are to provide a comprehensive review on BM alterations in ischemic stroke, stimulate novel hypotheses in the field, and promote the development of innovative therapies for ischemic stroke.
Structure and Function of the BM
The BBB is a dynamic structure composed of specialized ECs, pericytes, astrocytes, and the BM (Figure 1). These components play important roles in maintaining BBB integrity. Since the cellular constituents of the BBB are well-studied and many excellent reviews already exist, we predominantly focus on the BM in this article.
Figure 1.
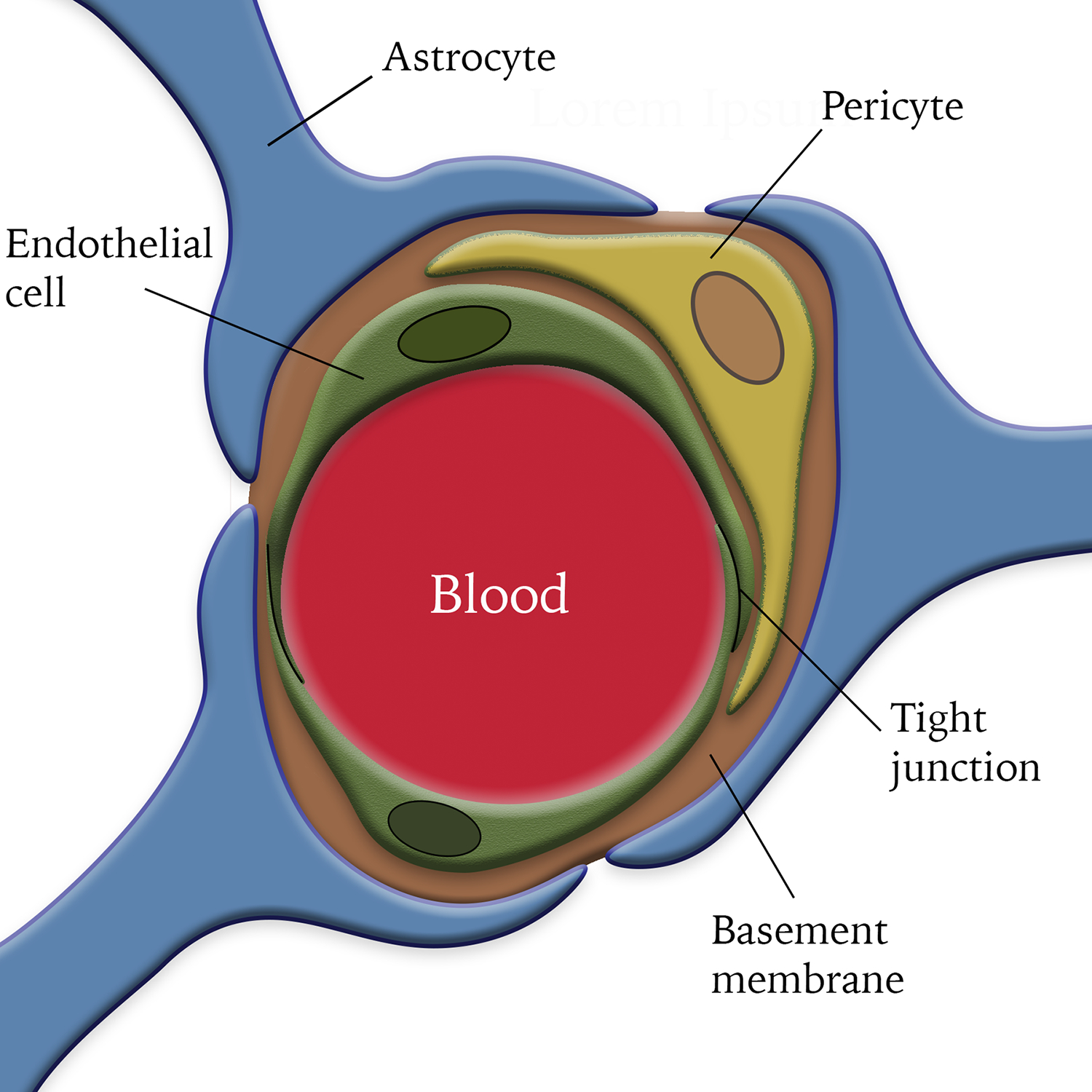
Diagram illustration of the blood-brain barrier. Endothelial cells connect with each other via tight junctions. Pericytes are embedded in the basement membrane and cover endothelial cells. Astrocytes wrap endothelial cells and pericytes with their endfeet.
Basement membrane
The BM is a sheet-like extracellular matrix (ECM) complex beneath epithelium and endothelium.8 At the BBB, the BM encircles the abluminal side of blood vessels and is located at the interface of the circulation system and CNS. Brain BM consists of five major proteins: collagen IV, laminin, nidogen, perlecan, and agrin. The critical roles of these components in BBB integrity are discussed below in detail.
Collagen IV
Collagens are triple-helix proteins composed of three polyproline α-chains. Among 29 different collagen isoforms found so far, collagen IV is the major one that makes up the BM.9, 10 A recent single cell-RNAseq analysis shows that Col4a1 and Col4a2, which make α1-α1-α2 collagen IV, are synthesized by brain endothelial cells, mural cells (pericytes and vascular smooth muscle cells [vSMCs]), and astrocytes.11 Consistent with this finding, Col4a1 and Col4a2 were detected in both endothelial cells and pericytes at mRNA and protein levels in vitro.12
It has been reported that Col4a1/Col4a2-deficient mice show embryonic lethality at E10.5–11.5.13 Interestingly, structural defects of BM are observed in the mutants at E10.5–11.5 but not before E9.5,13 strongly suggesting that collagen IV is not required for initial BM formation but contributes to its stabilization. Similarly, the Col4a1Δex41/+ mutants that skip Col4a1 exon 41 show disorganized vascular BM and brain hemorrhage due to increased intracellular accumulation of mutant collagen IV.14–16 Further studies demonstrate that deletion of Col4a1 exon 41 in endothelial cells and pericytes, but not astrocytes, leads to brain hemorrhage,14 highlighting a critical role of endothelium- and pericyte-derived collagen IV in cerebrovascular function. In addition, mice with various point-mutations in Col4a1 and Col4a2 also develop brain hemorrhage, although not as severe as the Col4a1Δex41/+ mice.16, 17 More importantly, Col4a1/Col4a2 mutations result in BM defects and are associated with diverse disorders in humans, including stroke (hemorrhagic and ischemic) and small vessel disease.16, 18, 19 Genome-wide association studies have linked Col4a120, 21 and Col4a221, 22 with stroke. These results highlight an important role of collagen IV in the pathogenesis of stroke.
Laminin
Laminins are large glycoproteins that consist of one α chain, one β chain, and one γ chain. There are five, four, and three isoforms for α, β, and γ chains, respectively.17, 23, 24 Various combinations of these isoforms generate many different laminin isoforms. Interestingly, different cells synthesize distinct laminin isoforms. For example, astrocytes predominantly make laminin-211,25, 26 pericytes mainly express α4/α5- and γ1-containing laminins,12, 27 and ECs synthesize laminin-411 and −511.25, 28 There are also reports showing that astrocytes make laminin-α5 and pericytes produce laminin-α2. For example, immunohistochemistry revealed laminin-α5 expression in retinal astrocytes.29 Single cell-RNAseq analysis found laminin-α2 expression in brain pericytes11 and laminin-α2 level was significantly reduced in mice with decreased pericyte density.30 Additionally, there is also evidence suggesting the existence of laminin-β2 and -γ3 in the BM. Laminin-β231–33 and laminin-γ331, 32, 34 display a similar expression profile: in the pial membrane, a subset of cerebrovascular BM, and retinal BM. Compared to laminin-β1 or -γ1, laminin-β2 and -γ3 show a much restricted distribution pattern.34 Further studies demonstrate that these isoforms are mainly synthesized by retinal astrocytes and possibly endothelial cells.32, 35, 36
Using the Cre-Lox system, we have shown that ablation of astrocytic laminin-γ1 (laminin-211) leads to severe BBB disruption and age-dependent intracerebral hemorrhage.37 Consistent with our finding, laminin-α2 knockout mice display BBB breakdown,38 strongly indicating an indispensable role of astrocyte-derived γ1-containing laminins in BBB maintenance. Similarly, we also generated mice with laminin-γ1 deficiency in mural cells by using PDGFRβ-driven Cre. In C57Bl6 dominant background, these mutants are grossly normal at young age but start to show mild BBB compromise at old age.39 Given that BBB compromise is not observed in vSMC-specific laminin conditional knockout mice,27 we concluded that pericyte-derived γ1-containing laminins also contributes to BBB integrity but to a lesser extent compared to astrocytic laminin. In addition, we and others have demonstrated that deletion of laminin-α5 in ECs fails to affect BBB integrity,40–42 suggesting a dispensable role of endothelial laminin-α5 (laminin-511) in BBB maintenance and possible compensation by laminin-411. Laminin-α4 knockout mice, however, fail to show BBB disruption in adulthood (after laminin-511 expression43), although vascular leakage is observed at perinatal stage (before laminin-511 expression).44 This data suggests that loss of laminin-411 may be compensated by laminin-511. Due to this mutual compensation between laminin-411 and −511, the role of endothelium-derived γ1-containing laminins in BBB integrity remains unknown. We are currently investigating the function of endothelium-derived γ1-containing using mutants with simultaneous deletion of laminin-411 and −511. Unlike laminin-α2, -α4, -α5, and -γ1, the roles of laminin-β2 and -γ3 in BBB maintenance remain largely unknown. Although laminin-β2−/− mice die at P15-P30 due to defects in the kidney/retina/neuromuscular junctions45–49 and laminin-γ3−/− mice have a normal lifespan with minor abnormalities in the cerebellum and retina,34–36, 50 BBB integrity in these mutants has not been examined. Future research should answer this important question. Altogether, these findings suggest that laminin plays an important role in BBB maintenance under homeostatic condition.
Nidogen
Nidogens, glycoproteins with three globular domains, function as a bridge to connect laminins and collagen IV.51 Two nidogen isoforms (nidogen-1 and −2) have been identified in mammals.52 Single-cell RNAseq analysis demonstrates that nidogen-1 and −2 are mainly produced by mural cells and endothelial cells, and possibly by astrocytes.11 Consistent with this report, nidogen-1 and −2 are found in both endothelial cells and pericytes at the mRNA level in vitro, although they are detected in endothelial cells but not pericytes at the protein level.12
Genetic ablation of nidogen 1 fails to induce obvious abnormalities: the mutant mice are fertile and have normal BM structures in the kidney, skeletal muscle, and heart.53 Interestingly, abnormal movement of the hind legs and a thinner BM in brain capillaries are observed in a different nidogen 1 knockout mouse line.54 Like nidogen 1 mutants, mice lacking nidogen 2 fail to show obvious abnormalities, are fertile, and have normal BM structure.55 In sharp contrast to these single knockout mice, mutants with simultaneous abrogation of both nidogen isoforms die within a day after birth with disorganized BM structure and reduced BM deposition.56, 57 Based on these results, it is reasonable to hypothesize that functional compensation exists between the two isoforms of nidogen. Unfortunately, BBB integrity was not examined in neither the single mutants nor the double knockout mice. Future studies should address the functional significance of nidogen in BBB integrity.
Perlecan
Perlecan is a large heparan sulfate proteoglycan with five domains and three glycosaminoglycan chains in the N-terminus.58 Its expression is substantially increased in cerebrovasculature during embryogenesis,59–61 suggesting an important role of perlecan in brain development. Single cell-RNAseq analysis reveals that perlecan is mainly synthesized by endothelial and mural cells at the BBB.11 In vitro studies show that perlecan is predominantly generated by endothelial cells rather than pericytes, although co-culture of endothelial cells with pericytes induces perlecan expression in pericytes.12, 62 Furthermore, perlecan expression predominantly colocalizes with endothelial cells but not pericytes in the brain,62 indicating that perlecan is mainly synthesized by endothelial cells.
Echoed with the expression data, loss-of-function studies show that perlecan−/− mice die either at E10.5 (40%) or right after birth (60%) due to respiratory failure caused by cartilage defects.59, 63 These perlecan−/− mice display impaired brain development with normal BM structure.64 To avoid lethality and enable investigation of BBB integrity, conditional perlecan-deficient mice, which express perlecan in cartilage, were generated. These mutants have no detectable expression of perlecan in the brain, are grossly normal, and fail to show BBB or BM damage under homeostatic conditions,62 indicating a dispensable role of perlecan in BM formation and BBB maintenance. Similarly, the perlecan hypomorphic mutants (C1532Yneo), which produce approximately 10% of total perlecan, are viable with normal BM structure, although they have a smaller body size, show enhanced susceptibility to exencephaly, and develop skeletal muscle defects.65 BBB integrity in these hypomorphs, however, was not examined. It should be noted that an in vitro study suggests that perlecan may support BBB function via promoting basic fibroblast growth factor (bFGF) uptake into ECs.66 The exact role of perlecan in BBB maintenance needs further investigation.
Agrin
Agrin is a multidomain heparan sulfate proteoglycan. It exists in either a matrix-associated form found at cerebrovascular BM and neuromuscular junction, or a transmembrane form expressed mainly by neurons.67–70 In the brain, agrin is produced by neurons, glia, and vascular cells.67, 69, 71, 72 A recent single cell-RNAseq analysis reports that agrin is synthesized by endothelial cells, mural cells, and astrocytes at the BBB.11
Previous studies have demonstrated that agrin plays an important role in neuromuscular junction formation/maintenance and synaptogenesis.72–74 Echoed with these reports, agrin-deficient mice die at birth due to neuromuscular dysfunctions.73, 75 Recently, there is evidence suggesting that agrin contributes to BBB maintenance. For example, agrin accumulation in brain BM correlates with the development of barrier function in brain microvessels70, 72 and agrin stabilizes adherens junctions in brain endothelial cells in vitro.76 To overcome the perinatal lethality of agrin-deficient mice and enable in vivo BBB permeability study, a miniaturized form of agrin was expressed in muscles of these mutant mice.77 The resulting conditional agrin-deficient mice live to adulthood and lack agrin expression in the brain.77 Although failed to show enhanced BBB permeability, the conditional agrin-deficient mice displayed reduced levels of adhesion junction proteins and tight junction proteins in vitro and in vivo.76 Similarly, mice with endothelium-specific ablation of agrin fail to show BBB disruption or BM abnormalities, although AQP4 is reduced.68 These results suggest that loss of agrin weakens the barrier function but is not sufficient to disrupt it, possibly due to compensatory mechanisms.
Ischemic stroke
Stroke is a severe medical, social, and economic burden. It is estimated that 1 in every 20 deaths in the United States is from stroke, which adds up to about 140,000 deaths each year.78 In addition, stroke caused long-term disability costs about $34 billion every year in the United States.79 Stroke is broadly categorized into hemorrhagic stroke, which is caused by bleeding in or around the brain, and ischemic stroke, which is caused by blockage of blood flow to the brain. Ischemic stroke, which accounts for about 87% of all strokes,79 is the focus of this review.
Based on the cause of blood vessel blockage, ischemic stroke is often divided into three groups: atherosclerosis, cardio-embolism, and lacunar infarcts.80 At the onset of the ischemia, a series of pathological events occur. First, cerebral blood flow (CBF) and oxygen supply are reduced and ATP quickly becomes depleted in the ischemic brain.81 This leads to energy-dependent membrane pump failure, followed by excessive glutamate release and brain edema.82–84 Next, reactive oxygen species (ROS) generation is enhanced85 and BBB is disrupted,86 resulting in inflammatory response and neuronal injury.87
BBB breakdown is a major pathology in ischemic stroke. Time-course studies have demonstrated a biphasic BBB disruption after ischemic stroke in the transient middle cerebral artery occlusion (tMCAO) model, which involves both ischemia and reperfusion. The first BBB opening occurs at 6–12 hours after reperfusion, while the second BBB disruption occurs at 72 hours after reperfusion86, 88–90 (Figure 2A). Similarly, there is evidence supporting a biphasic BBB opening in the permanent middle cerebral artery occlusion (pMCAO) model, which involves ischemia without reperfusion91 (Figure 2B). The temporal changes of BBB permeability after ischemic stroke in both tMCAO and pMCAO models are summarized in Figure 2. This biphasic BBB opening phenomenon, however, is challenged by an observation that neutrophil infiltration displays a single peak after ischemic stroke.92 Similarly, there are many excellent reviews on how ECs, pericytes, and astrocytes alter in ischemic stroke. Thus, in this section, we mainly discuss how the BM and its individual components change in ischemic stroke. These changes are summarized in Table 1.
Figure 2.
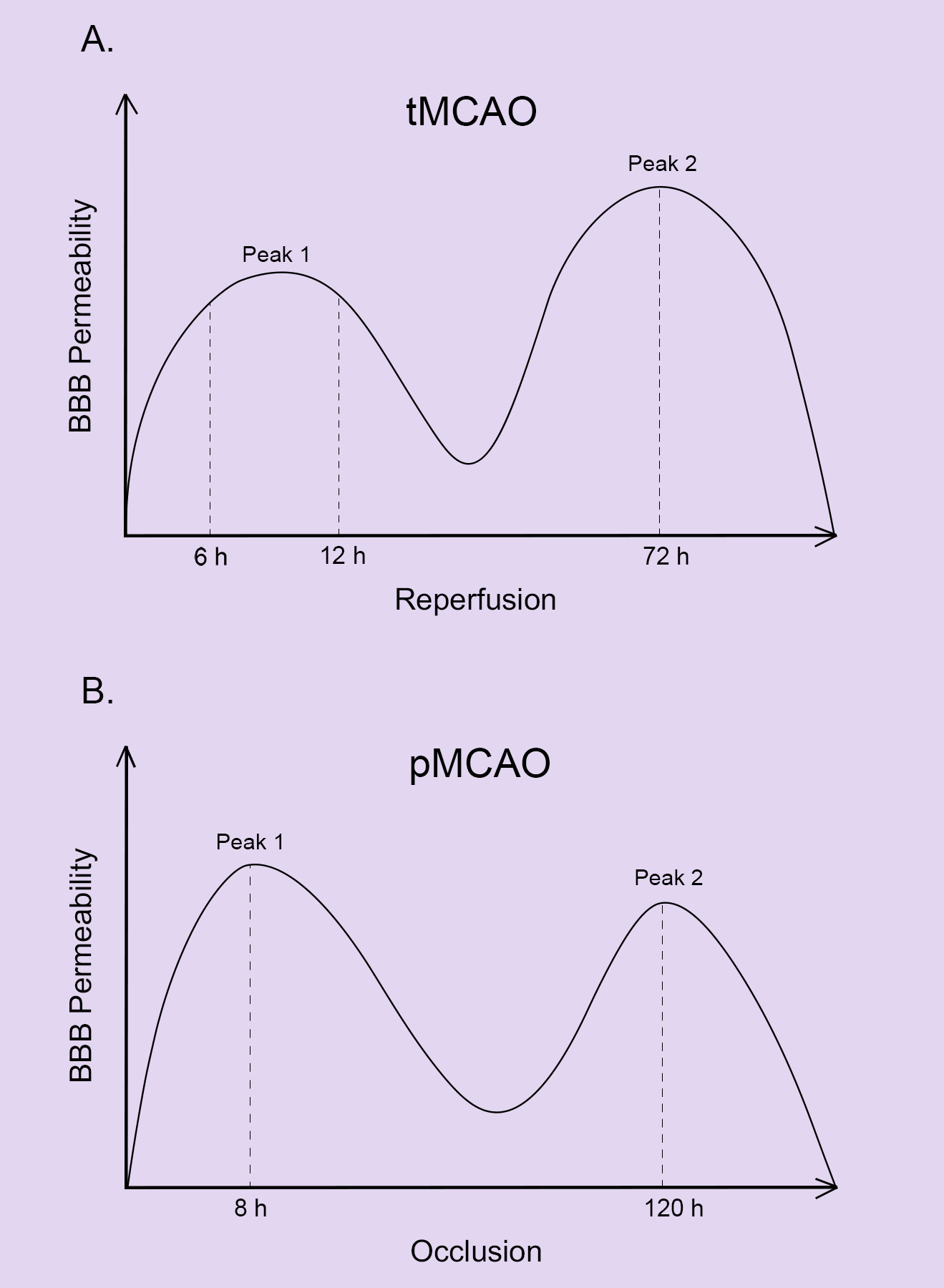
Temporal changes of BBB permeability after ischemic stroke. A. BBB permeability follows a biphasic pattern in the tMCAO model with the first peak at 6–12 hours after reperfusion and the second peak at 72 hours after reperfusion. B. BBB permeability follows a biphasic pattern in the pMCAO model with the first peak at 8 hours after occlusion and the second peak at 120 hours after occlusion.
Table 1.
Changes of BM components after ischemic stroke
| BM/BM Component | Changes after ischemic stroke | Time Course (post injury) | Model | Species | References |
|---|---|---|---|---|---|
| Basement Membrane | Dissolution | 24 hours | tMCAO | Rat | 93 |
| As early as 10 minutes | Focal cerebral ischemia/reperfusion | Rat | 94 | ||
| 12–48 hours | pMCAO | Rat | 96 | ||
| Thickening | 3–72 hours | Photothrombotic stroke | Mouse | 95 | |
| Collagen IV | Decrease | 24 hours | tMCAO | Rat | 104, 137 |
| 24 hours | tMCAO | Baboon | 106 | ||
| 1–6 hours | Thromboembolic model of MCAO | Rat | 105 | ||
| 4–24 hours | tMCAO with systemic inflammation | Mouse | 108 | ||
| 6 hours after death | Ischemic stroke: in regions with neutrophil infiltration | Human | 109 | ||
| Unaltered | 4–24 hours | tMCAO without systemic inflammation | Mouse | 108 | |
| 6 hours after death | Ischemic stroke: in regions without neutrophil infiltration | Human | 109 | ||
| Increase | 24 hours | Infrarenal aorta clamping/removal (spinal cord) | Rat | 107 | |
| Laminin | Decrease | 3 hours | tMCAO | Mouse | 110 |
| 24–72 hours | Transient forebrain ischemia | Mongolian gerbil | 111 | ||
| 1–24 hours | tMCAO | Baboon | 106 | ||
| 1–12 hours after death | - | Human | 112 | ||
| Unaltered | 4–24 hours | tMCAO | Mouse | 108 | |
| Increase | 32 days | Transient ischemia | Rat | 113 | |
| 6–24 hours | pMCAO | Mouse | 114 | ||
| Nidogen | Unknown | - | - | - | - |
| Perlecan | Decrease | 1 hour-7 days | tMCAO | Baboon | 118 |
| Fragmentation | 1–7 days | Focal cerebral ischemia | Rat | 119 | |
| 1–7 days | Transient tandem ipsilateral CCA and distal MCA occlusion | Mouse | 119 | ||
| Agrin | Decrease | 1–24 hours | Transient global cerebral ischemia | Rat | 123 |
| 1–7 days | tMCAO | Rat | 71 |
BM changes in ischemic stroke
Similar to the cellular components of the BBB, the BM also displays substantial changes upon the onset of ischemic stroke. Mounting studies have reported BM dissolution during ischemia. For example, loss of BM and BM degradation have been found to occur soon after ischemia.93, 94 Echoed with this observation, the BM becomes diffused, thickened, and electron-light after ischemia at ultrastructural levels.95, 96 Given that the levels and activities of various proteases, including the MMPs,97–102 are dramatically enhanced after ischemia, it is hypothesized that BM dissolution after stroke is mainly caused by increased degradation rather than reduced production. Below we discuss how each individual component of the BM changes after ischemic stroke.
Collagen IV
How collagen IV changes after ischemic stroke is controversial. On one hand, there is evidence suggesting that collagen IV is degraded. For example, collagen IV level has been found to decrease in rat brains after ischemic stroke in a variety of ischemic stroke models.103–105 Another study on the non-human primates has also shown that the ratio of collagen IV-containing vessels decreases from 1.02±0.03 to 0.57±0.10 in basal ganglia after 3 hours of ischemia followed by 24 hours of reperfusion.106 On the other hand, there is also evidence showing increased or unaltered collagen IV after ischemic stroke. One study reported increased collagen IV level in the spinal cord after ischemic stroke.107 In addition, it has been shown that reduction of collagen IV is not detected after ischemic stroke in a tMCAO model, although it is observed when these mice are further challenged with systemic inflammation.108 Similar to this finding, in a human case study of ischemic stroke with hemorrhagic complications, collagen IV degradation is found in brain regions with neutrophil infiltration but not those without neutrophil infiltration.109 These different findings may be due to distinct animals, ischemic models, and/or time points. Future research should address this discrepancy by utilizing both rodents and human samples, performing multiple ischemic models, and analyzing at a variety of time points after stroke.
Laminin
Like collagen IV, controversial findings exist on how laminin changes after ischemic stroke. On the one hand, there is evidence supporting a reduction of laminin after ischemic stroke. It has been shown that laminin expression is significantly reduced in tMCAO model at 24 hours after reperfusion in mice.110 A similar result is found in Mongolian gerbils after ischemia-reperfusion injury.111 In addition, laminin expression is also significantly decreased after tMCAO and reperfusion in non-human primates (baboons).106 Consistent with these reports, laminin level has been found to decrease after ischemic stroke in a clinical study involving 50 patients, although it is gradually recovered to control level by day 12 after stroke.112 On the other hand, there are also reports showing unaltered or up-regulated laminin level after ischemic stroke. For instance, laminin expression has been found unchanged in ischemic brains with or without systemic inflammation.108 Additionally, significantly elevated laminin level is observed in reactive astrocytes in the brain 32 days after ischemia.113 Furthermore, a transient up-regulation of laminin has been reported in both ischemic brain and in ECs after OGD.114 Again, these controversial results may be due to different animal models and distinct time points. In addition, laminin antibodies used in these studies may also contribute to this discrepancy. A pan-laminin antibody rather than subunit-specific laminin antibody was used in most previous studies. Thus, the changes of specific laminin isoforms after ischemic stroke remain elusive. Given that distinct cells express distinct laminin isoforms with possibly different functions,115, 116 it is important to understand how each laminin isoform changes after ischemic stroke. Future studies should answer this important question.
Nidogen
How nidogen changes after ischemic stroke remain unknown. One study reported that stroke patients had increased plasma nidogen levels, although the increase was not statistically significant.117 Additionally, an in vitro study showed that oxidative stress, a critical pathology of ischemic stroke, up-regulated nidogen level in human brain ECs.117 These findings suggest that nidogen expression may be increased after ischemic stroke. The underlying molecular mechanisms, however, need further investigation.
Perlecan
Perlecan is one of the most sensitive ECM proteins in ischemic brain. It has been demonstrated that there is a 37–61% reduction of perlecan within 2 hours after tMCAO in baboons.118 Similarly, using two different perlecan monoclonal antibodies, it has been shown that perlecan signal is significantly decreased in the striatum after tMCAO.118 Interestingly, unlike other ECM proteins, perlecan is not degraded by proteolysis. Instead, it is cleaved into small fragments. Perlecan domain V, one proteolytic fragment of perlecan, is significantly increased after ischemic stroke in both rodents and humans.119, 120 Functional studies support a beneficial role of perlecan, especially its domain V, in ischemic stroke. For example, larger infarct volume and exacerbated BBB damage were found in conditional perlecan-deficient mice.62 Subsequent studies demonstrated that perlecan exerts a beneficial role in ischemic brain by enhancing pericyte recruitment through cooperative action of PDGFRβ and integrin-α5β1.62 Additionally, recombinant perlecan domain V has been shown to reduce infarct volume, attenuate neuronal death, promote angiogenesis, and improve neurological function in ischemic stroke.62, 119, 121, 122 The therapeutic potential of perlecan domain V is currently under investigation.
Agrin
There is evidence showing that agrin is degraded after ischemic stroke. One study showed a gradual decline of agrin at both mRNA and protein levels at 1–24 hours after reperfusion in a transient global cerebral ischemia model.123 Another study reported that agrin was cleaved by matrix metalloproteinase-3 at 1–7 days after injury in a transient focal cerebral ischemia model.71 These findings suggest a negative correlation between agrin and ischemic stroke. The functional significance of agrin in ischemic stroke, however, remains unknown. Future studies should address this important question.
Targeting BBB in the therapies of ischemic stroke
So far, the only FDA-approved drug for ischemic stroke is tissue plasminogen activator (tPA). Although tPA can efficiently restore blood supply to the ischemic area by breaking down blood clots, its narrow therapeutic window greatly limits its clinical use.124–126 It is estimated that tPA helps less than 10% of stroke patients.127 Therefore, finding novel and effective treatments for ischemic stroke is urgent.
The BBB, a dynamic structure that actively regulates stroke pathogenesis and progression, may be targeted to treat ischemic stroke. First, preventing degradation of the BM is one strategy.128 Mounting evidence suggests that inhibiting MMP activity is able to improve stroke outcome. For example, it has been shown that both MMP-9-neutralizing antibody100 and MMP-9 knockdown129 reduce infarct volume after ischemic stroke. More importantly, MMP inhibitors display promising therapeutic effects in ischemic stroke in several clinical trials.130–133 Next, certain BM components may have a therapeutic effect in ischemic stroke. It has been shown that perlecan domain V plays a neuroprotective role in ischemic stroke and is able to improve stroke outcome.134 Thus, perlecan domain V may be used to treat ischemic stroke. Like perlecan, laminin also contributes to the pathogenesis of ischemic stroke. For instance, we recently reported that mice lacking mural cell-derived laminin-α5 demonstrated decreased infarct volume, less severe BBB disruption, and better neurological function after tMCAO.135 Similarly, mice deficient in integrin-α5, a receptor for laminin and other ECM proteins on ECs at the BBB,116 showed significantly smaller infarct volume and attenuated BBB damage following transient tandem ipsilateral common carotid artery/MCA occlusion.136 These findings suggest that laminin-α5 and integrin-α5 play a detrimental role in ischemic stroke, and that blocking this signaling pathway may have a therapeutic effect in ischemic stroke. Future studies should address: (1) what are the specific laminin (-α5β?γ?) and integrin (α5β?) isoforms involved in this process? and (2) how to target these laminin/integrin isoforms specifically without affecting other isoforms? Answering these important questions will enable us to develop innovative BM-based therapies for ischemic stroke.
Conclusions
BBB breakdown is not only a consequence, it also actively regulates the progression of ischemic stroke. Understanding how exactly the BM and its individual components change in ischemic stroke will enrich our knowledge in stroke pathogenesis, identify novel targets with therapeutic potential, and promote the development of innovative therapies for this devastating disorder. Compared to the cellular constituents of the BBB, its non-cellular component, the BM, is understudied. In this review, we introduce the structure & function of the BM and summarize the alteration of each individual BM component in ischemic stroke. Furthermore, we also discuss novel molecular targets with therapeutic potential and key questions in the field.
Acknowledgments
MK searched the literature and drafted the article. YY gave suggestions and edited the article. All authors read and approved the final article.
Sources of Funding
This work was partially supported by the NIH/NHLBI R01 HL146574 to YY and American Heart Association Scientist Development Grant 16SDG29320001 to YY.
Footnotes
Disclosures
None.
References
- 1.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics—2018 update: A report from the american heart association. Circulation. 2018;137:e67–e492 [DOI] [PubMed] [Google Scholar]
- 2.Goyal M, Menon BK, van Zwam WH, Dippel DW, Mitchell PJ, Demchuk AM, et al. Endovascular thrombectomy after large-vessel ischaemic stroke: A meta-analysis of individual patient data from five randomised trials. Lancet. 2016;387:1723–1731 [DOI] [PubMed] [Google Scholar]
- 3.Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: From physiology to disease and back. Physiol Rev. 2019;99:21–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G. Functional morphology of the blood-brain barrier in health and disease. Acta neuropathologica. 2018;135:311–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdullahi W, Tripathi D, Ronaldson PT. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. American journal of physiology. Cell physiology 2018;315:C343–C356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prakash R, Carmichael ST. Blood-brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr Opin Neurol. 2015;28:556–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann A, Dege T, Kunze R, Ernst AS, Lorenz H, Bohler LI, et al. Early blood-brain barrier disruption in ischemic stroke initiates multifocally around capillaries/venules. Stroke; a journal of cerebral circulation. 2018;49:1479–1487 [DOI] [PubMed] [Google Scholar]
- 8.LeBleu VS, Macdonald B, Kalluri R. Structure and function of basement membranes. Exp Biol Med (Maywood). 2007;232:1121–1129 [DOI] [PubMed] [Google Scholar]
- 9.Sorushanova A, Delgado LM, Wu Z, Shologu N, Kshirsagar A, Raghunath R, et al. The collagen suprafamily: From biosynthesis to advanced biomaterial development. Advanced Materials. 2019;31:1801651. [DOI] [PubMed] [Google Scholar]
- 10.Boudko SP, Danylevych N, Hudson BG, Pedchenko VK. Basement membrane collagen iv: Isolation of functional domains. Methods Cell Biol. 2018;143:171–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554:475–480 [DOI] [PubMed] [Google Scholar]
- 12.Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–5101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen iv is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–1628 [DOI] [PubMed] [Google Scholar]
- 14.Jeanne M, Jorgensen J, Gould DB. Molecular and genetic analyses of collagen type iv mutant mouse models of spontaneous intracerebral hemorrhage identify mechanisms for stroke prevention. Circulation. 2015;131:1555–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, et al. Mutations in col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171 [DOI] [PubMed] [Google Scholar]
- 16.Kuo DS, Labelle-Dumais C, Gould DB. Col4a1 and col4a2 mutations and disease: Insights into pathogenic mechanisms and potential therapeutic targets. Human molecular genetics. 2012;21:R97–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao Y Basement membrane and stroke. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2019;39:3–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lanfranconi S, Markus HS. Col4a1 mutations as a monogenic cause of cerebral small vessel disease: A systematic review. Stroke; a journal of cerebral circulation. 2010;41:e513–518 [DOI] [PubMed] [Google Scholar]
- 19.Federico A, Di Donato I, Bianchi S, Di Palma C, Taglia I, Dotti MT. Hereditary cerebral small vessel diseases: A review. J Neurol Sci. 2012;322:25–30 [DOI] [PubMed] [Google Scholar]
- 20.Malik R, Rannikmae K, Traylor M, Georgakis MK, Sargurupremraj M, Markus HS, et al. Genome-wide meta-analysis identifies 3 novel loci associated with stroke. Annals of neurology. 2018;84:934–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nature genetics. 2018;50:524–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rannikmae K, Davies G, Thomson PA, Bevan S, Devan WJ, Falcone GJ, et al. Common variation in col4a1/col4a2 is associated with sporadic cerebral small vessel disease. Neurology. 2015;84:918–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao Y Laminin: Loss-of-function studies. Cellular and molecular life sciences : CMLS. 2017;74:1095–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nirwane A, Yao Y. Laminins and their receptors in the cns. Biological reviews of the Cambridge Philosophical Society. 2019;94:283–306 [DOI] [PubMed] [Google Scholar]
- 25.Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in t cell recruitment across the blood–brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol. 2001;153:933–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jucker M, Tian M, Norton DD, Sherman C, Kusiak JW. Laminin α2 is a component of brain capillary basement membrane: Reduced expression in dystrophic dy mice. Neuroscience. 1996;71:1153–1161 [DOI] [PubMed] [Google Scholar]
- 27.Gautam J, Zhang X, Yao Y. The role of pericytic laminin in blood brain barrier integrity maintenance. Sci Rep. 2016;6:36450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorokin LM, Pausch F, Frieser M, Kröger S, Ohage E, Deutzmann R. Developmental regulation of the laminin α5 chain suggests a role in epithelial and endothelial cell maturation. Dev Biol. 1997;189:285–300 [DOI] [PubMed] [Google Scholar]
- 29.Stenzel D, Franco CA, Estrach S, Mettouchi A, Sauvaget D, Rosewell I, et al. Endothelial basement membrane limits tip cell formation by inducing dll4/notch signalling in vivo. EMBO Rep. 2011;12:1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561 [DOI] [PubMed] [Google Scholar]
- 31.Radner S, Banos C, Bachay G, Li YN, Hunter DD, Brunken WJ, et al. Beta2 and gamma3 laminins are critical cortical basement membrane components: Ablation of lamb2 and lamc3 genes disrupts cortical lamination and produces dysplasia. Dev Neurobiol. 2013;73:209–229 [DOI] [PubMed] [Google Scholar]
- 32.Gnanaguru G, Bachay G, Biswas S, Pinzón-Duarte G, Hunter DD, Brunken WJ. Laminins containing the β2 and γ3 chains regulate astrocyte migration and angiogenesis in the retina. Development. 2013;140:2050–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunter DD, Llinas R, Ard M, Merlie JP, Sanes JR. Expression of s-laminin and laminin in the developing rat central nervous system. The Journal of comparative neurology. 1992;323:238–251 [DOI] [PubMed] [Google Scholar]
- 34.Li YN, Radner S, French MM, Pinzon-Duarte G, Daly GH, Burgeson RE, et al. The gamma3 chain of laminin is widely but differentially expressed in murine basement membranes: Expression and functional studies. Matrix Biol. 2012;31:120–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biswas S, Bachay G, Chu J, Hunter DD, Brunken WJ. Laminin-dependent interaction between astrocytes and microglia: A role in retinal angiogenesis. The American journal of pathology. 2017;187:2112–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biswas S, Watters J, Bachay G, Varshney S, Hunter DD, Hu H, et al. Laminin-dystroglycan signaling regulates retinal arteriogenesis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2018;32:6261–6273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao Y, Chen Z-L, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun. 2014;5:3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menezes MJ, McClenahan FK, Leiton CV, Aranmolate A, Shan X, Colognato H. The extracellular matrix protein laminin α2 regulates the maturation and function of the blood-brain barrier. J Neurosci. 2014;34:15260–15280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gautam J, Cao Y, Yao Y. Pericytic laminin maintains blood-brain barrier integrity in an age-dependent manner. Translational stroke research. 2019; doi: 10.1007/s12975-019-00709-8 [DOI] [PubMed] [Google Scholar]
- 40.Gautam J, Miner JH, Yao Y. Loss of endothelial laminin α5 exacerbates hemorrhagic brain injury. Transl Stroke Res. 2019;10:705–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song J, Zhang X, Buscher K, Wang Y, Wang H, Di Russo J, et al. Endothelial basement membrane laminin 511 contributes to endothelial junctional tightness and thereby inhibits leukocyte transmigration. Cell Rep. 2017;18:1256–1269 [DOI] [PubMed] [Google Scholar]
- 42.Song J, Lokmic Z, Lämmermann T, Rolf J, Wu C, Zhang X, et al. Extracellular matrix of secondary lymphoid organs impacts on b-cell fate and survival. Proc Natl Acad Sci U S A. 2013;110:E2915–E2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, et al. Endothelial basement membrane laminin α5 selectively inhibits t lymphocyte extravasation into the brain. Nature medicine. 2009;15:519–527 [DOI] [PubMed] [Google Scholar]
- 44.Thyboll J, Kortesmaa J, Cao R, Soininen R, Wang L, Iivanainen A, et al. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol Cell Biol. 2002;22:1194–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature. 1995;374:258–262 [DOI] [PubMed] [Google Scholar]
- 46.Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP. The renal glomerulus of mice lacking s-laminin/laminin beta 2: Nephrosis despite molecular compensation by laminin beta 1. Nat Genet. 1995;10:400–406 [DOI] [PubMed] [Google Scholar]
- 47.Miner JH, Go G, Cunningham J, Patton BL, Jarad G. Transgenic isolation of skeletal muscle and kidney defects in laminin beta2 mutant mice: Implications for pierson syndrome. Development. 2006;133:967–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jarad G, Cunningham J, Shaw AS, Miner JH. Proteinuria precedes podocyte abnormalities inlamb2−/− mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest. 2006;116:2272–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Libby RT, Lavallee CR, Balkema GW, Brunken WJ, Hunter DD. Disruption of laminin beta2 chain production causes alterations in morphology and function in the cns. J Neurosci. 1999;19:9399–9411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Denes V, Witkovsky P, Koch M, Hunter DD, Pinzon-Duarte G, Brunken WJ. Laminin deficits induce alterations in the development of dopaminergic neurons in the mouse retina. Vis Neurosci. 2007;24:549–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yurchenco PD. Basement membranes: Cell scaffoldings and signaling platforms. Cold Spring Harbor Perspectives in Biology. 2011;3:a004911–a004911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mokkapati S, Fleger-Weckmann A, Bechtel M, Koch M, Breitkreutz D, Mayer U, et al. Basement membrane deposition of nidogen 1 but not nidogen 2 requires the nidogen binding module of the laminin gamma1 chain. J Biol Chem. 2011;286:1911–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murshed M, Smyth N, Miosge N, Karolat J, Krieg T, Paulsson M, et al. The absence of nidogen 1 does not affect murine basement membrane formation. Mol Cell Biol. 2000;20:7007–7012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong L, Chen Y, Lewis M, Hsieh J-C, Reing J, Chaillet JR, et al. Neurologic defects and selective disruption of basement membranes in mice lacking entactin-1/nidogen-1. Lab Invest. 2002;82:1617–1630 [DOI] [PubMed] [Google Scholar]
- 55.Schymeinsky J, Nedbal S, Miosge N, Poschl E, Rao C, Beier DR, et al. Gene structure and functional analysis of the mouse nidogen-2 gene: Nidogen-2 is not essential for basement membrane formation in mice. Mol Cell Biol. 2002;22:6820–6830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bader BL, Smyth N, Nedbal S, Miosge N, Baranowsky A, Mokkapati S, et al. Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol Cell Biol. 2005;25:6846–6856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bose K, Nischt R, Page A, Bader BL, Paulsson M, Smyth N. Loss of nidogen-1 and −2 results in syndactyly and changes in limb development. J Biol Chem. 2006;281:39620–39629 [DOI] [PubMed] [Google Scholar]
- 58.Roberts J, Kahle MP, Bix GJ. Perlecan and the blood-brain barrier: Beneficial proteolysis? Front Pharmacol. 2012;3:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, et al. Perlecan maintains the integrity of cartilage and some basement membranes. The Journal of cell biology. 1999;147:1109–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Handler M, Yurchenco PD, Iozzo RV. Developmental expression of perlecan during murine embryogenesis. Dev Dyn. 1997;210:130–145 [DOI] [PubMed] [Google Scholar]
- 61.Edwards DN, Bix GJ. Roles of blood-brain barrier integrins and extracellular matrix in stroke. Am J Physiol Cell Physiol. 2019;316:C252–C263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakamura K, Ikeuchi T, Nara K, Rhodes CS, Zhang P, Chiba Y, et al. Perlecan regulates pericyte dynamics in the maintenance and repair of the blood-brain barrier. The Journal of cell biology. 2019;218:3506–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nature genetics. 1999;23:354–358 [DOI] [PubMed] [Google Scholar]
- 64.Costell M, Gustafsson E, Aszódi A, Mörgelin M, Bloch W, Hunziker E, et al. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rodgers KD, Sasaki T, Aszodi A, Jacenko O. Reduced perlecan in mice results in chondrodysplasia resembling schwartz-jampel syndrome. Hum Mol Genet. 2007;16:515–528 [DOI] [PubMed] [Google Scholar]
- 66.Deguchi Y, Okutsu H, Okura T, Yamada S, Kimura R, Yuge T, et al. Internalization of basic fibroblast growth factor at the mouse blood-brain barrier involves perlecan, a heparan sulfate proteoglycan. J Neurochem. 2002;83:381–389 [DOI] [PubMed] [Google Scholar]
- 67.Thomsen MS, Birkelund S, Burkhart A, Stensballe A, Moos T. Synthesis and deposition of basement membrane proteins by primary brain capillary endothelial cells in a murine model of the blood-brain barrier. Journal of neurochemistry. 2017;140:741–754 [DOI] [PubMed] [Google Scholar]
- 68.Rauch SM, Huen K, Miller MC, Chaudry H, Lau M, Sanes JR, et al. Changes in brain beta-amyloid deposition and aquaporin 4 levels in response to altered agrin expression in mice. J Neuropathol Exp Neurol. 2011;70:1124–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burgess RW, Skarnes WC, Sanes JR. Agrin isoforms with distinct amino termini: Differential expression, localization, and function. The Journal of cell biology. 2000;151:41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barber AJ, Lieth E. Agrin accumulates in the brain microvascular basal lamina during development of the blood-brain barrier. Developmental dynamics : an official publication of the American Association of Anatomists. 1997;208:62–74 [DOI] [PubMed] [Google Scholar]
- 71.Sole S, Petegnief V, Gorina R, Chamorro A, Planas AM. Activation of matrix metalloproteinase-3 and agrin cleavage in cerebral ischemia/reperfusion. J Neuropathol Exp Neurol. 2004;63:338–349 [DOI] [PubMed] [Google Scholar]
- 72.Kroger S, Schroder JE. Agrin in the developing cns: New roles for a synapse organizer. News Physiol Sci. 2002;17:207–212 [DOI] [PubMed] [Google Scholar]
- 73.Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, et al. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535 [DOI] [PubMed] [Google Scholar]
- 74.Hilgenberg LG, Ho KD, Lee D, O’Dowd DK, Smith MA. Agrin regulates neuronal responses to excitatory neurotransmitters in vitro and in vivo. Molecular and cellular neurosciences. 2002;19:97–110 [DOI] [PubMed] [Google Scholar]
- 75.Serpinskaya AS, Feng G, Sanes JR, Craig AM. Synapse formation by hippocampal neurons from agrin-deficient mice. Developmental biology. 1999;205:65–78 [DOI] [PubMed] [Google Scholar]
- 76.Steiner E, Enzmann GU, Lyck R, Lin S, Ruegg MA, Kroger S, et al. The heparan sulfate proteoglycan agrin contributes to barrier properties of mouse brain endothelial cells by stabilizing adherens junctions. Cell and tissue research. 2014;358:465–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin S, Maj M, Bezakova G, Magyar JP, Brenner HR, Ruegg MA. Muscle-wide secretion of a miniaturized form of neural agrin rescues focal neuromuscular innervation in agrin mutant mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11406–11411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang Q, Tong X, Schieb L, Vaughan A, Gillespie C, Wiltz JL, et al. Vital signs: Recent trends in stroke death rates - united states, 2000–2015. MMWR Morb Mortal Wkly Rep. 2017;66:933–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: A report from the american heart association. Circulation. 2017;135:e146–e603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sommer CJ. Ischemic stroke: Experimental models and reality. Acta Neuropathologica. 2017;133:245–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xing C, Arai K, Lo EH, Hommel M. Pathophysiologic cascades in ischemic stroke. International Journal of Stroke. 2012;7:378–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Patel RAG, McMullen PW. Neuroprotection in the treatment of acute ischemic stroke. Progress in Cardiovascular Diseases. 2017;59:542–548 [DOI] [PubMed] [Google Scholar]
- 83.Kostandy BB. The role of glutamate in neuronal ischemic injury: The role of spark in fire. Neurol Sci. 2012;33:223–237 [DOI] [PubMed] [Google Scholar]
- 84.Von Kummer R, Dzialowski I. Imaging of cerebral ischemic edema and neuronal death. Neuroradiology. 2017;59:545–553 [DOI] [PubMed] [Google Scholar]
- 85.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: Mechanisms in search of treatments. Neuron. 2010;67:181–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sifat AE, Vaidya B, Abbruscato TJ. Blood-brain barrier protection as a therapeutic strategy for acute ischemic stroke. The AAPS Journal. 2017;19:957–972 [DOI] [PubMed] [Google Scholar]
- 87.Yang C, Hawkins KE, Doré S, Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. American Journal of Physiology-Cell Physiology. 2019;316:C135–C153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hone E, Hu H, Sprowls S, Farooqi I, Grasmick K, Lockman P. Biphasic blood-brain barrier openings after stroke. Neurol Disord Stroke Int. 2018;1:1011 [Google Scholar]
- 89.Yang Y, Rosenberg GA. Blood–brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219 [DOI] [PubMed] [Google Scholar]
- 91.Liu P, Zhang R, Liu D, Wang J, Yuan C, Zhao X, et al. Time-course investigation of blood-brain barrier permeability and tight junction protein changes in a rat model of permanent focal ischemia. The journal of physiological sciences : JPS. 2018;68:121–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang RL, Chopp M, Chen H, Garcia JH. Temporal profile of ischemic tissue damage, neutrophil response, and vascular plugging following permanent and transient (2h) middle cerebral artery occlusion in the rat. Journal of the neurological sciences. 1994;125:3–10 [DOI] [PubMed] [Google Scholar]
- 93.Hamann GF, Burggraf D, Martens HK, Liebetrau M, Jager G, Wunderlich N, et al. Mild to moderate hypothermia prevents microvascular basal lamina antigen loss in experimental focal cerebral ischemia. Stroke. 2004;35:764–769 [DOI] [PubMed] [Google Scholar]
- 94.Yepes M, Sandkvist M, Wong MK, Coleman TA, Smith E, Cohan SL, et al. Neuroserpin reduces cerebral infarct volume and protects neurons from ischemia-induced apoptosis. Blood. 2000;96:569–576 [PubMed] [Google Scholar]
- 95.Nahirney PC, Reeson P, Brown CE. Ultrastructural analysis of blood-brain barrier breakdown in the peri-infarct zone in young adult and aged mice. J Cereb Blood Flow Metab. 2016;36:413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kwon I, Kim EH, del Zoppo GJ, Heo JH. Ultrastructural and temporal changes of the microvascular basement membrane and astrocyte interface following focal cerebral ischemia. J Neurosci Res. 2009;87:668–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood–brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100 [DOI] [PubMed] [Google Scholar]
- 98.Gasche Y, Fujimura M, Morita-Fujimura Y, Copin J-C, Kawase M, Massengale J, et al. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: A possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 1999:1020–1028 [DOI] [PubMed] [Google Scholar]
- 99.Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, Del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999:624–633 [DOI] [PubMed] [Google Scholar]
- 100.Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC, Dawson VL. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats : Inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030 [DOI] [PubMed] [Google Scholar]
- 101.Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279–291 [DOI] [PubMed] [Google Scholar]
- 102.Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab. 1996:360–366 [DOI] [PubMed] [Google Scholar]
- 103.Hamann GF, Liebetrau M, Martens H, Burggraf D, Kloss CUA, B??Ltemeier G, et al. Microvascular basal lamina injury after experimental focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 2002:526–533 [DOI] [PubMed] [Google Scholar]
- 104.Trinkl A, Vosko MR, Wunderlich N, Dichgans M, Hamann GF. Pravastatin reduces microvascular basal lamina damage following focal cerebral ischemia and reperfusion. Eur J Neurosci. 2006;24:520–526 [DOI] [PubMed] [Google Scholar]
- 105.Vosko MR, Busch E, Burggraf D, Bültemeier G, Hamann GF. Microvascular basal lamina damage in thromboembolic stroke in a rat model. Neurosci Lett. 2003;353:217–220 [DOI] [PubMed] [Google Scholar]
- 106.Hamann GF, Okada Y, Fitridge R, del Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke; a journal of cerebral circulation. 1995;26:2120–2126 [DOI] [PubMed] [Google Scholar]
- 107.Anik I, Kokturk S, Genc H, Cabuk B, Koc K, Yavuz S, et al. Immunohistochemical analysis of timp-2 and collagen types i and iv in experimental spinal cord ischemia–reperfusion injury in rats. J Spinal Cord Med. 2011;34:257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:9451–9462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rosell A, Cuadrado E, Ortega-Aznar A, Hernandez-Guillamon M, Lo EH, Montaner J. Mmp-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type iv collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. 2008;39:1121–1126 [DOI] [PubMed] [Google Scholar]
- 110.Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, et al. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zalewska T, Ziemka-Nalęcz M, Sarnowska A, Domańska-Janik K. Transient forebrain ischemia modulates signal transduction from extracellular matrix in gerbil hippocampus. Brain Res. 2003;977:62–69 [DOI] [PubMed] [Google Scholar]
- 112.Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S. Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: Influence of different therapies. Stroke. 2003;34:2165–2170 [DOI] [PubMed] [Google Scholar]
- 113.Jucker M, Bialobok P, Kleinman HK, Walker LC, Hagg T, Ingram DK. Laminin-like and laminin-binding protein-like immunoreactive astrocytes in rat hippocampus after transient ischemia. Antibody to laminin-binding protein is a sensitive marker of neural injury and degeneration. Annals of the New York Academy of Sciences. 1993;679:245–252 [DOI] [PubMed] [Google Scholar]
- 114.Ji K, Tsirka SE. Inflammation modulates expression of laminin in the central nervous system following ischemic injury. Journal of Neuroinflammation. 2012;9:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yao Y Basement membrane and stroke. J Cereb Blood Flow Metab. 2019;39:3–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nirwane A, Yao Y. Laminins and their receptors in the cns. Biological Reviews. 2019;94:283–306 [DOI] [PubMed] [Google Scholar]
- 117.Ning M, Sarracino DA, Kho AT, Guo S, Lee SR, Krastins B, et al. Proteomic temporal profile of human brain endothelium after oxidative stress. Stroke. 2011;42:37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fukuda S, Fini CA, Mabuchi T, Koziol JA, Eggleston LL Jr., del Zoppo GJ. Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke; a journal of cerebral circulation. 2004;35:998–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee B, Clarke D, Al Ahmad A, Kahle M, Parham C, Auckland L, et al. Perlecan domain v is neuroprotective and proangiogenic following ischemic stroke in rodents. J Clin Invest. 2011;121:3005–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kahle MP, Lee B, Pourmohamad T, Cunningham A, Su H, Kim H, et al. Perlecan domain v is upregulated in human brain arteriovenous malformation and could mediate the vascular endothelial growth factor effect in lesional tissue. Neuroreport. 2012;23:627–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Al-Ahmad AJ, Lee B, Saini M, Bix GJ. Perlecan domain v modulates astrogliosis in vitro and after focal cerebral ischemia through multiple receptors and increased nerve growth factor release. Glia. 2011;59:1822–1840 [DOI] [PubMed] [Google Scholar]
- 122.Bix GJ, Gowing EK, Clarkson AN. Perlecan domain v is neuroprotective and affords functional improvement in a photothrombotic stroke model in young and aged mice. Translational stroke research. 2013;4:515–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Baumann E, Preston E, Slinn J, Stanimirovic D. Post-ischemic hypothermia attenuates loss of the vascular basement membrane proteins, agrin and sparc, and the blood-brain barrier disruption after global cerebral ischemia. Brain research. 2009;1269:185–197 [DOI] [PubMed] [Google Scholar]
- 124.Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. New England Journal of Medicine. 2008;359:1317–1329 [DOI] [PubMed] [Google Scholar]
- 125.Catanese L, Tarsia J, Fisher M. Acute ischemic stroke therapy overview. Circulation Research. 2017;120:541–558 [DOI] [PubMed] [Google Scholar]
- 126.Lin L, Wang X, Yu Z. Ischemia-reperfusion injury in the brain: Mechanisms and potential therapeutic strategies. Biochem Pharmacol (Los Angel). 2016;5:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Adeoye O, Hornung R, Khatri P, Kleindorfer D. Recombinant tissue-type plasminogen activator use for ischemic stroke in the united states: A doubling of treatment rates over the course of 5 years. Stroke. 2011;42:1952–1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chaturvedi M, Kaczmarek L. Mmp-9 inhibition: A therapeutic strategy in ischemic stroke. Molecular Neurobiology. 2014;49:563–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hu Q, Chen C, Khatibi NH, Li L, Yang L, Wang K, et al. Lentivirus-mediated transfer of mmp-9 shrna provides neuroprotection following focal ischemic brain injury in rats. Brain Res. 2011;1367:347–359 [DOI] [PubMed] [Google Scholar]
- 130.Switzer JA, Hess DC, Ergul A, Waller JL, Machado LS, Portik-Dobos V, et al. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke; a journal of cerebral circulation. 2011;42:2633–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Malhotra K, Chang JJ, Khunger A, Blacker D, Switzer JA, Goyal N, et al. Minocycline for acute stroke treatment: A systematic review and meta-analysis of randomized clinical trials. Journal of Neurology. 2018;265:1871–1879 [DOI] [PubMed] [Google Scholar]
- 132.Sheng Z, Liu Y, Li H, Zheng W, Xia B, Zhang X, et al. Efficacy of minocycline in acute ischemic stroke: A systematic review and meta-analysis of rodent and clinical studies. Frontiers in Neurology. 2018;9:1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lakhan SE, Kirchgessner A, Tepper D, Leonard A. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol. 2013;4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bix GJ, Gowing EK, Clarkson AN. Perlecan domain v is neuroprotective and affords functional improvement in a photothrombotic stroke model in young and aged mice. Translational Stroke Research. 2013;4:515–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nirwane A, Johnson J, Nguyen B, Miner JH, Yao Y. Mural cell-derived laminin-α5 plays a detrimental role in ischemic stroke. Acta Neuropathologica Communications. 2019;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Roberts J, De Hoog L, Bix GJ. Mice deficient in endothelial α5 integrin are profoundly resistant to experimental ischemic stroke. J Cereb Blood Flow Metab. 2017;37:85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hamann GF, Liebetrau M, Martens H, Burggraf D, Kloss CUA, Bultemeier G, et al. Microvascular basal lamina injury after experimental focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 2002:526–533 [DOI] [PubMed] [Google Scholar]