

Abstract
Rationale:
Chronic thromboembolic pulmonary hypertension (CTEPH) is characterized by defective thrombus resolution, pulmonary artery obstruction and vasculopathy. Transforming growth factor-beta (TGFβ) signaling mutations have been implicated in pulmonary arterial hypertension, whereas TGFβ’s role in the pathophysiology of CTEPH is unknown.
Objective:
To determine whether defective TGFβ signaling in endothelial cells contributes to thrombus non-resolution and fibrosis.
Methods and Results:
Venous thrombosis was induced by inferior vena cava ligation in mice with genetic deletion of TGFβ1 in platelets (Plt.TGFβ-KO) or TGFβ type II receptors in endothelial cells (End.TGFβRII-KO). Pulmonary endarterectomy specimens from CTEPH patients were analyzed using immunohistochemistry. Primary human and mouse endothelial cells were studied using confocal microscopy, quantitative PCR and western blot. Absence of TGFβ1 in platelets did not alter platelet number or function, but was associated with faster venous thrombus resolution, whereas endothelial TGFβRII deletion resulted in larger, more fibrotic and higher vascularized venous thrombi. Increased circulating active TGFβ1 levels, endothelial TGFβRI/ALK1 and TGFβRI/ALK5 expression were detected in End.TGFβRII-KO mice, and activated TGFβ signaling was present in vessel-rich areas of CTEPH specimens. CTEPH-ECs and murine endothelial cells lacking TGFβRII simultaneously expressed endothelial and mesenchymal markers and transcription factors regulating endothelial-to-mesenchymal transition, similar to TGFβ1-stimulated endothelial cells. Mechanistically, increased endothelin-1 levels were detected in TGFβRII-KO endothelial cells, murine venous thrombi or endarterectomy specimens and plasma of CTEPH patients, and endothelin-1 overexpression was prevented by inhibition of ALK5, and to a lesser extent of ALK1. ALK5 inhibition and endothelin receptor antagonization inhibited mesenchymal lineage conversion in TGFβ1-exposed human and murine endothelial cells and improved venous thrombus resolution and pulmonary vaso-occlusions in End.TGFβRII-KO mice.
Conclusions:
Endothelial TGFβ1 signaling via type I receptors and endothelin-1 contribute to mesenchymal lineage transition and thrombofibrosis, which were prevented by blocking endothelin receptors. Our findings may have relevant implications for the prevention and management of CTEPH.
Keywords: Endothelial cells, endothelin-1, fibrosis, pulmonary hypertension, remodeling, thrombosis
Subject Terms: Basic Science Research, Cell Signaling-Signal Transduction, Fibrosis, Vascular Biology
Graphical Abstract
INTRODUCTION
Pulmonary hypertension encompasses a broad spectrum of cardiac and pulmonary diseases that lead to progressive right heart failure and ultimately death. Chronic thromboembolic pulmonary hypertension (CTEPH) is clinically classified as group 4 of pulmonary hypertension.1 Histologically, CTEPH can be distinguished from other forms of pulmonary hypertension by the occlusion of pulmonary arteries by unresolved thrombofibrotic material partly recanalized by large and irregular neovessels.2, 3
Previous studies suggested that CTEPH represents the result of an inadequate healing response to acute pulmonary embolism and incomplete resolution of thrombi.4 Because the pathomechanisms underlying CTEPH are largely unknown and no therapy is available to reverse this process, surgical removal of the thrombofibrotic material obliterating the pulmonary arteries currently represents the only ‘curative’ approach.
Endothelial cells are important during venous thrombus resolution, as suggested by findings in mice.5-7 Supporting a role for an imbalanced and defective angiogenesis in the pathogenesis of CTEPH, a higher expression of angiostatic growth factors has been reported in endothelial cells isolated from pulmonary endarterectomy (PEA) specimens,8 whereas others found that they possess a highly increased in vitro angiogenic potential.9 Endothelial cells may undergo phenotypic conversion into mesenchymal cells, a process termed endothelial-to-mesenchymal transition (EndMT). EndMT was shown to contribute to tissue fibrosis, including the lung,10, 11 but endothelial phenotype switching may also generate migratory, invasive and sprouting cells required for new vessel formation.12
Transforming Growth Factor-beta (TGFβ) is a multifunctional cytokine affecting many cell types and tissue remodeling processes, including angiogenesis and organ fibrosis. In humans, the TGFβ family consists of more than 30 members, including the isoforms TGFβ1, β2 and β3. Binding of TGFβ ligands assembles a heteromeric receptor complex consisting of two type I (TGFβRI, also called activin receptor-like kinases or ALK1 and ALK5) and two type II (TGFβRII) receptor components, serine-threonine protein kinases which regulate gene expression by activation of SMAD transcription factors.13 TGFβRI/ALK5 receptors are broadly expressed and propagate the signal via phosphorylation of SMAD2 and SMAD3. In endothelial cells, TGFβ can also activate ALK1, which induces phosphorylation of SMAD1 and SMAD5. The diversity of the endothelial response to TGFβ is facilitated, at least in part, via the accessory TGFβ receptor endoglin.14 TGFβ type III receptors (TGFβRIII or betaglycan) are ubiquitously expressed and present TGFβ ligands to type II receptors.15
Fibrotic lesions containing large, irregular vessels are typical histological findings in CTEPH. Interestingly, gene mutations of TGFβ signaling molecules, such as the bone morphogenetic protein receptor type 2, endoglin, ALK1 or SMAD9 have been reported in patients belonging to group 1 of pulmonary hypertension.16 TGFβ expression and an important role of TGFβ signaling pathways have been demonstrated in pulmonary hypertension experimentally induced by hypoxia17 or monocrotaline,18 while TGFβ ligand trapping improved the hemodynamics, remodeling and survival in preclinical pulmonary hypertension models.19 However, the role of TGFβ signaling in venous thrombus resolution and thrombofibrosis in CTEPH has never been directly addressed. Using whole genome microarray analysis of human endothelial cells outgrown from CTEPH PEA specimens, we observed a significant upregulation of TGFβ and associated signaling molecules (M.L.B., unpublished data). The present study was undertaken to examine the hypothesis that overexpression of TGFβ1 and activated TGFβ1 signaling in endothelial cells is causally involved in venous thrombus non-resolution and the development of thrombofibrosis in CTEPH.
METHODS
A detailed description of all materials and methods is provided in the Online Supplemental Information and are available from the corresponding author upon reasonable request.
Studies involving experimental animals.
Generation of mice:
Mice with platelet-specific deletion of transforming growth factor beta 1 (Plt.TGFβ-KO) were generated, as described.20 To generate mice with inducible, endothelial-specific deletion of TGFβRII, mice with loxP-flanked (floxed, flox/flox) TGFβRII locus were mated with mice expressing a tamoxifen-inducible Cre recombinase-estrogen receptor ERT2 fusion protein under control of the endothelial receptor tyrosine kinase (Tie2) promoter. Mice expressing a green fluorescent reporter gene under control of the Tie2.CreERT2 promoter were described earlier.21 Genotyping was performed using primer pairs shown in Online Table I. Cre recombinase activity was induced with tamoxifen citrate containing rodent chow (TD55125; Harlan Teklad). Age- and sex-matched littermates were used throughout the study. Mice were assigned a numerical code to ensure that experiments and analyses were carried out in a blinded manner.
Induction of venous thrombosis and pulmonary embolism:
Venous thrombosis was induced in mice by subtotal ligation of the inferior Vena cava (IVC) over a spaceholder (5-0 Prolene suture; Ethicon). Only male mice, aged between 12 and 14 weeks, were examined to minimize any effects of sex-specific differences in the susceptibility to venous thrombosis, as described before.22 Surgical IVC ligation was performed by the same operateur blinded to the mouse genotype/treatment group. In a subset of mice, bosentan (Tocris) was used to antagonize endothelin-1 signaling using littermate mice randomly assigned to the control or treatment group. Non-invasive hemodynamic measurements were performed to determine blood flow velocity and the extent of venous thrombosis using high frequency ultrasound (Vevo 3100; VisualSonics). Mice that did not develop a thrombus on day 1 after surgery were excluded from the study. Three weeks after surgery, lungs and the IVC were either paraffin embedded and processed for histological analysis or digested and prepared for flow cytometry. Paraffin-embedded IVC segments were cut into 5 μm-thick serial longitudinal sections and out of them, five sections (equally spaced through the thrombus) were stained using Carstairs’ to simultaneously detect fibrin and fibrosis and to select the maximal thrombus area (Online Figure IA and IB). Paraffin-embedded lung tissue was cut into 5 μm-thick serial cross sections and stained using Carstairs’ or rabbit monoclonal antibodies against fibrinogen (abcam; ab189490), CD41 (exbio; 11-763-C100) or TGFβ1 (Novus Biologicals; MAB240). All experiments involving animals had been approved by the Animal Research Committee of the University of Mainz and the authorities of Rhineland-Palatine and complied with national guidelines for the care and use of laboratory animals.
Enzyme-linked immunoassay:
Plasma levels of active TGFβ1, plasminogen activator inhibitor-1 (PAI-1), platelet-factor 4 (PF4) and endothelin-1 (ET-1) were determined using enzyme-linked immunoassay (TGFβ1 from R&D Systems; PAI-1, PF4 and ET-1 from abcam).
Aortic ring assay:
Murine aortas cut into 2 mm-sized pieces were embedded in either matrigel™ or collagen type I (both Corning) and cultivated in Endothelial Cell Growth Medium MV2 kit (PromoCell). In some experiments, SB431542 (10 μM; to block ALK5; Tocris), K02288 (10 μM; to block ALK1; Selleckchem) or bosentan (10 μM; to block endothelin receptors; Tocris) were added.
Studies using primary endothelial cells.
Isolation and cultivation:
Human Umbilical Vein Endothelial Cells (HUVECs, PromoCell), Human Pulmonary Arterial Endothelial Cells (HPAECs, ATCC), Human Pulmonary Vein Endothelial Cells (HPVECs, CellBiologics) and endothelial cells outgrown from PEA tissue (CTEPH-ECs) were cultivated, as suggested by the supplier or as published.2 Mouse primary endothelial cells (mPECs) were isolated from lungs of male and female mice using magnetic beads (Miltenyi).
Immunofluorescence confocal microscopy:
Cells were plated onto gelatin-coated coverslips, fixed with 4% PFA and permeabilized using 0.2% Triton X-100 (in PBS; Roth). After blocking of unspecific antigen binding, primary antibodies were added followed by incubation with fluorescence-conjugated secondary antibodies. Images were collected on a Leica LSM710 confocal microscope and analyzed with Leica software (LAS X). Isotype IgG controls (Dako; X0944 for mouse, X0936 for rabbit and Santa Cruz, sc-2026 for rat) were used to exclude unspecific background staining (Online Figure IIA and IIB).
Studies using murine platelets.
Platelet counts in anticoagulated whole blood were determined using an Automated Hematology Analyzer (KX-21N; Sysmex). PRP was obtained by centrifugation of citrated blood and processed for flow cytometry analysis of activated integrin αIIbβ3 expression or light transmission aggregometry in response to ADP (Sigma-Aldrich, A2754), thrombin (Sigma-Aldrich; T6624) or convulxin (EnzoLifeSciences, ALX-350-100-C050), respectively.
Studies involving human tissue.
Patient recruitment:
Tissue specimens were obtained from patients diagnosed with CTEPH who underwent PEA surgery at the Kerckhoff Clinic, Department of Thoracic Surgery, Bad Nauheim, Germany. All patients with confirmed CTEPH transferred for PEA were eligible for the study; only patients who refused or withdrew consent were excluded. Plasma samples used in the study were obtained from the Mainz Registry for Pulmonary Hypertension (PHYREM) at the University Medical Center Mainz, Germany. Out of 150 consecutive patients who had been prospectively enrolled in the registry at the time that plasma for the present study was requested, we selected samples (n=45 in total), which met specific requirements, such as: (i) plasma availability from three follow-up appointments; (ii) absence of HIV infection; (iii) absence of inflammatory disease; and (iv) absence of congenital heart disease. Of these patients, the diagnosis was chronic thromboembolic pulmonary hypertension (CTEPH) in 19 and pulmonary arterial hypertension in 26. Both studies were conducted in accordance with the amended Declaration of Helsinki and approved by the local ethics committee, and all patients gave written informed consent.
Histology:
Serial cross sections and microarrays through PEA tissue specimens were stained using modified Carstairs’ stain and classified into five distinct regions containing primarily fresh or organized thrombus, myofibroblasts, vessels or fibrosis, as described.2 Interstitial collagen was detected using picrosirius red staining followed by microscopy under polarized light.
Flow cytometry.
Cells were fixed in 0.1% PFA and permeabilized using 0.1% Triton X-100. Unspecific binding was blocked using FcR blocking reagent (Miltenyi Biotech; 130-092-575) followed by incubation with fluorescence-labeled primary antibodies.
Quantitative real time polymerase chain reaction.
Total RNA was isolated using Trizol® reagent (Ambion), treated with DNase I (Promega) and reverse transcribed into cDNA using iScript cDNA Synthesis Kit (BioRad). Primer sequences and quantitative real time PCR conditions are shown in Online Table II (human) and Online Table III (mouse).
Western blot.
Cells were lysed in RIPA buffer (Cell Signaling Technology). Proteins were fractionated by gel electrophoresis and transferred to nitrocellulose membranes (Protran®, Whatman). After blocking, membranes were incubated overnight with primary antibodies against ET-1 (ThermoFisher; PA3-067). Antibodies against glyceraldehyde-3-phosphate dehydrogenase (GAPDH; HyTest; 5G4 Mab 6C5) were used to ensure total protein loading. Protein bands were visualized using horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences; NA934V for rabbit and NA931V for mouse) and detected with SuperSignal® West Pico Substrate (Pierce).
Immunohistochemistry.
Immunohistochemistry was performed on 4% zinc formalin-fixed paraffin sections, as described.2 For mouse thrombi, serial sections were used; for PEA specimens, tissue microarrays were prepared. Sections were photographed on an Olympus BX51 microscope and analyzed using analysis software (Image ProPlus). Isotype IgG controls were used to exclude unspecific background staining (Online Figure IIC and IID).
Statistical analysis.
Quantitative data are presented as mean±standard error of the mean (SEM). Normal distribution was examined using the D’Agostino-Pearson omnibus normality test. For comparison of two groups and normal distribution, Student’s t-test was performed. If more than two groups and/or different time points were compared, One- resp. Two-way ANOVA followed by Bonferroni’s multiple comparisons test were performed. Multiple test correction across experiments was not performed. Non-parametric tests were used if normal distribution was not present. All analyses were performed using GraphPad Prism version 7.04 for Windows (GraphPad Prism Software).
RESULTS
Platelet-specific deletion of TGFβ1 results in faster venous thrombus resolution.
TGFβ1, its receptors (TGFβRII, TGFβRI/ALK1, TGFβRI/ALK5) and phospho-SMAD2/3 or phospho-SMAD1/5 were expressed in resolving venous thrombi obtained from C57BL/6 wild-type mice at day 21 after IVC ligation (Online Figure IIIA). To study the role of TGFβ during venous thrombus resolution, mice with genetic deletion of TGFβ1 in platelets (Plt.TGFβ-KO), the main source of circulating TGFβ1,23 were subjected to IVC ligation and venous thrombus resolution followed over 21 days. Of note, we and others had previously shown that absence of TGFβ in platelets does not affect platelet function24,25 and that platelets contribute to venous thrombus progression in this model.26 The presence and co-localization of TGFβ1 and CD41-positive platelets in 2 days-old venous thrombi is shown in Online Figure IIIB. Non-invasive ultrasound imaging revealed that the percentage of mice forming a venous thrombus (76 vs. 82% in Plt.TGFβ-WT; P=0.3856; Figure 1A) was similar in the Plt.TGFβ-WT and Plt.TGFβ-KO group. In those mice that formed a thrombus and were included in the study, the thrombus area at day 1 after IVC ligation also did not differ (P=0.6818; Online Figure IVA). Circulating platelet numbers (P=0.8359 and P=0.7046, respectively; Online Figure VA) and plasma PF4 levels (P>0.9999 for both; Online Figure VB) did not differ between Plt.TGFβ-WT and Plt.TGFβ-KO mice, uninjured or at day 21 after IVC ligation. Analysis of activated integrin αIIbβ3 expression, without and in response to ADP and thrombin (Online Figure VE and VF) and platelet aggregation in response to thrombin (Online Figure VIA and VIB) also revealed no differences between Plt.TGFβ-WT and Plt.TGFβ-KO mice. Venous thrombi resolved significantly faster in Plt.TGFβ-KO mice compared to Plt.TGFβ-WT controls (P=0.0156 at day 21; Figure 1B) resulting in significantly smaller thrombi at day 21 (2.91±0.33 vs. 5.95±0.62 mm2 in Plt.TGFβ-WT mice, P=0.0004; Figure 1C and 1D; 2.31±0.65 vs. 6.11±0.67 mm3, P=0.0067; not shown). Carstairs’ staining revealed no differences in the fibrotic area as proportion of the total thrombus area between Plt.TGFβ-KO and Plt.TGFβ-WT mice (22.7±2.02 vs. 26.4±2.06% in Plt.TGFβ-WT mice, P=0.2115; Figure 1E and 1F). Similar findings were obtained following immunohistochemistry of smooth muscle α-actin (2.78±0.28 vs. 2.76±0.69% in Plt.TGFβ-WT mice, P=0.9838; not shown) and sirius red staining of interstitial collagen (6.72±1.83 vs. 5.81±1.16% in Plt.TGFβ-WT mice, P=0.6615; Figure 1G and 1H).
Figure 1. Platelet-specific deletion of TGFβ1 and venous thrombus resolution.

The percentage of mice that formed a venous thrombus on day 1 after IVC ligation is shown (A). Thrombus resolution (B) and thrombus area at day 21 after IVC ligation (C, D) were measured by ultrasound in Plt.TGFβ-WT (n=10) and Plt.TGFβ-KO (n=9) mice. In panel B, thrombus area at day 21 is expressed relative to thrombus area at day 1 after surgery. In panel C, representative ultrasound snapshots of thrombosed IVC segments in Plt.TGFβ-WT and Plt.TGFβ-KO mice at day 21 after IVC ligation are shown (with 3-dimensional thrombus reconstruction as insert). Representative pictures of longitudinal cross-sections through the thrombosed IVC at day 21 after ligation following Carstairs’ staining (E) and quantification of thrombofibrosis (blue signal) (n=10 Plt.TGFβ-WT and n=9 Plt.TGFβ-KO mice; F). Representative pictures after Sirius red stain (G) and quantification of interstitial collagen (red signal) (n=15 for Plt.TGFβ-WT and n=9 for Plt.TGFβ-KO mice; H). Exact p-values, as determined by χ2 test in (A), Two-Way ANOVA followed by Bonferroni’s multiple comparisons test (3 comparisons per genotype) in (B; non-significant p-values are not shown), Mann-Whitney test in (D) and Student’s t-test in (F) and (H), are shown in the graphs. Scale bars in (E) and (G) represent 200 μm.
Deletion of TGFβRII in endothelial cells is associated with increased expression of TGFβ type I receptors and activated TGFβ1 signaling.
To examine the importance of TGFβ signaling in endothelial cells for thrombus (non)-resolution, we generated mice in which TGFβRII was specifically deleted in endothelial cells (End.TGFβRII-KO). Confocal microscopy analysis confirmed lower TGFβRII expression in endothelial cells lining the IVC of End.TGFβRII-KO mice compared to End.TGFβRII-WT controls (Online Figure VIIA). Flow cytometry analysis of CD31-positive cells from mouse lungs demonstrated significantly reduced numbers of TGFβRII-positive cells in End.TGFβRII-KO mice compared to cells from End.TGFβRII-WT mice (0.25-fold, P<1×10−4), but also revealed a significant increase in the number of CD31-positive cells expressing ALK1 (2.95-fold, P=0.0074), ALK5 (11.3-fold, P=0.0199), phospho-SMAD2/3 (1.89-fold, P=0.0008) or phospho-SMAD5 (1.85-fold, P=0.0424), whereas the number of CD31/TGFβRIII-positive cells did not differ (1.08-fold; P=0.6189; Online Figure VIIB). Confocal microscopy analysis confirmed higher TGFβRI/ALK1 and TGFβRI/ALK5 expression in mPECs isolated from End.TGFβRII-KO mice compared to End.TGFβRII-WT controls (Online Figure VIIC).
Total (i.e. latent) TGFβ1 levels in plasma did not differ, however endothelial-specific deletion of TGFβRII was associated with significantly increased plasma levels of activated TGFβ1 (604±1.5 vs. 148±3.2 pg/mL in End.TGFβRII-WT mice; P=0.0050). In this regard, mPECs from End.TGFβRII-KO mice were found to overexpress factors involved in TGFβ activation, such as matrix metalloproteinase-9 (MMP9; 3.7-fold, P=0.0202) or thrombospondin-1 (TSP1; 2.9-fold, P=0.0089; Online Figure VIIIA). Activated TGFβ1 plasma levels further increased at day 21 after IVC ligation in End.TGFβRII-WT (25.8-fold), and to a greater extent in End.TGFβRII-KO mice (27.4-fold; P=0.0377).
Activated TGFβ signaling in endothelial cells delays thrombus resolution and promotes the formation of fibrotic, vessel-rich thrombi.
Next, End.TGFβRII-WT and End.TGFβRII-KO mice were subjected to IVC ligation. The percentage of mice developing a venous thrombus (73 vs. 66% in End.TGFβRII-WT; P=0.3569; Figure 2A) and the thrombus area at day 1 after surgery (P=0.3539; Online Figure IVB) did not significantly differ between genotypes. Similar to Plt.TGFβ mice, plasma PF4 levels (Online Figure VC), platelet activated integrin αIIbβ3 expression (Online Figure VG and VH) and platelet aggregation in response to thrombin (Online Figure VIC and VID) did not differ in End.TGFβRII-WT and End.TGFβRII-KO mice, uninjured or at day 21 after IVC ligation. Preincubation of C57BL6/J mouse platelets with TGFβ1 (10 ng/mL) did not alter their response to the platelet agonists thrombin (Online Figure VIE) or convulxin (Online Figure VIF). Endothelial deletion of TGFβRII was associated with significantly slower thrombus resolution (P<1×10−4 vs. End.TGFβRII-WT mice at day 14 and 21 after IVC ligation; Figure 2B) resulting in increased venous thrombus size at day 21 (3.71±1.05 vs. 0.85±0.38 mm2 in End.TGFβRII-WT mice, P=0.0187; Figure 2C and 2D; 16.63±0.41 vs. 5.23±1.05 mm3, P=0.0005; not shown). Analysis of mouse lungs revealed significantly higher numbers of vessels occluded with fibrinogen-positive, unresolved thrombotic material in End.TGFβRII-KO mice that had formed a thrombus compared to End.TGFβRII-WT mice (6.6±0.53 vs. 1.0±0.49 occluded arteries in End.TGFβRII-WT mice, P<1×10−4; Figure 3A-3C). In these areas, CD41-positive platelets and strong TGFβ1 immunosignals could also be observed (Figure 3D). Of note, clinical signs of respiratory insufficiency or increased mortality (less than 1% in both groups) were not observed in End.TGFβRII-KO mice. Pulmonary artery obstructions were less frequent in mice that did not develop venous thrombosis, irrespective of their genotype (2.7±0.57 vs. 2.0±0.65 in End.TGFβRII-WT mice, P=0.9368), and also not observed in Plt.TGFβ mice (P>0.9999; Online Figure IXA-IXC). Venous thrombi in End.TGFβRII-KO mice contained significantly higher amounts of fibrotic material (27.6±2.5 vs. 16.0±2.8% in End.TGFβRII-WT mice; P=0.0311; Figure 2E and 2F) and interstitial collagen (28.2±2.4 vs. 17.9±2.6%; P=0.0262; Figure 2G and 2H) compared to End.TGFβRII-WT animals. The SMA-positive area was also significantly increased (6.9±0.5 vs. 3.9±1.4% in End.TGFβRII-WT mice; P=0.0465; Figure 4A and 4C). Furthermore, SMA immunosignals frequently co-localized with CD31-positive cells lining vascular channels, which were increased in thrombi of End.TGFβRII-KO (P=0.0029 vs. End.TGFβRII-WT mice; Figure 4D), a finding reminiscent of vessels in patients with CTEPH (Figure 4B).
Figure 2. Endothelial-specific deletion of TGFβRII and venous thrombus remodeling.

The percentage of mice that formed a venous thrombus on day 1 after IVC ligation is shown (A). Thrombus resolution (B) and thrombus area (C, D) measured by ultrasound at day 21 after IVC ligation in End.TGFβRII-WT and End.TGFβRII-KO mice (n=11 mice per group). In panel B, thrombus area at day 14 and 21 is expressed relative to thrombus area at day 1. In panel C, representative ultrasound snapshots of thrombosed IVC segments in End.TGFβRII-WT and End.TGFβRII-KO mice at day 21 after IVC ligation shown (with 3-dimensional thrombus reconstruction as insert). Representative pictures of longitudinal cross-sections through the thrombosed IVC at day 21 after ligation following Carstairs’ staining (E) and quantification of thrombofibrosis (blue signal) (n=7 End.TGFβRII-WT and n=9 End.TGFβRII-KO mice; F). Representative pictures after Sirius red stain (G) and quantification of interstitial collagen (red signal) (n=7 mice per group; H). Exact p-values, as determined by χ2 test in (A), Two-Way ANOVA followed by Bonferroni’s multiple comparisons test (3 comparisons per genotype) in (B; non-significant p-values are not shown), and Mann-Whitney test in (D, F and H) are shown in the graphs. Scale bars represent 200 μm.
Figure 3. Detection of pulmonary vascular occlusions following venous thrombosis.

Representative images after analysis of pulmonary artery obstructions in PBS- and formalin-infused paraffin-embedded mouse lungs from End.TGFβRII-WT and End.TGFβRII-KO mice at day 21 after IVC ligation using Carstairs’ staining (A; arrows) or immunostaining of fibrinogen (B). Mice that developed a venous thrombus at day 1 and mice without a thrombus were examined separately to distinguish pulmonary embolization and in situ thrombosis (n=7 mice per group). End.TGFβRII-KO mice treated with bosentan over 3 weeks were also examined (marked in green; n=5 End.TGFβRII-WT and n=7 End.TGFβRII-KO mice). Scale bars represent 10 μm. The results of the quantitative analysis of red, fibrin-rich material (arrows in A) in the pulmonary parenchyma are shown (C). Exact p-values, as determined by One-Way ANOVA followed by Bonferroni’s multiple comparisons test (7 comparisons) are shown in panel C. Non-significant p-values are not shown. Representative images of lungs from End.TGFβRII-WT and End.TGFβRII-KO mice at day 21 after IVC ligation showing results after immunostaining for the platelet marker CD41 and TGFβ1. Scale bar represent 10 μm.
Figure 4. Presence of myofibroblasts and endothelial cells in chronic venous thrombi of End.TGFβRII mice.

Representative images and quantitative analysis of the SMA-immunopositive area (A, C) and vascular channels lined by CD31-immunopositive cells (A, D) in 21-days-old venous thrombi of End.TGFβRII-WT (n=6 in C and n=7 in D) and End.TGFβRII-KO mice (n=6). Data shown in panel D represent the area lined by CD31-positive channels per total thrombus area (%). Scale bars represent 10 μm. Cells expressing endothelial and myofibroblast markers are highlighted by black arrows. Representative images of SMA- and CD31-immunopositive cells in vessel-rich regions of PEA tissue specimens from patients with CTEPH are shown in (B). Scale bars represent 100 μm. Representative flow cytometry dot blots after analysis of 21-days-old venous thrombi for the number of cells double-positive for CD31 and FSP1 (n=4 mice per group; E, F) or CDH5 and SMA (n=4 mice per group; G, H). Exact p-values, as determined by Mann-Whitney test, are shown in panels C, D, F and H.
Flow cytometry analysis of venous thrombi 21 days after IVC ligation confirmed higher numbers of CD31-positive cells also expressing FSP1 (65.9±3.2 vs. 39.9±5.3% in End.TGFβRII-WT mice; P=0.0286; Figure 4E and 4F) or CDH5-positive cells also expressing SMA (23.6±1.7 vs. 13.2±2.0% in End.TGFβRII-WT mice; P=0.0286; Figure 4G and 4H) in End.TGFβRII-KO mice. Lineage tracing employing endothelial reporter animals and confocal microscopy confirmed that (GFP-positive) endothelial cells expressed the mesenchymal markers SMA, FSP1 or DDR2 at day 21 (Online Figure XA), in line with endothelial-to-mesenchymal phenotype conversion during venous thrombus remodeling. On the other hand, and regarding the possible expression of FSP1 in myelomonocytic cells, a cell type also involved in thrombus resolution, the number of Mac2-positive macrophages was decreased within organizing thrombi of End.TGFβRII-KO mice (7.5±0.15 vs. 13.0±1.7% in thrombi of End.TGFβRII-WT mice; P=0.0174; not shown).
Quantitative PCR analysis of CD31-positive mPECs revealed significantly higher mRNA levels of mesenchymal markers and transcription factors involved in regulating endothelial-to-mesenchymal transition (SNAIL; 3.0-fold, P=0.0079), TWIST (7.0-fold, P=0.0119), ZEB1 (3.6-fold, P=0.0049) upon genetic deletion of TGFβRII, whereas mRNA expression of the endothelial marker Cdh5 was significantly reduced (0.37-fold, P<1×10−4; Online Figure VIIIA). Interestingly, mRNA levels of FGFR1, an antagonist of TGFβ signaling involved in EndMT,27 were significantly lower in endothelial cells lacking TGFβ type II receptors (0.16-fold, P=0.0001; Online Figure VIIIA), and decreased numbers of CD31/FGFR1-positive cells were confirmed using flow cytometry analysis of mouse lungs (3.0-fold, P<1×10−4; Online Figure VIIB). At the functional level, a higher number of endothelial lectin-positive cells co-expressing SMA or FSP1 (44.3±2.0 vs. 1.0±1.0% and 49.7±1.2 vs. 0.0±0% in End.TGFβRII-WT, respectively, P<1×10−4 for both; Online Figure XB and XC) and an increased sprouting into matrigel™ or collagen (342±42 vs. 105±11 μm total sprout length in End.TGFβRII-WT, P<1×10−4, and 258±24 vs. 94±15 μm, P<1×10−4; Online Figure XD) was observed in aortic rings from End.TGFβRII-KO mice, whereas dual-positive cells were rarely detected in their End.TGFβRII-WT counterparts. These findings in transgenic mice suggested that increased TGFβ1 signaling via type I receptors in endothelial cells impairs venous thrombus resolution and that enhanced endothelial-to-mesenchymal transition may play a role in this process.
Evidence of overactive TGFβ signaling and endothelial-to-mesenchymal transition in patients with chronic thromboembolic pulmonary hypertension.
To examine the importance of our findings in mice for thrombus non-resolution in humans, tissue microarrays of PEA specimens from CTEPH patients were immunostained for components of the TGFβ signaling pathway. Expression of TGFβ1 (Figure 5A and 5B) and its receptors TGFβRII (Figure 5A and 5C), TGFβRI/ALK1 (Figure 5A and 5D) and TGFβRI/ALK5 (Figure 5A and 5E) was significantly increased in vessel-rich regions, and TGFβRII- and ALK5-immunopositive cells were also observed at significantly increased numbers in areas containing myofibroblasts. Phospho-SMAD2/3 (Figure 5A and 5F) and phospho-SMAD5-positive cells (Figure 5A and 5G) were also significantly elevated in areas containing vessels compared to all other regions.
Figure 5. Expression of TGFβ1, its receptors and downstream signaling mediators in pulmonary endarterectomy specimens of patients with CTEPH.

Representative images in regions primarily containing fresh or organized thrombus, myofibroblasts, vessels or fibrosis (A) and the results of the quantitative analysis after immunohistochemical analysis of the expression of TGFβ1 (B), TGFβRII (C), TGFβRI/ALK1 (D), TGFβRI/ALK5 (E), p-SMAD2/3 (F) and p-SMAD5 (G) in PEA samples from n=9 CTEPH patients are shown. Scale bars represent 100 μm. Exact p-values, as determined by One-Way ANOVA followed by Bonferroni’s multiple comparisons test (10 comparisons), are shown in panels B-G. Non-significant p-values are not shown.
Immunohistochemical analysis of PEA tissue microarrays suggested co-localization of immunosignals for CD31 and CDH5 with FSP1, DDR2 or SMA in vessel-rich regions (Online Figure XIA-XIE). Confocal microscopy analysis confirmed that CDH5-positive cells also express SMA, as shown in PEA specimens and primary endothelial cells isolated from CTEPH tissue (Figure 6A). HPAECs examined in parallel were found to express endothelial, but not mesenchymal markers. HPAECs were used as controls in these experiments because preliminary analyses confirmed significantly higher expression of TGFβRII (P=0.0316 vs. HUVECs), ALK1 (P=0.0079 vs. HPVECs and P=0.0047 vs. HUVECs), ALK5 (P=0.0024 vs. HPVECs and P=0.0012 vs. HUVECs) and endoglin (P=0.0126 vs. HPVECs and P=0.0088 vs. HUVECs) (Online Figure XIIA-XIID). Quantitative real time PCR analysis of CTEPH-ECs demonstrated that CDH5 mRNA expression levels were significantly reduced (0.36-fold, P=0.0012; Figure 6C) and SMA (ACTA2) mRNA levels significantly increased (23.6-fold, P<1×10−4; Figure 6D) compared to HPAECs, and increased SMA expression was also observed in HPAECs following exposure to TGFβ1, whereas HUVECs (lacking TGFβ receptor type I-III isoforms) did not respond to TGFβ1 (Figure 6B).
Figure 6. Expression of endothelial and mesenchymal markers in primary human pulmonary endothelial cells from patients with CTEPH: comparison to TGFβ1 stimulation.

Representative pictures after immunofluorescence confocal microscopy for VE-cadherin (CDH5) and SMA in CTEPH tissue specimens, endothelial cells outgrown from CTEPH tissue (CTEPH-ECs) and HPAECs (A). Representative confocal pictures for CDH5 and SMA in HPAECs and HUVECs with and without TGFβ1 stimulation (10 ng/ml for 7 days) to induce EndMT (B). Quantitative analysis of CDH5 (n=5 biological replicates; C), SMA (ACTA2; n=4 for CTEPH and n=6 for all others; D), SNAIL (n=3; E), ZEB1 (n=3; F), TWIST (n=5; G) and FGFR1 (n=4 for CTEPH and n=5 for all others; H) mRNA expression in HPAECs with and without TGFβ1 stimulation (10 ng/mL for 7 days) and CTEPH-ECs. Scale bars represent 10 μm. Exact p-values, as determined by One-Way ANOVA followed by Bonferroni’s multiple comparisons test (3 comparisons) and by Mann-Whitney test for the comparison of HPAECs with and without TGFβ1 stimulation, are shown in panels C-H. Non-significant p-values are not shown.
Previous studies have shown that TGFβ1 is a major inducer of EndMT.27 To examine the importance of TGFβ1 for EndMT in CTEPH, the expression of endothelial and mesenchymal markers and transcription factors regulating EndMT was examined in CTEPH-ECs and compared to those in HPAECs with and without TGFβ1 stimulation. Highly elevated mRNA levels of transcription factors involved in EndMT, such as SNAIL (8.0-fold, P<1×10−4; Figure 6E), ZEB1 (2.3-fold, P=0.0043; Figure 6F) and TWIST (73.4-fold, P=0.0241; Figure 6G) were observed in CTEPH-ECs, similar to HPAECs stimulated with TGFβ1. FGFR1 mRNA levels were significantly lower in CTEPH-ECs and TGFβ1-stimulated HPAECs (0.36-fold, P=0.0042, and 0.12-fold, P=0.0002, respectively; Figure 6H). Taken together, increased TGFβ1 expression and signaling as well as endothelial-to-mesenchymal transition were observed in vessel-rich areas of CTEPH tissue and primary endothelial cells outgrown from it, supporting a role during the development of thrombofibrotic lesions, similar to our findings in mice.
Endothelin-1 is induced by TGFβ1, elevated in human and murine thrombofibrosis and endothelin receptor antagonization prevents EndMT.
Endothelin receptor antagonists are one of the main therapeutic components in the management of patients with PAH.1 Stimulation of HPAECs with TGFβ1 significantly increased ET-1 mRNA (11.5-fold, P<1×10−4 vs. HPAECs; Figure 7A) and protein (2.1-fold, P<1×10−4; Figure 7B-7D) expression, and this effect could be prevented by coincubation with the ALK5 inhibitor SB431542 (P<1×10−4 vs. TGFβ1-stimulated HPAECs for mRNA levels in Figure 7A; P=0.0023 vs. TGFβ1-stimulated HPAECs for protein levels in Figure 7C) and, although to a lesser extent, by the ALK1 inhibitor K02288 (P<1×10−4; Figure 7A, 7C and 7D). ET-1 was most prominently expressed in vessel-rich regions in CTEPH PEA specimens (P<1×10−4 vs. all others; Figure 7E and 7F), and significantly increased ET-1 level were detected in plasma of CTEPH patients (12.6±2.29 pg/mL) compared to healthy controls (2.92±0.26 pg/mL; P=0.0022; mean age, 56 years; 40% male; Figure 7G), and ET-1 plasma levels in CTEPH patients were similar to those in patients with PAH (9.2±0.54 pg/mL; P>0.9999). Although platelets are present in CTEPH PEA lesions,2 plasma levels of platelet factor 4 (PF4) did not differ between patients with CTEPH (224±19.6 pg/mL) or PAH (250±22.4 pg/mL) and those from healthy controls (269±22.3 pg/mL, P=0.9902 vs. CTEPH and P>0.9999 vs. PAH; Online Figure VD). The clinical characteristics of both patient groups are shown in Online Table IV.
Figure 7. Endothelin-1 expression in primary human pulmonary endothelial cells and in patients with CTEPH: effect of TGFβ1.

Quantitative real time PCR (n=4 biological replicates; A) and western blot analysis (n=3 biological replicates; C) of endothelin-1 (EDN1) mRNA or protein expression in HPAECs treated with TGFβ1 (10 ng/mL for 7 days). A representative western blot membrane is shown in (D). In some experiments, SB431542 (10 μM) was added to block ALK5 or K02288 (10 μM) to block ALK1 dependent signaling. Exact p-values, as determined by One-Way ANOVA followed by Bonferroni’s multiple comparisons test (4 comparisons), are shown in panels A and C. Data are normalized to HPRT1 and are expressed as -fold change vs. HPAECs (set at 1). Representative confocal pictures of HPAECs treated with TGFβ1 after immunodetection of endothelin-1 (ET-1; B). Scale bars represent 10 μm. Analysis of ET-1 expression in human n=6 PEA specimens primarily containing fresh or organized thrombus, myofibroblasts, vessels or fibrosis. Representative pictures (E) and the results of the quantitative analysis (F) are shown. Scale bar represents 100 μm. Exact p-values, as determined by One-Way ANOVA followed by Bonferroni’s multiple comparisons test (10 comparisons), are shown in panel F. Plasma levels of ET-1 in patients with CTEPH (n=19) or PAH (n=26) and healthy individuals (n=5; G). Patients who received endothelin receptor antagonists are marked in green, patients who received soluble guanylate cyclase stimulators are marked in red. Exact p-values, as determined by Kruskall-Wallis test followed by Dunn’s multiple comparisons test (3 comparisons), are shown in panel G. Non-significant p-values are not shown.
Elevated ET-1 mRNA (8.9-fold, P=0.0022; Figure 8A) and protein (Figure 8B) expression was also observed in primary ECs isolated from End.TGFβRII-KO mice compared to those from End.TGFβRII-WT mice. Plasma ET-1 levels in End.TGFβRII-KO animals were significantly elevated compared to End.TGFβRII-WT mice at baseline (18.2±2.09 vs. 7.2±0.71 pg/mL in End.TGFβRII-WT mice, P=0.0074) and further increased to a significantly greater extent 21 days after IVC ligation (32.1±4.6 vs. 12.6±3.2 pg/mL in End.TGFβRII-WT mice, P=0.0002 vs. End.TGFβRII-WT and P=0.0030 vs. End.TGFβRII-KO at baseline; Figure 8C). At this time point, a strong ET-1 immunosignal was observed in resolving murine thrombi, particularly in those of End.TGFβRII-KO mice (Figure 8D). Of note, ET-1 plasma levels and immunosignals in thrombi of Plt.TGFβ-KO mice did not differ from those in their Plt.TGFβ-WT littermates (P>0.9999; Online Figure XIIIA and XIIIB). Moreover, preincubation of C57BL6/J wild-type platelets with ET-1 (1 μM) did not alter their response to thrombin or convulxin stimulation (Online Figure VIE and VIF). Antagonization of endothelin receptors using bosentan reduced the outgrowth (P=0.0347 in End.TGFβRII-WT and P<1×10−4 in End.TGFβRII-KO mice; Online Figure XIVA and XIVB) and mesenchymal marker expression (Online Figure XIVC) of endothelial sprouts from aortic rings of End.TGFβRII-KO mice. Importantly, administration of bosentan following IVC ligation significantly reduced venous thrombus size at day 21 in End.TGFβRII-KO mice (P=0.0079 vs. control-treated mice) to values similar to those observed in End.TGFβRII-WT mice (1.4±0.10 vs. 2.3±0.09 mm2 in End.TGFβRII-WT mice, P>0.9999; Figure 8E and 8F). The number of occluded pulmonary arteries also significantly decreased compared to control-treated End.TGFβRII-KO mice (3.9±0.67 vs. 6.6±0.53 of occluded arteries per microscope field in End.TGFβRII-KO, P=0.0103; Figure 3A-3C). Flow cytometry analysis of 21 day-old venous thrombi showed that bosentan increased the relative number of CD31- or Cdh5-positive cells within thrombi (2.1- and 1.8-fold, respectively; P<1×10−4 and P<0.0007 vs. control-treated End.TGFβRII-KO mice), whereas the percentage of CD31- or CDH5-positive cells simultanously expressing FSP1 or SMA was significantly decreased compared to control-treated End.TGFβRII-KO mice (0.07- and 0.32-fold; P<1×10−4 and P=0.0006 vs. control-treated End.TGFβRII-KO mice; Figure 8G and 8H), confirming our in vitro data and underlining the importance of endothelin-1 signaling during thrombus non-resolution and endothelial-to-mesenchymal transition in vivo.
Figure 8. Endothelin-1 expression in mouse pulmonary endothelial cells or venous thrombi and effects of endothelin receptor antagonization on EndMT.

Quantitative real time PCR analysis (A) and representative confocal pictures (B) after analysis of endothelin-1 (EDN1) mRNA or protein expression in mPECs isolated from End.TGFβRII-WT and End.TGFβRII-KO mice and cultured in MV2 medium. Exact p-values, as determined by Mann-Whitney test are shown (n=6 biological replicates). Data are normalized to 18S and are expressed as -fold change vs. End.TGFβRII-WT mice (set at 1). Scale bars represents 10 μm. Plasma levels of ET-1 in samples from End.TGFβRII-WT and End.TGFβRII-KO mice uninjured (no IVC ligation; n=10 per group) and 21 days after IVC ligation (n=6 per group). Exact p-values, as determined by One-Way ANOVA followed by Bonferroni’s multiple comparisons test (4 comparisons), are shown. Non-significant p-values are not shown. Representative images of endothelin-1 protein expression in thrombosed vein wall segments 21 days after IVC ligation of End.TGFβRII-WT and End.TGFβRII-KO mice (D). Scale bars represent 10 μm. Quantitative analysis of thrombus area at day 21 after IVC ligation in End.TGFβRII-WT and End.TGFβRII-KO controls and mice treated with bosentan (n=5 per group; 5 mg/kg BW; E). Representative ultrasound images are shown in (F). Quantitative flow cytometry analysis of CD31- and CD31/FSP1 (n=4 per group; G) or CDH5- and CDH5/SMA (n=4 per group; H) double-positive cells residing within 21 day-old thrombi of End.TGFβRII-KO control- and bosentan-treated mice. Results in mice treated with bosentan are labeled in green. Exact p-values, as determined by Two-Way ANOVA followed by Bonferroni’s multiple comparisons test (6 comparisons; E) and multiple t test (G and H), are shown. Non-significant p-values are not shown.
Coincubation of HPAECs with bosentan also completely prevented the increased expression of SNAIL (P=0.0068; Online Figure XVA), ZEB1 (P=0.0005; Online Figure XVB), SMA (P=0.0013; Online Figure XVC) and COL1A1 (P=0.0009; Online Figure XVD) in HPAECs stimulated with TGFβ1, whereas it increased the expression of FGFR1, a major inhibitor of EndMT (P<1×10−4; Online Figure XVE). Of note, bosentan also normalized the TGFβ1-induced mRNA overexpression of ET-1 to levels observed in unstimulated human endothelial cells (P=0.0003; Online Figure XVF). Findings very similar to those after bosentan with regard to SMA, COL1A1 and FGFR1 mRNA expression were observed in TGFβ1-stimulated HPAECs treated with the ALK5 inhibitor SB431542 (P=0.0011, P=0.0049 and P<1×10−4, respectively), whereas inhibition of ALK1 using K02288 had no effect or increased the levels of the respective mRNAs (P=0.0232, P>0.9999 and P>0.9999, respectively; Online Figure XVIA-XVIC). Also, SB431542 and bosentan significantly increased the mRNA expression of ALK1 (P=0.0001 and P=0.0033, respectively; Online Figure XVIIA), whereas K02288 reduced the expression of ALK5 in HPAECs stimulated with TGFβ1 (P=0.0428; Online Figure XVIIB).
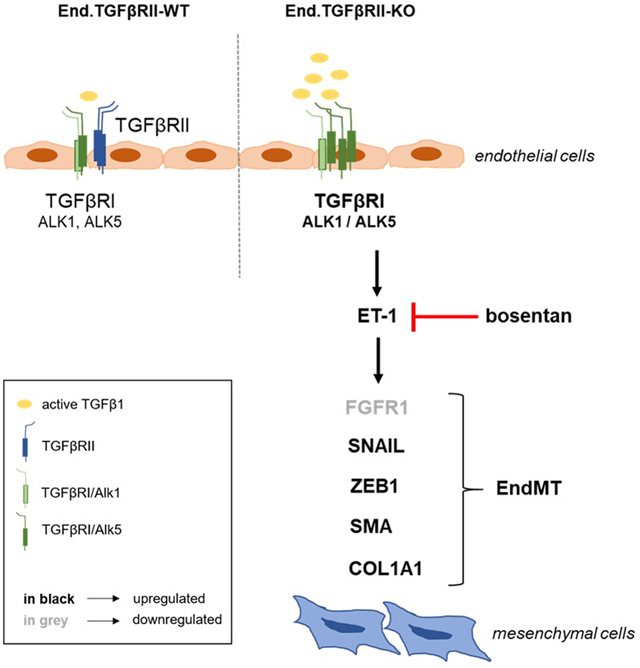
In summary, our findings in two transgenic mouse strains at late time points following experimental venous thrombosis, in PEA tissue microarrays of patients with CTEPH as well as in human and mouse pulmonary endothelial cells suggest that TGFβRI-mediated induction of ET-1 and signaling via ALK5 promotes endothelial-to-mesenchymal lineage conversion and thrombofibrosis and that these sequelae of increased circulating active TGFβ1 levels and TGFβ signaling in endothelial cells are reversible upon antagonization of endothelin receptors (Online Figure XVIII).
DISCUSSION
A distinctive feature of CTEPH is the obliteration of pulmonary arteries with thrombofibrotic material. TGFβ is a multifunctional growth factor regulating diverse biological processes including angiogenesis and fibrosis, whereas much less is known about its role during thrombus remodeling. Our group has recently shown that TGFβ1 released from activated platelets promotes neointima formation following carotid artery injury and thrombosis.25 Using a mouse model of venous thrombosis, we now demonstrate a major role of TGFβ signaling during postthrombotic vascular remodeling also in the venous system. More specifically, we showed that platelet-specific deletion of TGFβ1 did not alter platelet number or activation or their response to agonist stimulation, in line with previous reports,25,26 but accelerates the resolution of venous thrombi. On the other hand, increased levels of active TGFβ1 in plasma, as observed after endothelial-specific TGFβRII deletion possibly due to increased MMP9 and TSP1 expression and latent TGFβ1 activation, resulted in delayed venous thrombus resolution and persistent pulmonary vascular obstructions with platelet-/fibrin-rich material also containing TGFβ1. In these mice, overexpression of TGFβ type I receptors and endothelin-1 was observed, and activated TGFβ signaling and ET-1 overexpression was also present in plasma and PEA specimens of patients with CTEPH, particularly in areas containing vessels. Importantly, findings of enhanced endothelial sprouting, mesenchymal lineage transition and venous thrombus non-resolution could be reversed by endothelin receptor antagonization.
Defective angiogenesis and impaired thrombus revascularization may underlie the accumulation of thrombofibrotic material in pulmonary arteries of patients with CTEPH. Mice with inducible genetic deletion of TGFβRII in endothelial cells exhibited larger venous thrombi containing numerous enlarged vascular structures and increased amounts of myofibroblasts and collagen, thus recapitulating some of the typical features of the vascular lesions in CTEPH. Regarding the mechanisms underlying the delayed thrombus resolution in the presence of overactivated endothelial TGFβ signaling, differential expression of TGFβ target genes involved in vein wall remodeling, in particular uPA and MMP9,28 may have played a role, whereas plasma or endothelial mRNA levels of PAI-1 did not differ. In addition to fibrinolysis, blocking of ligand-induced VEGFR phosphorylation5 or deletion of VEGFR receptors in Tie2-positive cells7 was shown to delay the resolution of murine venous thrombi, whereas VEGF gene therapy enhanced this process.6 TGFβ1 downregulates the expression and binding capacity of VEGF receptors,29 and our analyses revealed significantly lower levels of VEGF and its main receptors in mPECs from End.TGFβRII-KO mice. On the other hand, mRNA expression of angiopoietin-2, a negative regulator of angiopoietin-1–Tie2 signaling and angiogenesis,30 was elevated in endothelial cells lacking TGFβRII. Plasma and tissue levels of angiopoeitin-2 are upregulated in patients with idiopathic PAH and have been proposed as biomarker of disease severity.31
Although the vasculopathy in CTEPH is believed to be the result of thrombus non-resolution,3 prothrombotic alterations such as highly activated platelets and increased spontaneous platelet aggregation have also been reported.32,33 In this regard, plasma PF4 levels were not found to be elevated in patients with CTEPH compared to those with non-thrombotic forms of pulmonary hypertension or healthy controls in our study. Importantly, deletion of TGFβ1 in platelets did not alter platelet number and activation or their aggregatory response to agonist stimulation, suggesting that the observed differences in vein thrombus size are not due to differences in the aggregatory response of platelets derived from Plt.TGFβ-KO mice, but a reduction in TGFβ released from activated platelets following IVC ligation.
Previous studies have shown that TGFβ is indispensable during angiogenesis, an integral part of almost every tissue remodeling process. Severe vascular defects and an embryonically lethal phenotype were observed in mice with global gene inactivation34 or constitutive deletion of TGFβ type II receptors in Tie1.Cre35 and Tie2.Cre36 expressing cells. Immature, highly dilated and fused yolk sac blood vessels and resorption at E11.5 were present in mice with systemic inactivation of type I TGFβ receptors.37 Endothelial-restricted deletion of TGFβRII in adult mice did not affect vessel morphogenesis.38 Also in our study, a vascular phenotype of endothelial TGFβRII deficiency was only observed following thrombotic vein injury. Interestingly, thrombin was shown to promote the internalization of TGFβ type II receptors in endothelial cells39 suggesting that downregulation of endothelial TGFβRII occurs in vivo in response to prothrombotic stimuli.
The angiogenic response to TGFβ depends on the balance between two distinct TGFβ type I receptor signaling pathways, with endothelial-restricted ALK1 promoting endothelial cell proliferation, migration and sprouting and the broadly expressed ALK5 favoring blood vessel maturation and extracellular matrix synthesis.40,41 TGFβ has been shown to possess a higher affinity for ALK5 than ALK1.42 Although basal endothelial sprouting and outgrowth activity was increased in End.TGFβRII-KO mice, endothelial cells lacking TGFβ type II receptors were less mature (as indicated by lower CDH5 expression) and also expressed mesenchymal markers. Co-localization with mesenchymal markers and increased expression of transcriptional regulators of EndMT were also observed in murine venous thrombi and in human CTEPH-ECs ex vivo or PEA specimens in situ. Expression of mesenchymal markers in TGFβ1-stimulated HPAECs could be inhibited blocking ALK5 signaling, whereas ALK1 inhibitors had no effect. Endothelial phenotype switching may be necessary to generate highly migratory and invasive cells and to initiate angiogenesis, as suggested by findings in brain-derived microvascular endothelial cells where stimulation with TGFβ1 was shown to simultaneously induce angiogenesis and EndMT.43
Increased plasma levels of TGFβ1 and the presence of EndMT has been reported in patients with PAH10,44 or experimental models of the disease.11 Despite the prominent presence of thrombofibrosis in CTEPH, few studies have examined EndMT in this subgroup of pulmonary hypertension. Ex vivo, ‘transitional’ cells co-expressing endothelial and smooth muscle cell markers were observed in endarterectomy material of patients with CTEPH,45 similar to our findings in PEA specimens. Moreover, conditioned medium from the myofibroblast-like cells was found to contain higher amounts of TGFβ1 and to induce smooth muscle α-actin expression in co-cultured endothelial cells.45 TGFβ signaling via SMAD2/3−SLUG was also shown to mediate EndMT during human interpositional vein graft remodeling.46 Although a recent study reported reduced tubulointerstitial renal fibrosis in mice with endothelium-specific heterozygous TGFβ type II receptor deletion,47 the remaining TGFBRII allele may have prevented the increase in TGFβ1 and TGFβRI expression observed in our model.
The findings of this study also suggest a role for ET-1 during endothelial-to-mesenchymal lineage conversion and thrombofibrosis downstream of TGFβ1 and its receptors. Previous studies have shown that TGFβ is a potent inducer of ET-1,48 and that TGFβ and ET-1 synergistically promote extracellular matrix production and fibrosis.49 Increased production of ET-1 has been observed in various human fibrotic diseases, including pulmonary hypertension,10,50 and ET-1 may potentiate the profibrotic effects of TGFβ via mechanisms involving EndMT.51 Increased ET-1 expression has been primarily reported in patients with non-thrombotic forms of pulmonary hypertension and shown to play a role in pulmonary artery vasoconstriction, pulmonary arterial smooth muscle cell proliferation and pulmonary hypertension.52 A previous study examining the plasma miRNA profile of patients with CTEPH found miRNA let-7c to be downregulated and identified TGFβ1 and ET-1 as its main targets.53 In this and another study,54 significantly higher plasma ET-1 levels were detected in patients with CTEPH, similar to our findings. Beyond the well-known vasoactive and mitogenic effects of ET-1, a direct role for ET-1 during thrombus resolution has never been shown. Of note, we also could not detect any effects of ET-1 (and of TGFβ1) on wild-type platelet response to agonist stimulation. Here we demonstrate that endothelin receptor antagonization prevented the TGFβ1-induced expression of receptors and transcriptional regulators involved in EndMT, SMA and collagen expression and also restored venous thrombus size and the presence of pulmonary vaso-occlusions in End.TGFβRII-KO mice to findings in End.TGFβRII-WT mice. Although a causal role of endothelin-1 in CTEPH has not been directly demonstrated, ET receptor antagonists are prescribed in patients with inoperable CTEPH or persistent/recurrent pulmonary after PEA and have been shown to improve hemodynamic parameters and survival.55,56 Our findings of increased ET-1 expression in chronic murine venous thrombi, human CTEPH endothelial cells or PEA specimens and reversal of overactivated endothelial TGFβ signaling following endothelin receptor antagonization provide a pathophysiological basis of this current clinical practice and also support the usefulness of blocking endothelin-1 signaling beyond its vasoactive effects.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Chronic thromboembolic pulmonary hypertension (CTEPH) is a life-threatening cardiovascular disease characterized by obstruction of pulmonary artery branches by unresolved thrombofibrotic material.
Currently, there is no therapy to “prevent” development of CTEPH after venous thromboembolism, and treatment consists mostly of surgical extraction of the thrombofibrotic material.
Mutations of genes encoding TGFβ signaling molecules have been implicated in the pathophysiology of other forms of pulmonary arterial hypertension.
What New Information Does This Article Contribute?
We show that increased TGFβ1 signaling in endothelial cells via ALK5/TGFβRI receptors delays the resolution of venous thrombi in mice and results in chronic pulmonary thromboembolism.
We identified activated TGFβ signaling and plasma levels of endothelin-1 (ET-1) as a potential pathomechanism underlying venous thrombus non-resolution and thromboembolism.
Inhibition of ET-1 with bosentan, an ET-1 receptor antagonist, reversed the conversion of endothelial cells to myofibroblasts and improved venous thrombus resolution but also inhibited thrombotic pulmonary obstructions.
Our findings in two transgenic mouse strains following experimental thrombosis of the inferior vena cava are supported by PEA tissue microarrays of patients with CTEPH and analysis of human and mouse pulmonary endothelial cells. Taken together, our results suggest that ALK5/TGFβRI-mediated induction of ET-1 promotes endothelial-to-mesenchymal lineage conversion and thrombofibrosis, and that these sequelae of increased circulating active TGFβ1 levels and TGFβ signaling in endothelial cells are reversible upon antagonization of endothelin receptors. They may thus have clinically relevant implications for patients with, or at risk of developing CTEPH.
ACKNOWLEDGMENTS
The authors would like to thank Marina Janocha (Center for Cardiology, Cardiology I, University Medical Center Mainz, Germany) and Kathrin Groß (Center for Thrombosis and Hemostasis, University Medical Center Mainz, Germany) for excellent technical assistance. We are also grateful to Sylvia Hoffmann (Center for Thrombosis and Hemostasis) for the transportation of human tissue samples. Results shown in this study are part of the medical thesis of CL.
SOURCE OF FUNDING
This study was supported by the Bundesministerium für Bildung und Forschung (BMBF 01E01003; Virchow fellowship and TRP X15 to MLB and TRP X8 to KS), the Deutsches Zentrum für Herz-Kreislauf-Forschung (DZHK e.V.; DZHK Doktorandenstipendium to NSR), the Robert-Müller-Stiftung (Universität Mainz; Doktorandenstipendium to CL), the Deutsche Forschungsmeinschaft (DFG; INST 371/47-1 FUGG; and BOS 3482/3-1, BOS 3482/4 to MB) and the National Institutes of Health (1R01HL141513, 1R01HL139641 to MB). The authors are responsible for the content of this publication.
Nonstandard Abbreviations and Acronyms:
- ALK
activin receptor-like kinase
- CTEPH
chronic thromboembolic pulmonary hypertension
- CTEPH-ECs
endothelial cells outgrown from CTEPH tissue
- EndMT
endothelial-to-mesenchymal transition
- ET-1
endothelin-1
- FGFR1
fibroblast growth factor receptor 1
- FSP-1
fibroblast-specific protein-1
- HPAECs
human pulmonary arterial endothelial cells
- HPVECs
human pulmonary vein endothelial cells
- HUVECs
human umbilical vein endothelial cells
- IVC
inferior Vena cava
- mPECs
mouse pulmonary endothelial cells
- PAH
pulmonary arterial hypertension
- PAI-1
plasminogen activator inhibitor-1
- PEA
pulmonary endarterectomy
- PF4
platelet factor 4
- PFA
paraformaldehyde
- PRP
platelet-rich plasma
- SMA
smooth muscle alpha-actin
- SMAD
small mothers against decapentaplegic homolog
- SNAIL
snail family zinc finger 1
- TGFβ
transforming growth factor-beta
- Tie2
endothelial receptor tyrosine kinase
- TMA
tissue microarrays
- TWIST1
Twist-related protein 1
- ZEB1
zinc finger E-box binding homeobox 1
Footnotes
DISCLOSURE
None declared.
REFERENCES
- 1.Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, Aboyans V, Vaz Carneiro A, Achenbach S, Agewall S, Allanore Y, Asteggiano R, Paolo Badano L, Albert Barbera J, Bouvaist H, Bueno H, Byrne RA, Carerj S, Castro G, Erol C, Falk V, Funck-Brentano C, Gorenflo M, Granton J, Iung B, Kiely DG, Kirchhof P, Kjellstrom B, Landmesser U, Lekakis J, Lionis C, Lip GY, Orfanos SE, Park MH, Piepoli MF, Ponikowski P, Revel MP, Rigau D, Rosenkranz S, Voller H, Luis Zamorano J. 2015 esc/ers guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the european society of cardiology (esc) and the european respiratory society (ers): Endorsed by: Association for european paediatric and congenital cardiology (aepc), international society for heart and lung transplantation (ishlt). Eur Heart J. 2016;37:67–119. [DOI] [PubMed] [Google Scholar]
- 2.Bochenek ML, Rosinus NS, Lankeit M, Hobohm L, Bremmer F, Schütz E, Klok FA, Horke S, Wiedenroth CB, Münzel T, Lang IM, Mayer E, Konstantinides S, Schäfer K. From thrombosis to fibrosis in chronic thromboembolic pulmonary hypertension. Thromb Haemost. 2017;117:769–783. [DOI] [PubMed] [Google Scholar]
- 3.Simonneau G, Torbicki A, Dorfmuller P, Kim N. The pathophysiology of chronic thromboembolic pulmonary hypertension. Eur Respir Rev. 2017;26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma S, Lang IM. Current understanding of the pathophysiology of chronic thromboembolic pulmonary hypertension. Thromb Res. 2018;164:136–144. [DOI] [PubMed] [Google Scholar]
- 5.Evans CE, Grover SP, Humphries J, Saha P, Patel AP, Patel AS, Lyons OT, Waltham M, Modarai B, Smith A. Antiangiogenic therapy inhibits venous thrombus resolution. Arterioscler Thromb Vasc Biol. 2014;34:565–570. [DOI] [PubMed] [Google Scholar]
- 6.Modarai B, Humphries J, Burnand KG, Gossage JA, Waltham M, Wadoodi A, Kanaganayagam GS, Afuwape A, Paleolog E, Smith A. Adenovirus-mediated vegf gene therapy enhances venous thrombus recanalization and resolution. Arterioscler Thromb Vasc Biol. 2008;28:1753–1759. [DOI] [PubMed] [Google Scholar]
- 7.Alias S, Redwan B, Panzenboeck A, Winter MP, Schubert U, Voswinckel R, Frey MK, Jakowitsch J, Alimohammadi A, Hobohm L, Mangold A, Bergmeister H, Sibilia M, Wagner EF, Mayer E, Klepetko W, Hoelzenbein TJ, Preissner KT, Lang IM. Defective angiogenesis delays thrombus resolution: A potential pathogenetic mechanism underlying chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2014;34:810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zabini D, Nagaraj C, Stacher E, Lang IM, Nierlich P, Klepetko W, Heinemann A, Olschewski H, Balint Z, Olschewski A. Angiostatic factors in the pulmonary endarterectomy material from chronic thromboembolic pulmonary hypertension patients cause endothelial dysfunction. PLoS One. 2012;7:e43793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naito A, Sakao S, Lang IM, Voelkel NF, Jujo T, Ishida K, Sugiura T, Matsumiya G, Yoshino I, Tanabe N, Tatsumi K. Endothelial cells from pulmonary endarterectomy specimens possess a high angiogenic potential and express high levels of hepatocyte growth factor. BMC Pulm Med. 2018;18:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, Dorfmuller P, Remy S, Lecerf F, Plante S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. [DOI] [PubMed] [Google Scholar]
- 11.Qiao L, Nishimura T, Shi L, Sessions D, Thrasher A, Trudell JR, Berry GJ, Pearl RG, Kao PN. Endothelial fate mapping in mice with pulmonary hypertension. Circulation. 2014;129:692–703. [DOI] [PubMed] [Google Scholar]
- 12.Sun JX, Chang TF, Li MH, Sun LJ, Yan XC, Yang ZY, Liu Y, Xu WQ, Lv Y, Su JB, Liang L, Han H, Dou GR, Wang YS. Snai1, an endothelial-mesenchymal transition transcription factor, promotes the early phase of ocular neovascularization. Angiogenesis. 2018;21:635–652. [DOI] [PubMed] [Google Scholar]
- 13.Shi Y, Massague J. Mechanisms of tgf-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. [DOI] [PubMed] [Google Scholar]
- 14.Goumans MJ, Lebrin F, Valdimarsdottir G. Controlling the angiogenic switch: A balance between two distinct tgf-b receptor signaling pathways. Trends Cardiovasc Med. 2003;13:301–307. [DOI] [PubMed] [Google Scholar]
- 15.Wang XF, Lin HY, Ng-Eaton E, Downward J, Lodish HF, Weinberg RA. Expression clonng and characterization of the TGF-β type III receptor. Cell 1991;76:797–805. [DOI] [PubMed] [Google Scholar]
- 16.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res. 2014;115:189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gore B, Izikki M, Mercier O, Dewachter L, Fadel E, Humbert M, Dartevelle P, Simonneau G, Naeije R, Lebrin F, Eddahibi S. Key role of the endothelial tgf-beta/alk1/endoglin signaling pathway in humans and rodents pulmonary hypertension. PLoS One. 2014;9:e100310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaiman AL, Podowski M, Medicherla S, Gordy K, Xu F, Zhen L, Shimoda LA, Neptune E, Higgins L, Murphy A, Chakravarty S, Protter A, Sehgal PB, Champion HC, Tuder RM. Role of the tgf-beta/alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med. 2008;177:896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yung LM, Nikolic I, Paskin-Flerlage SD, Pearsall RS, Kumar R, Yu PB. A selective transforming growth factor-beta ligand trap attenuates pulmonary hypertension. Am J Respir Crit Care Med. 2016;194:1140–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Azhar M, Yin M, Bommireddy R, Duffy JJ, Yang J, Pawlowski SA, Boivin GP, Engle SJ, Sanford LP, Grisham C, Singh RR, Babcock GF, Doetschman T. Generation of mice with a conditional allele for transforming growth factor beta 1 gene. Genesis. 2009;47:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jäger M, Hubert A, Gogiraju R, Bochenek ML, Münzel T, Schäfer K. Inducible knockdown of endothelial protein tyrosine phosphatase-1b promotes neointima formation in obese mice by enhancing endothelial senescence. Antioxid Redox Signal. 2018. [DOI] [PubMed] [Google Scholar]
- 22.Alvarado CM, Diaz JA, Hawley AE, Wroblewski SK, Sigler R, Myers DD Jr. Male Mice have increased thrombotic potential: sex differences in a mouse model of venous thrombosis. Thromb Res. 2011;127(5):478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grainger DJ, Wakefield L, Bethell HW, Farndale RW, Metcalfe JC. Release and activation of platelet latent tgf-beta in blood clots during dissolution with plasmin. Nat Med. 1995;1:932–937. [DOI] [PubMed] [Google Scholar]
- 24.Meyer B, Wang W, Qu J, Croft L, Degen JL, Coller BS, Ahamed J. Platelet TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood. 2012;119:1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schütz E, Bochenek ML, Riehl DR, Bosmann M, Munzel T, Konstantinides S, Schäfer K. Absence of transforming growth factor beta 1 in murine platelets reduces neointima formation without affecting arterial thrombosis. Thromb Haemost. 2017;117:1782–1797. [DOI] [PubMed] [Google Scholar]
- 26.von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen PY, Qin L, Tellides G, Simons M. Fibroblast growth factor receptor 1 is a key inhibitor of tgfbeta signaling in the endothelium. Sci Signal. 2014;7:ra90. [DOI] [PubMed] [Google Scholar]
- 28.Deatrick KB, Eliason JL, Lynch EM, Moore AJ, Dewyer NA, Varma MR, Pearce CG, Upchurch GR, Wakefield TW, Henke PK. Vein wall remodeling after deep vein thrombosis involves matrix metalloproteinases and late fibrosis in a mouse model. J Vasc Surg. 2005;42:140–148. [DOI] [PubMed] [Google Scholar]
- 29.Mandriota SJ, Menoud PA, Pepper MS. Transforming growth factor beta 1 down-regulates vascular endothelial growth factor receptor 2/flk-1 expression in vascular endothelial cells. J Biol Chem. 1996;271:11500–11505. [DOI] [PubMed] [Google Scholar]
- 30.Felcht M, Luck R, Schering A, Seidel P, Srivastava K, Hu J, Bartol A, Kienast Y, Vettel C, Loos EK, Kutschera S, Bartels S, Appak S, Besemfelder E, Terhardt D, Chavakis E, Wieland T, Klein C, Thomas M, Uemura A, Goerdt S, Augustin HG. Angiopoietin-2 differentially regulates angiogenesis through tie2 and integrin signaling. J Clin Invest. 2012;122:1991–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumpers P, Nickel N, Lukasz A, Golpon H, Westerkamp V, Olsson KM, Jonigk D, Maegel L, Bockmeyer CL, David S, Hoeper MM. Circulating angiopoietins in idiopathic pulmonary arterial hypertension. Eur Heart J. 2010;31:2291–2300. [DOI] [PubMed] [Google Scholar]
- 32.Yaoita N, Shirakawa R, Fukumoto Y, Sugimura K, Miyata S, Miura Y, Nochioka K, Miura M, Tatebe S, Aoki T, Yamamoto S, Satoh K, Kimura T, Shimokawa H, Horiuchi H. Platelets are highly activated in patients of chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2014;34:2486–2494. [DOI] [PubMed] [Google Scholar]
- 33.Remková A, Šimková I, Valkovičová T. Platelet abnormalities in chronic thromboembolic pulmonary hypertension. Int J Clin Exp Med. 2015;8:9700–9707. [PMC free article] [PubMed] [Google Scholar]
- 34.Oshima M, Oshima H, Taketo MM. Tgf-beta receptor type ii deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol. 1996;179:297–302. [DOI] [PubMed] [Google Scholar]
- 35.Carvalho RL, Itoh F, Goumans MJ, Lebrin F, Kato M, Takahashi S, Ema M, Itoh S, van Rooijen M, Bertolino P, Ten Dijke P, Mummery CL. Compensatory signalling induced in the yolk sac vasculature by deletion of tgfbeta receptors in mice. J Cell Sci. 2007;120:4269–4277. [DOI] [PubMed] [Google Scholar]
- 36.Shang Y, Doan CN, Arnold TD, Lee S, Tang AA, Reichardt LF, Huang EJ. Transcriptional corepressors hipk1 and hipk2 control angiogenesis via tgf-beta-tak1-dependent mechanism. PLoS Biol. 2013;11:e1001527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larsson J, Goumans MJ, Sjostrand LJ, van Rooijen MA, Ward D, Leveen P, Xu X, ten Dijke P, Mummery CL, Karlsson S. Abnormal angiogenesis but intact hematopoietic potential in tgf-beta type i receptor-deficient mice. The EMBO Jl. 2001;20:1663–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL, Oh SP. Alk5- and tgfbr2-independent role of alk1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood. 2008;111:633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang H, Low B, Rutherford SA, Hao Q. Thrombin induces endocytosis of endoglin and type-ii tgf-beta receptor and down-regulation of tgf-beta signaling in endothelial cells. Blood. 2005;105:1977–1985. [DOI] [PubMed] [Google Scholar]
- 40.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct tgf-beta type i receptors. The EMBO J. 2002;21:1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (alk)1 is an antagonistic mediator of lateral tgfbeta/alk5 signaling. Mol Cell. 2003;12:817–828. [DOI] [PubMed] [Google Scholar]
- 42.ten Dijke P, Yamashita H, Ichijo H, Franzen P, Laiho M, Miyazono K, Heldin CH. Characterization of type i receptors for transforming growth factor-beta and activin. Science. 1994;264:101–104. [DOI] [PubMed] [Google Scholar]
- 43.Krishnan S, Szabo E, Burghardt I, Frei K, Tabatabai G, Weller M. Modulation of cerebral endothelial cell function by tgf-beta in glioblastoma: Vegf-dependent angiogenesis versus endothelial mesenchymal transition. Oncotarget. 2015;6:22480–22495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan Y, Wang XJ, Li SQ, Yang SH, Lv ZC, Wang LT, He YY, Jiang X, Wang Y, Jing ZC. Elevated levels of plasma transforming growth factor-β1 in idiopathic and heritable pulmonary arterial hypertension. Int J Cardiol. 2016;222:368–374. [DOI] [PubMed] [Google Scholar]
- 45.Sakao S, Hao H, Tanabe N, Kasahara Y, Kurosu K, Tatsumi K. Endothelial-like cells in chronic thromboembolic pulmonary hypertension: Crosstalk with myofibroblast-like cells. Respir Res. 2011;12:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cooley BC, Nevado J, Mellad J, Yang D, St Hilaire C, Negro A, Fang F, Chen G, San H, Walts AD, Schwartzbeck RL, Taylor B, Lanzer JD, Wragg A, Elagha A, Beltran LE, Berry C, Feil R, Virmani R, Ladich E, Kovacic JC, Boehm M. Tgf-beta signaling mediates endothelial-to-mesenchymal transition (endmt) during vein graft remodeling. Sci Transl Med. 2014;6:227ra234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xavier S, Vasko R, Matsumoto K, Zullo JA, Chen R, Maizel J, Chander PN, Goligorsky MS. Curtailing endothelial tgf-beta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in ckd. J Am Soc Nephrol: JASN. 2015;26:817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodriguez-Pascual F, Reimunde FM, Redondo-Horcajo M, Lamas S. Transforming growth factor-beta induces endothelin-1 expression through activation of the smad signaling pathway. J Cardiovasc Pharmacol. 2004;44 Suppl 1:S39–42. [DOI] [PubMed] [Google Scholar]
- 49.Lambers C, Roth M, Zhong J, Campregher C, Binder P, Burian B, Petkov V, Block LH. The interaction of endothelin-1 and tgf-beta1 mediates vascular cell remodeling. PLoS One. 2013;8:e73399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swigris JJ, Brown KK. The role of endothelin-1 in the pathogenesis of idiopathic pulmonary fibrosis. BioDrugs. 2010;24:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wermuth PJ, Li Z, Mendoza FA, Jimenez SA. Stimulation of transforming growth factor-beta1-induced endothelial-to-mesenchymal transition and tissue fibrosis by endothelin-1 (et-1): A novel profibrotic effect of et-1. PLoS One. 2016;11:e0161988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shao D, Park JE, Wort SJ. The role of endothelin-1 in the pathogenesis of pulmonary arterial hypertension. Pharmacol Res. 2011;63:504–511. [DOI] [PubMed] [Google Scholar]
- 53.Guo L, Yang Y, Liu J, Wang L, Li J, Wang Y, Liu Y, Gu S, Gan H, Cai J, Yuan JX, Wang J, Wang C. Differentially expressed plasma micrornas and the potential regulatory function of let-7b in chronic thromboembolic pulmonary hypertension. PLoS One. 2014;9:e101055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reesink HJ, Meijer RC, Lutter R, Boomsma F, Jansen HM, Kloek JJ, Bresser P. Hemodynamic and clinical correlates of endothelin-1 in chronic thromboembolic pulmonary hypertension. Circ J. 2006;70:1058–1063. [DOI] [PubMed] [Google Scholar]
- 55.Nishimura R, Tanabe N, Sugiura T, Shigeta A, Jujo T, Sekine A, Sakao S, Kasahara Y, Tatsumi K. Improved survival in medically treated chronic thromboembolic pulmonary hypertension. Circ J. 2013;77:2110–2117. [DOI] [PubMed] [Google Scholar]
- 56.Jais X, D’Armini AM, Jansa P, Torbicki A, Delcroix M, Ghofrani HA, Hoeper MM, Lang IM, Mayer E, Pepke-Zaba J, Perchenet L, Morganti A, Simonneau G, Rubin LJ. Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: Benefit (bosentan effects in inoperable forms of chronic thromboembolic pulmonary hypertension), a randomized, placebo-controlled trial. J Am Coll Cardiol. 2008;52:2127–2134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.