

Abstract
BACKGROUND
The relative stabilities of the intact and defective HIV genomes over time during effective antiretroviral therapy (ART) have not been fully characterized.
METHODS
We used the intact proviral DNA assay (IPDA) to estimate the rate of change of intact and defective proviruses in HIV-infected adults on ART. We used linear spline models with a knot at seven years and a random intercept and slope up to the knot. We estimated the influence of covariates on rates of change.
RESULTS
We studied 81 individuals for a median of 7.3 (IQR 5.9-9.6) years. Intact genomes declined more rapidly from initial suppression through seven years (15.7% per year decline; 95% CI -22.8%, -8.0%) and more slowly after seven years (3.6% per year; 95% CI -8.1%, +1.1%). The estimated half-life of the reservoir was 4.0 years (95% CI 2.7-8.3) until year seven and 18.7 years (95% CI 8.2-infinite) thereafter. There was substantial variability between individuals in the rate of decline until year seven. Intact provirus declined more rapidly than defective provirus (P < 0.001) and showed a faster decline in individuals with higher CD4+ T cell nadirs.
CONCLUSION
The biology of the replication-competent (intact) reservoir differs from that of the replication-incompetent (non-intact) pool of proviruses. The IPDA will likely be informative when investigating the impact of interventions targeting the reservoir.
FUNDING
Delaney AIDS Research Enterprise, UCSF/Gladstone Institute of Virology & Immunology CFAR, CFAR Network of Integrated Systems, amfAR Institute for HIV Cure Research, I4C and Beat-HIV Collaboratories, Howard Hughes Medical Institute, Gilead Sciences, Bill and Melinda Gates Foundation.
Keywords: AIDS/HIV
Keywords: Molecular biology
The biology of the replication-competent (intact) reservoir differs from that of the replication-incompetent (nonintact) pool of proviruses in HIV-1–infected individuals on suppressive ART.
Introduction
The latent HIV-1 reservoir is established early in the course of infection and persists despite suppressive antiretroviral therapy (ART) (1–4). Strategies under development to cure HIV infection often involve targeting the latent viral reservoir with the goal of decreasing the number of replication-competent proviruses that contribute to viral rebound (5).
The latent reservoir is not static (6). In addition to accurately and precisely quantifying the reservoir, it is crucial to understand how its size changes over time in individuals on prolonged suppressive ART. Understanding variability in the rate of change of the reservoir size, correlates of this variability, and factors associated with rapid decay is likely to be useful in the design and interpretation of HIV cure interventions.
Current assays have well-described limitations in their ability to quantify the replication-competent reservoir. Quantitative viral outgrowth assays (QVOA) measure reactivated virus capable of infecting cells in culture. These assays directly measure the replication-competent population, but their scalability is limited (7). They are time and labor intensive, with a slow turnaround time to detect outgrowth. In addition, they are relatively insensitive because not all latently infected cells will be induced by stimulation, underestimating the reservoir size (8, 9). In contrast, PCR-based assays are easier to perform. They have a larger dynamic range but are unable to differentiate between defective and replication-competent virus. These assays vastly overestimate the size of the replication-competent reservoir (7, 10).
The intact proviral DNA assay (IPDA) was recently developed to address these issues by separately quantifying intact and defective proviruses (11). The IPDA uses droplet digital PCR (ddPCR) to simultaneously analyze the packaging signal and env regions of proviruses. Intact proviruses demonstrate amplification at both regions, while defective proviruses demonstrate amplification at a single region or do not amplify (11). This assay has the potential to provide a more useful estimate of the replication-competent reservoir by detecting a greater number of intact proviruses than QVOA, while distinguishing intact sequences from those defective ones that are unlikely to be clinically relevant. However, the performance of this assay in clinical cohorts remains unknown.
Using the IPDA, we analyzed proviruses in CD4+ T cells purified from longitudinal peripheral blood mononuclear cell (PBMC) samples from highly characterized HIV-infected individuals on suppressive ART to identify changes in intact and defective provirus over time. Based on recent work using this assay (11), we hypothesized that intact and defective provirus would demonstrate different rates of change. We further hypothesized that the rate of decline would correlate with markers of immune status, such as proximal CD4+ T lymphocyte count and CD4+ T cell nadir.
Results
Characteristics of study participants.
Eighty-one individuals were studied (Table 1). Most were male (95.1%), and the median age was 49 years. The median nadir CD4+ T cell count was 183 cells/mm3 (IQR 60–326), median proximal CD4+ T cell count at the first time point sampled was 584 cells/mm3 (IQR 444–751), and median proximal CD4/CD8 ratio was 0.64 (IQR 0.41–1.01). Individuals had been on suppressive ART for a median of 617 days (IQR 84–1369) at the time of the first PBMC sample included in the IPDA analysis. Individuals were studied for a median of 7.3 years (IQR 5.9–9.6). A total of 216 measurements across the cohort were performed. On average, 2.7 samples were studied per subject. At the first visit, 39 individuals were on a regimen including a protease inhibitor (PI), 49 were on a regimen including a nonnucleoside reverse transcriptase inhibitor (NNRTI), and 9 were on a regimen including an integrase inhibitor (these were not mutually exclusive).
Table 1. Characteristics of study participants at earliest IPDA study time point.

Baseline HIV-1 provirus measures.
Intact proviral DNA levels were measured with the IPDA as previously described (11). A detailed description is given in Methods. Representative assay output, positive and negative controls, gating, and procedures for dealing with polymorphisms are described in Supplemental Figures 1–4 (Supplemental material available online with this article; https://doi.org/10.1172/jci.insight.132997DS1).
The median intact HIV proviral DNA level at first visit was 151 copies/1 × 106 CD4+ T cells (IQR 40–398; Figure 1). The median frequency of provirus containing defects in the 3′ and 5′ regions were 574 and 404 copies/ 1 × 106 cells, respectively. The median ratio of intact/defective genomes was 0.15 (IQR 0.05–0.33).
Figure 1. Baseline proviral DNA in participants at the first study time point.
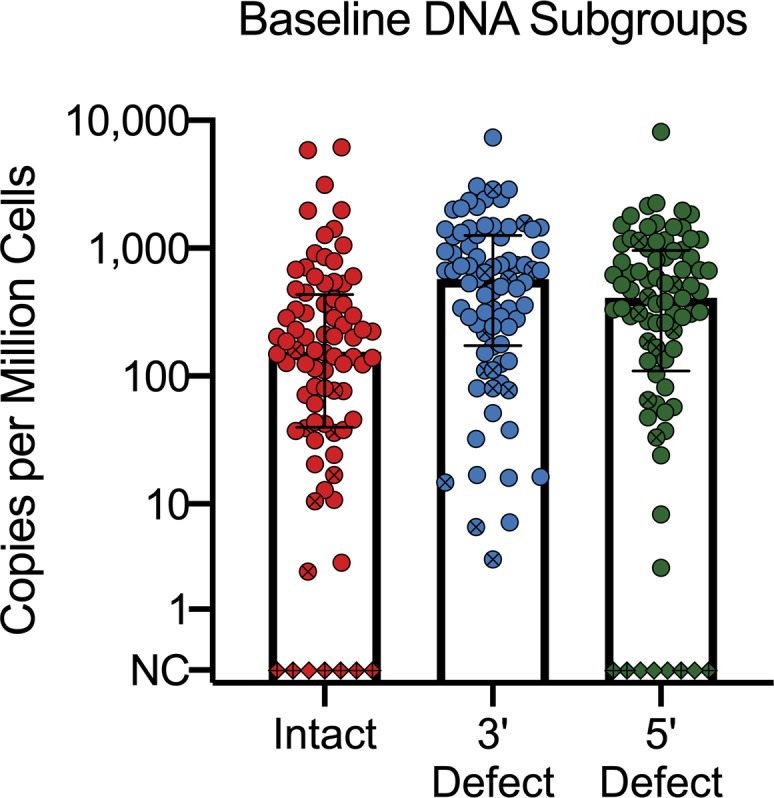
Note that the preceding duration of suppressive ART differs between participants. NC; no copies detected. Circles indicate participants with detectable provirus. Diamonds indicate participants without detectable provirus. Crossed circles and diamonds indicate participants who did not exhibit decay of intact provirus over the first 7 years in longitudinal analyses.
In models estimating the effect of covariates on starting levels of intact provirus, there was a statistically significant effect of CD4+ T cell nadir (0.65-fold change for every 100-cell increase; 95% CI, 0.53–0.80; P < 0.0001) and proximal CD4/CD8 ratio (0.49-fold change for every twofold increase of the ratio; 95% CI, 0.34–0.70; P = 0.0001). CD4+ T cell nadir and CD4/CD8 ratio also had a statistically significant effect on starting levels of 3′ defective provirus (0.75-fold change for every 100-cell increase; 95% CI, 0.62–0.92; P = 0.0064; and 0.67-fold effect for every 2-fold increase of the ratio; 95% CI, 0.54–0.83; P = 0.0006) and 5′ defective provirus (0.52-fold change for every 100-cell increase; 95% CI, 0.42–0.64; P < 0.0001; and 0.40-fold effect for every twofold increase of the ratio; 95% CI, 0.28–0.59; P < 0.0001). Age, race, sex, protective HLA alleles, and CCR5 heterozygosity did not have a substantial effect on starting levels of intact or defective proviruses.
Intact HIV-1 decay kinetics.
We first modeled the decay of the intact provirus. Our initial model assumed a constant rate of decline throughout the measured interval, but we found strong evidence that this assumption was not realistic (P = 1.6 × 10–7).
In a model allowing for a change in the rate of decline, the best fit was achieved with a cut point of 7 years of effective ART (Figure 2A). In this model, there was a 15.7% per-year decline (95% CI, –22.8% to –8.0%; P = 0.0002) in the size of the intact reservoir from initial suppression through year 7 of suppression. Following year 7, there was a 3.6% (95% CI, –8.1% to +1.1%; P = 0.13) per-year decline in the size of the intact reservoir. The estimated half-life of the reservoir was 4.0 years (95% CI, 2.7–8.3) until year 7. This agrees with the half-life of latently infected cells determined using the QVOA (3.7 years; ref. 12). After year 7, the half-life was 18.7 years (95% CI, 8.2–infinite).
Figure 2. Longitudinal dynamics of proviral populations measured using IPDA.

Light gray lines indicate each participant; dashed lines denote participants who did not exhibit decay of intact provirus over the first 7 years in longitudinal analyses. Thick, colored lines indicate the fitted model. Note the knot (cut point where slope can change) at 7 years in the fitted models. (A) Intact virus declines at –15.7% (95% CI, –22.8, –8.0) per year to year 7 (P = 0.0002), after which it declines at –3.6% per year (95% CI, –8.1, +1.1; P = 0.13). (B) Combined defective virus declines at –4.0% (95% CI, –7.0, –0.9) per year to year 7 (P = 0.013), after which it declines at –1.5% per year (95% CI, –3.2, +0.2; P = 0.085). (C) The 3′ defective virus declines at –5.9% (95% CI, –9.3, –2.3) per year to year 7 (P = 0.0018), after which it declines at –0.9% per year (95% CI, –2.8, +1.1; P = 0.37). (D) The 5′ defective virus increases at +1.2% (95% CI, –6.4, +9.3) per year to year 7 (P = 0.76), after which it declines at –1.5% per year (95% CI, –7.1, +4.5; P = 0.62).
There was substantial variability between individuals in the estimated rate of decline until year 7 (Figure 3A). No patterns emerged when this was examined according to whether individuals had been on suppressive ART for greater than or less than 1 year. Although most individuals exhibited a decline in the intact reservoir, a subset demonstrated little change or expansion (13 of 69, 18.8%) over time.
Figure 3. Proviral decay rates in study participants.

(A) Percent change per year in intact and defective provirus measured in study participants using IPDA. Note these slopes correspond to the first 7 years of virologic suppression. Values are estimated person-specific random slope from a mixed effects model plus the overall fixed-effect slope. (B) Intact proviral decay rate as a function of intact provirus at baseline. Note that intact provirus at baseline is the fitted random intercept plus the overall fixed-effect intercept, corresponding to the level at the start of viral suppression. (C) Intact proviral decay rate does not substantially correlate with 3′ provirus decay rate. (D) Intact proviral decay rate does not substantially correlate with 5′ provirus decay rate. Crossed circles indicate participants who did not exhibit decay of intact provirus over the first 7 years in longitudinal analyses.
Defective HIV-1 decay kinetics.
Figure 2, B–D, displays models for provirus with 3′ and 5′ defects, as well as the combined pool of defective provirus. In a model allowing for a change in the rate of decline at 7 years, there was a 4.0% per–year decline (95% CI, –7.0 to –0.9; P = 0.013) in combined defective provirus through year 7. This was followed by a 1.5% per-year decline (95% CI, –3.2 to +0.2; P = 0.085) in combined defective provirus after year 7. The estimated half-life of the combined defective proviral DNA was 17.1 years (95% CI, 9.6–78.2) to year 7 and 45.1 years (95% CI, 21.1–infinite) after year 7.
Provirus with 3′ defects declined at 5.9% per year (95% CI, –9.3 to –2.3; P = 0.0018) until year 7. Seventeen of 69 (24.6%) individuals exhibited an expansion in 3′ defective provirus over the first 7 years. After year 7, this subset of defective provirus declined at 0.9% per year (95% CI, –2.8, +1.1; P = 0.37). The estimated half-life of the 3′ defective proviral DNA was 11.4 years (95% CI, 7.1–29.7) to year 7 and 77.4 years (95% CI, 24.1–infinite) after year 7.
Notably, 5′ defective provirus appeared to be stable and even increased 1.2% per year (95% CI, –6.4 to +9.3; P = 0.76) through year 7 of suppression. Twenty-eight of 69 (40.5%) individuals exhibited an expansion in 5′ defective provirus during this period. Afterward, this subset of defective provirus declined at 1.5% per year (95% CI, –7.1 to +4.5; P = 0.62). After year 7, the estimated half-life of the 5′ defective proviral DNA was 47.3 years (95% CI, 9.4–infinite).
A paired two-tailed t test comparing the individuals’ fitted slopes during the first 7 years of suppression for intact versus defective provirus had P < 0.0001.
Correlates of provirus rate of decline.
We also estimated factors associated with the rate of the decline in the intact and nonintact populations over the first 7 years of suppression (Figure 4 and Supplemental Table 1).
Figure 4. Estimated effects of covariates on the rate of decline of intact and combined defective provirus during the first 7 years of suppression.

(A) Effect of demographic characteristics on rate of decline. (B) Effect of immunologic parameters on rate of decline. Note that these effects are from models that do not include the effect of the covariate on the level of provirus at the start of viral suppression. NNRTI, nonnucleoside reverse transcriptase inhibitor; PI, protease inhibitor; INSTI, integrase strand transfer inhibitor.
Individuals with higher CD4+ T cell count nadir values had a faster rate of decline in cells containing intact genomes. For every 100 cells higher the nadir CD4, there was an estimated additional 5.4% per-year decline. There was also evidence of an association of proximal CD4+ T cell values with the rate of decline of intact provirus, along with proximal CD8+ T cells (when log transformed) and proximal CD4/CD8 ratio (when not log transformed).
In models that included both CD4+ T cell nadir and 1 of the 3 concurrent T cell measures (Supplemental Table 2), nadir CD4 remained associated with the intact proviral decay rate (estimates –4.0% to –5.3%, P = 0.0001 to P = 0.0066). The estimates for the concurrent measures remained similar when controlled for nadir CD4, except the estimate for CD4/CD8 ratio, which decreased to –6.4% (P = 0.20).
The estimates described above are from models that assume no influence of nadir CD4+ T cell count on the initial level at the beginning of viral suppression. Models that allowed nadir CD4+ T cell count to be associated with both the initial level and the subsequent rate of decline provided strong evidence of association with at least one but could not provide strong evidence for which.
The rate of decline of defective provirus also correlated with CD4+ T cell nadir. In models that included both nadir and 1 of the 3 concurrent measures, CD4+ T cell nadir again remained associated (estimates –1.3% to –2.2%, P = 0.0031 to P = 0.0084).
As compared with NNRTI- and integrase strand transfer inhibitor–based (INSTI-based) regimens, PI-based regimens were associated with slower decline of the intact proviral reservoir, although this association was not statistically significant.
There was a statistically significant correlation between the rate of decline and starting level of intact provirus (Figure 3B). The rate of decline of intact provirus did not substantially correlate with the rate of decline of 3′ or 5′ defective provirus (Figures 3, C and D).
A subset of individuals appeared to have very rapid declines in intact provirus. Of the 7 individuals in the top decile of intact proviral decline (rates faster than –33% per year), only 1 had a protective HLA allele. The baseline levels of intact and 3′ defective genomes in these rapid decliners was higher than that observed in the remainder of the cohort (median 1989 vs. 140 copies, P = 0.005; 1464 vs. 531, P = 0.006). There was little difference in the median baseline level of 5′ defective genomes. The age, CD4+ T cell nadir, and CD4/CD8 ratio were similar in those with rapid declines as compared with the remainder of the cohort.
Discussion
Using an established and well-characterized cohort of treated HIV-infected adults, we characterized the intact and nonintact genomes over several years, using the IPDA to estimate the size of these 2 populations. We found that both populations declined over time, with a more rapid decline found during the first 7 years of ART. Importantly, we found that cells containing intact genomes decayed faster than those containing nonintact or defective genomes. The rate of decay correlated with the CD4+ T cell nadir, with higher nadir values associated with a higher rate of decay of both intact and defective provirus. These findings provide strong evidence that the biology of the replication-competent (intact) reservoir likely differs from that of the replication-incompetent (nonintact) pool of persistent proviruses. Assays that provide a more direct estimate of the intact population will likely be more useful and informative when investigating the impact of interventions targeting the reservoir.
Intact and defective proviruses exhibited different rates of decay during the first 7 years following ART initiation. Intact proviruses decayed at a faster rate. A similar finding was suggested in a smaller number of participants with 2 time points analyzed in the development of the IPDA (11). In addition, a recent study using single-genome, near–full-length sequencing techniques suggested differences in the rate of decline between cells containing intact proviruses (13). There are several potential mechanisms that could explain this. Cells containing intact virus may be more likely to produce proteins and be more effectively cleared by the host immune response, even as it has been shown that cells containing defective genomes may still be targeted (9, 14). Cells harboring replication-competent virus may be more likely to produce virions, resulting in a higher frequency of cell death by direct cytotoxic effects.
The estimated half-life of the latent reservoir, particularly after 7 years, continues to be a significant obstacle to HIV eradication. Prior work using longitudinal QVOA estimated the half-life of the reservoir as approximately 44 months, similar to the 48 months estimated here for the first 7 years of viral suppression (12, 15). These prior studies had not included observations beyond 7 years.
We observed a nonconstant decay over time, and the decay rate slowed substantially after longer duration of suppression. Our model estimates that 226 years of effective ART would be necessary to achieve a 4 log10 decrease in intact proviral DNA levels. This suggests once more that ART alone will be insufficient to clear the HIV reservoir and further supports that novel agents will be needed for eradication. Although this study did not investigate the mechanism through which this nonconstant decay occurs, 1 possibility is that it reflects the dynamics of different populations of CD4+ T cells, with the initial steeper slope associated with transcriptionally active, shorter-lived cells carrying intact proviruses followed by a slower slope reflective of a second cell population of infected CD4+ T cells that is longer lived or replenished.
Changes in T cell subset frequencies might also contribute if cells that are infected at a lower rate (e.g., naive T cells) expand over time and dilute the reservoir, although this has yet to be confirmed as a mechanism governing reservoir dynamics (16, 17). Furthermore, if changing T cell subset frequencies were alone responsible for decay, this would not explain the variable decay between intact and defective provirus unless certain subsets are enriched for certain proviruses.
Our analysis only provides perspectives on the rates at which intact and defective genomes change over time. Notably, we did not measure the composition of the reservoir, which is known to be dynamic. HIV is maintained, in large part, due to clonal expansion of infected cells, a process that presumably reflects the effect of homeostatic cytokines and antigen on maintenance of T cell memory (6, 18–23). These clones are known to wax and wane over time (24). During long-term ART, the reservoir tends to become more clonal in nature (25). Most clones contain defective genomes, but clonal populations containing intact genomes have been reported (19, 20, 26–28). Our findings are consistent with a recent small study (n = 4) (13), which found that proviruses with a 5′ deletion tended to expand over time. The authors postulated that this could be due to a lack of negative selection pressure related to inefficient protein expression in these clones or due to enhanced immune evasion related to Nef (13). Although we evaluated fewer time points per individual, our study lends additional support to this observation within a larger sample. Additional studies are clearly needed.
Individuals with higher nadir CD4+ T cell values exhibited a faster rate of decay of intact provirus in the first 7 years, suggesting that this marker of immune status is important in determining reservoir dynamics. A similar association has been described with total viral DNA (29). Net decay in intact proviruses includes rates of expansion (e.g., homeostatic proliferation) and decay (e.g., immune-mediated clearance, memory T cell deletion). If a lower nadir favors the former, the slower decay could be driven by higher amounts of homeostatic proliferation. Similarly, those who start ART earlier will theoretically have more robust clearance mechanisms of virus-infected cells. This observation continues to lend support to the benefit of early ART initiation and suggests that those who have initiated ART early in the course of their infection may be optimal participants for cure-related interventions.
We also observed a potentially differential effect of ART agents, with individuals on PI-based regimens exhibiting slower decay dynamics than those not on such regimens. Notably, in 2 intensification studies in which episomal DNA (2-LTR circles) were measured prospectively, those individuals on PI regimens were more likely to exhibit a transient increase in DNA levels (a marker of residual virus replication) (30, 31). The clinical significance of these observations remains uncertain.
We did not encounter an individual for whom amplification consistently failed for either the Psi or Env reaction across all time points tested. However, intact proviruses and/or 5′ defective proviruses were not detectable in a small number of individuals at baseline. One explanation for this observation is that intact or 5′ defective proviruses could have been present at levels below the detection limit of the assay. We anticipate that, in such scenarios, analysis of a larger number of CD4+ T cells would likely enable detection of intact proviruses. Total proviral DNA levels were very low in rare individuals, such that even defective proviruses were difficult to detect. In some of these cases, dual-positive droplets (intact proviruses) comprised a higher percentage of the overall infected cell population than expected, and there were lower levels of single-positive droplets (defective proviruses). These findings are potentially consistent with a recent period of viremia or expansion of a T cell clone harboring an intact provirus. It is also possible that some individuals may harbor a polymorphism in an IPDA primer or probe binding site that might interfere with amplification and result in inability to detect Env- or Psi-positive droplets. Because the IPDA employs duplex PCR reactions, complete amplification failures for a single amplicon are readily apparent (Supplemental Figure 4; ref. 11). In this sense, the IPDA has a distinct advantage over single amplicon PCR assays for which the absence of signal can be due either to a low frequency of target template or polymorphisms that preclude amplification. When only 1 of the IPDA reactions gives positive droplets, a polymorphism may be present and require the use of alternative primers or probes in order to detect intact proviruses.
This study has several limitations. First, while all participants had been on ART for at least 1 year at the first IPDA time point, there was variability in the preceding duration of documented suppression because of sample availability. This meant that we were able to evaluate intact proviral measurements at the initial point of suppression for only 20 participants (25% of the cohort). Importantly, this prevented our data alone from being able to determine whether CD4+ T cell nadir influences the rate of decline during suppression or the initial level of provirus subtypes at the start of suppression (or both), but we believe that the former is biologically plausible. Second, our study cohort was primarily composed of white men due to sample availability, and we were therefore unable to explore with much precision sex- or race-based differences. Although a recent study did not show sex-based differences in HIV-1 DNA levels in CD4+ T cells (32), previous studies have shown differences in PBMCs; further work should aim to include a higher proportion of women. Third, we chose to study individuals who initiated ART during chronic infection. Those who started therapy during acute infection likely have different dynamics, as has been suggested by prior studies (33–35). Fourth, our models were based on a median 2.7 time points per individual. This limits the precision of estimates of half-life and relationships with covariates. If possible, future studies could include a greater number of time points per participant. Fifth, the direction of causation could be reversed or bidirectional for some covariates, such as concurrent CD4 and CD8.
There are also limitations with respect to assay performance. The IPDA was developed as a high-throughput molecular assay aimed at quantifying the intact proviruses. Because the IPDA uses ddPCR for proviral discrimination, the assay cannot be used to separately quantify clonal proviruses. Therefore, deconvolution of the dynamics of clonal expansion and/or decay of intact and defective proviruses was not possible in this longitudinal analysis. As described (11), proviruses with defects affecting both proviral discrimination reactions are not quantified by the IPDA. Previous analysis indicates that such proviruses comprise a small fraction of the total population of persistent proviruses (11). Nevertheless, the dynamics of these proviruses remain unknown. In addition, polymorphisms affecting the IPDA primer or probe binding sites may lead to an underestimate of intact proviruses in some situations. Polymorphisms precluding amplification in 1 of the IPDA reactions at all time points were not observed in this cohort but have been observed in other patient populations and are readily discernible (our unpublished observations). In these situations, the use of alternative primers is required. When both IPDA reactions give positive droplets for a given individual, it remains possible that a subset of proviruses in that individual harbor polymorphisms that interfere with amplification. However, as we have previously shown (11), the frequencies of intact proviruses detected by the IPDA are, on average, about 100-fold higher than those obtained with the QVOA, likely because it detects many proviruses that are not induced by a single round of T cell activation used in the QVOA (9, 19). In any event, because IPDA values greatly exceed QVOA values, these undetected variants, if present, are likely to make only a minor contribution to the reservoir. It is important to note that the IPDA primers and probes bind to highly conserved regions of the viral genome. In addition, the digital droplet PCR format of the assay is tolerant of minor polymorphisms in primer and probe binding sites owing to its use of endpoint PCR amplification. Importantly, despite this tolerance for minor polymorphisms, the IPDA has essentially no background, as demonstrated by uninfected donor control material processed and analyzed in parallel with every IPDA batch (Supplemental Figure 1). Polymorphisms reducing PCR efficiency can result in droplets with slightly lower amplitude, but these are still correctly scored as positive droplets so long as the droplet amplitude exceeds the gates set with DNA from uninfected donors. It is also worth noting that all molecular assays employed in the HIV persistence field — both absolutely quantitative (e.g., IPDA, cell-associated RNA [CA-RNA], cell-associated DNA [CA-DNA]) and relatively quantitative (e.g., near–full-length proviral sequencing and related approaches) — are sensitive to polymorphisms at primer and/or probe binding sites. Defining specific polymorphisms that affect amplification, their frequency across people infected with HIV-1, and approaches for mitigating these effects for any molecular assay for persistent HIV-1 will require the analysis of large and diverse cohorts of infected individuals. With respect to the IPDA, this is an area of active study.
Finally, while near–full-length proviral sequencing might have provided more accurate discrimination of intact from defective proviruses, this method is not currently feasible to use in large studies such as this and, importantly, does not directly provide absolute quantitation of proviral frequencies per measured input cell. Furthermore, near–full-length sequencing and related approaches rely on the long-range amplification of proviruses. Although the overall technical limitations of long-range PCR amplification are well known, it is conceivable that there are differences in the amplification efficiency of intact versus defective proviruses due to differences in length (as a result of large deletions) or nucleotide content (as a result of extensive hypermutation). These sources of bias are under active investigation and should be considered when interpreting proviral sequencing data sets.
In conclusion, we found a differential rate of decay for intact and defective provirus in a cohort of HIV-infected individuals and confirmed several prior observations about reservoir dynamics and their correlates that used fewer optimal assays. The IPDA, despite potential limitations, could be a useful tool in evaluating the impact of upcoming curative interventions for HIV infection.
Methods
Study participants and samples.
Study participants had been enrolled in the UCSF SCOPE cohort between 2001 and 2017. SCOPE is a large, longitudinal cohort of HIV-infected individuals with detailed characterization of clinical and virologic outcomes. From this cohort, we identified individuals who had been on ART for at least 1 year, had plasma HIV RNA levels that were below the level of quantification at the baseline visit for this analysis, and subsequently maintained viral suppression for at least 2 years, allowing for isolated HIV RNA levels that were above the limit of quantification but below 200 copies RNA/mL. We prioritized those participants who had the longest duration of viral suppression and who had stored PBMCs that spanned this period; samples for subsequent analysis were then selected that spanned this period of suppression. We calculated the duration of suppression as the time between the first documented suppressed plasma HIV RNA and the IPDA sample. In some cases, a participant had been on ART for a year but had not had confirmation of plasma HIV RNA suppression before the IPDA time point. In these cases, we took a conservative approach and used the time point for the IPDA sample as the date of first suppression. Basic HIV parameters, including plasma HIV RNA and CD4+ T lymphocyte count, were measured using clinical assays at the time of specimen collection.
IPDA measurements.
An in-depth description of the IPDA rationale and procedure is available in Bruner et al. (11). In this study, the IPDA was performed by Accelevir Diagnostics under company standard operating procedures. Cryopreserved PBMCs from each participant were viably thawed, and total CD4+ T cells were obtained via immunomagnetic selection (EasySep Human CD4+ T cell Enrichment Kit, Stemcell Technologies), with cell count, viability, and purity assessed by flow cytometry both before and after selection. An average of 2.5 million untouched CD4+ T cells were recovered for each sample. Genomic DNA was isolated using the QIAamp DNA Mini Kit (Qiagen). DNA concentrations were determined by fluorometry (Qubit dsDNA BR Assay Kit, Thermo Fisher Scientific), and DNA quality was determined by ultraviolet-visible (UV/VIS) spectrophotometry (QIAxpert, Qiagen). Genomic DNA was then analyzed by IPDA.
As described previously (11), each IPDA consists of 2 multiplex ddPCR reactions performed in parallel: (a) the HIV-1 Proviral Discrimination reaction, which distinguishes intact from defective proviruses; and (b) the Copy Reference/Shearing reaction, which quantifies DNA shearing and input diploid cell equivalents. The HIV-1 Proviral Discrimination reactions employ 2 independent hydrolysis probe reactions that simultaneously interrogate the packaging signal and Rev-response element on individual proviruses to discriminate intact from defective proviruses (11). ddPCR droplets containing intact proviruses exhibit probe signal from both discriminatory amplicons, while droplets containing defective proviruses exhibit only 1 probe signal from a single discriminatory amplicon. The Copy Reference/Shearing reactions employ 2 independent hydrolysis probe reactions targeting the human RPP30 gene (chromosome 10: 90,880,081 on GRCh38). As described and demonstrated previously (11), assay results are affected by DNA shearing between IPDA amplicons, which artificially reduces intact provirus counts while increasing defective provirus counts. The amount of intraamplicon shearing is directly measured by the Copy Reference/Shearing reaction for each sample. The rationale for the design of this duplex ddPCR reaction was previously described in depth (11). For each sample, the DNA shearing index (DSI) is calculated based on the ratio of dual fluorescent to single fluorescent droplets to determine the frequency of intraamplicon shearing, which occurred during DNA isolation and assay setup. The average DSI measured for the samples was 0.36. Shearing correction is applied to both IPDA component reactions to account for the measured intraamplicon shearing using the DSI, and final IPDA results are reported as proviral frequencies per million CD4+ T cells. ddPCR reactions were assembled via automated liquid handlers to maximize reproducibility. As described previously (11), ddPCR was performed on the Bio-Rad QX200 AutoDG Digital Droplet PCR system using the appropriate manufacturer-supplied consumables and the ddPCR Supermix for probes (no dUTPs) (Bio-Rad). HIV-1 Proviral Discrimination reactions were performed using the primer and probe sequences as described (11). Copy Reference/DNA Shearing reactions were performed with the DNA Shearing Control for IPDA Multiple digital PCR primer/probe mix (20×) (Accelevir Diagnostics, catalog 19-1001). As described previously (11), up to 700 ng of genomic DNA was analyzed in each HIV-1 proviral discrimination reaction to ensure optimal droplet occupancy. For Copy Reference/DNA Shearing reactions, the input genomic DNA was diluted 1:100 such that up to 7 ng of genomic DNA was analyzed per reaction. Final input DNA concentrations were dependent upon the concentration of recovered DNA. Samples were batch processed and analyzed. For each batch, CD4+ T cells from uninfected donors were parallel processed and analyzed as negative controls (Supplemental Figure 1). Gates were established using these negative control reactions. J-Lat full-length clone 6.3 cells were used as positive controls (Supplemental Figure 1). The J-Lat full-length clone 6.3 from E. Verdin (Gladstone Institutes and UCSF, San Francisco, CA, USA) was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. Cells were authenticated through short tandem repeat analysis and tested negative for mycoplasma. Operators were blinded to subject identity and time point. Typical patient sample results are shown in Supplemental Figure 2. Atypical patterns possibly reflecting polymorphisms are illustrated in Supplemental Figures 3 and 4.
Statistics.
We sought to estimate the rate of change over time in log(copies per million cells), along with the influence of covariates on that rate of change, both for the primary outcome of intact provirus and the secondary outcomes of defective provirus (total combined defective provirus and its 2 subtypes). Our primary predictor was, therefore, the duration of viral suppression at the time of each sample collection, which is a time-varying predictor that changes within each person across their different measurements, permitting longitudinal estimation of rates of change. We used mixed-effects models for repeated measures and did not calculate differences or rates of change between observations before modeling. Because some observations were zero (no copies detected), we initially evaluated mixed effects negative binomial regression models, which permit inclusion of observations of zero without modifications (36). In simple preliminary models, however, we encountered a convergence problem for 1 type of defective provirus, and a modified linear regression approach (see below) produced very similar results for the models that converged without issues. In addition, observations of zero were infrequent for intact provirus (7%), with 84% of observations having > 10 copies measured, and even less frequent for defective provirus. We therefore applied random-intercept, random-slope linear regression to loge(0.5 + copies per million cells), allowing a covariance term between the 2 random effects. The addition of 0.5 permits log transformation when no copies were detected, but it results in very little change to most observations, approximately preserving the interpretation of back-transformed regression coefficients as multiplicative effects. For rates of change and the influence of covariates on it, we present percent effects as 100 × (exp[coefficient] – 1).
We initially modeled intact provirus over time using a linear effect of duration of suppression, implying a constant rate of change regardless of how long viral load has been suppressed, but there was evidence of nonlinearity, implying that a single, global rate of decline would not be meaningful. We therefore evaluated linear spline models with 1 knot, which allow the rate of change to slow after a specified duration of suppression (the knot). We evaluated different placements for the knot at 3–9 years, and we selected 7 years as the value that optimized the Akaike Information Criterion (AIC) (37). A likelihood ratio test of this versus the model with constant rate of decline at all times produced P = 1.6 × 10–7. These models included a random intercept and a random slope up to the knot. Additionally, allowing for random person-to-person variation in the slope after the knot did not improve the model (the estimated random effect variance was zero, corresponding to no person-to-person variation in the rate of decline after 7 years). Our “base model,” therefore, estimated a rate of decline up to 7 years of viral suppression and a rate after 7 years, while allowing for random person-to-person variation in levels at the start of suppression and in the rate of change over the first 7 years of suppression. We also attempted to fit 2-compartment (or “2-phase”) models that have previously been used in similar applications (22, 38), but these did not fit as well as the simpler linear spline model. Adding an additional knot before or after 7 years also did not improve the AIC. We obtained P values for the different rates of decline for intact versus defective virus by applying a paired two-tailed t test to the individuals’ fitted slopes up to 7 years (fixed plus random effects). In plots of fitted random slopes, we excluded individuals with no data on the rate of decline over 7 years (n = 11), due to a single time point being studied or the first sample timed after year 7 of suppression. To compare rates of decline in intact versus defective virus, we applied paired two-tailed t tests to fitted slopes from the base model with no covariates.
For the primary analysis, we estimated the influence of covariates on rates of change over the first 7 years of suppression. To do so, we used interaction terms for the covariate with duration of suppression, which were calculated for each observation based on the covariate’s history up to the time of the observation. We included covariates that change over time by making 2 assumptions: (a) the influence of the covariate between the start of suppression and the first observation in this study is absorbed into the random effects terms and so is not explicitly modeled; and (b) the influence of a covariate during an interval between observations is determined by the average of its values at the beginning and end of the interval. For example, a covariate X that, for a given person, takes on values x1, x2, x3, and x4 at times t1, t2, t3, and t4 after the start of suppression would have an interaction term that takes on the values: 0 at time t1; (t2 – t1) × (x1 + x2)/2 at time t2; (t2 – t1) × (x1 + x2)/2 + (t3 – t2) × (x2 + x3)/2 at time t3; and (t2 – t1) × (x1 + x2)/2 + (t3 – t2) × (x2 + x3)/2 + (t4 – t3) × (x3 + x4)/2 at time t4. The interaction terms reflect the cumulative effect that the influence of X on the rate of change has had up to each time. For each interval between time points, the influence of X on the rate of change is reflected in the measurement at the end of the interval, with the influence during the interval equal to the duration of the interval (the first term in each of the of the expressions given above) times the average value of the covariate in the interval (the second term in the expressions above). The measurements at the ends of the later intervals reflect the influence from all the previous intervals. Because the estimated rate of change in the base model was much slower after 7 years, with an estimate of no person-to-person variation, we did not evaluate the influence of covariates on the rate of change after 7 years of suppression.
As a secondary analysis, we estimated the effects of covariates on levels of intact and defective virus (rather than rates of change), again using random-intercept, random-slope linear regression models with the same linear spline model for the effect of duration of suppression. In these models, fixed covariates can be interpreted as influencing the level at the start of viral suppression. This simple interpretation is less clear for covariates that change over time, so we recommend consideration of the plausibility of such an influence and the alternative possibilities of reverse or bidirectional causation. For the effects of covariates on levels, we report fold-effects defined as exp(coefficient). Models allowing fixed covariates to influence both initial levels and rates of decline were evaluated as an additional secondary analysis.
Study approval.
The SCOPE protocol was approved by the UCSF IRB. All individuals provided informed consent at the time of enrollment.
Author contributions
MJP, PB, RFS, GML, and SGD designed the study. KDR, SB, JL, and GML performed the assays. MJP and PB performed statistical analyses. MJP, PB, GML, JNM, PWH, TJH, JDS, RFS, GML, and SGD interpreted the results. MJP, PB, GML, and SGD wrote the manuscript, which was edited by PWH, TJH, JDS, and RFS. All authors read and approved the final manuscript.
Supplementary Material
Acknowledgments
This work was supported the Delaney AIDS Research Enterprise (DARE; AI096109, A127966). The SCOPE cohort receives support from the UCSF/Gladstone Institute of Virology & Immunology CFAR (P30 AI027763), the CFAR Network of Integrated Systems (R24 AI067039), and the amfAR Institute for HIV Cure Research (amfAR 109301). Additional support was provided by the I4C (UM1 AI126603) and Beat-HIV (UM1 AI126620) Collaboratories, the Howard Hughes Medical Institute, Gilead Sciences (unrestricted research grant), and the Bill and Melinda Gates Foundation (OPP1115715). MJP receives funding on a training grant NIH/NIAID T32 AI60530-12. Accelevir Diagnostics acknowledges support from SBIR grants from NIH (R44AI124996) and NSF (1738428).
Version 1. 02/11/2020
In-Press Preview
Version 2. 02/27/2020
Electronic publication
Version 3. 02/27/2020
Figure 3 corrected; Conflict of interest statement, Acknowledgments, and Table 1 clarified.
Footnotes
Conflict of interest: AccelevirDx is a for-profit company; aspects of the IPDA are the subject of patent application PCT/US16/28822 filed by Johns Hopkins University and AccelevirDx holds an exclusive license for this patent application. MJP, SB, and JL have nothing to disclose. PB, JNM, and SGD report grants from the NIH during the conduct of the study. KDR is an employee of AccelevirDx. PWH reports grants and/or personal fees from Gilead Sciences, Viiv, Janssen, and Biotron, outside the submitted work. TJH reports grants and/or personal fees from Merck & Co. and Gilead Sciences, outside the submitted work. JDS reports personal fees from Gilead Sciences, outside the submitted work. RFS is an inventor on the IPDA patent application. He holds no equity interest in AccelevirDx. GML is an employee of and equity holder in AccelevirDx. SGD reports grants and/or personal fees from Gilead Sciences, Merck & Co., Viiv, AbbVie, Eli Lilly, ByroLogyx, and Enochian Biosciences outside the submitted work.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: JCI Insight. 2020;5(4):e132997.https://doi.org/10.1172/jci.insight.132997.
Contributor Information
Michael J. Peluso, Email: michael.peluso@ucsf.edu.
Peter Bacchetti, Email: PBacchetti@epi.ucsf.edu.
Kristen D. Ritter, Email: kritter@accelevir.com.
Jun Lai, Email: jlai5@jhmi.edu.
Jeffrey N. Martin, Email: Martin@psg.ucsf.edu.
Peter W. Hunt, Email: peter.hunt@ucsf.edu.
Timothy J. Henrich, Email: timothy.henrich@ucsf.edu.
Janet D. Siliciano, Email: Jsilicia@jhmi.edu.
Robert F. Siliciano, Email: rsiliciano@jhmi.edu.
Gregory M. Laird, Email: greg.laird@accelevir.com.
Steven G. Deeks, Email: sdeeks@php.ucsf.edu.
References
- 1.Chun TW, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA. 1997;94(24):13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finzi D, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278(5341):1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 3.Wong JK, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278(5341):1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 4.Finzi D, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5(5):512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 5.Deeks SG, et al. International AIDS Society global scientific strategy: towards an HIV cure 2016. Nat Med. 2016;22(8):839–850. doi: 10.1038/nm.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohn LB, et al. HIV-1 integration landscape during latent and active infection. Cell. 2015;160(3):420–432. doi: 10.1016/j.cell.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siliciano JD, Siliciano RF. Assays to Measure Latency, Reservoirs, and Reactivation. Curr Top Microbiol Immunol. 2018;417:23–41. doi: 10.1007/82_2017_75. [DOI] [PubMed] [Google Scholar]
- 8.Barton KM, Palmer SE. How to Define the Latent Reservoir: Tools of the Trade. Curr HIV/AIDS Rep. 2016;13(2):77–84. doi: 10.1007/s11904-016-0304-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho YC, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155(3):540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massanella M, Richman DD. Measuring the latent reservoir in vivo. J Clin Invest. 2016;126(2):464–472. doi: 10.1172/JCI80567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruner KM, et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature. 2019;566(7742):120–125. doi: 10.1038/s41586-019-0898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siliciano JD, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. 2003;9(6):727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 13.Pinzone MR, et al. Longitudinal HIV sequencing reveals reservoir expression leading to decay which is obscured by clonal expansion. Nat Commun. 2019;10(1):728. doi: 10.1038/s41467-019-08431-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imamichi H, et al. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc Natl Acad Sci USA. 2016;113(31):8783–8788. doi: 10.1073/pnas.1609057113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crooks AM, et al. Precise Quantitation of the Latent HIV-1 Reservoir: Implications for Eradication Strategies. J Infect Dis. 2015;212(9):1361–1365. doi: 10.1093/infdis/jiv218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robbins GK, et al. Incomplete reconstitution of T cell subsets on combination antiretroviral therapy in the AIDS Clinical Trials Group protocol 384. Clin Infect Dis. 2009;48(3):350–361. doi: 10.1086/595888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zerbato JM, McMahon DK, Sobolewski MD, Mellors JW, Sluis-Cremer N. Naive CD4+ T Cells Harbor a Large Inducible Reservoir of Latent, Replication-competent Human Immunodeficiency Virus Type 1. Clin Infect Dis. 2019;69(11):1919–1925. doi: 10.1093/cid/ciz108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chomont N, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15(8):893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hosmane NN, et al. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J Exp Med. 2017;214(4):959–972. doi: 10.1084/jem.20170193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee GQ, et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J Clin Invest. 2017;127(7):2689–2696. doi: 10.1172/JCI93289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maldarelli F, et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014;345(6193):179–183. doi: 10.1126/science.1254194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer S, et al. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci USA. 2008;105(10):3879–3884. doi: 10.1073/pnas.0800050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner TA, et al. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 2014;345(6196):570–573. doi: 10.1126/science.1256304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, et al. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc Natl Acad Sci USA. 2018;115(11):E2575–E2584. doi: 10.1073/pnas.1720665115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hiener B, et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4+ T Cells from Effectively Treated Participants. Cell Rep. 2017;21(3):813–822. doi: 10.1016/j.celrep.2017.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bui JK, et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Pathog. 2017;13(3):e1006283. doi: 10.1371/journal.ppat.1006283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorenzi JC, et al. Paired quantitative and qualitative assessment of the replication-competent HIV-1 reservoir and comparison with integrated proviral DNA. Proc Natl Acad Sci USA. 2016;113(49):E7908–E7916. doi: 10.1073/pnas.1617789113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simonetti FR, et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc Natl Acad Sci USA. 2016;113(7):1883–1888. doi: 10.1073/pnas.1522675113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Besson GJ, et al. HIV-1 DNA decay dynamics in blood during more than a decade of suppressive antiretroviral therapy. Clin Infect Dis. 2014;59(9):1312–1321. doi: 10.1093/cid/ciu585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buzón MJ, et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat Med. 2010;16(4):460–465. doi: 10.1038/nm.2111. [DOI] [PubMed] [Google Scholar]
- 31.Hatano H, et al. Increase in 2-long terminal repeat circles and decrease in D-dimer after raltegravir intensification in patients with treated HIV infection: a randomized, placebo-controlled trial. J Infect Dis. 2013;208(9):1436–1442. doi: 10.1093/infdis/jit453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scully EP, et al. Sex-Based Differences in Human Immunodeficiency Virus Type 1 Reservoir Activity and Residual Immune Activation. J Infect Dis. 2019;219(7):1084–1094. doi: 10.1093/infdis/jiy617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hocqueloux L, et al. Long-term antiretroviral therapy initiated during primary HIV-1 infection is key to achieving both low HIV reservoirs and normal T cell counts. J Antimicrob Chemother. 2013;68(5):1169–1178. doi: 10.1093/jac/dks533. [DOI] [PubMed] [Google Scholar]
- 34.Chun TW, et al. Decay of the HIV reservoir in patients receiving antiretroviral therapy for extended periods: implications for eradication of virus. J Infect Dis. 2007;195(12):1762–1764. doi: 10.1086/518250. [DOI] [PubMed] [Google Scholar]
- 35.Jain V, et al. Antiretroviral therapy initiated within 6 months of HIV infection is associated with lower T-cell activation and smaller HIV reservoir size. J Infect Dis. 2013;208(8):1202–1211. doi: 10.1093/infdis/jit311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bacchetti P, et al. Statistical analysis of single-copy assays when some observations are zero. J Virus Erad. 2019;5(3):167–173. doi: 10.1016/S2055-6640(20)30047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akaike H. A new look at the statistical model identification. IEEE Transactions on Automatic Control. 1974;19(6):716–723. doi: 10.1109/TAC.1974.1100705. [DOI] [Google Scholar]
- 38.Wu H, Ding AA. Population HIV-1 dynamics in vivo: applicable models and inferential tools for virological data from AIDS clinical trials. Biometrics. 1999;55(2):410–418.rics. doi: 10.1111/j.0006-341X.1999.00410.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.