

Platelet-mimicking nanoparticles enable the targeted delivery of small interfering RNA to tumors for anticancer therapy.
Abstract
Small interfering RNA (siRNA) is a powerful tool for gene silencing that has been used for a wide range of biomedical applications, but there are many challenges facing its therapeutic use in vivo. Here, we report on a platelet cell membrane–coated metal-organic framework (MOF) nanodelivery platform for the targeted delivery of siRNA in vivo. The MOF core is capable of high loading yields, and its pH sensitivity enables endosomal disruption upon cellular uptake. The cell membrane coating provides a natural means of biointerfacing with disease substrates. It is shown that high silencing efficiency can be achieved in vitro against multiple target genes. Using a murine xenograft model, significant antitumor targeting and therapeutic efficacy are observed. Overall, the biomimetic nanodelivery system presented here provides an effective means of achieving gene silencing in vivo and could be used to expand the applicability of siRNA across a range of disease-relevant applications.
INTRODUCTION
RNA interference (RNAi) is a naturally occurring mechanism for gene down-regulation that, since its first discovery in the late 1990s, has been widely leveraged as a tool for biological studies (1). Through a robust process mediated by the RNA-induced silencing complex present within the cytosol, target genes can be posttranscriptionally silenced via degradation of the corresponding mRNA (2). Small interfering RNAs (siRNAs) are short and well-defined double-stranded RNA molecules that can be synthetically manufactured to take advantage of the RNAi pathway (3). Over time, siRNAs have become an indispensable tool for validating gene function (4). They have also been widely explored as therapeutics for human disease (5), and an siRNA-based treatment for transthyretin-mediated amyloidosis was recently approved by the U.S. Food and Drug Administration (6). Despite their great promise, there are several hurdles that have prevented the more widespread approval of siRNA therapies, and these include their inherent vulnerability in biological environments, suboptimal uptake and knockdown efficiency, rapid blood clearance, and possible immunogenicity (7).
The formulation of siRNA into nanoscale delivery vehicles represents an effective strategy for enhancing bioavailability and biocompatibility (8). This general approach has helped to greatly improve therapeutic outcomes for RNAi therapies (6, 9). Overall, there are several key design considerations when it comes to the nanodelivery of siRNA. First, the payload needs to be incorporated in a manner that protects it from the surrounding environment after in vivo administration (10). siRNA is subject to degradation by endogenous ribonucleases (RNases), and strategies to prevent this from happening are thus required. Localization to a tissue of interest is also important, and this can be highly challenging for RNAi delivery platforms to achieve. To address this, a number of different strategies can be used to decrease nonspecific interactions while improving targeting efficiency (11). Last, endosomal escape of siRNA payloads is a major barrier for siRNA therapeutics (12). To associate with the RNAi machinery, internalized siRNAs need to reach the cytosolic compartment upon internalization (13). The development of formulations that are structurally stable outside of the target cell, but rapidly disassociate and disrupt endosomes upon uptake, is thus highly desirable.
Here, we report on a biomimetic approach for the targeted delivery of siRNA payloads in vivo (Fig. 1). Specifically, synthetic siRNAs were loaded inside porous metal-organic framework (MOF) nanoparticles through a facile one-pot protocol with high loading efficiency. The zeolitic imidazolate framework-8 (ZIF-8) MOFs used in this work have minimal toxicity and have previously been used for the delivery of biomolecules (14). The structural integrity of the MOF scaffolds is pH dependent (15, 16), a property that we leveraged to traffic siRNA payloads to the cytosol. The siRNA-loaded MOFs were further coated by a layer of naturally derived platelet membrane, which has been shown to interact with a number of different disease-relevant substrates, including atherosclerotic plaque and bacteria (17, 18). In particular, platelet membrane–based nanoparticles have excelled at anticancer therapy due to their tumor-targeting capabilities (19, 20). Using the final nanoformulation, siRNA delivery and gene silencing efficiency were evaluated in vitro. In the end, a murine breast cancer tumor model was used to assess tumor targeting and therapeutic efficacy. Although the present work used siRNAs to address tumor-relevant genes, it is envisioned that the reported hybrid nanodelivery platform could be used to achieve targeted silencing for a wide range of therapeutic applications.
Fig. 1. Platelet membrane–coated siRNA-loaded MOFs (P-MOF-siRNA) for gene silencing.
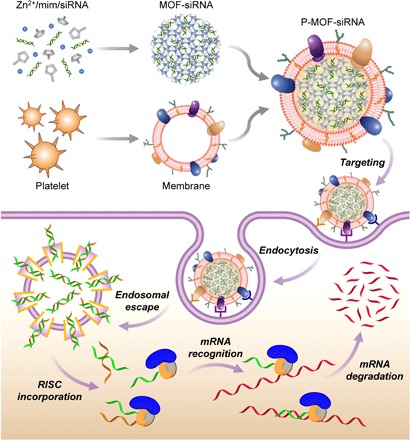
To fabricate the P-MOF-siRNA formulation, siRNA-loaded MOF (MOF-siRNA) cores are generated by mixing the siRNA payload with Zn2+ and 2-methylimidazole (mim), followed by coating with natural cell membrane derived from platelets. When the P-MOF-siRNA nanoparticles are endocytosed by a target cell, the low pH of the endosomes causes escape of the siRNA into the cytosol. Upon incorporation with RNA-induced silencing complex (RISC), the target mRNA is then recognized and degraded, leading to gene silencing.
RESULTS AND DISCUSSION
To encapsulate siRNA within the nanoscale MOFs, a modified in situ biomineralization method was used (21, 22). Specifically, siRNA was first premixed with 2-methylimidazole, followed by addition of a solution containing zinc ions, initiating the ZIF-8 self-assembly process and causing the solution to become turbid. The resulting siRNA-loaded MOF (MOF-siRNA) nanoparticles were then allowed to mature in solution for several hours. For the coating process, purified platelet membrane was mixed with the MOF-siRNA nanoparticles, and the two components were physically co-extruded through porous membranes of decreasing size (23). Last, the platelet membrane–coated MOF-siRNA nanoparticles (P-MOF-siRNA) were isolated by centrifugation. The MOF-siRNA nanoparticles were slightly larger than unloaded MOFs, and membrane coating further increased their size, with the final P-MOF-siRNA formulation exhibiting an average diameter of approximately 175 nm (Fig. 2A). Moreover, while loading of the negatively charged siRNA payload slightly decreased the surface zeta potential compared with unloaded MOFs, the membrane coating had a profound impact on shielding the positively charged MOF core, resulting in P-MOF-siRNA nanoparticles that were considerably negative in charge (Fig. 2B). Both the size and surface zeta potential data suggested successful coating of the membrane around the MOF-siRNA nanoparticles, and the core-shell morphology of the P-MOF-siRNA formulation was confirmed by visualizing negatively stained samples under transmission electron microscopy (Fig. 2C). In terms of siRNA loading, the encapsulation efficiency was quantified by incorporating a fluorescently labeled payload at increasing concentrations (Fig. 2D). It was demonstrated that siRNA could be incorporated with high efficiency over a wide range of inputs, and P-MOF-siRNA nanoparticles were fabricated using 500 nM siRNA, 20 mg/ml 2-methylimidazole, and 1 mg/ml zinc nitrate hexahydrate for subsequent studies. The favorable loading exhibited by the MOFs was likely attributed to strong electrostatic interactions between the framework’s metal nodes and the siRNA’s backbone phosphates, as well as physical confinement within the nanoporous structure (16, 24).
Fig. 2. Formulation and characterization.
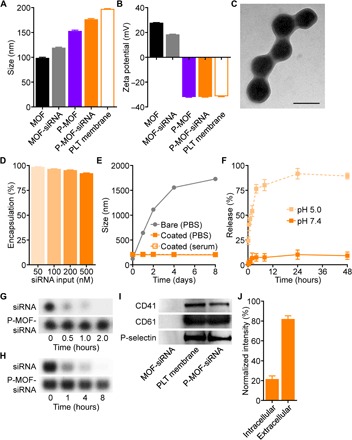
(A) Diameter of pristine MOF, MOF-siRNA, P-MOF, P-MOF-siRNA, and platelet (PLT) membrane vesicles after formulation (n = 3, mean + SD). (B) Zeta potential of pristine MOF, MOF-siRNA, P-MOF, P-MOF-siRNA, and platelet membrane vesicles after formulation (n = 3, mean + SD). (C) Transmission electron microscopy image of P-MOF-siRNA negatively stained with uranyl acetate (scale bar, 200 nm). (D) Encapsulation efficiency of siRNA inside P-MOF-siRNA at various siRNA inputs (n = 3, mean + SD). (E) Stability of MOF-siRNA and P-MOF-siRNA over time in PBS or serum-containing medium (n = 3, mean ± SD). (F) siRNA release from P-MOF-siRNA at pH 5.0 or pH 7.4 over time (n = 3, mean ± SD). (G and H) Degradation of siRNA, either in free form or in P-MOF-siRNA, when exposed to purified RNase (G) or serum-containing medium (H) for increasing amounts of time. (I) Western blots for three characteristic platelet surface markers (CD41, CD61, and P-selectin) in MOF-siRNA, platelet membrane vesicles, and P-MOF-siRNA. (J) Dot blot intensity of P-MOF-siRNA probed with antibodies against the intracellular or extracellular domains of CD47 (n = 3, mean + SD).
To further characterize the final formulation, long-term stability was evaluated in phosphate-buffered saline (PBS) and serum-containing medium over the course of 1 week (Fig. 2E). While P-MOF-siRNA nanoparticles exhibited minimal change in size over this period, uncoated MOF-siRNA particles grew considerable, highlighting the benefit of using a cell membrane coating for stabilizing nanoparticles under physiological conditions (25). Release of the siRNA payload from P-MOF-siRNA nanoparticles was measured over time in PBS at different pH values (Fig. 2F). Whereas a minimal amount of release was observed at a near-neutral pH 7.4, considerable burst release in the first few hours was observed at a more acidic pH 5.0. This pH-dependent property of the formulation suggested that the siRNA could be protected within the nanoparticulate structure until after cellular internalization, upon which the mildly acidic environment of the endosomes would trigger siRNA release (15). To confirm the utility of nanoencapsulation for preserving the integrity of siRNA, P-MOF-siRNA nanoparticles were incubated with RNase or serum-containing medium for varying amounts of time (Fig. 2, G and H). Because of protection by the MOF scaffold (26, 27), little degradation of the payload was observed for P-MOF-siRNA nanoparticles; this was in stark contrast to free siRNA, which was rapidly degraded by RNase either in purified form or within serum. Protein analysis was used to confirm the successful translocation of the platelet membrane onto the surface of the P-MOF-siRNA nanoparticles, which could be used to bestow tumor-targeting properties (28, 29). Using gel electrophoresis to first separate proteins based on molecular weight, important binding markers, including CD41, CD61, and P-selectin, were confirmed through Western blotting analysis to be present on the P-MOF-siRNA nanoparticles (Fig. 2I). The sidedness of the membrane coating was characterized by performing dot blots for different CD47 domains, and it was determined that approximately 80% of the platelet membrane was presented in a right-side-out orientation (Fig. 2J), which is essential for maintaining disease-targeting properties.
Next, we sought to characterize the impact of membrane coating on nanoparticle interaction with macrophages. To conduct the study, either bare MOF-siRNA or membrane-coated MOF-siRNA nanoparticles were incubated with murine J774 macrophages. Flow cytometric analysis revealed that the platelet membrane coating was able to significantly reduce interaction of the MOF-siRNA nanoparticles with the macrophages, and the level of uptake was consistent with nanoparticles coated with red blood cell (RBC) membrane (R-MOF-siRNA) (Fig. 3A). Further, the secretion of frontline proinflammatory cytokines by macrophages was measured to evaluate the potential short-term immunogenicity of the platform. Whereas incubation with bare MOF-siRNA led to significantly elevated levels of both interleukin-6 (IL-6) and tumor necrosis factor–α (TNFα), the membrane coating was able to abrogate this effect (Fig. 3, B and C). To confirm the cancer-targeting ability of P-MOF-siRNA, fluorescently labeled nanoparticles were incubated with SK-BR-3 cells. Fluorescence imaging demonstrated that the platelet membrane coating greatly improved the affinity of the nanoparticles toward SK-BR-3 cells in comparison to RBC membrane–coated nanoparticles (Fig. 3D). Because the SK-BR-3 cell line is known to express CD24 (30), which is a counter receptor to P-selectin found on platelets (31), we also studied the effect of inhibiting this interaction. Antibody blocking significantly attenuated binding of P-MOF-siRNA to the tumor cells. The data confirmed that platelet membrane, which displays specificity toward a number of different surface markers commonly expressed by cancer cells (32), has utility for tumor targeting.
Fig. 3. In vitro cellular uptake and GFP knockdown.
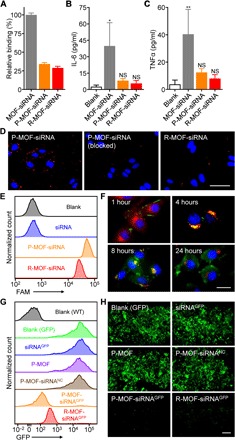
(A) Uptake of MOF-siRNA, P-MOF-siRNA, or R-MOF-siRNA by J774 macrophages after 24 hours of incubation (n = 3, mean + SD). (B and C) Secretion of proinflammatory cytokines IL-6 (B) or TNFα (C) by J774 macrophages after 24 hours of incubation with MOF-siRNA, P-MOF-siRNA, or R-MOF-siRNA (n = 3, mean + SD). *P < 0.05, **P < 0.01; NS, not significant; one-way analysis of variance (ANOVA). (D) Binding of fluorescently labeled P-MOF-siRNA, P-MOF-siRNA preblocked with anti–P-selectin, or R-MOF-siRNA to SK-BR-3 cells after incubation for 30 min (scale bar, 50 μm; nuclei, blue; nanoparticles, red). (E) Uptake of siRNA in SK-BR-3 cells 24 hours after incubation with free siRNA, P-MOF-siRNA, or R-MOF-siRNA. (F) Fluorescent visualization of siRNA localization in SK-BR-3 cells 1, 4, 8, and 24 hours after incubation with P-MOF-siRNA (scale bar, 20 μm; siRNA, green; nuclei, blue; endosomes, red). (G) Fluorescence of GFP-transduced SK-BR-3 cells after incubation with siRNAGFP, P-MOF, P-MOF-siRNANC, P-MOF-siRNAGFP, or R-MOF-siRNAGFP for 48 hours; wild-type (WT) cells were used to establish the baseline. (H) Visualization of gene knockdown in GFP-transduced SK-BR-3 cells after incubation with siRNAGFP, P-MOF, P-MOF-siRNANC, P-MOF-siRNAGFP, or R-MOF-siRNAGFP for 48 hours (scale bar, 200 μm; GFP, green).
After confirming successful targeting of the tumor cells, we then assessed whether the P-MOF-siRNA formulation could be used to facilitate internalization of the siRNA payload. To this end, we used fluorescently labeled siRNA, in either free form, P-MOF-siRNA, or R-MOF-siRNA. After 24 hours of incubation with SK-BR-3 breast cancer cells, the amount of uptake was quantified by flow cytometry (Fig. 3E). Free siRNA exhibited a negligible amount of uptake due to its inability to cross the cell membrane. On the other hand, cells incubated with the two nanoformulations, which are generally taken up via endocytosis (33), displayed a substantial amount of uptake. Because of the specific interactions of cancer cells with platelets, the P-MOF-siRNA nanoparticles were better at delivering the siRNA compared with R-MOF-siRNA nanoparticles. Afterward, we sought to demonstrate that the internalized payloads could be released from the endosomal compartment, which is essential for the silencing effect of siRNA given that RNAi machinery is located within the cytosol (34). To accomplish this, fluorescently labeled siRNA was delivered via P-MOF-siRNA, and the intracellular localization was visualized by fluorescence microscopy over the course of 24 hours (Fig. 3F). At 1 hour after the start of incubation, most of the payload could still be found on the periphery of the cells, but a notable portion had already been endocytosed after 4 hours, as indicated by the colocalization of the dye-labeled siRNA with LysoTracker. At 8 hours, the siRNA signal could be seen throughout the cell, and this effect was even more pronounced after one full day of incubation. Overall, these data indicated that the siRNA could successfully escape or release into the cytosol after dissociation of the MOF scaffold in the more acidic pH of the endosomes, which facilitates endosomal disruption (15).
After confirming the cytosolic delivery of the siRNA payload by P-MOF-siRNA, the gene silencing efficiency of the nanoformulation was evaluated. To accomplish this, we first chose green fluorescent protein (GFP) as a model target. Various samples, including free siRNA against GFP (siRNAGFP), empty P-MOF, P-MOF-siRNA loaded with a negative control siRNA (P-MOF-siRNANC), P-MOF-siRNA loaded with siRNAGFP (P-MOF-siRNAGFP), and R-MOF-siRNA loaded with siRNAGFP (R-MOF-siRNAGFP), were incubated with GFP-expressing SK-BR-3 cells. After 48 hours, the GFP expression of the cells was assessed by flow cytometry (Fig. 3G). At this time point, there was little reduction in GFP expression observed for the siRNAGFP, P-MOF, and P-MOF-siRNANC groups. While both the P-MOF-siRNAGFP and R-MOF-siRNAGFP groups showed gene silencing effects, the effect of the former was considerably stronger than the latter. The P-MOF-siRNAGFP formulation reduced the level of expression to near the baseline established by wild-type SK-BR-3 cells. The same study was repeated using fluorescence imaging, and the results corroborated that P-MOF-siRNAGFP was better at knocking down gene expression compared with R-MOF-siRNAGFP (Fig. 3H). The results from these studies validated the utility of MOF-based nanoparticulate delivery for improving siRNA activity, and the increased cancer cell–specific interactions afforded by the platelet membrane helped to further boost silencing efficiency.
Next, we sought to evaluate the potential of the platform for the targeted silencing of a therapeutically relevant target. For this purpose, we chose survivin, which can play an essential role in tumor progression and metastasis (35). Survivin is overexpressed in most breast carcinomas and is highly correlated with human epidermal growth factor receptor 2 (HER2) gene expression (36). Knockdown of survivin mRNA in HER2-positive SK-BR-3 cells has previously been shown to induce cell apoptosis (37). To test the effect of targeting survivin using our platform, we incubated SK-BR-3 cells with a number of different samples, including siRNA against survivin (siRNASur), P-MOF, P-MOF-siRNANC, P-MOF-siRNA loaded with siRNASur (P-MOF-siRNASur), and R-MOF-siRNA loaded with siRNASur (R-MOF-siRNASur), and tracked cell viability over the course of 3 days (Fig. 4A). Free siRNASur, P-MOF, and P-MOF-siRNANC had no impact on the health of the cancer cells, whereas both P-MOF-siRNASur and R-MOF-siRNASur exhibited cytotoxic activity. Because of the increased interactions of platelets with the cancer cells, treatment with P-MOF-siRNASur had the most profound effect, and this was most evident at 48 hours. It should be noted that the increase in viability at 72 hours was likely due to the transient nature of siRNA knockdown. Western blot analysis confirmed that survivin protein expression was the lowest in the cells treated with P-MOF-siRNASur at 48 hours (Fig. 4B), and the same trend was observed when visualizing protein expression by fluorescence microscopy after immunofluorescence staining (Fig. 4C). Notably, the cells treated with P-MOF-siRNASur had highly irregular nuclei in comparison with the other control groups. Last, the levels of survivin mRNA, which is the most immediate target of siRNA, were quantified by polymerase chain reaction (PCR), and it was shown that P-MOF-siRNASur treatment resulted in an approximately 80% reduction (Fig. 4D). Of the other control groups, only the R-MOF-siRNASur group had any appreciable knockdown effect, causing an approximately 40% reduction in survivin gene expression.
Fig. 4. In vitro survivin knockdown.
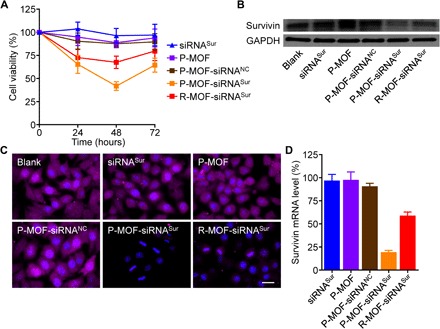
(A) Viability of SK-BR-3 cells after incubation with siRNASur, P-MOF, P-MOF-siRNANC, P-MOF-siRNASur, or R-MOF-siRNASur for 0, 24, 48, and 72 hours (n = 6, mean ± SD). (B) Western blot for survivin in SK-BR-3 cells after incubation with siRNASur, P-MOF, P-MOF-siRNANC, P-MOF-siRNASur, or R-MOF-siRNASur for 48 hours; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. (C) Fluorescent visualization of survivin protein expression in SK-BR-3 cells after incubation with siRNASur, P-MOF, P-MOF-siRNANC, P-MOF-siRNASur, or R-MOF-siRNASur for 48 hours (scale bar, 20 μm; survivin, purple; nuclei, blue). (D) Relative survivin mRNA expression in SK-BR-3 cells after incubation with siRNASur, P-MOF, P-MOF-siRNANC, P-MOF-siRNASur, or R-MOF-siRNASur for 48 hours (n = 3, mean + SD).
Upon confirming in vitro siRNA delivery and activity, we next sought to evaluate the in vivo behavior of the nanoformulation. First, fluorescently labeled P-MOF-siRNASur and R-MOF-siRNASur were administered into nude mice bearing subcutaneous SK-BR-3 tumors. After 1 hour, the animals were euthanized and the distribution of the nanoparticles in all of the major organs was quantified after homogenization (Fig. 5, A and B). Most of the particles for both groups were found in the liver and the spleen, which is characteristic of most nanoformulations (17, 23). Notably, the platelet membrane coating had a profound impact on tumor localization, as the P-MOF-siRNASur nanoparticles had a sixfold higher accumulation compared with R-MOF-siRNASur. This trend was also visualized by ex vivo macroscopic fluorescence imaging of the excised tumors (Fig. 5C). The quick distribution and targeting is a phenomenon that has previously been observed for similar human platelet membrane–coated nanoparticle platforms (17). Following the biodistribution study, we then assessed the therapeutic benefits of the nanoformulations using the same SK-BR-3 tumor model. After the tumors were allowed to grow to a palpable size, P-MOF-siRNASur was administered intravenously every 3 days for a total of four administrations, and the relative change in tumor growth was monitored (Fig. 5D). R-MOF-siRNASur was selected as a control to highlight the advantage of the platelet membrane coating, whereas other controls were omitted given their lack of potency in the in vitro studies. It was evident that both nanoformulations could deliver sufficient siRNA payload to significantly affect tumor growth kinetics, with the strongest effect observed for the P-MOF-siRNASur group given its targeting properties. This was also reflected in the overall survival, where both treatments were able to significantly delay the time it took for the tumors to reach endpoint (Fig. 5E). Whereas untreated mice exhibited a median survival of 50 days, R-MOF-siRNASur and P-MOF-siRNASur treatment prolonged the value to 66 and 92 days, respectively.
Fig. 5. In vivo delivery and antitumor efficacy.
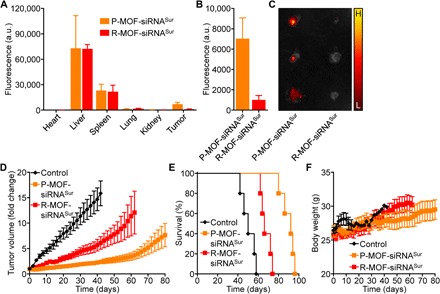
(A) Nanoparticle biodistribution in the heart, liver, spleen, lungs, kidneys, and tumor 1 hour after intravenous administration with fluorescently labeled P-MOF-siRNASur or R-MOF-siRNASur (n = 3, mean + SD). a.u., arbitrary units. (B) Quantification of nanoparticle localization within the tumor 1 hour after intravenous administration with fluorescently labeled P-MOF-siRNASur or R-MOF-siRNASur (n = 3, mean + SD). (C) Ex vivo fluorescent imaging of tumors 1 hour after intravenous administration with fluorescently labeled P-MOF-siRNASur or R-MOF-siRNASur (H, high signal; L, low signal). (D) Growth kinetics of SK-BR-3 tumors implanted subcutaneously into nu/nu mice and treated intravenously with P-MOF-siRNASur or R-MOF-siRNASur every 3 days for a total of four administrations (n = 5; mean ± SEM). (E) Survival of the mice in (D) over time (n = 5). (F) Body weight of the mice in (D) over time (n = 5; mean ± SD).
Regarding the safety of the nanoformulations, none of the treated mice exhibited any significant weight loss over the course of treatment (Fig. 5F). The biosafety was also evaluated by histological analysis of major organs taken 24 hours after intravenous administration of a high dose of P-MOF-siRNASur to healthy mice. From hematoxylin and eosin (H&E) staining, it could be seen that the overall structure, integrity, and immune infiltrate levels in heart, liver, spleen, lung, and kidney tissues were near identical to the same samples from healthy controls (Fig. 6A). Terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling (TUNEL) staining confirmed that the levels of apoptosis in the nanoparticle-treated mice were similar to those of healthy mice (Fig. 6B).
Fig. 6. In vivo safety.
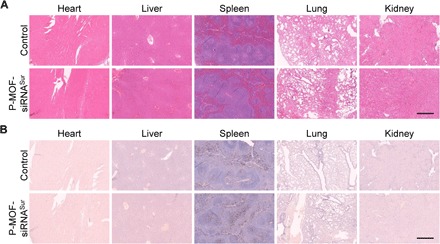
H&E (A) and TUNEL (B) staining of histological sections from major organs, including the heart, liver, spleen, lungs, and kidneys, 24 hours after intravenous administration with a high dose of P-MOF-siRNASur (scale bars, 500 μm).
In conclusion, we have successfully fabricated a biomimetic nanoparticle platform for the effective delivery of siRNA payloads. The formulation was synthesized by first encapsulating siRNA within a nanoscale MOF structure, followed by coating with a natural layer of cell-derived membrane. The MOF component not only enabled high loading efficiency but also facilitated release of siRNA into the cytosol upon dissociation within the endosomal compartment. Using a platelet membrane coating, cancer cell–specific binding was achieved. The final nanoformulation was able to effectively silence gene expression and could be used to achieve strong control of tumor growth when designed against a therapeutically relevant target. The approach presented in this work is highly generalizable, as any kind of RNAi molecule can be facilely incorporated. Multiple payloads could also be encapsulated together within the same MOF to concurrently knock down different targets for improved efficacy. Nucleic acid payloads for other applications, including gene delivery and immune modulation, could be explored. In addition, the membrane component can be easily modulated, and coatings derived from different immune-compatible cell sources can be used to provide application-specific benefits (38–40). Regarding eventual use in human patients, using membrane sourced from universal donors would largely mitigate concerns about the long-term immunogenicity of the platform. Ultimately, the use of cell membrane–coated MOFs as nanodelivery vehicles could help to greatly expand the utility of nucleic acid–based therapies, facilitating their downstream clinical translation.
MATERIALS AND METHODS
Nanoparticle preparation
To prepare the MOF nanoparticles, solutions of zinc nitrate hexahydrate (Sigma-Aldrich) and 2-methylimidazole (Sigma-Aldrich) were mixed together at final concentrations of 1 and 20 mg/ml, respectively. The resulting suspension was vortexed for 30 s and left undisturbed for 3 hours. Loading of the payload was achieved by premixing an appropriate amount of siRNA with the 2-methylimidazole solution to achieve siRNA input concentrations ranging from 50 to 500 nM. Unless otherwise stated, studies were conducted with formulations made using siRNA at an input concentration of 500 nM. Silencer 5′-carboxyfluorescein (FAM)–labeled negative control #1 siRNA (Invitrogen) was used for characterization studies, and unlabeled silencer negative control #1 siRNA (siRNANC; Invitrogen), silencer siRNA against GFP (siRNAGFP; Invitrogen), and siRNA against survivin (siRNASur; Santa Cruz Biotechnology) were used in the functional studies. For membrane coating, human platelet membrane was derived from type O+ platelet-rich plasma (San Diego Blood Bank) by a freeze-thaw process and suspended at 2 mg/ml in water (17). The platelet membrane solution was then added to an equal volume of MOF or MOF-siRNA nanoparticles for 30 min, followed by sequential extrusion through 1000-, 400-, and 200-nm polycarbonate porous membranes (Whatman) using an Avanti mini extruder. Nanoparticles were isolated by centrifugation at 10,000g for 5 min and then resuspended in water for further use. Platelet membrane vesicles were prepared by extruding purified platelet membrane through the same set of porous membranes. RBC membrane–coated samples were prepared by a similar approach using cell membrane that was derived from purified human RBCs (BioreclamationIVT).
Nanoparticle characterization
Nanoparticle size and surface zeta potential were measured by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS. To visualize nanoparticle morphology, samples were adsorbed onto a carbon-coated 400-mesh copper grid (Electron Microscopy Sciences) and stained with 1 weight % uranyl acetate (Electron Microscopy Sciences), followed by imaging on a JEOL 1200 EX II transmission electron microscope. For the stability study, samples were stored at room temperature in PBS or Dulbecco’s modified Eagle’s medium (DMEM; HyClone) supplemented with 10% fetal bovine serum (FBS; HyClone), and size was measured periodically by DLS. To quantify siRNA release, nanoparticles were resuspended in PBS at pH 5.0 or pH 7.4. At predetermined time points, aliquots from each group were centrifuged to pellet the nanoparticles, and the fluorescence of the FAM dye (excitation/emission = 494/520 nm) in the supernatant was measured using a BioTek Synergy Mx microplate reader. For the siRNA degradation study in purified RNase, a working solution of an RNase cocktail enzyme mix (Invitrogen) was prepared by diluting 1:1000 with distilled water. Then, 10 μl of the diluted enzyme mix was added into 100-μl aliquots containing 50 pmol of siRNA in free form or in P-MOF-siRNA, and the mixtures were incubated at 37°C for increasing amounts of time. Each sample was then prepared with DNA loading buffer (Lamda Biotech) and loaded into a 1.5% agarose gel (Apex Bioresearch Products) containing GelRed nucleic acid stain (Biotium). The agarose gel was run in 1× tris-acetate-EDTA buffer (Invitrogen) at 120 V for 30 min and imaged using a Bio-Rad Gel Doc XR system. For the siRNA degradation study in serum-containing medium, 100 μl of DMEM containing 10% FBS was added into 10-μl aliquots containing 50 pmol of siRNA in free form or in P-MOF-siRNA, and the mixtures were incubated at 37°C for increasing amounts of time. Samples were then prepared and run as described above. For the protein characterization, samples were normalized to 1 mg/ml of protein content or an equivalent amount of MOF-siRNA, and they were prepared in NuPAGE lithium dodecyl sulfate sample loading buffer (Novex). The samples were then run on 12-well Bolt 4 to 12% bis-tris minigels (Invitrogen) in 3-(N-morpholino)propanesulfonic acid (MOPS) running buffer (Novex). For Western blot analysis, the protein was transferred to 0.45-μm nitrocellulose membranes (Pierce) in Bolt transfer buffer (Novex) at 10 V for 60 min. The membranes were then blocked with 2% bovine serum albumin (BSA; Sigma-Aldrich) in PBS with 0.05% Tween 20 (National Scientific). The blots were incubated with primary antibodies specific for human P-selectin (AK4; BioLegend), CD41 (HIP8; BioLegend), or CD61 (VI-PL2; BioLegend), followed by the appropriate horseradish peroxidase (HRP)–conjugated secondary antibodies (BioLegend). Development was done using enhanced chemiluminescence (ECL) Western blotting substrate (Pierce) in an ImageWorks Mini-Medical/90 developer. For the membrane sidedness study, dot blot analysis was conducted on P-MOF-siRNA in nondenatured form to maintain its structure. Denatured platelet ghosts, on which the intracellular and extracellular portions of CD47 protein could both be accessed, were used as standards. The ghosts were serially diluted and prepared in a 4× detergent solution made with 37% glycerol (Fisher Chemical), 55% 1 M tris-HCl (pH 8) (Mediatech), and 8% Triton X-100 (EMD Millipore). Standards were spotted twice, and P-MOF-siRNA was spotted three times onto nitrocellulose membrane. After air-drying, the membrane was blocked with 5% BSA prepared in PBS with 0.05% Tween 20. Blots were then probed using antibodies against the extracellular domain of CD47 (ARP63284_P050; Aviva Systems Biology) or the intracellular domain of CD47 (GTX80538; GeneTex), followed by the corresponding HRP-conjugated secondary antibody. The membrane was then incubated with ECL Western blotting substrate and developed. ImageJ software was used to quantify the signal from each individual spot.
In vitro interaction with macrophages
To assess nanoparticle uptake by macrophages, murine J774 cells (TIB-67; American Type Culture Collection) were cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin (Gibco) and seeded in six-well plates at 2 × 105 cells per well. The cells were then incubated with FAM-labeled siRNA, encapsulated within MOF-siRNA, P-MOF-siRNA, or R-MOF-siRNA, at an siRNA concentration of 50 nM for 24 hours, followed by fixation with 4% paraformaldehyde in PBS (Affymetrix). Flow cytometry data were collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed with FlowJo software. To evaluate cytokine secretion, MOF-siRNA, P-MOF-siRNA, or R-MOF-siRNA was incubated with J774 cells at an siRNA concentration of 50 nM. At 24 hours, the culture medium was collected, and cytokine concentrations were assayed using mouse IL-6 and TNFα enzyme-linked immunosorbent assay (ELISA) kits (BioLegend) per the manufacturer’s instructions.
In vitro targeting and intracellular localization
Human SK-BR-3 breast cancer cells (HTB-30; American Type Culture Collection) were maintained in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. To characterize the targeting mechanism, SK-BR-3 cells were seeded in Thermo Fisher Scientific Nunc Lab-Tek four-well chambered coverglass at 5 × 104 cells per well overnight before fixation with 4% paraformaldehyde in PBS. The fixed cells were then stained with Hoechst 33342 (Invitrogen) and blocked with 4% BSA in PBS. The cells were incubated with P-MOF-siRNA or R-MOF-siRNA, fluorescently labeled using 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD; excitation/emission = 644/665 nm; Invitrogen), in 1% BSA in PBS at an siRNA concentration of 50 nM for 30 min. P-MOF-siRNA preblocked by anti–P-selectin antibody was used as an additional control in the study. Imaging was conducted on a Thermo Fisher Scientific EVOS FL cell imaging system. For the payload targeting study, SK-BR-3 cells were seeded in six-well plates at 2 × 105 cells per well. The cells were then incubated with FAM-labeled siRNA in free form, P-MOF-siRNA, or R-MOF-siRNA at an siRNA concentration of 50 nM for 24 hours, followed by fixation with 4% paraformaldehyde in PBS. Flow cytometry data were collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed with FlowJo software. To visualize endosomal escape, SK-BR-3 cells were seeded in Thermo Fisher Scientific Nunc Lab-Tek 4-well chambered coverglass at 5 × 104 cells per well and incubated with P-MOF-siRNA loaded with FAM-labeled siRNA at a concentration of 50 nM for 1, 4, 8, and 24 hours. The cells were then washed with PBS before staining with Hoechst 33342 and LysoTracker Red DND-99 (Invitrogen), and imaging was conducted using a Keyence BZ-X710 fluorescence microscope.
In vitro GFP knockdown
GFP-transduced SK-BR-3 breast cancer cells were maintained in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and 0.1% hygromycin B (InvivoGen). For flow cytometry, the cells were seeded in six-well plates at 2 × 105 cells per well. The cells were then incubated with siRNAGFP, P-MOF, P-MOF-siRNANC, P-MOF-siRNAGFP, or R-MOF-siRNAGFP at an siRNA concentration of 50 nM for 48 hours, followed by fixation with 4% paraformaldehyde in PBS. Flow cytometry data were collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed with FlowJo software. Wild-type SK-BR-3 cells were used to establish the baseline signal. For fluorescence imaging, the cells were seeded in four-well chambered coverglass at 5 × 104 cells per well and incubated with the same samples for 48 hours. The cells were then washed with PBS, and imaging was conducted using a Keyence BZ-X710 fluorescence microscope.
In vitro survivin knockdown
To evaluate cytotoxicity, SK-BR-3 cells were seeded in 96-well plates at 1 × 104 cells per well. The cells were then incubated with siRNASur, P-MOF, P-MOF-siRNANC, P-MOF-siRNASur, or R-MOF-siRNASur at an siRNA concentration of 50 nM for 24, 48, or 72 hours. Cell viability was quantified using a CellTiter AQueous One Solution cell proliferation assay (Promega) following the manufacturer’s instructions. For Western blotting analysis, the cells were incubated with the above samples for 48 hours, followed by detachment and lysis in a Thermo Fisher Scientific FS30D bath sonicator. The lysed cells were centrifuged at 14,000g, after which the supernatant was collected. Protein concentrations were normalized to 1 mg/ml, and each sample was prepared in NuPAGE lithium dodecyl sulfate sample loading buffer before running on 12-well Bolt 4 to 12% bis-tris minigels in MOPS running buffer. After transferring to nitrocellulose membrane and blocking, the blots were probed with antibody against human survivin (D-8; Santa Cruz Biotechnology), followed by the appropriate HRP-conjugated secondary antibody. Development was done using ECL Western blotting substrate in an ImageWorks Mini-Medical/90 developer. Anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Poly6314; BioLegend) was used as the loading control. For immunofluorescence, the cells were seeded in four-well chambered coverglass at 5 × 104 cells per well and incubated with the same samples for 48 hours. After washing with PBS and fixation with 4% paraformaldehyde in PBS, the cells were permeabilized using 0.1% Triton X-100 in PBS. The cells were then blocked with 2% BSA in PBS. After labeling with anti-human survivin overnight, the cells were stained with the appropriate Alexa Fluor 647–conjugated secondary antibody (BioLegend) and Hoechst 33342. Imaging was conducted using a Keyence BZ-X710 fluorescence microscope. For quantitative PCR analysis, the cells were seeded in six-well plates at 2 × 105 per well and incubated with the same samples for 48 hours. Total RNA was obtained using an RNeasy Mini kit (Qiagen) following the manufacturer’s instructions. Samples were prepared with a Quant-X One-Step qRT-PCR SYBR kit (Takara Bio) and human survivin PCR primer pairs (Sino Biological). The samples were analyzed with a Bio-Rad CFX96 real-time PCR detection system.
In vivo tumor studies and biosafety
All animal experiments were performed in accordance with National Institutes of Health (NIH) guidelines and approved by the Institutional Animal Care and Use Committee of the University of California San Diego. For the targeting studies, 5 × 106 SK-BR-3 cells were subcutaneously inoculated into the right flank of female nude (nu/nu) mice (Charles River Labs) and allowed to grow to an average size of 500 mm3. Mice were then intravenously administered with P-MOF-siRNASur or R-MOF-siRNASur labeled with 10 μg of DiD at an siRNA dosage of 2 nmol per mouse. After 1 hour, the mice were euthanized, and their tumors and major organs were excised for analysis. Ex vivo fluorescence images of the tumors were obtained using a Xenogen IVIS 200 system. All organ samples were subsequently homogenized in PBS using a Biospec Mini-Beadbeater-16, and fluorescence values were quantified using a BioTek Synergy Mx microplate reader. For the efficacy study, 5 × 106 SK-BR-3 breast cancer cells were subcutaneously inoculated into the right flank of female nude mice and allowed to grow to a size of 50 to 100 mm3. The mice were then treated by intravenous injection with PBS, P-MOF-siRNASur, or R-MOF-siRNASur at an siRNA dosage of 2 nmol per mouse every 3 days for a total of four administrations. Tumor volume, calculated as (length × width2)/2, and body weight were measured over time. Tumor sizes were individually normalized to the volume at the start of treatment, and the survival endpoint was predefined as tumor size >1200 mm3. To study the biocompatibility of P-MOF-siRNASur, the nanoparticles were administered at an siRNA dose of 8 nmol to healthy nude mice. After 24 hours, the major organs were collected, histologically sectioned, and stained with H&E (Leica Biosystems) or an HRP-DAB TUNEL assay kit (Abcam). Images were obtained using a Hamamatsu NanoZoomer 2.0-HT slide scanning system.
Acknowledgments
Funding: This work was supported by the NIH under award number R01CA200574. J.Zho. was supported by an NIH 5T32CA153915 training grant from the National Cancer Institute. Author contributions: J.Zhu., R.H.F., and L.Z. conceived and designed the experiments. J.Zhu., H.G., J.Zho., and Q.Z. performed all experiments. All authors analyzed and discussed the data. J.Zhu., R.H.F., and L.Z. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability statement: All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Hannon G. J., RNA interference. Nature 418, 244–251 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Hannon G. J., Rossi J. J., Unlocking the potential of the human genome with RNA interference. Nature 431, 371–378 (2004). [DOI] [PubMed] [Google Scholar]
- 3.McManus M. T., Sharp P. A., Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 3, 737–747 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Dorsett Y., Tuschl T., siRNAs: Applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov. 3, 318–329 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Wittrup A., Lieberman J., Knocking down disease: A progress report on siRNA therapeutics. Nat. Rev. Genet. 16, 543–552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams D., Gonzalez-Duarte A., O’Riordan W. D., Yang C.-C., Ueda M., Kristen A. V., Tournev I., Schmidt H. H., Coelho T., Berk J. L., Lin K.-P., Vita G., Attarian S., Planté-Bordeneuve V., Mezei M. M., Campistol J. M., Buades J., Brannagan T. H., Kim B. J., Oh J., Parman Y., Sekijima Y., Hawkins P. N., Solomon S. D., Polydefkis M., Dyck P. J., Gandhi P. J., Goyal S., Chen J., Strahs A. L., Nochur S. V., Sweetser M. T., Garg P. P., Vaishnaw A. K., Gollob J. A., Suhr O. B., Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 11–21 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Castanotto D., Rossi J. J., The promises and pitfalls of RNA-interference-based therapeutics. Nature 457, 426–433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin H., Kanasty R. L., Eltoukhy A. A., Vegas A. J., Dorkin J. R., Anderson D. G., Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 15, 541–555 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Kim H. J., Kim A., Miyata K., Kataoka K., Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug Deliv. Rev. 104, 61–77 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Higuchi Y., Kawakami S., Hashida M., Strategies for in vivo delivery of siRNAs: Recent progress. BioDrugs 24, 195–205 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Lorenzer C., Dirin M., Winkler A.-M., Baumann V., Winkler J., Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 203, 1–15 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Varkouhi A. K., Scholte M., Storm G., Haisma H. J., Endosomal escape pathways for delivery of biologicals. J. Control. Release 151, 220–228 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Dominska M., Dykxhoorn D. M., Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 123, 1183–1189 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Zhuang J., Young A. P., Tsung C.-K., Integration of biomolecules with metal–organic frameworks. Small 13, 1700880 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Zhuang J., Kuo C.-H., Chou L.-Y., Liu D.-Y., Weerapana E., Tsung C.-K., Optimized metal-organic-framework nanospheres for drug delivery: Evaluation of small-molecule encapsulation. ACS Nano 8, 2812–2819 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Zhang H., Chen W., Gong K., Chen J., Nanoscale zeolitic imidazolate framework-8 as efficient vehicles for enhanced delivery of CpG oligodeoxynucleotides. ACS Appl. Mater. Interfaces 9, 31519–31525 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Hu C.-M. J., Fang R. H., Wang K.-C., Luk B. T., Thamphiwatana S., Dehaini D., Nguyen P., Angsantikul P., Wen C. H., Kroll A. V., Carpenter C., Ramesh M., Qu V., Patel S. H., Zhu J., Shi W., Hofman F. M., Chen T. C., Gao W., Zhang K., Chien S., Zhang L., Nanoparticle biointerfacing by platelet membrane cloaking. Nature 526, 118–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang R. H., Kroll A. V., Gao W., Zhang L., Cell membrane coating nanotechnology. Adv. Mater. 30, e1706759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Q., Sun W., Qian C., Wang C., Bomba H. N., Gu Z., Anticancer platelet-mimicking nanovehicles. Adv. Mater. 27, 7043–7050 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Q., Qian C., Sun W., Wang J., Chen Z., Bomba H. N., Xin H., Shen Q., Gu Z., Engineered nanoplatelets for enhanced treatment of multiple myeloma and thrombus. Adv. Mater. 28, 9573–9580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang K., Ricco R., Doherty C. M., Styles M. J., Bell S., Kirby N., Mudie S., Haylock D., Hill A. J., Doonan C. J., Falcaro P., Biomimetic mineralization of metal-organic frameworks as protective coatings for biomacromolecules. Nat. Commun. 6, 7240 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alsaiari S. K., Patil S., Alyami M., Alamoudi K. O., Aleisa F. A., Merzaban J. S., Li M., Khashab N. M., Endosomal escape and delivery of CRISPR/Cas9 genome editing machinery enabled by nanoscale zeolitic imidazolate framework. J. Am. Chem. Soc. 140, 143–146 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Hu C.-M. J., Zhang L., Aryal S., Cheung C., Fang R. H., Zhang L., Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. U.S.A. 108, 10980–10985 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z., Fu Y., Kang Z., Liu X., Chen N., Wang Q., Tu Y., Wang L., Song S., Ling D., Song H., Kong X., Fan C., Organelle-specific triggered release of immunostimulatory oligonucleotides from intrinsically coordinated DNA–metal–organic frameworks with soluble exoskeleton. J. Am. Chem. Soc. 139, 15784–15791 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Luk B. T., Jack Hu C.-M., Fang R. H., Dehaini D., Carpenter C., Gao W., Zhang L., Interfacial interactions between natural RBC membranes and synthetic polymeric nanoparticles. Nanoscale 6, 2730–2737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shieh F.-K., Wang S.-C., Yen C.-I., Wu C.-C., Dutta S., Chou L.-Y., Morabito J. V., Hu P., Hsu M.-H., Wu K. C. W., Tsung C.-K., Imparting functionality to biocatalysts via embedding enzymes into nanoporous materials by a de novo approach: Size-selective sheltering of catalase in metal–organic framework microcrystals. J. Am. Chem. Soc. 137, 4276–4279 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Liao F.-S., Lo W.-S., Hsu Y.-S., Wu C.-C., Wang S.-C., Shieh F.-K., Morabito J. V., Chou L.-Y., Wu K. C. W., Tsung C.-K., Shielding against unfolding by embedding enzymes in metal–organic frameworks via a de novo approach. J. Am. Chem. Soc. 139, 6530–6533 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Schlesinger M., Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 11, 125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meikle C. K. S., Kelly C. A., Garg P., Wuescher L. M., Ali R. A., Worth R. G., Cancer and thrombosis: The platelet perspective. Front. Cell Dev. Biol. 4, 147 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W., Ma H., Zhang J., Zhu L., Wang C., Yang Y., Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 7, 13856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aigner S., Sthoeger Z. M., Fogel M., Weber E., Zarn J., Ruppert M., Zeller Y., Vestweber D., Stahel R., Sammar M., Altevogt P., CD24, a mucin-type glycoprotein, is a ligand for P-selectin on human tumor cells. Blood 89, 3385–3395 (1997). [PubMed] [Google Scholar]
- 32.Amo L., Tamayo-Orbegozo E., Maruri N., Eguizabal C., Zenarruzabeitia O., Rinon M., Arrieta A., Santos S., Monge J., Vesga M. A., Borrego F., Larrucea S., Involvement of platelet-tumor cell interaction in immune evasion. Potential role of podocalyxin-like protein 1. Front. Oncol. 4, 245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oh N., Park J.-H., Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomed. 9 (suppl. 1), 51–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pratt A. J., MacRae I. J., The RNA-induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 284, 17897–17901 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ambrosini G., Adida C., Altieri D. C., A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 3, 917–921 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Jha K., Shukla M., Pandey M., Survivin expression and targeting in breast cancer. Surg. Oncol. 21, 125–131 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Altieri D. C., Targeting survivin in cancer. Cancer Lett. 332, 225–228 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang R. H., Hu C.-M., Luk B. T., Gao W., Copp J. A., Tai Y., O’Connor D. E., Zhang L., Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 14, 2181–2188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thamphiwatana S., Angsantikul P., Escajadillo T., Zhang Q., Olson J., Luk B. T., Zhang S., Fang R. H., Gao W., Nizet V., Zhang L., Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management. Proc. Natl. Acad. Sci. U.S.A. 114, 11488–11493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Q., Dehaini D., Zhang Y., Zhou J., Chen X., Zhang L., Fang R. H., Gao W., Zhang L., Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol. 13, 1182–1190 (2018). [DOI] [PubMed] [Google Scholar]