

Abstract
Ebola virus (EBOV) causes severe human disease with a high case fatality rate. The balance of evidence implies that the virus circulates in bats. The molecular basis for host–viral interactions, including the role for phosphorylation during infections, is largely undescribed. To address this, and to better understand the biology of EBOV, the phosphorylation of EBOV proteins was analyzed in virions purified from infected monkey Vero-E6 cells and bat EpoNi/22.1 cells using high-resolution mass spectrometry. All EBOV structural proteins were detected with high coverage, along with phosphopeptides. Phosphorylation sites were identified in all viral structural proteins. Comparison of EBOV protein phosphorylation in monkey and bat cells showed only partial overlap of phosphorylation sites, with shared sites found in NP, VP35, and VP24 proteins, and no common sites in the other proteins. Three-dimensional structural models were built for NP, VP35, VP40, GP, VP30 and VP24 proteins using available crystal structures or by de novo structure prediction to elucidate the potential role of the phosphorylation sites. Phosphorylation of one of the identified sites in VP35, Thr-210, was demonstrated to govern the transcriptional activity of the EBOV polymerase complex. Thr-210 phosphorylation was also shown to be important for VP35 interaction with NP. This is the first study to compare phosphorylation of all EBOV virion proteins produced in primate versus bat cells, and to demonstrate the role of VP35 phosphorylation in the viral life cycle. The results uncover a novel mechanism of EBOV transcription and identify novel targets for antiviral drug development.
Electronic supplementary material
The online version of this article (10.1007/s00018-019-03303-1) contains supplementary material, which is available to authorized users.
Keywords: Ebola virus, Phosphorylation, Transcription, Replication
Introduction
The 2013–2016 epidemic of Ebola virus (EBOV) in West Africa resulted in more than 28,000 human infections and over 11,000 deaths [1]. The current EBOV outbreak in the eastern part of the Democratic Republic of the Congo started in 2018, and has already resulted in over 2600 infections and over 1700 deaths (as of July 28, 2019) [2]. At present, there are no approved vaccines or treatments for EBOV infection. The suspected natural host and reservoir of EBOV is Old World fruit bats, some species of which were found to be seropositive or PCR positive for the virus [3–5]. EBOV belongs to the family Filoviridae within the order Mononegavirales whose members typically possess enveloped, non-segmented, negative strand RNA genomes [6]. The 19-kb-long genomic RNA of EBOV contains seven genes in a linear order, and code, from 3′ to 5′ end, for nucleoprotein NP (739 amino acids, aa), polymerase cofactor VP35 (340 aa) which is similar to phosphoproteins (P proteins) of other non-segmented negative strand RNA viruses, matrix protein VP40 (326 aa), the sole envelope glycoprotein GP (676 aa), the second nucleoprotein VP30 (288 aa), minor matrix protein VP24 (251 aa), and the large polymerase component L (2212 aa) [6]. Additionally, two soluble nonstructural proteins, secreted or soluble glycoprotein sGP (364 aa) and small secreted glycoprotein ssGP (298 aa) are encoded by alternative mRNAs transcribed from GP gene as a result of transcriptional editing [7–9]. NP, VP35, VP30 and L form the ribonucleoprotein [6]. After EBOV nucleocapsid enters the cytoplasm, polyadenylated messenger RNAs are sequentially transcribed from the 3′ end to the 5′ end of the genomic RNA template. Alternatively, genomic RNA serves as a template for the antigenomic (complementary) positive strand RNA, which in turn serves as a template for genomic RNA in genome replication [10]. While replication of the EBOV genome is mediated by the polymerase complex (comprising NP, VP35 and L), transcription of viral genes additionally requires VP30 [11]. VP24 has also been shown to be important for the normal formation of the nucleoprotein complex [12–14]. The ribonucleoprotein complex is transported by the matrix protein VP40 to the plasma membrane where the components assemble into an enveloped virus particle [12, 15].
We and others previously showed that dephosphorylation of the N-terminal and central parts of VP30 impairs EBOV transcription and shifts the balance of the transcription–replication activity of the polymerase complex toward replication [16–18]. We demonstrated that host protein phosphatase-1 (PP1) dephosphorylates VP30 in cultured cells and shifts polymerase activity toward transcription [17]. Dynamic phosphorylation of VP30 at Ser-29 was shown to be important for VP30 transport with the nucleocapsid to the site of viral mRNA synthesis [19]. Recently, NP was shown to recruit the host PP2A-B56 protein phosphatase for VP30 dephosphorylation and upregulation of viral transcription [20]. Thus, targeting phosphorylation of EBOV proteins can be adapted for therapeutics aimed at inhibiting EBOV transcription. Correspondingly, we identified a small molecule inhibitor of PP1, 1E7-03, which increased EBOV VP30 phosphorylation and effectively suppressed replication of EBOV particles in cultured cells [17]. We also showed that phosphorylation of VP30 at Thr-143 and Thr-146 leads to strong induction of VP30 phosphorylation in the N-terminal serine cluster, suggesting a ‘cascade phosphorylation’ regulation [17]. ‘Cascade phosphorylation’ of the phosphoproteins (P proteins) of two other non-segmented negative strand viruses, the rhabdovirus vesicular stomatitis virus and the paramyxovirus respiratory syncytial virus, regulates the transcriptional activity of their polymerase complexes [21, 22]. Also, specific inhibition of PP2A-B56 through the expression of a PP2A-binding peptide led to strong suppression of EBOV transcription and infection suggesting that PP2A/NP interaction is a plausible target for EBOV inhibition [20]. Previous mass spectrometry analysis of plasmid-expressed EBOV NP identified phosphorylation of Ser-281, Thr-563, Ser-587, and Ser-647 in the hydrophilic C-terminal half of the protein [23]. This part of NP is critical for incorporating nucleocapsids into virions [24]. Thus, phosphorylation is likely to play a critical role in the regulation of polymerase activity and viral assembly of EBOV and other non-segmented negative strand viruses. However, a lack of detailed data on phosphorylation of EBOV proteins, as well as that of non-segmented negative strand viruses in general, severely limits our understanding of EBOV biology and hinders the development of phosphorylation-targeted therapeutics. Furthermore, previous studies that characterized phosphorylation of EBOV VP30 and NP were performed with virus or plasmid-expressed proteins produced in cells derived from primates, which are accidental hosts of EBOV; while no such studies were performed with virus propagated in cells of bats, the presumed natural reservoir of the virus.
In the present study, we used high-resolution liquid chromatography-linked tandem mass spectrometry (nano LC–MS/MS) to identify and map specific phosphorylation sites in all viral proteins present in EBOV particles propagated in primate and bat cells. We then applied principles of secondary and tertiary protein structure, solvent accessibility criteria, and conformational dynamics to assess candidate modification sites to elucidate their potential function. As only partial structures are experimentally determined for NP, VP35 and VP30, we resorted to de novo molecular modeling using the Rosetta/Robetta protocols [25]. For each protein, we selected the top scoring model. After conditioning, energy minimization and manual inspection, we tested the obtained models for their stability using molecular dynamics (MD). We carried out 100-ns simulations for each protein model in a fully solvated water box with periodic boundary conditions.
We focused on VP35 to determine the role of the identified phosphorylation sites in genome transcription and replication. We identified a novel VP35 phosphorylation site, Thr-210, and showed that its phosphorylation governs the transcriptional activity of the EBOV polymerase complex. We also demonstrated that VP35 Thr-210 phosphorylation is important for VP35 interaction with NP. This is the first study to compare phosphorylation of all EBOV virion proteins produced in primate versus bat cells. It is also the first to demonstrate the role of VP35 phosphorylation in the viral life cycle. Our results uncover a new mechanism of filovirus transcription and identify novel targets for antiviral drug development.
Results
Phosphoproteomic analysis of EBOV virions
The goal of our study was to analyze the phosphorylation of EBOV proteins packaged in viral particles propagated in Vero-E6 cells in comparison to virus propagated in EpoNi/22.1 cells. Vero-E6 was derived from monkey renal epithelia, and is widely used as a model of human infection [10]; while EpoNi/22.1 cells were obtained from the kidney of the African fruit bat Epomops buettikoferi, a species closely related to Epomops franqueti, one of the species that has been heavily implicated as a potential reservoir [3, 26]. Purified virions were isolated by ultracentrifugation of supernatants from infected cells in sucrose gradients and heated at 95 °C in 4% SDS for 15 min to inactivate the virus (Fig. 1a). EBOV proteins were resolved on 10% SDS-PAGE (Fig. 1b). While EBOV purified from Vero-E6 cells showed distinct bands corresponding to viral proteins, EpoNi/22.1 cell-produced EBOV contained many more non-viral protein bands likely reflecting less efficient replication of EBOV in bat cells, resulting in co-purification of host proteins (Fig. 1b). The protein gel was cut into equal fragments, and peptides were extracted after in-gel trypsinization and analyzed by high-resolution LC–MS/MS. Viral proteins were detected with high coverage (see details in the following sections). Post-translational modifications (PTMs) were analyzed using Proteome Discover 1.4.
Fig. 1.

Global phosphorylation analysis of EBOV propagated in primate and bat cells. a Vero-E6 cells infected with EBOV expressing GFP: live virus (left) or virus inactivated by heat treatment in the presence of SDS (right) on days 3 and 6 post infection. GFP fluorescent images showing GFP expression, BF bright field images showing cell monolayers. b Purified EBOV preparations propagated in Vero-E6 and EpoNi/22.1 cells resolved on 10% SDS-PAGE and stained with Coomassie blue. Approximate positions of individual EBOV proteins are indicated at right. c Heatmap of the identified EBOV phosphorylated amino acids in the viruses propagated in Vero-E6 (EBOV V) and EpoNi/22.1 (EBOV E) cells in all seven viral EBOV proteins shown in blue. Phosphorylated amino acids identified in virus propagated in one type of cells, but not covered by MS/MS analysis of virus from another type of cells are marked by “X”. The identified phosphorylated amino acids that are conserved in other filoviruses are shown in gray. The amino acids found to be phosphorylated in virus grown in at least one type of cells but lacking in corresponding amino acids of identified peptides in virus grown in another type of cells or amino acids which are not conserved in additional filoviruses are shown in white. d Venn diagrams showing host-specific conservation of identified phosphorylation sites in both Vero-E6- and EpoNi/22.1-grown EBOV
Host-specific phosphorylation of the viral proteins
We compared the conservation of the identified phosphorylation sites between the viruses grown in Vero-E6 cells versus in EpoNi/22.1 cells. All phosphorylation sites identified in three viral proteins (seven sites in VP35 and one each in NP and VP24) originating from Vero-E6 produced virus were conserved in EpoNi/22.1-derived virus (Fig. 1c, d). Additionally, 19, 3 and 2 more sites were phosphorylated, respectively, in NP, VP35 and VP24 from Vero-E6-derived virus only. The greatest discordance was found in VP30, which showed no overlap between the four sites identified in the two cell lines. Likewise, the only phosphorylation site identified in the L protein of EpoNi/22.1 derived virus was also not conserved. Both GP (5 sites) and VP40 (1 site) were only phosphorylated in Vero-E6-derived virus. Overall, more sites were identified in primate cell-derived virions than in bat cell-derived virions. Novel phosphorylation sites were identified in VP30 and L of EpoNi/22.1-derived virions that were lacking in Vero-E6-grown virus. These data point to the importance of host cell kinases and phosphatases that influence the phosphorylation status of EBOV proteins, a likely difference of their activities in different host species, and their possible preference for specific residues and motifs [27].
The identified phosphorylation sites are conserved across filovirus species
Next, we compared the presence of the identified phosphorylation sites across all eight known members of the family Filoviridae in the context of the overall amino acid similarity between these proteins (Fig. 1c, Table S1). Sequence-wise, the two matrix proteins, VP24 and VP40, show the highest similarity among the five Ebolavirus species [28, 29]. Correspondingly, the three identified phosphorylated residues in VP24 from Vero-E6-grown virus (one of which was also identified in EpoNi/22.1-grown virus) were found to be fully conserved in the ebolaviruses Bundibugyo virus (BDBV), Sudan virus (SUDV), and Tai Forest virus (TAFV). The only phosphorylation site found in VP40 of Vero-E6-derived virus (Thr-277) was present in BDBV, TAFV, and in SUDV, but not in Reston virus (RESTV). The large polymerase protein, L, which is the most conserved protein among many non-segmented negative-strand viruses, demonstrated a high conservation of phosphorylation sites identified in Vero-E6-grown virus across the ebolaviruses, but a much lesser degree of conservation across the two marburgviruses, Marburg virus (MARV) and Ravn virus (RAVV). The single unique phosphorylation site (Ser-1544) identified in the L protein of EpoNi/22.1-derived virions was conserved across all filoviruses except Lloviu virus (LLOV). GP, the least conserved filovirus protein, demonstrated a high degree of conservation of phosphorylation sites: 80% for BDBV, SUDV, TAFV and 60% for RESTV. The least conservation of identified phosphorylation sites in Vero-E6-derived virions was found in VP30 (60%) followed by NP (69%) and VP35 (70%) among ebolaviruses. The phosphorylation sites identified in EpoNi/22.1-derived virus were similarly highly conserved for L, and less conserved in VP30. However, a high level of conservation was observed for the seven phosphorylated residues identified in VP35 from EpoNi/22.1-derived virions: 100% in TAFV and RESTV, and 71% for BDBV and SUDV. These data demonstrate that the canonical phosphorylation sites conserved among multiple filovirus species include both the phosphorylation sites experimentally identified only in Vero-E6-grown virus, and those identified only in EpoNi/22.1-grown virus. These data also show that in general, the level of conservation of the identified phosphorylation sites corresponds to the overall similarity of the proteins across filovirus species with two exceptions: the disproportional conservation of the phosphorylation sites in GP from Vero-E6-grown virus, and the low conservation of the phosphorylation sites in VP30 from both Vero-E6- and EpoNi/22.1-derived virions.
To visualize our results and to obtain additional insight into the biological role of the identified phosphorylation sites in Vero-E6 cells for individual proteins and in protein–protein interactions, we built three-dimensional models of VP24, VP40 and GP from available coordinates. For VP30, VP35 and NP, we developed full-length structures using de novo modeling approaches, and analyzed conformational stability using molecular dynamics (MD) simulations.
Phosphorylation of the envelope glycoprotein (GP)
GP is the only virally encoded protein present on the surface of virions. It is the sole determinant of viral entry [30], and a virulence factor [31]. This type I transmembrane protein is cleaved by furin-like proteases into disulphide-bridged GP1 and membrane-anchored GP2 [32]. No phosphorylation sites have previously been reported in EBOV GP. However, phosphorylation of serine residues between amino acids 260 and 273 on the ectodomain of plasmid-expressed GP of MARV have been described [33]. In our mass spectrometry analysis, we detected EBOV GP with 45% coverage (Fig. 2a) and identified five novel phosphorylated residues (Figs. 2b, S1, S2). From the crystal structure of GP, three GP1 subunits were demonstrated to form a chalice-like globular structure, which is encased by the three GP2 subunits to form a cradle [34–36]. In Fig. 2c, which shows the structure of GP (PDB ID: 3CSY), all identified phosphorylation sites are localized in the GP1 subunit; specifically, Ser-267, Thr-269, Thr-270, Thr-307 and Thr-309 are located on a loop in a long coil region of highly conserved stretches of the glycan cap (Fig. 2c). Intriguingly, these phosphorylation sites were also located in the epitopes of multiple antibodies isolated from BDBV survivors [37]. Thus, the identified GP1 phosphorylation sites are located in critical regions and are likely to be functional.
Fig. 2.

Phosphorylation of EBOV GP from Vero-E6-grown virus. a MS/MS analysis of GP conducted with Proteome Discoverer 2.1 and SEQUEST search engine. Peptides were in-gel digested with trypsin, eluted and subjected to MS analysis on Thermo LTQ Orbitrap XL mass spectrometer. Peptides identified with high, median and low probability are shown in green, red and blue, respectively. Phosphorylated residues are underscored. b Identified GP phosphopeptides. c Structure of the GP trimer with the identified phosphorylation sites indicated (viewed from the top). Shown is a GP trimeric complex consisting of 3 GP1-GP2 disulphide-linked monomers that latch together to form a chalice-like structure. Coordinates were taken from the crystal structure (PDB ID: 3CSY) and modeled in UCSF Chimera
Phosphorylation of the matrix proteins
Phosphorylation of VP40 matrix protein
VP40 is the major EBOV matrix protein and the most abundant virion component. It associates with the lipid bilayer and is important for EBOV budding and nucleocapsid recruitment, as well as virus structure and stability [38–40]. VP40 exhibits great structural plasticity and forms distinct structural assemblies including a butterfly-shaped dimer, a linear hexamer, and an arrangement of dimers forming an octameric pore-like structure that binds RNA [41, 42]. We detected VP40 with 78.5% coverage (Fig. 3a). Only one phosphorylation site was identified in VP40 (Figs. 3b, S3, S4). In Fig. 3c, which shows the butterfly-shaped VP40 dimer of dimers (PDB ID: 4LDB [41]), the phosphorylation site (shown in molecules A and B) is located on a short loop (Thr-277). This site is located on the outward face of the quaternary structure, making it accessible to interacting kinases and phosphatases, and therefore is likely to be functional.
Fig. 3.

Phosphorylation of EBOV VP40 from Vero-E6-grown virus. a MS/MS analysis of VP40 conducted as described in legend for Fig. 2a. b Identified VP40 phosphopeptides. c Structure of VP40 with the identified phosphorylation sites indicated shown as a dimer of VP40 homodimers. The three phosphorylation sites are located on the loops of the outer region of the quaternary structure. Coordinates were taken from crystal structure PDB ID: 4LDB (residues 44–326) and modeled in UCSF Chimera
Phosphorylation of VP24 secondary matrix protein
VP24, the minor matrix protein, is involved in nucleocapsid formation and regulation of replication [13, 43]. It also interacts with the nuclear transport protein karyopherin to subvert the host interferon response [44–47]. VP24 forms a single domain structure with a compact pyramidal fold. There is a ‘top’ subdomain that consists of a collection of six alpha helices and three antiparallel beta sheets and a ‘central/bottom’ subdomain formed by a five-stranded antiparallel beta sheet and four helices [47, 48]. We detected VP24 with 84.9% coverage (Figs. 4a, S5, S6) and identified three phosphorylated residues (Fig. 4b). Figure 4c shows a model of the VP24 dimer from the VP24 dimeric structure (PDB ID: 4M0Q, [49]). Ser-12 is located within the N-terminal loop (residues 1–20) which is not seen in the crystal structure, likely because it is highly flexible; it is also likely solvent-exposed. Ser-146 is located on a connecting domain with a turn and a half helical element that leads into an alpha-helical domain. The Ser-151 residue is located on the exposed face of a helix (Fig. 4c). One of the three identified residues, Ser-12, was also phosphorylated in EpoNi/22.1-derived virus. Since most of the identified phosphorylation sites are exposed, they are likely to be involved in interaction with GP on one side, and the VP40 matrix or the internal RNA-binding proteins on the other side.
Fig. 4.

Phosphorylation of EBOV VP24 from Vero-E6-grown virus. a MS/MS analysis of VP24 performed as described in legend for Fig. 2a. b Identified VP24 phosphopeptides. c The identified phosphorylation sites indicated on the 3D structure of VP24 shown as a dimer (from PDB ID: 4M0Q). The KPNA5 binding loop is also indicated. The molecular structural image was created in UCSF Chimera
Phosphorylation of the ribonucleoprotein complex
Phosphorylation of the nucleoprotein (NP)
NP, together with the L and VP35 proteins, plays a central role in replication and transcription of EBOV genome. Post-translational modifications of NP, namely O-glycosylation and sialylation, are required for its interaction with VP35 [13]. Furthermore, the first 450 amino acids of the N-terminal part are important for NP–NP interaction and, together with the next 150 amino acid residues, are necessary for nucleocapsid formation and viral replication [50]. A prior study using plasmid-expressed NP identified several phosphorylation sites in the C-terminus [23]. These residues are highly conserved across filovirus species. We detected NP with 75.2% coverage (Fig. 5a), and identified 20 potential phosphorylation sites (Figs. 5b, S7, S8). The existing crystal structures of EBOV NP capture only parts of the N-terminal region, residues 33–367 (PDB ID: 4ZTA and 4ZTG; PDB ID: 4Z9P, [51]), residues 36–351 [52] (PDB ID: 4YPI, [49), residues 38–385 [53], and a C-terminal domain, residues 641–739 (PDB ID: 4QB0 and 4QAZ [54]). To gain insight into the potential role of the phosphorylation sites we discovered, we constructed a full-length model of NP by de novo prediction [25] (see details in “Materials and methods”) (Fig. 5c). According to the Rosetta protocol, NP is made of four domains: domain 1 (residues 1–367), domain 2 (residues 368–436), domain 3 (residues 437–645), and domain 4 (residues 646–739). Domain 1 is largely ordered, as observed in the experimental structures. Domains 2 and 3 are inherently unstructured, from disorder prediction. Domain 4 is folded, as inferred from the crystal structures.
Fig. 5.

Phosphorylation of EBOV NP from Vero-E6-grown virus. a MS/MS analysis of NP performed as described in legend for Fig. 2a. b Identified NP phosphopeptides. c Modeled full length structure of NP with the phosphorylation sites indicated. The majority of phosphorylated sites are in the unstructured region, on loops. The existing crystal structure of the structured N-terminal domain is shown in yellow. The model was created in UCSF Chimera. d RMSD (conformational stability) of NP structure over time determined in AMBER 14
To test the conformational stability of the modeled NP structure, an equilibrium MD simulation was conducted in periodic water box using the model as a starting point. From the MD trajectories, the model had highly stable globular-like ‘structured’ sections (N-terminal part) and highly flexible ‘unstructured’ C-terminal sections (Fig. 5d). Within Domain 1, there were two well-structured regions (residues 1–241 and 242–353) that moved relative to each other and preserved their secondary and tertiary structures. A 3-helix bundle element (residues 354–411) that connects domain 1 and domain 2 maintained its stability through 100 ns MD time. The two C-terminal regions of NP (residues 412–645 and 646–739) were highly flexible. The general pattern of NP protein flexibility was complex and heterogeneous as typically observed for intrinsically disordered proteins and support NP’s role in forming the ribonucleoprotein complex.
Six of the twenty identified phosphorylation sites reside in domain 1 (Fig. 5c). Of these residues, Ser-125, Ser-126, Thr-206 and Thr-270 lie on a loop in this structured domain, while Ser-285 and Ser-286 are located on exposed helix surfaces (Fig. 5c). Interestingly, Thr-270 located in domain 1 was phosphorylated in bat cells as well. The stretch of fourteen phosphorylated residues Thr-536, Ser-541, Thr-545, Thr-563, Thr-597, Ser-598, Thr-601, Thr-603, Tyr-686, Thr-687, Tyr-688, Ser-691, Tyr-696 and Thr-701 lie within the large unstructured tract in domain 3 (Fig. 5c). Site Thr-563 was detected in the previous mass spectrometry study that additionally identified the phosphorylation of Ser-581, Ser-587 and Ser-647 in plasmid-expressed NP [23]. The phosphorylated residues we identified are likely to be functional because of their respective location: within the domain 1, which is responsible for NP dimerization, and domain 3, which is important for viral capsid formation and replication [50].
Phosphorylation of the major polymerase subunit L
We detected L polymerase with 67.9% coverage (Fig. S9) and identified 17 phosphorylation sites consisting of 7 serine, 7 threonine and 3 tyrosine residues (Figs. 1c, S9). The phosphopeptides are shown in Fig. S10. Additionally, we identified one novel phosphorylation residue in EpoNi/22.1-derived virus (Figs. 1c, S11). There are no experimental structures available for EBOV L protein. Because of its large size (2212 amino acids), we did not attempt to model it.
Phosphorylation of transcription factor VP30
EBOV VP30 is a zinc finger nucleoprotein and a component of the polymerase protein complex. VP30 is required for transcription but not replication of the viral genome [11, 16]. VP30 is phosphorylated at its N-terminus within two serine clusters, 29–31 and 40–46, and at Thr-52 [18], and, as we recently showed with plasmid-expressed VP30, also on Thr-143 and Thr-146 [17]. We detected VP30 with 64.2% coverage (Fig. 6a) and identified two phosphorylated threonine residues with high confidence (Fig. 6b). The MS/MS spectra of peptides containing phosphorylated residues Thr-52 and Thr-146 are shown in Fig. 6c, d. Two additional residues Thr-119 and Ser-124 were phosphorylated in EpoNi/22.1 derived virus (Figs. 1c, S12). Coordinates exist only for the C-terminal domain of EBOV VP30 (residues 142–272; (PDB ID: 2I8B [55] and PDB ID: 3V7O [56]). Thus, we constructed a full-length model of VP30 de novo as described above (Fig. 6e). The model consists entirely of helices with large stretches of loops. MD simulation analysis demonstrated that the global structure of VP30 contains two structural regions exhibiting distinct dynamical characteristics (Fig. 6f). The C-terminal part of the protein (Fig. 6f, shown in blue) maintained its globular-like structure during the entire 100-ns MD trajectory. In contrast, the N-terminal part (Fig. 6f, shown in red) is unfolded and highly flexible, as is typical for inherently unstructured proteins. The phosphorylated residue Thr-52 is localized on the N-terminal loop (amino acids 1–67) (Fig. 6e). The phosphorylated residue Thr-146 is located at the beginning of a short helix in the ordered domain which was found in the VP30 crystal structure. This residue lies on the outward face of the domain and is solvent accessible (Fig. 6e). This finding confirms our previous observation with plasmid-derived VP30 showing that it is phosphorylated at the N-terminal serine clusters as well as on Thr-143 and Thr-146. The identified phosphorylation sites are solvent accessible and likely to play a role in VP30 binding to RNA and interaction with VP35.
Fig. 6.

Phosphorylation of EBOV VP30 from Vero-E6-grown virus. a MS/MS analysis of VP30 performed as described in legend for Fig. 2a. b Identified VP30 phosphopeptides. MS/MS spectra of the VP30 peptide 50–57 (c) and peptide 88–98 (d). The colored peaks indicate matched MS/MS fragments. Green color indicates precursors, as outlined in the figure; blue and red colors indicate y and b ions, respectively. The spectrum gives positive identification of peptides VPTVFHKK (c) and KDHQLESLTDR (d) with the indicated phosphorylation sites. e Modeled full-length structure of VP30 with the phosphorylation sites indicated. The model was created in UCSF Chimera. f RMSD (conformational stability) of VP30 structure over time determined in AMBER 14
Phosphorylation of polymerase cofactor VP35
VP35 is a multifunctional nucleoprotein: it is involved in the assembly of the EBOV nucleocapsid, serves as a co-factor of the viral polymerase complex, and is an interferon (IFN) antagonist [57–60]. IFN antagonism of VP35 is associated with the interferon-inhibiting domain (IID), which binds dsRNA, centered on Arg-312 within the central basic patch [57]. We detected VP35 with 93.2% coverage (Fig. 7a) and identified 10 phosphorylated amino acid residues (Fig. 7b). Seven of the ten residues (Ser-205, Thr-206, Thr-207, Ser-208, Thr-210, Ser-310 and Ser-317) were also phosphorylated in EpoNi/22.1-derived virus (Fig. S13). The MS/MS spectra of peptides containing representative phosphorylated residues Thr-191 and Thr-210 are shown in Fig. 7c, d (see remaining spectra in Fig. S14). The available VP35 structures (PDB ID 3FKE [61]; 3L26, 3L2A [57]; 4GHL [62]; and 4GHA [63], and others) cover only the C-terminal interferon inhibitory domain (residues 215–340), with only two identified phosphorylation sites (Ser-310 and Ser-317) within the IID that map to these partial structures. However, structures of the oligomerization domain of Marburg virus have recently become available (PDB ID: 5TOH and PDB ID: 5TOI) [64].
Fig. 7.

Phosphorylation of EBOV VP35 from Vero-E6-grown virus. a MS/MS analysis of VP35 performed as described in legend for Fig. 2a. b Identified VP35 phosphopeptides. MS/MS spectra of the VP35 peptide 189–197 (c) and peptide 198–222 (d). The colored peaks indicate matched MS/MS fragments. Green color indicates precursors, as outlined in the figure; blue and red colors indicate y and b ions, respectively. The spectrum gives positive identification of peptides DETVPQSVR (c) and EAFNNLNSTTSLT EENFGKPDISAK (d) with the indicated phosphorylation sites. e The identified phosphorylation sites indicated on the modeled full length 3D structure of VP35. The model was created in UCSF Chimera. f RMSD (conformational stability) of VP35 structure over time determined in AMBER 14. g VP35 phosphorylation in Vero E6 and 293T cells. Flag-tagged EBOV VP35-expressing plasmid was transfected in Vero E6 or 293T cells, which were untreated (control) or treated with 0.1 μM okadaic acid (OA) for 2 h. VP35 was immunoprecipitated with anti-Flag antibodies from cellular lysates, resolved on 10% SDS PAGE and analyzed by LC–MS analysis. Peptides identified with Proteome Discoverer 2.1, and SEQUEST search engine are shown. Peptides identified with high, median and low probability are shown in green, red and blue, respectively. Phosphorylated residues are underscored. h Quantitative analysis of VP35 phosphorylation. Peptides expression (from g) was quantified using SIEVE 2.1. Ion elution profiles (left) and integrated intensities (right) are shown in blue for control samples and in red for the cells treated with okadaic acid. The 188DETVPQSV197 peptide was used for normalization. Phosphorylation of 198EAFNNLNSTTSLTEENFGKPDISAK222 peptide was increased in the okadaic acid-treated cells. Mean values ± SD based on three samples. Peaks were integrated and p value was calculated using SIEVE 2.1
To gain insight into the role of phosphorylation at the sites we identified, we modeled the full-length structure of EBOV VP35 ab initio (Fig. 7e). The initial step detected four domains corresponding to residues 1–64, 65–167, 168–208 and 209–340. Domain 1 was predicted to be highly unstructured and to contain an RNA binding fragment. Domain 2 was expected to be a coiled coil oligomerization domain. Domain 3 was predicted to be highly unstructured connecting region. Domain 4 corresponded to the known experimental structure of the C-terminal fragment [65] which has IFN inhibiting activity. MD characteristics of the full-length VP35 model (Fig. 7f) were very similar to that of VP30. Two separate sections of the polypeptide located in its central and C-terminal parts (shown in blue) were found to be conformationally stable. In contrast, the N-terminal section and another central section (shown in red and green, respectively) were found to be unstructured and highly flexible, in agreement with the model prediction.
Phosphorylated sites 310 and 317 (both serines) fall into domain 4 of our structural model, which has been well defined by crystallography (PDB ID: 3FEK) [65]. Phosphorylated residue Ser-310 sits on a short loop within the beta sheet subdomain of the structure, and Ser-317 is located on a longer loop that connects the beta sheet subdomain to the 4-helix bundle subdomain (Fig. 7e). We evaluated these new phosphorylated sites by solvent accessibility: the C-terminal domain (domain 4 in our model) matches the experimental crystal structures, for example, structures PDB ID 3FKE [61]; 3L26, 3L2A [57]; 4GHL [62]; and 4GHA [63], and is well characterized. We proceeded to estimate the relative solvent-exposed surface area (rSASA) values for these two phosphorylated sites. The calculations were performed using Parameter Optimized Surfaces (POPS) [66, 67] (see “Materials and methods”). Residue Ser-317 is well exposed (rSASA = 0.528), while Ser-310 is moderately exposed (rSASA = 0.217, Table S2). Specifically, the two phosphorylation sites lie at the beginning of a polyproline type II segment formed by a linker that connects beta strand 2 and beta strand 3, residues 312–325 [57, 61, 68, 69]. As a structural feature, polyproline II promotes favorable protein–protein and protein–nucleic acid interactions [70]. These residues (Ser-310 and Ser-317) are well exposed to solvent and a potential kinase. The rest of the putative phosphorylation sites lie in the predominantly intrinsically unstructured domains (Fig. 7e) in the modeled VP35 structure. Because of the highly flexible nature of these intrinsically unstructured regions, these phosphorylation sites are also expected to be solvent accessible, and are consistent with their availability for phosphorylation (Table S2). Ser-187 and Thr-191 are located on loops connecting short helical elements surrounded by disordered and flexible regions (Fig. 7e). Ser-195 lies in a helix (Fig. 7e). Phosphorylation sites located in residues 205–210, Ser-205, Thr-206, Thr-207, Ser-208 and Thr-210, reside in a conserved STTSLT motif (Fig. 7a, d). These residues sit in an unstructured region (Fig. S15) shown as a loop that connects the N-terminal domains to the C-terminus, domain 4 (IID) (Figs. S15, 7e). Their location in a loop region is expected to make them flexible and accessible.
To confirm that VP35 is phosphorylated in human cells used for the functional studies (below), we expressed Flag-tagged VP35 in Vero-E6 cells and 293T cells. The cells were also mock-treated or treated with okadaic acid to induce VP35 phosphorylation. VP35 was immunoprecipitated with anti-Flag antibodies and its phosphorylation was analyzed by MS/MS. VP35 expressed in both Vero-E6 cells and 293T cells was found to be phosphorylated at the N-terminus and Ser-205/Thr-210 cluster (Fig. 7g, h). In addition, VP35 in Vero-E6 was also phosphorylated on Ser-187, Thr-191, and the protein in 293T cells was phosphorylated on Ser-310/Ser-317 (Fig. 7g, h). Treatment with okadaic acid induced phosphorylation of Ser-205/Thr-210 cluster in VP35 in both Vero-E6 and 293T cells (11-fold and 4-fold, respectively, Fig. 7h).
Phosphorylation of VP35 T210 is required for EBOV transcriptional activity
To further delineate the role of the identified VP35 phosphorylation sites in polymerase activity, we utilized an EBOV minigenome system, which consists of the viral polymerase complex reconstituted in vitro by intracellular co-expression of the L, NP, VP35 and VP30 proteins [11]. We disabled eight of the ten phosphorylation sites identified in virus purified from infected Vero-E6 cells by replacement of their serine or threonine residues with alanine. These substitutions did not affect VP35 expression in 293T, Vero-E6, or EpoNi/22.1 cells except for the mutants T206A, T207A and S208A, which were expressed at lower levels in Vero-E6 cells (Fig. 8a–c, bottom panels). Interestingly, VP35 T210A mutation almost completely abrogated EBOV transcriptional activity in 293T cells and to a lower extent (20 and 50% of WT) in Vero-E6 and EpoNi/22.1 cells, respectively (Fig. 8a–c). In contrast, the phosphorylation-mimicking mutation T210D had only a marginal effect on transcription (Fig. 8a–c). Consistent with its potential biological role, the Thr-210 residue is conserved in all ebolaviruses except SUDV (Fig. 1c). The mutation S310A also significantly reduced transcription in all three cell lines, while the T206A mutation reduced transcription in 293T cells only (Fig. 8a–c). However, the phosphorylation-mimicking mutation S310D also caused a similar decrease in transcription (Fig. 8a–c).
Fig. 8.

Phosphorylation of VP35 T210 is required for EBOV transcriptional activity. Analysis of the mutations in the mini-genome system: 293T (a) or Vero-E6 (b) or EpoNi/22.1 (c) cells were transfected with the indicated EBOV VP35 constructs, along with plasmids encoding NP, L, VP30 proteins, and T7 polymerase, a plasmid expressing EBOV minigenome containing a firefly luciferase reporter gene, and a control Renilla luciferase plasmid. Firefly luciferase activity from EBOV minigenome was normalized to Renilla luciferase and the minigenome activity of WT VP35. Bottom: Western blots for VP35 and GAPDH or alpha-tubulin proteins. d Effect of amino acid substitutions on the level of phosphorylation: 293T cells were transfected with plasmids encoding VP35 or its mutated forms and pulsed with 32P-orthophosphate with or without treatment with okadaic acid. Top: Western blot analysis with VP35-specific antibodies and analysis of 32P phosphorylation by phosphor imager. analysis of selected VP35 mutants in the mini-genome system as described for b and c in 293T cells: Q-RT-PCR analysis of luciferase mRNA (e) and anti-mini-genome (f). a–f Mean values based on triplicate samples ± SE. Comparison of the mutants to VP35 WT: *p < 0.05 (one way ANOVA and Dunnett’s test). g Detailed view of phosphorylation candidate Thr-210 in the context of modeled full length structure of VP35. Thr-210 resides in an unstructured region modeled as a loop with helical propensity (shown in yellow) that connects the N-terminal region including the oligomerization domain (shown partially, in green) and the C-terminal interferon inhibitory domain (IID, orange and red). The image was created in PyMOL. h. A detailed view of phosphorylated residues Ser-310 and Ser-317 shown in the context of EBOV VP35 IID in complex with an 8-bp double-stranded RNA (dsRNA) (from PDB ID: 3L25). Four molecules of IID bind dsRNA, which is depicted in orange red (bases are colored blue). Ser-310 of two IID molecules (molecules a and d shown in light gray) make direct contact (~ 3 Å) with dsRNA but their corresponding Ser-317 residues (out of view in this image) do not make contact. Neither Ser 310 of IID molecules (b, e colored gold) nor their corresponding Ser-317 make any contact with dsRNA. The model was created in Chimera
To confirm that VP35 is phosphorylated in cultured cells, 293T cells were transfected with VP35 expressing plasmids, and the cells were treated with 32P orthophosphate and 100 nM okadaic acid to induce VP35 phosphorylation. Phosphorylation of WT VP35 was increased more than threefold in cells treated with okadaic acid (Fig. 8d), confirming that the protein undergoes dynamic phosphorylation likely controlled by a cellular phosphatase. The mutation T210A, but not the T191A or S310A mutations, reduced VP35 phosphorylation (Fig. 8d). VP35 phosphorylation was also reduced in the combination S205A/T206A/T207A/S208A/T210A (205-210A) mutant (Fig. 8d). To determine if phosphorylation of VP35 affects the balance of genome transcription and replication in a manner similar to the effect of VP30 phosphorylation [16, 17], we compared the effect of the VP35 mutants T210A and T210D on the relative abundance of minigenome-produced mRNA and antigenomic RNA by strand-specific qRT-PCR. Consistent with the luciferase-based minigenome data, T210A almost completely abrogated the synthesis of mRNA Fig. 8e). However, unlike VP30, we observed an inhibiting effect on the synthesis of antigenome as well (Fig. 8f). Phosphorylation-mimicking mutation T210D partially reversed the inhibiting effects of T210A (Fig. 8e, f). As illustrated in Fig. 7e, Thr-210 is located in an unstructured region, which encompasses a loop with some helical propensity. This loop connects the three-stranded oligomerization domain from our model with the structured C-terminal IID (Fig. 8g). In contrast, Ser-310 and Ser-317 lie within the IID domain that binds to double-stranded RNA. From the crystal structure of the IID domain bound to an 8-bp double-stranded RNA fragment (PDB ID: 3L25) [57], Ser-310 appears to interact with an RNA cytidine base (Fig. 8h). Thr-210 lies in an unstructured and flexible region and has the conformational freedom to interact with the replicative and transcriptional machinery. Importantly, phosphorylation of the cluster containing Thr-210 was demonstrated not only in the virus purified from Vero-E6 and EpoNi/22.1 cells, but also in plasmid-expressed VP35 in Vero-E6 and 293T cells (Fig. 7g). Collectively, our data identify Thr-210 phosphorylation as a requirement for EBOV transcription and genome replication.
The VP35 Thr-210 is not involved in IFN antagonism
As noted above, in addition to playing a role in EBOV replication and transcription, VP35 also serves as an antagonist of type I IFN signaling by binding double-stranded RNA to prevent virus-induced activation of host interferon regulatory factor 3 [60, 71]. The latter activity is associated with the IID, in particular with basic residues R305, K309, R312 and R322 [57, 68, 72]. Thus, we tested whether the reduced transcriptional activity of VP35 T210 mutants was also accompanied by a loss of IFN antagonism. We therefore analyzed the effect of T210A and T210D on the ability of VP35 to antagonize type I IFN. Despite the lack of transcriptional activity by VP35 T210A (contrasted with full activity by T210D), these mutants retained equal ability to inhibit Sendai virus mediated IFN-β reporter activation (Fig. 9a). In contrast, the R312A mutant displayed the expected partial loss of inhibitory activity (Fig. 9b). When tested for the ability to inhibit activation of the IFN-induced interferon stimulated gene 54 (ISG54) promoter, both T210 mutants demonstrated full inhibitory activity equal to WT VP35, while no inhibition was detected for VP35 R312 mutant (Fig. 9b). These results support previously published data demonstrating that only the C-terminus of VP35 is involved in IFN antagonism [57, 73], and suggest that the involvement of T210 phosphorylation in polymerase activity is not related to IFN antagonism.
Fig. 9.

VP35 T210 is not involved in IFN antagonism. 293T cells were transfected with an IFN-β promoter-firefly luciferase construct or an ISG54 firefly luciferase construct, a Renilla luciferase expression plasmid, three different concentrations of either empty pCAGGS vector (EV) or the indicated EBOV VP35 constructs, and R312A mutant. Cells were infected or mock-infected with Sendai virus (SeV) (a) or treated or mock-treated with universal IFN (b), lysates were analyzed by dual luciferase assay, and luciferase activity was normalized to that with the empty vector after SeV infection or UIFN treatment (100%). Mean values based on triplicate samples ± SE. Comparison of the mutants to VP35 WT: ****p < 0.0001 and *p < 0.05 (Student’s one-tailed t test)
Phosphorylation of VP35 Thr-210 is required for binding to NP
The interaction between VP35 and NP is critical for EBOV polymerase activity [58]. To determine whether the lack of polymerase activity in the T210A mutant can be attributed to the loss of VP35–NP interaction, NP was co-expressed with WT VP35, VP35 T210A or VP35 T210D mutant in 293T cells (Fig. 10a). The interaction of NP with VP35 was analyzed in co-immunoprecipitation assays (Fig. 10b, c). The VP35 T210A mutation markedly reduced VP35 association with NP. Thus, the lack of transcriptional activity in T210A might be due to reduced VP35–NP interaction. To test whether the overall phosphorylation level of VP35 contributed to NP binding, 293T cells were pretreated with okadaic acid prior to immunoprecipitation of NP (Fig. 10d). In lysates obtained from okadaic acid-treated cells, VP35 T210A reduced interaction with NP, whereas T210D restored binding (Fig. 10e, f). Importantly, treatment with potato acid phosphatase (PAPP) completely abrogated binding of VP35 to NP (Fig. 10e, f), suggesting that VP35 phosphorylation is critical for this interaction. Together, these data indicate that Thr-210 phosphorylation contributes to VP35 binding to NP, but that additional phosphorylation sites are likely to contribute to this interaction.
Fig. 10.
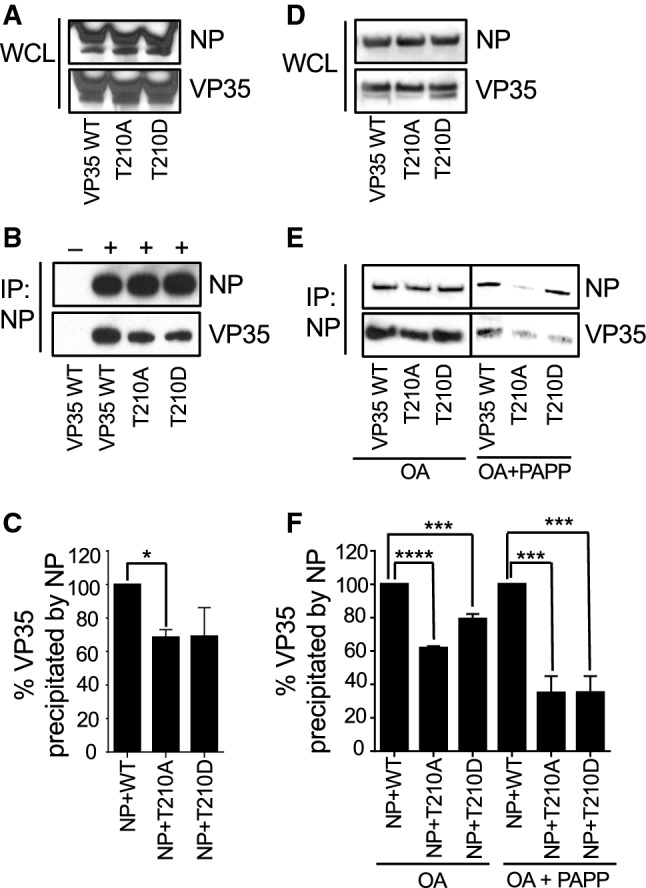
VP35 T210 is involved in interaction with NP. a–c Co-immunoprecipitation of VP35 and NP. 293T cells were co-transfected with NP and either WT or mutant VP35. a Whole cell lysates (WCL) analyzed by Western blotting with antibodies specific to NP or VP35. b NP was precipitated from the lysates with anti-NP antibody cross-linked to magnetic beads. NP and VP35 were detected with anti-NP and anti-VP35 antibodies. + or − at the top indicates the presence of NP antibody used for immunoprecipitation. c Densitometry analysis of the immunoprecipitation analyses depicted on above panel (lanes 2–4), amount of VP35 mutants immunoprecipitated with NP normalized to WT VP35 (100%). The experiments shown on a–c were performed two times with similar results. d–f Effect of okadaic acid and protein phosphatase treatment on VP35–NP interaction. 293T cells were co-transfected with NP and either WT or mutant VP35 and treated with okadaic acid. Where indicated, cell lysates were treated with potato acid phosphatase (PAPP) prior to immunoprecipitation with anti-NP antibody. d Western blot analysis of whole cell lysates. e NP was precipitated from the lysates with anti-NP antibody cross-linked to magnetic beads. NP and VP35 were detected with anti-NP and anti-VP35 antibodies. f Densitometry analysis of the immunoprecipitation analysis depicted on above e. The experiment was performed at least three times with similar results. Comparison of the mutants to VP35 WT: *p < 0.05 (one-way ANOVA and Dunnett’s test)
Discussion
EBOV is dependent upon host enzymes such as kinases and phosphatases for infection, pathogenesis, and viral replication. The stability, dynamics, signaling, cellular sub-location, and regulation of the intricate complex of viral and host proteins are affected through reversible post-translational modifications including protein phosphorylation. Targeting EBOV phosphorylation can serve as a potential therapeutic mechanism. Yet to date, little information is available on EBOV protein phosphorylation. Here, we successfully discovered and mapped phosphorylation sites in all seven structural proteins of EBOV. To our knowledge, this is the first comprehensive mapping of phosphorylation in all EBOV proteins. The majority of the phosphorylation sites described in this study have not been previously reported. Furthermore, the current study includes the first comparison of global phosphorylation profiles of EBOV particles propagated in primate and bat cells, representing accidental and natural hosts of the virus, respectively. Remarkably, the phosphorylation profiles appear to be distinct for EBOV derived from primate and bat cells (Fig. 1c). Many residues that were phosphorylated in primate-derived EBOV were not phosphorylated in bat-derived virions and vice versa. Analysis of the conservation of phosphorylated Thr, Ser and Tyr residues across filovirus species demonstrated their partial conservation. The different phosphorylation profiles of viral particles derived from the two hosts suggest that the role of phosphorylation in EBOV replication may be species specific and indeed may play a role in the drastically different pathogenesis of EBOV in primates and bats. We cannot, however, entirely dismiss the possibility that some of the identified modifications may represent sulfurylation rather than phosphorylation. To achieve a more advanced understanding of the functional importance of the phosphorylated residues, we constructed molecular models of EBOV proteins using existing crystallographic data. Where such data were not available, we used ab initio modeling, or comparative modeling when feasible, and mapped the phosphorylation sites identified in Vero-E6-grown virions on the generated models to get the structural context of phosphorylation and discern their functional role.
GP, the only EBOV envelope protein is an important factor in EBOV pathogenesis: it is involved in cell fusion, immune evasion, and contributes to host cell death [74]. GP is phosphorylated by ERK MAPK kinase [74] and is required for viral entry [75]. The five new phosphorylation sites we identified in GP lie on the surface of GP1, which forms spikes, and are likely to be important in membrane fusion and pathogenesis in general.
VP24 is a major viral virulence factor and directly subverts the immune response [46]. VP24 is active in the cellular MAPK kinase signaling pathway and prevents phosphorylation of p38 for IFN activation [76]. Three clusters of amino acid residues in VP24 interact with karyopherin α: residues 134–139 (cluster 1), 184–186 (cluster 2) and 201–207 (cluster 3) [47]. Intriguingly, most of the identified phosphorylation sites are located close to clusters 2 and 3. The proximity of the phosphorylation sites strongly implicates them in EBOV pathogenesis: phosphorylation site Ser-12 lies on the N-terminal loop, Ser-146 is adjacent to VP24’s karyopherin α5 binding site; and Ser-146 and Ser-151 border clusters 2 and 3.
EBOV nucleoprotein NP was found to be phosphorylated at multiple positions. A prior study with plasmid-expressed NP identified 4 sites (Thr-563, Ser-581, Ser-587 and Ser-647), all in the acidic and disordered C-terminal half, but found no phosphopeptides in the N-terminal (1–450) half [23]. Consistent with this previous observation, our data also show phosphorylation of Thr-563 but not Ser-581, Ser-587 or Ser-647. The 20 sites that we discovered in EBOV NP are located in both N- and C-terminal halves, and 19 of them are novel. With the exception of three phosphotyrosines, the phosphorylation sites are either serines (7 sites) or threonines (10 sites). In contrast to the acidic C-terminal half, the N-terminal half is mostly hydrophobic and engages in NP–NP binding [50]. Both the N-terminal and C-terminal halves participate in the recruitment and assembly of the nucleocapsid [13]. NP’s N-terminal residues 2–150 and C-terminal residues 601–739 bind VP40 [77].
VP30 phosphorylation controls the balance of EBOV transcription/replication activities [78]. Thr-146 was localized in the ordered domain of VP30 at the beginning of a short helix, possibly representing a molecular recognition element. The other phosphorylation site is located in the disordered region, similar to NP.
VP35 has been shown to undergo phosphorylation through its trafficking and colocalization with cellular kinases IKKε and TBK-1 [59]. Interaction of VP35 with IKKε and TBK-1 was shown to block the interaction of interferon regulatory factor 3 (IRF-3) with these host kinases, preventing phosphorylation necessary for activation of type I interferon genes [59]. In the current study, we identified 10 new VP35 phosphorylation sites. All ten phosphorylation sites we found in VP35 lie in flexible or disordered regions of the N-terminal half of the protein. We tested functionality of the identified VP35 phosphorylation sites using the minigenome assay. Specifically, we examined the role of the VP35 phosphorylated residues in EBOV transcription and replication. We found that phosphorylation of two residues, Thr-210 and Ser-310, altered EBOV transcription. Mutation of both residues to alanine caused a significant reduction in minigenome transcription. However, only alanine mutation of Thr-210, but not Ser-310 reduced VP35 phosphorylation. Mutation of Ser-310 to alanine significantly diminished transcription in a minigenome assay in 293T, Vero-E6, and EpoNi/22.1 cells. By contrast, mutation of Ser-317 to alanine did not have the effect (Fig. 8a–c).
The de novo assembled full-length models of NP, VP30, and VP35 were tested for their stability by MD. The results confirmed that these proteins contain significant flexible as well as well-folded domains. VP30 and VP35 behaved similarly. Their ordered or folded regions retained their structure during the 100-ns MD simulation, while the predicted unstructured regions were very flexible, transversing diverse conformational states. In NP, the protein’s two central regions preserved their compact initial conformations relative to the N-terminal domain and each other as rigid bodies. Similarly to the unstructured regions of the VP30 and VP35 proteins, the C-terminal part of NP was also predicted to be unfolded and very flexible.
Studies of the P protein of parainfluenza virus type V, a member of the family Paramyxoviridae, order Mononegavirales, demonstrated that phosphorylation of Ser-157 and Ser-308 negatively affects replication of the viral genome due to interaction with polo-like kinase 1 [79, 80], whereas phosphorylation of Thr-286 upregulates mRNA transcription [81]. A study with vesicular stomatitis virus, another mononegavirus, and a member of the family Rhabdoviridae, demonstrated that alanine substitutions of three phosphorylation sites: Ser-60, Thr-62 and Ser-64, reduce viral transcription but do not significantly affect replication [82]. In our study, dephosphorylation of VP35’s Thr-210 residue inhibited both viral transcription and genome replication suggesting that as EBOV’s P analog VP35 has a distinct mechanism of action.
The loss of polymerase activity due to substitution of Thr-210 to alanine was not associated with a loss of interferon antagonist function. This was not surprising, as only the basic residues within the central basic patch of the C-terminal interferon inhibitory domain are critical for dsRNA binding and IFN inhibition [57, 61]. Three of the four first basic patch residues, Arg-225, Lys-248, and Lys-251, are also critical for VP35’s polymerase cofactor function, most likely due to interaction with NP [58]. In two recent structural studies, the N-terminus of VP35 was also found to be necessary for VP35–NP interaction [51, 53]. We observed a reduced interaction of NP with VP35 T210A but not VP35 T210D, suggesting that Thr-210 phosphorylation facilitates the binding of VP35 and NP. This is the first report demonstrating involvement of phosphorylation of VP35, or any EBOV protein in transcription and in interaction with NP.
This study identifies multiple phosphorylation sites in all structural proteins of EBOV, identifies differences in phosphorylation profiles of EBOV particles propagated in primate versus bat cells, maps the identified phosphorylation sites on modeled 3D structures on the proteins, and demonstrates that phosphorylation of VP35 Thr-210 is required for viral polymerase activity and for interaction with NP. Follow-up studies should focus on investigation of the role of the identified phosphorylation sites in the function of additional proteins, particularly GP, which is the sole envelope protein. Furthermore, studies on additional types of cells relevant to EBOV infection such as dendritic cells and macrophages would broaden the importance of these findings.
Materials and methods
Cell lines and viruses
293T and Vero-E6 cells were obtained from ATCC. EpoNi/22.1 cells derived from a kidney of adult Epomops buettikoferi bats [26] were kindly provided by Dr. Christian Drosten, University of Bönn, Germany. 293T cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM), high glucose, supplemented with 10% fetal calf serum (FCS) and 0.1% gentamicin sulfate (Corning), and plated on PureCoat amine flat bottom multi-well plates (Corning). Vero-E6 cells were maintained in Modified Eagle Medium (MEM), supplemented with 10% FBS, 1% sodium pyruvate, 1% MEM non-essential amino acids (Sigma) and 0.1% gentamicin sulfate, and plated on tissue culture-treated multi well plates. EpoNi/22.1 cells were maintained in DMEM–F-12 GlutaMAX medium, supplemented with 10% FBS and 0.1% gentamicin sulfate. Recombinant EBOV, strain Mayinga, expressing GFP [83] was propagated in Vero-E6 and EpoNi/22.1 cells at a multiplicity of infection 0.1 PFU/cell and incubated for 5 days. Supernatants of infected cells were clarified from cell debris by low-speed centrifugation, and then subjected to ultracentrifugation through 25% sucrose cushion (2 h, + 4 ºC, 175,000×g). Pellets were re-suspended in 1× STE buffer (10 mM Tris, 1 mM EDTA, 0.1 M NaCl) and further purified by ultracentrifugation in 20–60% sucrose gradient (1.5 h, + 4 ºC, 288,000×g). The virus-containing band was harvested, and EBOV virions were washed from sucrose by final ultracentrifugation in 1× STE buffer (1 h, + 4 ºC, 175,000×g). The obtained viral particles were suspended in 1× STE buffer with 4% SDS and heat-inactivated at 95 ºC for 15 min. Virus inactivation was confirmed by inoculation of Vero-E6 cell monolayers with an aliquot of inactivated virus preparation or live virus control, and by microscopic observation of cytopathic effect and GFP fluorescence for up to 7 days. All works with EBOV were performed within the BSL-4 facilities of the Galveston National Laboratory. Sendai virus (SeV) strain Cantell was provided by Dr. Christopher Basler (Georgia State University).
VP35 mutagenesis
The phosphorylation sites identified in VP35 by mass spectrometry analysis were mutated to alanine using ‘QuikChange II’ site-directed mutagenesis kit (Agilent Technologies); primer sequences can be provided upon request. Aspartate substitutions of T210 and S310 were introduced by overlap extension PCR using Pfx polymerase (Thermo Fisher Scientific) with two sets of external primers (5′ and 3′ ends of pCEZ-VP35 sequence) and two internal primers containing the mismatched bases. After sequence confirmation, the mutated plasmids were digested with SacI and BglII endonucleases and cloned back into pCEZ-VP35 vector. Mutated VP35 plasmids were transfected in 293T cells using TransIT-LT1 Transfection Reagent (Mirus) at 3 µl per µg of plasmid DNA, and cell lysates were harvested 48 h posttransfection by suspending in 5× passive lysis buffer (Promega).
Analysis of VP35 protein by Western blotting
Cell lysates were denatured at 70 °C for 10 min and separated in NuPAGE 4–12% Bis–Tris gel along with Novex Sharp Pre-Stained Protein Standard, and proteins were transferred to nitrocellulose membrane using the iBlot Gel transfer system (Thermo Fisher Scientific). Membranes were blocked with 5% milk in PBST for 1 h and incubated with primary rabbit polyclonal antibodies against EBOV VP35 at 1:1000 dilution (IBT Bioservices) and secondary anti-rabbit IgG antibodies conjugated to horseradish peroxidase at 1:3000 dilution (Cell Signaling Technology). Protein bands were visualized using the chemiluminescent substrate—Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific), scanned and quantified using Image Studio Lite Version 5.2 software (LI-COR Biotechnology).
Minigenome experiments
The plasmids pCEZ-NP, pCEZ-VP35, pCEZ-VP30, pCEZ-L, pC-T7 and monocistronic EBOV minigenome expressing firefly luciferase reporter gene [84, 85] were kindly provided by Dr. Yoshihiro Kawaoka (University of Wisconsin). The pRL-TK Renilla luciferase expression plasmid was purchased from Promega. Experiments involving the EBOV minigenome were performed essentially as previously described [17]. Confluent monolayers were transfected with the following amount of plasmids: 50 ng of pCEZ-NP; 50 ng of pCEZ-VP35 or its mutants; 30 ng of pCEZ-VP30; 400 ng of pCEZ-L; 100 ng of pC-T7; 100 ng of EBOV minigenome, and 4 ng of pRL-TK control reporter plasmid. The plasmids were transfected with TransIT-LT1 Transfection Reagent. At 48 h post-transfection, cells were lysed with either passive lysis buffer to measure transcription by the dual luciferase reporter assay (Promega), or with Buffer RLT Plus (Qiagen) for the analysis of replication of the minigenome by strand-specific qRT-PCR. Firefly luciferase activity was normalized to Renilla luciferase activity. Minigenome reporter activation was expressed as percent activity relative to the positive control reaction (50 ng of WT VP35 plasmid), which was set to 100%.
Hot-start reverse transcription with a tagged primer
RNA isolation was performed using RNeasy Plus Mini Kit (Qiagen) as per manufacturer’s instructions. First-strand synthesis was performed using SuperScript III reverse transcriptase (Thermo Fisher Scientific) essentially as described in [86] with some modifications as follows. Reverse transcription primers that distinguishes the cRNA and mRNA strands of the EBOV minigenome were designed with a unique 18–20 nucleotide tag (highlighted in bold) besides the strand-specific sequence. The following reverse transcription primers were used—cRNA RT primer: GCTAGCTTCAGCTAGGCATCGTGCGACCATTTTTCCAGGAATCCT; mRNA RT primer: CCAGATCGTTCGAGTCGTTTTTTTTTTTTTTTTTATAATCTGTAT. A mixture containing 200 ng of RNA and 10 pmol of tagged primer was heated for 10 min at 65 °C, chilled immediately on ice for 5 min, and again heated to 60 °C for 5 min. Subsequently, 15 µl of preheated RT master mix containing 4 µl of 5× first strand buffer, 1 µl 0.1 M dithiothreitol, 1 µl dNTP mix (10 mM each), 1 µl Superscript III reverse transcriptase (200 U/µl, Invitrogen), 1 µl RNasin Plus RNase inhibitor (40 U/µl, Promega) and 6.5 µl saturated trehalose (Life Sciences Advanced Technologies, St. Petersburg, FL, USA) were added and incubated at 60 °C for 1 h. Then, 10 units of exonuclease I (Thermo Fisher Scientific) were added to remove RT primer, incubated at 37 °C for 30 min and heat-inactivated at 85 °C for 15 min. All cDNA reactions were diluted 10× with nuclease-free water and stored at − 20 °C.
Droplet digital PCR
The unique tags used in the RT primers were used as forward primers in qPCR, whereas both probes and reverse primers were specific to the EBOV minigenome. To increase the fluorescent signal of quantitative PCR reactions, a 12-nucleotide AT-rich flap (shown in lower case letters) was added to the 5′ end of tagged primers [87]. The primers used in qPCR are as follows:
cRNA forward—aataaatcataaGCTAGCTTCAGCTAGGCATC;
cRNA probe—GCTCGCCAGAATAAACGTTGCA;
cRNA reverse—aataaatcataaCATTGACCACGCTCATCAGAA;
mRNA forward—aataaatcataaCCAGATCGTTCGAGTCGT;
mRNA probe—CCGTGTAAGCGGCCGCATAGT;
mRNA reverse—aataaatcataaTCCTCATAAAGGCCAAGAAGGG.
Quantitative PCR was performed with Taqman (Applied Biosystems) primer–probe mix using the QX200 Droplet Digital PCR System (BioRad, Hercules, CA). Four microliters of a 10-fold dilution of the cDNA were added to the ddPCR reaction mixture [10 µl of 2× ddPCR Supermix for Probes (No dUTP), 1 µl of 20× Taqman custom assay and 5 µl of nuclease-free water]. ddPCR reaction mixtures were loaded onto cartridges to create droplets on a QX200 Droplet Generator (BioRad). The droplets were transferred onto 96-well Twin. Tec PCR Plates (Eppendorf, Hauppauge, NY) and amplified on a C1000 Thermal Cycler with 96-Deep Well Reaction Module (BioRad). The cycle conditions of qPCR were 95 °C for 10 min, followed by 39 cycles of 94 °C for 30 s and 60 °C for 1 min, and a final enzyme deactivation step of 98 °C for 10 min. Finally, the PCR plates were loaded onto a Droplet Reader, which quantifies the number of positive and negative droplets in each sample. Analysis was performed using QuantaSoft software to get the final concentrations in each sample.
IFN antagonism assays
Both ISG54 promoter-firefly luciferase reporter plasmid and an IFN-β promoter-firefly luciferase reporter were kindly provided by Dr. Christopher Basler. 293T cells (5 × 104) were transfected with 0.1 µg of either reporter plasmid, 0.1 µg of a constitutively expressing Renilla luciferase reporter construct to serve as a transfection control, and three different concentrations (250, 25 or 2.5 ng/µl) of pCEZ-EV (empty vector) or pCEZ-VP35 or the mutants (T210A, T210D and R312A). Trans-IT LT1 was used as a transfection reagent. Twenty-four h post-transfection with ISG54 promoter, cells were treated with 1000 U/ml of universal IFN; 24 h post transfection with IFN-β promoter, cells were infected with 150 hemagglutinin activity units of SeV. Eighteen h later, cells were harvested and analyzed for luciferase activity using dual luciferase reporter assay as described above. Firefly luciferase activity was normalized to Renilla luciferase activity. The results are presented as percent induction of the positive control (SeV infected or IFN stimulated, empty vector transfected).
Transfections
293T and Vero-E6 cells were seeded in 6-well plates to achieve 50% confluence on the day of transfection. The cells were transfected with indicated plasmids using Lipofectamine and Plus reagents (Life Technologies) following manufacturer’s protocol. The efficiency of transfection was verified using a plasmid encoding green fluorescent protein. The cells were cultured for 48 h post-transfection and analyzed.
Co-immunoprecipitation assays
293T cells co-transfected with 2 µg each of pCEZ-NP and either pCEZ-VP35 or mutants or empty vector were collected 48 h later by lysing cells in 10× Cell Lysis Buffer (Cell Signaling Technology) and Halt Protease and Phosphatase Inhibitor Cocktail, EDTA-free (100×) (Thermo Fisher Scientific). Lysates were clarified by low-speed centrifugation, and immunoprecipitation of NP or VP35 proteins was performed using a Pierce Direct Magnetic IP/coIP kit (Thermo Fisher Scientific) as per manufacturer’s protocols. Total protein in lysates was quantified by Pierce BCA Protein Assay kit (Thermo Fisher Scientific). Briefly, 2 µg of rabbit-anti VP35 or rabbit-anti NP antibody (IBT Bioservices) was coupled to the NHS-activated magnetic beads for 1 h and incubated with the clarified cell lysates overnight at 4 °C. Thereafter, beads were washed twice with IP lysis/wash buffer, and once with ultrapure water, and the bound antigen was eluted. IP lysates, as well as whole cell lysates, were denatured and analyzed by western blotting as described above.
VP35 phosphorylation in cell culture
The sequences of WT VP35 and mutants with C-terminal Flag sequence were synthesized by GenScript and cloned in pcDNA3.1(−) vector at NotI and KpnI sites. 293T cells were seeded in 6-well plates and transfected with 2.5 µg/well of VP35 Flag plasmids. At 48 h post transfection, media was changed for a phosphate-free DMEM (Life Technologies) for 1 h. Then the media was changed to phosphate-free DMEM supplemented with 0.5 mCi/ml of (32P)-orthophosphate and cells were further incubated for 2 h at 37 °C. Cells were also treated with 1 μM okadaic acid (Sigma) to block cellular PPP-phosphatases. Cells were washed with PBS and lysed in whole cell lysis buffer (50 mM Tris–HCl, pH 7.5, 0.5 M NaCl, 1% NP-40, 0.1% SDS) supplemented with protease cocktail (Sigma). After 10 min on ice, cellular material was scraped and then centrifuged at 20,800×g, 4 °C for 30 min. The supernatant was recovered and immediately used for immunoprecipitation. VP35 was precipitated with anti-Flag monoclonal antibodies (Sigma) coupled to protein A/G agarose (Santa Cruz biotechnologies) for 2 h at 4 °C in a TNN Buffer containing 50 mM Tris–HCl, pH 7.5, 0.15 M NaCl, and 1% NP-40. The immunoprecipitated VP35 was recovered by heating for 2 min at 100 °C in Tricine SDS-loading buffer, and resolved on 10% Tris-Tricine SDS-PAGE. Dried gels were exposed to Phosphor Imager screen and analyzed Packard Phosphor Imager (Packard Instruments).
Mass spectrometry
Heat-inactivated EBOV viral particles were resolved on 10% Tris-Tricine SDS-PAGE. Proteins were in-gel digested and processed for mass spectrometry analysis as previously described [17]. Tryptic peptides were purified by ZipTip C18 (Merc Millipore Ltd) and resuspended in 50 μl of water with 0.1% formic acid (v/v). The high-resolution mass spectrometry (HR-MS) analysis was performed on an LTQ XL Orbitrap mass spectrometer (Thermo Fisher Scientific) using the installed Xcalibur software (version 2.1.0, Thermo Fisher Scientific) after the separation of LC-20AD nano HPLC system (Shimadzu Corporation, Columbia, MD, USA). A total of 10 μl of sample was loaded and washed for 6 min on a C18-packed precolumn (1 cm × 150 μm, 5 μm, 200 Å, Michrom Bioresources, Auburn, CA, USA) with a solvent of A:B = 99:1 (A, 0.1% formic acid aqueous solution; B, 0.1% formic acid acetonitrile solution) at a constant flow of 12 μl/min. Peptides were transferred onward to an in-house C18-packed analytical column (25 cm × 150 μm, 5 μm, 200 Å, Michrom Bioresources, Auburn, CA, USA) and separated with a linear gradient of 6–55 min, 2–40% B, 55–62 min, 40–80% B, 62–70 min, 80% B (v/v) at the flow rate of 600 nl/min. Mobile phase A was 0.1% formic acid in water and mobile phase B—0.1% formic acid in acetonitrile. The Orbitrap was operated under data-dependent acquisition mode. The spray voltage, capillary temperature and capillary voltage were set to 2.0 kV, 200 °C, and 39.5 V, respectively. Full-scan mass spectra were acquired in Orbitrap over 300–2000 m/z with a resolution of 30,000, followed by MSn scans by CID activation mode. The three most intense ions were selected for fragmentation using collision-induced dissociation (CID) in the LTQ (normalized collision energy of 35, parent mass selection window of 2.5 Da, activation time of 30 ms, and minimum signal threshold for MS/MS scans set to 500 counts). Charge state rejection (charge state 1 was rejected) as well as dynamic exclusion (repeat counts, 2; repeat duration, 10 s; exclusion duration, 10 s) was enabled.
LC–MS/MS raw data were searched by Proteome Discoverer 1.4 (PD 1.4) using SEQUEST search engine (Thermo Fisher Scientific), against a combined FASTA database (20,657 sequences) containing a UniProt EBOV database (97 sequences) that was concatenated with common contaminant proteins and African green monkey (Chlorocebus aethiops) proteome from UniProt, as Vero-E6 cells used for propagation of EBOV were derived from this species of monkeys. The search was performed at a false discovery cut-off ≤ 1%. A maximum of two missed cleavage sites was allowed. The mass tolerance for the precursor ion was 30 ppm and for the fragment—0.1 Da. These high-resolution MS/MS settings significantly improved signal–noise ratio as opposed to a typical fragment tolerance of 0.7 Da. Phosphorylation of serine, threonine and tyrosine were enabled as dynamic modifications, while carbamidomethylation of cysteine was set as fixed modification. Statistical filter settings (Xcorr) for peptides with different charges are: charge 2 = 1.5, charge 3 = 2.0 and charges > 4 = 2.5 for high confidence peptides; charge 2 = 0.5, charge 3 = 0.8 and charges > 4 = 1 for modest confidence peptides. We followed the previously published guidelines to repot MS/MS spectra [88]. The label-free quantification of phosphopeptides eluting between 10 and 80 min was performed with SIEVE 2.1 software (Thermo Fisher Scientific). Briefly, the chromatographic peaks detected by Orbitrap were aligned and the peptides peaks were detected with a minimum signal intensity of 1 × 105; quantitative frames were determined based on m/z (width: 30 ppm) and retention time (width: 2.5 min). The identified phosphopeptides by PD 1.4 were uploaded as framing seeds. Statistical filters were set to assess the quality of the data. The CV raw MS intensities of the triplicates had to be within 30%. This helped to minimize the effect of run-to-run variability.
Reconstruction of 3D protein structures
We assembled EBOV protein structures using existing PDB structures for GP (PDB ID: 3CSY), VP40 (PDB ID: 4LDB) and VP24 (dimeric structure, PDB ID: 4M0Q). At present, there is no available full-length structure for EBOV RNA-directed RNA polymerase L and we did not attempt to model its structure. For VP30, VP35 and NP, only partial structures exist. To construct full-length protein structures, we used the Rosetta ab initio prediction protocols [25]. The Rosetta protocol first determines domains that form the full-length protein. This involves massive sequence alignment using to detect distant evolutionary relationships, secondary structure information and domain boundaries using validated NCBI tools such as BLAST and PSI-BLAST [89], 3D-Jury [90] and Pfam [91]. The assumption is that the protein domain constitutes the evolutionary and independent folding unit if it is homologous to sequences with known structure that domain is modeled by comparative modeling. Domains without a match after an iterative search procedure were modeled using a de novo fragment assembly method that uses the insertion and evaluation of several thousands of small peptide fragments using conformational sampling in a Monte Carlo strategy. The domains obtained were joined by linker regions. Rosetta relies on an accurate knowledge-based force field derived from experimental structures in PDB, and minimization and all-atom refinement. [25]. The top-5 predicted structures were examined manually, refined and dihedral values (φ, ψ) were evaluated using the Ramachandran diagram [92]. Molecular graphics images were produced in the package UCSF Chimera from the Computer Graphics Laboratory, University of California, San Francisco (http://www.cgl.ucsf.edu/chimera) [93] or in PyMOL, using The PyMOL Molecular Graphics System, version 1.8 (Schrödinger, LLC).
Molecular dynamics (MD)
The modeled structures were tested for stability in long-time MD simulation. All-atom models of the proteins under investigation were solvated in TIP3P water molecules within a cube box, ensuring a solvent shell of at least 12 Å around the solute. The solute was neutralized with Na+ Cl− ion pairs at approximate salt concentration of 150 mM. The ions were initially placed at random, but at least 5 Å away from protein and 3.5 Å away from one another. MD simulations were performed with the AMBER 14 suite (University of California, San-Francisco) [94] of programs using ff14SB force field parameters [95] and Joung/Cheatham ion parameters for the surrounding ions running on the multiprocessor cluster of SPbPU (“Tornado”). MD simulations involved several standard steps: (a) creation of topology file for all proteins and preparation of input data for AMBER 14 using the TLEAP tool; (b) construction of hydration models for the proteins under investigation in a periodic water box with a minimal distance to the water box border of 12 Å; (c) energy minimization and thermodynamic equilibration of the hydrated peptide–DNA complexes and surrounding solvent; (d) MD simulations at constant temperature.
Thermodynamically equilibrated systems were used to perform MD simulations at 310 °K using the Langevin thermostat with constant pressure of 1 atm and the MD duration of 100 ns with time steps of 2 fs. The states of the molecular systems were recorded after every 10 ps of MD time for analysis. Neighbor searching was performed at every 10 MD steps. The PME algorithm was used for electrostatic interactions with the cutoff of 1.0 nm as implemented in AMBER. The cut-off of 1.0 nm was used for van der Waals interactions. SHAKE algorithm was used to constrain the bonds involving hydrogen. For analyzing protein stability, protein conformations were fitted to its initial MD conformation using only protein structural domains maintaining its stability during 100-ns MD simulations and time dependences of the no fit root mean square deviation (RMSD) of the C-alpha atoms were used.
Solvent accessibility
Solvent accessibility values were obtained using POPS* [67] which is an implementation of the method of Lee and Richards [96] further optimized using parameterization derived from a curated set of high-resolution crystallographic structures (at least 1.8 Å). Coordinates of the protein were submitted to the server at http://mathbio.crick.ac.uk/wiki/POPS. The output is a file that lists the total surface area and the solvent accessible area computed at the atomic level for each residue along with measures of hydrophobicity and hydrophilicity. The relative solvent accessible surface area is calculated as the ratio of the solvent-exposed area to the total surface area.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by NIH Research Grants U19AI109664 (AB and SN), 1P50HL118006 (SN), 1R01HL125005 (SN), 5G12MD007597 (SN) and P30AI117970 (SN). We thank Dr. Ryan Leib (Stanford University) for generating and providing FASTA database containing EBOV proteins that was concatenated with proteins of African green monkey and common contaminants. We thank Dr. Christian Drosten (University of Bönn, Germany) for providing EpoNi/22.1 bat cells. We thank Dr. Yoshihiro Kawaoka (University of Wisconsin) for providing the plasmids expressing EBOV NP, VP35, L, VP30, and the T7 polymerase and Dr. Christopher Basler (Georgia State University) for providing Sendai virus, ISG54 promoter-firefly luciferase reporter plasmid, and IFN-ß promoter firefly luciferase reporter plasmid. We also thank Mr. Adam Rink for editing the manuscript.
Author contributions
AI, PR, PAI, XL and TA performed experiments and analyzed the data. CP modeled protein structures and MP conducted MD simulations. AI, PR, CP, AB and SN wrote the manuscript. GKA contributed interpretation of the data. AB and SN conceived the project. All authors read and approved the final manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Andrey Ivanov and Palaniappan Ramanathan contributed equally to this work.
Contributor Information
Alexander Bukreyev, Email: abukreye@utmb.edu.
Sergei Nekhai, Email: snekhai@howard.edu.
References
- 1.CDC (2016) Ebola outbreak in West Africa—case counts. http://www.cdc.gov/vhf/ebola/outbreaks/2014-west-africa/case-counts.html
- 2.WHO (2019) Ebola situation reports: Democratic Republic of the Congo. http://www.who.int/ebola/situation-reports/drc-2018/en
- 3.Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, et al. Fruit bats as reservoirs of Ebola virus. Nature. 2005;438(7068):575–576. doi: 10.1038/438575a. [DOI] [PubMed] [Google Scholar]
- 4.Pigott DM, Golding N, Mylne A, Huang Z, Henry AJ, Weiss DJ, et al. Mapping the zoonotic niche of Ebola virus disease in Africa. Elife. 2014;3:e04395. doi: 10.7554/elife.04395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pigott DM, Millear AI, Earl L, Morozoff C, Han BA, Shearer FM, et al. Updates to the zoonotic niche map of Ebola virus disease in Africa. Elife. 2016 doi: 10.7554/elife.16412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhn JH. Filoviruses: a compendium of 40 years of epidemiological, clinical, and laboratory studies. Vienna, Austria: Springer; 2008. Molecular characteristics of filoviruses; pp. 175–264. [PubMed] [Google Scholar]
- 7.Volchkov VE, Becker S, Volchkova VA, Ternovoj VA, Kotov AN, Netesov SV, et al. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology. 1995;214(2):421–430. doi: 10.1006/viro.1995.0052. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez A, Ksiazek TG, Rollin PE, Miranda ME, Trappier SG, Khan AS, et al. Detection and molecular characterization of Ebola viruses causing disease in human and nonhuman primates. J Infect Dis. 1999;179(Suppl 1):S164–S169. doi: 10.1086/514282. [DOI] [PubMed] [Google Scholar]
- 9.Mehedi M, Falzarano D, Seebach J, Hu X, Carpenter MS, Schnittler HJ, et al. A new Ebola virus nonstructural glycoprotein expressed through RNA editing. J Virol. 2011;85(11):5406–5414. doi: 10.1128/jvi.02190-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldmann H, Sanchez A, Geisbert TW. Filoviridae: Marburg and Ebola viruses. In: Knipe DM, Howley PM, editors. Fields virology. 6. Philadelphia: Lippincott Williams & Wilkins; 2013. pp. 923–956. [Google Scholar]
- 11.Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J Virol. 1999;73(3):2333–2342. doi: 10.1128/JVI.73.3.2333-2342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bharat TA, Noda T, Riches JD, Kraehling V, Kolesnikova L, Becker S, et al. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc Natl Acad Sci USA. 2012;109(11):4275–4280. doi: 10.1073/pnas.1120453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Xu L, Sun Y, Nabel GJ. The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol Cell. 2002;10(2):307–316. doi: 10.1016/S1097-2765(02)00588-9. [DOI] [PubMed] [Google Scholar]
- 14.Mateo M, Carbonnelle C, Martinez MJ, Reynard O, Page A, Volchkova VA, et al. Knockdown of Ebola virus VP24 impairs viral nucleocapsid assembly and prevents virus replication. J Infect Dis. 2011;204(Suppl 3):S892–S896. doi: 10.1093/infdis/jir311. [DOI] [PubMed] [Google Scholar]
- 15.Nanbo A, Watanabe S, Halfmann P, Kawaoka Y. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci Rep. 2013;3:1206. doi: 10.1038/srep01206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biedenkopf N, Hartlieb B, Hoenen T, Becker S. Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex: impact on viral transcription and replication. J Biol Chem. 2013;288(16):11165–11174. doi: 10.1074/jbc.m113.461285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ilinykh PA, Tigabu B, Ivanov A, Ammosova T, Obukhov Y, Garron T, et al. Role of protein phosphatase 1 in dephosphorylation of Ebola virus VP30 protein and its targeting for the inhibition of viral transcription. J Biol Chem. 2014;289(33):22723–22738. doi: 10.1074/jbc.m114.575050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Modrof J, Muhlberger E, Klenk HD, Becker S. Phosphorylation of VP30 impairs Ebola virus transcription. J Biol Chem. 2002;277(36):33099–33104. doi: 10.1074/jbc.m203775200. [DOI] [PubMed] [Google Scholar]
- 19.Biedenkopf N, Lier C, Becker S. Dynamic phosphorylation of VP30 is essential for Ebola virus life cycle. J Virol. 2016;90(10):4914–4925. doi: 10.1128/jvi.03257-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kruse T, Biedenkopf N, Hertz EPT, Dietzel E, Stalmann G, Lopez-Mendez B, et al. The Ebola virus nucleoprotein recruits the host PP2A-B56 phosphatase to activate transcriptional support activity of VP30. Mol Cell. 2018;69(1):136–145.e6. doi: 10.1016/j.molcel.2017.11.034. [DOI] [PubMed] [Google Scholar]
- 21.Barik S, Banerjee AK. Sequential phosphorylation of the phosphoprotein of vesicular stomatitis virus by cellular and viral protein kinases is essential for transcription activation. J Virol. 1992;66(2):1109–1118. doi: 10.1128/JVI.66.2.1109-1118.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barik S, McLean T, Dupuy LC. Phosphorylation of Ser232 directly regulates the transcriptional activity of the P protein of human respiratory syncytial virus: phosphorylation of Ser237 may play an accessory role. Virology. 1995;213(2):405–412. doi: 10.1006/viro.1995.0013. [DOI] [PubMed] [Google Scholar]
- 23.Peyrol J, Thizon C, Gaillard JC, Marchetti C, Armengaud J, Rollin-Genetet F. Multiple phosphorylable sites in the Zaire Ebolavirus nucleoprotein evidenced by high resolution tandem mass spectrometry. J Virol Methods. 2013;187(1):159–165. doi: 10.1016/j.jviromet.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Shi W, Huang Y, Sutton-Smith M, Tissot B, Panico M, Morris HR, et al. A filovirus-unique region of Ebola virus nucleoprotein confers aberrant migration and mediates its incorporation into virions. J Virol. 2008;82(13):6190–6199. doi: 10.1128/jvi.02731-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rohl CA, Strauss CE, Chivian D, Baker D. Modeling structurally variable regions in homologous proteins with rosetta. Proteins. 2004;55(3):656–677. doi: 10.1002/prot.10629. [DOI] [PubMed] [Google Scholar]
- 26.Kuhl A, Hoffmann M, Muller MA, Munster VJ, Gnirss K, Kiene M, et al. Comparative analysis of Ebola virus glycoprotein interactions with human and bat cells. J Infect Dis. 2011;204(Suppl 3):S840–S849. doi: 10.1093/infdis/jir306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keck F, Ataey P, Amaya M, Bailey C, Narayanan A. Phosphorylation of single stranded rna virus proteins and potential for novel therapeutic strategies. Viruses. 2015;7(10):5257–5273. doi: 10.3390/v7102872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cong Q, Pei J, Grishin NV. Predictive and comparative analysis of Ebolavirus proteins. Cell Cycle. 2015;14(17):2785–2797. doi: 10.1080/15384101.2015.1068472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jun SR, Leuze MR, Nookaew I, Uberbacher EC, Land M, Zhang Q, et al. Ebolavirus comparative genomics. FEMS Microbiol Rev. 2015;39(5):764–778. doi: 10.1093/femsre/fuv031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofmann-Winkler H, Kaup F, Pohlmann S. Host cell factors in filovirus entry: novel players, new insights. Viruses. 2012;4(12):3336–3362. doi: 10.3390/v4123336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groseth A, Marzi A, Hoenen T, Herwig A, Gardner D, Becker S, et al. The Ebola virus glycoprotein contributes to but is not sufficient for virulence in vivo. PLoS Pathog. 2012;8(8):e1002847. doi: 10.1371/journal.ppat.1002847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volchkov VE, Feldmann H, Volchkova VA, Klenk HD. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc Natl Acad Sci USA. 1998;95(10):5762–5767. doi: 10.1073/pnas.95.10.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanger C, Muhlberger E, Lotfering B, Klenk HD, Becker S. The Marburg virus surface protein GP is phosphorylated at its ectodomain. Virology. 2002;295(1):20–29. doi: 10.1006/viro.2002.1374. [DOI] [PubMed] [Google Scholar]
- 34.Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454(7201):177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JE, Fusco ML, Abelson DM, Hessell AJ, Burton DR, Saphire EO. Techniques and tactics used in determining the structure of the trimeric Ebolavirus glycoprotein. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 11):1162–1180. doi: 10.1107/s0907444909032314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, Bowden TA, et al. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature. 2016;535(7610):169–172. doi: 10.1038/nature18615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flyak AI, Shen X, Murin CD, Turner HL, Fusco ML, Lampley R, et al. Cross-reactive and potent neutralizing antibody responses in human survivors of natural Ebolavirus infection. Cell. 2016;164(3):392–405. doi: 10.1016/j.cell.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jasenosky LD, Neumann G, Lukashevich I, Kawaoka Y. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J Virol. 2001;75(11):5205–5214. doi: 10.1128/jvi.75.11.5205-5214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noda T, Ebihara H, Muramoto Y, Fujii K, Takada A, Sagara H, et al. Assembly and budding of Ebolavirus. PLoS Pathog. 2006;2(9):e99. doi: 10.1371/journal.ppat.0020099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomis-Ruth FX, Dessen A, Timmins J, Bracher A, Kolesnikowa L, Becker S, et al. The matrix protein VP40 from Ebola virus octamerizes into pore-like structures with specific RNA binding properties. Structure. 2003;11(4):423–433. doi: 10.1016/S0969-2126(03)00050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bornholdt ZA, Noda T, Abelson DM, Halfmann P, Wood MR, Kawaoka Y, et al. Structural rearrangement of ebola virus VP40 begets multiple functions in the virus life cycle. Cell. 2013;154(4):763–774. doi: 10.1016/j.cell.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radzimanowski J, Effantin G, Weissenhorn W. Conformational plasticity of the Ebola virus matrix protein. Protein Sci. 2014;23(11):1519–1527. doi: 10.1002/pro.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoenen T, Jung S, Herwig A, Groseth A, Becker S. Both matrix proteins of Ebola virus contribute to the regulation of viral genome replication and transcription. Virology. 2010;403(1):56–66. doi: 10.1016/j.virol.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 44.Shabman RS, Gulcicek EE, Stone KL, Basler CF. The Ebola virus VP24 protein prevents hnRNP C1/C2 binding to karyopherin alpha1 and partially alters its nuclear import. J Infect Dis. 2011;204(Suppl 3):S904–S910. doi: 10.1093/infdis/jir323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol. 2007;81(24):13469–13477. doi: 10.1128/jvi.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, et al. Ebola virus VP24 binds karyopherin α1 and blocks STAT1 nuclear accumulation. J Virol. 2006;80(11):5156–5167. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, et al. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe. 2014;16(2):187–200. doi: 10.1016/j.chom.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang AP, Bornholdt ZA, Liu T, Abelson DM, Lee DE, Li S, et al. The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog. 2012;8(2):e1002550. doi: 10.1371/journal.ppat.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edwards MR, Johnson B, Mire CE, Xu W, Shabman RS, Speller LN, et al. The Marburg virus VP24 protein interacts with Keap1 to activate the cytoprotective antioxidant response pathway. Cell Rep. 2014;6(6):1017–1025. doi: 10.1016/j.celrep.2014.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watanabe S, Noda T, Kawaoka Y. Functional mapping of the nucleoprotein of Ebola virus. J Virol. 2006;80(8):3743–3751. doi: 10.1128/jvi.80.8.3743-3751.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirchdoerfer RN, Abelson DM, Li S, Wood MR, Saphire EO. Assembly of the Ebola virus nucleoprotein from a chaperoned VP35 complex. Cell Rep. 2015;12(1):140–149. doi: 10.1016/j.celrep.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dong S, Yang P, Li G, Liu B, Wang W, Liu X, et al. Insight into the Ebola virus nucleocapsid assembly mechanism: crystal structure of Ebola virus nucleoprotein core domain at 1.8 A resolution. Protein Cell. 2015;6(5):351–362. doi: 10.1007/s13238-015-0163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leung DW, Borek D, Luthra P, Binning JM, Anantpadma M, Liu G, et al. An intrinsically disordered peptide from Ebola virus VP35 controls viral RNA synthesis by modulating nucleoprotein-RNA interactions. Cell Rep. 2015;11(3):376–389. doi: 10.1016/j.celrep.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dziubanska PJ, Derewenda U, Ellena JF, Engel DA, Derewenda ZS. The structure of the C-terminal domain of the Zaire Ebolavirus nucleoprotein. Acta Crystallogr D Biol Crystallogr. 2014;70(Pt 9):2420–2429. doi: 10.1107/s1399004714014710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hartlieb B, Muziol T, Weissenhorn W, Becker S. Crystal structure of the C-terminal domain of Ebola virus VP30 reveals a role in transcription and nucleocapsid association. Proc Natl Acad Sci USA. 2007;104(2):624–629. doi: 10.1073/pnas.0606730104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clifton MC, Kirchdoerfer RN, Atkins K, Abendroth J, Raymond A, Grice R, et al. Structure of the Reston ebolavirus VP30 C-terminal domain. Acta Crystallogr F Struct Biol Commun. 2014;70(Pt 4):457–460. doi: 10.1107/s2053230x14003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leung DW, Prins KC, Borek DM, Farahbakhsh M, Tufariello JM, Ramanan P, et al. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat Struct Mol Biol. 2010;17(2):165–172. doi: 10.1038/nsmb.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prins KC, Binning JM, Shabman RS, Leung DW, Amarasinghe GK, Basler CF. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J Virol. 2010;84(20):10581–10591. doi: 10.1128/jvi.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prins KC, Cardenas WB, Basler CF. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol. 2009;83(7):3069–3077. doi: 10.1128/jvi.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Basler CF, Wang X, Muhlberger E, Volchkov V, Paragas J, Klenk HD, et al. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc Natl Acad Sci USA. 2000;97(22):12289–12294. doi: 10.1073/pnas.220398297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leung DW, Ginder ND, Fulton DB, Nix J, Basler CF, Honzatko RB, et al. Structure of the Ebola VP35 interferon inhibitory domain. Proc Natl Acad Sci USA. 2009;106(2):411–416. doi: 10.1073/pnas.0807854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramanan P, Edwards MR, Shabman RS, Leung DW, Endlich-Frazier AC, Borek DM, et al. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc Natl Acad Sci USA. 2012 doi: 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bale S, Dias JM, Fusco ML, Hashiguchi T, Wong AC, Liu T, et al. Structural basis for differential neutralization of ebolaviruses. Viruses. 2012;4(4):447–470. doi: 10.3390/v4040447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bruhn JF, Kirchdoerfer RN, Urata SM, Li S, Tickle IJ, Bricogne G, et al. Crystal structure of the Marburg virus VP35 oligomerization domain. J Virol. 2017 doi: 10.1128/jvi.01085-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leung DW, Ginder ND, Fulton DB, Nix J, Basler CF, Honzatko RB, et al. Structure of the Ebola VP35 interferon inhibitory domain. Proc Natl Acad Sci USA. 2009;106(2):411–416. doi: 10.1073/pnas.0807854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Somavarapu AK, Balakrishnan S, Gautam AK, Palmer DS, Venkatraman P. Structural interrogation of phosphoproteome identified by mass spectrometry reveals allowed and disallowed regions of phosphoconformation. BMC Struct Biol. 2014;14:9. doi: 10.1186/1472-6807-14-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cavallo L, Kleinjung J, Fraternali F. POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 2003;31(13):3364–3366. doi: 10.1093/nar/gkg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kimberlin CR, Bornholdt ZA, Li S, Woods VL, Jr, Macrae IJ, Saphire EO. Ebolavirus VP35 uses a bimodal strategy to bind dsRNA for innate immune suppression. Proc Natl Acad Sci USA. 2010;107(1):314–319. doi: 10.1073/pnas.0910547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leung DW, Borek D, Farahbakhsh M, Ramanan P, Nix JC, Wang T, et al. Crystallization and preliminary X-ray analysis of Ebola VP35 interferon inhibitory domain mutant proteins. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66(Pt 6):689–692. doi: 10.1107/s1744309110013266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adzhubei AA, Sternberg MJ, Makarov AA. Polyproline-II helix in proteins: structure and function. J Mol Biol. 2013;425(12):2100–2132. doi: 10.1016/j.jmb.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 71.Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, et al. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol. 2003;77(14):7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schumann M, Gantke T, Muhlberger E. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J Virol. 2009;83(17):8993–8997. doi: 10.1128/jvi.00523-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hartman AL, Towner JS, Nichol ST. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology. 2004;328(2):177–184. doi: 10.1016/j.virol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 74.Zampieri CA, Fortin JF, Nolan GP, Nabel GJ. The ERK mitogen-activated protein kinase pathway contributes to Ebola virus glycoprotein-induced cytotoxicity. J Virol. 2007;81(3):1230–1240. doi: 10.1128/JVI.01586-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson JC, Martinez O, Honko AN, Hensley LE, Olinger GG, Basler CF. Pyridinyl imidazole inhibitors of p38 MAP kinase impair viral entry and reduce cytokine induction by Zaire Ebolavirus in human dendritic cells. Antiviral Res. 2014;107:102–109. doi: 10.1016/j.antiviral.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Halfmann P, Neumann G, Kawaoka Y. The Ebolavirus VP24 protein blocks phosphorylation of p38 mitogen-activated protein kinase. J Infect Dis. 2011;204(Suppl 3):S953–S956. doi: 10.1093/infdis/jir325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Noda T, Watanabe S, Sagara H, Kawaoka Y. Mapping of the VP40-binding regions of the nucleoprotein of Ebola virus. J Virol. 2007;81(7):3554–3562. doi: 10.1128/JVI.02183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Modrof J, Moritz C, Kolesnikova L, Konakova T, Hartlieb B, Randolf A, et al. Phosphorylation of Marburg virus VP30 at serines 40 and 42 is critical for its interaction with NP inclusions. Virology. 2001;287(1):171–182. doi: 10.1006/viro.2001.1027. [DOI] [PubMed] [Google Scholar]
- 79.Timani KA, Sun D, Sun M, Keim C, Lin Y, Schmitt PT, et al. A single amino acid residue change in the P protein of parainfluenza virus 5 elevates viral gene expression. J Virol. 2008;82(18):9123–9133. doi: 10.1128/jvi.00289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun D, Luthra P, Li Z, He B. PLK1 down-regulates parainfluenza virus 5 gene expression. PLoS Pathog. 2009;5(7):e1000525. doi: 10.1371/journal.ppat. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sun D, Luthra P, Xu P, Yoon H, He B. Identification of a phosphorylation site within the P protein important for mRNA transcription and growth of parainfluenza virus 5. J Virol. 2011;85(16):8376–8385. doi: 10.1128/jvi.00618-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pattnaik AK, Hwang L, Li T, Englund N, Mathur M, Das T, et al. Phosphorylation within the amino-terminal acidic domain I of the phosphoprotein of vesicular stomatitis virus is required for transcription but not for replication. J Virol. 1997;71(11):8167–8175. doi: 10.1128/JVI.71.11.8167-8175.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lubaki NM, Ilinykh P, Pietzsch C, Tigabu B, Freiberg AN, Koup RA, et al. The lack of maturation of Ebola virus-infected dendritic cells results from the cooperative effect of at least two viral domains. J Virol. 2013;87(13):7471–7485. doi: 10.1128/jvi.03316-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neumann G, Feldmann H, Watanabe S, Lukashevich I, Kawaoka Y. Reverse genetics demonstrates that proteolytic processing of the Ebola virus glycoprotein is not essential for replication in cell culture. J Virol. 2002;76(1):406–410. doi: 10.1128/JVI.76.1.406-410.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Watanabe S, Noda T, Halfmann P, Jasenosky L, Kawaoka Y. Ebola virus (EBOV) VP24 inhibits transcription and replication of the EBOV genome. J Infect Dis. 2007;196(Suppl 2):S284–S290. doi: 10.1086/520582. [DOI] [PubMed] [Google Scholar]
- 86.Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, et al. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J Virol Methods. 2011;173(1):1–6. doi: 10.1016/j.jviromet.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Afonina I, Ankoudinova I, Mills A, Lokhov S, Huynh P, Mahoney W. Primers with 5′ flaps improve real-time PCR. Biotechniques. 2007;43(6):770–774. doi: 10.2144/000112631. [DOI] [PubMed] [Google Scholar]
- 88.Binz PA, Barkovich R, Beavis RC, Creasy D, Horn DM, Julian RK, Jr, et al. Guidelines for reporting the use of mass spectrometry informatics in proteomics. Nat Biotechnol. 2008;26(8):862. doi: 10.1038/nbt0808-862. [DOI] [PubMed] [Google Scholar]
- 89.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ginalski K, Rychlewski L. Detection of reliable and unexpected protein fold predictions using 3D-Jury. Nucleic Acids Res. 2003;31(13):3291–3292. doi: 10.1093/nar/gkg503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963;7:95–99. doi: 10.1016/S0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- 93.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 94.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11(8):3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55(3):379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.