

Abstract
Genes that are mutated in Autism Spectrum Disorders can be classified broadly as either synaptic or developmental. But what if this is a false distinction? A recent spate of publications has provided evidence for developmental mechanisms that rely on neural activity for proper cortical development. Conversely, a growing body of evidence indicates a role for developmental mechanisms, particularly chromatin remodeling, during learning, in response to neural activity. Here, we review these recent publications and propose a model in which genes that confer ASD risk operate in signal transduction networks critical for both cortical development and synaptic homeostasis.
Keywords: Autism, Signal transduction, Synapse, Neural activity, Chromatin, Cortical development, Sensory integration, E/I balance, Neuron migration, Genetics, Genomics, Precision medicine
ASD defines a group of individually rare disorders
The past two decades have seen significant advances in our understanding of conditions falling under the label Autism Spectrum Disorder (ASD) (reviewed in [1]). According to the DSM-V, a diagnosis of ASD must meet two main clinical criteria -- social communication deficits and repetitive behaviors or interests -- and these symptoms must be present early in life and interfere significantly with social and/or occupational relationships [2]. In addition, individuals with ASD also often suffer from one or more comorbidities, which may include intellectual disability (ID), epilepsy, motor and sensory abnormalities, and/or attention-deficit/hyperactivity disorder (reviewed in [3]). This great phenotypic heterogeneity is reflected in the corresponding genetic heterogeneity discovered in the past decade (Reviewed in [4]). Critically, any given ASD risk gene explains only a small percentage (<1%) of all ASD cases, and there is no identifiable genetic cause for >50% of individual ASD cases; the remaining cases could represent undiscovered genes, or the underlying risk could involve gene-environment interactions, non-coding mutation(s), and/or the collective impact of many common variants rather than the main effect of a single likely-gene-disrupting mutation [5–7]. Diverse environmental factors, such as maternal immune activation [8], maternal autoantibodies [9], or certain chemical exposures, including valproic acid and pesticides (reviewed in [6]), also contribute to risk. Given the relative ease, feasibility, and accessibility of exome and whole genome sequencing, the identification of genes and genomic regions hypothesized to contribute to ASD risk will likely continue to rise.
Collectively, this phenotypic and genetic diversity suggests that the term ‘ASD’ defines a group of individually rare disorders with collectively common phenotypes. Despite the high prevalence of ASD -- as many as 1 in 59 children aged 8 are diagnosed [10] -- each individual gene typically accounts for a vanishingly small number of total patients. ‘ASD’ is therefore an umbrella term much like ‘cancer’ or ‘epilepsy’, describing the clinical behavioral phenotype of many rare disorders with different etiologies.
Common biological pathways in ASD
Different genetic or environmental insults may converge on a common biological pathway to cause the behavioral outcomes that fall under the umbrella term ‘ASD’. This concept first arose out of genetic linkage studies that identified multiple genomic regions of interest that failed to cross-replicate at the level of genome-wide significance in different patient cohorts [11]. The large-scale gene identification studies that followed noted common features of ASD-implicated genes: neuron-enriched, particularly at the synapse, or involved in transcription/chromatin remodeling [12–15]. Transcriptomic analyses pointed to modules of co-expressed genes associated with neuron function and immune activation; the former correlated with genetic variants while the latter did not, suggesting a cause-effect relationship [16]. Interestingly, many neuronally expressed genes identified by genetic linkage, and gene expression modules identified by transcriptomics, are activated most strongly during midgestation [17]. This observation, combined with increased ASD risk from mid-gestational environmental exposures, histopathological evidence for disrupted brain development [18], and mutations in a large number of genes sharing the ontology term ‘development,’ led to a widely accepted theory of ASD as a neurodevelopmental disorder. According to this hypothesis, diverse genetic and environmental risk factors converge on biological processes occurring at a critical point during mid-gestation, when forebrain progenitor cells are differentiating into cortical neurons and migrating to their final positions in the cortical plate. Cytoarchitectural and circuitry deficits resulting from perturbed mid-gestational development would therefore set the brain onto a divergent developmental trajectory, in which deficits compound over time to produce the behavioral characteristics of ASD.
This development-focused hypothesis, however, does not adequately account for the fact that a substantial number of ASD genes are expressed at the excitatory synapse throughout life and are involved in critical synaptic processes such as long term potentiation (LTP), long term depression (LTD), and homeostatic plasticity [19]. In fact, many of the most studied ASD-linked genes encode synaptic adhesion molecules (Neuroligins, Neurexins), neurotransmitter receptors (GluN2B), scaffolding proteins (Shanks), and regulators of local dendritic protein synthesis (FMRP) that play critical roles in activity-dependent signal transduction [20]. According to the “Autism as a Signalopathy” hypothesis, homeostatic signaling that maintains the synapse over time and allows for functional plasticity is altered in ASD (reviewed in [21]). Convergence among risk genes could therefore arise through multiple ways of disrupting a specific homeostatic biochemical process, such as synaptic calcium regulation or local protein synthesis.
Here, we propose that these two conceptual models might not be separate entities at all. At the core of our argument is a growing body of evidence showing that mid-gestational development and lifelong activity-dependent synaptic homeostasis are largely controlled by the same sets of critical genes. In fact, recent work has highlighted crucial and unexpected roles for excitatory glutamatergic synapse signaling in specific cortical developmental processes, including radial migration of progenitor cells, wiring of sensory maps, and determination of inhibitory neuron number. Conversely, studies of signal transduction downstream of synaptic activity have revealed critical roles for genes involved in chromatin modification, transcription, translation, and splicing -- processes traditionally labeled ‘developmental’. In the following sections, we review these studies in the context of ASD with the goal of establishing a unified model that incorporates both developmental and synaptic signaling genes into a single overarching paradigm.
Glutamate transmission is required for cortical migration
Excitatory neurons in the cortex develop in a stereotypical inside-out pattern: newly generated neurons migrate away from progenitor cells in the ventricular zone and pass through established cells on their way to their final positions in the cortical plate. These multipolar migrating neurons become bipolar near the subplate, where they associate with the radial glial scaffold and accelerate--an important developmental transition (reviewed in [22]). A recent study found that migrating multipolar neurons form transient glutamatergic synapses with presynaptic subplate neurons, and NMDAR-mediated synaptic transmission is required for the migrating neurons to become bipolar and accelerate [23]. Moreover, knockdown of Dlg4, which encodes the synaptic scaffolding protein PSD95, knockout of Grin1, which encodes an NMDA glutamate receptor subunit, or simply blocking the activity of subplate neurons themselves, impairs migration. Thus, in order to initiate radial glia-guided migration, young cortical neurons must be capable of excitatory synaptic transmission just after exiting the cell cycle (Figure 1A,B).
Figure 1: Glutamatergic neurotransmission is required for cortical migration.

A) Cortical excitatory neurons arise from neural progenitor cells (NPCs) and exit the ventricular zone as migrating multipolar neurons. As they cross the subplate, they change into a bipolar morphology for their final migration into the cortical plate. B) This multipolar-to-bipolar transition is dependent on NMDAR- and PSD95-dependent excitatory synapses forming between subplate neurons and migrating multipolar cells. C) In Fragile X, and potentially other models of ASD, disrupted glutamatergic signal transduction pathways lead to an accumulation of multipolar neurons below the subplate, and delayed cortical migration. D) In the early postnatal period, BrdU or electroporation-labeling studies in mice revealed incomplete migration compared to age-matched controls (E14: embryonic day 14). E) However, by adulthood, these differences normalize, leaving no cytoarchitectural trace of developmental delay.
What might happen to migration if synaptic signal transduction pathways were perturbed by an ASD-linked mutation? Intriguingly, in the mouse model of Fragile X Syndrome (Fmr1−/y), migrating neurons accumulate below the subplate and exhibit delayed multipolar-to-bipolar transition (Figure 1C) [24]. Importantly, there are no gross abnormalities in cortical layering postnatally, suggesting that delays in cortical migration resolve over time. Similarly, in maternal immune activation (MIA) models (reviewed in [8]), the migration of later-born cortical cells also appears delayed [25], without long-lasting changes in cortical layering [26], suggesting that early lamination defects in the developing ASD brain may be undetectable in adulthood. Delayed migration due to ASD risk factors (reviewed in [27]) could therefore affect neuron numbers in early development, while leaving no long-term signature in cortical layering (Figure 1 D,E). The downstream effects of this delayed migration on cellular or circuit-level development, however, may persist beyond the time at which the excitatory architecture normalizes, as we discuss in the next section.
Excitatory signaling controls inhibitory cell numbers and E/I balance
Following their radial migration, excitatory neurons form circuits that include the appropriate numbers of inhibitory neurons, which migrate into the cortex tangentially, primarily from their origin in the medial ganglionic eminence. Recent work in mice found that between postnatal days 5 and 10, interneurons undergo waves of programmed cell death (apoptosis), which are regulated by PI3K/AKT/mTOR and its upstream regulator PTEN [28], a pathway commonly involved in ASD pathogenesis (reviewed in [29]). Importantly, these waves of apoptosis are controlled by the local activity of excitatory neurons. The resulting cortical circuits therefore integrate the appropriate numbers of inhibitory neurons relative to the number of excitatory neurons present between postnatal day (P) 5 and P10 (Figure 2 A,B). Modulating activity levels during this stage adds support for the role of glutamatergic transmission in establishing the appropriate ratio of excitatory to inhibitory cells: Increasing the activity of excitatory neurons between P5 and P8 with the excitatory DREADD hM3Dq led to increased numbers of interneurons on P21, while decreasing activity with the DREADD hM4Di led to reduced numbers of interneurons on P21 [28]. Thus, synaptic transmission between P5 and P10 determines the final numbers of interneurons and sets the foundation for excitatory/inhibitory (E/I) balance in the cortex.
Figure 2: Glutamatergic neurotransmission is required for GABA neuron survival.
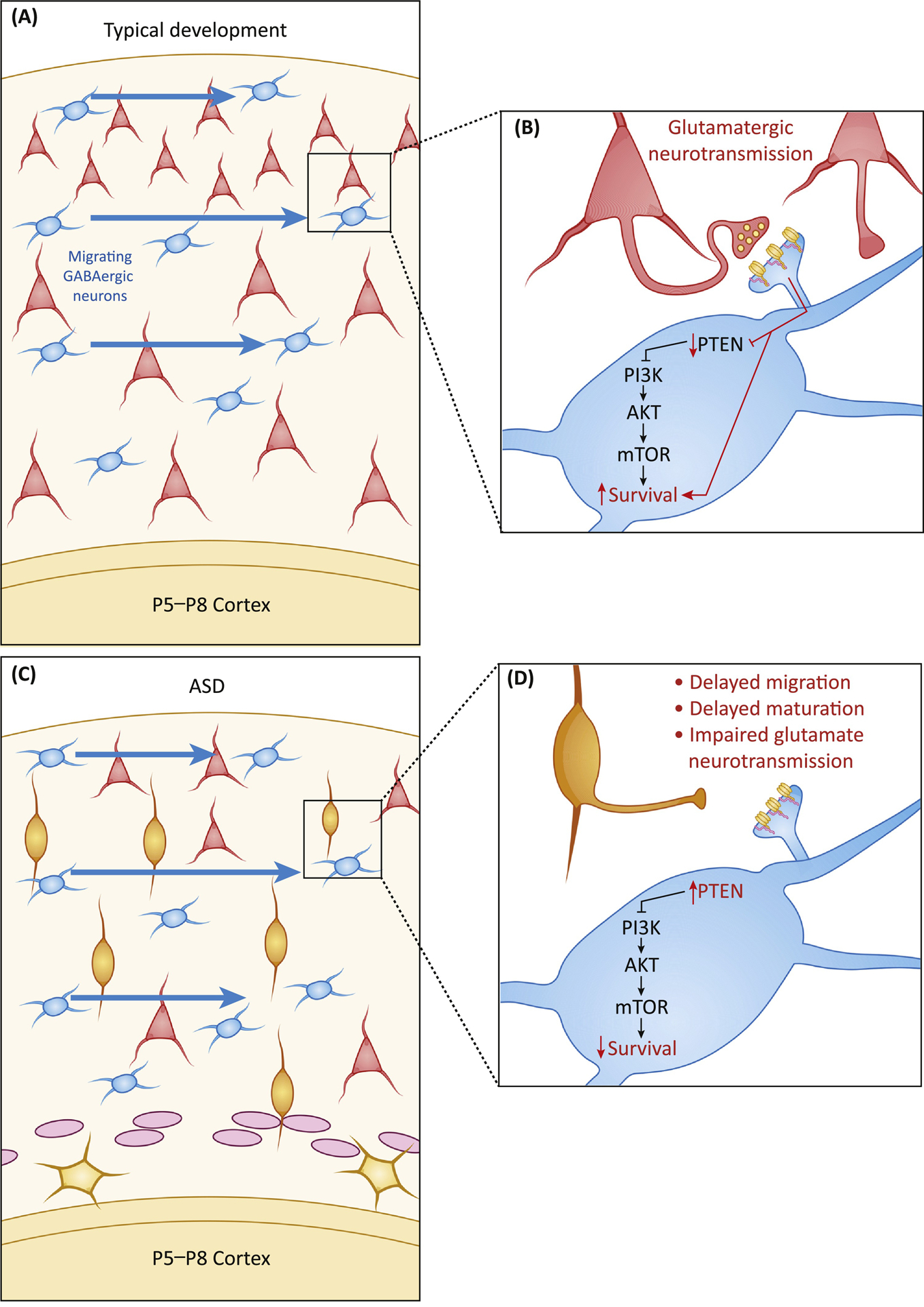
A) GABAergic neurons migrate tangentially, from their origin in the medial ganglionic eminence to their final locations in the cortex, arriving in the early postnatal period (illustration timing refers to mice; P5–8: postnatal days 5 to 8). B) Upon arrival, GABAergic neurons must form excitatory glutamatergic synapses with local cortical neurons. Transmission through these synapses promotes survival in a PTEN-dependent manner. C) If the timing of excitatory neuron migration is disrupted by an ASD-linked risk factor, local cortical neurons may not have yet reached their final position, or they may be too immature to form functional synapses. D) If local pyramidal cells are unable to form synapses with GABAergic neurons, PTEN activity increases, leading to reduced mTOR pathway activation and increased apoptosis of GABAergic neurons.
Disruptions to E/I balance are thought to be a central feature of ASD (reviewed in [30]). In the Fmr1−/y mouse model of Fragile X, parvalbumin (PV+) interneuron development in the auditory cortex is delayed, with only 50% of the expected number of PV+ neurons present on P14, but normal numbers by P21 [31]. Conversely, electrophysiological responses to auditory stimuli are normal on P14, but enhanced on P21, reflecting a lasting consequence of early E/I disruption despite normalization of cell numbers [31]. Similarly, in mice lacking the PDZ domain of Shank3 (Shank3B−/−), early hyperexcitability in cortico-striatal projection neurons at P14 leads to premature circuit maturation and hypoexcitability in the same circuits by P60 [32]. This early cortico-striatal hyperexcitability is driven by a more general cortical hyperexcitability, measurable as early as P0, which increases cortico-striatal connectivity. In fact, decreasing cortical layer 5 activity in Shank3B−/− mice using an inhibitory DREADD between P10 and P13 normalizes cortico-striatal hyperexcitability [32].
Lastly, in an MIA mouse model, patches of disorganization were identified in the upper cortical layers of adults that lack expression of the layer-enriched markers SATB2 and TBR1 [33]. These areas are reminiscent of the patches of abnormal laminar cytoarchitecture observed in postmortem ASD human brains characterized by prominent abnormal expression of excitatory cortical neuron markers and milder, inconsistently abnormal expression of select autism genes and interneuron markers, including PV [18]. Similarly, within the MIA model patches, the number of PV+ neurons is reduced, which causes local cortical circuit hyperexcitability. Increasing the activity of small cortical patches using optogenetics in wildtype mice demonstrated that this local cortical hyperexcitability is both necessary and sufficient to produce the behavioral abnormalities characteristic of the MIA model [33].
Collectively, the forementioned studies demonstrate that early disruptions in cortical activity can have long-lasting secondary effects on circuit development and function (Figure 2 C–E). These secondary effects often involve the early incorporation of inhibitory interneurons, a critical step in establishing the circuitry for more complex cortical functions, such as multisensory integration (reviewed in [34]). For example, in the mouse insular cortex, coincident auditory and tactile stimulation are integrated, producing super-additive responses in the region where the auditory and tactile response fields overlap [35]. This multisensory integration (MSI) is dependent on the maturation and activity of sufficient numbers of interneurons -- particularly PV+ interneurons -- and (in mice) the proper pruning of multisensory circuits during the third and fourth postnatal weeks. In four different mouse models of autism (BTBR/acallosal, Shank3B−/−, Mecp2-null, Gad65-null), a lack of sufficient inhibition during early postnatal development leads to impaired MSI [35]. Remarkably, these four ASD models arrive at a convergent point in circuit dysfunction by presumably vastly different developmental mechanisms: the inbred BTBR strain lacks a corpus callosum and harbors many homozygous mutations; the Gad65-null mouse is deficient in GABA (inhibitory) production; scaffolding of glutamate receptors is impaired in the Shank3B−/− mouse, and the Mecp2-null mouse lacks an important nucleic acid binding protein. However, in each model, abnormalities in cortical inhibitory neurotransmission present by P15 led to disrupted MSI. By this time in development, an earlier critical period has passed in which interactions between excitatory and inhibitory neurons is required to establish E/I balance and lay the foundations for proper sensory integration.
What about the sensory inputs themselves? One of the more intriguing findings to appear in the past several years is that perturbation of ASD-linked genes exclusively in the peripheral nervous system of mice is sufficient to drive at least some ASD-like behaviors. Using specific CRE drivers, Orefiche et al. [36] knocked out either Mecp2 or Gabrb3 exclusively in sensory neurons, causing abnormal social behavior. Remarkably, re-expression of Mecp2 exclusively in developing sensory neurons was sufficient to mitigate social behavior impairments in Mecp2−/y mice. Thus, abnormal sensory input during development is another potential mechanism for E/I imbalance and disrupted sensory integration.
Abnormal development produces ongoing deficits in glutamate tone
Abnormal glutamatergic tone has been observed consistently in ASD patients (e.g. [37], reviewed in [38]) and mouse models (reviewed in [39,40]). All three major classes of glutamate receptors—AMPA, NMDA and mGluR—have been implicated, and each receptor class shows complex developmental interactions that lead to model-, brain region- and age-specific changes in receptor tone.
AMPA-mediated neurotransmission has been implicated widely in ASD mouse models, including CNTNAP2 [41], Shank3 [42], Neuroligin3-R704C-knockin [43], 16p11.2 deletion [44] and Ube3a-overexpressing (a model of 15q duplication) [45]. Interestingly, both up- and downregulation of AMPA receptor (AMPAR) tone negatively impacts social behavior: bidirectional deficits have been reported in different models, and acute treatment of adolescent (P28–32) wildtype mice with agonists or antagonists of AMPARs results in impaired social behavior [41]. However, disruption of AMPAR-mediated neurotransmission can have a variety of downstream effects, making it difficult to pinpoint which receptors are causative. For example, knockout of the glutamate receptor interacting proteins (GRIP) 1/2 specifically in cerebellar purkinje cells, which disrupts trafficking of AMPARs, causes ASD-like behaviors in mice but also increases mGluR5 levels and signaling intermediates downstream of mGluR5 [46].
Bidirectional NMDA receptor (NMDAR) dysfunction has been implicated in many ASD models (reviewed in [39]), including Shank2 [47], Shank3 [48,49], CDKL5 [50], and Fragile X [51]. Moreover, both NMDAR2A [52] and NMDAR2B [53] are ASD-linked genes. Intriguingly, in the Shank2ΔExon 6−7 ASD model mouse, NMDAR hyperfunction is observed in both the cortex and hippocampus pre-weaning, which rapidly shifts to NMDAR hypofunction after weaning. Chronic treatment with an NMDAR antagonist between P7 and P21 prevents both NMDAR hypofunction and social behavior deficits in adults, demonstrating that pre-weaning NMDAR tone can induce long-term changes in adult function [54]. These data are reminiscent of the developmental switch between hyperactive (P14) and hypo-active (P60) cortico-striatal circuits in Shank3B−/− mice ([32], discussed above), but seem to involve cell-intrinsic receptor-mediated, rather than circuit-based, mechanisms.
Metabotropic glutamate neurotransmission has received considerable attention, originally due to its hypothesized role in Fragile X syndrome [55], but more recently due to its involvement in a variety of ASD models, including Shank3 [56], TSC1/2 [57], PTEN [58], 16p11.2 deletion [59] and GPRASP2 [60]. As with AMPA and NMDA receptors, mGluRs interact with other receptor systems over developmental time to modulate both synaptic plasticity [61] and cortical development. For example, knockout of mGluR5 specifically in PV+ fast spiking interneurons leads to reduced cell numbers and decreased inhibitory currents in adult mice, which correlates with abnormal behaviors, demonstrating that mGluRs are critical for setting cortical E/I balance [62].
Similarly, mGluRs play a crucial role in the wiring of cortical sensory fields. The establishment of functional cortical areas in the mouse begins during late embryogenesis with the emergence of thalamic activity (reviewed in [63]). A recent study demonstrated that patterned thalamocortical activity from as early as E17.5 is required for the initial wiring of the topographic map in mouse primary somatosensory cortex (S1) [64]. In S1, shifting the pattern of thalamic spontaneous activity from synchronized waves to asynchronous activity, using thalamus-specific expression of an inwardly rectifying potassium channel (ThKir), disrupted the formation of the stereotyped shape and distribution of whisker barrel columns in S1. Instead, the area of cortical activation expands, and barrel-specific fields spread out and overlap with each other, such that stimulation of multiple whiskers results in activation of the same neuron populations. Specifically, synaptic signaling through mGluR5, whose mRNA expression is ~3-fold higher in the barrel cortex of ThKir animals compared with controls, is required for this receptive field spreading: Acute blockade of mGluR5 in adult ThKir mice restricts the area of cortical activation to the immediate area of thalamic innervation, more closely resembling a typical barrel field [64]. These results indicate that mGluR tone is established by excitatory activity during development and is critical for ongoing maintenance of proper cortico-thalamic transmission in adulthood.
Collectively, these data demonstrate that glutamatergic tone during development is critical for establishing balanced excitatory and inhibitory integration in primary and multisensory cortical areas, which in turn affects ASD-relevant behaviors in adult animals.
Neuronal activity reactivates developmental pathways
While synaptic genes are critical during development, genes typically associated with neural development are likewise critical for synaptic processes during learning and memory (Figure 3, Key Figure). In hippocampal neurons, for example, following fear conditioning, hundreds of regions of chromatin become accessible, or open to binding by transcriptional regulators. A recent high-throughput sequencing study found that, of the 2365 regions that changed accessibility, a significant portion overlap bivalent promoters that are active during development [65]. These data suggest a re-activation of developmental genes following a learning event, and reignite an older theory that posited that, in Aplysia, processes controlling development and learning share striking mechanistic similarities [66].
Figure 3, Key Figure: ASD-linked ‘synaptic’ and ‘developmental’ proteins involved in synaptic homeostasis.

This cartoon view of a neuron, with an expanded synaptic spine to show proteins, illustrates some of the many ASD-linked proteins involved in synaptic homeostasis and plasticity. At the synapse, neurotransmitter receptors, tethered in place by synaptic adhesion molecules and intracellular scaffolding proteins, transmit molecular cues to kinases and other effectors. Signal transduction cascades carry these signals to the nucleus, where histone modifying enzymes and helicases regulate chromatin accessibility. RNA is then spliced and further processed into export granules, which are transported into the dendrite for local protein synthesis. Disruption of any protein in this complex and spatially distributed process could alter the overall homeostatic balance of the synapse. ASD-linked genes are involved at each step.
Gene/protein acronyms, ordered according to the two “key” insets: DLGs: Disks large homologs; AMPAR: AMPA-selective glutamate receptor; NMDAR: N-methyl-D-aspartate receptor; mGluRs: metabotropic glutamate receptors; CaV42.1: Voltage-gated calcium channel 2.1; Shanks: SH3 and multiple ankyrin repeat domains proteins; SynGAP: Synaptic Ras GTPase-activating protein; CaMKII: Calcium/calmodulin-dependent protein kinase type II; PI3K: Phosphatidylinositol 4,5-bisphosphate 3-kinase; PTEN: Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase; FMR1: Fragile X mental retardation protein 1; EIF4: Eukaryotic translation initiation factor 4; CHD8: Chromodomain-helicase-DNA-binding protein 8; HDACs: Histone deacetylases; DNAMT: DNA methyltransferase.
Indeed, chromatin modifiers that have long been associated with development are now widely acknowledged to play essential roles in cognition throughout life [67]. Neuron-specific overexpression of the histone deacetylase HDAC2, which occupies promoters and represses expression of genes implicated in memory, reduces spine density, synaptic plasticity, and memory in mice. Conversely, chronic treatment with HDAC inhibitors or deletion of Hdac2 improves memory function [68]. Moreover, in postmortem human ASD brains, both DNA acetylation [69] and methylation [70] are strikingly different from controls but show convergence among different types of idiopathic and genetically defined ASDs.
Many ASD genes involved in gene regulation are members of the ATPase-dependent chromatin remodeling complex, which uses the energy from ATP hydrolysis to move nucleosomes (see Table 1). The BRG1/BRM-associated (BAF) complex influences DNA methylation patterns by regulating the expression of DNA methylases and their recruitment to chromatin. As a group, these genes have been associated with several developmental syndromes characterized as “BAFopathies” (or “SWI/SNF-related intellectual disability syndromes”) (Reviewed in [71]). In adult animals, the BAF complex is directly involved in memory consolidation in the hippocampus [72], nucleus accumbens [73], and amygdala [74], indicating an important role in synaptic plasticity.
Table 1:
Features of ASD chromatin modification genes with dual functions in developing and mature neurons.
| CHD8 | CHD7 | CHD3 | CHD4 | ARID1B | SMARCA4 / BRG1 | SMARCA2 / BRM | ADNP | |
|---|---|---|---|---|---|---|---|---|
| Syndromic | X | X | X | X | X | X | X | X |
| ASD | X | X | X | X | X | X | X | X |
|
Intellectual Disability |
X | X | X | X | X | X | X | X |
| Distinct facial features | X | X | X | X | X | X | X | X |
| Sleep problems | X | X | NR | NR | NR | NR | NR | X |
| Seizures | X | X | NR | NR | NR | X | X | X |
| GIa complications | X | X | NR | NR | X | NR | X | NR |
| Mouse cortical neuron migration, lamination, interneuron # defect | X[94] | NRb | X[95] | X[95] | X[96] | Early lethality[97] | X[98] | Early lethality[99] |
| Molecular biology in the mature neuron | Activates BAFc Complex genes [75,100] |
CoIPsd with topoisomerase important for expression of extremely long ASD genes [101] | In complex with CHD4 [95] | Represses activity- dependent gene expression [78,102] | Mediates interaction between HATse and promoters, regulates Grin2b, cFos, Arc, Pvalb, Dlg4, Slc17a7 [96,103,104] | Regulates activity- dependent expression of neuronal genes, such as Grin2b [105] | Regulates dendritic spine number [106] | Not confined to nucleus; controls PSD95 accumulation in dendritic spines [107] |
GI: Gastrointestinal
NR: Not reported
BAF: BRG1-associated SWI/SNF
CoIPs: Co-immunoprecipitates
HATs: Histone acetyltransferases; X indicates that the feature has been observed in cases in which the gene is mutated.
Another family of ATPase-dependent transcriptional regulators critical for neural development and function is the chromodomain helicase DNA-binding family (CHD) (reviewed in [75]). CHD8 is a high-confidence syndromic ASD gene that is significantly co-expressed throughout cortical development with other ASD risk genes, many of which are known to regulate transcription and/or modify chromatin, including the histone methyltransferase SETD2, itself a probable ASD gene [50]. Knockdown of a Drosophila CHD homolog, kismet, causes morphological abnormalities and reduced glutamatergic synapse function at the neuromuscular junction. Treatment with HDAC inhibitors does not rescue these morphological deficits, but does normalize excitatory neurotransmission, suggesting an ongoing pathogenic mechanism involving chromatin remodeling and synaptic function [77]. Similarly, CHD4 [78] and CHD1 [79] each play important roles in memory consolidation.
In fact, even in ASD models with primarily synaptic mutations, chromatin remodeling can play an important role. In the Shank3 exon 21 deletion (Shank3+/ΔC) mouse model, Hdac2 is upregulated in the frontal cortex, leading to reduced H3K9 acetylation. Acute treatment with the HDAC inhibitor romidepsin normalizes acetylation and social behavior for ~3 weeks [80]. Conversely, in the same model, histone H3K9 methylation is increased, and treatment with an inhibitor of the histone methyltransferases EHMT1/2 rescues social deficits and normalizes NMDAR-mediated synaptic function [81]. In addition to highlighting the potential of drugs targeting chromatin modifiers as experimental therapeutics for ASD, these studies demonstrate the interconnectedness of synaptic activity and pathogenic mechanisms involving chromatin modification.
In fact, might it be possible that the association between ASD-linked chromatin modifiers and the gene ontology term ‘neurodevelopment’ is a consequence of synchronized neural activity during development, rather than something intrinsic to the developmental process itself? In line with this idea, RNA co-expression studies identify ‘neurodevelopmental’ modules by the coordinated expression of hundreds of individual mRNAs using correlation network analysis [82]. Development is the only time during which activity-dependent processes are widely synchronized in the brain, which broadly engages activity-dependent programs. By contrast, in the adult, only a small percentage of synapses on a small percentage of neurons are active at any given time. Hayashi-Takagi et al. estimated that, following rotarod learning, only ~2% of spines in ~16% of neurons in upper layers of the motor cortex express a transgene controlled by the activity-dependent Arc promoter, suggesting sparse expression of activity-dependent genes, even following an intense learning paradigm [83]. In the context of ASD, such activity-dependent genes may serve similar functions in development and learning. For example, the activity-dependent expression of more than 200 genes is altered in neurons differentiated from induced pluripotent stem cells derived from fibroblasts of Timothy Syndrome (TS) cases, a result that was linked to sustained increased intracellular calcium after global membrane depolarization [84]. In the developing mouse cortex, manipulating exon usage of CACNA1C, the gene mutated in TS and which encodes the L-type calcium channel Cav1.2, causes a calcium-dependent shift in cell fate specification [85]. Thus, it is possible that many ‘developmental’ genes are not strictly developmental per se, but rather activity-dependent genes that cross a certain signal-to-noise threshold only during widely synchronized synaptic activity characteristic of embryonic and early postnatal development.
Implications for subgrouping and treatment beyond developmental critical periods
As we approach a decade since the development of the first ASD-specific animal models, the collective data point to several nodes of convergence in developmental and synaptic processes. Some of these processes will be clinically useful, others mechanistically illuminating but likely of limited clinical utility. Given that most children present in the clinic long after the closure of the developmental critical periods that determine E/I ratios and cortical patterning, it is likely too late to alter these early developmental setpoints. It may be possible, however, to normalize ongoing abnormalities in synaptic signaling and see behavioral improvements despite abnormal circuitry. Indeed, many drug rescue or gene restoration experiments in mouse models have produced promising behavioral results for a subset of ASD-like behaviors (see Box 1).
Box 1: Adult restoration of function in mouse models of ASD.
The two hypothesized functions of ASD genes, in early neural development and in ongoing synaptic signaling, clearly diverge at one critical area: that of clinical treatment options. In the developmental model, correcting a genetic deficit after a critical period of development would not be able to alter behavioral phenotypes. By contrast, correcting ongoing deficits in synaptic signaling should improve outcomes throughout life. For children presenting in the clinic in early-to-mid childhood, the results of the mouse restoration-of-function experiments discussed below may predict the feasibility of drug treatment strategies.
In fact, adult mouse restoration of function is sufficient to rescue ASD-like behaviors in multiple models of ASD, including Mecp2 duplication [108], Shank3 deletion [109], Ube3a loss-of-function/Angelman Syndrome [110], Mecp2 loss-of-function/Rett syndrome [111], and Syngap1 haploinsufficiency [112]. In addition, several drug treatments have been effective in mouse models (reviewed in [113]), including mGluR5 antagonism in Fragile X and BTBR [114,115], rapamycin treatment in tuberous sclerosis [116], HDAC inhibition in Shank3+/ΔC [80], and manipulations of gut microbiota [117,118]. Remarkably, in most of these studies, the animal’s interest in social interactions improved, suggesting that the key diagnostic behavior of ASD is acutely modifiable in the adult rodent brain.
Three major caveats with regards to treatment studies in animal models should be noted. First, in almost all cases, not all abnormal behaviors are improved. Typically, social behavior is modifiable, but repetitive behaviors, such as over-grooming, are not, suggesting that only some behavioral circuits remain plastic or amenable to the treatment strategies in these studies. Second, most of the genes ‘rescued’ fall into the category of ‘synaptic homeostasis’ rather than ‘chromatin modification.’ It is unclear yet whether abnormal behaviors can be modified if rescue experiments were attempted for chromatin binding genes such as Chd8, Auts2, or Tbr1, although results from rescue experiments in Drosophila for Chd7/kismet [77] and in mice for Brg1-associated BAF53b [72] have been promising. Third, in human clinical trials, adolescent and adult rescue experiments using mGluR5 antagonists in Fragile X [119,120] or rapamycin analogs in tuberous sclerosis [121] failed, despite being effective in mouse models. While several reasons have been offered for these failures [86], including rapid development of tolerance, trial design flaws, and the relatively late developmental stage at the time of treatment, they nevertheless cast doubt on whether adult restoration of behavioral functions will be possible in humans, as observed in animal models of these disorders.
The diagnostic category ‘autism’, however, is biologically too broad to expect wide success from one single drug or therapy. Indeed, no drugs targeting core ASD behaviors have passed early-phase clinical trials (reviewed in [86]). Conversely, most individual ASD mutations are likely too rare to support commercially viable drug development. As an alternative, perhaps the hundreds of ASD-linked mutations could be placed into a few subgroups that share enough biological similarity to allow for common intervention strategies, customized for each subtype, making drug development more feasible. Yet classifying ASD subtypes suffers from the dual challenges of ‘one-to-many’ and ‘many-to-one’: different mutations in a given gene can be associated with different mechanistic outcomes (e.g., Shank3 [49] and Neuroligin-3 [87]), while mutations in many different genes can lead to similar behavioral phenotypes [4]. So, how can such groups be identified?
While effective solutions to this question remain elusive, at least there is currently some clarity on strategies that do not seem to work effectively. Subgrouping based on behavioral presentation, EEG patterns, or MRIs has not yielded actionable treatment groups, despite 20+ years of effort [88]. The most successful grouping strategy to-date involves monogenic ASD-associated syndromes, like Fragile X, Timothy, and Rett syndromes -- cases that meet the diagnostic criteria for ASD and show additional categorical evidence, such as distinctive dysmorphology [4] [89]. More recently, the identification of rare mutations in ASD patients traditionally labeled non-syndromic, including CHD8 [76], ARID1B [90], GRIN2B [91,92], and TRIO [93], highlights groups of individuals with common features (the gene-first approach). Given sufficient time, research on these rare genetic ASDs could result in specific intervention strategies for each one of them, but each intervention will only apply to a tiny percentage of the ASD population.
Subgrouping based on shared biology may be more amenable to precision medicine. But given the limited utility of the mouse to predict human-specific neuropsychiatric disorders, subtyping using mouse data alone may be unreliable. With the advent of induced pluripotent stem (iPS) cell technology and direct differentiation of fibroblasts or blood cells to neurons in vitro, we now have the opportunity to access primary, patient-derived neurons to address questions of biological similarity, an important step towards answering questions about shared mechanisms of ASD. Given the dual role of many ASD genes in development and synaptic activity as outlined here, one could envision synaptic signal transduction as an accessible means of characterizing, subtyping, and potentially modifying the clinical outcome of children with ASD.
Concluding Remarks
While ASD risk genes have traditionally been classified as acting at the synapse or acting during development, recent work has highlighted the interconnectedness of these two classifications. Synaptic genes such as neurotransmitter receptors or synaptic scaffolds are critical during activity-dependent developmental checkpoints, while chromatin modifiers traditionally labeled ‘developmental’ are crucial to lifelong synaptic homeostasis. Moving forward, models of ASD gene function should incorporate both developmental and homeostatic features while attempting to map the pathways from gene mutation to behavioral outcome. Formidable challenges, however, include: 1) the observation that a single mutation can lead to a wide range of age- and circuit-specific phenotypes, making it difficult to know which deficits are relevant to the disorder, and 2) the fact that many different genetic variants underlie similar phenotypes (see Outstanding Questions). Even with extensive characterization of multiple specific variants, it remains unclear which of the many shared biological processes are ASD-relevant. Perhaps by focusing on the most basic processes of signal transduction downstream of synaptic activity, we can begin to understand the neurodevelopmental roots of E/I imbalance, circuit deficits, and ongoing synaptic abnormalities characteristic of ASD.
Outstanding Questions Box.
How do deficits in excitatory synapse structure or synaptic signal transduction manifest as changes in cortical neuron migration? Birth-dating and live imaging of cortical development in additional mouse models of ASD with mutations in excitatory synapse genes (e.g. Shank3, Grin2b) could identify specific deficits in the kinetics of neuron migration due to synaptic gene mutations.
How do deficits in neuron migration manifest as changes in sensory processing or integration in the cortex? Studies examining multisensory integration in additional models of ASD, or the effects of disrupted E/I balance on the development of cortical or subcortical functions, may be productive avenues for future research.
Is disrupted sensory input a primary driver of the autistic phenotype? Additional animal-model studies of abnormal sensory inputs to a wild-type brain could shed light on this important question.
To what degree are different genetic ASDs alike on a molecular level? Can one use shared molecular signatures to identify ASD subtypes? Studies of individual ASDs should gradually give way to studies examining similar features of multiple genetic ASDs, using both mouse and human iPS cell systems.
Highlights Box.
Autism Spectrum Disorder (ASD) risk genes are often described as either developmental or synaptic. But a growing body of evidence shows substantial overlaps and links between these categories, raising the question of how useful the distinction between the two is, in the context of ASD.
Developmental processes, such as the radial migration of cortical excitatory neurons and apoptosis of inhibitory neurons, depend on intact excitatory signal transduction.
Conversely, genes typically thought of as developmental play important roles in activity-dependent plasticity of excitatory synapses.
By homing in on specific biological processes that are disrupted in different ASD models, one may be able to identify biologically relevant subtypes among heterogeneous patient cohorts. We argue that for much of the ASD patient population, this subgrouping could be more amenable to precision medicine than subgrouping based on behavioral or genetic approaches.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de la Torre-Ubieta L et al. (2016) Advancing the understanding of autism disease mechanisms through genetics. Nat. Med 22, 345–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Psychiatric Association (2013) Neurodevelopmental Disorders In Diagnostic and Statistical Manual of Mental Disorders 0 s American Psychiatric Association [Google Scholar]
- 3.Gillberg C and Fernell E (2014) Autism plus versus autism pure. J. Autism Dev. Disord 44, 3274–3276 [DOI] [PubMed] [Google Scholar]
- 4.Sestan N and State MW (2018) Lost in Translation: Traversing the Complex Path from Genomics to Therapeutics in Autism Spectrum Disorder. Neuron 100, 406–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.An J-Y et al. (2018) Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362, eaat6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hertz-Picciotto I et al. (2018) Understanding environmental contributions to autism: Causal concepts and the state of science. Autism Res. Off. J. Int. Soc. Autism Res 11, 554–586 [DOI] [PubMed] [Google Scholar]
- 7.Turner TN et al. (2016) Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am. J. Hum. Genet 98, 58–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Careaga M et al. (2017) Maternal Immune Activation and Autism Spectrum Disorder: From Rodents to Nonhuman and Human Primates. Biol. Psychiatry 81, 391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones KL and Van de Water J (2019) Maternal autoantibody related autism: mechanisms and pathways. Mol. Psychiatry 24, 252–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christensen DL et al. (2018) Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. Morb. Mortal. Wkly. Rep. Surveill. Summ. Wash. DC 2002 65, 1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geschwind DH (2008) Autism: many genes, common pathways? Cell 135, 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Rubeis S et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilman SR et al. (2011) Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glessner JT et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satterstrom FK et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell DOI: 10.1016/j.cell.2019.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voineagu I et al. (2011) Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willsey AJ et al. (2013) Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoner R et al. (2014) Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med 370, 1209–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peça J et al. (2011) SnapShot: Autism and the synapse. Cell 147, 706, 706.e1 [DOI] [PubMed] [Google Scholar]
- 20.Lautz JD et al. (2018) Synaptic activity induces input-specific rearrangements in a targeted synaptic protein interaction network. J. Neurochem 146, 540–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mullins C et al. (2016) Unifying Views of Autism Spectrum Disorders: A Consideration of Autoregulatory Feedback Loops. Neuron 89, 1131–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohtaka-Maruyama C and Okado H (2015) Molecular Pathways Underlying Projection Neuron Production and Migration during Cerebral Cortical Development. Front. Neurosci 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohtaka-Maruyama C et al. (2018) Synaptic transmission from subplate neurons controls radial migration of neocortical neurons. Science 360, 313–317 [DOI] [PubMed] [Google Scholar]
- 24.La Fata G et al. (2014) FMRP regulates multipolar to bipolar transition affecting neuronal migration and cortical circuitry. Nat. Neurosci 17, 1693–1700 [DOI] [PubMed] [Google Scholar]
- 25.Soumiya H et al. (2011) Prenatal immune challenge compromises the normal course of neurogenesis during development of the mouse cerebral cortex. J. Neurosci. Res 89, 1575–1585 [DOI] [PubMed] [Google Scholar]
- 26.Smith SEP et al. (2012) Maternal immune activation increases neonatal mouse cortex thickness and cell density. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol 7, 529–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiner O et al. (2016) Regulation of neuronal migration, an emerging topic in autism spectrum disorders. J. Neurochem 136, 440–456 [DOI] [PubMed] [Google Scholar]
- 28.Wong FK et al. (2018) Pyramidal cell regulation of interneuron survival sculpts cortical networks. Nature 557, 668–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huber KM et al. (2015) Dysregulation of Mammalian Target of Rapamycin Signaling in Mouse Models of Autism. J. Neurosci. Off. J. Soc. Neurosci 35, 13836–13842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson SB and Valakh V (2015) Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 87, 684–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen TH et al. (2018) Genetic Reduction of Matrix Metalloproteinase-9 Promotes Formation of Perineuronal Nets Around Parvalbumin-Expressing Interneurons and Normalizes Auditory Cortex Responses in Developing Fmr1 Knock-Out Mice. Cereb. Cortex N. Y. N 1991 28, 3951–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peixoto RT et al. (2016) Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(−/−) mice. Nat. Neurosci 19, 716–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin Yim Y et al. (2017) Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 549, 482–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isaacson JS and Scanziani M (2011) How inhibition shapes cortical activity. Neuron 72, 231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gogolla N et al. (2014) Sensory Integration in Mouse Insular Cortex Reflects GABA Circuit Maturation. Neuron 83, 894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orefice LL et al. (2019) Targeting Peripheral Somatosensory Neurons to Improve Tactile-Related Phenotypes in ASD Models. Cell 178, 867–886.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horder J et al. (2018) Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models. Transl. Psychiatry 8, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rojas DC (2014) The role of glutamate and its receptors in autism and the use of glutamate receptor antagonists in treatment. J. Neural Transm. Vienna Austria 1996 121, 891–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee E-J et al. (2015) NMDA receptor dysfunction in autism spectrum disorders. Curr. Opin. Pharmacol 20, 8–13 [DOI] [PubMed] [Google Scholar]
- 40.Moretto E et al. (2018) Glutamatergic synapses in neurodevelopmental disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 84, 328–342 [DOI] [PubMed] [Google Scholar]
- 41.Kim J-W et al. (2019) Pharmacological modulation of AMPA receptor rescues social impairments in animal models of autism. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol 44, 314–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bidinosti M et al. (2016) CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science 351, 1199–1203 [DOI] [PubMed] [Google Scholar]
- 43.Etherton MR et al. (2011) An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. EMBO J. 30, 2908–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Portmann T et al. (2014) Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep. 7, 1077–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith SEP et al. (2011) Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med 3, 103ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mejias R et al. (2019) Purkinje cell-specific Grip1/2 knockout mice show increased repetitive self-grooming and enhanced mGluR5 signaling in cerebellum. Neurobiol. Dis 132, 104602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Won H et al. (2012) Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265 [DOI] [PubMed] [Google Scholar]
- 48.Duffney LJ et al. (2015) Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep. 11, 1400–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y et al. (2016) Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 89, 147–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang S et al. (2019) Altered NMDAR signaling underlies autistic-like features in mouse models of CDKL5 deficiency disorder. Nat. Commun 10, 2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lundbye CJ et al. (2018) Inhibition of GluN2A NMDA receptors ameliorates synaptic plasticity deficits in the Fmr1-/y mouse model. J. Physiol 596, 5017–5031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barnby G et al. (2005) Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am. J. Hum. Genet 76, 950–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Roak BJ et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chung C et al. (2019) Early Correction of N-Methyl-D-Aspartate Receptor Function Improves Autistic-like Social Behaviors in Adult Shank2−/− Mice. Biol. Psychiatry 85, 534–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bear MF et al. (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377 [DOI] [PubMed] [Google Scholar]
- 56.Wang X et al. (2016) Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun 7, 11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Auerbach BD et al. (2011) Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takeuchi K et al. (2013) Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc. Natl. Acad. Sci. U. S. A 110, 4738–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian D et al. (2015) Contribution of mGluR5 to pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat. Neurosci 18, 182–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edfawy M et al. (2019) Abnormal mGluR-mediated synaptic plasticity and autism-like behaviours in Gprasp2 mutant mice. Nat. Commun 10, 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chokshi V et al. (2019) Input-Specific Metaplasticity in the Visual Cortex Requires Homer1a-Mediated mGluR5 Signaling. Neuron DOI: 10.1016/j.neuron.2019.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barnes SA et al. (2015) Disruption of mGluR5 in parvalbumin-positive interneurons induces core features of neurodevelopmental disorders. Mol. Psychiatry 20, 1161–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martini FJ et al. (2018) Impact of thalamocortical input on barrel cortex development. Neuroscience 368, 246–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Antón-Bolaños N et al. (2019) Prenatal activity from thalamic neurons governs the emergence of functional cortical maps in mice. Science 364, 987–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koberstein JN et al. (2018) Learning-dependent chromatin remodeling highlights noncoding regulatory regions linked to autism. Sci. Signal 11, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marcus EA et al. (1994) Chapter 23 A comparison of the mechanistic relationships between development and learning in Aplysia In Progress in Brain Research 100 (Bloom FE, ed), pp. 179–188, Elsevier; [DOI] [PubMed] [Google Scholar]
- 67.Day JJ and Sweatt JD (2011) Epigenetic mechanisms in cognition. Neuron 70, 813–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guan J-S et al. (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun W et al. (2016) Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell 167, 1385–1397.e11 [DOI] [PubMed] [Google Scholar]
- 70.Wong CCY et al. (2019) Genome-wide DNA methylation profiling identifies convergent molecular signatures associated with idiopathic and syndromic autism in post-mortem human brain tissue. Hum. Mol. Genet 28, 2201–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aref-Eshghi E et al. (2018) BAFopathies’ DNA methylation epi-signatures demonstrate diagnostic utility and functional continuum of Coffin–Siris and Nicolaides–Baraitser syndromes. Nat. Commun 9, 4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vogel-Ciernia A et al. (2013) The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat. Neurosci 16, 552–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.White AO et al. (2016) BDNF rescues BAF53b-dependent synaptic plasticity and cocaine-associated memory in the nucleus accumbens. Nat. Commun 7, 11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoo M et al. (2017) BAF53b, a Neuron-Specific Nucleosome Remodeling Factor, Is Induced after Learning and Facilitates Long-Term Memory Consolidation. J. Neurosci 37, 3686–3697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goodman JV and Bonni A (2019) Regulation of neuronal connectivity in the mammalian brain by chromatin remodeling. Curr. Opin. Neurobiol 59, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bernier R et al. (2014) Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Latcheva NK et al. (2018) Epigenetic crosstalk: Pharmacological inhibition of HDACs can rescue defective synaptic morphology and neurotransmission phenotypes associated with loss of the chromatin reader Kismet. Mol. Cell. Neurosci 87, 77–85 [DOI] [PubMed] [Google Scholar]
- 78.Yang Y et al. (2016) Chromatin remodeling inactivates activity genes and regulates neural coding. Science 353, 300–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schoberleitner I et al. (2019) Role for Chromatin Remodeling Factor Chd1 in Learning and Memory. Front. Mol. Neurosci 12, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qin L et al. (2018) Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci 21, 564–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Z-J et al. (2019) Amelioration of autism-like social deficits by targeting histone methyltransferases EHMT1/2 in Shank3-deficient mice. Mol. Psychiatry DOI: 10.1038/s41380-019-0351-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Langfelder P and Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hayashi-Takagi A et al. (2015) Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525, 333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Paşca SP et al. (2011) Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med 17, 1657–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Panagiotakos G et al. (2019) Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy Syndrome. eLife 8, e51037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Berry-Kravis EM et al. (2018) Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat. Rev. Drug Discov 17, 280–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tabuchi K et al. (2007) A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Loth E et al. (2016) Identification and validation of biomarkers for autism spectrum disorders. Nat. Rev. Drug Discov 15, 70–73 [DOI] [PubMed] [Google Scholar]
- 89.Zoghbi HY and Bear MF (2012) Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol 4, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoyer J et al. (2012) Haploinsufficiency of ARID1B, a Member of the SWI/SNF-A Chromatin-Remodeling Complex, Is a Frequent Cause of Intellectual Disability. Am. J. Hum. Genet 90, 565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lemke JR et al. (2014) GRIN2B Mutations in West Syndrome and Intellectual Disability with Focal Epilepsy. Ann. Neurol 75, 147–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Platzer K et al. (2017) GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet 54, 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sadybekov A et al. (2017) An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun 8, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu Q et al. (2018) Autism-associated CHD8 deficiency impairs axon development and migration of cortical neurons. Mol. Autism 9, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nitarska J et al. (2016) A Functional Switch of NuRD Chromatin Remodeling Complex Subunits Regulates Mouse Cortical Development. Cell Rep. 17, 1683–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jung E-M et al. (2017) Arid1b haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat. Neurosci 20, 1694–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bultman S et al. (2000) A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell 6, 1287–1295 [DOI] [PubMed] [Google Scholar]
- 98.Nott A et al. (2013) S-nitrosylation of HDAC2 regulates the expression of the chromatin-remodeling factor Brm during radial neuron migration. Proc. Natl. Acad. Sci. U. S. A 110, 3113–3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mandel S et al. (2007) Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev. Biol 303, 814–824 [DOI] [PubMed] [Google Scholar]
- 100.Zhao C et al. (2018) Dual Requirement of CHD8 for Chromatin Landscape Establishment and Histone Methyltransferase Recruitment to Promote CNS Myelination and Repair. Dev. Cell 45, 753–768.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Feng W et al. (2017) Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat. Commun 8, 14758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weiss K et al. (2016) De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am. J. Hum. Genet 99, 934–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ka M et al. (2016) Essential Roles for ARID1B in Dendritic Arborization and Spine Morphology of Developing Pyramidal Neurons. J. Neurosci. Off. J. Soc. Neurosci 36, 2723–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Celen C et al. (2017) Arid1b haploinsufficient mice reveal neuropsychiatric phenotypes and reversible causes of growth impairment. eLife 6, e25730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Qiu Z and Ghosh A (2008) A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron 60, 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Loe-Mie Y et al. (2010) SMARCA2 and other genome-wide supported schizophrenia-associated genes: regulation by REST/NRSF, network organization and primate-specific evolution. Hum. Mol. Genet 19, 2841–2857 [DOI] [PubMed] [Google Scholar]
- 107.Oz S et al. (2014) The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol. Psychiatry 19, 1115–1124 [DOI] [PubMed] [Google Scholar]
- 108.Sztainberg Y et al. (2015) Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature DOI: 10.1038/nature16159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mei Y et al. (2016) Adult Restoration of Shank3 Expression Rescues Selective Autistic-Like Phenotypes. Nature 530, 481–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rotaru DC et al. (2018) Adult Ube3a Gene Reinstatement Restores the Electrophysiological Deficits of Prefrontal Cortex Layer 5 Neurons in a Mouse Model of Angelman Syndrome. J. Neurosci. Off. J. Soc. Neurosci 38, 8011–8030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guy J et al. (2007) Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. Science 315, 1143–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Creson TK et al. (2019) Re-expression of SynGAP protein in adulthood improves translatable measures of brain function and behavior. eLife 8, e46752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kazdoba TM et al. (2016) Translational Mouse Models of Autism: Advancing Toward Pharmacological Therapeutics. Curr. Top. Behav. Neurosci 28, 1–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Michalon A et al. (2012) Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Burket JA et al. (2011) Complex effects of mGluR5 antagonism on sociability and stereotypic behaviors in mice: possible implications for the pharmacotherapy of autism spectrum disorders. Brain Res. Bull 86, 152–158 [DOI] [PubMed] [Google Scholar]
- 116.Ehninger D et al. (2008) Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med 14, 843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sgritta M et al. (2019) Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 101, 246–259.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hsiao EY et al. (2013) Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Youssef EA et al. (2018) Effect of the mGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacology 43, 503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Berry-Kravis E et al. (2016) Mavoglurant in fragile X syndrome: Results of two randomized, double-blind, placebo-controlled trials. Sci. Transl. Med 8, 321ra5–321ra5 [DOI] [PubMed] [Google Scholar]
- 121.Krueger DA et al. (2017) Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann. Clin. Transl. Neurol 4, 877–887 [DOI] [PMC free article] [PubMed] [Google Scholar]