

Abstract
Morphine and structurally-derived compounds are µ opioid receptor (µOR) agonists, and the most effective analgesic drugs. However, their usefulness is limited by serious side effects, including dependence and abuse potential. The N-substituent in morphinans plays an important role in opioid activities in vitro and in vivo. This study presents the synthesis and pharmacological evaluation of new N-phenethyl substituted 14-O-methylmorphinan-6-ones. Whereas substitution of the N-methyl substituent in morphine (1) and oxymorphone (2) by an N-phenethyl group enhances binding affinity, selectivity and agonist potency at the µOR of 1a and 2a, the N-phenethyl substitution in 14-methoxy-N-methylmorphinan-6-ones (3 and 4) converts selective µOR ligands into dual µ/δOR agonists (3a and 4a). Contrary to N-methylmorphinans 1–4, the N-phenethyl substituted morphinans 1a–4a produce effective and potent antinociception without motor impairment in mice. Using docking and molecular dynamics simulations with the µOR, we establish that N-methylmorphinans 1–4 and their N-phenethyl counterparts 1a–4a share several essential receptor-ligand interactions, but also interaction pattern differences related to specific structural features, thus providing a structural basis for their pharmacological profiles. The emerged structure-activity relationships in this class of morphinans provide important information for tuning in vitro and in vivo opioid activities towards discovery of effective and safer analgesics.
Subject terms: Drug discovery, Medicinal chemistry, Pharmacology, Preclinical research, Drug development, G protein-coupled receptors, Molecular modelling, Cheminformatics, Medicinal chemistry
Introduction
Morphine (1, Fig. 1), the prototypical opioid, has been used for decades for pain relief, and its addictive properties are long and well recognized. Over the years, numerous semisynthetic and synthetic investigations were reported aiming at optimizing morphine’s biological actions, especially its safety profile1–3. These studies have resulted in clinically useful drugs for the treatment of pain and other human disorders (drug abuse, alcohol abuse, and gastrointestinal motility dysfunction), as well as in research tools1–5. Morphine and structurally-derived compounds (e.g. oxycodone, oxymorphone, hydromorphone) are agonists at the µ opioid receptor (µOR), a G protein-coupled receptor (GPCR), and the opioid receptor subtype that primarily mediates desirable (analgesia) but also undesirable effects (i.e. constipation, respiratory depression, sedation, analgesic tolerance and dependence) of opioids4–6. Moreover, the number of people misusing opioids, as well as of opioid-related deaths have increased dramatically during the past years7.
Figure 1.
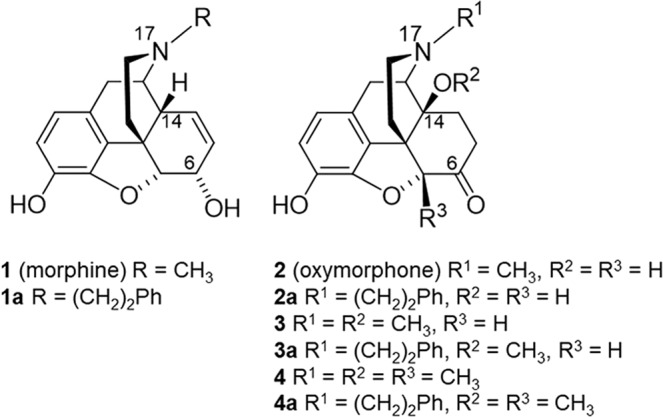
Structures of N-methylmorphinans 1–4 and their N-phenethyl counterparts 1a–4a. Ph, phenyl.
The N-phenethyl substituted derivative of morphine (1a, Fig. 1) was prepared by Clark et al. and Small et al. in the 1950s8,9, and the N-phenethyl substituted derivative of oxymorphone (2a, Fig. 1) was prepared by Seki et al. in the 1960s10, with both morphinan opioids having increased antinociceptive potency in mice than their N-methyl analogues11,12. We have reported the in vitro profile of 1a and 2a, including binding and activation of the µOR, and were first to show that the presence of an N-phenethyl group at position 17 is highly favorable in terms of improved affinity and selectivity at the µOR and potent µOR agonism in vitro13.
The hydroxyl group at position 14 is known to play a critical role in the agonist activity in vitro and in vivo of N-methylmorphinan-6-ones2,3,14,15. The 14-methoxy substituted analogues of oxymorphone (2), namely 14-O-methyloxymorphone (14-OMO, 3, Fig. 1)16 and 14-methoxymetopon (14-MM, 4, Fig. 1)17 show increased µOR affinity and agonism, efficacy and potency than oxymorphone (2)16–18. Whereas 3 induces the usual morphine-like adverse effects, 4 has a superior benefit/risk ratio15,18. In this study, we describe the synthesis and pharmacological evaluation of N-phenethyl substituted derivatives 3a and 4a (Fig. 1). We have also aimed to investigate the effect of the replacement of the N-methyl group in 14-OMO (3) and 14-MM (4) by an N-phenethyl substituent in 3a and 4a, respectively, on in vitro profiles (opioid receptor binding and functional activities), and in vivo behavioural properties (nociception and motor function) in mice. Furthermore, the current work was undertaken to understand the consequences of the substitution of the N-methyl group in N-methylmorphinans 1–4 by an N-phenethyl group in 1a–4a on their pharmacological activities using molecular docking and molecular dynamics (MD) simulations, to gain insights on their binding and subtype profile for opioid receptors. The emerged structure-activity relationships (SARs) in this class of opioid morphinans provide essential information for tuning functional in vitro and in vivo activities towards discovery of effective and safer analgesics for the pain treatment.
Results and Discussion
Chemistry
The new N-phenethylmorphinans 3a and 4a were prepared from their precursors 519 and 6, respectively, by N-alkylation with phenethyl bromide as presented in Scheme 1. The synthesis of N-phenethylmorphinans 1a and 2a has been earlier reported8–10, with some modifications as described13.
Scheme 1.
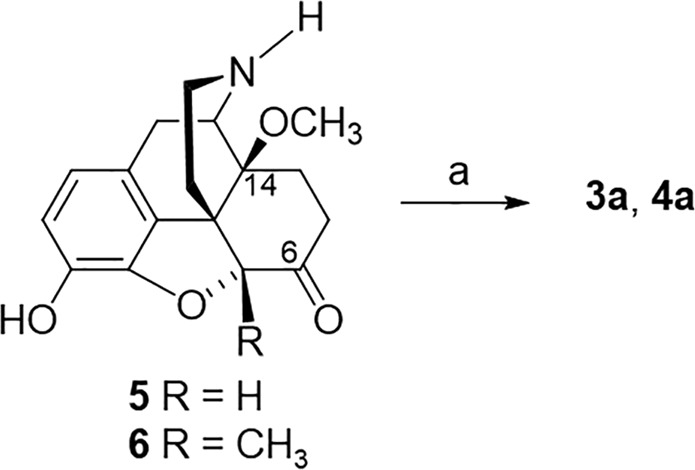
Synthesis of Compounds 3aª and 4aa aReagents and conditions: (a) phenethyl bromide, NaHCO3, DMF, 80 °C, 48 h.
Pharmacological evaluation
Binding affinities at the human µOR, δ (δOR) and κ (κOR) opioid receptors were first determined in in vitro competition binding assays using membranes from Chinese hamster ovary (CHO) cells stably transfected with one of the recombinant human opioid receptors (CHO-hµOR, CHO-hδOR and CHO-hκOR cells) as previously described13,20. We have reported earlier that N-phenethyl substituted morphinans 1a and 2a have higher affinities at the µOR in the rat brain than their parent N-methylmorphinans morphine (1) and oxymorphone 213. Herein, we have evaluated their binding profile at the human µOR expressed in CHO cells, and made similar observations with 1a and 2a displaying ca. 13- and 12-fold increased affinity at the human µOR than 1 and 2, respectively (Table 1). In this study, comparison of the in vitro binding at the µOR of 14-methoxy-N-methylmorphinan-6-ones to their N-phenethyl analogues revealed that introduction of a phenethyl group at position 17 does not influence affinity at the µOR when relating 14-OMO (3) vs. 3a, and 14-MM (4) vs. 4a (P > 0.05, t-test). Furthermore, all N-phenethyl derivatives 1a–4a have higher affinities (5- to 13-fold) than their N-methyl counterparts 1–4 at the human δOR (P < 0.05, t-test). Particularly, 3a and 4a showed very low nanomolar affinities at the δOR (Ki = 1.81 nM and 1.45 nM, respectively). While affinities of 1a, 2a and 3a at the human κOR were in the range of their parent molecules 1–3, a ca. 3-fold decrease in the κOR affinity was noted for 4a vs. 4 (Table 1). We have also observed that replacement of the N-methyl group in 1 and 2 with an N-phenethyl group enhanced not only µOR affinity but also µOR selectivity vs. δOR and vs. κOR of 1a and 2a. In the case of 14-methoxy-N-phenethylmorphinan-6-ones 3a and 4a, a reduction in µOR vs. δOR selectivity was noticed, while selectivity for µOR vs. κOR was higher than that of 14-OMO (3) and 14-MM (4), respectively (Table 1).
Table 1.
Comparison of Binding Affinities at Opioid Receptors of N-Methylmorphinans 1–4 and Their N-Phenethyl Analogues 1a–4a.
| Compound | Ki (nM)a | ||||
|---|---|---|---|---|---|
| µOR | δOR | κOR | Ki ratio δOR/µOR | Ki ratio κOR/µOR | |
| Morphine (1) | 3.35 ± 0.30 | 195 ± 26 | 96.4 ± 0.5 | 58 | 29 |
| 1a | 0.25 ± 0.09*** | 24.5 ± 8.7* | 93.5 ± 2.9 | 98 | 374 |
| Oxymorphone (2) | 1.41 ± 0.30 | 79.1 ± 9.3 | 32.6 ± 9.8 | 56 | 23 |
| 2a | 0.12 ± 0.07* | 10.7 ± 5.1* | 42.2 ± 7.3 | 89 | 352 |
| 14-OMO (3) | 0.27 ± 0.09 | 9.08 ± 0.31 | 10.3 ± 1.8 | 34 | 38 |
| 3a | 0.19 ± 0.02 | 1.81 ± 0.68*** | 15.8 ± 8.8 | 9.5 | 83 |
| 14-MM (4) | 0.25 ± 0.04 | 18.6 ± 0.98 | 12.8 ± 1.5 | 74 | 51 |
| 4a | 0.24 ± 0.03 | 1.45 ± 0.31*** | 35.3 ± 5.5** | 6.0 | 147 |
aDetermined in competition binding assays using membranes from CHO cells stably expressing the human opioid receptors. Values represent the mean ± SEM (n = 3–4). *P < 0.05, **P < 0.01 and ***P < 0.001 for N-methylmorphinans vs. respective N-phenethyl analogues (unpaired t-test).
In vitro opioid activities of targeted compounds at the human µOR and δOR were determined in the guanosine-5′-O-(3-[35S]thio)-triphosphate ([35S]GTPγS) binding (Table 2) and forskolin-induced cAMP accumulation assays (Table 3), performed as described13,21. The κOR-mediated G protein activation was assessed using [35S]GTPγS binding assays with CHO cell membranes expressing the human κOR (Table 2). Previous work from our laboratory on the introduction of a phenethyl group at the nitrogen in morphine (1) and oxymorphone (2) showed an increase in agonist potency by 2- to 3-fold and full efficacy for 1a and 2a in inducing µOR-mediated G protein signaling as assessed by [35S]GTPγS binding and calcium mobilization assays13. Similarly, enhanced µOR agonist potencies by 4- to 5-fold were measured for N-phenethyl analogues 1a and 2a as compared to morphine and oxymorphone, respectively, in the cAMP accumulation assay, while the δOR agonism remained unchanged (Table 3). In the series of 14-O-methylmorphinan-6-ones, exchanging the N-methyl by an N-phenethyl substituent did not largely influence agonist potency and full efficacy at the µOR of 14-OMO (3) vs. 3a, and 14-MM (4) vs. 4a (P > 0.05, t-test). These findings reveal that the N-phenethyl substitution in 14-methoxy-N-methylmorphinan-6-ones does not cause any change in binding affinity nor in vitro agonism at the µOR. All compounds displayed full efficacies at the δOR with different levels of potencies, while at the κOR a partial agonist profile with very low potencies was noted (Tables 2 and 3). In the [35S]GTPγS binding assay, the 14-methoxy-N-phenethylmorphinan-6-ones 3a and 4a displayed the highest agonist potencies at the δOR (EC50 = 9.34 nM and 9.54 nM, respectively), which were higher (4-fold) than potencies of their N-methyl counterparts 14-OMO (3) and 14-MM (4). The same observation was made when comparing agonist activity of 3 vs. 3a and 4 vs. 4a at the δOR in the cAMP accumulation assay, with a significant increase in potency (P < 0.05, t-test) for the N-phenethylmorphinan-6-ones 3a and 4a (Table 3). Thus, the outcomes from the [35S]GTPγS functional and cAMP accumulation assays are in agreement with the results on increased binding affinity at the δOR of 3a and 4a compared to 3 and 4, respectively. Additionally, 3a and 4a have a functional profile in vitro as dual µ/δOR full agonists, a class of ligands nowadays targeted as new analgesics with reduced unwanted side effects. Numerous pharmacological and biochemical reports and studies with opioid receptor knockout mice have provided evidence on the modulatory interactions between the µOR and δOR systems22–26. While the mechanisms are still unknown, several studies have established that the therapeutic profile of opioids could be improved by simultaneous modulation of the µOR and δOR, with compounds designed to target both receptors based on peptidic structures, non-peptidic structures or utilize the morphinan scaffold25–32.
Table 2.
Comparison of Functional Activities at Opioid Receptors of N-Methylmorphinans 1–4 and Their N-Phenethyl Analogues 1a–4a.
| Compound | µORa | δORa | κORa | |||
|---|---|---|---|---|---|---|
| EC50 (nM) | % stim. | EC50 (nM) | % stim. | EC50 (nM) | % stim. | |
| Morphine (1) | 34.4 ± 5.1b | 89 ± 17b | 668 ± 65b | 109 ± 14b | 710 ± 23b | 76 ± 2b |
| 1a | 10.3 ± 0.9b* | 113 ± 8b | 712 ± 86b | 138 ± 17b | 1049 ± 29b | 19 ± 2b*** |
| Oxymorphone (2) | 7.80 ± 1.61b | 92 ± 5b | 259 ± 33b | 87 ± 40b | 463 ± 116b | 48 ± 11b |
| 2a | 2.67 ± 1.06b* | 97 ± 3b | 131 ± 60b | 101 ± 9b | 225 ± 74b | 7.5 ± 0.01b* |
| 14-OMO (3) | 1.21 ± 0.48 | 95 ± 5 | 38.5 ± 6.9 | 102 ± 4 | 135 ± 29 | 65.9 ± 6.5 |
| 3a | 1.26 ± 0.63 | 98 ± 10 | 9.34 ± 0.60* | 107 ± 5 | 144 ± 9 | 35.4 ± 7.5* |
| 14-MM (4) | 2.66 ± 0.58 | 99 ± 5 | 36.8 ± 12.4 | 100 ± 9 | 181 ± 9 | 68.9 ± 9.2 |
| 4a | 1.86 ± 0.84 | 102 ± 13 | 9.54 ± 2.33* | 103 ± 2 | 334 ± 114 | 51.3 ± 10.4 |
aDetermined in [35S]GTPγS binding assays using membranes from CHO cells stably expressing the human opioid receptors. Percentage stimulation (% stim.) relative to the agonist DAMGO (µOR), DPDPE (δOR) or U69,593 (κOR). Values represent the mean ± SEM (n = 3–4). bData from ref. 13. *P < 0.05, **P < 0.01 and ***P < 0.001 for N-methylmorphinans vs. respective N-phenethyl analogues (unpaired t-test).
Table 3.
Agonist potencies and efficacies at the human µOR and δOR of N-methylmorphinans 1–4 and their respective N-phenethy analogues 1a–4a in the cAMP accumulation assay.
| Compound | µORa | δORa | ||
|---|---|---|---|---|
| EC50 (nM) | % stim. | EC50 (nM) | % stim. | |
| Morphine (1) | 13.5 ± 2.83 | 106 ± 9 | 374 ± 24 | 94 ± 18 |
| 1a | 2.87 ± 0.91* | 107 ± 5 | 315 ± 110 | 105 ± 8 |
| Oxymorphone (2) | 2.48 ± 0.79 | 109 ± 2 | 56.9 ± 5.1 | 87 ± 16 |
| 2a | 0.59 ± 0.04* | 97 ± 6 | 50.1 ± 16.2 | 99 ± 18 |
| 14-OMO (3) | 0.19 ± 0.14 | 90 ± 8 | 5.42 ± 0.93 | 98 ± 3 |
| 3a | 0.078 ± 0.004 | 88 ± 12 | 0.60 ± 0.07** | 93 ± 2 |
| 14-MM (4) | 0.31 ± 0.07 | 93 ± 5 | 4.06 ± 0.89 | 99 ± 8 |
| 4a | 0.15 ± 0.07 | 98 ± 12 | 0.55 ± 0.16* | 96 ± 4 |
aDetermined in the forskolin-induced cAMP accumulation assay using CHO cells co-expressing the human opioid receptors and the cAMP biosensor GloSensor-22F (CHO-hµOR-p22F or CHO-hδOR-p22F cells). Percentage stimulation (% stim.) relative to the agonist DAMGO (µOR) or DPDPE (δOR). Values represent the mean ± SEM (n = 3–4). *P < 0.05 and **P < 0.01 for N-methylmorphinans vs. respective N-phenethyl analogues (unpaired t-test).
We13 and others11,12 have reported that the N-phenethyl substituted morphinans 1a and 2a exhibit increased antinociceptive potencies than their respective N-methyl analogues morphine (1) and oxymorphone (2) in mouse models of acute thermal nociception after subcutaneous (s.c.) administration, which is in line with findings from binding and functional in vitro assays. We have shown that 1a was 22- and 28-fold more effective than morphine (1) in the hot-plate and tail-flick tests, respectively13. Further, the N-phenethyl analogue of oxymorphone (2a) was found to be highly active with about 2-fold higher potency than oxymorphone (1) (Table 4)13. In this study, we have also evaluated if targeted structural changes in 14-OMO (3) and 14-MM (4) also affects antinociceptive activities. Antinociceptive effects of N-phenethyl substituted 3a and 4a were assessed in the tail-flick assay in mice after s.c. administration as described13. Antinociceptive potencies (ED50 values) were determined at the peak of action and compared to those of N-methyl counterparts 3 and 4 (Table 4). All compounds increased tail-withdrawal latencies to thermal stimulation in a time- and dose-dependent manner with a peak effect generally occurring at 30 min (Fig. 2). As shown in Table 4 and Fig. 3, 3a and 4a display similar antinociceptive activities to their analogues 14-OMO (3) and 14-MM (4), respectively (P > 0.05, two-way ANOVA), indicating that exchanging the N-methyl by an N-phenethyl group in 14-O-methylmorphinan-6-ones does not affect the in vivo agonism.
Table 4.
Antinociceptive Potencies of N-Methylmorphinans 1–4 and Their N-Phenethyl Analogues 1a–4a in the Tail-Flick Test in Mice.
| Compound | ED50 (mg/kg, s.c.) (95% CL)a |
|---|---|
| Morphine (1) | 3.06 (1.76–5.31)b |
| 1a | 0.11 (0.027–0.40)b |
| Oxymorphone (2) | 0.35 (0.16–0.77)b |
| 2a | 0.15 (0.058–0.40)b |
| 14-OMO (3) | 0.014 (0.0051–0.037) |
| 3a | 0.014 (0.0086–0.023) |
| 14-MM (4) | 0.024 (0.0093–0.062) |
| 4a | 0.024 (0.0091–0.062) |
aED50 values and 95% confidence intervals (CL in parentheses) were calculated at 30 min (peak effect) from dose-response curves (n = 5–6 mice per group). bData from ref. 13.
Figure 2.

Time- and dose-dependent antinociceptive effects of 14-methoxy-N-methylmorphinan-6-ones 14-OMO (3) and 14-MM (4) and their respective N-phenethyl analogues 3a and 4a in the tail-flick assay in mice after s.c. administration. Data are shown as the mean %Maximum Possible Effect (%MPE) ± SEM (n = 5–6 mice per group).
Figure 3.

Comparison of dose-dependent antinociceptive effects of 14-methoxy-N-methylmorphinan-6-ones 14-OMO (3) and 14-MM (4) and their respective N-phenethyl analogues 3a and 4a in the tail-flick assay in mice at 30 min after s.c. administration. Data are shown as mean %Maximum Possible Effect (%MPE) ± SEM (n = 5–6 mice per group). P > 0.05 for 3 vs. 3a, and 4 vs. 4a (two-way ANOVA).
Clinically used opioid analgesics, such as morphine, oxycodone or fentanyl, are known to produce sedation and motor dysfunction, side effects that limits their clinical usefulness33–35. With literature evidence that mixed µOR/δOR agonists are efficacious analgesics with reduced side effects25–32, we have evaluated the effect of 3a and 4a as mixed µ/δOR agonists, and behavioral consequences of the replacement of the N-methyl group in N-methylmorphinans 1–4 by an N-phenethyl group in 1a–4a on motor coordination in mice using the rotarod assay, a well-established model for evaluating loss of coordinated locomotion36. The first behavioral data on motor function following systemic s.c. administration of N-phenethyl substituted derivatives of morphine and oxymorphone, 1a and 2a, respectively are presented. Mice were s.c. treated with the respective compound at doses equivalent to a 3- to 4-fold the antinociceptive ED50 dose. Rotarod experiments demonstrate the lack of the mixed µ/δOR agonists 3a and 4a to induce motor dysfunction, having an improved profile than their parent µOR selective agonists 14-OMO (3) and 14-MM (4), respectively (Fig. 4). Whereas morphine (1) and oxymorphone (2) caused a significant deficit in rotarod performance, their N-phenethyl substituted 1a and 2a did not affect the evoked locomotor activity of mice (Fig. 4). In this study, we show that N-phenethyl substituted morphinans 1a–4a elicit effective and potent antinociception without motor impairment in mice.
Figure 4.

Effect of N-methylmorphinans 1–4 and their respective N-phenethyl analogues 1a–4a in the mouse rotarod assay. Mice were tested 30 min after s.c. administration of control (saline) or test compounds. Data depicts latencies to fall from the rotarod as the mean percent changes from baseline performance ± SEM (n = 6 mice per group). *P < 0.05, **P < 0.01 and ***P < 0.001 vs. saline group; #P < 0.05 and ###P < 0.001 vs. N-methylmorphinan treated group; one-way ANOVA followed by Tukey’s post hoc test.
Molecular modeling
The µOR was the first opioid receptor type resolved in an inactive (PDB ID: 4DKL)37 and an active conformation (PDB ID: 5C1M)38. The access to crystal structures of the µOR provides essential knowledge on key aspects of the µOR pharmacology and its function37–39. All investigated morphinans (Fig. 1) bind and are agonists at the µOR. The observed similarities or differences in their in vitro and in vivo activity profiles incited exploration of their binding modes at the µOR. Molecular docking investigations were performed with N-methylmorphinans 1–4 and their N-phenethyl counterparts 1a–4a, where a 3D-pharmacophore approach based on the LigandScout program29 was applied to analyze shared and distinct receptor-ligand interactions. Docking studies using the active conformation of the µOR (PDB ID: 5C1M)38 revealed comparable binding orientations for all targeted morphinans, which are in accordance with BU7227 in its co-crystallized conformation. An overview of detected interactions is presented in Figs. 5 and S1.
Figure 5.

Predicted binding modes at the µOR (PDB ID: 5C1M) and receptor-ligand interaction patterns of N-methylmorphinans 1–4 and their respective N-phenethyl analogues 1a–4a. (A) Morphine (1) and 1a, (B) oxymorphone (2) and 2a, (C) 14-OMO (3) and 3a, and (D) 14-MM (4) and 4a. Yellow spheres indicate lipophilic contacts, red arrows hydrogen bond acceptors, green arrows hydrogen bond donors and positively charged centers are shown as blue spheres.
Although all investigated compounds show a comparable binding mode to the µOR in which the morphinan moiety was found to adopt a similar orientation and to share several essential receptor-ligand interactions, we have also observed interaction pattern differences related to specific structural features (Fig. 5). The tertiary amine forms an essential charge interaction with D147 and a π–cation interaction with Y148 residue. The crucial role of D147 for the binding to the µOR of morphinan ligands, as well as other chemotypes (i.e. mitragynine pseudoindoxyl) and peptides (i.e. DAMGO) has been described37,38,41–44. The interaction with Y148 is also recognized as an important requirement for ligands (small molecules and peptides) to bind to the µOR37,38,41–44. In this study, the oxygen of the partially saturated furan ring (E-ring) of the morphinan system serves as a hydrogen bond acceptor for Y148 in both series of N-methyl (1–4) and N-phenethyl substituted morphinans (1a–4a). The 14-O-methylmorphinan-6-ones 3, 4, 3a and 4a show a lipophilic contact of the 14-methoxy group with I322 (Fig. 5C,D). The phenolic substructure lies opposite to M151, V236 and V300 residues. The 14-hydroxyl group of oxymorphone (2) and its N-phenethyl analogue 2a forms hydrogen bonds to both D147 and Y326 residues (Fig. 5B). Compounds 1 and 1a exhibited the same interaction pattern with the only difference in the additional lipophilic contacts of the N-phenethyl moiety of 1a with a lipophilic subpocket (Fig. 5A). For all ligands with an N-phenethyl group, this moiety is embedded in a lipophilic pocket formed by A117, W293 and Y326 residues.
The recent crystal structure of the δOR (PDB ID: 6PT3)45 with the co-crystallized agonist DPI-287 supports our proposed binding mode of 4a at the δOR, since DPI-287 also has a phenyl ring which is filling the beforementioned lipophilic subpocket (Fig. 6). The active κOR structure (PDB ID: 6B73) was also determined in complex with the epoxymorphinan agonist MP110446. Compared to the µOR, the size and shape of this subpocket was found to be highly similar for the δOR (Figs. 6A and S2), but different for the κOR (Figs. 6 and S2). Whereas µOR and δOR have an alanine residue in position 2.53 (according to Ballesteros-Weinstein nomenclature), a valine residue at the same position requires more space in the κOR resulting in a smaller lipophilic subpocket (Fig. S2). We suggest that this structural difference is important for the subtype selectivity of targeted N-phenethyl substituted morphinans and the resulting dual µ/δOR activity of 3a and 4a.
Figure 6.

Structural comparison of the orthosteric binding pocket of the µOR (PDB ID: 5C1M; dark grey), δOR (PDB ID: 6PT3; light grey) and κOR (PDB ID: 6B73, green), with 4a in its µOR bound conformation (A). Size and shape of the lipophilic subpocket formed by A2.53, M3.36 and W6.48, which hosts the N-phenethyl substituent of 4a is different at the κOR (green), due to a valine instead of an alanine residue. (B) The proposed binding mode of 4a (blue) at the δOR (PDB ID: 6PT3) shows a similar orientation of the N-phenethyl substituent compared to the phenyl ring of the co-crystallized agonist DPI-287 (salmon).
Several studies have evidenced MD simulations as an effective approach to examine binding modes between opioid receptors and their ligands39,41–44. In order to further validate the binding modes of morphinans 1–4 and 1a–4a depicted using molecular docking, we performed all-atoms MD simulations of the µOR as described47. The analysis of MD simulations support the docking results and show that the binding location, the major ligand orientation in the binding pocket and the key interactions reported from the docking experiments remain firm over 100 ns of MD simulations (Figs. 7 and S3).
Figure 7.

MD simulations of the µOR (PDB ID: 5C1M) support the docking results. (A) The binding mode of 4a is stabilized by a lipophilic contact I322 with the 14-O-methyl group (black circle) and furthermore the N-phenethyl moiety. (B) 1 shows more fluctuations in its binding mode, due to the absence of these interactions.
All investigated ligands are full agonists at the µOR, in accordance with the observation that all structures provide a full constriction of the orthosteric binding site as a key feature for receptor activation. The additional lipophilic contacts of the N-phenethyl substituent deep in the core region of the receptor are supposed to enhance ligand binding. This effect is less prominent for morphinans in which the parent compound already has a binding affinity in the subnanomolar range to the µOR (Table 1). This may be explained by additional interactions with the receptor, such as additional hydrogen bonds for the 14-hydroxymorphinans 2 and 2a, or an additional lipophilic contact with I322 residue for the 14-O-methylmorphinans, 14-OMO (3), 14-MM (4), 3a and 4a. The latter interaction is of particular importance for orientation of the ligand in the binding site, and is visualized in Fig. 7. Interestingly, N-methyl substituted 1 and 2 show a slightly different orientation compared with their related N-phenethyl analogues (Fig. S4). This effect is not observable for 3, 3a, 4 and 4a, which suggests that an increased µOR affinity of morphinan analogs can be achieved by either a methoxy substitution at position 14 or by an N-phenethyl group. Since the combination of the two strategies does not show additive effects, we assume that the optimal orientation can be sufficiently triggered by only one of the two substitutions. Furthermore, a direct comparison of the active δOR45 and the µOR38 crystal structures unveils the high similarity of their binding pockets. While all residues forming key interactions are identical, a major difference was observed at the beginning of helix seven at position 7.35 (Fig. S5). The tryptophan residue in µOR was found to be optimal in forming lipophilic contacts with the morphinan moiety, whereas this receptor-ligand contact is missing for the δOR, due to a leucine at this position. This might explain why all studied compounds are have higher affinity for the µOR compared to the δOR.
Conclusions
The results of the present study provide SAR evidence on the consequences of an N-methyl substitution in morphinan opioids 1–4 by an N-phenethyl in 1a–4a on in vitro and in vivo activities, with molecular docking and MD simulations studies offering a structural basis for the observed pharmacological profiles at the opioid receptors. Pharmacological findings are supported by docking and MD simulations analysis with N-methyl substitute morphine (1) and oxymorphone (2) showing a slightly different orientation in the binding pocket of the µOR compared to their related N-phenethyl analogues, 1a and 2a, respectively. This was not noticed for 14-OMO (3) vs. 3a, and 14-MM (4) vs. 4a, indicating that an increased µOR affinity can be achieved by either a 14-methoxy or by an N-phenethyl substitution, as key sites to be targeted in modulating the binding affinity and efficacy of morphinans to the µOR. Whereas replacement of the N-methyl substituent in morphine (1) and oxymorphone (2) by an N-phenethyl group enhanced binding affinity, selectivity and agonist potency at the µOR of 1a and 2a, the N-phenethyl substitution in 14-methoxy-N-methylmorphinan-6-ones (3 and 4) turned selective µOR ligands into dual µ/δOR agonists (3a and 4a), a profile that currently emerges as a promising approach to opioid analgesic drug discovery26–32. Furthermore, we have demonstrated that the N-phenethyl substituted morphinans 1a–4a are effective and potent antinociception agents without causing unwanted motor impairment in mice after s.c. administration. Altogether, these data offer important insights on the SARs in the morphinan class of opioid ligands, by increasing the current understanding of the impact of different substituents at the nitrogen and position 14 on ligand-µOR binding, receptor activation and the link between antinociception and side effects (i.e. motor function).
Materials and Methods
Chemistry
General chemical and analytical methods were performed according to protocols as described previously20. All chemicals were of reagent grade and obtained from standard commercial sources. Melting points were determined on a Kofler melting point microscope and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker Avance II + spectrometer operating at 600 MHz and equipped with a Prodigy TCI probe. IR spectra were taken on a Bruker Alpha FT-IR spectrometer (for detection, an ATR sensor was used). Mass spectra were recorded on a Varian MAT 44 S apparatus. Elemental analyses were performed at the Microanalytic Laboratory of the University of Vienna, Austria. For column chromatography (MPLC), silica gel 60 (0.040–0.063 mm, Fluka, Switzerland) was used. Compounds 3a and 4a were used as bases for testing. The combustion analysis values were found to be within ± 0.4% of the calculated values, confirming a purity of the tested compounds of >95%.
Synthesis of 4,5α-Epoxy-3-hydroxy-14-methoxy-N-phenethylmorphinan-6-one (3a)
A mixture 4,5α-epoxy-3-hydroxy-14-methoxymorphinan-6-one hydrochloride (5 ∙ HCl) (100 mg, 0.296 mmol), prepared according to the described procedure19, phenethyl bromide (76.7 mg, 0.40 mmol), and NaHCO3 (67.3 mg, 0.8 mmol) in 3 mL DMF was stirred at 80 °C for 48 h. The cooled mixture was filtered, the filtrate evaporated to dryness and purified by column chromatography (CH2Cl2/MeOH/NH4OH 97.5/1.5/1) to yield 40 mg (47%) of compound 3a. Mp 180–182 °C. IR (ATR) 2929 (OH), 1718 (CO) cm−1; 1H NMR (CDCl3): δ 7.25–7.14 (m, 5 arom. H), 6.73 (d, J = 8.0 Hz, H-C(1)), 6.62 (d, J = 8.0 Hz, H-C(2)), 4.68 (s, H-C(5)), 3.13 (s, CH3O); 13C NMR (CDCl3): δ 209.54, 143.40, 140.37, 138.68, 129.34, 128.83, 128.37, 126.10, 125.08, 119.85, 117.57, 90.56, 75.61, 56.98, 56.33, 51.03, 47.96, 44.23, 35.50, 34.42, 29.17, 24.97, 23.44; MS (ESI) m/z 406.3 [M + 1]+. Anal. (C25H27NO4 ∙ 0.2CH2Cl2 ∙ 0.1MeOH) C, H, N.
Synthesis of 4,5α-Epoxy-3-hydroxy-14-methoxy-5-methyl-N-phenethylmorphinan-6-one (4a)
A mixture 4,5α-epoxy-3-hydroxy-14-methoxy-5-methylmorphinan-6-one hydrobromide (6 ∙ HBr) (100 mg, 0.23 mmol), phenethyl bromide (74.3 mg, 0.41 mmol), NaHCO3 (66.8 mg, 0.8 mmol), in 2.5 mL DMF was stirred at 80 °C for 48 h. The mixture was cooled and filtered, the filtrate evaporated to dryness and the crude product purified by column chromatography (CH2Cl2/MeOH/NH4OH 97/2/1) to yield 46 mg (38%) of compound 4a. Mp 179–180 °C. IR (ATR) 2920 (OH), 1718 (CO) cm−1. 1H NMR (CDCl3): δ 7.31–7.25 (m, 5 arom. H), 6.70 (d, J = 8.1 Hz, H-C(1)), 6.57 (d, J = 8.1 Hz, H-C(2)), 3.17 (s, CH3O), 1.55 (s, CH3-C(5)); 13C NMR (CDCl3): δ 213.01, 143.11, 140.37, 138.36, 128.80, 128.39, 126.10, 119.47, 117.35, 96.48, 76.20, 56.98, 56.38, 51.39, 47.50, 43.91, 34.56, 34.38, 25.86, 25.15, 23.44, 17.43; MS (ESI) m/z 420.3 [M + 1]+. Anal. (C26H29NO4 ∙ 0.5CH2Cl2) C, H, N.
Synthesis of 4,5α-Epoxy-3-hydroxy-14β-methoxy-5-methylmorphinan-6-one hydrobromide (6 ∙ HBr)
A solution of 3,14-dimethoxy-4,5α-epoxy-5β-methylmorphinan-6-one hydrochloride (1.0 g, 2.73 mmol), prepared according to the described procedure48, in 3.5 ml of 48% HBr was refluxed for 15 min. After cooling, the brownish solution was evaporated, the residue treated with MeOH and again evaporated. The oily residue was crystallized from MeOH to yield 713 mg (66%) of colorless 6 ∙ HBr. Mp> 230 °C. (dec.). IR (KBr): 3545 and 3495 (+NH, OH), 1720 (CO) cm−1. 1H-NMR (DMSO-d6): δ 9.37 (s, OH), 8.65 (broad s, +NH2), 6.64 (dd, J = 8.2, 8.2 Hz, 2 arom. H), 3.36 (s, CH3O), 1.48 (s, CH3-C(5)). MS (ESI) m/z 316 [M + 1]+. Anal. (C18H21NO4 ∙ HBr∙MeOH) C, H, N.
Pharmacology. drugs and chemicals
Cell culture media and supplements were obtained from Sigma-Aldrich Chemicals (St. Louis, MO), or Life Technologies (Carlsbad, CA). Radioligands [3H][D-Ala2,N-Me-Phe4,Gly-ol5]enkephalin ([3H]DAMGO, 50 Ci/mmol), [3H]Diprenorphine (37 Ci/mmol), and [35S]GTPγS (1250 Ci/mmol) were purchased from PerkinElmer (Boston, MA). [3H]HS665 (30.65 Ci/mmol) was prepared by Dr. Geza Toth (Institute of Biochemistry, Biological Research Centre, Hungarian Academy of Sciences, Szeged, Hungary) as previously described49. DAMGO, [D-Pen2,D-Pen5]enkephalin (DPDPE), U69,593, Diprenorphine, Tris(hydroxymethyl) aminomethane (Tris), 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), Hank’s Balanced Salt Solution (HBSS), unlabeled GTPγS, guanosine diphosphate (GDP) and forskolin were obtained from Sigma-Aldrich Chemicals (St. Louis, MO). Morphine hydrochloride was obtained from Gatt-Koller GmbH (Innsbruck, Austria). Compounds 1a and 2a were synthesized according to the described procedure13, and 14-OMO (3) and 14-MM (4) were prepared as earlier described16,50. All other chemicals were of analytical grade and obtained from standard commercial sources.
In Vitro Assays
Cell cultures
CHO cells stably expressing the human opioid receptors, µOR, δOR or κOR (CHO-hµOR, CHO-hδOR and CHO-hκOR cell lines), were kindly provided by Dr. Lawrence Toll (SRI International, Menlo Park, CA). The CHO-hµOR and CHO-hδOR cell lines were maintained in Dulbecco’s Minimal Essential Medium (DMEM)/Ham’s F-12 medium supplemented with fetal bovine serum (FBS, 10%), Penicillin/Streptomycin (0.1%), L-Glutamine (2 mM) and Geneticin (400 µg/ml). The CHO-hκOR cell line was maintained in DMEM supplemented with FBS (10%), Penicillin/Streptomycin (0.1%), L-Glutamine (2 mM) and Geneticin (400 µg/ml). CHO-hµOR or CHO-hδOR cells were stably transfected with the cAMP biosensor GloSensor-22F (Promega, Madison, WI), according to the previously described protocol21. Transfection was performed using the Viafect Transfection Reagent (Promega), according to the manufacturer’s instructions, and positive clones were selected with Hygromycin B (400 μg/mL). All cell cultures were maintained at 37 °C in 5% CO2 humidified air.
Competition binding assays
In vitro binding assays were conducted on human opioid receptors stably transfected into CHO cells according to the published procedures20. Briefly, CHO-hµOR, CHO-hδOR and CHO-hκOR cells grown at confluence were removed from the culture plates by scraping, homogenized in 50 mM Tris-HCl buffer (pH 7.4), using a Polytron homogenizer, then centrifuged once and washed by an additional centrifugation at 27,000 × g for 15 min, at 4 °C. The final pellet was resuspended in 50 mM Tris-HCl buffer (pH 7.4), and cell membranes (15–20 µg) were incubated with various concentrations of test compound and the appropriate radioligand [3H]DAMGO or [3H]Diprenorphine for 60 min at 25 °C, or [3H]HS665 for 30 min at 0 °C. Non-specific binding was determined using 1–10 µM of the unlabeled counterpart of each radioligand. Reactions were terminated by rapid filtration through Whatman glass GF/C fiber filters. Filters were washed three times with 5 mL of ice-cold 50 mM Tris-HCl buffer (pH 7.4) using a Brandel M24R cell harvester (Gaithersburg, MD). Radioactivity retained on the filters was counted by liquid scintillation counting using a Beckman Coulter LS6500 (Beckman Coulter Inc., Fullerton, CA). All experiments were performed in duplicate, and repeated at least three times with independently prepared samples.
[35S]GTPγS Functional Assays. Binding of [35S]GTPγS to membranes from CHO stably expressing the human opioid receptors was conducted according to the published procedures13,20. Cell membranes were prepared in Buffer A (20 mM HEPES, 10 mM MgCl2 and 100 mM NaCl, pH 7.4) as described for competitive radioligand binding assays. Cell membranes (5–10 µg) in Buffer A were incubated with 0.05 nM [35S]GTPγS, 10 µM GDP and various concentrations of test peptides in a final volume of 1 mL, for 60 min at 25 °C. Non-specific binding was determined using 10 µM GTPγS, and the basal binding was determined in the absence of test ligand. Samples were filtered over Whatman glass GF/B fiber filters and counted as described for competitive binding assays. All experiments were performed in duplicate, and repeated at least three times with independently prepared samples.
cAMP accumulation assay
Inhibition of the forskolin-stimulated intracellular cAMP accumulation in CHO cells co-expressing the hµOR and the cAMP biosensor GloSensor-22F (CHO-hµOR-p22F cells) and CHO cells co-expressing the hδOR and the cAMP biosensor GloSensor-22F (CHO-hδOR-p22F cells) was performed using the Glo-Sensor cAMP assay (Promega) according to the published procedure21. Cells were seeded in growth medium into 384-well plates at a density of 5,000 cells in 30 μL per well and incubated overnight. On the day of assay, culture media was removed, and cells were pre-equilibrated for 90 min with 4% v/v of the GloSensor cAMP reagent in reaction medium (20 mM HEPES, 1 x HBSS, pH 7.4) at 37 °C and 5% CO2. Cells were then treated with various concentrations of test compounds for 15 min at room temperature. Forskolin (10 μM) was added to each well, and luminescence was measured after 20 min using PerkinElmer Wallac Victor 1420 Mulitlable Counter. All experiments were performed in triplicate, and repeated at least three times with independently prepared samples.
In Vivo assays
Animals and Drug Administration. In vivo studies were performed as described previously20. Male CD1 mice (30–35 g, 7–8 weeks old) were obtained from the Center of Biomodels and Experimental Medicine (CBEM) (Innsbruck, Austria). Mice were group-housed in a temperature controlled room with a 12 h light/dark cycle and with free access to food and water. All animal studies were conducted in accordance with ethical guidelines and animal welfare standards according to Austrian regulations for animal research, and were approved by the Committee of Animal Care of the Austrian Federal Ministry of Science and Research. Test compounds or vehicle (saline) were administered by s.c. route in a volume of 10 µL/1 g of body weight. Each experimental group included five to six animals. Separate groups of mice received the respective dose of compound, and individual mice were only used once for behavioral testing.
Tail-flick assay
The radiant heat tail-flick test was used to assess antinociceptive effects of test compounds after s.c. administration in mice, according to the original procedure of D’Amour and Smith51. The tail-flick test was performed using an UB 37360 Ugo Basile analgesiometer (Ugo Basile s.r.l., Varese, Italy. The reaction time required by the mouse to remove its tail after application of the radiant heat was measured and defined as the tail-flick latency (in seconds). Tail-flick latencies were measured before (basal latency, BL) and after drug or saline (control) s.c. administration (i.e. 30, 60 and 120 min) and (test latency, TL). A cut-off time of 10 s was used in order to minimize tissue damage.
Rotarod assay
Possible motor dysfunction or sedative effects of test compounds were assessed in mice using the rotarod test, as earlier described52,53. The accelerating rotarod treadmill (Acceler Rota-Rod 7650, Ugo Basile s.r.l., Varese, Italy) for mice (diameter 3.5 cm) was used. Animals were habituated to the equipment in two training sessions (30 min apart) one day before testing. On the experimental day, mice were placed on the rotarod, and treadmill was accelerated from 4 to 40 rpm over a period of 5 min. The time spent on the drum was recorded for each mouse before (baseline) and at 30 min after s.c. administration of saline (control) or test compound. Decreased latencies to fall in the rotarod test indicate impaired motor performance. A 300 s cut-off time was used.
Data analysis
Data were analysed and graphically processed using GraphPad Prism 5.0. software (GraphPad Prism Software Inc., San Diego, CA, USA) and are presented as mean ± SEM. The Ki (nM), potency EC50 (nM), and efficacy Emax (%) values were determined from concentration-response curves by nonlinear regression analysis. The Ki values were determined by the method of Cheng and Prusoff54. In the [35S]GTPγS binding assays, efficacy was determined relative to the reference full opioid agonists, DAMGO (µOR), DPDPE (δOR), and U69,593 (κOR). In the cAMP accumulation assay, efficacy was determined relative to the reference µOR agonist DAMGO. The antinociceptive effect (as percentage of Maximum Possible Effect, %MPE) was calculated according to the formula = [(TL – BL)/(cut-off time – BL)] × 100, and the dose necessary to produce a 50% MPE (ED50) and 95% confidence limits (95% CL) were determined using the method of Litchfield and Wilcoxon55. In the rotarod test, percentage (%) changes from the rotarod latencies obtained before (baseline, B) and after drug administration (test, T) were calculated as: 100 × (T/B). Data were statistically evaluated using unpaired t-test, one-way ANOVA with Tukey’s multiple comparison post hoc test, or two-way ANOVA with significance set at P < 0.05.
Molecular modeling
The structure of the human µOR was remodeled based on the crystal structure of the murine µOR (PDB ID: 5C1M)38 by using the mutation tool of MOE (Molecular Operating Environment), 2014.09; Chemical Computing Group Inc.) with subsequent sidechain optimization. We used the active crystal structures of the δOR (PDB ID: 6PT3)45 and κOR (PDB ID: 6B73)46 for docking experiments. All receptor-ligand docking experiments were performed with the CCDCs software GOLD version 5.156. Water molecules and ligands were removed and correct protonation states were assigned. All residues of the inner receptor core region and the C-terminal domain were defined as potential binding site (12 Å around the γ-carbon atom of D147; PDB ID: 5C1M). For receptor-ligand docking default settings were applied and GoldScore served as scoring function. All obtained docking poses and receptor-ligand interactions were analyzed using LigandScout 4.240 using a 3D-pharmacophore approach. All-atoms MD simulations were performed in triplicates with Desmond version 2018-3 on the Curta compute cluster of the Freie Universität Berlin. All conditions and settings used for system building and simulation were chosen based on a previously reported protocol47.
Supplementary Information
Acknowledgements
This work was supported by the Tyrolean Research Fund (UNI-0404/1596), the Förderungsbeiträge Aktion D. Swarovski KG 2014, the Bilateral Cooperation Program Austria-France “Amadée” (FR 12/2016), the Austrian Science Fund (FWF: TRP19-B18 and I 2463-B21) and the German Research Foundation (DFG: 407626949). Tanila Ben Haddou was supported by the University of Innsbruck PhD stipend program.
Author contributions
M.D. and M.B. contributed equally to the work. M.S., G.W., H.S. and D.M. conceived and designed the study; M.D., M.B., T.B.H., E.G., L.S., A.R., S.H., A.L. and C.K. performed the research; All authors contributed to the data analyses; M.D., M.B., H.S., G.W. and M.S. wrote the manuscript with comments from S.H. and D.M. All authors have given approval to the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Maria Dumitrascuta and Marcel Bermudez.
Supplementary information
is available for this paper at 10.1038/s41598-020-62530-w.
References
- 1.Fürst S, Hosztafi S. The Chemical and Pharmacological Importance of Morphine Analogues. Acta Physiol. Hung. 2008;95:3–44. doi: 10.1556/APhysiol.95.2008.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Schmidhammer, H. & Spetea, M. Synthesis of 14-Alkoxymorphinans and Their Pharmacological Activities. In Chemistry of Opioids. Nagase, H., Ed.; Springer: Berlin Heidelberg, Top Curr Chem. 299, 63−91 (2011). [DOI] [PubMed]
- 3.Lewis, J. W. & Husbands, S. M. 14-Amino-4,5-Epoxymorphinan Derivatives and Their Pharmacological Actions. In Chemistry of Opioids. Nagase, H., Ed.; Springer: Berlin Heidelberg, Top Curr Chem. 299, 93−119 (2011). [DOI] [PubMed]
- 4.Pasternak GW, Pan YX. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013;65:1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spetea M, Asim MF, Wolber G, Schmidhammer H. The µ Opioid Receptor and Ligands Acting at the µ Opioid Receptor, as Therapeutics and Potential Therapeutics. Curr. Pharm. Des. 2013;19:7415–7434. doi: 10.2174/13816128113199990362. [DOI] [PubMed] [Google Scholar]
- 6.Benyamin R, et al. Opioid Complications and Side Effects. Pain Physician. 2008;11:S105–S120. [PubMed] [Google Scholar]
- 7.Volkow N, Benveniste H, McLellan AT. Use and Misuse of Opioids in Chronic Pain. Annu. Rev. Med. 2018;69:451–465. doi: 10.1146/annurev-med-011817-044739. [DOI] [PubMed] [Google Scholar]
- 8.Clark RL, Pessolano AA, Weijlard J, Pfister K. N-substituted Epoxymorphinans. J. Am. Chem. Soc. 1953;75:4963–4967. doi: 10.1021/ja01116a024. [DOI] [Google Scholar]
- 9.Small LF, Eddy NB, Ager JH, May EL. An Improved Synthesis of N-Phenethylnormorphine and Analogs. J. Org. Chem. 1958;23:1387–1388. doi: 10.1021/jo01103a615. [DOI] [Google Scholar]
- 10.Seki I, Takagi H, Kobayashi S. Pharmacological Studies on Morphine Derivatives. 3. On the Chemical Structure-Activity Relationships of 14-Hydroxymorphine Derivatives. Yakugaku Zasshi. 1964;84:280–286. doi: 10.1248/yakushi1947.84.3_280. [DOI] [PubMed] [Google Scholar]
- 11.Winter CA, Orahovats PD, Lehman EG. Analgesic Activity and Morphine Antagonism of Compounds Related to Nalorphine. Arch. Int. Pharmacodyn. 1957;110:186–202. [PubMed] [Google Scholar]
- 12.Loew GW, Berkowitz DS. Quantum Chemical Studies of N-Substituent Variation in the Oxymorphone Series of Opiate Narcotics. J. Med. Chem. 1978;21:101–106. doi: 10.1021/jm00199a018. [DOI] [PubMed] [Google Scholar]
- 13.Ben Haddou T, et al. Pharmacological Investigations of N-Substituent Variation in Morphine and Oxymorphone: Opioid Receptor Binding, Signaling and Antinociceptive Activity. PLoS ONE. 2014;9:e99231. doi: 10.1371/journal.pone.0099231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stavitskaya L, Coop A. Most Recent Developments and Modifications of 14-Alkylamino and 14-Alkoxy-4,5-Epoxymorphinan Derivatives. Mini Rev. Med. Chem. 2011;11:1002–1008. doi: 10.2174/138955711797247752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spetea M, Schmidhammer H. Recent Advances in the Development of 14-Alkoxy Substituted Morphinans as Potent and Safer Opioid Analgesics. Curr. Med. Chem. 2012;19:2442–2457. doi: 10.2174/092986712800269308. [DOI] [PubMed] [Google Scholar]
- 16.Schmidhammer H, et al. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 1. Highly Potent Opioid Agonists in the Series of (–)-14-Methoxy-N-Methylmorphinan-6-ones. J. Med. Chem. 1984;27:1575–1579. doi: 10.1021/jm00378a009. [DOI] [PubMed] [Google Scholar]
- 17.Schmidhammer H, Schratz A, Mitterdorfer J. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 8. 14-Methoxymetopon, an Extremely Potent Opioid Agonist. Helv. Chim. Acta. 1990;73:1784–1787. doi: 10.1002/hlca.19900730623. [DOI] [Google Scholar]
- 18.Schmidhammer, H. & Spetea, M. Development of 5-Substituted N-Methylmorphinan-6-ones as Potent Opioid Analgesics with Improved Side-Effect Profile. Int. J. Med. Chem. 1−12 (2012). [DOI] [PMC free article] [PubMed]
- 19.Schmidhammer H, et al. (−)-N,N′-But-2-ene-1,4-Diylbimorphinans. Pharmazie. 1991;46:101–102. [PubMed] [Google Scholar]
- 20.Dumitrascuta M, et al. Synthesis, Pharmacology, and Molecular Docking Studies on 6-Desoxo-N-Methylmorphinans as Potent μ-Opioid Receptor Agonists. J. Med. Chem. 2017;60:9407–9412. doi: 10.1021/acs.jmedchem.7b01363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaserer T, Lantero A, Schmidhammer H, Spetea M, Schuster D. μ Opioid Receptor: Novel Antagonists and Structural Modeling. Sci. Rep. 2016;6:21548. doi: 10.1038/srep21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi JN, Mosberg HI, Porreca F. Modulation of the Potency and Efficacy of Mu-Mediated Antinociception by Delta Agonists in the Mouse. J. Pharmacol. Exp. Ther. 1990;254:683–689. [PubMed] [Google Scholar]
- 23.Malmberg AB, Yaksh TL. Isobolographic and Dose-Response Analyses of the Interaction Between Intrathecal Mu and Delta Agonists: Effects of Naltrindole and its Benzofuran Analog (NTB) J. Pharmacol. Exp. Ther. 1992;263:264–275. [PubMed] [Google Scholar]
- 24.Zhu Y, et al. Retention of Supraspinal Delta-Like Analgesia and Loss of Morphine Tolerance in Delta Opioid Receptor Knockout Mice. Neuron. 1999;24:243–252. doi: 10.1016/S0896-6273(00)80836-3. [DOI] [PubMed] [Google Scholar]
- 25.Massotte D. In vivo opioid receptor heteromerization: where do we stand? Br J Pharmacol. 2015;172(2):420–34. doi: 10.1111/bph.12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujita W, Gomes I, Devi LA. Heteromers of μ-δ Opioid Receptors: New Pharmacology and Novel Therapeutic Possibilities. Br. J. Pharmacol. 2015;172:375–387. doi: 10.1111/bph.12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ananthan S. Opioid Ligands with Mixed Mu/Delta Opioid Receptor Interactions: An Emerging Approach to Novel Analgesics. AAPS J. 2006;8:E118–E125. doi: 10.1208/aapsj080114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ananthan S, et al. 14-Alkoxy- and 14-Acyloxypyridomorphinans: μ Agonist/δ Antagonist Opioid Analgesics with Diminished Tolerance and Dependence side Effects. J. Med. Chem. 2012;55:8350–8363. doi: 10.1021/jm300686p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Podolsky AT, et al. Novel Fentanyl-Based Dual μ/δ-Opioid Agonists for the Treatment of Acute and Chronic Pain. Life Sci. 2013;93:1010–1016. doi: 10.1016/j.lfs.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsumoto K, et al. Orally Active Opioid Mu/Delta Dual Agonist MGM-16, a Derivative of the Indole Alkaloid Mitragynine, Exhibits Potent Antiallodynic Effect on Neuropathic Pain in Mice. J. Pharmacol. Exp. Ther. 2014;348:383–392. doi: 10.1124/jpet.113.208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevenson. GW, et al. The Mixed-Action Delta/Mu Opioid Agonst MMP-2200 Does not Produce Conditioned Place Preference but Does Maintain Drug Self-Administration in Rats, and Induces in Vitro Markers of Tolerance and Dependence. Pharmacol. Biochem. Behav. 2015;132:49–55. doi: 10.1016/j.pbb.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lei, W., Vekariya, R. H., Ananthan, S., Streicher, J. M. A Novel Mu-Delta Opioid Agonist Demonstrates Enhanced Efficacy With Reduced Tolerance and Dependence in Mouse Neuropathic Pain Models. J. Pain., 10.1016/j.jpain.2019.05.017 (2019). [DOI] [PMC free article] [PubMed]
- 33.Hayes AG, Tyers MB. Determination of Receptors that Mediate Opiate Side Effects in the Mouse. Br. J. Pharmacol. 1983;7:731–736. doi: 10.1111/j.1476-5381.1983.tb10011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowdle TA. 1998. Adverse Effects of Opioid Agonists and Agonist-Antagonists in Anaesthesia. Drug Saf. 1998;19:173–189. doi: 10.2165/00002018-199819030-00002. [DOI] [PubMed] [Google Scholar]
- 35.Meert TF, Vermeirsch HA. 2005. A Preclinical Comparison between Different Opioids: Antinociceptive Versus Adverse Effects. Pharmacol. Biochem. Behav. 2005;80:309–326. doi: 10.1016/j.pbb.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Kieffer BL. Opioids: First Lessons from Knockout Mice. Trends Pharmacol. Sci. 1999;20:19–26. doi: 10.1016/S0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- 37.Manglik A, et al. Crystal Structure of the µ-Opioid Receptor Bound to a Morphinan Antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang W, et al. Structural Insights into μ-Opioid Receptor Activation. Nature. 2015;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marino KA, Shang Y, Filizola M. Insights into the Function of Opioid Receptors from Molecular Dynamics Simulations of Available Crystal Structures. Br. J. Pharmacol. 2018;175:2834–2845. doi: 10.1111/bph.13774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolber G, Langer T. LigandScout: 3-D Pharmacophores Derived from Protein-bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 41.Váradi A, et al. Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit β-Arrestin-2. J. Med. Chem. 2016;59:8381–8397. doi: 10.1021/acs.jmedchem.6b00748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manglik A, et al. Structure-based Discovery of Opioid Analgesics with Reduced Side Effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koehl A, et al. Structure of the µ-Opioid Receptor-Gi Protein Complex. Nature. 2018;558:547–552. doi: 10.1038/s41586-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Obeng S, et al. Structure-Activity Relationship Studies of 6α- and 6β-Indolylacetamidonaltrexamine Derivatives as Bitopic Mu Opioid Receptor Modulators and Elaboration of the “Message-Address Concept” To Comprehend Their Functional Conversion. ACS Chem. Neurosci. 2019;10:1075–1090. doi: 10.1021/acschemneuro.8b00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Claff T, et al. Elucidating the Active δ-Opioid Receptor Crystal Structure with Peptide and Small-Molecule agonists. Sci. Adv. 2019;5:eaax9115. doi: 10.1126/sciadv.aax9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Che T, et al. Structure of the Nanobody-stabilized Active State of the κ Opioid Receptor. Cell. 2018;172:55–67. doi: 10.1016/j.cell.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bock A, et al. Ligand Binding Ensembles Determine Graded Agonist Efficacies at a G Protein-Coupled Receptor. J. Biol. Chem. 2016;291:16375–16389. doi: 10.1074/jbc.M116.735431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidhammer H, Nussbaumer C, Patel D, Traynor JR. 14-O-Methyl Derivatives of 5-Methylnaltrexone and 5-Methylnaloxone. Helv. Chim. Acta. 1994;77:1585–1589. doi: 10.1002/hlca.19940770614. [DOI] [Google Scholar]
- 49.Guerrieri E, Mallareddy JR, Tóth G, Schmidhammer H, Spetea M. Synthesis and Pharmacological Evaluation of [3H]HS665, a Novel, Highly Selective Radioligand for the Kappa Opioid Receptor. ACS Chem. Neurosci. 2015;6:456–463. doi: 10.1021/cn5002792. [DOI] [PubMed] [Google Scholar]
- 50.Fürst S, Búzás B, Friedmann T, Schmidhammer H, Borsodi A. Highly Potent Novel Opioid Receptor Agonist in the 14-Alkoxymetopon Series. Eur. J. Pharmacol. 1993;236:209–215. doi: 10.1016/0014-2999(93)90591-5. [DOI] [PubMed] [Google Scholar]
- 51.D’Amour FE, Smith DL. A Method for Determining Loss of Pain Sensation. J. Pharmacol. Exp. Ther. 1941;72:74–79. [Google Scholar]
- 52.Jones BJ, Roberts DJ. The Quantitative Measurement of Motor incoordination in Naive Mice using an Accelerating Rotarod. J. Pharm. Pharmacol. 1968;20:302–304. doi: 10.1111/j.2042-7158.1968.tb09743.x. [DOI] [PubMed] [Google Scholar]
- 53.Spetea M, Bohotin CR, Asim MF, Stübegger K, Schmidhammer H. In Vitro and In Vivo Pharmacological Profile of the 5-Benzyl Analogue of 14-Methoxymetopon, a Novel µ Opioid Analgesic with Reduced Propensity to Alter Motor Function. Eur. J. Pharm. Sci. 2010;41:125–135. doi: 10.1016/j.ejps.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng Y, Prusoff WH. Relationship Between the Inhibition Constant (K1) and the Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 55.Litchfield JT, Jr., Wilcoxon F. A Simplified Method of Evaluating Dose-Effect Experiments. J. Pharmacol. Exp. Ther. 1949;96:99–113. [PubMed] [Google Scholar]
- 56.Jones G, Willet P, Glen RC, Leach AR, Taylor R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.