

Abstract
In past decades, interdisciplinary research has been of great interest for scholars. Thiazolidine motifs behave as a bridge between organic synthesis and medicinal chemistry and compel researchers to explore new drug candidates. Thiazolidine motifs are very intriguing heterocyclic five-membered moieties present in diverse natural and bioactive compounds having sulfur at the first position and nitrogen at the third position. The presence of sulfur enhances their pharmacological properties, and, therefore, they are used as vehicles in the synthesis of valuable organic combinations. They show varied biological properties viz. anticancer, anticonvulsant, antimicrobial, anti-inflammatory, neuroprotective, antioxidant activity and so on. This diversity in the biological response makes it a highly prized moiety. Based on literature studies, various synthetic approaches like multicomponent reaction, click reaction, nano-catalysis and green chemistry have been employed to improve their selectivity, purity, product yield and pharmacokinetic activity. In this review article, we have summarized systematic approaches for the synthesis of thiazolidine and its derivatives, along with their pharmacological activity, including advantages of green synthesis, atom economy, cleaner reaction profile and catalyst recovery which will help scientists to probe and stimulate the study of these scaffolds.
Keywords: Thiazolidine, Thiazolidinone, Synthesis, Biological profile, Anticancer, Antimicrobial, Antitubercular activity
Article Highlights
Thiazolidine derivatives have diverse therapeutic and pharmaceutical activity and are used in probe design.
The novel synthesis of thiazolidine derivatives using various agents is discussed with respect to yield, purity, selectivity and pharmacokinetic activity.
The accessible clinical applications in various biological targets are critically reviewed.
These data provide useful information for designing next-generation drug candidates.
Developing multifunctional drugs and improving their activity should be a focus of research.
Introduction
Heterocyclic composites are an important class of organic compounds possessing broad applications in various fields of science [1–7]. One of the most eminent heterocyclic motifs, thiazolidine, is a five-membered heterocycle system having the formula C3H7NS containing one nitrogen and one sulfur atom, and which exhibits notable medicinal and pharmaceutical properties. In the thiazolidine nucleus, a large number of substitutions are possible on 2, 4 and 5 positions responsible for enhancing the compound's pharmaceutical importance. Thiazolidine and its composites are key components of many natural products and drugs (Fig. 1), and are also present in many synthetic compounds such as anticancer [8–11], antimicrobial [12–14], antitumor [15, 16], antidiabetic [17], antiparasitic [18, 19], anti-inflammatory [20–22], antitubercular [23], antifungal [24], antiviral [25, 26], anti-HIV [27–29], cytotoxicity [30], antitrypanosomal [31], antinociceptive and anti-hypernociceptive compounds [32]. In addition, the use of thiazolidines as an inhibitor of tyrosyl-DNA phosphodiesterase I [33] and influenza neuraminidase [34], pro-drugs for the treatment of cystinosis [35], radioprotective against γ-irradiation [36] and as S1P1 receptor agonists [37, 38] has also been reported. They are also used in peptide and protein modification [39], protein chemical synthesis [40], as activators to innate immunity [41] and also act as immunostimulating agents [42].
Fig. 1.

Some commercially available thiazolidine-based drugs
These derivatives are also involved in various syntheses as synthetic precursors, potential biomarkers for oxidative stress and formaldehyde exposure [43], heterogeneous catalysts [44, 45], free radicals, superoxide anion radical and hydroxyl radical scavengers [46–48], inducible nitric oxide synthase (iNOS) inhibitors [49], to construct non-fullerene small molecules [50] and on and on. Green chemistry was formulated in the 1990s and includes 12 principles. The green processes diminish the adverse effects of any chemical reaction by following certain criteria viz. catalyst- and solvent-free synthesis and designing of biodegradable and less toxic products with high efficiency. In this review article, we have compiled researches which step ahead toward green chemistry. In the past few years, divergent synthetic strategies [51] have been introduced aimed at efficient and green synthesis using inexpensive reactants, nontoxic solvents, reusable catalysts, nanoparticle-catalyzed synthesis and solvent-free synthesis with high yields using different techniques such as microwave irradiation (MWI), sonochemistry, surface chemistry and others (Fig. 2). Multicomponent reactions (MCRs) (Strecker, Ugi, Bucherer-Bergs, Biginelli) are excellent pathways for the synthesis of heterocycles [52] as they have all the features for ideal synthesis like simple procedure for generating complex hybrid molecules in fewer steps which possess excellent pharmaceutical activity with high atom economy and eco-friendliness [53, 54]. The notable features of thiazolidine scaffolds compel us to study the literature and outline the current status of thiazolidine, its derivatives and also their biological significance. Recently, Jain et al. [55] reviewed the biological activity of thiazolidinone derivatives from 2000 to 2011. In 2013, Jain and coauthors [56] reviewed the multifarious applications of various thiazolidine-2,4-dione derivatives. Nanjan et al. [57] reviewed the antidiabetic activity of thiazolidinone. But there is no detailed review of the synthesis and biological activity of thiazolidines to date. So here, we have summarized a literature survey of different strategies developed for the synthesis of thiazolidines and their analogs, highlighting their activity and their use as starting materials in the synthesis of various heterocyclic systems with potent pharmaceutical properties during the period of 2014–2019, hoping to inspire new and even more creative approaches for researchers (Fig. 3). We have also included a comparative study of various synthetic protocols of thiazolidine derivatives with their advantages (+) and disadvantages (−) (Figs. 4, 5, 6, 7). In this review, an endeavor has been made to underline the recent issues in thiazolidine synthesis like environmental, health, cost and energy issues. By using truly greener pathways, we can overcome these factors for a promising future of thiazolidine derivatives.
Fig. 2.
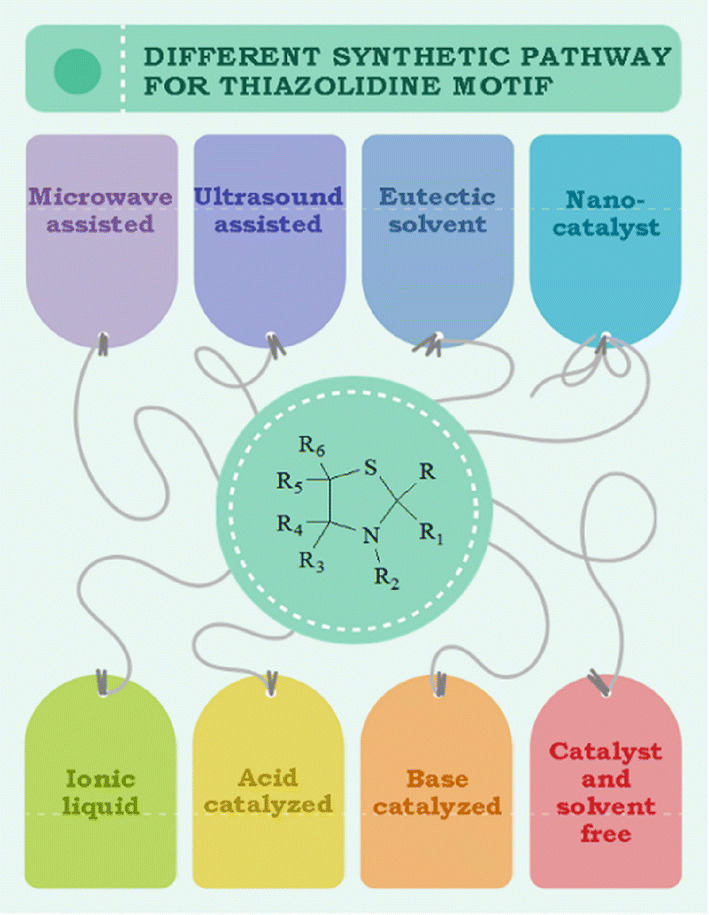
Different synthetic pathways for the thiazolidine motif
Fig. 3.
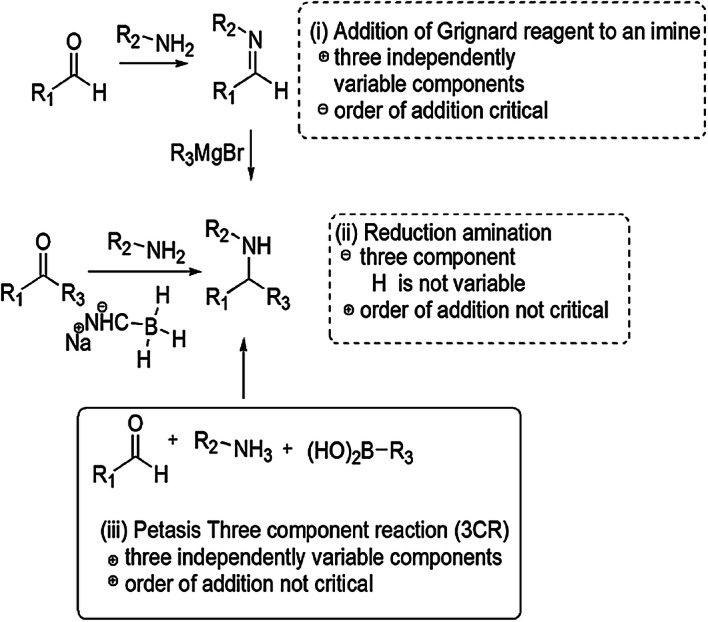
Synthesis of secondary amines via three different pathways; (iii) represents the efficiency and diversity of MCRs.
Modified from Ref. [61]
Fig. 4.

Fig. 5.

Fig. 6.

Fig. 7.

Synthesis of Thiazolidine Derivatives
Acid-Catalyzed Synthesis
Shelke and his coworkers [58] described a simple, more proficient, less wasteful, cost-effective, one-pot mechanism for the formation of bioactive 2-((substituted)-2-chloroquinolin-3-yl)-3-((substituted) phenyl) thiazolidin-4-ones (4) from 2-chloro quinoline-3-carbaldehyde (1), aniline (2) and thioglycolic acid (TGA) (3) with β-cyclodextrin-SO3H catalyst (Scheme 1). The authors studied this procedure in different conditions like various solvents (isopropanol, methanol, acetic acid, toluene, ethanol and DMF) and varied catalysts (p-TSA, sulfamic acid, hydrochloric acid and sulfanilic acid). In this synthesis, β-cyclodextrin-SO3H was the most favorable with water as its heterogeneous biopolymer-based solid acid catalyst and was reusable without any vital changes. A safe and greener reaction pathway, low toxicity, high yield of product and easy isolation method were advantages of this protocol. The reaction mechanism is depicted in Scheme 2. The mechanism shows that β-cyclodextrin-SO3H might first activate the aldehyde by enhancing the electrophilic character of its carbonyl moiety which formed an imine intermediate and β-cyclodextrin-SO3H anion, and this anion further increased the nucleophilic character of thioglycolic acid which reacted with the imine intermediate and formed an adduct, which was further activated by β-cyclodextrin-SO3H anion, followed by ring closure and final dehydration, resulting in formation of novel products.
Scheme 1.
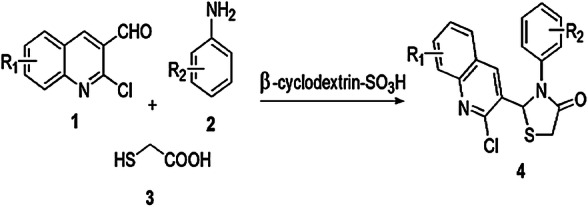
Synthesis of 1-[(substituted)-2-chloroquinolin-3-yl]-3-[(substituted) phenyl] thiazolidin-4-one 4
Scheme 2.

Possible mechanism for the synthesis of thiazolidinone derivative 4.
Modified from Ref. [58]
Bhattacharyya and coauthors [59] investigated an ingenious, catalytic, regio- and stereoselective methodology for the preparation of various 2-iminothiazolidines from racemic and non-racemic activated aziridines and substituted isothiocyanates using Lewis acid-catalyzed DROC. A general mechanism for DROC-mediated synthesis explained that initially N-arylsulfonylaziridine, activated by the Lewis acid (BF3·OEt2) with TBAHS, gave an intermediate which was highly reactive and showed SN2-type ring-opening reaction with isothiocyanates to furnish a new intermediate with an inverted configuration. This intermediate underwent 5-exo-dig cyclization to afford 2-iminothiazolidine (7) derivatives, which have high yields and excellent enantioselectivity (Scheme 4). In this synthetic strategy, different stereoisomers under different conditions gave enantio- and diastereospecific products. Firstly, enantiopure (R)-2-phenyl-N-tosylaziridine (R)-5a reacted with phenyl isothiocyanates 6a, using scandium(III) triflate as a catalyst in dichloromethane at 0 °C, to form (S,Z)-N,5-diphenyl-3-tosylthiazolidin-2-imine ((S)-7a) single regioisomer (52%) with low enantiomeric excess (34%) (Scheme 3). Various solvents, catalyst and temperatures were applied to obtain the best reaction conditions. When the reaction was catalyzed by BF3·OEt2 and TBAHS (tetrabutylammonium hydrogen sulfate) at −30 °C, high yields were obtained (97%) with excellent enantiospecificity (98% ee). The authors also studied the effect of different substituents on yield and stereoselectivity. This study continued in substituted (R)-2-phenyl-N-arylsulfonylaziridines and phenyl isothiocyanate under previous reaction conditions and formed corresponding 2-iminothiazolidines with good enantiomeric excess. When 2-aryl-N-tosylaziridines cyclized with phenyl isothiocyanate using BF3.OEt2 and TBAHS in CH2Cl2 at 0 °C, regioisomers of 2-iminothiazolidines were produced in high yields (up to 99%). With the same previous reaction conditions, except the amount of catalyst, trans-2, 3-disubstituted aziridines gave single diastereomers (de, ee > 99%) related 2-iminothiazolidines in 90% yield (Scheme 4).
Scheme 4.

Possible mechanism for the synthesis of thiazolidine derivatives 7 using Lewis acid-catalyzed domino ring-opening cyclization
Scheme 3.

Synthesis of thiazolidine derivatives using domino ring-opening cyclization
Multicomponent reactions are used for synthesis of various thiazolidine derivatives under different environmental conditions, some of which are summarized in Table 1. The comparison in Fig. 3 is illustrated in two points, concerning the addition of more than two components in a single step without considering their sequence [61]. In past years, researchers reviewed the mechanistic and chemical properties of MCRs, their sustainability and relation with biological activity [60, 61].
Table 1.
Different multicomponent reactions for the synthesis of thiazolidine derivatives

The anti-glioma activity and cytotoxicity of the thiazolidin-4-ones were evaluated by Da Silva and coworkers [62]. In this procedure, primary amines, aldehyde and mercaptoacetic acid were reacted via one-pot MCR in the presence of BF3 and p-toluenesulfonic acid (PTSA) and formed derivatives of thiazolidin-4-one (Scheme 5). Pyridine containing thiazolidinones gave good antitumor activity through decrease in cell feasibility of glioblastoma multiform cells. Among all synthesized derivatives, 9b, 9e, 9g and 10e exhibited potent antitumor effect against reference cells.
Scheme 5.
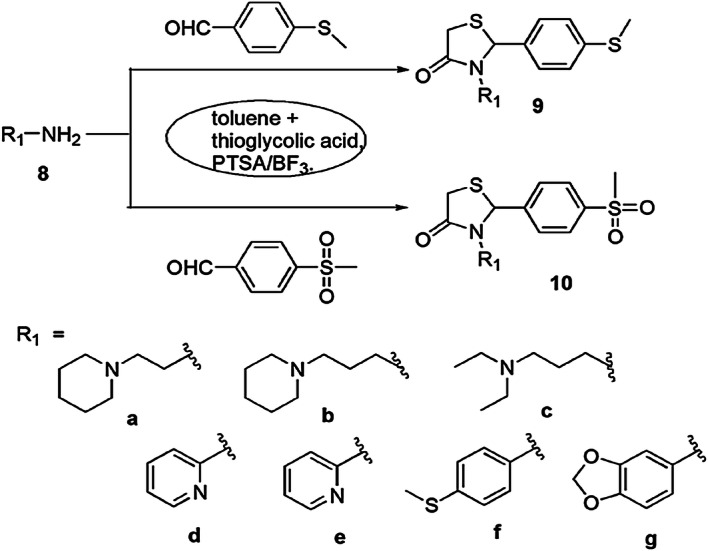
Synthesis of thiazolidinone derivatives 9 and 10 in the presence of PTSA/BF3
Recently, Rahim et al. [63] reported a novel and efficient method for the synthesis of arylhydrazide bearing thiazolidinone and analyzed them for α-amylase and urease inhibitors. In this protocol, hydrazine hydrate and 4-cyanobenzoate were refluxed with substituted aldehydes/acetophenone, resulting in corresponding Schiff base 11, which further reacted with thioglycolic acid using acetic acid to give the final product thiazolidinone 12 (Scheme 6). A screening of 11a–r and 12a–o was conducted for α-amylase and urease inhibitory processes, respectively. Among all compounds, six derivatives, 11b, 11e, 11f, 11j, 11k, 11r, and nine derivatives, 12a, 12b, 12e, 12f, 12g, 12h, 12j, 12k and 12l, were highly potent inhibitors as compared to reference drug acrobase (α-amylase inhibitor) and thiourea (urease inhibitory), respectively.
Scheme 6.

Synthesis of substituted thiazolidinone derivatives
Base-Catalyzed Synthesis
Kaboudin and colleagues [64] investigated a novel, facile, one-pot and high-yielding protocol for thiazolidin-4-one derivatives from one-pot, four-component condensation and cyclization of various benzaldehydes 13 and hydrazine with α-haloketones and allyl isothiocyanate (Scheme 7). In order to determine the optimal reaction conditions, the reaction was carried out using different catalysts like NaOH, pyridine, DBU and Et3N at different temperatures, and the best results were achieved in the presence of the Et3N catalyst with 7 h of reflux in methanol. The plausible reaction mechanism was decoded as the hydrazine reacting with allyl isothiocyanate and aldehyde to afford an intermediate, which showed nucleophilic substitution with α-chloroacetylchloride and underwent intramolecular cyclization, generating novel thiazolidin-4-one 14 (Scheme 8). Easy accessibility of reactant, operation simplicity, mild reaction conditions, clean reaction profile, high yields and purity of products were the notable advantages of this protocol.
Scheme 7.

Synthesis of thiazolidine derivatives 14(a–r) using basic catalyst
Scheme 8.

Putative mechanism for the synthesis of compound 14.
Modified from Ref. [64]
An efficient and greener one-pot method for the synthesis of a range of bioactive tetrahydrothiazolo[3,2-a]quinolin-6-ones was employed using cysteamine hydrochloride, 1,1-bis(methylthio)-2-nitro ethylene, trimethylamine with aromatic aldehyde and dimedone in H2O/EtOH and triethylamine by Bayat and coworkers [65] (Scheme 9). According to the investigation of the reaction mechanism, cysteamine hydrochloride (15) initially reacted with 1,1-bis(methylthio)-2-nitroethene (16) in H2O/EtOH to furnish 2-(nitromethylene)thiazolidine (18). Subsequently, aromatic aldehyde (19) on condensation with dimedone (17) formed an adduct. Further Michael addition of 2-(nitromethylene)thiazolidine 16 and the adduct resulted in the formation of an intermediate, followed by imine–enamine tautomerization, then nucleophilic addition of an amino moiety to the carbonyl group, and, finally, intramolecular cyclization produced desired products 20(a–p) in 75–94% yields (Scheme 10). The remarkable features of this new protocol were operational simplicity, mild reaction conditions, easily available substrates, easy workup, high yield in short time periods and a highly environmentally benign method.
Scheme 9.

Synthesis of thiazolidine derivatives 20a–p using Et3N
Scheme 10.

Possible mechanism for the synthesis of thiazolidine derivatives 20.
Modified from Ref. [65]
Santeusanio et al. [66] reported a domino reaction, also known as a cascade reaction, consisting of at least two subsequent reactions where segregation of the intermediate is not required and occurs intramolecularly with advantages like less waste generation, fast reaction and high atom economy. In the multifarious thiazolylidene ring congregation, ATAs (β-amidothioamides) can act as hetero-mononucleophiles or heterodinucleophiles. A library of thiazolylidene derivatives 24 and 25 was synthesized by the reaction of ATAs 21 and DDs 22 and 23 (Scheme 11). As a result, two types of products were formed with different pathways. A variety of solvents such as MeOH, THF and DMF were used for the completion of reaction without using a base, but these conditions did not give satisfactory results. Further study showed that when the reaction was performed with an organic base in CHCl3 at ambient temperature, the best results were obtained.
Scheme 11.

Synthesis of thiazolidine derivatives 24 and 25 using different ring closure pathways.
Modified from Ref. [66]
A simple, more efficient, cost-effective and convenient synthetic procedure for the synthesis of 2-heteroaryliino-1,3-thiazolidin-4-ones using 3-aminothiophenes, chloroacetyl chloride and ammonium thiocyanate along with different solvents and catalysts was reported by Revelant et al. [67] (Scheme 12). Compounds 26 and 27 were found effective in antiproliferative activity against five cancer cell lines, i.e. MDA-MB-231, HCT116, HT29, MCF7 and SW620.
Scheme 12.

Synthesis of compounds 26 and 27 under basic conditions
Yahiaoui and coauthors [68] described a reliable and simple protocol for the preparation of 2-thioxo-thiazolidin-4-ones 28 by condensation of aromatic amines and CS2 using NH4OH and chloroacetic acid, and the compounds were further treated with CH3CN under basic conditions (Scheme 13). The authors also developed a combined experimental and theoretical approach on the molecular structure of 2-thioxo-3N-(2-methoxyphenyl)-5 [4′-methyl-3′N-(2′-methoxyphenyl) thiazol-2′(3′H)-ylidene] thiazolidin-4-one.
Scheme 13.

Synthesis of 2-thioxo-3N-(2-methoxyphenyl)-5[4′-methyl-3′N-(2′-methoxyphenyl) thiazol-2′(3′H)-ylidene] thiazolidin-4-one
Hassan and colleagues [69] synthesized new thiazolidine derivatives 30(a–d) from hydrazide 29, ethyl bromoacetate and anhydrous sodium acetate using ethanol, and also evaluated their anticancer activity against HepG2, PC-3, MCF-7 and A549 human cancer cell lines (Scheme 14). However, these compounds did not show good anticancer activity.
Scheme 14.

Synthesis of substituted thiazolidine derivatives under basic conditions
Nanoparticle-Catalyzed Synthesis
Nanoparticles have a large surface-to-volume ratio and active coordination parts, compared to bulk materials. Nano-heterogeneous catalyst shows high activity, low energy consumption and 100% selectivity; therefore, these are suitable for the preparation of bioactive heterocyclic compounds [70, 71]. Recently, magnetic nanoparticles have received great attention because of their enormous benefits such as cost-efficiency, good stability, easy separation and high surface area [72]. Coating of nanoparticles with various materials like copper [73], chitosan [74] and l-proline [75] improves their activity [76].
Safaei-Ghomi and his group [77] developed a novel, efficient, pseudo-five-component preparation of bis-thiazolidinones 31(a–j) from substituted benzaldehyde, ethylenediamine and TGA (3) with nano-CdZr4(PO4)6 as a retrievable and robust catalyst (Scheme 15). The 0.6 mol% catalyst loading was sufficient for the good yield of products. It was observed that the best results were obtained with aromatic aldehydes with electron-withdrawing groups (EWG) as compared to electron-donating groups (EDG). The catalyst CdZr4(PO4)6 provided the surface to carry out the reaction. Primarily imine formed as an intermediate by the reaction of benzaldehyde and ethylene diamine on the surface of the catalyst, which activated the C=O, C=N and S–H groups. Then, attack by the sulfur atom on the activated imine group occurred, followed by intramolecular cyclization to give thiazolidinones (Scheme 16).
Scheme 15.

Synthesis of bis-thiazolidinones using nano-CdZr4(PO4)6
Scheme 16.

Mechanism for the synthesis of bis-thiazolidinones using nano-CdZr4(PO4)6.
Modified from Ref. [77]
Nano-CoFe2O4@SiO2/PrNH2, an appropriate addition in the field of synthetic chemistry, was achieved by Safaei-Ghomi and coworkers [78]. The treatment of CoFe2O4@SiO2 with APTES in refluxing toluene led to the concept of CoFe2O4@SiO2/PrNH2 nanoparticles as a peculiar magnetically recyclable, green and efficient catalyst, and the compound was found to be effective in the synthesis of 1,3-thiazolidin-4-ones 32 via MCRs of aniline, chloro benzaldehyde and TGA with different solvents (Scheme 17). The catalyst was reusable for seven progressive cycles. This novel reaction was carried out on different aromatic aldehydes and aniline, and it was concluded that the EWG in aldehydes reacted faster as compared to EDGs. Two mechanisms have been proposed for the reaction. In the first mechanism, aldehyde and amine interacted to form imine intermediate, then a sulfur atom of mercaptoacetic acid attacked the carbon atom of imine which was catalyzed by CoFe2O4@SiO2/PrNH2 NPs followed by intramolecular cyclization to give thiazolidinones (Pathway A). In the second mechanism, initially, formation of amide took place from aldehyde, thioglycolic acid and amine followed by cyclocondensation of a carbonyl group of aldehyde with sulfur and a nitrogen motif catalyzed by the same NPs to give cyclic compounds, thiazolidinones (Pathway B) (Scheme 18).
Scheme 17.
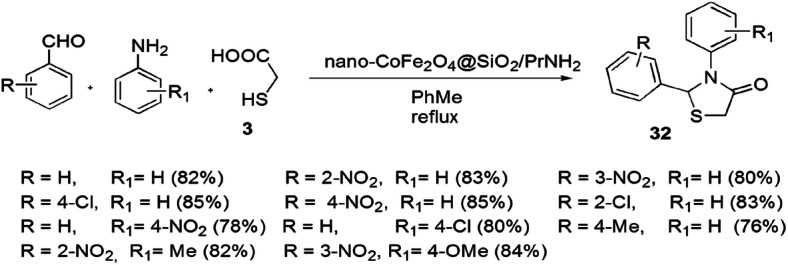
Synthesis of 1,3-thiazolidin-4-ones using nano-CoFe2O4@SiO2/PrNH2
Scheme 18.

Mechanism for the synthesis of 1,3-thiazolidin-4-ones using nano-CoFe2O4@SiO2/PrNH2.
Modified from Ref. [78]
The convenient and green FeNi3-ionic liquid (IL) magnetic nanoparticle (MNP)-promoted solvent-free, one-pot synthesis of varied 1,3-thiazolidin-4-ones from amine, aldehyde and TGA in excellent yields was established by Sadeghzadeh et al. [79] (Scheme 19). The use of robust, stable, easily recoverable, inexpensively synthesized and reusable magnetic ILs supported on FeNi3 nano-catalyst made the reaction protocol eco-friendly and of high synthetic utility. This protocol has significant additional advantages such as being solvent-free, an immobilization technique and a fast reaction procedure. The authors applied various conditions like different solvents and catalyst to obtain the sustainable reaction conditions, and it was observed that 0.001 g of FeNi3-IL MNPs at 50 °C without solvent was suitable for the reaction. The predicated mechanism for this synthesis was explained as the condensation of aldehyde and amine to form an imine intermediate which combined with thioglycolic acid to furnish the intermediate that consequently cyclized to afford product 33 (Scheme 20).
Scheme 19.
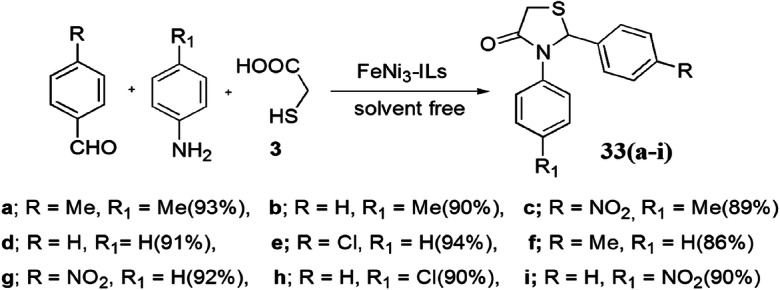
Synthesis of 1,3-thiazolidin-4-one using FeNi3-IL MNPs
Scheme 20.

Mechanism for the synthesis of 1,3-thiazolidin-4-one using FeNi3-IL MNPs.
Modified from Ref. [79]
An adept, feasible, one-pot synthesis of 1,3-thiazolidin-4-one derivatives was introduced by Azgomi et al. [80] using MNPs@SiO2-IL (nano-Fe3O4@SiO2-supported ionic liquid). The reaction was derived from the condensation of arylaldehyde, thioglycolic acid and anilines in solvent-free conditions with high to excellent yields (Scheme 21). The pathway of reaction involved MNPs@SiO2-IL, which activated oxygen of the carbonyl group and amine for the formation of an imine intermediate via enhancing the electrophilicity of the carbonyl group. The thioglycolic acid attacked the activated imine intermediate and formed a new intermediate, which, upon activation by catalyst MNPs@SiO2-IL, produced 34(a–j) (Scheme 22). The authors studied the effect of various catalysts such as dicarbethoxydihydrocollidine (DDC), Baker’s yeast, SiO2, HClO4-SiO2, H2SO4-SiO2, TfOH-SiO2 (trifluoromethenesulfonic acid-SiO2), Bi(SCH2COOH)3 and MNPs@SiO2-IL in different solvents like EtOH, H2O, CH2Cl2, EtOAc, DMF, CH3CN, THF and toluene and also under solvent-free conditions. The use of 0.0007 g of MNPs@SiO2-IL at 70 °C without solvent exhibited the best results. The catalyst was reused for ten runs without considerable loss in its activity.
Scheme 21.
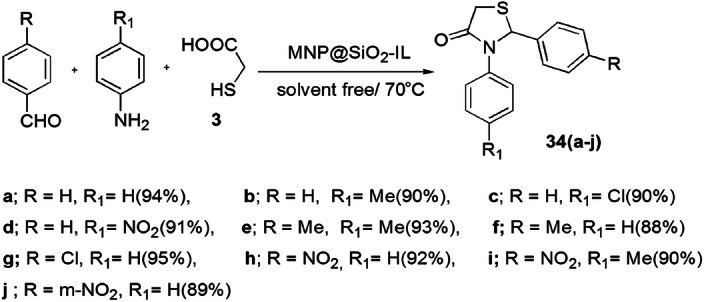
Synthesis of 1,3-thiazolidin-4-one 34 using MNP@SiO2-IL
Scheme 22.

Mechanism for the synthesis of 1,3-thiazolidin-4-one 34 using MNP@SiO2-IL.
Modified from Ref. [80]
Ionic Liquid-Assisted Synthesis
Ionic liquids are powerful solvents and catalysts, which have various advantages as they are nonvolatile, have low vapor pressure, minimize chemical waste, have a wide range of anion and cation combinations, have high chemical and thermal stability and are easily recyclable.
Subhedar et al. [81] observed one-pot, multicomponent reaction (MCR) using [Et3NH][HSO4] as a catalyst, and the products were found in excellent yields with a high purity, whereas lower yields were obtained in the absence of the catalyst. An environmentally and highly efficient protocol for 1,3-thiazolidin-4-ones was prepared from cyclocondensation of aniline, aromatic aldehyde and thioglycolic acid with [Et3NH][HSO4] (Scheme 23). The catalyst was reused five times, and high yields (80%) were obtained at 80 °C when 25 mol% of the catalyst was used. 1,3-Thiazolidin-4-ones were tested in vitro for their antimycobacterial activity, antimicrobial activity and cytotoxicity. This analysis showed that these derivatives displayed excellent selectivity towards dormant M. Bovis BCG and MTB H37Ra strains.
Scheme 23.
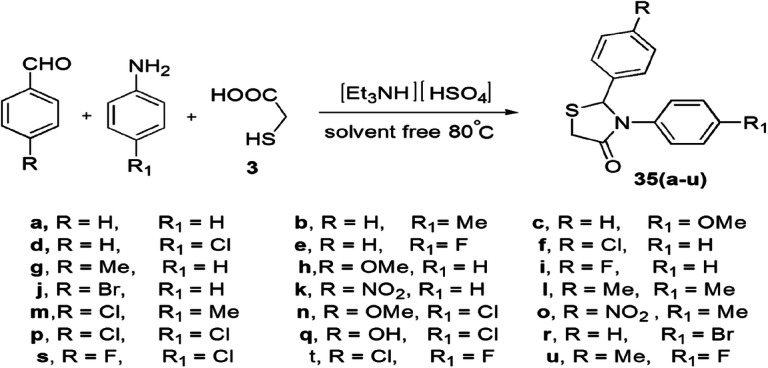
Synthesis of 1,3-thiazolidin-4-ones using [Et3NH][HSO4]
Chen et al. [82] reported a one-pot, three-component condensation reaction of substituted 2-aminobenzimidazoles, isothiocyanate and triethylamine using ethylene dichloride (EDC) as a solvent and formed 2-imino-1,3-thiazolidines and 2-imino-1,3-thiazolines (Scheme 24). In this protocol, 2-aminobenzimidazole adhered on ionic liquid (IL), then isothiocyanate proceeded with IL-anchored 2-aminobenzimidazole, yielding isothiourea which combined with 1,2-dichloroethene by inter- and intramolecular processes and generated 2-imino-1,3-thiazolidines. ILs provided high atom economy and simplicity in product isolation (Scheme 25).
Scheme 24.

Synthesis of thiazolidine derivatives using ionic liquids
Scheme 25.

Mechanism for the synthesis of thiazolidines 36 using ionic liquids.
Modified from Ref. [82]
Malla and colleagues [83] investigated an ingenious, greener, solvent-free, high-yielding, one-pot, three-component synthesis of thiazolidine derivatives from 1,3-diketones, cyanates and ethylchloroacetate using [Et3NH][HSO4] IL as a catalyst, which afforded good yields (92–98%) with high purity (Scheme 26). Here, [Et3NH][HSO4] is an inexpensive, eco-friendly catalyst, stable in water and air, exhibited both catalytic and medium engineering capability, is recyclable up to five runs without any significant loss of catalytic activity and also eliminated the additional use of the solvent. According to the possible reaction mechanism, initially, ionic liquid protonated the cyanates to furnish an intermediate, which underwent nucleophilic addition with 1,3-diketones and formed a new intermediate, which further reacted with ethylchloroacetate with consequent expulsion of HCl. The nitrogen of the compound attacked the carbonyl group and eliminated ethanol to form a C–N bond and finally formed the products 37 (Scheme 27). The authors applied diverse catalysts like [Et3NH][HSO4], [Me3NH][HSO4], [Et2NH2][H2PO4] and [Me3NH][CH3COO], and various solvents such as dimethyl sulfoxide (DMSO), EtOH, DMF, CH3NO2, toluene and [Et3NH][HSO4] in different amounts at varied temperature for optimization of the reaction conditions w.r.t. good yields and time (Table 2). The solvent also played a crucial role in the yields of reaction; i.e. nonpolar solvent (85%) ˃ polar-aprotic solvent (55–74%) ˃ polar-protic solvent (52%). However, the best results were obtained at 120 °C in 20 mol% of IL as a reaction media under solvent-free conditions. High atom economy, operational simplicity, an environmentally friendly nature, easy catalyst synthesis, low waste material, mild conditions and shorter reaction time are the notable advantages of this procedure.
Scheme 26.
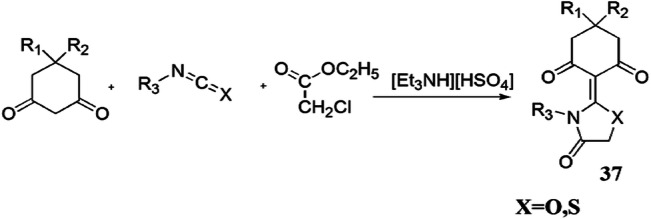
Synthesis of thiazolidine derivatives 37 using [Et3NH][HSO4]
Scheme 27.

Possible mechanism for the synthesis of thiazolidinones 37.
Modified from Ref. [83]
Table 2.
Synthesis of thiazolidinone derivatives using different substituents (37a–37n)

Novel thiazolidinone derivatives 38 were synthesized by Sadeghzadeh and coauthors, [84] in which aldehyde, amine and thioglycolic acid were reacted using heterogeneous catalyst Fe3O4/SiO2/Salen/Mn/IL MNPs under solvent-free conditions at ambient temperature with good to excellent results (Scheme 28). Here, the catalyst offered several advantages viz. ease of synthesis, easy recovery by an external magnet, operational simplicity and reusability up to six runs without any significant loss of activity. The authors applied various catalysts such as SiO2/Salen/Fe3O4/Mn/IL MNPs, phosphotungstic acid, NbCl5 [niobium(v)chloride], PEG–SO3H (sulfonated polyethylene glycol), InCl3 (indium chloride), Pd(PPh3)4, cerium(IV) ammonium nitrate and nano-SiO2/TiO2/RuO2/Pd/FeNi3 in different solvents (H2O, EtOH, THF, CH2Cl2, n-hexane) at different temperatures for optimization of the reaction condition w.r.t. yields and time. By the study, they concluded that Fe3O4/SiO2/Salen/Mn/IL MNP without solvent at room temperature gave the best results. This method has many merits, including high atom economy, synthetic efficiency, solvent-free conditions and shorter reaction time.
Scheme 28.
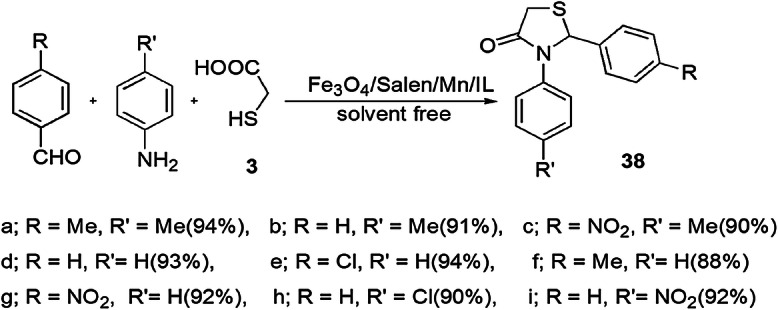
Synthesis of 1,3-thiazolidin-4-one 38 using Fe3O4/Salen/Mn/IL MNPs
Eutectic Solvent-Assisted Synthesis
Yedage and his group [85] synthesized 1,3-thiazolidin-4-ones by MCRs with aniline, benzaldehyde and TGA (3) in the presence of choline chloride:glycerol (1:2) as DESs (Scheme 29). DESs are the particular type of ionic solvents formed by combination of a Lewis acid, Brønsted acid and a base. These are used as an eco-friendly and reusable solvents or catalysts due to many characteristics like lower melting point as compared to individual components because of delocalization of charge on the whole mixture, high viscosity and inadequate density, so they can be liquid at the broad range of atmospheric conditions and incombustible. The mechanism of the reaction showed that DES firstly interacted with the oxygen of the carbonyl compound, and, simultaneously, the nitrogen atom of aniline attacked the carbon atom; then, imine was formed as an intermediate, which further reacted with TGA to form the product. At last, DES was removed and reused at least five times continuously without any damage.
Scheme 29.
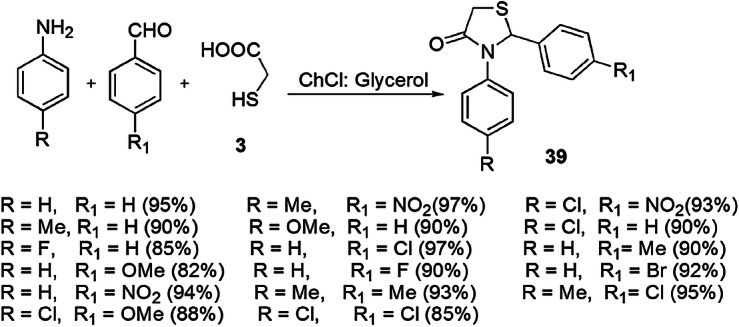
Synthesis of 1,3-thiazolidin-4-ones using DES
Catalyst- and Solvent-Free Synthesis
Dahiya et al. [86] investigated a series of 2-iminothiazolidines via DROC reaction without use of any catalyst and solvent (Scheme 30). An ecologically benign process containing inactive aziridine and aroyl isothiocyanates under the solvent-free conditions at room temperature produced 2-iminothiazolidines. Here, aroyl isothiocyanates played a role either as an ambient nucleophile (–SCN or –NCS) or electrophile. When this reaction was accomplished with solvent, the yield was comparatively low. This process was examined with substituted aziridine and aroyl isothiocyanates, and the authors demonstrated that the synthesis was facile when aziridine bearing electron-donating groups and aroyl isothiocyanates having electron-withdrawing substituents were taken. The mechanism of the reaction showed that this methodology involved [3+2] cycloaddition reaction which occurred by the SN2 pathway. This was confirmed by the reaction of optically active aziridine and aryl isothiocyanate, and it gave optically active product. The lone pair on the nitrogen of aziridine made it a nucleophile, and its attack on the sp hybrid carbon of heterocumulene NCS formed a thiourea intermediate, whose sulfur atom attacked the benzylic site of aziridine, and ring opening occurred (Scheme 31).
Scheme 30.

Synthesis of 2-iminiothiazolidine via [3+2] cycloaddition reaction
Scheme 31.
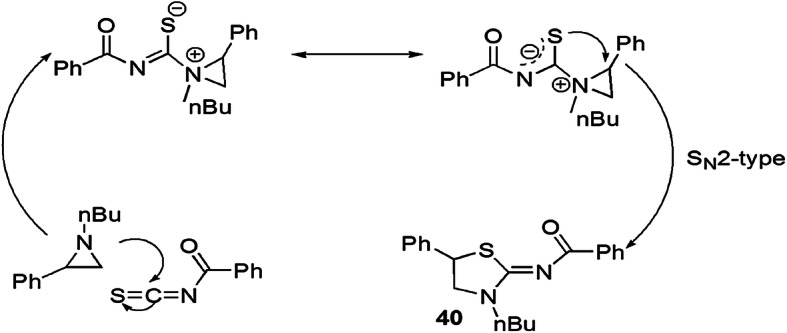
Possible mechanism for the synthesis of 2-imino thiazolidine 40 via [3+2] cycloaddition.
Modified from Ref. [86]
Microwave-Assisted Synthesis
Microwave treatment promotes better thermal management of reactions which are temperature-sensitive, and produces uniform heating and increase product yields using mild conditions. Mahmoodi et al. [87] reported an appropriate, one-pot, three-component reaction for the preparation of thiazolidinones using KSF@Ni as a heterogeneous catalyst by the reaction of aldehydes, thiosemicarbazide and maleic anhydride under MWI (Scheme 32). Microwave MCRs have several advantages such as shorter reaction time, high product yields, cleaner reaction profile, minimal by-products, and greater efficiency and cost-effectiveness. Heterogeneous catalyst has special features such as a long life span, easy removal and recovery, an innoxious nature, and is safe for storage. When the reaction was performed in the polar solvent, e.g. EtOH, DMF and MeOH, the reaction proceeded slowly. This method worked successfully under MWI at 70 °C and 50 psi in the mixture of DMF:toluene (1:1) in short reaction time (5 min) to give high yield (60%). The catalyst was recyclable for seven runs and gave almost the same yield of products. These products showed in vitro antibacterial activity against Gram-positive and Gram-negative bacterial strains E. coli, P. aeruginosa, S. aureus and M. luteus.
Scheme 32.
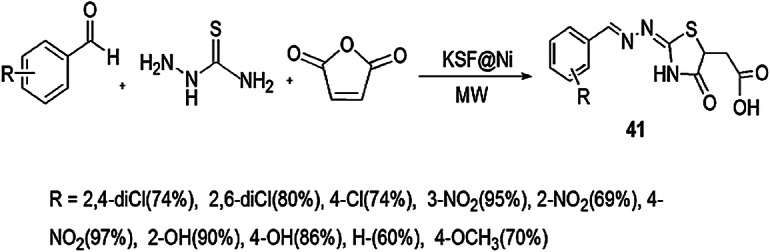
Synthesis of thiazolidinone 41 using MW irradiation
Sangeetha and coworkers [88] disclosed microwave-assisted, solvent-free synthesis of thiazolidine derivatives (42) using Amberlite IR-120H resin as a greener and reusable catalyst. 3/5-Alkyl-2,6-diarylpiperidin-4-ones were condensed with thiosemicarbazide and ethyl-2-bromopropionate under 250 W MWI, which afforded high yield of 2,6-diarylpiperidin-4-ylidene thiazolidin-4-one derivatives within 3–6 min. In contrast, in the conventional method, it took more time and gave comparatively low yields (Scheme 33). All the synthesized compounds were evaluated for in vitro antibacterial activity against five different bacterial strains S. aureus, S. pyogenes, P. aeruginosa, K. pneumoniae and E. coli and antitubercular activity against M. tuberculosis H37Rv, using streptomycin and isoniazid (INH) as standard drugs, respectively. Among all compounds, most displayed good antibacterial potency, and 42c and 42d showed high antitubercular activity.
Scheme 33.

Synthesis of new thiazolidine derivatives under MWI
Ultrasound-Assisted Synthesis
Ultrasound irradiation is used as an alternative source of energy, providing a green pathway for various reaction pathways [89] and is based on the acoustic cavitation phenomenon. Safaei-Ghomi and coauthors [90] designed a facile, effective, one-pot, three-component synthesis of thiazolidinones from aldehyde, aniline and TGA using nano-CdZr4(PO4)6 as an effective heterogeneous catalyst applying 60 W ultrasonic irradiation (Scheme 34). In this method, initially carbonyl groups of aldehyde and amine reacted to form an imine intermediate, which was further attacked by the sulfur atom of thioglycolic acid and underwent intramolecular cyclization by removal of water to furnish 1,3-thiazolidin-4-ones with 88% yield in 25 min. According to the reaction mechanism, it was suggested that nano-CdZr4(PO4)6 activated the carbonyl moiety of aldehyde and acid via coordinating with the carbonyl oxygen (Scheme 35). For comparative study, the authors employed diverse solvents, e.g. H2O, EtOH, PhMe, THF and DMF, and catalysts like H2SO4 and nano-SnO/ZrO2/CdZr4(PO4)6. However, PhMe and nano-CdZr4(PO4)6 were proved best as they gave high yields in short reaction time. The authors also studied the reaction under reflux conditions, but it displayed low yields in long reaction time as compared to ultrasonic irradiations. The use of nano-CdZr4(PO4)6 as an efficient, highly active, easily recovered, eco-friendly catalyst with notable reusability without loss of its activity is the superior feature of this method. Simple reaction procedure, easily available reactants, use of sonochemical technique, high yields, synthetic efficiency, easy workup and short reaction time are the additional benefits of this protocol.
Scheme 34.

Synthesis of 1,3-thiazolidin-4-ones by nano-CdZr4(PO4)6
Scheme 35.

Mechanism for the synthesis of thiazolidine derivatives using nano-CdZr4(PO4)6.
Modified from Ref. [90]
Shaabani et al. [91] reported novel MCRs for synthesis of pseudopeptide-linked rhodanine scaffolds using combination of tandem Michael/domino cycloaddition/Ugi reaction strategy. In this diversity-oriented synthesis, initially, primary amines, carbon disulfide and maleic anhydride were condensed to form thiazolidine 44, which further reacted with benzaldehydes, aniline and isocyanides using water under ultrasound irradiations to produce final product 45 in good yields (Scheme 36). While using conventional stirring, the conversion rate was very slow. The use of easily available substrate, green solvent, ultrasound radiations and without use of any catalyst make the reaction protocol inexpensive and eco-friendly. The authors have also investigated the effect of solvents like DMF, THF, EtOH and CH3CN on the reaction.
Scheme 36.

Synthesis of pseudopeptides containing rhodanine scaffolds
Catalyst-Free Synthesis
Salehitabar et al. [92] demonstrated the feasible methodology for the synthesis of ethyl 5-(alkylimino)-4-hydroxy-2-(arylimino)-3-alkylthiazolidine-4-carboxylates 46 from ethyl 3-chloro-3-(alkylimino)-2-oxopropanoate with 1-alkyl-3-arylthioureasin MeCN (acetonitrile) at room temperature via one-pot synthesis to afford various derivatives in good yields. The authors applied various solvents such as acetone, dry MeCN, tetrahydrofuran or toluene to achieve the best reaction conditions and concluded that dry MeCN was suitable and gave 73% yield at room temperature. Ease of operation, broad range of substrate, high synthetic utilit, and good yields are the main advantages of this procedure (Scheme 37).
Scheme 37.

Putative mechanism for the synthesis of compound 46.
Modified from Ref. [92]
Halimehjani et al. [93] proposed the rapid, one-pot, three-component synthesis of thiazolidine-2-thiones using nitroepoxide, primary amine and carbon disulfide in H2O at ambient temperature with good yield of products (Scheme 38). Simplicity, efficiency, mild conditions, high yield of products and short reaction times are the attracting features of this protocol.
Scheme 38.

Synthesis of thiazolidine-2-thiones without using catalyst
A novel catalyst-free, one-step synthesis of versatile thiazolidine derivative 48 has been developed by Aly and coworkers [94] using bisthiosemicarbazones and DMAD in methanol to afford good yields (Scheme 39). All synthesized compounds were screened for antagonist of the leukotriene B4 (LTB4) receptor. Some molecules were found potently active towards BLT1 and BLT2 receptors.
Scheme 39.

Synthesis of thiazolidine derivatives under catalyst-free conditions
Solvent-Free Synthesis
Ebrahimi and coworkers [95] reported the synthesis of 1,3-thiazolidin-4-one using APS (ammonium persulfate) as a catalyst under solvent-free conditions. The use of an economical catalyst, high yield and high atom economy are the benefits of this reaction. The synthesis involved cyclocondensation reaction of substituted aniline, benzaldehyde and thioglycolic acid (Scheme 40). The reaction was tested under different conditions viz. solvents, temperature and catalysts. The suitable conditions were solvent-free synthesis at 90 °C in the presence of 10 mol% of APS which yielded 84% product. To explore the synthetic utility of the protocol, different aldehydes were taken, and the yield of products was not sensitive to electronic variations and steric factor.
Scheme 40.

APS-catalyzed synthesis of thiazolidinon-4-ones 49
Metal-Catalyzed Synthesis
A novel approach for the construction of [60]-fullerothiazolidinethiones from C60, aliphatic amines and CS2 via Cu-catalyzed aerobic oxidative reaction was developed by Wu et al. [96] (Scheme 41). The authors applied various catalysts viz. CuSO4·5H2O, CuCl2, CuBr2, CuCl, CuBr, CuI and CuCl2·2H2O in different solvents like DMF/o-DCB, o-DCB and DMF for good yields and reaction time. From the study, they concluded that the best results were obtained in the presence of Cu(OAc)2·H2O (20 mol%) with DMF/o-DCB (4:1). The suggested reaction mechanism showed that primary amine and carbon disulfide reacted in DMF and o-DCB in aerobic conditions to produce dithiocarbamic acid and further reacted with C60 to form aminium radical cation and C60 radical anion via a SET mechanism. Oxidative amination reaction took place in the C60 radical anion, O2 and aminium radical cation to form a C60–N bond, followed by Cu-catalyzed aerobic oxidation to produce the C60–S bond (Scheme 42).
Scheme 41.

Synthesis of [60]fullerothiazolidinethiones 50 by DMF
Scheme 42.

Mechanism for the synthesis of [60]fullerothiazolidinethiones 50.
Modified from Ref. [96]
Shehzadi et al. [97] developed an ingenious, high-yielding, one-pot four-component strategy for the synthesis of a library of thiazolidine-2-imines from aldehydes, alkynes, amines and isothiocyanates using Cu(I) and Zn(II) as composed catalyst, and examined their inhibitory activity against acetylcholinesterase (AChE) (Scheme 43). The authors applied different catalysts, solvents and temperature range using various ratios of reactants to optimize reaction parameters. According to the possible reaction mechanism, initially, metal acetylide attacked imine and generated an alkyn-amide nucleophile, which further reacted with isothiocyanate as an electrophile to form an intermediate. This intermediate has two nucleophilic atoms, thus two possibilities occur: nitrogen attack gave S-cyclized product thiazolidine-2-imine; and sulfur attack produced N-cyclized product imidazolidine-2-thione 51. Single-crystal X-ray analysis confirmed the existence of 2-imino thiazolidine. All analogs demonstrated good inhibition activity, but derivative 51 s exhibited 88-fold stronger inhibition (IC50, 0.0023 ± 0.0002 μM) than neostigmine methyl sulfate (standard drug).
Scheme 43.

Synthesis of thiazolidin-2-imines 51 using metal catalyst
Multistep Synthesis
A series of novel 4-thiazolidinone containing 1,8-dioxo-octahydroxanthene derivatives were prepared by Robati and colleagues [98] by the reaction of 1,3-dicarbonyl cyclic compounds, nitro benzaldehyde and isothiocyanate with dialkylacetylene dicarboxylates in ethanol at ambient temperature via four-component reaction. The reaction was accompanied by an economical catalyst and less harmful solvent under mild reaction conditions, with larger product yields and fewer waste products. This procedure consisted of sequential addition of 1,3-cyclohexanedione and nitro benzaldehydes to form nitro 1,8-dioxo-octahydroxanthene derivatives, which underwent reduction by SnCl2·2H2O to produce the amino octahydroxanthene, that, upon refluxing with isothiocyanates, formed thioureas, which were then subjected to further reaction with dialkylacetylene dicarboxylate (DAAD) to form the final products 52 (Scheme 44).
Scheme 44.

Synthesis of 1,8-dioxo-octahydroxanthene derivatives containing 4-thiazolidinone 52(a–f)
A nitrogen equivalent of an aldehyde or ketone where the carbonyl group is exchanged by an imine or azomethine group is termed as a Schiff base. This is the most consequential moiety in the medicinal and pharmaceutical fields. Kandile et al. [99] developed a novel synthesis by reacting a Schiff base and TGA with anhydrous ZnCl2 or DMF, and the product, bis spiro[(5-methylindoline3,2-(4 H) thiazolidine)-2,40(1H)-dione]1,10-biphenyl, was obtained (Scheme 45) and further scrutinized for antimicrobial activity.
Scheme 45.

Synthesis of bis spiro[(5-methylindoline3,2-(4H)thiazolidine)-2,40(1H)-dione]1,10-biphenyl derivatives 53
The novel synthesis of 4-thiazolidinones was reported by Cihan-Üstündag et al. [100]. The protocol followed multistep synthesis as initially p-fluoro aniline reacted with NaNO2 and converted into the respective diazonium salt, which combined with ethyl-2-benzyl-3-oxobutanoate, which was further refluxed in concentrated HCl to produce ethyl 5-fluoro-3-phenyl-1H-indole-2-carboxylate. Ethyl 5-fluoro-3-phenyl-1H-indole-2-carboxylate was converted to respective hydrazide in the presence of hydrazine and was attacked by isothiocyanates to form substituted thiosemicarbazides, further cyclized using ethyl bromoacetate and fused sodium acetate to give desired products, 5-fluoro-N′-(4-oxo-3-substituted-1,3-thiazolidinon-2-ylidene)-3-phenyl-1H-indol-2-carbohydrazides 54(a–g) (Scheme 46).
Scheme 46.

Synthesis of thiazolidine derivatives 54(a–g) via multistep synthesis
On-Surface Synthesis
The convenient, rapid, highly reproducible, one-pot, three-component synthesis of 4-thiazolidinone derivatives by the condensation of aldehyde, amine and TGA (3) at room temperature with silica gel as promoter was established by Thakare and coworkers [101] (Schemes 47, 48). The possible reaction pathway of the reaction indicated that aldehyde along with amine produced an imine intermediate which combined with thioglycolic acid to form another intermediate. Silica gel activated the carbonyl group by increasing their electrophilicity, and, consequently, intramolecular addition took place, and a new intermediate was obtained. On further removal of H2O molecule, the cyclized product of substituted 4-thiazolidinone was obtained. The authors investigated this reaction in various solvents like THF, Et2O, acetonitrile, dichloromethane and 1,2-dichloroethene, but dichloromethane was found to be the most suitable. The products were obtained without any workup procedure. Immediate synthesis and use of inexpensive and nontoxic chemicals make this protocol green and eco-friendly.
Scheme 47.
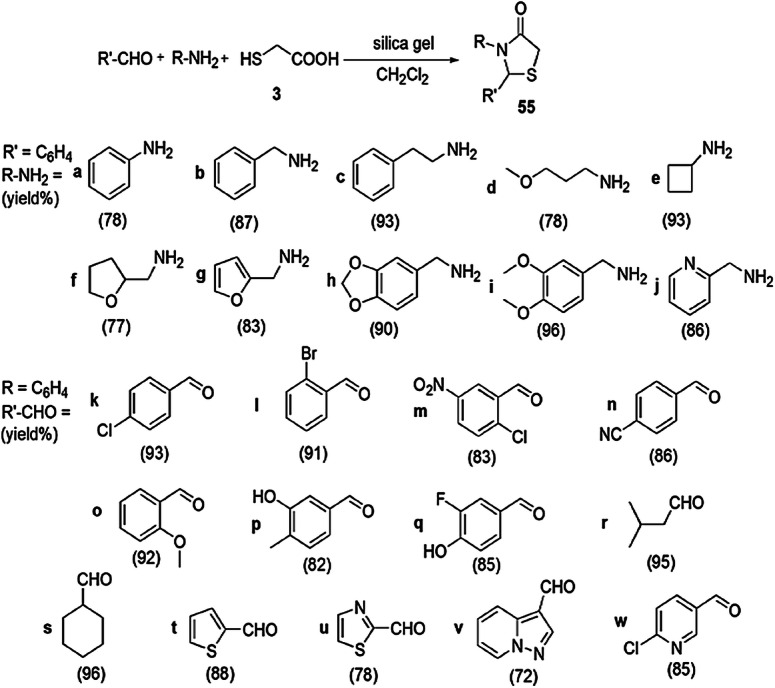
On-surface synthesis of thiazolidinone derivatives 55(a–w)
Scheme 48.

Silica gel-mediated synthesis of thiazolidininone 55.
Modified from Ref. [101]
A simple and efficient silica gel-mediated synthesis of thiazolidine derivatives was developed by Singh and colleagues [102]. The derivatives were also examined as antiproliferative agents. In this method, propargyl amine and isothiocyanate reacted in CH2Cl2 that underwent slow autoxidation at room temperature using CDCl3 to afford thiazolidin-4-ones 56 in good to high yields (Scheme 49). The antiproliferative properties were investigated against HCT-116 (colon) and MCF7 (breast) cancer cell lines using MTT growth assay. Some of the synthesized analogs showed good activity without affecting normal cell lines.
Scheme 49.

Synthesis of (Z)-2-imino-5-(Z)ylidene-N-substituted thiazolidines 56
Biological Activity of Thiazolidine Derivatives
Antimicrobial Activity
Abdel Hafez and coworkers [103] developed a series of 2-(2-(1-(3,4-dimethoxyphenyl)ethylidine)hydrazono)-substituted thiazolidinone derivatives and tested them for antimicrobial activity against fungal strains (A. niger and A. flavus) and yeast strains (S. cerevisiae, C. albicans). Thiosemicarbazone derivatives (57a and 57b) were refluxed with appropriate α-halo carbonyl compounds such as chloroacetone and ethyl 2-bromopropanoate using anhydrous sodium acetate in ethanol to produce corresponding thiazolidine analogs 58 and 59 (Scheme 50). Among all compounds, 57a, 57b, 58 and 59b showed similar antifungal activity as compared to reference drug, ketoconazole. Compound 59b was found the most potent for antifungal activity.
Scheme 50.

Synthesis of thiazolidine derivatives 57(a–b), 58 and 59(a–b)
Obydennov et al. [104] synthesized 1,3-thiazolidin-4-one and pyrrolidine-2,3,5-trione motifs linked to (5Z)-[2-(2,4,5-trioxopyrrolidin-3-ylidene)-4-oxo-1,3-thiazolidin-5-ylidene]acetate derivatives via an exocyclic C=C bond, explained their isomeric form and also evaluated their in vitro fungicidal activity on R. solani, A. solani, F. solani, C. coccodes and P. infestans fungi, by the poisoned food technique. The reaction of thioamides and acetylene dicarboxylate in ethanol at ambient temperature gave 2-methylidene-1,3-thiazolidin-4-one derivatives, which reacted with oxalyl chloride in dry MeCN at 70 °C to produce 4-(4-oxo-1,3-thiazolidin-2-ylidene)pyrrolidine-2,3,5-trione derivatives 61(a–f) in 57–87% yield. Here, compounds 60(a–f) have a Z configuration of exocyclic bond in the C-5 position. In the C-2 position, configuration of the bond was affected by the solvent system, steric hindrance and intramolecular interaction such as hydrogen bonding. The authors also explained the pathway of isomerization of compounds 61(a–f) as illustrated in Scheme 51. Compound 61b was found to be most potent toward all the strains with EC50 values of 0.052–0.445 mg/mL using Consento as the standard drug.
Scheme 51.

Synthesis of 4-(4-oxo-1,3-thiazolidin-2-ylidene)pyrrolidine-2,3,5-trione derivatives 61(a–f)
Trotsko and coauthors [105] synthesized (2,4-dioxothiazolidin-5-yl/ylidene)acetic acid derivatives using thiourea and maleic anhydride as starting materials under different reaction conditions, as illustrated in Scheme 52, and evaluated them for antibacterial activity using broth microdilution method. Cefuroxime and oxacillin were used as standard drugs. For reference strain, American Type Culture Collection (ATCC) B. subtilis, M. luteus, B. cereus, S. aureus and S. epidermidis for Gram-positive bacteria and E. coli, K. pneumoniae, Proteus mirabilis and Pseudomonas aeruginosa for Gram-negative bacteria were used. The researchers showed that the presence of an EWG at the phenyl ring was more potent as compared to an EDG; the geometry of the molecule did not affect the activity. Compound 64 was proved to be the most active compound with an MIC of 3.91 mg/L.
Scheme 52.

Synthesis of 1,3-thiazolidin-4-ones 62 and 63
Trotsko and his group [106] synthesized thiazolidine-2,4-dione-based chlorophenyl thiosemicarbazone hybrids, which behaved as “hybrid pharmacophores” and were screened for antibacterial activity by broth microdilution technique using cefuroxime, ciprofloxacin and oxacillin as standard drugs. The authors combined two active moieties TZD (thiazolidine-2,4-dione) and thiosemicarbazides, which contain an N–N–C(=S)–N structural fragment, and they exhibited good potency of antibacterial activity at low nontoxic concentrations. The antibacterial activity was performed on 12 strains of Gram-positive and Gram-negative bacteria. Five derivatives (65a, 65b, 66a, 66b, 68g) were most potent against all used Gram-positive bacterial strains (Table 3). The pathway of the reaction is outlined in Scheme 53.
Table 3.
The activity of most potent chlorophenylthiosemicarbazones against Gram-positive bacteria determined on the basis of MIC (minimal inhibitory concentration, in mg/L and µM) and MBC (minimal bactericidal concentration, in mg/L and µM) with cytotoxic effect on human embryonic kidney cells (HEK-293) after 24 h of incubation
| Compound | S. aureus | S. aureus | B. subtilis | EC70 ±SD (mg/L) (toxicity threshold) | |||
|---|---|---|---|---|---|---|---|
| ATCC 6538 | ATCC 25923 | ATCC 6633 | |||||
| MIC | MBC | MIC | MBC | MIC | MBC | ||
| 76a | 3.91 | 3.91 | 3.91 | 62.5 | 31.25 | 125 | 30.82 ±3.73 |
| 8.4 | 8.4 | 8.4 | 135 | 67.5 | 270 | ||
| 76b | 3.91 | > 1000 | 3.91 | > 1000 | 7.81 | 62.5 | 21.28 ±3.84 |
| 7.9 | > 2010.6 | 7.9 | > 2010.6 | 15.7 | 125.7 | ||
| 77a | 7.81 | 250 | 7.81 | 250 | 7.81 | 250 | 14.86 ±1.12 |
| 16.9 | 542.4 | 16.9 | 542.4 | 16.9 | 524.4 | ||
| 77b | 7.81 | 500 | 7.81 | 500 | 7.81 | 500 | 10.94 ±1.35 |
| 16.9 | 1084.8 | 16.9 | 1084.8 | 16.9 | 1084.8 | ||
| 79g | 31.25 | > 1000 | 7.81 | > 1000 | 3.91 | 62.5 | 19.01 ±3.80 |
| 63.7 | > 2036.9 | 15.9 | > 2036.9 | 8 | 127.3 | ||
| Cefuroxime | 0.98 | – | 0.49 | – | 15.63 | – | |
| 2.3 | 1.2 | 36.8 | |||||
Scheme 53.

Synthesis of thiazolidine derivatives with highly active compounds
Xianga and colleagues [107] investigated a novel synthesis of thiazolidin-2-cyanamide derivatives, and their antimicrobial activity was determined against the T3SS of Xanthomonas oryzae on rice. The title compounds were developed in multistep synthesis, i.e. firstly Meerwein arylation took place in substituted aniline and formed an intermediate, 5-substituted phenyl-2-furancarboxylic acid, which on reaction with thionyl chloride, 2-cyanoiminoradical-1,3-thiazolidine and K2CO3 in acetonitrile solution furnished the desired compounds in good yields (Scheme 54). The authors concluded that the new compounds reduced the symptoms of disease, displayed antivirulence effects and did not affect bacterial growth.
Scheme 54.

Synthesis of (thiazolidin-2-ylidene) cyanamide derivatives 69(a–n)
Abdel-Galil et al. [108] synthesized thiazolidinone derivatives bearing a phenyl benzoate nucleus. The derivatives were screened for in vitro antibacterial activity against two types of bacterial strains, Gram-positive bacteria, E. coli and Gram-negative bacteria, S. aureus, and was compared with a standard chemotherapeutic drug (ampicillin) using disc diffusion method (Scheme 55). Knoevenagel condensation of thiazolidin-5-one derivatives with 4-formylphenyl benzoate in the presence of piperidine afforded arylidine products 70(a–c). Compounds 70(a–c) showed good to moderate activity against reference strains.
Scheme 55.

Synthesis of thiazolidin-5-one derivatives 70(a–c)
Barakat et al. [109] described a scheme for the preparation of new derivatives of pyrrolidine/thioxothiazolidin-4-ones containing spiro-oxindole. The condensation reaction between isatin and l-proline generated azomethine ylide in situ, which underwent 1,3-dipolar cycloaddition with 5-arylidene-2-thioxothiazolidin-4-one (71a and 71b) (prepared by a previously reported method by Knoevenagel condensation) to afford spiro-oxindole/pyrrolidine/thioxothiazolidin-4-one derivatives (72a, 72b) in excellent yields (Scheme 56). These compounds were tested against two Gram-positive bacteria, S. pneumonia and B. subtilis and two Gram-negative bacteria, P. aeruginosa and E. coli, and they showed significant antibacterial properties. The antifungal activity of compounds was elucidated against A. fumigates, S. racemosum, G. candidum and C. albicans with the diffusion and serial dilution method. Fluconazole and amphotericin B were used as standard drugs. The structure of compounds 72a and 72b were determined with X-ray single-crystal diffraction, Hirshfeld surface analysis and DFT studies at the B3LYP/6-311G (d,p) level of theory. The 3D structures of both compounds were different, due to presence of diverse substituents at the phenyl ring and co-crystallization with solvent molecules. Compound 72a docked with hydrophobic–hydrophobic interaction, but compound 72b docked with both hydrophobic–hydrophobic and H-bonding interactions. Aminoglycoside phosphotransferase and lanosterol 14 α-demethylase were selected as target proteins for antibacterial and antifungal activity, respectively. Both compounds exhibited more antimicrobial activity against standard drugs. Compound 72b gave better activity than 72a against B. subtilis, S. pneumonia and E. coli and also showed high potency against antifungal treatment. The molecular docking study revealed that substitution on phenyl rings plays an important role in the geometry of compounds, which controls its behavior and mode of interaction. The study established that hybrid compounds 72a and 72b showed good results against skin infection and wound infection.
Scheme 56.

Synthesis of compounds 71 and 72
(Z)-5-Benzylidene-3-(5-heptadecyl-1,3,4-thiadiazol-2-yl) imino thiazolidin-4-one derivatives were prepared by Abdelmajeid et al. [110] and were further evaluated for their antimicrobial activity against E. coli (Gram-negative bacterial strain), S. aureus (Gram-positive bacterial strain) and A. flavus and C. albicans (two fungal species) by using modified Kirby–Bauer disc diffusion technique and Mueller–Hinton agar method. Equimolar quantities of stearic acid and thiosemicarbazide in the presence of POCl3 produced 5-heptadecyl-1,3,4-thiadiazole-2-amine, which on chlorination with chloroacetylchloride formed 2-chloro-N-(5-heptadecyl-1,3,5 thiadiazole-2-yl) acetamide, which further reacted with KSCN to give 3-(5-heptadecyl-1,3,4-thiadiazol-2-yl)-2 imino thiazolidin-4-one (73), and 73 further combined with benzaldehyde to produce (Z)-5-benzylidene-3-(5-heptadecyl-1,3,4 thiadiazol-2-yl) imino thiazolidin-4-one (74) (Scheme 57). Compound 74 was more potent for antimicrobial activity as compared to compound (73). For positive control of antibacterial and antifungal activity, ampicillin and amphotericin B were used as standard drugs, respectively. Both products are safe for the environment and humans and are widely used in beauty products, fabric and dye industries as an emulsifier and wetting agent.
Scheme 57.

Synthesis of compounds 73 and 74
A class of novel analogs, thiazolidinediones (TZD), were prepared by Trotsko and coworkers [111], and the compounds were scrutinized for in vitro antiproliferative and antibacterial activity. The synthesis of target compounds by 2-(2,4-dioxothiazolidin-5-yl/-ylidene)acetic chloride and salicylaldehyde through an efficient synthetic route is illustrated in Scheme 58. They were screened for antiproliferative activity by using normal human skin fibroblasts (BJ) and tumor cell lines, namely A549, HepG2 and MCF-7, were assessed by colorimetric MTT assay, and the antibacterial activity was determined in vitro against three Gram-positive and Gram-negative bacteria by broth microdilution. Compounds 75(a, e, f, g, i, j) and 76(a, b) showed antiproliferative activity against tumor cell lines. In the MCF-7 cells, the IC50 value of compound 18 was 1.59 mg/mL, which was 13 times lower then irinotecan (reference drug) and the safety index (SI) value was threefold higher than the reference strain. Compounds 75f, 75g and 76b showed high potency against Gram-positive bacteria, and compound 75g seemed to be a promising agent for anticancer treatment.
Scheme 58.

Synthesis of 2-(2,4-dioxothiazolidin-5-yl/ylidene)acetic acid derivatives 75 and 76
Pânzariu and coworkers [112] demonstrated the synthesis of thiazolidine-4-one derivatives bearing a nitro-l-arginine methyl ester (NO2-Arg-OMe) and screened them for their antioxidant and antimicrobial activity. Novel arginine-linked thiazolidinone derivatives 77(a–j) were synthesized in two steps; initially, one-pot condensation and cyclization reaction took place between ethyl 3-aminopropionate hydrochloride, aromatic aldehydes and TGA to form thiazolidine derivatives, which further reacted with Nω-nitro-l-arginine methyl ester hydrochloride in the presence of HOBt and EDC.HCl to furnish final products 77(a–j) (Scheme 59). The in vitro antioxidant activity was evaluated by ferric/phosphomolybdenum reducing antioxidant power assays and DPPH/ABTS scavenging assays. For investigation of antibacterial activity, two Gram-positive (S. aureus, S. lutea) and Gram-negative (E. coli, P. aeruginosa) bacterial strains were used, and for antifungal activity, Candida spp. (C. glabrata, C. albicans, C. parapsilosis) were used. Among all compounds, 77g and 77h displayed high free radical scavenging ability for DPPH and ABTS radicals, whereas 77j and 77e exhibited the highest activity for ABTS scavenging and ferric and phosphomolybdenum reducing antioxidant power, respectively. Significant antimicrobial activity was shown by 77j which was found most potent against P. aeruginosa, S. aureus and S. lutea strains. These properties were stimulated in the presence of bromo and nitro groups at the phenyl ring of thiazolidinone.
Scheme 59.

Synthesis of thiazolidine-4-one derivatives 77(a–j)
Khan et al. [113] prepared a range of biphenyl tetrazole-thiazolidinedione derivatives and checked them as PDF enzyme inhibitors and for in vitro antibacterial activity. The preparation of biphenyl tetrazole-thiazolidinediones 78(a–j) was carried out via condensation of biphenyl trityltetrazole-thiazolidinediones and THF using sodium hydroxide (Scheme 60). The antibacterial activity was screened against bacterial strains E. coli and B. subtilis using a twofold serial dilution method, and dimethyl sulfoxide was used for solvent control. PDF enzyme was taken out from E. coli (NCIM-2931). All derivatives 78(a–j) showed good to high activity against E. coli PDF-Ni enzyme and exhibited good binding properties. Among all synthesized compounds, 78b, 78c and 78h were the most potent with IC50 values of 16.25 mM, 18.00 mM and 17.25 mM, respectively. Compounds 78b, 78c and 78h also showed antibacterial activity with an MIC range of 8.00–26.00 mg/mL, compared with standard ciprofloxacin. The SAR studies concluded that all reactants, thiazolidinedione (head group), acidic group (tetrazole) and the biaryl group played an important role in the inhibitory process, and the activity was also affected by substitution on the head group.
Scheme 60.

Synthesis of biphenyl tetrazole-thiazolidineones 78(a–j)
Gilani and coauthors [114] synthesized a series of unique thiazolidinones from N-(6-chlorobenzo[d]thiazol-2-yl)hydrazine carboxamide derivatives and evaluated them for in vitro antimicrobial activity against four bacterial strains, namely, S. aureus (Gram-positive bacteria) or E. coli, P. aeruginosa, K. pneumoniae (Gram-negative bacteria) and five fungal species, namely, C. albicans, A. niger, A. flavus, M. purpureus and P. citrinum, via the serial plate dilution method. The compounds displayed good antimicrobial activity at 12.5–200 µg/mL in DMSO. The products were obtained from the reaction of 2-amino-6-chloro-benzothiazole and sodium cyanate which further reacted with hydrazine hydrate solution in alcohol followed by ring closure between carboxamide and aromatic aldehydes. The authors reported that the most active compounds against all bacterial strains had methyl 2,4-dichloro and 4-nitro at the phenyl ring attached to the thiazolidinones ring, and on substituting the above groups with a chloro and acetyl group, their activity was decreased. When these groups were replaced by dichloro, methyl and phenoxy substituents, they showed high potency against all tested fungal strains. Here, the Gram-positive strain showed less activity as compared to Gram-negative strain (Scheme 61).
Scheme 61.

Synthesis of benzothiazole incorporated thiazolidin-4-one 79
Nastasa et al. [115] designed a range of new 5-(chromene-3-yl)methylene-2,4-thiazolidinone derivatives and tested them for antimicrobial activity against two Gram-positive bacteria (L. monocytogenes, S. aureus), two Gram-negative bacteria (E. coli, S. typhimurium) and one fungus strain (C. albicans) with the agar diffusion method. Gentamicin and fluconazole were used as reference for antibacterial and antifungal activity, respectively. The Knoevenagel condensation reaction of 6,8-dichloro-4-oxo-4H-chromene-3-carbaldehyde, 2,4-thiazolidinedione and anhydrous sodium acetate in acetic acid, when treated with α-bromoalkylarylketones, dimethylformamide (DMF) and anhydrous potassium hydroxide produced N-substituted 5-(chromene-3-yl) methylene-2,4-thiazolidinediones (Scheme 62). All compounds showed low to high growth inhibitory effect against the tested strains, in which compound 80g was found most potent at all used concentrations, while compound 80f gave better effect, and compounds 80a and 80e exhibited similar activity as the standard drug gentamicin.
Scheme 62.

Synthesis of N-substituted 5-(chromene-3-yl) methylene-2,4-thiazolidinediones 80a–g
Novel N-substituted-1,3-thiazolidinone derivatives were synthesized by De Monte and coworkers [116], and their in vitro antifungal activity was determined against various phytopathogenic fungi, 22 Candida spp. (C. tropicalis, C. albicans, C. krusei, C. glabrata, C. parapsilosis and C. sake), and clotrimazole, ketoconazole, miconazole, fluconazole, tioconazole and amphotericin B were used as standard drugs. The thiazolidinones were designed via a contained N1 hydrogen moiety substituted with aromatic, heteroaromatic, cyclic and bicyclic structure, and on this basis it was classified into three series. Thiosemicarbazides were condensed with various carbonyl compounds using acetic acid and formed a thiosemicarbazone intermediate, which cyclized with ethyl bromoacetate in methanol and sodium acetate to form the thiazolidinone derivatives and further reacted with 4-nitrobenzyl bromide and (chloromethyl)naphthalene and N-(chloromethyl)phthalimide via condensation to afford the desired products 81 (A1–A26, B1–B26 and C1–C26) (Scheme 63). The SAR study showed that the presence of aliphatic and aromatic (both homo and hetero) moieties affected the inhibition activity of the compounds. The final compounds did not show satisfactory inhibition for bacterial activity, but compounds 81(A6, A7, A10, B6, C1 and C6) exhibited high antifungal activity against Candida spp. on Hep2 cells (human laryngeal epidermoid carcinoma). The molecular docking study explained the mechanism of the desired compounds and revealed that these compounds mainly showed Van der Waals interactions with the hydrophobic binding pocket of active cells similar to the reference drug lanosterol.
Scheme 63.

Synthesis of the N-substituted-1,3-thiazolidinone derivatives 81
Pansare and coworkers [117] introduced a one-pot three-component synthesis of 3-(4-chloro-2-hydroxyphenyl)-2-(substituted) thiazolidin-4-one using 2-amino-5-chlorophenol with different aldehydes in N,N-dimethylformamide (DMF) and glacial AcOH as a catalyst. The synthesis was carried out by both conventional and microwave heating. The compounds were further screened for antimicrobial activity (Scheme 64), and microwave-assisted synthesis afforded a high yield of the products. According to the mechanism, initially 2-amino-5-chlorophenol and benzaldehyde reacted to give imine intermediate. The imine reacted with mercaptoacetic acid and underwent intramolecular cyclization to form thiazolidinones (Scheme 65). All compounds were studied for in vitro antimicrobial activity against two Gram-positive bacteria (B. subtilis and S. aureus), two Gram-negative bacteria (E. coli and S. typhimurium) and four fungal strains (C. albicans, A. flavus, A. niger and C. neoformans) using a serial macrodilution method. Ciprofloxacin and ampicillin were used as reference antibacterial drugs, and fluconazole and miconazole were used as reference antifungal drugs. Among all the tested derivatives, 82f, 82g, 82l and 82m exhibited broad-spectrum activity, i.e. growth inhibitor against reference drug excluding fungus A. niger. Compounds 82g and 82m were more potent than the reference antifungal drug. All compounds also showed cytotoxicity on two cell lines, HeLa and MCF-7, using sulforhodamine B (SRB) assay. The SAR study revealed that structural variation and molecular strain affected the activity of the products. The activity of the compounds was controlled by substituents, i.e. an EDG on the phenyl ring enhanced the activity, whereas an EWG decreased activity. The remarkable features of this protocol were high yields with shorter reaction time, a unique, rapid and convenient synthesis, and all the compounds were non-cytotoxic.
Scheme 64.

Synthesis of 3-(4-chloro-2-hydroxyphenyl)-2-substituted thiazolidin-4-ones 82(a–n)
Scheme 65.
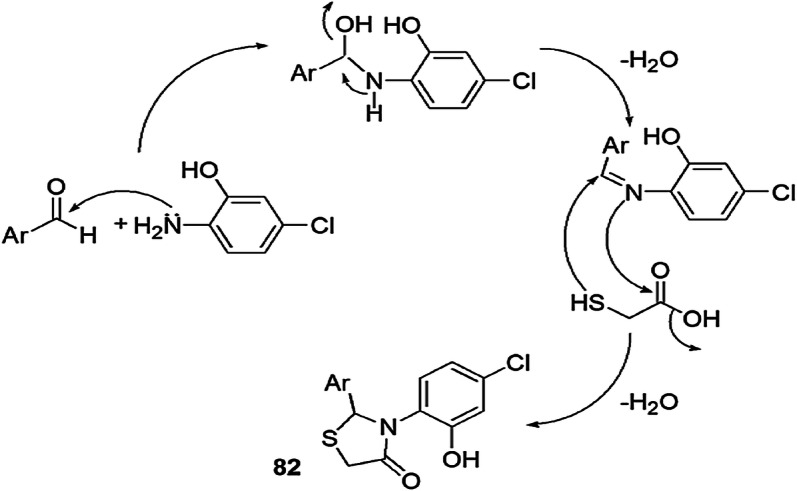
Possible mechanism for the synthesis of 3-(4-chloro-2-hydroxyphenyl)-2-substituted thiazolidin-4-one 82.
Modified from Ref. [117]
A series of new 4-thiazolidinones linked with a pyrone moiety were synthesized and screened for in vitro antimicrobial activity by Nechak et al. [118]. All compounds were screened against five microorganisms, Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus (ATCC 43300), Staphylococcus aureus (ATCC 25923) and Candida albicans. The 4-thiazolidinones 83(a–d) were synthesized from thiosemicarbazones and ethyl 2-bromopropionate by refluxing in acetonitrile in high yields. Thiosemicarbazones and appropriate maleimide were refluxed with acetonitrile to produce thiazolidinones 83(a–h). The mechanism showed that in the synthesis of 83(a–d), thiosemicarbazones attacked bromoester, which underwent intramolecular cyclization (Scheme 66), while in the synthesis of 84(a–h), thiosemicarbazone attacked the double bond of maleimide, followed by ring opening and nucleophilic attack on the nitrogen atom (Scheme 67). Among all the compounds, 83(a–d) and 84a–e (except 84d) showed high antibacterial activity against Pseudomonas aeruginosa. Compound 83d exhibited antifungal activity against Candida albicans. Compounds 83c and 84(a–h) (except 84f) exhibited significant antibacterial activity against S. aureus (ATCC43300), and compounds 83a–d (except 83b) displayed the highest activity against S. aureus (ATCC 25923). The SAR study revealed that substitution of the R group from –CH3, C2H5 and H on compounds 83c and 84e, respectively, gave a broad spectrum of active drugs against S. aureus (Gram-positive bacteria) and P. aeruginosa (Gram-negative bacteria). In the inhibitory process, these compounds avert some intracellular and extracellular enzyme functions and microbial metabolism like DNA, RNA and protein synthesis of microorganisms.
Scheme 66.

Proposed mechanism for the synthesis of pyrone-linked 4-thiazolidinones compounds 83(a–d)
Scheme 67.

Possible mechanism for the synthesis of 4-thiazolidinones 84(a–h)
Kunzler and colleagues [119] proposed a novel one-pot, two-step synthesis of 2-aryl-3-((piperidin-1-yl)ethyl)thiazolidinones 85(a–p) from amine and arene aldehydes under refluxing toluene, and evaluated their antifungal and cytotoxic properties (Scheme 68). The in vitro antifungal activity was evaluated against Candida albicans, Candida guilliermondii, Candida parapsilosis, Cryptococcus laurentii, Rhodotorula sp., Geotrichum sp. and Trichosporon asahii using fluconazole as reference drug, and the cytotoxic properties were evaluated by Vero cells. The MIC and MFC values were found to be same for all synthesized compounds. The results showed that compounds 85h and 85i were 1.6 times more potent than fluconazole, whereas compounds 85b, 85e, 85g and 85k showed the same activity toward Rhodotorula spp., and all compounds, especially 85g, were found less toxic in cytotoxicity assay. These compounds worked best in growth inhibition against Rhodotorula sp. yeast. The SAR study explained that the change of electronic substituents did not directly affect growth inhibition against tested phytopathogens. Developments in recent decades in the medicinal field have generated serious problems in which pathogenic bacteria continuously develop resistance to the currently used antibacterial drugs. Discovery of agents which will block pathogenic mechanisms rather than killing the infecting microbe will help overcome bacterial resistance and will aid in the development of effective treatments.
Scheme 68.

Synthesis of 2-aryl-3-((piperidin-1-yl)ethyl)thiazolidinones 85(a–p)
Anticancer Activity
A series of novel thiazolidinones with a 3-hydroxy-2-naphthoic motif was synthesized and tested for in vitro cytotoxicity against both HepG2 and human renal cell adenocarcinoma (769-P). The in vivo impact of the compounds on the CNS of mice was studied by Popiołeka and coauthors [120]. 1,3-Thiazolidin-4-one derivatives (86a–c) were prepared via reaction of 3-hydroxy-2-naphthoic acid hydrazide with aldehydes followed by cyclization with thioglycolic acid in dioxane (Scheme 69). For reference cell lines, rat cardiac myoblasts (H9c2) and GMK were used. Compound 86c was most potent and selective against 769-P cell lines at the IC25 concentration. The in vivo study revealed that compounds 86a–c were nontoxic against the CNS of Swiss mice, showing the highest biological activity, and compound 86c showed significant anodyne activity due to blocking of the cell cycle at the G2/M phase and stimulation of apoptosis. The authors assumed that the study of compounds (86a–c) as an anticancer drug could be linked with the cyclooxygenase-2 (COX-2) enzyme, which is associated with apoptosis, angiogenesis and metastasis effects in reference cell lines. These results confirmed significant antitumor activity of compounds (86a–c).
Scheme 69.
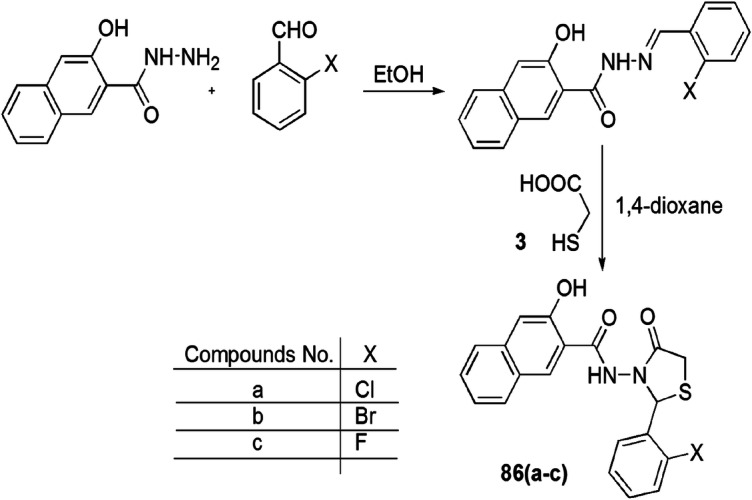
Synthesis of 1,3-thiazolidin-4-one derivatives 86a–c
Yakaiah and coworkers [121] described a proficient one-pot multicomponent synthesis of pyrazolo-oxothiazolidine derivatives 87(a–j) and evaluated their antiproliferative activity against A549 cell lines. Pyrazolo-oxothiazolidine derivatives were obtained in high yields (91%) from 1-(benzofuran-2-yl)-3-(substituted)-arylprop-2-en-1-ones, dialkyl acetylenedicarboxylates and thiosemicarbazide reaction at different reaction conditions and catalyst and 20 mol% of NaOH and ethanol at 80 °C (Scheme 70). For the study of antiproliferative activity of pyrazolo-oxothiazolidine derivatives, the catalytic site of receptors EGFR 14 and VEGFR2 were used, and sorafenib (IC50, 3.779 μg/mL) was used as a standard drug. The results of in vitro inhibition showed that the IC50 value of compounds 87a (0.930 μg/mL), 87e (1.207 μg/mL), 87f (0.808 μg/mL), 87g (1.078 μg/mL), 87h (0.967 μg/mL) and 87j (2.445 μg/mL) displayed good potency (Table 4).
Scheme 70.

Synthesis of pyrazolo-oxothiazolidine derivatives 87(a–j)
Table 4.
Antiproliferative activity of pyrazolo-oxothiazolidine derivatives 87a–j on A549 (lung cancer) cell line
| S. no. | Name of compound | IC50 on A549 cell line (μg/mL) |
|---|---|---|
| 1 | 87a | 0.930 |
| 2 | 87e | 1.207 |
| 3 | 87f | 0.808 |
| 4 | 87g | 1.078 |
| 5 | 87h | 0.967 |
| 6 | 87j | 2.445 |
| 7 | Sorafenib | 3.779 |
Rodrigues and his group [122] synthesized benzylidene-2,4-thiazolidinedione derivatives 88(a–d) which were found to display selective cytotoxic and genotoxic activity and were screened on the NCI-H292 (human lung carcinoma), MCF-7 (breast adenocarcinoma), HEp-2 (cervix carcinoma), K562 (leukemia) and HT29 (colon adenocarcinoma) cell lines using MTT assay and non-tumor cells (human peripheral blood mononuclear cells, PBMC) using the alamarBlue assay. The aldehydes reacted with thiourea and monochloroacetic acid in an aqueous medium to produce thiazolidine-2,4-dione derivatives 88(a–d) (Scheme 71). The highest genotoxicity and cytotoxicity was found for compound 5-(2-bromo-5-methoxybenzylidene)-thiazolidine-2,4-dione (88d) with the lowest IC50 value of 1.26 μg/mL for NCI-H292 which did not affect normal cells.
Scheme 71.

Synthesis of thiazolidine derivatives 88a–d
Synthesis, cytotoxicity and anticancer activity of pyridine-thiazolidinones were reported by Ansari et al. [123]. To obtain unique human CAIX inhibitors, the synthesis was carried out with the mixture of 3-(furan-2-ylmethyl)-2-(phenylimino)-1,3-thiazolidin-4-one, aldehydes and hexahydropyridine using ethanol (Scheme 72). The cell cytotoxicity was identified by standard MTT assay and CAIX (PDB ID: 3IAI) was used for molecular docking study. The derivatives of 89 showed low to moderate inhibition against CAIX. Compounds 89 (c, d, f, g, j, m, n and q) (IC50 values = 50.92 μM, 57.31 μM, 40.41 μM, 40.18 μM, 63.11 μM, 60.92 μM, 38.40 μM and 43.52 μM, respectively) containing alkoxy and chloro groups displayed low inhibitory activity, and compounds 89 (l, o, r, s) (IC50 values = 20.92 μM, 16.68 μM, 6.64 μM and 10.04 μM, respectively) having a disubstituted methoxy group or heterocyclic substitution showed good inhibitory effect. Compounds 89e, h (1.61 μM), k (1.84 μM), and r (6.64 μM) with a nitro or hydroxyl group exhibited outstanding inhibitory activity. It was concluded that inhibition of CAIX not only depends on substituents but also on the molecular skeleton. HEK cell lines were used as reference for cytotoxicity. All compounds gave satisfactory activity for cancer against MCF-7 and HepG2 cell lines. Compounds 89h and 89k showed the most promising activity for cancer treatment.
Scheme 72.

Synthesis of pyridine-thiazolidinone derivatives 89(a–r)
The synthesis and anticancer activity of various thiazolidine-2,4-dione derivatives were evaluated by Asati et al. [124]. The thiazolidine derivatives were obtained from 4-((2,4-dioxothiazolidin-5-ylidene)methyl)benzohydrazide and aliphatic or aromatic acids in the presence of POCl3 (Scheme 73) and showed anticancer activity against MCF-7 with the SRB method. The H-bonding interaction of the oxygen atom at the second and fourth position of thiazolidinedione derivatives with ASP186 and LYS67, respectively, played a vital role in the activity and nature of the substituents, which was responsible for the activity. Among the tested compounds, compound 90x demonstrated the most marked effect in the MCF-7 cell lines (GI50 value 0.004 µM) and displayed a −6.68 docking score against PIM-1 kinase. Compounds 90c (GI50 values 0.028), 90d (0.012), 90e (0.097), 90f (0.055), 90h (0.087), 90l (0.087), 90o (0.031) and 90t (0.019) showed potent activity, while compounds 90 (g, i–k, m, q, r, s, u and v) showed intermediate inhibition effect. When EWGs like Cl, Br and I were used, the activity increased, and the activity decreased in the presence of EDGs like 2-methyl. The SAR studies indicated that the presence of a phenyl ring was crucial for anticancer activity.
Scheme 73.

Synthesis of thiazolidine derivatives 90(a–x)
A range of 4-hydroxy-thiazolidine-2-thione derivatives 91a–91z were synthesized and screened for antiproliferative activity by Li and coworkers [125]. For the study of the 4-hydroxy-thiazolidine-2-thione moiety as a drug, it was divided into two subunits, subunit A (the phenyl group) and subunit B (the pyridin-3-ylmethyl moiety), on the basis of their structure. The modification in subunit A was carried out by the reaction of substituted sulfonyl chlorides and 4-aminoacetophenone, and they formed intermediates which further reacted with phenyltrimethylammonium tribromide, pyridin-3-ylmethenamine and carbon disulfide (Scheme 74). This compound was more potent as pyruvate kinase M2 isoform (PKM2) activators. The SARs study using fluorescent PK-LDH coupled assay showed that the electronic and steric effect of substituents at the benzene ring affected activation activity of PKM2. Compounds 91t and 91y (AC50 value 0.52 µM, 0.74 µM respectively) showed higher potency than other derivatives (AC50 = 2.96 µM). The EWG (91h–91m), multi-substituents (91d and 91e) and large groups which showed steric hindrance (91n–91p) on the benzene ring deactivated PKM2, and when the ring was substituted with –CH3 and –OCH3 groups at the meta and para positions, the activity was decreased, whereas a methyl group at the ortho position at nanomolar concentration increased the activity. The PKM2 activity was affected when the benzene ring was replaced with different rings (91q, 91r, 91z), replacement of the N-containing bicyclic ring with other rings, and length of carbon linker between the 4-hydroxy-thiazolidine-2-thione framework and the pyridine framework due to hydrogen bonding interaction with PKM2. The previous study showed that 91w arrested the G2/M phase of the cell cycle in HCT116 cell lines. Here, compound 91w (IC50 values 0.46–0.81 µM) was the most active antitumor agent against HCT116, Hela, H1299 and PC3 cell lines.
Scheme 74.

Synthesis of novel 4-hydroxy-thiazolidine-2-thione derivatives 91(a–z)
Kulabaş et al. [126] synthesized 4-thiazolidinone derivatives from thiourea and ethyl bromoacetate and screened them for anticancer and antiviral activity. Enterovirus, Yellow fever virus, Murine norovirus and Chikungunya virus strains were used for screening of antiviral potency. 4-Thiazolidinone derivatives were obtained in two different ways; firstly, cyclization of chloroacetamide with NH4SCN, and secondly, cyclization of thiourea and ethyl bromoacetate with sodium acetate. In this method, five tautomeric forms were obtained (Scheme 75). All synthesized compounds have EC50 value higher than 0.3 µM. The anticancer activity was evaluated against K562, MCF-7, A549, PC-3, HT-29, HeLa and SJSA1 cell lines at a dose of 10 µM. Compounds 92 and 93 showed significant inhibition effect, i.e. 32.29% inhibition in HT29 and 35.82% inhibition in HeLa cell lines, respectively. Compound 94 was identified as the most active substance (38.92% inhibition vs. HT-29 and 35.96% inhibition vs. SJSA1) and has a high survival rate of 92.01% from NIH3T3 cell lines (Table 5).
Scheme 75.

Synthesis of 1,3-thiazolidin-4-one compounds 94 and 95
Table 5.
Cytotoxicity and anticancer activity of some active compounds in selected cancer cell lines [126]
| Compound | % Proliferation (at 10 µM dose) | ||||
|---|---|---|---|---|---|
| NIH3T3 | A549 | HeLa | HT-29 | MCF-7 | |
| 92 | 78.12 | 89.29 | 78.77 | 67.71 | 91.61 |
| 93 | 92.01 | 76.26 | 80.89 | 61.08 | 79.60 |
| 94 | 98.19 | 74.46 | 64.18 | 81.81 | 97.97 |
Bataille and coworkers [127] synthesized novel 4-thiazolidinone derivatives and evaluated their anticancer activity for cancer in blood and lymph tissue via inhibition of the PIM kinase family by differential scanning fluorimetry (DSF). The in vitro antiproliferative activity was evaluated in two cancer cell lines (MV4-11 and K562) against selected inhibitors. They used mainly two approaches; Firstly, they synthesized fused tricyclic series through hydrolysis followed by conjugate addition, which showed significant improvement in both solubility and metabolic stability. Another approach showed improvement in metabolic activity by addition of pseudothiohydantoin in place of the rhodanine head group. A compound of series 96 was formed by Knoevenagel condensation of carbonyl compounds and rhodanine using piperidine in EtOH (Scheme 76). The other derivatives were synthesized by Suzuki–Miyaura coupling, using RB(OH)2 and Pd(PPh3)4 in Na2CO3, which further underwent Knoevenagel condensation with rhodanine using piperidine as catalyst to obtain tricyclic compounds. The compounds were divided into 11 classes on the basis of their structure. These series possessed better selectivity for pan-PIM kinases than other kinases. All tested compounds displayed good activity, and derivative 98b exhibited high activity against the K562 cell line, with an IC50 value of 0.75lM. Compound 97a has a phenyl group, and displayed a 90-fold increase in activity, with an IC50 value of 6.7 ± 3.1 nM, and the in silico study revealed that their rhodanine head group showed H-bonding interaction with water which was connected with the Lys67 residue, and this interaction was found to be sandwich-like interaction between the products and the lipophilic area of the PIM binding pocket. The SAR study indicated that an electronic effect did not play a significant role in inhibition activity. Compound 98a containing a sulfonamide group showed excellent PIM1 inhibitor activity (IC50 25 ± 4 nM) and also provided better kinetic solubility than other compounds.
Scheme 76.

Synthesis of 4-thiazolidinone derivatives 96, 97 and 98 and their most biologically active compounds
Sharma and coworkers [128] synthesized novel benzimidazole nucleus-linked thiazolidinedione hybrids and screened them as potential cytotoxic and apoptosis-inducing agents against human cancer cell lines, prostate (PC-3 and DU-145), breast (MDA-MB-231), A549 and MCF10A using MTT assay, and 5-FU was used as the standard drug. Novel benzimidazole-thiazolidinedione hybrids were synthesized by Knoevenagel condensation in substituted thiazolidinedione and 1-alkyl-1H-benzo[d]imidazole-2-carbaldehydes in moderate to high yields. The pathway of the reaction is outlined in Scheme 77. All compounds have good growth inhibition against the tested cancer cell lines. Compounds 99j, 99p and 99r showed good antitumor effects with IC50 ≤ 15 μM against A549 cell lines and were safe to MCF10A (Table 6). The contact of compound 99p with the A549 cell showed arrest of the G2/M phase of the cell cycle, inhibited in vitro cell migration (using wound-healing assay) via disruption of F-actin assembly and decreased F-actin extension, and apoptotic activity such as cell contraction, cell wall distortion and decreased viable cell numbers. For induction of apoptosis acridine orange-ethidium bromide (AO-EB), annexin V-FITC/propidium iodide staining, DAPI, MitoSOX and rhodamine-123 assays were used in A549 cells for compound 99p. According to SAR studies, the substitution on the head position gave high bioactive compounds as compared to tail substitution.
Scheme 77.

Synthesis of (Z)-5-[(1-alkyl-1H-benzo[d]imidazole-2-yl)methylene]-3-(2-oxoethyl)thiazolidine-2,4-dione derivatives 99(a–t)
Table 6.
IC50 values (µM) of the most active compounds against selected human cancer cell lines [128]
| Compound | PC-3 | DU-145 | MDA-MB-231 | A549 |
|---|---|---|---|---|
| 99j | 41.10 ± 3.08 | 37.52 ± 1.45 | 36.03 ± 1.53 | 13.35 ± 1.26 |
| 99p | 39.87 ± 3.16 | 31.41 ± 1.52 | 29.18 ± 0.9 | 11.46 ± 1.46 |
| 99r | > 50 | 31.36 ± 1.12 | 33.82 ± 1.37 | 15.30 ± 0.92 |
| 5-FU | 45.32 ± 2.08 | 40.58 ± 1.83 | 35.98 ± 1.52 | 30.47 ± 1.09 |
Kumar and coworkers [129] developed a simple and direct synthesis of 4-thiazolidinone derivatives in the presence of propylphosphonic anhydride (T3P)-DMSO media as a cyclodehydrating agent and evaluated their cytotoxicity both in vitro and in vivo against leukemic cell lines (Reh and Nalm6). 4-Thiazolidine derivatives 100(a–j) were produced from primary and secondary alcohols and aryl amines using T3P-DMSO via one-pot synthesis at high yields (Scheme 78). The relative study displayed that the compounds exhibited better activity on Reh cell lines than Nalm6 cell lines. Among all tested compounds, 3-(4-bromophenyl)-2-(4-(dimethylamino) phenyl)thiazolidin-4-one (100e) showed potent activity against Reh cells and Nalm6 cells with IC50 values of 11.9 μM and 13.5 μM, respectively. The cell cycle analysis showed that 100e was able to aggregate cells in cell cycle subG1 phase, and also decreased the mitochondrial membrane potential and increased cell death in tested cell lines via apoptotic cell death. The EAC tumor model in a Swiss albino mouse revealed a significant reduction in tumor cell volume and did not affect the other organs and cells.
Scheme 78.
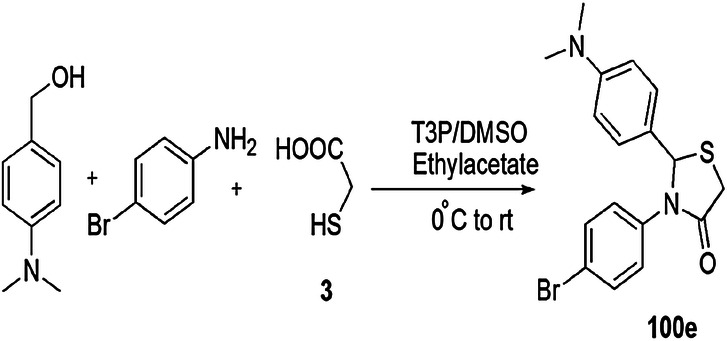
Synthesis of 3-(4-bromophenyl)-2-(4-(dimethylamino)phenyl)thiazolidin-4-one (100e)
Anh and colleagues [130] synthesized a new range of hybrid thiazolidine compounds attached with the naturally occurring paeonol. The compounds exhibited selective anticancer activity against eight cancer cell lines, HepG2, acute promyeloidleukemia, KB, LU-1 (human lung cancer), LLC, SW480 (colon adenocarcinoma), hormone-dependent prostate cancer and MCF7 (breast cancer) using MTT assay. The predicted reaction mechanism for this protocol was decoded as paeonol reacting with dimethylformamide (DMF) via Vilsmeier–Haack reaction to afford 3-formyl-7-methoxychromone, which underwent Knoevenagel condensation with 2,4-thiazolidinedione using base catalyst (piperidine or sodium acetate), to give chromonylthiazolidines 101(a–i) (Scheme 79). All semi-synthesized compounds showed low cytotoxic effect against all cell lines; however, this cytotoxic effect was highly selective. Compounds 101a and 101b showed highly selective cytotoxicity in this series against KB (IC50 44.1 ± 3.6 μg/mL) and MCF7 (IC50 32.8 ± 1.4 μg/mL) cell lines, respectively. The advantages of this research are the use of easily available, affordable and natural starting material paeonol and hybrid chromonylthiazolidines as selective antitumor agents.
Scheme 79.

Synthesis of the chromonylthiazolidine derivatives 101(a–i)
A series of novel hybrid compounds 2, 4-thiazolidinedione incorporated pyrazole were produced and introduced for anticancer activity by Kumar and coworkers [131]. 3-(Substituted aryl)-1-phenyl-1H-pyrazolyl-2,4-thiazolidinediones 102(a–h) were prepared by the cyclization of substituted phenyl hydrazones via Vilsmeier–Hack reaction followed by condensation with 2, 4-thiazolidinedione employing piperidine as a catalyst in glacial acetic acid (Scheme 80). The synthesized moieties were tested for their anticancer potency against three cancer cell lines, A549, MCF-7 and DU145 using MTT based cytotoxic assay and Doxil as a reference drug. Among all the hybrids, compound 102b emerged as a highly active cytotoxic agent for all three cancer lines, A549, MCF-7, DU145 (IC50 values 4.63, 1.32 and 5.25 μg) respectively. Compounds 102c and 102h showed significant activity against A549 and MCF-7 with IC50 values ranging from 4.44 to 9.16 μg.
Scheme 80.

Synthesis of 3-(substituted aryl)-1-phenyl-1H-pyrazole-2,4-thiazolidinones derivatives 102(a–h)
The series of substituted thiazolidinediones have been prepared and their anticancer activity and cytotoxicity were evaluated in vitro by Akshaya et al. [132] 5-(4-substituted)-3-{[(4-substituted) amino]methyl}-1,3-thiazolidine-2,4-diones were obtained from reflux of 5-arylidene thiazolidine-2,4diones, formaldehyde and aromatic amine in the presence of methanol. The reaction proceeded via two-step mechanism involving Knoevenagel condensation of thiazolidinediones with aromatic aldehydes using l-tyrosine as a catalyst which further underwent Mannich reaction with aromatic amines and formed Mannich bases (Scheme 81). All derivatives showed good cytotoxic effect on both PPAR-γ using wet lab synthesis and MCF-7 cell lines by MTT assay. The outcomes of SARs revealed that presence of electron withdrawing groups like bromine, chlorine, fluorine enhanced the activity of drugs. Compounds 103a and 103b showed high potency against MCF-7, due to electronegativity of substituents and non-bonding valence electron, whereas presence of electron releasing group decreased activity of product against MCF-7 cell lines.
Scheme 81.

Synthesis of 5-(4-substituted)-3-{[(4-substituted)amino]methyl}1,3-thiazolidine-2,4-diones 103(a–e)
Anti-inflammatory Activity
Yeh and coworkers [133] synthesized a large panel of thiazolidinedione derivatives and evaluated them for in vitro and in vivo activity against glutaminase gene transcripts such as kidney isoform (KGA), GAC, liver isoform and GAB, and also investigated them in relation to chemical structure, activity and their selectivity. The compounds were synthesized via multistep synthesis of diketone, amine and p-toluenesulfonic acid. The intermediate obtained was further reacted with POCl3 in DMF to obtain bromoarene which was refluxed with corresponding arylboronic acid, sodium bicarbonate and Pd(PPh3)4 as a catalyst in the mixture of ethanol/water/DME to form an adduct, which reacted with thiazolidinedione, AcOH and piperidine using toluene to produce compound 104. Most of the compounds exhibited high inhibition against KGA and GAC and good inhibition against GAB. The synthesized compounds inhibited cell growth, glutamate production, clonogenicity and decreased the size of tumor of xenografted human pancreatic AsPC-1 carcinoma cells in mice (Scheme 82).
Scheme 82.

Synthesis of fused thiazolidine-2,4-dione derivative 104
Ashour and coworkers [134] synthesized substituted thiazolidinones possessing pyrazolyl benzenesulfonamides and evaluated them as anti-inflammatory active drugs by applying formalin-induced paw edema bioassay, and the analgesic activity was studied from the rat tail withdrawal technique, using diclofenac sodium as a standard drug. For the preparation of 4-[5-amino-4[[(2E)-2-[(5Z)-5-substituted-4-oxo-1,3-thiazolidin-2ylidene]hydrazinyl]carbonyl]-1H-pyrazol-1yl]benzenesulfonamides (105, 106), firstly, ethyl 5-amino-1-(4-sulfamoylphenyl)-1H-pyrazole-4-carboxylate and chloroacetyl chloride were refluxed in dry DMF, which cyclized with ammonium thiocyanate in ethanol to produce an adduct in high yields and purity, followed by Knoevenagel condensation with aldehydes or 3-aryl-1-phenyl-1H-pyrazole-4-carboxaldehydes and piperidine in dry DMF/absolute EtOH mixture (Scheme 83). All compounds showed effective anti-inflammatory and analgesic activity. Compounds 105a, 105b and 106a (45–48%) displayed superior activity than the diclofenac sodium (38%), and compounds 105a and 106a (22–42%) exhibited high activity as compared to the reference drug (16%). All active compounds showed minimal ulcerogenic effects. According to SAR study, after 4-h intervals, the substituted ylidene derivatives (105b, 106b) were more active than the unsubstituted phenyl analogs (105a, 106a). The in vitro COX inhibition assay results showed that all thiazolidinones were more selective to COX-2 than COX-1, and the motif 105b exhibited the maximum potency for both COX-1 and COX-2 (IC50 values of 4.51 µM and 1.06 µM, respectively).
Scheme 83.

Synthesis of pyrazolylbenzenesulfonamides-linked thiazolidinones 105 and 106
A novel series of thiazolidinyl esters were produced from benzoic acid ester and screened as potent in vitro phosphodiesterase 4 (PDE4) inhibitors, in chronic obstructive pulmonary diseases (COPD) and related inflammatory diseases by Carzaniga et al. [135]. According to the mechanism, initially alcohol coupled with the appropriate thiazolidine-carboxylic acid isomers in EDC and DMAP, which further showed Boc cleavage to obtain corresponding esters, which when sulfonylated in pyridine gave final sulfonamide products. Some synthesized compounds reacted with amine in CDI and DMF to prepare appropriate amides. Compounds (S*,S**)-107 demonstrated ideal in vitro ADME and pharmacological properties with minimum risk of side effects, and these were highly effective in asthma and pulmonary inflammation (Fig. 8).
Fig. 8.
(S*,S**)-107 3,5-Dichloro-4-((S)-2-(3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl)-2-((S)-3-(3-dimethylcarbamoyl)-phenylsulfonyl)thiazolidine-2-carbonloxy)ethyl)pyridine 1-oxide as PDE4 inhibitor
Novel thiazolidinone derivatives were designed and produced, and their potency evaluated against diabetes mellitus by Maccari and colleagues [136]. Different hydroxybenzaldehydes, 2-chloroacetamide and acetonitrile were refluxed in potassium carbonate to afford 2-(formylaryloxy)acetamides. The Knoevenagel condensation of 2,4-thiazolidinedione and arylaldehydes provided 108(a–d), whereas in different conditions, 110(a–d) were obtained. Pure 2-[(4-oxo-2-thioxothiazolidin-5-ylidenemethyl)phenoxy] acetamides and (5-arylidene-4-oxo-2-thioxothiazolidin-3-yl)acetic acids were obtained by the reaction of 2-thioxo-4-thiazolidinone and (4-oxo-2-thioxothiazolidin-3-yl)acetic acid, respectively, with aldehydes by refluxing in a mixture of glacial acetic acid and sodium acetate (Scheme 84). The synthesized compounds showed anti-inflammatory activity for diabetes mellitus and other diseases via inhibited aldose reductase (AR) enzyme. The in vitro inhibition study displayed that compounds 108–111 exhibited good activity against reference drugs sorbinil and epalrestat. Compounds (5-arylidene-2,4-dioxothiazolidin-3-yl)acetic acids 110(a–d) and corresponding 2-thioxo isosteres 111(a–d) proved to be the most potent for AR inhibition. The SAR study displayed that compounds 108a, 108c, 109a and 109c were more potent than their relative para-substituted compounds 97b, 97d, 98b and 98d, and the presence of the m-methoxy group or p-carboxymethoxy group induced inhibition of AR enzyme. Some compounds also reduced both NF-kB activation and iNOS expression. The binding interaction between final compounds and the active site of AR was investigated by molecular docking study. The enzymatic inhibition results revealed that the fundamental 2-thioxo-4-thiazolidinone ring (109a–d and 111a–d) showed more significant interaction than the isosteric 2,4-thiazolidinedione ring (108a–d and 110a–d).
Scheme 84.

Synthesis of 5-arylidene-4-thiazolidinone derivatives 108–111(a–d)
Antiparasitic Activity
Oderinlo and coworkers [137] synthesized new thiazolidinedione derivatives possessing organometallic ferrocene and evaluated them for antiparasitic activity. Ferrocene-containing derivatives 112(a–d) and 114 were prepared by ferrocene carboxaldehyde, thiazolidinedione derivatives or 2-(phenylimino)thiazolidin-4-ones using piperidine in ethanol, whereas derivatives 113(a–b) were prepared by solution of (Z)-(5-((ferrocenyl)methylene)-2,4-dioxothiazolidin-3-yl)acetic acid and 1-methylpiperazine or phenylpiperazine with dry DMF using EDC·HCl, HOBt·H2O and DIPEA (Scheme 85). All compounds were screened in vitro for antitrypanosomal activity against Trypanosoma brucei brucei 427 trypomastigotes, antiplasmodial activity against the chloroquine-resistant Dd2 strain of P. falciparum malaria parasite and cytotoxicity against the HeLa cell lines. Pentamidine (PMD), CQ and EMT were used as standard drugs. By the result of growth inhibition assay, it was concluded that the synthesized compounds exhibited higher selectivity against T. b. brucei than P. falciparum, and compounds 113a and 113b displayed high antitrypanosomal activity with IC50 values of 1.94 μM and 3.31 μM, respectively, when compared to TZD-4 (IC50 = 7.51 μM). In case of P. falciparum, none of the molecules gave better potency compared to TZD-4. All tested compounds showed non-toxicity and > 90% HeLa cell viability, which confirmed the specificity of compounds against protozoan parasites.
Scheme 85.

Synthesis of ferrocene carboxaldehyde thiazolidine derivatives 112–114
A series of quinoline–thiazolidinone hybrids containing arylidene derivatives was developed as antimalarial agents via in vitro, in vivo and in silico study by Jain and coworkers [138]. For the synthesis of novel compounds, firstly, a mixture of 2-hydrazino-4-methylquinoline reacted with various aromatic aldehydes in glacial acetic acid, and refluxed with mercaptoacetic acid using anhydrous ZnCl2 in 1,4 dioxane to obtain respective thiazolidinones, which reacted with aryl aldehyde in glacial acetic acid and sodium acetate and produced 115a–e in good yields (Scheme 86). All the effective compounds were evaluated by in vitro schizont maturation inhibition (SMI) assay against chloroquine-sensitive strain 3D7 and chloroquine-resistant strain RKL9 strain of Plasmodium falciparum. Compound 115g displayed the most potent activity against both 3D7 and RKL9 strains with EC50 (3D7/RKL-9) values of 0.423/0.824 mg/mL via in vitro study and also showed the highest parasitemia inhibition (73.38%) against P. berghei from in vivo study. Among all compounds, 115b, 115e, 115g, 115j and 115n displayed significant activity from in vitro study and in vivo study against P. berghei in Swiss albino mice. The SAR studies concluded that the presence of a p-chloro group in one phenyl ring along with a p-methyl group in another ring gave the most potent derivative 115g among the series. For the inhibition activity, the p-methyl group was more potent than the p-methoxy group at the phenyl ring, and the size of the ring also affected the activity of compounds. The in silico study explained the possible pathway for antimalarial activity of synthesized compounds, and the docking study proved the synthesized compounds as antimalarial agents.
Scheme 86.

Synthesis of quinoline–thiazolidinone hybrids containing arylidene derivatives 115(a–r)
Antiviral Activity
HIV-1 RT acts as a prime target for synthesis of inhibitors for HIV/AIDS treatment. The thiazolidine nucleus contains essential pharmacophoric elements like a benzene ring, a nitrogen atom, substitution at C-2 and N-3, lipophilic characters and a biophoric space of various heterocycles around the N-3 position, which is necessary for HIV inhibition. A series of allophenylnorstatine-based peptidomimetics with various P1, P2, P3 and P2′ moieties (Fig. 9) was synthesized and screened as HIV-1 protease inhibitors by Hidaka et al. [139]. All compounds with P2 tetrahydrofuranylglycine (Thfg) exhibited significant activity against wild-type HIV-1 protease and the virus against lopinavir/ritonavir- or darunavir-resistant strains. Among all derivatives, compound 116d (KNI-1657) displayed strong activity against the reference strain. Compound 116a including P2 (2S, 3′R)-Thfg showed 96% inhibition against HIV-1 protease at 1-nM concentration. Compound 116b showed 98% inhibition at 60 nM, and 116c in human serum displayed the most potent inhibition among the series. The derivatives with P3 modifications including chromonylcarbonyl, benzofurancarbonyl and 7-methoxybenzofurancarbonyl were found to be highly active against HIV protease, whereas selected P2′ derivatives displayed inhibition activity in a 93–97% range at 1 nM.
Fig. 9.

Ligand selections for combinational modifications and some fused thiazolidine derivatives as HIV protease inhibitors 116(a–d)
Antidiabetic Activity
Wang and colleagues [140] designed and produced a range of novel thiazolidinedione derivatives and explained their inhibitor activity on alpha glucosidase using acarbose as the standard drug. The desired compounds 117 and 118 were synthesized via the following method: Firstly, phthalimide reacted with HCHO and formed 2-(hydroxymethyl)isoindoline-1,3-dione, which on refluxing with thionyl SOCl2 afforded 2-(chloromethyl)isoindoline-1,3-dione. The treatment of 2-(chloromethyl)isoindoline-1,3-dione with diverse hydroxyl-substituted aromatic aldehydes using anhydrous K2CO3 in dry acetone provided an adduct, which further underwent Knoevenagel condensation with rhodanine or thiazolidine-2,4-dione using β-alanine. It was previously observed that in the final compounds, a high yield of Z-stereoisomer was obtained (Scheme 87). Among all the synthesized derivatives, most compounds were potent inhibitors as compared to the reference drug. Compounds 117k, 118a, 118b, 118e, 118h and 118k have IC50 values of 20.95 ± 0.21, 16.11 ± 0.19, 7.72 ± 0.16, 7.91 ± 0.17, 6.59 ± 0.15 and 5.44 ± 0.13 μM, respectively, which represent their high inhibition activity. Compounds 117(a, b, e, h, j) and 107(c, g, i, j) were equipotent to reference drug acarbose. The derivative 118k with chloro and rhodanine groups at the 2- and 4-positions of the phenyl ring, respectively, exhibited the highest inhibition activity against α-glucosidase, with an IC50 value of 5.44 ± 0.13 μM. The SAR study showed that substitution of oxygen with sulfur in final compounds displayed remarkable increase in activity. It was also observed that EWGs chloro (117k, 118k) and nitro (117h, 118h) at the ortho-position of the phenyl ring increased inhibition against the reference drug than electron-donating substituents (–OCH3). The binding interaction between the most active compounds and the active site of α-glucosidase was confirmed by molecular docking studies. In the binding place of α-glucosidase, compounds changed in “L-shaped” conformation, showed hydrogen bond interaction and displayed CH–π interactions with the residue of Phe-157, Phe-177 and Phe-300.
Scheme 87.

Synthesis of thiazolidine-2,4-dione derivatives 117 and 118
Novel substituted benzylideneamino-benzylidene-thiazolidinones were prepared and further studied for their utilization on PPAR-γ, hyperglycemia and hypoglycemia for control of diabetes mellitus by Chhajed et al. [141]. Thiazolidinone was reacted with 4-nitrobenzaldehyde using sodium acetate and glacial AcOH to produce (E)-5-(4-nitrobenzyliden)thiazolidine-2,4-diones, which on reduction by SnCl2, gave (E)-5-(4-aminobenzyl)t hiazolidine-2,4-dione, which combined with substituted aldehyde in concentrated sulfuric acid to form (E)-5-((E)-4-((E)-4-benzylideneamino)benzylidene)thiazolidine-2,4-diones (Scheme 88). All compounds were tested using glucose uptake assay in 3T3-L6 cell lines with rosiglitazone as a reference drug. Among all tested compounds, 119(a–h) exhibited strong binding with the PPAR-γ binding site, and hyperglycemic control was shown by 119a, 119b and 119g. The SAR study revealed that the activity of the compounds was controlled by the benzylidine moiety’s substitution. The activity was increased when a molecule substituted at the para position in place of the meta position. In the glucose uptake assay, the substituted analog with a methyl group showed 1.9-fold activity, while the ethyl group showed 0.9-fold activity.
Scheme 88.

Synthesis of substituted bezylideneamino-benzylidene-thiazolidine-2,4-diones 119
Kumar and group [142] developed a novel synthesis of 5-((3-(aryl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino)thiazolidin-4-ones 120(a–g) and evaluated them for in vitro α-amylase inhibition. The compound 120(a–g) were obtained in the form of a mixture of 2E,5Z (37.1–42.0%) and 2Z,5Z isomer (58.4–62.8%). p-Tolyl thiourea and ethyl bromoacetate were reacted to form 2-(p-tolylimino) thiazolidin-4-one, that was refluxed with 3-(aryl)-1-phenyl-1H-pyrazole-4-carbaldehyde using piperidine in ethanol to produce desired products 120(a–g) (Scheme 89). Compound 120a showed 90.04% inhibition, and the docking studies concluded that interaction between 120a and human pancreatic alpha-amylase were the same as acarbose.
Scheme 89.

Synthesis of 5-((3-(aryl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-(p-tolylimino) thiazolidin-4-ones 120(a–g)
Patel and coworkers [143] designed a large panel of 5-[4-(substituted) benzylidine]thiazolidine-2,4-diones and screened them for antidiabetic activity by OGTT methods using pioglitazone as reference on male Wistar rats. The compounds were synthesized via 1,3 dipolar cycloaddition using chloroacetic acid and thiourea and water, which further underwent Knoevenagel condensation with 4-Cl benzaldehyde to furnish 5-(4-chlorobenzylidene)-2,4-thiazolidinedione. Thiazolidinedione was refluxed with primary aniline using acetonitrile and K2CO3 in microwave irradiation of 200 W to produce 5-[4-(substituted) benzylidene]thiazolidine-2,4-diones 121(a–j) (Scheme 90). The thiazolidinedione ring showed strong interaction with the binding site of the receptors and showed more potency for antidiabetic activity. The presence of a lipophilic group and EDG at the second and fourth position of the aromatic ring enhanced their activity. Compound 121e displayed high antidiabetic activity, whereas 121g and 121h showed moderate antidiabetic activity.
Scheme 90.

Synthesis of 5-[4-(substituted) benzylidene]thiazolidine-2,4-diones 121(a–j)
Badiger et al. [144] synthesized 5-{[2-(4-fluorobenzyl)-6-arylimidazo[2,1-b] [1,3,4] thiadiazol-5-yl]methylene}thiazolidine-2,4-diones and tested them in male Wistar rats for their antidiabetic activity (in vivo hypoglycemic and hypolipidemic activity). Imidazo[2,1-b] [1,3,4] thiadiazoles underwent Vilsmeier–Haack reaction and formed imidazo[2,1-b] [1,3,4] thiadiazole-5-carbaldehydes. The mechanism involved the reaction of 2-amino-1,3,4-thiadiazole and α-haloketones (phenacylbromides) using dry ethanol to give products that were neutralized with the help of aqueous sodium carbonate solution, and the respective free bases imidazo[2,1-b] [1,3,4] thiadiazoles were obtained in high yields, which further displayed Vilsmeier–Haack reaction to give imidazo[2,1-b] [1,3,4] thiadiazole-5-carbaldehydes which further underwent Knoevenagel condensation with thiazolidinone in piperidine to yield the targeted products (Scheme 91). Compounds 122c and 122d exhibited good hypoglycemic and hypolipidemic activity.
Scheme 91.

Synthesis of substituted 5-{[2-(4-fluorobenzyl)-6-arylimidazo[2,1-b] [1,3,4] thiadiazol-5-yl]methylene}thiazolidine-2,4-diones 122(a–d)
Mahapatra and coworkers [145] reported the fragment-based synthesis of aryl/alkylsulfonyloxy-5-(3-methoxybenzylidene)thiazolidine-2,4-dione derivatives and evaluated their biological activity for inhibition of PTP1B and anti-hyperglycemic activity. Thiazolidinones were formed by condensation of thiourea and chloroacetic acid in concentrated H2SO4, and consequently, aryl/alkylsulfonate esters were obtained by o-sulfonylation of 4-hydroxy-3-methoxybenzaldehyde in basic medium followed by Knoevenagel condensation, and TZD and aryl/alkylsulfonate esters furnished the final products, aryl/alkylsulfonyloxy-5-(3-methoxybenzylidene)thiazolidine-2,4-diones 123(a–j) (Scheme 92). The in silico study revealed that compounds 123b and 123e interacted with both catalyst active sites and other aryl phosphate binding sites and also concluded that H-bonding interaction was absent. The SAR study explained that the presence of phenyl/methyl sulfonate enhanced their activity as compared to other bulky substituents, and the larger arylidene motif displayed better inhibition. Compounds 123b (IC50 value 7.31) and 123e (IC50 value 8.73) were potent inhibitors for in vitro PTP1B activity and in vivo anti-hyperglycemic activity. Compounds 123e and 123i were also good PPARγ agonists, insulin sensitizers and PTP1B inhibitors, and all the newly synthesized compounds showed potent activity for diabetes.
Scheme 92.

Synthesis of aryl/alkylsulpfonyloxy-5-(3-methoxybenzylidene)thiazolidine-2,4-diones 123(a–j)
Alemán-González-Duhart and coworkers [146] designed a new synthetic approach to synthesize thiazolidinedione (TZD) derivatives 124a and 124b and tested them on the healthy animals for their nontoxic usage. Compound 124b was synthesized by 1,3-thiazolidine-2,4-dione and salicylaldehyde in solvent-free conditions at 120 °C, and 124a was prepared by 1,3-thiazolidine-2,4-dione and 3-chloro-2-fluorobenzaldehyde in water at 145 °C (Scheme 93). The authors investigated that the existence of EW heteroatom on aromatic ring, showed better interaction due to hydrogen bonding between the TZD head and binding site of LBD of PPARγ. Both compounds showed activity similar to other reported thiazolidinediones (rosiglitazone, pioglitazone and troglitazone).
Scheme 93.

Synthesis of 1,3-thiazolidine-2,4-dione derivatives 124(a, b)
Antioxidant Activity
Constantin et al. [147] developed a novel, selective, nontoxic synthesis of thiazolidine-4-one derivatives containing a xanthine structure and evaluated them for antioxidant and antidiabetic activity. Initially, theophylline (1,3-dimethylxanthine) reacted with sodium methoxide and furnished the salt, which combined with ethyl chloroacetate and gave theophylline ethyl acetate. The condensation of theophylline ethyl acetate with hydrazine hydrate led to corresponding hydrazide, which when refluxed with various aromatic aldehydes, theophyllineacethydrazide derivatives were obtained and further cyclized with mercaptoacetic acid to produce theophyllinyl-acetamido-thiazolidin-4-one derivatives 125(a–k) (Scheme 94). The in vitro antioxidant activity was done using DPPH and ABTS radical scavenging assay and phosphomolybdenum reducing antioxidant power assays. Among all synthesized compounds, 125f, 125d, 125c and 125k exhibited good activity for antiradical scavenging, and 125c and 125k displayed potent activity in phosphomolybdenum reducing antioxidant power. The radical scavenging activity was enhanced in the presence of methyl, methoxy and halogens (fluoro, chloro) substituents at the para position of the phenyl ring in final compounds.
Scheme 94.

Synthesis of theophenyllinyl-acetamido-thiazolidin-4-one derivatives 125(a–k)
Secci et al. [148] reported the design and synthesis of 36 new thiazolidinone derivatives and evaluated them as potent antioxidants; the compounds also displayed chelating properties. For in vitro antifungal activity, all synthesized compounds were screened against six topical drugs of Candida spp. (C. glabrata, C. tropicalis, C. albicans, C. krusei, C. parapsilosis and C. sakè) and reference drugs (fluconazole, clotrimazole, ketoconazole, amphotericin B, miconazole and tioconazole) using broth microdilution method. Carbonyl compounds and thiosemicarbazide reacted in ethanol and acetic acid to form thiosemicarbazone, which underwent cyclization with ethyl bromoacetate using sodium acetate in methanol to produce 1,3-thiazolidine-4-one derivatives 126G (1 and 2). The resulting compounds were refluxed with 2-/3-/4-chloro and fluorobenzyl bromide and 2-/3-nitrobenzyl bromide using anhydrous acetone and potassium carbonate to prepare N-substituted thiazolidinone derivatives 126(A–H). p-Nitrobenzyl-N-substituted thiazolidinone derivatives reacted with sodium dithionite in DMF in basic aqueous solution which resulted in synthesis of 4-aminobenzyl derivatives 126I (Scheme 95). Compound 126G3 was the most active against all tested fungal species, due to the substitution of the benzyl group with -Cl at the meta position with an MIC value of 2 μg/mL. Compound 126B1 also had the same effect against all species, except C. parapsilosis. Compounds 126(A3, C3, D3 and E3) presented moderate activity (MICs = 2 μg/mL) against C. albicans. The synthesized compounds have the same activity as the reference when the benzyl group was substituted with the EWG. Compounds 126(A3, B1, C2, C3, D3, E3, E4, G3, G4, H2 and I1) displayed the highest antifungal activity against Candida spp. All compounds were tested for antioxidant and metal chelating assays, reducing power from CUPRAC, free radical scavenging ability by DPPH and ABTS assay and phosphomolybdenum assays. The chelating property and antioxidant activity was estimated via EDTA and Trolox. Among the synthesized compounds, most compounds showed similar antioxidant effects and cytotoxicity as the reference compounds.
Scheme 95.

Synthesis of N-substituted thiazolidinone derivatives 126(A–I series)
Anticonvulsant Activity
Faizi and coworkers [149] synthesized new BZD derivatives and examined the compounds for biological evaluation. The novel compounds were synthesized by reaction of hydrazide, aldehydes and thioglycolic acid using anhydrous ZnCl2 in dry toluene. The synthesized compounds consisted of the functional group which easily combined with benzodiazepine receptors and 4-thiazolidinone ring as a pharmacophore moiety. Here, the mechanism showed that, firstly, an aromatic nucleophilic substitution reaction occurred in 2,4-dichlorobenzoic acid with phenol to form 2,4-chloro-2-phenoxybenzoic acid, followed by esterification, and then combined with hydrazine hydrate to yield 4-chloro-2-phenoxybenzohydrazide. The products 127(a–j) were synthesized by treatment of 4-chloro-2-phenoxy-benzoic acid hydrazide with different benzaldehydes and were further refluxed with thioglycolic acid (Scheme 96). Compound 127i displayed significant anticonvulsant effect in PTZ test and showed good hypnotic effect, whereas compounds 127a, 127b, 127e, 127f, 127h and 127i revealed good activity in the MES model. Flumazenil worked to antagonize the sedative-hypnotic effect of compounds which contain benzodiazepine. However, 127i did not display any change in the anterograde memory and also did not show neurotoxicity. Some of the tested compounds exhibited antiepileptic effects by PTZ and MES models.
Scheme 96.

Synthesis of 4-chloro-N-(2-(substituted-phenyl)-4-oxothiazolidin-3-yl)-2-phenoxybenzamide (BZD) derivatives 127(a–j)
Nikalje and coworkers [150] reported the synthesis of novel N-(2-oxo-2-(4-oxo-2-substituted thiazolidin-3ylamino)ethyl)benzamide derivatives under MW conditions. The compounds 128(a–j) were prepared by multistep synthesis. In the first step, benzoyl chloride reacted with glycine in aq. NaOH to form 2-benzamidoacetic acid, which showed esterification and gave ethyl 2-benzamido acetate. Ethyl 2-benzamido acetate and hydrazine hydrate were refluxed in ethanol to obtain (2-hydrazinyl-2-oxoethyl)benzamide, which was further refluxed with different heterocyclic and aromatic aldehydes using glacial acetic acid in ethanol to obtain Schiff bases. Compounds 128(a–j) were prepared under MWI for 12–17 min by cyclization of the Schiff base and thioglycolic acid, using anhydrous ZnCl2 and DMF (Scheme 97). The synthesis of final compounds was based on a four-point pharmacophoric HBD model, electron donor moiety (D), distal aryl domain (A) and distal hydrophilic-hydrophobic aryl ring (C) for validation using computational parameters like molecular docking, pharmacokinetic properties (ADME) and log P and for anticonvulsant evaluation, maximal electroshock seizure and subcutaneous pentylenetetrazole, and chemical testing in a mouse model. The neurotoxicity was evaluated by calculating minimal motor impairment via rotarod test. Compound 128h showed highly potent and long-acting protection against a 30-mg/kg dose at 0.5-h and 4-h intervals, whereas compound 128f also showed high potency only at a 0.5-h interval. But compound 128d showed potent and long-acting inhibition against 100 mg/kg at both time intervals. Compounds 128a, 128g and 128h displayed shielding of 100 mg/kg at a 0.5-h time interval in sc-PTZ screening. The SAR study manifested that the presence of small electron-donating polar groups at the phenyl ring and the presence of a heterocyclic ring (-indolyl) in the place of the aromatic ring exhibited good anticonvulsant effect. The docking studies concluded that all the newly synthesized compounds acted by inhibition voltage-gated ion channels (generally sodium channels).
Scheme 97.

Synthesis of thiazolidine-benzamide derivatives 128(a–j)
Miscellaneous Activity
Sabbua and colleagues [151] synthesized a new amino alcohol and thiazolidinedione hybrid, inspired by aegeline, and also evaluated their anti-adipogenic activity. Compounds (Z)-N-(3-(4-((2,4-dioxothiazolidin-5-ylidene)methyl) phenoxy)-2-hydroxypropyl)benzamides were produced from Knoevenagel condensation of N-(3-(4-formylphenoxy)-2-hydroxypropyl)benzamide with methanol and piperidine. Other thiazolidine-2,4-dione derivatives were prepared through ring-opening reaction of (Z)-3-benzyl-5-(4-(oxiran-2-ylmethoxy)benzylidene)thiazolidine-2,4-dione with substituted benzylamines and picolylamines, respectively, in isopropanol under different conditions. Among all the synthesized compounds, 129 was the potent inhibitor of adipocyte differentiation, which stopped adipogenesis and lipidosis in 3T3–L1 preadipocyte cells by blocking of the S-phase of the cell cycle, prevented mitotic clonal development and decreased two vital transcription factors, PPARg and C/EBPa, and correlated genes like aP2 and FAS of adipocyte differentiation. The authors concluded that the final compounds effectively worked on obesity and related metabolic disorders (Fig. 10).
Fig. 10.
Structure of the most anti-adipogenic compound 129 [3-benzyl-5-(4-{2-hydroxy-3-[(pyridin-2-ylmethyl)-amino]-propoxy}-benzylidene)-thiazolidine-2,4-dione]
Sousa et al. [152] investigated Se-phenyl-thiazolidine-4-carboselenoate (130) (Se-PTC) as a protective agent against oxidative and behavioral stress in a mania model induced by ouabain (OUA), which is an extremely hazardous substance, in male rats (Fig. 11). The authors concluded that Se-PTC showed anti-manic-like action and protection against hyperlocomotion and brain oxidative stress in rats, prevented increased locomotor activity, and normalized the activity of Na+/K+-ATPase, while the LiCl (lithium chloride) blocked the increased crossing induced by OUA and also managed bipolar disorder (BD).
Fig. 11.
Chemical structure of (R)-Se-phenyl thiazolidine-4-carboselenoate (Se-PTC)
Rahim and coauthors [153] synthesized 4-thiazolidinone analogs and investigated them for urease inhibition. The final compounds were synthesized in two steps: initially, substituted carbonyl compounds were refluxed with thiosemicarbazide and HCl in EtOH, and Schiff bases were formed, which further reacted with chloroacetic acid and CH3COONa using glacial AcOH, to furnish 4-thiazolidinone derivatives 131(a–t) (Scheme 98). All synthesized molecules gave moderate to excellent inhibition, with IC50 values in the range of 1.73–69.65 as compared to standard thiourea. Among all newly synthesized compounds, 131(c, f, h, j, o, p, q, s and t) showed excellent inhibition activity with IC50 values of 9.34 ± 0.02, 14.62 ± 0.03, 8.43 ± 0.01, 7.3 ± 0.04, 2.31 ± 0.002, 5.75 ± 0.003, 8.81 ± 0.005 and 1.73 ± 0.001, respectively. The pharmacological activity of compounds was affected by the steric effect, different substituents and their positions (Table 7).
Scheme 98.

Synthesis of 4-thiazolidinone derivatives 131(a–t)
Table 7.
Different substituents of thiazolidine analogs 131(a–t)

Yadav et al. [154] investigated an efficient, less toxic synthesis of CBTMT and MBTMT and evaluated their corrosion inhibition activity on mild steel in 15% HCl sol. using potentiodynamic polarization, weight loss measurements and EIS techniques (Fig. 12). The concentration of inhibitor and temperature affected their efficiency of reaction: with increase in concentration, efficiency increased, but increased temperature decreased their efficiency. The weight loss measurement revealed that MBTMT (97.5%) showed higher efficiency than CBTMT (95.8%) at 303 K. This inhibition process was based mainly on adsorption, with both inhibitors obeying the Langmuir adsorption isotherm, and it was concluded that their inhibition depended on the covered surface area and number of active sites, i.e. N and S atoms. The higher value of µ (dipole moment) and lower value of ΔE (energy gap) enhanced compound inhibition efficiency.
Fig. 12.

Structure of inhibitors CBTMT (132) and MBTMT (133)
Discussion
Green chemistry is a fascinating area in the field of sustainable synthesis. Green solvents and catalysts play a significant role in synthetic organic chemistry. Today’s chemistry researchers need to explore new avenues of synthetic methodologies and biological applications of heterocyclic scaffolds. Though various developments have occurred in the synthetic and biological study of thiazolidine and its derivatives, there is a need for further development of the chemistry to enhance discoveries in this field. During the extensive review of the literature review on thiazolidine and its derivatives, several strengths and weaknesses were noted. For example, solvents used in the process of synthesis are hazardous to the environment, and therefore solvent-free synthesis has proven to be a boon for the environment and society, as it reduces waste and energy consumption. Solvents also serve as a heat transfer medium, and thus detailed study of their thermodynamics is required. A major drawback regarding the purification procedures (column chromatography) is that they require a large amount of solvents for purification, so extensive research is needed to avoid flaws. Ionic liquids are used as green solvents due to their recyclability, low volatility and so forth, but their high cost, safety issues and lack of generalization limit their use. In the recent scenario, crucial improvements in issues such as energy efficiency, by-product generation, impurity removal procedures, release of toxic products, whole life-cycle study and following common basic procedures with some modifications are necessary in the context of cost, population, environment, energy, health and resources. For industrial-level applications, synthesis methods could fulfill parameters including atom economy, environmental factor (e-factor), ecoscale, process profile, life-cycle analysis, environmental quotient and effective mass yield. Regarding pharmacological significance, thiazolidine-containing drugs show a variety of biological activity due to interactions such as hydrogen bonding, ion–dipole interaction, hydrophobic action, π–π interaction, Van der Waal forces and bonding with various enzymes and pharmacophores.
Besides all these concerns, several drugs containing thiazolidine derivatives are banned due to their side effects, fluid retention properties and idiosyncratic hepatotoxicity, and several are associated with risk for bladder cancer, edema, liver failure and heart failure. Therefore, critical study is needed with special emphasis on SAR and the role of PPARs in molecular mechanisms and biological activity. A better understanding of the structure of ligands and receptors and their interaction needs to be achieved. Keeping all these facts in view, thiazolidine motifs will prove to be valuable candidates for designing more effective and potent drugs by reducing their side effects. This review article includes a detailed study of thiazolidine and its derivatives, and their varied synthesis and pharmaceutical applications, and will pave a better pathway for future directions and challenges.
Conclusion
This review article summarizes the major developments in the fabrication of the thiazolidine nucleus and its hybrids via molecular hybridization techniques that proceed through fusion of active motifs and pharmacophore units with advantages including dual action, change in selectivity profile, lower toxicity, and the design of new scaffolds in environmentally benign conditions such as microwave- and ultrasound-mediated synthesis, reusable catalysts, easy workup and solvent-free techniques with high atom economy. The application of MW-, nanoparticle- and ionic liquid-mediated synthesis in such processes has opened a new avenue for thiazolidine formation. It is likely that in the future, many more powerful and benign methodologies will emerge in this field. Furthermore, attention should be paid to modification and substitution in thiazolidine nuclei and also toward SAR and docking studies for improving the design strategies for drug targets. On a global scale, the opportunity in the field of thiazolidine is broad for synthetic and medical discovery, and this knowledge has been applied in the synthesis of multi-target hybrid drugs, which control diseases such as diabetes, cancer, multiple sclerosis and lupus. We trust that this review will serve as an update for researchers focused on the synthesis of thiazolidine derivatives and will encourage further growth in this field.
Acknowledgements
Nusrat Sahiba is thankful to CSIR-HRDG for providing a junior research fellowship as financial support [09/172(0088)/2018-EMR-I]. The authors are grateful to the Department of Chemistry, M.L.S.U. Udaipur (Raj.), India, for providing necessary laboratory and library facilities. We would also like to express sincere thanks to the anonymous reviewers for their beneficial comments.
Abbreviations
- iNOS
Inducible nitric oxide synthase
- MWI
Microwave irradiation
- DMF
Dimethylformamide
- TGA
Thioglycolic acid
- p-TSA
p-Toluenesulfonic acid
- DROC
Domino ring-opening cyclization
- TBAHS
Tetrabutylammonium hydrogen sulfate
- DBU
1,8-Diazabicyclo[5.4.0]undec-7-ene
- ATAs
β-Amidothioamides
- DDs
1,2-Diaza-1,3-dienes
- THF
Tetrahydrofuran
- HT29
Human colon cancer cell line
- MCF-7
Human breast cancer cell line
- EWG
Electron-withdrawing group
- EDG
Electron-donating group
- APTES
Aminopropyltriethoxysilane
- MCR
Multicomponent reaction
- MNP
Magnetic nanoparticle
- IL
Ionic liquid
- DDC
Dicarbethoxydihydrocollidine
- EDC·HCl
N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride
- DMSO
Dimethyl sulfoxide
- DES
Deep eutectic solvent
- APS
Ammonium persulfate
- FG
Functional group
- o-DCB
Ortho-dichlorobenzene
- SET
Single-electron transfer mechanism
- DAAD
Dialkylacetylene dicarboxylate
- EC50
Half maximal effective concentration
- Thfg
Tetrahydrofuranyl glycine
- MIC
Minimal inhibitory concentration
- ee
Enantiomeric excess
- MCF10A
Normal breast epithelial cells
- TZD
Thiazolidine-2,4-dione
- T3SS
Type III secretion system
- Xoo
Xanthomonas oryzae pv. oryzae
- DFT
Density functional theory
- HOBt
1-Hydroxybenzotriazole
- KSCN
Potassium thiocyanate
- HepG2
Hepatocellular carcinoma
- KB
Epidermoid carcinoma
- LLC
Lewis lung carcinoma
- A549
Human lung cancer cell line
- DU145
Human prostate cancer cell line
- HeLa
Human cervical cell line
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay
- GAC
Glutaminase C
- GAC
Glutaminase B
- SI
Safety index
- DPPH
2,2-Diphenyl-1-picrylhydrazyl
- ABTS
2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid
Peptide deformylase
- ATCC
American Type Culture Collection
- SAR
Structure–activity relationship
- CAIX
Carbonic anhydrase IX
- SRB
Sulforhodamine B
- MFC
Minimum fungicidal concentration
- GMK
Green monkey kidney
- CNS
Central nervous system
- IC50
Half maximal inhibitory concentration
- COX-2
Cyclooxygenase-2
- G2
Gap 2 phase
- M phase
Mitosis phase
- PBMC
Human peripheral blood mononuclear cell
- GI
Glycemic index
- PK-LDH
Pyruvate kinase lactate dehydrogenase coupled assay
- PKM2
Pyruvate kinase M2 isoform
- DSF
Differential scanning fluorimetry assay
- AO-EB
Acridine orange-ethidium bromide
- GAB
Glutaminase B
- DMAP
4-Dimethylaminopyridine
- TZD
Thiazolidinedione
- CDI
1,1′-carbonyldiimiadazole
- COPD
Chronic obstructive pulmonary disease
- DIPEA
N,N-diisopropylethylamine
- PDE4
Phosphodiesterase 4
- CQ
Chloroquine
- CQR
Chloroquine-resistant
- ADME
Absorption, distribution, metabolism, excretion
- EMT
Emetine
- AR
Aldose reductase
- HIV
Human immunodeficiency virus
- C/EBPa
Enhancer-binding protein alpha
- PPARγ
Peroxisome proliferative activator receptor gamma
- SMI
Schizont maturation inhibition
- OGTT
Oral glucose tolerance test
- PTP1B
Protein-tyrosine phosphatase 1B
- CUPRAC
Cupric ion reducing method
- LBD
Ligand-binding domain
- EDTA
Ethylenediaminetetraacetate
- HBD
Hydrogen-binding domain
- MES
Maximal electroshock seizure
- PMD
Pentamidine
- OUA
Ouabain
- BD
Bipolar disorder
- Se-PTC
Se-phenyl-thiazolidine-4-carboselenoate
- CBTMT
(Z)-5-(4-Chlorobenzylidene)-3-(benzo[d]thiazol-2-yl)-2-(4-methoxyphenyl)thiazolidine-4-one
- MBTMT
(Z)-5-(4-Methoxybenzylidene)-3-(benzo[d]thiazol-2-yl)-2-(4-methoxyphenyl)thiazolidine-4-one
- EIS
Electrochemical impedance spectroscopy
- EAC
Ehrlich ascites carcinoma
Compliance with Ethical Standards
Conflict of interest
The authors declare no conflict of interest, financial or otherwise.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Nusrat Sahiba, Email: sahibanusrat786@gmail.com.
Shikha Agarwal, Email: shikha_urj@yahoo.com.
References
- 1.Guo L, Zhang D. Cyclic poly(α-peptoid)s and their block copolymers from N-heterocyclic carbene-mediated ring-opening polymerizations of N-substituted N-carboxylanhydrides. J Am Chem Soc. 2009;131:18072–18074. doi: 10.1021/ja907380d. [DOI] [PubMed] [Google Scholar]
- 2.Butuc E, Gherasim GM. Ordered heterocyclic copolymers. Polyamide-imides with S-triazine rings. J Polym Sci. 1984;22:503–507. [Google Scholar]
- 3.Stead CV. Halogenated heterocycles in reactive dyes. Dyes Pigments. 1982;3:161–171. [Google Scholar]
- 4.Sniady A, Morreale MS, Wheeler KA, Dembinski R. Room-temperature electrophilic 5-endo-dig chlorocyclization of alk-3-yn-1-ones with the use of pool sanitizer: synthesis of 3-chlorofurans and 5-chlorofuropyrimidine nucleosides. Eur J Org Chem. 2008 doi: 10.1002/ejoc.200800397. [DOI] [Google Scholar]
- 5.Sarvary A, Maleki A. A review of syntheses of 1,5-disubstituted tetrazole derivatives. Mol Divers. 2014;19:189–212. doi: 10.1007/s11030-014-9553-3. [DOI] [PubMed] [Google Scholar]
- 6.Shaabani A, Maleki A, Rezayan AH, Sarvary A. Recent progress of isocyanide-based multicomponent reactions in Iran. Mol Divers. 2011;15:41–68. doi: 10.1007/s11030-010-9258-1. [DOI] [PubMed] [Google Scholar]
- 7.Maleki A, Sarvary A. Synthesis of tetrazoles via isocyanide-based reactions. RSC Adv. 2015;5:60938–60955. [Google Scholar]
- 8.Zhang Q, Zhou H, Zhai S, Yan B. Natural product-inspired synthesis of thiazolidine and thiazolidinone compounds and their anticancer activities. Curr Pharm Des. 2010;16:1826–1842. doi: 10.2174/138161210791208983. [DOI] [PubMed] [Google Scholar]
- 9.Havrylyuk D, Zimenkovsky B, Vasylenko O, Lesyka R. Synthesis and anticancer and antiviral activities of new 2-pyrazoline-substituted 4-thiazolidinones. J Heterocycl Chem. 2013;50:E55–E62. [Google Scholar]
- 10.Osmaniye D, Levent S, Ardıç CM, Atlı Ö, Özkay Y, Kaplancıklı ZA. Synthesis and anticancer activity of some novel benzothiazole-thiazolidine derivatives. Phosphorus Sulfur Silicon Relat Elem. 2017;193:249–256. [Google Scholar]
- 11.El-Gaby MSA, Ismail ZH, Abdel-Gawad SM, Aly HM, Ghorab MM. Synthesis of thiazolidine and thiophene derivatives for evaluation as anticancer agents. Phosphorus Sulfur Silicon Relat Elem. 2009;184:2645–2654. [Google Scholar]
- 12.Agarwal S, Agarwal DK, Gautam N, Agarwal K, Gautam DC. Synthesis and in vitro antimicrobial evaluation of benzothiazole incorporated thiazolidin-4-ones derivatives. J Korean Chem Soc. 2014;58:33–38. [Google Scholar]
- 13.Sankar PS, Divya K, Padmaja A, Padmavathi V. Synthesis and antimicrobial activity of azetidinone and thiazolidinone derivatives from azolylindolyl Schiff’s bases. Med Chem. 2017;7:340–347. [Google Scholar]
- 14.Deep A, Kumar P, Narasimhan B, Ramasamy K, Mani V, Mishra RK, Majeed ABA. Synthesis, antimicrobial, anticancer evaluation of 2-(aryl)-4- thiazolidinone derivatives and their QSAR studies. Curr Top Med Chem. 2015;15:990–1002. doi: 10.2174/1568026615666150317221849. [DOI] [PubMed] [Google Scholar]
- 15.Gouda MA, Abu-Hashem AA. Synthesis, characterization, antioxidant and antitumor evaluation of some new thiazolidine and thiazolidinone derivatives. Arch Pharm Chem Life Sci. 2011;11:170–177. doi: 10.1002/ardp.201000165. [DOI] [PubMed] [Google Scholar]
- 16.Nishida S, Maruoka H, Yoshimura Y, Goto T, Tomita R, Masumoto E, Okabe F, Yamagata K, Fujioka T. Synthesis and biological activities of some new thiazolidine derivatives containing pyrazole ring system. J Heterocycl Chem. 2012;49:303–309. [Google Scholar]
- 17.Datar PA, Aher SB. Design and synthesis of novel thiazolidine-2,4-diones as hypoglycemic agents. J Saudi Chem Soc. 2016;20:S196–S201. [Google Scholar]
- 18.Moreira DRM, Costa SPM, Hernandes MZ, Rabello MM, de Oliveria Filho GB, de Melo Cristiane ML, da Rocha LF, de Simone CA, Ferreira RS, Fradico JRB, Meira CS, Guimarães ET, Srivastava RM, Pereira VRA, Soares MBP, Leite ACL. Structural investigation of anti-Trypanosoma cruzi 2-iminothiazolidin-4-ones allows the identification of agents with efficacy in infected mice. J Med Chem. 2012;55:10918–10936. doi: 10.1021/jm301518v. [DOI] [PubMed] [Google Scholar]
- 19.Zehetmeyr FK, Pereira da Silva MAM, Pereira KM, Berne MEA, Cunico W, Júnior JCC, Gouvea DP, da Silva Nascente P, de Oliveria Hübner S, Siqueira GM. Ovicidal in vitro activity of 2-aryl-3-(2-morpholinoethyl)thiazolidin-4-ones and 2-aryl-3-(3-morpholinopropyl)thiazolidin-4-ones against Fasciola hepatica (Linnaeus, 1758) Exp Parasitol. 2018;192:60–64. doi: 10.1016/j.exppara.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 20.Geronikaki AA, Lagunin AA, Hadjipavlou-Litina DI, Eleftheriou PT, Filimonov DA, Poroikov VV, Alam I, Saxena AK. Computer-aided discovery of anti-inflammatory thiazolidinones with dual cyclooxygenase/lipoxygenase inhibition. J Med Chem. 2008;51:1601–1609. doi: 10.1021/jm701496h. [DOI] [PubMed] [Google Scholar]
- 21.Ma L, Xie C, Ma Y, Liu J, Xiang M, Ye X, Zheng H, Chen Z, Xu Q, Chen T, Chen J, Yang J, Qiu N, Wang G, Liang X, Peng A, Yang S, Wei Y, Chen L. Synthesis and biological evaluation of novel 5-benzylidenethiazolidine-2,4-dione derivatives for the treatment of inflammatory diseases. J Med Chem. 2011;54:2060–2068. doi: 10.1021/jm1011534. [DOI] [PubMed] [Google Scholar]
- 22.Barros CD, Amato AA, de Oliveira TB, Iannini KBR, da Silva AL, da Silva TG, Leite ES, Hernandes MZ, de Lima MCA, Galdino SL, de Asis Rocha Neves F, da Rocha Pitta I. Synthesis and anti-inflammatory activity of new arylidene-thiazolidine-2,4-diones as PPARγ ligands. Bioorg Med Chem. 2010;18:3805–3811. doi: 10.1016/j.bmc.2010.04.045. [DOI] [PubMed] [Google Scholar]
- 23.Chilamakuru NB, Shankaranath V, Rajasekhar KK, Singirisetty T. Synthesis, characterisation and anti-tubercular activity of some new 3,5-disubstituted-2,4-thiazolidinediones. Asian J Pharm Clin Res. 2013;6:29–33. [Google Scholar]
- 24.Siddiqui IR, Singh PK, Singh J, Singh J. Synthesis and fungicidal activity of novel 4,4′-bis(2″-aryl-5″-methyl/unsubstituted-4″-oxo-thiazolidin-3″-yl) bibenzyl. J Agric Food Chem. 2003;51:7062–7065. doi: 10.1021/jf0342324. [DOI] [PubMed] [Google Scholar]
- 25.Nitsche C, Schreier VN, Behnam MAM, Kumar A, Bartenschlager R, Klein CD. Thiazolidinone-peptide hybrids as dengue virus protease inhibitors with antiviral activity in cell culture. J Med Chem. 2013;56:8389–8403. doi: 10.1021/jm400828u. [DOI] [PubMed] [Google Scholar]
- 26.Romine JL, St. Laurent DR, Leet JE, Martin SW, Serrano-Wu MH, Yang F, Gao M, O’Boyle DR, Lemm JA, Sun JH, Nower PT, Huang X, Deshpande MS, Meanwell NA, Snyder LB. Inhibitors of HCV NS5A: from iminothiazolidinones to symmetrical stilbenes. ACS Med Chem Lett. 2011;2:224–229. doi: 10.1021/ml1002647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang S, Tala SR, Lu H, Abo-Dya NE, Avan I, Gyanda K, Lu L, Katritzky AR, Debnath AK. Design, synthesis, and biological activity of novel 5-((arylfuran/1H-pyrrol-2-yl)methylene)-2-thioxo-3-(3-(trifluoromethyl)phenyl)thiazolidin-4-ones as HIV-1 fusion inhibitors targeting gp41. J Med Chem. 2011;54:572–579. doi: 10.1021/jm101014v. [DOI] [PubMed] [Google Scholar]
- 28.Barreca ML, Balzarini J, Chimirri A, De Clercq E, De Luca L, Holtje HD, Holtje M, Monforte AM, Monforte P, Pannecouque C, Rao A, Zappala M. Design, synthesis, structure-activity relationships, and molecular modeling studies of 2,3-diaryl-1,3-thiazolidin-4-ones as potent anti-HIV agents. J Med Chem. 2002;45:5410–5413. doi: 10.1021/jm020977+. [DOI] [PubMed] [Google Scholar]
- 29.Rawal RK, Tripathi R, Katti SB, Pannecouquec C, De Clercq E. Design, synthesis, and evaluation of 2-aryl-3-heteroaryl-1,3- thiazolidin-4-ones as anti-HIV agents. Bioorg Med Chem. 2007;15:1725–1731. doi: 10.1016/j.bmc.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Janovec L, Sabolová D, Kožurková M, Paulíková H, Kristian P, Ungvarský J, Moravčíková E, Bajdichova M, Podhradsky D, Imrich J. Synthesis, DNA interaction, and cytotoxic activity of a novel proflavine-dithiazolidinone pharmacophore. Bioconjugate Chem. 2007;18:93–100. doi: 10.1021/bc060168v. [DOI] [PubMed] [Google Scholar]
- 31.Patrick DA, Gillespie JR, McQueen J, Hulverson MA, Ranade RM, Creason SA, Herbst ZM, Gelb MH, Buckner FS, Tidwell RR. Urea derivatives of 2-aryl-benzothiazol-5-amines: a new class of potential drugs for human African trypanosomiasis. J Med Chem. 2017;60:957–971. doi: 10.1021/acs.jmedchem.6b01163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pavin NF, Donato F, Cibin FW, Jesse CR, Schneider PH, de Salles HD, Amaral Soares L, Alves D, Savegnago L. Antinociceptive and anti-hypernociceptive effects of Se-phenyl thiazolidine-4-carboselenoate in mice. Eur J Pharmacol. 2011;668:169–176. doi: 10.1016/j.ejphar.2011.06.038. [DOI] [PubMed] [Google Scholar]
- 33.Sirivolu VR, Vernekar SKV, Marchand C, Naumova A, Chergui A, Renaud A, Stephen AG, Chen F, Sham YY, Pommier Y, Wang Z. 5-Arylidenethioxothiazolidinones as inhibitors of tyrosyl-DNA phosphodiesterase I. J Med Chem. 2012;55:8671–8684. doi: 10.1021/jm3008773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Jing F, Xu F, Xie Y, Shi F, Fang H, Li M, Xu W. Design, synthesis and biological activity of thiazolidine-4-carboxylic acid derivatives as novel influenza neuraminidase inhibitors. Bioorg Med Chem. 2011;19:2342–2348. doi: 10.1016/j.bmc.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 35.Ramazani Y, Levtchenko EN, Heuvel LVD, Schepdael AV, Paul P, Ivanova EA, Pastore A, Hartman TM, Price NPJ. Evaluation of carbohydrate-cysteamine thiazolidines as pro-drugs for the treatment of cystinosis. Carbohydr Res. 2017;439:9–15. doi: 10.1016/j.carres.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Hosseinimehr SJ, Shafiee A, Mozdarani H, Akhlagpour S, Froughizadeh M. Radioprotective effects of 2-imino-3-[(chromone-2-yl)carbonyl] thiazolidines against γ-irradiation in mice. J Radiat Res. 2002;43:293–300. doi: 10.1269/jrr.43.293. [DOI] [PubMed] [Google Scholar]
- 37.Bolli MH, Abele S, Binkert C, Bravo R, Buchmann S, Bur D, Gatfield J, Hess P, Kohl C, Mangold C, Mathys B, Menyhart K, Muller C, Nayler O, Scherz M, Schmidt G, Sippel V, Steiner B, Strasser D, Treiber A, Weller T. 2-Imino-thiazolidin-4-one derivatives as potent, orally active S1P1 receptor agonists. J Med Chem. 2010;53:4198–4211. doi: 10.1021/jm100181s. [DOI] [PubMed] [Google Scholar]
- 38.Urbano M, Guerrero M, Velaparthi S, Crisp M, Chase P, Hodder P, Schaeffer MT, Brown S, Rosen H, Roberts E. Discovery, synthesis and SAR analysis of novel selective small molecule S1P4-R agonists based on a (2Z,5Z)-5-((pyrrol-3-yl)methylene)-3-alkyl-2-(alkylimino)thiazolidin-4-one chemotype. Bioorg Med Chem Lett. 2011;21:6739–6745. doi: 10.1016/j.bmcl.2011.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bi X, Pasunooti KK, Lescar J, Liu CF. Thiazolidine-masked α-oxo aldehyde functionality for peptide and protein modification. Bioconjugate Chem. 2017;28:325–329. doi: 10.1021/acs.bioconjchem.6b00667. [DOI] [PubMed] [Google Scholar]
- 40.Katayama H, Morisue S. A novel ring opening reaction of peptide N-terminal thiazolidine with 2, 2′-dipyridyl disulfide (DPDS) efficient for protein chemical synthesis. Tetrahedron. 2017;73:3541–3547. [Google Scholar]
- 41.Oh J, Patel J, Park HB, Crawford JM. β-Lactam biotransformations activate innate immunity. J Org Chem. 2018;83:7173–7179. doi: 10.1021/acs.joc.8b00241. [DOI] [PubMed] [Google Scholar]
- 42.Chen H, Yin Q, Li C, Wang E, Gao F, Zhang X, Yin Z, Wei S, Li X, Meng M, Zhang P, Li N, Zhang J. Synthesis of C-pseudonucleosides bearing thiazolidin-4-one as a novel potential immunostimulating agent. ACS Med Chem Lett. 2011;2:845–848. doi: 10.1021/ml200155k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J, Chan KKJ, Chan W. Identification of protein thiazolidination as a novel molecular signature for oxidative stress and formaldehyde exposure. Chem Res Toxicol. 2016;29:1865–1871. doi: 10.1021/acs.chemrestox.6b00271. [DOI] [PubMed] [Google Scholar]
- 44.Bivona LA, Giacalone F, Vaccaro L, Aprile C, Gruttadauria M. Cross-linked thiazolidine network as support for palladium: a new catalyst for Suzuki and Heck reactions. ChemCatChem. 2015;7:2526–2533. [Google Scholar]
- 45.Kozell V, McLaughlin M, Strappaveccia G, Santoro S, Bivona LA, Aprile C, Gruttadauria M, Vaccaro L. Sustainable approach to waste-minimized Sonogashira cross-coupling reaction based on recoverable/reusable heterogeneous catalytic/base system and acetonitrile azeotrope. ACS Sustain Chem Eng. 2016;4:7209–7216. [Google Scholar]
- 46.Bozdag-dündar O, Ertan R, Aboul-Enein HY, Kładna A, Kruk I. Free radical scavenging abilities of flavonylthiazolidine-2,4-dione compounds. Luminescence. 2011;26:10–16. doi: 10.1002/bio.1176. [DOI] [PubMed] [Google Scholar]
- 47.Bozdag-dündar O, Gürkan S, Aboul-Enein HY, Kruk I, Kładna A. Scavenging of superoxide anion radical and hydroxyl radical by novel thiazolylthiazolidine-2,4-dione compounds. Luminescence. 2009;24:194–201. doi: 10.1002/bio.1095. [DOI] [PubMed] [Google Scholar]
- 48.De B, Adhikari I, Nandy A, Saha A, Goswami BB. In silico modelling of thiazolidine derivatives with antioxidant potency: models quantify the degree of contribution of molecular fragments towards the free radical scavenging ability. J Mol Struct. 2017;1138:17–26. [Google Scholar]
- 49.Ueda S, Terauchi H, Yano A, Matsumoto M, Kubo T, Kyoya Y, Suzuki K, Ido M, Kawasaki M. 4,5-Dialkylsubstituted 2-imino-1,3-thiazolidine derivatives as potent inducible nitric oxide synthase inhibitors. Bioorg Med Chem. 2004;12:4101–4116. doi: 10.1016/j.bmc.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 50.Xiao B, Tang A, Yang J, Wei Z, Zhou E. P3HT-based photovoltaic cells with a high Voc of 1.22 V by using a benzotriazole-containing nonfullerene acceptor end-capped with thiazolidine-2,4-dione. ACS Macro Lett. 2017;6:410–414. doi: 10.1021/acsmacrolett.7b00097. [DOI] [PubMed] [Google Scholar]
- 51.Thyagarajan BS, Simon Nee Glowienka JA. Novel synthesis of thiazolidine derivatives. Synth Commun. 1988;18:197–203. [Google Scholar]
- 52.Maleki A, Ghassemi M, Firouzi-Haji R. Green multicomponent synthesis of four different classes of six-membered N-containing and O-containing heterocycles catalyzed by an efficient chitosan-based magnetic bionanocomposite. Pure Appl Chem. 2014 doi: 10.1515/pac-2017-0702. [DOI] [Google Scholar]
- 53.Maleki A. One-pot multicomponent synthesis of diazepine derivatives using terminal alkynes in the presence of silica-supported superparamagnetic iron oxide nanoparticles. Tetrahedron Lett. 2013;54:2055–2059. [Google Scholar]
- 54.Maleki A. One-pot three-component synthesis of pyrido [2′,1′:2,3]imidazo[4,5-c]isoquinolines using Fe3O4@SiO2–OSO3H as an efficient heterogeneous nanocatalyst. RSC Adv. 2014;4:64169–64173. [Google Scholar]
- 55.Jain AK, Vaidya A, Ravichandran V, Kashaw SK, Agrawal RK. Recent developments and biological activities of thiazolidinone derivatives: a review. Bioorg Med Chem. 2012;20:3378–3395. doi: 10.1016/j.bmc.2012.03.069. [DOI] [PubMed] [Google Scholar]
- 56.Jain VS, Vora DK, Ramaa CS. Thiazolidine-2,4-diones: progress towards multifarious applications. Bioorg Med Chem. 2013;21:1599–1620. doi: 10.1016/j.bmc.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 57.Nanjan MJ, Mohammed M, Kumar BRP, Chandrasekar MJN. Thiazolidinediones as antidiabetic agents: a critical review. Bioorg Chem. 2018;77:548–567. doi: 10.1016/j.bioorg.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 58.Shelke RN, Pansare DN, Pawar CD, Deshmukh AC, Pawar RP, Bembalkar SR. Synthesis of 2-((substituted)-2-chloroquinolin-3-yl)-3-((substituted) phenyl) thiazolidin-4-one with β-cyclodextrin-SO3H catalyst under solvent-free condition. J Chem. 2017;6:24–33. [Google Scholar]
- 59.Bhattacharyya A, Kavitha CV, Ghorai MK. Stereospecific synthesis of 2-iminothiazolidines via domino ring- opening cyclization of activated aziridines with aryl- and alkyl isothiocyanates. J Org Chem. 2016;81:6433–6443. doi: 10.1021/acs.joc.6b01551. [DOI] [PubMed] [Google Scholar]
- 60.Domling A, Wang W, Wang K. Chemistry and biology of multicomponent reactions. Chem Rev. 2012;112:3083–3135. doi: 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Biggs-Houck JE, You-nai A, Shaw JT. Recent advances in multicomponent reactions for diversity-oriented synthesis. Curr Opin Chem Biol. 2010;14:371–382. doi: 10.1016/j.cbpa.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 62.da Silva DS, da Silva CEH, Soares MSP, Azambuja JH, da Rosa Carvalho T, Zimmer GC, Frizzo CP, Braganhol E, Spanevello RM, Cunico W. Thiazolidin-4-ones from 4-(methylthio)benzaldehyde and 4(methylsulfonyl)benzaldehyde: synthesis, antiglioma activity and cytotoxicity. Eur J Med Chem. 2016;124:574–582. doi: 10.1016/j.ejmech.2016.08.057. [DOI] [PubMed] [Google Scholar]
- 63.Rahim F, Taha M, Ullah H, Wadood A, Selvaraj M, Rab A, Sajid M, Shah SAA, Uddin N, Gollapalli M. Synthesis of new arylhydrazide bearing Schiff bases/thiazolidinone: α-amylase, urease activities and their molecular docking studies. Bioorg Chem. 2019 doi: 10.1016/j.bioorg.2019.103112. [DOI] [PubMed] [Google Scholar]
- 64.Kaboudin B, Shiran JA. Novel one-pot four-component condensation cyclization reactions for the synthesis of thiazolidine-4-one and 3H-thiazoles. J Sulfur Chem. 2018 doi: 10.1080/17415993.2018.1497168. [DOI] [Google Scholar]
- 65.Bayat M, Hosseini FS, Nasri S. An efficient one-pot synthesis of tetrahydrothiazolo[3,2-a]quinolin-6-one derivatives. J Sulfur Chem. 2018;39:99–111. [Google Scholar]
- 66.Santeusanio S, Majer R, Perrulli FR, de Crescentini L, Favi G, Giorgi G, Mantellini F. Divergent approach to thiazolylidene derivatives: a perspective on the synthesis of a heterocyclic skeleton from β-amidothioamides reactivity. J Org Chem. 2017;82:9773–9778. doi: 10.1021/acs.joc.7b02135. [DOI] [PubMed] [Google Scholar]
- 67.Revelant G, Huber-Villaumje S, Dunand S, Kirsch G, Schohn H, Hesse S. Synthesis and biological evaluation of novel 2-heteroarylimino-1,3-thiazolidin-4-ones as potential anti-tumor agents. Eur J Med Chem. 2015;94:102–112. doi: 10.1016/j.ejmech.2015.02.053. [DOI] [PubMed] [Google Scholar]
- 68.Yahiaoui S, Moliterni A, Corriero N, Cuocci C, Toubal K, Chouaih A, Djafri A, Hamzaoui F. 2-thioxo-3N-(2-methoxyphenyl)-5[4′-methyl-3′N-(2′-methoxyphenyl) thiazol-2′(3′H)-ylidene] thiazolidin-4-one: synthesis, characterization, X-ray single crystal structure investigation and quantum chemical calculations. J Mol Struct. 2018 doi: 10.1016/j.molstruc.2018.09.052. [DOI] [Google Scholar]
- 69.Hassan RM, Abd-Allah WH, Salman AM, Abdel-Sattar El-Azzouny A, Aboul-Enein MN. Design, synthesis and anticancer evaluation of novel 1,3-benzodioxoles and 1,4-benzodioxines. Eur J Pharm Sci. 2019;139:105045. doi: 10.1016/j.ejps.2019.105045. [DOI] [PubMed] [Google Scholar]
- 70.Maleki A, Jafari AA, Yousefi S. Green cellulose-based nanocomposite catalyst: design and facile performance in aqueous synthesis of pyranopyrimidines and pyrazolopyranopyrimidines. Carbohydr Polym. 2017;175:409–416. doi: 10.1016/j.carbpol.2017.08.019. [DOI] [PubMed] [Google Scholar]
- 71.Gandhi D, Agarwal S. MgO NPs catalyzed eco-friendly reaction: a highly effective and green approach for the multicomponent one-pot synthesis of polysubstituted pyridines using 2-aminobenzothiazole. J Heterocycl Chem. 2018;55:2977–2984. [Google Scholar]
- 72.Maleki A, Movahed H, Ravaghi P. Magnetic cellulose/Ag as a novel eco-friendly nanobiocomposite to catalyze synthesis of chromene-linked nicotinonitriles. Carbohydr Polym. 2017;156:259–267. doi: 10.1016/j.carbpol.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 73.Maleki A, Eskandarpour V, Rahimi J, Hamidi N. Cellulose matrix embedded copper decorated magnetic bionanocomposite as a green catalyst in the synthesis of dihydropyridines and polyhydroquinolines. Carbohydr Polym. 2018;208:251–260. doi: 10.1016/j.carbpol.2018.12.069. [DOI] [PubMed] [Google Scholar]
- 74.Maleki A, Firouzi-Haji R, Hajizadeh Z. Magnetic guanidinylated chitosan nanobiocomposite: a green catalyst for the synthesis of 1,4-dihydropyridines. Int J Biol Macromol. 2017;116:320–326. doi: 10.1016/j.ijbiomac.2018.05.035. [DOI] [PubMed] [Google Scholar]
- 75.Maleki A, Firouzi-Haji R. l-Proline functionalized magnetic nanoparticles: a novel magnetically reusable nanocatalyst for one-pot synthesis of 2,4,6-triarylpyridines. Sci Rep. 2018;8:17303. doi: 10.1038/s41598-018-35676-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maleki A, Hajizadeh Z, Sharifi V, Emdadi Z. A green, porous and eco-friendly magnetic geopolymer adsorbent for heavy metals removal from aqueous solutions. J Clean Prod. 2019 doi: 10.1016/j.jclepro.2019.01.084. [DOI] [Google Scholar]
- 77.Safaei-Ghomi J, Nazemzadeh SH, Shahbazi-Alavi H. Nano-CdZr4(PO4)6 as a reusable and robust catalyst for the synthesis of bis-thiazolidinones by a multicomponent reaction of aldehydes, ethylenediamine and thioglycolic acid. J Sulfur Chem. 2017;38:195–205. [Google Scholar]
- 78.Safaei-Ghomi J, Navvab M, Shahbazi-Alavi H. CoFe2O4@SiO2/PrNH2 nanoparticles as highly efficient and magnetically recoverable catalyst for the synthesis of 1,3-thiazolidin-4-ones. J Sulfur Chem. 2016;37:601–612. [Google Scholar]
- 79.Sadeghzadeh SM, Daneshfar F. Ionic liquid immobilized on FeNi3 as catalysts for efficient, green, and one-pot synthesis of 1,3-thiazolidin-4-one. J Mol Liq. 2014;199:440–444. [Google Scholar]
- 80.Azgomi N, Mokhtary M. Nano-Fe3O4@SiO2supported ionic liquid as an efficient catalyst for the synthesis of 1,3-thiazolidin-4-ones under solvent-free conditions. J Mol Catal A Chem. 2015;398:58–64. [Google Scholar]
- 81.Subhedar DD, Shaikh MH, Arkile MA, Yeware A, Sarkar D, Shingate BB. Facile synthesis of 1,3-thiazolidin-4-ones as antitubercular agents. Bioorg Med Chem Lett. 2016;26:1704–1708. doi: 10.1016/j.bmcl.2016.02.056. [DOI] [PubMed] [Google Scholar]
- 82.Chen CY, Barve IJ, Sun CM. One-pot three-component synthesis of 2-imino-1,3-thiazolines on soluble ionic liquid support. ACS Combin Sci. 2016;18:638–643. doi: 10.1021/acscombsci.6b00106. [DOI] [PubMed] [Google Scholar]
- 83.Malla AM, Parveen M, Ahmad F, Azaz S, Alam M. [Et3NH][HSO4]-catalyzed eco-friendly and expeditious synthesis of thiazolidine and oxazolidine derivatives. RSC Adv. 2015;5:19552–19569. [Google Scholar]
- 84.Sadeghzadeh SM, Malekzadeh M. Synthesis of 1,3-thiazolidin-4-one using ionic liquid immobilized onto Fe3O4/SiO2/Salen/Mn. J Mol Liq. 2015;202:46–51. [Google Scholar]
- 85.Yedage DB, Patil DV. Environmentally benign deep eutectic solvent for synthesis of 1,3-thiazolidin-4-ones. Chem Select. 2018;3:3611–3614. [Google Scholar]
- 86.Dahiya A, Ali W, Patel BK. Catalyst and solvent free domino ring opening cyclization: a greener and atom economic route to 2-iminothiazolidines. ACS Sustain Chem Eng. 2018;6:4272–4281. [Google Scholar]
- 87.Mahmoodi NO, Mohammadgholipour S, Pirbasti FG. Microwave-assisted one-pot three-component synthesis of thiazolidinones using KSF@Ni as an efficient heterogeneous catalyst. J Sulfur Chem. 2017;38:668–678. [Google Scholar]
- 88.Sangeetha P, Sankar C, Tharini K. New thiazoldinone substituted 2,6-diarypiperid in-4-one: synthesis, crystal structure, spectral characterization, binding mode with calf thym us DNA. J Mol Struct. 2019;1198:126899. [Google Scholar]
- 89.Maleki A. Green oxidation protocol: selective conversions of alcohols and alkenes to aldehydes, ketones and epoxides by using a new multiwall carbon nanotube-based hybrid nanocatalyst via ultrasound irradiation. Ultrason Sonochem. 2017;40:460–464. doi: 10.1016/j.ultsonch.2017.07.020. [DOI] [PubMed] [Google Scholar]
- 90.Safaei-Ghomi J, Navvab M, Shahbazi-Alavi H. One-pot sonochemical synthesis of 1,3-thiazolidin-4-ones using nano-CdZr4(PO4)6 as a robust heterogeneous catalyst. Ultrason Sonochem. 2015;31:102–104. doi: 10.1016/j.ultsonch.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 91.Shaabani A, Hooshmand SE. Diversity-oriented catalyst-free synthesis of pseudopeptides containing rhodanine scaffolds via a one-pot sequential isocyanide-based six-component reactions in water using ultrasound irradiation. Ultrason Sonochem. 2017 doi: 10.1016/j.ultsonch.2017.06.030. [DOI] [PubMed] [Google Scholar]
- 92.Salehitabar L, Yavari I. A synthesis of functionalized 2,5-diiminothiazolidines from Nef-isocyanide adduct and 1-alkyl-3-arylthioureas. Phosphorus Sulfur Silicon Relat Elem. 2017;192:1195–1200. [Google Scholar]
- 93.Halimehjani AZ, Nosood YL. Synthesis of N, S-heterocycles and dithiocarbamates by the reaction of dithiocarbamic acids and S-alkyl dithiocarbamates with nitroepoxides. Org Lett. 2017;19:6748–6751. doi: 10.1021/acs.orglett.7b03501. [DOI] [PubMed] [Google Scholar]
- 94.Aly AA, Ibrahim MAA, Shehata EM, Hassan AAM, Brown AB. Prospective new amidinothiazoles as leukotriene B4 inhibitors. J Mol Struct. 2018 doi: 10.1016/j.molstruc.2018.07.085. [DOI] [Google Scholar]
- 95.Ebrahimi S. One-pot synthesis of 1,3-thiazolidin-4-one using ammonium persulfate as catalyst. J Sulfur Chem. 2016;37:587–592. [Google Scholar]
- 96.Wu SL, Gao X. Copper-catalyzed aerobic oxidative reaction of C60 with aliphatic primary amines and CS2. J Org Chem. 2018;83:2125–2130. doi: 10.1021/acs.joc.7b03061. [DOI] [PubMed] [Google Scholar]
- 97.Shehzadi SA, Khan I, Saeed A, Larik FA, Channar PA, Hassan M, Raza H, Abbas Q, Seo SY. One-pot four-component synthesis of thiazolidin-2-imines using CuI/ZnII dual catalysis: a new class of acetylcholinesterase inhibitors. Bioorg Chem. 2019;84:518–528. doi: 10.1016/j.bioorg.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 98.Robati M, Ghazanfari D, Islami MR, Saidi K (2017) Synthesis of new 1, 8-dioxo-octahydroxanthene derivatives containing 4-thiazolidinone moiety. Iran J Chem Chem Eng, p 36
- 99.Kandile NG, Mohamed MI, Ismaeel HM. Synthesis of new Schiff bases bearing 1,2,4-triazole, thiazolidine and chloroazetidine moieties and their pharmacological evaluation. J Enzym Inhib Med Chem. 2017;32:119–129. doi: 10.1080/14756366.2016.1238365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cihan-Üstündag G, Gürsoy E, Naesens L, Ulusoy-Güzeldemirci N, Çapan G. Synthesis and antiviral properties of novel indole-based thiosemicarbazides and 4-thiazolidinones. Bioorg Med Chem. 2016;24:240–246. doi: 10.1016/j.bmc.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Thakare MP, Kumar P, Kumar N, Pandey SK. Silica gel promoted environment-friendly synthesis of 2,3-disubstituted 4-thiazolidinones. Tetrahedron Lett. 2014;55:2463–2466. [Google Scholar]
- 102.Abdel Hafez NA, Elsayed MA, El-Shahawi M, Awad GEA, Ali KA. Synthesis and antimicrobial activity of new thiazolidine-based heterocycles as rhodanine analogues. J Heterocycl Chem. 2018;55:685–691. [Google Scholar]
- 103.Singh RP, Aziz MN, Gout D, Fayad W, ElManawaty MA, Lovely CJ. Novel thiazolidines: synthesis, antiproliferative properties and 2D-QSAR studies. Bioorg Med Chem. 2019 doi: 10.1016/j.bmc.2019.115047. [DOI] [PubMed] [Google Scholar]
- 104.Obydennov KL, Khamidullina LA, Galushchinskiy AN, Shatunova SA, Kosterina MF, Kalinina TA, Fan Z, Glukhareva TV, Morzherin YY. Discovery of methyl (5Z)-[2-(2,4,5-trioxopyrrolidin-3-ylidene)-4-oxo-1,3-thiazolidin-5-ylidene]acetates as antifungal agents against potato diseases. J Agric Food Chem. 2018;66:6239–6245. doi: 10.1021/acs.jafc.8b02151. [DOI] [PubMed] [Google Scholar]
- 105.Trotsko N, Kosikowska U, Paneth A, Wujec M, Malm A. Synthesis and antibacterial activity of new (2,4-dioxothiazolidin-5-yl/ylidene)acetic acid derivatives with thiazolidine-2,4-dione, rhodamine and 2-thiohydantoin moieties. Saudi Pharm J. 2018;26:568–577. doi: 10.1016/j.jsps.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Trotsko N, Kosikowska U, Paneth A, Plech T, Malm A, Wujec M. Synthesis and antibacterial activity of new thiazolidine-2,4-dione-based chlorophenyl thiosemicarbazone hybrids. Molecules. 2018;23:1023. doi: 10.3390/molecules23051023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xiang X, Tao H, Jiang S, Zhang LH, Cui ZN. Synthesis and bioactivity of thiazolidin-2-cyanamide derivatives against type III secretion system of Xanthomonas oryzae on rice. Pestic Biochem Phys. 2018;149:89–97. doi: 10.1016/j.pestbp.2018.06.011. [DOI] [PubMed] [Google Scholar]
- 108.Abdel-Galil E, Moawad EB, El-Mekabaty A, Said GE. Synthesis, characterization and antibacterial activity of some new thiazole and thiazolidinone derivatives containing phenyl benzoate moiety. Synth Commun. 2018;149:2083–2092. [Google Scholar]
- 109.Barakat A, Soliman SM, Al-Majid AM, Ali M, Islam MS, Elshaier YAMM, Ghabbour HA. New spiro-oxindole constructed with pyrrolidine/thioxothiazolidin-4-one derivatives: regioselective synthesis, x-ray crystal structures, Hirshfeld surface analysis, DFT, docking and antimicrobial studies. J Mol Struct. 2018;1152:101–114. [Google Scholar]
- 110.Abdelmajeid A, Amine MS, Hassan RA. Fatty acids in heterocyclic synthesis. Part XVII: synthesis of non-ionic surfactants containing piperidine, piperazine, imidazole based on thiadiazole and microbiological activities evaluation. Int J Org Chem. 2017;7:346–368. [Google Scholar]
- 111.Trotsko N, Przekora A, Zalewska J, Ginalska G, Paneth A, Wujec M. Synthesis and in vitro antiproliferative and antibacterial activity of new thiazolidine-2,4-dione derivatives. J Enzym Inhib Med Chem. 2018;33:17–24. doi: 10.1080/14756366.2017.1387543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pânzariu AT, Apotrosoaei M, Vasincu IM, Drăgan M, Constantin S, Buron F, Routier S, Profire L, Tuchilus C. Synthesis and biological evaluation of new 1,3-thiazolidine-4-one derivatives of nitro-l-arginine methyl ester. Chem Cent J. 2016;10:6. doi: 10.1186/s13065-016-0151-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Khan FAK, Jadhav KS, Patil RH, Shinde DB, Arote RB, Sangshetti JN. Biphenyl tetrazole-thiazolidinediones as novel bacterial peptide deformylase inhibitors: synthesis, biological evaluations and molecular docking study. Biomed Pharmacother. 2016;83:1146–1153. doi: 10.1016/j.biopha.2016.08.036. [DOI] [PubMed] [Google Scholar]
- 114.Gilani SJ, Nagarajan K, Dixit SP, Taleuzzaman M, Khan SA. Benzothiazole incorporated thiazolidin-4-ones and azetidin-2-ones derivatives: synthesis and in vitro antimicrobial evaluation. Arab J Chem. 2016;9:S1523–S1531. [Google Scholar]
- 115.Nastasă CM, Duma M, Pîrnău A, Vlase L, Tiperciuc B, Oniga O. Development of new 5-(chromene-3-yl) methylene-2,4-thiazolidinediones as antimicrobial agents. Clujul Med. 2016;89:122–127. doi: 10.15386/cjmed-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.De Monte C, Carradori S, Bizzarri B, Bolasco A, Caprara F, Mollica A, Rivanera D, Mari E, Zicari A, Akdemir A, Secci D. Anti-candida activity and cytotoxicity of a large library of new N-substituted-1,3-thiazolidin-4-one derivative. Eur J Med Chem. 2016;107:82–96. doi: 10.1016/j.ejmech.2015.10.048. [DOI] [PubMed] [Google Scholar]
- 117.Pansare DN, Mulla NA, Pawar CD, Shende VR, Shinde DB. One pot three components microwave assisted and conventional synthesis of new 3-(4-chloro-2-hydroxyphenyl)-2-(substituted) thiazolidin-4-one as antimicrobial agent. Bioorg Med Chem Lett. 2017;24:3569–3573. doi: 10.1016/j.bmcl.2014.05.051. [DOI] [PubMed] [Google Scholar]
- 118.Nechak R, Bouzroura SA, Benmalek Y, Salhi L, Martini SP, Morizur V, Dunach E, Kolli BN. Synthesis and antimicrobial activity evaluation of novel 4-thiazolidinones containing a pyrone moiety. Synth Commun. 2015;45:262–272. [Google Scholar]
- 119.Kunzler A, Neuenfeldt PD, des Neves AM, Pereira CMP, Marques GH, Nascente PS, Fernandes MHV, Hübner SO, Cunico W. Synthesis, antifungal and cytotoxic activities of 2-aryl-3-((piperidin-1-yl)ethyl)thiazolidinones. Eur J Med Chem. 2013;64:74–80. doi: 10.1016/j.ejmech.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 120.Popiołeka Ł, Piątkowska-Chmiel I, Gawrońska-Grzywacz M, Biernasiuk A, Izdebskab M, Herbet M, Sysa M, Malm A, Dudka J, Wujec M. New hydrazide-hydrazones and 1,3-thiazolidin-4-ones with 3-hydroxy-2naphthoic moiety: synthesis, in vitro and in vivo studies. Biomed Pharmacother. 2018;103:1337–1347. doi: 10.1016/j.biopha.2018.04.163. [DOI] [PubMed] [Google Scholar]
- 121.Yakaiah S, Kumar PSV, Rani PB, Prasad KD, Aparna P. Design, synthesis and biological evaluation of novel pyrazolooxothiazolidine derivatives as antiproliferative agents against human lung cancer cell line A549. Bioorg Med Chem Lett. 2018;28:630–636. doi: 10.1016/j.bmcl.2018.01.027. [DOI] [PubMed] [Google Scholar]
- 122.MdoD Rodrigues, Santiago Priscila BGS, Marques KMR, Pereira VRA, de Castro Maria CAB, Cantalice JCLL, da Silva TG, Adam ML, do Nascimento SC, de Albuquerque JFC, Militao GCG. Selective cytotoxic and genotoxic activities of 5-(2-bromo-5-methoxybenzylidene)-thiazolidine-2,4-dione against NCI-H292 human lung carcinoma cells. Pharm Rep. 2018;70:446–454. doi: 10.1016/j.pharep.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 123.Ansari F, Idrees D, Hassan I, Ahmad K, Avecilla F, Azam A. Design, synthesis and biological evaluation of novel pyridine-thiazolidinone derivatives as anticancer agents: targeting human carbonic anhydrase IX. Eur J Med Chem. 2018;144:544–556. doi: 10.1016/j.ejmech.2017.12.049. [DOI] [PubMed] [Google Scholar]
- 124.Asati V, Bharti SK. Design, synthesis and molecular modeling studies of novel thiazolidine-2,4-dione derivatives as potential anti-cancer agents. J Mol Struct. 2018;1154:406–417. [Google Scholar]
- 125.Li R, Ning X, Zhou S, Lin Z, Wu X, Chen H, Bai X, Wang X, Ge Z, Li R, Yin Y. Discovery and structure-activity relationship of novel 4-hydroxy-thiazolidine-2-thione derivatives as tumor cell specific pyruvate kinase M2 activators. Eur J Med Chem. 2017;143:48–65. doi: 10.1016/j.ejmech.2017.11.023. [DOI] [PubMed] [Google Scholar]
- 126.Kulabaş N, Özakpınar ÖB, Özsavcı D, Leyssen P, Neyts J, Küçükgüzel İ. Synthesis, characterization and biological evaluation of thioureas, acylthioureas and 4-thiazolidinones as anticancer and antiviral agents. J Res Pharm. 2017;21:371–384. [Google Scholar]
- 127.Bataille CJR, Brennan MB, Byrne S, Davies SG, Durbin M, Fedorov O, Huber KVM, Jones AM, Knapp S, Liu G, Nadali A, Quevedo CE, Russell AJ, Walker RG, Westwood R, Wynne GM. Thiazolidine derivatives as potent and selective inhibitors of the PIM kinase family. Bioorg Med Chem. 2017;25:2657–2665. doi: 10.1016/j.bmc.2017.02.056. [DOI] [PubMed] [Google Scholar]
- 128.Sharma P, Reddy TS, Thummuri D, Senwar KR, Kumar NP, Naidu VGM, Bharghava SK, Shankaraiah N. Synthesis and biological evaluation of new benzimidazole-thiazolidinedione hybrids as potential cytotoxic and apoptosis inducing agents. Eur J Med Chem. 2016;124:608–621. doi: 10.1016/j.ejmech.2016.08.029. [DOI] [PubMed] [Google Scholar]
- 129.Kumar KSS, Hanumappa A, Vetrivel M, Hegde M, Girish YR, Byregowda TR, Rao S, Raghavan SC, Rangappa KS. Antiproliferative and tumor inhibitory studies of 2,3 disubstituted 4-thiazolidinone derivatives. Bioorg Med Chem Lett. 2015;25:3616–3620. doi: 10.1016/j.bmcl.2015.06.069. [DOI] [PubMed] [Google Scholar]
- 130.Anh HLT, Cuc NT, Tai BH, Yen PH, Nhiem NX, Thao DT, Nam NH, Minh CV, Kiem PV, Kim YH. Synthesis of chromonylthiazolidines and their cytotoxicity to human cancer cell lines. Molecules. 2015;20:1151–1160. doi: 10.3390/molecules20011151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kumar KS, Reddy BM, Babu VH. Synthesis of some novel 2,4-thiazolidinedione incorporated pyrazole derivatives as anti-cancer agents. Int J Pharm Pharm Sci. 2014;6:831–834. [Google Scholar]
- 132.Akshaya TT, Arunlal VB, Babu G, Biju CR. Synthesis, characterisation and in vitro anticancer activity of thiazolidine-2,4-dione derivatives. J Drug Discov Ther. 2014;2:42–47. [Google Scholar]
- 133.Yeh TK, Kuo CC, Lee YZ, Ke YY, Chu KF, Hsu HY, Chang HY, Liu YW, Song JS, Yang CW, Lin LM, Sun M, Wu SH, Kuo PC, Shih C, Chen CT, Tsou LK, Lee SJ. Design, synthesis, and evaluation of thiazolidine-2,4-dione derivatives as a novel class of glutaminase inhibitors. J Med Chem. 2017;60:5599–5612. doi: 10.1021/acs.jmedchem.7b00282. [DOI] [PubMed] [Google Scholar]
- 134.Ashour HMA, El-Ashmawy IM, Bayad AE. Synthesis and pharmacological evaluation of new pyrazolyl benzenesulfonamides linked to polysubstituted pyrazoles and thiazolidinones as anti-inflammatory and analgesic agents. Monatsh Chem. 2016;147:605–618. [Google Scholar]
- 135.Carzaniga L, Amari G, Rizzi A, Capaldi C, De Fanti R, Ghidini E, Villetti G, Carnini C, Moretto N, Facchinetti F, Caruso P, Marchini G, Battipaglia L, Patacchini R, Cenacchi V, Volta R, Amadei F, Pappani A, Capacchi S, Bagnacani V, Delcanale M, Puccini P, Catinella S, Civelli M, Armani E. Discovery and optimization of thiazolidinyl and pyrrolidinyl derivatives as inhaled PDE4 inhibitors for respiratory diseases. J Med Chem. 2017;60:10026–10046. doi: 10.1021/acs.jmedchem.7b01044. [DOI] [PubMed] [Google Scholar]
- 136.Maccari R, Vitale RM, Ottanà R, Rocchiccioli M, Marrazzo A, Cardile V, Graziano ACE, Amodeo P, Mura U, Corso AD. Structure activity relationships and molecular modelling of new 5-arylidene-4-thiazolidinone derivatives as aldose reductase inhibitors and potential anti-inflammatory agents. Eur J Med Chem. 2014;81:1–14. doi: 10.1016/j.ejmech.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 137.Oderinlo OO, Tukulula M, Isaacs M, Hoppe HC, Taylor D, Smith VJ, Khanye SD. New thiazolidine-2,4-dione derivatives combined with organometallic ferrocene: Synthesis, structure and antiparasitic activity. Appl Organomet Chem. 2018 doi: 10.1002/aoc.4385. [DOI] [Google Scholar]
- 138.Jain S, Kumar A, Saini D. Novel arylidene derivatives of quinoline based thiazolidinones: synthesis, in vitro, in vivo and in silico study as antimalarials. Exp Parasitol. 2018;185:107–114. doi: 10.1016/j.exppara.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 139.Hidaka K, Kimura T, Sankaranarayanan R, Wang J, McDaniel KF, Kempf DJ, Kameoka M, Adachi M, Kuroki R, Nguyen JT, Hayashi Y, Kiso Y. Identification of highly potent human immunodeficiency virus type-1 protease inhibitors against lopinavir and darunavir resistant viruses from allophenylnorstatine-based peptidomimetics with P2 tetrahydrofuranylglycine. J Med Chem. 2018;61:5138–5153. doi: 10.1021/acs.jmedchem.7b01709. [DOI] [PubMed] [Google Scholar]
- 140.Wang GC, Peng YP, Xie ZZ, Wang J, Chen M. Synthesis, α-glucosidase inhibition and molecular docking studies of novel thiazolidine-2,4-dione or rhodanine derivatives. Med Chem Commun. 2017;8:1477–1484. doi: 10.1039/c7md00173h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chhajed SS, Chaskar S, Kshirsagar SK, Haldar GMA, Mahapatra DK. Rational design and synthesis of some PPAR-γ agonists: substituted benzylideneaminobenzylidene- thiazolidine-2,4-diones. Comput Biol Chem. 2017;67:260–265. doi: 10.1016/j.compbiolchem.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 142.Kumar P, Duhan M, Kadyan K, Sindhu J, Kumar S, Sharma H. Synthesis of novel inhibitors of α-amylase based on thiazolidine-4-one skeleton containing pyrazole moiety and their configurational studies. Med Chem Commun. 2017;8:1468–1476. doi: 10.1039/c7md00080d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Patel KD, Patel CN, Patel GM. Microwave assisted synthesis and antidiabetic activity of novel 5-[4-(substituted) benzylidine]thiazolidine-2,4-dione. Med Chem. 2016;6:647–651. [Google Scholar]
- 144.Badiger NP, Shashidhar N, Vaidya PN. Synthesis of novel 5-{[2-(4-fluorobenzyl)-6- arylimidazo[2,1-b] [1,3,4]thiadiazol-5-yl]methylene}thiazolidine-2,4-diones as potent antidiabetic agents. Int J Sci Eng Appl. 2015;4:2319–7560. [Google Scholar]
- 145.Mahapatra MK, Kumar R, Kumar M. Synthesis, biological evaluation and in silico studies of 5-(3-methoxybenzylidene)thiazolidine-2,4-dione analogues as PTP1B inhibitors. Bioorg Chem. 2017;71:1–9. doi: 10.1016/j.bioorg.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 146.Alemán-González-Duhart D, Tamay-Cach F, Correa-Basurto J, Padilla-Martínez, Álvarez-Almazán S, Mendieta-Wejebe JE. In silico design, chemical synthesis and toxicological evaluation of 1,3-thiazolidine-2,4-dione derivatives as PPARγ agonists. Regul Toxicol Pharmacol. 2017;86:25–32. doi: 10.1016/j.yrtph.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 147.Constantin S, Lupascu FG, Apotrosoaei M, Vasincu IM, Lupascu D, Buron F, Routier S, Profire L. Synthesis and biological evaluation of the new 1,3-dimethylxanthine derivatives with thiazolidine-4-one scaffold. Chem Cent J. 2017;11:12. doi: 10.1186/s13065-017-0241-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Secci D, Carradori S, Bizzarri B, Chimenti P, de Monte C, Mollica A, Rivanera D, Zicari A, Mari E, Zengin G, Aktumsek A. Novel 1,3-thiazolidin-4-one derivatives as promising anti-Candida agents endowed with anti-oxidant and chelating properties. Eur J Med Chem. 2016;117:144–156. doi: 10.1016/j.ejmech.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 149.Faizi M, Jahani R, Ebadi SA, Tabatabai SA, Rezaee E, Lotfaliei M, Amini M, Almasirad A. Novel 4-thiazolidinone derivatives as agonists of benzodiazepine receptors: design, synthesis and pharmacological evaluation. EXCLI J. 2017;16:52–62. doi: 10.17179/excli2016-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nikalje APG, Shaikh AN, Shaikh SI, Khan FAK, Sangshetti JN, Shind DB. Microwave assisted synthesis and docking study of N-(2-oxo-2-(4-oxo-2-substituted thiazolidin-3ylamino)ethyl)benzamide derivatives as anticonvulsant agents. Bioorg Med Chem Lett. 2014;21:444–448. doi: 10.1016/j.bmcl.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 151.Sabbua S, Srivastava A, Yadav P, Varshney S, Choudhary R, Balaramnavar VM, Tadigoppulaa N, Gaikwad AN. Aegeline inspired synthesis of novel amino alcohol and thiazolidinedione hybrids with antiadipogenic activity in 3T3-L1 cells. Eur J Med Chem. 2018;143:780–791. doi: 10.1016/j.ejmech.2017.11.041. [DOI] [PubMed] [Google Scholar]
- 152.Sousa FSS, Seus N, Alves D, Salles HD, Schneider PH, Savegnago L. Evaluation of Se-phenyl-thiazolidine-4-carboselenoate protective activity against oxidative and behavioral stress in the maniac model induced by ouabain in male rats. Neurosci Lett. 2017;651:182–187. doi: 10.1016/j.neulet.2017.04.030. [DOI] [PubMed] [Google Scholar]
- 153.Rahim F, Zaman K, Ullah H, Taha M, Wadood A, Javed MT, Rehman W, Ashraf M, Uddin R, Uddin I, Asghar H, Khan AA, Khan MK. Synthesis of 4-thiazolidinone analogs as potent in vitro anti-urease agents. Bioorg Chem. 2015;63:123–131. doi: 10.1016/j.bioorg.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 154.Yadav M, Kumar S, Kumari N, Bahadur I, Ebenso EE. Experimental and theoretical studies on corrosion inhibition effect of synthesized benzothiazole derivatives on mild steel in 15% HCl solution. Int J Electrochem Sci. 2015;10:602–624. [Google Scholar]
- 155.Alizadeh A, Rostamnia S, Zohreh N, Hosseinpour R. A simple and effective approach to the synthesis of rhodanine derivatives via three-component reactions in water. Tetrahedron Lett. 2009;50:1533–1535. [Google Scholar]
- 156.Attanasi OA, De Crescentini L, Favi G, Filippone P, Giorgi G, Mantellini F, Moscatelli G, Behalo MS. An efficient one-pot, three-component synthesis of 5-hydrazinoalkylidene rhodanines from 1,2-diaza-1,3-dienes. Org Lett. 2009;11:2265–2268. doi: 10.1021/ol900545v. [DOI] [PubMed] [Google Scholar]
- 157.Attanasi OA, Bartoccini S, Favi G, Giorgi G, Perrulli FR, Santeusanio S. Powerful approach to heterocyclic skeletal diversity by sequential three-component reaction of amines, isothiocyanates, and 1,2-diaza-1,3-dienes. J Org Chem. 2012;77:1161–1167. doi: 10.1021/jo2021949. [DOI] [PubMed] [Google Scholar]
- 158.Dalmal T, Appalanaidu K, Kosurkar UB, Babu NJ, Kumbhare RM. One-pot synthesis of 2-imino-4-(trifluoromethyl)thiazolidin-4-ol derivatives in a three-component reaction: application to structurally diverse scaffolds of biological interest through subsequent reactions. Eur J Org Chem. 2014 doi: 10.1002/ejoc.201301710. [DOI] [Google Scholar]
- 159.Gan SF, Wan JP, Pan YJ, Sun CR. Water-mediated multicomponent reaction: a facile and efficient synthesis of multisubstituted thiazolidine-2-thiones. Synlett. 2010;6:0973–0975. [Google Scholar]
- 160.Kasmi-Mir S, Djafri A, Paquin L, Hamelin J, Rahmouni M. One-pot synthesis of 5-arylidene-2-imino-4-thiazolidinones under microwave irradiation. Molecules. 2006;11:597–602. doi: 10.3390/11080597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schlüter T, Frerichs N, Schmidtmann M, Martens J. Consecutive multicomponent reactions: synthesis of 3-Acyl-4-alkynyl-substituted 1,3-thiazolidines. Synthesis. 2018;50:1123–1132. [Google Scholar]
- 162.Dandia A, Singh R, Bhaskarana S, Samant SD. Versatile three component procedure for combinatorial synthesis of biologically relevant scaffold spiro[indole-thiazolidinones] under aqueous conditions. Green Chem. 2011;13:1852–1859. [Google Scholar]