

Abstract
One of the important factors and consequences in persistent asthma is the change in the vasculature of the airways and lung parenchyma. These changes could contribute to worsening asthma control and predispose asthmatics to critical asthma syndromes. For many years, the contribution of vasculature to severe asthma was limited to discussion of small and medium vessel vasculitis commonly referred to as Churg − Strauss syndrome. This comprehensive review will explore the known mechanisms that are associated with remodeling of the vasculature in a variety of critical asthma presentations. Inflammation of pulmonary and bronchial small blood vessels may contribute significantly but silently to asthma pathobiology. Inflammation in the vasculature of the lung parenchyma can decrease lung capacity while inflammation in airway vasculature can decrease airflow. This review will provide a modern perspective on Churg–Strauss syndromes with a focus on phenotyping, mechanism, and ultimately modern therapeutic approaches. Vascular remodeling and airway remodeling are not mutually exclusive concepts in understanding the progression of asthma and frequency of acute exacerbations. Furthermore, the contribution of vascular leak, particularly in the parenchymal vasculature, has become an increasingly recognized component of certain presentations of poorly controlled, severe persistent asthmatic and during exacerbations. We highlight how these mechanisms can contribute to some the severe presentations of influenza infection in patients with a history of asthma. The ultimate aim of this review is to summarize the current literature concerning vasculitis and the contribution of airway and parenchymal vascular remodeling to presentation of persistent asthma and its consequences during acute exacerbations and critical asthma syndromes.
Keywords: Asthma, Churg–Strauss syndromes, Vasculature, Eosinophilic granulomatosis polyangiitis
Introduction
Asthma is a chronic inflammatory airway disease that is characterized by a specific inflammatory cell response resulting in a swollen, edematous, hyper-reactive airway. For certain patients, asthma is punctuated by frequent acute exacerbations and some patients deteriorate alarmingly into one of the critical asthma syndromes (CAS). Modern therapy and research has focused on the trigger of the inflammatory response in asthma but few treatments are directed at vasculature of the airway and parenchyma, most notably diuretics. Blood vessels in the airway wall and lung parenchyma serve as the highways for inflammatory cells. These highways can be more abundant in the airways of chronic asthma patients and they may also express adhesion molecules that allow for easier translocation of eosinophils, lymphocytes, and in certain cases neutrophils. In fact, primary inflammation of the vasculature with vascular leak can present as a severe asthma phenotype. More recently, certain viral syndromes have triggered acute, non-cardiogenic pulmonary edema in asthma patients, highlighting the potential role of vascular leak from parenchymal vasculature in the pathogenesis of CAS.
We begin with a discussion of the most severe vascular abnormalities in CAS, vasculitis, and the Churg–Strauss syndrome, and then highlight lesser forms of vascular injury in CAS including vascular remodeling and vascular leak with pulmonary edema.
Churg–Strauss Syndrome Vasculitis: Small Vessel Vasculitis in CAS
Drs. Jacob Churg and Lotte Strauss of Sweden reported the distinct clinical and pathological findings of 13 asthma patients in 1951 as cases of eosinophilic granulomatosis with polyangiitis (EGPA) or previously Churg–Strauss syndrome (CSS) [1]. They initially evaluated 23 patients that had died from asthma in an attempt to identify patients with a triad of asthma, hyper-eosinophilia, and evidence of vasculitis, and ultimately identified 13 patients with the clinical syndrome and pathologic evidence of necrotizing vasculitis. Eleven of the 13 described (nine females, four males) patients died and had necrotizing vasculitis involving the heart, lungs, skin, nervous system, and kidneys, and interestingly all had evidence of bacterial sinus infection. Slowly progressive asthma or an acute asthma exacerbation immediately preceded their demise, and by our definition this was a study of patients who presented with a CAS. Autopsy revealed eosinophilic and neutrophilic vasculitis in the lungs involving the pulmonary arteries with only one patient having pulmonary veins involvement. The bronchial arteries were not described as having vasculitic changes. In addition to vasculitic changes, most of the patients had patchy eosinophilic inflammation involving the lung parenchyma. Although there was evidence of asthma with hyalanized basement membrane and mucous cell hypertrophy, these changes were relatively mild. This striking observation identified a high degree of vascular inflammation in the setting of mild airway changes that resulted in presentation of acute severe asthma accompanied with systemic organ involvement. The causes of death for the 11 patients included cardiac and renal failure (four patients), perforated ulcer (one patient), cerebral hemorrhage (three patients), and one patient with status asthmaticus.
Since the original description of EGPA, the understanding of the natural history and manifestation of the disease has evolved. Unique biologic imprints in EGPA have been uncovered since 1951. One example is the presence of anti-neutrophil cytoplasmic antibodies (ANCA). ANCAs were discovered in 1991 and now serve as the principle serologic test to identify small vessel vasculitides [2]. The presence of ANCA in patients with EGPA has been estimated to be around 70 % but recent studies have identified that ANCA are increased during disease activity [3–5]. In contrast, others have used the presence of ANCA to identify distinct forms of EGPA; in ANCA-positive patients the disease is more centered on small vessel vasculitis, while ANCA-negative patients have more eosinophilic infiltration of the epineurium and tissue eosinophilic granulomas [6].
Contrary to popular opinion, formal allergy testing of EGPA patients has confirmed atopy in only 30 % [7, 8]. Acute infection triggering the vasculitic response is hypothesized but not yet proven. So how the transition from stable asthma to CAS in EGPA patients remains unknown. The introduction of leukotriene receptor antagonists (LTRA) appeared to be associated with an increased number of CAS cases with vasculitis and was felt to be associated with reduction of inhaled or oral steroids [9]. However a recent report using the FDA Adverse Event Reporting System failed to identify a clear temporal relationship between starting of LTRA with decrease in steroid dosing and subsequent EGPA diagnosis [10].
The biology in the ANCA-positive CAS patient is potentially different than ANCA-negative patients. Clinical outcomes studies have identified that ANCA-positive patients are more likely to have vasculitic presentation and relapse than ANCA-negative patients. However a pathologic role for ANCA in EGPA has not been established. Understanding the relationship between eosinophils and ANCA is important in order to identify the mechanism of disease persistence and progression.
Recent data has confirmed that the long-held belief that EGPA is associated with Th2 cytokine production is true [11, 12]. Cytokines such as IL-4, IL-5, and IL-13 are increased in the circulation and tissues of EGPA patients [13]. Examination of the circulating lymphocytes in patients with EGPA suggests an imbalance of clonal expansion of unique T cells [14]. A simplified scheme of the sequence of events is that a unique allergen–antigen presenting cell interaction triggers the unique oligoclonal expansion of Th2 and Th17 T cells that can go on to propagate (a) granulomatous vasculitis, (b) B cell-mediated production of ANCA, and/or (c) direct tissue damage from eosinophil infiltration [15]. An individual patient can manifest all of these findings or only a portion, resulting in variable phenotypic presentation.
There is very little agreement on the best treatment fort patients with EGPA. In principle, the goal of therapy is to induce remission and then to maintain remission of their inflammatory process. Patient’s risk for aggressive disease can be stratified using the Five-Factor Score (FFS) that assigns a point for any of the following: cardiac involvement, CNS involvement, gastrointestinal involvement, proteinuria >1 g/24 h, and creatinine >140 μM/l [15]. When the FFS score is greater than 1, the patient is considered to be at higher risk for relapse and severity. The choice to only use glucocorticoids versus adding an additional immune suppressant therapy is controversial. One trial reported that patients who have an FFS score of 0 achieve remission 90 % of time but had as many as one third of patients relapse [15, 16]. Most experts would agree that patients in a high-risk category should be treated with combination of glucocorticoids and immune suppressants. Therapy that is more specifically targeted to the unique biology of CAS with vasculitis has recently been described in small series of patients. The production of ANCA is suggestive of an active B cell involvement and the potential therapeutic efficacy of anti-B cell antibody therapy with rituximab was evaluated in a single center cohort of EGPA patients. Nine patients (six ANCA-positive and three ANCA-negative patients) refractory to traditional treatment were treated with rituximab. Clinical follow-up at 3 and 9 months demonstrated no relapse, decline in ANCA and eosinophilia, normalization of C-reactive protein, and tapering of prednisone in all of the patients [17]. The authors suggested that a more robust clinical trial of this approach is warranted and larger randomized controlled trials of these CAS patients testing rituximab, anti IL-5 antibody therapy, and plasma exchange is under way. A comprehensive review of clinical trials utilizing traditional and novel treatment strategies for EGPA has recently been published [15].
Exactly how acute onset of vascular inflammation can transition controlled asthma to EGPA and a CAS remains unclear. The anatomical location of the EGPA vasculitis is in the pulmonary circulation leading to pulmonary manifestations that include alveolar hemorrhage, nodules (often migratory), and pleural effusion. All of these manifestations involve the lung parenchyma and the small arteries and arterioles. EGPA is almost always associated with an elevated peripheral eosinophil count, and release of major basic protein and eosinophil cationic protein, which leads to tissue damage [18, 19]. As previously mentioned, bacterial infection has been proposed as a trigger of the vasculitic phase of inflammation but prospective studies are not available to confirm this association. Furthermore, there are no studies however that have established if the number, surface area, or distribution of the pulmonary and bronchial circulation is different in EGPA patients compared to patients without asthma. Moreover, there are no studies that have established that the endothelium is more activated in EGPA patients or that vascular inflammation leads to airway inflammation.
A recently published retrospective study of the French Vasculitis Study Group represents one on the most comprehensive examination of clinical outcomes associated with ANCA. This study confirmed patterns that had previously been recognized, namely that a majority of patients diagnosed with EGPA had a pre-existing diagnosis of asthma (91 %) and that only a minority of patients were ANCA-positive (31 %) [20]. Multivariate analysis identified cardiac involvement, age, and diagnosis before 1996 as risk factors for death. The clinical features of EGPA stratified by ANCA status is represented in Table 1. Interestingly, ANCA-negative patients are more likely to have the more serious cardiac involvement as compared to ANCA-positive patients while also less likely to have evidence of vasculitis or renal involvement.
Table 1.
Typical clinical features of EGPA stratified by ANCA presence
| Clinical feature | ANCA positive | ANCA negative |
|---|---|---|
| Asthma | ++++ | ++++ |
| Lung involvement | ++ | +++ |
| Cardiac involvement | + | ++ |
| Skin involvement | ++ | ++ |
| Renal involvement | ++ | + |
| CNS involvement | + | + |
Key Points
Eosinophilic granulomatosis with polyangiitis (EGPA) or the Churg–Strauss syndrome should be considered in any patient with critical asthma syndrome.
EGPA patients often have a pre-existing diagnosis of severe persistent asthma and present acutely at onset with a systemic vasculitis flare
ANCA are often elevated during a flare but are not present in all patients with EGPA
Vasculitis typically involves small and medium vessels including arteries, veins, and capillaries.
Vascular Remodeling of the CAS Airway
The term airway remodeling is a collective term that describes the presumed simultaneous changes that are seen in chronic asthma. These pathologic features include [1] mucous cell hyperplasia, [2] basement membrane thickening, [3] airway smooth muscle hypertrophy, [4] inflammatory cell infiltrate, and added a short time ago [5] vascular remodeling. The significance of vascular remodeling has only recently been appreciated [21–24]. The first description by Dunnill documented an increased prominence of the vascular bed in 20 fatal asthma patients [25]. Evans and colleagues were the first to describe how the features of airway remodeling were interdependent, coining the phrase epithelial mesenchymal trophic unit (EMTU) [26]. Conceptually, the EMTU will respond as biologic unit to an outside stimulus, such as an allergen. This is illustrated in Fig. 1, which demonstrates the multicellular response of asthmatic airway.
Fig. 1.
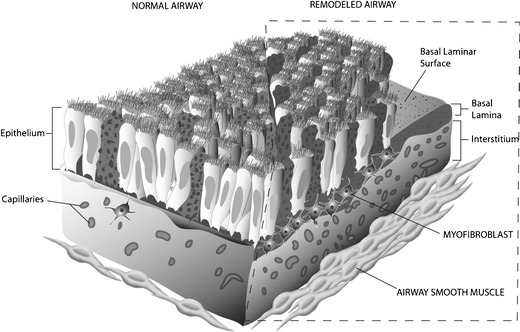
The epithelial mesenchymal trophic unit (EMTU). A representation of the multicellular morphologic changes that represent airway remodeling. The normal airway is shown on the left and the characteristic mucous cell hypertrophy, thickened basement membrane, myofibroblast proliferation, increased vascular density, and airway smooth muscle hypertrophy is represented on the right
The supply of circulating blood in the lung arises from the pulmonary circulation and the bronchial circulation. The pulmonary circulation originates from the pulmonary artery and eventually terminates in a capillary bed that surrounds the alveoli while the bronchial circulation originates off the aorta and divides into a capillary plexus that essentially envelopes the airway smooth muscle layer. An increase in the size, number, density, and rate of vascular leak of these airway capillaries can contribute to the development of CAS. Recent work using a rhesus macaque model of asthma has demonstrated that the number and density of blood vessels is increased in the airway walls of asthmatic subjects and related work has also shown that the airway epithelium produces more vascular endothelial growth factor (VEGF) in response to allergen stimulation [24].
The concept that allergic inflammation can drive angiogenesis and that mediators of angiogenesis can drive asthmatic inflammation is gaining momentum. Several researchers have demonstrated that the airway epithelium can produce growth factors and mediators of vascular inflammation. Our laboratory at UC Davis has shown an increase in VEGF expression at the gene and protein level in vivo and in allergic animal exposed to house dust mite allergen (HDMA). Using an ex vivo air–liquid culture system for airway epithelial cell, we demonstrated that HDMA exposure directly stimulates the increase in gene expression of VEGF [24]. An environment that promotes angiogenesis can drive allergic inflammation. Lee and colleagues used a transgenic mouse model to overexpress VEGF165 in the airway epithelium and found marked angiogenesis, an increase in IL-13, mucous production, leukocyte infiltration of the airway, and smooth muscle hyperplasia through a VEGF dependent mechanism [27]. Over production of VEGF clearly contributed to vascular remodeling.
We hypothesized that Th2 cytokines (IL-4, IL-5, and IL13) could promote expression of VEGF while Th1 cytokines (γ-interferon) can block this expression. Our laboratory has reported that air–liquid interface cultures that are treated with Th2 vs. Th1 cytokines promote VEGF protein and gene expression. CXC chemokines associated with angiogenesis also increase VEGF expression. Others have reported that mast cells and basophils can secrete VEGF and that VEGF secretion by these cells is increased by IgE and prostaglandin stimulation [28, 29]. Eosinophils can also directly promote angiogenesis through secretion of proangiogenic mediators [30]. These studies confirm that resident cells (epithelial, endothelial, and matrix associated cells) interact with cells of the allergic immune response from asthmatic patients and together promote proliferation of blood vessels in the airway wall, and allergic airway inflammation and persistence of the airway-remodeling paradigm. The positive biofeedback of vascular remodeling in the airway is illustrated in Fig. 2.
Fig. 2.

Biofeedback loop of angiogenesis in the asthmatic airway. The biofeedback loop of angiogenesis is shown. Allergen stimulates the resident cells of the EMTU to produce angiogenic growth factors, which promotes vascular growth as well as recruitment, and survival of characteristic inflammatory cells of asthma. The resulting Th2 cytokine inflammation positively feeds back to the EMTU to increase production of angiogenic growth factors
The presence of increased vascular density and increase in vascular remodeling in persistent asthma is well accepted but the specific role of vascular remodeling in severe asthma exacerbations and CAS is less clear. Other than the original work by Dunnill, few studies have quantitatively examined the degree of vascularity found in patients with severe or fatal asthma. One exception is the work by Carroll and colleagues who quantified the size and number of vessels in the submucosa of patients that died from asthma and compared that to stable mild asthmatics and controls. They found that distribution of large caliber vessels was much greater in the airways of fatal asthmatics. They conclude that the presence of these large dilated submucosal vessels may contribute to an a increase in mucosal edema in CAS [31].
In an effort to correlate vascular mediators with airway hyper-responsiveness and airway permeability, Tseliou and colleagues recruited 38 subjects with severe refractory asthma (SRA), 35 patients with stable moderate asthma, and 20 controls, and they quantitated the presence of several different vascular mediators from their induced sputum. The mediators were then analyzed by logistic regression and the researchers found that angiogenic mediators angiopoetin 1 and 2 were increased in SRA patients and these mediators correlated with VEGF levels and measures of airway vascular permeability (AVP) [32]. Papadaki and colleagues took a similar approach and found that sputum cysteinyl leukotrienes in sputum correlated strongly with sputum VEGF in patents with more severe asthma [33]. Hossny and colleagues measured sputum VEGF in children with acute asthma and followed their sputum VEGF to recovery. They found that sputum VEGF correlated very strongly with the clinical picture, being higher during the acute phase and decreasing as symptoms decreased [34]. Abdel-Rahman and colleagues demonstrated a positive correlation between VEGF and acute asthma as well as negative correlation with pulmonary function [35]. A separate study documented that nitric oxide (NO) is a key mediator of VEGF-induced effect in the developing lung [36]. Given that multiple cell types in the lung can produce VEGF, it is not clear that the studies outlined above are reflective of vascular changes or inflammation or markers of epithelial injury. To summarize, CAS may have long-term sequelae including VEGF-mediated structural airway changes that in turn predispose the patient to future critical asthma events.
Lastly, rodent models of asthma almost always have parenchymal inflammation but this pattern occurs infrequently in humans. Acute atypical or viral pneumonia should include the differential diagnosis of severe asthma exacerbations that can progress into one of the CAS. EGPA as previously discussed is a potential albeit rare cause of parenchymal inflammation and should always be considered when an asthmatic patient presents with radiographic evidence of air-space densities or consolidation suggesting lung parenchymal involvement.
Mycoplasma pneumoniae and Chlamydia pneumoniae are known to exacerbate asthma but may rarely result in presentations with lobar pneumonia. Regardless of cause, parenchymal inflammation can accompany asthma exacerbations and presentations can be severe. The combination of airflow limitation and pulmonary edema results in a drastic drop in both vital capacity (FVC) and functional residual capacity (FRC) leaving the patient at greater risk for hypercapnia and hypoxemia, respectively.
Key Points
The airways of chronic asthma and critical asthma patients have a higher density and number of sub-mucosal blood vessels
Allergic inflammation promotes the growth of airway vasculature and vascular growth factors promote allergic inflammation
Increased vascular remodeling likely promotes bronchoconstriction
Pulmonary Edema Complicating CAS
Negative-pressure pulmonary edema may be a significant factor in some CAS presentations. During an acute asthma attack, patients generate very powerful inspiratory efforts. In a landmark study, Stalcup and Mellins established that in the setting of CAS a very large negative pleural pressure is generated that could lead to non-cardiogenic pulmonary edema [37]. These observations led to investigation of the use of diuretics in acute asthma as an adjunct to bronchodilator therapy [38]. The degree to which negative pressure pulmonary edema may occur in asthmatics is not clear, it is fair to say that pulmonary edema is an uncommon presentation of CAS. It is possible that large shifts in negative pleural pressures may lead to mild increases in airway blood flow and contribute to a persistence of airway hyper-reactivity.
In 2009, a strain of influenza (H1N1) caused a pandemic. People at highest risk for serious complications including death tended to be younger individuals particularly those that were pregnant or had asthma or both. Asthma was the most common comorbidity among those afflicted with the infection [39]. Although having asthma seemed to predispose to getting H1N1 infection, it also appeared to be a protective feature in terms of mortality. Features associated with poor outcome among those with asthma were presence of pneumonia and ARDS on admission. Autopsy examination of patient who died due to H1N1 infection demonstrated diffuse alveolar damage, necrosis of the bronchioles, and alveolar hemorrhage [40]. The airway epithelium is the site of primary infection and viral replication for influenza. It is not clear based on the available evidence if the immune reactions that are generating in the airway epithelium are different for asthmatic patients.
Key Points
Common causes of pulmonary edema complicating critical asthma syndrome include viral infections
Asthmatic patients appear to be at higher risk for certain strains of influenza infection, the presentation of which may include pulmonary edema
As in vasculitis, pulmonary edema will decrease the FVC and FRC thereby worsening peak air flow and oxygenation, respectively
Summary
The structure, growth, and proliferation of blood vessels in the lung can be associated with CAS in a variety of ways. The blood vessels can be the primary target of the disease as is seen in EGPA. Patient with EGPA often present clinically as a CAS. In EGPA, the pulmonary circulation is the primary target of disease activity and may be associated with parenchymal lung involvement as well as non-pulmonary involvement. Similarly, certain viral syndromes may affect the lung parenchyma and pulmonary circulation, and present as a CAS. This is perhaps best illustrated by the cohort of patients that were infected by the H1N1 virus resulting in an ARDS-like presentation. As in EGPA patients, patient with severe H1N1 complicating their asthma often presented with severe gas exchange abnormalities, reflecting the large surface area of the lung affected. In contrast, chronic asthma is associated with a distinct remodeling of the conducting airway, including an increase in density of the sub-mucosal vasculature. Patients presenting with a CAS have been demonstrated to have an increased density of airway vasculature as well as an increase in biomarkers of vascular remodeling. What percentage of CAS is secondary to remodeling of airway or parenchymal vasculature is not known. Finally, the role of vascular remodeling in shaping the cluster phenotypes of asthma is also not clear, and presents and excellent opportunity for further research.
References
- 1.Churg J, Strauss L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol. 1951;27:277–301. [PMC free article] [PubMed] [Google Scholar]
- 2.Tervaert JW, Limburg PC, Elema JD, Huitema MG, Horst G, The TH, Kallenberg CG. Detection of autoantibodies against myeloid lysosomal enzymes: a useful adjunct to classification of patients with biopsy-proven necrotizing arteritis. Am J Med. 1991;91:59–66. doi: 10.1016/0002-9343(91)90074-8. [DOI] [PubMed] [Google Scholar]
- 3.Sinico RA, Di Toma L, Maggiore U, Bottero P, Radice A, Tosoni C, Grasselli C, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg–Strauss syndrome. Arthritis Rheum. 2005;52:2926–2935. doi: 10.1002/art.21250. [DOI] [PubMed] [Google Scholar]
- 4.Sable-Fourtassou R, Cohen P, Mahr A, Pagnoux C, Mouthon L, Jayne D, Blockmans D, et al. Antineutrophil cytoplasmic antibodies and the Churg–Strauss syndrome. Ann Intern Med. 2005;143:632–638. doi: 10.7326/0003-4819-143-9-200511010-00006. [DOI] [PubMed] [Google Scholar]
- 5.Keogh KA, Specks U. Churg–Strauss syndrome: clinical presentation, antineutrophil cytoplasmic antibodies, and leukotriene receptor antagonists. Am J Med. 2003;115:284–290. doi: 10.1016/S0002-9343(03)00359-0. [DOI] [PubMed] [Google Scholar]
- 6.Ramentol-Sintas M, Martinez-Valle F, Solans-Laque R. Churg–Strauss syndrome: an evolving paradigm. Autoimmun Rev. 2012;12:235–240. doi: 10.1016/j.autrev.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg–Strauss syndrome. Medicine (Baltimore) 1984;63:65–81. doi: 10.1097/00005792-198403000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Hayakawa H, Sato A, Yagi T, Shimizu T, Miyajima H, Taniguchi M, Akiyama J. Clinical features and prognosis of Churg–Strauss syndrome. Nihon Kyobu Shikkan Gakkai Zasshi. 1993;31:59–64. [PubMed] [Google Scholar]
- 9.Knoell DL, Lucas J, Allen JN. Churg–Strauss syndrome associated with zafirlukast. Chest. 1998;114:332–334. doi: 10.1378/chest.114.1.332. [DOI] [PubMed] [Google Scholar]
- 10.Hauser T, Mahr A, Metzler C, Coste J, Sommerstein R, Gross WL, Guillevin L, et al. The leucotriene receptor antagonist montelukast and the risk of Churg–Strauss syndrome: a case-crossover study. Thorax. 2008;63:677–682. doi: 10.1136/thx.2007.087825. [DOI] [PubMed] [Google Scholar]
- 11.Kiene M, Csernok E, Muller A, Metzler C, Trabandt A, Gross WL. Elevated interleukin-4 and interleukin-13 production by T cell lines from patients with Churg–Strauss syndrome. Arthritis Rheum. 2001;44:469–473. doi: 10.1002/1529-0131(200102)44:2<469::AID-ANR66>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 12.Jakiela B, Sanak M, Szczeklik W, Sokolowska B, Plutecka H, Mastalerz L, Musial J, et al. Both Th2 and Th17 responses are involved in the pathogenesis of Churg–Strauss syndrome. Clin Exp Rheumatol. 2011;29:S23–S34. [PubMed] [Google Scholar]
- 13.Dallos T, Heiland GR, Strehl J, Karonitsch T, Gross WL, Moosig F, Holl-Ulrich C, et al. CCL17/thymus and activation-related chemokine in Churg–Strauss syndrome. Arthritis Rheum. 2010;62:3496–3503. doi: 10.1002/art.27678. [DOI] [PubMed] [Google Scholar]
- 14.Muschen M, Warskulat U, Perniok A, Even J, Moers C, Kismet B, Temizkan N, et al. Involvement of soluble CD95 in Churg–Strauss syndrome. Am J Pathol. 1999;155:915–925. doi: 10.1016/S0002-9440(10)65191-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaglio A, Buzio C, Zwerina J. Eosinophilic granulomatosis with polyangiitis (Churg–Strauss): state of the art. Allergy. 2013;68:261–273. doi: 10.1111/all.12088. [DOI] [PubMed] [Google Scholar]
- 16.Ribi C, Cohen P, Pagnoux C, Mahr A, Arene JP, Lauque D, Puechal X, et al. Treatment of Churg–Strauss syndrome without poor-prognosis factors: a multicenter, prospective, randomized, open-label study of seventy-two patients. Arthritis Rheum. 2008;58:586–594. doi: 10.1002/art.23198. [DOI] [PubMed] [Google Scholar]
- 17.Thiel J, Hassler F, Salzer U, Voll RE, Venhoff N. Rituximab in the treatment of refractory or relapsing eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome) Arthritis Res Ther. 2013;15:R133. doi: 10.1186/ar4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, Hagen EC, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–192. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 19.Zwerina J, Axmann R, Jatzwauk M, Sahinbegovic E, Polzer K, Schett G. Pathogenesis of Churg–Strauss syndrome: recent insights. Autoimmunity. 2009;42:376–379. doi: 10.1080/08916930902832348. [DOI] [PubMed] [Google Scholar]
- 20.Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, Maurier F, et al. Eosinophilic granulomatosis with polyangiitis (Churg–Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum. 2013;65:270–281. doi: 10.1002/art.37721. [DOI] [PubMed] [Google Scholar]
- 21.Hashimoto M, Tanaka H, Abe S. Quantitative analysis of bronchial wall vascularity in the medium and small airways of patients with asthma and COPD. Chest. 2005;127:965–972. doi: 10.1378/chest.127.3.965. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Wilson JW. Increased vascularity of the bronchial mucosa in mild asthma. Am J Respir Crit Care Med. 1997;156:229–233. doi: 10.1164/ajrccm.156.1.9607066. [DOI] [PubMed] [Google Scholar]
- 23.Salvato G. Quantitative and morphological analysis of the vascular bed in bronchial biopsy specimens from asthmatic and non-asthmatic subjects. Thorax. 2001;56:902–906. doi: 10.1136/thorax.56.12.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avdalovic MV, Putney LF, Schelegle ES, Miller L, Usachenko JL, Tyler NK, Plopper CG, et al. Vascular remodeling is airway generation-specific in a primate model of chronic asthma. Am J Respir Crit Care Med. 2006;174:1069–1076. doi: 10.1164/rccm.200506-848OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunnill MS. The pathology of asthma, with special reference to changes in the bronchial mucosa. J Clin Pathol. 1960;13:27–33. doi: 10.1136/jcp.13.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans MJ, Van Winkle LS, Fanucchi MV, Plopper CG. The attenuated fibroblast sheath of the respiratory tract epithelial–mesenchymal trophic unit. Am J Respir Cell Mol Biol. 1999;21:655–657. doi: 10.1165/ajrcmb.21.6.3807. [DOI] [PubMed] [Google Scholar]
- 27.Lee CG, Link H, Baluk P, Homer RJ, Chapoval S, Bhandari V, Kang MJ, et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat Med. 2004;10:1095–1103. doi: 10.1038/nm1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Textor B, Licht AH, Tuckermann JP, Jessberger R, Razin E, Angel P, Schorpp-Kistner M, et al. JunB is required for IgE-mediated degranulation and cytokine release of mast cells. J Immunol. 2007;179:6873–6880. doi: 10.4049/jimmunol.179.10.6873. [DOI] [PubMed] [Google Scholar]
- 29.Abdel-Majid RM, Marshall JS. Prostaglandin E2 induces degranulation-independent production of vascular endothelial growth factor by human mast cells. J Immunol. 2004;172:1227–1236. doi: 10.4049/jimmunol.172.2.1227. [DOI] [PubMed] [Google Scholar]
- 30.Hoshino M, Takahashi M, Aoike N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J Allergy Clin Immunol. 2001;107:295–301. doi: 10.1067/mai.2001.111928. [DOI] [PubMed] [Google Scholar]
- 31.Carroll NG, Cooke C, James AL. Bronchial blood vessel dimensions in asthma. Am J Respir Crit Care Med. 1997;155:689–695. doi: 10.1164/ajrccm.155.2.9032214. [DOI] [PubMed] [Google Scholar]
- 32.Tseliou E, Bakakos P, Kostikas K, Hillas G, Mantzouranis K, Emmanouil P, Simoes D, et al. Increased levels of angiopoietins 1 and 2 in sputum supernatant in severe refractory asthma. Allergy. 2012;67:396–402. doi: 10.1111/j.1398-9995.2011.02768.x. [DOI] [PubMed] [Google Scholar]
- 33.Papadaki G, Bakakos P, Kostikas K, Hillas G, Tsilogianni Z, Koulouris NG, Papiris S, Loukides S. Vascular endothelial growth factor and cysteinyl leukotrienes in sputum supernatant of patients with asthma. Respir Med. 2013;107(9):1339–1345. doi: 10.1016/j.rmed.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 34.Hossny E, El-Awady H, Bakr S, Labib A. Vascular endothelial growth factor overexpression in induced sputum of children with bronchial asthma. Pediatr Allergy Immunol. 2009;20:89–96. doi: 10.1111/j.1399-3038.2008.00730.x. [DOI] [PubMed] [Google Scholar]
- 35.Abdel-Rahman AM, el-Sahrigy SA, Bakr SI. A comparative study of two angiogenic factors: vascular endothelial growth factor and angiogenin in induced sputum from asthmatic children in acute attack. Chest. 2006;129:266–271. doi: 10.1378/chest.129.2.266. [DOI] [PubMed] [Google Scholar]
- 36.Bhandari V, Choo-Wing R, Lee CG, Yusuf K, Nedrelow JH, Ambalavanan N, Malkus H, et al. Developmental regulation of NO-mediated VEGF-induced effects in the lung. Am J Respir Cell Mol Biol. 2008;39:420–430. doi: 10.1165/rcmb.2007-0024OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stalcup SA, Mellins RB. Mechanical forces producing pulmonary edema in acute asthma. N Engl J Med. 1977;297:592–596. doi: 10.1056/NEJM197709152971107. [DOI] [PubMed] [Google Scholar]
- 38.Pendino JC, Nannini LJ, Chapman KR, Slutsky A, Molfino NA. Effect of inhaled furosemide in acute asthma. J Asthma. 1998;35:89–93. doi: 10.3109/02770909809055409. [DOI] [PubMed] [Google Scholar]
- 39.McKenna JJ, Bramley AM, Skarbinski J, Fry AM, Finelli L, Jain S. Asthma in patients hospitalized with pandemic influenza A(H1N1)pdm09 virus infection-United States, 2009. BMC Infect Dis. 2013;13:57. doi: 10.1186/1471-2334-13-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mauad T, Hajjar LA, Callegari GD, da Silva LF, Schout D, Galas FR, Alves VA, et al. Lung pathology in fatal novel human influenza A (H1N1) infection. Am J Respir Crit Care Med. 2010;181:72–79. doi: 10.1164/rccm.200909-1420OC. [DOI] [PubMed] [Google Scholar]