

Abstract
Diabetic nephropathy (DN) is a secondary complication of both type 1 and type 2 diabetes, resulting from uncontrolled high blood sugar. 30–40 % of diabetic patients develop DN associated with a poor life expectancy and end-stage renal disease, causing serious socioeconomic problems. Although an exact pathogenesis of DN is still unknown, several factors such as hyperglycemia, hyperlipidemia, hypertension and proteinuria may contribute to the progression of renal damage in diabetic nephropathy. DN is confirmed by measuring blood urea nitrogen, serum creatinine, creatinine clearance and proteinuria. Clinical studies show that intensive control of hyperglycemia and blood pressure could successfully reduce proteinuria, which is the main sign of glomerular lesions in DN, and improve the renal prognosis in patients with DN. Diabetic rodent models have traditionally been used for doing research on pathogenesis and developing novel therapeutic strategies, but have limitations for translational research. Diabetes in animal models such as rodents are induced either spontaneously or by using chemical, surgical, genetic, or other techniques and depicts many clinical features or related phenotypes of the disease. This review discusses the merits and demerits of the models, which are used for many reasons in the research of diabetes and diabetic complications.
Keywords: Diabetes mellitus, Diabetic nephropathy, Rodent models, Streptozotocin, Genetic models, Non-obese models
Introduction
Diabetes mellitus (DM) is a complex metabolic disorder of the endocrine system resulting from a defect in insulin secretion, insulin action, or a mixture of both (Lachin and Reza 2012). DM is the most common cause of chronic kidney disease (CKD) and end-stage renal disease (ESRD) in the industrialized world. The secondary complications of DM are neuropathy, retinopathy and nephropathy (Bhatti et al. 2013). Diabetic nephropathy (DN), a long-term major microvascular complication of DM type 1 and type 2, affects a large population worldwide (Kong et al. 2013). Glomerular endothelia, mesangial cells, podocytes and tubular epithelia are the cellular elements of the kidney which are targets of hyperglycemic injury (Arora and Singh 2013). DN is characterized by glomerulosclerosis, thickening of the glomerular basement membrane, glomerular and renal cell, mesangial cell expansion, podocyte loss and tubulointerstitial fibrosis, which ultimately result in progressive albuminuria, reduction in glomerular filtration rate, elevation of arterial blood pressure and fluid retention (Mason and Wahab 2003; Arora and Singh 2013). DN is also characterized by an increased urinary albumin excretion (UAE) and is categorized into stages: microalbuminuria (UAE >20 and ≤99 μg/min) and macroalbuminuria (UAE ≥200 μg/min) (Gross et al. 2005). The exact pathogenesis of DN is still not clear (Kong et al. 2013). Furthermore, risk factors for the development of DN include older age, overweight, smoking, non-Caucasian race, male sex and poor glycemic, lipid and blood pressure controls (Ritz and Orth 1999).
DN has since metamorphosed from a clinical rarity to a single major cause of kidney failure in the industrialized world. Clinical evidence has reported that intensive control of glycemia and blood pressure could successfully reduce proteinuria, which is the main sign of glomerular lesions in DN, and improve the renal prognosis in patients with DN (Kume et al. 2014).
However, the impact of these interventions will fail to stem the increased prevalence of renal failure projected over the next decade (Gilbertson et al. 2005). As the total number of people with diabetes is projected to increase substantially by 2050, the prevalence of DN will rise dramatically (Reutens and Atkins 2011).
Diabetes is a complex metabolic disorder; it involves many pathways which ultimately lead to β-cell destruction or insulin resistance to receptors. Till date, scientists have tried to study the exact pathogenesis or down-regulatory pathways involved in disease progression. Therefore, to study the exact pathogenesis of DM there is a need to develop animal models, in which pathological changes are produced as observed in humans. In scientific literature, there are a number of models available for DN. In the present review, we have tried to collect all the models and discussed the mechanism, merits and demerits of each model.
Rodent animal models for DN
Rodents are most frequently used as animal models for understanding the complex pathogenesis of DN (Wolf et al. 2014). They provide opportunities to the researchers to explore the genetic and environmental factors that may influence the development of the disease (Chatzigeorgiou et al. 2009). There are a number of rodent models available which are classified as in Table 1.
Table 1.
Classification of models
| Category of diabetic nephropathy | Models |
|---|---|
| Chemically induced | |
| STZ-induced type-1 DN | |
| STZ-induced type 1 DN in rats | |
| STZ-induced type-1 DN in mice | |
| Low-dose STZ-induced type 2 DN in high-fat diet (HFD)-fed heminephrectomized rats | |
| STZ-induced advanced DN in 5/6 nephrectomized rats | |
| STZ-induced DN in uninephrectomized animals | |
| Alloxan-induced DN | |
| Surgically induced | |
| Renal ischemia-induced advanced DN in rats | |
| Genetically induced | |
| Insulin-2 Akita mice | |
| Non-obese diabetic mouse | |
| LepRdb/LepRdb(db/db) mouse | |
| C57BL6 | |
| ROP mouse | |
| FVB mouse | |
| KK mice | |
| Transgenically induced | |
| Inducible cAMP early repressor transgenic (ICER 1γ Tg) mouse model of DN | |
| Human RAGE gene overexpressed mouse for advanced DN | |
| MafA−/-MafK+ overexpressing hybrid transgenic mouse model of severe DN | |
STZ-induced type-1 diabetic nephropathy in rats
Chemical constitution
Streptozotocin (2-deoxy-2-(3-methyl-3-nitrosoureido)-d-glucopyranose (C18H15N3O7) was discovered from the strain of soil microbes, Streptomyces Achromogens, as a new antibiotic in 1956 and developed as a therapeutic agent for the treatment of metastatic insulin-producing islet cell tumor (Szkudelski 2001). STZ has been determined to be the nitrosoamide methylnitrosourea (MNU) linked to the C2 position of d-glucose (Bennett and Pegg 1981). The nitrosamide MNU contributes to its alkylation properties, and glucose moiety directs its selective pancreatic β-cell uptake via the glucose transporter (GLUT-2) (Elsner et al. 2000). Once STZ enters the β-cells, it is then cleaved between the 2′-carbon and methyl nitrogen to form carbamoylating and alkylating reactive species (Schnedl et al. 1994; Thulesen et al. 1997; Elsner et al. 2000).
Dose
Streptozotocin-induced DM in rats produces all the secondary complications in rodents including DN (Wei et al. 2003). A single dose administration of STZ (40, 50, 55, 60 and 65 mg/kg i.p.) in Sprague–Dawley (SD), Wistar–Kyoto (WKY) or spontaneously hypertensive (SHR) rats has been shown to induce DN after 4–8 weeks, as assessed in terms of serum creatinine (SC), blood urea nitrogen (BUN), proteinuria, creatinine clearance (CC), which is often associated with extracellular matrix deposition, dyslipidemia and development of glomerulosclerosis and tubulointerstitial fibrosis (Casey et al. 2005; Shah and Singh 2006; Gojo et al. 2007; Budhiraja and Singh 2008; Haidara et al. 2009). Following the STZ injection, rats should be given drinking water with glucose to limit early mortality due to hypoglycemia. Generally, 3 days or 1 week after STZ, animal should be screened and those with fasting blood glucose above 240 mg/dL are generally included in the studies of DN (Casey et al. 2005; Gojo et al. 2007). It has been demonstrated that animals showing excessive hyperglycemia are vulnerable to the development of ketonuria and mortality. Thus, in long-term studies of DN, the blood glucose levels are maintained in a desirable range (16–33, 300–600 mg/dL) by a daily subcutaneous injection of long-acting insulin (2–4 U/rat) (Davis and Alonso 2004). Further, it is generally supposed that STZ may induce pancreatic damage selectively due to specific expression of GLUT-2 in the pancreas. However, it has been observed that STZ causes direct DNA damage and cell proliferation in rat kidney, which last for at least for 3 weeks. Therefore, drug treatment should not be started at least 3 weeks after STZ administration to allow kidneys to recover from the acute and mild nephrotoxic effects of STZ (Kraynak et al. 1995).
Mechanism of action
The underlying mechanisms of STZ-induced DM is the following: (a) DNA strand breakage in pancreatic islets and consequent activation of nuclear poly (ADP-ribose) synthetase, which results in the depletion of intracellular nicotinamide adenine dinucleotide (NAD) and adenosine triphosphate (ATP) levels; (b) excessive generation of reactive oxygen species (ROS), such as superoxide (O2−), hydrogen peroxide and hydroxyl radicals (Takasu et al. 1991); (c) STZ being a nitric oxide (NO) donor partially mediates restriction of mitochondrial ATP generation. Further, NO also binds and inhibits the activation of iron-containing aconitase (Welsh et al. 1994); (d) augmented ATP dephosphorylation increases the supply of substrate for xanthine oxidase and enhances the production of uric acid, the final product of ATP degradation (Nukatsuka et al. 1988; Nukatsuka et al. 1990a; Nukatsuka et al. 1990b). The overactivation of protein kinase C (PKC) and increased renal RAAS have been well implicated in the pathogenesis of STZ-induced DN (Brownlee et al. 1988; Chang et al. 2003; Chung and Chung 2003; Asaba et al. 2005; Navaneethan et al. 2005; Tojo et al. 2005) (Fig. 1).
Fig. 1.
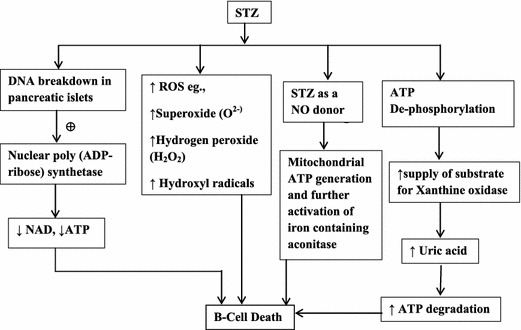
Mechanism of action of STZ
It has been revealed that altered dynamic changes in gene expression of CD-1 (ICR) mouse kidney in early phases of DN are related to glucose and lipid metabolism, protein synthesis and degradation, signal transduction, ion transport and extracellular matrix and ultimately leads to glomerulosclerosis, mesangial cell expansion and glomerular hypertrophy (Wada and Yagihashi 2010). Further, reports proposed that mice that received high-dose STZ developed more albuminuria than mice that received a low-dose STZ regimen, although they showed similar levels of hyperglycemia (Hackbarth et al. 1981; Hackbarth and Hackbarth 1981).
To exclude the nonspecific cytotoxicity of high-dose STZ, multiple injections of sub-diabetogenic doses of STZ (40–50 mg/kg) daily i.p. were given for five consecutive days in CD-1 mice. The mice had mild initial hyperglycemia during the initial 5–6 days with a subsequent complete diabetic syndrome observed on the 8th and 11th day, which was accompanied by pronounced insulinitis with infiltrating lymphocytes and macrophages and pancreatic β-cell necrosis (Rossini et al. 1977; Leiter 1982, 1985). The lower levels of albuminuria with low-dose STZ than with high-dose STZ showed reduced direct renal toxicity (Hackbarth et al. 1981; Hackbarth and Hackbarth 1981; Susztak et al. 2004).
Low-dose STZ-induced type 2 diabetic nephropathy in high-fat diet (HFD)-fed heminephrectomized rats
Unlike high-dose STZ, which induces type 1 DM (T1DM) by causing severe pancreatic damage, an administration of low-dose STZ has been shown to produce partial pancreatic damage and mild glucose intolerance which are important characteristics of the late stage of type 2 DM (Kelly et al. 2003). Further, administration of STZ (40 mg/kg, i.v.) to heminephrectomized rats followed by feeding with a high-fat diet presented a significant increase in plasma glucose levels after 15 weeks and decreased plasma insulin levels, increased plasma total cholesterol and triglyceride levels and increased blood pressure at 25 weeks (Kelly et al. 2003). These animals displayed microalbuminuria and increased creatinine clearance at the age of 15 weeks, subsequently followed by overt proteinuria and mesangial expansion after the age of 25 weeks (Sugano et al. 2006). In this model, either by manipulating dietary formula or by pharmacological intervention, it would be possible to normalize blood pressure, hyperlipidemia and hyperglycemia to analyze how different treatment modalities retard the progression of DN.
STZ-induced advanced diabetic nephropathy in 5/6 nephrectomized rats
It has been investigated that a single injection of STZ (35 mg/Kg, i.p.) in five/six nephrectomized rats results in the development of hyperglycemia, hypoinsulinemia, hypertriglyceridemia and hypercholesterolemia along with increased serum glycosylated proteins, a metabolic abnormality resembling DN in humans (Yokozawa et al. 2001). Increased levels of serum creatinine, urinary protein and decreased creatinine clearance were also observed, leading to the development of advanced DN and overt albuminuria within 80 days, as this animal model was associated with severe lesions of the glomerular capillary loops, mesangial area and Bowman’s capsule, coagulation in the glomerular capillary loops and azotemia (Yokozawa et al. 2001). The major limitation of this animal model is that the development of glomerulopathy was not purely due to hyperglycemia. The development of elevated blood pressure and hyperlipidemia may independently influence the progression of DN. Further, the bilateral surgical manipulation in STZ-treated nephrectomies rats may produce artifacts in histological analyses (Yokozawa et al. 2001).
STZ-induced diabetic nephropathy in uninephrectomized animals
Uninephrectomy was reported to accelerate the progression of renal injury observed in STZ-induced DN in different rat strains including SD rats, Wistar and SHR, which may be a consequence of increased glomerular capillary pressure (Kang et al. 2000; Utimura et al. 2003). Utimura et al. (2003) showed that if Wistar rats were first uninephrectomized and 3 weeks after surgery made diabetic by a single injection of STZ (65 mg/kg i.v.), the blood glucose was then maintained between 16 and 22 mmol/L (300–400 mg/dL) for the next 8 months with partial insulin treatment. These uninephrectomized diabetic rats achieved a urine albumin excretion rate (UAER) of 60 mg/24 h at 8 months, which was nearly three times higher than non-diabetic control rats (Utimura et al. 2003). The uninephrectomy (UNX) per se produced elevated levels of serum creatinine and urinary total protein, along with uremia in rats (Zhao et al. 2008). In addition, UNX rats shows increased serum total cholesterol, triglycerides and low-density lipoprotein (LDL) cholesterol 10 months after uninephrectomy. Further, fat redistribution and increased renal accumulation of fats and lipid peroxides were observed in UNX rats. Pharmacological treatment with ACE inhibitors such as lisinopril and enalapril prevents the development of chronic renal dysfunction by reducing hypercholesterolemia, fat redistribution or transformation and lipid peroxidation in UNX rats (Zhao et al. 2008).
Merits and demerits
STZ is mostly used to induce experimental DM, since it has some advantages over alloxan such as prolonged hyperglycemia, longer half-life (15 min) and the development of well-characterized diabetic complications such as DN with fewer incidences of ketosis as well as mortality. Both alloxan and STZ diabetic animals are commonly used for screening the compounds including natural products for their insulinomimetic, insulinotropic and other hypoglycemic/antihyperglycemic activities (Young et al. 1990; Katovitch et al. 1991). But nowadays, alloxan is almost replaced by STZ for induction of diabetes in laboratory animals (Ozturk et al. 1996).
Alloxan-induced diabetic nephropathy
Chemical constitution
Alloxan (2,4,5,6-tetraoxypyrimidine;2,4,5,6-pyrimidinetetrone) is an oxygenated pyrimidine derivative and also a barbituric acid derivative that can induce diabetes in animals due to the specific necrosis of pancreatic β-cells (Peschke et al. 2000; Szkudelski 2001). Brugnatelli originally isolated alloxan in 1818 (McLetchie 1982). Alloxan was firstly produced by the oxidation of uric acid by nitric acid (Bailley et al. 1946). Alloxan is considered a strong oxidizing agent in the form of dialuric acid. Dialuric acid, a reduction product of alloxan, has also been shown to be diabetogenic in animals (Bailley et al. 1946; Brückmann and Wertheimer 1947) and cause ultrastructural changes similar to those observed in response to alloxan (Szkudelski 2001). This drug can be administered parenterally, i.e., intravenously, intraperitoneally or subcutaneously to exert its diabetogenic action. The induction of insulinopenia due to alloxan causes a state of experimental DM called ‘alloxan diabetes’ (Dunn and Letchie 1943; Goldner 1945; McLetchie 1982).
Dose
The dose of alloxan needed for developing diabetes depends on the animal species, route of administration and nutritional status (Federiuk et al. 2004). Single-dose administration of alloxan 120 mg/kg body weight i.p. results in the development of diabetes in rats after 72 h. Alloxan induced diabetes characterized by a significant increase in plasma glucose, enhanced levels of renal damage markers, plasma creatinine, urea nitrogen and urinary albumin. Diabetic renal injury is associated with increased kidney weight to body weight ratio and glomerular hypertrophy (Da and Sil 2012).
Mechanism of action
Alloxan shows two distinct pathological effects: selective inhibition of glucose-induced insulin secretion through specific inhibition of glucokinase, the glucose sensor of the beta cell, and it causes a state of insulin-dependent diabetes through its ability to induce ROS formation, resulting in the selective necrosis of β-cells. These two effects are due to specific chemical properties of alloxan-like selective cellular uptake and accumulation of alloxan in pancreas. Alloxan is an unstable chemical compound having molecular shape similar to glucose (Weaver et al. 1979; Lenzen and Munday 1991; Gorus et al. 1982). Both alloxan and glucose are hydrophilic and do not penetrate the lipid bilayer of the plasma membrane. Due to structural similarity of alloxan to glucose molecule, the GLUT2 glucose transporter in the beta cell plasma membrane accepts this glucomimetic and transports it into the cytosol (Weaver et al. 1978; Gorus et al. 1982). Alloxan does not inhibit the function of GLUT2 transporter (Elsner et al. 2002), and therefore selectively enters beta cells in an unrestricted manner (Boquist et al. 1983; Malaisse et al. 2001). It is not toxic to insulin-producing cells that do not express this GLUT2 transporter (Elsner et al. 2002; Bloch et al. 2003).
Alloxan generates reactive oxygen species (ROS) in a cyclic reaction with its reduction product, dialuric acid (Winterbourn and Munday 1989). In beta cells, the toxic effect of alloxan is initiated by free radicals formed during cyclic redox reaction. Autoxidation of dialuric acid generates free radicals such as superoxide radicals and hydrogen peroxide, hydroxyl radicals as well as intermediate of the alloxan radical (Munday 1988; Winterbourn et al. 1989; Winterbourn and Munday 1989). Alloxan generates ROS by reacting with thiol groups on proteins such as enzymes and albumin (Sakurai and Miura 1989; Lenzen and Mirzaie-Petri 1992) (Fig. 2).
Fig. 2.
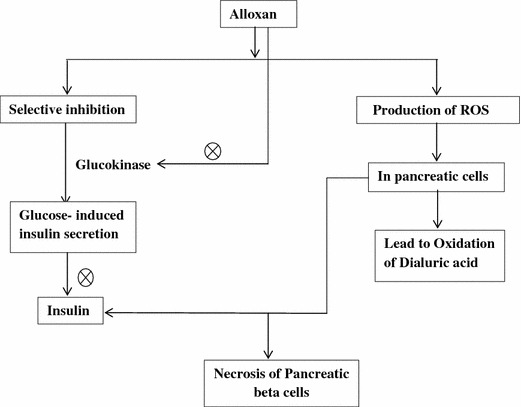
Mechanism of action of alloxan
Merits and demerits
Alloxan has low stability and very short half-life (<1 min). Because of the acidic nature of the solution, intravenous route of its administration is preferred. The hypoglycemic phase is severe, due to this alloxan should not be given to fasted animals. Alloxan-treated animals show severe polydipsia, hyperglycemia, glucosuria, hyperlipidemia, polyphagia and also develop various complications of uncontrolled diabetes such as DN, neuropathy, cardiomyopathy, well-marked retinopathy and others. Alloxan is disadvantageous as the percentage incidence of diabetes is quite variable. Further, the incidence of ketosis and mortality is very high. Alloxan-treated animals show reversal of hyperglycemia in which pancreatic regeneration is early and common. Because of these limitations, Alloxan is less commonly used as compared with the STZ (Bailey and Day 1989; Pelé-Tounian et al. 1998; Young et al. 1990; Katovitch et al. 1991).
Renal ischemia-induced advanced diabetic nephropathy in rats
It has been reported that unilateral renal ischemia of 30 min in T1DM rats causes a severe progressive renal injury leading to ESRD, whereas in non-DM rats ischemia of the same duration causes a reversible injury (Melin et al. 1997). Ischemia–reperfusion (I/R) with hyperglycemia leads to the development of DN (Ziyadeh 2004). It is also reported that in diabetic rats, renal ischemia produces progressive kidney damage which resembles DN in humans. Animals exhibit early signs of diabetic renal injury, such as mesangial cell expansion and basement membrane thickening (Hirose et al. 1982; Evan et al. 1984; Evan and Tanner 1986; Yong and Bleasel 1986; Steffes et al. 1989).
Renal fibrosis and tubulointerstitial inflammation, the prominent pathological features of human DN, have been proposed as a consequence of renal ischemia (Ziyadeh 2004). Recently, it has been demonstrated that apoptosis in tubular cells, especially in outer medulla, is induced by renal I/R injury (Daemen et al. 1999; Chien et al. 2001). Insulin-like growth factor 1 (IGF-1) shows some structural homologies with insulin and is known as an anti-apoptotic agent that decreases the injury after renal I/R (Goes et al. 1996; Daemen et al. 1999). Insulin has anti-apoptotic effects on epithelial cells from mammary glands (Farrelly et al. 1999). Treatment with insulin for 1 week before renal I/R decreases the structural and functional damage to the kidney caused by 30 min of arterial clamping in the DM rat associated with renal lactate accumulation (Podrazik et al. 1989). There is elevation of reactive oxygen species (ROS) in rat kidneys subjected to I/R, and treatment with superoxide dismutase decreases apoptosis in proximal tubular cells (Chien et al. 2001). Hyperglycemia also induces oxidative stress and increases lipid peroxidation in cultured tubular cells (Kuramochi and Homma 1993; Han et al. 2000). In DM rats, treatment with anti-oxidants such as vitamin C and E decreases renal expression of TGF-β, and vitamin C inhibits renal hypertrophy (Craven et al. 1997). Like other chronic renal diseases, DN is associated with an infiltration of inflammatory cells. Bohle and co-workers found T lymphocytes, fibroblasts, monocytes/macrophages and a few B-cells in kidneys with DN (Bohle et al. 1991). Mostly, renal I/R injury in non-DM rats describes a transient inflammation (Forbes et al. 2000). Combination of I/R with DM resulted in a severe inflammatory response that persisted for at least 8 weeks (Melin et al. 1997). However, good glycemic control before and during I/R prevented deterioration of renal function despite uncontrolled hyperglycemia during the first 4 weeks (Melin et al. 1997).
Diabetic kidney has also been associated with an increased ratio of free NADH/NAD+, similar to that found in hypoxia. This is called ‘‘pseudohypoxic state’’ (Williamson et al. 1993). It has been shown that TNF-α expression in kidney mediates neutrophil infiltration and injury after renal I/R (Donnahoo et al. 1999). Also, it is well known that renal I/R induces inflammatory response, which is exacerbated in the diabetic kidney (Eddy and Giachelli 1995: Panés et al. 1996) and concern with the degree of arterial obliteration (Ziyadeh 2004). Further, an impaired response to nitric oxide might be responsible for the more pronounced decrease in renal function in the early post-ischemic phase in DM rats. It has been found that diabetic rats subjected to 1 h of bilateral renal artery clamping died within 48 h, while most non-diabetic animals survived after the ischemic injury (Wald et al. 1990). In addition, renal ischemia of 30 min duration only produces transient reversible renal injury, but the same extent of renal ischemia causes a rapidly progressive DN and end-stage renal failure, along with severe tubulointerstitial inflammation and renal fibrosis within 8 weeks in the diabetic rat (Melin et al. 1997; Mu et al. 1999; Melin et al. 2002; Mills et al. 2004).
Merits and demerits
This model is widely used to induce DN in diabetic rats. It is reported that unilateral renal ischemia of 30 min produces progressive kidney damage which resembles DN in humans (Evan and Tanner 1986; Steffes et al. 1989). But still this model is not fully established, because the pathophysiology of I/R injury is multifactorial and the exact sequence of events in I/R injury remains unknown. However, the role of inflammation has clear and several other important events resulting in tissue damage and kidney failure (De Vries et al. 2012).
Genetic model of diabetic nephropathy
Chemical or surgical maneuvers for diabetes induction might, however, cause some diversity among individual animals in terms of the extent of severity and the onset of diabetes. Accordingly, by use of a genetic approach, a diabetic state and particular pathogenic molecular culprit would be most stably induced. Thus, genetic models serve as an essential experimental tool for investigating the molecular mechanisms and genetic susceptibility in the development of DN. There are numerous genetic models that mimic human diseases.
Insulin-2 Akita mice
Insulin-2 Akita (Ins2Akita) is a mouse mutant model of type 1 diabetes. It has been reported that this model is insulin responsive and represents a model of maturity-onset diabetes of the young with insulin resistance. These mice are commercially available through the Jackson Laboratories (Mathews 2002). The Ins2 gene is the mouse homolog of human preproinsulin gene that causes misfolding of the insulin protein. Mice possess another active insulin gene, Ins1, which lacks an intron present in the C-polypeptide-encoding region. The Akita (Ins2Akita) spontaneous mutation (commonly referred as Mody) is an autosomal dominant mutation in the insulin II gene (Ins2). Mice heterozygous for the Akita spontaneous mutation (Ins2Akita) are viable and fertile, whereas mice homozygous for the Ins2Akita allele die within 1–2 months (Ron 2002; Srinivasan and Ramarao 2007). Ins2Akita mutation disrupts normal insulin processing and causes a failure in secretion of mature insulin, which results in early development of hyperglycemia, characterized by hyperglycemia, hypoinsulinemia, polydipsia and polyuria, beginning at approximately 3–4 weeks of age. Obesity or insulitis does not accompany diabetes. Histologically, there is a reduction in active pancreatic β-cell density without insulitis and islets release very little mature insulin at 4–35 weeks of age. These mutant mice respond well to exogenously administered insulin. Thus, Insulin-2 Akita (Ins2Akita) mouse shows development of pancreatic β-cell damage, with little release of mature insulin from viable β-cells as a result of β-cell-selective proteotoxicity resulting from misfolding of insulin 2 (Ron 2002).
It has been demonstrated that there is a twofold increase in the urinary albumin excretion rate in Ace2−/y Ins2WT/C96Y mice as compared to Ace2+/y Ins2WT/C96Y mice [both produced when Ace2−/− mice crossed with Akita mice (Ins2WT/C96Y)] due to similar blood glucose level (Haseyama et al. 2002). Ace2−/y Ins2WT/C96Y mice reveal increased mesangial matrix scores and glomerular basement membrane thickness as compared to Ace2+/y Ins2WT/C96Y mice. Kidney levels of angiotensin II were not increased in the diabetic mice and treatment with an Ang II receptor blocker decreased urinary albumin excretion rate in Ace2−/y Ins2WT/C96Y mice, suggesting that the acceleration of kidney injury in mice is Ang II mediated (Wang and Li 2008). Renal immunopathologic studies demonstrated significant deposition of IgA in the glomeruli of Akita mice (Haseyama et al. 2002). The severity of DN in the C57BL/6J Ins2Akita (hybrid strain of C57BL/6J and Ins2Akita) mouse does not seem to be robust.
Meanwhile, the Ace-2/Ang-(1-7)/Mas axis functions as a negative regulator of the RAS. It was demonstrated that Ace2 knockout (ACE2-/y) mice show impaired glucose tolerance. Now, it has been proven that the Ace2/Ang-(1-7)/Mas axis can ameliorate insulin resistance in the liver. Activation of the Ace2/Ang-(1-7)/Mas axis increases glucose uptake and decreases glycogen synthesis in the liver by increased expression of glucose transporters and insulin receptor substrates and decreased expression of enzymes for glycogen synthesis. Ace2 knockout mice presented elevated levels of oxidative stress; exposure to Ang-(1-7) reduced the stress in hepatic cells, and activation of the Ace2/Ang-(1-7)/Mas axis led to improved hepatic insulin resistance through the Akt/PI3 K/IRS-1/JNK insulin signaling pathway. It has been documented for the first time that the Ace2/Ang-(1-7)/Mas axis can ameliorate insulin resistance in the liver, which is considered to be the primary cause of the development of T2DM (Cao et al. 2014).
Merits and demerits
This model is insulin responsive, causing severe DN. It also shows histological changes at 4–35 weeks (Mathews 2002). Ins2Akita mice could be used as a substitute for mice that are rendered insulin-dependent diabetic by treatment with alloxan or STZ (Ron 2002). This model is also considered as a new diabetes target (Cao et al. 2014) and has rarely any disadvantages, but even then it is not used commonly.
Non-obese diabetic mouse
The non-obese diabetic (NOD) mouse was obtained by selectively breeding offspring from the cataract-prone ICL-ICR mouse. Insulinitis is present at the age of 4–5 weeks followed by partial β-cell destruction and decreased insulin level in circulation in these mice. Frank diabetes typically appears at the age between the 12th and 30th week. These animals can survive for weeks without administration of insulin, since keto acidosis is mild in these for any extended time after the onset of hyperglycemia, indicating more complete insulin deficiency.
In addition, the inheritance of specific MHC class II alleles and many non-MHC loci as polygenic susceptibility loci, transmission of the disease by hematopoietic stem cells, the development of an intra-islet inflammatory infiltrate (insulinitis) with anti-islet cell antibodies and a strict dependence of disease on T cells showed that autoimmune disease would lead to β-cell failure in NOD mice. The model presents a number of similarities with the features of human type 1 diabetes (Leiter et al. 1987; Atkinson and Leiter 1999; Cameron et al. 2008). Furthermore, studies of DN using this model also have supported roles for TGF-β and advanced glycosylation end products (AGE) in the pathogenesis of mesangial proliferation and sclerosis (Sakurai and Miura 1989; Ritz and Orth 1999).
LepRdb/LepRdb(db/db)mouse
The db/db mutation on the C57BLKS background has been examined intensively and shows features similar to human DN. The diabetic gene (db) is transmitted as an autosomal recessive trait, and it has been reported that long intracellular domain form of OB-R is crucial for initiating intracellular signal transduction. As a corollary, the inability to produce this form of OB-R, which leads to the severely obese phenotype found in db/db mice, and weight-reducing effects of leptin being mediated by signal transduction through a leptin receptor in the hypothalamus showed that the db gene encodes for a G-to-T point mutation of the leptin receptor, leading to abnormal splicing and defective signaling of the adipocyte-derived hormone leptin (Stephens and Caro 1998). This obese and diabetic mutant (db) was recognized in the C57BLKS/J strain and was subsequently also backcrossed to a pure C57BL/6J background. The C57BLKS/J mouse shares 84 % of its alleles with the common C57BL/6 strain and 16 % with DBA/2 J strain (Naggert et al. 1995). The C57BLKS/J db/db mouse exhibits hyperinsulinemia by 10 days of age, and blood glucose levels are elevated at (7.2 ± 2.3 mM) 1 month of age (Lee and Bressler 1981), while the db/db mouse develops frank hyperglycemia with glucose values of 9.7 ± 1.6 and 15.7 ± 4.3 mM by 8 week and at 10 week of age. Progressive hyperglycemia (28.6 ± 13.2 mM) is noted at 16 weeks of age (Like et al. 1972; Lee and Bressler 1981).
Besides this, increased level of urinary collagen IV excretion, glomerular mesangial cell expansion and increased albumin secretion also occur in diabetic db/db mice that showed similarities to the changes found in human DN (Gartner 1978; Cohen et al. 2001). After 16 weeks of age, there is a very consistent threefold increase in mesangial matrix expansion based on several independent studies (Sharma et al. 2003). However, the degree of albuminuria does not consistently increase with the duration of diabetes, as there are similar levels of albuminuria between 8 and 25 weeks (Koya et al. 2000; Cohen et al. 2001; Sharma et al. 2003). The severity of diabetes in db/db mice depends on the C57BL/6 background in the diabetic phenotype and is less severe than that in C57BLKS/J; as these mice age, plasma glucose seems to normalize (Koenig et al. 1976; Leiter et al. 1981; Meade et al. 1981; Leiter et al. 1987, 1989).
More recently, some investigators have investigated that a subset of approximately 50 % of C57BL/6J db/db mice developed persistent hyperglycemia (Chow et al. 2004; Zheng et al. 2004). In these mice, more robust albuminuria, glomerular lesion and mesangial cell expansion have been described (Chow et al. 2004).
Susceptibility of inbred mice strain to diabetic nephropathy
Studies have identified certain strains (e.g., 129, ROP, NON, KK/HIJ) as more prone to glomerulosclerosis than other commonly studied strains (e.g., C57BL/6, FVB/NJ) that seem relatively resistant to renal disease. It is noteworthy that studies of DN have been performed in fewer than 5 % of the available mouse strains.
C57bl/6
C57BL/6 is a widely used inbred strain; it has been reported that urine protein excretion in C57BL/6 mice increased at 4 weeks after five/six nephrectomies and was back to normal at 8 and 12 weeks in remnant kidney model, which proposed that kidney disease in this strain is resistant to renal injury (Zheng et al. 1998; Ma and Fogo 2003).
It has been revealed that infusion of DOCA salt and angiotensin II leads to hypertensive glomerular damage, increased expression of profibrotic and inflammatory genes, albuminuria, tubular casts, increased plasma cholesterol, cardiac hypertrophy and fibrosis in C57BL/6 mice (Kirchhoff et al. 2007). Further, studies indicate that on the C57BLKS background, type 2 diabetic db/db mice have lesions consistent with DN (thickened basement membrane, mesangial expansion), whereas they are resistant to DN on a C57Bl/6 background. Moreover, these models have not been completely designated; this difference proposed two possibilities: either the worse severity of hyperglycemia that develops in C57BLKS causes this (111) (a combination of peripheral insulin resistance and insulin deficiency develops in C57BLKSLepr db, whereas C57BL/6Lepr db have peripheral insulin resistance) or C57BLKS expresses modifier genes that predispose to DN (Noonan and Banks 2000; Tikellis et al. 2008).
ROP mouse
The ROP [Ra/+ (ragged), Os/+ (oligosyndactyly) and Pt/+ (pintail)] strain of mouse obtained from a heterogeneous stock were heterozygous for three mutant alleles, viz. Ra/+ (ragged), Os/+ (osteosyndactylism) and Pt/+ (pintail). Estimation of reduced renal mass in kidney of Os/+ ROP mice showed approximately 50 % fewer nephrons in the Os/+ mice than in the +/+mice (Zalups 1993). Further, decreased renal mass was responsible for more glomerulosclerosis in wild-type mice (Os+/+, Ra+/+, and Pt+/+) of ROP background than C57BL/6 mice (Esposito et al. 1999; Cornacchia et al. 2001; Fornoni et al. 2003). Mice on the ROP background, in the absence of Os, Ra, or Pt mutant alleles, may show increased propensity for glomerulosclerosis and hyperglycemia after combined nephron mass reduction and diabetes (Zheng et al. 1998).
FVB mouse
FVB/NJ was obtained from outbred Swiss mice at NIH inbred for the fv1b allele and shows sensitivity to the Friend leukemia virus B strain. FVB/NJ is commonly used for transgenic injection because of the prominent pronuclei in the fertilized eggs and the large litter size. FVB/N mice are transgenic for a calmodulin mini gene regulated by the rat insulin II promoter, which has shown fivefold increase in calmodulin and its mRNA in beta cells and insulin secretory defect in mice leading to the development of T1DM (Epstein et al. 1989, 1992). They develop glomerular capillary basement membrane thickening, not albuminuria (Carlson et al. 1997). The LepRdb mutation has been backcrossed onto the FVB/NJ strain, and the kidneys from these mice show mesangial sclerosis, but not albuminuria, and GFR has been described (Chua et al. 2002).
KK mice
These mice were originally established from inbreeding a Japanese mouse (Kondo et al. 1957). KK mice exhibit only mild insulin resistance and seem to be predisposed to the development of renal lesions, very reminiscent of DN (Reddi et al. 1977; Reddi and Camerini-Davalos 1989). The severity of hyperglycemia, insulin resistance and impaired glucose tolerance is exacerbated further by initiation of the agouti (Ay) allele into the KK (Suto et al. 1998). Pharmacological treatment with candesartan has been shown to reduce urinary type IV collagen and albumin excretion and attenuate glomerular hypertrophy and mesangial matrix accumulation possibly by suppression of TGF-betaS/Smad signaling pathway in KK/Ta mice with DN (Liao et al. 2003). Further, glipizide treatment has been demonstrated to improve glucose intolerance, decrease kidney size, reduce kidney glycoprotein and mesangial matrix accumulation, and proteinuria in type II diabetic KK mice (Reddi et al. 1990).
Merits and demerits
The NOD mouse model of T1DM has contributed greatly to our understanding of disease pathogenesis and has facilitated the development and testing of therapeutic strategies to battle the diseases in humans. In LepRdb/LepRdb(db/db) mouse, the db/db mutation on the C57BLKS background has been examined intensively and shows features similar to human DN (Stephens and Caro 1998). Approximately, 50 % of C57BL/6J db/db mice developed more persistent hyperglycemia (Chow et al. 2004; Zheng et al. 2004). C57BL/6, ROP, FVB and KK are commonly used strains of mouse to induce complications of DM such as DN, retinopathy, neuropathy and cardiomyopathy. Moreover, these models have not been completely designated; due to this, these models are less commonly used as compared to the chemical models.
Inducible cAMP early repressor transgenic (ICER 1γ Tg) mouse model of diabetic nephropathy
ICER 1γ Tg mouse has been reported to exhibit persistent hyperglycemia due to low production of insulin and insulin-producing β-cells (Inada et al. 1999). ICER Iγ is a transcriptional repressor transcribed from an alternative intronic promoter of the CRAM gene and competes with transcriptional activators activated by insulin for binding to the DNA and cyclineA gene transcription (Foulkes et al. 1991; Inada et al. 1998, 2008). This results in suppression of insulin synthesis, β-cell proliferation and development of severe diabetes as early as 2 weeks of age, but with an excellent survival rate without insulin therapy (Inada et al. 1999; Kajihara et al. 2003).
Human RAGE gene overexpressed mouse for advanced diabetic nephropathy
When transgenic mice with overexpressed human RAGE in vascular cells (RAGETg) are cross bred with another transgenic mouse deficient in the islet production of insulin, the resultant double transgenic animals develop exhibiting renal insufficiency and advanced glomerulosclerosis that resemble human DN, implicating the functional importance of AGE-RAGE system in the development of DN (Takamura et al. 1998). The RAGE-overexpressing IDDM mice are the first animal model in which the process of diabetes-induced kidney changes lead to ESRD. The administration of AGE formation inhibitors such as (±)-2-isopropylidenehydrazono-4-oxo-thiazolidin- 5-ylacetanilide (OPB-9195) has been shown to produce beneficial effects in this model (Takamura et al. 1997).
MafA−/−MafK+ overexpressing hybrid transgenic mouse model of severe diabetic nephropathy
MafA is a transcription factor belonging to Maf family and contains a C-terminal basic leucine zipper domain that binds to specific DNA sequences that are named Maf recognition elements (MAREs). They are divided into two subgroups, i.e., large and small Maf proteins. Small Maf protein family includes MafK, MafF and MafG, which do not possess the transactivation domain (Kataoka et al. 2002), whereas the large Maf proteins possess an N-terminal activation domain and stimulate the transcription of target genes. MafA plays an important role in insulin gene transcription and MafA-deficient (MafA−/−) mice develop diabetes mellitus due to decrease in insulin gene transcription, impaired glucose-stimulated insulin secretion and mild abnormalities of the pancreatic islets (Zhang et al. 2005). This mild diabetic phenotype, due to the presence of other large Maf proteins, MafB and/or c-Maf can stimulate the expression of insulin genes and other MafA target genes (Kataoka et al. 1995; Inada et al. 1998; Olbrot et al. 2002; Kajihara et al. 2003; Matsuoka et al. 2003; Kataoka et al. 2004). Therefore, MafA−/− transgenic mice that overexpressed MafK, specifically MafK+ mice in pancreatic beta cells, had impairment of glucose-stimulated insulin secretion. MafK is another small Maf protein that acts as a dominant negative protein to suppress the effect of large Maf proteins in pancreatic beta cells (Kataoka et al. 1995, 2004). MafK+ mice has been shown to exhibit reversible hyperglycemia, which appears at younger age and spontaneously disappears at older age without development of overt diabetes, due to compensatory upregulation of endogenous Maf A. Further, transgenic mice with MafA−/− knockout and overexpression of MafK+ in pancreatic beta cells overexpressed MafK in pancreatic β-cells. (MafA−/-MafK+) mice in order to depress MafA protein and transcriptional activity of other large Maf proteins in mice have been evaluated. The resultant transgenic (MafA−/-MafK+) mice developed overt severe diabetes mellitus at the age of 5 weeks (Shimohata et al. 2006, 2009) (Fig. 3; Table 2).
Fig. 3.

Mechanism of action of human RAGE and MafA−/-MafK+
Table 2.
Some animal models of types 1 and 2 diabetes mellitus studied for experimental DN
| Type of model | Type of DM | Advantages | Limitations |
|---|---|---|---|
| STZ-induced DN | Type 1 | Well established, reproducible, easily done in the laboratory | Insulin resistance does not occur |
| High-fat diet, low-dose STZ heminephrectomized rats | Type 2 | Occurrence of overt DB, hypertension, hyperlipidemia | Developed mesangial cell expansion after 25 weeks, thus time consuming |
| STZ-induced advanced DN in 5/6 nephrectomized rats | Type1 | Occurrence of azotemia, hyperglycemia and glomerular lesions, thus advanced DN | Development of glomerulopathy was not due to hyperglycemia |
| STZ-induced DN in uninephrectomized rats | Type 1 | Well established | Not characterized |
| Renal ischemia-induced DN in STZ-treated rats | Type 1 | Advanced DN occurrence in 6–8 weeks, occurrence of renal fibrosis, tubular inflammation and DN is similar to human DN | Not fully characterized |
| Genetic model of DN | Types 1 and 2 | Animals are commercially available, spontaneous development of β-cell failure may mimic disease | Biohazard, strain-dependent dosing, potential for non-specific renal effects, autosomal recessive, mutation in gene |
Merits and demerits
Transgenic mice are useful as animal models for human disease. They are also useful in cases where we are unsure of the role of a particular protein product. ICER 1γ Tg mouse shows sustained hyperglycemia (Inada et al. 1999). RAGE-overexpressing IDDM mice exhibit renal insufficiency and advanced glomerulosclerosis seen in human DN (Takamura et al. 1998), whereas MafK+ mice display reversible hyperglycemia, which appears at younger age and disappears at older age without overt diabetes (Shimohata et al. 2009). It is a costly exercise to produce transgenic animals and a special animal license must be applied before making transgenic and also time consuming, 2 generation times are required before results are known. In some countries transgenic animals are banned since the techniques involved germline gene therapy. Animal rights activists argue that to produce and breed animals manipulated to suffer from disease is painful and unwarranted.
Conclusion
Nowadays, a number of rodent animal models are being developed to better understand disease pathogenesis and develop new drugs for DN. The therapeutic agents currently available are limited; morbidity and mortality due to nephropathy are continuously increasing worldwide. Most experiments are carried out on rodents, even though other species with human-like biological characteristics are also used. But there is a long tradition of using these rodent animals as models of human DN, in which diabetes is developed either spontaneously or by using chemical, surgical, genetic or other techniques, to depict many clinical features or related phenotypes of the disease.
Acknowledgments
We wish to express our gratitude to Shri. Parveen Garg, Chairman, ISF College of Pharmacy, Moga, Punjab for his inspiration and constant support.
References
- Arora MK, Singh UK. Molecular mechanisms in the pathogenesis of diabetic nephropathy: an update. Vascul Pharmacol. 2013;58:259–271. doi: 10.1016/j.vph.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Asaba K, Toj A, Onozato ML, Goto A, Quinn MARK T, Fujita T, Wilcox CS. Effects of NADPH oxidase inhibitor in diabetic nephropathy. Kidney Int. 2005;67:1890–1898. doi: 10.1111/j.1523-1755.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med. 1999;5:601–604. doi: 10.1038/9442. [DOI] [PubMed] [Google Scholar]
- Bailey CJ, Day C. Traditional plant medicines as treatments for diabetes. Diabetes Care. 1989;12:553–564. doi: 10.2337/diacare.12.8.553. [DOI] [PubMed] [Google Scholar]
- Bailley C, Bailley O, Leech R. Diabetes mellitus in rabbits injected with dialuric acid. Proc Soc Exp Biol Med. 1946;63:502–505. doi: 10.3181/00379727-63-15651. [DOI] [PubMed] [Google Scholar]
- Bennett RA, Pegg AE. Alkylation of DNA in rat tissues following administration of streptozotocin. Cancer Res. 1981;41:2786–2790. [PubMed] [Google Scholar]
- Bhatti R, Sharma S, Singh J, Singh A, Ishar MPS. Effect of Aegle marmelos (L.) Correa on alloxan induced early stage diabetic nephropathy in rats. Indian J Exp Biol. 2013;51:464–469. [PubMed] [Google Scholar]
- Bloch KO, Vorobeychik M, Yavrians K, Vardi P. Selection of insulin-producing rat insulinoma (RINm) cells with improved resistance to oxidative stress. Biochem Pharmacol. 2003;65:1797–1805. doi: 10.1016/s0006-2952(03)00198-9. [DOI] [PubMed] [Google Scholar]
- Bohle AMBC, Wehrmann M, Bogenschütz O, Batz C, Müller GA, Müller GA. The pathogenesis of chronic renal failure in diabetic nephropathy: investigation of 488 cases of diabetic glomerulosclerosis. Pathol Res Pract. 1991;187:251–259. doi: 10.1016/s0344-0338(11)80780-6. [DOI] [PubMed] [Google Scholar]
- Boquist L, Nelson L, Lorentzon R. Uptake of labeled alloxan in mouse organs and mitochondria in vivo and in vitro. Endocrinology. 1983;113:943–948. doi: 10.1210/endo-113-3-943. [DOI] [PubMed] [Google Scholar]
- Brownlee M, Cerami A, Vlassara H. Advanced products of nonenzymatic glycosylation and the pathogenesis of diabetic vascular disease. Diabetes Metab Rev. 1988;4:437–451. doi: 10.1002/dmr.5610040503. [DOI] [PubMed] [Google Scholar]
- Brückmann G, Wertheimer E. Alloxan studies: the action of alloxan homologues and related compounds. J Biol Chem. 1947;168:241–256. [PubMed] [Google Scholar]
- Budhiraja S, Singh J. Protein kinase C beta inhibitors: a new therapeutic target for diabetic nephropathy and vascular complications. Fun Clin Pharmacol. 2008;22:231–240. doi: 10.1111/j.1472-8206.2008.00583.x. [DOI] [PubMed] [Google Scholar]
- Cameron AJ, Boyko EJ, Sicree RA, Zimmet PZ, Söderberg S, Alberti KGMM, Tuomilehto J, Chitson P, Shaw JE. Central obesity as a precursor to the metabolic syndrome in the AusDiab study and Mauritius. Obesity. 2008;16:2707–2716. doi: 10.1038/oby.2008.412. [DOI] [PubMed] [Google Scholar]
- Cao X, Yang FY, Xin Z, Xie RR, Yang JK. The ACE2/Ang-(1-7)/Mas axis can inhibit hepatic insulin resistance. Mol Cell Endocrinol. 2014;393:30–38. doi: 10.1016/j.mce.2014.05.024. [DOI] [PubMed] [Google Scholar]
- Carlson EC, Audette JL, Klevay LM, Nguyen H, Epstein PN. Ultrastructural and functional analyses of nephropathy in calmodulin-induced diabetic transgenic mice. Anat Rec. 1997;247:9–19. doi: 10.1002/(SICI)1097-0185(199701)247:1<9::AID-AR2>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Casey RG, Joyce M, Roche-Nagle G, Chen G, Bouchier-Hayes D. Pravastatin modulates early diabetic nephropathy in an experimental model of diabetic renal disease. J Surg Res. 2005;123:176–181. doi: 10.1016/j.jss.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Chang HR, Cheng CH, Shu KH, Chen CH, Lian JD, Wu MY. Study of the polymorphism of angiotensinogen, angiotensin-converting enzyme and angiotensin receptor in type II diabetes with end-stage renal disease in Taiwan. J Chin Med Assoc. 2003;66:51–56. [PubMed] [Google Scholar]
- Chatzigeorgiou A, Halapas A, Kalafatakis K, Kamper E. The use of animal models in the study of diabetes mellitus. In Vivo. 2009;23:245–258. [PubMed] [Google Scholar]
- Chien CT, Lee PH, Chen CF, Ma MC, Lai MK, Hsu SM. De novo demonstration and co-localization of free-radical production and apoptosis formation in rat kidney subjected to ischemia/reperfusion. J Am Soc Nephrol. 2001;12:973–982. doi: 10.1681/ASN.V125973. [DOI] [PubMed] [Google Scholar]
- Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transpl. 2004;19:2987–2996. doi: 10.1093/ndt/gfh441. [DOI] [PubMed] [Google Scholar]
- Chua S, Liu SM, Li Q, Yang L, Thassanapaff V, Fisher P. Differential beta cell responses to hyperglycaemia and insulin resistance in two novel congenic strains of diabetes (FVB-Lepr db) and obese (DBA-Lep ob) mice. Diabetologia. 2002;45:976–990. doi: 10.1007/s00125-002-0880-z. [DOI] [PubMed] [Google Scholar]
- Chung SSM, Chung SK. Genetic analysis of aldose reductase in diabetic complications. Curr Med Chem. 2003;10:1375–1387. doi: 10.2174/0929867033457322. [DOI] [PubMed] [Google Scholar]
- Cohen MP, Lautenslager GT, Shearman CW. Increased urinary type IV collagen marks the development of glomerular pathology in diabetic < i > d/db </i > mice. Metabolism. 2001;50:1435–1440. doi: 10.1053/meta.2001.28074. [DOI] [PubMed] [Google Scholar]
- Cornacchia F, Fornoni A, Plati AR, Thomas A, Wang Y, Inverardi L, Striker LJ, Striker GE. Glomerulosclerosis is transmitted by bone marrow–derived mesangial cell progenitors. J Clin Invest. 2001;108:1649–1656. doi: 10.1172/JCI12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven PA, DeRubertis FR, Kagan VE, Melhem M, Studer RK. Effects of supplementation with vitamin C or E on albuminuria, glomerular TGF-beta, and glomerular size in diabetes. J Am Soc Nephrol. 1997;8:1405–1414. doi: 10.1681/ASN.V891405. [DOI] [PubMed] [Google Scholar]
- Da J, Sil PC. Taurine ameliorates alloxan-induced diabetic renal injury, oxidative stress-related signaling pathways and apoptosis in rats. Amino Acids. 2012;43:1509–1523. doi: 10.1007/s00726-012-1225-y. [DOI] [PubMed] [Google Scholar]
- Daemen MA, van‘t Veer C, Denecker G, Heemskerk VH, Wolfs TG, Clauss M, Buurman WA. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J Clin Invest. 1999;104:541–549. doi: 10.1172/JCI6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Alonso MD. Hypoglycemia as a barrier to glycemic control. J Diabetes Complicat. 2004;18:60–68. doi: 10.1016/S1056-8727(03)00058-8. [DOI] [PubMed] [Google Scholar]
- De Vries DK, Schaapherder AF, Reinders ME. Mesenchymal stromal cells in renal ischemia/reperfusion injury. Alloimmun Transplant. 2012;3:162. doi: 10.3389/fimmu.2012.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnahoo KKL, Meng X, Ayala A, Cain MP, Harken AH, Meldrum DR. Early kidney TNF-alpha expression mediates neutrophil infiltration and injury after renal ischemia–reperfusion. Am J Physiol. 1999;277:922–929. doi: 10.1152/ajpregu.1999.277.3.R922. [DOI] [PubMed] [Google Scholar]
- Dunn J, Letchie N. Experimental alloxan diabetes in the rats. Lancet. 1943;2:384. [Google Scholar]
- Eddy AA, Giachelli CM. Renal expression of genes that promote interstitial inflammation and fibrosis in rats with protein-overload proteinuria. Kidney Int. 1995;47:1546–1557. doi: 10.1038/ki.1995.218. [DOI] [PubMed] [Google Scholar]
- Elsner M, Guldbakke B, Tiedge M, Munday R, Lenzen S. Relative importance of transport and alkylation for pancreatic beta-cell toxicity of streptozotocin. Diabetologia. 2000;43:1528–1533. doi: 10.1007/s001250051564. [DOI] [PubMed] [Google Scholar]
- Elsner M, Tiedge M, Guldbakke B, Munday R, Lenzen S. Importance of the GLUT2 glucose transporter for pancreatic beta cell toxicity of alloxan. Diabetologia. 2002;45:1542–1549. doi: 10.1007/s00125-002-0955-x. [DOI] [PubMed] [Google Scholar]
- Epstein PN, Overbeek PA, Means AR. Calmodulin-induced early-onset diabetes in transgenic mice. Cell. 1989;58:1067–1073. doi: 10.1016/0092-8674(89)90505-9. [DOI] [PubMed] [Google Scholar]
- Epstein PN, Boschero AC, Atwater I, Cai X, Overbeek PA. Expression of yeast hexokinase in pancreatic beta cells of transgenic mice reduces blood glucose, enhances insulin secretion, and decreases diabetes. Proc Natl Acad Sci USA. 1992;89:12038–12042. doi: 10.1073/pnas.89.24.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito C, He CJ, Striker GE, Zalups RK, Striker LJ. Nature and severity of the glomerular response to nephron reduction is strain-dependent in mice. Am J Pathol. 1999;154:891–897. doi: 10.1016/S0002-9440(10)65336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan AP, Tanner GA. Proximal tubule morphology after single nephron obstruction in the rat kidney. Kidney Int. 1986;30:818–827. doi: 10.1038/ki.1986.261. [DOI] [PubMed] [Google Scholar]
- Evan AP, Mong SA, Gattone VH, Connors BA, Aronoff GR, Luft FC. The effect of streptozotocin and streptozotocin-induced diabetes on the kidney. Ren Physiol. 1984;7:78–89. doi: 10.1159/000172927. [DOI] [PubMed] [Google Scholar]
- Farrelly N, Lee YJ, Oliver J, Dive C, Streuli CH. Extracellular matrix regulates apoptosis in mammary epithelium through a control on insulin signaling. J cell biol. 1999;144:1337–1348. doi: 10.1083/jcb.144.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federiuk IF, Casey HM, Quinn MJ, Wood MD, Ward KW. Induction of type-1 diabetes mellitus in laboratory rats by use of alloxan: route of administration, pitfalls, and insulin treatment. Comp Med. 2004;54:252–257. [PubMed] [Google Scholar]
- Forbes JM, Hewitson TD, Becker GJ, Jones CL. Ischemic acute renal failure: long-term histology of cell and matrix changes in the rat. Kidney Int. 2000;57:2375–2385. doi: 10.1046/j.1523-1755.2000.00097.x. [DOI] [PubMed] [Google Scholar]
- Fornoni A, Lenz O, Striker LJ, Striker GE. Glucose induces clonal selection and reversible dinucleotide repeat expansion in mesangial cells isolated from glomerulosclerosis-prone mice. Diabetes. 2003;52:2594–2602. doi: 10.2337/diabetes.52.10.2594. [DOI] [PubMed] [Google Scholar]
- Foulkes NS, Laoide BM, Schlotter F, Sassone-Corsi P. Transcriptional antagonist cAMP-responsive element modulator (CREM) down-regulates c-fos cAMP-induced expression. Proc Natl Acad Sci USA. 1991;88:5448–5452. doi: 10.1073/pnas.88.12.5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner K. Glomerular hyperfiltration during the onset of diabetes mellitus in two strains of diabetic mice (C57BL/6Jdb/db and C57BL/KsJdb/db) Diabetologia. 1978;15:59–63. doi: 10.1007/BF01219330. [DOI] [PubMed] [Google Scholar]
- Gilbertson DT, Liu J, Xue JL, Louis TA, Solid CA, Ebben JP, Collins AJ. Projecting the number of patients with end-stage renal disease in the United States to the year 2015. J Am Soc Nephrol. 2005;16:3736–3741. doi: 10.1681/ASN.2005010112. [DOI] [PubMed] [Google Scholar]
- Goes N, Urmson J, Vincent D, Ramassar V, Halloran PF. Effect of recombinant human insulin-like growth factor-1 on the inflammatory response to acute renal injury. J Am Soc Nephrol. 1996;7:710–720. doi: 10.1681/ASN.V75710. [DOI] [PubMed] [Google Scholar]
- Gojo A, Utsunomiya K, Taniguchi K, Yokota T, Ishizawa S, Kanazawa Y, Kurata H, Tajima N. The Rho-kinase inhibitor, fasudil, attenuates diabetic nephropathy in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2007;568:242–247. doi: 10.1016/j.ejphar.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Goldner MG. Alloxan diabetes-its production and mechanism. Bull N Y Acad Med. 1945;21:44. [PMC free article] [PubMed] [Google Scholar]
- Gorus FK, Malaisse WJ, Pipeleers DG. Selective uptake of alloxan by pancreatic B-cells. Biochem J. 1982;208:513. doi: 10.1042/bj2080513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 2005;28:164–176. doi: 10.2337/diacare.28.1.164. [DOI] [PubMed] [Google Scholar]
- Hackbarth H, Hackbarth D. Genetic analysis of renal function in mice. 1. Glomerular filtration rate and its correlation with body and kidney weight. Lab Anim. 1981;15:267–272. doi: 10.1258/002367781780893731. [DOI] [PubMed] [Google Scholar]
- Hackbarth H, Baunack E, Winn M. Strain differences in kidney function of inbred rats: 1. Glomerular filtration rate and renal plasma flow. Lab Anim. 1981;15:125–128. doi: 10.1258/002367781780959080. [DOI] [PubMed] [Google Scholar]
- Haidara MA, Mikhailidis DP, Rateb M, Ahmed ZA, Yassin HZ, Ibrahim IM, Rashed LA. Evaluation of the effect of oxidative stress and vitamin E supplementation on renal function in rats with streptozotocin-induced Type 1 diabetes. J Diabetes Complicat. 2009;23:130–136. doi: 10.1016/j.jdiacomp.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Han HJ, Choi HJ, Park SH. High glucose inhibits glucose uptake in renal proximal tubule cells by oxidative stress and protein kinase C. Kidney Int. 2000;57:918–926. doi: 10.1038/sj.ki.4491414. [DOI] [PubMed] [Google Scholar]
- Haseyama T, Fujita T, Hirasawa F, Tsukada M, Wakui H, Komatsuda A, Ohtani HIROSHI, Miura AB, Imai H, Koizumi AKIO. Complications of IgA nephropathy in a non-insulin-dependent diabetes model, the Akita mouse. Tohoku J Exper Med. 2002;198:233–244. doi: 10.1620/tjem.198.233. [DOI] [PubMed] [Google Scholar]
- Hirose K, Osterby R, Nozawa M, Gundersen HJ. Development of glomerular lesions in experimental long-term diabetes in the rat. Kidney Int. 1982;21:689–695. doi: 10.1038/ki.1982.82. [DOI] [PubMed] [Google Scholar]
- Inada A, Yamada Y, Someya Y, Kubota A, Yasuda K, Ihara Y, Kagimoto S, Kuroe A, Tsuda K, Seino Y. Transcriptional repressors are increased in pancreatic islets of type 2 diabetic rats. Biochem Biop Res Co. 1998;253:712–718. doi: 10.1006/bbrc.1998.9833. [DOI] [PubMed] [Google Scholar]
- Inada A, Someya Y, Yamada Y, Ihara Y, Kubota A, Ban N, Watanabe R, Tsuda K, Seino Y. The cyclic AMP response element modulator family regulates the insulin gene transcription by interacting with transcription factor IID. J Biol Chem. 1999;274:21095–21103. doi: 10.1074/jbc.274.30.21095. [DOI] [PubMed] [Google Scholar]
- Inada A, Kanamori H, Arai H, Akashi T, Araki M, Weir GC, Fukatsu A. A model for diabetic nephropathy: advantages of the inducible cAMP early repressor transgenic mouse over the streptozotocin-induced diabetic mouse. J Cell Physiol. 2008;215:383–391. doi: 10.1002/jcp.21316. [DOI] [PubMed] [Google Scholar]
- Kajihara M, Sone H, Amemiya M, Katoh Y, Isogai M, Shimano H, Yamada N, Satoru Takahashi S. Mouse MafA, homologue of zebrafish somite Maf 1, contributes to the specific transcriptional activity through the insulin promoter. Biochem Biop Res Co. 2003;312:831–842. doi: 10.1016/j.bbrc.2003.10.196. [DOI] [PubMed] [Google Scholar]
- Kang MJ, Ingram A, Ly H, Thai K, Scholey JW. Effects of diabetes and hypertension on glomerular transforming growth factor-&bgr; receptor expression. Kidney Int. 2000;58:1677–1685. doi: 10.1046/j.1523-1755.2000.00328.x. [DOI] [PubMed] [Google Scholar]
- Kataoka K, Igarashi K, Itoh K, Fujiwara KT, Noda M, Yamamoto M, Nishizawa M. Small Maf proteins heterodimerize with Fos and may act as competitive repressors of the NF-E2 transcription factor. Mol cell boil. 1995;15:2180–2190. doi: 10.1128/mcb.15.4.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H. MafA is a glucose-regulated and pancreatic β-cell-specific transcriptional activator for the insulin gene. J Biol Chem. 2002;277:49903–49910. doi: 10.1074/jbc.M206796200. [DOI] [PubMed] [Google Scholar]
- Kataoka K, Shioda S, Ando K, Sakagami K, Handa H, Yasuda K. Differentially expressed Maf family transcription factors, c-Maf and MafA, activate glucagon and insulin gene expression in pancreatic islet alpha-and beta-cells. J Mol Endocrinol. 2004;32:9–20. doi: 10.1677/jme.0.0320009. [DOI] [PubMed] [Google Scholar]
- Katovitch MJ, Meldrum MJ, Vasselli JR. Beneficial effects of dietary acarbose in the streptozotocin induced diabetic rat. Metabolism. 1991;40:1275–1282. doi: 10.1016/0026-0495(91)90028-u. [DOI] [PubMed] [Google Scholar]
- Kelly DJ, Zhang Y, Hepper C, Gow RM, Jaworski K, Kemp BE, Wilkinson-Berka JL, Gilbert RE. Protein kinase C β inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes. 2003;52:512–518. doi: 10.2337/diabetes.52.2.512. [DOI] [PubMed] [Google Scholar]
- Kirchhoff F, Krebs C, Abdulhag UN, Meyer-Schwesinger C, Maas R, Helmchen U, Hilgers KF, Wolf G, Stahl RAK, Wenzel U. Rapid development of severe end-organ damage in C57BL/6 mice by combining DOCA salt and angiotensin II. Kidney Int. 2007;73:643–650. doi: 10.1038/sj.ki.5002689. [DOI] [PubMed] [Google Scholar]
- Koenig RJ, Araujo DC, Cerami A. Increased hemoglobin AIc in diabetic mice. Diabetes. 1976;25:1–5. doi: 10.2337/diab.25.1.1. [DOI] [PubMed] [Google Scholar]
- Kondo K, Nozawa K, Tomita T, Ezaki K. Inbred strains resulting from Japanese mice. Bull Exp Anim. 1957;6:107–112. [Google Scholar]
- Kong LL, Wu H, Cui WP, Zhou WH, Luo P, Sun J, Yuan H, Miao LN. Advances in murine models of diabetic nephropathy. J Diabetes Res. 2013;2013:1–10. doi: 10.1155/2013/797548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya D, Haneda M, Nakagawa H, Isshiki K, Sato H, Maeda S, TOSHIRO Sugimoto T. Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC β inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J. 2000;14:439–447. doi: 10.1096/fasebj.14.3.439. [DOI] [PubMed] [Google Scholar]
- Kraynak AR, Storer RD, Jensen RD, Kloss MW, Soper KA, Clair JH, DeLuca JG, Nichols WW, Eydelloth RS. Extent and persistence of streptozotocin-induced DNA damage and cell proliferation in rat kidney as determined by in vivo alkaline elution and BrdUrd labeling assays. Toxicol Appl Pharmacol. 1995;135:279–286. doi: 10.1006/taap.1995.1234. [DOI] [PubMed] [Google Scholar]
- Kume S, Yamahara K, Yasuda M, Maegawa H, Koya D. Autophagy: emerging therapeutic target for diabetic nephropathy. Semin Nephrol. 2014;34:9–16. doi: 10.1016/j.semnephrol.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Kuramochi G, Homma S. Postischemic recovery process of renal oxygen consumption in normal and streptozotocin diabetic rats. Ren Fail. 1993;15:587–594. doi: 10.3109/08860229309069408. [DOI] [PubMed] [Google Scholar]
- Lachin T, Reza H. Anti diabetic effect of cherries in alloxan induced diabetic rats. Recent Pat Endocr Metab Immune Drug Discov. 2012;6:67–72. doi: 10.2174/187221412799015308. [DOI] [PubMed] [Google Scholar]
- Lee SM, Bressler R. Prevention of diabetic nephropathy by diet control in the db/db mouse. Diabetes. 1981;30:106–111. doi: 10.2337/diab.30.2.106. [DOI] [PubMed] [Google Scholar]
- Leiter EH. Multiple low-dose streptozotocin-induced hyperglycemia and insulitis in C57BL mice: influence of inbred background, sex, and thymus. Proc Natl Acad Sci USA. 1982;79:630–634. doi: 10.1073/pnas.79.2.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiter EH. Differential susceptibility of BALB/c sublines to diabetes induction by multi-dose streptozotocin treatment. Curr Top Microbiol Immunol. 1985;122:78. doi: 10.1007/978-3-642-70740-7_12. [DOI] [PubMed] [Google Scholar]
- Leiter EH, Coleman DL, Eisenstein AB, Strack I. Dietary control of pathogenesis in C57BLKsJdbdb diabetes mice. Metabolis. 1981;30:554–562. doi: 10.1016/0026-0495(81)90130-x. [DOI] [PubMed] [Google Scholar]
- Leiter EH, Prochazka M, Coleman DL. The non-obese diabetic (NOD) mouse. Am J Pathol. 1987;128:380. [PMC free article] [PubMed] [Google Scholar]
- Leiter EH, Chapman HD, Coleman DL. The influence of genetic background on the expression of mutations at the diabetes locus in the mouse. V. Interaction between the db gene and hepatic sex steroid sulfotransferases correlates with gender-dependent susceptibility to hyperglycemia. Endocrinology. 1989;124:912–922. doi: 10.1210/endo-124-2-912. [DOI] [PubMed] [Google Scholar]
- Lenzen S, Mirzaie-Petri M. Inhibition of aconitase by alloxan and the differential modes of protection of glucose, 3-O-methylglucose, and mannoheptulose. Naunyn Schmiedebergs Arch Pharmacol. 1992;346:532–536. doi: 10.1007/BF00169009. [DOI] [PubMed] [Google Scholar]
- Lenzen S, Munday R. Thiol-group reactivity, hydrophilicity and stability of alloxan, its reduction products and its < i > N </i > -methyl derivatives and a comparison with ninhydrin. Biochem Pharmacol. 1991;42:1385–1391. doi: 10.1016/0006-2952(91)90449-f. [DOI] [PubMed] [Google Scholar]
- Liao J, Kobayashi M, Kanamuru Y, Nakamura S, Makita Y, Funabiki K, Horikoshi S, Tomino Y. Effects of candesartan, an angiotensin II type 1 receptor blocker, on diabetic nephropathy in KK/Ta mice. J Nephrol. 2003;16:841. [PubMed] [Google Scholar]
- Like AA, Lavine RL, Poffenbarger PL, Chick WL. Studies in the diabetic mutant mouse: VI. Evolution of glomerular lesions and associated proteinuria. Am J Pathol. 1972;66:193. [PMC free article] [PubMed] [Google Scholar]
- Ma LJ, Fogo AB. Model of robust induction of glomerulosclerosis in mice: importance of genetic background. Kidney Int. 2003;64:350–355. doi: 10.1046/j.1523-1755.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- Malaisse WJ, Doherty M, Ladriere L, Malaisse-Lagae F. Pancreatic uptake of [2-14C] alloxan. Int J Mol Med. 2001;7:311. [PubMed] [Google Scholar]
- Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol. 2003;14:1358–1373. doi: 10.1097/01.asn.0000065640.77499.d7. [DOI] [PubMed] [Google Scholar]
- Mathews C. Rodent models for the study of type 2 diabetes in children (juvenile diabesity) Pediatr Diabetes. 2002;3:163–173. doi: 10.1034/j.1399-5448.2002.30307.x. [DOI] [PubMed] [Google Scholar]
- Matsuoka TA, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, Stein R. Members of the large Maf transcription family regulate insulin gene transcription in islet β cells. Mol Cell Biol. 2003;23:6049–6062. doi: 10.1128/MCB.23.17.6049-6062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLetchie NGB. Alloxan diabetes: the sorcerer and his apprentice. Diabetologia. 1982;23:72–75. doi: 10.1007/BF00257736. [DOI] [PubMed] [Google Scholar]
- Meade CJ, Brandon DR, Smith W, Simmonds RG, Harris SHEILA, Sowter C. The relationship between hyperglycaemia and renal immune complex deposition in mice with inherited diabetes. Clin Exp Immunol. 1981;43:109. [PMC free article] [PubMed] [Google Scholar]
- Melin J, Hellberg O, Akyürek LM, Källskog Ö, Larsson E, Fellström BC. Ischemia causes rapidly progressive nephropathy in the diabetic rat. Kidney Int. 1997;52:985–991. doi: 10.1038/ki.1997.420. [DOI] [PubMed] [Google Scholar]
- Melin J, Hellberg O, Larsson E, Zezina L, Fellström BC. Protective effect of insulin on ischemic renal injury in diabetes mellitus. Kidney Int. 2002;61:1383–1392. doi: 10.1046/j.1523-1755.2002.00284.x. [DOI] [PubMed] [Google Scholar]
- Mills GW, Avery PJ, McCarthy MI, Hattersley AT, Levy JC, Hitman GA, Sampson M, Walker M. Heritability estimates for beta cell function and features of the insulin resistance syndrome in UK families with an increased susceptibility to type 2 diabetes. Diabetologia. 2004;47:732–738. doi: 10.1007/s00125-004-1338-2. [DOI] [PubMed] [Google Scholar]
- Mu JL, Naggert JK, Svenson KL, Collin GB, Kim JH, McFarland C, Nishina PM, Levine DM, Williams KJ, Paigen B. Quantitative trait loci analysis for the differences in susceptibility to atherosclerosis and diabetes between inbred mouse strains C57BL/6J and C57BLKS/J. J Lipid Res. 1999;40:1328–1335. [PubMed] [Google Scholar]
- Munday R. Dialuric acid autoxidation: effects of transition metals on the reaction rate and on the generation of “active oxygen” species. Biochem Pharmacol. 1988;37:409–413. doi: 10.1016/0006-2952(88)90207-9. [DOI] [PubMed] [Google Scholar]
- Naggert JK, Fricker LD, Varlamov O, Nishina PM, Rouille Y, Steiner DF, Carroll RJ, Paigen BJ, Leiter EH. Hyperproinsulinaemia in obese fat/fat mice associated with a carboxypeptidase E mutation which reduces enzyme activity. Nat Genet. 1995;10:135–142. doi: 10.1038/ng0695-135. [DOI] [PubMed] [Google Scholar]
- Navaneethan SD, Singh S, Choudhry W. Nodular glomerulosclerosis in a non-diabetic patient: case report and review of literature. J Nephrol. 2005;18:613–615. [PubMed] [Google Scholar]
- Noonan WT, Banks RO. Renal function and glucose transport in male and female mice with diet-induced type ii diabetes mellitus. Proc Soc Exp Biol Med. 2000;225:221–230. doi: 10.1046/j.1525-1373.2000.22528.x. [DOI] [PubMed] [Google Scholar]
- Nukatsuka M, Sakurai H, Yoshimura Y, Nishida Kawada J. Enhancement by streptozotocin of O < sup > − </sup > < sub > 2 </sub > radical generation by the xanthine oxidase system of pancreatic β-cells. FEBS Lett. 1988;239:295–298. doi: 10.1016/0014-5793(88)80938-4. [DOI] [PubMed] [Google Scholar]
- Nukatsuka M, Yoshimura Y, Nishida M, Kawada J. Allopurinol protects pancreatic β cells from the cytotoxic effect of strepto-zotocin: in vitro study. J Pharmacobiodyn. 1990;13:259–262. doi: 10.1248/bpb1978.13.259. [DOI] [PubMed] [Google Scholar]
- Nukatsuka M, Yoshimura Y, Nishida M, Kawada J. Importance of the concentration of ATP in rat pancreatic β cells in the mechanism of streptozotocin-induced cytotoxicity. J Endocrinol. 1990;127:161–165. doi: 10.1677/joe.0.1270161. [DOI] [PubMed] [Google Scholar]
- Olbrot M, Rud J, Moss LG, Sharma A. Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci USA. 2002;99:6737–6742. doi: 10.1073/pnas.102168499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozturk Y, Atlan VM, Yildizoglu-Ari N. Effects of experimental diabetes and insulin on smooth muscle functions. Pharmacol Rev. 1996;48:69–112. [PubMed] [Google Scholar]
- Panés J, Kurose I, Rodriguez-Vaca MD, Anderson DC, Miyasaka M, Tso P, Granger DN. Diabetes exacerbates inflammatory responses to ischemia-reperfusion. Circulation. 1996;93:161–167. doi: 10.1161/01.cir.93.1.161. [DOI] [PubMed] [Google Scholar]
- Pelé-Tounian A, Wang X, Rondu F, Lamouri A, Touboul E, Marc S, Ktorza A. Potent antihyperglycaemic property of a new imidazoline derivative S-22068 (PMS 847) in a rat model of NIDDM. Br J Pharmacol. 1998;124:1591–1596. doi: 10.1038/sj.bjp.0701989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschke E, Ebelt H, Brömme HJ, Peschke D. ‘Classical’and ‘new’diabetogens—comparison of their effects on isolated rat pancreatic islets in vitro. Cell Mol Life Sci. 2000;57:158–164. doi: 10.1007/s000180050505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podrazik RM, Natale JE, Zelenock GB, D’Alecy LG. Hyperglycemia exacerbates and insulin fails to protect in acute renal ischemia in the rat. J Surg Res. 1989;46:572–578. doi: 10.1016/0022-4804(89)90022-x. [DOI] [PubMed] [Google Scholar]
- Reddi AS, Camerini-Davalos RA. Hereditary diabetes in the KK mouse: an overview. Adv Exp Med Biol. 1989;246:7–15. doi: 10.1007/978-1-4684-5616-5_2. [DOI] [PubMed] [Google Scholar]
- Reddi AS, Oppermann W, Velasco CA, Camerini-Davalos RA. Diabetic microangiopathy in KK mice: II. Suppression of glomerulosclerosis by pyridinolcarbamate. Exp Mol Pathol. 1977;26:325–339. doi: 10.1016/0014-4800(77)90060-0. [DOI] [PubMed] [Google Scholar]
- Reddi AS, Velasco CA, Reddy PR, Khan MY, Camerini-Davalos RA. Diabetic microangiopathy in KK mice: VI. Effect of glycemic control on renal glycoprotein metabolism and established glomerulosclerosis. Exp Mol Pathol. 1990;53:140–151. doi: 10.1016/0014-4800(90)90038-f. [DOI] [PubMed] [Google Scholar]
- Reutens AT, Atkins RC. Epidemiology of diabetic nephropathy. Contrib Nephrol. 2011;170:1–7. doi: 10.1159/000324934. [DOI] [PubMed] [Google Scholar]
- Ritz E, Orth SR. Nephropathy in patients with type 2 diabetes mellitus. N Engl J Med. 1999;341:1127–1133. doi: 10.1056/NEJM199910073411506. [DOI] [PubMed] [Google Scholar]
- Ron D. Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J Clin Invest. 2002;109:443–445. doi: 10.1172/JCI15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossini AA, Like AA, Chick WL, Appel MC, Cahill GF. Studies of streptozotocin-induced insulitis and diabetes. Proc Natl Acad Sci USA. 1977;74:2485–2489. doi: 10.1073/pnas.74.6.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai K, Miura T. Generation of free radicals by alloxan in the presence of bovine serum albumin: a role of protein sulfhydryl groups in alloxan cytotoxicity. Biochem Int. 1989;19:405–412. [PubMed] [Google Scholar]
- Schnedl WJ, Ferber S, Johnson JH, Newgard CB. STZ transport and cytotoxicity: specific enhancement in GLUT2-expressing cells. Diabetes. 1994;43:1326–1333. doi: 10.2337/diab.43.11.1326. [DOI] [PubMed] [Google Scholar]
- Shah DI, Singh M. Possible role of exogenous cAMP to improve vascular endothelial dysfunction in hypertensive rats. Fundam Clin Pharmacol. 2006;20:595–604. doi: 10.1111/j.1472-8206.2006.00449.x. [DOI] [PubMed] [Google Scholar]
- Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/dbmouse. Am J Physiol Renal Physiol. 2003;284:F1138–F1144. doi: 10.1152/ajprenal.00315.2002. [DOI] [PubMed] [Google Scholar]
- Shimohata H, Yoh K, Morito N, Shimano H, Kudo T, Takahashi S. MafK overexpression in pancreatic β-cells caused impairment of glucose-stimulated insulin secretion. Biochem Biophys Res Commun. 2006;346:671–680. doi: 10.1016/j.bbrc.2006.05.184. [DOI] [PubMed] [Google Scholar]
- Shimohata H, Yoh K, Fujita A, Morito N, Ojima M, Tanaka H, Hirayama K. MafA-deficient and beta cell-specific MafK-overexpressing hybrid transgenic mice develop human-like severe diabetic nephropathy. Biochem Biophys Res Commun. 2009;389:235–240. doi: 10.1016/j.bbrc.2009.08.124. [DOI] [PubMed] [Google Scholar]
- Srinivasan K, Ramarao P. Animal models in type 2 diabetes research: an overview. Ind J Med Res. 2007;125:451–472. [PubMed] [Google Scholar]
- Steffes MW, Østerby R, Chavers B, Mauer SM. Mesangial expansion as a central mechanism for loss of kidney function in diabetic patients. Diabetes. 1989;38:1077–1081. doi: 10.2337/diab.38.9.1077. [DOI] [PubMed] [Google Scholar]
- Stephens TW, Caro JF. To be lean or not to be lean. Is leptin the answer? Exp Clin Endocrinol Diabetes. 1998;106:1–15. doi: 10.1055/s-0029-1211943. [DOI] [PubMed] [Google Scholar]
- Sugano M, Yamato H, Hayashi T, Ochiai H, Kakuchi J, Goto S, Nishijima F. High-fat diet in low-dose-streptozotocin-treated heminephrectomized rats induces all features of human type 2 diabetic nephropathy: a new rat model of diabetic nephropathy. Nutr Metab Cardiovasc Dis. 2006;16:477–484. doi: 10.1016/j.numecd.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Susztak K, Böttinger E, Novetsky A, Liang D, Zhu Y, Ciccone E, Wu D, Dunn S, McCue P, Sharma K. Molecular profiling of diabetic mouse kidney reveals novel genes linked to glomerular disease. Diabetes. 2004;53:784–794. doi: 10.2337/diabetes.53.3.784. [DOI] [PubMed] [Google Scholar]
- Suto JI, Matsuura S, Imamura K, Yamanaka H, Sekikawa K. Genetic analysis of non-insulin-dependent diabetes mellitus in KK and KK-Ay mice. Eur J Endocrinol. 1998;139:654–661. doi: 10.1530/eje.0.1390654. [DOI] [PubMed] [Google Scholar]
- Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. 2001;50:537–546. [PubMed] [Google Scholar]
- Takamura N, Maruyama T, Ahmed S, Suenaga A, Otagiri M. Interactions of aldosterone antagonist diuretics with human serum proteins. Pharm Res. 1997;14:522–526. doi: 10.1023/a:1012168020545. [DOI] [PubMed] [Google Scholar]
- Takamura T, Kato I, Kimura N, Nakazawa T, Yonekura H, Takasawa S, Okamoto H. Transgenic mice overexpressing type 2 nitric-oxide synthase in pancreatic β cells develop insulin-dependent diabetes without insulitis. J Biol Chem. 1998;273:2493–2496. doi: 10.1074/jbc.273.5.2493. [DOI] [PubMed] [Google Scholar]
- Takasu N, Komiya I, Asawa T, Nagasawa Y, Yamada T. Streptozocin-and alloxan-induced H2O2 generation and DNA fragmentation in pancreatic islets: H2O2 as mediator for DNA fragmentation. Diabetes. 1991;40:1141–1145. doi: 10.2337/diab.40.9.1141. [DOI] [PubMed] [Google Scholar]
- Thulesen J, Ørskov C, Holst JJ, Poulsen SS. Short term insulin treatment prevents the diabetogenic action of streptozotocin in rats. Endocrinology. 1997;138:62–68. doi: 10.1210/endo.138.1.4827. [DOI] [PubMed] [Google Scholar]
- Tikellis C, Bialkowski K, Pete J, Sheehy K, Su Q, Johnston C, Cooper ME, Thomas MC. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018–1025. doi: 10.2337/db07-1212. [DOI] [PubMed] [Google Scholar]
- Tojo A, Onozato ML, Kobayashi N, Goto A, Matsuoka H, Fujita T. Antioxidative effect of p38 mitogen-activated protein kinase inhibitor in the kidney of hypertensive rat. J Hypertens. 2005;23:165–174. doi: 10.1097/00004872-200501000-00027. [DOI] [PubMed] [Google Scholar]
- Utimura R, Fujihara CK, Mattar AL, Malheiros DMAC, Noronha IDL, Zatz R. Mycophenolate mofetil prevents the development of glomerular injury in experimental diabetes1. Kidney Int. 2003;63:209–216. doi: 10.1046/j.1523-1755.2003.00736.x. [DOI] [PubMed] [Google Scholar]
- Wada R, Yagihashi S. Pathology of diabetic neuropathy. Nihon Rinsho. 2010;68:542. [PubMed] [Google Scholar]
- Wald H, Markowitz H, Zevin S, Popovtzer M. Opposite effects of diabetes on nephrotoxic and ischemic acute tubular necrosis. Proc Soc Exp Biol Med. 1990;195:51–56. doi: 10.3181/00379727-195-43117. [DOI] [PubMed] [Google Scholar]
- Wang Z, Li L. Adenovirus-mediated RNA interference against collagen-specific molecular chaperone 47-kDa heat shock protein suppresses scar formation on mouse wounds. Cell Biol Int. 2008;32:484–493. doi: 10.1016/j.cellbi.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Weaver DC, McDaniel ML, Naber SP, Barry CD, Lacy PE. Alloxan stimulation and inhibition of insulin release from isolated rat islets of Langerhans. Diabetes. 1978;27:1205–1214. doi: 10.2337/diab.27.12.1205. [DOI] [PubMed] [Google Scholar]
- Weaver DC, Barry CD, Mcdaniel ML, Marshall GR, Lacy PE. Molecular requirements for recognition at a glucoreceptor for insulin release. Mol Pharmacol. 1979;16:361–368. [PubMed] [Google Scholar]
- Wei M, Ong L, Smith MT, Ross FB, Schmid K, Hoey AJ, Burstow D, Brown L. The streptozotocin-diabetic rat as a model of the chronic complications of human diabetes. Heart Lung Circ. 2003;12:44–50. doi: 10.1046/j.1444-2892.2003.00160.x. [DOI] [PubMed] [Google Scholar]
- Welsh N, Eizirik DL, Sandler S. Nitric oxide and pancreaticbeta-cell destruction in insulin dependent diabetes mellitus:don’t take NO for an answer. Autoimmunity. 1994;18:285–290. doi: 10.3109/08916939409009530. [DOI] [PubMed] [Google Scholar]
- Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, Nyengaard JR, Enden MD, Kilo C, Tilton RJ. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801–813. doi: 10.2337/diab.42.6.801. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, Munday R. Glutathione-mediated redox cycling of alloxan: mechanisms of superoxide dismutase inhibition and of metal-catalyzed OH formation. Biochem Pharmacol. 1989;38:271–277. doi: 10.1016/0006-2952(89)90037-3. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, Cowden WB, Sutton HC. Auto-oxidation of dialuric acid, divicine and isouramil: superoxide dependent and independent mechanisms. Biochem Pharmacol. 1989;38:611–618. doi: 10.1016/0006-2952(89)90206-2. [DOI] [PubMed] [Google Scholar]
- Wolf E, Braun-Reichhart C, Streckel E, Renner S. Genetically engineered pig models for diabetes research. Transgenic Res. 2014;23:27–38. doi: 10.1007/s11248-013-9755-y. [DOI] [PubMed] [Google Scholar]
- Yokozawa T, Nakagawa T, Wakaki K, Koizumi F. Animal model of diabetic nephropathy. Exp Toxicol Pathol. 2001;53:359–363. doi: 10.1078/0940-2993-00203. [DOI] [PubMed] [Google Scholar]
- Yong LCJ, Bleasel AF. Pathological changes in streptozotocin induced diabetes mellitus in the rat. Exp Pathol. 1986;30:97–107. doi: 10.1016/s0232-1513(86)80067-6. [DOI] [PubMed] [Google Scholar]
- Young DA, Ho RS, Bell PA, Cohen DK, McIntosh RH, Nadelson J. Inhibition of hepatic glucose production by SDZ 51641. Diabetes. 1990;39:1408–1413. doi: 10.2337/diab.39.11.1408. [DOI] [PubMed] [Google Scholar]
- Zalups RK. The Os, + mouse a genetic animal model of reduced renal mass. Am J Physiol. 1993;264:53–60. doi: 10.1152/ajprenal.1993.264.1.F53. [DOI] [PubMed] [Google Scholar]
- Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol. 2005;25:4969–4976. doi: 10.1128/MCB.25.12.4969-4976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HL, Sui Y, Guan J, He L, Zhu X, Fan RR, Xu G. Fat redistribution and adipocyte transformation in uninephrectomized rats. Kidney Int. 2008;74:467–477. doi: 10.1038/ki.2008.195. [DOI] [PubMed] [Google Scholar]
- Zheng F, Striker GE, Esposito C, Lupia E, Striker LJ. Strain differences rather than hyperglycemia determine the severity of glomerulosclerosis in mice. Kidney Int. 1998;54:1999–2007. doi: 10.1046/j.1523-1755.1998.00219.x. [DOI] [PubMed] [Google Scholar]
- Zheng S, Noonan WT, Metreveli NS, Coventry S, Kralik PM, Carlson EC, Epstein PN. Development of late-stage diabetic nephropathy in OVE26 diabetic mice. Diabetes. 2004;53:3248–3257. doi: 10.2337/diabetes.53.12.3248. [DOI] [PubMed] [Google Scholar]
- Ziyadeh FN. Mediators of diabetic renal disease: the case for TGF-β as the major mediator. J Am Soc Nephrol. 2004;15:55–57. doi: 10.1097/01.asn.0000093460.24823.5b. [DOI] [PubMed] [Google Scholar]