

Abstract
Sepsis is a clinical syndrome with no effective protective or therapeutic treatments. Acacetin, a natural flavonoid compound, has anti-oxidative and anti-inflammatory effects which can potentially work to reduce sepsis. We investigated the potential protective effect of acacetin on sepsis-induced acute lung injury (ALI) ALI and dissect out the underlying mechanisms. Mice were divided into five groups: a sham group, a sepsis-induced ALI group, and three sepsis groups pre-treated with 20, 40, and 80 mg/kg body weight of acacetin. We found that acacetin significantly attenuated sepsis-induced ALI, in histological examinations and lung edema. Additionally, acacetin treatment decreased protein and inflammatory cytokine concentration and the number of infiltrated inflammatory cells in BALF compared with that in the non-treated sepsis mice. Pulmonary myeloperoxidase (MPO) activity was lower in the acacetin-pre-treated sepsis groups than in the sepsis group. The mechanism underlying the protective effect of acacetin on sepsis is related to the regulation of certain antioxidation genes, including inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), superoxide dismutases (SODs), and heme oxygenase 1 (HO-1).Taken together, our results indicate that acacetin pre-treatment inhibits sepsis-induced ALI through its anti-inflammatory and antioxidative activity, suggesting that acacetin may be a potential protective agent for sepsis-induced ALI.
Keywords: Acacetin, Sepsis, COX-2, SODs, iNOS
Introduction
Acute lung injury (ALI) and its severest form, acute respiratory syndrome (ARDS), lead to the development of multiple organ dysfunction syndrome (MODS)characterized by hypoxemia, pulmonary infiltrates, and the absence of an elevated pulmonary capillary wedge pressure (Matthay et al. 2011, 2012). Sepsis is a potentially fatal whole-body inflammation (a systemic inflammatory response syndrome or SIRS) caused by severe infection (Bone et al. 1992; Levy et al. 2003). Sepsis develops when the initial host response to an infection is amplified and becomes damaging to the host (Weber and Swirski 2014). Some structural components of bacteria (pathogen-associated molecular patterns-PAMPs), are recognized by pattern recognition receptors (PRRs) expressed in phagocytes and other cell types (Hargreaves et al. 2005) and are responsible for the initiation of the septic process. The process can lead to activation of NF-κB and transcription of several pro-inflammatory genes, including TNF-α, IL-6, and IL-1β.
Recent evidence suggests that nitric oxide (NO) is an important endogenous regulatory molecule, implicated in both pro- and anti-inflammatory processes in the lung (Sharma et al. 2007; Hamza et al. 2010; Hossain et al. 2012). Additionally, it is well known that administration of LPS increases the expression and activity of inducible nitric oxide synthase (iNOS) which, consequently, increases nitric oxide (NO) generation (Parratt 1997). Cyclooxygenase (COX), the prostaglandin H synthase enzyme, is involved in a wide variety of inflammatory diseases, including lung injury (Fukunaga et al. 2005). COX-2 is an inducible enzyme catalyzing the conversion of arachidonic acid to prostaglandins. Prostaglandin (PG) E2 is a potent inflammatory mediator. The expression of COX-2, the inducible isoform of COX, increases markedly during inflammation (Zhang et al. 2011), mainly in response to various proinflammatory stimuli, such as tumor necrosis factor alpha (TNF-α), interleukin (IL) -1β, IL-6, and LPS (Serou et al. 1999; Huang et al. 2000). In LPS-induced ALI, endotoxin administration disrupts the alveolocapillary barrier because of the overproduction of NO and PGE2 through the induction of iNOS and COX-2, respectively (Mann et al. 2005; Grommes et al. 2011).
Nuclear factor-κB (NF-κB) is a key transcription factor that regulates the expression of iNOS, COX-2, and various proinflammatory cytokines such as TNF-α and IL-1β (Tak et al. 2001). NF-κB is a dimer consisting of p65, a transcription-activating component, or p50. In resting cells, NF-κB exists in an inactive state in the cytoplasm, coupled with an inhibitory protein called IκB. Upon activation, IκB undergoes phosphorylation and degradation, and NF-κB is translocated into the nucleus where it binds to DNA and activates gene transcription (Kretz-Remy et al. 2001).
Acacetin (5,7-dihydroxy-4′-methoxyflavone) is an O-methylated flavone and is naturally present in plants such as chrysanthemum (Chatterjee et al. 1981) and safflower (Roh et al. 2004), and in Calamintha (Marin et al. 2001) and Linaria species (Smirnova 1974). Acacetin possesses anti-peroxidative, anti-inflammatory, and anti-plasmodial activity (Y. H. Liao et al. 1999; Kraft et al. 2003; Pan et al. 2006), as well as anticancer activity (Singh et al. 2005). In addition, acacetin strongly inhibits the expression of proinflammatory cytokines, iNOS, and COX-2 in LPS-induced RAW 264.7 cells (Pan et al. 2006).
In this study, we evaluated the protective effect of acacetin on sepsis-induced ALI in mice. Our results demonstrated that acacetin treatment could improve symptoms of sepsis-induced ALI, including a reduction of histological changes, lung edema, protein concentration in bronchoalveolar lavage fluid (BALF), pulmonary myeloperoxidase (MPO) activity, number of infiltrating inflammatory cells, and proinflammatory cytokine production. Our findings suggest that acacetin my attenuate sepsis-induced ALI through its anti-inflammatory and anti-oxidative activity.
Materials and methods
Mice
Fifty adult (6 week-old) specific pathogen-free female C57BL/6 mice were obtained from the Shanghai Laboratory Animal Center (SLAC; Shanghai, China). All animals were housed in a room with temperature at 24 ± 1 °C, a 12 h light–dark cycle, and a relative humidity of about 40–80%. Animals had access to tap water and normal chow ad libitum. All experimental protocols were performed in accordance with the Declaration of the National institutes of Health Guide for Care and Use of Laboratory Animals.
Sepsis-induced ALI model
The animals were randomly divided to five groups (10 mice/group): (1) Sham-operated animals (Sham group) underwent the same procedure with the exception that the cecum was neither ligated nor punctured; (2) Cecum-induced sepsis group (CLP group); (3) 20 mg/kg acacetin pre-treated sepsis group (CLP + 20 mg/kg group; treatment with 20 mg/kg acacetin followed by treatment with CLP); (4) 40 mg/kg acacetin pre-treated sepsis group (CLP + 40 mg/kg. group; treatment with 40 mg/kg acacetin followed by CLP induction); and (5) 80 mg/kg acacetin pre-treated sepsis group (CLP + 80 mg/kg group; treatment with 80 mg/kg acacetin followed by CLP induction). The animals were treated orally with acacetin at the respective concentrations (Jung et al. 2014; Kim et al. 2014; Wenjun Zeng et al. 2017) 2 days before sepsis induction. After2 days of acacetin treatment, mice were anesthetized using sodium pentobarbital (intraperitoneally, 40 mg/kg). Next, the ventral neck, abdomen, and groin were shaved and washed with 10% povidoneiodine. Sepsis was induced by cecal ligation and puncture as previously described (Rittirsch et al. 2009). Briefly, the lower abdomen area was shaved and disinfected, a median 0.5–1.0 cm incision was made in the lower abdomen. After careful dissection, the cecum was ligated below the ileocecal valve, followed by a single’ through and through’ perforation (21-gauge needle); The caecum was replaced in the abdomen, and the incision was closed. After surgery, the animals were returned to their cages and were allowed access to food and water ad libitum.
Preparation of lung samples
The lung tissue of the mice was collected for histological analysis of lung edema and measurement of MPO activity. The lungs were collected on ice, weighed, and homogenized in ice-cold PBS. The resultant homogenates were centrifuged at 8000×g for 10 min at 4 °C. The supernatants were stored at − 80 °C until analysis.
BALF collection, cell counting, and protein concentration detection in BALF
BALF was prepared as previously described (Kuo et al. 2011). Briefly, the lungs were flushed three times with sterile saline via a tracheal cannula. After centrifugation, the supernatant was stored at − 20 °C for assessment of inflammatory cytokines and macrophage inflammatory protein-2 (MIP-2). The sedimented cells were resuspended in saline, and total cell count was determined using a hemocytometer. The percentage of neutrophils was determined using the Wright–Giemsa staining. The protein concentration of the BALF was determined using a BCA protein assay kit (Beyotime, Jiang Su, China).
Pathological examination of the lung tissue
The right lower lung lobes were immersed in 4% paraformaldehyde and fixed for 48 h. The lung lobes were subsequently embedded in paraffin, and tissue blocks were cut into 5 μm sections, mounted on glass slides, and stained with hematoxylin and eosin (H&E). Blinded morphologic examinations of the lung lobes were performed using light microcopy. To determine the extent of lung injury, the following five pathological features were considered: (1) the presence of exudates, (2) hyperemia/congestion, (3) intra-alveolar hemorrhages/debris, (4) cellular infiltration, and (5) cellular hyperplasia (Nishina et al. 1997). For each mouse, lung injury severity was examined independently by three pathologists.
Lung wet-to-dry weight ratio measurement
The wet-to-dry weight ratio of the lung samples collected from the upper and middle lobes of the right lung in each mouse was examined to assess lung tissue edema. The lung samples were obtained after the mice were euthanized, and weighed immediately after procurement. Subsequently, the samples were desiccated in an oven at 70 °C until a stable dry weight was achieved. The wet-to-dry weight ratio was calculated by dividing the wet weight by the dry weight.
Measurement of MPO activity
After 24 h of sepsis induction, mice were anesthetized and the lung tissues were dissected, weighed, and homogenized in 0.5% HTAB buffer (hexadecyltrimethylammonium bromide in 50 mM potassium phosphate buffer) to obtain a 10% homogenate. After centrifugation at 10,000×g for 2 min, the resultant supernatant fractions were assayed for MPO activity using a test kit purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, Jiangsu, China). The supernatant samples were diluted in phosphate citrate buffer (pH 5.0) and samples absorbance was measured at 460 nm using a microplate reader. The specific activity of MPO in the lung was expressed as unit/mg lung tissue.
Cytokine measurement in BALF
The concentration of the pro-inflammatory cytokines and chemokines TNF-α, IL-1β, IL-6, and MIP-2 in the BALF supernatants was measured with a commercially available enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems; Minneapolis, MN, USA) per the manufacturer’s instructions.
Western blot analysis
The lung tissues harvested from mice that underwent different treatments were homogenized in PBS and radioimmunoprecipitation assay (RIPA) buffer. The homogenates were centrifuged at 12,000×g for 15 min to obtain the respective supernatants. Protein samples (50 μg) were resolved using denaturing 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using standard methods, and were after transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were subsequently incubated with a blocking solution of 5% non-fat milk for 1.5 h at room temperature. The blotted proteins were separately probed with the indicated primary antibodies at 4 °C overnight or at room temperature for 2 h. The membranes were subsequently incubated with horseradish peroxidase-conjugated (HRP) secondary antibody at room temperature for 1 h, and the immuno-labeled proteins were visualized using enhanced chemiluminescence reagents (Bio-Rad Laboratories). The following antibodies were used: rabbit anti-mouse iNOS polyclonal antibody (ab3523, Abcam), rabbit anti-mouse COX-2 polyclonal antibody (ab15191, Abcam), rabbit anti-mouse beta-actin antibody (ab189073, Abcam), rabbit anti-mouse NF-κB p65 polyclonal antibody (ab16502, Abcam), rabbit anti-mouse IκB alpha polyclonal antibody (ab32518, Abcam). HRP-conjugated goat anti-rabbit IgG (ab6721, Abcam) was used as secondary antibody.
Isolation of alveolar macrophages
Alveolar macrophages were isolated from lung BALF as previously described (Lavnikova et al. 1993). Briefly, BALF was collected as described above and centrifuged at 400×g at 4 °C for 10 min. The cells were then incubated in 100 mm sterilized polystyrene Petri dishes at 37 °C for 2 h. Cells that had adhered to the bottom of the dish were harvested and replated for further experiments. Phycoerythrin-conjugated anti-CD11b and fluorescein isothiocyanate-conjugated anti-F4/80 (both purchased from eBioscience, San Diego, CA, USA) were used to confirm the > 95% purity of isolated macrophages by flow cytometric analysis. Viability was determined to be > 98% by trypan blue (Sigma) exclusion.
Determination of ROS production
Levels of ROS in lung tissues and cells were measured by the oxidative conversion of 2′,7′-dichlorofluorescein diacetate (DCFH-DA) to the fluorescent compound dichlorofluorescin (DCF). In brief, lung homogenates or alveolar macrophages were incubated with PBS containing 15 μM 2′,7′-DCFH-DA (Nanjing Jiancheng Bioengineering Institute) for 30 min at 37 °C to label intracellular ROS. The cells were then washed with PBS, and cellular fluorescence was determined using a microplate reader (Promega, Madison, WI, USA) at 490 and 520 nm.
Cell culture and treatments
RAW264.7 murine macrophage-like cells were maintained at sub-confluence under a 95% air/5% CO2 humidified atmosphere at 37 °C. Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/mL), and streptomycin (100 or 100 μL/mL) was used as cell culture medium. The cells were pretreated with different concentrations of acacetin (10, 50, or 100 μg/mL) for 4 h before LPS (1 μg/mL) stimulation. The 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (Promega; Madison, WI, USA) was used to assess cell viability.
NF-κB p65 DNA-binding activity assay
The NF-κB p65 DNA-binding activity was measured using the TransAM™ NFκB p65 Chemi Transcription Factor Assay Kit (Active Motif; Carlsbad, CA, USA).
Statistical analyses
Statistical analyses were performed using SPSS software, version 17.0 (SPSS, Inc.; Chicago, IL, USA). All data were presented as the mean ± SD. One-way analysis of variance (ANOVA) followed by a Student’s t test were used for statistical tests. P < 0.05 was considered statistically significant.
Results
Acacetin attenuates sepsis-induced pulmonary inflammation in ALI mice
To assess the effect of acacetin on sepsis-induced acute lung injury (ALI) in mice, we first measured pathological changes in sepsis mice by using H&E staining. Histological evaluation of the lungs by light microscopy revealed that sepsis caused severe ALI as characterized by edema formation, inflammatory cell infiltration, interalveolar septal thickening, and patchy intra-alveolar and interstitial hemorrhages (Fig. 1b) when compared with the non-treated sham group mice (Fig. 1a). Acacetin treatment alleviated the pathological changes in the lung tissues (Fig. 1c–e). Our findings were consistent with the data of lung injury scores (Fig. 1f). Taken together, these results indicate that acacetin could attenuate the degree of pathologic pulmonary inflammation in sepsis–induced ALI.
Fig. 1.
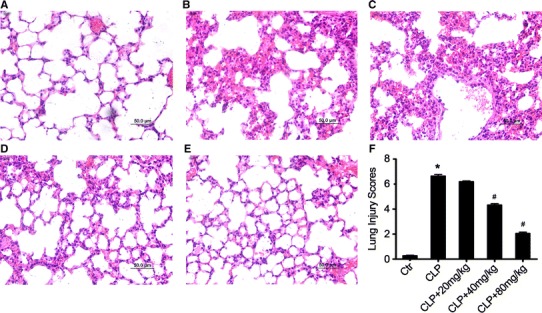
Acacetin attenuates sepsis-induced ALI in vivo. Twenty-four hours after sepsis induction in the presence or absence of acacetin pre-treatment, sepsis mice were exsanguinated and their right lower lungs were fixed. Subsequently, the lung tissue sections were stained with hematoxylin and eosin (H&E). Histologic studies of representative lung sections in the control group (a), sepsis-induced ALI group (b), 20 mg/kg acacetin-pre-treated sepsis group (c), 40 mg/kg acacetin-pre-treated sepsis group (d), and 80 mg/kg acacetin-pre-treated sepsis group (e). f Representative lung injury scores of the acacetin-pre-treated mice are shown. The image (200×magnification) is representative for each different treatment group. The values represent the mean ± SD (n = 10/group). *P < 0.05, compared to the non-treated control group; # P < 0.05, compared to the sepsis-induced ALI group
Acacetin treatment alleviates sepsis-induced lung edema and protein leakage in lung tissue of ALI mice
To further evaluate the protective effect of acacetin on sepsis-induced acute lung injury we measured lung edema, an indicator of sepsis-induced ALI due to changes in barrier permeability (Fig. 2a), and the protein concentration in BALF, an indicator of the state of the pulmonary permeability barrier (Fig. 2b). The protein concentration in BALF (Fig. 2b) and lung wet-to-dry ratio (Fig. 2a) were significantly increased in the sepsis-induced ALI mice when compared to the non-treated control group mice. However, pre-treatment with acacetin dramatically reduced the sepsis-induced lung edema and protein concentration in BALF in a dose-dependent manner when compared to the mice in sepsis group (Fig. 2a, b).
Fig. 2.
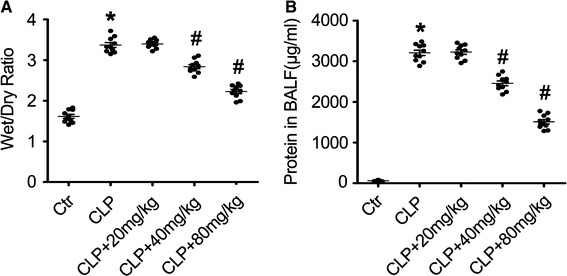
Acacetin effect on the lung wet-to-dry weight ratio and protein concentration in the BALF of LPS-induced sepsis mice. Mice received an oral pre-treatment of acacetin (20, 40, or 80 mg/kg) 2 days before the induction of sepsis via CLP surgery. The lung wet-to-dry weight ratio (a) and the protein concentration in the bronchoalveolar lavage fluid (BALF) (b) were determined at 24 h after sepsis induction. The values represent the mean ± SD (n = 10/group). *P < 0.05, compared to the non-treated control group; #P < 0.05, compared to the sepsis-induced ALI group
The effect of acacetin pre-treatment on pulmonary MPO activity and inflammatory cell infiltration in sepsis-induced ALI mice
Tissue damage in ALI is related to pulmonary MPO activity, and neutrophil extravasation is one of the major histological markers of inflammatory and immunological responses in injured lung tissue (Abraham 2003; Zhou et al. 2012). Furthermore, pulmonary MPO activity is also a reliable marker of pulmonary neutrophil infiltration (McCabe et al. 2001). Therefore, we determined the MPO activity in the lung tissue homogenates and the number of infiltrated neutrophils in the BALF of mice in the different treatment groups at 24 h after sepsis induction. As shown in Fig. 3, sepsis induction significantly increased the pulmonary MPO activity in the sepsis-induced ALI group when compared to the non- sepsis-induced, non-treated sham group mice. Furthermore, pulmonary MPO activity was dramatically lower in the acacetin-treated mice than in the sepsis group mice (Fig. 3a). These results were further supported by the data of neutrophil infiltrates in BALF (Fig. 3b).
Fig. 3.
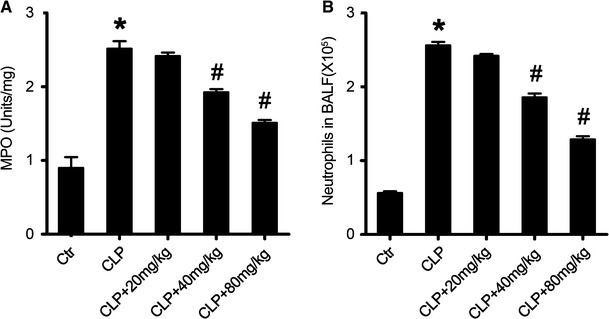
Acacetin effect on sepsis-induced pulmonary MPO activity and neutrophil infiltration. a The myeloperoxidase (MPO) activity in the lung homogenates from sepsis mice and in the different treatment groups. b The number of infiltrated neutrophils in the bronchoalveolar lavage fluid (BALF) from mice treated with acacetin at the indicated concentrations. The values represent the mean ± SD (n = 10/group). *P < 0.05, compared to the non-treated control group; #P < 0.05, compared to the sepsis-induced ALI group
The effect of acacetin on the inflammatory cytokine concentration in the BALF of sepsis mice
LPS is a major stimulator for inducing production of several inflammatory and chemotactic cytokines involved in the inflammatory processes in sepsis, including TNF-α, IL-1β, IL-6, and MIP-2 (Goodman et al. 2003; Bhatia et al. 2004; Cribbs et al. 2010). To analyze the effect of acacetin on sepsis-induced inflammatory cytokine production, we determined the concentrations of TNF-α, IL-1β, IL-6, and MIP-2 in the BALF of mice in the different treatment groups using an ELISA assay. As shown in Fig. 4, sepsis induction significantly increased the concentration of TNF-α (Fig. 4a), IL-1β (Fig. 4b), IL-6 (Fig. 4c), and MIP-2 (Fig. 4d) in the BALF of sepsis-induced mice when compared to the sham group mice. Acacetin pre-treatment inhibited the elevation of these pro-inflammatory cytokines, as observed in the acacetin-treated sepsis mice. These results suggest that acacetin could inhibit sepsis-induced inflammatory responses in ALI mice.
Fig. 4.
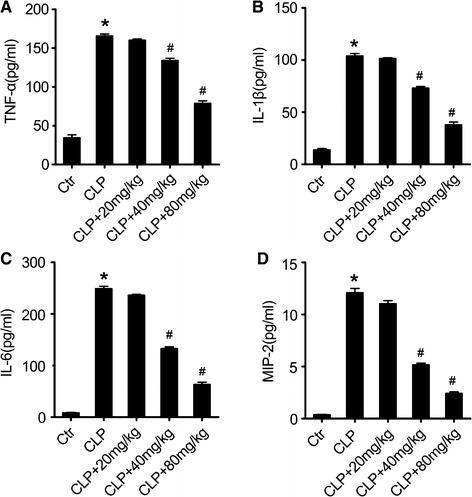
Acacetin effect on the concentration of inflammatory cytokines in the bronchoalveolar lavage fluid. Twenty-four hours after sepsis induction by CLP, the lungs of the sepsis mice were flushed with saline and the bronchoalveolar lavage fluid (BALF) was collected. The concentration of the inflammatory cytokines tumor necrosis factor alpha (TNF-α,) (a), interleukin- (IL) 1β (b), IL-6 (c), and macrophage inflammatory protein-2 (MIP-2), (d) was determined using an ELISA assay. The values represent the mean ± SD (n = 10/group). *P < 0.05, compared to the non-treated control group; #P < 0.05, compared to the sepsis-induced ALI group
The effect of acacetin on sepsis-induced iNOS and COX-2 expression in lung tissue of sepsis mice
Previous studies have reported that iNOS and COX-2 play a critical role in acute lung injury and suppression of iNOS and COX-2 expression protects against sepsis (Speyer et al. 2003; Fukunaga et al. 2005; Peng et al. 2005; Jinzhou et al. 2008; Kung et al. 2011). To investigate the possible mechanism underlying the protective effect of acacetin in sepsis mice in vivo, we measured the effect of acacetin on iNOS and COX-2 expression in the different treatment groups. The expression of iNOS and COX-2 in the lung tissues was markedly increased after sepsis induction, and was significantly inhibited by acacetin pre-treatment (Fig. 5a, b).
Fig. 5.
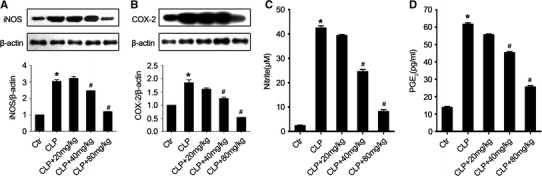
Acacetin effect on iNOS and COX-2 expression. Twenty-four hours after sepsis induction by CLP, sepsis mice in the different treatment groups were exsanguinated and their lung tissues were removed. The protein level of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) in the lung homogenates was determined using Western blotting (Individual sample in each group was used in WB data and the combined group was used in bar graphs). a The protein level of iNOS in the lung tissues from mice in the different treatment groups. b The protein level of COX-2 in the lung tissues from mice in the different treatment groups. c The level of nitrite in the lung homogenates. d The level of prostaglandin E2 (PGE2) in the lung homogenates. The values represent the mean ± SD (n = 10/group). *P < 0.05, compared to the non-treated control group; #P < 0.05, compared to the sepsis-induced ALI group
iNOS and COX-2 mediate inflammatory processes through NO and prostaglandin release, respectively, after sepsis induction (Wilgus et al. 2000; FitzGerald 2003; Fukunaga et al. 2005). As shown in Fig. 5c, d, treatment with acacetin in a dose-dependent reduced the level of NO and PGE2 in the lung tissues of sepsis-induced ALI mice. These results were consistent with the data of iNOS and COX-2 expression in the acacetin-treated sepsis mice.
The effect of acacetin on the expression of antioxidative enzymes and HO-1 in sepsis mice
Sepsis causes oxidative damage to lung tissues though uncontrolled pathophysiological reactions. Previous studies have reported that antioxidative enzymes (AOEs), such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx), could protect tissue against oxidative injury (Bhaskaran et al. 2013). In our study we determined the expression of certain AOEs and the production of ROS in sepsis mice in the different treatment groups. Sepsis induction treatment led to reduced AOE expression of catalase, MnSOD, CuZnSOD, and GPx-1 (Fig. 6a) and higher production of ROS (Fig. 6b) in lung tissues of sepsis mice when compared to the non-treated sham group. However, treatment with acacetin significantly restored the expression of these AOEs and reduced production of ROS in lung tissues (Fig. 6a, b).
Fig. 6.
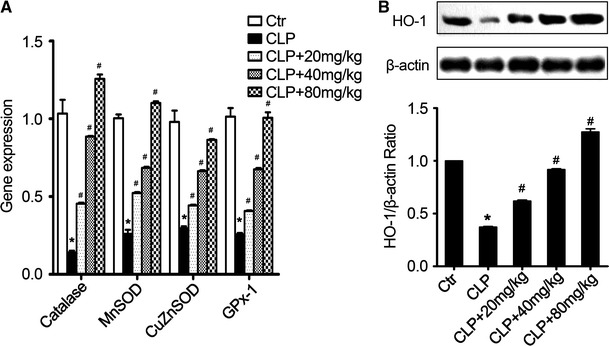
Acacetin effect on the expression of antioxidative enzymes in the lung tissues. a The mRNA level of the indicated antioxidative enzymes (AOEs) in sepsis mice in the different treatment groups. b The production of ROS in lung tissues from different treated sepsis mice. c The protein level of heme oxygenase 1 (HO-1) in the lung tissues from sepsis mice in the different treatment groups. d The production of ROS in alveolar macrophages with the indicated treatment. (Individual sample in each group was used in WB data and the combined group was used in bar graphs). The values represent the mean ± SD (n = 10/group). *P < 0.05, compared to the non-treated control group; #P < 0.05, compared to the sepsis-induced ALI group
HO-1 is another antioxidative protein that can ameliorate sepsis-induced ALI (Gong et al. 2008). Here, sepsis induction inhibited the expression of HO-1 in the lung tissues of sepsis mice when compared to the non-treated control group mice (Fig. 6c). Acacetin not only inhibited sepsis-induced decrease of HO-1 expression, but also increased the HO-1 expression in a concentration-dependent manner (Fig. 6c). Next, to confirm the anti-oxidative protective activity of acacetin on sepsis-induced ALI, we measured ROS production in alveolar macrophages under different conditions. As shown in Fig. 6d, LPS stimulation significantly increased ROS production in alveolar macrophages, however, acacetin treatment dramatically decreased LPS-induced ROS production in alveolar macrophages. Taken together, these results indicate that the antioxidative activity of acacetin could reduce sepsis-induced oxidative damage to lung tissue by increasing the expression of AOEs and HO-1.
The effect of acacetin on NF-κB activation in lungs of sepsis mice of different treatments
Although inflammation elicited through the activation of innate immune pathways is critical for effective host responses to infection, uncontrolled inflammation can contribute to tissue injury (Wallach et al. 2014). The NF-κB pathway, which regulates transcription of a variety of proinflammatory mediators, such as TNF-α, iNOS, and COX-2 (Surh et al. 2001), is involved in eliciting pulmonary inflammation. We measured the activation of NF-κB in lung tissues of sepsis mice in the different treatment groups. Sepsis induction increased the level of NF-κB p65, which plays an important role in activation of various pro-inflammatory genes (Fig. 7). Acacetin treatment inhibited the expression of NF-κB p65 in lung tissues of sepsis mice when compared to the non-treated sepsis group (Fig. 7a).
Fig. 7.
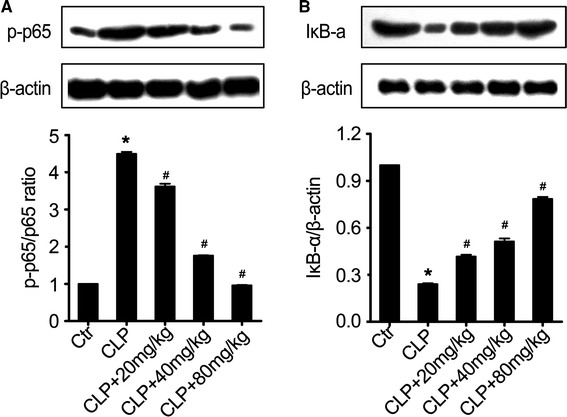
Acacetin effect on the activation of the NF-κB pathway. a The expression of NF-κB p65 in the lung tissues of sepsis mice in the different treatment groups. b The phosphorylation of IκB-α in the lung tissues of sepsis mice in the different treatment groups. Individual sample in each group was used in WB data and the combined group was used in bar graphs. The values represent the mean ± SD (n = 10/group). *P < 0.05, compared to the non-treated control group; #P < 0.05, compared to the sepsis-induced ALI group
We also determined if treatment with acacetin inhibited sepsis-induced NF-κB activation by inhibiting IκB-α phosphorylation or degradation (Fig. 7b). Acacetin treatment significantly inhibited IκB-α phosphorylation when compared to the non-treated sepsis group mice. These results suggest that the anti-inflammatory activity of acacetin might also be related to the NF-κB pathway inhibition in our in vivo model of ALI.
The effect of acacetin on the NF-κB p65 DNA-binding activity
The NF-κB signaling pathway is essential for the regulation of sepsis-induced inflammation and injury (Z. Liao et al. 2012). To further assess the effect of acacetin on NF-κB activation in vitro, we used RAW264.7 cells to investigate the effect of acacetin on the NF-κB p65 DNA-binding activity.Cell viability was evaluated using the MTT assay (Fig. 8a). Acacetin was found to be non-toxic at concentrations up to 100 μg (Fig. 8a). Acacetin treatment significantly inhibited the LPS-induced DNA-binding activity of NF-κBp65. These data suggest that acacetin might suppress LPS-induced inflammatory damage to lung tissues by inhibiting the DNA-binding activity of NF-κB p65.
Fig. 8.
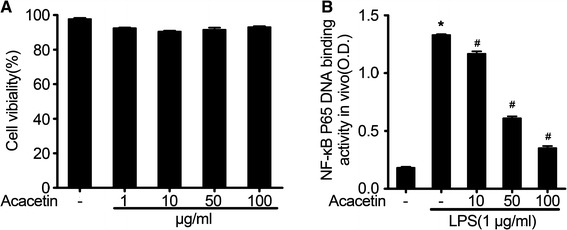
Acacetin effect on the DNA-binding activity of NF-κB in vitro. a The effect of acacetin on cell viability in lipopolysaccharide- (LPS) induced RAW264.7 cells. b The effect of acacetin on the DNA-binding activity of NF-κB in LPS-induced RAW264.7 cells. The values represent the mean ± SD. *P < 0.05, compared to the non-treated control group cells; #P < 0.05, compared to only LPS-stimulated group cells
Acacetin treatment improved the survival rate of sepsis mice
Survival rate is a key indicator of the protective effect of acacetin on sepsis-induced ALI in mice. To further examine the protective effects of acacetin, we examined the survival rate in different treated septic mice according to a previous report (Cheng et al. 2007). As shown in Fig. 9, we observed that acacetin treatment significantly improved the survival rate of sepsis-induced ALI mice when compared to untreated mice with sepsis (Fig. 9).
Fig. 9.
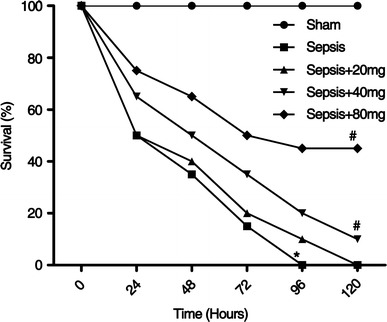
Astilbin effect on sepsis-induced mortality in mice. Sepsis was induced by CLP surgery in mice with or without different dosages of acacetin treatment (20, 40 and 80 mg/kg) respectively. Survival rates were determined at 0, 24, 48, 72, 96, and 120 h. *P < 0.05 compared with sham group mice; #P < 0.05 compared with CLP-induced sepsis group mice
Discussion
Our study showed that acacetin treatment significantly inhibited sepsis-induced ALI, and reduced iNOS and COX-2 expression. Furthermore, our results indicated that the protective effect of acacetin against sepsis-induced ALI was primarily mediated by its antioxidative and anti-inflammatory activity, which could enhance the expression of certain key AOEs and inactivate the NF-κB signaling pathway in our in vivo model.
The breakdown of the alveolar–capillary barrier causes obstruction of the pulmonary gas exchange in ALI. Neutrophil activation, which increases sepsis-induced alveolar–capillary barrier permeability and results in the generation of reactive oxygen species (ROS), is one of the mechanisms underlying the barrier’s breakdown. In addition, it has been proposed that sepsis can induce pulmonary ROS production, and that ROS could increase local inflammation and thereby contribute to pulmonary tissue damage (Geerts et al. 2001). Although a moderate amount of ROS is required for the innate immune system to kill pathogens, excessive amounts of ROS can cause harmful effects, including pulmonary tissue damage, apoptosis, and necrosis (Grommes et al. 2011). Therefore, the injection of SOD, a specific superoxide radical scavenger, could protect mice from virus infection- or sepsis-induced ALI (Maeda et al. 1991). Additionally,microvascular endothelial injury could lead to higher capillary permeability. The increased capillary permeability enhances the entrance of protein-rich fluid into the peribrochovascular intersititium, leading to lung edema (Johnson et al. 2010; Antoine Roch et al. 2011). We demonstrated that acacetin treatment significantly alleviated sepsis and the concomitant pathological changes in the sepsis-induced ALI mouse model (Fig. 1), the wet-to-dry weight ratio (Fig. 2a), protein concentration in the BALF (Fig. 2b), MPO activity (Fig. 3a), inflammatory cell infiltration (Fig. 3b), and pro-inflammatory cytokine production (Fig. 4) in the lungs of ALI mice. These results strongly indicate that acacetin might have a potential protective effect in sepsis-induced ALI.
Previous studies have reported that excessive and uncontrolled oxidative stress also plays an important role in sepsis-induced ALI (Shah et al. 2011). Partly because of its antioxidative activity, acacetin might attenuate the severity of sepsis-induced ALI given that acacetin decreased the expression of iNOS (Fig. 5a) and COX-2 (Fig. 5b). Furthermore it increased the expression of HO-1 (Fig. 6b) and that of the AOEs catalase, MnSOD, CuZnSOD, and GPx-1 (Fig. 6a). The expression of these genes might decrease the oxidative stress-induced pulmonary damage.
Inhibition of pulmonary inflammation may be critical for treating sepsis-induced pulmonary injury. Pulmonary inflammatory responses and edema are positively correlated with pulmonary function, including airway pressure and the oxygenation index. Additionally, TNF-α, IL-1β, IL-6, and MIP-2 are potent pro-inflammatory cytokines that play a role in the initiation and amplification of inflammatory responses during ALI (Cannizzaro et al. 2011). As shown in the present study, inhibiting the overproduction of these pro-inflammatory cytokines decreases pulmonary injury in the sepsis-induced ALI mouse model. Moreover, NF-κB is an important transcription factor that mediates the secretion of pro-inflammatory cytokines, such as TNF-α and IL-6, by inflammatory cells. Intranuclear blockage of NF-κB has been shown to suppress the expression of iNOS (Hatano et al. 2001) and COX-2 (Ke et al. 2007). Therefore, several current anti-inflammatory therapies target NF-κB activity. In our study, we demonstrated that acacetin treatment markedly blocked LPS-stimulated NF-κB activation in vivo and in vitro (Figs. 7 and 8).
In summary, we provide evidence that acacetin may have a protective effect on sepsis-induced ALI by reducing oxidative stress and inflammatory responses in the lung tissue. The protective effect of acacetin may be due to o NF-κB pathway inhibition and the increase of antioxidant-related gene expression. Although the exact mechanism underlying the protective role of acacetin against sepsis-induced ALI needs further investigation, our results suggest that acacetin may be a potent protective agent for pulmonary injury.
Acknowledgements
The study was supported by China-Japan Friendship Hospital project (No. 2015-1-QN-22), National Natural Science Foundation of China (No.81601725) and National key clinical key construction project.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31:S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- Bhaskaran N, Srivastava JK, Shukla S, Gupta S. Chamomile confers protection against hydrogen peroxide-induced toxicity through activation of Nrf2-mediated defense response. Phytother Res. 2013;27:118–125. doi: 10.1002/ptr.4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- Cannizzaro V, Hantos Z, Sly PD, Zosky GR. Linking lung function and inflammatory responses in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2011;300:L112–L120. doi: 10.1152/ajplung.00158.2010. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Sarkar S, Saha SK. Acacetin 7-O-β-d-galactopyranoside from Chrysanthemum indicum. Phytochemistry. 1981;20:1760–1761. doi: 10.1016/S0031-9422(00)98580-7. [DOI] [Google Scholar]
- Cheng PY, Lee YM, Wu YS, Chang TW, Jin JS, Yen MH. Protective effect of baicalein against endotoxic shock in rats in vivo and in vitro. Biochem Pharmacol. 2007;73:793–804. doi: 10.1016/j.bcp.2006.11.025. [DOI] [PubMed] [Google Scholar]
- Cribbs SK, Matthay MA, Martin GS. Stem cells in sepsis and acute lung injury. Crit Care Med. 2010;38:2379–2385. doi: 10.1097/CCM.0b013e3181f96f5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzGerald GA. COX-2 and beyond: approaches to prostaglandin inhibition in human disease. Nat Rev Drug Discov. 2003;2:879–890. doi: 10.1038/nrd1225. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol. 2005;174:5033–5039. doi: 10.4049/jimmunol.174.8.5033. [DOI] [PubMed] [Google Scholar]
- Geerts L, Jorens PG, Willems J, De Ley M, Slegers H. Natural inhibitors of neutrophil function in acute respiratory distress syndrome. Crit Care Med. 2001;29:1920–1924. doi: 10.1097/00003246-200110000-00012. [DOI] [PubMed] [Google Scholar]
- Gong Q, Yin H, Fang M, Xiang Y, Yuan CL, Zheng GY, Zheng F. Heme oxygenase-1 upregulation significantly inhibits TNF-alpha and Hmgb1 releasing and attenuates lipopolysaccharide-induced acute lung injury in mice. Int Immunopharmacol. 2008;8:792–798. doi: 10.1016/j.intimp.2008.01.026. [DOI] [PubMed] [Google Scholar]
- Goodman RB, Pugin J, Lee JS, Matthay MA. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003;14:523–535. doi: 10.1016/S1359-6101(03)00059-5. [DOI] [PubMed] [Google Scholar]
- Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza M, Wang XM, Wu T, Brahim JS, Rowan JS, Dionne RA. Nitric oxide is negatively correlated to pain during acute inflammation. Mol Pain. 2010;6:55. doi: 10.1186/1744-8069-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Medzhitov R. Innate sensors of microbial infection. J Clin Immunol. 2005;25:503–510. doi: 10.1007/s10875-005-8065-4. [DOI] [PubMed] [Google Scholar]
- Hatano E, Bennett BL, Manning AM, Qian T, Lemasters JJ, Brenner DA. NF-kappaB stimulates inducible nitric oxide synthase to protect mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis. Gastroenterology. 2001;120:1251–1262. doi: 10.1053/gast.2001.23239. [DOI] [PubMed] [Google Scholar]
- Hossain M, Qadri SM, Liu L. Inhibition of nitric oxide synthesis enhances leukocyte rolling and adhesion in human microvasculature. J Inflamm (Lond) 2012;9:28. doi: 10.1186/1476-9255-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZF, Massey JB, Via DP. Differential regulation of cyclooxygenase-2 (COX-2) mRNA stability by interleukin-1 beta (IL-1 beta) and tumor necrosis factor-alpha (TNF-alpha) in human in vitro differentiated macrophages. Biochem Pharmacol. 2000;59:187–194. doi: 10.1016/S0006-2952(99)00312-3. [DOI] [PubMed] [Google Scholar]
- Jinzhou Z, Tao H, Wensheng C, Wen W, Jincheng L, Qin C, Dinghua Y. Cyclooxygenase-2 suppresses polymorphonuclear neutrophil apoptosis after acute lung injury. J Trauma. 2008;64:1055–1060. doi: 10.1097/TA.0b013e318047c07c. [DOI] [PubMed] [Google Scholar]
- Johnson Elizabeth R, Matthay Michael A. Acute Lung Injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv. 2010;23:243–252. doi: 10.1089/jamp.2009.0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SK, Kim JE, Lee SY, Lee MH, Byun S, Kim YA, Lim TG, Reddy K, Huang Z, Bode AM, Lee HJ, Lee KW, Dong Z. The P110 subunit of PI3-K is a therapeutic target of acacetin in skin cancer. Carcinogenesis. 2014;35:123–130. doi: 10.1093/carcin/bgt266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke J, Long X, Liu Y, Zhang YF, Li J, Fang W, Meng QG. Role of NF-kappaB in TNF-alpha-induced COX-2 expression in synovial fibroblasts from human TMJ. J Dent Res. 2007;86:363–367. doi: 10.1177/154405910708600412. [DOI] [PubMed] [Google Scholar]
- Kim HR, Park CG, Jung JY. Acacetin (5,7-dihydroxy-4′-methoxyflavone) exhibits in vitro and in vivo anticancer activity through the suppression of NF-κB/Akt signaling in prostate cancer cells. Int J Mol Med. 2014;33:317–324. doi: 10.3892/ijmm.2013.1571. [DOI] [PubMed] [Google Scholar]
- Kraft C, Jenett-Siems K, Siems K, Jakupovic J, Mavi S, Bienzle U, Eich E. In vitro antiplasmodial evaluation of medicinal plants from Zimbabwe. Phytother Res. 2003;17:123–128. doi: 10.1002/ptr.1066. [DOI] [PubMed] [Google Scholar]
- Kretz-Remy C, Arrigo AP. Selenium: a key element that controls NF-kappa B activation and I kappa B alpha half life. BioFactors. 2001;14:117–125. doi: 10.1002/biof.5520140116. [DOI] [PubMed] [Google Scholar]
- Kung CW, Lee YM, Cheng PY, Peng YJ, Yen MH. Ethyl pyruvate reduces acute lung injury via regulation of iNOS and HO-1 expression in endotoxemic rats. J Surg Res. 2011;167:e323–e331. doi: 10.1016/j.jss.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Kuo MY, Liao MF, Chen FL, Li YC, Yang ML, Lin RH, Kuan YH. Luteolin attenuates the pulmonary inflammatory response involves abilities of antioxidation and inhibition of MAPK and NFkappaB pathways in mice with endotoxin-induced acute lung injury. Food Chem Toxicol. 2011;49:2660–2666. doi: 10.1016/j.fct.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Lavnikova N, Prokhorova S, Helyar L, Laskin DL. Isolation and partial characterization of subpopulations of alveolar macrophages, granulocytes, and highly enriched interstitial macrophages from rat lung. Am J Respir Cell Mol Biol. 1993;8:384–392. doi: 10.1165/ajrcmb/8.4.384. [DOI] [PubMed] [Google Scholar]
- Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Ramsay G. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–1256. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- Liao YH, Houghton PJ, Hoult JR. Novel and known constituents from Buddleja species and their activity against leukocyte eicosanoid generation. J Nat Prod. 1999;62:1241–1245. doi: 10.1021/np990092+. [DOI] [PubMed] [Google Scholar]
- Liao Z, Dong J, Wu W, Yang T, Wang T, Guo L, Wen F. Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARgamma/NF-kappaB pathway. Respir Res. 2012;13:110. doi: 10.1186/1465-9921-13-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H, Akaike T. Oxygen free radicals as pathogenic molecules in viral diseases. Proc Soc Exp Biol Med. 1991;198:721–727. doi: 10.3181/00379727-198-43309C. [DOI] [PubMed] [Google Scholar]
- Mann JR, Backlund MG, DuBois RN. Mechanisms of disease: inflammatory mediators and cancer prevention. Nat Clin Pract Oncol. 2005;2:202–210. doi: 10.1038/ncponc0140. [DOI] [PubMed] [Google Scholar]
- Marin PD, Grayer RJ, Veitch NC, Kite GC, Harborne JB. Acacetin glycosides as taxonomic markers in Calamintha and Micromeria. Phytochemistry. 2001;58:943–947. doi: 10.1016/S0031-9422(01)00352-1. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe AJ, Dowhy M, Holm BA, Glick PL. Myeloperoxidase activity as a lung injury marker in the lamb model of congenital diaphragmatic hernia. J Pediatr Surg. 2001;36:334–337. doi: 10.1053/jpsu.2001.20709. [DOI] [PubMed] [Google Scholar]
- Nishina K, Mikawa K, Takao Y, Maekawa N, Shiga M, Obara H. ONO-5046, an elastase inhibitor, attenuates endotoxin-induced acute lung injury in rabbits. Anesth Analg. 1997;84:1097–1103. doi: 10.1213/00000539-199705000-00026. [DOI] [PubMed] [Google Scholar]
- Pan MH, Lai CS, Wang YJ, Ho CT. Acacetin suppressed LPS-induced up-expression of iNOS and COX-2 in murine macrophages and TPA-induced tumor promotion in mice. Biochem Pharmacol. 2006;72:1293–1303. doi: 10.1016/j.bcp.2006.07.039. [DOI] [PubMed] [Google Scholar]
- Parratt JR. Nitric oxide. A key mediator in sepsis and endotoxaemia? J Physiol Pharmacol. 1997;48:493–506. [PubMed] [Google Scholar]
- Peng X, Abdulnour RE, Sammani S, Ma SF, Han EJ, Hasan EJ, Hassoun PM. Inducible nitric oxide synthase contributes to ventilator-induced lung injury. Am J Respir Crit Care Med. 2005;172:470–479. doi: 10.1164/rccm.200411-1547OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roch Antoine, Guervilly Christophe, Papazian Laurent. Fluid management in acute lung injury and ards. Ann Intensive Care. 2011;1:16. doi: 10.1186/2110-5820-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JS, Han JY, Kim JH, Hwang JK. Inhibitory effects of active compounds isolated from safflower (Carthamus tinctorius L.) seeds for melanogenesis. Biol Pharm Bull. 2004;27:1976–1978. doi: 10.1248/bpb.27.1976. [DOI] [PubMed] [Google Scholar]
- Serou MJ, DeCoster MA, Bazan NG. Interleukin-1 beta activates expression of cyclooxygenase-2 and inducible nitric oxide synthase in primary hippocampal neuronal culture: platelet-activating factor as a preferential mediator of cyclooxygenase-2 expression. J Neurosci Res. 1999;58:593–598. doi: 10.1002/(SICI)1097-4547(19991115)58:4<593::AID-JNR12>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Shah NR, Iqbal MB, Barlow A, Bayliss J. Severe physical exertion, oxidative stress, and acute lung injury. Clin J Sport Med. 2011;21(6):537–538. doi: 10.1097/JSM.0b013e318235151e. [DOI] [PubMed] [Google Scholar]
- Sharma JN, Al-Omran A, Parvathy SS. Role of nitric oxide in inflammatory diseases. Inflammopharmacology. 2007;15:252–259. doi: 10.1007/s10787-007-0013-x. [DOI] [PubMed] [Google Scholar]
- Singh RP, Agrawal P, Yim D, Agarwal C, Agarwal R. Acacetin inhibits cell growth and cell cycle progression, and induces apoptosis in human prostate cancer cells: structure-activity relationship with linarin and linarin acetate. Carcinogenesis. 2005;26:845–854. doi: 10.1093/carcin/bgi014. [DOI] [PubMed] [Google Scholar]
- Smirnova LP, KI B, Ban’kovskii AI. Acacetin and its glycosides in plants of the genus Linaria. Chem Nat Comp. 1974;10:96–97. doi: 10.1007/BF00568246. [DOI] [Google Scholar]
- Speyer CL, Neff TA, Warner RL, Guo RF, Sarma JV, Riedemann NC, Ward PA. Regulatory effects of iNOS on acute lung inflammatory responses in mice. Am J Pathol. 2003;163:2319–2328. doi: 10.1016/S0002-9440(10)63588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh YJ, Chun KS, Cha HH, Han SS, Keum YS, Park KK, Lee SS. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat Res. 2001;480–481:243–268. doi: 10.1016/S0027-5107(01)00183-X. [DOI] [PubMed] [Google Scholar]
- Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Kang TB, Kovalenko A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat Rev Immunol. 2014;14:51–59. doi: 10.1038/nri3561. [DOI] [PubMed] [Google Scholar]
- Weber GF, Swirski FK. Immunopathogenesis of abdominal sepsis. Langenbecks Arch Surg. 2014;399:1–9. doi: 10.1007/s00423-013-1129-7. [DOI] [PubMed] [Google Scholar]
- Wilgus TA, Ross MS, Parrett ML, Oberyszyn TM. Topical application of a selective cyclooxygenase inhibitor suppresses UVB mediated cutaneous inflammation. Prostaglandins Other Lipid Mediat. 2000;62:367–384. doi: 10.1016/S0090-6980(00)00089-7. [DOI] [PubMed] [Google Scholar]
- Zeng Wenjun, Zhang Chunyun, Cheng Hongwei, Yun-Long Wu, Liu Jie, Chen Zekun, Huang Jian-gang, Ericksen Russell Erick, Chen Liqun, Zhang Haiping, Wong Alice Sze Tsai, Zhang Xiao-kun, Han Weiping, Zeng Jin-Zhang. Targeting to the non-genomic activity of retinoic acid receptor-gamma by acacetin in hepatocellular carcinoma. Sci Rep. 2017;7:348. doi: 10.1038/s41598-017-00233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LN, Zheng JJ, Zhang L, Gong X, Huang H, Wang CD, Wan JY. Protective effects of asiaticoside on septic lung injury in mice. Exp Toxicol Pathol. 2011;63:519–525. doi: 10.1016/j.etp.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Zhou X, Dai Q, Huang X. Neutrophils in acute lung injury. Front Biosci (Landmark Ed) 2012;17:2278–2283. doi: 10.2741/4051. [DOI] [PubMed] [Google Scholar]