

Abstract
Progressive hepatic fibrosis is the final common pathway for most chronic liver injuries, leading to cirrhosis with risk of liver failure and hepatocellular carcinoma. It is now recognized that fibrosis is a dynamic process, and may be reversible prior to the establishment of advanced architectural changes to the liver. The most effective antifibrotic strategy is to cure the underlying disease process before advanced fibrosis has developed. Unfortunately, this is often not possible, and specific antifibrotic therapies are needed. Advances in the understanding of the pathogenesis of liver fibrosis have identified several potential novel therapeutic targets, but unfortunately clinical development has been disappointing. One major limitation has been the often prolonged natural history of fibrosis compared to experimental models, and difficulties in accurate noninvasive fibrosis assessment, thus making clinical trial design difficult. In this review, we highlight the most promising current antifibrotic strategies.
Keywords: Liver, Fibrosis, Hepatic stellate cell, Antifibrotic, Therapy
Introduction
In the United States, more than 20,000 deaths per year are attributed to cirrhosis and its complications [1]. A further 16,000 patients are currently on the waiting list for a liver transplant [1]. End-stage liver disease from chronic hepatitis C (CHC) infection will continue to pose a significant health and economic burden over the next decade. Despite significant advances in antiviral therapies, many CHC patients are ineligible or nonresponders to current treatment. The development of effective treatment to slow progression of hepatic fibrosis is therefore a healthcare priority.
At present, the most effective therapies for fibrosis are disease-specific and aim to eliminate the inciting agent. As yet, fibrosis-specific therapies are not available and clinical development has proven challenging. One of the great advances in our understanding of liver disease over the past decade has been that fibrogenesis is a dynamic process that may be reversible. Progress in our understanding of the molecular pathogenesis of hepatic fibrosis has made it increasingly possible to target key molecules or pathways involved in fibrogenesis. It is hoped that this approach will lead to therapeutic breakthroughs in the near future. In this review, we discuss the antifibrotic strategies that have been studied in clinical trials, as well as the most promising of the preclinical candidates, in the context of current understanding of the mechanisms of hepatic fibrogenesis.
Hepatic Fibrogenesis
Fibrogenesis
Fibrosis is a dynamic process that represents the wound healing response to injury, and is dependent on a balance between fibrogenesis and fibrolysis. Fibrogenesis is a chronic, typically indolent process, characterized by a complex array of biologic processes that lead to the increased production of extracellular matrix (ECM), resulting in deposition of fibrous scar tissue and of the potential to develop cirrhosis. The key effector cell type involved in this process is the hepatic stellate cell (HSC).
Liver injury triggers the activation of quiescent HSCs via initiating and subsequent perpetuating signals (Fig. 1). Initiating signals include oxidative stress signals [2], apoptotic bodies [3], TLR4 ligands (lipopolysaccharide) [4•] and paracrine stimuli from neighboring Kupffer cells, sinusoidal epithelial cells and hepatocytes, driven by the ongoing liver insult. Subsequent autocrine and paracrine perpetuating signals maintain and transdifferentiate the activated HSC into a contractile myofibroblast. This process involves the increased secretion of soluble mediators and their receptors, including cytokines, chemokines and growth factors. Key mediators include transforming growth factor-β (TGF-β) [5, 6], platelet-derived growth factor (PDGF) [7], angiotensin II [8, 9], leptin [10], and signaling via the cannabinoid receptor-1 (CB1R) [11]. Recently, expression of αvβ6 integrin on activated cholangiocytes has been recognized to promote activation of latent TGF-β1 and be a potent fibrogenic stimulus in models of cholestatic liver disease [12]. As a result, the myofibroblast remains in a state of enhanced proliferation, contractility, chemotaxis, and fibrogenesis. Fibrogenesis has two key features. The excess secretion and deposition of extracellular matrix proteins, including type I and III collagen (vs type IV), is compounded by an imbalance between increased myofibroblast secretion of tissue inhibitors of metalloproteinases (primarily TIMP-1/2) and decreased production of the fibrinolytic matrix metalloproteinases (MMP-2/3/9/1) [2].
Fig. 1.
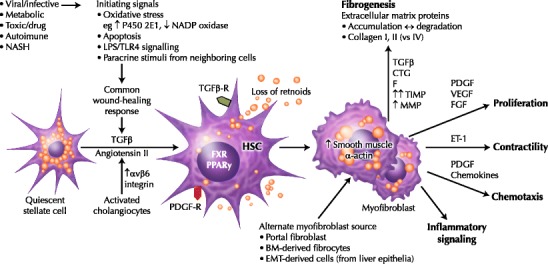
Hepatic fibrogenesis. Hepatic injury of many causes leads to a common wound healing response, which induces activation of quiescent hepatic stellate cells (HSCs). Activated HSCs are characterized by a loss of intracellular retinoids, increased proliferation, changes in cellular morphology, and increased synthesis and secretion of cytokines and chemokines, transforming into the contractile myofibroblast cell. The most striking biologic consequence of HSC activation is increased extracellular matrix protein secretion and deposition. Over time, other cell lineages may contribute to the fibrogenic cell population, including portal fibroblasts, bone marrow (BM)-derived fibrocytes, and liver epithelial cells (via epithelial-mesenchymal transition, EMT). CTGF—connective tissue growth factor; ET-1—endothelin-1; FGF—fibroblast growth factor; LPS—lipopolysaccharide (endotoxin); MMP—matrix metalloproteinases; NASH—non-alcoholic steatohepatitis; PDGF—platelet-derived growth factor; PPAR—peroxisome proliferator-activated receptor; TGF—transforming growth factor; TIMP—tissue inhibitors of metalloproteinases; TLR—Toll-like receptor; VEGF—vascular endothelial growth factor
Counter-regulatory mechanisms that have been identified in vitro to down-regulate HSC activation and decrease fibrogenesis in cell culture models include the cytokines interferon-γ (IFN-γ) [13] and adiponectin [14, 15], the nuclear receptors peroxisome proliferators activated receptor (PPAR) γ nuclear receptor [16, 17] and farnesoid X receptor (FXR) [18], and cannabinoid receptor-2 (CB2R) signaling [11].
Over time, other cell populations may contribute to myofibroblast proliferation (Fig. 1), although it is likely that HSCs remain the dominant source. These include portal fibroblasts, bone-marrow derived fibrocytes and liver epithelial cells (including hepatocytes, cholangiocytes, via epithelial-mesenchymal transition [EMT]). The extent to which these alternate cell types contribute to fibrogenesis and the timing of their recruitment may vary by disease etiology. A key role for hedgehog-pathway mediated EMT of ductular-type liver progenitor cells in the pathogenesis of nonalcoholic fatty liver disease (NASH)-cirrhosis was recently identified [19].
Regression of Fibrosis
The dynamic nature of hepatic injury is reflected by reversibility in even advanced cases of fibrosis [20]. Reversion of HSC activation appears to play an important role in this regard. Less is known about this process. Apoptosis of HSCs may account for the decrease in activated stellate cells during resolution of hepatic fibrosis. Successful fibrosis reversion must also include restoration of liver architecture through matrix degradation, requiring dominant expression and secretion of the MMPs over the TIMPs [2].
Antifibrotic Therapies
Antifibrotic treatment strategies can be considered as either disease-specific or fibrosis-specific. The best antifibrotic therapy is to remove the underlying source of liver injury. It is now well-established that elimination of the fibrogenic stimulus can lead to regression of accumulated fibrosis, even in the setting of early cirrhosis. Examples include sustained virologic clearance in CHC infection [21, 22], durable viral suppression in patients with chronic hepatitis B [23–25], venesection for hemochromatosis [26, 27], chelation for Wilson’s disease, immunosuppression for auto-immune hepatitis [28, 29] and weight loss for nonalcoholic steatohepatitis [30, 31].
Unfortunately, curative therapy is not possible in many patients either because disease-specific therapy may not be available, available disease-specific therapies may have failed, or due to late presentation with established cirrhosis. These patients have limited therapeutic options to reduce risk of developing complications from end-stage liver disease. Fortunately, a number of promising targeted approaches are in development (Fig. 2).
Fig. 2.

Current status of targeted antifibrotic therapies in preclinical or clinical development. CB1R—cannabinoid receptor-1; CB2R—cannabinoid receptor-2; FXR—farnesoid-X receptor; IFN—interferon; pegIFN—pegylated interferon-α; PPAR—peroxisome proliferator-activated receptor; TGF—transforming growth factor; TIMP—tissue inhibitors of metalloproteinases; X—clinical development halted
Interferon-γ
IFN-γ is a potent inhibitor of TGF-β signaling in vitro, and has been shown to inhibit fibrogenesis and reduce the extent of histologic fibrosis in small animal models. A number of studies have been performed in patients with chronic viral hepatitis.
IFN-γ was first used in the early 1990s, when it was compared to IFN-α for antiviral effect in a small cohort of patients with CHC infection. Although a negative study, a trend towards reduction in fibrosis stage was noted in the IFN-γ arm [32]. A subsequent pilot study focused specifically on the antifibrotic effect of IFN-γ in patients with CHC infection. Although there was no overall reduction in fibrosis stage, six patients (30%) had more than 1% absolute reduction in morphometric fibrosis score, and four patients (20%) displayed improvement in Ishak fibrosis score [33]. Another small study suggested antifibrotic benefit for IFN-γ in the setting of chronic hepatitis B infection [34]. These data led to the performance of a large, double-blind, placebo-controlled multicenter trial investigating the antifibrotic efficacy of 48 weeks of therapy with IFN-γ [35•]. In this trial, 502 patients with CHC were randomly assigned to IFN-γ 100 μg, IFN-γ 200 μg, or placebo, 3 times weekly. Pre- and post-treatment biopsies were available from 420 patients. Unfortunately no improvement in Ishak score was observed between the 3 treatment groups (12.1% vs 12.4% vs 16% of patients with reduction ≥1 stage on the Ishak fibrosis score, P-value > 0.05). It is important to note that most patients enrolled in this study (84%) were cirrhotic at baseline, representing a difficult-to-treat group. Subgroup analysis suggested that patients with ≥60% induction of serum interferon-inducible T cell-α (ITAC) by 24 weeks’ therapy had a significantly better liver histology outcome, possibly identifying an IFN-γ responsive population. However, for now, clinical development of IFN-γ as an antifibrotic agent has been halted.
Maintenance-Dose Pegylated Interferon-α for CHC
Three large, randomized, controlled trials have studied the use of long-term, low-dose pegylated interferon (pegIFN) therapy for the prevention of disease progression in CHC patients with moderate-to-advanced fibrotic liver disease, most of whom were prior nonresponders to IFN-α-based regimens (Hepatitis C Antiviral Long-term Treatment Against Cirrhosis [HALT-C] [36•], Evaluation of Peg-Intron in Control of Hepatitis C Cirrhosis [EPIC-3] [37•], and Colchicine versus Peg-Intron Long-Term [COPILOT] [38•]). These large clinical trials recruited several hundred patients, and were well-designed with long-term follow-up (3.5–5 years) to evaluate important clinical outcomes (portal hypertensive complications, hepatocellular carcinoma, death, or transplantation) with or without histology as endpoints. Disappointingly, low-dose pegIFN therapy was ineffective and often poorly tolerated, and none of the studies showed overall clinical or histologic benefit. EPIC-3 and COPILOT did suggest a possible benefit from maintenance pegIFN in patients with baseline portal hypertension, although morbidity in this group was high and the results were not conclusive. In summary, there appears to be no place for low-dose, maintenance, antiviral therapy for CHC-related hepatic fibrosis. This situation may change with the clinical development of direct antivirals, if potent and durable viral suppression can be achieved in the absence of viral resistance.
Peroxisome Proliferator-Activated Receptor γ Ligands
PPAR-γ is a nuclear receptor that appears to play an important role in fibrotic liver injury. Activated HSCs are characterized by low level expression of PPAR-γ, and PPAR-γ ligands have been shown to reverse the activated phenotype of HSCs and improve fibrosis in experimental models [39]. Several PPAR-γ ligands, including rosiglitazone and pioglitazone, have been investigated for antifibrotic effect in patients with NASH. Because of their insulin-sensitizing effect, these compounds are particularly attractive in a disease state characterized by the metabolic syndrome. Larger studies are currently underway (see www.clinicaltrials.gov, NCT00063622, NCT00227110, NCT00699036).
A large, phase 2 study using farglitazar in 265 CHC patients with Ishak fibrosis stages 2 to 4 was recently completed [40]. Farglitazar is a third PPAR-γ agonist, shown to inhibit stellate cell activation in vitro. Patients were randomly assigned to 0.5 or 1.0 mg of farglitazar, or placebo, once daily, for 52 weeks. The primary endpoint was histologic assessment of paired pretreatment and post-treatment liver biopsies, by morphometric assessment of α-smooth muscle actin (α-SMA) and collagen, as well as ranked assessment of inflammation and fibrosis using the Ishak and METAVIR scoring systems. In these patients with moderate fibrosis at entry, no effect on α-SMA, collagen, or ranked assessment of the paired biopsies was seen. Clinical development has been halted.
Angiotensin II Antagonists
The renin-angiotensin system is recognized as an important stimulus for the development of hepatic fibrosis, as well as portal hypertension. Therefore, use of angiotensin II antagonists potentially offers dual clinical benefit. The key effector is downstream angiotensin II. Angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type-1 receptor antagonists (AIIRA) inhibit liver fibrosis in small-animal models. In addition, they are already widely used for cardiovascular and renal indications, where antifibrotic effects have been observed, and safety data are well-established, thus making them attractive candidates. A recently published pilot study evaluated the antifibrotic benefits of the AIIRA losartan in hepatitis C virus (HCV)-related hepatic fibrosis [41]. Fourteen patients were treated with losartan for 18 months and had paired liver biopsy assessment. Losartan therapy was associated with decreased hepatic expression of profibrogenic genes (including procollagen α1(I) and α1(IV), urokinase-type plasminogen activator, metalloproteinase type-2), as well as NADPH oxidase components, a key mediator of angiotensin II-induced oxidative stress. METAVIR fibrosis stage was observed to decrease in seven patients, although no significant change occurred in fibrosis overall. Phase 2/3 clinical trials investigating candesartan and irbesartan as antifibrotic agents in the setting of HCV infection are in progress (www.clinicaltrials.gov, NCT00930995 and NCT00265642, respectively);
Angiotensin 1–7 (Ang-[1–7]) is a peptide product of the recently described ACE homologue, ACE2, and signals via the mas receptor. Ang-(1–7) has been reported to be upregulated in human liver disease, and to have antifibrotic actions in a rat model of cirrhosis. Therefore, the ACE2/Ang-(1–7)/mas axis represents a potential target for antifibrotic therapy in humans.
Caspase Inhibitors
Apoptosis is a driving force for the initiation and perpetuation of HSC activation and fibrogenesis, and may be particularly relevant to patients with chronic hepatic inflammation from viral hepatitis. At the cellular level, the caspase family of cysteine proteases is the key inducer and effector of apoptotic cell death, and has an important role in HCV-related liver injury. Caspase inhibitors have entered early-phase human trials for the amelioration of inflammation and prevention of fibrosis in the setting of chronic hepatitis C. The first agent to enter human studies was IDN-6556 (PF-03491390). For this agent, 105 patients were enrolled in a phase 2, placebo-controlled, dose-ranging study of 14 days’ duration [42]. In this study, 80 patients had CHC, and 25 had other chronic liver diseases including chronic hepatitis B (CHB), NASH, primary biliary cirrhosis, and primary sclerosing cholangitis. In patients with CHC, significant reductions of serum aminotransaminases were observed at all except the lowest dose. Similar responses were observed in patients with CHB and NASH. No antiviral effect was noted and adverse events were similar to placebo. Hepatic fibrosis was not an appropriate endpoint for this 14-day study. Longer studies were planned, but development has since been halted. GS-9450 is a second caspase inhibitor currently in a phase 2 program. The primary endpoint is hepatic inflammation, but morphometric quantitation of hepatic collagen staining will be examined as a secondary endpoint (www.clinicaltrials.gov, NCT00874796). A third caspase inhibitor, VX-166, was shown to reduce hepatic fibrosis in an animal model of NASH [43]. One important concern with the use of caspase inhibitors is the risk of potentiating hepatocarcinogenesis, particularly if long-term therapy is required in patients with advanced fibrosis, itself a premalignant state.
Other Clinical Candidates
Activators of the FXR nuclear receptor were shown to have antifibrotic activity in rodent models of cirrhosis. A phase 2 study of INT-747, an agonist of the FXR nuclear receptor, in type 2 diabetics with NASH, was recently completed (www.clinicaltrials.gov, NCT 00501592). Although the primary objectives of this study were assessment of safety and tolerability, and effect on insulin resistance and markers of hepatic inflammation, INT-747 is a potential antifibrotic agent. Other agents for which an antifibrotic signal was observed include pentoxifylline and pirfenidone.
Preclinical Candidates
Multiple steps in the fibrogenic and fibrolytic pathways have been identified as possible therapeutic targets and investigated in experimental models. Notable findings reported recently include 1) the use of αvβ6 integrin inhibitors to retard fibrosis progression in animal models of biliary cirrhosis [44•]; 2) the identification of a key role for cannabinoid receptor signaling in fibrogenesis, with CB2R agonists significantly reducing hepatic collagen content in a rat model of cirrhosis [45] and the CB1R antagonist SR141716A also shown to be antifibrotic [46]; 3) the identification of stellate cell TLR4 signaling as a key profibrogenic modulator of TGF-β signaling, an effect driven by intestinal microflora-derived LPS stimulation, suggesting a protective role for molecular inhibition of TLR4 signaling and for modification of the intestinal microflora by antibiotics or probiotics [4]; and 4) the use of monoclonal antibody strategies to selectively target myofibroblasts [47].
Complementary Medical Strategies
Among the general population, interest in complementary or alternative medicine (CAM) is significant. Although convincing data for efficacy are lacking, CAM approaches are widely used and are generally thought to be safe. One of the interesting findings from the HALT-C study was that regular coffee intake (>3 cups/d) [48] was associated with lower rates of disease progression in CHC patients [47]. The active ingredient remains unclear; it is unlikely to be caffeine, because tea intake was not beneficial. Other potential antifibrotics include milk thistle (active compound = silymarin [silybinin-1/2]), TJ-9 (baicalein), TJ-135 (emodin), coptis (berberine), turmeric (curcumin), and red wine (trans-reservatrol). Many of these agents are believed to have antioxidant properties that may reduce inflammation.
Challenges for the Field
Thus far, the translation of promising preclinical candidates into effective clinical antifibrotic agents has been disappointing. Several possible explanations exist. The first may relate to limitations of the preclinical models for modeling complex human disease. It is particularly difficult to capture the complex interactions that occur between multiple cell populations in a cell culture model. Fibrogenic pathways in small animal models may not be relevant to human physiology. Beyond preclinical studies, clinical development also has significant challenges, not the least the indolent nature and often prolonged natural history of fibrosis progression, and perhaps regression. Several years of antifibrotic therapy may be required to determine clinical or histologic benefit, and these endpoints are difficult to capture within the time and economic constraints of most clinical trials. Further, accurate repeated measures of liver fibrosis besides liver biopsy are required. Current noninvasive tools (eg, serum biomarkers or transient elastography) are too insensitive for monitoring changes in fibrosis. These issues were recently reviewed in detail [49].
Conclusions
Despite great scientific advances providing novel insights into the molecular pathogenesis of hepatic fibrosis in recent years, effective antifibrotic therapies are not yet available. Although several emerging potential therapeutic targets exist, antifibrotic drug development is challenging, and significant hurdles need to be overcome at the preclinical and clinical stages. In the meantime, greater emphasis must be placed on the early identification of patients with potentially treatable chronic liver disease.
Acknowledgments
Disclosure
No potential conflicts of interest relevant to this article were reported.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
- 1.United Network for Organ Sharing: Online database system. Available at http://www.unos.org/data/. Accessed November 2009.
- 2.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaeschke H. Inflammation in response to hepatocellular apoptosis. Hepatology. 2002;35:964–966. doi: 10.1053/jhep.2002.0350964. [DOI] [PubMed] [Google Scholar]
- 4.Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 5.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura T, Sakata R, Ueno T, et al. Inhibition of transforming growth factor beta prevents progression of liver fibrosis and enhances hepatocyte regeneration in dimethylnitrosamine-treated rats. Hepatology. 2000;32:247–255. doi: 10.1053/jhep.2000.9109. [DOI] [PubMed] [Google Scholar]
- 7.Borkham-Kamphorst E, van Roeyen CR, Ostendorf T, et al. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J Hepatol. 2007;46:1064–1074. doi: 10.1016/j.jhep.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 8.Yokohama S, Yoneda M, Haneda M, et al. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004;40:1222–1225. doi: 10.1002/hep.20420. [DOI] [PubMed] [Google Scholar]
- 9.Yang L, Bataller R, Dulyx J, et al. Attenuated hepatic inflammation and fibrosis in angiotensin type 1a receptor deficient mice. J Hepatol. 2005;43:317–323. doi: 10.1016/j.jhep.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 10.Ikejima K, Takei Y, Honda H, et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122:1399–1410. doi: 10.1053/gast.2002.32995. [DOI] [PubMed] [Google Scholar]
- 11.Mallat A, Teixeira-Clerc F, Deveaux V, Lotersztajn S. Cannabinoid receptors as new targets of antifibrosing strategies during chronic liver diseases. Expert Opin Ther Targets. 2007;11:403–409. doi: 10.1517/14728222.11.3.403. [DOI] [PubMed] [Google Scholar]
- 12.Wang B, Dolinski BM, Kikuchi N, et al. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology. 2007;46:1404–1412. doi: 10.1002/hep.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397:710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- 14.Kamada Y, Tamura S, Kiso S, et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125:1796–1807. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 15.Adachi M, Brenner DA. High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology. 2008;47:677–685. doi: 10.1002/hep.21991. [DOI] [PubMed] [Google Scholar]
- 16.Marra F, Efsen E, Romanelli RG, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119:466–478. doi: 10.1053/gast.2000.9365. [DOI] [PubMed] [Google Scholar]
- 17.Galli A, Crabb DW, Ceni E, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–1940. doi: 10.1053/gast.2002.33666. [DOI] [PubMed] [Google Scholar]
- 18.Fiorucci S, Rizzo G, Antonelli E, et al. Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther. 2005;315:58–68. doi: 10.1124/jpet.105.085597. [DOI] [PubMed] [Google Scholar]
- 19.Syn WK, Jung Y, Omenetti A, et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology. 2009;137:1478–1488. doi: 10.1053/j.gastro.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poynard T, McHutchison J, Manns M, et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 2002;122:1303–1313. doi: 10.1053/gast.2002.33023. [DOI] [PubMed] [Google Scholar]
- 21.Bruno S, Stroffolini T, Colombo M, et al. Sustained virological response to interferon-alpha is associated with improved outcome in HCV-related cirrhosis: a retrospective study. Hepatology. 2007;45:579–587. doi: 10.1002/hep.21492. [DOI] [PubMed] [Google Scholar]
- 22.Veldt BJ, Heathcote EJ, Wedemeyer H, et al. Sustained virologic response and clinical outcomes in patients with chronic hepatitis C and advanced fibrosis. Ann Intern Med. 2007;147:677–684. doi: 10.7326/0003-4819-147-10-200711200-00003. [DOI] [PubMed] [Google Scholar]
- 23.Dienstag JL, Schiff ER, Wright TL, et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med. 1999;341:1256–1263. doi: 10.1056/NEJM199910213411702. [DOI] [PubMed] [Google Scholar]
- 24.Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology. 2006;131:1743–1751. doi: 10.1053/j.gastro.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 25.Dienstag JL, Goldin RD, Heathcote EJ, et al. Histological outcome during long-term lamivudine therapy. Gastroenterology. 2003;124:105–117. doi: 10.1053/gast.2003.50013. [DOI] [PubMed] [Google Scholar]
- 26.Falize L, Guillygomarc’h A, Perrin M, et al. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology. 2006;44:472–477. doi: 10.1002/hep.21260. [DOI] [PubMed] [Google Scholar]
- 27.Powell LW, Dixon JL, Ramm GA, et al. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch Intern Med. 2006;166:294–301. doi: 10.1001/archinte.166.3.294. [DOI] [PubMed] [Google Scholar]
- 28.Dufour JF, DeLellis R, Kaplan MM. Reversibility of hepatic fibrosis in autoimmune hepatitis. Ann Intern Med. 1997;127:981–985. doi: 10.7326/0003-4819-127-11-199712010-00006. [DOI] [PubMed] [Google Scholar]
- 29.Cotler SJ, Jakate S, Jensen DM. Resolution of cirrhosis in autoimmune hepatitis with corticosteroid therapy. J Clin Gastroenterol. 2001;32:428–430. doi: 10.1097/00004836-200105000-00014. [DOI] [PubMed] [Google Scholar]
- 30.Dixon JB, Bhathal PS, Hughes NR, O’Brien PE. Nonalcoholic fatty liver disease: Improvement in liver histological analysis with weight loss. Hepatology. 2004;39:1647–1654. doi: 10.1002/hep.20251. [DOI] [PubMed] [Google Scholar]
- 31.Mummadi RR, Kasturi KS, Chennareddygari S, Sood GK. Effect of bariatric surgery on nonalcoholic fatty liver disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2008;6:1396–1402. doi: 10.1016/j.cgh.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 32.Saez-Royuela F, Porres JC, Moreno A, et al. High doses of recombinant alpha-interferon or gamma-interferon for chronic hepatitis C: a randomized, controlled trial. Hepatology. 1991;13:327–331. doi: 10.1002/hep.1840130220. [DOI] [PubMed] [Google Scholar]
- 33.Muir AJ, Sylvestre PB, Rockey DC. Interferon gamma-1b for the treatment of fibrosis in chronic hepatitis C infection. J Viral Hepat. 2006;13:322–328. doi: 10.1111/j.1365-2893.2005.00689.x. [DOI] [PubMed] [Google Scholar]
- 34.Weng HL, Wang BE, Jia JD, et al. Effect of interferon-gamma on hepatic fibrosis in chronic hepatitis B virus infection: a randomized controlled study. Clin Gastroenterol Hepatol. 2005;3:819–828. doi: 10.1016/S1542-3565(05)00404-0. [DOI] [PubMed] [Google Scholar]
- 35.Pockros PJ, Jeffers L, Afdhal N, et al. Final results of a double-blind, placebo-controlled trial of the antifibrotic efficacy of interferon-gamma1b in chronic hepatitis C patients with advanced fibrosis or cirrhosis. Hepatology. 2007;45:569–578. doi: 10.1002/hep.21561. [DOI] [PubMed] [Google Scholar]
- 36.Di Bisceglie AM, Shiffman ML, Everson GT, et al. Prolonged therapy of advanced chronic hepatitis C with low-dose peginterferon. N Engl J Med. 2008;359:2429–2441. doi: 10.1056/NEJMoa0707615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poynard T, Colombo M, Bruix J, et al. Peginterferon alfa-2b and ribavirin: effective in patients with hepatitis C who failed interferon alfa/ribavirin therapy. Gastroenterology. 2009;136:1618–1628. doi: 10.1053/j.gastro.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 38.Afdhal N, Levine R, Brown RJ, et al. E. Colchicine vs peg-interferonalfa 2B long term therapy: results of the 4 year COPILOT trial. J Hepatol. 2008;48(Suppl 2):S4 (A3). [Google Scholar]
- 39.Yang L, Chan CC, Kwon OS, et al. Regulation of peroxisome proliferator-activated receptor-gamma in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G902–G911. doi: 10.1152/ajpgi.00124.2006. [DOI] [PubMed] [Google Scholar]
- 40.McHutchison J, Goodman Z, Makhlouf H, et al. Double-blind, randomized, placebo-controlled, multi-center, phase II dose-ranging study to assess the antifibrotic activity of farglitazar in chronic hepatitis C infection. Hepatology. 2008;48(4 Suppl):1139A. [Google Scholar]
- 41.Colmenero J, Bataller R, Sancho-Bru P, et al.: Effects of losartan on hepatic expression of non-phagocytic NADPH oxidase and fibrogenic genes in patients with chronic hepatitis C. Am J Physiol Gastrointest Liver Physiol 2009 (Epub ahead of print). [DOI] [PMC free article] [PubMed]
- 42.Pockros PJ, Schiff ER, Shiffman ML, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46:324–329. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 43.Witek RP, Stone WC, Karaca FG, et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology. 2009;50:1421–1430. doi: 10.1002/hep.23167. [DOI] [PubMed] [Google Scholar]
- 44.Patsenker E, Popov Y, Stickel F, et al. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology. 2008;135:660–670. doi: 10.1053/j.gastro.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munoz-Luque J, Ros J, Fernandez-Varo G, et al. Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J Pharmacol Exp Ther. 2008;324:475–483. doi: 10.1124/jpet.107.131896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teixeira-Clerc F, Julien B, Grenard P, et al. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med. 2006;12:671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 47.Douglass A, Wallace K, Parr R, et al. Antibody-targeted myofibroblast apoptosis reduces fibrosis during sustained liver injury. J Hepatol. 2008;49:88–98. doi: 10.1016/j.jhep.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 48.Freedman ND, Everhart JE, Lindsay KL, et al. Coffee intake is associated with lower rates of liver disease progression in chronic hepatitis C. Hepatology. 2009;50:1360–1369. doi: 10.1002/hep.23162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Popov Y, Schuppan D. Targeting liver fibrosis: Strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50:1294–1306. doi: 10.1002/hep.23123. [DOI] [PubMed] [Google Scholar]