

Abstract
A novel avian influenza A (H7N9) virus caused 5-10 % mild and 30.5 % fatal human infections as of December 10, 2015. In order to investigate the reason for the higher rate of fatal outcome of this infection, this study compared the molecular epidemiology and virology of avian influenza A (H7N9) viruses from mild (N = 14), severe (N = 50) and fatal (N = 35) cases, as well as from non-human hosts (N = 73). The epidemiological results showed that the average age of the people in the mild, severe and fatal groups was 27.6, 52 and 62 years old, respectively (p < 0.001). Males accounted for 42.9 % (6/14), 58.0 % (29/50), and 74.3 % (26/35) of cases in the mild, severe and fatal group respectively (p = 0.094). Median days from onset to start of antiviral treatment were 2, 5 and 7 days in the mild, severe and fatal group, respectively (p = 0.002). The median time from onset to discharge/death was 12, 40 and 19 days in the mild, severe and fatal group, respectively (p < 0.001). Analysis of whole genome sequences showed that PB2 (E627K), NA (R294K) and PA (V100A) mutations were markedly associated with an increased fatality rate, while HA (N276D) and PB2 (N559T) mutations were clearly related to mild cases. There were no differences in the genotypes, adaptation to mammalian hosts, and genetic identity between the three types of infection. In conclusion, advanced age and delayed confirmation of diagnosis and antiviral intervention were risk factors for death. Furthermore, PB2 (E627K), NA (R294K) and PA (V100A) mutations might contribute to a fatal outcome in human H7N9 infection.
Electronic supplementary material
The online version of this article (doi:10.1007/s00705-016-2781-3) contains supplementary material, which is available to authorized users.
Keywords: Avian Influenza, H7N9 Virus, Oseltamivir, Human Isolate, Fatal Group
Introduction
Since the identification of the first human infection with a novel avian influenza A (H7N9) virus in China in 2013 [6], 669 confirmed cases have been reported worldwide up to 03 June 2015, according to the China annonmanet for the legislated Infectious diseases (http://www.nhfpc.gov.cn/jkj/s3578/201509/a67694612ff243a6a0788ad6a949ade0.shtml). The majority of cases were identified in the south and east of China [1, 12]. Almost all infected individuals were hospitalised, and 36 % (158/444) died [12]. The fatality rate was much higher than that observed for seasonal influenza in the United States (∼0.04 %) [12] and a previous H7N7 outbreak, but it was lower than that of H5N1 (70.0 %) in China [2]. Numerous studies have indicated that the higher mortality rate is related to delays in diagnosis and antiviral intervention, as well as old age and chronic diseases [17, 19].
Accumulating evidence has indicated that the infection source of H7N9 was presumably poultry and related products; however, human-to-human transmission was implicated in 20.0 % of confirmed cases [3, 10, 21, 26, 36, 38]. Nonetheless, evidence of sustained human-to-human transmission is very limited. Gene sequencing, receptor-binding-site (RBS) binding assays, and a series of animal studies have revealed that the H7N9 virus has an increased capacity to bind to mammalian respiratory cells. For example, substitutions in the HA at amino acid residues Q226L and G186V, and mutations in PA, PB1 and PB2 (D701N and E627K) are associated with higher virus replication rates, severity of illness and transmissibility [4, 23, 39, 44]. In accordance with this, Wang et al. reported that PB2 E627K mutation slightly increased the case fatality rate [33]. Additional studies by Hai et al. and Sleeman et al. showed that that the emergence of the Arg292Lys mutation in the neuraminidase (NA) gene appears to be associated with failure of response to NA inhibitors and to adverse clinical outcomes [9, 29]. A further important study indicated that H7N9 and H9N2 viruses are co-circulating in some live-bird markets, which might alter host adaptation and virus pathogenicity [14]. This raises concerns regarding the potential for H7N9 to adapt and lead to an infection pandemic.
The present study was an epidemiological investigation of 99 avian influenza A (H7N9) cases that occurred from 2013 to 2015. We compared the epidemiological features and signature amino acids, evolution and reassortments of the H7N9 virus with full genome sequences from mild, severe and fatal cases. We attempted to find a potential association between H7N9 mutations and reassortments and clinical outcome. The results might help in improving understanding of the epidemiology of this H7N9 strain, thereby helping to decrease the disease burden and to address the H7N9 challenge.
Materials and methods
Ethics statement
The present study was approved by the Medical Ethical Committee and the National Health and Family Planning Commission. Written informed consent was obtained from all subjects who participated in the study and/or their families. The activities regarding the collection of human samples and the study protocol were approved by ethics committee of the local hospital and local institutional review board located in Zhejiang, Guangdong Province, and Shanghai, China.
Case definitions and category
(1) Definition of a confirmed H7N9 case
The case definitions were established on the basis of ‘The Diagnosis and Treatment Programs of Human Infections with H7N9 Virus’, issued by the National Health and Family Planning Commission of the People’s Republic of China [34]. A confirmed H7N9 case was defined as a case in which a patient had influenza-like illness or in which respiratory specimens had tested positive for H7N9 virus via either of the following: isolation of H7N9 virus or positive results by rRT-PCR assay for H7N9, or a fourfold or greater increase in antibody titer for H7N9 virus based on testing of an acute serum specimen (collected 7 days or less after symptom onset) and a convalescent serum specimen collected at least 2 weeks later [27].
(2) Definition of a mild H7N9 case
A case in which an individual with confirmed H7N9 virus infection met the respiratory infection criteria was classified as a mild case, presenting with mild respiratory symptoms that did not have any complications throughout the clinical course, such as acute respiratory distress syndrome (ARDS), multi-organ failure, hypoxaemia, etc. [5].
(3) Definition of a severe H7N9 case
A case in which an individual with confirmed H7N9 virus infection met any one of the following criteria was classified as a severe case: presenting with severe respiratory symptoms with any complications including ARDS, shock, multi-organ failure, hypoxemia, etc., requiring hospitalization or intensive care unit admission or mechanical ventilation for medical reasons. The objective index was as follows: 1) X-ray showed lesions in multiple lobes or disease progression >50 % within 48 hours; 2) dyspnea with a respiratory rate > 24 breaths per minute; 3) hypoxemia with oxygen saturation ≤ 92 % on oxygen at a flow rate of 3 to 5 litres/min; 4) shock, ARDS or multiple organ dysfunction syndrome [5, 27, 41].
(4) Definition of a fatal H7N9 case
A case in which an individual with confirmed H7N9 virus infection died of this infection was classified as a fatal case.
Exposure definitions
People were predominantly exposed to the virus in two ways. The first of these was via exposure to poultry: individuals who came into direct or indirect contact with, or within proximity of, healthy, sick or dead poultry (including all types of poultry or birds, e.g., chickens, ducks, geese, pet birds and pigeons), having poultry in the neighborhood and eating poultry products that were not properly processed, were all included. The second manner of exposure was human-to-human transmission. This refers to whether infected individuals had had close contact with a confirmed or probable human case (any time from the day before the onset of illness to death or during the period of hospitalization) in the 2 weeks before the onset of his/her symptoms [27].
Data source
A total of 552 confirmed cases of avian H7N9 influenza were analyzed. However, only 99 of the 552 confirmed cases were selected for further analysis, as the clinical outcomes of the remaining 453 cases were not known. The selected cases included 14 mild, 50 severe and 35 fatal infections, and whole-genome sequences were generated from all 99 cases. Of these, 84 sequences with numbers are detailed in supplementary Table 1. However, the remaining 15 sequences do not have a Global Initiative on Sharing Avian Influenza Data (GISAID) number. An additional 44 sequences were generated from chickens, of which 43 are listed in Supplementary Table 2, while the remaining sequence is without a GISAID number. Furthermore, we analyzed 30 sequences from environmental isolates, of which 28 are listed in Supplementary Table 3. The remaining two sequences are without a GISAID number. All samples from patients, the environment, and chickens were collected between 31 March, 2013 and 31 December, 2014. Reference sequences (38 strains) were downloaded from a database (GISAID [http://platform.gisaid.org/epi3/frontend] and GenBank [http://www.ncbi.nlm.nih.gov/nuccore/?term=H7N9]). Details of the sequences are listed in Supplementary Table 4.
Epidemiological and clinical investigation
When a suspected case of H7N9 virus infection had been confirmed, the provincial epidemiologists and local public health doctors conducted the initial field investigations using a standard questionnaire. Field investigators interviewed the patient in confirmed case(s) and/or the relatives of the patient to determine exposure history (poultry exposure and human-to-human transmission), including the dose, duration and intensity of the exposure during the 2 weeks prior to the onset of illness. We calculated the incubation period as the number of days since the date of last exposure to the onset of illness. The questionnaire included data regarding dates, times, frequency and patterns of exposure to poultry and/or other animals, as well as bird environments. All available medical records, including the date of onset of illness, visits to clinical facilities, hospitalization, antiviral treatment, and clinical outcomes, were provided by local clinical health workers.
Sample collection, transportation and storage
A total of 99 respiratory specimens, including 80 throat swabs and 19 sputum specimens, from patients admitted to different hospitals (located in Zhejiang and Guangdong Province) were collected and shipped at 4 °C to the local hospitals and local CDCC for H7N9 laboratory testing by rRT-PCR assay. All of the surveyors and laboratory technicians were strictly trained to ensure that the interviews and laboratory investigations were conducted according to uniform standards.
Rapid viral RNA testing and sequencing
Total viral RNA was extracted from the collected samples using a QIAGEN RNeasy Mini Kit according to the manufacturer’s instructions. The specific primer and probe sets were provided by the Chinese CDC. The detection limits of the RT-PCR assays were approximately 100 copies/ml. Whole-genome sequencing of the isolated H7N9 samples was carried out by amplification and sequencing of the eight genomic fragments. Nucleotide sequences of the amplified products were determined directly by dideoxy sequencing, using an ABI PRISM BigDye Terminator Cycle Sequencing Kit [6].
Data analysis and statistics
Evolutionary relationships of the generated sequences were identified using the maximum-likelihood method, as implemented in MEGA 6.0 software (http://www.megasoftware.net). The current addresses of all confirmed cases were geocoded using the Google Map geocoding service (https://googledevelopers.appspot.com/maps/documentation/javascript/examples/geocoding-simple). After obtaining the X (longitude) and Y (latitude) coordinates, we used ArcGIS (ArcGIS, version10.2). The world basemap used was the publically available map maintained by ESRI (http://www.arcgis.com/home/item.html?id=3864c63872d84aec91933618e3815dd2). For spatial analysis, a spatial distribution map of the cases was constructed.
All statistical analyses were conducted using Statistical Analysis System, version 9.2 (SAS Institute, Cary, NC, USA). Quantitative measurements are presented as the median and range of the observed values, and qualitative measurements are presented as relative and absolute frequencies. An analysis of variance (F test) was applied to the measurement data. Chi-square tests (x 2) were used to compare the distribution of the different variables of qualitative measurements between the three groups. Fisher’s exact test was used in the analysis of contingency tables when the sample sizes were small (the expected values in any of the cells of a contingency table were below 5; the number of total samples was no more than 40; the data were very unequally distributed among the cells of the table). All p-values given are two-sided and were considered statistically significant at 0.05.
Results
Epidemiological characteristics in mild, severe and fatal cases
As of 31 January 2015, H7N9 had resulted in 552 confirmed infections, with a fatality rate of 37.7 %. The epidemiological distribution of mild, severe and fatal cases is shown in Fig. 1 and virtual features are shown in Fig. 2. A total of 42.9 % (6/14) of the subjects with mild disease were males and 57.1 % (8/14) were females, 58.0 % (29/50) of the severe cases were males and 42.0 % (21/50) were females, and 74.3 % (26/35) of the subjects who died were males and 25.7 % (9/35) were females. There was no difference in the gender distribution between the three groups (p = 0.094). However, the average age in the mild cases was 27.6 years (range, 5 months–82 years), versus 52 years (range 1.5–85 years) for the severe cases and 62 years (range 27–91 years) for cases of death. There was a significant difference in the age distribution between the three groups (p < 0.001). The most predominant age group was 0-, 60- and 60- in the mild, severe and fatal group, respectively (Fig. 3).
Fig. 1.
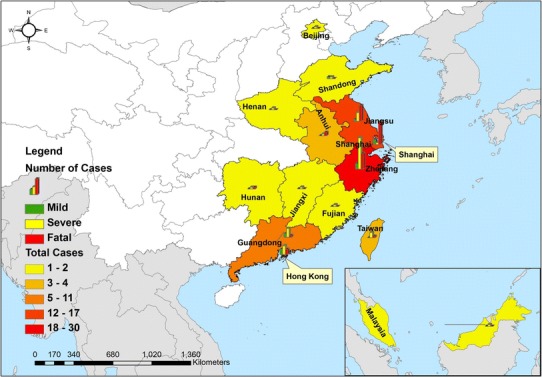
Geographical distribution of mild, severe and fatal cases of infection with the novel avian influenza A (H7N9) viruses between March 2013 and December 2014 (N = 99)
Fig. 2.
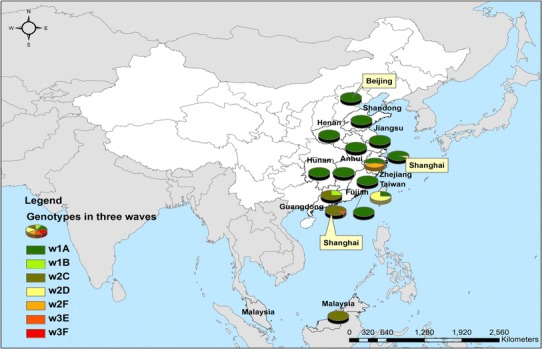
Geographical distribution of six different subtypes of the novel avian influenza A(H7N9) viruses between March 2013 and December 2014 (N = 99). w1, wave 1 (from 31 March 2013 to 30 September 2013); w2, wave 2 (from 01 October 2013 to 30 September 2014); w3, wave 3 (from 01 October 2014 to 30 September 2015)
Fig. 3.
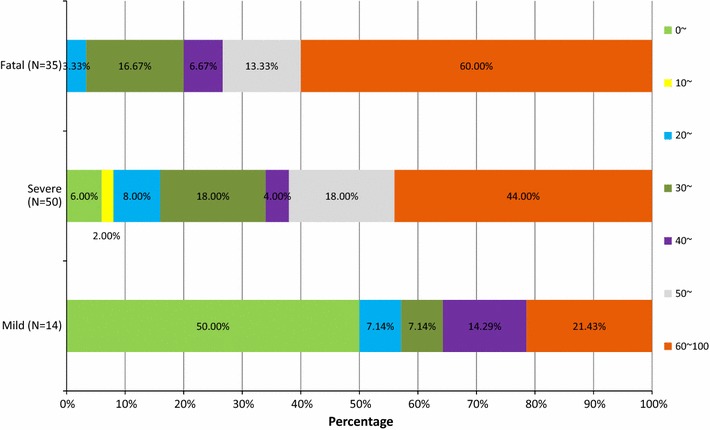
Age distribution (%) of mild, severe and fatal cases of infection with the novel avian influenza A (H7N9) viruses from March 2013 to December 2014 (N = 99)
The median time from exposure to onset was 4 (1–7), 2 (0–6) and 5 (1–13) days (p = 0.037) in the mild, severe and fatal group, respectively. The median time from onset to consultation was 1 (0–5), 3 (0–13) and 3 (0–7) days (p = 0.059); from onset to laboratory confirmation, it was 3 (0–8), 8 (1-–26) and 11 (0–41) days (p = 0.001); from onset to beginning antiviral treatment, it was 2 (0–5), 5 (0–10) and 7 (1–12) days (p = 0.002); and from onset to discharge/death, it was 12 (0–32), 40 (3–126), and 19 (3–85) days (p < 0.001) in the mild, severe and fatal group, respectively (Table 1). With the exception of the period from onset to consultation, all time relationships were statistically significantly different, as shown in Table 1 and Fig. 4.
Table 1.
Epidemiologic and clinical characteristics of mild, severe and fatal cases confirmed as avian H7N9 virus infection from March 31 of 2013 to December 31 of 2014
| Characteristic | Mild (N = 14) | Severe (N = 50) | Fatal (N = 35) | p-value |
|---|---|---|---|---|
| Population distribution | ||||
| Average age (years) | 27.6 (5 months-82 years) | 52 (1.5-85) | 62 (27-91) | <0.001 |
| Gender (% male) | 42.9 % (6/14) | 58.0 % (29/50) | 74.3 % (26/35) | 0.094 |
| Illness progress | ||||
| Median days from exposure to onset | 4 (1-7) | 2 (0-6) | 5 (1-13) | 0.037 |
| Median days from onset to consultation | 1 (0-5) | 3 (0-13) | 3 (0-7) | 0.059 |
| Median days from onset to confirmation | 3 (0-8) | 8 (1-26) | 11 (0-41) | 0.001 |
| Median days from onset to antiviral therapy | 2 (0-5) | 5 (0-10) | 7 (1-12) | 0.002 |
| Median days from onset to outcome | 12 (0-32) | 40 (3-126) | 19 (3-85) | <0.001 |
The p -value pertains to the comparison of the confirmed cases from mild, severe and fatal cases
We used an F-test to analyze the average age and median days for a three-group comparison
Chi-square (χ2) tests were applied to compare the distribution of the different variables of qualitative measurements, such as the gender distribution
Fig. 4.
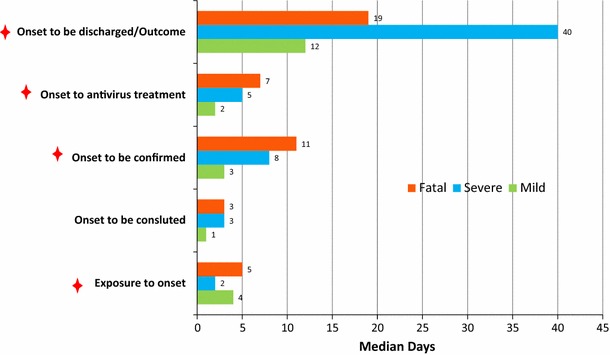
The median time (days) from onset to outcome/discharge in mild, severe and fatal cases of infection with the novel (H7N9) avian influenza A viruses between March 2013 and December 2014 (N = 99)
Phylogenetic analysis in the mild, severe and fatal groups
Whole genome sequences were generated from 99 H7N9 human isolates and 73 non-human isolates and aligned against 38 reference sequences from databases. Multiple sequence alignments showed 99.1–99.9 % sequence identity between isolates from the three clinical groups and non-human isolates. From 31 March 2013 to 01 October 2015, the H7N9 outbreak occurred in three waves. The first of these lasted from 31 March 2013 to 30 September 2013, the second occurred between 01 October and 30 September 2014, and the third wave lasted from 01 October 2014 to 30 September 2015. Phylogenetic analysis based on the HA segments revealed that the sequences from the mild, severe and fatal groups all clustered in five clades and that clade A varied between the waves and the reported areas. Clade A included the 2014 early-stage isolates from the region of the Yangtze River (Jiangsu, Zhejiang, Fujian, Jiangxi, Shandong and Anhui province, as well as Shanghai). Clade B included the 2013 isolates from China, with the exception of regions of the Pearl River (Guangdong and Hong Kong). Clade C included 2014 late-stage isolates from regions of the Yangtze River. Clades D and E included isolates from the 2013 and 2014 outbreak, respectively, from the Pearl River areas, including Guangdong and Hong Kong (Fig. 5a-h), as shown in Fig. 2.
Fig. 5.
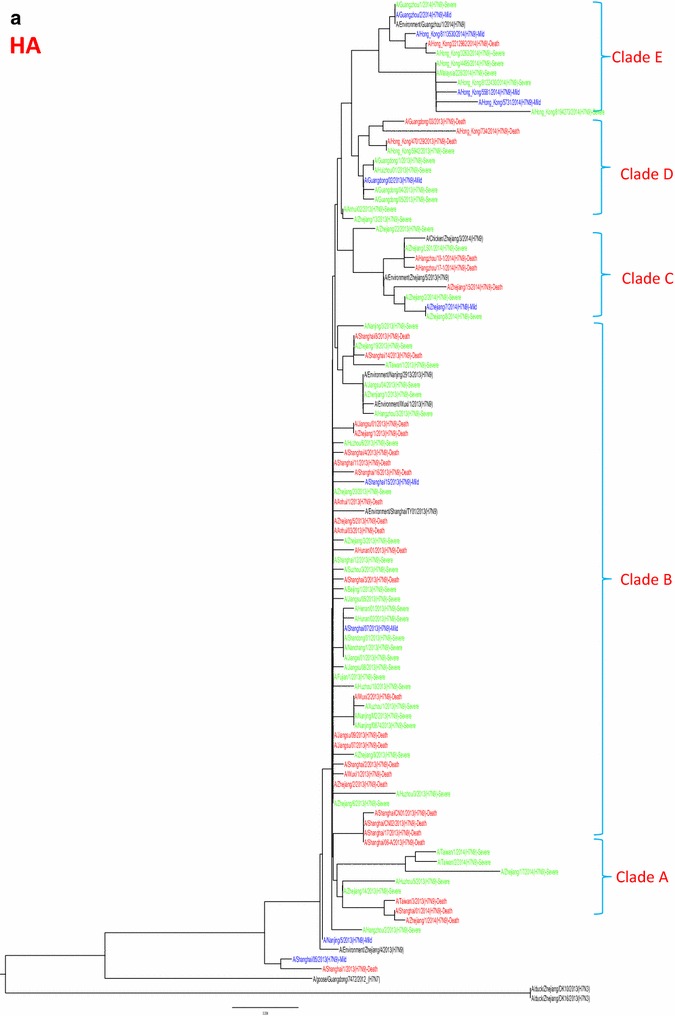
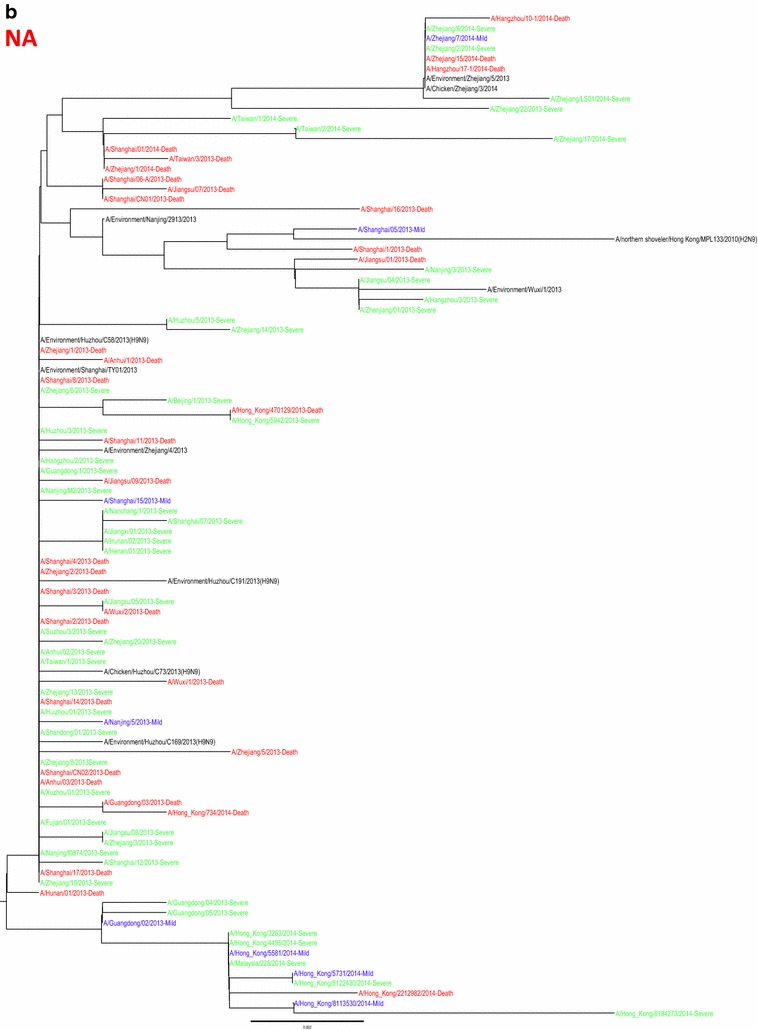
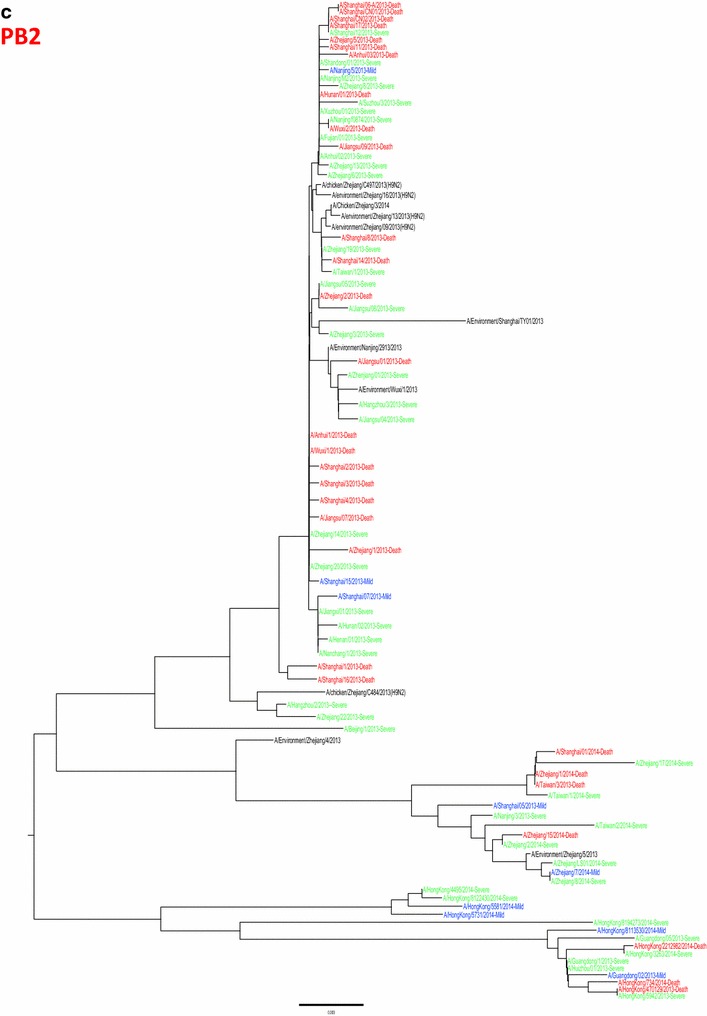
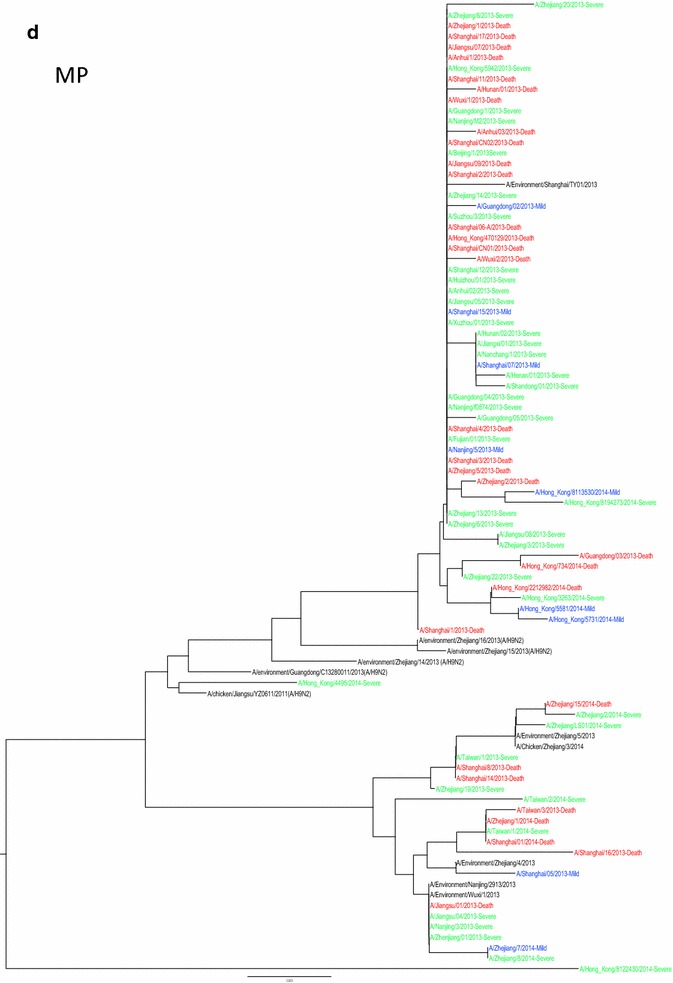

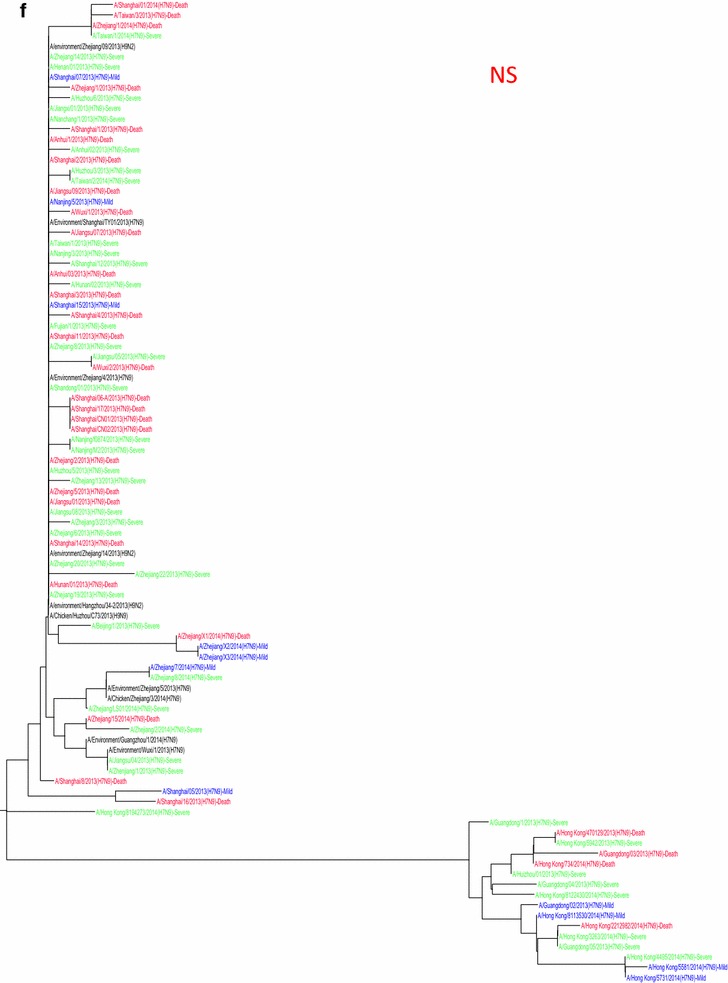
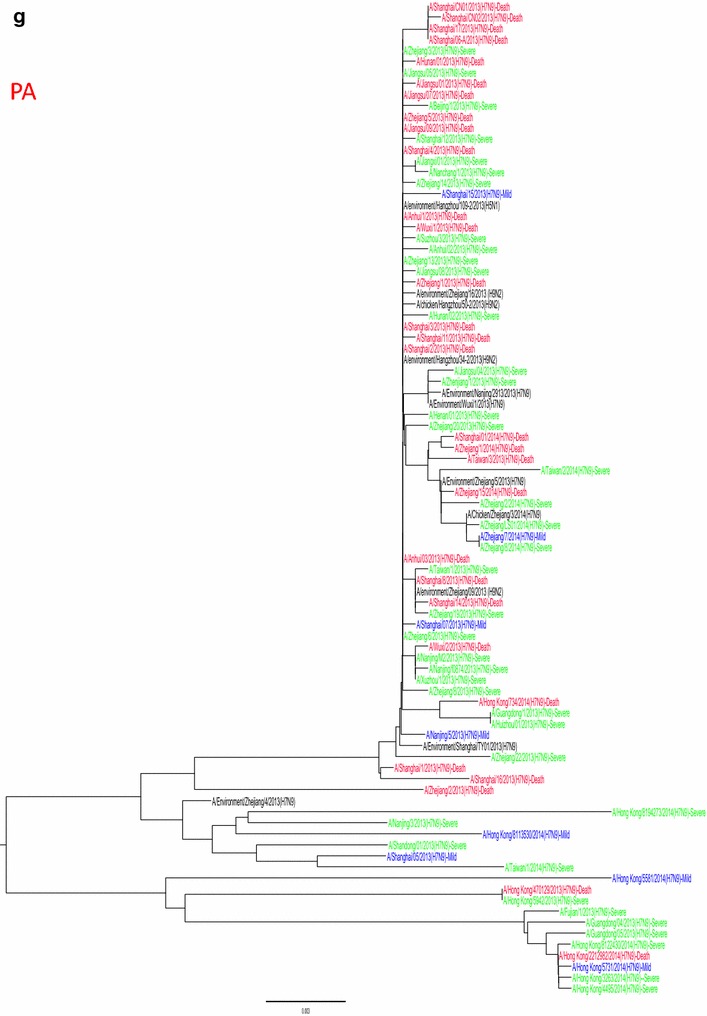
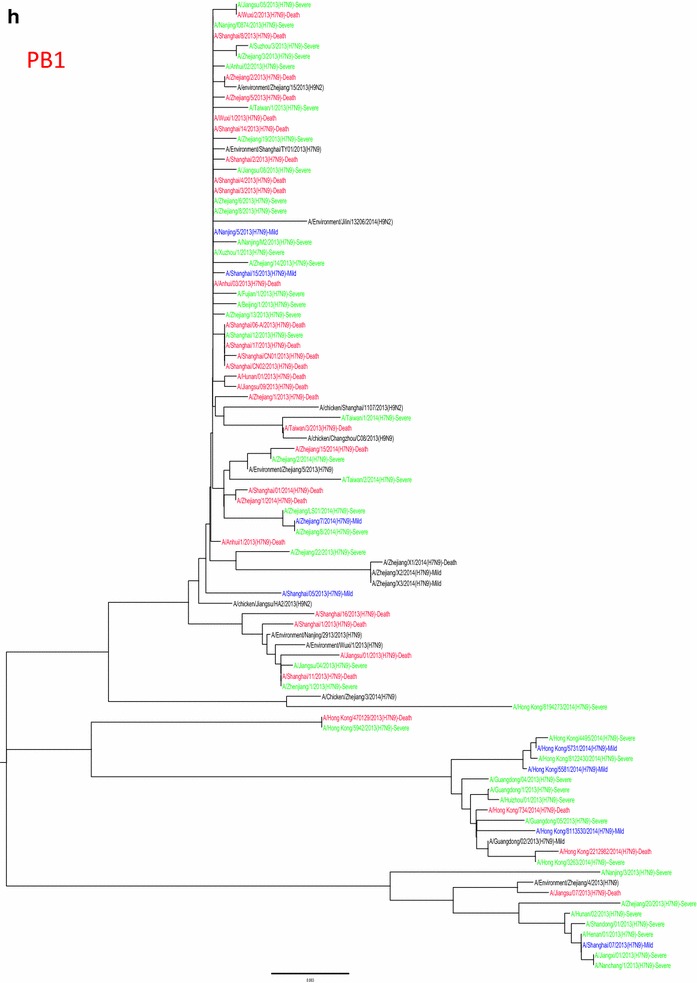
Phylogenetic trees based on sequences of HA (a), NA (b), PB2 (c), MP (d), NP (e), NS (f), PA (g) and PB1 (h) generated from isolates from mild, severe and fatal cases of infection with the novel avian influenza A (H7N9) viruses between March 2013 and December 2014 (N = 99). Blue, mild isolates; green, severe isolates; red, fatal isolates
Mutation analysis in the mild, severe and fatal groups
Analysis of mutations in HA sequences indicated that there was no statistical difference in G186V among the three clinical groups and non-human isolates. Q226L was more often found in the severe and fatal groups than in the mild group, but there was no statistical difference (p = 0.080). However, Q226L was identified as the amino acid difference that distinguishes human isolates from non-human isolates (p < 0.001). All of these mutations were associated with increased affinity for α-2,6-linked sialic acid receptors. In contrast, the G228S mutation was not found in any of the isolates from non-human or human cases, irrespective of whether they were mild, severe or fatal. All non-human isolates retained the amino acid ‘G’, which means they had high affinity for human receptors. Conversely, there was an obvious statistical difference between activated C antigen sites (N276D), related to antibody binding sites, among the three clinical groups (p = 0.046) and human vs. non-human isolates (p < 0.001). However, no difference was found between the other antigen sites, including R57M, R57K, E114K, T133A and G186V, and the three clinical groups and non-human isolates. L168I was identified as statistically different among the mild, severe and fatal groups (p = 0.050) and between the non-human isolates and human isolates (p = 0.001), and this mutation had not been reported to date. The HA cleavage site from mild, severe and fatal human cases and non-human isolates possessed only a single amino acid R (arginine), indicating that it still had characteristics of a low-pathogenic avian influenza (LPAI) virus (Table 2).
Table 2.
Key genetic mutations in mild, severe and fatal cases of confirmed infection with avian H7N9 virus from March 31 of 2013 to December 31 of 2014
| Segment | Mutation | Vaccine candidate isolate | Human isolates | p1 value | Non-human isolates | p2 value | |||
|---|---|---|---|---|---|---|---|---|---|
| A/Anhui/1/2013/H7N9 | Mild (N = 14) | Severe (N = 50) | Fetal (N = 35) | Chicken (N = 43) | Environmental (N = 30) | ||||
| HA | RBS positions (H3 numbering) | ||||||||
| Q226L | L | 8/10 (80.0 %) | 49/50 (98.0 %) | 31/34 (91.2 %) | 0.080 | 37/43 (86.0 %) | 19/30 (63.3 %) | <0.001 | |
| Q226I | 0/10 (0.0 %) | 0/50 (0.0 %) | 2/34 (5.9 %) | 0.165 | 0/43 (0.0 %) | 0/30 (0.0 %) | – | ||
| Q226P | 0/10 (0.0 %) | 1/50 (2.0 %) | 0/34 (0.0 %) | 0.641 | 0/43 (0.0 %) | 0/25 (0.0 %) | – | ||
| G228S | G | 0/10 (0.0 %) | 0/50 (0.0 %) | 0/34 (0.0 %) | – | 0/43 (0.0 %) | 0/30 (0.0 %) | – | |
| Antigenic site (H3 numbering) | |||||||||
| R57K (antigenic site E) | R | 6/10 (60.0 %) | 15/50 (30.0 %) | 6/34 (17.6 %) | 0.234 | 4/43 (9.30 %) | 5/30 (16.7 %) | 0.052 | |
| R57M (antigenic site E) | 0/10 (0.0 %) | 4/50 (8.0 %) | 0/34 (0.0 %) | 5/43 (11.6 %) | 1/30 (3.3 %) | ||||
| T133A (antigenic site A) | T | 1/10 (10.0 %) | 5/50 (10.0 %) | 3/34 (8.8 %) | 0.263 | 0/43 (0.0 %) | 1/30 (3.3 %) | 0.09 | |
| G186V (antigenic site B) | V | 9/10 (90.0 %) | 49/50 (98.0 %) | 33/34 (97.1 %) | 0.419 | 43/43 (100.0 %) | 30/30 (100.0 %) | 0.247 | |
| N276D (antigenic site C) | N | 3/10 (30.0 %) | 6/50 (12.0 %) | 1/34 (2.9 %) | 0.046 | 0/43 (0.0 %) | 0/30 (0.0 %) | <0.001 | |
| No report (H3 numbering) | |||||||||
| E114K | E | 2/10 (20.0 %) | 4/50 (8.0 %) | 1/34 (2.9 %) | 0.191 | 1/43 (2.3 %) | 0/30 (0.0 %) | 0.073 | |
| L168I | L | 2/10 (20.0 %) | 7/50 (14.0 %) | 0/34 (0.0 %) | 0.050 | 0/43 (0.0 %) | 0/30 (0.0 %) | 0.001 | |
| NA | Related to drug resistance | ||||||||
| E120V | E | 0/10 (0.0 %) | 0/48 (0.0 %) | 0/33 (0.0 %) | – | 0/43 (0.0 %) | 0/30 (0.0 %) | – | |
| V248I | V | 1/10 (10.0 %) | 4/48 (8.3 %) | 3/33 (9.1 %) | 0.983 | 1/43 (2.3 %) | 1/30 (3.3 %) | 0.614 | |
| V251I | I | 10/10 (100.0 %) | 47/48 (97.9 %) | 32/33 (97.0 %) | 0.846 | 43/43 (100.0 %) | 30/30 (100.0 %) | 0.695 | |
| H276Y | H | 0/10 (0.0 %) | 0/48 (0.0 %) | 0/33 (0.0 %) | – | 0/43 (0.0 %) | 0/30 (0.0 %) | – | |
| R294K | R | 0/10 (0.0 %) | 1/48 (2.1 %) | 4/33 (12.1 %) | 0.01 | 0/43 (0.0 %) | 0/30 (0.0 %) | 0.01 | |
| N296S | N | 0/10 (0.0 %) | 0/48 (0.0 %) | 0/33 (0.0 %) | – | 0/43 (0.0 %) | 0/30 (0.0 %) | – | |
| N337T | N | 1/10 (10.0 %) | 7/41 (17.1 %) | 3/33 (9.1 %) | 0.571 | 1/43 (2.3 %) | 1/30 (3.3 %) | 0.125 | |
| PB1 | Increased H5 virus transmission among ferrets | ||||||||
| I368V | V | 11/11 (100.0 %) | 39/41 (95.1 %) | 27/31 (87.1 %) | 0.261 | 35/35 (100.0 %) | 30/30 (100.0 %) | 0.045 | |
| Increased replication in mammalian cells | |||||||||
| L598P/M | L | 0/11 (0.0 %) | 0/41 (0.0 %) | 1/31 (3.2 %) | 0.428 | 0/35 (0.0 %) | 0/30 (0.0 %) | 0.434 | |
| PB1-F2 | Increased pathogenicity in mice | ||||||||
| 25AA | 90AA | 1/11 (9.1 %) | 6/41 (14.6 %) | 1/31 (3.2 %) | 0.267 | 9/35 (25.7 %) | 11/30 (36.7 %) | 0.03 | |
| 57AA | 2/11 (18.2 %) | 2/41 (4.9 %) | 0/31 (0.0 %) | 0.054 | 0/35 (0.0 %) | 0/30 (0.0 %) | 0.200 | ||
| 76AA | 0/11 (0.0 %) | 0/41 (0.0 %) | 2/31 (6.5 %) | 0.179 | 2/35 (5.7 %) | 0/30 (0.0 %) | 0.356 | ||
| 90AA | 90AA | 8/11 (72.7 %) | 33/41 (80.5 %) | 28/31 (90.3 %) | 0.333 | 24/35 (68.6 %) | 19/30 (63.3 %) | 0.05 | |
| PB2 | Enhanced polymerase activity and increased virulence in mice | ||||||||
| L89V | L | 9/9 (100.0 %) | 44/44 (100.0 %) | 30/30 (100.0 %) | – | 43/43 (100.0 %) | 30/30 (100.0 %) | – | |
| K191E | K | 6/9 (66.7 %) | 16/44 (36.4 %) | 9/30 (30.0 %) | 0.119 | 7/43 (16.3 %) | 6/30 (20.0 %) | 0.016 | |
| N559T | N | 6/9 (66.7 %) | 18/44 (40.9 %) | 7/30 (23.3 %) | 0.048 | 7/43 (16.3 %) | 8/30 (26.7 %) | 0.011 | |
| M570I | M | 5/9 (55.6 %) | 13/44 (29.5 %) | 7/30 (23.3 %) | 0.80 | 4/43 (9.3 %) | 7/30 (23.3 %) | 0.026 | |
| Q591K/L | Q | 1/9 (11.1 %) | 1/44 (2.3 %) | 1/30 (3.3 %) | 0.430 | 0/43 (0.0 %) | 0/30 (0.0 %) | 0.214 | |
| E627K | K | 3/9 (33.3 %) | 30/44 (68.2 %) | 26/30 (86.7 %) | 0.006 | 8/43 (18.6 %) | 0/30 (0.0 %) | <0.001 | |
| D701N | D | 1/9 (11.1 %) | 4/44 (9.1 %) | 2/30 (6.7 %) | 0.860 | 0/43 (0.0 %) | 0/30 (0.0 %) | 0.119 | |
| PA | Species-associated signature positions | ||||||||
| V100A | A | 4/8 (50.0 %) | 30/41 (73.2 %) | 27/30 (90.0 %) | 0.038 | 28/35 (80.0 %) | 24/30 (80.0 %) | 0.137 | |
| I308V | I | 0/8 (0.0 %) | 3/41 (7.3 %) | 0/30 (0.0 %) | 0.236 | 0/35 (0.0 %) | 1/30 (3.3 %) | 0.250 | |
| K356R | R | 6/8 (75.0 %) | 34/41 (82.9 %) | 28/30 (93.3 %) | 0.289 | 34/35 (97.1 %) | 30/30 (100.0 %) | 0.024 | |
| S409N | N | 7/8 (87.5 %) | 39/41 (95.1 %) | 29/30 (96.7 %) | 0.574 | 31/35 (88.6 %) | 25/28 (89.3 %) | 0.635 | |
| T618K | T | 0/8 (0.0 %) | 0/41 (0.0 %) | 1/30 (3.3 %) | 0.437 | 0/35 (0.0 %) | 0/30 (0.0 %) | 0.430 | |
| NP | Increased virulence in mice | ||||||||
| N321S | N | 0/11 (0.0 %) | 1/41 (2.4 %) | 1/32 (3.1 %) | 0.842 | 1/34 (2.9 %) | 1/30 (3.3 %) | 0.983 | |
| D375E | D | 4/11 (36.4 %) | 7/41 (17.1 %) | 4/32 (12.5 %) | 0.200 | 1/34 (2.9 %) | 4/30 (13.3 %) | 0.071 | |
| NS1 | Increased virulence in mice | ||||||||
| P42S | S | 11/11 (100.0 %) | 42/42 (100.0 %) | 32/32 (100.0 %) | – | 36/36 (100.0 %) | 30/30 (100.0 %) | – | |
| T92A | T | 1/11 (9.1 %) | 0/42 (0.0 %) | 1/32 (3.1 %) | 0.195 | 0/36 (0.0 %) | 0/30 (0.0 %) | 0.119 | |
| PDZ binding motif | Deletion | Deletion | Deletion | Deletion | – | Avian type | Avian type | – | |
| M1 | Increased virulence in mice | ||||||||
| N30D | D | 9/9 (100.0 %) | 44/44 (100.0 %) | 31/31 (100.0 %) | – | 36/36 (100.0 %) | 30/30 (100.0 %) | – | |
| T215A | A | 9/9 (100.0 %) | 44/44 (100.0 %) | 31/31 (100.0 %) | – | 36/36 (100.0 %) | 30/30 (100.0 %) | – | |
| M2 | Antiviral resistance (amantadine) | ||||||||
| S31N | N | 9/9 (100.0 %) | 44/44 (100.0 %) | 31/31 (100.0 %) | – | 36/36 (100.0 %) | 30/30 (100.0 %) | – | |
Single letters refer to the amino acid (aa) found in the indicated protein at a specific site. The numbering starts with the first methionine codon for these proteins
p1 value: comparison of the mutation frequency among the isolates from mild, severe and fatal cases
p2 value: comparison of the mutation frequency between human and non-human isolates
Qualitative measurements are presented as relative and absolute frequencies. Chi-square (χ2) tests were applied to compare the distribution of the different variables of qualitative measurements. Fisher’s exact test was used in the analysis of contingency tables when the sample sizes were small
“-” denotes no difference between the groups
The amino acid sequences from the three clinical groups and non-human isolates indicated that aa 69–73 were deleted in the stalk region of the NA protein, which potentially influences virus replication. Although E120V, H276Y and N296S substitutions were absent from sequences from clinical and non-clinical isolates, R294K was found in 2.1 % and 12.1 % of isolates from the severe and fatal cases, respectively. This suggested that resistance to the antiviral NA inhibitor oseltamivir was most often found in the fatal group (p =0.01). Compared to the non-human isolates, the R294K mutation rate was much higher in the human isolates (p = 0.01). Of the 35 NA sequences from the patients treated with oseltamivir, three (8.6 %) showed an R294K mutation, while five (14.3 %) exhibited V248I, V251I and N337T substitutions.
The E627K substitution in the PB2 gene was more frequently found in the severe (68.2 %) and fatal (86.7 %) groups than in the mild group (33.3 %) (p = 0.006) and non-human isolates (18.6 %) (p < 0.001). In contrast, N559T was the major mutation in the mild group compared to the other two groups (p = 0.048). No differences were observed among the sequences of the three clinical groups with regard to other site mutations, including L89V, K191E, M570I, Q591K/L and D701N. However, the mutation rates of K191E (p = 0.016), N559T (p = 0.011), M570I (p = 0.026) and E627K (p < 0.001) differed between the non-human and human isolates, as shown in Table 2.
No significant difference between the three groups was observed in I368V and L598P/M in the PB1 segment. However, the non-human isolates had a higher rate of I368V than the human isolates (p = 0.045), as shown in Table 2. Full-length PB1-F2 protein (87–90 amino acids) has been associated with increased virulence in mice, and PB1-F2 protein with 90 amino acids was detected in 72.7 %, 80.5 % and 90.3 % of the mild, severe and fatal human cases, respectively, with no statistical difference (p = 0.333). A total of 25.7 % of the chicken isolates and 36.7 % of the environmental virus isolates had PB1-F2 protein with 25 amino acid mutations, which was higher than that observed in the human isolates (p = 0.03). In contrast, 68.6 % of the chicken isolates and 63.3 % of the environmental virus isolates had the 90-aa variant of the PB1-F2 protein. The mutation rates of non-human isolates were much lower than those of human isolates (p = 0.05), as shown in Table 2.
Potential human-like signatures in the PA gene have been reported previously. In the present study, the PA genes from 8 mild, 41 severe and 30 fatal cases, as well as 65 non-human viruses, were analyzed. V100A was a significantly predominant mutation in the fatal group, compared to the mild and severe cases (p = 0.038). However, the other mutations, I308V, K356R, S409N and T618K, were not statistically significantly different among the three groups. The K356R mutation rate in the non-human isolates was higher than in the human isolates (p = 0.024) (Table 2).
A total of 11 mild, 41 severe and 32 fatal isolates were analysed at the NP segment. A substitution of N321S and D375E remained conserved in the three clinical groups and the non-human isolates (Table 2).
Similarly, NS segments from 11 mild, 42 severe and 32 fatal isolates were analysed. No differences in P42S and T92A were observed in the three clinical groups and non-human isolates. In addition, all H7N9 viruses had a truncated NS gene with a PDZ motif deletion. PDZ deletion might influence the pathogenicity of H7N9 viruses in human or avian hosts (Table 2).
It was found that the M1 protein from all of the human and non-human isolates had virulence sites (N30D and T215A) and that the M2 protein from all of the human and non-human isolates had an S31N substitution, indicating resistance to adamantine (amantadine and rimantadine), as shown in Table 2.
Discussion
Since the spring of 2013, a novel avian-origin influenza A (H7N9) virus has emerged and spread among humans in China, resulting in a high fatality rate [6, 15]. The risk factors that might contribute to a fatal outcome regarding human H7N9 infection include old age, a history of smoking, chronic lung disease, immunosuppressive disorders, chronic drug use, and delayed oseltamivir intervention [19]. The present study described the major differences in epidemiological and viral features between mild, severe and fatal cases during the three epidemic waves of the worldwide H7N9 outbreak. The age and gender characteristics of H7N9 cases are unusual compared to those of H5N1 cases. In H5N1, an average age of 26 years (interquartile range: 19–25) and a more equal gender distribution were the prominent features [12]. However, the present study indicated that the average age in the mild group (27.6 years) was far younger than that in the severe (52 years) and fatal groups (62 years) [2, 32], with 50 % of mild cases being found in children <10 years of age. However, the gender distribution has no statistical difference among three groups.
Mild cases were frequently identified in family clusters and secondary cases, permitting early detection due to monitoring of the close contacts of index cases, as well as a prompt start to antiviral treatment once symptoms began. However, the predominance of older people among the severe and fatal cases is likely to be due to their having had underlying lung conditions and impaired immune functions, as well as a greater chance and high level of exposure [13, 17]. The age and gender findings were consistent across the first, second and third waves of the epidemic. In accordance with the results reported herein, a strong association has been shown between old age and a fatal outcome in (H1 and H3) seasonal influenza (>65 years old), which is different from what has been observed in H5N1 avian influenza [40].
The median incubation period was 4, 2 and 5 days in the mild, severe and fatal group, respectively, which is comparable to what has been reported previously (3.1 days) [2, 15]. The median number of days from onset to confirmation (11) and onset to start of antiviral treatment (7 days) found in the fatal group was relatively long compared to that observed in the mild and severe groups, and was also in accordance with the findings of Li et al. [15] and Liu et al [20], which might indicate that delayed time of case confirmation and antiviral intervention are closely associated with a fatal outcome. In particular, the median number of days from the onset to outcome in fatal cases (19 days) was lower than that of onset to outcome or discharge in the severe cases (40 days), but higher than that in the mild cases (12 days). However, it remains comparable to that of patients infected with H5N1, which is 18.7 days [2].
Detailed characterisation of the genes and protein structure of H7N9 has revealed a number of important features associated with the transmissibility, pathogenicity and severity of this virus in humans [12]. For the HA segment, the H7N9 hemagglutinin outer-surface proteins, from all isolates of the three clinical groups lacked a multi-basic cleavage site. Despite the severity of symptoms and fatality rate in human infections, the absence of the multi-basic cleavage site in HA indicates that H7N9 would be considered an LPAI virus [30]. Analysis of the protein sequence of the H7N9 viruses infecting humans showed substitutions in the HA at amino acid residues Q226L and G186V. These substitutions have been associated with increased affinity for α-2,6-linked sialic acid receptors, indicating that the virus may adapt more rapidly to infect mammals via an enhanced capacity for mutations [4, 18]. The increased binding affinity of H7N9 compared to that of H5N1, raises concerns regarding a potential adaptation of the virus that would allow it to cause a pandemic [12]. R57K, related to the antigenic site E, and N276D, related to the antigenic site C, were the most frequently observed mutations in mild cases, but R57M, related to the antigenic site E, was the major mutation in severe cases. Interestingly, the present study is the first report of the occurrence of the novel mutations L168I and E114K, near the antibody accessible site. These new mutations were detected in isolates from mild cases; however, their significance remains unclear.
Given the severity of the infection in humans, virus susceptibility to antiviral drugs is vitally important for treatment [18, 31]. Genomic sequences of virus isolates showed that 12.1 % of the fatal cases, 2.1 % of the severe cases and none of the mild cases had an R294K mutation, which affects NA inhibitors - oseltamivir, zanamivir, peramivir and laninamivir [9, 11, 29, 31]. In contrast, all H7N9 viruses assessed to date have a mutation in the M gene (S31N), which confers resistance to the commercially available adamantine drugs amantadine and rimantadine [12].
The PB2, PA and PB1 genes code for proteins that form the polymerase enzyme complex, which is necessary for viral replication [7, 12, 16]. Genomic sequencing of H7N9 isolates indicated that the substitution E627K in PB2 was more frequent in the fatal group than in the severe and mild groups, whereas N559T was found in the mild group. The PB2 amino acid change increases virulence and enhances polymerase activity in mice [16]. This, along with other factors, likely contributes to the increased severity of the disease in humans with H7N9 infection. The PB2 from H7N9 isolates from birds retained Glu at position 627 and Asp at 701, strongly suggesting that the mutation is positively selected upon replication in the human host, as reported previously for zoonotic A (H7N7) and A(H5N1) infections [1]. For PA, over 90 % of fatal cases have V100A and K356R mutations, which were present in species-associated signature positions. Interestingly, the PA-356R signature is associated with increased H5N1virulence [37]. Additional markers of adaptation to non-avian hosts or virulence were noted in the PB1-F2 gene, where 90.3 % of fatal isolates showed the 90 amino acid mutations in the PB1 gene – higher than that of non-human isolates and the mild and severe cases. There were no obvious differences in the NS1 and M1 genes among the three clinical groups.
All identified virus genotypes contained at least one difference in an internal gene compared with the genotypes detected in the delta of the Yangtze River and Pearl River regions during the past three epidemic waves, indicating that those genotypes may have been generated through inter-provincial poultry trade and further reassortment with local H9N2 viruses [22, 25]. Phylogenetic analysis indicated that a total of six genotypes were circulating in human cases worldwide, but no difference in distribution of genotypes was observed among the three clinical groups in the present study [24, 42]. In the first epidemic wave of 2013, genotype A was detected in all Chinese areas, with the exception of the Pearl River areas (Guangdong Province and Hong Kong), particularly in the Yangtze River delta, including Zhejiang, Jiangsu, Jiangxi, Shandong, Fujian and Anhui Provinces, as well as Shanghai. These findings support the hypothesis that H7N9 infection originated from this area. Furthermore, some external and internal genes exchanged and generated F genotypes in these areas in the second and third epidemic waves. In contrast, genotypes B, C and E were predominant in the Pearl River (Guangdong Province and Hong Kong) in the first, second and third epidemic waves, which might indicate that other subtypes were circulating in the human and poultry populations. The infectivity, transmissibility and pathogenicity of the different genotypes has not yet been characterised. New genotypes with higher fitness may be generated by genetic tuning in the future [33, 45].
The present study had several limitations, including the small number of sequences from mild cases (14 sequences), the small number of sequences from cases of the third epidemic wave, and the failure to find a statistical association between identified genotypes and clinical outcome due to the limited number of mild cases for each genotype. Therefore, investigations are ongoing, with eyes fully open to any new cases or episodes, in order to cover all cases not included in the present study to analyze, monitor and control this serious challenge.
In conclusion, advanced age, as well as a delay in confirmation of diagnosis and start of antiviral treatment, were the greatest contributory factors to a high risk of death. Furthermore, PB2 (E627K), NA(R294K) and PA (V100A) mutations were slightly associated with an increased fatality rate. However, HA (N276D) and PB2 (N559T) were obviously related to a mild outcome. Although H7N9 isolates from all over the world presented with a variety of genotypes, there were no sequence differences in the mild, severe and fatal cases. However, there was an obvious difference in the distribution of genotypes in the regions of the Pearl and Yangtze rivers and between the three different epidemic waves. There was also evidence to suggest that genetic tuning not only mediated species switching but may also have allowed the virus to increase its adaptation to more efficiently infect humans and enable person-to-person transmission [28, 43]. However, the mechanism driving genetic tuning has not yet been fully elucidated, and no vaccine for the prevention of influenza A (H7N9) virus infection is available to date. Thus, an effective avian influenza surveillance network for discovering new infections and monitoring any genetic changes and potential adaptations in the H7N9 virus at the early stages is essential. Canada recently reported two confirmed family cases of influenza A (H7N9), imported from China. This showed that it is possible to transfer the H7N9 virus from China to the rest of the world via travel and transportation [35]. Close cooperation between the different sectors related to human health, poultry and wild birds is necessary to respond to the potential pandemic risk posed by the novel avian influenza A(H7N9) viruses [8].
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by grants from Provincial Medical Research Fund of Zhejiang, China (2014C13G2010106). We acknowledge the authors who submitted the sequences used in this study to GenBank and GISAID.
Abbreviations
- ARDS
Acute respiratory distress syndrome
- HA
Haemagglutinin
- HPAI
Highly pathogenic avian influenza
- LPAI
Low-pathogenic avian influenza
- ICU
Intensive care unit
- NA
Neuraminidase
- PA, PB1, PB2
Polymerase subunits
- RBS
Receptor-binding site
Compliance with ethical standards
Conflict of interest
None declared.
Contributor Information
Zhiruo Zhang, Phone: (+86) 15618977655, Email: zhangzhiruo@sjtu.edu.cn.
Shelan Liu, Phone: (+86) 571-87115137, Email: liushelan@126.com, Email: liushelan@gmail.com.
References
- 1.Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, Yao H, Wo J, Fang Q, Cui D, Li Y, Yao X, Zhang Y, Wu H, Zheng S, Diao H, Xia S, Zhang Y, Chan KH, Tsoi HW, Teng JL, Song W, Wang P, Lau SY, Zheng M, Chan JF, To KK, Chen H, Li L, Yuen KY. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet. 2013;381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cowling BJ, Jin L, Lau EH, Liao Q, Wu P, Jiang H, Tsang TK, Zheng J, Fang VJ, Chang Z, Ni MY, Zhang Q, Ip DK, Yu J, Li Y, Wang L, Tu W, Meng L, Wu JT, Luo H, Li Q, Shu Y, Li Z, Feng Z, Yang W, Wang Y, Leung GM, Yu H. Comparative epidemiology of human infections with avian influenza A H7N9 and H5N1 viruses in China: a population-based study of laboratory-confirmed cases. Lancet. 2013;382:129–137. doi: 10.1016/S0140-6736(13)61171-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding H, Chen Y, Yu Z, Horby PW, Wang F, Hu J, Yang X, Mao H, Qin S, Chai C, Liu S, Chen E, Yu H. A family cluster of three confirmed cases infected with avian influenza A (H7N9) virus in Zhejiang Province of China. BMC Infect Dis. 2014;14:3846. doi: 10.1186/s12879-014-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dortmans JC, Dekkers J, Wickramasinghe IN, Verheije MH, Rottier PJ, van Kuppeveld FJ, de Vries E, de Haan CA. Adaptation of novel H7N9 influenza A virus to human receptors. Sci Rep. 2013;3:3058. doi: 10.1038/srep03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feng L, Wu JT, Liu X, Yang P, Tsang TK, Jiang H, Wu P, Yang J, Fang VJ, Qin Y, Lau EH, Li M, Zheng J, Peng Z, Xie Y, Wang Q, Li Z, Leung GM, Gao GF, Yu H, Cowling BJ (2014) Clinical severity of human infections with avian influenza A(H7N9) virus, China, 2013/14. Euro Surveill 19(49) [DOI] [PMC free article] [PubMed]
- 6.Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K, Xu X, Lu H, Zhu W, Gao Z, Xiang N, Shen Y, He Z, Gu Y, Zhang Z, Yang Y, Zhao X, Zhou L, Li X, Zou S, Zhang Y, Li X, Yang L, Guo J, Dong J, Li Q, Dong L, Zhu Y, Bai T, Wang S, Hao P, Yang W, Zhang Y, Han J, Yu H, Li D, Gao GF, Wu G, Wang Y, Yuan Z, Shu Y. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 7.Gao Y, Zhang Y, Shinya K, Deng G, Jiang Y, Li Z, Guan Y, Tian G, Li Y, Shi J, Liu L, Zeng X, Bu Z, Xia X, Kawaoka Y, Chen H. Identification of amino acids in HA and PB2 critical for the transmission of H5N1 avian influenza viruses in a mammalian host. PLoS Pathog. 2009;5:e1000709. doi: 10.1371/journal.ppat.1000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu H, Chen B, Zhu H, Jiang T, Wang X, Chen L, Jiang Z, Zheng D, Jiang J. Importance of Internet surveillance in public health emergency control and prevention: evidence from a digital epidemiologic study during avian influenza A H7N9 outbreaks. J Med Int Res. 2014;16:e20. doi: 10.2196/jmir.2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hai R, Schmolke M, Leyva-Grado VH, Thangavel RR, Margine I, Jaffe EL, Krammer F, Solorzano A, Garcia-Sastre A, Palese P, Bouvier NM. Influenza A(H7N9) virus gains neuraminidase inhibitor resistance without loss of in vivo virulence or transmissibility. Nat Commun. 2013;4:2854. doi: 10.1038/ncomms3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu J, Zhu Y, Zhao B, Li J, Liu L, Gu K, Zhang W, Su H, Teng Z, Tang S, Yuan Z, Feng Z, Wu F (2014) Limited human-to-human transmission of avian influenza A(H7N9) virus, Shanghai, China, March to April 2013. Euro Surveill 19(25) [DOI] [PubMed]
- 11.Hu Y, Lu S, Song Z, Wang W, Hao P, Li J, Zhang X, Yen HL, Shi B, Li T, Guan W, Xu L, Liu Y, Wang S, Zhang X, Tian D, Zhu Z, He J, Huang K, Chen H, Zheng L, Li X, Ping J, Kang B, Xi X, Zha L, Li Y, Zhang Z, Peiris M, Yuan Z. Association between adverse clinical outcome in human disease caused by novel influenza A H7N9 virus and sustained viral shedding and emergence of antiviral resistance. Lancet. 2013;381:2273–2279. doi: 10.1016/S0140-6736(13)61125-3. [DOI] [PubMed] [Google Scholar]
- 12.Jernigan DB, Cox NJ. H7N9: preparing for the unexpected in influenza. Ann Rev Med. 2015;66:361–371. doi: 10.1146/annurev-med-010714-112311. [DOI] [PubMed] [Google Scholar]
- 13.Ji H, Gu Q, Chen LL, Xu K, Ling X, Bao CJ, Tang FY, Qi X, Wu YQ, Ai J, Shen GY, Dong DJ, Yu HY, Huang M, Cao Q, Xu Y, Zhao W, Xu YT, Xia Y, Chen SH, Yang GL, Gu CL, Xie GX, Zhu YF, Zhu FC, Zhou MH. Epidemiological and clinical characteristics and risk factors for death of patients with avian influenza A H7N9 virus infection from Jiangsu Province, Eastern China. PLOS One. 2014;9:e89581. doi: 10.1371/journal.pone.0089581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ke C, Lu J, Wu J, Guan D, Zou L, Song T, Yi L, Zeng X, Liang L, Ni H, Kang M, Zhang X, Zhong H, He J, Lin J, Smith D, Burke D, Fouchier RA, Koopmans M, Zhang Y. Circulation of reassortant influenza A(H7N9) viruses in poultry and humans, Guangdong Province, China, 2013. Emerg Infect Dis. 2014;20:2034–2040. doi: 10.3201/eid2012.140765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Q, Zhou L, Zhou M, Chen Z, Li F, Wu H, Xiang N, Chen E, Tang F, Wang D, Meng L, Hong Z, Tu W, Cao Y, Li L, Ding F, Liu B, Wang M, Xie R, Gao R, Li X, Bai T, Zou S, He J, Hu J, Xu Y, Chai C, Wang S, Gao Y, Jin L, Zhang Y, Luo H, Yu H, He J, Li Q, Wang X, Gao L, Pang X, Liu G, Yan Y, Yuan H, Shu Y, Yang W, Wang Y, Wu F, Uyeki TM, Feng Z. Epidemiology of human infections with avian influenza A(H7N9) virus in China. N Engl J Med. 2014;370:520–532. doi: 10.1056/NEJMoa1304617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, Chen H, Jiao P, Deng G, Tian G, Li Y, Hoffmann E, Webster RG, Matsuoka Y, Yu K. Molecular basis of replication of duck H5N1 influenza viruses in a mammalian mouse model. J Virol. 2005;79:12058–12064. doi: 10.1128/JVI.79.18.12058-12064.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu B, Havers F, Chen E, Yuan Z, Yuan H, Ou J, Shang M, Kang K, Liao K, Liu F, Li D, Ding H, Zhou L, Zhu W, Ding F, Zhang P, Wang X, Yao J, Xiang N, Zhou S, Liu X, Song Y, Su H, Wang R, Cai J, Cao Y, Wang X, Bai T, Wang J, Feng Z, Zhang Y, Widdowson MA, Li Q. Risk factors for influenza A(H7N9) disease–China, 2013. Clin Infect Dis. 2014;59:787–794. doi: 10.1093/cid/ciu423. [DOI] [PubMed] [Google Scholar]
- 18.Liu D, Shi W, Shi Y, Wang D, Xiao H, Li W, Bi Y, Wu Y, Li X, Yan J, Liu W, Zhao G, Yang W, Wang Y, Ma J, Shu Y, Lei F, Gao GF. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: phylogenetic, structural, and coalescent analyses. Lancet. 2013;381:1926–1932. doi: 10.1016/S0140-6736(13)60938-1. [DOI] [PubMed] [Google Scholar]
- 19.Liu S, Sun J, Cai J, Miao Z, Lu M, Qin S, Wang X, Lv H, Yu Z, Amer S, Chai C. Epidemiological, clinical and viral characteristics of fatal cases of human avian influenza A (H7N9) virus in Zhejiang Province, China. J Infect. 2013;67:595–605. doi: 10.1016/j.jinf.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 20.Liu SL, Zhang ZR, Wang C, Dong Y, Cui LB, Yang XH, Sun Z, Wang J, Chen J, Huang RJ, Miao F, Ruan B, Xie L, He HX, Deng J. 2009 pandemic characteristics and controlling experiences of influenza H1N1 virus 1 year after the inception in Hangzhou, China. J Med Virol. 2010;82:1985–1995. doi: 10.1002/jmv.21964. [DOI] [PubMed] [Google Scholar]
- 21.Liu T, Bi Z, Wang X, Li Z, Ding S, Bi Z, Wang L, Pei Y, Song S, Zhang S, Wang J, Sun D, Pang B, Sun L, Jiang X, Lei J, Yuan Q, Kou Z, Yang B, Shu Y, Yang L, Li X, Lu K, Liu J, Zhang T, Xu A. One family cluster of avian influenza A(H7N9) virus infection in Shandong, China. BMC Infect Dis. 2014;14:98. doi: 10.1186/1471-2334-14-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu J, Wu J, Zeng X, Guan D, Zou L, Yi L, Liang L, Ni H, Kang M, Zhang X, Zhong H, He X, Monagin C, Lin J, Ke C. Continuing reassortment leads to the genetic diversity of influenza virus H7N9 in Guangdong, China. J Virol. 2014;88:8297–8306. doi: 10.1128/JVI.00630-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mok CK, Lee HH, Lestra M, Nicholls JM, Chan MC, Sia SF, Zhu H, Poon LL, Guan Y, Peiris JS. Amino acid substitutions in polymerase basic protein 2 gene contribute to the pathogenicity of the novel A/H7N9 influenza virus in mammalian hosts. J Virol. 2014;88:3568–3576. doi: 10.1128/JVI.02740-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pu J, Wang S, Yin Y, Zhang G, Carter RA, Wang J, Xu G, Sun H, Wang M, Wen C, Wei Y, Wang D, Zhu B, Lemmon G, Jiao Y, Duan S, Wang Q, Du Q, Sun M, Bao J, Sun Y, Zhao J, Zhang H, Wu G, Liu J, Webster RG. Evolution of the H9N2 influenza genotype that facilitated the genesis of the novel H7N9 virus. Proc Natl Acad Sci USA. 2015;112:548–553. doi: 10.1073/pnas.1422456112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi W, Shi W, Li W, Huang L, Li H, Wu Y, Yan J, Jiao P, Zhu B, Ma J, Gao GF, Liao M, Liu D. Continuous reassortments with local chicken H9N2 virus underlie the human-infecting influenza A (H7N9) virus in the new influenza season, Guangdong, China. Protein Cell. 2014;5:878–882. doi: 10.1007/s13238-014-0084-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi X, Qian YH, Bao CJ, Guo XL, Cui LB, Tang FY, Ji H, Huang Y, Cai PQ, Lu B, Xu K, Shi C, Zhu FC, Zhou MH, Wang H. Probable person to person transmission of novel avian influenza A (H7N9) virus in Eastern China, 2013: epidemiological investigation. Bmj. 2013;347:f4752. doi: 10.1136/bmj.f4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qin Y, Horby PW, Tsang TK, Chen E, Gao L, Ou J, Nguyen TH, Duong TN, Gasimov V, Feng L, Wu P, Jiang H, Ren X, Peng Z, Li S, Li M, Zheng J, Liu S, Hu S, Hong R, Farrar JJ, Leung GM, Gao GF, Cowling BJ, Yu H. Differences in the epidemiology of human cases of avian influenza A(H7N9) and A(H5N1) viruses infection. Clin Infect Dis. 2015;61:563–571. doi: 10.1093/cid/civ345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ren L, Yu X, Zhao B, Wu F, Jin Q, Zhang X, Wang J. Infection with possible precursor of avian influenza A(H7N9) virus in a child, China, 2013. Emerg Infect Dis. 2014;20:1362–1365. doi: 10.3201/eid2008.140325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sleeman K, Guo Z, Barnes J, Shaw M, Stevens J, Gubareva LV. R292K substitution and drug susceptibility of influenza A(H7N9) viruses. Emerg Infect Dis. 2013;19:1521–1524. doi: 10.3201/eid1909.130724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suguitan AL, Jr, Matsuoka Y, Lau YF, Santos CP, Vogel L, Cheng LI, Orandle M, Subbarao K. The multibasic cleavage site of the hemagglutinin of highly pathogenic A/Vietnam/1203/2004 (H5N1) avian influenza virus acts as a virulence factor in a host-specific manner in mammals. J Virol. 2012;86:2706–2714. doi: 10.1128/JVI.05546-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.To KK, Chan JF, Chen H, Li L, Yuen KY. The emergence of influenza A H7N9 in human beings 16 years after influenza A H5N1: a tale of two cities. Lancet Infect Dis. 2013;13:809–821. doi: 10.1016/S1473-3099(13)70167-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang C, Yu H, Horby PW, Cao B, Wu P, Yang S, Gao H, Li H, Tsang TK, Liao Q, Gao Z, Ip DK, Jia H, Jiang H, Liu B, Ni MY, Dai X, Liu F, Van Kinh N, Liem NT, Hien TT, Li Y, Yang J, Wu JT, Zheng Y, Leung GM, Farrar JJ, Cowling BJ, Uyeki TM, Li L. Comparison of patients hospitalized with influenza A subtypes H7N9, H5N1, and 2009 pandemic H1N1. Clin Infect Dis. 2014;58:1095–1103. doi: 10.1093/cid/ciu053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang D, Yang L, Gao R, Zhang X, Tan Y, Wu A, Zhu W, Zhou J, Zou S, Li X, Sun Y, Zhang Y, Liu Y, Liu T, Xiong Y, Xu J, Chen L, Weng Y, Qi X, Guo J, Li X, Dong J, Huang W, Zhang Y, Dong L, Zhao X, Liu L, Lu J, Lan Y, Wei H, Xin L, Chen Y, Xu C, Chen T, Zhu Y, Jiang T, Feng Z, Yang W, Wang Y, Zhu H, Guan Y, Gao GF, Li D, Han J, Wang S, Wu G, Shu Y (2014) Genetic tuning of the novel avian influenza A(H7N9) virus during interspecies transmission, China, 2013. Euro Surveill 19(25) [DOI] [PubMed]
- 34.WHO (2013) Overview of the emergence and characteristics of the avian influenza A(H7N9) virus. http://www.who.int/influenza/human_animal_interface/influenza_h7n9/WHO_H7N9_review_31May13.pdf
- 35.William T, Thevarajah B, Lee SF, Suleiman M, Jeffree MS, Menon J, Saat Z, Thayan R, Tambyah PA, Yeo TW. Avian influenza (H7N9) virus infection in Chinese tourist in Malaysia, 2014. Emerg Infect Dis. 2015;21:142–145. doi: 10.3201/eid2101.141092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao XC, Li KB, Chen ZQ, Di B, Yang ZC, Yuan J, Luo HB, Ye SL, Liu H, Lu JY, Nie Z, Tang XP, Wang M, Zheng BJ (2014) Transmission of avian influenza A(H7N9) virus from father to child: a report of limited person-to-person transmission, Guangzhou, China, January 2014. Euro Surveill 19(25) [DOI] [PubMed]
- 37.Xu W, Sun Z, Liu Q, Xu J, Jiang S, Lu L. PA-356R is a unique signature of the avian influenza A (H7N9) viruses with bird-to-human transmissibility: potential implication for animal surveillances. J Infect. 2013;67:490–494. doi: 10.1016/j.jinf.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Yi L, Guan D, Kang M, Wu J, Zeng X, Lu J, Rutherford S, Zou L, Liang L, Ni H, Zhang X, Zhong H, He J, Lin J, Ke C. Family clusters of avian influenza A H7N9 virus infection in Guangdong Province, China. J Clin Microbiol. 2015;53:22–28. doi: 10.1128/JCM.02322-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yiu Lai K, Wing Yiu Ng G, Fai Wong K, Fan Ngai Hung I, Kam Fai Hong J, Fan Cheng F, Kwok Cheung Chan J. Human H7N9 avian influenza virus infection: a review and pandemic risk assessment. Emerg Microb Infect. 2013;2:e48. doi: 10.1038/emi.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu H, Gao Z, Feng Z, Shu Y, Xiang N, Zhou L, Huai Y, Feng L, Peng Z, Li Z, Xu C, Li J, Hu C, Li Q, Xu X, Liu X, Liu Z, Xu L, Chen Y, Luo H, Wei L, Zhang X, Xin J, Guo J, Wang Q, Yuan Z, Zhou L, Zhang K, Zhang W, Yang J, Zhong X, Xia S, Li L, Cheng J, Ma E, He P, Lee SS, Wang Y, Uyeki TM, Yang W. Clinical characteristics of 26 human cases of highly pathogenic avian influenza A (H5N1) virus infection in China. PloS One. 2008;3:e2985. doi: 10.1371/journal.pone.0002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu H, Cowling BJ, Feng L, Lau EH, Liao Q, Tsang TK, Peng Z, Wu P, Liu F, Fang VJ, Zhang H, Li M, Zeng L, Xu Z, Li Z, Luo H, Li Q, Feng Z, Cao B, Yang W, Wu JT, Wang Y, Leung GM. Human infection with avian influenza A H7N9 virus: an assessment of clinical severity. Lancet. 2013;382:138–145. doi: 10.1016/S0140-6736(13)61207-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu X, Jin T, Cui Y, Pu X, Li J, Xu J, Liu G, Jia H, Liu D, Song S, Yu Y, Xie L, Huang R, Ding H, Kou Y, Zhou Y, Wang Y, Xu X, Yin Y, Wang J, Guo C, Yang X, Hu L, Wu X, Wang H, Liu J, Zhao G, Zhou J, Pan J, Gao GF, Yang R, Wang J. Influenza H7N9 and H9N2 viruses: coexistence in poultry linked to human H7N9 infection and genome characteristics. J Virol. 2014;88:3423–3431. doi: 10.1128/JVI.02059-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeng H, Belser JA, Goldsmith CS, Gustin KM, Veguilla V, Katz JM, Tumpey TM. A(H7N9) virus results in early induction of proinflammatory cytokine responses in both human lung epithelial and endothelial cells and shows increased human adaption compared with avian H5N1 virus. J Virol. 2015;89(8):4655–4667. doi: 10.1128/JVI.03095-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu W, Li L, Yan Z, Gan T, Li L, Chen R, Chen R, Zheng Z, Hong W, Wang J, Smith DK, Guan Y, Zhu H, Shu Y. Dual E627K and D701N mutations in the PB2 protein of A(H7N9) influenza virus increased its virulence in mammalian models. Sci Rep. 2015;5:14170. doi: 10.1038/srep14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu W, Shu Y. Genetic tuning of avian influenza A (H7N9) virus promotes viral fitness within different species. Microb Infect Inst Pasteur. 2015;17:118–122. doi: 10.1016/j.micinf.2014.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.