

Abstract
H7N9 influenza A virus (IAV)-infected human cases are increasing and reported over 200 mortalities since its first emergence in 2013. Host inflammatory response contributes to the clearance of influenza virus; meanwhile, the induced “cytokine storm” also leads to pathological lesions. However, what inflammation-related response of the host for H7N9 influenza A virus infection to survival from injures of exuberant cytokine release is still obscure. In this research, expression pattern and histological distribution of inflammation-related genes, RIP3, NLRP3, IL-1β, TNF-α, Slit2 and Robo4 in the lung of BALB/c mice infected with two H7N9 IAV strains with only a PB2 residue 627 difference were investigated, as well as the histopathological injury of the lung. Results showed that significantly higher expression level of NLRP3, RIP3, IL-1β and TNF-α in H7N9-infected groups compared with the control would play a key role in driving lung pathological lesion. While the expression level of Slit2 and Robo4 in H7N9 rVK627E group had significantly increased trend than VK627 which might be the main factor to inhibit the interstitial pneumonia and infiltration. Also, H7N9 induced the histopathological changes in the lung of infected mice, and RIP3, NLRP3, IL-1β, TNF-α, Slit2 and Robo4 showed cell-specific distribution in the lung. The results will provide basic data for further research on the mechanism of inflammatory response and understanding of the role of site 627 in PB2 in H7N9 IAVs infection. In addition, enhancing the resilience of the host vascular system to the inflammatory response by regulation of Slit2–Robo4 signaling pathway might provide a novel strategy for H7N9 IAVs infection.
Keywords: H7N9, Influenza A virus, RIP3, NLRP3, IL-1β, TNF-α, Slit2, Robo4
Introduction
Since the first case of human infection in March 2013, continuous H7N9 influenza A viruses (IAV) infection cases were reported in both humans and birds, and more than 500 laboratory-confirmed human cases with over 30 % fatality highlight the potential pandemic threat [1]. Though H7N9 IAV is avian origin, it shows low pathogenicity in birds but with signs of adaptation to mammalian species, including increased ability to bind with mammalian cell receptors, and replicate at temperature close to the normal body temperature of mammals which is lower than that of birds. Almost all H7N9 IAVs isolated from human have an amino acid change at site 226 in HA which contributed to dual receptor-binding property [2]. A H7N9 IAV strain isolated from Anhui, China, can bind human receptors while it still retains the avian receptor-binding property [3]. An E-to-K amino acid change at residue 627 of polymerase basic protein 2 (PB2) occurred frequently in the H7N9 IAV isolates obtained from human, but not in viruses isolated from poultry. Mok et al. [4] tested the importance of PB2-627K using A/Shanghai/2/2013 as backbone and found that this amino acid change contributed to mammalian adaptation. Mice infected with the H7N9 IAV mutant containing the avian signature protein PB2-627E showed a marked decrease in weight loss and pathological lesions compared to mice infected with the wild-type strain (PB2-627K) [4]. H7N9 can induce pneumonia and hypoxic hepatitis [5], leading to systemic immune response [6]. As to human beings, clinical presentation of H7N9 IAV infection is characterized by rapidly progressing severe pneumonia and induced complications such as acute respiratory distress syndrome (ARDS), septic shock and multi-organ failure [7]. Host clearance of IAV requires the development of host’s inflammation-related responses [8, 9], especially associated with exuberant pulmonary inflammation characterized by influx of inflammatory cells and production of proinflammatory cytokines including IL-1β, TNF-α and so forth. However, the detailed systematic data of H7N9 IAVs inducing the inflammatory responses of host are still obscure.
Pattern recognition receptors (PRRs) are responsible for identifying pathogen-associated molecular patterns which promote the initiation and orchestration of innate and adaptive immune response [10]. At the early stage of IAVs infection, the viral RNA, which is present in infected cells, is recognized as foreign by various PRRs, which leads to the secretion of type I interferon (IFNs), pro-inflammatory cytokines, eicosanoids and chemokines. Nod-like receptors (NLRs), one of pathogen-associated molecular patterns (PAMPs), play a central role in inflammation development in virus infection and regulate the host response to adaptive immunity [11]. Among various types of inflammasome, the NLRP3 inflammasome is well characterized in a variety of mammalian cells and contributes to regulate the cleavage and maturation of pro-IL-1β, the inactive precursor of IL-1β [12]. IL-1β binding with its receptors induces the activation of multiple cytokines such as TNF-α [13], playing the vital role during the early stage of host defense against IAVs infection [14]. With the disease development, IAVs-infected dead cells release intracellular components which stimulate the immune cells. This is dependent on the activity of the receptor-interacting protein 3(RIP3) which could switch TNF-induced cell death from apoptosis to necrosis by regulating TNF-induced reactive oxygen species production [15, 16] and accelerate systematic inflammation and thus lead to mortality [17]. In addition, RIP3 initiates mitochondrial fission to active NLRP3 inflammasome [18] which would accelerate the inflammatory response. Though these cytokines and chemokines could result in infiltration of leukocytes to the site of infection, which would be beneficial, the resulting “cytokine storm” also contributes to pathogenesis and mortality [19]. The inflammatory cytokines, such as IL-1β and TNF-α, which directly combat the infectious agent and activate additional immune and inflammation response, injure the host by triggering leakage from capillaries, tissue edema and organ failure. This is one of the major reasons that lead to the pathogenesis and the poor medical outcome during the late stage of IAV infection. Therefore, for survival of the infected patients or animals, a balance must be struck between the protective response and the excessive inflammatory response.
Previous reports showed that limiting the disruptive effects of proinflammatory mediators on the vasculature is vital to limit the injury of host, which is associated with the Slit2 and its receptor, the transmembrane roundabout receptor 4 (Robo4) [20, 21]. Slit2 is a secreted protein, which was previously known for its role of repulsion in axon guidance and neuronal migration, can also inhibit leukocyte chemotaxis induced by chemotactic factors [22]. During IAVs infection, cytokines produced in host inflammatory response lead to internalization of VE-cadherin and disruption of barrier function, which results in vascular leak and accumulation of protein-rich edema fluid in the alveolar space. Binding with Robo4, Slit2 enhances vascular barrier function against multiple cytokines by enhancing VE-cadherin, a primary molecular determinant of intact barrier function in the endothelium, at the cell surface [19]. Thus, this strengthened barrier can protect host from the lethal effects of inflammatory response induced by influenza infection.
How the host inflammation-related genes expressed in H7N9 IAVs infection? Does Slit2–Robo4 pathway play any role to protect host from the effects of inflammatory response induced by H7N9 IAV infection? Is there any innate relationship between the pathogenicity of H7N9 IAV and the host inflammatory response? Here, we detected and analyzed the expression profile and histological distribution of RIP3, NLRP3, IL-1β, TNF-α, Slit2, Robo4 in the lung at different time points during infection in BALB/c mice by H7N9 IAV strains only with difference at site 627 in PB2. The results will provide basic data for further study of the mechanism of inflammatory response and understanding of the role of site 627 of PB2 in H7N9 IAVs infection.
Materials and methods
Ethics statement
The animal experiments were approved by the Institutional Animal Care and Use Committee at the South China Agricultural University (Certification Number: CNAS BL0011) and performed in accordance with the “Guidelines for Experimental Animals” of the Ministry of Science and Technology (Beijing, China) and in association with Assessment and Accreditation of Laboratory Animal Care International-accredited facility.
Viruses and BALB/c mice infection by H7N9 influenza A virus
Two strains of H7N9 IAVs were used in this research. A/Chicken/Guangdong/G1/2013 (VK627) was isolated from live poultry markets of Guangdong Province. The other was site-mutation virus A/Chicken/Guangdong/G1-pB2627E, with PB2-627K (wild-type strain) to PB2-627E (rVK627E). Our previous research showed that VK627 did not induce serious weight loss or clinical signs while rVK627E showed a marked decrease in weight loss compared to mice infected with the wild-type strain [23, 24].
Thirty-six SPF female BALB/c mice (18.0–20.0 g, 5 weeks) from the Guangdong Experimental Animal Centre (Guangzhou, China) were randomly divided into three groups and set for sampling three every group every time point. After being anesthetized with CO2, the mice were inoculated intranasally with 106 EID50 of VK627 or rVK627E influenza virus; the mice in control were treated only by PBS. All mice were weighed before challenging and surveyed for daily weight loss as a measure of morbidity. At 1, 3, 5 and 7 days post-inoculation (dpi), the lungs of three euthanized mice in each groups, respectively, were collected promptly at the same time point of each day. Each lung was divided into two parts. One part was frozen in liquid nitrogen for 2 h before storage at −86 °C for RNA extraction, and the other was fixed in 10 % neutralized buffered formalin for histopathological and immunohistochemical detection.
RNA extraction, cDNA preparation and real-time PCR
Total RNA of all samples was extracted, treated, quality determined, stored and reverse-transcribed to cDNA according to our previous reports [24–27]. Relative expression levels of RIP3, NLRP3, IL-1β, TNF-α, Slit2 and Robo4 were determined by real-time PCR using DNA Engine 7500 Continuous Fluorescence Detection System (Applied Biosystems, CA, USA) and SYBR® Premix Ex TaqTM Kit (Takara Biotech, Dalian, China). Primers used for qRT-PCR (Table 1) were designed with Oligo 7 software (Molecular Biology Insights Inc., Cascade, CO, USA).The melting curves for each PCR were carefully analyzed to avoid non-specific amplifications in PCR products. The relative expression levels of these genes were calculated by normalizing the levels of gene transcripts to that of β-actin transcripts using a relative standard curve method through 2−ΔΔCt formula. The data were confirmed with three repeat experiments. The statistical analysis was performed by SPSS software (version 20.0), and the figure was made by GraphPad Prism (GraphPad Software, La Jolla, CA). One-way ANOVA and t test were used to determine statistical significance between samples in relative expression levels of RIP3, NLRP3, IL-1β, TNF-α, Slit2 and Robo4 among different groups at different time point, respectively.
Table 1.
Primers used for quantitative real-time PCR
| Genes | Forward primer (5′–3′) | Forward primer (5′–3′) |
|---|---|---|
| Rip3 | CCAGAGAGCCAAGCCAAAGAG | AGCCACGGGGTCAGAAGATGT |
| Nlrp3 | TGAAGTGGATTGAAGTGAAAGCC | TGTGAAAAAAACCCAGGGAAAGC |
| IL-1β | CCTGTGTCTTTCCCGTGGAC | CATCTCGGAGCCTGTAGTGC |
| TNF-α | CTTCCAGAACTCCAGGCGGT | CACTTGGTGGTTTGCTACGACG |
| Slit2 | GTTTCATAGGACTCGGCTCTGT | GGAAGTAGGGTTTTTGGCATCG |
| Robo4 | CTCATGGTGGAAAGACGGGAAA | TGAATGCGAACAGCCAGAAG |
| β-Actin | GGTGGGAATGGGTCAGAAGGA | TGGCTGGGGTGTTGAAGGTC |
Histopathological and immunohistochemical detection
Serial sections were taken from the same samples for histopathological observation using the routine HE staining and immunohistochemical detection. Immunohistochemical detection was performed as described in our previous reports [25–27]. Briefly, the slices were then incubated with the corresponding antibodies (antibody for RIP3, abcam, Cat. No., ab152130; antibody for NLRP3, abcam, Cat. No., ab4207; antibody for IL-1β, santa crus biotechnology, Cat. No., sc-7884; antibody for TNF-α, abcam, Cat. No., ab6671; antibody for Slit2, abcam, Cat. No., ab134166; antibody for Robo4, santa crus biotechnology, Cat. No., sc-166872) for 1.5 h at 37 °C according to the manufactures’ guidelines. Based on the origin of primary antibody, appropriate HRP-conjugated IgG originated from sheep or horse was selected as secondary antibody. The secondary antibodies were obtained from ZSBG-BIO (Beijing, China) except for RIP3 which obtained from Cell Signaling Technology (Beverly, MA, USA). After incubation with the secondary antibody for 1.5 h at 37 °C, the slices were washed with PBS at 5-min intervals a total of three times. DAB (3, 3′-diaminobenzidine tetrahydrochloride) Kit (ZSGQ-BIO, China) was used for coloration. Slices were re-dyed with hematoxylin for 1 min and washed with running water for 10 min. Finally, the slices were dehydrated using graded ethanol, vitrified by dimethylbenzene and deposited in neutral balsam.
Results
Virulence of H7N9 IAVs in BALB/c mice
The weight loss, clinical signs and virus isolation from tissues after inoculation with VK627 and rVK627E H7N9 IAV in mice were similar to our previous reports [23, 24]. The histopathological changes in lungs of mice in different groups are different between the two infected groups at different time points. At 1 dpi, VK627 group showed serious diffuse venous hyperemia in alveolar wall while rVK627E showed only slightly and locally venous hyperemia in alveolar wall. At 3 dpi, VK627 showed liquid infiltration in some alveolar and still serious venous hyperemia, while rVK627E still showed slightly and locally venous hyperemia in alveolar wall. At 5–7 dpi, VK627 virus induced slightly interstitial pneumonia and infiltration of inflammatory cells, while rVK627E still showed slightly and locally venous hyperemia in alveolar wall.
Statistic analysis of expression level of RIP3 and NLRP3 in the lung of BALB/c mice
In this research, RIP3 and NLRP3 were chosen as inflammatory-activation relative genes and detected their expression level by real-time PCR (Fig. 1), considering about their vital role in the activation of inflammatory response. As to the expression level of Rip3 (Fig. 1a), comparing with the control group, both of the H7N9 IAV-infected groups showed significantly increased. In the VK627 group, the expression level of RIP3 increased from 1 to 3 dpi and reached its peak at 5 dpi while decreased to the level similar to 1 dpi at 7 dpi. It is noticeable that the level of RIP3 kept significantly higher (P < 0.01) than other two groups from 1 to 7 dpi. Differently, in the rVK627E group, the expression of RIP3 level was significantly higher (P < 0.01) than that of the control group at the start and end, which was different with that in 3 dpi and 5 dpi (P < 0.05). For the expression level of NLRP3 (Fig. 1b), both of the H7N9 IAV-infected groups showed significantly higher compared with the control group. In the VK627 group, the expression level increased from 1 to 3 dpi and reached the maximum and then continuously decreased at 5 and 7 dpi. In the rVK627E group, the expression level exhibited similar trend despite the maximum at 5 dpi. Interestingly, the expression level of NLRP3 in the VK627 group was significantly higher than the other two groups (P < 0.01) and that in the rVK627E group was just significantly higher than in the control groups (P < 0.01). To sum up, both of the strains of virus can induce higher expression level of RIP3 and NLRP3 in the lung of BALB/c mice.
Fig. 1.
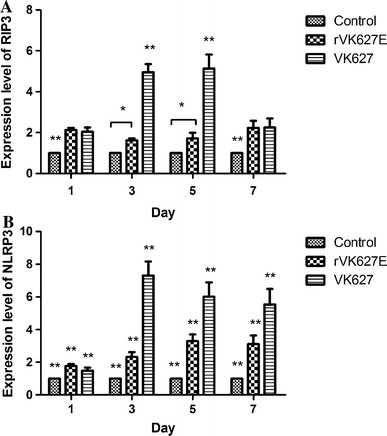
Relative mRNA expression levels of RIP3 (a) and NLRP3 (b) in the lung of H7N9 AIV-infected and control BALB/c mice at different time points. *P < 0.05; **P < 0.01. Data are represented as mean ± standard deviation (SD)
Statistic analysis of expression level of IL-1β and TNF-α in the lung of BALB/c mice
IL-1β binding with its receptors induces the activation of multiple cytokines such as TNF-α [13], playing the vital role during the early stage of host defense against IAVs infection [14] and acting as the downstream cytokines of NLRP3 inflammasome. In this research, IL-1β and TNF-α were chosen as inflammation relative cytokines and detected their expression level by real-time PCR. As to the expression level of IL-1β (Fig. 2a), the trend was similar to that of the NLRP3 that both of the H7N9 IAV-infected groups showed significantly higher compared with the control group. In the VK627 group, the expression level of IL-1β increased from 1 to 3 dpi and reached the maximum and then continuously decreased at 5 and 7 dpi, though it kept the highest level (P < 0.01) during the whole period compared with the other two groups. Differently, in the rVK627E group, the expression level reached the maximum at 3 dpi. Though much lower than the VK627 group (P < 0.01), the expression level of IL-1β in rVK627E group was significantly higher than that in control group during this period (P < 0.01) except at 3 dpi (P < 0.05). Differently, though the expression level of TNF-α in H7N9 IAV-infected groups showed significantly higher compared with the control group, the trend of each group was diverse from that of the IL-1β (Fig. 2b). In the VK627 group, the expression level of TNF-α showed highest level at 1 and 5 dpi and the lowest level at 3 dpi. Interestingly, the expression in the rVK627E group showed gentle decrease during the whole period with the highest level at 1 dpi and a stable low level at 5 and 7 dpi. Overall, the expression level of TNF-α in the VK627 group was significantly higher than those of the other two groups (P < 0.01) and that in the rVK627E group was just significantly higher than in the control groups (P < 0.01). Both of the strains of virus can induce higher expression level of IL-1β and TNF-α in the lung of BALB/c mice.
Fig. 2.
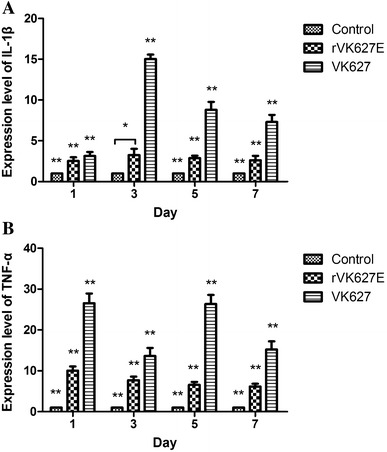
Relative mRNA expression levels of IL-1β (a) and TNF-α (b) in the lung of H7N9 IAV-infected and control BALB/c mice at different time points *P < 0.05; **P < 0.01. Data are represented as mean ± standard deviation (SD)
Statistic analysis of expression level of Slit2 and Robo4 in the lung of BALB/c mice
As to the expression level of Slit2, different infected groups showed various trends compared with that of control (Fig. 3a). In the VK627 group, the expression level of Slit2 showed a stable increase during the whole period and with the lowest level at 1 dpi, which was significantly lower than the other two groups (P < 0.01). Similar trend also showed that in the rVK627E group the level increased from 1 to 7 dpi with the minimum at 1 dpi which was significantly lower than that in control group (P < 0.01), and the maximum at 7 dpi, significantly higher than that in the other two groups (P < 0.01). It was noticeable that the expression level of Slit2 in both VK627 and rVK627E groups was significantly lower (P < 0.01) than that in control group at 1 dpi and increased at 3 dpi. Then, expression level of Slit2 in rVK627E group was significantly higher than that in control group at 5 and 7 dpi while that in VK627 showed no significant difference compared with that of control. Similarly, the expression level of Robo4 (Fig. 3b) exhibited a parallel trend compared with that of Slit2. In the VK627 group, initiated with a continuous increase from 1 to 5 dpi, with the lowest level at 1 dpi compared with the other two groups (P < 0.01), the expression level of Robo4 gently declined at 7 dpi. Differently, the expression in rVK627E group increased from 1 to 7 dpi with the minimum at 1 dpi, which was significantly lower than that in control (P < 0.01), and the maximum at 7 dpi, significantly higher than that in the other two groups (P < 0.01). The expression level of Robo4 in both VK627 and rVK627E group was significantly lower (P < 0.01) than that in control group at 1 dpi and increased to the same level as control group at 3 dpi. Then, expression level of Robo4 in the rVK627E group was significantly higher than that in control group at 5 and 7 dpi while that in VK627 showed no significant difference compared with the control group. These results showed that both of the VK627 and rVK627E strains of H7N9 IAV inhibited the expression of Slit2 and Robo4 in lung at the early stage during H7N9 infection (from 1 to 3 dpi). After that, expression level of Slit2 and Robo4 increased dramatically in rVK627E group, while it remained similar level as non-infected in VK627 group.
Fig. 3.

Relative mRNA expression levels of Slit2 (a) and Robo4 (b) in the lung of H7N9 IAV-infected and control BALB/c mice at different time points. *P < 0.05; **P < 0.01. Data are represented as mean ± standard deviation (SD)
Immunohistochemical detection of RIP3, NLRP3, IL-1β, TNF-α, Slit2 and Robo4
Immunohistochemical detection was performed to show the cell-specific distribution of the protein of RIP3, NLRP3, IL-1β, TNF-α, Slit2 and Robo4 in the lung of BALB/c mice. RIP3 protein can be detected in all groups. For the control, the epithelial cell of alveolar wall exhibited mild positive staining, which was similar to that in pulmonary arterial vascular endothelial cells. Smooth muscle cells of blood vessel and tracheal submucosal layer are medium positive (Fig. 4a), while they are medium to strong positive of the same cells in rVK627E and VK627 groups (Fig. 4b, c). As to the NLRP3 protein, for the control, the positive cells and distribution of NLRP3 in lung were consistent with our previous study [26, 27] (Fig. 4d). The infiltrated inflammatory cells and exudation were positive in the lung of infected groups (Fig. 4e, f). IL-1β in the lung of the control showed some alveolar epithelial cells of the pulmonary alveoli and bronchiole were very weak positive (Fig. 4g) according to our previous research [26], while they were mild to strong positive of the same cells in rVK627E and VK627 groups (Fig. 4h, i). In terms of the TNF-α, the positive cells were mainly the epithelial cells of the pulmonary alveoli in mice of the control (Fig. 4j) according to our previous research [26], while stronger positives were in the epithelial cells of the pulmonary alveoli as well as the epithelial cells and the submucosa of bronchi and bronchioles in both VK627- and rVK627E-infected mice (Fig. 4k, l). The alveolar epithelial cells and mucosal epithelial cells of trachea were medium to strong positive staining for Slit2 protein expression in control and rVK627E groups (Fig. 4m, n), while they were weak positive of the same cells in VK627 groups (Fig. 4o). Similarly, for the Robo4, the location of positive cells in the lung had consistency to that of Slit2 with different positive intensity, which was weak to mild positive staining in the control and VK627 group (Fig. 4p, r) and mild to medium positive staining in the rVK627E group (Fig. 4q).
Fig. 4.
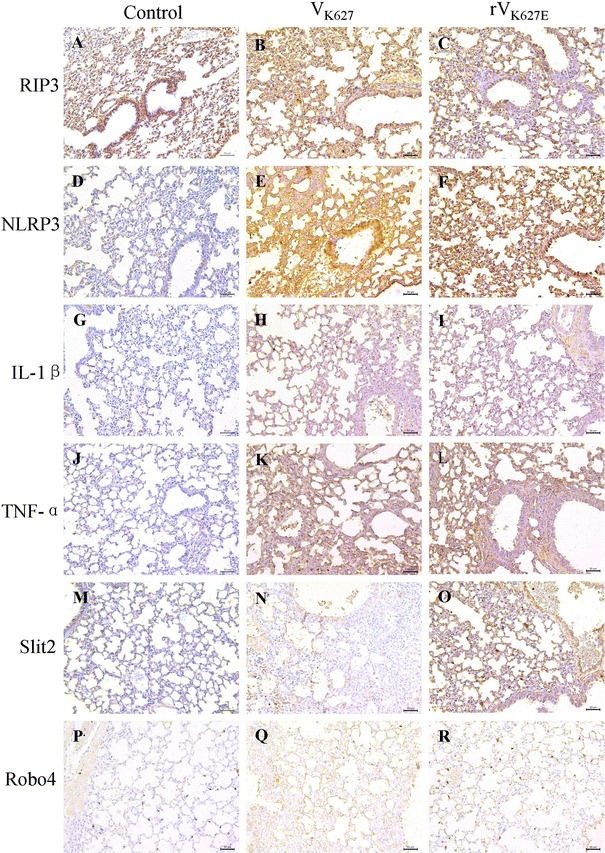
Distribution of RIP3 (a–c), NLRP3 (d–f), IL-1β (g–i), TNF-α (j–l), Slit2 (m–o), Robo4 (p–r) in the lung of H7N9 AIV-infected BALB/c mice and the controls at 5 dpi. Scale bar 50 μm
Discussion
Novel avian-origin H7N9 influenza A virus has emerged in China since the first human case reported in 2013, and there were 655 laboratory-confirmed cases reported in mainland China by 15 June 2015 [28]. The clinical characteristics of these patients often included fever and cough, pneumonia and acute respiratory distress syndrome (ARDS) [29, 30], and human H7N9 IAV infection cases showed the significant association between a longer incubation period and a greater risk of death [28]. Researches about host responses induced by influenza virus showed that through the recognition of pathogen-associate molecular patterns (PAMPs) by various pattern recognition receptors (PRRs) [31, 32], mainly containing the Toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I) and NOD-like receptor family members (NLRs) [33–37], the host can block virus replication and promote viral clearance. Among these PRRs, NLRP3 is vital and efficient for cell-intrinsic recognition [38] and its activation and function is closely associated with RIP3. Activation of NLRP3 inflammasome complexes results in the autocatalytic processing of pro-caspase 1 into its active from and then cleaves pro-IL-1β into IL-1β. Then mediated by TLR, IL-1 receptor (IL-1R) and tumor necrosis factor receptor (TNFR) signaling, cytokine production and secretion are triggered including IL-1β and TNF-α [39], which contributes to the defense against IAVs infection of host [14, 40].
Activation of inflammatory response to infection facilitates virus clearance and produces excessive pulmonary inflammation and tissue damage leading to high pathogenicity and lethality [9, 41]. In this research, we considered the mRNA expression and protein distribution of Slit2 and Robo4 during H7N9 IAV infection, which could limit the disruptive effects of proinflammatory mediators on the vasculature. Previous reports showed that by mounting a protective adaptive immune response, the host could survive from the infection of sub-lethal doses of influenza A virus or with inactive virus [9, 42]. In this research, we choose the H7N9 IAV strains of A/Chicken/Guangdong/G1/2013 (VK627) and its site-mutation virus (rVK627E) with low pathogenic, which would be suitable for the research of the potential mechanism of inflammatory response induced by IAV infection in mice.
Result of the virulence of H7N9 IAV in BALB/c mice demonstrated that VK627 caused more serious histopathological changes than that in rVK627E group. It is noticeable that after 5 dpi, VK627 virus induced continuously slightly interstitial pneumonia with infiltration of inflammatory cells, whereas the rVK627E virus showed only slightly and locally venous hyperemia in alveolar wall. Coincidently, mRNA expression level of RIP3, NLRP3, IL-1β and TNF-α continuously significantly higher in VK627 group than that in rVK627E, which might contribute to the more serious interstitial pneumonia in VK627 group. This result was similar to that of Mok et al. [4]. In return, the dramatic increase expression level of Slit2 and Robo4 after 3 dpi might be the main factor to inhibit the interstitial pneumonia and infiltration in rVK627E group, while interstitial pneumonia and infiltration in the VK627 group maintained, in which the expression of Slit2 and Robo4 exhibited similar level to control group. However, it is interesting that from 1 to 3 dpi, the expression of Slit2 and Robo4 in both infected groups was significantly lower than in control, and it seems that there would be a potential mechanism of IAVs to inhibit the Slit2–Robo4 signaling pathway which deserves further detailed research. Similarly, immunohistochemistry showed that stronger stain of RIP3, NLRP3, IL-1β and TNF-α protein was detected in the lung tissues of VK627 group than that in rVK627E group at 5 dpi, while stronger stain of Slit2 and Robo4 protein was detected in rVK627E group than that in VK627 group at the same time point. This implied that both VK627 and rVK627E H7N9 IAV could induce the host response in the lung of BALB/c mice, while more serious tissue lesion was companied with higher level of cytokines including IL-1β and TNF-α during VK627 infection compared with rVK627E. However, BALB/c mice infected with rVK627E might be associated with the function of Slit2–Robo4 signaling pathway.
In summary, both VK627 and rVK627E H7N9 IAV can activate host inflammation responses in the lung of BALB/c mice. NLRP3, RIP3, IL-1β and TNF-α would play a key role in driving lung lesions while Slit2-Robo4 signal pathway might contribute to protect the host form the excessive pulmonary inflammation. The results will provide basic data for further study of the mechanism of immune and inflammation response and further understanding of the role of site 627 in PB2 in infection induced by H7N9 IAVs. In addition, the vascular system in the lung which regulated by Slit2–Robo4 signaling pathway might provide a potential important role for host survival from inflammatory lesion induced by H7N9 IAVs infection.
Acknowledgments
This work was partially supported by Science and Technology nova Program of Pearl River of Guangzhou (2014J2200072), Development Program for Excellent Young Teachers in Guangdong Province (2013), Program for National Broiler Industry (nycytx-42-G3-03) and Science and Technology Planning Project of Guangdong Province (20140224, 2013B020224003).
Compliance with ethical standards
Conflict of interest
The authors have declared that no competing interests exist.
Contributor Information
Ming Liao, Email: mliao@scau.edu.cn.
Zhangyong Ning, Email: ningzhyong@scau.edu.cn.
References
- 1.Zhu H, Lam TT, Smith DK, Guan Y. Emergence and development of H7N9 influenza viruses in China. Curr Opin Virol. 2016;16:106–113. doi: 10.1016/j.coviro.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 2.Yamayoshi S, Fukuyama S, Yamada S, Zhao D, Murakami S, et al. Amino acids substitutions in the PB2 protein of H7N9 influenza A viruses are important for virulence in mammalian hosts. Sci Rep. 2015;5:8039. doi: 10.1038/srep08039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi Y, Zhang W, Wang F, Qi J, Wu Y, et al. Structures and receptor binding of hemagglutinins from human-infecting H7N9 influenza viruses. Science. 2013;342:243–247. doi: 10.1126/science.1242917. [DOI] [PubMed] [Google Scholar]
- 4.Mok CK, Lee HH, Lestra M, Nicholls JM, Chan MC, et al. Amino acid substitutions in polymerase basic protein 2 gene contribute to the pathogenicity of the novel A/H7N9 influenza virus in mammalian hosts. J Virol. 2014;88:3568–3576. doi: 10.1128/JVI.02740-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Liu J, Yu L, Zhou N, Ding W, et al. Prevalence and characteristics of hypoxic hepatitis in the largest single-centre cohort of avian influenza A(H7N9) virus-infected patients with severe liver impairment in the intensive care unit. Emerg Microbes Infect. 2016;5:e1. doi: 10.1038/emi.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng Y, Hu L, Lu S, Chen Q, Zheng Y, et al. Molecular pathology analyses of two fatal human infections of avian influenza A(H7N9) virus. J Clin Pathol. 2015;68:57–63. doi: 10.1136/jclinpath-2014-202441. [DOI] [PubMed] [Google Scholar]
- 7.Cao HF, Liang ZH, Feng Y, Zhang ZN, Xu J, et al. A confirmed severe case of human infection with avian-origin influenza H7N9: a case report. Exp Ther Med. 2015;9:693–696. doi: 10.3892/etm.2014.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braciale TJ, Sun J, Kim TS. Regulating the adaptive immune response to respiratory virus infection. Nat Rev Immunol. 2012;12:295–305. doi: 10.1038/nri3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 11.Carneiro LA, Magalhaes JG, Tattoli I, Philpott DJ, Travassos LH. Nod-like proteins in inflammation and disease. J Pathol. 2008;214:136–148. doi: 10.1002/path.2271. [DOI] [PubMed] [Google Scholar]
- 12.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang N, Zhang X, Liu X, Wang H, Xue J, et al. Chrysophanol inhibits NALP3 inflammasome activation and ameliorates cerebral ischemia/reperfusion in mice. Mediat Inflamm. 2014;2014:370530. doi: 10.1155/2014/370530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao G, Liu C, Kou Z, Gao T, Pan T, et al. Differences in the pathogenicity and inflammatory responses induced by avian influenza A/H7N9 virus infection in BALB/c and C57BL/6 mouse models. PLoS ONE. 2014;9:e92987. doi: 10.1371/journal.pone.0092987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 16.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 17.Meng L, Jin W, Wang X. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci USA. 2015;112:11007–11012. doi: 10.1073/pnas.1514730112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rayamajhi M, Miao EA. The RIP1–RIP3 complex initiates mitochondrial fission to fuel NLRP3. Nat Immunol. 2014;15:1100–1102. doi: 10.1038/ni.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.London NR, Zhu W, Bozza FA, Smith MC, Greif DM, et al. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010;2:19r–23r. doi: 10.1126/scitranslmed.3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim R, Lappas M. Slit2 exerts anti-inflammatory actions in human placenta and is decreased with maternal obesity. Am J Reprod Immunol. 2015;73:66–78. doi: 10.1111/aji.12334. [DOI] [PubMed] [Google Scholar]
- 21.Chaturvedi S, Robinson LA. Slit2–Robo signaling in inflammation and kidney injury. Pediatr Nephrol. 2015;30:561–566. doi: 10.1007/s00467-014-2825-4. [DOI] [PubMed] [Google Scholar]
- 22.Wu JY, Feng L, Park HT, Havlioglu N, Wen L, et al. The neuronal repellent Slit inhibits leukocyte chemotaxis induced by chemotactic factors. Nature. 2001;410:948–952. doi: 10.1038/35073616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qi W, Shi W, Li W, Huang L, Li H, et al. Continuous reassortments with local chicken H9N2 virus underlie the human-infecting influenza A (H7N9) virus in the new influenza season, Guangdong, China. Protein Cell. 2014;5:878–882. doi: 10.1007/s13238-014-0084-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao C, Ma W, Sun N, Huang L, Li Y, et al. PB2-588 V promotes the mammalian adaptation of H10N8, H7N9 and H9N2 avian influenza viruses. Sci Rep. 2016;6:19474. doi: 10.1038/srep19474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu M, Qi W, Huang Z, Zhang K, Ye J, et al. Expression profile and histological distribution of IFITM1 and IFITM3 during H9N2 avian influenza virus infection in BALB/c mice. Med Microbiol Immunol. 2015;204:505–514. doi: 10.1007/s00430-014-0361-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu M, Zhang K, Qi W, Huang Z, Ye J, et al. Expression pattern of NLRP3 and its related cytokines in the lung and brain of avian influenza virus H9N2 infected BALB/c mice. Virol J. 2014;11:229. doi: 10.1186/s12985-014-0229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Z, Yu M, Tong S, Jia K, Liu R, et al. Tissue-specific expression of the NOD-like receptor protein 3 in BALB/c mice. J Vet Sci. 2014;15:173–177. doi: 10.4142/jvs.2014.15.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Virlogeux V, Yang J, Fang VJ, Feng L, Tsang TK, et al. Association between the severity of influenza A (H7N9) virus infections and length of the incubation period. PLoS ONE. 2016;11:e148506. doi: 10.1371/journal.pone.0148506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y, Liang W, Yang S, Wu N, Gao H, et al. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet. 2013;381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen Z, Chen Z, Li X, Xu L, Guan W, et al. Host immunological response and factors associated with clinical outcome in patients with the novel influenza A H7N9 infection. Clin Microbiol Infect. 2014;20:O493–O500. doi: 10.1111/1469-0691.12505. [DOI] [PubMed] [Google Scholar]
- 31.Janeway CJ. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):1–13. doi: 10.1101/SQB.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 33.Koyama S, Ishii KJ, Kumar H, Tanimoto T, Coban C, et al. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J Immunol. 2007;179:4711–4720. doi: 10.4049/jimmunol.179.7.4711. [DOI] [PubMed] [Google Scholar]
- 34.Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heer AK, Shamshiev A, Donda A, Uematsu S, Akira S, et al. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J Immunol. 2007;178:2182–2191. doi: 10.4049/jimmunol.178.4.2182. [DOI] [PubMed] [Google Scholar]
- 36.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Jiang F, Ramanathan A, Miller MT, Tang GQ, Gale MJ, et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pang IK, Iwasaki A. Control of antiviral immunity by pattern recognition and the microbiome. Immunol Rev. 2012;245:209–226. doi: 10.1111/j.1600-065X.2011.01073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 40.Pang IK, Ichinohe T, Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat Immunol. 2013;14:246–253. doi: 10.1038/ni.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.García-Ramírez RA, Ramírez-Venegas A, Quintana-Carrillo R, Camarena ÁE, Falfán-Valencia R, et al. TNF, IL6, and IL1B polymorphisms are associated with severe influenza A (H1N1) virus infection in the Mexican population. PLoS ONE. 2015;10:e144832. doi: 10.1371/journal.pone.0144832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol. 2006;7:517–523. doi: 10.1038/ni1322. [DOI] [PubMed] [Google Scholar]