

Abstract
The purpose of this study was to investigate whether atrial overexpression of angiotensin-converting enzyme 2 (ACE2) by homogeneous transmural atrial gene transfer can reverse atrial remodeling and its mechanisms in a canine atrial-pacing model. Twenty-eight mongrel dogs were randomly divided into four groups: Sham-operated, AF-control, gene therapy with adenovirus-enhanced green fluorescent protein (Ad-EGFP) and gene therapy with Ad-ACE2 (Ad-ACE2) (n = 7 per subgroup). AF was induced in all dogs except the Sham-operated group by rapid atrial pacing at 450 beats/min for 2 weeks. Ad-EGFP and Ad-ACE2 group then received epicardial gene painting. Three weeks after gene transfer, all animals except the Sham group underwent rapid atrial pacing for another 3 weeks and then invasive electrophysiological, histological and molecular studies. The Ad-ACE2 group showed an increased ACE2 and Angiotensin-(1–7) expression, and decreased Angiotensin II expression in comparison with Ad-EGFP and AF-control group. ACE2 overexpression attenuated rapid atrial pacing-induced increase in activated extracellular signal-regulated kinases and mitogen-activated protein kinases (MAPKs) levels, and decrease in MAPK phosphatase 1(MKP-1) level, resulting in attenuation of atrial fibrosis collagen protein markers and transforming growth factor-β1. Additionally, ACE2 overexpression also modulated the tachypacing-induced up-regulation of connexin 40, down-regulation of connexin 43 and Kv4.2, and significantly decreased the inducibility and duration of AF. ACE2 overexpression could shift the renin–angiotensin system balance towards the protective axis, attenuate cardiac fibrosis remodeling associated with up-regulation of MKP-1 and reduction of MAPKs activities, modulate tachypacing-induced ion channels and connexin remodeling, and subsequently reduce the inducibility and duration of AF.
Electronic supplementary material
The online version of this article (doi:10.1007/s00395-015-0499-0) contains supplementary material, which is available to authorized users.
Keywords: Atrial fibrillation, Angiotensin-converting enzyme 2, Gene therapy, Atrial remodeling, Renin–angiotensin system
Introduction
Atrial fibrillation (AF) is the most common clinical arrhythmia associated with cardiovascular morbidity and increased mortality. Substantial evidence have demonstrated that activation of the local renin–angiotensin system (RAS), especially angiotensin II (Ang II), plays an important role in atrial remodeling characterized by interstitial fibrosis, and contributes to the onset and maintenance of AF in paced animal models [10, 13, 22, 32, 34, 42]. Angiotensin-converting enzyme (ACE)-dependent increase in the levels of activated extracellular signal-regulated kinases (ERKs) and mitogen-activated protein kinases (MAPKs) have been reported in atrial tissues of AF patients and animal models [1, 10, 13, 18, 22, 32, 36]. Experimental studies have proven that this increase in MAPKs, which has been suggested to have a crucial role in atrial fibrosis, can be reduced by treatment with ACE inhibitors [1, 18, 24, 36, 37]. MAPK phosphatase 1 (MKP-1), an important member of the dual-specificity phosphatase family, is a critical counteracting phosphatase that directly regulates the magnitude and duration of p38-MAPK and ERK auto-phosphorylation and activation [5]. MKP-1 is also expressed in cardiomyocytes and is involved in cardiomyocyte apoptosis and cardiac hypertrophy [6]. Caunt et al. [5] found that MKP-1 negatively regulated the inflammatory response of the innate immune system by inhibiting MAPK activation. However, whether the dynamic regulation between MAPKs and MKP-1 participates in the pathophysiologic mechanism of cardiac remodeling, especially for atrial fibrotic remodeling, has not been established.
Angiotensin-converting enzyme 2 (ACE2) is a monocarboxypeptidase that metabolizes vasoconstrictive octopeptide Ang II into vasodilative heptapeptide angiotensin-(1–7) [Ang-(1–7)], thereby functioning as a negative regulator of the renin–angiotensin system. Recent studies [15, 43] have shown that ACE2 overexpression could suppress Ang II-mediated myocardial hypertrophy and fibrosis, and prevent cardiac dysfunction. However, the detailed signal transduction mechanism following the activation of Mas receptor induced by ACE2/ANG-(1–7) is still poorly understood; and the effects of ACE2/Ang-(1–7) on atrial structural remodeling are yet to be determined.
Meanwhile, on the electrical remodeling, several lines of evidence have suggested that Ang II had direct electrophysiological effects via modification of ion channels related to atrial repolarization, such as transient outward potassium current (Ito), L-type calcium current (ICaL) and connexins [19, 29]. Furthermore, the direct electrophysiological effects were antagonized by ACE inhibitors or Ang II receptor blockers in vitro and vivo studies [19, 29, 38, 40]. Of note, Ferreira et al. [14] found that Ang-(1–7) might have some antiarrhythmogenic effects during myocardial ischemic reperfusion. On the contrary, Donoghue et al. [12] reported that ACE2 overexpression by generating transgenic mice induces connexin dysregulation and results in profound electrophysiological disturbances. In fact, few studies investigated the effects of ACE2/Ang-(1–7) axis on ion channels remodeling, especially Ang II-related adverse electrical remodeling. With the deepened understanding of ACE2/Ang-(1–7) axis on cardiac structural remodeling, to explore the effects of ACE2/Ang-(1–7) axis on atrial ion channels remodeling and its detailed mechanism has important theoretical and practical value.
Some studies have demonstrated that epicardial gene painting causes a homogeneous and complete transmural atrial gene transfer [21]. Therefore, the purpose of the present study was to investigate whether successful atrial overexpression of ACE2 by homogeneous transmural atrial gene transfer can help to reverse AF-induced atrial remodeling, and to elaborate the dynamic regulation between ACE/Ang II and Ang-(1–7) axis, as well as its downstream mechanisms in a canine atrial-pacing model.
Methods
Animal model and gene transfer
Twenty-eight mongrel dogs of either gender, weighing 20–30 kg, were randomized into 4 groups: Sham-operated (Sham), AF-Control, Ad-EGFP (adenovirus-enhanced green fluorescent protein) and Ad-ACE2 groups (n = 7 per subgroup). A programmable pacemaker (Huanan Medical Technology Co., Ltd. Henan, China) was fixed on the back of dogs and attached to a pacing lead (St. Jude Medical) in the right atrial through external jugular vein. All dogs in the AF-Control, Ad-EGFP and Ad-ACE2 group were paced at 450 beats/min with the use of 0.2 ms square-wave pulses at twice-threshold current for a period of 14 days. The dogs in Sham group were instrumented without pacing. The surface electrocardiogram (ECG) was verified every other day to ensure continuous 1:1 atrial capture.
After atrial burst pacing for 2 weeks, all dogs received median sternotomy operation and the pericardium was incised, but the gene transfer procedure was performed in the Ad-EGFP and Ad-ACE2 groups only. An invasive electrophysiology (EP) study was performed as described below. After EP study, the gene-painting procedure was performed as previously described [2, 8, 21]. In brief, the virus/trypsin/poloxamer gel was painted onto every accessible area of the atrial epicardium with a rounded-bristle flat paintbrush composed of camel hair. Each atrium was coated twice for 60 s each time, and approximately 5 min was allowed between paint coatings to permit absorption. After it was painted, the heart was left exposed to air for 10 min to allow virus penetration. Subsequently, all dogs except the Sham group underwent atrial burst pacing for another 3 weeks.
The animals in the present study were maintained in accordance with the guiding principles of the NIH Guide for the Care and Use of Laboratory Animals. The experimental protocol was approved by the Chongqing Medical University Animal Care and Use Committee.
In vivo electrophysiological studies
All dogs underwent an invasive EP study as previously described [30]. In brief, atrial effective refractory periods (AERPs) at the left and right atrium were measured at 3 basic cycle lengths B (BCL) (250, 300 and 350 ms). Eight basic drive stimuli were given, followed by a single premature stimulus, and all stimuli were set at twice the diastolic threshold. The S1S2 interval was increased in steps of 2 ms, and AERP was determined to be the shortest S1S2 interval that resulted in a propagated atrial response. The mean of three ERPs values at each BCL was used for data analysis. The inducibility of AF was evaluated by premature atrial stimulation during ERP measurement and atrial burst pacing lasting for 30 s at pacing cycle length of 100 ms. AF was considered inducible if the burst stimulus or S1–S2 stimulus produced a rapid irregular rhythm faster than 400 beats/min lasting more than 10 s and associated with irregular atrioventricular conduction. To estimate the mean AF duration, AF was induced 5 times and the duration of induced AF was recorded.
Clinical observations and ECG recordings were performed every other day during feeding in awake, alert animals. On post-gene transfer day 21, all dogs underwent terminal EP study, and were then euthanized by intracardiac KCl overdose in fully anesthetized dogs. The hearts were immediately removed for histological and molecular studies.
Histopathology and immunohistochemistry
Histologic studies were performed to identify the potential pathologic substrate underlying conduction abnormalities in rapid atrial-pacing dogs. Atrial tissue was fixed in 10 % formalin, embedded in paraffin, cut to 5-μm thickness and stained with hematoxylin and eosin or picrosirius red. The immunoperoxidase method was used for staining of Ang II and Ang-(1–7). The tissue samples were reacted with rabbit anti-Ang II antibody and rabbit anti-Ang-(1–7) antibody (1:100 dilution; Phoenix Pharmaceuticals), respectively. All histopathological sections were analyzed by an investigator blinded to animal grouping using a customized imaging analysis system (Image-Pro Plus 6.0, Media Cybernetics, USA).
Cryosections (5 µm thickness) of atrial tissue were prepared. The sections were incubated overnight with anti-connexin 40 (CX40) antibody (1:100 dilution) and anti-connexin 43 (CX43) antibody (1:100 dilution; Santa Cruz Biotechnology, USA), and then incubated with TRITC-conjugated rabbit anti-goat IgG or FITC-conjugated goat anti-mouse IgG (1:75 dilution; CWBIO, China). Confocal images were collected using an LSM-510 laser scanning microscope (Carl Zeiss Ltd). The degree of confocality was kept constant for each experiment to minimize overlap of the CX43/CX40 label.
Western blot analysis
Immunoblotting was performed as described previously. Briefly, tissues from left atrial appendage (≈100 mg) were homogenized in lysis buffer and purified by centrifugation (12,000g for 15 min at 4 °C). Protein concentration was determined with the BCA Protein Assay Kit (Beyotime, China) using bovine serum albumin as the standard. Solubilized protein was separated by electrophoresis and transferred to nitrocellulose membranes. Nonspecific binding was blocked by incubation in 5 % milk or bovine serum albumin in tris-buffered saline Tween 20. Membranes were probed with the specific antibodies, followed by incubation with horseradish peroxidase-conjugated secondary antibodies. The immunoreactive bands were detected by electrochemiluminescence and quantified by densitometry using a UMAX Astra 2200 scanner and ImageJ 1.33 software. The data were quantified by the densitometry using UMAX Astra 2200 scanner and Quantity-One software (Bio-Rad), and were normalized to GAPDH expression.
Ang II and Ang-(1–7) levels by ELISA
The concentration of Ang II and Ang-(1–7) in the atrial tissue was measured by a commercial enzyme-linked immunosorbent assay (ELISA) kit (Bachem, USA). In brief, the myocardium was mechanically homogenized on ice, using a homogenizer. Homogenized samples were centrifuged at 12,000 rpm for 15 min at 4 °C. Each supernatant was then transferred into a fresh test-tube and stored at −80 °C, and the levels of Ang II and Ang-(1–7) were determined by ELISA.
RNA isolation and real-time reverse transcription-polymerase chain reaction (RT-PCR)
RNA was isolated from atrial tissue with the use of the TRIzol reagent (Takara, Dalian, China) as directed by the manufacturer. Real-time RT-PCR was performed on complementary DNA generated with the PrimeScript® RT reagent Kit (Takara) with the SYBR® Premix Ex Taq™ II kit (Takara) in a MX3005 RT-PCR machine (Stratagene). The mixtures were heated at 50 °C for 2 min, at 95 °C for 90 s followed by 40 cycles at 95 °C for 15 s and 60 °C for 30 s. All reactions were performed in triplicate, with GAPDH (glyceraldehyde phosphate dehydrogenase) serving as an internal control. The primer sequences of all genes are presented in Supplement Table 1. The results were quantified as Ct values, where Ct is defined as the threshold cycle of PCR at which amplified product is first detected, and expressed as the ratio of target/control. The relative expression ratio of the target genes was calculated using 2(−ΔΔCt) method.
Statistical evaluation
Data were expressed as mean values ± SD for continuous variables, and frequencies were measured for categorical variables. Variables were compared between groups with the use of t test for continuous measures and Chi-square test for categorical variables. ANOVA was performed to analyze the differences in means among the 4 groups. Statistical significance was defined as 2 tailed 2p < 0.05. All statistical analyses were performed using SPSS statistical software (version 17.00, Chicago, IL, USA).
Results
Changes in electrophysiological variables
The general conditions of the experimental dogs are shown in Table 1. No difference was found in body weight, temperature and heart rate at baseline among 4 groups. Two AF-control dogs and 3 Ad-EGFP dogs developed spontaneous paroxysmal atrial fibrillation during the 5-weeks follow-up. AF can be induced after 2 weeks of rapid atrial pacing in 2 AF-control dogs, 4 Ad-EGFP dogs and 4 Ad-ACE 2 dogs respectively, but none of the 21 dogs developed any persistent AF. Atrial effective refractory period (AERP) at various basic cycle lengths in the right and left atrium for all groups are shown in Table 2. Except in Sham group, after 3 weeks atrial tachypacing, AERP at all cycle lengths 3 weeks post-gene transfer at either site decreased significantly (p < 0.05). As illustrated in Table 3, after 3 weeks of gene transfer, AF became more inducible in the AF-control and Ad-EGFP group, with the inducibility and duration of AF increased dramatically compared with baseline and Sham dogs (p < 0.01), whereas the inducibility and duration of AF were found to be markedly lower in the Ad-ACE2 group than baseline and those in the AF-control and Ad-EGFP groups (p < 0.01).
Table 1.
Demographic features of the experimental dogs
| Sham (n = 7) | AF-Control (n = 7) | Ad-EGFP (n = 7) | Ad-ACE2 (n = 7) | p value△ | |
|---|---|---|---|---|---|
| Male | 4 | 3 | 5 | 4 | 0.876 |
| SPAF | 0 | 2 | 3 | 0 | 0.245 |
| Weight (kg) | 23.7 ± 3.4 | 24.2 ± 1.4 | 23.3 ± 2.4 | 23.8 ± 3.4 | 0.957 |
| Heart rate (beat/min) | |||||
| Baseline | 150 ± 5 | 152 ± 6 | 154 ± 7 | 155 ± 8 | 0.633 |
| 3 weeks | 151 ± 4 | 167 ± 4* | 166 ± 7# | 161 ± 5 | <0.001 |
| Temperature (°C) | |||||
| Baseline | 38.3 ± 0.4 | 38.3 ± 0.5 | 38.2 ± 0.4 | 38.2 ± 0.4 | 0.930 |
| 3 weeks | 38.4 ± 0.4 | 38.3 ± 0.5 | 38.3 ± 0.5 | 38.1 ± 0.5 | 0.779 |
△The p value of one-way ANOVA comparing the four groups.; baseline vs. 3 weeks: * p = 0.002; # p = 0.019, n = 7
SPAF spontaneous paroxysmal atrial fibrillation in each group
Table 2.
Change in mean AERP (ms) before and 3 weeks after gene transfer
| BCL (ms) | Time | Right atria | Left atria | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sham | AF-Control | Ad-EGFP | Ad-ACE2 | Sham | Control | Ad-EGFP | Ad-ACE2 | ||
| 350 | Baseline | 121 ± 7 | 123 ± 9 | 124 ± 7 | 123 ± 5 | 117 ± 4 | 121 ± 8 | 119 ± 6 | 122 ± 9 |
| 3 weeks | 120 ± 8 | 107 ± 7* | 106 ± 8* | 108 ± 6* | 117 ± 8 | 102 ± 8* | 104 ± 5* | 108 ± 6* | |
| 300 | Baseline | 116 ± 6 | 119 ± 5 | 116 ± 3 | 117 ± 6 | 113 ± 6 | 115 ± 4 | 115 ± 5 | 114 ± 8 |
| 3 weeks | 114 ± 7 | 103 ± 4* | 104 ± 6* | 103 ± 7* | 102 ± 10 | 100 ± 5* | 102 ± 5* | 104 ± 5* | |
| 250 | Baseline | 107 ± 6 | 108 ± 7 | 112 ± 5 | 109 ± 6 | 107 ± 9 | 109 ± 8 | 111 ± 7 | 107 ± 10 |
| 3 weeks | 107 ± 5 | 97 ± 4* | 99 ± 4* | 100 ± 5* | 100 ± 10 | 97 ± 7* | 99 ± 5* | 100 ± 5* | |
* Baseline vs. 3 weeks, p < 0.05
BCL basic cycle length (n = 7 per group); ms millisecond
Table 3.
Changes in inducibility and duration of AF before and after gene transfer
| AF cases | AF times | AF inducibility (%) | Mean AF duration (s) | |
|---|---|---|---|---|
| Sham | ||||
| Baseline | 2 (28.6 %) | 6 | 28.6 | 35.1 ± 62.8 |
| 3 weeks | 3 (42.9 %) | 9 | 42.9 | 66.7 ± 86.1 |
| AF-control | ||||
| Baseline | 2 (28.6 %) | 19 | 28.6 | 274.4 ± 213.9 |
| 3 weeks | 7 (100 %) | 9 | 100 | 850.1 ± 225.2* |
| Ad-EGFP | ||||
| Baseline | 4 (57.1 %) | 20 | 57.1 | 227.3 ± 226.8 |
| 3 weeks | 7 (100 %) | 10 | 100 | 944.6 ± 256.0* |
| Ad-ACE2 | ||||
| Baseline | 4 (57.1 %) | 15 | 57.1 | 192.6 ± 190.6 |
| 3 weeks | 3 (42.9 %) | 13 | 42.9 | 89.1 ± 92.3**,# |
AF cases, the number of dogs having spontaneous or induced AF by procedure stimulation; AF times, the times of induced AF by procedure stimulation
* Compared with baseline and sham dogs: p < 0.01
** Compared with baseline: p = 0.097
#Compared with Ad-EGFP and control: p < 0.01
Histological evaluation
Serious pericardial inflammation, effusion, and hemorrhage were not observed in any of the dogs. Representative histological sections from each group are presented in Fig. 1. Atrial myocyte from Sham dogs showed a normal composition of sarcomeres distributed throughout the cell, and the intra-cellular space also appeared normal. In contrast, atrial myocytes of AF-control and Ad-EGFP dogs showed a loss of some contractile materials and abnormal sarcomeres. In addition, extensive interstitial fibrosis evidenced by picrosirius red staining was found in these tissues. Thick layers of fibrous tissue were observed in the endocardium and epicardium. Furthermore, the amount of connective tissue was increased, and this extended around the parenchymal cells. In contrast, these pathologic abnormalities of atrial tissues were attenuated in the Ad-ACE2 dogs. The percentage of fibrosis in all atrial regions in the Ad-ACE2 dogs was markedly lower than those in the AF-control and Ad-EGFP dogs (5.8 ± 2.4 vs. 11.9 ± 2.3 % and 14.3 ± 3.4 % at the right atrial appendage, p < 0.001), and was comparable with that in the Sham dogs (5.2 ± 2.5 %, p = 0.614).
Fig. 1.
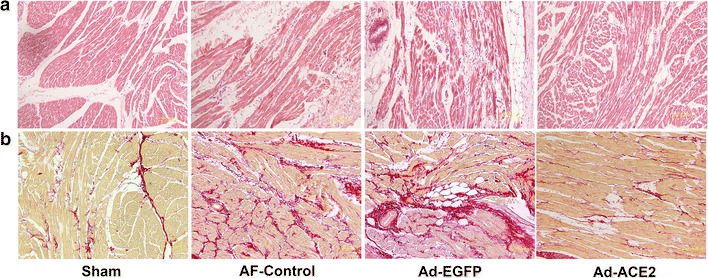
Representative images of atrial tissues stained with hematoxylin and eosin (a) and picrosirius red staining (b). Atrial myocyte from sham dogs showed normal composition of sarcomeres distributed throughout the cell, while those from AF-control and Ad-EGFP dogs showed a loss of some contractile materials and abnormal sarcomeres. In addition, extensive interstitial fibrosis, evidenced by picrosirius red stain was found in these tissues. In contrast, these pathologic abnormalities of atrial tissues were attenuated in the Ad-ACE2 group (n = 7)
ACE2 overexpression and RAS components
The ACE2 mRNA expression in the Ad-EGFP and AF-control dogs was lower than those in the Sham dogs, with no significant difference between the two formers; but it was fivefold higher in Ad-ACE2 dogs than that in the Sham dogs. Similarly, western blot analysis showed 1.84-fold increase in ACE2 in the Ad-ACE2 dogs compared with the sham dogs, but substantial decrease in the Ad-EGFP or AF-control dogs 3 weeks after gene transfer (Fig. 2; Table 4). Semi-quantitative immunohistochemical analysis showed that ACE2 stained in the myocardium, epicardium, and endocardium, and located mainly in the atrial myocytes. Weak ACE2 staining was observed in atrial tissues from dogs in the AF-control and Ad-EGFP groups, whereas marked ACE2 staining was depicted in atrial tissues in the Ad-ACE2 group (Fig. 3).
Fig. 2.
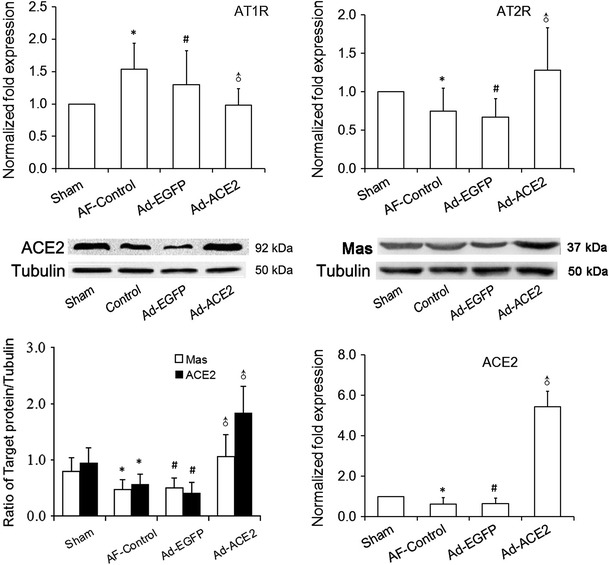
Western blot analysis of ACE2 and Mas protein levels and real-time RT-PCR analysis of the mRNA expression of RAS components from atrial tissues. Compared with sham dogs, ACE2 protein expression in the AF-control and Ad-EGFP dogs was significantly decreased compared with that in the sham dogs, but was increased twofold in the Ad-ACE2 dogs. Similarly, ACE2 gene and Mas protein expression showed the same statistical trend as its protein expression. Real-time PCR showed up-regulation of AT1R and down-regulation of AT2R induced by rapid atrial pacing, which were attenuated by overexpression of ACE2. *p < 0.05 versus sham; # p < 0.05 versus sham; ♂ p < 0.001 versus AF-control and Ad-EGFP (n = 7)
Table 4.
Western blot analysis of ACE2 protein levels and real-time RT-PCR analysis of the mRNA expression of RAS components from atrial tissues
| Sham | AF-Control | Ad-EGFP | Ad-ACE2 | p* | p # | p ♂ | |
|---|---|---|---|---|---|---|---|
| mRNA ACE2 | 1.00 ± 0.00 | 0.53 ± 0.28 | 0.51 ± 0.12 | 5.43 ± 0.77 | 0.042 | 0.034 | <0.001 |
| mRNA AT1R | 1.00 ± 0.00 | 1.54 ± 0.40 | 1.58 ± 0.09 | 0.98 ± 0.26 | <0.001 | <0.001 | <0.001 |
| mRNA AT2R | 1.00 ± 0.00 | 0.65 ± 0.22 | 0.60 ± 0.18 | 1.28 ± 0.55 | 0.048 | 0.026 | <0.001 |
| Protein ACE2 | 0.94 ± 0.27 | 0.56 ± 0.18 | 0.41 ± 0.19 | 1.84 ± 0.47 | 0.028 | 0.003 | <0.001 |
| Protein Mas | 0.80 ± 0.24 | 0.47 ± 0.18 | 0.50 ± 0.20 | 1.06 ± 0.39 | 0.032 | 0.048 | <0.001 |
* Sham vs. AF-control; # Sham vs. Ad-EGFP; ♂ Ad-ACE2 vs. Ad-EGFP (or AF-control), n = 7
Fig. 3.
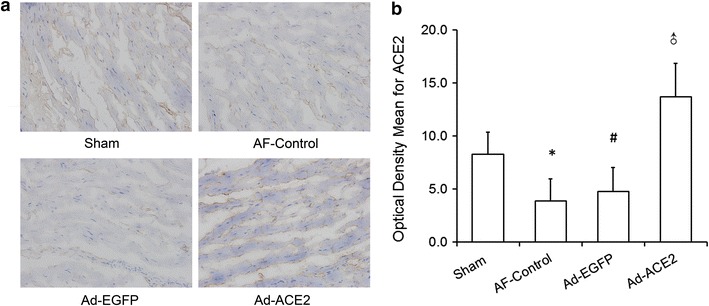
ACE2 expression staining after gene transfer in 4 groups of dogs. a Semi-quantitative immunohistochemical staining of atrial myocytes. Marked ACE2 staining was present in Ad-ACE2 dogs compared with other dogs. b Semi-quantitative immunohistochemical analysis of the expression of ACE2. In comparison with Sham group, the expression levels of ACE2 were significantly higher in the Ad-ACE2 group, while that was lower in the Ad-EGFP and AF-Control group. *p < 0.01 versus sham; # p < 0.001 versus sham; ♂ p < 0.001 versus Sham, AF-Control and Ad-EGFP, n = 7
In comparison with the sham and Ad-ACE2 group, the expression levels of Ang II by semi-quantitative immunohistochemical analysis were significantly higher in the Ad-EGFP and AF-control group (p < 0.01 to 0.05). In contrast, the expression of Ang-(1–7) were lower in the Ad-EGFP and AF-control group than in the sham and Ad-ACE2 group (p < 0.01). Corresponding to that, the changing trend of both Ang II and Ang-(1–7) in atrial tissue detected by ELISA was similar to the results of semi-quantitative analysis of immunohistochemistry (Figs. 4, 5; Tables 5, 6).
Fig. 4.
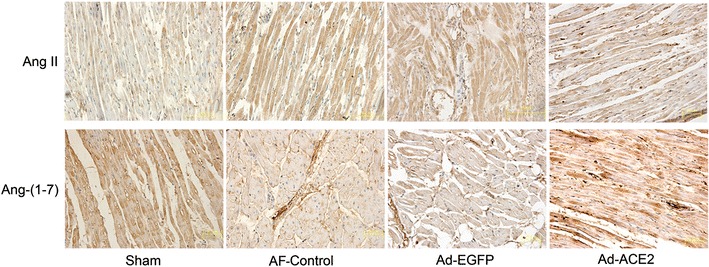
Representative images of Ang II and Ang-(1–7) immunohistochemistry in paraffin sections
Fig. 5.
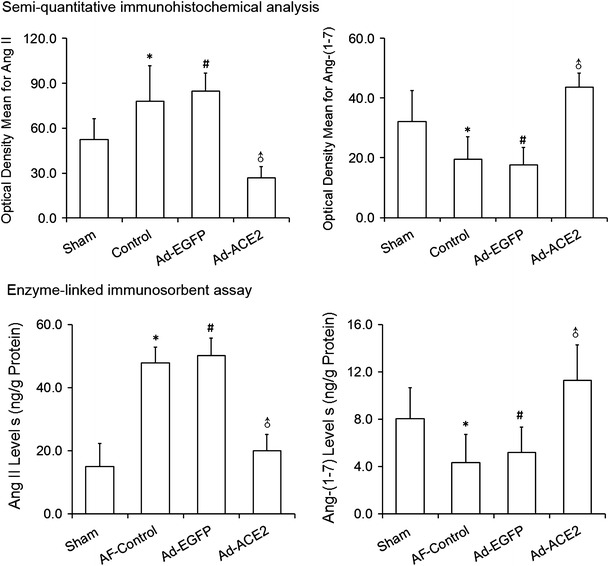
Semi-quantitative immunohistochemical and enzyme-linked immunosorbent assay analysis of Ang II and Ang-(1–7). Based on optical density and immunosorbent assays, the levels of Ang II were significantly decreased while the levels of Ang-(1-7) were significantly increased in Ad-ACE2. *p < 0.05 versus sham; # p < 0.05 versus sham; ♂ p < 0.001 versus AF-control and Ad-EGFP (n = 7)
Table 5.
Semi-quantitative immunohistochemical analysis of the expression of Ang II and Ang-(1–7)
| Optical density mean | Sham | AF-Control | Ad-EGFP | Ad-ACE2 | p* | p # | p ♂ |
|---|---|---|---|---|---|---|---|
| Ang II | 52.6 ± 13.9 | 78.1 ± 23.7 | 84.6 ± 12.3 | 27.0 ± 7.3 | <0.01 | <0.001 | <0.001 |
| Ang-(1–7) | 32.2 ± 10.2 | 19.6 ± 7.4 | 17.6 ± 5.9 | 43.6 ± 4.8 | 0.004 | <0.001 | < 0.001 |
* Sham vs. AF-control; # Sham vs. Ad-EGFP; ♂ Ad-ACE2 vs. Ad-EGFP (or AF-control); Ang II, Sham vs. Ad-ACE2, p < 0.001; Ang-(1–7), Sham vs. Ad-ACE2, p < 0.01, n = 7
Table 6.
Enzyme-linked immunosorbent assay of Ang II and Ang-(1–7)
| Protein (ng/g) | Sham | AF-Control | Ad-EGFP | Ad-ACE2 | p* | p # | p ♂ |
|---|---|---|---|---|---|---|---|
| Ang II | 15.0 ± 7.2 | 47.8 ± 5.0 | 50.2 ± 5.5 | 20.0 ± 5.2 | <0.001 | <0.001 | <0.001 |
| Ang-(1–7) | 8.0 ± 2.6 | 4.3 ± 2.4 | 5.2 ± 2.1 | 11.3 ± 3.0 | 0.012 | 0.05 | <0.001 |
* Sham vs. AF-control; # Sham vs. Ad-EGFP; ♂ Ad-ACE2 vs. Ad-EGFP (or AF-control); Ang II, Sham vs. Ad-ACE2, p = 0.123; Ang-(1–7), Sham vs. Ad-ACE2, p = 0.026, n = 7
Real-time RT-PCR showed that AT1R (Angiotensin type-1 receptor) gene expression was higher in the atrial myocardium of the AF-control and Ad-EGFP dogs compared with the Sham group (p < 0.001), with no significant difference between the two groups, but was down-regulated significantly in the Ad-ACE2 group (p < 0.001). In contrast, AT2R (Angiotensin type-2 receptor) gene expression was markedly reduced in the AF-control and Ad-EGFP group in comparison with that in the sham and Ad-ACE2 group (p < 0.05) 3 weeks after gene transfer. It is worth noting that protein expression of Mas, the receptor of Ang-(1–7), quantified by western blot analysis, was also down-regulated in the AF-control and Ad-EGFP group but was up-regulated in the sham and Ad-ACE2 group (Fig. 2).
Our data indicated that rapid atrial pacing significantly decreased ACE2, Ang-(1–7), Mas and AT2R expression levels, but increased Ang II and AT1R expression levels, when compared with Sham group, which were reversed by ACE2 overexpression in effectively infected atrial myocardium of Ad-ACE2 dogs.
MAPKs and MKP-1 protein expression
To investigate the potential mechanism of ACE2 improving atrial structural remodeling, the dynamic balance between MAPKs and MKP-1 was evaluated. The amount of p38MAPK, ERK1/2 and p-ERK1/2 protein were significantly increased in the AF-control and Ad-EGFP group compared with Sham and Ad-ACE2 group (p < 0.05), with no difference between the latter two (Fig. 6; Table 7). In contrast, the amount of MKP-1 protein was significantly lower in AF-control and Ad-EGFP group in comparison with the Sham and Ad-ACE2 group (p < 0.01), suggesting that Ad-ACE2 overexpression not only attenuates the increase of MAPKs protein expression induced by rapid atrial pacing, but also up-regulates the protein expression of MKP-1.
Fig. 6.
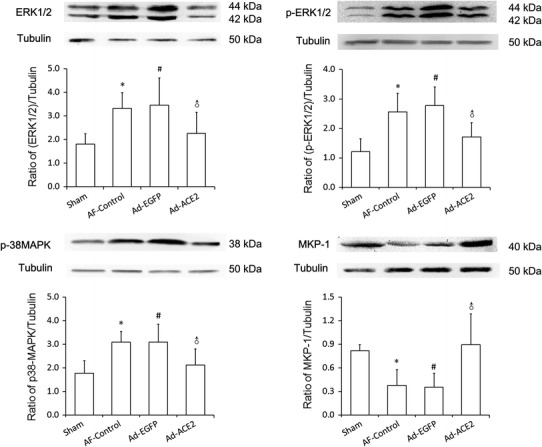
Western blot analysis of MAPKs and MPK-1 protein levels from atrial tissues. Compared to Sham and Ad-ACE2 group, the protein expression level of p38, ERK1/2 and p-ERK1/2 in AF-control and Ad-EGFP group were significantly higher, but the protein expression level of MKP-1 was lower. *p < 0.05 versus sham; # p < 0.05 versus sham; ♂ p < 0.05 versus AF-control and Ad-EGFP (n = 7)
Table 7.
Western blot analysis of MAPKs and MPK-1 protein levels from atrial tissues
| Sham | AF-Control | Ad-EGFP | Ad-ACE2 | p* | p # | p ♂ | |
|---|---|---|---|---|---|---|---|
| ERK1/2 | 1.81 ± 0.43 | 3.33 ± 0.67 | 3.46 ± 1.15 | 2.26 ± 0.89 | <0.01 | <0.01 | <0.05 |
| p-ERK1/2 | 1.21 ± 0.43 | 2.56 ± 0.63 | 2.78 ± 0.63 | 1.71 ± 0.69 | <0.001 | <0.001 | <0.05 |
| P38MAPK | 1.77 ± 0.54 | 3.10 ± 0.45 | 3.10 ± 0.75 | 2.13 ± 0.68 | <0.01 | 0.011 | <0.05 |
| MKP-1 | 0.82 ± 0.08 | 0.38 ± 0.20 | 0.35 ± 0.18 | 0.90 ± 0.39 | <0.01 | <0.01 | <0.001 |
* Sham vs. AF-control; # Sham vs. Ad-EGFP; ♂ Ad-ACE2 vs. Ad-EGFP (or AF-control), n = 7
Collagen, fibronectin and TGF-β1 protein expression
As shown in Fig. 8, compared with Sham-operated and Ad-ACE2 dogs, the protein expression levels of Col I, Col III, TGF-β1 and fibronectin by western blot analysis increased dramatically in the AF-control and Ad-EGFP dogs (p < 0.05), while there were no significant differences between the Sham and Ad-ACE2 group. Corresponding to that, compared to the Sham and Ad-ACE2 group, the mRNA expression levels of TGF-β1, Col1 α2 and Col 4α5 were also significantly higher in the AF-control and Ad-EGFP group (p < 0.05), indicating that Ad-ACE2 overexpression can antagonize rapid atrial pacing-induced increase in TGF-β1, fibronectin and collagens protein and gene expression (Fig. 7; Tables 8, 9).
Fig. 8.
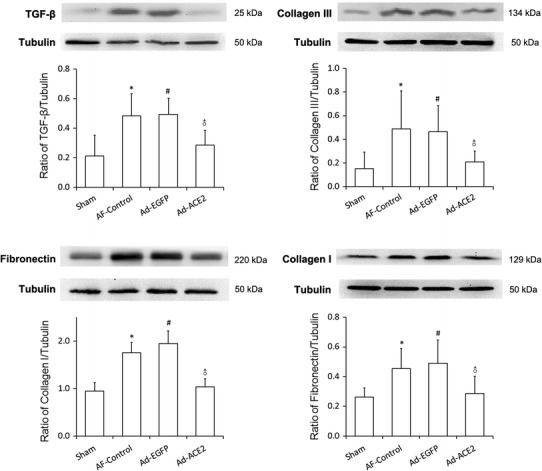
Western blot analysis of Collagen, Fibronectin and TGF-β1 protein levels from atrial tissues. Compared to Sham and Ad-ACE2 group, the protein expression level of Col I, Col III, TGF-β1 and fibronectin in the AF-control and Ad-EGFP group were significantly higher.*p < 0.05 versus sham; # p < 0.05 versus sham; ♂ p < 0.05 versus AF-control and Ad-EGFP, n = 7
Fig. 7.
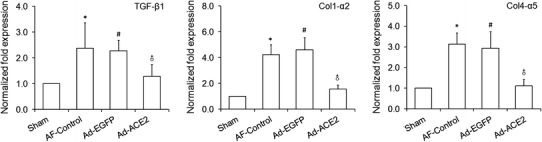
Real-time quantitative reverse transcription-polymerase chain reaction (RT-PCR) analysis of fibrosis-related indicators. Note: Including transforming growth factor-β1 (TGF-β1), Collagen, type I, alpha 2 (COL1α2) and Collagen, type IV, alpha 5 (COL4α5) mRNA expression. Compared to the Sham group, the mRNA expression levels of TGF-β1, Col I and Col IV were also significantly higher in the AF-Control and Ad-EGFP dogs, while they were significantly lower in the Ad-ACE2 group in comparisons with latter two groups. *p < 0.001 versus sham; # p < 0.001 versus sham; ♂ p < 0.01 versus AF-Control and Ad-EGFP, n = 7
Table 8.
Western blot analysis of TGF-β1 and Collagen III protein levels from atrial tissues
| Sham | AF-Control | Ad-EGFP | Ad-ACE2 | *p | #p | ♂p | |
|---|---|---|---|---|---|---|---|
| TGF-β1 | 0.21 ± 0.14 | 0.48 ± 0.15 | 0.49 ± 0.11 | 0.29 ± 0.10 | 0.004 | 0.003 | <0.05 |
| Collagen III | 0.15 ± 0.14 | 0.49 ± 0.32 | 0.47 ± 0.22 | 0.21 ± 0.09 | 0.012 | 0.018 | <0.05 |
| Collagen I | 0.95 ± 0.18 | 1.75 ± 0.21 | 1.95 ± 0.26 | 1.03 ± 0.18 | <0.001 | <0.001 | <0.001 |
| Fibronectin | 0.26 ± 0.06 | 0.46 ± 0.13 | 0.49 ± 0.16 | 0.29 ± 0.12 | 0.007 | 0.002 | 0.016/0.005 |
* Sham vs. AF-Control; # Sham vs. Ad-EGFP; ♂ Ad-ACE2 vs. Ad-EGFP (or AF-Control), n = 7
Table 9.
Real-time quantitative reverse transcription-polymerase chain reaction (RT-PCR) analysis of fibrosis-related indicators
| Sham | AF-control | Ad-EGFP | Ad-ACE2 | p* | p # | p ♂ | |
|---|---|---|---|---|---|---|---|
| TGF-β1 | 1.00 ± 0.00 | 2.37 ± 0.98 | 2.27 ± 0.41 | 1.29 ± 0.45 | <0.001 | <0.001 | <0.01 |
| Col1-α2 | 1.00 ± 0.00 | 4.23 ± 0.77 | 4.60 ± 0.93 | 1.56 ± 0.31 | <0.001 | <0.001 | <0.001 |
| Col4-α5 | 1.00 ± 0.00 | 3.14 ± 0.54 | 2.93 ± 0.81 | 1.11 ± 0.31 | <0.001 | <0.001 | <0.001 |
* Sham vs. AF-control; # Sham vs. Ad-EGFP; ♂ Ad-ACE2 vs. Ad-EGFP (or AF-control), n = 7
Ion channel protein and gene expression
To further explore the effects of ACE2 overexpression on atrial ion channels, some ion channel proteins related to atrial repolarization were evaluated by real-time RT-PCR and western blot. Compared with Sham-operated group, both Cav1.2 and Kv4.2 mRNA expression were lower in the myocardium of AF-control and Ad-EGFP group (p < 0.01), but was up-regulated significantly in Ad-ACE2 group (Fig. 9). Furthermore, the protein expression of Kv 4.2 (Kv 4.3) and L-type Ca++ CP α1C, which was coded by mRNA of Kv4.2 (Kv4.3) and Cav1.2, respectively, showed similar trends among the 4 groups (Fig. 10). No significant difference was found in terms of Kv4.3 and KChiP2 mRNA expressions among the four groups (Table 10).
Fig. 9.
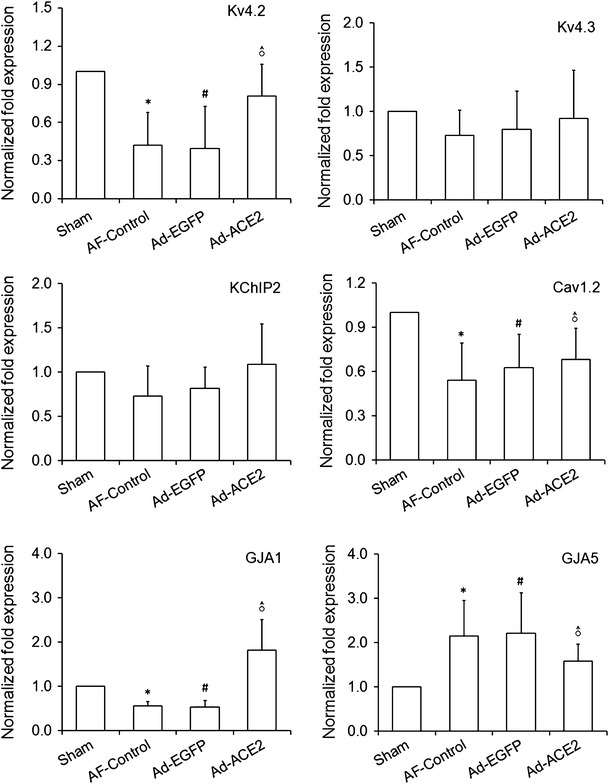
Real-time RT-PCR analysis of the mRNA expression of repolarization-related ion channel proteins. Compared with sham-operated dogs, both Cav1.2 and Kv4.2 mRNA abundance were lower in the myocardium of AF-control and Ad-EGFP dogs (p < 0.01), but was up-regulated significantly in Ad-ACE2 dogs. No significant difference was found in terms of Kv4.3 and KChiP2 mRNA abundance among the four groups. Kv4.3, Kv4.2 and KChIP2 are the core components of Ito channel. Cav1.2, GJA1 and GJA5 is the main component of ICaL channel, connexin 43 and connexin 40, respectvely. *p < 0.05 versus sham; # p < 0.05 versus sham; ♂ p < 0.05 versus AF-control and Ad-EGFP; Cav1.2: Sham vs. Ad-ACE2, p = 0.012; GJA5: Sham vs. Ad-ACE2, p = 0.130, n = 7
Fig. 10.
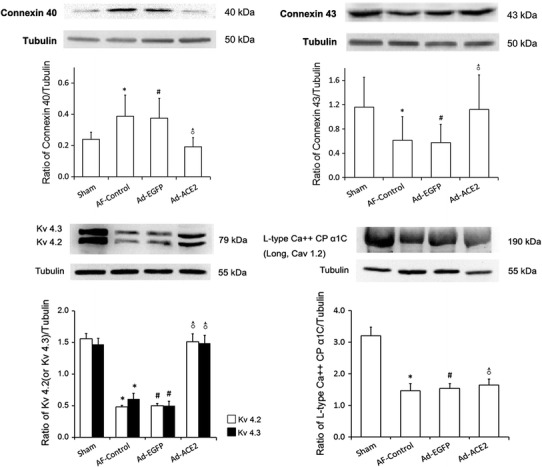
Western blot analysis of ion channel and connexins protein levels from atrial tissues. Compared to Sham and Ad-ACE2 group, the protein expression levels of Kv4.2/4.3 and Cx43 in AF-control and Ad-EGFP groups were significantly lower, but the protein expression levels of L-type Ca++ CP α1C protein and Cx40 in Sham group were higher than other 3 groups. Kv4.2/4.3: *p < 0.01 versus sham; # p < 0.01 versus sham; ♂ p < 0.01 versus AF-control and Ad-EGFP. L-type Ca++ CP α1C protein: *p < 0.01 versus sham; # p < 0.01 versus sham; ♂ p < 0.01 versus Sham, n = 7. Cx43/Cx40: *p < 0.05 versus sham; # p < 0.05 versus sham; ♂ p < 0.05 versus AF-control and Ad-EGFP, n = 7
Table 10.
Real-time RT-PCR analysis of the mRNA expression of repolarization-related ion channel proteins
| Sham | AF-Control | Ad-EGFP | Ad-ACE2 | p* | p # | p ♂ | |
|---|---|---|---|---|---|---|---|
| Kv4.2 | 1.00 ± 0.00 | 0.42 ± 0.26 | 0.40 ± 0.33 | 0.81 ± 0.25 | <0.001 | <0.001 | <0.05 |
| Kv4.3 | 1.00 ± 0.00 | 0.73 ± 0.28 | 0.80 ± 0.43 | 0.92 ± 0.55 | 0.223 | 0.364 | 0.38/0.58 |
| KChIP2 | 1.00 ± 0.00 | 0.73 ± 0.34 | 0.81 ± 0.24 | 1.09 ± 0.46 | 0.146 | 0.310 | 0.06/0.144 |
| Cav1.2 | 1.00 ± 0.00 | 0.54 ± 0.25 | 0.63 ± 0.23 | 0.68 ± 0.21 | <0.001 | 0.004 | 0.239/0.659 |
| GJA1 | 1.00 ± 0.00 | 0.56 ± 0.10 | 0.53 ± 0.15 | 1.82 ± 0.69 | 0.045 | 0.035 | <0.001 |
| GJA5 | 1.00 ± 0.00 | 2.15 ± 0.80 | 2.21 ± 0.91 | 1.58 ± 0.39 | 0.005 | 0.004 | 0.138/0.104 |
* Sham vs. AF-control; * Sham vs. Ad-EGFP; ♂ Ad-ACE2 vs. Ad-EGFP (or AF-control); Cav1.2, Sham vs. Ad-ACE2, p = 0.012; GJA5: Sham vs. Ad-ACE2, p = 0.130, n = 7
Compared with Sham and Ad-ACE2 group, the protein expression level of CX40 was significantly increased in the AF-control and Ad-EGFP groups (p < 0.05). In contrast, the protein expression levels of CX43 were higher in the AF-control and Ad-EGFP groups were significantly lower (p < 0.05). No significant differences were observed in the expression of CX40 and CX43 between Sham and Ad-ACE2 groups (Fig. 10). Moreover, the mRNA expression levels of CX40 and CX43 was significantly higher in the AF-control and Ad-EGFP groups than those in Sham and Ad-ACE2 group (Table 11).
Table 11.
Western blot analysis of connexin 40 and connexin 43 protein levels from atrial tissues
| Sham | AF-Control | Ad-EGFP | Ad-ACE2 | *p | #p | ♂p | |
|---|---|---|---|---|---|---|---|
| Connexin 40 | 0.24 ± 0.05 | 0.39 ± 0.14 | 0.38 ± 0.13 | 0.19 ± 0.16 | 0.011 | 0.018 | <0.01 |
| Connexin 43 | 1.16 ± 0.49 | 0.61 ± 0.39 | 0.58 ± 0.30 | 1.12 ± 0.57 | 0.031 | 0.022 | 0.044/0.031 |
| Kv 4.2 protein | 1.56 ± 0.09 | 0.49 ± 0.03 | 0.50 ± 0.02 | 1.51 ± 0.12 | <0.001 | <0.001 | <0.001 |
| Kv 4.3 protein | 1.47 ± 0.10 | 0.61 ± 0.09 | 0.50 ± 0.07 | 1.49 ± 0.12 | <0.001 | <0.001 | <0.001 |
| L-type Ca++ CP α1C | 3.20 ± 0.27 | 1.47 ± 0.23 | 1.54 ± 0.15 | 1.65 ± 0.19 | <0.001 | <0.001 | <0.001& |
* Sham vs. AF-Control; * Sham vs. Ad-EGFP; ♂ Ad-ACE2 vs. Ad-EGFP (or AF-control); & Ad-ACE2 vs. Sham, n = 7
We evaluated connexin localization using confocal immunohistochemistry (Fig. 11). In longitudinally sectioned atrial myocytes from all groups, CX43 was located mainly at the intercalated discs, with little signal at the lateral sarcolemma. In contrast, CX40 was located predominantly at the lateral sarcolemma, but there was still a certain amount of signal at the intercalated discs.
Fig. 11.
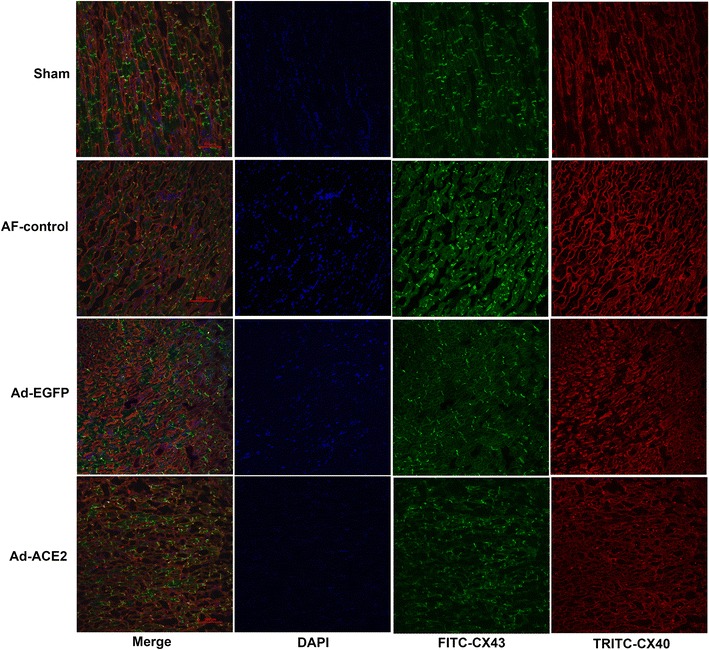
Representative confocal immunohistochemistry images of CX40 (red) and CX43 (green). CX43 was located mainly at the intercalated discs, while CX40 was located predominantly at the lateral sarcolemma, n = 7
Discussion
Main findings
We have previously demonstrated 100 % gene transfer rate in atrial muscle after epicardia gene painting. In this study, we investigated the effects of atrial overexpression of ACE2 by homogeneous transmural atrial gene transfer on atria structural and electrical remodeling in a canine atrial-pacing model. Our data indicated that rapid atrial pacing significantly decreased ACE2, Ang-(1–7) and AT2R expression levels, but increased Ang II and AT1R expression levels, when compared with Sham group, which were reversed by ACE2 overexpression in effectively infected atrial myocardium of Ad-ACE2 dogs.
The present study made a full demonstration and the salient finding are as follows: (1) ACE2 overexpression suppressed local activation of RAS induced by atrial rapid pacing and reversed the imbalance between Ang II and Ang-(1–7); (2) for the detailed signal transduction mechanism following ACE2/Ang-(1–7), our experiment found that ACE2 overexpression attenuated cardiac fibrosis remodeling associated with up-regulation of MKP-1 and reduction of MAP kinase activities; (3) for the first time, our study demonstrated that ACE2 overexpression ameliorated rapid atrial pacing-induced ion channels and connexin remodeling, and reduced the inducibility and duration of AF.
Structural and electrical remodeling
Extensive evidence confirm that structural remodeling, particularly interstitial fibrosis, is an important contributor to the AF substrate [13, 22, 32, 34, 42]. Our present experimental study indicated that compared with Sham atrial tissues, atrial tissues from AF-control and Ad-EGFP dogs showed a severely reduced number of sarcomeres and extensive interstitial fibrosis after atrial pacing for 5 weeks. In contrast, ACE2 overexpression significantly reversed these changes. This was in agreement with previous studies using viral vectors in various models [11, 17, 26, 27, 43], showing that overexpression of ACE2 by homogeneous transmural atrial gene transfer suppressed interstitial fibrosis.
Both animal and human studies have demonstrated that prolonged episodes of AF-induced AERP shortening and a loss of rate-related AERP shortening, which in term promoted the occurrence and persistence of AF [13]. Recent data suggested that ACE inhibitors or Ang II receptor blockers had no protective effect against AERP shortening and did not abolish the rate adaptation in dogs receiving chronic atrial tachypacing, but attenuated the inducibility and duration of AF [22]. The latter was attributed to improvement of hemodynamic status and structural remodeling. Consistent with previous results [22, 26], our experiment also found that ACE2 overexpression had no preventive effect on the AERP shortening after 5 weeks of rapid atrial pacing, but decreased the AF inducibility and shortened AF duration, as well as significantly reducing atrial interstitial fibrosis. Therefore, the improvement of atrial interstitial fibrosis by ACE2 overexpression may be one of the important mechanisms of its therapeutic effects.
ACE2 overexpression and RAS components
A wealth of evidence suggests that local activation of RAS, especially Ang II, is involved in atrial electrical and structural remodeling that perpetuates AF [10, 13, 22, 32, 34, 42]. Elevated levels of ACE and Ang II and up-regulation of profibrotic AT1R in the atrial myocardium have been reported in AF patients and animal models [13, 16]. For the endogenous negative regulation factors, Pan et al. [31] reported that ACE2 gene and protein expression in the fibrillating atria of pigs were significantly decreased compared with those in the sinus rhythm subjects. In the present study, local atrial RAS was found to be markedly activated in atrial-pacing dogs, as manifested by the increased expression levels of Ang II and AT1R, and decrease of ACE2, AT2R, Ang-(1–7), as well as the target of Ang-(1–7), Mas receptor; while atrial ACE2 overexpression could reverse these changes above. Together, these results suggest that atrial rapid pacing gives rise to local activation of RAS in atrial tissues and tips the balance between the protective axis of Ang-(1–7)/Mas and detrimental axis of Ang II/AT1R, while ACE2 overexpression in atrial tissue leads to a shift of the RAS balance towards the protective axis. We acknowledge that this study does not investigate the detailed molecular mechanism of the imbalance of RAS components, particularly the up-regulated or down-regulated mechanisms of those receptors, which might involve p38MAPK and ERK1/2 [20, 25], and needs to be further elucidated.
Antifibrotic mechanism of ACE2 overexpression
Several lines of evidence showed that Ang II binding to AT1R stimulates proliferation of fibroblasts, accumulation of extracellular matrix via activation of MAPK and up-regulation of TGF-β contributing to cardiac fibrosis [1, 32, 34]. Inhibitions of generations of Ang II can attenuate rapid atrial pacing or heart failure-induced MPAKs activation and atrial fibrosis [23]. Consistent with prior studies, the present study confirmed that ACE2 overexpression could suppress ERK1/2 and p38MAPK activation caused by rapid atrial pacing, followed by down-regulation of TGF-β and collagen fibers, and subsequently ameliorate interstitial fibrosis. Furthermore, this beneficial effect of ACE2 overexpression was again associated with shifting of the RAS balance towards the protective axis.
MAPKs are activated by phosphorylation on both threonine and tyrosine residues by MAPK kinases and are inactivated through dephosphorylation, by either serine/threonine and tyrosine protein phosphatases or dual-specificity phosphatases. MKP-1 is a critical counteracting phosphatase that directly regulates the magnitude and duration of p38MAPK, as well as auto-phosphorylation and activation of Jun N-terminal kinase and ERK [5, 6, 27, 28]. Recently, McCollum et al. [27, 28] reported that Ang-(1–7) participated in maintaining cardiac homeostasis by reducing proliferation and collagen production by cardiac fibroblasts, associated with up-regulation of MKP-1 and down-regulation of MAPK phosphorylation. In agreement with these, our results for the first time indicated that ACE2 overexpression reversed the imbalance between MAPKs and MKP-1 induced by rapid atrial pacing, as manifested by down-regulation of MAPKs and up-regulation of MKP-1, and directly mediated downstream protective antifibrotic signals. Therefore, reversal of this imbalance may be a new therapeutic target of cardiac remodeling.
Effects of ACE2/Ang-(1–7) on ion channels and connexins
Atrial tachycardia remodeling alters ionic currents and gene expression of ion channels in a manner which plays a critical role in shaping the atrial action potential and promotes the occurrence of AF [29]. Reductions in atrial myocyte I CaL and Ito current resulting in action potential duration shortening and loss of its rate adaptation have been reported in both human and animal tachycardia-induced atrial remodeling [4, 29, 41]. A reduction in potassium channels gene expression has been reported as an adaptation mechanism serving to prolong the initially reduced AERP and action potential duration [34]. From gene and protein expression profile, the present results firstly demonstrated a significant reduction of mRNA expression of Cav1.2 and Kv4.2 in atrial tachypacing dogs, as well as their corresponding coding protein L-type Ca++ CP α1C and Kv4.2/4.3 protein, respectively. Meanwhile, ACE2 overexpression could reverse decreased Kv4.2 expression level induced by atrial tachypacing (AF-control and Ad-EGFP group), but had no changes on the expression level of Cav1.2mRNA and its coding protein when compared with AF-control dogs. In another canine model of AF after atrial tachycardia, Liu et al. reported that Ang-(1–7) prevented atrial tachycardia-induced decrease in current density of Ito and I CaL, and shortened 90 % action potential duration [35]. However, similar to our findings, Ang-(1–7) did not prevent the decrease of Cav1.2 mRNA level. In fact, it is generally known that I CaL down-regulation is a major contributor to the decrease in action potential duration and refractoriness linked with AF [9]. Therefore, these findings might be able to explain no significant difference in terms of AERP observed between AF-control and Ad-ACE2 group. Of note, other Ca2+ handling proteins and ion channel proteins, such as RyR2, Serca2a, Calsequestrin, NCX1, can be implicated in atrial repolarization and electrical remodeling except I CaL [9, 39]; moreover, above ion channel current density were not evaluated; and all of them were not addressed in this paper and are worth to more work for this subject.
To our best knowledge, this study firstly focused on the effects of ACE2/Ang-(1–7) on expression and distribution of CX43 and CX40. As part of electrical remodeling, changes in gap junctions and connexins in AF have been reported. Previous work on connexin expression and function in animal models and humans primarily points towards AF-associated remodeling in which there is a decrease in Cx43 expression but an increase in Cx40 expression [33]. The present study once again confirmed the previous reports, and further demonstrated that ACE2 overexpression reversed connexin remodeling and ultimately reduced AF inducibility. Of note, in the AF-control and Ad-EGFP group, reduced CX43 were mainly found at the cell poles, i.e., at the intercalated discs interconnecting the ends of the myocytes, while increased CX40 were located at the lateral borders, which has been interpreted as the basis of the anisotropy in atrial tissues. Furthermore, these changes would lead to an increase of non-uniform anisotropy and local conduction heterogeneity facilitating reentry in the dilated atria, resulting in the increase of AF inducibility [33]. However, we have not observed any major changes in the ratio of phosphorylated and nonphosphorylated forms of CX40 and CX43 in association with the change of CX40 and CX43 expression levels. Further experiments are necessary to elucidate the mechanisms involved in ACE2-induced changes of these two cardiac connexins.
Previous studies have demonstrated that Ang II had direct electrophysiologic effects via modification of ion channels related to atrial repolarization, such as Ito and ICaL, and connexins [13, 29]. Some evidence suggested that activated MAPKs by Ang II also had a certain effects on ion channel and connexin [3, 35]. Therefore, the amelioration of ion channels (Cav1, 2, Kv4.2/Kv4.3, KChiP2), and connexins remodeling by ACE2 overexpression in this study, as well as Ca2+ handling remodeling in previous study, might be related to its inhibition of MAPKs through up-regulation of MKP-1. Conceivably more detailed molecular mechanisms are awaited with great interest. Additionally, for the effects of ACE2/Ang-(1–7) axis on cardiac electrical remodeling, a few studies yielded discrepant results [7, 12, 14], with antiarrhythmic or proarrhythmic effects. More intensive researches are needed to illuminate on this crucial issue.
Conclusion
In conclusion, ACE2 overexpression could shift the RAS balance towards the protective axis, and attenuate cardiac fibrosis remodeling associated with reversed imbalance between MAPKs and MKP-1, ameliorate tachypacing-induced ion channels and connexins remodeling, and subsequently reduce the inducibility and duration of AF. Therefore, ACE2 overexpression by homogeneous transmural atrial gene transfer could improve atria electrical and structural remodeling in a canine atrial-pacing model.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Grant no. 81170166 and no. 31871075, Yin YH), the Research Fund for the Doctoral Program of Higher Education (Grant no. 20095503110002, Yin YH), Key projects of Bureau of Health of Chongqing (Grant no. 2010-1-67, Su L and Grant no. 2010-1-67, Lan XB), and the Program for Innovative Research Team of Chongqing Kuanren Hospital.
Conflict of interest
There is no conflict of interest.
Footnotes
J. Fan and L. Zou contributed equally to this paper.
References
- 1.Adam O, Lohfelm B, Thum T, Gupta SK, Puhl SL, Schafers HJ, Bohm M, Laufs U. Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res Cardiol. 2012;107:278. doi: 10.1007/s00395-012-0278-0. [DOI] [PubMed] [Google Scholar]
- 2.Amit G, Kikuchi K, Greener ID, Yang L, Novack V, Donahue JK. Selective molecular potassium channel blockade prevents atrial fibrillation. Circulation. 2010;121:2263–2270. doi: 10.1161/CIRCULATIONAHA.109.911156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolderman RW, Bruin P, Hermans JJ, Boerakker MJ, Dias AA, van der Veen FH, Maessen JG. Atrium-targeted drug delivery through an amiodarone-eluting bilayered patch. J Thorac Cardiovasc Surg. 2010;140:904–910. doi: 10.1016/j.jtcvs.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 4.Bosch RF, Scherer CR, Rub N, Wohrl S, Steinmeyer K, Haase H, Busch AE, Seipel L, Kuhlkamp V. Molecular mechanisms of early electrical remodeling: transcriptional downregulation of ion channel subunits reduces I(Ca, L) and I(to) in rapid atrial pacing in rabbits. J Am Coll Cardiol. 2003;41:858–869. doi: 10.1016/S0735-1097(02)02922-4. [DOI] [PubMed] [Google Scholar]
- 5.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choudhary R, Palm-Leis A, Scott RR, Guleria RS, Rachut E, Baker KM, Pan J. All-trans retinoic acid prevents development of cardiac remodeling in aortic banded rats by inhibiting the renin–angiotensin system. Am J Physiol Heart Circ Physiol. 2008;294:H633–H644. doi: 10.1152/ajpheart.01301.2007. [DOI] [PubMed] [Google Scholar]
- 7.Coutinho DCO, Monnerat-Cahli G, Ferreira AJ, Medei E. Activation of angiotensin-converting enzyme 2 improves cardiac electrical changes in ventricular repolarization in streptozotocin-induced hyperglycaemic rats. EUROPACE. 2014;16:1689–1696. doi: 10.1093/europace/euu070. [DOI] [PubMed] [Google Scholar]
- 8.Cui K, Fan J, Kao G, Ling Z, Yin Y, Su L. A Method for Highly Effective Gene Transfer into Atrial Myocardium by “Painting” on Atrial Epicardial in Canine. Chinese Journal of Biochemistry and Molecular Biology. 2011;27:1167–1173. [Google Scholar]
- 9.Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. NAT REV DRUG DISCOV. 2012;11:275–291. doi: 10.1038/nrd3682. [DOI] [PubMed] [Google Scholar]
- 10.Dobrev D, Nattel S. New insights into the molecular basis of atrial fibrillation: mechanistic and therapeutic implications. Cardiovasc Res. 2011;89:689–691. doi: 10.1093/cvr/cvr021. [DOI] [PubMed] [Google Scholar]
- 11.Dong B, Yu QT, Dai HY, Gao YY, Zhou ZL, Zhang L, Jiang H, Gao F, Li SY, Zhang YH, Bian HJ, Liu CX, Wang N, Xu H, Pan CM, Song HD, Zhang C, Zhang Y. Angiotensin-converting enzyme-2 overexpression improves left ventricular remodeling and function in a rat model of diabetic cardiomyopathy. J Am Coll Cardiol. 2012;59:739–747. doi: 10.1016/j.jacc.2011.09.071. [DOI] [PubMed] [Google Scholar]
- 12.Donoghue M, Wakimoto H, Maguire CT, Acton S, Hales P, Stagliano N, Fairchild-Huntress V, Xu J, Lorenz JN, Kadambi V, Berul CI, Breitbart RE. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J Mol Cell Cardiol. 2003;35:1043–1053. doi: 10.1016/S0022-2828(03)00177-9. [DOI] [PubMed] [Google Scholar]
- 13.Ehrlich JR, Hohnloser SH, Nattel S. Role of angiotensin system and effects of its inhibition in atrial fibrillation: clinical and experimental evidence. Eur Heart J. 2006;27:512–518. doi: 10.1093/eurheartj/ehi668. [DOI] [PubMed] [Google Scholar]
- 14.Ferreira AJ, Santos RA, Almeida AP. Angiotensin-(1–7): cardioprotective effect in myocardial ischemia/reperfusion. Hypertension. 2001;38:665–668. doi: 10.1161/01.HYP.38.3.665. [DOI] [PubMed] [Google Scholar]
- 15.Ferreira AJ, Shenoy V, Qi Y, Fraga-Silva RA, Santos RA, Katovich MJ, Raizada MK. Angiotensin-converting enzyme 2 activation protects against hypertension-induced cardiac fibrosis involving extracellular signal-regulated kinases. Exp Physiol. 2011;96:287–294. doi: 10.1113/expphysiol.2010.055277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goette A, Lendeckel U. Expression of angiotensin II receptors in human left and right atrial tissue in atrial fibrillation with and without underlying mitral valve disease. J Am Coll Cardiol. 2004;43(2363):2363–2364. doi: 10.1016/j.jacc.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 17.Hall MC, Kirubakaran S, Chowdhury R, Abidin N, Peters NS, Garratt CJ. Effects of angiotensin receptor blockade on atrial electrical remodeling and the ‘second factor’ in a goat burst-paced model of atrial fibrillation. J Renin Angiotensin Aldosterone Syst. 2010;11:222–233. doi: 10.1177/1470320310369604. [DOI] [PubMed] [Google Scholar]
- 18.He X, Gao X, Peng L, Wang S, Zhu Y, Ma H, Lin J, Duan DD. Atrial fibrillation induces myocardial fibrosis through angiotensin II type 1 receptor-specific Arkadia-mediated downregulation of Smad7. Circ Res. 2011;108:164–175. doi: 10.1161/CIRCRESAHA.110.234369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hussain W, Patel PM, Chowdhury RA, Cabo C, Ciaccio EJ, Lab MJ, Duffy HS, Wit AL, Peters NS. The Renin–Angiotensin system mediates the effects of stretch on conduction velocity, connexin43 expression, and redistribution in intact ventricle. J Cardiovasc Electrophysiol. 2010;21:1276–1283. doi: 10.1111/j.1540-8167.2010.01802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ichiki T, Takeda K, Tokunou T, Funakoshi Y, Ito K, Iino N, Takeshita A. Reactive oxygen species-mediated homologous downregulation of angiotensin II type 1 receptor mRNA by angiotensin II. Hypertension. 2001;37:535–540. doi: 10.1161/01.HYP.37.2.535. [DOI] [PubMed] [Google Scholar]
- 21.Kikuchi K, McDonald AD, Sasano T, Donahue JK. Targeted modification of atrial electrophysiology by homogeneous transmural atrial gene transfer. Circulation. 2005;111:264–270. doi: 10.1161/01.CIR.0000153338.47507.83. [DOI] [PubMed] [Google Scholar]
- 22.Kumagai K, Nakashima H, Urata H, Gondo N, Arakawa K, Saku K. Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation. J Am Coll Cardiol. 2003;41:2197–2204. doi: 10.1016/S0735-1097(03)00464-9. [DOI] [PubMed] [Google Scholar]
- 23.Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S. Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation. 2001;104:2608–2614. doi: 10.1161/hc4601.099402. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Li WM, Gong YT, Li BX, Liu W, Han W, Dong D, Sheng L, Xue JY, Zhang L, Chu S, Yang BF. The effects of cilazapril and valsartan on the mRNA and protein expressions of atrial calpains and atrial structural remodeling in atrial fibrillation dogs. Basic Res Cardiol. 2007;102:245–256. doi: 10.1007/s00395-007-0641-8. [DOI] [PubMed] [Google Scholar]
- 25.Liang B, Wang X, Zhang N, Yang H, Bai R, Liu M, Bian Y, Xiao C, Yang Z. Angiotensin-(1–7) Attenuates Angiotensin II-Induced ICAM-1, VCAM-1, and MCP-1 Expression via the MAS Receptor Through Suppression of P38 and NF-kappaB Pathways in HUVECs. Cell Physiol Biochem. 2015;35:2472–2482. doi: 10.1159/000374047. [DOI] [PubMed] [Google Scholar]
- 26.Liu E, Yang S, Xu Z, Li J, Yang W, Li G. Angiotensin-(1–7) prevents atrial fibrosis and atrial fibrillation in long-term atrial tachycardia dogs. Regul Pept. 2010;162:73–78. doi: 10.1016/j.regpep.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 27.McCollum LT, Gallagher PE, Ann TE. Angiotensin-(1–7) attenuates angiotensin II-induced cardiac remodeling associated with upregulation of dual-specificity phosphatase 1. Am J Physiol Heart Circ Physiol. 2012;302:H801–H810. doi: 10.1152/ajpheart.00908.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCollum LT, Gallagher PE, Tallant EA. Angiotensin-(1–7) abrogates mitogen-stimulated proliferation of cardiac fibroblasts. Peptides. 2012;34:380–388. doi: 10.1016/j.peptides.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michael G, Xiao L, Qi XY, Dobrev D, Nattel S. Remodelling of cardiac repolarization: how homeostatic responses can lead to arrhythmogenesis. Cardiovasc Res. 2009;81:491–499. doi: 10.1093/cvr/cvn266. [DOI] [PubMed] [Google Scholar]
- 30.Nakashima H, Kumagai K, Urata H, Gondo N, Ideishi M, Arakawa K. Angiotensin II antagonist prevents electrical remodeling in atrial fibrillation. Circulation. 2000;101:2612–2617. doi: 10.1161/01.CIR.101.22.2612. [DOI] [PubMed] [Google Scholar]
- 31.Pan CH, Lin JL, Lai LP, Chen CL, Stephen HS, Lin CS. Downregulation of angiotensin converting enzyme II is associated with pacing-induced sustained atrial fibrillation. FEBS Lett. 2007;581:526–534. doi: 10.1016/j.febslet.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 32.Pellman J, Lyon RC, Sheikh F. Extracellular matrix remodeling in atrial fibrosis: mechanisms and implications in atrial fibrillation. J Mol Cell Cardiol. 2010;48:461–467. doi: 10.1016/j.yjmcc.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polontchouk L, Haefliger JA, Ebelt B, Schaefer T, Stuhlmann D, Mehlhorn U, Kuhn-Regnier F, De Vivie ER, Dhein S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J Am Coll Cardiol. 2001;38:883–891. doi: 10.1016/S0735-1097(01)01443-7. [DOI] [PubMed] [Google Scholar]
- 34.Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011;91:265–325. doi: 10.1152/physrev.00031.2009. [DOI] [PubMed] [Google Scholar]
- 35.Sovari AA, Iravanian S, Dolmatova E, Jiao Z, Liu H, Zandieh S, Kumar V, Wang K, Bernstein KE, Bonini MG, Duffy HS, Dudley SC. Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J Am Coll Cardiol. 2011;58:2332–2339. doi: 10.1016/j.jacc.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai CT, Lai LP, Hwang JJ, Chen WP, Chiang FT, Hsu KL, Tseng CD, Tseng YZ, Lin JL. Renin-angiotensin system component expression in the HL-1 atrial cell line and in a pig model of atrial fibrillation. J Hypertens. 2008;26:570–582. doi: 10.1097/HJH.0b013e3282f34a4a. [DOI] [PubMed] [Google Scholar]
- 37.Tsai CT, Lai LP, Kuo KT, Hwang JJ, Hsieh CS, Hsu KL, Tseng CD, Tseng YZ, Chiang FT, Lin JL. Angiotensin II activates signal transducer and activators of transcription 3 via Rac1 in atrial myocytes and fibroblasts: implication for the therapeutic effect of statin in atrial structural remodeling. Circulation. 2008;117:344–355. doi: 10.1161/CIRCULATIONAHA.107.695346. [DOI] [PubMed] [Google Scholar]
- 38.Tsai CT, Wang DL, Chen WP, Hwang JJ, Hsieh CS, Hsu KL, Tseng CD, Lai LP, Tseng YZ, Chiang FT, Lin JL. Angiotensin II increases expression of alpha1C subunit of L-type calcium channel through a reactive oxygen species and cAMP response element-binding protein-dependent pathway in HL-1 myocytes. Circ Res. 2007;100:1476–1485. doi: 10.1161/01.RES.0000268497.93085.e1. [DOI] [PubMed] [Google Scholar]
- 39.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XH, Nattel S, Dobrev D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129:145–156. doi: 10.1161/CIRCULATIONAHA.113.006641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.von LD, Kockskamper J, Rubertus SU, Zhu D, Schmitto JD, Schondube FA, Hasenfuss G, Pieske B. Direct pro-arrhythmogenic effects of angiotensin II can be suppressed by AT1 receptor blockade in human atrial myocardium. EUR J HEART FAIL. 2008;10:1172–1176. doi: 10.1016/j.ejheart.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 41.Yue L, Melnyk P, Gaspo R, Wang Z, Nattel S. Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ Res. 1999;84:776–784. doi: 10.1161/01.RES.84.7.776. [DOI] [PubMed] [Google Scholar]
- 42.Zhao J, Xu W, Yun F, Zhao H, Li W, Gong Y, Yuan Y, Yan S, Zhang S, Ding X, Wang D, Zhang C, Dong D, Xiu C, Yang N, Liu L, Xue J, Li Y. Chronic obstructive sleep apnea causes atrial remodeling in canines: mechanisms and implications. Basic Res Cardiol. 2014;109:427. doi: 10.1007/s00395-014-0427-8. [DOI] [PubMed] [Google Scholar]
- 43.Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z, Oudit GY. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation. 2010;122(717–728):18–728. doi: 10.1161/CIRCULATIONAHA.110.955369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.