

Abstract
Excessive autophagic activity of alveolar type II epithelial (AT-II) cells is one of the main causes of acute lung injury (ALI); however, the underlying molecular mechanism remains to be determined. The microRNAs (miRNAs) are involved with autophagy in many diseases. The objective of this study was therefore to investigate the relationship between the miRNA expression and the autophagic activity of the AT-II cells in the pathogenesis of ALI and its molecular mechanism. A mouse model of ALI and AT-II cell injury was induced using lipopolysaccharide (LPS) in vivo and in vitro, and the expression of miR-34a and the autophagy-related proteins LC3 II/I and p62 were determined. Moreover, the autophagic activity was investigated after miR-34a overexpression and inhibition. The effects of miR-34a on its target gene, FoxO3, in regulating autophagic activity in AT-II cells were also determined. LPS induced autophagic activity and increased the expression of miR-34a in lung tissues and in AT-II cells. The in vitro results showed that the upregulation of miR-34a suppressed, whereas the inhibition of miR-34a promoted, autophagy in AT-II cells. Moreover, miR-34a could directly bind to the 3′-untranslated region of the autophagy-related gene, FoxO3, to decrease its expression. In addition, the knockdown of FoxO3 expression inhibited the autophagic activity in AT-II cells. Together, this study suggested that miR-34a might suppress the excessive autophagic activity in AT-II cells via targeting FoxO3 to reduce the damage of LPS-induced ALI.
KEY WORDS: acute lung injury, alveolar type II epithelial cells, autophagy, miR-34a, FoxO3
INTRODUCTION
Autophagy is an evolutionarily conserved lysosomal degradation pathway [14, 15]. It has been shown to be both protective and damaging in a variety of different models, suggesting that its role in human diseases is complex. Considerable evidence has shown the role of autophagy and selective autophagy in cell survival, cell death, and immune and inflammatory responses related to the pathogenesis of complex lung disease [14]. Previous studies have reported that autophagy is an induced reaction in response to various injuries such cigarette smoke, oxidative stress, and treatment with TNF-alpha or lipopolysaccharide (LPS), and different types of cell autophagy may have protective or adverse effects on acute lung injury (ALI) [7]. Accumulating evidence has reported that the excessive autophagic activity of alveolar type II epithelial cells plays an important pathogenic role in ALI [7, 20], and its inhibition is better to ALI [7, 14].
Lo et al. reported that autophagy in the septic lung resulting from cecal ligation and puncture (CLP) represented a protective response [12]. However, autophagy, by virtue of excessive autophagosome accumulation in alveolar type II epithelial cells, may play a maladaptive role in the late stages of sepsis, leading to ALI. Two studies [13, 19] independently reported that excessive autophagic activity of alveolar type II epithelial cells may contribute to the development of ARDS (acute respiratory distress syndrome) in H5N1 influenza patients. Inhibition of autophagy could be used as a novel strategy for the treatment of H5N1 infection, and studies have suggested that autophagy blocking agents (e.g., beclin1 and 3-methyladenine) might be useful for decreasing the incidence of ALI during the infection of H5N1 virus [17, 19]. Previous studies [9, 10] also reported that inhalation of nanomaterials, including starburst polyamidoamine dendrimer (PAMAM) and COOH-CNT (a functionalized single-walled carbon nanotube), induced ALI by deregulating the Akt-TSC2-mTOR signaling pathway in vivo. Additional in vitro studies reported that treatment with PAMAM or COOH-CNT resulted in autophagosome aggregation in alveolar type II epithelial cells. The autophagy inhibitor, 3-methyladenine, rescued the nanoparticle-induced excessive autophagy and ameliorated ALI in mice. Smoke exposure also caused ALI, and smoke exposure can lead to excessive autophagy in alveolar type II epithelial cells [2].
The excessive autophagic activity of alveolar type II epithelial cells could lead to increased secretion of inflammatory factors, cell death, and various dysfunctions, which resulting in aggravation of ALI. Autophagy inhibitors can reduce alveolar type II epithelial cell autophagic activity and can inhibit the development of ALI. It is therefore important to study the autophagic regulation mechanism of alveolar type II epithelial cells during ALI.
MicroRNAs are small non-coding RNAs that negatively regulate gene expression by binding to the 3′-UTR of their various target mRNAs to promote mRNA degradation or to inhibit translation. Recently, studies to determine the genetic components of ALI/ARDS pathogenesis have investigated the involvement of miRNAs in this process. The microRNA-34a (miR-34a) is a multifunctional regulator involved in cell proliferation, apoptosis, growth, and autophagy. It has been reported that miR-34a suppressed autophagic activity in angiotensin II-treated cardiomyocytes [8] and tubular epithelial cells during acute kidney injury [11].
The miR-34a plays an important role in the development of the heart and lung in mammals. It has been reported that miR-34a expression was significantly increased in neonatal lungs in response to hypoxia [1], bleomycin-induced pulmonary fibrosis [22], and in Staphylococcal enterotoxin B-induced ALI [18]. A previous study also reported that miR-34a modulated the autophagy activity via the direct inhibition of ATG9A and ATG4B expression [8, 24].
In this study, we characterized miR-34a expression in ALI mouse lung tissues and in alveolar type II epithelial cells induced by LPS and investigated the effects of miR-34a on alveolar type II epithelial cell autophagy in ALI. The results data showed that miR-34a targeted the 3′-UTR sequence of FoxO3 mRNA and modulated its expression, suggesting that miR-34a might suppress alveolar type II epithelial cell autophagy by targeting FoxO3.
MATERIALS AND METHODS
Animals and the ALI Model
Healthy male C57BL/6 mice aged 8–10 weeks and weighing 17.6–25.4 g purchased from the Experimental Animal Center of the Hunan University of Chinese Medicine (Changsha, China) were used for experimentation and allowed to acclimate for 3 days before experimentation. The animals were fed rodent chow and water ad libitum and were randomly divided into different groups: an ALI group with intratracheal instillation of 3 mg/kg LPS (Escherichia coli 0111:B4, Sigma, St. Louis, Missouri, USA) and a control group with intratracheal instillation of equal volume of normal saline. The mice were anesthetized by an intraperitoneal injection of 10% chloral hydrate (QingDao YuLong Algae CO. LTD., QingDao, China) and kept in a supine position while spontaneous breathing was monitored. Mice of ALI group sacrificed at the indicated times (6, 12, 24 h) after injury, and those of control group were sacrificed at 24 h after intratracheal instillation of normal saline. After the experimental protocol was completed, lung tissue from animals (n = 6 per time point) was rapidly obtained. ALI induction was verified by pathological examination of the lung.
Lung Histopathology
At necropsy, the left lung was excised and fixed with 4% paraformaldehyde for 24 h, and then the lung tissue was dehydrated with graded alcohol and embedded in paraffin at 52 °C. Sections were prepared and stained with hematoxylin and eosin. For evaluation of the severity of lung injury, each lung section was blindly assigned a lung injury score (LIS) by two pathologists using the method described by Nishina et al. [16]. Briefly, lung tissue sections were assessed for alveolar congestion, hemorrhage, infiltration or aggregation of neutrophils in the airspace or vessel wall, and thickness of the alveolar wall/hyaline membrane. The degree of lung injury was scored as follows: 0, minimum; 1, mild; 2, moderate; 3, severe; and 4, maximum damage. For each animal, six high-magnification fields were randomly selected for grading and an average LIS score was calculated.
Isolation of Murine Alveolar Type II Epithelial Cells and Induction of Cell Injury
Alveolar type II epithelial cells were isolated at 90–95% purity from 6-week-old mice following the procedure described by Corti and colleagues [3]. Briefly, mice were killed, the pulmonary artery was cannulated, and the lungs were perfused with normal saline in situ to flush out blood. The trachea was cannulated, and 2 ml dispase II (5 U/ml in PBS; Becton-Dickinson, San Jose, CA) were injected in the lungs followed by 0.3 ml warmed low-melting-point agarose (1% in PBS) to prevent the isolation of Clara cells and upper airway epithelial cells. The lungs were cooled on ice, dissected free, rinsed with saline, and placed in 5 ml dispase to digest at room temperature for 60 min with gentle rocking. Pancreatic DNase (0.01% in DMEM; Sigma-Aldrich) was added for the final 5 min of incubation. Lung tissue was teased apart, and the resulting cell suspension was filtered sequentially through 100-, 40-, and 21-μm sterile nylon meshes. Leukocytes were removed by panning with rat polyclonal anti-murine CD45 and anti-murine CD16/CD32 antibodies (both Becton-Dickinson) for 2 h at 37 °C. Nonadherent cells were collected, pelleted by centrifugation, resuspended in normal saline, and counted using a hemocytometer. Purity of isolated AT-II cell preparations was determined by visualization of lamellar bodies in modified Papanicolaou-stained cytospins. For the incubation experiments, alveolar type II epithelial cells were induced with LPS (1 μg/ml). A dose of LPS was chosen that was consistent with previous reports and with our pilot study [23].
Transfection of miR-34a Mimics or Inhibitor
MiR-34a mimics or a miR-34a inhibitor and their negative control oligonucleotides were obtained from GenePharma (Shanghai, China). The transfection was performed using Lipofectamine™ 2000 (Invitrogen, Carlsbad, California, USA) according to the instructions provided by the manufacturer. Briefly, approximately 5 × 105 cells were seeded to flasks containing 5 ml of appropriate complete growth medium and incubated at 37 °C with 5% CO2 until the cells were 70 to 80% confluent (24 h). After rinsing with serum-free, antibiotic-free medium, cells were transfected with either miR-34a mimics or a miR-34a inhibitor and their negative control oligonucleotides in 20 μL lipofectamine, followed by incubation at 37 °C in a CO2 incubator for 6 h. The transfected cells were resuspended and cultured in regular culture medium for 48–72 h before analyses, and cells were treated with LPS (1 μg/ml) to induce injury.
siRNA Transfection
FoxO3 siRNA and control siRNA were purchased from Cell Signaling Technology (Beverly, MAass., USA). Approximately 5 × 104 cells were seeded in each well of a 24-well microplate and grown for 24 h to reach 30–50% confluency. The cells were then incubated with a mixture of siRNA and Lipofectamine™ 2000 (Invitrogen) in 100 μl of serum-free Opti-MEM® according to the manufacturer’s instructions. Medium may be changed after 4–6 h. Incubate the cells at 37 °C in a CO2 incubator for 48 h. The expression of FoxO3 was detected using a real-time polymerase chain reaction (RT-PCR) and western blotting.
RNA Extraction and RT-PCR
Total RNA was extracted using the TRIzol® reagent (Invitrogen) according to the manufacturer’s instructions. Five micrograms of total RNA was then used as a template to synthesize cDNA using the First Strand Synthesis Kit (Invitrogen). The cDNA from this synthesis was then used in quantitative RT-PCR analyses using the TaqMan system (ABI-Prism 7700 Sequence Detection System, Biosystems, Bethesda, Maryland, USA) using SYBR® Green dye. The following primer pairs were used: mouse FoxO3; forward, 5′-GTGGACCGACTTCCGCTCGC-3′ and reverse, 5′-GCTTGCCAGGATGGGCGACA-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH); forward, 5′-TGGTATCGTGGAAGGACTC-3′ and reverse, 5′-AGTAGAGGCAGGGATGATG-3′. The RT-PCR data were normalized by measuring the average cycle threshold (Ct) ratios between candidate genes and the control gene, GAPDH. The formula, 2Ct(candidate)/2Ct(control), was used to calculate the normalized ratios.
TaqMan RT-PCR for the Quantitation of miRNA
Total RNA was isolated from frozen lung tissue and cell lines using Trizol™ (Invitrogen), then reverse transcribed using the Taqman™ microRNA reverse transcription kit, and subjected to RT-PCR using the TaqMan™microRNA assay kit (Biosystems) according to the manufacturer’s instructions. The reactions were performed in triplicate using a Stratagene Mx3000 instrument, and the miRNA expression was normalized to U6.
Western Blot Analysis
Proteins were isolated and separated by 12% SDS–PAGE and transferred onto PVDF membranes (Schleicher & Schuell, Germany). The membranes were blocked overnight in phosphate-buffered saline (PBS) containing 10% nonfat dry milk and 0.5% Tween-20 and then incubated with primary antibodies for 2 h. Horseradish peroxidase-conjugated IgG was used as secondary antibodies. The immunoreactive bands were visualized using diaminobenzidine (DAB; Boster Biological Technology, Wuhan, China), and the protein expression was normalized to GAPDH. The following antibodies were used: rabbit polyclone anti-LC3B (1:1000; Novus Biologicals, Littleton, Colorado, USA),anti-p62 monoclonal antibody (BD Transduction Laboratories, Lake Franklin, New Jersey, USA), rabbit anti-FoxO3 polyclonal antibody (Abcam, Cambridge Science Park, UK), goat GAPDH monoclonal antibody (Sigma), HRP-conjugated anti-goat, and anti-rabbit IgG (Boster Biological Technology).
Construction of the Plasmid Vector
To determine whether miR-34a directly bound to the 3′-UTR of FoxO3, we utilized a 3′-UTR luciferase reporter assay. The sequence of 443 nucleotides (including the binding sites for miR-34a) of the 3′-UTR of FoxO3 were amplified by PCR using the following primers: forward, 5′-ACACAAGCTTCTTATCTTGTATTTCCAT-3′ and reverse,5′-ACACGAGCTCAGCTGTGAACACCAACCC-3′ and inserted downstream of the luciferase gene in the pLuc luciferase vector (Ambion, Austen, Texas, USA). Site-directed mutagenesis of the miR-34a target site at the 3′-UTR of FoxO3 was performed using the QuickChange mutagenesis kit (Stratagene, Heidelberg, Germany). The resulting constructs were sequenced and named pLuc-FoxA1-wt or pLuc-FoxA1-mut.
Luciferase Assay
For reporter assays, exponentially growing alveolar type II epithelial cells were cultured in 24-well plates and transfected with 100 ng of pLuc-FoxO3-wt or pLuc-FoxO3-mut and 50 nM of miR-34a mimic or miR-34a inhibitor using Lipofectamine™ 2000 (Invitrogen). Forty-eight hours after transfection, the cells were harvested and assayed with the Dual-Luciferase Reporter assay kit (Promega, Madison, Wisconsin, USA) according to the manufacturer’s instructions. All transfections were performed in triplicate from at least three independent experiments.
Statistical Analysis
Data in the figures and text were expressed as the mean ± standard error of the mean (SEM). Each experiment was performed at least three times, and statistical analyses were performed using one-way analysis of variance. Otherwise, representative data were shown. A value of P < 0.05 was considered statistically significant.
RESULTS
LPS Induced ALI in Mice
Histological examination by light microscopy revealed that in the control lungs, the alveoli were fully distended and no alveolar wall edema or congestion was found. Occasionally, inflammatory cells were scattered throughout the lung. In contrast, in the lung tissue from LPS-treated animals, the histological features were consistent with ALI, including alveolar hemorrhage, destruction of alveolar attachment points, an increased number of interstitial cells, and extensive neutrophil infiltration. The LIS significantly increased in the LPS group (Fig. 1a). These results showed that LPS successfully induced ALI in mice.
Fig. 1.
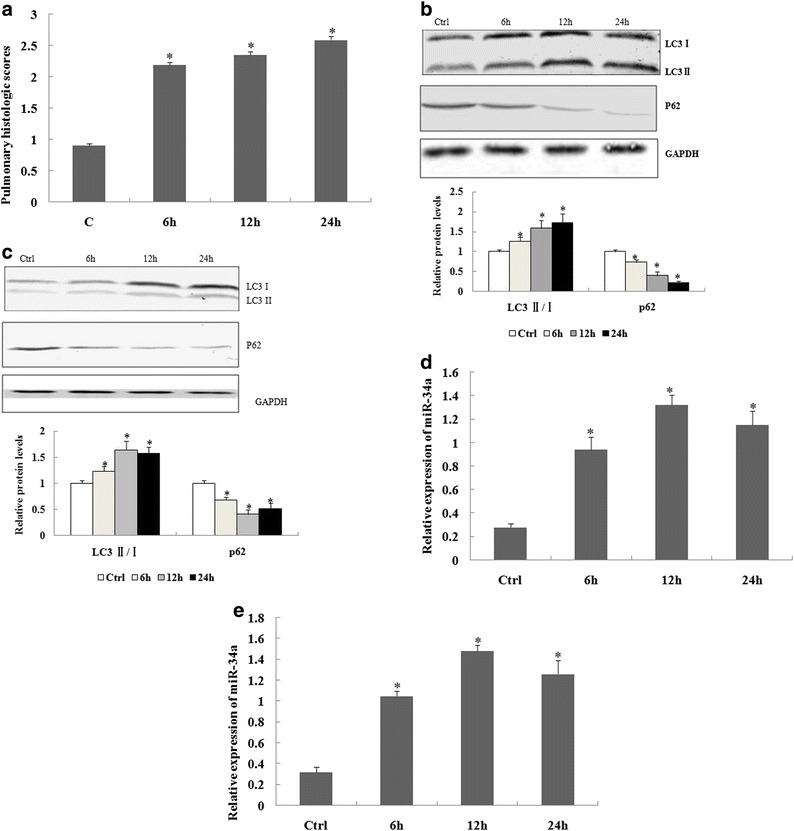
Change of pulmonary histological scores, autophagy, and miR-34a expression in lung tissue of acute lung injury (ALI) mice and alveolar type II epithelial cells induced by lipopolysaccharide (LPS). a The changes of pulmonary histological scores in different groups. b The levels of autophagy-related protein expression were determined by western blotting in lung tissues of an ALI model induced by LPS or in alveolar type II epithelial cells of mice induced by LPS (c) (d) The levels of miR-34a were determined by real-time polymerase chain reaction in lung tissues of an ALI model induced by LPS or in alveolar type II epithelial cells of mice induced by LPS (e). The relative values of all results were determined and expressed as the mean ± standard error of the mean of three experiments performed in duplicate. *Statistically significant difference versus the control group (Ctrl), p < 0.05.
LPS Induced Autophagy and miR-34a Expression in Lung Tissue of Mice and in Alveolar Type II Epithelial Cells
Accumulating evidence has shown that excessive autophagic activity occurs in alveolar type II epithelial cells during ALI. We therefore characterized the levels of autophagy-related proteins LC3 II/Iand p62 expression in ALI mouse lung tissues and in LPS-induced alveolar type II epithelial cells. The results showed that the levels of LC3 II/I expression were elevated at 6, 12, and 24 h in lung tissue of ALI mice and in LPS-induced alveolar type II epithelial cells. In contrast, the levels of p62 protein were repressed at 6, 12, and 24 h after LPS induction (Fig. 1b, c).
A previous study reported that miR-34a is involved in the process of autophagy. We therefore determined the expression of miR-34a in ALI mice and alveolar type II epithelial cells. The results showed that miR-34a expression was significantly increased at 6, 12, and 24 h (Fig. 1d, e), suggesting that LPS cause autophagy in the lungs of mice and alveolar type II epithelial cells and induce miR-34a expression.
The miR-34a Regulated Autophagy in Murine Alveolar Type II Epithelial Cells
We then determined the effect of miR-34a on autophagic activity in mouse alveolar type II epithelial cells. The miR-34a expression was upregulated by miR-34a mimcs, while it was downregulated by transfection of a miR-34a inhibitor in alveolar type II epithelial cells (Fig. 2a). Transfection with miR-34a mimics decreased the levels of LC3 II/Iin alveolar type II epithelial cells treated with LPS at 12 h. However, treatment with the miR-34a inhibitor increased the levels of LC3 II/I in alveolar type II epithelial cells. In contrast, the levels of p62 protein were induced in the miR-34a mimic group but suppressed in the miR-34a inhibitor group (Fig. 2b). Taken together, the results showed that miR-34a suppressed LPS-induced autophagic activity in alveolar type II epithelial cells.
Fig. 2.
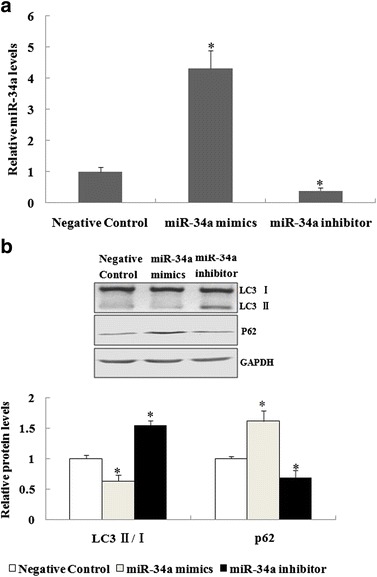
The miR-34a suppressed autophagic activity in mouse alveolar type II epithelial cells. a Forty-eight hours after alveolar type II epithelial cells were transfected with miR-34a mimics, miR-34a inhibitor, or the negative control, Taqman real-time quantitative polymerase chain reaction analyses were performed to assess miR-34a expression. b Forty-eight hours after transfection with miR-34a mimics, a miR-34a inhibitor, or a negative control, alveolar type II epithelial cells were incubated with LPS, and the autophagy-related protein expression determined by western blot analyses. The relative values of all results were expressed as the mean ± standard error of the mean of three experiments performed in duplicate. *A statistically significant difference versus the negative control, p < 0.05.
The miR-34a Directly Targeted FoxO3 in Murine Alveolar Type II Epithelial Cells
It has been reported that miRNAs post-transcriptionally reduced the protein expression of specific target mRNAs. Bioinformatic analyses showed that there was one binding site of miR-34a in the 3′-UTR region of FoxO3. FoxO3 is an important protein in autophagy, so we proposed that the mechanism of miR-34 that suppressed autophagy could be partly due to the downregulation of FoxO3 during ALI.
The role of miR-34a in the regulation of the FoxO3 gene expression was therefore further investigated. To confirm the role of miR-34a in the regulation of FoxO3 expression, quantitative RT-PCR and western blotting were performed. The results showed that miR-34a mimics or the miR-34a inhibitor had no significant effect on the mRNA levels of FoxO3 when compared with a negative control (Fig. 3a). Western blot analyses showed that the miR-34a mimics suppressed FoxO3 protein expression, whereas transfection of the miR-34a inhibitor promoted FoxO3 protein expression compared with the negative control (Fig. 3b). To further confirm that miR-34a targets FoxO3, the miR-34a binding sequence present at the 3′-UTR of the FoxO3 mRNA was subcloned downstream of the firefly luciferase reporter gene in pLUC vectors and then co-transfected with either the negative control, miR-34a mimics, or the miR-34a inhibitor into alveolar type II epithelial cells. The relative luciferase activity of the reporter containing the wild-type 3′-UTR of FoxO3 was significantly suppressed when the miR-34a mimics were co-transfected, but not when it was co-transfected with a mutant sequence with the miR-34a binding site (Fig. 3c). In addition, there was a significant increase in luciferase activity for the reporter containing the wild-type 3′-UTR of FoxO3 when the miR-34a inhibitor was co-transfected, but not when it contained a mutant sequence with a miR-34a binding site. Together, the results showed that miR-34a bound directly to a specific site on the 3′-UTR of FoxO3 to regulate its expression.
Fig. 3.
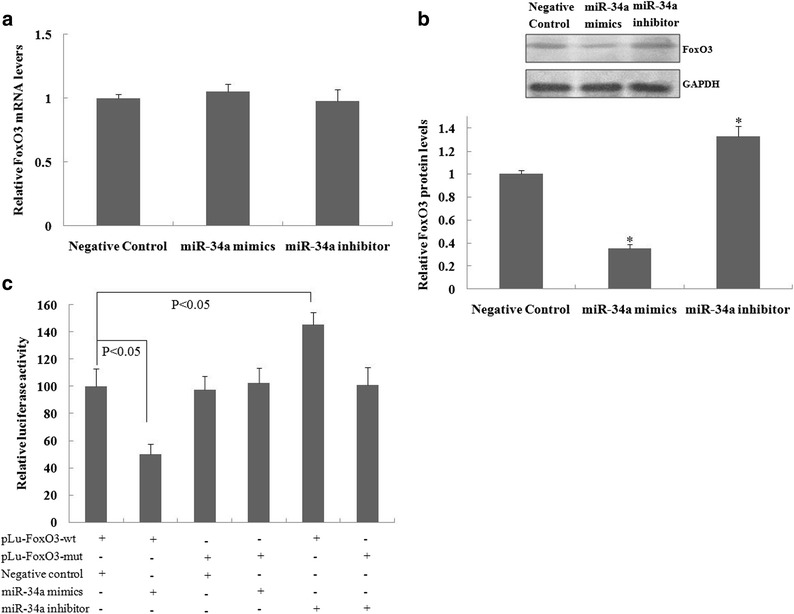
The miR-34a suppressed FoxO3 protein expression in murine alveolar type II epithelial cells. Forty-eight hours after alveolar type II epithelial cells were transfected with miR-34a mimics, a miR-34a inhibitor, or a negative control, Taqman real-time quantitative polymerase chain reaction or western blot analyses were performed to assess FoxO3 mRNA (a) and protein (b) expression. * A statistically significant difference versus the negative control, p < 0.05. (c) The alveolar type II epithelial cells were co-transfected with miR-34a mimics or a miR-34a inhibitor and pLu-FoxO3. After 48 h, the luciferase activity was performed. The values are expressed as the relative luciferase activity after normalization to Renilla luciferase activity. The relative values of all results were determined and expressed as the mean ± SEM of three experiments performed in duplicate.
FoxO3 Knockdown Inhibited Autophagic Activity in Murine Alveolar Type II Epithelial Cells
We then characterized the effect of FoxO3 inhibition on autophagy in alveolar type II epithelial cells. After treating murine alveolar type II epithelial cells with FoxO3 siRNA, the FoxO3 mRNA and protein expression were significantly suppressed (Fig. 4a, b). Figure 4c shows that FoxO3 siRNA treatment significantly decreased the ratio of LC3 II/I, whereas it induced p62 expression when compared to the control siRNA group when alveolar type II epithelial cells were incubated with LPS for 12 h.
Fig. 4.
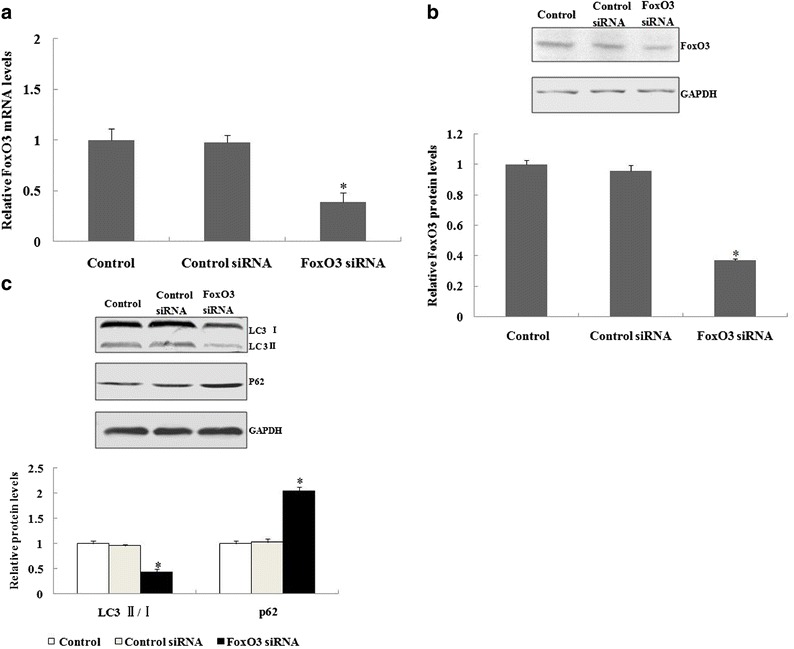
Autophagy was suppressed in murine alveolar type II epithelial cells when FoxO3 was inhibited by siRNA. a, b After 48 h of transfection with control siRNA or FoxO3 siRNA, the real-time polymerase chain reaction or western blotting was used to analyzethe FoxO3 mRNA and protein expression in murine alveolar type II epithelial cells. c After 48 h of transfection with control siRNA or FoxO3 siRNA, alveolar type II epithelial cells were incubated with lipopolysaccharide, and autophagy-related protein expression was determined by western blot analyses. *A statistically significant difference versus the control group, p < 0.05.
DISCUSSION
The role of autophagy involves maintenance of normal cell and tissue homeostasis by elimination of damaged proteins and organelles in cells. However, excessive or deficient autophagy can contribute to disease pathogenesis. In the stage of ALI induced by H5N1 influenza, CLP or nanomaterial particles, past studies have reported that alveolar type II epithelial cell autophagy was excessively activated [12, 13, 17, 19]. Further studies confirmed that excessive autophagy activation of alveolar type II epithelial cells was a key feature of aggravated ALI. The present study showed that the ratio of LC3 II/Iincreased, whereas the level of p62 was reduced in lung tissues of LPS-induced ALI mice and LPS-induced alveolar type II epithelial cells, to simulate alveolar type II epithelial cell injury during ALI. This suggested that the autophagic activity of mouse alveolar type II epithelial cells also increased in LPS-induced ALI.
Significant progress has been made towards understanding the contribution of miRNAs, including miR-34a in autophagy [5]. The miR-101 inhibits autophagy in the MCF-7 cell line [6], and the miRNA-212/132 family regulates cardiomyocyte autophagy [21]. The miR-34a suppressed autophagy by suppression of ATG4B in tubular epithelial cells in acute kidney injury [11]. Another study reported that miR-34a played an important role in the regulation of Ang II-induced cardiomyocyte hypertrophy by the inhibition of ATG9A expression and autophagic activity [8]. Previous studies have also reported that miR-34a is also involved in various lung diseases and could regulate senescence in alveolar type II epithelial cells of patients with idiopathic pulmonary fibrosis [4]. Our study showed a significant upregulation of miR-34a in LPS-induced mouse lung tissue and alveolar type II epithelial cells. Furthermore, overexpression of miR-34a by miR-34a mimics suppressed LPS-induced autophagy of alveolar type II alveolar epithelial cells, but inhibition of the expression of miR-34a by a miR-34a inhibitor significantly increased the LPS-induced autophagy of alveolar type II epithelial cells. These results suggested that inhibiting the excessive autophagic activity of type II alveolar epithelial cells was a mechanism involved in the increased miR-34a expression and protection against ALI. However, the mechanisms involving a miR-34a decrease in autophagic activity of alveolar type II epithelial cells during ALI still remain poorly understood.
Bioinformatic analyses showed that FoxO3, an autophagy-related gene, was a candidate target of miR-34a. Thus, we predicted that miR-34a regulates alveolar type II epithelial cells autophagy by regulation of FoxO3 expression. Thus, the possibility of a regulatory mechanism involving miR-34a and FoxO3 was further explored. Alveolar type II epithelial cells of mice were isolated and incubated with LPS to simulate alveolar type II epithelial cell injury in ALI, and then transfected with miR-34a mimics or a miR-34a inhibitor. FoxO3 expression significantly decreased posttransfection after treatment with miR-34a mimics compared with a negative control in alveolar type II epithelial cells, while FoxO3 expression significantly increased posttransfection after treatment with the miR-34a inhibitor. The luciferase reporter assay was then used to characterize the mechanism of miR-34a as a regulator of FoxO3 expression during ALI. The results showed that miR-34a suppressed FoxO3 protein expression by binding to the 3′-UTR region. We therefore concluded that FoxO3 is a target gene of miR-34a. We also transfected FoxO3 siRNA into alveolar type II epithelial cells to show that FoxO3 siRNA treatment significantly decreased the autophagic activity when alveolar type II epithelial cells were incubated with LPS.
The miR-34a suppresses autophagy that might be beneficial or prejudiced for pathogenesis, depending on the disorder. However, the function of miR-34a and its targets have not been reported in ALI and alveolar type II epithelial cells. Autophagy can be harmful or beneficial in ALI. Moderate autophagy protects cells against various kinds of injury. However, excessive autophagy, especially for alveolar type II epithelial cells, plays an important pathogenic role in ALI, and its inhibition may prevent ALI [7, 14, 20]. In the present study, the autophagy activity of lung tissue increased in an LPS-induced ALI model and in LPS-induced alveolar type II epithelial cells that induced injury to alveolar type II epithelial cells during ALI. The miR-34a was upregulated in ALI, which inhibited LPS-induced autophagy of alveolar type II epithelial cell injury during ALI. In addition, the results showed that targeting of FoxO3, an autophagy-related gene, involved miR-34a, and silencing of FoxO3 expression inhibited LPS-induce autophagy.
In conclusion, this study provided the first evidence that miR-34a suppresses the autophagic activity of alveolar type II epithelial cells during LPS-induced ALI by inhibiting FoxO3 expression. Because of the important roles of excessive autophagic activity of alveolar type II epithelial cells in the pathological process of ALI, we predict that the induction of miR-34a is a protective factor that could be used as a novel therapeutic approach for the treatment of ALI.
We therefore propose that the autophagy activity of alveolar type II epithelial cells and the expression of miR-34a increase in LPS-induced ALI. The miR-34a could suppress this excessive autophagic activity of alveolar type II epithelial cells by inhibition of FoxO3 expression, resulting in protection against ALI.
Acknowledgements
This work was supported by funding from the Natural Science Foundation of Hunan Province, China (No. 2014JJ7062), Research Foundation of Traditional Chinese medicine of Hunan Province, China (No. 201696), The Open Research Foundation of Key Laboratory of traditional Chinese medicine in Hunan, Hunan University of Chinese Medicine China (No. ZYNK201506), the Research Foundation of Education Bureau of Hunan Province, China (No. 13C691), the 58 batch Postdoctoral Science Foundation funded, China (No.2015M580690), the Research Foundation of Stem Cell Regulation and Application, Hunan University of Chinese Medicine, China (No. 2013GXB02).
Compliance with Ethical Standards
Animal experimentations were conducted according to the National Institute of Health Guide for the Care and Use of Laboratory Animals and received the approval of the Animal Care and Use Committee of Hunan University of Chinese Medicine, Changsha China.
Footnotes
Lan Song and Fangliang Zhou contributed equally to this work.
Contributor Information
Duanfang Liao, Email: dfliao66@aliyun.com.
Zhaojun Xu, Email: Xuzj1492@163.com.
References
- 1.Bhaskaran M, Xi D, Wang Y, Huang C, Narasaraju T, Shu W, Zhao C, et al. Identification of microRNAs changed in the neonatal lungs in response to hyperoxia exposure. Physiological Genomics. 2012;44(20):970–980. doi: 10.1152/physiolgenomics.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bodas M, Min T, Vij N. Critical role of CFTR-dependent lipid rafts in cigarette smoke-induced lung epithelial injury. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2011;300(6):L811–L820. doi: 10.1152/ajplung.00408.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corti M, Brody AR, Harrison JH. Isolation and primary culture of murine alveolar type II cells. American Journal of Respiratory Cell and Molecular Biology. 1996;14(4):309–315. doi: 10.1165/ajrcmb.14.4.8600933. [DOI] [PubMed] [Google Scholar]
- 4.Disayabutr S, Kim EK, Cha SI, Green G, Naikawadi RP, Jones KD, Golden JA, et al. miR-34 miRNAs regulate cellular senescence in Type II alveolar epithelial cells of patients with idiopathic pulmonary fibrosis. PloS One. 2016;11(6):e0158367. doi: 10.1371/journal.pone.0158367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frankel LB, Lund AH. MicroRNA regulation of autophagy. Carcinogenesis. 2012;33(11):2018–2025. doi: 10.1093/carcin/bgs266. [DOI] [PubMed] [Google Scholar]
- 6.Frankel LB, Wen J, Lees M, Hoyer-Hansen M, Farkas T, Krogh A, Jaattela M, Lund AH. microRNA-101 is a potent inhibitor of autophagy. EMBO Journal. 2011;30(22):4628–4641. doi: 10.1038/emboj.2011.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Y, Liu J, Wu YF, Lou J, Mao YY, Shen HH, Chen ZH. mTOR and autophagy in regulation of acute lung injury: a review and perspective. Microbes and Infection. 2014;16(9):727–734. doi: 10.1016/j.micinf.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Huang J, Sun W, Huang H, Ye J, Pan W, Zhong Y, Cheng C, et al. miR-34a modulates angiotensin II-induced myocardial hypertrophy by direct inhibition of ATG9A expression and autophagic activity. PloS One. 2014;9(4):e94382. doi: 10.1371/journal.pone.0094382. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Li C, Liu H, Sun Y, Wang H, Guo F, Rao S, Deng J, et al. PAMAM nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. Journal of Molecular Cell Biology. 2009;1(1):37–45. doi: 10.1093/jmcb/mjp002. [DOI] [PubMed] [Google Scholar]
- 10.Liu HL, Zhang YL, Yang N, Zhang YX, Liu XQ, Li CG, Zhao Y, et al. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt-TSC2-mTOR signaling. Cell Death & Disease. 2011;2:e159. doi: 10.1038/cddis.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu XJ, Hong Q, Wang Z, Yu YY, Zou X, Xu LH. MicroRNA-34a suppresses autophagy in tubular epithelial cells in acute kidney injury. American Journal of Nephrology. 2015;42(2):168–175. doi: 10.1159/000439185. [DOI] [PubMed] [Google Scholar]
- 12.Lo S, Yuan SS, Hsu C, Cheng YJ, Chang YF, Hsueh HW, Lee PH, Hsieh YC. Lc3 over-expression improves survival and attenuates lung injury through increasing autophagosomal clearance in septic mice. Annals of Surgery. 2013;257(2):352–363. doi: 10.1097/SLA.0b013e318269d0e2. [DOI] [PubMed] [Google Scholar]
- 13.Ma J, Sun Q, Mi R, Zhang H. Avian influenza A virus H5N1 causes autophagy-mediated cell death through suppression of mTOR signaling. Journal of Genetics and Genomics. 2011;38(11):533–537. doi: 10.1016/j.jgg.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Mizumura K, Cloonan SM, Haspel JA, Choi AM. The emerging importance of autophagy in pulmonary diseases. Chest. 2012;142(5):1289–1299. doi: 10.1378/chest.12-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Nishina K, Mikawa K, Takao Y, Maekawa N, Shiga M, Obara H. ONO-5046, an elastase inhibitor, attenuates endotoxin-induced acute lung injury in rabbits. Anesthesia & Analgesia. 1997;84(5):1097–1103. doi: 10.1213/00000539-199705000-00026. [DOI] [PubMed] [Google Scholar]
- 17.Pan H, Zhang Y, Luo Z, Li P, Liu L, Wang C, Wang H, Li H, Ma Y. Autophagy mediates avian influenza H5N1 pseudotyped particle-induced lung inflammation through NF-kappaB and p38 MAPK signaling pathways. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2014;306(2):L183–L195. doi: 10.1152/ajplung.00147.2013. [DOI] [PubMed] [Google Scholar]
- 18.Rao R, Nagarkatti P, Nagarkatti M. Role of miRNA in the regulation of inflammatory genes in staphylococcal enterotoxin B-induced acute inflammatory lung injury and mortality. Toxicological Sciences. 2015;144(2):284–297. doi: 10.1093/toxsci/kfu315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Y, Li C, Shu Y, Ju X, Zou Z, Wang H, Rao S, et al. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Science Signaling. 2012;5(212):ra16. doi: 10.1126/scisignal.2001931. [DOI] [PubMed] [Google Scholar]
- 20.Tang PS, Mura M, Seth R, Liu M. Acute lung injury and cell death: how many ways can cells die? American Journal of Physiology - Lung Cellular and Molecular Physiology. 2008;294(4):L632–L641. doi: 10.1152/ajplung.00262.2007. [DOI] [PubMed] [Google Scholar]
- 21.Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, Dangwal S, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nature Communications. 2012;3:1078. doi: 10.1038/ncomms2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie T, Liang J, Guo R, Liu N, Noble PW, Jiang D. Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiological Genomics. 2011;43(9):479–487. doi: 10.1152/physiolgenomics.00222.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Z, Zhang C, Cheng L, Hu M, Tao H, Song L. The microRNA miR-17 regulates lung FoxA1 expression during lipopolysaccharide-induced acute lung injury. Biochemical and Biophysical Research Communications. 2014;445(1):48–53. doi: 10.1016/j.bbrc.2014.01.108. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, Chen D, He Y, Melendez A, Feng Z, Hong Q, Bai X, et al. MiR-34 modulates Caenorhabditis elegans lifespan via repressing the autophagy gene atg9. Age (Dordrecht, Netherlands) 2013;35(1):11–22. doi: 10.1007/s11357-011-9324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]