

Abstract
We report on 12 patients with chronic granulomatous disease transplanted with hematopoietic stem cells from matched unrelated (n = 9) or matched sibling donors (n = 3). The most common infectious complication was pulmonary aspergillosis, which nine patients had previously developed. Only 5 of 12 individuals had normal lung function prior to transplantation. At a mean follow-up of 53 months 9 of the 12 patients are alive including 7 of 9 following matched unrelated donor (MUD) transplantation. One patient died from ARDS, another from systemic BK virus infection, the third from complications of chronic graft-versus-host disease. Seven of nine surviving patients have normal lung function now. HSCT from a MUD is an option worth considering when no matched family donor is available. Restricted lung function prior to HSCT does not appear to be a limiting factor for such treatment.
Keywords: Chronic granulomatous disease, Stem cell transplantation, Matched unrelated donor, Outcome
Introduction
Life expectancy in patients with chronic granulomatous disease (CGD) has been improved with the use of systematic antibiotic prophylaxis and aggressive treatment by antimicrobial and antimycotic agents [1]. Nevertheless life threatening infections are not always prevented and the annual mortality rate is estimated at around 5%, with invasive aspergillosis being the most common cause of death [2]. Furthermore complications arise from granuloma formation leading to organ obstruction and restrictive pulmonary function. Hematopoietic stem cell transplantation (HSCT) from matched sibling donors (MSD) has been successfully performed for a number of years [3]. If no MSD is available, HSCT from a matched unrelated donor (MUD) has become an alternative, but so far there are only few cases published. Here, we report our recent experience of HSCT in CGD which includes nine cases undergoing this treatment from MUD.
Patient characteristics
All 12 patients reviewed in this series were transplanted and followed in a single center over a period of 15 years. Charts were reviewed retrospectively with analysis of the clinical, immunologic, laboratory features and outcome. Nine patients grafted from a MUD were compared to three patients transplanted from a MSD.
All patients (n = 12) were male and between 4 and 20 years of age (mean 9.5 years). Most patients (n = 11) had X-linked CGD, two of whom also had McLeod phenotype (Table 1). Nine patients had previously suffered from lung aspergillosis. Other complications included lymph node or liver abscesses (n = 9), osteomyelitis (n = 2) and granuloma formation involving the lungs (n = 5) and the central nervous system (n = 1). Notably lung function prior to HSCT was normal in only five individuals. Conditioning regimens differed (Table 2). The three patients transplanted from MSD received busulfan (16 mg/kg) and cyclophosphamide (200 mg/kg). Six of the patients transplanted from MUD received busulfan (16 mg/kg), cyclophosphamide (120 mg/kg) and fludarabine (160 mg/m2) in addition to antithymocyte globuline or anti-CD52 monoclonal antibody. Two patients were conditioned with fludarabine (160 mg/m2), melphalane (70 mg/m2) in addition to treatment with radioimmunotherapy (UPN 614, 621). The one remaining patient was conditioned with total body irradiation and fludarabine (UPN 544). Three of nine children tranplanted from a MUD (UPN 508, 494, 513) received granulocyte transfusions in aplasia as infectious prophylaxis.
Table 1.
Patient characteristics
| UPN | Donor | Age at diagnosis | Genetics | Clinical complications prior to HSCT | Lung function prior to HSCT |
|---|---|---|---|---|---|
| 190 | MSD | 1.5 | XL (gp91phox) | Lung aspergillosis, suppurative lymphadenitis, liver and lung abscesses | Normal |
| 504 | MSD | 2 | XL (gp91phox) | Suppurative lymphadenitis, no pulmonary complications | Normal |
| 521 | MSD | 13 | XL (gp91phox) | Lung aspergillosis, skin abscesses, osteomyelitis | Restricted |
| 461 | MUD | 2 | XL (gp91phox) McLeod | Lung aspergillosis, granulomas (lungs and CNS) | Restricted |
| 483 | MUD | 3 | XL (gp91phox) | Lung aspergillosis, perianal abscess, granulomas (lungs and GIT), segmental lung resection | Restricted |
| 494 | MUD | 2 | XL (gp91phox) | Lung aspergillosis, liver abscess, granulomas (lungs) | Restricted |
| 508 | MUD | 0.3 | XL (gp91phox) | Skin, liver and spleen abscesses, granulomas (lungs) | Normal |
| 513 | MUD | 1 | XL (gp91phox) | Lung aspergillosis, suppurative lymphadenitis, osteomyelitis, glomerulonephritis | Restricted |
| 544 | MUD | 2 | XL (gp91phox) McLeod | Lung aspergillosis, BCGitis, segmental lung resection | Normal |
| 603 | MUD | 3 | XL (gp91phox) | Lung aspergillosis, suppurative lymphadenitis, perianal abscess | Normal |
| 614 | MUD | 3 | AR (p47phox) | Lung aspergillosis, skin abscess, granulomas (lungs, skin) | Restricted |
| 621 | MUD | 5.6 | XL (gp91phox) | Lung nocardiosis | Severely restricted |
UPN unique patient number, MSD matched sibling donor, MUD matched unrelated donor, HSCT hematopoietic stem cell transplantation, GIT gastrointestinal tract, CNS central nervous system, XL X-linked inheritance, McLeod phenotype: deletion of the Xk-gene next to the gp91phox-gene leads to reduced expression of Kell-antigens
Table 2.
Characteristics of HSCT and outcome
| UPN | Age at HSCT (years) | Donor | Graft | Conditioning | Antibody | Engraftment/chimerism | Graft failure | GvHD | Outcome (follow-up period) | Lung function after HSCT |
|---|---|---|---|---|---|---|---|---|---|---|
| 190 | 8 | MSD | BM | Bu/Cy | None | Complete | No | No | a/w >15.5 years | Normal |
| 504 | 5 | MSD | BM | Bu/Cy | None | Complete | No | No | a/w >64 months | Normal |
| 521 | 20 | MSD | BM | Bu/Cy | None | Complete | No | aGvHD (°2) | Death on day +77 (fatal systemic BK virus infection) | NA |
| 461 | 5 | MUD | BM | Bu/Cy | ATG | Complete | No | aGvHD (°2) | a/w >79 months | Pulmonary restriction |
| 483 | 7 | MUD | BM | Bu/Flu/Cy | Campath | Complete | No | No | a/w >71 months | Normal |
| 494 | 14 | MUD | BM | Bu/Cy | ATG | Complete | No | No | Death on day +28 (fatal ARDS) | NA |
| 508 | 4 | MUD | PBSC | Bu/Flu/Cy | Campath | Complete | No | aGvHD (°1) | a/w >62 months | Normal |
| 513 | 15 | MUD |
1st HSCT: BM 2nd HSCT: PBSC |
Bu/Flu/Cy | Campath | Complete (following retransplantation) | +day 20 | aGvHD (°2), cGvHD | Death on day +532 (fatal encephalitis under immunosuppression for cGvHD) | NA |
| 544 | 14 | MUD | PBSC | TBI/Flu | ATG | Complete | No | No | a/w >49 months | Normal |
| 603 | 10 | MUD | BM | Bu/Flu/Cy | ATG | Complete | No | No | a/w >24 months | Normal |
| 614 | 4 | MUD | BM | Flu/Mel/RIT | ATG | Autologous | Progressive autologous reconstitution | No | a/w >22 months | Normal |
| 621 | 9 | MUD | BM | Flu/Mel/RIT | ATG | Complete | No | aGvHD (°2) | a/w >20 months | Pulmonary restriction |
Bu busulfan, Cy cyclophosphamide, Flu Fludarabine, Mel melphalane, ATG antithymocyte globuline, Campath anti-CD52 monoclonal antibody, RIT radioimmunotherapy, TBI total body irradiation, BM bone marrow, PBSC peripheral blood stem cells, +day day after HSCT, ARDS acute respiratory failure, cGvHD chronic graft-versus-host disease, NA not applicable
Radioimmunotherapy (RIT) was performed with Yttrium-90 nuclide-coupled anti-CD66 monoclonal antibody. This therapy is targeted to CD66 + myeloid precursor cells in the marrow [4]. In this non-malignant setting the dosage was 17 Gy for two patients (UPN 614, UPN 621). RIT was performed in the Department of Nuclear Medicine, Ulm University (Prof. SN. Reske).
Results
Nine of the 12 patients survive at a mean follow-up time of 53 months (20–188 months) free of infectious complications and with stable graft function (see Fig. 1 and Table 3). One patient died on day +28 from acute respiratory failure following HSCT from a MUD (UPN 494, Table 2). No pathogen was found on lung biopsy and the etiology remained unclear. Another patient grafted from MSD died from systemic BK virus infection at 8 weeks post HSCT (UPN 521). The third patient suffered from therapy-resistant chronic graft-versus-host disease (GvHD). He succumbed to the sequelae of an encephalitis with cerebrovascular stroke 18 months after HSCT from a MUD (UPN 513). Chimerism is complete in 8 of 9 surviving patients (Table 3). Graft failure occurred in 2 patients, both transplanted from a MUD. One of these patients rejected the first transplant early and engrafted after receiving a second transplant on day +35, but died from chronic GvHD (UPN 513). The other patient who was conditioned with RIT in addition to fludarabine melphalane and ATG had progressive graft failure with recurrence of disease (UPN 614). He is scheduled for retransplantion. Two patients developed chronic GvHD of whom one died (UPN 513) and the other requires intermittent oxygen for GvHD of the lungs (UPN 461). At present lung function remains pathologic in two individuals, while two patients recovered normal lung function (Table 2).
Fig. 1.
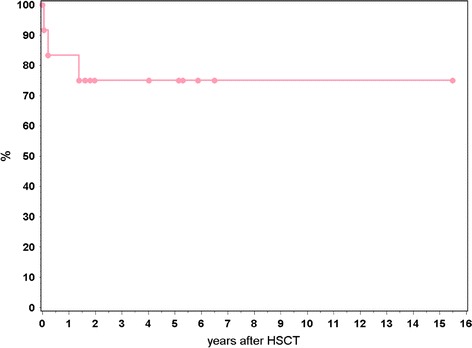
Kaplan-Meier estimates of survival in 12 patients with CGD transplanted from MUD (n = 9) and MSD (n = 3)
Table 3.
Outcome and complications following HSCT from MUD vs. MSD
| Outcome/complications | MUD (n = 9) | MSD (n = 3) |
|---|---|---|
| Mean follow-up time in months | 46.5 (21.5−79) | 126 (64−188) |
| Survival | 7/9 | 2/3 |
| Mixed chimerism | 1/7 | 0/2 |
| Graft failure | 2/8* | 0/3 |
| Chronic GvHD | 1/7 | 0/2 |
| Normal lung function post HSCT | 5/7 | 2/2 |
* One engrafted after retransplantation, but died at 18 months post HSCT
Discussion
Patients with CGD suffer from recurrent pyogenic and fungal infections. Furthermore they develop complications from granuloma formation. Both infections and granuloma formation often result in permanent lung damage. Control of chronic inflammation by granulomas requires long-term systemic immunosuppression which often aggravates growth failure in CGD patients. With the introduction of antibiotic prophylaxis, survival improved to over 90% in patients under the age of 10 years [5]. Despite an increase in the mean age of survivors from 8 to 16 years, in a Japanese cohort the overall mortality remained unchanged at around 20% [6]. Survival data of the United States CGD registry indicate that mortality in patients with XL-CGD is about 5% per year [2]. One-third of deaths in this registry were due to aspergillus infection. Other reasons for this limited prognosis are due to progressive pulmonary restriction and also non-compliance with medication in adolescents.
At present guidelines for using HSCT to treat CGD include patients suffering from one or more life-threatening infections, from severe complication of granuloma formation with organ dysfunction including lung restriction, or patients being non-compliant with antibiotic prophylaxis [1]. However, in the absence of a family donor, the use of alternative donors for HSCT is only rarely considered so far due to the concern of increased transplantation associated risks such as higher toxicity and more severe GvHD. Published reports with successful HSCT from a MUD in CGD patients between the ages of 8 months and 39 years are few and mostly single case reports [7–14].
Our experience reported here in 12 patients with CGD, the majority of whom were transplanted from a MUD, shows an overall survival rate of 75% (9/12). In a European survey of 23 patients (age range 3–20 years) receiving bone marrow from MSDs after myeloablative conditioning, survival was 82% [3].
Interestingly in our experience the use of MUD did not result in more complications or worse outcome as compared to results in our smaller cohort of three patients given MSD transplants. As opposed to the above-mentioned European cohort neither exacerbation of infection during aplasia nor inflammatory flares during neutrophil engraftment were observed. Incidence of chronic GvHD is similar, with 8% in our patients compared to 11% in the European cohort [3]. Stable engraftment with full donor chimerism was observed in 90% of our patients, with a median follow-up time of 5 years (20 months to 15.5 years). Graft failure occurred in two patients transplanted from a MUD, retransplantation from the same donor was successful in one. The other patient had progressive autologous reconstitution with recurrence of disease 1.5 years after HSCT. In the European cohort stable engraftment was observed in all survivors with a median follow-up time of 2 years (4 months to 12 years).
There are two important prognostic parameters for HSCT in CGD: the presence of active infections at the time of transplantation, and a poor general clinical condition in particular a restricted lung function. In our experience HSCT should be seriously considered after the first life-threatening infection, and in the absence of a MSD, HSCT from a MUD donor is an option worth considering. Optimal conditioning regimens need to be investigated in order to reduce toxicity and lower the risk of GvHD without compromising stable engraftment.
Acknowledgement
We thank Sandra Steinmann for excellent help in the preparation of the manuscript.
References
- 1.Seger RA. Modern management of chronic granulomatous disease. Br J Haematol. 2008;140:255–66. doi: 10.1111/j.1365-2141.2007.06880.x. [DOI] [PubMed] [Google Scholar]
- 2.Winkelstein JA, Marino MC, Johnston RB, Jr, et al. Chronic granulomatous disease: report on a national registry of 368 patients. Medicine. 2000;79:155–69. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Seger RA, Gungor T, Belohradsky BH, Blanche S, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985–2000. Blood. 2002;100:4344–50. doi: 10.1182/blood-2002-02-0583. [DOI] [PubMed] [Google Scholar]
- 4.Kotzerke J, Bunjes D, Scheinberg DA. Radioimmunoconjugates in acute leukemia treatment: the future is radiant. Bone Marrow Transplant. 2005;36:1021–6. doi: 10.1038/sj.bmt.1705182. [DOI] [PubMed] [Google Scholar]
- 5.Mouy R, Fischer A, Vilmer E, Seger R, Griscelli C. Incidence, severity, and prevention of infections in chronic granulomatous disease. J Pediatr. 1989;114:555–60. doi: 10.1016/S0022-3476(89)80693-6. [DOI] [PubMed] [Google Scholar]
- 6.Hasui M. Chronic granulomatous disease in Japan: incidence and natural history. The Study Group of Phagocyte Disorders of Japan. Pediatr Int. 1999;41:589–93. doi: 10.1046/j.1442-200x.1999.01129.x. [DOI] [PubMed] [Google Scholar]
- 7.The Westminster Hospitals Bone-Marrow Transplant Team Bone-marrow transplant from an unrelated donor for chronic granulomatous disease. Lancet. 1977;1:210–3. [PubMed] [Google Scholar]
- 8.Hobbs JR, Monteil M, McCluskey DR, Jorges E, el Tumi M. CGD 100% corrected by displacement bone marrow transplantation from a volunteer unrelated donor. Eur J Pediatr. 1992;151:806–10. doi: 10.1007/BF01957929. [DOI] [PubMed] [Google Scholar]
- 9.Porta F, Mazzolari E, Verzeri U, et al. MUD transplant in an infant affected by chronic granulomatous disease. Bone Marrow Transplant. 2000;25:S190. [Google Scholar]
- 10.Watanabe C, Yajima S, Taguchi T, Toya K, Fujii Y, Hongo T, Ohzeki T. Successful unrelated bone marrow transplantation for a patient with CGD and associated resistant pneumonitis and Aspergillus osteomyelitis. Bone Marrow Transplant. 2001;28:83–7. doi: 10.1038/sj.bmt.1703086. [DOI] [PubMed] [Google Scholar]
- 11.Gungor T, Halter J, Klink A, Junge S, Stumpe KDM, Seger R, Schanz U. Successful low toxicity hematopoietic stem cell transplantation for high-risk adult chronic granulomatous disease patients. Transplantation. 2005;79:1596–606. doi: 10.1097/01.TP.0000163466.73485.5E. [DOI] [PubMed] [Google Scholar]
- 12.Schuetz C, Hoenig M, Schulz A, Lee-Kirsch MA, Roesler J, Friedrich W, von Bernuth H. Successful unrelated bone marrow transplantation in a child with chronic granulomatous disease complicated by pulmonary and cerebral granuloma formation. Eur J Pediatr. 2007;166:785–8. doi: 10.1007/s00431-006-0317-7. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki N, Hatakeyama N, Yamamoto M, Mizue N, Kuroiwa Y, Yoda M, Takahashi J, Tani Y, Tsutsumi H. Treatment of McLeod phenotype chronic granulomatous disease with reduced-intensity conditioning and unrelated-donor umbilical cord blood transplantation. Int J Hematol. 2007;85:70–2. doi: 10.1532/IJH9706129. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharya A, Slatter M, Curtis A, Chapman CE, Barge D, Jackson A, Flood TJ, Abinun M, Cant AJ, Gennery AR. Successful umbilical cord blood stem cell transplantation for chronic granulomatous disease. Bone Marrow Transplant. 2003;31:403–5. doi: 10.1038/sj.bmt.1703863. [DOI] [PubMed] [Google Scholar]