

Abstract
Primary immunodeficiency disorders are heterogeneous group of illnesses that predispose patients to serious complications. Registries for these disorders have provided important epidemiological data and shown both racial and geographical variations. The clinical features of 76 patients with primary immunodeficiency disorders registered in Kuwait National Primary Immunodeficiency Registry from 2004 to 2006 were recorded. Ninety-eight percent of the patients presented in childhood. The prevalence of these disorders in children was 11.98 in 100,000 children with an incidence of 10.06 in 100,000 children. The distribution of these patients according to each primary immunodeficiency category is: combined T and B cell immunodeficiencies (21%), predominantly antibody immunodeficiency (30%), other well defined immunodeficiencies (30%), diseases of immune dysregulation (7%), congenital defects of phagocyte number, function or both (8%), and complement deficiencies (4%). The consanguinity rate within the registered patients was 77%. The patients had a wide range of clinical features affecting different body systems. Primary immunodeficiency disorders are prevalent in Kuwait and have a significant impact into the health system.
Keywords: Immunodeficiency, registry, epidemiology, Arab, Kuwait
Introduction
Primary immunodeficiency disorders (PID) are heterogeneous group of illnesses not only in their clinical and immune expression but also in their basic pathophysiologic mechanisms. Patients with PID are prone to develop recurrent, serious, and unusual infections. The patients are also prone to develop non-infectious presentations. These include, but not limited to, neutropenia, autoimmunity, absence lymphoid tissues or lymphadenopathy, enteropathy, failure to thrive, dermatitis, malignancies, and atopy [1–4]. Recent advances in molecular biology have helped identify the primary biologic defect in a growing number of these disorders. PID are classified into eight categories according to the International Union of Immunological Societies (IUIS) Primary Immunodeficiency Diseases Classification Committee [5]:
Combined T and B cell immunodeficiencies
Predominantly antibody immunodeficiency
Other well-defined immunodeficiencies
Diseases of immune dysregulation
Congenital defects of phagocyte number, function, or both
Defects in innate immunity
Autoinflammatory disorders
Complement deficiencies
Unfortunately, PID are frequently misdiagnosed. Many physicians do not often consider the possibility of immunodeficiency early enough in the differential diagnosis or they consider themselves inadequate to evaluate the immune system [6]. Currently, no screening is performed for these defects at birth, during childhood, or in adulthood. Therefore, PID are usually detected only after the individual has experienced recurrent infections that may or may not have caused permanent organ damage [7–9]. Early diagnosis by increasing awareness of physicians about these disorders is important so life saving treatment and precautions can be implemented to decrease mortality and morbidity and to improve the quality of life [10–12].
There are several registries for primary immunodeficiency disorders [13–25]. These registries helped in determining the frequency of PID in these countries. They also helped in characterization of PID patients with emphasis on clinical presentations and the significant morbidity caused by and associated with PID. Data from these registries have shown both racial and geographical variations in the prevalence and pattern of PID.
Kuwait is a small country (17,820 km2) in the Arabian Peninsula with a population of 3,051,845. The crude birth rate is 16.8 in 1,000, crude death rate is 1.4 in 1,000, natural increase rate is 16.1 in 1,000, and infant mortality rate is 8.1 in 1,000 live births [26]. To participate in the global effort in providing epidemiological data and to have better knowledge about PID, Kuwait National Primary Immunodeficiency Registry (KNPIDR) was established in 2007 after being approved by the Research and Ethics Committee of the Ministry of Health. The objectives of the KNPIDR are:
Determine the prevalence and frequency of different PID in Kuwait
Identify clinical presentation patterns for PID in Kuwait
Identify natural history of PID in Kuwait
Help to asses epidemiology of PID in Kuwait
Determine particularities about PID affecting the population in Kuwait
Determine the health impact of PID in Kuwait
Enhance the knowledge of PID among physicians in Kuwait
Coordinate clinical research into these disorders
Development of strategies to improve the care and the quality of life of patients with PID
This is the first report of PID in Kuwait obtained from KNPIDR.
Method
Data Collection
A data form was developed in collaboration with US immunodeficiency net (USIDNET). The charts of the patients registered into Kuwait National Primary Immunodeficiency Registry from 2004 to 2006 were reviewed, and the obtained information was recorded into the data form and divided into five sections: sociodemographic data, diagnosis, clinical presentation, laboratory tests, and treatment.
Patients Diagnosis and Classification
The patients were diagnosed and classified according to both the clinical and laboratory criteria of PID reported by IUIS Primary Immunodeficiency Diseases Classification Committee [5]. Secondary immunodeficiencies (drug-induced, virus-induced, and immunodeficiency associated with metabolic disorders, etc.) were ruled out by obtaining detailed history and by performing appropriate testing when these disorders were suspected. The immunological tests performed for our patients were done using the standard techniques and included complete blood count with peripheral blood smear evaluation, serum immunoglobulins, antibody response to previous vaccines, lymphocyte phenotype (T, B, and NK cells) by flow cytometry, lymphocyte stimulation test, autoantibodies, nitro blue tetrazolium dye test, and complement hemolytic activity (CH50) with specific complement component when needed. Genetic testing was done only for a group of patients.
Registry Database Computer Program
A computerized database software program was designed based on the collected data using Rapid Application Development (RAD) tool Microsoft Access 2003 with Visual Basic for Application (VBA) code, and OLE control extension (OCX) component technology. All database design is based on Relational Database Management System (RDMS) techniques. Forms and dialogs were designed in Microsoft Access, whereas some advanced controls like Microsoft Flexgrid, Rich Text, and Common Dialog were used wherever needed by empowering component OCX technology.
The registry program allows patient’s data entry from the data form and also capable of performing basic statistical analysis. Furthermore, the registry data can be loaded into Microsoft Excel Program, and specialized reporting of these data was created and implemented using Microsoft Access Report Writer. Each patient was given a specific code to ensure confidentiality.
Statistical Analysis
The registry database program is capable of performing basic statistical analysis. SPSS statistical software package (version 12.0) was used to calculate the mean, incidence, and prevalence.
Results
Frequency and Distribution of PID
A total of 76 patients (47 males and 29 females) with PID were registered in KNPIDR and presented in this report. The distribution of these patients according to PID categories is: combined T and B cell immunodeficiencies (16 patients, 21%), predominantly antibody immunodeficiency (23 patients, 30%), other well-defined immunodeficiencies (23 patients, 30%), diseases of immune dysregulation (five patients, 7%), congenital defects of phagocyte number, function or both (six patients, 8%), and complement deficiencies (three patients, 4%; Fig. 1). No patients with either defects in innate immunity or autoinflammatory disorders were registered. The frequency of each immunodeficiency phenotype in each category is shown in Table I. Three patients presented as others (one in the first category and two in the fourth category) have PID but could not be categorized according to the IUIS classification system. Of the combined T and B cell immunodeficiencies, Omenn syndrome was the most commonly reported (six patients). Patients with agammaglobulinemia and absent B cells were the most common among the predominantly antibody immunodeficiency group (eight patients). Within the other well-defined immunodeficiencies, DiGeorge syndrome (eight patients) was the most common followed by Hyper IgE syndrome (seven patients).
Fig. 1.
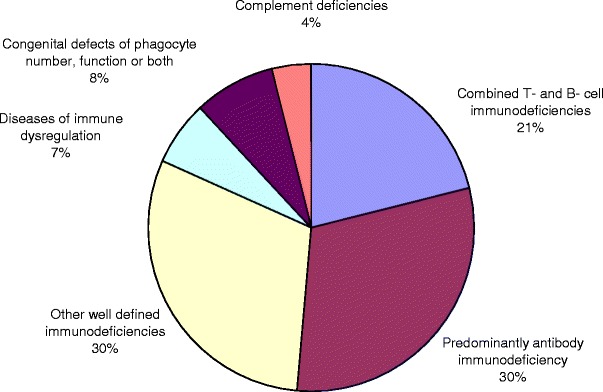
Distribution of primary immunodeficiency disorders in Kuwait.
Table I.
Frequency of Each PID Phenotype and Characteristics of Patients from KNPIDR
| Category | Diagnosis | Number of patients (males/females) | Onset age (months)a | Diagnosis age (months)a | Diagnosis delay (months)a | Consanguinity | Family history of immunodeficiency | Use of prophylaxis antimicrobials | Use of IVIG | Deaths |
|---|---|---|---|---|---|---|---|---|---|---|
| Combined T and B cell immunodeficiency | 16 (6/10) | 2.5 (0–9) | 9 (0–60) | 7.5 (0–58) | 100% | 75% | 15 | 15 | 11 | |
| T-B+ SCID | 3 (2/1) | |||||||||
| T-B− SCID | 2 (0/2) | |||||||||
| Omenn syndrome | 6 (3/3) | |||||||||
| MHC II deficiency | 4 (1/3) | |||||||||
| Others | 1 (0/1) | |||||||||
| Predominantly antibody deficiency | 23 (18/5) | 30 (4–312) | 56 (7–384) | 27 (1–114) | 70% | 26% | 8 | 16 | 1 | |
| Severe reduction in all serum Ig isotypes with absent B cells | 8 (7/1) | |||||||||
| Severe reduction in at least 2 serum Ig isotypes with normal or low number of B cells | 2 (2/0) | |||||||||
| Severe reduction in serum IgG and IgA with increased IgM and normal number of B cells | 5 4/1 | |||||||||
| Selective IgA deficiency | 4 (2/2) | |||||||||
| Specific antibody deficiency with normal Ig concentrations and numbers of B cells | 1 (1/0) | |||||||||
| Transient hypogammaglobulinemia of infancy | 3 (2/1) | |||||||||
| Other well-defined immunodeficiency syndromes | 23 (16/7) | 7 (0–36) | 34 (0–120) | 27 (0–108) | 61% | 39% | 13 | 2 | 2 | |
| DiGeorge syndrome | 8 (5/3) | |||||||||
| Hyper IgE syndrome | 7 (5/2) | |||||||||
| Ataxia-telangiectasia | 5 (3/2) | |||||||||
| Wischott–Aldrich syndrome | 3 (3/0) | |||||||||
| Diseases of immune dysregulation | 5 (2/3) | 11 (5–18) | 51 (18–156) | 40 (0–151) | 100% | 20% | 2 | 2 | 0 | |
| Autoimmune lymphoproliferative syndrome | 1 (0/1) | |||||||||
| Chediak–Higashi syndrome | 2 (1/1) | |||||||||
| Others | 2 (1/1) | |||||||||
| Congenital defect in phagocyte number, function or both | 6 (4/2) | 2 (0–3) | 8 (1–14) | 7 (1–12) | 83% | 50% | 4 | 0 | 1 | |
| Chronic granulomatous disease | 4 (4/0) | |||||||||
| Cyclic neutropenia | 2 (0/2) | |||||||||
| Complement deficiency | 3 (1/2) | 100% | 66% | 1 | 0 | |||||
| Hereditary angioedema | 2 (1/1) | |||||||||
| Others | 1 (0/1) |
aMean
Patient Characteristics
The mean age of the patients at onset of symptoms was 15 months (0 month–26 years), while the mean age at diagnosis was 43 months (0 month–38 years). The mean delay in diagnosis, which is defined as the time between the initial presentation and the time of definite diagnosis, was 29 months (0 month–31 years; Table I). Fifty-nine patients (78%) presented before the age of 1 year, nine patients (12%) presented between 1 and 3 years, and only one patient presented in adulthood. The consanguinity rate within the registered patients was 77% with variation among different categories reaching up to 100% in some groups (Table I). A positive family history of PID was noticed in 33 patients (43%), with the highest frequency in the combined T and B cell immunodeficiencies (75%).
Clinical Features
The patients had a wide range of clinical features involving different systems (Fig. 2). Infectious complications were the most common affecting 64 patients (84%) followed by hematologic-lymphoid (e.g., lymphadenopathy, absent lymphoid tissues, and bleeding) 45 patients (60%), skin (e.g. dermatitis, alopecia, and telangiectasia) 32 patients (42%), and gastrointestinal tract (e.g., chronic diarrhea and abnormal teeth) 29 patients (38%). The other systems involved were growth-development-endocrine 22 patients (28%), cardiovascular-renal 22 patients (22%), hepatobiliary 16 patients (21%), neuropsychiatry 16 patients (21%), atopy 16 patients (21%), sinopulmonary 12 patients (15%), autoimmune six patients (8%), musculoskeletal five patients (7%), and neoplastic complication in one patient with ataxia-telangiectasia. Only one patient with Omenn syndrome had vaccination complication presented as Polioviremia with cardiomyopathy and one patient who has family history of MHC II deficiency was screened after birth and was asymptomatic at the time of diagnosis.
Fig. 2.
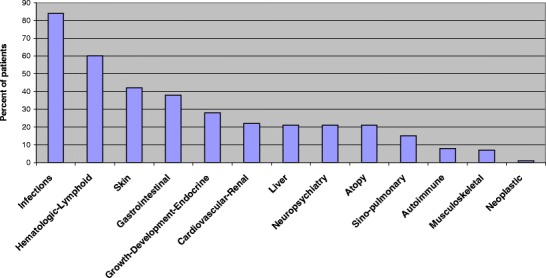
Frequency of system involvement in patients with primary immunodeficiency in Kuwait.
Analysis of systems involvement in each PID category (Fig. 3) shows that infectious complications and hematologic-lymphoid involvement were prominent in all categories. Skin manifestations were most prominent in combined T and B cell immunodeficiencies in eight patients (50%) and other well-defined immunodeficiencies in 15 patients (65%) and not prominent in predominantly antibody immunodeficiency in three patients only (13%). Atopic disorders and autoimmune manifestations were more relevant in patients with predominantly antibody immunodeficiency (34 and 13%, respectively), while gastrointestinal manifestations were more prominent in patients with combined T and B cell immunodeficiencies (50%).
Fig. 3.
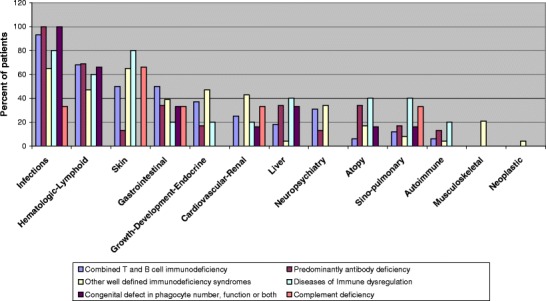
System involvement per category of patients with primary immunodeficiency.
Pneumonia was the most common infectious complications affecting 41 patients (54%), followed by otitis media in 28 patients (37%), skin infections in 19 patients (25%), sepsis in 14 patients (18%), urinary tract infections in ten patients (13%), infective gastroenteritis in seven patients (9%), sinusitis in six patients (8%), and lymphadenitis in three patients (4%).
Genetic Testing and Management
Genetic testing was performed in 29 patients (38%), and a genetic defect was identified in 25 patients (32%). The identified mutated genes were RAG1: five patients, BTK: one patient, AICD: four patients, 22q11.2: eight patients, WAS: two patients, ATM: two patients, and CYBA: three patients. Prophylaxis antimicrobial therapy was used in 43 patients (56%), while antibody replacement therapy in the form of intravenous immunoglobulins was used in 35 patients (46%; Table I). Bone marrow transplantation was performed in eight patients (10%), of whom, five patients had combined immunodeficiency, and three patients had chronic granulomatous disease.
Mortality
There were 15 deaths among the registered patients (Table I). Eleven deaths (73%) were in the combined T and B cell immunodeficiencies group, with a mean age of death in this group of 12 months (2–30 months). The causes of death in these patients were respiratory failure with ARDS secondary to pneumonia, sepsis, multiorgan failure, and renal failure.
Discussion
Primary immunodeficiency disorders are heterogeneous group of illnesses of the immune system. In this article, the first report of PID in Kuwait obtained from KNPIDR is presented. As 98% of the registered patients presented in childhood (0–14 years), the prevalence of PID was calculated in children and was found to be 11.98 in 100,000 children, with an incidence of 10.06 in 100,000 children. This corresponds to an estimated occurrence of PID in 1 of 1,000 live births. The prevalence of combined T and B cell immunodeficiencies in children was found to be 2.56 in 100,000 children, with an incidence of 1.92 in 100,000 children and an estimated occurrence of 1 in 3,000 live births. It should be emphasized that these figures do not represent the true prevalence and incidence because of either under-diagnosis of milder forms of PID or under-reporting of serious PID due to early deaths in undiagnosed patients.
The distribution of PID in this report shows a high frequency of combined T and B cell immunodeficiencies (21%) which is almost double the frequency of similar disorders reported in other registries [13–23]. In Sweden, combined T and B cell immunodeficiencies were reported to affect 21% of the registered patients [24]. However, a critical review of the cases according to the current classification shows that only 8% of their registered patients are affected by combined T and B cell immunodeficiencies.
The presented data also show a relatively lower frequency of predominantly antibody immunodeficiency (30%) compared to 40–71% reported by other registries. Interestingly, this finding is consistent with a previous report about PID in Kuwait [27]. These differences could be due to racial background. This was clearly documented in multiple reports: higher frequency of selective IgA deficiency in Latin Americans compared to both the Japanese and Australian population [14, 20, 23]; elevated number of ataxia-telangiectasia in Costa Ricas compared to Switzerland [14, 21]; high prevalence of complement deficiencies in Ireland and North Africa [17, 28, 29]; and increased prevalence of humoral immunodeficiency in Arabs compared to the western population [19]. The very high rate of consanguinity in our registry (77%) could also account for these differences, with a particular increase in the autosomal recessive diseases. Under-diagnosis of some milder forms of PID like selective IgA deficiency and IgG subclasses deficiency may also play a role in these differences. Common variable immunodeficiency, which presents later in life compared to other PID, is the most common PID reported in Iran, Ireland, Australia, and Norway, with a frequency of about 25% of all PID patients [13, 17, 20, 25]. In our registry where the mean age of presentation was 15 months and 90% of the patients presented before the age of 3 years, only two patients with common variable immunodeficiency (3%) were registered. This could also be the cause of lower frequency of predominantly antibody immunodeficiency.
The mean delay in diagnosis of PID was almost 2 1/2 years. The delay in diagnosis was most properly due to lack of awareness of physicians about PID. This delay has definitely caused a significant effect on the quality of life of patients with PID before diagnosis. Our data show a male predominance (62%) similar to observations noted in other registries [13, 15–17, 24, 25]. Forty-eight percent of the patients were treated with immunoglobulins, which is close to the data from European Society for Immunodeficiencies registry of 41% [15].
In this report, it is clearly documented that PID can affect different systems in the body. Of interest is the variation in systems involvement in different PID categories. Examples are the higher occurrence of atopic and autoimmune disorders in patients with predominantly antibody immunodeficiency, while skin and gastrointestinal manifestations were more prominent in patients with combined T and B cell immunodeficiencies. This finding is in agreement with previous reports [1–4]. Early diagnosis by increasing the index of suspicion of physicians and subspecialists about these disorders is important so life saving interventions can be done to decrease mortality and morbidity and to improve the quality of life [30–34].
Seventy-three percent of our patients with combined T and B cell immunodeficiencies died. The reason for this high death rate is that immune reconstitution in the form of bone marrow/stem cell transplant is not available in Kuwait for pediatrics patients. Establishment of such service should be a priority of the health authorities to prevent such mortalities.
Infectious complications were the most common clinical features in our patients. None of the patients developed significant complications to BCG vaccine compared to the reports by others [13, 24]. The practice in Kuwait until 2005 is to vaccinate children with BCG at school age when all forms of serious PID like combined T and B cell immunodeficiencies, Wischott–Aldrich syndrome, and chronic granulomatous disease have presented and fortunately been diagnosed. The vaccination policy has changed, and BCG vaccine is currently given at 3 months of age; it will be interesting to review the effect of this change on the occurrence of complications related to BCG vaccine after few years. It is important to emphasize that patients with recurrent, serious, and unusual infections should be screened for PID. Physicians should be educated about the ten warning signs of PID so early interventions with intravenous immunoglobulins and immune reconstitution can be used to prevent significant tissue damage, morbidity, and mortality [30, 35–38].
Conclusions
The development of KNPIDR has provided a substantial epidemiological data about PID in Kuwait and helped to determine the prevalence of these disorders. These data will hopefully help the health authorities to develop strategies to improve the care and the quality of life of patients with PID in the country. The illustration of high mortality rate in the combined T and B cell immunodeficiencies should stimulate immunologist to develop newborn screening protocols so immune reconstitution can be implemented early in life.
Acknowledgments
Kuwait National Primary Immunodeficiency Registry (KNPIDR) was established by a fund from Kuwait Foundation for the Advancement of Science (KFAS). The author would like to thank Dr. Hans Ochs and Dr. Mike Blaese from USIDNET for their collaboration in the development of the data form, Dr. Haitham Al-Khayat and Dr. Ali Sadek for their help in statistical analysis, and all physicians who suspected immunodeficiency and referred the patients to the clinical immunology service.
References
- 1.Ochs HD, Stiehm R, Winkelstein J. Antibody deficiencies. In: Stiehm ER, Ochs HD, Winkelstein JA, editors. Immunologic disorders in infants & children. 5. Philadelphia: Elsevier Saunders; 2004. pp. 356–426. [Google Scholar]
- 2.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. J Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 3.Woroniecka M, Ballow M. Office evaluation of children with recurrent infections. Pediatr Clin North Am. 2000;47(6):1211–1223. doi: 10.1016/S0031-3955(05)70268-6. [DOI] [PubMed] [Google Scholar]
- 4.Buckley RH. Primary immunodeficiency diseases due to defects in lymphocytes. N Engl J Med. 2000;343(18):1313–1323. doi: 10.1056/NEJM200011023431806. [DOI] [PubMed] [Google Scholar]
- 5.Notarangelo L, Casanova JL, Conley ME, Chapel H, Fischer A, Puck J, et al. Primary immunodeficiency diseases: an update from international union of immunological societies primary immunodeficiency diseases classification committee meeting in Budapest, 2005. J Allergy Clin Immunol. 2006;117:883–896. doi: 10.1016/j.jaci.2005.12.1347. [DOI] [PubMed] [Google Scholar]
- 6.Tangsinmankong N, Bahna SL, Good RA. The immunologic workup of the child suspected of immunodeficiency. Ann Allergy Asthma Immunol. 2001;87:362–370. doi: 10.1016/S1081-1206(10)62915-8. [DOI] [PubMed] [Google Scholar]
- 7.Immune Deficiency Foundation Diagnosting & Clinical Care Guidelines for Primary immunodeficiency Diseases: http://www.primaryimmune.org (July 1, 2007).
- 8.Stiem ER, Chin TW, Hass A, Peerless AG. Infectious complications of primary immunodeficiencies. Clin Immunol Immunopathol. 1986;40:69–89. doi: 10.1016/0090-1229(86)90070-X. [DOI] [PubMed] [Google Scholar]
- 9.Webster ABD. Infection in primary immunodeficiency syndrome. Curr Opin Infect Dis. 1994;7:444–449. doi: 10.1097/00001432-199408000-00005. [DOI] [Google Scholar]
- 10.Knutsen AP, Wall DA. Umbilical cord blood transplantation in severe T-cell immunodeficiency disorders: two-year experience. J Clin Immunol. 2000;20:466–476. doi: 10.1023/A:1026463900925. [DOI] [PubMed] [Google Scholar]
- 11.Cavazzana-Calvo M, Hacein-Bay S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 12.Buckley RH. A historical review of bone marrow transplantation for primary immunodeficiencies. J Allergy Clin Immunol. 2004;113(4):793–800. doi: 10.1016/j.jaci.2004.01.764. [DOI] [PubMed] [Google Scholar]
- 13.Rezaei N, Aghamohammadi A, Moin M, Pourpak Z, Movahedi M, Gharazlou M, et al. Frequency and clinical manifestations of patients with primary immunodeficiency disorders in Iran: update from the Iranian primary immunodeficiency registry. J Clin Immunol. 2006;26(6):519–532. doi: 10.1007/s10875-006-9047-x. [DOI] [PubMed] [Google Scholar]
- 14.Leiva LE, Zelazco M, Oleastro M, Carneiro-Sampaio M, Condino-Neto A, Costa-Caravalho BT, et al. Primary immunodeficiency diseases in Latin America: The second report of the LAGID registry. J Clin Immunol. 2007;27(1):101–108. doi: 10.1007/s10875-006-9052-0. [DOI] [PubMed] [Google Scholar]
- 15.The European Society for Immunodeficiencies: http://www.esid.org/statistics.php (July 15, 2007).
- 16.Lee WI, Kuo ML, Huang JL, Lin SJ, Wu CJ. Distribution and clinical aspects of primary immunodeficiencies in a Taiwan pediatric tertiary hospital during a 20-year period. J Clin Immunol. 2005;25(2):162–173. doi: 10.1007/s10875-005-2822-2. [DOI] [PubMed] [Google Scholar]
- 17.Abuzakouk M, Feighery C. Primary immunodeficiency disorders in the Republic of Ireland: first report of the national registry in children and adults. J Clin Immunol. 2005;25(1):73–77. doi: 10.1007/s10875-005-0360-9. [DOI] [PubMed] [Google Scholar]
- 18.Mila LIambi J, Etxagibel Galdos A, Matamoros Flori N. The Spanish Registry of Primay Immunodeficiencies (REDIP) Allergol Immunopathol. 2001;29(3):122–125. doi: 10.1016/s0301-0546(01)79031-3. [DOI] [PubMed] [Google Scholar]
- 19.Al-Attas RA, Rahi AH. Primary antibody deficiency in Arabs: first report from eastern Saudi Arabia. J Clin Immunol. 1998;18(5):368–371. doi: 10.1023/A:1023247117133. [DOI] [PubMed] [Google Scholar]
- 20.Baumgart KW, Britton WJ, Kemp A, French M, Roberton D. The spectrum of immunodeficiency disorders in Australia. J Allergy Clin Immunol. 1997;100(3):415–423. doi: 10.1016/S0091-6749(97)70257-4. [DOI] [PubMed] [Google Scholar]
- 21.Ryser O, Morell A, Hitzig WH. Primary immunodeficiencies in Switzerland: first report of the national registry in adults and children. J Clin Immunol. 1988;8(6):479–485. doi: 10.1007/BF00916954. [DOI] [PubMed] [Google Scholar]
- 22.Luzi G, Pesce AM, Rinaldi S. Primary immunodeficiencies in Italy. Data revised from the Italian Register of Immunodeficiencies-IRID (1977–88) Allergol Immunopathol. 1991;19(2):53–57. [PubMed] [Google Scholar]
- 23.Hayakawa H, Iwata T, Yata J, Kobayashi N. Primay immunodefiedncy syndrome in Japan. Overview of a nationwide survey on primary immunodeficiency syndrome. J Clin Immunol. 1981;1(1):31–39. doi: 10.1007/BF00915474. [DOI] [PubMed] [Google Scholar]
- 24.Fasth A. Primary immunodeficiency disorders in Sweden: cases among children 1974–1979. J Clin Immunol. 1982;2:86–92. doi: 10.1007/BF00916891. [DOI] [PubMed] [Google Scholar]
- 25.Stary-Pedersen A, Abrahamsen TG, Froland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol. 2000;20(6):477–484. doi: 10.1023/A:1026416017763. [DOI] [PubMed] [Google Scholar]
- 26.Kuwait Health 2004–2006. Health & Vital Statistics Division, Department of Statistics & Medical Records. Ministry of Health, State of Kuwait.
- 27.White AG, Raju KT, Abouna GM. A six year experience with recurrent infections and immunodeficiency in children in Kuwait. J Clin Lab Immunol. 1988;26(2):97–101. [PubMed] [Google Scholar]
- 28.Zimran A, Rudensky B, Kramer MR. Hereditary complement deficiency in survivors of meningococcal disease. High prevalence of C7/C8 deficiency in Sphardic (Moroccan) Jews. Q I Med. 1987;63:349–358. [PubMed] [Google Scholar]
- 29.Schlesinger M, Nave Z, Levi Y, Fishelson Z. Prevalence of hereditary properdin, C7 and C8 deficiencies in patients with meningococcal infections. Clin Exp Immunol. 1990;81:423–427. doi: 10.1111/j.1365-2249.1990.tb05350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elder ME. T-cell immunodeficiencies. Pediatr Clin North Am. 2000;47(6):1253–1273. doi: 10.1016/S0031-3955(05)70270-4. [DOI] [PubMed] [Google Scholar]
- 31.Quartier P, Debre M, De Blic J, de Sauverzac R, Sayegh N, Jabado N, et al. Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia: a retrospectic survey of 31 patients. J Pediatr. 1999;134:589–596. doi: 10.1016/S0022-3476(99)70246-5. [DOI] [PubMed] [Google Scholar]
- 32.Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2002;109:1001–1004. doi: 10.1067/mai.2002.124999. [DOI] [PubMed] [Google Scholar]
- 33.Stadtmauer G, Cunningham-Rundles C. Outcome analysis and cost assessment in immunologic disorders. JAMA. 1997;278:2018–2023. doi: 10.1001/jama.278.22.2018. [DOI] [PubMed] [Google Scholar]
- 34.Conley ME, Howard V. Clinical findings leading to the diagnosis of X-linked agammaglobulinemia. J Pediatr. 2002;141:566–571. doi: 10.1067/mpd.2002.127711. [DOI] [PubMed] [Google Scholar]
- 35.Jeffrey Modell Foundation: http://www.jmfworld.com/ (July 15, 2007).
- 36.Buckley RH, Schiff SE, Schiff RI, Market L, Williams LW, Roberts JL, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med. 1999;340:508–516. doi: 10.1056/NEJM199902183400703. [DOI] [PubMed] [Google Scholar]
- 37.Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood. 2002;99:872–878. doi: 10.1182/blood.V99.3.872. [DOI] [PubMed] [Google Scholar]
- 38.Antoine C, Muller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J, et al. European Group for Blood and Marrow Transplantatoin; European Society for Immunodeficiency. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968–99. Lancet. 2003;361:553–560. doi: 10.1016/S0140-6736(03)12513-5. [DOI] [PubMed] [Google Scholar]