

Abstract
This study investigated the effects of peptide apelin-12 (H-Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Met-Pro-Phe-OH, A12) and its novel structural analog (H-(NαMe)Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Nle-Pro-Phe-OH, AI) on myocardial antioxidant enzyme activities, lipid peroxidation, and reactive oxygen species formation in ex vivo and in vivo models of myocardial ischemia/reperfusion (I/R) injury. Isolated working rat hearts were subjected to global ischemia and reperfusion. Infusion of 140 μM A12 or AI before global ischemia improved cardiac function recovery; increased the activity of Cu,Zn superoxide dismutase (Cu,Zn SOD), catalase (CAT), and glutathione peroxidase (GSH-Px); decreased malondialdehyde (MDA) content in reperfused heart; and reduced the formation of hydroxyl radical adduct of the spin trap 5,5-dimethyl-1-pyrroline-N-oxide in the myocardial effluent during early reperfusion compared with these indices in control. Anesthetized open-chest rats were subjected to the left anterior descending coronary artery occlusion and coronary reperfusion. Peptide A12 or its analog AI was injected intravenously at the onset of reperfusion at a dose of 0.35 μmol/kg. Treatment with A12 or AI significantly limited infarct size and reduced the activity of lactate dehydrogenase and creatine kinase MB isoenzyme in blood plasma at the end of reperfusion compared with control. These effects were accompanied by complete recovery of Cu,Zn SOD, CAT, and GSH-Px activities; and decrease in MDA content in the area at risk by the end of reperfusion. The study concluded that C-terminal fragment of native peptide apelin-12 and its synthesized analog is involved in the upregulation of cardiac antioxidant defense systems and attenuation of lipid peroxidation in myocardial I/R injury.
Keywords: Apelin-12, Its structural analog, Antioxidant defense, Myocardial ischemia/reperfusion injury, Rat heart
Introduction
Myocardial ischemia/reperfusion (I/R) injury involves complex pathophysiologic events that contribute to cardiac dysfunction and cardiomyocyte death. Several therapeutic methods such as induction of preconditioning, postconditioning, and pharmacological agents have been examined to prevent or limit the I/R injury; however, the suitable treatment modalities have not been fully proved yet [1]. A promising approach to solving this issue is the application of natural biomolecules or their synthetic analogs that may trigger mechanisms of endogenous cardioprotection. One of these compounds is a new adipocytokine apelin, the endogenous ligand for the G-protein-coupled APJ receptor [2]. Apelin and APJ receptor are widely expressed in various organs and tissues, such as the heart, adipose tissue, lung, kidney, and adrenal glands [3]. Apelin and APJ receptor have a functional role in cardiovascular development and may also participate in cardiovascular pathological processes [4–7].
The apelin precursor is translated as a 77-amino-acid pre-proprotein which is processed to C-terminal bioactive fragments including apelin-36, -19, -17, -13 (or its pyroglutamate analog [Pyr]1-apelin-13), and -12 [8]. Apelin-12 (A12), one of the most potent apelin peptides, corresponds to the sequence 66–77. A12 is involved in the regulation of body fluid homeostasis and in the central control of feeding [9, 10]. Administration of exogenous A12 reduces arterial blood pressure in anesthetized rats due to activation of endothelial nitric oxide synthase [11] and exerts a positive inotropic action in failing myocardium of rodents [12]. APJ receptor is identified as a co-receptor for human immunodeficiency virus type I (HIV-I) and A12 blocks HIV-1 entry through the receptor [13, 14]. There is a growing body of evidence indicating protective effects of A12 in experimental myocardial ischemia and reperfusion. A12 administration improves postischemic recovery of isolated rat heart after global ischemia [15]. Intravenous A12 injection after regional myocardial ischemia limits infarct size and reduces cardiomyocyte membrane damage in rats in vivo [16]. Recent clinical study revealed that plasma A12 concentration is reduced early after acute myocardial infarction and remains significantly below baseline at 24 weeks [17]. These facts suggest that A12/APJ system may be a promising tool in the treatment of coronary heart disease. However, apelin peptides are rapidly cleared from the circulation with a half-life of no longer than 5 min because of hydrolysis by various peptidases including angiotensin-converting enzyme 2 (ACE2) [18, 19]. We synthesized structural analogs of A12 which were more resistant to enzymatic degradation and evaluated their efficacy in rat heart models of I/R injury. Metabolic and functional protection afforded by A12 analogs appeared to be comparable with the effects of the native peptide A12 [17, 20].
Mechanisms of action of apelin peptides in I/R injury are not well understood. Traditionally they are attributed to mobilization of the PI3 K–Akt and MEK1/2–ERK1/2 salvage kinases, and inhibition of the mitochondrial permeability transition pore (mPTP) opening [7]. Phosphorylation and activation of endothelial nitric oxide synthase (eNOS) are also implicated in myocardial protection afforded by apelins [21, 22]. Antioxidant properties of A12 and its analogs and ability of these peptides to scavenge reactive oxygen species (ROS) have not been studied so far. However, some studies imply possibility of using apelin-13 and [Pyr] 1-apelin-13 to reduce oxidative stress [23–25]. This work was designed to examine the effects of A12 and its synthetic structural analog AI on activities of Cu,Zn superoxide dismutase (Cu,Zn SOD), catalase (CAT), glutathione peroxidase (GSH-Px), a secondary product of lipoperoxidation, malondialdehyde (MDA), and ROS formation in reperfused rat heart. We used ex vivo and in vivo model of myocardial I/R injury to assess a link between a potential antioxidant defense and cardiac function recovery or infarct size limitation.
Materials and methods
Chemicals
Peptides A12 and analog AI were synthesized by the automatic solid-phase method using an Applied BioSystems 431A peptide synthesizer (Germany) and Fmoc technology (Table 1). They were purified by preparative HPLC and identified by 1H-NMR spectroscopy and mass spectrometry [16]. Enzymes and chemicals were purchased from Sigma Chemical Co. (St Louis, MO, USA). Solutions were prepared using deionized water (Millipore Corp. Bedford, MA, USA).
Table 1.
Structure of apelin-12 and its analog AI
| H-Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Met-Pro-Phe-OH | Apelin-12 |
|---|---|
| H-(N α Me)Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Nle-Pro-Phe-OH | Analog AI |
The substitutions are shown in bold
Animals
Male Wistar rats weighing 290–340 g were housed in cages in groups of three and maintained at 20–30 °C with a natural light–dark cycle. All animals had free access to standard pelleted diet (Aller Petfood, St. Petersburg, Russia) and tap water. The care and use of the animals were conducted in accordance with the European Convention for the Protection of Vertebrate Animals Used for Experimental and other Scientific Purposes (No. 123 of 18 March 1986).
Isolated perfused hearts
Rats were heparinized by intraperitoneal injection (1,600 IU/kg body weight) and anesthetized with urethane (1.3 g/kg body weight). Hearts were excised and immediately placed into ice-cold Krebs–Henseleit bicarbonate buffer (KHB) until contraction stopped. The aorta was then cannulated and Langendorff perfusion was performed at a constant pressure equivalent to 75 cm H2O for 15 min. Working perfusion was performed according to a modified method of Neely under constant left atrium pressure and aortic pressure of 20 and 100 cm H2O, respectively. KHB containing (in mM) NaCl 118, KCl 4.7, CaCl2 3.0, Na2EDTA 0.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25.0, and glucose 11.0 was saturated with a mixture of 95 % O2 and 5 % CO2; pH was 7.4 ± 0.1 at 37 °C; it was passed through a 5 μm Millipore filter (Bedford, MA, USA) before use. A needle was inserted into the left ventricular (LV) cavity to register LV pressure via a Gould Statham P50 transducer, SP 1405 monitor, and a Gould Brush SP 2010 recorder (Gould, Oxnard, CA, USA). The contractile function intensity index was calculated as the LV developed pressure–heart rate product (LVDP × HR), where LVDP is the difference between LV systolic and LV end-diastolic pressure. Cardiac pump function was assessed by cardiac output, the sum of aortic output and coronary flow.
The steady-state values of cardiac function were recorded after preliminary 20 min of perfusion in working mode. The peptides were dissolved in KHB immediately prior to the experiments. Isolated hearts were randomly assigned into one of three groups:
A12 (n = 12). After preliminary working perfusion, a 5-min infusion of 140 μM A12 at a constant flow rate of 4 ml/min was applied. Then the hearts were subjected to 35 min of normothermic global ischemia followed by 5 min of Langendorff perfusion at a flow rate of 4 ml/min with subsequent 25 min of working reperfusion. Previously we have shown that such way of A12 administration is optimal for cardiac function recovery in this experimental protocol [20].
Analog AI (n = 12). A 5-min infusion of 140 μM analog AI at a flow rate of 4 ml/min was used prior to global ischemia. Further protocol was the same as for the A12 group.
Control (n = 12). After preliminary working perfusion, infusion of KHB without peptides was applied at the same rate for 5 min before global ischemia. Then the hearts underwent reperfusion procedure as in the A12 group.
The hearts were freeze-clamped in liquid nitrogen for biochemical studies at the end of reperfusion. In separate series, the hearts were freeze-clamped after preliminary working perfusion for determination of antioxidant enzymes activity and MDA content.
Spin trap measurements in perfusate
100 mM 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was added to aliquots of the coronary effluents collected at the end of the steady state and at 1, 3, and 5 min of the reperfusion. The effluent samples were rapidly frozen and stored in liquid nitrogen until electron paramagnetic resonance (EPR) measurements. Addition of DMPO to the effluent perfusate was used to avoid improvement of cardiac function and coronary flow by the spin trap during reperfusion [26]. Varian E-109 E X-band electron spin resonance spectrometer (USA) was used for registration of EPR spectra of perfusate samples. EPR signals of DMPO spin adducts were recorded in a glass capillary tube at room temperature. Spectrometer settings were the following: magnetic field modulation of 0.1 mT, modulation of frequency of 100 kHz, microwave power of 10 mW, and microwave frequency of 9.15 GHz. DMPO-OH spin adduct concentrations were calculated as described previously using a standard solution of the spin label TEMPO (2,2,6-tetramethylpiperidine-N-oxyl).
Anesthetized rats in vivo
Rats were anesthetized with 20 % urethane (120 mg/kg body wt i.p.) and artificially ventilated with a KTR-5 animal respirator (Hugo Sacks Electronik) with a volume of 2–3 ml at a rate of 70–75 breaths/min. The right jugular vein was catheterized for drug administration. The left carotid artery was cannulated for monitoring arterial blood pressure and blood sampling. Electrocardiogram (ECG) leads were placed to record HR. The chest was opened by a left thoracotomy in the fifth intercostal space, and the heart was exposed by removing the pericardium. After pericardiotomy, a 5–0 prolene ligature was placed under the left anterior descending (LAD) coronary artery; where it emerges from beneath the left atrial appendage and the ends were threaded through a small plastic tube to form a snare for reversible LAD coronary artery occlusion. Complete LAD coronary artery occlusion was confirmed by observing cyanosis of the myocardium as well as the ST-segment elevation and immediate fall in the mean arterial pressure (MAP) by 15–30 mm Hg. Arterial blood pressure was recorded with a pressure transducer (Statham p23Db, Oxnard, USA) using a polygraph Biograph-4 (St. Petersburg, Russia). The MAP, HR, and standard lead II ECG were recorded on a computer using a LabView 7.1 data acquisition system (National Instruments, USA). Arterial blood pH, partial CO2 pressure, and O2 saturation values were monitored with an ABL-30 acid–base gas analyzer (Radiometer, Denmark) and maintained at the physiological level throughout the experiment.
After 30-min stabilization of hemodynamic parameters (initial state), LAD coronary artery was occluded for 40 min to simulate regional ischemia; the duration of subsequent reperfusion was 1 h. The prepared animals were randomly assigned into one of three groups: control, A12, or analog AI. After the period of LAD coronary artery occlusion, 0.5 ml of saline was administrated by i.v. bolus injection at the onset of reperfusion in control. A12 or analog AI was administrated by i.v. bolus injection at the onset of reperfusion at a dose of 0.35 μmol/kg. Previously we have found that this dose is optimal for A12 and AI [16]. The peptides were dissolved in saline before administration; the volume of injected solution was 0.5 ml.
At the end of the reperfusion, LAD coronary artery was reoccluded and 2 ml of 2 % Evans Blue solution was injected through the jugular vein to distinguish the myocardial non-ischemic area from the area at risk (AAR). In separate series of experiments, the LV area with LAD coronary artery to be occluded was freeze-clamped in liquid nitrogen in the steady state; at the end of reperfusion the AAR was freeze-clamped in liquid nitrogen for biochemical studies.
Determination of infarct size
After staining with Evans Blue, the heart was excised and the LV was transversely cut into 1.5-mm-thick slices which were incubated in 0.1 M sodium phosphate buffer pH 7.4, containing 1 % 2,3,5-triphenyl-tetrazolium chloride (TTC, Sigma, USA) for 10 min at 37 °C. The non-infarcted AAR was stained deep red, infarct tissue gray white, and non-ischemic area blue. The slices were fixed in 10 % formalin for 5 min. Then they were placed between two transparent glasses and captured using a scanner at 600 d.p.i. resolution; the saved images were analyzed by computerized planimetry using Imagecal software. The slices were then weighed for determination of LV weight. The AAR was expressed as a percentage of LV weight, the infarct size (IS) was expressed as a percentage of the AAR in each group [16].
Determination of necrosis markers
At the end of the steady state and reperfusion, blood samples were collected for plasma separation and stored at −70 °C for further analysis. Plasma lactate dehydrogenase (LDH) was determined enzymatically with pyruvate as substrate using standard kits from BioSystems S.A. (Barcelona, Spain). Plasma creatine kinase MB (CK-MB) activity was assessed by an immunoinhibition method using standard kits from BioSystems S.A. (Barcelona, Spain) from the rate of NADPH formation by means of the hexokinase and glucose-6-phosphate dehydrogenase coupled reactions.
Preparation of tissue homogenate
Isolated perfused hearts or areas at risk excised from the LV were freeze-clamped in liquid nitrogen. The frozen tissue samples were homogenized in 50 mM Na phosphate buffer, pH 7.4 (1:10 wt/vol) using an Ultra-Turrax T-25 homogenizer (IKA-Labortechnik, Staufen, Germany). The homogenates were centrifuged at 1,000 g for 10 min at 4 °C in a Joan MR-23 centrifuge (France). The supernatants thus obtained contained the cytosolic fraction and were used for the estimation of Cu,Zn SOD, CAT, and GSH-Px activities; MDA content; and protein. The measurements were done using a Hitachi 557 spectrophotometer (Japan). Protein in the supernatants was estimated by the method of Lowry et al. [27].
Determination of antioxidant enzymes activity and MDA
SOD activity was measured at 560 nm as the rate of suppression of nitrotetrazolium blue reduction when superoxide anion radical was generated during oxidation of xanthine by xanthine oxidase [28]. For 1 unit of activity, the amount of protein was taken which provided 50 % inhibition of nitrotetrazolium blue reduction under standard conditions.
Catalase activity was determined at 240 nm by measuring the rate of H2O2 utilization as described elsewhere [29] using the molar extinction coefficient for H2O2 equal to 43.6 M−1·cm−1 for the calculations. The amount of the enzyme utilizing 1 μmol H2O2 per min was taken as 1 activity unit. GSH-Px activity was measured by the modified method [30] at 340 nm in the reaction mixture containing 0.85 mM glutathione reduced (GSH), 0.12 mM NADPH, 0.5 unit/ml yeast glutathione reductase, 1 mM EDTA, and 0.2 mM tret-butyl hydroperoxide as a substrate in 50 mM K,Na phosphate buffer (pH 7.4 at 30 °C). The amount of enzyme converting 1 μmol GSH per minute was taken as 1 activity unit. MDA was estimated by the standard method in the reaction with 2-thiobarbituric acid in acidic media by a formation of trimetin complex having an absorption maximum at 532 nm [31].
Statistical analysis
All data are presented as mean ± SEM. Results were analyzed by one-way ANOVA followed by Bonferroni multiple range test post-hoc analysis for calculation differences between more than two groups. Comparisons between two groups involved use of the Student’s unpaired t test. A p < 0.05 was considered statistically significant.
Results
Effects of apelins on postischemic recovery of isolated perfused rat heart
In the control group, contractile and pump function indices of isolated perfused rat heart at the end of reperfusion were considerably reduced compared with the steady-state values (Table 2). Infusion of A12 or its structural analog AI significantly improved cardiac function recovery compared with control group. Augmented recovery of cardiac function indices in the A12 and AI groups was associated with better myocardial relaxation, as evidenced by decrease in LV diastolic pressure rise during reperfusion compared with that one in control. Additionally, treatment with peptide AI enhanced restoration of coronary flow compared with control and significantly increased LVDPxHR product recovery compared with A12.
Table 2.
Effects of infusion of A12 or its analog AI before global ischemia on functional recovery of isolated perfused rat heart at the end of reperfusion
| Steady state | Control | A12 | AI | |
|---|---|---|---|---|
| LV systolic pressure (mm Hg) | 99 ± 1 | 71 ± 2a | 86 ± 2ab | 87 ± 4ab |
| LV diastolic pressure (mm Hg) | −3 ± 1 | 8 ± 1a | 4 ± 1ab | 2 ± 1ab |
| LV developed pressure (mm Hg) | 102 ± 1 | 62 ± 2a | 82 ± 3ab | 85 ± 5ab |
| Heart rate, beat (min) | 303 ± 1 | 242 ± 6a | 276 ± 6ab | 279 ± 9ab |
| LVDP × HR (mm Hg/min) | 31,301 ± 368 | 15,337 ± 932a | 23,476 ± 125ab | 24,102 ± 219abc |
| Aortic output (ml/min) | 26 ± 3 | 2 ± 1a | 14 ± 1ab | 16 ± 2ab |
| Coronary flow (ml/min) | 18 ± 2 | 14 ± 1 | 16 ± 1 | 17 ± 1b |
| Cardiac output (ml) | 44 ± 4 | 15 ± 1a | 30 ± 2ab | 33 ± 3ab |
| Stroke volume (μl) | 144 ± 1 | 62 ± 4a | 106 ± 6ab | 112 ± 6ab |
The values are expressed as means + SEM. Each value represents a mean of ten experiments
a p < 0.05 versus steady state; b p < 0.05 versus control; and c p < 0.05 versus A12
Effects of apelins on activities of Cu,Zn SOD, CAT, and GSH-Px; and MDA content in isolated perfused rat heart
A significant decrease in myocardial activities of Cu,Zn SOD, CAT, and GSH-Px; and an increase in MDA content were observed in the control group as compared to the steady-state values (Fig. 1). A complete recovery of Cu,Zn SOD activity, and a significant increase in CAT activity was observed in the A12-treated group as compared to control group. Pre-ischemic A12 infusion, however, slightly restored myocardial GSH-Px activity and did not significantly reduce MDA level in myocardial tissue as compared to control group. Treatment with analog AI completely restored the activities of Cu,Zn SOD, CAT, and GSH-Px in reperfused hearts to the steady-state values. AI infusion also markedly reduced lipid peroxidation as evidenced by reduction in MDA content as compared to control group.
Fig. 1.

Effects of A12 or AI infusion before ischemia on antioxidant enzyme activities and MDA content in isolated perfused rat heart. The values are expressed as means + SEM. Each value represents a mean of eight experiments. St. st. (steady state): 20-min preliminary working perfusion; Cont. (control): 5-min Langendorff perfusion + 35-min global ischemia + 5-min Langendorff perfusion + 25-min working reperfusion; A12: 5-min Langendorff perfusion with 140 μM A12 before global ischemia; AI: 5-min Langendorff perfusion with 140 μM AI before global ischemia. * p < 0.05 versus steady state; # p < 0.05 versus control; and + p < 0.05 versus A12
Effects of apelins on DMPO-OH adduct formation in coronary effluent of isolated perfused rat heart
EPR spectra of the effluents consisted of the four-component narrow signals with the ratio of the intensities of 1:2:2:1 belonging to DMPO-OH adduct (Fig. 2a). The effluent DMPO-OH concentrations did not differ significantly between the groups in the steady state (Fig. 2b). A significant increase in DMPO-OH concentration was observed on early reperfusion in the control group as compared to the steady-state value. Pre-ischemic administration of A12 or AI substantially decreased DMPO-OH formation during reperfusion compared with control. In this case DMPO-OH concentrations did not differ from the steady-state values. As is known DMPO-OH adduct could arise from a decomposition of superoxide radical adduct DMPO-OOH as well as from direct trapping of OH. radicals generated in the Haber–Weiss and the Fenton reactions [32]. Since the detection of DMPO-OH adduct in the coronary effluent does not directly reflect ROS formation in the heart, the obtained data indicate that both peptides reduced the release of ROS-generating systems and hydrogen peroxide from myocardial tissue.
Fig. 2.
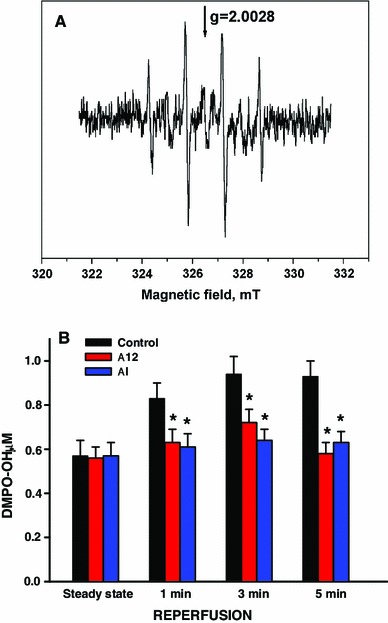
a EPR spectrum of myocardial effluent of isolated perfused rat heart containing hydroxyl radical spin adducts DMPO-OH. DMPO: 5,5-dimethyl-1pyrroline-N-oxide. b Effects of A12 or AI infusion before ischemia on DMPO-OH adduct concentrations in myocardial effluent of isolated perfused rat heart. The values are expressed as means + SEM. Each value represents a mean of six experiments. Steady state: the last minute of 20-min preliminary working perfusion; Reperfusion: 5-min Langendorff reperfusion after 35-min global ischemia; Control: 5-min Langendorff perfusion + 35-min global ischemia + 5-min Langendorff reperfusion; A12: 5-min Langendorff perfusion with 140 μM A12 before global ischemia + 35-min global ischemia + 5-min Langendorff reperfusion; AI: 5-min Langendorff perfusion with 140 μM AI before global ischemia + 35-min global ischemia + 5-min Langendorff reperfusion. * p < 0.05 versus control
Effects of apelins on myocardial infarction and CK-MB and LDH activities in plasma in anesthetized rats
The percentage ratios of AAR/LV did not differ significantly between the groups (Table 3). In control group, the percentage ratio of IS/AAR was 40.5 ± 2.1 %. Administration of A12 or its analog AI at onset of reperfusion significantly reduced this index as compared to control group (by 40 and 30 %, respectively), thus indicating limitation of infarct size. There was no significant difference in the percentage ratio of IS/AAR between A12 and AI groups.
Table 3.
Effects of A12 and analog AI administration at the onset of reperfusion on myocardial infarct size and activities of cardiac biomarkers in plasma of anesthetized rats
| Steady state | Control | A12 | AI | |
|---|---|---|---|---|
| AAR/LV wt. (%) | 38.6 ±1.6 | 41.5 ± 2.7 | 37.2 ± 1.5 | |
| IS/AAR (%) | 40.5 ± 2.1 | 24.2 ± 1.9a | 27.6 ± 1.8a | |
| CK-MB (IU/L) | 274.3 ± 27.3 | 2,173.4 ± 71.0 | 915.1 ± 131.2ab | 1,078.1 ± 53.5ab |
| LDH (IU/L) | 91.7 ± 20.5 | 1,521.4 ± 134.4 | 704.3 ± 133.5ab | 718.3 ± 103.4ab |
In control group, the activity of CK-MB in plasma increased almost 8-fold by the end of reperfusion compared with the steady-state value. Treatment with A12 or analog AI reduced the activity of CK-MB isoenzyme as compared to control group by 58 and 51 %, respectively. In control group, LDH activity in plasma was 16 times higher than in the steady state. Administration of peptides caused significant decrease in LDH activity as compared to control group on average by 53 %.
The values are expressed as means + SEM. Each value represents a mean of ten experiments. AAR: area at risk; LV: left ventricle; IS: infarct size; CK-MB: creatine kinase MB fraction; and LDH: lactate dehydrogenase. a p < 0.05 versus steady state and b p < 0.05 versus control.
Effects of apelins on activities of Cu,Zn SOD, CAT, and GSH-Px; and MDA content in the area at risk in anesthetized rats
A significant decrease in myocardial activities of Cu,Zn SOD, and GSH-Px, a slight reduction of CAT activity, and a concomitant increase in myocardial MDA content were observed in the AAR in the control group at the end of reperfusion compared with the values in steady state (Fig. 3). Intravenous administration of A12 at the onset of reperfusion completely restored activities of Cu,Zn SOD, CAT, and GSH-Px, and reduced myocardial MDA content in the AAR to the initial value. Treatment with analog AI had the similar effects on antioxidant enzyme activities and significantly decreased lipid peroxidation in the AAR at the end of reperfusion.
Fig. 3.

Effects of A12 or AI administration at the onset of reperfusion on antioxidant enzyme activities and MDA content in the area at risk in anesthetized rats. The values are expressed as means + SEM. Each value represents a mean of eight experiments. St. st. (steady state): 30-min stabilization of hemodynamic parameters; Cont. (control): 40-min LAD occlusion + i.v. saline bolus injection + 1 h reperfusion; A12: i.v. A12 bolus injection (0.35 μmol/kg) at the onset of reperfusion; AI: i.v. AI bolus injection (0.35 μmol/kg) at the onset of reperfusion. * p < 0.05 versus steady state; # p < 0.05 versus control
Discussion
It is well recognized that mechanisms of I/R injury include an overproduction of ROS [33]. The interaction of ROS with cell membrane lipids and essential proteins contribute to myocardial cell damage, leading to depressed cardiac function and irreversible tissue injury with concomitant depletion of certain key endogenous antioxidant enzymes, e.g., SOD, CAT, and GSH-Px [34]. Elevated ROS increases the probability of the mPTP opening, which is followed by bioenergetic collapse and ultimately cell death [35]. The therapeutic strategies to attenuate myocardial injury induced by oxidative stress include controlled reperfusion, cardioprotective phenomena, and administration of various interventions [36]. The present study demonstrates, for the first time, that effects of exogenous peptide A12 and its structural analogs AI are associated with enhanced antioxidant defense against myocardial I/R injury. This is manifested by enhancing the enzymatic antioxidant activity and inhibiting the lipid peroxidation in both experimental models (Figs. 1, 3). In addition, the peptides are capable to decrease the generation of short-lived ROS as proved by attenuation of the DMPO-OH adduct formation in myocardial effluent of isolated rat heart (Fig. 2b). Importantly, preventing the oxidative stress by A12 and its analog AI improves cardiac function recovery or limits infarct size and promotes membrane integrity at the end of reperfusion. Therefore, these novel peptides may be considered as therapeutic interventions to diminish the progression of myocardial damage during I/R injury.
The obtained results are principally consistent with the previously described antioxidant properties of some unmodified C-terminal fragments of apelin. Apelin-13 treatment ameliorated the inhibited SOD activity and reduced ROS formation in cultured cardiomyocytes subjected to hypoxia/reoxygenation [23]. In isoproterenol-treated rats, administration of exogenous apelin-13 decreased LDH activity in plasma and myocardial MDA content [24]. Activation of CAT by [Pyr1]apelin-13 prevented oxidative stress-linked hypertrophy in cultured rat cardiomyocytes [25]. Chronic treatment of mice with [Pyr1]apelin-13 attenuated pressure-overload-induced LV hypertrophy and decreased plasma lipid hydroperoxide concentration. These effects were associated with increased myocardial CAT activity. Reasons for the increase in antioxidant enzyme activities under the influence of apelin peptides remain unexplored. To our knowledge, there is only mention of the fact that [Pyr1]apelin-13 induced CAT mRNA but not GSH-Px expression in cultured rat cardiomyocytes [25].
The enhancement of intracellular Cu,Zn SOD activity is one of the essential components of myocardial protection afforded by A12 and its analog AI. Indeed, large amounts of superoxide anion (O2-*) formed during I/R injury may be diminished by the upregulation of SOD. Hydrogen peroxide (H2O2), the less toxic product of the SOD reaction, can be transformed then into water and O2 by CAT or into H2O as a result of the oxidization of reduced glutathione (GSH) into glutathione disulfide (GSSG) by GSH-Px [37]. It is well documented that apelin administration leads to NO production due to activation of eNOS and upregulation of eNOS gene expression [5, 6, 8, 11]. During reperfusion, NO can be scavenged by O2-* with formation of a highly reactive peroxynitrite (ONOO-) [34, 36]. Removing a large fraction of O2-* by the increased activity of SOD may also reduce the yield of ONOO-, thus limiting nitrosyl stress. In addition, the opening of mPTP in I/R injury results in a further rise in of ROS generation [35]. Probably A12 or the analog AI may attenuate this stage of oxidative stress due to the activation of Cu,Zn SOD. Thus, enhanced Cu,Zn SOD activity may play a key role in controlling levels of reactive oxygen and nitrogen species in apelin-treated hearts.
It is likely that A12 and its analog AI can reduce oxidative stress due to the antioxidant properties of NO. As is known, NO prevents mitochondrial oxygen damage and lipid peroxidation [38]. The protective role of NO also includes the reduction of ROS production by neutrophils due to the inhibition of their adherence to vascular endothelia [39]. Moreover, endogenous NO is involved in the upregulation of the key antioxidant enzymes, Cu,Zn SOD, CAT, and GSH-Px, which represents adaptive protective mechanism to prevent the formation of toxic quantities of reactive oxygen and nitrogen species [40, 41]. NO is a mediator of beneficial effects of apelin peptides consisting in a limitation of infarct size, reduction of cell membrane damage, and improvement of postischemic functional and metabolic recovery. Such interconnection is confirmed by a significant reduction or abolition of apelin action in the presence of L-NNA and L-NAME, the inhibitors of NOS, in different models of myocardial I/R injury [21, 22]. This approach will be useful for assessing the role of NO in the antioxidant effects of these peptides.
The fact that the modification of the natural peptide A12 does not reduce its antioxidant capacity seems important. Both peptides enhance the antioxidant enzymes activity and attenuate hydroxyl radical adducts formation during reperfusion almost equally. However, advantages of analog AI may be more apparent in situations with a prolonged exposure to peptides. Due to higher proteolytic stability, the half-life of AI in human blood plasma is ten times longer than that of the natural peptide (unpublished data). Additionally, this peptide reduces the MAP and HR to a significantly lesser degree than A12 when administered intravenously [16]. We believe that the use of analog AI may be promising in clinical settings, coronary angioplasty, cardiac surgery (as an additive to cardioplegic and reperfusion solutions), and treatment of cardiac failure.
Conclusions
Natural peptide A12 and its synthesized analog AI reduce I/R injury when administrated before ischemia or at the onset of reperfusion acting as a pharmacological pre- or post-conditioning tools. Cardioprotective effects of the peptides include prevention or attenuation of oxidative stress by increasing the activity of antioxidant enzymes in postischemic heart, which leads to reduction of ROS formation and lipid peroxidation. Further well-controlled studies are required to assess their potential therapeutic utility in human beings.
Acknowledgments
This study was supported by a grant from The Russian Foundation for Basic Research No. 11-04-00078a. The authors are grateful to Dr. M.V. Sidorova for synthesis of the peptides and discussion of the results.
References
- 1.Ferdinandy P, Schulz R, Baxter G. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;9:418–458. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- 2.Kleinz MJ, Davenport AP. Emerging roles of apelin in biology and medicine. Pharmacol Therap. 2005;107:198–211. doi: 10.1016/j.pharmthera.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, Liu Y, Osmond DH, George SR, O’Dowd BF. Characterization of apelin, the ligand for the APJ receptor. J Neurochem. 2000;74:34–41. doi: 10.1046/j.1471-4159.2000.0740034.x. [DOI] [PubMed] [Google Scholar]
- 4.Kunduzova O, Alet N, Delesque-Touchard N, Millet L, Castan-Laurell I, Muller C, et al. Apelin/APJ signaling system: a potential link between adipose tissue and endothelial angiogenic processes. FASEB J. 2008;22:4146–4153. doi: 10.1096/fj.07-104018. [DOI] [PubMed] [Google Scholar]
- 5.Japp AG, Cruden NL, Amer DA, Li VK, Goudie EB, Johnston NR, et al. Vascular effects of apelin in vivo in man. J Am Coll Cardiol. 2008;52:908–913. doi: 10.1016/j.jacc.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Szokodi I, Tavi P, Foldes G, Voutilainen-Myllyla S, Ilves M, Tokola H, et al. Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ Res. 2002;91:434–440. doi: 10.1161/01.RES.0000033522.37861.69. [DOI] [PubMed] [Google Scholar]
- 7.Simpkin JC, Yellon DM, Davidson SM, Lim SY, Wynne AM, Smith CC. Apelin-13 and apelin-36 exhibit direct cardioprotective activity against ischemia-reperfusion injury. Bas Res Cardiol. 2007;102:518–528. doi: 10.1007/s00395-007-0671-2. [DOI] [PubMed] [Google Scholar]
- 8.Pitkin SL, Maguire JJ, Bonner TI, Davenport AP. International union of basic and clinical pharmacology. LXXIV. Apelin receptor nomenclature, distribution, pharmacology, and function. Pharmacol Rev. 2010;62:331–342. doi: 10.1124/pr.110.002949. [DOI] [PubMed] [Google Scholar]
- 9.Lee DK, George SR, O’Dowd BF. Unravelling the roles of the apelin system: prospective therapeutic applications in heart failure and obesity. Trends Pharmacol Sci. 2006;27:190–194. doi: 10.1016/j.tips.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Japp AG, Newby DE. The apelin-APJ system in heart failure: pathophysiologic relevance and therapeutic potential. Biochem Pharmacol. 2008;75:1882–1892. doi: 10.1016/j.bcp.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 11.Jia YX, Lu ZF, Zhang J, Pan CS, Yang JH, Zhao J, Yu F, Duan XH, Tang CS, Qi YF. Apelin activates l-arginine/nitric oxide synthase/nitric oxide pathway in rat aortas. Peptides. 2007;28:2023–2029. doi: 10.1016/j.peptides.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 12.Dai T, Ramirez-Correa G, Gao WD. Apelin increases contractility in failing cardiac muscle. Eur J Pharmacol. 2006;553:222–228. doi: 10.1016/j.ejphar.2006.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zou MX, Liu HY, Haraguchi Y, Soda Y, Tatemoto K, Hoshino H. Apelin peptides block the entry of human immunodeficiency virus (HIV) FEBS Lett. 2000;473:15–18. doi: 10.1016/S0014-5793(00)01487-3. [DOI] [PubMed] [Google Scholar]
- 14.Cayabyab M, Hinuma S, Farzan M, Choe H, Fukusumi S, Kitada C, Nishizawa N, Hosoya M, Nishimura O, Messele T, Pollakis G, Goudsmit J, Fujino M, Sodroski J. Apelin, the natural ligand of the orphan seven-transmembrane receptor APJ, inhibits human immunodeficiency virus type 1 entry. J Virol. 2000;4:11972–11976. doi: 10.1128/JVI.74.24.11972-11976.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pisarenko OI, Shulzhenko VS, Pelogeykina YuA, Studneva IM, Khatri DN. Apelin-12 improves metabolic and functional recovery of rat heart after global ischemia. Health. 2010;2:927–934. doi: 10.4236/health.2010.28137. [DOI] [Google Scholar]
- 16.Pisarenko OI, Serebryakova LI, Studneva IM, Pelogeykina YuA, Tskitishvili OV, Bespalova ZhD, Sidorova MV, Az’muko AA, Khatri DN, Pal’keeva ME, Molokoedov AS. Effects of structural analogues of apelin 12 in acute myocardial infarction in rats. J Pharmacol Pharmacother. 2013;4:198–203. doi: 10.4103/0976-500X.114600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weir RA, Chong KS, Dalzell JR, Petrie CJ, Murphy CA, Steedman T, et al. Plasma apelin concentration is depressed following acute myocardial infarction in man. Eur J Heart Fail. 2009;11:551–558. doi: 10.1093/eurjhf/hfp043. [DOI] [PubMed] [Google Scholar]
- 18.Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J, et al. Acute cardiovascular effects of apelin in humans: potential role in patients with chronic heart failure. Circulation. 2010;121:1818–1827. doi: 10.1161/CIRCULATIONAHA.109.911339. [DOI] [PubMed] [Google Scholar]
- 19.Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 20.Pisarenko OI, Shulzhenko VS, Pelogeykina YuA, Studneva IM. Attenuation of myocardial ischemia and reperfusion injury by novel analogues of apelin-12. Int J Pharmaceut Biomed Res. 2012;3:16–21. [Google Scholar]
- 21.Rastaldo R, Cappello S, Folino A, Berta GN, Sprio AE, Losano G, et al. Apelin-13 limits infarct size and improves cardiac postischemic mechanical recovery only if given after ischemia. Am J Physiol Heart Circ Physiol. 2011;300:H2308–H2315. doi: 10.1152/ajpheart.01177.2010. [DOI] [PubMed] [Google Scholar]
- 22.Pisarenko OI, Pelogeykina YA, Shulzhenko VS, Studneva IM. Nitric oxide synthase mediates the apelin-induced improvement of myocardial postischemic metabolic and functional recovery. Open J Mol Integ Physiol. 2012;2:1–7. doi: 10.4236/ojmip.2012.21001. [DOI] [Google Scholar]
- 23.Zeng XJ, Zhang LK, Wang HX, et al. Apelin protects heart against ischemia/reperfusion injury in rat. Peptides. 2009;30:1144–1152. doi: 10.1016/j.peptides.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Jia YX, Pan CS, Zhang J, et al. Apelin protects myocardial injury induced by isoproterenol in rats. Regul Pept. 2006;133:147–154. doi: 10.1016/j.regpep.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 25.Foussal C, Lairez O, Calise D, et al. Activation of catalase by apelin prevents oxidative stress-linked cardiac hypertrophy. FEBS Lett. 2010;584:2363–2370. doi: 10.1016/j.febslet.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 26.Tosaki A, Blasig IE, Pali T, Ebert B. Heart protection and radical trapping by DMPO during reperfusion in isolated working rat hearts. Free Radic Biol Med. 1990;8:363–372. doi: 10.1016/0891-5849(90)90102-O. [DOI] [PubMed] [Google Scholar]
- 27.Lowry OH, Rosebrough NJ, Farr AI, Randall RJ. Protein measurements with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 28.Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Analyt Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 29.Beers RF, Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem. 1952;95:133–140. [PubMed] [Google Scholar]
- 30.Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte peroxidase. J Lab Clin Med. 1967;2:158–169. [PubMed] [Google Scholar]
- 31.Lankin VZ, Konovalova GG, Tikhaze AK, Nedosugova LV. The effect of natural dicarbonyls on activity of antioxidant enzymes in vitro and in vivo. Biomed Chem. 2012;6:81–86. doi: 10.18097/pbmc20125806727. [DOI] [PubMed] [Google Scholar]
- 32.Kappusamy P, Zwier JL. Characterization of free-radical generation by xanthine oxidase. Evidence for hydroxyl radical generation. J Biol Chem. 1989;264:9880–9884. [PubMed] [Google Scholar]
- 33.Murphy E, Steenbergen Ch. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dhalla NS, Elmoselhi AB, Hata T, Makino N. Status of myocardial antioxidants in ischemia–reperfusion injury. Cardiovasc Res. 2000;47:446–456. doi: 10.1016/S0008-6363(00)00078-X. [DOI] [PubMed] [Google Scholar]
- 35.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787:1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 36.Rohilla A, Kushnoor N, Kushnoor A. Myocardial ischemia reperfusion injury-pathogenesis and prevention. Int J Res Pharmaceut Biomed Sci. 2012;3:929–934. [Google Scholar]
- 37.Wink DA, Hanbauer I, Krishna MC, DeGraff W, Gamson J, Mitchell JB. Nitric oxide protects against cellular damage and cytotoxicity from reactive oxygen species. Proc Natl Acad Sci USA. 1993;90:9813–9817. doi: 10.1073/pnas.90.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Donnell VB, Chumley PH, Hogg N, Bloodsworth A, Darley-Usmar VM, Freeman BA. Nitric oxide inhibition of lipid peroxidation: kinetics of reaction with lipid peroxyl radicals and comparison with alpha tocopherol. Biochemistry. 1997;36:15216–15223. doi: 10.1021/bi971891z. [DOI] [PubMed] [Google Scholar]
- 39.Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 40.Frank S, Zacharowski K, Wray GM, Thiemermann CH, Pfeilschifter J. Identification of copper/zinc superoxide dismutase as a novel nitric oxide-regulated gene in rat glomerular mesangial cells and kidneys of endotoxemic rats. FASEB J. 1999;13:869–882. doi: 10.1096/fasebj.13.8.869. [DOI] [PubMed] [Google Scholar]
- 41.Husain K, Hazelrigg SR. Oxidative injury due to chronic nitric oxide synthase inhibition in rat: effect of regular exercise on the heart. Biochim Biophys Acta. 2002;1587:75–82. doi: 10.1016/S0925-4439(02)00070-4. [DOI] [PubMed] [Google Scholar]