

Abstract
The cytotoxic granzyme B (GrB)/perforin pathway has been traditionally viewed as a primary mechanism that is used by cytotoxic lymphocytes to eliminate allogeneic, virally infected and/or transformed cells. Although originally proposed to have intracellular and extracellular functions, upon the discovery that perforin, in combination with GrB, could induce apoptosis, other potential functions for this protease were, for the most part, disregarded. As there are 5 granzymes in humans and 11 granzymes in mice, many studies used perforin knockout mice as an initial screen to evaluate the role of granzymes in disease. However, in recent years, emerging clinical and biochemical evidence has shown that the latter approach may have overlooked a critical perforin-independent, pathogenic role for these proteases in disease. This review focuses on GrB, the most characterized of the granzyme family, in disease. Long known to be a pro-apoptotic protease expressed by cytotoxic lymphocytes and natural killer cells, it is now accepted that GrB can be expressed in other cell types of immune and nonimmune origin. To the latter, an emerging immune-independent role for GrB has been forwarded due to recent discoveries that GrB may be expressed in nonimmune cells such as smooth muscle cells, keratinocytes, and chondrocytes in certain disease states. Given that GrB retains its activity in the blood, can cleave extracellular matrix, and its levels are often elevated in chronic inflammatory diseases, this protease may be an important contributor to certain pathologies. The implications of sustained elevations of intracellular and extracellular GrB in chronic vascular, dermatological, and neurological diseases, among others, are developing. This review examines, for the first time, the multiple roles of GrB in disease pathogenesis.
Keywords: apoptosis, chronic disease, extracellular matrix, granzyme B, immunity, inflammation
INTRODUCTION
Granzymes
Granzymes are a family of conserved serine proteases stored within the cytotoxic granules of cytotoxic lymphocytes (CLs) whose functions were once believed to be primarily involved in immune-targeted cell death. There are 5 granzymes expressed in humans: granzymes A, B, H, K, and M, and 11 in mice (A, B, C, D, E, F, G, K, L, M, and N).1 Granzymes A and B are the most abundant granzymes and for this reason have been the most studied. Granzyme B (GrB), which is the primary focus of this review, has received much attention over the past two decades.
GrB was first discovered in the mid-1980s, where several groups reported the presence of the protease in granules within CLs.2, 3, 4, 5 Also known as cytotoxic T lymphocyte-associated serine esterase 1 or granzyme 2, GrB is a 32-kDa serine protease resembling chymotrypsin, and has homologues expressed in a number of different species. The gene product encoding GrB is ∼3500 bp long, contains five exons and four introns, and maps to chromosome 14 on the human genome.6 Similar to caspases, GrB has a preference for cleaving peptides immediately adjacent to aspartate (Asp) residues.7, 8 This specificity is due to the structure of the GrB active site, which contains an arginine (Arg) residue positioned at the side of the active site pocket.9 An interaction between an Asp residue at the P1 position of the substrate and the Arg residue within the active site is key for enzyme–substrate interaction.9
Although once believed to be expressed exclusively by natural killer (NK) cells and cytotoxic T cells (CTLs), recent reports have shown that GrB can be expressed by various additional cell types. Under certain pro-inflammatory conditions, GrB can be expressed by CD4+ cells, mast cells, activated macrophages, neutrophils, basophils, dendritic cells (DCs), T regulatory cells, and nonimmune cell types such as smooth muscle cells (SMCs), chondrocytes, keratinocytes, type II pneumocytes, Sertoli cells, primary spermatocytes, granulosa cells, and syncytial trophoblasts.10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20
Granzyme expression is regulated at both the transcriptional and translational levels, and is influenced by many of the same factors that stimulate immune cell activation. Transcriptional activation of GrB within T lymphocytes involves activation of the T cell receptor and co-stimulation with cytokines.21 The promoter region upstream of the GrB transcription start site contains binding sites for two transcription factors, activating transcription factor/cyclic AMP-responsive element-binding protein (ATF/CREB) and activator protein-1 (AP-1), and two lymphoid specific-factors, Ikaros and core-binding factor (CBF/PEBP2).22, 23, 24, 25 All transcription factors act together to regulate GrB expression, and mutations to any of the transcription factor-binding sites will abrogate GrB expression.24, 26 Most lymphocytes constitutively express GrB transcripts and upregulate transcription when the lymphocyte has been activated. In T lymphocytes and NK cells, many of the extracellular factors that stimulate T cell activation will also augment GrB expression, including the composition of the cytokine milieu, the nature of receptor engagement, and the presence of helper or regulatory CD4+ T cell populations.21
The post-transcriptional regulation of GrB is evident in many cells types, although the mechanisms involved in this regulation are not fully understood. Comparable levels of GrB transcripts are detected in resting and activated plasma DCs, but significantly higher levels of GrB protein is evident in the activated cells.15 Resting mouse NK cells have an abundance of GrB transcripts, but not of GrB protein or cytotoxicity.27 However, once the cells have been activated, there is a significant increase in GrB protein levels, with relatively little change in GrB transcript levels.27 In contrast, human mast cells express high levels of GrB transcripts and relatively low levels of GrB protein.28
The post-translational regulation of GrB is accomplished through several mechanisms that include the synthesis of GrB as a propeptide requiring proteolytic cleavage for activation and the tagging of GrB with a mannose-6-phosphate receptor (MPR) used to target the protease to the acidic lytic granule.29 These mechanisms will be discussed in more detail in the Granzyme synthesis, storage and exocytosis section of this review.
Specific inhibitors regulate the activity of GrB to minimize accidental GrB-mediated apoptosis. The only known endogenous inhibitor of GrB in humans is protease inhibitor-9 (PI-9), which is a potent inhibitor of GrB and is expressed by immune cells as protection against accidental cytosolic GrB leakage.30, 31 Endothelial cells, vascular SMCs, and hepatocytes have also shown an ability to express PI-9 as a means of protection from GrB-mediated cytotoxicity.32, 33, 34, 35 High levels of PI-9 expression can be found in DCs, T cells, and endothelial cells of lymphoid and non-lymphoid tissue, as well as in cells of immune-privileged tissues, including the eyes, testes, ovaries, and placenta.36 In mice, serine protease inhibitor 6 shares homology with human PI-9, in which it functions to regulate GrB activity.37 Recently, Sipione et al38 discovered an inhibitor of mouse and human GrB, serpina3n, expressed by mouse Sertoli cells. Serpina3n shares homology with the human α-1-antichymotrypsin, which interestingly does not appear to show any inhibitory effect on GrB in humans. GrB activity may also be indirectly influenced by the actions of granzyme H (GrH) and granzyme M (GrM). GrM may inhibit the action of human PI-9, thereby promoting GrB activity,39 whereas GrH has the ability to cleave the adenoviral protein Ad5-100K in an effort to counter viral defense against GrB.40
Given its role in immune-mediated cytotoxicity, increased attention has been devoted toward GrB in an effort to elucidate the mechanisms of CTLs and NK cell-mediated elimination of cells. Traditional views on the function of GrB have, for the most part, been limited to its intracellular pro-apoptotic role. However, as discussed below, recent reports suggest that there may also be perforin- and/or apoptosis-independent roles for GrB in disease progression. Apoptosis and extracellular matrix (ECM) modification are hallmarks of many, if not most, chronic inflammatory disorders and the involvement of GrB in a number of inflammatory diseases is becoming evident and will be the focus of this review.
GrB-INDUCED CELL DEATH
Granzyme Synthesis, Storage and Exocytosis
A wide variety of immune cells possess cytotoxic capabilities, which are generally mediated by two pathways: the death receptor-mediated pathway involving the engagement of cell surface receptors with death ligands41, 42, 43, 44, 45 and the granule pathway mediated by the granzyme family of proteins.21, 46 The granule pathway is the primary pathway used for the clearance of pathogen-infected cells and the eradication of tumors (reviewed in Russell and Ley47). The key effectors of the granule pathway are the granzymes. Granzymes are expressed with a signal sequence that directs them to the endoplasmic reticulum. Cleavage of this signal peptide produces an inactive pro-enzyme that contains an N-terminal dipeptide and requires cleavage to produce an active protease.48, 49 In the golgi, granzymes are tagged with a mannose 6-phosphate used to target the granzymes to the lytic granule.29 Once inside the granule, granzymes are activated by removal of the N-terminal dipeptide by cathepsin C and stored on a scaffold of the chondroitin sulfate proteoglycan, serglycin.48, 49, 50, 51, 52, 53, 54 Storage on this scaffolding, in combination with the acidic pH of the lytic granules, acts to minimize the proteolytic activity of granzymes55 (reviewed in Chowdhury and Lieberman21).
Upon engagement, activated granzymes are delivered to target cells after the rapid polarization of lytic granules toward the immunological synapse (IS).56 The IS functions as a conduit for the transfer of lytic granules and other soluble factors between the CLs and the target cell.57 The movement of lytic granules from CLs to the target cell is directional and depends on an underlying microtubule cytoskeleton, known as the microtubule-organizing center.58, 59, 60 Once delivered to the site of secretion, lytic granules fuse with the plasma membrane and release their contents into a secretory cleft formed between the CL and the target cell.
Granzyme Entry into Target Cells
Delivery of granzymes into the cytoplasm of target cells induces cell death. The major lytic proteins packaged within the granules are granzymes and the pore-forming protein perforin. Traditional models indicate that GrB delivery into a target cell is mediated by perforin; however, the mechanism by which this is accomplished remains an active area of investigation. Perforin is a Ca2+-dependent pore-forming protein that multimerizes in the target cell's plasma membrane, forming 5–20 nm pores.61, 62 Early models suggested that perforin facilitates the movement of granzymes into the cytoplasm of the target cell through the formation of pores; however, this model was challenged when it was discovered that granzymes could be endocytosed without perforin.63, 64, 65, 66 More recent models show that GrB binds to the MPR and is rapidly endocytosed by the target cell.67 This process is likely dynamin-dependent and results in the formation of endosomes containing GrB and perforin; however, a dynamin-independent role has been proposed.67, 68, 69, 70, 71 GrB is then released from the endosomes into the cytosol, using a perforin-dependent mechanism where it can target cellular substrates and initiate cell death.69, 72, 73 However, although increasing evidence supports a role for MPR in GrB uptake, controversy does exist with regard to the dynamin/MPR mechanism as some evidence suggests this may not be required for GrB entry. It should also be noted that transfer of GrB to the endosome can occur in a perforin-independent manner.69, 71, 72, 74 Although a role beyond facilitating granzyme entry into cells is not known, perforin is required for CL-driven GrB-mediated apoptosis, as perforin deficiencies are associated with impaired lymphocyte-mediated cytotoxicity. Perforin-deficient mice are highly susceptible to viral infection75, 76 and cancer.77, 78 In humans, perforin deficiencies are associated with familial hemophagocytic lymphohistocytosis, an autosomal-recessive disorder resulting in uncontrolled T and NK cell activation and proliferation.79, 80, 81
Although MPR and perforin are believed to be important for GrB delivery, several groups have reported alternative mechanisms for GrB entry. First, serglycin–GrB complexes, which interact with perforin, can incorporate into target cell membranes in vitro and deliver GrB to the cytoplasm without the formation of perforin-induced pores.52, 53, 68, 82 This process was shown by Veugelers et al67 to be enhanced by the cell surface receptor, heparan sulfate. GrB entry may also be facilitated by other cell-surface-bound proteins, such as heat shock protein-70 (Hsp-70).83 Hsp-70 present on the surface of tumor cells not only facilitates the movement of GrB into the tumor cell but also stimulates the production and delivery of GrB by NK cells.84
GrB-induced Apoptosis
GrB delivery and substrate identification in apoptotic cell death has been the subject of much research. Once released into the cytoplasm, GrB can target substrates in the cytosol and the nucleus, and induce apoptosis through multiple pathways, as illustrated in Figure 1.
Figure 1.
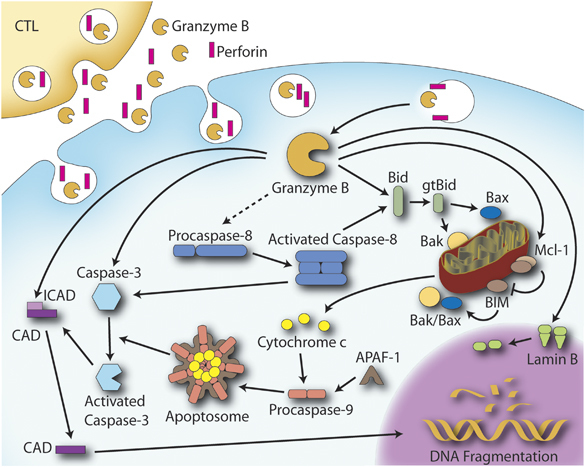
Classical granzyme B (GrB)/perforin-mediated apoptosis pathway. GrB internalization is facilitated by perforin. Upon internalization, GrB initiates apoptosis primarily through the cleavage of Bid into a truncated form (gtBid) that triggers mitochondrial cytochrome c release and apoptosome formation leading to caspase activation and manifestation of the apoptosis phenotype. GrB can also bypass the mitochondrial pathway and initiate caspase activation directly and/or cleave caspase substrates such as the inhibitor of caspase-activated deoxyribonuclease (ICAD), thereby allowing CAD to translocate to the nucleus to fragment DNA. GrB also cleaves the nuclear membrane protein lamin B, resulting in a loss of integrity of the nuclear membrane.
GrB shows broad substrate specificity, but preferentially cleaves after Asp residues. One of the first substrates to be identified for GrB was pro-caspase-3.85 GrB-activated caspase-3 results in the processing of several cellular substrates integral to eliciting the apoptotic phenotype, including the inhibitor of caspase-activated deoxyribonuclease, the DNA damage sensor, poly(ADP ribose) polymerase (PARP), nuclear lamins, and many others (reviewed in Hengartner86). Several other caspases, including caspase-2, -6, -7, -8, and -10, are reported to serve as direct substrates for GrB in vitro; however, only cleavage of caspase-3, -7 and -8 has been established in vivo.87, 88, 89
A major mechanism by which GrB is believed to induce cell death is through the cleavage of the BH3-only protein Bid into a truncated form, gtBid, which then translocates to the mitochondrion and disrupts mitochondrial membrane integrity through interactions with the pro-apoptotic proteins Bax and/or Bak.90, 91, 92 Disruption of the mitochondrial membrane integrity increases membrane permeability, leading to the release of apoptogenic factors, such as cytochrome c, smac/DIABLO, Omi/HtrA2, and apoptosis-inducing factor (AIF).93, 94 Cytochrome c release stimulates the formation of a macromolecular complex consisting of cytochrome c, dATP, apaf-1, and pro-caspase-9 known as the apoptosome, which results in the activation of caspase-9 and the subsequent activation of caspases-3 and -7. GrB also cleaves the anti-apoptotic protein Mcl-1, which results in the release of the pro-apoptotic Bcl-2 family member Bim, followed by mitochondrial outer membrane permeabilization and cytochrome c release.95
Several other intracellular substrates have been identified including of PARP, the nuclear mitotic apparatus protein, cytoskeletal components such as α-tubulin, the nuclear-envelope intermediate filament protein (lamin B), and ROCKII.96, 97, 98, 99, 100, 101, 102, 103, 104 GrB has also been shown to target proteins involved in cellular homeostasis and the stress response, including Hsp-70 and Hsp-90 from the heat shock family of proteins, and also the heat-shock-associated proteins Hip, Hop, and Bag1-L.105, 106, 107, 108
Although the mechanisms of CL/NK-mediated GrB-induced apoptosis have been studied for some time, the relative contribution of these pathways to disease pathogenesis is less understood. With the discovery of GrB expression in nonimmune cell types such as chondrocytes and keratinocytes, the effect of apoptosis (if any) mediated by these cells in disease has yet to be determined and presents a new role for GrB in disease pathogenesis. However, as several GrB-expressing cells may not co-express perforin and/or form IS with target cells, it is likely that these cells would exert more of an extracellular impact on disease. A role of GrB in chronic inflammation and disease will be described in more detail in the following sections of this review.
EXTRACELLULAR GrB ACTIVITY
Until recently, GrB was largely studied in its intracellular capacity, specifically in the context of apoptosis. However, the granzymes were originally identified as both intracellular and extracellular proteases, and over the past few years, increased research has focused on extracellular GrB activity.109, 110, 111, 112, 113 Several groups have reported that GrB is present in the ECM of tissue and can be found extracellularly in bodily fluids.54, 111, 114 Several novel extracellular substrates for GrB have been identified, potential implications of ECM cleavage have been described, and extracellular GrB activity has been linked to arthritis, vascular pathologies, and other diseases. Although cell types such as keratinocytes, chondrocytes, and neutrophils can express both GrB and peforin simultaneously, other cell types such as mast cells express GrB in the absence of perforin and GrA, suggesting that GrB may act exclusively extracellularly in these cells.11, 20, 115, 116, 117 This is probably due to the GrB gene localization to a cluster of genes along with mast cell proteases (separate from the perforin and GrA genes). As a result, GrB can be expressed by myeloid cells (DCs, granulocytes) and others upon activation, independently of perforin.15, 17, 118
GrB is released from cytotoxic granules upon target cell recognition. Upon reaching the neutral pH of the extracellular environment, GrB is instantly active and can readily cleave susceptible extracellular substrates. The stimuli involved in GrB release have not been fully elucidated; however, several mechanisms have been described to date. GrB may leak into the ECM from the IS during target cell engagement, it may be released non-specifically upon TCR signaling or after prolonged IL-2 stimulation, and it is likely released after other unidentified stimuli.119, 120 Recently, Prakash et al121 found that GrB is constitutively released from CTLs and NK cells in vivo and that GrB release can occur in the absence of target cell engagement. GrB is released in both active and inactive forms, suggesting that there may be an extracellular GrB activator for the zymogen form of the enzyme.121 The pro-form of GrB may be regulated outside cells in a process similar to the extracellular regulation of other ECM proteases, such as the pro-forms of matrix metalloproteases (MMPs), although this has yet to be defined. Besides NK cells and CTLs, other immune and nonimmune cell types also express and secrete GrB; however, the stimuli and signaling pathways regulating GrB release are largely unknown in these cell types.
GrB is present in the plasma of healthy individuals with median levels of approximately 20–40 pg/ml reported in the literature.54, 111 Serum levels of GrB are elevated in several diseases such as human immunodeficiency virus-1 infection, Epstein–Barr virus infection, arthritis, and others.54, 111, 114 Apart from the potential blood clotting implications that will be described later, it is worth noting that although circulating GrB may be useful as a biomarker for several diseases, it may not have a large effect on disease progression and may be present in the serum due to leakage from tissues where it is more abundant. In diseased tissues, particularly in areas of inflammation, extracellular granzyme concentration would be expected to be much higher than that in the blood. In these focal areas, GrB will cause the most damage due to the abundance of ECM substrates and the associated network of susceptible cells in tissue. In addition to plasma, GrB is also present in the synovial fluid (SF) of rheumatoid arthritis (RA) patients, the cerebrospinal fluid (CSF) of multiple sclerosis (MS) and Rasmussen encephalitis patients, as well as the bronchoalveolar lavage (BAL) in chronic obstructive pulmonary disease (COPD) and lung inflammation.122, 123, 124, 125 Although the GrB inhibitor PI-9 is present in normal human plasma, GrB retains 70% of its activity in the plasma, suggesting that PI-9 does not efficiently inhibit GrB activity in the blood.126, 127 There is a lack of evidence for an endogenous extracellular GrB inhibitor that is physiologically effective, thus its extracellular activity may be largely unregulated in contrast to other ECM proteases such as MMPs, which are tightly regulated by the tissue inhibitors of metalloproteases. This lack of extracellular regulation of GrB activity may have important implications with respect to a potential degenerative role for GrB in disease.
ECM Substrates
Granzymes were initially discovered as both intracellular and extracellular proteases, and since early the 1990's, several ECM substrates for GrB such as aggrecan, fibronectin, vitronectin, and laminin have been identified (Table 1).109, 110, 113 In terms of cleavage site identification, the lone cleavage site described thus far is for vitronectin.134 The cleavage site is in the RGD domain of vitronectin, which is an integrin-binding motif. GrB-mediated cleavage of this domain disrupts cellular–vitronectin interactions and influences cell adhesion and migration properties.134
Table 1.
Extracellular GrB substrates and receptors: implications of proteolysis
| Protein | Implications |
|---|---|
| Proteoglycans | |
| Aggrecan128 | Disruption of structural integrity in cartilage |
| Cartilage proteoglycans128 | Disruption of structural integrity in cartilage |
| Blood proteins/clotting | |
| von Willebrand factor129 | GrB cleavage site in the domain of platelet interaction, prevention/delay of thrombosis |
| Plasminogen130 | Cleavage yields angiostatin, which is anti-angiogenic. Implications in angiogenesis |
| Plasmin130 | As plasmin is pro-angiogenic, cleavage results in the reduction of angiogenesis |
| Cell receptors | |
| Neuronal glutamate receptor131 | GrB cleaves the non-glycosylated form of the receptor into an autoantigenic fragment |
| FGFR1132 | Cleavage activates pro-cell death functions as well as inactivates pro-growth signals |
| Notch1132 | Cleavage results in cell signaling affecting tumor survival and antiviral activities |
| Acetylcholine receptor133 | Cleavage results in a reduction of the receptor in neuromuscular junctions and yields an autoantigenic fragment |
| Other ECM components | |
| Vitronectin134 | GrB cleavage site in integrin-binding domain, implications in cell adhesion, migration, and anoikis |
| Fibronectin134, 135 | Cell adhesion, migration, and anoikis |
| Fibrinogen129 | Matrix form of fibrinogen is cleaved. The uncleaved protein responsible for platelet adhesion and thrombus growth. Cleavage results in anti-thrombosis implications |
| Laminin134 | Cell adhesion, anoikis |
| Smooth muscle cell matrix135 | Cell adhesion, anoikis |
In addition to ECM proteolysis, GrB can act on extracellular substrates involved in the clotting cascade such as plasmin, plasminogen, von Willebrand Factor (VWF), and the matrix form of fibrinogen.129, 130 GrB cleaves VWF in domains that are necessary for platelet interaction and cleavage prevents platelet aggregation, spreading, tethering, and adhesion to the VWF multimer.129 As GrB has a high affinity for ECM binding, Buzza et al129 suggest that it would accumulate in areas of inflammation and prevent/delay thrombosis in these areas. GrB-dependent cleavage of plasmin and plasminogen has implications outside clotting as degradation of these proteins may also inhibit angiogenesis.130
GrB can cleave cell surface receptors such as the neuronal glutamate receptor, Notch1, and FGFR1.131, 132 The cleavage of these cell surface receptors may have implications in cell signaling affecting tumor survival and antiviral activities. Cleavage fragments of receptors can also have physiological activities such as seen with the FGFR1 cleavage fragment, which can activate pro-cell death functions and inactivate pro-growth signals facilitated by FGFR1 in cancer.
Consequences of ECM Degradation
GrB-mediated ECM cleavage may contribute to disease not only through mechanical damage but also through other mechanisms (Figure 2). One consequence of ECM cleavage is anoikis, a form of cell death similar to apoptosis that is caused by a loss of cell–matrix interaction. Fibronectin, laminin, and vitronectin are adhesive proteins that are involved in connecting cells to the surrounding insoluble matrix. As all of these proteins are substrates for GrB, GrB-mediated cleavage may induce anoikis in various cell types. Choy et al135 described anoikis in cultured SMCs when treated with extracellular GrB that cleaved fibronectin in the matrix. They135 also showed that ∼30% of SMC death induced by CLs was perforin-independent but GrB-dependent, pointing toward anoikis as the mechanism of cell death. Buzza et al134 further investigated this by seeding endothelial cells on pure fibronectin, laminin, and vitronectin matrices and reported cell detachment and death by anoikis.
Figure 2.
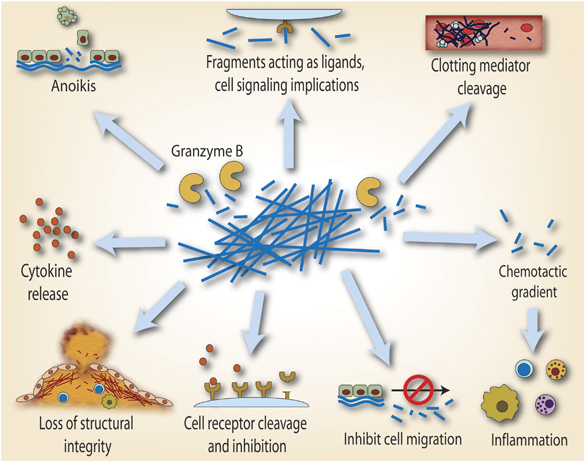
Putative extracellular (perforin-independent) roles for GrB in age-related chronic inflammatory disorders. During a number of chronic inflammatory conditions, GrB accumulates extracellularly in the tissues, blood stream, and other bodily fluids. GrB retains its activity in the blood, suggesting that, unlike MMPs and cathepsins, extracellular inhibitors of GrB activity may be limited. GrB can cleave proteins involved in structural integrity and wound healing such as fibronectin. GrB can also cleave proteins related to clotting (fibrinogen, vWF, plasminogen). GrB can induce detachment-mediated cell death (anoikis) through the cleavage of ECM. Although yet to be shown for granzymes, MMP-mediated fragments of fibronectin and elastin show chemotactic properties and may enhance the immune response in atherosclerosis. Fragments may also exhibit bioactive properties and may be able to release cytokines from the matrix. Granzymes may also have a role in the cleavage of cell surface receptors as seen with Notch1 and FGFR1.
GrB-dependent cleavage of vitronectin and fibronectin can also inhibit cell motility and migration.134 UVA stimulates keratinocyte motility; however, GrB expression during exposure inhibits this process, possibly protecting against carcinogenesis and the normal skin structure disruption that results from keratinocyte migration.136 An area poorly understood is whether GrB-generated ECM fragments can elicit chemotactic properties. GrB ECM cleavage fragments could possess chemotactic properties, leading to the recruitment of immune cells and promotion of inflammation.137 Indeed, fibronectin fragments can attract both neutrophils and monocytes.137, 138, 139, 140 Fibronectin fragments also have other properties such as inducing MMP expression by chondrocytes.140 ECM fragments may also act as signaling molecules in neighboring resident cells, as mentioned previously with the FGFR1 fragment.132 As the ECM has affinity for and serves as a reservoir for many growth factors and cytokines, the disruption of the ECM by GrB could induce the release of these growth factors and influence surrounding cells in an indirect manner.
GrB IN DISEASE
GrB has the capacity to act on both intracellular and extracellular substrates. Recently, with the discovery of new immune and nonimmune cell sources of the protease, its importance in infection and cancer is evolving while its contribution to other chronic diseases is emerging. Infiltrating immune cells during chronic inflammation results in elevated levels of GrB to diseased tissue and induces apoptosis in damaged and inflamed areas. Extracellular concentrations of GrB in bodily fluids are elevated in various diseases, and the extracellular activity of this protease in chronic inflammation is an emerging area of research. The remainder of the review will summarize and critically review the current knowledge pertaining to the intracellular and extracellular roles for GrB in inflammatory diseases. As there have been many excellent reviews written pertaining to the role of GrB activity in infection and cancer,21, 141, 142 we will focus on chronic diseases where the pathogenic role of GrB is emerging (Table 2 and Figure 3).
Table 2.
GrB in disease
| Condition | Intracellular versus extracellular | Description |
|---|---|---|
| Lung diseases | ||
| Chronic obstructive pulmonary disease (COPD)18, 143, 144 | Intracellular/ extracellular | Increased CTLs and NK cells expressing GrB in the blood and BAL of patients with COPD. Type II pneumocytes and alveolar macrophages in the lung express GrB. Increased perforin expression by CD8+ cells in the lung of smoking subjects with COPD |
| Asthma17, 122 | Intracellular/ extracellular | Increase in lymphocytes expressing GrB in the BAL fluid of patients suffering from allergic asthma after allergen challenge. Induction of GrB expression by basophils upon stimulation with IL-3 released by mast cells |
| Acute respiratory distress syndrome (ARDS)145 | Intracellular/ extracellular | GrB and perforin mRNA levels are upregulated in the BAL of patients in the acute phase of ARDS |
| Pulmonary sarcoidosis146, 147 | Intracellular/ extracellular | GrB and perforin are expressed by CD8+ and some CD4+ T cells in the BAL fluid. Serum levels of GrB are decreased in patients with sarcoidosis |
| Hypersensitivity pneumonitis125 | Extracellular | Granzyme B is increased in the BAL fluid of patients with hypersensitivity pneumonitis |
| Oral | ||
| Chronic obstructive sialadenitis/ sialolithiasis148 | Intracellular(?) | GrB is expressed by periductal and periacinar lymphocytes in patients with chronic sialadenitis |
| Papillon–Lefèvre syndrome (PLS)149, 150 |
NK cells in patients with PLS fail to induce the caspase cascade in target cells because of an inactive form of GrB as a result of a mutation in cathepsin C Reduced active GrB in cytotoxic cells in patients with PLS compared with controls |
|
| Blood disorders | ||
| Aplastic anemia151, 152 | Intracellular/ extracellular |
No difference in GrB-expressing cytotoxic effector cells in disease patients compared with controls Increase in perforin but no increase in GrB in bone marrow clot sections of disease patients compared with controls |
| Idiopathic neutropenia153 | No difference in GrB-expressing CD16+ cells in the blood between patients with chronic idiopathic neutropenia | |
| Chronic idiopathic thrombocytopenic purpura (ITP)154, 155, 156 | Intracellular(?) |
Increased GrB-expressing T cells in the blood of patients with ITP compared with controls Increased GrB mRNA levels in CD8+ cells in patients with ITP compared with controls |
| Skin diseases | ||
| Alopecia157, 158, 159 | Intracellular | GrB-expressing CTLs are closely associated with hair follicles and may damage follicles. Substance P increases CD8+ T cells expressing GrB, which may cause hair follicle regression |
| UV photoaging11, 136, 160 | Intracellular/ extracellular | GrB is expressed by keratinocytes in response to confluence, UVA, and UVB. UVB-treated keratinocytes have cytotoxic potential against co-cultured cells and GrB from UVA-treated keratinocytes can cleave fibronectin |
| Acne161 | GrB is upregulated in acne lesions | |
| Atopic dermatitis/allergic contact dermatitis162, 163, 164, 165 | Intracellular | GrB-expressing CD4+ and CD8+ T cells are observed in the perivascular infiltrate and focally at spongiosis sites. In contact dermatitis, keratinocytes neighboring GrB-expressing cells are damaged |
| Vitiligo166, 167 | Intracellular | GrB-expressing CTLs cluster around disappearing melanocytes and may induce apoptosis in these cells |
| Lichen planus165, 168, 169, 170, 171, 172 | Intracellular | GrB-expressing cells are found in close proximity to apoptotic keratinocytes. DCs expressing GrB are found in lesions |
| Lichen sclerosus173, 174, 175 | GrB is expressed in dermal infiltrate close to keratinocytes. Vasculitis associated with the disease contains GrB-positive cells in the perivascular infiltrate | |
| Stevens–Johnson syndrome/ toxic epidermal necrolysis176, 177, 178, 179, 180 | Intracellular/ extracellular | CTLs expressing GrB may induce apoptosis in keratinocytes. GrB-positive lymphocytes in blister fluid. GrB upregulation correlated to disease severity |
| Pityriasis rosea162 | GrB is expressed by immune cells in pityriasis rosea lesions | |
| Psoriasis163, 164 | GrB is expressed by some lymphocytes in psoriasis lesions | |
| Bullous blistering skin lesions181 | GrB is expressed in bullous lesions by T cells | |
| Discoid lupus erythematosus165, 182 | GrB is expressed on lesional lymphocytes expressing the skin-homing proteins CLA and MxA. GrB-positive cells are perivascular and located in the dermoepidermal junction | |
| Pemphigus vulgaris (PV)183 | Decreased ex vivo expression of GrB by circulating NK cells in patients with PV compared with controls | |
| Bones and joints | ||
| Rheumatoid arthritis (RA)13, 14, 54, 114, 115, 128, 184, 185, 186, 187, 188, 189, 190, 191 | Extracellular/ intracellular | GrB cleaves aggrecan and other cartilage components. Levels of GrB are markedly elevated in the synovial fluid and plasma of patients. All GrB-expressing cell types may also contribute to RA through GrB-mediated apoptosis, including chondrocytes that show the surface antigens of NK cells |
| Osteoarthritis187 | mRNA and protein expression of GrB in the synovium of joints | |
| Reactive arthritis192 | Extracellular | GrB expressed in the synovial tissue |
| Neurological disorders | ||
| Rasmussen's encephalitis (RE)124, 131, 193, 194, 195, 196 | Intracellular/ extracellular/ autoimmunity | GrB-expressing CTLs described in RE brains. GrB from CTLs is polarized toward neurons and astrocytes that express MHC I. Extracellular GrB levels in cerebrospinal fluid (CSF) are elevated. GrB cleaves the GLUR-3 receptor yielding an autoantigenic fragment |
| Multiple sclerosis (MS)123, 197, 198, 199 | Intracellular/ extracellular | CTLs are involved in neuronal toxicity and TH17 cells, which cross the blood–brain barrier, express GrB, and can kill neurons in vitro. Increase in extracellular GrB levels in the CSF in relapsing remitting MS |
| Guillain–Barré syndrome200 | Intracellular | GrB-expressing CTLs are increased and MHC I-expressing Schwann cells may be GrB targets. Implications in myelin sheath damage |
| Vasculitic neuropathy201 | GrB is expressed in the peri-vascular infiltrate | |
| Sensory perineuritis202 | Intracellular | GrB-expressing CTLs contribute to perineurial cell apoptosis |
| Ischemic stroke203 | Intracellular | GrB from CTLs and NK cells induce apoptosis of brain cells |
| Spinal cord injury204 | Intracellular | GrB levels are elevated and CTLs in close proximity to neurons in regions of damage |
| Myesina gravis133 | Extracellular/ autoantigen | GrB cleaves the autoantigen AChR. GrB is present in myasthenia gravis thymus glands but absent in controls |
| Autoimmune disease | ||
| Systemic lupus erythematosus (SLE)205, 206, 207, 208, 209 | Intracellular/ autoimmunity | Frequency of GrB-expressing CTLs coincides with disease progression. GrB is involved in autoantigen processing of XRCC4 and other potential SLE autoantigens |
| Neonatal lupus erythematosus210 | GrB expression in the left ventricle of hearts from fetuses/infants with complete atrioventricular block | |
| Scleroderma (SSc)130, 205, 211, 212 | Autoimmunity/ extracellular | GrB cleaves the autoantigens topoisomerase I, NOR-90, fibrillarin, B23, and others. SSc patients with ischemic digital loss have autoantibodies for CENP-C, which may be useful as biomarkers for IDL. The GrB cleavage product angiostatin inhibits angiogenesis and may be responsible for the poor circulation in SSc |
| Sjögren syndrome (SS)205, 213, 214, 215, 216, 217, 218, 219, 220 | Intracellular/ autoimmunity | GrB cleaves the autoantigens SS-B (La) autoantigen, α-fodrin, β-fodrin, type 3 muscarinic acetylcholine receptor, and others. CD4+ and CD8+ T cells induce apoptosis of epithelial cells through the granule pathway and these cells are only present in SS glands |
| Myositis205, 221, 222, 223, 224 | Intracellular/ autoimmunity | GrB cleaves autoantigens such as PMS-1 and HisRS. GrB-expressing cells are found in the endomysial sites of polymyositis and are proposed to cause muscle cell damage |
| Type 1 diabetes225 | Intracellular | Human and mouse β-cells undergo apoptosis in the presence of GrB, which correlates with a loss in islet insulin secretion capacity |
| Bile/liver/intestinal diseases | ||
| Lymphocytic gastritis (LG)226, 227, 228 | Intracellular? |
Intraepithelial CD8+ cells from LG children with celiac disease lack GrB Increase in GrB expressing intraepithelial lymphocytes in patients with acute gastric mucosal lesions compared with controls Increased GrB expressing intraepithelial lymphocytes in patients with non-celiac disease associated LG compared with patients with celiac disease associated LG |
| Autoimmune cholangitis (AC) and primary biliary cirrhosis (PBC)229 | GrB expressing T cells found in the bile duct epithelium. No difference in the number of GrB-expressing lymphocytes between patients with AC versus PBC | |
| Nodular regenerative hyperplasia (NRH) of the liver230 | Increased CD8+ lymphocytes expressing GrB in liver biopsy samples from patients with NRH compared with controls. | |
| Inflammatory bowel disease231 | Increased GrB-expressing intraepithelial lymphocytes in patients with Crohn's disease and ulcerative colitis | |
| Vascular diseases | ||
| Atherosclerosis10, 135, 189, 232, 233, 234, 235, 236 | Extracellular/ intracellular |
GrB levels increase with increased disease severity. Present in high levels in advanced atherosclerotic and TVD plaques. Elevated in lipid-rich regions Granzyme B in the blood is significantly higher in patients with unstable atherosclerotic plaques. The study also showed that raised plasma levels of GrB in unstable carotid plaques were associated with an increased frequency of cerebrovascular events (ie, strokes), suggesting that GrB may be a marker of plaque instability GrB expressed in macrophages in atherosclerotic plaques Perforin deficiency in LDLR-KO mice does not affect atherosclerosis. Supports role for extracellular GrB or its role in late-stage/advanced atherosclerosis GrB in the absence of perforin can induce smooth muscle cell apoptosis through the cleavage of extracellular matrix proteins. Fibronectin identified as a substrate |
| GrB production from PBMCs of unstable angina pectoris (UAP) patients was significantly higher than those with stable angina (SAP). GrB production from PBMCs increased with the increasing TIMI risk score in UAP patients. The percentage of GrB-positive lymphocytes to CD3-positive lymphocytes in UAP patients was significantly higher than in SAP | ||
| Acute transplant rejection237, 238, 239, 240, 241, 242, 243, 244, 245 | Intracellular | GrB-mediated apoptosis that occurs during the recruitment of inflammatory cells after the nonspecific injury to graft vessels. GrB can contribute to lesion formation through processes that include GrB-mediated apoptosis, and the promotion of EC activation and SMC migration |
| Allograft vasculopathy10, 246, 247 | Intracellular | GrB/perforin pathway involved in endothelial and smooth muscle cell apoptosis |
| Kawasaki disease248, 249, 250 | Extracellular | Children with Kawasaki's show elevated vascular inflammation and often die from fatal aortic dissections or aneurysms. Elevated levels observed in lesions, aneurysms, and plasma of Kawasaki's patients; however, its involvement of GrB disease pathogenesis requires further elucidation |
| Kidney diseases | ||
| Crescentic glomerulonephritis (CG)251 | Intracellular | Perforin-neutralizing antibody protects against the progression of CG in rats |
| Goodpasture's disease (GD)252 | Glomerular GrB expression and GD pathogenesis is reduced upon administration of anti-CD8+ antibody | |
| Esophagus | ||
| Achalasia253 | The inflammatory infiltrate found in the myenteric plexus contains cytotoxic T cells, some of which express GrB | |
| Esophagitis254 | Significant increase in GrB-expressing intraepithelial lymphocytes in biopsy specimens from patients with esophagitis compared with controls | |
| Crohn's disease255 | Increase in GrB-expressing cells in esophagus biopsy specimens taken from patients with CD compared with controls | |
| Other | ||
| Eosinophilic fasciitis256 | The inflammatory infiltrate in eosinophilic fasciitis contains some CD8+ cells expressing GrB, suggesting a cytotoxic immune response | |
| Cryptorchidism257 | Lymphocytes expressing GrB are decreased in the testis of patients with cryptorchidism | |
| Histiocytic-necrotizing lymphadenitis (HNL)/Kikuchi disease258, 259 | Intracellular/ apoptotic necrosis |
GrB-expressing cells found in necrotizing lesions of patients with Kikuchi disease The majority of lymphocytes found in the necrotic foci in HNL are GrBexpressing CD8+ cells |
| Chediak–Higashi syndrome260 | CTL granules are unable to release their contents upon recognition of a T-cell receptor, express normal levels of GrB | |
| Duchenne muscular dystrophy (DMD)/facioscapulohumeral dystrophy (FSHD)261 | Intracellular | GrB expression detected in muscle biopsy specimens from patients with DMD and FSHD but absent in control samples |
Figure 3.
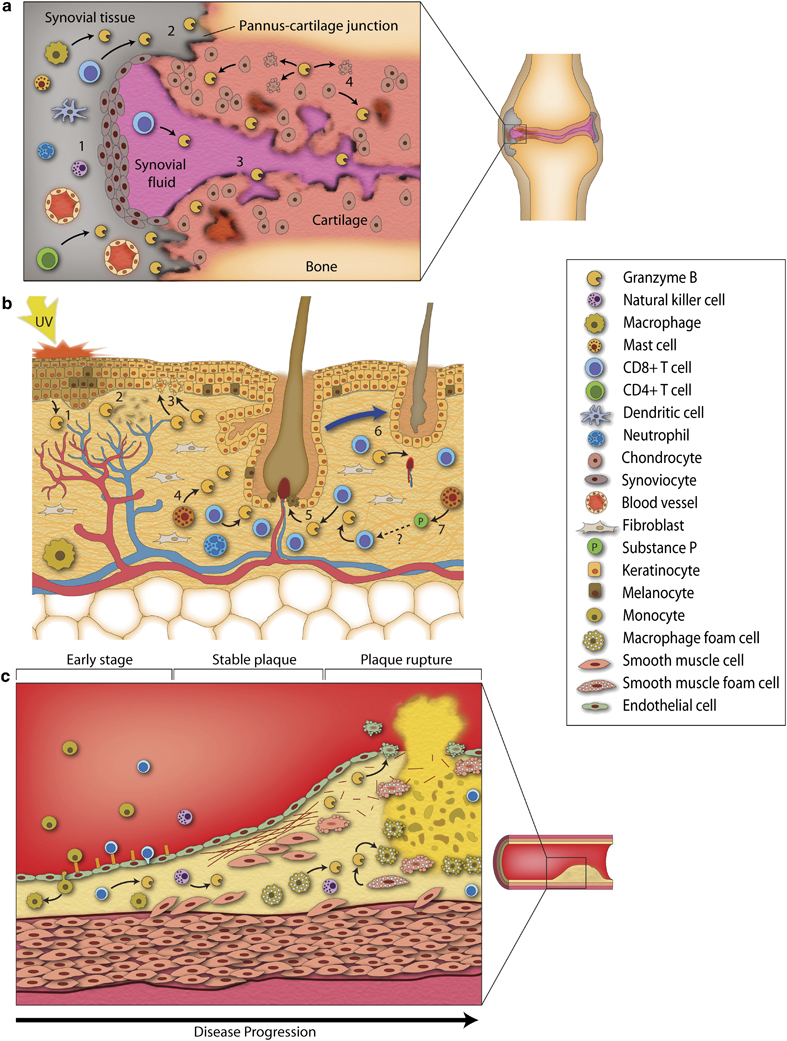
Granzyme B in the pathogenesis of rheumatoid arthritis, skin disease, and atherosclerosis. (a) Infiltrating immune cells in RA (CTLs, macrophages, NK cells, and T-helper cells) express and release GrB in joints and induce apoptosis in resident cells (1). GrB may contribute to proteoglycan degradation as GrB-positive cells are found at the pannus–cartilage junction, an area of cartilage destruction (2). Extracelllular GrB levels are elevated in the synovial fluid of RA joints and are believed to further degrade matrix (3). Chondrocytes express GrB in RA and are capable of inducing apoptosis in neighboring cells and secreting GrB into the extracellular milieu, which causes further extracellular damage (4). (b) GrB may contribute to skin aging, alopecia, and disease through various intracellular and extracellular pathways. UVA light, which is believed to be responsible for visible aging that occurs in the skin, induces reactive oxygen species production, which leads to GrB expression in keratinocytes (1). GrB from keratinocytes cleaves the ECM protein fibronectin (2), inhibits cell migration, and can also induce apoptosis in neighboring cells (3). Mast cells from the skin are another cell type that express GrB (4). GrB from CTLs has been proposed to induce melanocyte apoptosis in vitiligo (5). Invading CTLs express GrB in alopecia areata and influence hair follicle regression (6). Substance P secreted by skin mast cells increases GrB-positive CTLs in the skin, further promoting AA (7). Whether substance P induces mast cell degranulation leading to the release of GrB is unknown. (c) At the early stages of atherosclerosis, after endothelial dysfunction and intimal lipid retention, CTLs, and monocytes infiltrate the vessel wall and migrate into the intima. SMCs and macrophages engulf oxidized lipoproteins and become lipid-laden foam cells, leading to GrB expression in these cells. The latter may promote foam cell apoptosis in developing plaques. GrB can also cleave various extracellular matrix proteins that maintain fibrotic cap stability. In addition to a loss in matrix integrity, ECM cleavage will also result in a loss of SMC–ECM interactions, which may promote apoptosis. SMC expression of PI-9 is decreased in unstable plaques, rendering SMCs susceptible to GrB-dependent apoptosis and further promoting plaque instability and rupture.
GrB in Autoantigen Production
Autoimmunity results when the body does not recognize its own proteins as self, and as a result, it mounts an immune response against itself. The process of apoptosis has been intricately described as key initiator of autoimmune responses and autoimmune disease. During apoptosis, autoantigens from various subcellular compartments are clustered in surface blebs and apoptotic bodies of dead cells, in which they are highly accessible to the immune system.262 These peptides are captured and presented to immune cells by antigen-presenting cells, which drive the autoimmune response.
This process may occur during immune cell-mediated apoptosis, in which enzymes such as caspases and GrB are responsible for uncovering cryptic epitopes of intracellular proteins from the proteolytic cleavage of cellular substrates. Some of these hidden epitopes are not present during immune cell development and as a result the immune system may not always tolerate them (ie, recognize them as self-proteins), thus they become autoantigenic and are identified as foreign by the immune system.
Recently, it has been shown that some of these intracellular-derived autoantigens are cleaved by GrB during CTL-induced apoptosis. Furthermore, many of these cleavage fragments are unique to GrB and are not synthesized in other forms of cell death. Casciola-Rosen et al205 have described some of these GrB-specific autoantigens in great detail and several disease-specific examples are provided in this review. Of 29 well-defined autoantigens, 21 in systemic lupus erythematosus (SLE), scleroderma (SSc), Sjögren syndrome (SS), and myositis are directly cleaved by GrB into unique autoantigenic fragments. Non-autoantigens were either not cleaved by GrB or cleaved into fragments identical to those cleaved during other forms of apoptosis and as a result are not unique. This suggests these non-unique fragments were already tolerized during immune cell development and are not autoantigenic, whereas the uniquely cleaved autoantigens would have escaped tolerance.205 As GrB appears to selectively and uniquely generate a wide variety of autoantigens in several autoimmune diseases and as there is a high-titer autoantibody response to most of these antigens in autoimmune patients, GrB has been identified as a critical enzyme in autoimmune disease pathogenesis.205, 263
SLE
SLE is a disease involving a specific autoimmune response toward nuclear autoantigens. High amounts of soluble nucleosomes have been found in SLE serum and SLE macrophages have reduced phagocytic activity, suggesting there is a defect in the clearance of apoptotic cell material.264, 265 This may allow for a high availability of autoantigens for DCs to process and present, as this cell type is believed to be a driving force in SLE pathogenesis.266 In addition to DCs, CTLs are an important cell type in SLE autoimmunity. Effector CTLs from patients with SLE disease flares show greater perforin/GrB immunopositivity and the frequency of these cells coincides with disease progression. These CTLs also generate soluble nucleosomes and GrB-specific autoantigens, suggesting they contribute to the initiation of autoimmunity. Interestingly, in the presence of SLE serum, DCs can facilitate CTL differentiation into active effector cells, which are no different from those in SLE patients, providing further evidence that DCs are capable of presenting autoantigens to CTLs and inducing their activation in an autoimmune response.206
GrB has been implicated as a key protease in autoantigen processing in SLE and generates fragments unique to those of other autoimmunity-associated proteases such as the caspases. For example, GrB is capable of producing autoantigens from the non-homologous end-joining pathway of DNA double-strand break repair. Sera from SLE patients contained antibodies against XRCC4, and three of these patients recognized the specific GrB cleavage epitope, suggesting GrB may produce these fragments in vivo.207 However, in a mouse model of autoimmunity in which GrB-deficient mice were injected with the mineral oil pristane, GrB knockout mice still produced autoantibodies and had a higher mortality than mice expressing GrB, suggesting that GrB may not be a critical enzyme in autoantigen synthesis. However, the latter study would not rule out the possibility that the other granzymes may be contributing to autoantigen formation in this model. In addition, the authors suggest that GrB may be protective because of its role in viral clearance, as viral infection is believed to promote autoimmunity.208, 267
SSc
SSc or systemic sclerosis is an inflammatory connective tissue disease that affects the vasculature and results in blood flow reduction, tissue ischemia, and wound healing defects. GrB cleaves several SSc autoantigens including topoisomerase I, NOR-90, fibrillarin, and others.205 Some SSc patients also develop complications from the disorder, such as ischemic digital loss (IDL).211 Serum samples from SSc patients with IDL contain autoantibodies for the specific GrB cleavage fragments of CENP-C, pointing toward GrB as a mediator of IDL.211 Moreover, as there is a need for predictive biomarkers for digital loss in SSc, specific IDL autoantigens cleaved by GrB maybe a useful tool for diagnosis.
In addition to autoimmunity, extracellular cleavage fragments created by GrB also have other functional roles in SSc. The reduction of blood flow characteristic of SSc may be due to an imbalance between proangiogenic and antiangiogenic factors that control new vessel growth. Plasma from SSc patients shows reduced endothelial cell migration and tube formation in vitro. The serum sample was found to contain higher levels of the antiangiogenic protein angiostatin, which Mulligan-Kehoe et al130 found to be synthesized by GrB through the cleavage of plasminogen (in the presence of another granule protein) as well as from plasmin. As plasmin is proangiogenic in nature, GrB may also be responsible for decreasing levels of plasmin in SSc serum, thereby decreasing angiogenesis through the synthesis of angiostatin and the degradation of plasmin.130
Interestingly, autoantigen release may be dependent on cell type in SSc. B23 is resistant to GrB-mediated cleavage in many cell types but appears to be exclusively cleaved in vascular SMCs.212 Whether this cell-type exclusiveness in SMCs is due to a cell-specific abnormality of this protein (ie, post-translational modification, etc.) is unknown. Regardless, the selective production of this autoantigen and potentially others in vascular cells may contribute to the extensive vascular pathogenesis apparent in SSc.
SS
SS is a systemic autoimmune disease characterized by chronic inflammation in the salivary and lacrimal glands with focal infiltrate around ductal and acinar epithelial cells. This infiltrate interacts with epithelial cells and induces apoptosis. CD4+ and CD8+ T cells induce apoptosis in epithelial cells through the Fas/FasL and perforin/GrB pathways.213 In support of this notion, perforin/GrB-expressing infiltrate was found only in SS salivary glands and not in controls, suggesting that GrB is a culprit in epithelial cell death.214 The majority of cells surrounding apoptotic acinar epithelial cells are CD8+ T cells expressing the integrin αEβ7, suggesting that GrB from these integrin-expressing T cells may be involved in the pathogenesis of SS.215
During epithelial cell apoptosis, GrB may cleave several autoantigens in SS, namely the SS-B (La) autoantigen, α-fodrin, β-fodrin, and type 3 muscarinic acetylcholine receptor.216, 217, 218 GrB-mediated cleavage of the La protein resulted in its translocation from the nucleus into the cytoplasm, where the autoantigen is easily accessible to antigen-presenting cells and can initiate an immune response.219 As such, GrB may not only synthesize autoantigens in SS but may also render them more readily accessible for immunorecognition.
Myositis
Myositis is an autoimmune inflammatory skeletal muscle disease resulting in muscle weakness. Areas of muscle damage show a characteristic mononuclear cell infiltrate consisting of T lymphocytes and NK cells, which express major histocompatability complex II (MHC-II) molecules. These cells invade and destroy muscle fibers, leading to muscle fiber necrosis, potentially through muscle cell MHC-I. GrB- and perforin-expressing cells were found in the endomysial sites of polymyositis (PM) and may cause muscle cell damage, apoptosis, and necrosis in PM.221 However, although there is a significant amount of inflammation and cytotoxic molecules in PM, muscle cells are generally resistant to apoptosis, which may be linked to their expression of human IAP-like protein, a cell death repressor.222
In addition to its potential role in muscle cell integrity, GrB may be a source of autoantigen production in myositis through the processing of the DNA mismatch repair enzyme PMS-1 protein fragment, as 7.5% of myositis patient serum tests positive were for the autoantibody.268 The sera from myositis patients also recognized other unidentified protein fragments, suggesting there may be several more unidentified autoantigens derived from GrB cleavage in myositis.
Myositis patients can also develop interstitial lung disease and Levine et al223 showed that this lung pathogenesis may be due to a difference in the protein structure of the autoantigen histidyl-transfer RNA synthetase (HisRS). The conformation of the lung-specific HisRS makes it more susceptible to GrB cleavage compared with the normal HisRS conformation present in the rest of the body. This lung-specific conformation exposes a cryptic GrB cleavage site, resulting in HisRS fragmentation and GrB-dependent synthesis of an autoantigenic peptide, specific to the lung.223 It is interesting to speculate that GrB-specific autoantigens are cleaved when protein conformation is altered, thereby uncovering otherwise hidden GrB cleavage sites. Protein conformational changes due to post-translational modifications, pH changes, mechanical damage, dissociation from binding partners, changes in subcellular localization, and other factors may have a role in the susceptibility to GrB cleavage of not just autoantigens but also to intracellular and extracellular substrates involved in a vast array of activities, having endless potential implications in signaling, apoptosis, and extracellular activities in vivo.
RA
RA is a chronic autoimmune inflammatory disease involving tissue destruction in joints and tendons. GrB has been implicated intracellularly and extracellularly in this disease through apoptosis and cartilage matrix destruction (Figure 3a). GrB is present in several cell types in areas of inflammation as well as in regions of matrix disruption, making it a potentially important mediator of RA pathogenesis.
The expression of GrB by activated CTLs in the SF from RA patients was first described by Young et al,184 with ∼15% of SF lymphocytes expressing GrB. Since then, soluble extracellular GrB has been reported as increased in the SF and plasma of patients with RA, with levels averaging as high as 3 and 1 ng/ml, respectively.54, 114 The presence of extracellular GrB in the SF of RA complements the study by Froelich et al128 showing that GrB degrades the ECM synthesized by chondrocytes in vitro. One of these substrates was identified as aggrecan, a major constituent of cartilage; however, no other substrates have been identified to date. Fibronectin, as well as bioactive fibronectin fragments, has also been reported in inflammatory arthritis SF.137, 140, 269, 270 Fibronectin fragments are chemotactic for macrophages and neutrophils, and these fragments can release proteoglycans and cause chondrolysis by binding to cartilage.271, 272 However, despite the fact that fibronectin is a well-established substrate of GrB, no one has reported whether extracellular GrB activity contributes to the synthesis of these fragments.
Froelich et al128 proposed that CTLs secrete GrB extracellularly in the joint, which degrades and remodels the interstitial ECM. Complementary to this study is that of Ronday el al185 showing that GrB cleaves proteoglycans from bovine cartilage explants, resulting in glycosaminoglycan release. GrB-positive cells were identified at the invasive front of the inflamed synovial tissue (pannus) and in the cartilage–pannus junction, suggesting that GrB may be responsible for the cartilage invasion and destruction that occurs in this junction.185, 186
In addition to CTLs, other cell types may act as sources of GrB in RA. GrB-expressing NK cells are present in the synovial tissue of RA patients and these cells are the main GrB-expressing cell type in the RA joint.187, 188 Macrophages express GrB in the lining and perivascular areas of the synovial tissues of RA joints. Macrophage numbers are increased in RA compared with normal joints, suggesting that macrophages are also a significant source of GrB, and may contribute to cartilage destruction.189 Although CD4+ T cells are believed to act largely as T-helper cells in RA, CD4+CD28− cells have been suggested to show cytotoxicity. Unlike their CD28+ counterparts, which express GrB but not perforin, CD4+CD28− T cells express both perforin and GrB and show cytotoxic activity in vitro. CD4+CD28− perforin-expressing T cells account for up to 50% of CD4+ T cells in synovial tissue, suggesting they may be a significant source of GrB in RA.14 Mast cells, DCs, and neutrophils are other cell types upregulated in RA, which express GrB under specific circumstances.15, 20, 28 However, it is unknown whether these cell types express GrB in RA.
GrB and perforin are also expressed by articular chondrocytes as GrB and perforin transcript/protein levels are increased in diseased RA cartilage, as detected by semiquantitative RT-PCR and immunostaining.13, 115 Increased GrB and perforin levels in these cells correspond to chondrocyte apoptosis, particularly in the pannus region invading the cartilage, suggesting that self-synthesized GrB may induce self-regulated apoptosis. Cultured chondrocytes also express the surface antigens of NK cells and show cytotoxicity against co-cultured cells, suggesting that in addition to having a self-inducing-apoptosis capacity, chondrocytes may also act as resident cytotoxic cells in the cartilage of RA patients.115 Finally, secreted GrB from chondrocytes may have cartilage degradation potential as well; suggesting that GrB from this cell source has both intracellular and extracellular implications.13
Despite the fact that RA is an autoimmune disease and that increased levels of GrB are associated with rheumatoid factor autoantibody-positive patients, little is known regarding the GrB generation of RA-specific autoantigens.190 A rat chondrocyte cDNA library was immunoscreened with serum from an RA patient and the DNA-binding protein AHNAK was detected as a potential autoantigen. This protein is cleaved in vitro by GrB, suggesting that it may be involved in the autoantigen processing of this protein.209 GrB-mediated autoantigen processing could be another mechanism by which GrB may contribute to the pathogenesis of RA, as little study has been done thus far in this area.
Type I Diabetes
Type I diabetes results from the selective destruction of the insulin-producing β-cells in pancreatic islets, and is primarily a T-cell-mediated autoimmune disease directed against one or more β-cell autoantigens.273 Lymphocyte infiltration (termed insulitis) occurs early in models of type I diabetes and is necessary but not sufficient to cause β-cell destruction, insulin deficiency, and hyperglycemia.274 It is now accepted that β-islet destruction is caused by islet antigen-specific autoreactive T cells and that both CD4+ and CD8+ T cells are required for the disease to occur (reviewed in refs. 273,275,276) Although still an area of active investigation, β-cells are believed to be destroyed primarily by granule-mediated apoptosis.274, 276, 277, 278, 279, 280, 281
Mouse models of diabetes have provided support for T-cell-induced cell death with β-cell apoptosis observed in both spontaneous diabetes in non-obese diabetic mice277, 279 and in more accelerated diabetes models.277, 278 Furthermore, cell death mediators, including perforin, cytokines, and death ligands, have been identified in the insulitis lesions of NOD mice.282 Both CD4+ and CD8+ T-cell subsets are required for the development of diabetes in the NOD mouse; however, CD8+ T effector cells have a fundamental role in the destruction of pancreatic β-cells and contribute to sustaining islet inflammation.283, 284 Pancreatic β-cells express MHC-I molecules that present antigenic peptides to CD8+ T cells,282 and NOD mice lacking MHC-I expression and CD8+ T cells are resistant to type I diabetes.285, 286 Perforin has a key role in β-cell apoptosis in the NOD model, as perforin-deficient NOD mice are reported to develop insulitis but have a markedly reduced incidence of diabetes.75, 287 This implies that CTLs destroy β-cells, at least in part, by granule-mediated apoptosis. Support for GrB in β-cell damage is provided by in vitro studies indicating that both human and mouse β-cells undergo apoptosis in the presence of GrB, which correlates with a loss in the islet insulin secretion capacity.225
Lastly, there is evidence that GrB has a predominant role in the destruction of β-cells after pancreatic islet transplantation, with several recent studies showing elevated GrB levels in plasma preceding islet graft rejection.288, 289 This suggests that GrB may be a reliable indicator of ongoing graft loss after β-islet transplantation. Evidence from both human and mouse models has shown that β-cell death observed in diabetes occurs through apoptosis and that granule-mediated cytotoxicity is involved in β-cell loss. In summary, although studies to date would suggest that the GrB/perforin pathway contributes to β-cell apoptosis, it remains to be determined whether there is an extracellular role for GrB in diabetes. Whether GrB has a role in type 2 diabetes remains unclear.
Neurological Disorders
The central nervous system is regarded as an immune-privileged site because of the blood–brain barrier and an immunosuppressive environment. However, in many neurological conditions, there is a loss of immune privilege and, as a result, GrB has been implicated in the pathogenesis of a number of neurological disorders through mechanisms of immune cell-mediated apoptosis, receptor cleavage, autoantigen synthesis, and potential extracellular activities, as described below.
There are several neurological diseases where the role of GrB is just emerging, largely in an immune cell-mediated, pro-apoptotic role. In vasculitic neuropathy, the characteristic perivascular infiltrate expresses GrB and GrB-positive cells are significantly upregulated.201 In sensory perineuritis, GrB-expressing infiltrates were detected and T cells are believed to contribute to perineural cell apoptosis.202 During cerebral ischemia, in a rat model of ischemic stroke, GrB secreted from CTLs and NK cells results in the breakdown of Hsp-70 and AIF translocation from the mitochondria to the nucleus, ultimately resulting in apoptosis of brain cells.203 Finally, in a rat model of spinal cord injury, GrB levels were elevated and CTLs were found in close proximity to neurons in damaged regions of the spinal cord. Many of the neurons undergoing death were positive for GrB, suggesting that GrB is responsible for neuron cell death in spinal cord injury.204
In addition to inducing apoptosis intracellularly, GrB may contribute to neurological disease through other mechanisms, such as through receptor signaling or through direct cleavage of receptors. In myasthenia gravis (MG), a decrease in acetylcholine receptors (AChR) at neuromuscular junctions is observed as a result of autoimmune attack. Furthermore, GrB and the autoantigen fragments of AChR are observed in the thymus of MG patients but are not present in healthy controls. Casciola-Rosen et al133 showed that GrB cleaves AChR, suggesting that GrB-mediated cleavage of this protein exposes cryptic antigens that facilitate an autoimmune response in the disease and results in a loss of functional AChR. G-protein-coupled receptors are also linked to GrB and neurotoxicity. The addition of recombinant GrB to neuronal cell cultures results in neurotoxicity, even in the absence of perforin through the Giα-coupled receptor.290 Whether GrB facilitates cytotoxicity by cellular interaction or uptake, by cleaving the receptor itself, or by creating an ECM fragment that may act as a ligand is not known.
Rasmussen's encephalitis
Rasmussen's encephalitis (RE) is an autoimmune chronic inflammatory disease occurring mainly in childhood, characterized by seizures and a loss of motor skills and speech. Immune infiltrates consisting mainly of GrB-positive CTLs are found in the brains of these patients. Using confocal microscopy, Bien et al193 found that GrB was polarized toward neurons, some of which expressed MHC-I. This suggests that MHC-I-initiated GrB-mediated cytotoxicity is responsible for the loss of neurons in this disease. In similar studies, Bauer et al described GrB-positive lymphocytes in close proximity to astrocytes on the border of astrocyte-deficient lesions. Some of these cytotoxic cells showed GrB polarized toward astrocytic membranes. Similarly to neurons, astrocytes express MHC-I, suggesting that GrB is contributing to astrocyte loss in RE.194 In another study, Vβ T-cells expressing GrB were found in close apposition to neurons and astrocytes in patients, further supporting the aforementioned studies.195
Extracellular GrB activity has also been linked to RE as extracellular GrB levels in CSF are elevated in patients with RE, with the original cellular source believed to be CTLs.124 It is possible that an elevation in extracellular GrB could be used as a biomarker for RE, as it is present at earlier stages of the disease and remains slightly elevated as the disease progresses. Extracellular GrB is capable of cleaving the neuronal glutamate receptor subunit 3 (GLUR3) and producing an autoantigenic fragment. Antibodies against GLUR3 are present in the serum of children with this disease. However, GrB can only cleave the non-glycosylated form of the receptor, suggesting that normal tolerance is escaped when neurons produce the unglycosylated form of the protein. The change in glycosylation of this protein makes it more susceptible to GrB cleavage, possibly by exposing a cryptic cleavage site not present in the glycosylated form of the receptor.131 This suggests that GrB cleavage of ECM and receptors may also be regulated by post-translational modifications, such as glycosylation, an area that has been scarcely described and may be significant in disease pathogenesis in general.
Demyelination diseases: MS and Guillain–Barré syndrome
MS is a complex autoimmune disease of the central nervous system resulting in demyelination of nerves and GrB has been implicated in MS pathogenesis. In patients with relapsing remitting MS, GrB levels in peripheral T cells decrease, whereas plasma GrB levels do not change. However, there is an increase in extracellular GrB in the CSF as quantified by ELISA, suggesting that extracellular granzyme may contribute to the disease. There was no correlation between the CSF cell count and levels, suggesting that this is not due to an increase in cell number and that there is localized GrB release in the CSF/central nervous system of MS patients.123 As there are low numbers of NK cells in the CSF of MS patients, it is believed that the cellular source of GrB is CTLs. Although it has yet to be shown, the extracellular localization of GrB in CSF implies that the enzyme has access to the ECM in the spinal cord and brain, suggesting that it may be actively cleaving this matrix, perhaps similar to what has been described in arthritis.185
There have been several studies reporting an intracellular pro-apoptotic role for GrB in MS. As oligodendrocytes and neurons express MHC-I, GrB-expressing CTLs may be responsible for the induction of cytotoxicity resulting in axonal damage and demyelination.197 In addition, neurons expressing MHC-1 resist Fas-mediated killing but are susceptible to granule-mediated apoptosis.198 γδ T cells have also been suggested to direct cytotoxicity to the myelin-oligodendrocyte unit; however, inhibition of GrB in these cells only partially reduced cytolysis.291 TH17 lymphocytes crossing the blood–brain barrier express GrB and have the capacity to kill human fetal neuron cultures in vitro, suggesting that this cell type is also a source of cytotoxic GrB in MS.199
In Guillain–Barré syndrome, an inflammatory demyelinating disease of the peripheral nervous system, GrB-expressing CD8+ T cells were increased and localized close to neurons of the dorsal root ganglion. MHC-I molecules were also detected on Schwann cells and myelin sheaths, suggesting that these T cells may be responsible for myelin damage in both the peripheral nervous system and the central nervous system.200 This could be intracellularly and/or extracellularly mediated and more studies are required to determine the role of GrB in demyelination.
Skin Disorders
GrB expression in the skin
The role of GrB in dermatological conditions is emerging and the contribution of GrB to skin aging, alopecia, and disease is illustrated in Figure 3b. There are several potential sources of GrB in the skin, namely CLs, DCs, macrophages, mast cells, and keratinocytes.117, 160 Keratinocytes express GrB in vitro after treatment with UVA, UVB, and at high confluence.11, 136, 160 Interestingly, UVB and high confluence induce both GrB and perforin expression, whereas GrB is expressed without perforin in response to UVA. UVA induced GrB expression in human skin in vivo.136 In the latter study, UVA treatment of cultured keratinocytes caused a reactive oxygen species (ROS)-dependent release of macrophage migration inhibitor factor (MIF), and MIF induced GrB expression by keratinocytes. This GrB expression was p38 mitogen-activated protein kinase (MAPK)-dependent and resulted in the phosphorylation of its subsequent substrate MAPKAPK2. During UVB treatment, ROS production results in signaling through EGFR, MAPK activation, and subsequent MAPKAPK-2 phosphorylation.11 It should be noted that it is unclear as to whether these pathways are involved in the induction of GrB expression in other cell types.
Perforin was expressed in parallel with GrB in response to UVB and increased confluence, and UVB-irradiated HaCaT keratinocytes showed cytotoxic activity against various skin cell types in vitro, suggesting that GrB in this cell type shows cytotoxic potential.11 GrB from keratinocytes decreased Staphylococcus epidermidis growth, suggesting that keratinocyte GrB may be protective in vivo against skin bacterial infection.160 This is only the second report of GrB-mediated cytotoxicity by a nonimmune cell type (the first was described in chondrocytes), as most cell types do not express perforin simultaneously.115 Despite the evidence of UV induction of GrB, it remains unclear whether GrB from keratinocytes contributes to UV-dependent photoaging or wrinkling, as GrB should have ECM cleavage potential if released extracellularly.
GrB is also expressed by mast cells in mouse skin in vivo, as well as in cultured skin mast cells after IgE treatment.28, 117 Mast cells do not express perforin and GrB that is released extracellularly from these cells induces fibroblast anoikis, suggesting that it has a role in ECM degradation in the skin. No follow-up studies have been published describing GrB from mast cells or keratinocytes contributing to skin diseases in vivo, and the majority of dermatological studies to date exclusively examine CTLs as the sole source of GrB in the skin. As mast cells do not express perforin and keratinocytes express GrB (but not perforin) in response to stimuli such as UVA, there is clearly a potential extracellular, perforin-independent capacity for GrB in the skin. As the skin contains a high proportion of ECM such as collagen, elastin, and ground substance, future extracellular studies will be useful in further examining the extracellular activity of GrB and its role in disease.
Alopecia areata
Alopecia areata (AA) is an inflammatory condition that results in non-scarring patchy hair loss. Perifollicular and intrafollicular CTLs have been implicated in AA, suggesting that there is an immune cell attack on the normally immune privileged hair follicle during the growth phase (anagen) of the hair cycle.292, 293 GrB-expressing cytotoxic cells were found closely associated with human hair follicles in chronic AA patients, suggesting that GrB may contribute to CTL-mediated follicular damage.157 However, in a contradictory study, Sato-kawamura et al158 did not see GrB-positive cells around follicles in lesions of AA. In a C3H/HeJ mouse model of AA, GrB expression was described in immune cells in the intrafollicular dermis, although few of these cells expressed CD8.159 Interestingly, supplying the neuropeptide substance P to the skin of AA-affected mice led to an increase in CD8+ cells expressing GrB, possibly leading to an increase in cytotoxicity in the skin. Substance P also resulted in regression of hair follicles out of the growth phase into catagen, suggesting this could be related to a substance P-dependent increase in CD8+ GrB-expressing cells.159 Although more studies are required to determine causation and to define the mechanism by which GrB contributes to AA, there is an association between the disease and GrB expression.
Other skin disorders
GrB has been linked to other skin conditions such as acne, vitiligo, atopic dermatitis, allergic contact dermatitis, psoriasis, lichen planus (LP), lichen sclerosus, Stevens–Johnson syndrome, and toxic epidermal necrolysis.161, 162, 163, 164, 166, 168, 173, 176 GrB expression in most of these diseases was found to be localized to CTLs and GrB-specific disease implications were almost exclusively believed to be through intracellular CTL-mediated apoptosis of keratinocytes and other resident cell types. As more cellular sources of GrB have been identified as of late and the emerging role of extracellular GrB activity gains traction, determining other GrB sources and examining the importance of extracellular GrB activity will be a necessity in dermatological research involving GrB.
Vitiligo is characterized by a loss of pigmentation in the skin due to a progressive loss of melanocytes, the cell type that produces the pigment melanin. In the perilesional area of vitiligo biopsies, the border between the unpigmented lesion and normal pigmented skin, CD8+ CTLs were shown to cluster around disappearing melanocytes with ∼60% of these T cells expressing perforin and GrB, respectively. Furthermore, GrB-expressing T cells were not found elsewhere in vitiligo or in non-diseased control skin.166 As there have been several accounts that melanocytes are insensitive to FasL-mediated apoptosis, GrB from surrounding CTLs is believed to mediate the death of melanocytes in vitiligo and may be a major contributor to the vitiligo phenotype.167
GrB-expressing T cells have also been linked to atopic dermatitis and allergic contact dermatitis. In atopic dermatitis, a chronic inflammatory skin disease with eczematous lesions, an increase in GrB expression in CD4+ and CD8+ lymphocytes was described, particularly in the perivascular infiltrate and focally at spongiosis sites (areas of intracellular fluid accumulation in the dermis).164 In contact dermatitis, a skin disease caused by hapten exposure in sensitized individuals, a very similar GrB expression pattern was found, within CD4+ and CD8+ cells in perivascular infiltrate and spongiosis sites.163 Interestingly, keratinocytes in close proximity to GrB-positive lymphocytes showed signs of cell damage at sites of spongiosis. Weak GrB and perforin staining was described in these keratinocytes, suggesting they may be exposed to GrB from neighboring lymphocytes or may express the proteins themselves.
Lesions of the chronic inflammatory skin diseases, lichen sclerosis and LP, also showed abundant T cell expression of GrB. In lichen sclerosis, GrB mRNA expression was found in dermal infiltrate close to keratinocytes in the intra-epidermal area, was highly expressed at early stages of the disease, and levels were over 100 times greater than that of normal skin.173, 169 GrB-positive cells were found in close vicinity to apoptotic keratinocytes and through electron microscopy GrB was found to be transferred from a lymphocyte into an apoptotic keratinocyte, suggesting that GrB from lymphocytes is contributing to keratinocyte cell death in this disease.168 GrB-expressing plasmacytoid DCs usually found in blood were seen in oral LP lesions and may contribute to GrB-associated damage in this disorder.170
Stevens–Johnson syndrome and toxic epidermal necrolysis are rare and potentially life-threatening diseases involving keratinocyte apoptosis and epidermal necrosis, which are largely believed to be drug induced.176 GrB-positive lymphocytes were found in blister fluid and GrB upregulation correlated to increased disease severity. T cells in lesions express perforin and GrB, and it has been suggested that T cells may trigger keratinocyte cell death in these skin conditions.176, 177, 178, 179, 180
Vascular Diseases
Atherosclerosis
Atherosclerosis a lipid-driven inflammatory disease responsible for the majority of heart attacks, strokes, and lower limb loss, and, as such, is now the leading cause of death world-wide.294, 295, 296, 297 The most abundant inflammatory and immune cells in atherosclerotic lesions are T lymphocytes and macrophages, and their role in atherosclerosis is well documented.189, 298, 299, 300 Small populations of neutrophils, mast cells, DCs, and NK cells also are present.301, 302, 303 The role of GrB in atherosclerotic plaque progression is illustrated in Figure 3c. Until recently, GrB expression was believed to be limited to CTLs and NK cells. However, CD4+/CD8+ macrophages infiltrating inflammation sites and lipid-laden foam cells within atherosclerotic plaques also express GrB.10, 189, 304 In atherosclerotic lesions, GrB expression increases with disease severity and is observed in both lipid-rich areas and cellular regions surrounding the core of developing plaques, as well as in the shoulder regions of advanced plaques.10 Within the intima and media, GrB is found within SMCs, macrophages, and T cells, but can also be detected extracellularly. Furthermore, GrB is found to localize to TUNEL-positive foam cells, suggesting that it could be contributing to cell death in lipid-rich regions of developing plaques.10
The presence of GrB in advanced atherosclerotic lesions and its association with increased disease severity suggests that GrB may affect plaque stability. Indeed, elevated levels of GrB are detected in the plasma of patients with atherosclerosis, with the highest levels detected in patients with unstable plaques, lending support to the hypothesis that GrB influences plaque instability. In a study by Skjelland et al,232 increased GrB levels in the plasma corresponded to carotid plaque instability and increased incidence of cerebrovascular events in humans. Elevated GrB production was also observed in peripheral blood mononuclear cells isolated from patients with unstable angina pectoris compared with cells from patients with stable angina pectoris.233 Thus, evidence to date indicates that GrB might be involved in the initiation of plaque instability and rupture. In addition, the repercussions of elevated GrB levels may extend beyond plaque rupture and into post-infarct ventricular remodeling as Kondo et al recently reported elevated GrB levels in the plasma of patients with myocardial infarction (MI) in both the acute and subacute phases. They also found that sustained levels of GrB was an independent factor for late-stage left ventricular remodeling.234
The mechanisms by which GrB may contribute to plaque stability include ECM fragmentation, release of ECM-sequestered cytokines, and/or induction of macrophage or SMC apoptosis in the fibrous cap. To the latter point, Choy et al135 showed, using a cell culture model, that GrB could induce SMC apoptosis in the absence of perforin through the disruption of SMC–ECM interactions and induction of anoikis. Conversely, levels of the endogenous GrB inhibitor PI-9 in SMC markedly reduced in unstable plaques versus stable plaques, implying a role for GrB-induced SMC apoptosis. It should be noted, however, that although the GrB-inhibitory role of PI-9 is well-established, its effects on GrB as a potential mechanism were not considered nor discussed in the latter study. Rather, reduced PI-9 was associated with increased SMC caspase-1 activity and IL-1β production.35 With regard to its extracellular function in atherosclerosis, GrB is capable of cleaving multiple ECM proteins, including fibronectin, vitronectin, and laminin.134, 135 These data suggest that GrB may contribute to plaque instability through both intracellular and extracellular activities, leading to the formation of a necrotic core and thinning of the fibrous cap, and ultimately plaque rupture.10 Although ongoing, preliminary studies have been reported using apolipoprotein E × GrB double knockout mice in which GrB deficiency was found to be associated with reduced atherosclerosis, suggesting that this protease may be important in this disease.305
Although GrB appears to be involved in the pathogenesis of atherosclerosis, it should be noted that the precise effects of elevated GrB levels may depend on the stage of lesion development. During the early stages of lesion development, GrB-induced SMC apoptosis may be a beneficial way to reduce intimal hyperplasia,306 whereas in more advanced lesions, increased SMC apoptosis may decrease plaque stability through decreased cellularity and increased ECM proteolysis.
Cardiac allograft vasculopathy
Transplantation is the recommended therapy for end-stage organ disease. Although the prognosis for acute rejection has improved considerably with the introduction of immunosuppressive agents, long-term allograft survival has not improved appreciably since the early 1980s. Numerous studies have reported elevated GrB levels in the plasma and inflammatory infiltrate of biopsies from patients experiencing acute rejection, suggesting GrB may be a useful biomarker for the prediction of acute rejection.237, 238, 239, 240, 241, 242, 243, 244, 245 Although acute rejection constitutes a major challenge in the early postoperative period, the major impediment to long-term survival for solid organ transplant recipients is allograft vasculopathy (AV), which is an accelerated, immune-mediated form of occlusive arteriosclerotic vascular disease that affects vessels of solid organ allografts.307, 308 The pathogenesis of AV is complex and characterized by intimal accumulation of SMCs, T lymphocytes, and macrophages, as well as extensive medial thinning that is associated with increased SMC apoptosis.10, 308, 309
GrB is abundant in the intima, media, and adventitia of cardiac AV (CAV) vessels, and increased GrB corresponds to increased CAV disease severity.10 In the intima, GrB localizes to macrophage foam cells, whereas in the media and adventitia, GrB was found extracellularly, as well as in SMC and leukocyte infiltrates.10 Similar to developing atherosclerotic lesions, GrB within CAV lesions localizes to TUNEL-positive foam cells within the deep intima, suggesting that it contributes to cell death within the lesions.10 Roles for both GrB and perforin in CAV pathogenesis have been assessed using a mouse heterotopic cardiac transplant model of CAV.246, 247 In these studies, hearts transplanted into GrB-KO mice or perforin-KO mice showed smaller lesions, reduced luminal narrowing, and a marked reduction in endothelial cell apoptosis when compared with wild-type recipients.246, 247 Similar to its predicted role in atherosclerosis, GrB may contribute to CAV pathogenesis through apoptosis of SMC and endothelial cells, and through extracellular activities such as SMC anoikis and cleavage of ECM proteins. Both mechanisms are likely to contribute to the overall integrity of blood vessels and the development of CAV.
Other cardiovascular diseases
Inflammation, activated T cells, and SMC death are associated with other inflammatory vascular diseases such as Kawasaki disease, Takayasu myocarditis, and giant cell arteritis.248, 249, 310, 311, 312, 313 Kawasaki disease is a childhood form of vasculitis that affects the coronary arteries and is characterized histologically by inflammation and ECM destruction, resulting in the formation of coronary artery aneurysms.314 Elevated levels of CTLs, macrophages, and B cells have been noted in the lesions, aneurysms, and the plasma of Kawasaki's patients; however, the involvement of GrB in Kawasaki disease pathogenesis requires further elucidation.248, 249, 250 Increased plasma levels of GrB, in addition to GrB-positive CTL and macrophages, have been reported in Kawasaki disease; however, a more recent study showed elevated CTLs in acute Kawasaki disease in which neither GrB nor perforin could be detected249, 250 Whether the variations in GrB and perforin expression reflect alterations in differentiated T-cell populations, genetic differences in GrB/perforin production, or variation in preparation are unknown, but will prove to be an interesting area of future research.
Inflammatory Lung Diseases
COPD
COPD is an inflammatory lung disorder characterized by features of emphysema, chronic bronchitis, and small airways disease and is associated with chronic pulmonary and systemic inflammation.315 COPD patients who smoke have shown increased levels of CD8+ cells expressing perforin in the BAL fluid.143 An increase in the number of T cells and NK cells expressing GrB in the blood and BAL fluid of patients with COPD has also been reported.144 A plausible apoptotic role for GrB in COPD was suggested by Hodge et al,144 who showed a positive correlation between T cell-derived GrB in the BAL fluid and apoptosis of bronchial epithelial cells. CD8+ CTLs and NK cells infiltrating the lung in COPD patients are active and express GrB while GrB protein and mRNA were also detected in type II pneumocytes from patients with COPD.18 Although these findings raise the possibility of an interesting new source of GrB delivery into the parenchymal spaces of the diseased lung, more studies are needed to determine whether GrB release by these cells do in fact contribute significantly to COPD pathogenesis. Given the apoptotic and degenerative nature of COPD along with the pro-apoptotic and matrix-degrading ability of GrB, speculation of a major role for GrB in COPD pathogenesis continues to gain momentum.
Asthma
Asthma is a Th2-dependent inflammatory disease of the lung characterized by reversible airway obstruction, bronchial hyper-reactivity, and chronic inflammation leading to tissue remodeling. Little is known regarding the specific role of GrB in allergic asthma; however, Bratke et al122 were able to show an increase in GrB-expressing T cells and NK cells, as well as extracellular GrB in the BAL fluid of patients suffering from allergic asthma after allergen challenge, leading to speculation that GrB contributes to pathogenesis through matrix destruction and remodeling. These findings are supported by Tschopp et al,17 who reported an alteration in the granule content of basophils and the induction of GrB expression upon stimulation with IL-3 released by mast cells. Despite a lack of perforin expression, human basophils showed NK-like cytotoxicity, which was abolished after GrB inhibition further supporting a possible perforin-independent extracellular role of GrB in allergic asthma.
Other inflammatory lung diseases
Several other lung diseases are affected by GrB activity as well. It has been reported that GrB and perforin mRNA levels are increased in the BAL fluid of patients in the acute phase of acute respiratory distress syndrome (ARDS).145 A functioning and unbroken alveolar epithelium is critical for recovery from ARDS and increased granzyme-mediated apoptosis of alveolar epithelial cells could compromise the integrity of this barrier leading to disease progression. Furthermore, hypersensitivity pneumonitis has been shown to be associated with an increase in GrB protein levels in the BAL fluid supporting the notion that GrB contributes to alveolar distruction and matrix remodeling observed in this disease.125 GrB-expressing CD4+ and CD8+ T cells have also been detected in the BAL fluid of patients with pulmonary sarcoidosis, a disease characterized by a Th1-dominant immune response and the formation of granulomas often found in the lung.146 Reduced levels of GrB have also been detected in the serum of sarcoidosis patients, leading to speculation that the protease is increased in tissues elsewhere in the body where it participates in the cytotoxicity observed in pulmonary sarcoidosis.147 However, as with all inflammatory lung diseases, much work remains to be done to confirm a specific role for GrB in disease pathogenesis and to determine the intracellular and/or extracellular mechanisms that might be involved.
CONCLUSION
GrB is emerging as a multifunctional protease that may have important roles in a number of inflammatory and/or degenerative pathologies. The realization that this protease can be expressed and secreted by both immune and nonimmune cells, in addition to the fact that the function of this protease extends beyond the induction of CTL/NK-induced apoptosis suggests that there is much to be learned about this protease in disease. Nonetheless, it is becoming apparent that GrB may have a larger role in chronic inflammatory, autoimmune, and/or degenerative diseases than previously believed. Future studies may show GrB as a novel therapeutic target for such diseases.
Acknowledgements
We gratefully acknowledge funding from the Canadian Institutes of Health Research (DJG), the Michael Smith Foundation for Health Research (MSFHR)/British Columbia Transplant Research Foundation and the Heart and Stroke Foundation (HSFC) (DJG). WAB is supported by an Alexander Graham Bell Canadian Graduate Studentship from the Natural Sciences and Engineering Research Council of Canada and a graduate fellowship from the MSFHR. DMC is funded by post-doctoral fellowships from the HSFC and the CIHR IMPACT strategic training program.
Footnotes
Disclosure/conflict of interest
The authors declare no conflict of interest.
References
- 1.Bots M, Medema JP. Granzymes at a glance. J Cell Sci. 2006;119(Part 24):5011–5014. doi: 10.1242/jcs.03239. [DOI] [PubMed] [Google Scholar]
- 2.Bleackley RC, Duggan B, Ehrman N. Isolation of two cDNA sequences which encode cytotoxic cell proteases. FEBS Lett. 1988;234:153–159. doi: 10.1016/0014-5793(88)81323-1. [DOI] [PubMed] [Google Scholar]
- 3.Brunet JF, Dosseto M, Denizot F. The inducible cytotoxic T-lymphocyte-associated gene transcript CTLA-1 sequence and gene localization to mouse chromosome 14. Nature. 1986;322:268–271. doi: 10.1038/322268a0. [DOI] [PubMed] [Google Scholar]
- 4.Masson D, Nabholz M, Estrade C. Granules of cytolytic T-lymphocytes contain two serine esterases. EMBO J. 1986;5:1595–1600. doi: 10.1002/j.1460-2075.1986.tb04401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasternack MS, Eisen HN. A novel serine esterase expressed by cytotoxic T lymphocytes. Nature. 1985;314:743–745. doi: 10.1038/314743a0. [DOI] [PubMed] [Google Scholar]
- 6.Klein JL, Shows TB, Dupont B. Genomic organization and chromosomal assignment for a serine protease gene (CSPB) expressed by human cytotoxic lymphocytes. Genomics. 1989;5:110–117. doi: 10.1016/0888-7543(89)90093-1. [DOI] [PubMed] [Google Scholar]
- 7.Poe M, Blake JT, Boulton DA. Human cytotoxic lymphocyte granzyme B. Its purification from granules and the characterization of substrate and inhibitor specificity. J Biol Chem. 1991;266:98–103. doi: 10.1016/S0021-9258(18)52407-8. [DOI] [PubMed] [Google Scholar]
- 8.Murphy ME, Moult J, Bleackley RC. Comparative molecular model building of two serine proteinases from cytotoxic T lymphocytes. Proteins. 1988;4:190–204. doi: 10.1002/prot.340040306. [DOI] [PubMed] [Google Scholar]
- 9.Waugh SM, Harris JL, Fletterick R. The structure of the pro-apoptotic protease granzyme B reveals the molecular determinants of its specificity. Nat Struct Biol. 2000;7:762–765. doi: 10.1038/78992. [DOI] [PubMed] [Google Scholar]
- 10.Choy JC, McDonald PC, Suarez AC. Granzyme B in atherosclerosis and transplant vascular disease: association with cell death and atherosclerotic disease severity. Mod Pathol. 2003;16:460–470. doi: 10.1097/01.MP.0000067424.12280.BC. [DOI] [PubMed] [Google Scholar]
- 11.Hernandez-Pigeon H, Jean C, Charruyer A. Human keratinocytes acquire cellular cytotoxicity under UV-B irradiation. Implication of granzyme B and perforin. J Biol Chem. 2006;281:13525–13532. doi: 10.1074/jbc.M512694200. [DOI] [PubMed] [Google Scholar]
- 12.Hirst CE, Buzza MS, Sutton VR. Perforin-independent expression of granzyme B and proteinase inhibitor 9 in human testis and placenta suggests a role for granzyme B-mediated proteolysis in reproduction. Mol Hum Reprod. 2001;7:1133–1142. doi: 10.1093/molehr/7.12.1133. [DOI] [PubMed] [Google Scholar]
- 13.Horiuchi K, Saito S, Sasaki R. Expression of granzyme B in human articular chondrocytes. J Rheumatol. 2003;30:1799–1810. [PubMed] [Google Scholar]
- 14.Namekawa T, Wagner UG, Goronzy JJ. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. 1998;41:2108–2116. doi: 10.1002/1529-0131(199812)41:12<2108::AID-ART5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 15.Rissoan MC, Duhen T, Bridon JM. Subtractive hybridization reveals the expression of immunoglobulin-like transcript 7, Eph-B1, granzyme B, and 3 novel transcripts in human plasmacytoid dendritic cells. Blood. 2002;100:3295–3303. doi: 10.1182/blood-2002-02-0638. [DOI] [PubMed] [Google Scholar]
- 16.Sasson R, Dantes A, Tajima K. Novel genes modulated by FSH in normal and immortalized FSH-responsive cells: new insights into the mechanism of FSH action. FASEB J. 2003;17:1256–1266. doi: 10.1096/fj.02-0740com. [DOI] [PubMed] [Google Scholar]
- 17.Tschopp CM, Spiegl N, Didichenko S. Granzyme B, a novel mediator of allergic inflammation: its induction and release in blood basophils and human asthma. Blood. 2006;108:2290–2299. doi: 10.1182/blood-2006-03-010348. [DOI] [PubMed] [Google Scholar]
- 18.Vernooy JH, Moller GM, van Suylen RJ. Increased granzyme A expression in type II pneumocytes of patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:464–472. doi: 10.1164/rccm.200602-169OC. [DOI] [PubMed] [Google Scholar]
- 19.Grossman WJ, Verbsky JW, Tollefsen BL. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. 2004;104:2840–2848. doi: 10.1182/blood-2004-03-0859. [DOI] [PubMed] [Google Scholar]
- 20.Wagner C, Iking-Konert C, Denefleh B. Granzyme B and perforin: constitutive expression in human polymorphonuclear neutrophils. Blood. 2004;103:1099–1104. doi: 10.1182/blood-2003-04-1069. [DOI] [PubMed] [Google Scholar]
- 21.Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol. 2008;26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Babichuk CK, Duggan BL, Bleackley RC. In vivo regulation of murine granzyme B gene transcription in activated primary T cells. J Biol Chem. 1996;271:16485–16493. doi: 10.1074/jbc.271.28.16485. [DOI] [PubMed] [Google Scholar]
- 23.Fregeau CJ, Bleackley RC. Transcription of two cytotoxic cell protease genes is under the control of different regulatory elements. Nucleic Acids Res. 1991;19:5583–5590. doi: 10.1093/nar/19.20.5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wargnier A, Lafaurie C, Legros-Maida S. Down-regulation of human granzyme B expression by glucocorticoids. Dexamethasone inhibits binding to the Ikaros and AP-1 regulatory elements of the granzyme B promoter. J Biol Chem. 1998;273:35326–35331. doi: 10.1074/jbc.273.52.35326. [DOI] [PubMed] [Google Scholar]
- 25.Haddad P, Wargnier A, Bourge JF. A promoter element of the human serine esterase granzyme B gene controls specific transcription in activated T cells. Eur J Immunol. 1993;23:625–629. doi: 10.1002/eji.1830230307. [DOI] [PubMed] [Google Scholar]
- 26.Babichuk CK, Bleackley RC. Mutational analysis of the murine granzyme B gene promoter in primary T cells and a T cell clone. J Biol Chem. 1997;272:18564–18571. doi: 10.1074/jbc.272.30.18564. [DOI] [PubMed] [Google Scholar]
- 27.Fehniger TA, Cai SF, Cao X. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity. 2007;26:798–811. doi: 10.1016/j.immuni.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Strik MC, de Koning PJ, Kleijmeer MJ. Human mast cells produce and release the cytotoxic lymphocyte associated protease granzyme B upon activation. Mol Immunol. 2007;44:3462–3472. doi: 10.1016/j.molimm.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 29.Griffiths GM, Isaaz S. Granzymes A and B are targeted to the lytic granules of lymphocytes by the mannose-6-phosphate receptor. J Cell Biol. 1993;120:885–896. doi: 10.1083/jcb.120.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bird CH, Sutton VR, Sun J. Selective regulation of apoptosis: the cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol Cell Biol. 1998;18:6387–6398. doi: 10.1128/MCB.18.11.6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun J, Bird CH, Sutton V. A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J Biol Chem. 1996;271:27802–27809. doi: 10.1074/jbc.271.44.27802. [DOI] [PubMed] [Google Scholar]
- 32.Barrie MB, Stout HW, Abougergi MS. Antiviral cytokines induce hepatic expression of the granzyme B inhibitors, proteinase inhibitor 9 and serine proteinase inhibitor 6. J Immunol. 2004;172:6453–6459. doi: 10.4049/jimmunol.172.10.6453. [DOI] [PubMed] [Google Scholar]
- 33.Buzza MS, Hirst CE, Bird CH. The granzyme B inhibitor, PI-9, is present in endothelial and mesothelial cells, suggesting that it protects bystander cells during immune responses. Cell Immunol. 2001;210:21–29. doi: 10.1006/cimm.2001.1806. [DOI] [PubMed] [Google Scholar]
- 34.Kanamori H, Krieg S, Mao C. Proteinase inhibitor 9, an inhibitor of granzyme B-mediated apoptosis, is a primary estrogen-inducible gene in human liver cells. J Biol Chem. 2000;275:5867–5873. doi: 10.1074/jbc.275.8.5867. [DOI] [PubMed] [Google Scholar]
- 35.Young JL, Sukhova GK, Foster D. The serpin proteinase inhibitor 9 is an endogenous inhibitor of interleukin 1beta-converting enzyme (caspase-1) activity in human vascular smooth muscle cells. J Exp Med. 2000;191:1535–1544. doi: 10.1084/jem.191.9.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bladergroen BA, Strik MC, Bovenschen N. The granzyme B inhibitor, protease inhibitor 9, is mainly expressed by dendritic cells and at immune-privileged sites. J Immunol. 2001;166:3218–3225. doi: 10.4049/jimmunol.166.5.3218. [DOI] [PubMed] [Google Scholar]
- 37.Bots M, L VANB, Rademaker MT. Serpins prevent granzyme-induced death in a species-specific manner. Immunol Cell Biol. 2006;84:79–86. doi: 10.1111/j.1440-1711.2005.01417.x. [DOI] [PubMed] [Google Scholar]
- 38.Sipione S, Simmen KC, Lord SJ. Identification of a novel human granzyme B inhibitor secreted by cultured sertoli cells. J Immunol. 2006;177:5051–5058. doi: 10.4049/jimmunol.177.8.5051. [DOI] [PubMed] [Google Scholar]
- 39.Mahrus S, Kisiel W, Craik CS. Granzyme M is a regulatory protease that inactivates proteinase inhibitor 9, an endogenous inhibitor of granzyme B. J Biol Chem. 2004;279:54275–54282. doi: 10.1074/jbc.M411482200. [DOI] [PubMed] [Google Scholar]
- 40.Andrade F, Fellows E, Jenne DE. Granzyme H destroys the function of critical adenoviral proteins required for viral DNA replication and granzyme B inhibition. EMBO J. 2007;26:2148–2157. doi: 10.1038/sj.emboj.7601650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan G, O'Rourke K, Chinnaiyan AM. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111–113. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 42.Sheridan JP, Marsters SA, Pitti RM. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–821. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 43.Strater J, Wellisch I, Riedl S. CD95 (APO-1/Fas)-mediated apoptosis in colon epithelial cells: a possible role in ulcerative colitis. Gastroenterology. 1997;113:160–167. doi: 10.1016/S0016-5085(97)70091-X. [DOI] [PubMed] [Google Scholar]
- 44.Walczak H, Degli-Esposti MA, Johnson RS. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386–5397. doi: 10.1093/emboj/16.17.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu M, Das A, Tan Y. Induction of apoptosis in glioma cell lines by TRAIL/Apo-2l. J Neurosci Res. 2000;61:464–470. doi: 10.1002/1097-4547(20000815)61:4<464::AID-JNR14>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 46.Cullen SP, Martin SJ. Mechanisms of granule-dependent killing. Cell Death Differ. 2008;15:251–262. doi: 10.1038/sj.cdd.4402244. [DOI] [PubMed] [Google Scholar]
- 47.Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20:323–370. doi: 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- 48.Kummer JA, Kamp AM, Citarella F. Expression of human recombinant granzyme A zymogen and its activation by the cysteine proteinase cathepsin C. J Biol Chem. 1996;271:9281–9286. doi: 10.1074/jbc.271.16.9281. [DOI] [PubMed] [Google Scholar]
- 49.Smyth MJ, McGuire MJ, Thia KY. Expression of recombinant human granzyme B. A processing and activation role for dipeptidyl peptidase I. J Immunol. 1995;154:6299–6305. [PubMed] [Google Scholar]
- 50.Brown GR, McGuire MJ, Thiele DL. Dipeptidyl peptidase I is enriched in granules of in vitro- and in vivo-activated cytotoxic T lymphocytes. J Immunol. 1993;150:4733–4742. [PubMed] [Google Scholar]
- 51.Galvin JP, Spaeny-Dekking LH, Wang B. Apoptosis induced by granzyme B-glycosaminoglycan complexes: implications for granule-mediated apoptosis in vivo. J Immunol. 1999;162:5345–5350. [PubMed] [Google Scholar]
- 52.Grujic M, Braga T, Lukinius A. Serglycin-deficient cytotoxic T lymphocytes display defective secretory granule maturation and granzyme B storage. J Biol Chem. 2005;280:33411–33418. doi: 10.1074/jbc.M501708200. [DOI] [PubMed] [Google Scholar]
- 53.Metkar SS, Wang B, Aguilar-Santelises M. Cytotoxic cell granule-mediated apoptosis: perforin delivers granzyme B-serglycin complexes into target cells without plasma membrane pore formation. Immunity. 2002;16:417–428. doi: 10.1016/S1074-7613(02)00286-8. [DOI] [PubMed] [Google Scholar]
- 54.Spaeny-Dekking EH, Hanna WL, Wolbink AM. Extracellular granzymes A and B in humans: detection of native species during CTL responses in vitro and in vivo. J Immunol. 1998;160:3610–3616. [PubMed] [Google Scholar]
- 55.Balaji KN, Schaschke N, Machleidt W. Surface cathepsin B protects cytotoxic lymphocytes from self-destruction after degranulation. J Exp Med. 2002;196:493–503. doi: 10.1084/jem.20011836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bromley SK, Burack WR, Johnson KG. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 57.Huse M, Quann EJ, Davis MM. Shouts, whispers and the kiss of death: directional secretion in T cells. Nat Immunol. 2008;9:1105–1111. doi: 10.1038/ni.f.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geiger B, Rosen D, Berke G. Spatial relationships of microtubule-organizing centers and the contact area of cytotoxic T lymphocytes and target cells. J Cell Biol. 1982;95:137–143. doi: 10.1083/jcb.95.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kupfer A, Dennert G, Singer SJ. Polarization of the Golgi apparatus and the microtubule-organizing center within cloned natural killer cells bound to their targets. Proc Natl Acad Sci USA. 1983;80:7224–7228. doi: 10.1073/pnas.80.23.7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stinchcombe JC, Bossi G, Booth S. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001;15:751–761. doi: 10.1016/S1074-7613(01)00234-5. [DOI] [PubMed] [Google Scholar]
- 61.Sauer H, Pratsch L, Tschopp J. Functional size of complement and perforin pores compared by confocal laser scanning microscopy and fluorescence microphotolysis. Biochim Biophys Acta. 1991;1063:137–146. doi: 10.1016/0005-2736(91)90363-D. [DOI] [PubMed] [Google Scholar]
- 62.Tschopp J, Masson D, Schafer S. Inhibition of the lytic activity of perforin by lipoproteins. J Immunol. 1986;137:1950–1953. [PubMed] [Google Scholar]
- 63.Froelich CJ, Orth K, Turbov J. New paradigm for lymphocyte granule-mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J Biol Chem. 1996;271:29073–29079. doi: 10.1074/jbc.271.46.29073. [DOI] [PubMed] [Google Scholar]
- 64.Motyka B, Korbutt G, Pinkoski MJ. Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell. 2000;103:491–500. doi: 10.1016/S0092-8674(00)00140-9. [DOI] [PubMed] [Google Scholar]
- 65.Pinkoski MJ, Hobman M, Heibein JA. Entry and trafficking of granzyme B in target cells during granzyme B-perforin-mediated apoptosis. Blood. 1998;92:1044–1054. doi: 10.1182/blood.V92.3.1044. [DOI] [PubMed] [Google Scholar]
- 66.Shi L, Mai S, Israels S. Granzyme B (GraB) autonomously crosses the cell membrane and perforin initiates apoptosis and GraB nuclear localization. J Exp Med. 1997;185:855–866. doi: 10.1084/jem.185.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Veugelers K, Motyka B, Goping IS. Granule-mediated killing by granzyme B and perforin requires a mannose 6-phosphate receptor and is augmented by cell surface heparan sulfate. Mol Biol Cell. 2006;17:623–633. doi: 10.1091/mbc.e05-07-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raja SM, Metkar SS, Honing S. A novel mechanism for protein delivery: granzyme B undergoes electrostatic exchange from serglycin to target cells. J Biol Chem. 2005;280:20752–20761. doi: 10.1074/jbc.M501181200. [DOI] [PubMed] [Google Scholar]
- 69.Shi L, Keefe D, Durand E. Granzyme B binds to target cells mostly by charge and must be added at the same time as perforin to trigger apoptosis. J Immunol. 2005;174:5456–5461. doi: 10.4049/jimmunol.174.9.5456. [DOI] [PubMed] [Google Scholar]
- 70.Veugelers K, Motyka B, Frantz C. The granzyme B–serglycin complex from cytotoxic granules requires dynamin for endocytosis. Blood. 2004;103:3845–3853. doi: 10.1182/blood-2003-06-2156. [DOI] [PubMed] [Google Scholar]
- 71.Trapani JA, Sutton VR, Thia KY. A clathrin/dynamin- and mannose-6-phosphate receptor-independent pathway for granzyme B-induced cell death. J Cell Biol. 2003;160:223–233. doi: 10.1083/jcb.200210150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keefe D, Shi L, Feske S. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity. 2005;23:249–262. doi: 10.1016/j.immuni.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 73.Pipkin ME, Lieberman J. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol. 2007;19:301–308. doi: 10.1016/j.coi.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dressel R, Raja SM, Honing S. Granzyme-mediated cytotoxicity does not involve the mannose 6-phosphate receptors on target cells. J Biol Chem. 2004;279:20200–20210. doi: 10.1074/jbc.M313108200. [DOI] [PubMed] [Google Scholar]
- 75.Kagi D, Ledermann B, Burki K. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 76.Walsh CM, Matloubian M, Liu CC. Immune function in mice lacking the perforin gene. Proc Natl Acad Sci USA. 1994;91:10854–10858. doi: 10.1073/pnas.91.23.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smyth MJ, Street SE, Trapani JA. Cutting edge: granzymes A and B are not essential for perforin-mediated tumor rejection. J Immunol. 2003;171:515–518. doi: 10.4049/jimmunol.171.2.515. [DOI] [PubMed] [Google Scholar]
- 78.Smyth MJ, Thia KY, Street SE. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192:755–760. doi: 10.1084/jem.192.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Henter JI, Arico M, Elinder G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998;12:417–433. doi: 10.1016/S0889-8588(05)70520-7. [DOI] [PubMed] [Google Scholar]
- 80.Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6:940–952. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 81.Voskoboinik I, Trapani JA. Addressing the mysteries of perforin function. Immunol Cell Biol. 2006;84:66–71. doi: 10.1111/j.1440-1711.2005.01409.x. [DOI] [PubMed] [Google Scholar]
- 82.Bird CH, Sun J, Ung K. Cationic sites on granzyme B contribute to cytotoxicity by promoting its uptake into target cells. Mol Cell Biol. 2005;25:7854–7867. doi: 10.1128/MCB.25.17.7854-7867.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gross C, Schmidt-Wolf IG, Nagaraj S. Heat shock protein 70-reactivity is associated with increased cell surface density of CD94/CD56 on primary natural killer cells. Cell Stress Chaperones. 2003;8:348–360. doi: 10.1379/1466-1268(2003)008<0348:HSPRIA>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gross C, Koelch W, DeMaio A. Cell surface-bound heat shock protein 70 (Hsp70) mediates perforin-independent apoptosis by specific binding and uptake of granzyme B. J Biol Chem. 2003;278:41173–41181. doi: 10.1074/jbc.M302644200. [DOI] [PubMed] [Google Scholar]
- 85.Darmon AJ, Nicholson DW, Bleackley RC. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature. 1995;377:446–448. doi: 10.1038/377446a0. [DOI] [PubMed] [Google Scholar]
- 86.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 87.Adrain C, Murphy BM, Martin SJ. Molecular ordering of the caspase activation cascade initiated by the cytotoxic T lymphocyte/natural killer (CTL/NK) protease granzyme B. J Biol Chem. 2005;280:4663–4673. doi: 10.1074/jbc.M410915200. [DOI] [PubMed] [Google Scholar]
- 88.Medema JP, Toes RE, Scaffidi C. Cleavage of FLICE (caspase-8) by granzyme B during cytotoxic T lymphocyte-induced apoptosis. Eur J Immunol. 1997;27:3492–3498. doi: 10.1002/eji.1830271250. [DOI] [PubMed] [Google Scholar]
- 89.Talanian RV, Yang X, Turbov J. Granule-mediated killing: pathways for granzyme B-initiated apoptosis. J Exp Med. 1997;186:1323–1331. doi: 10.1084/jem.186.8.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heibein JA, Goping IS, Barry M. Granzyme B-mediated cytochrome c release is regulated by the Bcl-2 family members bid and Bax. J Exp Med. 2000;192:1391–1402. doi: 10.1084/jem.192.10.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pinkoski MJ, Waterhouse NJ, Heibein JA. Granzyme B-mediated apoptosis proceeds predominantly through a Bcl-2-inhibitable mitochondrial pathway. J Biol Chem. 2001;276:12060–12067. doi: 10.1074/jbc.M009038200. [DOI] [PubMed] [Google Scholar]
- 92.Sutton VR, Davis JE, Cancilla M. Initiation of apoptosis by granzyme B requires direct cleavage of bid, but not direct granzyme B-mediated caspase activation. J Exp Med. 2000;192:1403–1414. doi: 10.1084/jem.192.10.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jiang X, Wang X. Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J Biol Chem. 2000;275:31199–31203. doi: 10.1074/jbc.C000405200. [DOI] [PubMed] [Google Scholar]
- 94.Li P, Nijhawan D, Budihardjo I. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/S0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 95.Han J, Goldstein LA, Gastman BR. Degradation of Mcl-1 by granzyme B: implications for Bim-mediated mitochondrial apoptotic events. J Biol Chem. 2004;279:22020–22029. doi: 10.1074/jbc.M313234200. [DOI] [PubMed] [Google Scholar]
- 96.Adrain C, Duriez PJ, Brumatti G. The cytotoxic lymphocyte protease, granzyme B, targets the cytoskeleton and perturbs microtubule polymerization dynamics. J Biol Chem. 2006;281:8118–8125. doi: 10.1074/jbc.M509361200. [DOI] [PubMed] [Google Scholar]
- 97.Andrade F, Roy S, Nicholson D. Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity. 1998;8:451–460. doi: 10.1016/S1074-7613(00)80550-6. [DOI] [PubMed] [Google Scholar]
- 98.Froelich CJ, Hanna WL, Poirier GG. Granzyme B/perforin-mediated apoptosis of Jurkat cells results in cleavage of poly(ADP-ribose) polymerase to the 89-kDa apoptotic fragment and less abundant 64-kDa fragment. Biochem Biophys Res Commun. 1996;227:658–665. doi: 10.1006/bbrc.1996.1565. [DOI] [PubMed] [Google Scholar]
- 99.Goping IS, Sawchuk T, Underhill DA. Identification of {alpha}-tubulin as a granzyme B substrate during CTL-mediated apoptosis. J Cell Sci. 2006;119(Part 5):858–865. doi: 10.1242/jcs.02791. [DOI] [PubMed] [Google Scholar]
- 100.Sebbagh M, Hamelin J, Bertoglio J. Direct cleavage of ROCK II by granzyme B induces target cell membrane blebbing in a caspase-independent manner. J Exp Med. 2005;201:465–471. doi: 10.1084/jem.20031877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sharif-Askari E, Alam A, Rheaume E. Direct cleavage of the human DNA fragmentation factor-45 by granzyme B induces caspase-activated DNase release and DNA fragmentation. EMBO J. 2001;20:3101–3113. doi: 10.1093/emboj/20.12.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thomas DA, Du C, Xu M. DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity. 2000;12:621–632. doi: 10.1016/S1074-7613(00)80213-7. [DOI] [PubMed] [Google Scholar]
- 103.Zhang D, Pasternack MS, Beresford PJ. Induction of rapid histone degradation by the cytotoxic T lymphocyte protease granzyme A. J Biol Chem. 2001;276:3683–3690. doi: 10.1074/jbc.M005390200. [DOI] [PubMed] [Google Scholar]
- 104.Zhang D, Beresford PJ, Greenberg AH. Granzymes A and B directly cleave lamins and disrupt the nuclear lamina during granule-mediated cytolysis. Proc Natl Acad Sci USA. 2001;98:5746–5751. doi: 10.1073/pnas.101329598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bredemeyer AJ, Carrigan PE, Fehniger TA. Hop cleavage and function in granzyme B-induced apoptosis. J Biol Chem. 2006;281:37130–37141. doi: 10.1074/jbc.M607969200. [DOI] [PubMed] [Google Scholar]
- 106.Bredemeyer AJ, Lewis RM, Malone JP. A proteomic approach for the discovery of protease substrates. Proc Natl Acad Sci USA. 2004;101:11785–11790. doi: 10.1073/pnas.0402353101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Caruso JA, Reiners JJ., Jr Proteolysis of HIP during apoptosis occurs within a region similar to the BID loop. Apoptosis. 2006;11:1877–1885. doi: 10.1007/s10495-006-0083-z. [DOI] [PubMed] [Google Scholar]
- 108.Hostetter DR, Loeb CR, Chu F. Hip is a pro-survival substrate of granzyme B. J Biol Chem. 2007;282:27865–27874. doi: 10.1074/jbc.M704312200. [DOI] [PubMed] [Google Scholar]
- 109.Sayers TJ, Wiltrout TA, Sowder R. Purification of a factor from the granules of a rat natural killer cell line (RNK) that reduces tumor cell growth and changes tumor morphology. Molecular identity with a granule serine protease (RNKP-1) J Immunol. 1992;148:292–300. [PubMed] [Google Scholar]
- 110.Sower LE, Klimpel GR, Hanna W. Extracellular activities of human granzymes. Cell Immunol. 1996;171:159–163. doi: 10.1006/cimm.1996.0187. [DOI] [PubMed] [Google Scholar]
- 111.Buzza MS, Bird PI. Extracellular granzymes: current perspectives. Biol Chem. 2006;387:827–837. doi: 10.1515/BC.2006.106. [DOI] [PubMed] [Google Scholar]
- 112.Romero V, Andrade F. Non-apoptotic functions of granzymes. Tissue Antigens. 2008;71:409–416. doi: 10.1111/j.1399-0039.2008.01013.x. [DOI] [PubMed] [Google Scholar]
- 113.Kramer MD, Simon MM. Are proteinases functional molecules of T lymphocytes? Immunol Today. 1987;8:140–142. doi: 10.1016/0167-5699(87)90141-1. [DOI] [PubMed] [Google Scholar]
- 114.Tak PP, Spaeny-Dekking L, Kraan MC. The levels of soluble granzyme A and B are elevated in plasma and synovial fluid of patients with rheumatoid arthritis (RA) Clin Exp Immunol. 1999;116:366–370. doi: 10.1046/j.1365-2249.1999.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Saito S, Murakoshi K, Kotake S. Granzyme B induces apoptosis of chondrocytes with natural killer cell-like cytotoxicity in rheumatoid arthritis. J Rheumatol. 2008;35:1932–1943. [PubMed] [Google Scholar]
- 116.Froelich CJ, Pardo J, Simon MM. Granule-associated serine proteases: granzymes might not just be killer proteases. Trends Immunol. 2009;30:117–123. doi: 10.1016/j.it.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 117.Pardo J, Wallich R, Ebnet K. Granzyme B is expressed in mouse mast cells in vivo and in vitro and causes delayed cell death independent of perforin. Cell Death Differ. 2007;14:1768–1779. doi: 10.1038/sj.cdd.4402183. [DOI] [PubMed] [Google Scholar]
- 118.Caughey GH, Schaumberg TH, Zerweck EH. The human mast cell chymase gene (CMA1): mapping to the cathepsin G/granzyme gene cluster and lineage-restricted expression. Genomics. 1993;15:614–620. doi: 10.1006/geno.1993.1115. [DOI] [PubMed] [Google Scholar]
- 119.Isaaz S, Baetz K, Olsen K. Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway. Eur J Immunol. 1995;25:1071–1079. doi: 10.1002/eji.1830250432. [DOI] [PubMed] [Google Scholar]
- 120.Skold S, Zeberg L, Gullberg U. Functional dissociation between proforms and mature forms of proteinase 3, azurocidin, and granzyme B in regulation of granulopoiesis. Exp Hematol. 2002;30:689–696. doi: 10.1016/S0301-472X(02)00816-0. [DOI] [PubMed] [Google Scholar]
- 121.Prakash MD, Bird CH, Bird PI. Active and zymogen forms of granzyme B are constitutively released from cytotoxic lymphocytes in the absence of target cell engagement. Immunol Cell Biol. 2009;87:249–254. doi: 10.1038/icb.2008.98. [DOI] [PubMed] [Google Scholar]
- 122.Bratke K, Bottcher B, Leeder K. Increase in granzyme B+ lymphocytes and soluble granzyme B in bronchoalveolar lavage of allergen challenged patients with atopic asthma. Clin Exp Immunol. 2004;136:542–548. doi: 10.1111/j.1365-2249.2004.02468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Malmestrom C, Lycke J, Haghighi S. Relapses in multiple sclerosis are associated with increased CD8+ T-cell mediated cytotoxicity in CSF. J Neuroimmunol. 2008;196:159–165. doi: 10.1016/j.jneuroim.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 124.Takahashi Y, Mine J, Kubota Y. A substantial number of Rasmussen syndrome patients have increased IgG, CD4(+) T cells, TNFalpha, and Granzyme B in CSF. Epilepsia. 2009;50:1419–1431. doi: 10.1111/j.1528-1167.2008.01977.x. [DOI] [PubMed] [Google Scholar]
- 125.Tremblay GM, Wolbink AM, Cormier Y. Granzyme activity in the inflamed lung is not controlled by endogenous serine proteinase inhibitors. J Immunol. 2000;165:3966–3969. doi: 10.4049/jimmunol.165.7.3966. [DOI] [PubMed] [Google Scholar]
- 126.Kurschus FC, Kleinschmidt M, Fellows E. Killing of target cells by redirected granzyme B in the absence of perforin. FEBS Lett. 2004;562:87–92. doi: 10.1016/S0014-5793(04)00187-5. [DOI] [PubMed] [Google Scholar]
- 127.Rowshani AT, Strik MC, Molenaar R. The granzyme B inhibitor SERPINB9 (protease inhibitor 9) circulates in blood and increases on primary cytomegalovirus infection after renal transplantation. J Infect Dis. 2005;192:1908–1911. doi: 10.1086/497606. [DOI] [PubMed] [Google Scholar]
- 128.Froelich CJ, Zhang X, Turbov J. Human granzyme B degrades aggrecan proteoglycan in matrix synthesized by chondrocytes. J Immunol. 1993;151:7161–7171. [PubMed] [Google Scholar]
- 129.Buzza MS, Dyson JM, Choi H. Antihemostatic activity of human granzyme B mediated by cleavage of von Willebrand factor. J Biol Chem. 2008;283:22498–22504. doi: 10.1074/jbc.M709080200. [DOI] [PubMed] [Google Scholar]
- 130.Mulligan-Kehoe MJ, Drinane MC, Mollmark J. Antiangiogenic plasma activity in patients with systemic sclerosis. Arthritis Rheum. 2007;56:3448–3458. doi: 10.1002/art.22861. [DOI] [PubMed] [Google Scholar]
- 131.Gahring L, Carlson NG, Meyer EL. Granzyme B proteolysis of a neuronal glutamate receptor generates an autoantigen and is modulated by glycosylation. J Immunol. 2001;166:1433–1438. doi: 10.4049/jimmunol.166.3.1433. [DOI] [PubMed] [Google Scholar]
- 132.Loeb CR, Harris JL, Craik CS. Granzyme B proteolyzes receptors important to proliferation and survival, tipping the balance toward apoptosis. J Biol Chem. 2006;281:28326–28335. doi: 10.1074/jbc.M604544200. [DOI] [PubMed] [Google Scholar]
- 133.Casciola-Rosen L, Miagkov A, Nagaraju K. Granzyme B: evidence for a role in the origin of myasthenia gravis. J Neuroimmunol. 2008;201-202:33–40. doi: 10.1016/j.jneuroim.2008.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Buzza MS, Zamurs L, Sun J. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem. 2005;280:23549–23558. doi: 10.1074/jbc.M412001200. [DOI] [PubMed] [Google Scholar]
- 135.Choy JC, Hung VH, Hunter AL. Granzyme B induces smooth muscle cell apoptosis in the absence of perforin: involvement of extracellular matrix degradation. Arterioscler Thromb Vasc Biol. 2004;24:2245–2250. doi: 10.1161/01.ATV.0000147162.51930.b7. [DOI] [PubMed] [Google Scholar]
- 136.Hernandez-Pigeon H, Jean C, Charruyer A. UVA induces granzyme B in human keratinocytes through MIF: implication in extracellular matrix remodeling. J Biol Chem. 2007;282:8157–8164. doi: 10.1074/jbc.M607436200. [DOI] [PubMed] [Google Scholar]
- 137.Barilla ML, Carsons SE. Fibronectin fragments and their role in inflammatory arthritis. Semin Arthritis Rheum. 2000;29:252–265. doi: 10.1016/S0049-0172(00)80012-8. [DOI] [PubMed] [Google Scholar]
- 138.Norris DA, Clark RA, Swigart LM. Fibronectin fragment(s) are chemotactic for human peripheral blood monocytes. J Immunol. 1982;129:1612–1618. [PubMed] [Google Scholar]
- 139.Odekon LE, Frewin MB, Del Vecchio P. Fibronectin fragments released from phorbol ester-stimulated pulmonary artery endothelial cell monolayers promote neutrophil chemotaxis. Immunology. 1991;74:114–120. [PMC free article] [PubMed] [Google Scholar]
- 140.Stanton H, Ung L, Fosang AJ. The 45 kDa collagen-binding fragment of fibronectin induces matrix metalloproteinase-13 synthesis by chondrocytes and aggrecan degradation by aggrecanases. Biochem J. 2002;364(Part 1):181–190. doi: 10.1042/bj3640181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Trapani JA, Sutton VR. Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr Opin Immunol. 2003;15:533–543. doi: 10.1016/S0952-7915(03)00107-9. [DOI] [PubMed] [Google Scholar]
- 142.Bleackley RC. A molecular view of cytotoxic T lymphocyte induced killing. Biochem Cell Biol. 2005;83:747–751. doi: 10.1139/o05-146. [DOI] [PubMed] [Google Scholar]
- 143.Chrysofakis G, Tzanakis N, Kyriakoy D. Perforin expression and cytotoxic activity of sputum CD8+ lymphocytes in patients with COPD. Chest. 2004;125:71–76. doi: 10.1378/chest.125.1.71. [DOI] [PubMed] [Google Scholar]
- 144.Hodge S, Hodge G, Nairn J. Increased airway granzyme b and perforin in current and ex-smoking COPD subjects. COPD. 2006;3:179–187. doi: 10.1080/15412550600976868. [DOI] [PubMed] [Google Scholar]
- 145.Hashimoto S, Kobayashi A, Kooguchi K. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000;161:237–243. doi: 10.1164/ajrccm.161.1.9810007. [DOI] [PubMed] [Google Scholar]
- 146.Kurumagawa T, Seki S, Kobayashi H. Characterization of bronchoalveolar lavage T cell subsets in sarcoidosis on the basis of CD57, CD4 and CD8. Clin Exp Immunol. 2003;133:438–447. doi: 10.1046/j.1365-2249.2003.02228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dourado M, Bento J, Mesquita L. [Granzymes A and B in pulmonary sarcoidosis (experimental study)] Rev Port Pneumol. 2005;11:111–133. doi: 10.1016/S0873-2159(15)30495-5. [DOI] [PubMed] [Google Scholar]
- 148.Teymoortash A, Tiemann M, Schrader C. Characterization of lymphoid infiltrates in chronic obstructive sialadenitis associated with sialolithiasis. J Oral Pathol Med. 2004;33:300–304. doi: 10.1111/j.0904-2512.2004.00093.x. [DOI] [PubMed] [Google Scholar]
- 149.Meade JL, de Wynter EA, Brett P. A family with Papillon–Lefevre syndrome reveals a requirement for cathepsin C in granzyme B activation and NK cell cytolytic activity. Blood. 2006;107:3665–3668. doi: 10.1182/blood-2005-03-1140. [DOI] [PubMed] [Google Scholar]
- 150.Pham CT, Ivanovich JL, Raptis SZ. Papillon–Lefevre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. J Immunol. 2004;173:7277–7281. doi: 10.4049/jimmunol.173.12.7277. [DOI] [PubMed] [Google Scholar]
- 151.Kook H, Zeng W, Guibin C. Increased cytotoxic T cells with effector phenotype in aplastic anemia and myelodysplasia. Exp Hematol. 2001;29:1270–1277. doi: 10.1016/S0301-472X(01)00736-6. [DOI] [PubMed] [Google Scholar]
- 152.Xu JL, Nagasaka T, Nakashima N. Involvement of cytotoxic granules in the apoptosis of aplastic anaemia. Br J Haematol. 2003;120:850–852. doi: 10.1046/j.1365-2141.2003.04147.x. [DOI] [PubMed] [Google Scholar]
- 153.Papadaki HA, Coulocheri S, Xylouri I. Defective natural killer cell activity of peripheral blood lymphocytes correlates with the degree of neutropenia in patients with chronic idiopathic neutropenia of adults. Ann Hematol. 1998;76:127–134. doi: 10.1007/s002770050376. [DOI] [PubMed] [Google Scholar]
- 154.Olsson B, Andersson PO, Jernas M. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat Med. 2003;9:1123–1124. doi: 10.1038/nm921. [DOI] [PubMed] [Google Scholar]
- 155.Wang L, Zhang F, Zhu YY. [Mechanism of cell-mediated lysis of autologous platelets in chronic idiopathic thrombocytopenic purpura] Zhonghua Yi Xue Za Zhi. 2005;85:3048–3051. [PubMed] [Google Scholar]
- 156.Zhang F, Chu X, Wang L. Cell-mediated lysis of autologous platelets in chronic idiopathic thrombocytopenic purpura. Eur J Haematol. 2006;76:427–431. doi: 10.1111/j.1600-0609.2005.00622.x. [DOI] [PubMed] [Google Scholar]
- 157.Bodemer C, Peuchmaur M, Fraitaig S. Role of cytotoxic T cells in chronic alopecia areata. J Invest Dermatol. 2000;114:112–116. doi: 10.1046/j.1523-1747.2000.00828.x. [DOI] [PubMed] [Google Scholar]
- 158.Sato-Kawamura M, Aiba S, Tagami H. Strong expression of CD40, CD54 and HLA-DR antigen and lack of evidence for direct cellular cytotoxicity are unique immunohistopathological features in alopecia areata. Arch Dermatol Res. 2003;294:536–543. doi: 10.1007/s00403-002-0354-7. [DOI] [PubMed] [Google Scholar]
- 159.Siebenhaar F, Sharov AA, Peters EM. Substance P as an immunomodulatory neuropeptide in a mouse model for autoimmune hair loss (alopecia areata) J Invest Dermatol. 2007;127:1489–1497. doi: 10.1038/sj.jid.5700704. [DOI] [PubMed] [Google Scholar]
- 160.Berthou C, Michel L, Soulie A. Acquisition of granzyme B and Fas ligand proteins by human keratinocytes contributes to epidermal cell defense. J Immunol. 1997;159:5293–5300. [PubMed] [Google Scholar]
- 161.Trivedi NR, Gilliland KL, Zhao W. Gene array expression profiling in acne lesions reveals marked upregulation of genes involved in inflammation and matrix remodeling. J Invest Dermatol. 2006;126:1071–1079. doi: 10.1038/sj.jid.5700213. [DOI] [PubMed] [Google Scholar]
- 162.Hussein MR, Abdel-Magid WM, Saleh R. Phenotypical characteristics of the immune cells in allergic contact dermatitis, atopic dermatitis and pityriasis rosea. Pathol Oncol Res. 2009;15:73–79. doi: 10.1007/s12253-008-9103-3. [DOI] [PubMed] [Google Scholar]
- 163.Yawalkar N, Hunger RE, Buri C. A comparative study of the expression of cytotoxic proteins in allergic contact dermatitis and psoriasis: spongiotic skin lesions in allergic contact dermatitis are highly infiltrated by T cells expressing perforin and granzyme B. Am J Pathol. 2001;158:803–808. doi: 10.1016/S0002-9440(10)64027-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yawalkar N, Schmid S, Braathen LR. Perforin and granzyme B may contribute to skin inflammation in atopic dermatitis and psoriasis. Br J Dermatol. 2001;144:1133–1139. doi: 10.1046/j.1365-2133.2001.04222.x. [DOI] [PubMed] [Google Scholar]
- 165.Hussein MR, Aboulhagag NM, Atta HS. Evaluation of the profile of the immune cell infiltrate in lichen planus, discoid lupus erythematosus, and chronic dermatitis. Pathology. 2008;40:682–693. doi: 10.1080/00313020802320739. [DOI] [PubMed] [Google Scholar]
- 166.van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C. Local immune response in skin of generalized vitiligo patients. Destruction of melanocytes is associated with the prominent presence of CLA+ T cells at the perilesional site. Lab Invest. 2000;80:1299–1309. doi: 10.1038/labinvest.3780138. [DOI] [PubMed] [Google Scholar]
- 167.Oyarbide-Valencia K, van den Boorn JG, Denman CJ. Therapeutic implications of autoimmune vitiligo T cells. Autoimmun Rev. 2006;5:486–492. doi: 10.1016/j.autrev.2006.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shimizu M, Higaki Y, Higaki M. The role of granzyme B-expressing CD8-positive T cells in apoptosis of keratinocytes in lichen planus. Arch Dermatol Res. 1997;289:527–532. doi: 10.1007/s004030050234. [DOI] [PubMed] [Google Scholar]
- 169.Ammar M, Mokni M, Boubaker S. Involvement of granzyme B and granulysin in the cytotoxic response in lichen planus. J Cutan Pathol. 2008;35:630–634. doi: 10.1111/j.1600-0560.2007.00892.x. [DOI] [PubMed] [Google Scholar]
- 170.Santoro A, Majorana A, Roversi L. Recruitment of dendritic cells in oral lichen planus. J Pathol. 2005;205:426–434. doi: 10.1002/path.1699. [DOI] [PubMed] [Google Scholar]
- 171.Santoro A, Majorana A, Bardellini E. Cytotoxic molecule expression and epithelial cell apoptosis in oral and cutaneous lichen planus. Am J Clin Pathol. 2004;121:758–764. doi: 10.1309/GHY8AL2D45P2R234. [DOI] [PubMed] [Google Scholar]
- 172.Wenzel J, Scheler M, Proelss J. Type I interferon-associated cytotoxic inflammation in lichen planus. J Cutan Pathol. 2006;33:672–678. doi: 10.1111/j.1600-0560.2006.00527.x. [DOI] [PubMed] [Google Scholar]
- 173.Hunger RE, Bronnimann M, Kappeler A. Detection of perforin and granzyme B mRNA expressing cells in lichen sclerosus. Exp Dermatol. 2007;16:416–420. doi: 10.1111/j.1600-0625.2007.00543.x. [DOI] [PubMed] [Google Scholar]
- 174.Wenzel J, Wiechert A, Merkel C. IP10/CXCL10–CXCR3 interaction: a potential self-recruiting mechanism for cytotoxic lymphocytes in lichen sclerosus et atrophicus. Acta Derm Venereol. 2007;87:112–117. doi: 10.2340/00015555-0194. [DOI] [PubMed] [Google Scholar]
- 175.Regauer S, Liegl B, Reich O. Vasculitis in lichen sclerosus: an under recognized feature? Histopathology. 2004;45:237–244. doi: 10.1111/j.1365-2559.2004.01929.x. [DOI] [PubMed] [Google Scholar]
- 176.Chave TA, Mortimer NJ, Sladden MJ. Toxic epidermal necrolysis: current evidence, practical management and future directions. Br J Dermatol. 2005;153:241–253. doi: 10.1111/j.1365-2133.2005.06721.x. [DOI] [PubMed] [Google Scholar]
- 177.Nassif A, Moslehi H, Le Gouvello S. Evaluation of the potential role of cytokines in toxic epidermal necrolysis. J Invest Dermatol. 2004;123:850–855. doi: 10.1111/j.0022-202X.2004.23439.x. [DOI] [PubMed] [Google Scholar]
- 178.Borchers AT, Lee JL, Naguwa SM. Stevens–Johnson syndrome and toxic epidermal necrolysis. Autoimmun Rev. 2008;7:598–605. doi: 10.1016/j.autrev.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 179.Nassif A, Bensussan A, Boumsell L. Toxic epidermal necrolysis: effector cells are drug-specific cytotoxic T cells. J Allergy Clin Immunol. 2004;114:1209–1215. doi: 10.1016/j.jaci.2004.07.047. [DOI] [PubMed] [Google Scholar]
- 180.Posadas SJ, Padial A, Torres MJ. Delayed reactions to drugs show levels of perforin, granzyme B, and Fas-L to be related to disease severity. J Allergy Clin Immunol. 2002;109:155–161. doi: 10.1067/mai.2002.120563. [DOI] [PubMed] [Google Scholar]
- 181.Hussein MR, Ali FM, Omar AE. Immunohistological analysis of immune cells in blistering skin lesions. J Clin Pathol. 2007;60:62–71. doi: 10.1136/jcp.2006.037010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wenzel J, Uerlich M, Worrenkamper E. Scarring skin lesions of discoid lupus erythematosus are characterized by high numbers of skin-homing cytotoxic lymphocytes associated with strong expression of the type I interferon-induced protein MxA. Br J Dermatol. 2005;153:1011–1015. doi: 10.1111/j.1365-2133.2005.06784.x. [DOI] [PubMed] [Google Scholar]
- 183.Takahashi H, Amagai M, Tanikawa A. T helper type 2-biased natural killer cell phenotype in patients with pemphigus vulgaris. J Invest Dermatol. 2007;127:324–330. doi: 10.1038/sj.jid.5700527. [DOI] [PubMed] [Google Scholar]
- 184.Young LH, Joag SV, Lin PY. Expression of cytolytic mediators by synovial fluid lymphocytes in rheumatoid arthritis. Am J Pathol. 1992;140:1261–1268. [PMC free article] [PubMed] [Google Scholar]
- 185.Ronday HK, van der Laan WH, Tak PP. Human granzyme B mediates cartilage proteoglycan degradation and is expressed at the invasive front of the synovium in rheumatoid arthritis. Rheumatology (Oxford) 2001;40:55–61. doi: 10.1093/rheumatology/40.1.55. [DOI] [PubMed] [Google Scholar]
- 186.Smeets TJ, Kraan MC, Galjaard S. Analysis of the cell infiltrate and expression of matrix metalloproteinases and granzyme B in paired synovial biopsy specimens from the cartilage–pannus junction in patients with RA. Ann Rheum Dis. 2001;60:561–565. doi: 10.1136/ard.60.6.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kummer JA, Tak PP, Brinkman BM. Expression of granzymes A and B in synovial tissue from patients with rheumatoid arthritis and osteoarthritis. Clin Immunol Immunopathol. 1994;73:88–95. doi: 10.1006/clin.1994.1173. [DOI] [PubMed] [Google Scholar]
- 188.Tak PP, Kummer JA, Hack CE. Granzyme-positive cytotoxic cells are specifically increased in early rheumatoid synovial tissue. Arthritis Rheum. 1994;37:1735–1743. doi: 10.1002/art.1780371205. [DOI] [PubMed] [Google Scholar]
- 189.Kim WJ, Kim H, Suk K. Macrophages express granzyme B in the lesion areas of atherosclerosis and rheumatoid arthritis. Immunol Lett. 2007;111:57–65. doi: 10.1016/j.imlet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 190.Goldbach-Mansky R, Suson S, Wesley R. Raised granzyme B levels are associated with erosions in patients with early rheumatoid factor positive rheumatoid arthritis. Ann Rheum Dis. 2005;64:715–721. doi: 10.1136/ard.2003.007039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kraan MC, Haringman JJ, Weedon H. T cells, fibroblast-like synoviocytes, and granzyme B+ cytotoxic cells are associated with joint damage in patients with recent onset rheumatoid arthritis. Ann Rheum Dis. 2004;63:483–488. doi: 10.1136/ard.2003.009225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Smeets TJ, Dolhain RJ, Breedveld FC. Analysis of the cellular infiltrates and expression of cytokines in synovial tissue from patients with rheumatoid arthritis and reactive arthritis. J Pathol. 1998;186:75–81. doi: 10.1002/(SICI)1096-9896(199809)186:1<75::AID-PATH142>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 193.Bien CG, Bauer J, Deckwerth TL. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen's encephalitis. Ann Neurol. 2002;51:311–318. doi: 10.1002/ana.10100. [DOI] [PubMed] [Google Scholar]
- 194.Bauer J, Elger CE, Hans VH. Astrocytes are a specific immunological target in Rasmussen's encephalitis. Ann Neurol. 2007;62:67–80. doi: 10.1002/ana.21148. [DOI] [PubMed] [Google Scholar]
- 195.Schwab N, Bien CG, Waschbisch A. CD8+ T-cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain. 2009;132(Pt 5):1236–1246. doi: 10.1093/brain/awp003. [DOI] [PubMed] [Google Scholar]
- 196.Bauer J, Bien CG, Lassmann H. Rasmussen's encephalitis: a role for autoimmune cytotoxic T lymphocytes. Curr Opin Neurol. 2002;15:197–200. doi: 10.1097/00019052-200204000-00012. [DOI] [PubMed] [Google Scholar]
- 197.Pouly S, Antel JP. Multiple sclerosis and central nervous system demyelination. J Autoimmun. 1999;13:297–306. doi: 10.1006/jaut.1999.0321. [DOI] [PubMed] [Google Scholar]
- 198.Rensing-Ehl A, Malipiero U, Irmler M. Neurons induced to express major histocompatibility complex class I antigen are killed via the perforin and not the Fas (APO-1/CD95) pathway. Eur J Immunol. 1996;26:2271–2274. doi: 10.1002/eji.1830260945. [DOI] [PubMed] [Google Scholar]
- 199.Kebir H, Kreymborg K, Ifergan I. Human TH17 lymphocytes promote blood–brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wanschitz J, Maier H, Lassmann H. Distinct time pattern of complement activation and cytotoxic T cell response in Guillain–Barre syndrome. Brain. 2003;126(Part 9):2034–2042. doi: 10.1093/brain/awg207. [DOI] [PubMed] [Google Scholar]
- 201.Heuss D, Probst-Cousin S, Kayser C. Cell death in vasculitic neuropathy. Muscle Nerve. 2000;23:999–1004. doi: 10.1002/1097-4598(200007)23:7<999::AID-MUS1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 202.Oka N, Takahashi M, Kawasaki T. Apoptosis of perineurial cells in sensory perineuritis. Acta Neuropathol. 2000;99:317–320. doi: 10.1007/PL00007444. [DOI] [PubMed] [Google Scholar]
- 203.Chaitanya GV, Babu PP. Multiple apoptogenic proteins are involved in the nuclear translocation of apoptosis inducing factor during transient focal cerebral ischemia in rat. Brain Res. 2008;1246:178–190. doi: 10.1016/j.brainres.2008.09.075. [DOI] [PubMed] [Google Scholar]
- 204.Chaitanya GV, Kolli M, Babu PP. Granzyme-b mediated cell death in the spinal cord-injured rat model. Neuropathology. 2009;29:270–279. doi: 10.1111/j.1440-1789.2008.00980.x. [DOI] [PubMed] [Google Scholar]
- 205.Casciola-Rosen L, Andrade F, Ulanet D. Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J Exp Med. 1999;190:815–826. doi: 10.1084/jem.190.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Blanco P, Pitard V, Viallard JF. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52:201–211. doi: 10.1002/art.20745. [DOI] [PubMed] [Google Scholar]
- 207.Lee KJ, Dong X, Wang J. Identification of human autoantibodies to the DNA ligase IV/XRCC4 complex and mapping of an autoimmune epitope to a potential regulatory region. J Immunol. 2002;169:3413–3421. doi: 10.4049/jimmunol.169.6.3413. [DOI] [PubMed] [Google Scholar]
- 208.Graham KL, Thibault DL, Steinman JB. Granzyme B is dispensable for immunologic tolerance to self in a murine model of systemic lupus erythematosus. Arthritis Rheum. 2005;52:1684–1693. doi: 10.1002/art.21092. [DOI] [PubMed] [Google Scholar]
- 209.Skoldberg F, Ronnblom L, Thornemo M. Identification of AHNAK as a novel autoantigen in systemic lupus erythematosus. Biochem Biophys Res Commun. 2002;291:951–958. doi: 10.1006/bbrc.2002.6534. [DOI] [PubMed] [Google Scholar]
- 210.Nield LE, Silverman ED, Smallhorn JF. Endocardial fibroelastosis associated with maternal anti-Ro and anti-La antibodies in the absence of atrioventricular block. J Am Coll Cardiol. 2002;40:796–802. doi: 10.1016/S0735-1097(02)02004-1. [DOI] [PubMed] [Google Scholar]
- 211.Schachna L, Wigley FM, Morris S. Recognition of Granzyme B-generated autoantigen fragments in scleroderma patients with ischemic digital loss. Arthritis Rheum. 2002;46:1873–1884. doi: 10.1002/art.10407. [DOI] [PubMed] [Google Scholar]
- 212.Ulanet DB, Flavahan NA, Casciola-Rosen L. Selective cleavage of nucleolar autoantigen B23 by granzyme B in differentiated vascular smooth muscle cells: insights into the association of specific autoantibodies with distinct disease phenotypes. Arthritis Rheum. 2004;50:233–241. doi: 10.1002/art.11485. [DOI] [PubMed] [Google Scholar]
- 213.Tapinos NI, Polihronis M, Tzioufas AG. Sjogren's syndrome. Autoimmune epithelitis. Adv Exp Med Biol. 1999;455:127–134. doi: 10.1007/978-1-4615-4857-7_18. [DOI] [PubMed] [Google Scholar]
- 214.Polihronis M, Tapinos NI, Theocharis SE. Modes of epithelial cell death and repair in Sjogren's syndrome (SS) Clin Exp Immunol. 1998;114:485–490. doi: 10.1046/j.1365-2249.1998.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Fujihara T, Fujita H, Tsubota K. Preferential localization of CD8+ alpha E beta 7+ T cells around acinar epithelial cells with apoptosis in patients with Sjogren's syndrome. J Immunol. 1999;163:2226–2235. [PubMed] [Google Scholar]
- 216.Huang M, Ida H, Kamachi M. Detection of apoptosis-specific autoantibodies directed against granzyme B-induced cleavage fragments of the SS-B (La) autoantigen in sera from patients with primary Sjogren's syndrome. Clin Exp Immunol. 2005;142:148–154. doi: 10.1111/j.1365-2249.2005.02888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kuwana M, Okano T, Ogawa Y. Autoantibodies to the amino-terminal fragment of beta-fodrin expressed in glandular epithelial cells in patients with Sjogren's syndrome. J Immunol. 2001;167:5449–5456. doi: 10.4049/jimmunol.167.9.5449. [DOI] [PubMed] [Google Scholar]
- 218.Nagaraju K, Cox A, Casciola-Rosen L. Novel fragments of the Sjogren's syndrome autoantigens alpha-fodrin and type 3 muscarinic acetylcholine receptor generated during cytotoxic lymphocyte granule-induced cell death. Arthritis Rheum. 2001;44:2376–2386. doi: 10.1002/1529-0131(200110)44:10<2376::AID-ART402>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 219.Huang M, Ida H, Arima K. La autoantigen translocates to cytoplasm after cleavage during granzyme B-mediated cytotoxicity. Life Sci. 2007;81:1461–1466. doi: 10.1016/j.lfs.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 220.Rosen A, Casciola-Rosen L. Altered autoantigen structure in Sjogren's syndrome: implications for the pathogenesis of autoimmune tissue damage. Crit Rev Oral Biol Med. 2004;15:156–164. doi: 10.1177/154411130401500304. [DOI] [PubMed] [Google Scholar]
- 221.Cherin P, Herson S, Crevon MC. Mechanisms of lysis by activated cytotoxic cells expressing perforin and granzyme-B genes and the protein TIA-1 in muscle biopsies of myositis. J Rheumatol. 1996;23:1135–1142. [PubMed] [Google Scholar]
- 222.Li M, Dalakas MC. Expression of human IAP-like protein in skeletal muscle: a possible explanation for the rare incidence of muscle fiber apoptosis in T-cell mediated inflammatory myopathies. J Neuroimmunol. 2000;106:1–5. doi: 10.1016/S0165-5728(99)00162-9. [DOI] [PubMed] [Google Scholar]
- 223.Levine SM, Raben N, Xie D. Novel conformation of histidyl-transfer RNA synthetase in the lung: the target tissue in Jo-1 autoantibody-associated myositis. Arthritis Rheum. 2007;56:2729–2739. doi: 10.1002/art.22790. [DOI] [PubMed] [Google Scholar]
- 224.Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol. 2000;12:475–481. doi: 10.1097/00002281-200011000-00001. [DOI] [PubMed] [Google Scholar]
- 225.Estella E, McKenzie MD, Catterall T. Granzyme B-mediated death of pancreatic beta-cells requires the proapoptotic BH3-only molecule bid. Diabetes. 2006;55:2212–2219. doi: 10.2337/db06-0129. [DOI] [PubMed] [Google Scholar]
- 226.Drut R, Drut RM. Lymphocytic gastritis in pediatric celiac disease—immunohistochemical study of the intraepithelial lymphocytic component. Med Sci Monit. 2004;10:CR38–CR42. [PubMed] [Google Scholar]
- 227.Suzuki T, Ito M, Hayasaki N. Cytotoxic molecules expressed by intraepithelial lymphocytes may be involved in the pathogenesis of acute gastric mucosal lesions. J Gastroenterol. 2003;38:216–221. doi: 10.1007/s005350300039. [DOI] [PubMed] [Google Scholar]
- 228.Oberhuber G, Bodingbauer M, Mosberger I. High proportion of granzyme B-positive (activated) intraepithelial and lamina propria lymphocytes in lymphocytic gastritis. Am J Surg Pathol. 1998;22:450–458. doi: 10.1097/00000478-199804000-00010. [DOI] [PubMed] [Google Scholar]
- 229.Kaserer K, Exner M, Mosberger I. Characterization of the inflammatory infiltrate in autoimmune cholangitis. A morphological and immunhistochemical study. Virchows Arch. 1998;432:217–222. doi: 10.1007/s004280050158. [DOI] [PubMed] [Google Scholar]
- 230.Ziol M, Poirel H, Kountchou GN. Intrasinusoidal cytotoxic CD8+ T cells in nodular regenerative hyperplasia of the liver. Hum Pathol. 2004;35:1241–1251. doi: 10.1016/j.humpath.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 231.Mitomi H, Ohkura Y, Yokoyama K. Contribution of TIA-1+ and granzyme B+ cytotoxic T lymphocytes to cryptal apoptosis and ulceration in active inflammatory bowel disease. Pathol Res Pract. 2007;203:717–723. doi: 10.1016/j.prp.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 232.Skjelland M, Michelsen AE, Krohg-Sorensen K. Plasma levels of granzyme B are increased in patients with lipid-rich carotid plaques as determined by echogenicity. Atherosclerosis. 2007;195:e142–e146. doi: 10.1016/j.atherosclerosis.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 233.Tsuru R, Kondo H, Hojo Y. Increased granzyme B production from peripheral blood mononuclear cells in patients with acute coronary syndrome. Heart. 2008;94:305–310. doi: 10.1136/hrt.2006.110023. [DOI] [PubMed] [Google Scholar]
- 234.Kondo H, Hojo Y, Tsuru R. Elevation of plasma granzyme B levels after acute myocardial infarction. Circ J. 2009;73:503–507. doi: 10.1253/circj.CJ-08-0668. [DOI] [PubMed] [Google Scholar]
- 235.Schiller NK, Boisvert WA, Curtiss LK. Inflammation in atherosclerosis: lesion formation in LDL receptor-deficient mice with perforin and Lyst(beige) mutations. Arterioscler Thromb Vasc Biol. 2002;22:1341–1346. doi: 10.1161/01.ATV.0000024082.46387.38. [DOI] [PubMed] [Google Scholar]
- 236.Nakajima T, Schulte S, Warrington KJ. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation. 2002;105:570–575. doi: 10.1161/hc0502.103348. [DOI] [PubMed] [Google Scholar]
- 237.Altimari A, Gruppioni E, Capizzi E. Blood monitoring of granzyme B and perforin expression after intestinal transplantation: considerations on clinical relevance. Transplantation. 2008;85:1778–1783. doi: 10.1097/TP.0b013e318177dfe4. [DOI] [PubMed] [Google Scholar]
- 238.Cashion A, Sabek O, Driscoll C. Correlation of genetic markers of rejection with biopsy findings following human pancreas transplant. Clin Transplant. 2006;20:106–112. doi: 10.1111/j.1399-0012.2005.00450.x. [DOI] [PubMed] [Google Scholar]
- 239.Cashion AK, Sabek OM, Driscoll CJ. Serial peripheral blood cytotoxic lymphocyte gene expression measurements for prediction of pancreas transplant rejection. Transplant Proc. 2006;38:3676–3677. doi: 10.1016/j.transproceed.2006.10.113. [DOI] [PubMed] [Google Scholar]
- 240.Corti B, Altimari A, Gabusi E. Potential of real-time PCR assessment of granzyme B and perforin up-regulation for rejection monitoring in intestinal transplant recipients. Transplant Proc. 2005;37:4467–4471. doi: 10.1016/j.transproceed.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 241.D'Errico A, Corti B, Pinna AD. Granzyme B and perforin as predictive markers for acute rejection in human intestinal transplantation. Transplant Proc. 2003;35:3061–3065. doi: 10.1016/j.transproceed.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 242.Kummer JA, Wever PC, Kamp AM. Expression of granzyme A and B proteins by cytotoxic lymphocytes involved in acute renal allograft rejection. Kidney Int. 1995;47:70–77. doi: 10.1038/ki.1995.8. [DOI] [PubMed] [Google Scholar]
- 243.Li B, Hartono C, Ding R. Noninvasive diagnosis of renal-allograft rejection by measurement of messenger RNA for perforin and granzyme B in urine. N Engl J Med. 2001;344:947–954. doi: 10.1056/NEJM200103293441301. [DOI] [PubMed] [Google Scholar]
- 244.Simon T, Opelz G, Wiesel M. Serial peripheral blood perforin and granzyme B gene expression measurements for prediction of acute rejection in kidney graft recipients. Am J Transplant. 2003;3:1121–1127. doi: 10.1034/j.1600-6143.2003.00187.x. [DOI] [PubMed] [Google Scholar]
- 245.Yannaraki M, Rebibou JM, Ducloux D. Urinary cytotoxic molecular markers for a noninvasive diagnosis in acute renal transplant rejection. Transpl Int. 2006;19:759–768. doi: 10.1111/j.1432-2277.2006.00351.x. [DOI] [PubMed] [Google Scholar]
- 246.Choy JC, Cruz RP, Kerjner A. Granzyme B induces endothelial cell apoptosis and contributes to the development of transplant vascular disease. Am J Transplant. 2005;5:494–499. doi: 10.1111/j.1600-6143.2004.00710.x. [DOI] [PubMed] [Google Scholar]
- 247.Choy JC, Kerjner A, Wong BW. Perforin mediates endothelial cell death and resultant transplant vascular disease in cardiac allografts. Am J Pathol. 2004;165:127–133. doi: 10.1016/S0002-9440(10)63281-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Brown TJ, Crawford SE, Cornwall ML. CD8 T lymphocytes and macrophages infiltrate coronary artery aneurysms in acute Kawasaki disease. J Infect Dis. 2001;184:940–943. doi: 10.1086/323155. [DOI] [PubMed] [Google Scholar]
- 249.Kuijpers TW, Biezeveld M, Achterhuis A. Longstanding obliterative panarteritis in Kawasaki disease: lack of cyclosporin A effect. Pediatrics. 2003;112:986–992. doi: 10.1542/peds.112.4.986. [DOI] [PubMed] [Google Scholar]
- 250.Guzman-Cottrill JA, Garcia FL, Shulman ST. CD8 T lymphocytes do not express cytotoxic proteins in coronary artery aneurysms in acute Kawasaki disease. Pediatr Infect Dis J. 2005;24:382–384. doi: 10.1097/01.inf.0000157224.25722.76. [DOI] [PubMed] [Google Scholar]
- 251.Fujinaka H, Yamamoto T, Feng L. Anti-perforin antibody treatment ameliorates experimental crescentic glomerulonephritis in WKY rats. Kidney Int. 2007;72:823–830. doi: 10.1038/sj.ki.5002424. [DOI] [PubMed] [Google Scholar]
- 252.Reynolds J, Norgan VA, Bhambra U. Anti-CD8 monoclonal antibody therapy is effective in the prevention and treatment of experimental autoimmune glomerulonephritis. J Am Soc Nephrol. 2002;13:359–369. doi: 10.1681/ASN.V132359. [DOI] [PubMed] [Google Scholar]
- 253.Clark SB, Rice TW, Tubbs RR. The nature of the myenteric infiltrate in achalasia: an immunohistochemical analysis. Am J Surg Pathol. 2000;24:1153–1158. doi: 10.1097/00000478-200008000-00014. [DOI] [PubMed] [Google Scholar]
- 254.Resnick MB, Finkelstein Y, Weissler A. Assessment and diagnostic utility of the cytotoxic T-lymphocyte phenotype using the specific markers granzyme-B and TIA-1 in esophageal mucosal biopsies. Hum Pathol. 1999;30:397–402. doi: 10.1016/S0046-8177(99)90114-4. [DOI] [PubMed] [Google Scholar]
- 255.Oberhuber G, Puspok A, Peck-Radosavlevic M. Aberrant esophageal HLA-DR expression in a high percentage of patients with Crohn's disease. Am J Surg Pathol. 1999;23:970–976. doi: 10.1097/00000478-199908000-00016. [DOI] [PubMed] [Google Scholar]
- 256.Toquet C, Hamidou MA, Renaudin K. In situ immunophenotype of the inflammatory infiltrate in eosinophilic fasciitis. J Rheumatol. 2003;30:1811–1815. [PubMed] [Google Scholar]
- 257.Yakirevich E, Yanai O, Sova Y. Cytotoxic phenotype of intra-epithelial lymphocytes in normal and cryptorchid human testicular excurrent ducts. Hum Reprod. 2002;17:275–283. doi: 10.1093/humrep/17.2.275. [DOI] [PubMed] [Google Scholar]
- 258.Ohshima K, Shimazaki K, Kume T. Perforin and Fas pathways of cytotoxic T-cells in histiocytic necrotizing lymphadenitis. Histopathology. 1998;33:471–478. doi: 10.1046/j.1365-2559.1998.00532.x. [DOI] [PubMed] [Google Scholar]
- 259.Mori N, Yatabe Y, Asai J. Immunohistochemical study of necrotizing lymphadenitis: a possible mechanism for apoptosis involving perforin and granzyme-producing cytotoxic T cells. Pathol Int. 1997;47:31–37. doi: 10.1111/j.1440-1827.1997.tb04432.x. [DOI] [PubMed] [Google Scholar]
- 260.Baetz K, Isaaz S, Griffiths GM. Loss of cytotoxic T lymphocyte function in Chediak–Higashi syndrome arises from a secretory defect that prevents lytic granule exocytosis. J Immunol. 1995;154:6122–6131. [PubMed] [Google Scholar]
- 261.Sandri M, El Meslemani AH, Sandri C. Caspase 3 expression correlates with skeletal muscle apoptosis in Duchenne and facioscapulo human muscular dystrophy. A potential target for pharmacological treatment? J Neuropathol Exp Neurol. 2001;60:302–312. doi: 10.1093/jnen/60.3.302. [DOI] [PubMed] [Google Scholar]
- 262.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Ida H, Utz PJ, Anderson P. Granzyme B and natural killer (NK) cell death. Mod Rheumatol. 2005;15:315–322. doi: 10.3109/s10165-005-0426-6. [DOI] [PubMed] [Google Scholar]
- 264.Amoura Z, Piette JC, Chabre H. Circulating plasma levels of nucleosomes in patients with systemic lupus erythematosus: correlation with serum antinucleosome antibody titers and absence of clear association with disease activity. Arthritis Rheum. 1997;40:2217–2225. doi: 10.1002/art.1780401217. [DOI] [PubMed] [Google Scholar]
- 265.Licht R, Dieker JW, Jacobs CW. Decreased phagocytosis of apoptotic cells in diseased SLE mice. J Autoimmun. 2004;22:139–145. doi: 10.1016/j.jaut.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 266.Rosen A, Casciola-Rosen L. Clearing the way to mechanisms of autoimmunity. Nat Med. 2001;7:664–665. doi: 10.1038/89034. [DOI] [PubMed] [Google Scholar]
- 267.Graham KL, Utz PJ. Sources of autoantigens in systemic lupus erythematosus. Curr Opin Rheumatol. 2005;17:513–517. doi: 10.1097/01.bor.0000171215.87993.6b. [DOI] [PubMed] [Google Scholar]
- 268.Casciola-Rosen LA, Pluta AF, Plotz PH. The DNA mismatch repair enzyme PMS1 is a myositis-specific autoantigen. Arthritis Rheum. 2001;44:389–396. doi: 10.1002/1529-0131(200102)44:2<389::AID-ANR58>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 269.Carsons S, Mosesson MW, Diamond HS. Detection and quantitation of fibronectin in synovial fluid from patients with rheumatic disease. Arthritis Rheum. 1981;24:1261–1267. [PubMed] [Google Scholar]
- 270.Scott DL, Wainwright AC, Walton KW. Significance of fibronectin in rheumatoid arthritis and osteoarthrosis. Ann Rheum Dis. 1981;40:142–153. doi: 10.1136/ard.40.2.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Homandberg GA, Meyers R, Williams JM. Intraarticular injection of fibronectin fragments causes severe depletion of cartilage proteoglycans in vivo. J Rheumatol. 1993;20:1378–1382. [PubMed] [Google Scholar]
- 272.Homandberg GA, Meyers R, Xie DL. Fibronectin fragments cause chondrolysis of bovine articular cartilage slices in culture. J Biol Chem. 1992;267:3597–3604. doi: 10.1016/S0021-9258(19)50566-X. [DOI] [PubMed] [Google Scholar]
- 273.Baker C, Petrich de Marquesini LG, Bishop AJ. Human CD8 responses to a complete epitope set from preproinsulin: implications for approaches to epitope discovery. J Clin Immunol. 2008;28:350–360. doi: 10.1007/s10875-008-9177-4. [DOI] [PubMed] [Google Scholar]
- 274.Kawasaki E, Abiru N, Eguchi K. Prevention of type 1 diabetes: from the view point of beta cell damage. Diabetes Res Clin Pract. 2004;66(Suppl 1):S27–S32. doi: 10.1016/j.diabres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 275.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–229. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 276.Serreze DV, Leiter EH. Genes and cellular requirements for autoimmune diabetes susceptibility in nonobese diabetic mice. Curr Dir Autoimmun. 2001;4:31–67. doi: 10.1159/000060527. [DOI] [PubMed] [Google Scholar]
- 277.Augstein P, Stephens LA, Allison J. Beta-cell apoptosis in an accelerated model of autoimmune diabetes. Mol Med. 1998;4:495–501. doi: 10.1007/BF03401754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Kurrer MO, Pakala SV, Hanson HL. Beta cell apoptosis in T cell-mediated autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:213–218. doi: 10.1073/pnas.94.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.O'Brien BA, Harmon BV, Cameron DP. Apoptosis is the mode of beta-cell death responsible for the development of IDDM in the nonobese diabetic (NOD) mouse. Diabetes. 1997;46:750–757. doi: 10.2337/diab.46.5.750. [DOI] [PubMed] [Google Scholar]
- 280.Thomas HE, McKenzie MD, Angstetra E, et al. Beta cell apoptosis in diabetes. Apoptosis; 26 March 2009; e-pub ahead of print. [DOI] [PubMed]
- 281.Batarelo V, Durinovic-Bello I. The sentinel role of CD8 T cells in regulating CD4 T cell responses to proinsulin in beta-islet cell autoimmunity. Ann N Y Acad Sci. 2008;1150:270–272. doi: 10.1196/annals.1447.047. [DOI] [PubMed] [Google Scholar]
- 282.Thomas HE, Kay TW. Beta cell destruction in the development of autoimmune diabetes in the non-obese diabetic (NOD) mouse. Diabetes Metab Res Rev. 2000;16:251–261. doi: 10.1002/1520-7560(200007/08)16:4<251::AID-DMRR126>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 283.Kay TW, Chaplin HL, Parker JL. CD4+ and CD8+ T lymphocytes: clarification of their pathogenic roles in diabetes in the NOD mouse. Res Immunol. 1997;148:320–327. doi: 10.1016/S0923-2494(97)87241-0. [DOI] [PubMed] [Google Scholar]
- 284.Wong FS, Janeway CA., Jr The role of CD4 vs. CD 8T cells in IDDM. J Autoimmun. 1999;13:290–295. doi: 10.1006/jaut.1999.0322. [DOI] [PubMed] [Google Scholar]
- 285.Haskins K, Wegmann D. Diabetogenic T-cell clones. Diabetes. 1996;45:1299–1305. doi: 10.2337/diab.45.10.1299. [DOI] [PubMed] [Google Scholar]
- 286.Verdaguer J, Schmidt D, Amrani A. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med. 1997;186:1663–1676. doi: 10.1084/jem.186.10.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Amrani A, Verdaguer J, Anderson B. Perforin-independent beta-cell destruction by diabetogenic CD8(+) T lymphocytes in transgenic nonobese diabetic mice. J Clin Invest. 1999;103:1201–1209. doi: 10.1172/JCI6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Han D, Xu X, Baidal D. Assessment of cytotoxic lymphocyte gene expression in the peripheral blood of human islet allograft recipients: elevation precedes clinical evidence of rejection. Diabetes. 2004;53:2281–2290. doi: 10.2337/diabetes.53.9.2281. [DOI] [PubMed] [Google Scholar]
- 289.Han D, Xu X, Pastori RL. Elevation of cytotoxic lymphocyte gene expression is predictive of islet allograft rejection in nonhuman primates. Diabetes. 2002;51:562–566. doi: 10.2337/diabetes.51.3.562. [DOI] [PubMed] [Google Scholar]
- 290.Wang T, Allie R, Conant K. Granzyme B mediates neurotoxicity through a G-protein-coupled receptor. FASEB J. 2006;20:1209–1211. doi: 10.1096/fj.05-5022fje. [DOI] [PubMed] [Google Scholar]
- 291.Zeine R, Pon R, Ladiwala U. Mechanism of gammadelta T cell-induced human oligodendrocyte cytotoxicity: relevance to multiple sclerosis. J Neuroimmunol. 1998;87:49–61. doi: 10.1016/S0165-5728(98)00047-2. [DOI] [PubMed] [Google Scholar]
- 292.Hull SM, Nutbrown M, Pepall L. Immunohistologic and ultrastructural comparison of the dermal papilla and hair follicle bulb from “active” and “normal” areas of alopecia areata. J Invest Dermatol. 1991;96:673–681. doi: 10.1111/1523-1747.ep12470601. [DOI] [PubMed] [Google Scholar]
- 293.Ranki A, Kianto U, Kanerva L. Immunohistochemical and electron microscopic characterization of the cellular infiltrate in alopecia (areata, totalis, and universalis) J Invest Dermatol. 1984;83:7–11. doi: 10.1111/1523-1747.ep12261627. [DOI] [PubMed] [Google Scholar]
- 294.Lloyd-Jones D, Adams R, Carnethon M. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:e21–181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 295.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 296.Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- 297.Kuiper J, van Puijvelde GH, van Wanrooij EJ. Immunomodulation of the inflammatory response in atherosclerosis. Curr Opin Lipidol. 2007;18:521–526. doi: 10.1097/MOL.0b013e3282efd0d4. [DOI] [PubMed] [Google Scholar]
- 298.Daugherty A, Rateri DL. Atherosclerosis: cell biology and lipoproteins. Curr Opin Lipidol. 2008;19:328–329. doi: 10.1097/MOL.0b013e3282feec55. [DOI] [PubMed] [Google Scholar]
- 299.Daugherty A, Rateri DL, Lu H. As macrophages indulge, atherosclerotic lesions bulge. Circ Res. 2008;102:1445–1447. doi: 10.1161/CIRCRESAHA.108.178947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Hansson GK, Edfeldt K. Toll to be paid at the gateway to the vessel wall. Arterioscler Thromb Vasc Biol. 2005;25:1085–1087. doi: 10.1161/01.ATV.0000168894.43759.47. [DOI] [PubMed] [Google Scholar]
- 301.Bobryshev YV, Lord RS. S-100 positive cells in human arterial intima and in atherosclerotic lesions. Cardiovasc Res. 1995;29:689–696. doi: 10.1016/S0008-6363(96)88642-1. [DOI] [PubMed] [Google Scholar]
- 302.Jonasson L, Holm J, Skalli O. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131–138. doi: 10.1161/01.ATV.6.2.131. [DOI] [PubMed] [Google Scholar]
- 303.Kaartinen M, Penttila A, Kovanen PT. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation. 1994;90:1669–1678. doi: 10.1161/01.CIR.90.4.1669. [DOI] [PubMed] [Google Scholar]
- 304.Baba T, Ishizu A, Iwasaki S. CD4+/CD8+ macrophages infiltrating at inflammatory sites: a population of monocytes/macrophages with a cytotoxic phenotype. Blood. 2006;107:2004–2012. doi: 10.1182/blood-2005-06-2345. [DOI] [PubMed] [Google Scholar]
- 305.Boivin WA, Cruz RP, Zhao H. Abstract 5488: granzyme B contributes to extracellular matrix degradation and advanced atherosclerotic plaque formation. Circulation. 2008;118:S:559–S:55b. [Google Scholar]
- 306.Chamberlain CM, Granville DJ. The role of granzyme B in atheromatous diseases. Can J Physiol Pharmacol. 2007;85:89–95. doi: 10.1139/y06-090. [DOI] [PubMed] [Google Scholar]
- 307.Dong C, Redenbach D, Wood S. The pathogenesis of cardiac allograft vasculopathy. Curr Opin Cardiol. 1996;11:183–190. doi: 10.1097/00001573-199603000-00012. [DOI] [PubMed] [Google Scholar]
- 308.Rahmani M, Cruz RP, Granville DJ. Allograft vasculopathy versus atherosclerosis. Circ Res. 2006;99:801–815. doi: 10.1161/01.RES.0000246086.93555.f3. [DOI] [PubMed] [Google Scholar]
- 309.Mennander A, Paavonen T, Hayry P. Intimal thickening and medial necrosis in allograft arteriosclerosis (chronic rejection) are independently regulated. Arterioscler Thromb. 1993;13:1019–1025. doi: 10.1161/01.ATV.13.7.1019. [DOI] [PubMed] [Google Scholar]
- 310.Cid MC, Cebrian M, Font C. Cell adhesion molecules in the development of inflammatory infiltrates in giant cell arteritis: inflammation-induced angiogenesis as the preferential site of leukocyte–endothelial cell interactions. Arthritis Rheum. 2000;43:184–194. doi: 10.1002/1529-0131(200001)43:1<184::AID-ANR23>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 311.Cid MC, Font C, Oristrell J. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum. 1998;41:26–32. doi: 10.1002/1529-0131(199801)41:1<26::AID-ART4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 312.Seko Y, Minota S, Kawasaki A. Perforin-secreting killer cell infiltration and expression of a 65-kD heat-shock protein in aortic tissue of patients with Takayasu's arteritis. J Clin Invest. 1994;93:750–758. doi: 10.1172/JCI117029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Takeda N, Takahashi T, Seko Y. Takayasu myocarditis mediated by cytotoxic T lymphocytes. Intern Med. 2005;44:256–260. doi: 10.2169/internalmedicine.44.256. [DOI] [PubMed] [Google Scholar]
- 314.Baker AL, Newburger JW. Kawasaki disease. Circulation. 2008;118:e110–e112. doi: 10.1161/CIRCULATIONAHA.107.751404. [DOI] [PubMed] [Google Scholar]
- 315.Hogg JC, Chu F, Utokaparch S. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]