

Abstract
The Janus kinase/signal transduction and activator of transcription (JAK–STAT) signaling pathway is implicated in the pathogenesis of inflammatory and autoimmune diseases including rheumatoid arthritis, psoriasis, and inflammatory bowel disease. Many cytokines involved in the pathogenesis of autoimmune and inflammatory diseases use JAKs and STATs to transduce intracellular signals. Mutations in JAK and STAT genes cause a number of immunodeficiency syndromes, and polymorphisms in these genes are associated with autoimmune diseases. The success of small-molecule JAK inhibitors (Jakinibs) in the treatment of rheumatologic disease demonstrates that intracellular signaling pathways can be targeted therapeutically to treat autoimmunity. Tofacitinib, the first rheumatologic Jakinib, is US Food and Drug Administration (FDA) approved for rheumatoid arthritis and is currently under investigation for other autoimmune diseases. Many other Jakinibs are in preclinical development or in various phases of clinical trials. This review describes the JAK–STAT pathway, outlines its role in autoimmunity, and explains the rationale/pre-clinical evidence for targeting JAK–STAT signaling. The safety and clinical efficacy of the Jakinibs are reviewed, starting with the FDA-approved Jakinib tofacitinib, and continuing on to next-generation Jakinibs. Recent and ongoing studies are emphasized, with a focus on emerging indications for JAK inhibition and novel mechanisms of JAK–STAT signaling blockade.
Keywords: Rheumatoid Arthritis Patient, Plaque Psoriasis, Tofacitinib, Stat Pathway, Ruxolitinib
Key Points
| The Janus kinase/signal transduction and activator of transcription pathway transduces downstream of multiple cytokines critical to the pathogenesis of immune-mediated disease. |
| Janus kinase inhibitors are effective treatments for rheumatoid arthritis and are under investigation for many other immune-mediated diseases including psoriasis, systemic lupus erythematosus, inflammatory bowel disease, and rare autoinflammatory diseases with a type 1 interferon signature. |
| Second-generation Janus kinase inhibitors are more selective than currently approved drugs and are being studied for therapeutic efficacy and side-effect profile. |
Introduction
Cytokines are the soluble messengers that immune cells use to communicate, ultimately modulating protective responses against pathogens [1]. Yet, cytokines may also drive the dysregulated immune responses that characterize autoimmune diseases such as rheumatoid arthritis (RA). Inflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-6 maintain and aggravate inflammation in RA: they induce activation of leukocytes, endothelial cells, and fibroblasts like synoviocytes, promote differentiation of pathogenic immune cells, and promote synthesis of metalloproteinases that erode the joint [2]. The development over the past decade and a half of agents targeting cytokines or their receptors, denoted ‘biologics’, represents a landmark advancement in the treatment of autoimmune and inflammatory diseases. These agents include monoclonal antibodies and recombinant proteins that target cytokines or cytokine receptors, such as TNF-α, IL-6, and its receptor, IL-1, and others. However, despite the therapeutic success of biologics, it has become evident that targeting a single cytokine does not completely abrogate the pathology of rheumatologic disease for all patients. In addition, many of these agents lose efficacy over time owing to immunogenicity [3] and the intravenous or subcutaneous administration of these agents is an obstacle for some patients.
Over the past 20 years, our knowledge of the intracellular pathways downstream of cytokine receptors has greatly increased, and the inhibition of intracellular enzymes such as receptor-associated kinases represents a novel way to simultaneously inhibit multiple cytokines. Small orally available molecules can be passively transported through the cellular membrane and block the intracellular activity of their targets [3–6].
The type I/II cytokine receptor family is used by several cytokines implicated in the pathogenesis of rheumatologic disease [1, 5]. This family of cytokine receptors employs the Janus kinase-signal transduction and activation of transcription (JAK–STAT) pathway to effect signal transduction [7]. Upon binding of a type I/II cytokine to its cognate receptor, receptor-associated JAKs are activated and phosphorylate STATs, transcription factors critical for the activation of cytokine-specific genetic programs. Given the major role played by JAKs and STATs in the pathogenesis of autoimmunity [8, 9], it is perhaps no surprise that small molecules targeted against JAKs, or Jakinibs, represent an emerging treatment for autoimmune and inflammatory disease. In this review, we have described the JAK–STAT pathway and its role in human diseases, and then we have discussed the efficacy and safety of individual Jakinibs in different diseases. Electronic databases of EMBASE, PubMed, and SCOPUS were searched to identify all reports published in the English language that described ‘Janus kinase’, ‘JAK/STAT pathway’, ‘role of JAK/STAT pathway in human diseases’, and ‘JAK inhibitors’. We also searched relevant conferences for abstracts describing Jakinibs or the JAK–STAT pathway, searched Clinicaltrials.gov for individual Jakinibs, and reviewed press releases from Pfizer, Lilly, Galapagos, Incyte, and Abbvie.
Structure of JAKs and STATs, and Implications for Targeted Therapy
JAKs belong to the family of tyrosine kinases (TYKs). The basic structure of all JAKs consists of four structural domains composed of seven homologous regions [JH1–7] (Fig. 1). JH1 and JH2 denote the kinase and pseudokinase domains: the name Janus is an allusion to the double-faced Roman god of gates and doors owing to the presence of these two kinase domains. JH1 is the active catalytic phosphotransferase domain and the target of the Jakinibs developed so far, which compete with adenosine triphosphate at the catalytic site. Because the JH1 domains of the four JAKs exhibit a high degree of homology not only within the JAKs but also with other TYKs, development of a selective Jakinib has been challenging.
Fig. 1.
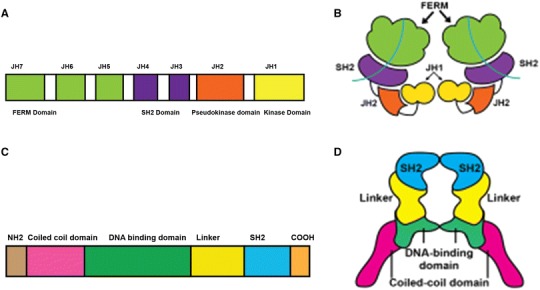
Structure of Janus kinase (JAK) and signal transduction and activation of transcription (STAT) molecules. a Linear structure of JAK molecule showing the different domains. JAKs have four functional domains: the kinase, pseudokinase, Four-point-one protein, Ezrin, Radixin and Moesin domain (FERM), and Src Homology 2 (SH2) domains. The kinase domain is the site of catalytic activity and inhibition by JAK inhibitors (Jakinibs). The FERM and pseudokinase domain interact with the kinase domain and primarily have regulatory functions. An alternative nomenclature for the domains based on their amino acid sequence classifies them as seven Janus homology (JH) domains. b Simplified three-dimensional image of JAK. The crystal structure of the FERM and SH2 domain was recently described and may contribute to receptor recognition. JH1 and JH2 are the kinase and pseudokinase domains respectively. c Linear structure of STAT molecule showing different domains. STAT proteins contain an amino terminal, a coiled coil, a DNA-binding domain (DBD), a linker, an SH2, and a transcriptional activation domain. The TAD domain is located at the C terminus and undergoes serine phosphorylation to recruit additional transcriptional activators. d Simplified three-dimensional image of STAT. Activated STAT dimers form a nutcracker-like structure as shown in this figure. The hinge of the nutcracker is formed by highly conserved SH2 domain and is commonly the target of STAT inhibitors. The linker region and DBD surround centrally located chromatin like the jaws of the nutcracker
The pseudokinase domain (JH2) was thought to have a regulatory function rather than a catalytic activity [5]: JH2 suppresses ligand-independent kinase activity through direct interactions with JH1 but is also required for ligand-induced JAK activation [10]. Recent work, however, has demonstrated that in JAK2, the JH2 has low-level catalytic activity [11, 12]. JH3 and JH4 are primarily involved in stabilizing the structural conformation of the enzyme whereas the JH5, JH6, and JH7/Four-point-one protein, Ezrin, Radixin, and Moesin domain (FERM) are critical for the association of the JAKs with their cognate receptors [13]. Notably, recent mapping of the FERM domains of receptor-bound JAK1 and TYK2 revealed striking differences in the structure that confers binding specificity, which might be exploited for the generation of selective inhibitors [5, 14–16].
The STAT transcription factors transmit type I/II cytokine signals downstream of the JAKs. STAT proteins contain an amino terminal, a coiled coil, a DNA-binding domain (DBD), a linker, a Src-homology2 (SH2), and a transcriptional activation domain (TAD) [17] (Fig. 1). Inactive cytoplasmic STATs exist primarily as monomers or preformed dimers [18]; upon activation, STAT dimers form a nutcracker-like structure. The highly conserved SH2 domain forms the hinge of this structure and is the target of most patented STAT inhibitors [19]. The linker region and DBD surround the centrally located chromatin like the jaws of the nutcracker, whereas the TAD is located at the C terminus and undergoes serine phosphorylation to recruit additional transcriptional activators and enhance transcriptional activity. These areas display the lowest sequence conservation [20]. Given the involvement of STATs in signaling events downstream of cytokine receptors as well as growth factors, STATs have long been considered as potential therapeutic targets for cancer and autoimmune disease [19]. Therefore, low-sequence conservation areas could represent a target for inhibitors, such as DNA-binding decoy oligonucleotides, with increased selectivity [21].
JAK–STAT Signaling
The JAK–STAT pathway has been used for over 500 million years as a means of intracellular signal transduction in response to cytokines and growth hormones, evolving before the divergence of protostomes from deuterostomes [22]. The canonical signaling cascade is initiated when type I/II cytokines bind to their cognate receptors (Fig. 2). Type I/II receptors are composed of distinct chains, which oligomerize upon binding of the cytokine. Oligomerization causes separation of the intracellular subunits of the cytokine receptor, which moves the receptor-associated JAKs apart from each other, relieving constitutive inhibition and resulting in their activation [1, 7, 23, 24]. The JAKs phosphorylate themselves and the intracellular portion of the receptors, which serve as docking sites for STAT transcription factors [25], which, in turn, are also phosphorylated. When phosphorylated by JAKs, inactive cytosolic STAT monomers undergo a conformational change that allows for the formation of active homodimers, heterodimers, or tetramers. The active STATs can then translocate into the nucleus where they act as transcription factors to regulate gene expression [1, 26, 27].
Fig. 2.
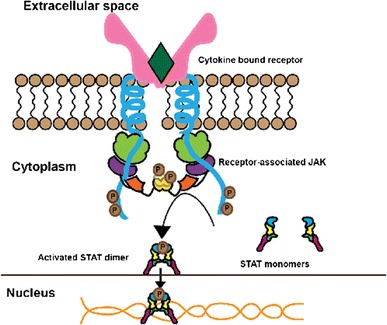
Cytokine signaling through the Janus kinase-signal transduction and activation of transcription (JAK–STAT) pathway. Binding of cytokine to the receptor leads to activation and phosphorylation of JAK and phosphorylation of the receptor. This in turn leads to phosphorylation and dimerization of STAT. Activated STAT dimer migrates to the nucleus and binds to specific DNA-binding sites regulating gene transcription. This culminates in alteration of cellular function
There are four JAK namely, JAK1, JAK2, JAK3, and TYK2 [5]. Different JAK-dependent cytokine receptors signal through different JAKs. Each receptor is composed of multiple subunits, and each subunit associates with a JAK (Fig. 3). Some receptor chains associate selectively with a specific JAK, whereas some are less selective. Thus, the extent to which a particular type I/II cytokine depends on a specific JAK to transduce signals is determined by the subunits of that cytokine’s receptor (Tables 1, 2). For example, the common γ-chain (γc), used by IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21, associates exclusively with JAK3 and is the only receptor subunit that uses JAK3 [28]. Other receptor chains are able to associate with more than one JAK isoform: for instance, the gp130 subunit can use JAK1, JAK2, and possibly TYK2 [16]. A further layer of specificity is conferred by the pairing of specific receptor chains with each other: thus, the γc pairs exclusively with JAK1-associated receptor subunits. Most type I/II cytokine receptors signal through multiple JAKs as a result of this pairing of distinct JAK-associated subunits. JAK2-associated growth factor and hormone-like cytokine receptors are the exception and their subunits have the unique property of self-pairing [5].
Fig. 3.
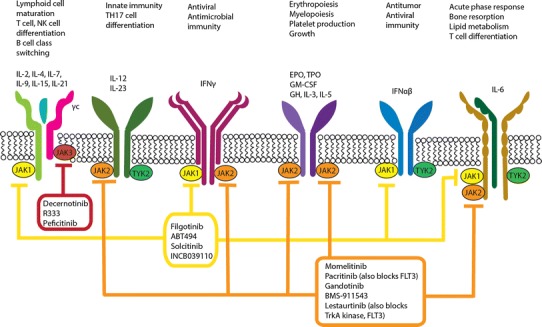
Physiological significance of Janus kinase (JAK) pathways and mechanism of action of new-generation, small-molecule JAK inhibitors (Jakinibs). Binding of various type I/II cytokines to specific receptor subunits leads to activation of specific JAK pathways. For example, γ-common chain (γc) associates only with JAK3 and mediates signaling of interleukin (IL)-2, IL-4, IL-7, IL-15, and IL-21. However, JAK1 has a broader role in cytokine signaling. Newer generation Jakinibs block specific JAK molecules compared with the first-generation Jakinibs that are non-selective. Thereby, the new-generation Jakinibs should have fewer side effects while maintaining similar efficacy as first-generation Jakinibs. However, some degree of off-target side effects such as cytopenias are seen even with selective Jakinibs such as decernotinib and ABT494. EPO erythropoietin, GH growth hormone, GM-CSF granulocyte macrophage-colony stimulating factor, IFN interferon, TH T-helper, TPO thrombopoietin, TYK tyrosine kinase
Table 1.
Type I cytokine receptors, ligands, and associated Janus kinase (JAK) and signal transduction and activator of transcription (STAT) molecules
| Common receptor chain | Ligand/cytokine | Associated JAK | Associated STAT |
|---|---|---|---|
| γ Chain | IL-2, IL-7, IL-9, IL-15, IL-21 | JAK1, JAK3 | STAT5, STAT3 |
| IL-4 | JAK1, JAK3 | STAT6 | |
| Shares IL-4Rα subunit | IL-13 | JAK1, JAK2, JAK3, TYK2 | STAT6 |
| βC | IL-3, IL-5 | JAK2 | STAT3, STAT5, STAT6 |
| GM-CSF | JAK2 | STAT3, STAT5 | |
| gp130 | IL-6, IL-11 | JAK1, JAK2, TYK2 | STAT1, STAT3 |
| IL-11 | JAK1, JAK2, TYK2 | STAT3 | |
| IL-12 | JAK 2, TYK2 | STAT4 | |
| IL-23 | TYK2, JAK2 | STAT3, STAT4 | |
| IL-27 | JAK1, JAK2, TYK2 | STAT1, STAT2, STAT3, STAT4, STAT5 | |
| GH | JAK 2 | STAT3, STAT5a | |
| EPO | JAK 2 | STAT5 | |
| TPO | JAK 2 | STAT1, STAT3, STAT5 | |
| Leptin | JAK 2 | STAT3, STAT5a | |
| G-CSF | JAK 2 | STAT5 |
G-CSF granulocyte-colony stimulating factor, IL interleukin, TPO , TYK tyrosine kinase
Table 2.
Type II cytokine receptors, ligands, and associated Janus kinase (JAK) and signal transduction and activator of transcription (STAT) molecules
| Ligand/cytokine | Associated JAK | Associated STAT |
|---|---|---|
| IFNα/β | JAK1, TYK2 | STAT1, STAT2, STAT4, sometimes STAT3 |
| IFNγ | JAK1, TYK2 | STAT1 |
| IL-10 | JAK1, JAK2, TYK2 | STAT135 |
| IL-19 | JAK1, JAK2, TYK2 | STAT3 |
| IL-20 | JAK1, JAK2, TYK2 | STAT3 |
| IL-22 | JAK1, JAK2, TYK2 | STAT1, STAT3, STAT5 |
| IL-24 | JAK1 | STAT3 |
| IL- 28 | JAK1, TYK2 | STAT1, STAT2, STAT3, STAT4, STAT5 |
| IL-29 | JAK1, TYK2 | STAT1, STAT2, STAT3, STAT4, STAT5 |
IFN interferon, IL interleukin, TYK tyrosine kinase
There are seven members of the mammalian STAT family: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. As mentioned above, upon activation of JAK-associated cytokine receptors, cytosolic STATs undergo tyrosine phosphorylation and dimerize. However, it is important to recognize a number of additional non-canonical roles for STATs. For example, STATs act not only as homodimers or heterodimers, but also as tetramers [29]. STATs can be phosphorylated by kinases other than JAKs, including Flt3R and pyruvate kinase [29]. In addition to tyrosine phosphorylation, STATs undergo serine phosphorylation in response to various external stimuli, which can augment transcriptional responses [30]. Serine phosphorylation also appears to be important for the ability of certain STATs to promote oxidative phosphorylation in mitochondria [29]. Finally, non-phosphorylated STATs are capable of dimerizing and acting as transcriptional regulators [31, 32]. STATs do not physically associate with a specific cytokine receptor but can be phosphorylated on specific tyrosine and serine residues. This results in a certain degree of functional overlap between STATs. Each member of the STAT family can be activated by multiple cytokines and their associated JAKs [33], and, in certain situations, one STAT protein can transmit signals that would normally be transduced by a different STAT.
Implications of the JAK–STAT Pathway in Inflammatory and Autoimmune Diseases
Mutations and polymorphisms in JAK and STAT genes have been linked with several human diseases, which is not surprising as a large number of cytokines and soluble factors signal through the JAK–STAT pathway [5]. Hematopoietic growth factors, including erythropoietin and thrombopoietin, signal through JAK2 [25], thus gain-of-function (GOF) mutations in JAK2 cause hematologic disorders. The most extensively described JAK2 mutation, V617F, causes polycythemia vera, essential thrombocythemia, and myelofibrosis [34, 35]. Somatic GOF mutations in JAK1 and JAK3 are also associated with hematologic malignancies such as T-cell acute lymphoblastic leukemia and solid organ malignancies such as breast cancer [36–38].
JAK1 and JAK2 deficiency phenotypes have not been described in humans, likely because the phenotype is incompatible with life: loss-of-function (LOF) mutations in either JAK is embryonically lethal in mice [39]. LOF TYK2 mutation causes a milder immunodeficiency characterized by susceptibility to viral infection [40, 41] because cells cannot respond to interferon (IFN)-γ or IFNα/β [42]. LOF mutations in JAK3 cause autosomal recessive severe combined immunodeficiency, which recapitulates the phenotype observed in patients with mutations in the γc subunit [41, 43]. T-cell and natural killer (NK) cell maturation are profoundly impaired given the importance of γc cytokines such as IL-7 and IL-15 in their development and B-cell functions are also affected. Patients present with severe recurrent infection, failure to thrive, atopic dermatitis, and chronic diarrhea. At this time, the only definitive treatment for this disease is hematopoietic stem cell transplantation. Because JAK3 is highly expressed in immune cells, patients with autosomal recessive severe combined immunodeficiency are spared from extra-immune disease manifestations. This observation formed the basis for interest in JAK3 blockade as a potential immunosuppressive therapy [5] with limited off-target effects.
STAT1 mutations resulting in either LOF or GOF have been described. Autosomal dominant LOF mutations cause Mendelian predisposition to mycobacterial diseases [33] because responses to IFNγ are impaired [44]: the mutation is dominant negative for type II IFN responses. However, signaling downstream of IFNα/β is unaffected because the mutation is autosomal recessive for type I IFN signaling. Therefore, heterozygous patients are not susceptible to viral infection. Complete biallelic STAT1 deficiency, by contrast, is purely autosomal recessive for type I and II IFN signaling. Affected patients therefore exhibit fatal susceptibility to viral disease in addition to mycobacterial infections [45]. GOF STAT1 mutations cause chronic mucocutaneous candidiasis because increased signaling downstream of IFNγ inhibits IL-17 production, ultimately causing defective responses to fungal infection. Chronic mucocutaneous candidiasis patients are also predisposed to autoimmunity, and GOF STAT1 mutations have been reported to cause a number of other autoimmune manifestations [46].
LOF mutation in the STAT2 gene causes increased susceptibility to viral infection and has also been described as a cause of sepsis-like syndrome following immunization with a live viral vaccine [47, 48]. This is consistent with the role of STAT2 in signaling downstream of type I IFNs, which are critical for immune responses to virus.
STAT3 signals downstream of IL-6 and is critical for the differentiation of T-helper (Th)17 cells, which secrete cytokines such as IL-17 and IL-22 [49, 50], among others. IL-17 is critical for immune responses to extracellular bacteria and fungi, and IL-22 promotes barrier integrity [51, 52]. Dominant-negative STAT3 mutations cause hyper immunoglobulin E syndrome, also known as Job’s syndrome, which is characterized by recurrent sinopulmonary infections, mucocutaneous candidiasis, dermatitis, elevated serum immunoglobulin E levels, and connective tissue abnormalities [53]. Activating mutations in STAT3, by contrast, cause early-onset autoimmune disease [54] with neonatal diabetes and autoimmune lymphoproliferative disease [55]. This phenotype is driven by increased STAT3 transcriptional activity, concomitant defective STAT1/5 phosphorylation, and reduced differentiation of regulatory T cells, which are important for immune tolerance [55]. Somatic STAT3 mutations are also associated with a broad range of hematologic and solid organ malignancies, which make STAT3 blockade an active area of research for therapeutic agents [56, 57].
There are two STAT5 genes, STAT5A and STAT5B. STAT5B signals downstream of many cytokines critical for immune cell growth and immune response and is particularly important for T and NK cells. As expected, LOF STAT5B mutations cause immunodeficiency [58]. However, STAT5 is also crucial for the differentiation and function of regulatory T cells, which constrain autoreactive immune responses. Thus, patients with STAT5B deficiency develop autoimmune disease. Moreover, because growth hormone signals through STAT5, the clinical spectrum of STAT5B deficiency also includes dwarfism [59].
The variety of pathology caused by JAK and STAT mutations dramatically illustrates the criticality of JAK–STAT signaling both for the normal and aberrant immune responses. Furthermore, a large body of genome-wide association studies also implicates the JAK–STAT pathway in the pathogenesis of common rheumatologic diseases [33]. For example, JAK2 polymorphisms are associated with Behçet’s disease, while single nucleotide polymorphisms in the TYK2 gene have been implicated in Crohn’s disease (CD) and lupus [25, 60, 61]. STAT3 polymorphisms are linked to CD, psoriasis, and Behcet’s disease [62, 63], whereas RA and systemic lupus erythematosus (SLE) are associated with STAT4 polymorphisms [64], and STAT6 polymorphisms have been linked to RA, atopy, and asthma [65].
Inhibitors of the JAK–STAT Pathway
What we have described so far illustrates the importance of JAKs and STAT in the homeostasis of the immune system and provides the rationale for targeting JAK–STAT signaling to treat autoimmune and inflammatory diseases. The potential of JAK inhibition as a therapeutic strategy was recognized in the 1990s [66] and fewer than 20 years later two small-molecule Jakinibs were approved by the US Food and Drug Administration (FDA): ruxolitinib for the treatment of myeloproliferative neoplasm and tofacitinib for the treatment of RA. Recently tofacitinib received positive opinion from European Medicine Agency (EMA) for its use in RA [67]. Current Jakinibs act by competitively blocking the adenosine triphosphate-binding site in the JH1 domain through non-covalent interactions [5]. Structural similarities of this binding site to the active domains of several other Tyrosine kinases presented a challenge for the development of a compound that would specifically block JAKs without off-target effects [5]. Furthermore, given the high conservation of the JH1 domain among JAKs, developing a Jakinib that would selectively block one JAK was even more challenging [5]. Despite these difficulties, multiple Jakinibs have been developed with reasonable specificity [5] and it has become apparent that pan-Jakinibs, with activity against multiple JAKs, are efficacious with an acceptable adverse-effect profile. It is still unclear if selective inhibition of a specific JAK translates into therapeutic specificity [16].
First-Generation Jakinibs
Tofacitinib
Tofacitinib was the first Jakinib approved for use in autoimmune diseases. It is a JAK1/JAK3 inhibitor with some activity against JAK2 [5, 16, 68] and negligible activity towards TYK2 [69].
Metabolism and pharmacokinetics The majority of the metabolism of tofacitinib occurs via cytochrome P450 (CYP) 3A4 and, to a lesser extent, CYP2C19 [70]. In vitro CYP3A4 inhibition with ketoconazole resulted in over 70% inhibition of metabolism [70], which supports in vivo data estimating a 103% increase in the tofacitinib area under the concentration-time curve (AUC) following ketoconazole administration [71]. Drug interaction studies with fluconazole also showed significant increases in the maximum plasma concentration (C max) and AUC of tofacitinib, resulting in dosing adjustment recommendations in the package insert for concomitant use with strong CYP3A4 inhibitors or moderate inhibitors of CYP3A4 and strong inhibitors of CYP2C19 [72, 73]. Interaction studies with rifampin, a CYP3A4 inducer, yielded lower C max and AUC values for tofacitinib; however, the pharmacodynamic and clinical significance of these changes are unknown [74]. Tofacitinib is not an inhibitor of CYP3A4 itself, as evidenced in a lack of alteration of midazolam pharmacokinetics, a CYP3A4 substrate, when co-administered with tofacitinib [75].
Tofacitinib extended release, the recently FDA-approved formulation suitable for once-daily dosing, relies on extrudable core system osmotic-delivery technology, which confers improved upper limits of drug loading vs. bilayer push-pull osmotic tablets [76]. The extended-release formulation provides equivalent total systemic exposure, C max, and minimum plasma concentration when compared with the immediate-release tablet dosed at 5 mg twice daily (BID) [76]. As expected, time to C max and elimination half-life are prolonged for the extended-release formulation [76]. Pharmacokinetic considerations and implications for drug interactions of the FDA-approved Jakinibs are shown in Table 3.
Table 3.
Pharmacokinetic properties of the selected Janus kinase (JAK) inhibitors
| Jakinib | Absorption (T max), h | Metabolism | Active metabolites | Elimination half-life, h | Excretion |
|---|---|---|---|---|---|
| Tofacitinib IR |
1 Gut bioavailability 93% |
CYP3A4 CYP2C19 |
Minimal; <10% drug-related activity 1/10 potency for JAK1 and JAK 3 vs. parent molecule |
~3 | ~30% renal excretion |
| Tofacitinib XR | 4 |
CYP3A4 CYP2C19 |
Same as IR formulation | ~5.9 | Same as IR formulation |
| Ruxolitinib | 2 |
CYP3A4 CYP2C9 |
Yes | ~3 | Negligible renal excretion |
| Baricitinib | 1.5 post-dose | ~8 | ~66% renal excretion |
CYP cytochrome P450, IR immediate releases, T max time to maximum plasma concentration, XR extended release
Topical tofacitinib ointment is currently being studied for plaque psoriasis. Pharmacokinetic data from a phase IIa trial showed quantifiable systemic concentration of the drug in 60% of the patients at one time point. The two different formulations of 2% tofacitinib ointment, composed of different vehicles, had time to maximum plasma concentration values of 0.5 and 2 h [77].
Efficacy of tofacitinib in rheumatoid arthritis Various phase II and phase III trials showed the safety and effectiveness of tofacitinib as monotherapy and in combination with other disease-modifying anti-rheumatic drugs (DMARDs) in the treatment of rheumatoid arthritis [78–89] (Table 4). The dose of tofacitinib in the reported phase II studies ranged between 1 and 30 mg BID. A dose-ranging study by Tanaka and colleagues showed incremental response in American College of Rheumatology 20% (ACR 20) with increasing doses of tofacitinib [79], but this observation was not confirmed in other clinical trials [80]. Such trials, including the phase III trials under the oral RA trials (ORAL) series, showed increased attainment of ACR 20 response with tofacitinib relative to placebo. Other measures of improvement as defined by the American College of Rheumatology (ACR 50, ACR 70), and functional status measured by the Health Assessment Questionnaire-Disability Index (HAQ-DI) and the 36-Item Short Form Health Survey were also improved (Table 4).
Table 4.
Phase II and III trials on tofacitinib in rheumatoid arthritis (RA)
| Study name | No. of subjects | Participants | Intervention | Study duration | Efficacy | Adverse events | Serious adverse events |
|---|---|---|---|---|---|---|---|
| Phase IIa, Kremer et al. [78] | 264 | Active RA, inadequate/toxic response to MTX, etanercept, infliximab or adalimumab | Tofacitinib 5, 15, and 30 mg twice daily or placebo × 6 weeks | 12 weeks | ACR 20 response rates 70.5, 81.2, and 76.8% in 5-, 15-, and 30-mg groups compared with 29.2% in placebo (p < 0.001) |
Infections (influenza, URI, UTI) 30.4% in 15-mg and 30-mg group vs. 26.2% in placebo Increase in mean LDL, HDL, and Cr (0.04–0.06 mg/dL). Sporadic neutropenia, anemia |
Gastroenteritis in 1 patient on tofacitinib 15 mg twice daily; severe leukopenia in 1 patient receiving tofacitinib 30 mg twice daily |
| Phase II, Tanaka et al. [79] | 140 | Active RA on stable MTX, inadequate response to MTX alone | Tofacitinib 1, 3, 5, and 10 mg twice daily or placebo × 12 weeks. MTX continued | 12 weeks |
ACR20 response rates: 1 mg twice daily, 64.3%; 3 mg twice daily, 77.8%; 5 mg twice daily, 96.3%; and 10 mg twice daily, 80.8% vs. placebo, 14.3%. (p < 0.0001) Significant improvement in ACR50, ACR70, HAQ-DI, and DAS28-CRP |
Nasopharyngitis, transaminitis, increase in Cr, LDL, HDL, total cholesterol | Foot deformity, osteoarthritis, femur fracture, cardiac failure, and acute dyspnea |
| Phase IIb, Kremer et al. [80] | 507 | Active RA on stable MTX, inadequate response to MTX alone | Tofacitinib (20 mg/day, 1 mg twice daily, 3 mg twice daily, 5 mg twice daily, 10 mg twice daily, or 15 mg twice daily). All patients continued stable MTX dose | 24 weeks |
ACR20 response on tofacitinib ≥3 mg BID significantly > placebo 52.9% for 3 mg, 50.7% for 5 mg, 58.1% for 10 mg, 56.0% for 15 mg, and 53.8% for 20 mg and 22% in placebo Improvements in ACR50, ACR70, HAQ-DI, DAS28-CRP |
>10% patients in tofacitinib group: diarrhea, URI, headache; transaminitis, increased cholesterol and serum creatinine, neutropenia, anemia | PNA, UTI, RTI; 1 death from PNA, severe anemia |
| Phase IIb, Fleischmann et al. [82] | 384 | Active RA, failure of at least one DMARD (lack of efficacy/toxicity), only anti malarials continued | Placebo, tofacitinib 1, 3, 5, 10, or 15 mg twice daily, or adalimumab 40 mg Q 2 weeks (total 6 injections) followed by tofacitinib 5 mg twice daily × 12 weeks | 24 weeks | ACR20 significantly improved in tofacitinib ≥3 mg groups compared with placebo. 39.2% (3 mg), 59.2% (5 mg), 70.5% (10 mg), 71.9% (15 mg) in tofacitinib group and 35.9% in adalimumab group, compared with 22.0% in placebo improvement in ACR50, and ACR70, DAS28-CRP/DAS28-ESR | UTI (7.7%), diarrhea (4.8%), headache (4.8%), and bronchitis (4.8%) | PNA, pneumococcal sepsis, acute pyelonephritis severe anemia |
| Phase III, Fleischmann et al. [83] (ORAL solo) | 611 | Active RA inadequate response to ≥1 DMARD non-biologic or biologic, off of all DMARD except antimalarialdrugs, NSAIDs, low-dose steroid permitted | Randomly assigned, 4:4:1:1, tofacitinib 5 mg twice daily × 6 months; Tofa 10 mg twice daily × 6 months; placebo × 3 months, then tofacitinib 5 or 10 mg twice daily × 3 months | 6 months; primary efficacy endpoints at 3 months | ACR 20 response significantly improved in tofacitinib groups vs. placebo (p < 0.001). 59.8% in the 5-mg tofacitinib group and 65.7% in the 10-mg tofacitinib group vs. 26.7% in placebo groups, Reductions in HAQ-DI (p < 0.001) | Headache, URI elevations in LDL, neutropenia | CCF, thrombocytopenia, cellulitis, lung Ca, uterine leiomyoma, COPD, pulmonary fibrosis, sleep apnea, PE, DVT, 1 death from heart failure |
| Phase III, Burmester et al. [84] (ORAL step) | 399 | Moderate-to-severe RA, inadequate response to TNF inhibitors. NSAIDS, low-dose steroid permitted | Randomly assigned 2:2:1:1 tofacitinib 5 mg twice daily; 10 mg twice daily; or placebo, MTX continued. At 3 months, placebo advanced to tofacitinib 5 mg twice daily or 10 mg twice daily | 6 months | ACR20 response rates 41.7% for tofacitinib 5 mg twice daily and 48.1% for tofacitinib 10 mg twice daily vs. 24.4% for placebo. Statistically significant improvements in HAQ-DI and DAS28 | Diarrhea nasopharyngitis, headache, and UTI, URI nasopharyngitis bronchitis | Panniculitis (n = 1); bronchopneumonia (n = 1) in 5 mg twice daily; pyelonephritis (n = 1) in 10 mg twice daily and diverticulitis (n = 1), aspiration PNA (n = 1) in 5 mg twice daily, pulmonary embolism (n = 1) in 10 mg twice daily |
| Phase III, Vollenhoven et al. [85] (ORAL-standard) | 717 | Active disease, inadequate response to MTX glucocorticoids (≤10 mg prednisone equivalent per day), NSAIDs permitted | Randomly assigned to tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, adalimumab 40 mg every 2 weeks, or placebo. At month 3, non-responders in placebo group switched to tofacitinib 5 mg or 10 mg; at month 6, all patients on placebo switched to tofacitinib | 12 months, results reported at 6 months | ACR 20 on 5 or 10 mg of tofacitinib were 51.5 and 52.6%, respectively, and 28.3% for placebo (p < 0.001). ACR20 47.2% for adalimumab greater reductions in HAQ-DI at month 3; higher % of patients with DAS28-ESR below 2.6 at month 6 in active-treatment groups than in the placebo group | Increase in LDL, HDL, neutropenia, anemia, transaminitis | AV block, MI, CCF, retinal detachment, GI bleed, cellulitis, herpes zoster, PNA, pulmonary TB, UTI, osteomyelitis, septic shock, fractures, cervical, ovarian and lung Ca, benign neuroma, cholesteatoma, salivary gland neoplasm |
| Phase III, Kremer et al. [86] (ORAL sync) | 792 | Active RA, inadequate response to ≥1 non-biologic or biologic DMARDs and continue background non-biologic DMARDs | Randomly assigned 4:4:1:1 to tofacitinib 5 or 10 mg twice daily, or placebo advanced to tofacitinib, 5 or 10 mg twice daily | 12 months | ACR20 response rates (month 6) for the tofacitinib 5-mg and 10-mg groups and placebo groups; were 52.1, 56.6 and 30.8%, respectively (p ≤ 0.001), HAQ-DI DAS28-ESR less than 2.6 response superior in tofacitinib groups versus placebo | Neutropenia, anemia, LDL, HDL, serum creatinine increased in tofacitinib groups | In tofacitinib groups, 2 cases of TB, 2 cases of other opportunistic infections, 3 cardiovascular events, and 4 deaths occurred |
| Phase III, van der Heijde et al. [87] (ORAL scan) | 797 |
Active RA, evidence of ≥3 joint erosions or, IgM RF or anti-CCP, on stable doses of MTX Low-dose corticosteroids and NSAIDs allowed Prior use of biologic or non-biologic DMARDs permitted |
Randomized 4:4:1:1 to tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, placebo to tofacitinib 5 mg twice daily, and placebo to tofacitinib 10 mg twice daily. At month 3, non-responder placebo-treated patients advanced to tofacitinib; remaining placebo-treated patients advanced at 6 months | 24 months (24-month interim analysis report) |
ACR20 improvement in tofacitinib 5 and 10 mg twice daily 51.5 and 61.8%, vs. 25.3% in placebo (p < 0.0001). At month 6, changes in total modified Sharp/van der Heijde score for tofacitinib 5 and 10 mg twice daily 0.12 and 0.06, respectively, vs. 0.47 for placebo (p = 0.0792 and p < 0.05, respectively) At month 3, changes in HAQ-DI score for tofacitinib was better than placebo At month 6, rates of remission for tofacitinib at 5 and 10 mg twice daily were 7.2 and 16.0% vs. 1.6% for placebo |
Lymph node TB (tofacitinib 10 mg twice daily) esophageal candidiasis (tofacitinib 5 mg twice daily and 10 mg twice daily), anemia, neutropenia, increased HDL, LDL, transaminitis | Pneumo-cystis jiroveci pneumonia (tofacitinib 5 mg twice daily), CMV sialadenitis (tofacitinib 10 mg twice daily), and CMV viremia (tofacitinib 10 mg twice daily), ARF, ARDS, cardiovascular events, malignancies |
| Phase III, Lee et al. [88] (ORAL start) | 958 | Active RA no exposure to therapeutic doses of MTX with ≥3 distinct joint erosions on hand and wrist or foot radiograph, or (+) IgM RF or anti-CCPAb | Randomly assigned to tofacitinib 5 or 10 mg of twice daily or MTX up to 20 mg per week over 8 weeks | 6 months |
ACR 20 response at 6 month: 71.3% in 5-mg group and 76.1% in 10-mg group 50.5% in the MTX group Mean changes in modified total Sharp score from baseline to month 6 were significantly smaller in tofacitinib groups than in the MTX group |
Increases in creatinine, LDL, HDL, neutropenia, lymphopenia, anemia | Herpes zoster, malignancy non-Hodgkin’s lymphoma, and chronic lymphocytic leukemia, prostate cancer, Burkitt’s B-cell lymphoma, and colon cancer |
ACR20/50/70 American College of Rheumatology 20, 50 and 70% improvement criteria, ARDS acute respiratory distress syndrome, ARF acute renal failure, AV block atrioventricular block, Ca carcinoma, CCF congestive cardiac failure, CCP cyclic citrullinated peptide, CMV cytomegalovirus, COPD chronic obstructive pulmonary disease, Cr serum creatinine, CRP C-reactive protein, DAS28 Disease Activity Score28, DMARDs disease-modifying anti-rheumatic drugs, DVT deep vein thrombosis, ESR erythrocyte sedimentation rate, GI bleed gastrointestinal bleed, HAQ-DI Health Assessment Questionnaire Disability Index, HDL high-density lipoprotein, LDL low-density lipoprotein, MI myocardial infarction, MTX methotrexate, NNT number needed to treat, NSAIDs non-steroidal anti-inflammatory drugs, PE pulmonary embolism, PNA pneumonia, PRO patient-reported outcome, PtGA patient global assessment, RF rheumatoid factor, RTI respiratory tract infections, SF-36 36-Item Short Form Health Survey, TNF tumor necrosis factor, TB tuberculosis, URI upper respiratory tract infections, UTI urinary tract infections
Several studies have examined the effect of tofacitinib on structural joint disease, assessed radiographically, with promising results. A phase II randomized controlled trial compared the effects of tofacitinib monotherapy, tofacitinib and methotrexate combination therapy, and methotrexate monotherapy on the musculoskeletal system. Magnetic resonance imaging (MRI) outcomes were reported as Outcome Measures in Rheumatology Clinical Trials RA MRI score (RAMRIS), quantitative RAMRIS, and dynamic contrast-enhanced MRI [90]. The study showed significant improvement in RAMRIS bone marrow edema at 6 months and improvement in synovitis scoring at 3 months in both tofacitinib monotherapy and tofacitinib with methotrexate combination therapy compared with methotrexate monotherapy. A significant difference was noted in synovitis scores by quantitative RAMRIS at 3 months. Erosive damage was significantly lower at 6 and 12 months in both the tofacitinib monotherapy and combination therapy groups compared with methotrexate monotherapy [91]. The phase III ORAL-Scan trial also used radiographic outcomes and showed slower rates of radiographic progression of disease in patients treated with tofacitinib at 5 or 10 mg BID with background methotrexate as measured by erosion score and joint space narrowing scores. The change in the joint space narrowing score was statistically significant at 12 months [88].
The long-term efficacy of tofacitinib in moderate to severe RA in 4000 patients was reported in the ORAL sequel study, which showed continued efficacy of the drug over 48 months as measured by ACR 20/50/70, Disease Activity Score 28-4-Erythrocyte Sedimentation Rate (DAS28-4-ESR), and HAQ-DI. [92]. Finally, a long-term extension trial collecting data on open-label tofacitinib following blinded treatment with adalimumab or tofacitinib for moderate to severe RA was reported recently, from patients in the ORAL standard and ORAL sequel trials. Results supported long-term efficacy of tofacitinib, with improved physical function and disease signs and symptoms as measured by ACR response criteria and DAS28-ESR in tofacitinib-treated patients [93].
Tofacitinib in psoriasis and psoriatic arthritis Many inflammatory cytokines critical to the pathogenesis of psoriasis signal through the JAK–STAT pathway, including type I/II IFNs, IL-12, IL-22, and IL-23 [94]. Thus, it is not unexpected that Jakinibs are an effective treatment for psoriatic skin and joint disease.
A phase I trial conducted on medically stable patients with mild to moderate psoriasis demonstrated the efficacy of tofacitinib at a dose of 10 mg BID or higher, as measured by patient global assessment and histology [95]. A subsequent phase II trial using a range of 2–15 mg BID doses of tofacitinib to treat more severe plaque disease also demonstrated statistically significant improvement in the Psoriasis Area and Severity Index (PASI) 75 and in other outcome measures including physician global assessment, PASI 50, and PASI 90 compared with placebo [96].
Phase III trials (OPT Pivotal 1 and OPT Pivotal 2 and Long Term Extension study) using oral tofacitinib at 5- and 10-mg BID dosing in patients with moderate to severe plaque psoriasis demonstrated efficacy of both doses over placebo as measured by standard criteria described above. The efficacy was maintained at 2 years in the long-term extension trial. The higher dose of 10 mg BID was found to be more efficacious [97, 98]. Indeed, comparison of both 5- and 10-mg BID doses with weekly etanercept established non-inferiority of only the 10-mg dose, whereas the 5-mg dose was inferior to TNF blockade [99, 100]. It was concluded from this result that the effective dose of tofacitinib in psoriasis would be 10 mg BID. Owing to potential safety concerns regarding the use of higher doses of tofacitinib, the drug failed to obtain FDA approval for the treatment of psoriasis. It remains to be seen whether this decision will be re-evaluated as more extensive safety data from long-term extension studies clarify the long-term risks of such treatment.
A 52-week, phase III, multisite, randomized, double-blind trial was conducted in 16 centers in Japan to study the efficacy, safety, and tolerability of tofacitinib in the treatment of psoriatic arthritis. The results showed that 62.8 and 72.7% of plaque psoriasis patients on 5 and 10 mg of tofacitinib, respectively, achieved a PASI 75. While only 12 patients had joint disease, 100% of these patients achieved ACR 20 response. Preliminary results from the larger phase III OPAL-Broaden and OPAL-Beyond studies were similarly encouraging, meeting their primary efficacy endpoints for DMARD-refractory and anti-TNF-refractory psoriatic arthritis [101]. These studies demonstrated that tofacitinib may represent a promising therapy for psoriatic arthritis, although more extensive results and future long-term studies may be needed to clarify the effects of tofacitinib on structural joint disease [102].
Topical tofacitinib in psoriasis A phase IIa, multi-center, double-blind, vehicle-controlled study was conducted to evaluate the efficacy, safety, tolerability, and systemic pharmacokinetics of topical tofacitinib in mild-to-moderate plaque psoriasis. Two different tofacitinib ointment formulations were assessed, both were administered BID for 4 weeks to a single fixed 300-cm2 area with one target plaque with or without one or more non-target plaques and normal skin. The primary endpoint, defined as the percentage change from baseline in the Target Plaque Severity Score, showed statistically significant improvement by about 50% only for one of the ointments at week 4 [77, 103].
Tofacitinib in inflammatory bowel disease Inflammatory bowel disease (IBD) comprises two major distinct entities: ulcerative colitis (UC) and CD. While the two forms of IBD exhibit many common clinical features, they are pathophysiologically distinct and may not respond to tofacitinib in the same way. Although a full understanding of IBD immunopathogenesis is lacking, it appears that both forms result from dysregulated immune responses. The role of type I/II cytokines in IBD is well established: IL-12, type II IFN, and IL-6 promote the function of pathogenic innate lymphoid cells and T cells [104]. However, patients are also immunodeficient: they are unable to constrain pathogenic microbiota. Moreover, anti-inflammatory cytokines such as IL-22 and IL-17A may have a protective role [104]. Thus, type I/II cytokine blockade may have unintended consequences.
Tofacitinib is being considered for the treatment of UC. After promising results from initial phase II trials [105–107], the efficacy of tofacitinib as induction therapy was assessed in two phase III trials in patients with moderate-to-severe UC (OCTAVE Induction 1 and 2), which were recently completed. Preliminary reports indicate that the trials met their primary and secondary endpoints, although full results are not yet available [108]. The OCTAVE Sustain trial, which examines the efficacy of tofacitinib as maintenance therapy, and the long-term extension OCTAVE open trial will provide more data regarding the efficacy of JAK inhibition in the treatment of UC. Approval of tofacitinib for UC will likely depend on the final outcomes of these studies [109].
Results for patients treated with tofacitinib for CD are less clear. A randomized, multi-center, phase II clinical trial including 139 patients with moderate-to-severe active CD receiving tofacitinib 1, 5, or 15 mg or placebo BID for 4 weeks did not show clinical efficacy. However, the study showed a statistically significant reduction in serum C-reactive protein (CRP) and fecal calprotectin levels in subjects receiving the 15-mg dose. The reasons for the negative results of the trial are unclear but may be related to the short study duration, possibly limiting the ability of tofacitinib to show any significant improvement in clinical response [109, 110]. Preliminary data from subsequent studies indicate a small effect for tofacitinib treatment in CD [111]; however, it is not clear whether such effects are clinically significant. Results also indicate that tofacitinib may be effective as maintenance therapy [112], although further data from an ongoing long-term clinical trial will answer this question more definitively (http://www.clinicaltrials.gov NCT01393626).
Ruxolitinib
The first FDA-approved Jakinib, ruxolitinib is a JAK1 and JAK2 inhibitor [5] with moderate inhibitory activity against Tyk2 [113]. As mentioned above, ruxolitinib was developed for the treatment of polycythemia vera and intermediate- and high-risk primary myelofibrosis [33, 114], where inappropriate activation of JAK2 underlies disease pathogenesis, and is FDA approved for these diseases. Ruxolitinib is also effective in the treatment of essential thrombocythemia [115] and has been granted Breakthrough Therapy Designation for the treatment of graft vs. host disease [116].
Metabolism and pharmacokinetics Ruxolitinib and tofacitinib have similar pharmacokinetic profiles but ruxolitinib has more active metabolites and lower renal excretion [70]. Ruxolitinib is metabolized primarily by CYP3A4 and to a lesser extent, CYP2C19 (Table 3). Pharmacokinetic and pharmacodynamic studies using ketoconazole and erythromycin, strong and intermediate inhibitors of CYP3A4 as well as rifampin, a CYP3A4 inducer, were conducted to determine the impact on the metabolism of ruxolitinib [117]. Co-administration of ketoconazole with single-dose ruxolitinib resulted in an increase in drug exposure for ruxolitinib of 91% and a prolongation of the elimination half-life of ruxolitinib of approximately 2 h. Co-administration of erythromycin, a moderate CYP3A4 inhibitor, with ruxolitinib exhibited much less significant impact on C max and drug exposure of ruxolitinib. Pharmacokinetic studies of ruxolitinib with rifampin resulted in a 52% decrease in C max of ruxolitinib with a decrease in terminal half-life of approximately 50% [117]. The pharmacodynamic impact of these interactions were assessed through an assay evaluating the extent of inhibition of STAT3 phosphorylation. Co-administration of ketoconazole resulted in a doubling of STAT3 phosphorylation inhibition, which was clinically and statistically significant. Co-administration of erythromycin and rifampin resulted in a 13% increase or a 10% decrease of pharmacodynamic activity respectively. These changes were not considered to be clinically significant [117]. Further pharmacokinetic modeling studies evaluated the effect of fluconazole, a moderate inhibitor of both CYP3A4 and CYP2C19, and estimated a two-fold increase in ruxolitinib AUC with fluconazole doses of the 100–200 mg total daily dose. This same modeling study also evaluated the impact of ruxolitinib on p-glycoprotein efflux pumps and did not predict a significant impact of ruxolitinib co-administration with p-glycoprotein substrates including digoxin [118].
Use of ruxolitinib in autoimmune diseases Studies using ruxolitinib to treat various inflammatory and autoimmune diseases have been promising. A phase IIa trial of ruxolitinib in RA showed encouraging results with improvement in ACR 20, 50, and 70 and HAQ-DI as compared with placebo after 28 days [113, 119]. A case of chilblain lupus erythematosus has been successfully treated with oral ruxolitinib [120]. Improvement in muscle strength and skin lesions was also reported in a patient with dermatomyositis and post-polycythemia vera JAK2 V617F-positive myelofibrosis [121]. Moreover, remarkable improvements in patients with alopecia areata treated with oral ruxolitinib for 3–5 months have been reported. Comparison of biopsy samples at baseline and after 12 weeks of treatment demonstrated decreased inflammation post-treatment [122].
Topical Ruxolitinib
Ruxolitinib has been reported to have significant cutaneous anti-inflammatory action [123], and effects on plaque psoriasis were investigated in a small placebo-controlled clinical trial (86). Topical ruxolitinib was well tolerated and superior to placebo in reducing the plaque area [124]. Topical ruxolitinib has also been reported to be useful in alopecia [125].
Baricitinib
Baricitinib is another selective JAK1/JAK2 inhibitor that inhibits intracellular signaling of multiple proinflammatory cytokines including IL-6, IL-12, IL-23, and IFNγ [126].
Metabolism and pharmacokinetics Renal clearance is the primary route of excretion for baricitinib; therefore, the role of CYP-mediated medication interactions are thought to be minimal for this drug [127]. It is speculated that the half-life would be prolonged in disease states with reduced renal function [127] (Table 3).
Efficacy of baricitinib in RA Baricitinib has progressed to phase III studies in RA. Phase IIb studies have demonstrated the efficacy of baricitinib at 4- and 8-mg dosing in RA unresponsive to methotrexate over a 12- to 24-week study period [128, 129]. Improvement in musculoskeletal MRI findings was demonstrated along with clinical response in a phase IIb substudy [130].
Table 5 summarizes important phase III trials on baricitinib in RA [131–134]. The trials unequivocally established the efficacy of baricitinib in active RA with improvement in all the measures of ACR response criteria. There was significant improvement in ACR 20/50/70, DAS28, and HAQ-DI in the subjects treated with baricitinib compared with placebo. The RA-BEAM (A Study in Moderate to Severe Rheumatoid Arthriris) study demonstrated superiority of baricitinib over adalimumab, a landmark finding not achieved with any other disease-modifying agent. The long-term extension of this study at 24 and 52 weeks showed prevention of progressive radiographic structural joint damage with baricitinib [134]. Similar radiographic improvement was shown in the RA-BUILD (A Study in Moderate to Severe Rheumatoid Arthritis Participants) study as well, where a change in the medial tibia stress syndrome score at week 24 was significantly lower in the baricitinib group compared with placebo [133]. An interesting finding in this study was the rapid improvement in ACR criteria within a week whereas, inability to respond to the drug within 4 weeks was predictive of future failure. This information could be used to prevent unnecessary drug exposure in non-responders [135].
Table 5.
Baricitinib in rheumatoid arthritis (RA)
| Study name | No.of subjects | Participants | Intervention | Study duration | Efficacy | Adverse events | Serious adverse events |
|---|---|---|---|---|---|---|---|
| Genovese et al. [132] (RA-BEACON) | 527 | Active RA with inadequate response/unacceptable side effects with ≥TNF inhibitors, other biologics DMARDs, or both | Randomly assigned in 1:1:1 baricitinib 2 or 4 mg daily or placebo for 24 weeks | 24 weeks | Baricitinib 4, 2 mg and placebo had ACR20 response at 12 weeks 55, 49 and 27% (p < 0.001). Significant difference in HAQ-DI and DAS28-CRP between higher-dose baricitinib group and placebo group | Mild neutropenia, increased serum creatinine, LDL, herpes zoster | Fatal stroke, MI, non melanomatous skin cancers |
| Dougados et al. [133] (RA-BUILD) | 684 | Active RA and inadequate response to conventional DMARDs | Randomized 1:1:1 to placebo or baricitinib (2 or 4 mg) daily with stable background treatment | 24 weeks | ACR20 response at week 12 was 62% with baricitinib 4 mg, 66% in 2 mg, and 40% with placebo (p ≤ 0.001). Improvements in ACR50, ACR70, DAS28, CDAI, SDAI, and HAQ-DI | Similar in all groups, no opportunistic infection, no GI perforation | Tuberculosis: 1 case, Non-melanomatous skin cancer: 1 case in baricitinib 4 mg |
| Fleischmann et al. [131] (RA-BEGIN) | 584 | Active RA, no previous DMARD other than ≤3 doses of MTX | Randomized 4:3:4 to MTX, baricitinib 4 mg once daily (baricitinib monotherapy), or baricitinib 4 mg QD + MTX for up to 52 weeks | Results reported at 24 weeks | ACR20 response higher with baricitinib 4 mg monotherapy vs. MTX (77% vs. 62%, p ≤ 0.01). Compared with MTX, baricitinib 4 mg monotherapy produced significantly greater improvements in ACR 50, 70, CDAI, SDAI, DAS28-CRP, HAQ-DI, MTX and baricitinib 4 mg combination was not superior to baricitinib monotherapy |
Similar in all groups Herpes zoster, anemia, leukopenia, transaminitis |
PJP, esophageal candidiasis in combination group |
| Taylor et al. [134] (RA-BEAM) | 1305 | Active RA on stable background MTX | Randomized 3:3:2 to placebo, baricitinib 4 mg once daily, or adalimumab 40 mg biweekly | 24 weeks, results reported at 12 and 24 weeks | ACR20 higher for baricitinib vs. placebo (70 vs. 40%, p ≤ 0.001). ACR20 with adalimumab 61%. Significant improvements in ACR 20/50/70 and HAQ-DI, DAS28, CDAI, and SDAI for baricitinib vs. placebo. Compared with ADA, baricitinib superior in ACR20 and DAS28-CRP. Diminished radiographic progression with Bari vs. placebo | Anemia, leukopenia, transaminitis, 3 opportunistic infections |
1 case of pneumonia and 1 duodenal ulcer hemorrhage 5 malignancies, 1 case of TB |
ACR20/50/70 American College of Rheumatology 20, 50 and 70% improvement criteria, ARDS acute respiratory distress syndrome, ARF acute renal failure, AV atrioventricular, Ca carcinoma, CCF congestive cardiac failure, CCP cyclic citrullinated peptide, CMV cytomegalovirus, COPD chronic obstructive pulmonary disease, Cr serum creatinine, CRP C-reactive protein, DAS28 Disease Activity Score 28, DMARD disease-modifying anti-rheumatic drugs, DVT deep vein thrombosis, ESR erythrocyte sedimentation rate, GI gastrointestinal, HAQ-DI Health Assessment Questionnaire-Disability Index, HDL high-density lipoprotein, LDL low-density lipoprotein, MI myocardial infarction, MTX methotrexate, NNT number needed to treat, NSAIDs non-steroidal anti-inflammatory drugs, PE pulmonary embolism, PJP pneumocystis jirovecii pneumonia, PNA pneumonia, PRO patient-reported outcome, PtGA patient global assessment, RF rheumatoid factor, RTI respiratory tract infections, SF-36 36-Item Short Form Health Survey, TB tuberculosis, TNF tumor necrosis factor, URI upper respiratory tract infections, UTI urinary tract infections
Following the RA-BUILD study, patient-reported outcome measures including pain, functional disability, and fatigue showed significant improvement with baricitinib therapy [136]. An Extension Study in Participants with Moderate to Severe Rheumatoid Arthritis (RA-BEYOND) is currently recruiting participants. The purpose of this study is to investigate the long-term safety and any side effects of baricitinib in participants who have completed a previous baricitinib RA study. The study will provide for 4 years of additional treatment with baricitinib (http://www.clinicaltrials.gov NCT01885078).
Baricitinib and other diseases Baricitinib has been shown to improve PASI 75 scores in plaque psoriasis in a phase IIb trial [137]. Baricitinib is also extremely effective in the treatment of autoinflammatory diseases characterized by an IFN gene signature, such as chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome [138]. Similar to ruxolitinib, baricitinib was found to be effective in alopecia areata in a patient who received this drug for the treatment of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome [139].
Oclacitinib
Oclatinib is a pan-Jakinib approved for canine eczema and atopic dermatitis [25, 140]. The efficacy of oclacitinib for canine atopic disease hints that Jakinibs may represent a promising therapeutic strategy for the treatment of allergic diseases in humans, and preliminary results indicate that tofacitinib may also be efficacious for atopic dermatitis [141]. Clinical trials are ongoing to further evaluate the efficacy of systemic and topical Jakinibs for this class of diseases.
Safety of Jakinibs: Lessons from Clinical Trials
Because Jakinibs simultaneously block signaling downstream of cytokines important for a range of physiological functions, their side effects can often be directly linked to their mechanism of action. Safety concerns include effects on hematopoiesis, innate and adaptive host defense, as well as growth. Because tofacitinib is the most extensively studied Jakinib, most available safety data are derived from clinical trials where this drug was used. These studies have demonstrated an acceptable safety profile [33], and the safety profiles of other Jakinibs appear comparable.
Infection secondary to immunosuppression represents a major concern in Jakinib-treated patients. Common side effects in RA clinical trials included infections such as nasopharyngitis or upper respiratory infections, bronchitis, and gastroenteritis. A number of opportunistic infections such as herpes zoster, tuberculosis, cellulitis, panniculitis, septic shock, and osteomyelitis were also reported [3, 142]. The observed risk was similar to that with other DMARDS, and a retrospective meta-analysis of pooled data from all the trials and extension studies indicated a lower risk of infection in tofacitinib-treated patients than for patients treated with biological DMARDs [142]. The exception to this is varicella zoster virus, for which the risk of reactivation is substantially higher in tofacitinib-treated patients [143, 144]. This increased risk may be in part owing to the importance of JAK3-dependent cytokines in driving the development and functions of NK cells, which are important for controlling viral infections such as herpes zoster [142]. However, NK cell counts are not markedly reduced in tofacitinib-treated patients; therefore, the etiology of zoster reactivation may be related to NK cell function or to effects on a different leukocyte population. Among other serious opportunistic infections, BK viremia and nephropathy have been reported in kidney transplant recipients treated with high-dose tofacitinib (30 and 15 mg BID) in combination with mycophenolate mofetil [145]. A larger multi-center clinical trial also showed a higher incidence (14–18%) of BK nephropathy in renal transplant recipients treated with tofacitinib compared with cyclosporine (6%) [146], also in combination with mycophenolate mofetil and at relatively high doses. Progressive multifocal leukoencephalopathy has been reported in a 75-year-old man treated with ruxolitinib for myelofibrosis [147].
Like infectious complications, Jakinib-driven cytopenias were expected because many hematopoietic growth factors including erythropoietin and granulocyte macrophage-colony stimulating factor signal through JAK2. Neutropenia and anemia were indeed observed albeit sporadically. A higher incidence of mild-to-moderate anemia, leukopenia, neutropenia, lymphopenia, and thrombocytopenia was observed in patients receiving tofacitinib 30 mg compared with patients on lower doses of tofacitinb [78]. In patients taking lower doses, cytopenias were typically mild and did not necessitate discontinuation of the drug.
One particular concern with long-term suppression of the JAK–STAT pathway is the possible development of malignancies. Both type I and II IFNs play an important role in the process of immunoediting, which is critical for the anti-tumor immune response [148]. In post-transplant patients treated with tofacitinib, the risk of lymphoproliferative malignancy was increased by JAK inhibition [149]. However, the phase II and III trials for autoimmune diseases have not shown an increased cancer risk associated with tofacitinib treatment [92]. The incidence of malignancy, including lymphoma and non-melanomatous skin cancer, is similar to that seen with other biologics.
The increase in low-density lipoprotein (LDL), high-density lipoprotein (HDL), and a few cardiovascular events such as atrioventricular block, congestive heart failure, and myocardial infarction were observed in tofacitinib-treated patients. However, long-term extension studies have not shown evidence of an increased rate of cardiovascular events [92]. Combined data from phase III trials on tofacitinib demonstrated stabilization of lipid levels after 3 months of treatment and the incidence of cardiovascular adverse events was similar to placebo [150]. The effect of tofacitinib on the lipid profile is similar to tocilizumab and may be secondary to blocking IL-6 signaling. The physiological role of IL-6 on hepatic lipid metabolism is complex and incompletely understood [151]. IL-6 is known to cause insulin resistance and high IL-6 levels have been noted in obesity. IL-6 also support the redistribution of fatty acids from the blood to peripheral tissues, which results in low serum levels of LDL, HDL, and triglycerides [25]. To date, long-term extension studies have not shown an increase in major cardiovascular events in tofacitinib-treated patients relative to those treated with placebo [150, 152]. Tofacitinib was actually shown to reduce vascular stiffness in RA patients in a small study of 18 patients [153]. Assessment of cholesterol and lipoprotein kinetics in RA patients before and after tofacitinib treatment in comparison to healthy volunteers revealed increased levels of cholesterol in RA patients after treatment and is secondary to reduced cholesterol ester catabolism and anti-atherogenic HDL level improvement [154].
Other changes in laboratory parameters included sporadic elevations in transaminases and creatinine. Clinically significant hepatic and renal compromise have not been reported; however, [92] a phase I randomized controlled trial assessed changes in serum creatinine and glomerular filtration rate in RA patients treated with tofacitinib and compared them with patients who received placebo. Tofacitnib treatment caused mild increases in creatinine (5%) and decreases in glomerular filtration rate (8%), which reversed rapidly upon drug discontinuation [155].
Next-Generation Jakinibs
Whereas first-generation Jakinibs including tofacitinib have shown efficacy in the treatment of inflammatory conditions like RA, nonselective pan-JAK blockade can be associated with unwanted adverse effects such as cytopenias. This raises the potential utility of next-generation Jakinibs with selective inhibitory activity for a specific JAK (Fig. 3), which, in principle might be used to treat selected autoimmune disorders with fewer adverse effects [156]. However, increased selectivity may also translate into reduced efficacy.
Decernotinib (VX-509)
Decernotinib is a next-generation Jakinib with in vitro kinase assays demonstrating five-fold selectivity towards JAK3 compared with JAK1, JAK2, and TYK2 [157]. Decernotinib showed promising results in animal models of autoimmune diseases [156] and was therefore moved into clinical trials for the treatment of RA.
Metabolism and pharmacokinetics Decernotinib possesses a unique pharmacokinetic profile with potential implications for medication interactions. The major metabolite of decernotinib, M3, acts as a potent inhibitor of CYP3A4 [158]. CYP3A4 is the most prevalent hepatic CYP enzyme and is implicated in metabolism for over 50% of currently marketed medications [159]. The clinical implications of M3’s inhibition of CYP3A4 may be far reaching, as one of the phase IIb dercernotinib studies excluded not only subjects taking moderate or strong inhibitors or inducers of CYP3A4 and p-glycoprotein, but also subjects taking any medication metabolized via CYP3A4 with the potential for toxicity at high levels of exposure [160]. In addition, adverse effects of this drug included elevations in lipid parameters [80, 132, 160]. Notably, several high-potency, commonly used hydroxymethylglutaryl Co-A reductase inhibitors (statins) including simvastatin and atorvastatin are metabolized via CYP3A4 [161]. Therefore, concomitant use with decernotinib could potentially increase the risk for statin-associated toxicity.
Decernotinib in RA Clinical data using decernotinib in RA have initially been promising. Phase II trials demonstrated efficacy at doses of 50–150 mg BID, with improvement of ACR response criteria and DAS28 joints using the CRP level (DAS28-CRP) compared with placebo. Adverse events reported were similar to first-generation Jakinibs such as infections, transaminitis, and hyperlipidemia [126, 160, 162]. Anemia was not observed, consistent with selectivity for JAK3 over JAK2. Surprisingly, neutropenia was seen in a large number of patients, which may indicate that the drug could have some off-target effects [157]. A phase IIb study recently demonstrated improvement in synovitis and osteitis with decernotinib along with conventional DMARDs in RA patients [163].
Filgotinib GLPG0634
Filgotinib inhibits both JAK1 and JAK2 in whole blood cell-based assays and kinase assays but displays an 30-fold selectivity towards JAK1 [126]. In vitro studies also demonstrated a dose-dependent inhibition of Th1 and Th2 and, to lesser extent, Th17 cell differentiation.
Metabolism and pharmacokinetics In vitro analysis of the impact of filgotinib and its active metabolite on CYP enzymes indicate that neither agent inhibits nor induces CYP activity at clinically relevant concentrations [164]. This conclusion was confirmed for CYP3A4 in a study on the impact of filgotinib on midazolam clearance in healthy volunteers, which showed no changes in midazolam metabolism [164]. The potential of filgotinib to impact cell-based transport systems including p-glycoprotein and breast cancer resistance protein were examined in vitro, with the authors concluding that filgotinib was unlikely to inhibit these transport systems. The effects of filgotinib on organic cation transporters including organic cation transporter 2 were also examined in vitro with observed inhibition, but this is of unknown clinical significance. The potential for filgotinib to affect organic anion transporters implicated in methotrexate (MTX) clearance was explored, with no changes in MTX clearance observed in doses of filgotinib up to 300 mg [164]. It appears that the unique pharmacokinetic profile of filgotinib may provide flexibility in dosing regimens (146).
Filgotinib in autoimmune diseases Filgotinib is currently is being investigated for the treatment of RA [165]. Phase IIA studies in patients with active RA and inadequate response to MTX showed efficacy of filgotinib over placebo at doses of 30 mg daily and higher [126, 166]. This was followed by two phase IIb trials: DARWIN1 and DARWIN 2. DARWIN1 is a study on 595 MTX-treated RA patients where filgotinib was added at a range of doses from 50 mg daily to 100 mg BID. The DARWIN2 study assessed filgotinib monotherapy in 280 RA patients with doses ranging from 50 to 200 mg daily [126]. In both studies, filgotinib was found to be superior to placebo in controlling disease activity as measured by ACR 20/50, DAS28-CRP, Simple Disease Activity Index (SDAI), and clinical disease activity index [167, 168].
Filgotinib is also being investigated in moderate to severe CD (FITZROY study) [109, 169]. Preliminary data from the trial showed significantly improved clinical outcomes and quality of life as measured by the clinical disease activity index and the inflammatory bowel disease questionnaire, respectively, with figlotinib compared with placebo [170].
Safety profiles in both DARWIN trials and the FITZROY trial were all favorable. In the FITZROY study, filgotinib showed a favorable lipid profile with an increase in HDL and no change in LDL, resulting in an improved atherogenic index. An increase in hemoglobin was also observed and no clinically significant changes from baseline in neutrophils or liver function tests were observed, consistent with intact signaling through JAK2. Notably, a trial in patients with SLE was closed for lack of efficacy [171].
ABT494
ABT-494 is a next-generation Jakinib with 74-fold selectivity for JAK1 over JAK2, based on the drug’s ability to bind JAK1 outside the adenosine triphosphate-binding site of JH1 in addition to the adenosine triphosphate binding site. Because the binding occurs with a less conserved domain, it is described as being specific for JAK1 [172]. Importantly, as JAK2 and JAK3 signaling remain unaffected, ABT-494 does not affect erythropoietin signaling or reduce peripheral NK cell counts at therapeutic doses [126].
Two multi-center, randomized, double-blind, placebo-controlled phase IIb studies (BALANCE I and II) were conducted in subjects with moderate to severe RA taking MTX who did not respond to either anti-TNF therapy (BALANCE I) or MTX (BALANCE II) (http://www.clinicaltrials.gov NCT01960855). Both studies demonstrated rapid improvement in ACR 20/50/70 and DAS28-CRP with ABT-494 compared with placebo. Improvements were observed as early as week 2 with ABT4-94 [173, 174]. Patient recruitment is currently ongoing for a phase III, double-blind, placebo-controlled study in RA with inadequate response to MTX, comparing ABT-494 with adalimumab on a stable background dose of MTX (http://www.clinicaltrials.gov NCT02629159).
Peficitinib (ASP015K)
Peficitinib (ASP015K) is a novel, orally bioavailable JAK inhibitor that inhibits JAK1, JAK2, JAK3, and TYK2 enzyme activities with moderate selectivity for JAK3 inhibition. Inhibition of JAK2 by peficitinib is relatively mild, which confers an acceptable safety profile [175] and some potential advantages over first-generation inhibitors.
Metabolism and pharmacokinetics Similar to other Jakinibs, peficitinib exhibits rapid oral absorption. Interestingly, in metabolic studies, peficitinib possessed no single dominant clearance pathway [176]. To date, clinically significant drug interactions with peficitinib have not been identified, and the major phase II trial with this agent lacked exclusion criteria based on potential drug interactions that may affect clearance of the Jakinib [175].
Peficitinib in autoimmune/inflammatory diseases Peficitinib reduced paw swelling and ankle bone destruction in a preclinical model of rat adjuvant-induced arthritis [177]. Early clinical studies have also been promising. A phase II trial in RA patients showed a statistically significant ACR 20 response compared with placebo at a range of doses from 25 to 150 mg [175]. A phase IIa trial in plaque psoriasis demonstrated dose-dependent efficacy of peficitinib in PASI, body surface area (BSA), Physician Static Global Assessment, and histological measures of severity after 6 weeks [178]. No major adverse events were reported by either of these trials.
Solcitinib (GSK2586184)
Solcitinib is another selective JAK1 inhibitor that has been evaluated for the treatment of moderate-to-severe plaque-type psoriasis. A 12-week, randomized, placebo-controlled clinical trial in moderate-to-severe plaque psoriasis revealed significant improvement in PASI 75 scores with solcitinib compared with placebo [179].
Because JAK1 transmits signals downstream of type I IFNs, and patients with SLE have evidence of aberrant type I IFN signaling, solcitinib was therefore assessed in a phase II, randomized, placebo-controlled study of patients with moderate-to-severe SLE. However, two cases of drug reaction with eosinophilia and systemic symptoms syndrome and severe but reversible liver function test abnormalities in six subjects were reported, necessitating early termination of the study [180, 181]. What this means for the use of other Jakinibs in SLE remains unclear.
INCB039110
INCB039110 is a next-generation JAK inhibitor with selective inhibitory action against JAK1. INCB-039110 was demonstrated as having a >20-fold selectivity for JAK1 over JAK2 and a >200-fold selectivity over JAK3. Preclinical studies supported its efficacy in mouse adjuvant arthritis models, at doses that did not inhibit the biological activity of erythropoietin. INCB039110 has also been shown to inhibit inflammatory pathways involved in the pathogenesis of psoriasis [182, 183].
A phase II, multi-center clinical trial using this drug to treat active RA demonstrated clinically significant improvement in ACR 20/50/70 and DAS28-CRP compared with placebo (http://www.clinicaltrials.gov NCT01626573) [126, 184], at doses ranging from 100 mg BID to 600 mg daily. A phase II clinical trial in plaque psoriasis was also encouraging, with improvement in PASI 50/75, PSGA, and affected body surface area in patients receiving INCB039110 compared with placebo [182]. Common adverse effects were similar to those seen with nonselective Jakinibs and included infectious nasopharyngitis, elevated transaminases, and hypertriglyceridemia [182].
Jakinibs in Preclinical Development
Several biotechnology companies are in the process of developing JAK inhibitors with the goal of creating a molecule with maximum efficacy and minimum off target effects [126]. The newer Jakinibs are isoform specific, which is postulated to diminish adverse events found with first generation non-selective Jakinibs. Some of the new-generation Jakinibs are covalently bound to specific sequences of the JAKs, resulting in better selectivity [126]. Table 6 summarizes additional JAK inhibitors in various stages of clinical and preclinical development.
Table 6.
Janus Kinase Inhibitors in preclinical and early clinical development
| Drug | Specificity | Clinical Status | Diseases |
|---|---|---|---|
| OP0155 [199] | JAK3 | Preclinical | Rat adjuvant induced arthritis |
| VR588 [200] | Pan JAK inhibitor (inhalational) | Preclinical | Asthma |
| SHR0302 [201] | JAK1, JAK2, JAK3 (strongest binding to JAK1) | Phase 1 | Rheumatoid arthritis |
| Pf-04965842 [202] | JAK1 | Phase 2b (ClinicalTrials.gov. NCT02780167) | Moderate To Severe Atopic Dermatitis |
| JTE-052 [203] | JAK1, 2, 3 and Tyk2 | Phase 2 (in Japan) | Atopic dermatitis, auoimmune disorders |
JAK Janus kinase
STAT Inhibition
As JAK substrates and key signaling molecules downstream of type I/II cytokine receptors, STATs have been investigated as an attractive target in the treatment of inflammation and autoimmunity, as well as malignancy. STAT3 is essential for signaling downstream of IL-6, which regulates the production of IL-17 by T cells and other immune cells, implicating STAT3 in the pathogenesis of many rheumatologic diseases [185, 186]. Moreover, constitutive activation of STAT3 and STAT5 has been observed in several human cancers and cancer cell lines [50].
Blocking the action of transcription factors, however, is much more challenging than inhibiting the activity of enzymes such as kinases. Challenges to the development of STAT inhibitors include issues with bioavailability and selectivity. For example, considerable homology exists between STAT1 and STAT3. STAT1 facilitates vital functions including cell death, apoptosis, and pathogen defense, thus off-target STAT1 blockade by a STAT3 inhibitor can lead to a host of undesired adverse effects such as increased survival of tumor cells [33, 187, 188]. Another challenge in developing STAT inhibitors is functional redundancy in the action of different STATs. Specifically, although STAT3 is critical for signaling downstream of IL-6, STAT3-deficient cells continue to respond to IL-6 stimulation through activation of STAT1 [189]. Thus, selective blockade of one STAT molecule may not be clinically useful, as another STAT might compensate for the inhibited protein.
Despite these limitations, several small-molecule inhibitors targeting the SH2 domain have been developed and tested in phase I and II clinical trials. One of these, OPB-31121, was studied in a phase I/II trial for hepatocellular carcinoma but had an unacceptable adverse effect profile, particularly peripheral neuropathy, and limited efficacy [190]. Another inhibitor, OPB-51602, appears safe and effective in the treatment of solid organ malignancies such as lung cancer [57] but is also associated with a high risk of peripheral neuropathy and has poor bioavailability, further limiting its tolerability [57, 191]. STAT6 plays an important role in allergic pathways acting downstream to IL-4 and IL-13. Phosphopeptides blocking the SH2 domain of STAT6 are being developed to inhibit phosphorylation and further downstream signaling of this pathway, which may be useful in allergic diseases such as bronchial asthma [192].
Because of difficulties with small-molecule inhibitors that target the SH2 domain, several other mechanisms of inhibition are being explored. STAT3 decoy oligonucleotides target the DNA-binding domain of the STAT and are much more selective than inhibitors that target the SH2 domain. However, phase 0 studies revealed that the first generation of decoys degraded quickly in vivo, limiting their effectiveness [19]. Development of intrabodies against phosphorylated forms of the STAT3 molecule have been shown to be effective in in vitro studies [193] and may represent another method of successfully targeting STATs therapeutically. Nonetheless, however promising STAT inhibition may be in the treatment of malignancy and autoimmunity, there are currently no clinical trials of STAT inhibitors actively recruiting patients.
Future Prospects for JAK–STAT Signaling Modulators in Inflammatory/Autoimmune Diseases
Given the number of cytokines that signal through the JAK–STAT pathway, it is no surprise that Jakinibs have become the first kinase inhibitors used successfully in the treatment of rheumatologic disease. At present, tofacitinib is the only Jakinib approved for autoimmune disease, but several more may soon follow as new data emerge and the development of novel agents continues. Clinical trials are ongoing with various Jakinibs in several autoimmune conditions ranging from rheumatoid arthritis to psoriasis. Similarly, diseases characterized by serum elevations of JAK-dependent cytokines could respond well to Jakinibs. These include diseases for which a type I IFN signature has been defined such as SLE, myositis, scleroderma, and primary Sjogren’s syndrome, as well as diseases driven by IL-6 such as relapsing polychondritis and large-vessel vasculitis. Several trials are indeed probing the efficacy of Jakinibs in the treatment of diseases characterized by an IFN signature, based on preclinical data [194] and on the observation that baricitinib is effective for the treatment of monogenic interferonopathies such as chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome and SAVI [stimulator of interferon genes (STING) associated vasculopathy with onset in infancy], which are also associated with a type I IFN signature [138]. Moreover, the preliminary success of Jakinibs in the treatment of alopecia and other skin-related diseases such as vitiligo is very exciting as no existing therapy has been effective in these relatively common conditions so far [195–197].
Optimal dosing strategies of Jakinibs also deserve further study, whereas currently approved regimens rely on a single dose for induction of remission and maintenance, it is not clear that this is the optimal strategy. It is possible that loading patients with high doses to induce remission, followed by a lower maintenance dose, would be a more effective strategy, as is the case for corticosteroids. Investigations of non-oral formulations of Jakinibs are also in the preliminary stages for diseases including psoriasis, allergic dermatitis, and ocular disease. Finally, biomarker development is ongoing to identify groups of patients with disease likely to respond to Jakinibs, as complex rheumatologic diseases may be driven by JAK-dependent cytokines to a different degree in different patients [198]. After the era of biologics, the development of Jakinibs and their successful use in autoimmune and inflammatory diseases heralds an exciting new chapter in rheumatology. These drugs are unique both structurally and functionally, as small molecules that can be administered orally rather than injected, and which simultaneously block multiple cytokines downstream of their receptors. In the coming years, Jakinibs are poised to change the field of autoimmune and rheumatologic diseases as ongoing basic, preclinical, and clinical research will surely determine the feasibility and benefits of selectivity, optimized dosing, formulation, and patient selection to minimize undesirable off-target effects and maximize clinical efficacy.
Acknowledgements
We are grateful to Dr. John O’Shea for his supervision and critical revision in the preparation of the manuscript.
Compliance with Ethical Standards
Funding
No sources of funding were used to support the writing of this article.
Conflict of interest
All the authors including S. Banerjee, A. Biehl, M. Gadina, S. Hasni, and D. Schwartz state that there are no conflicts of interest.
Footnotes
An erratum to this article is available at http://dx.doi.org/10.1007/s40265-017-0772-7.
An erratum to this article is available at http://dx.doi.org/10.1007/s40265-017-0736-y.
References
- 1.O’Shea JJ, Kontzias A, Yamaoka K, et al. Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis. 2013;72(Suppl. 2):ii111–5. doi:10.1136/annrheumdis-2012-202576. [DOI] [PMC free article] [PubMed]
- 2.Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2:16039. doi: 10.1038/nrdp.2016.39. [DOI] [PubMed] [Google Scholar]
- 3.Yamaoka K. Janus kinase inhibitors for rheumatoid arthritis. Current Opin Chem Biol. 2016;32:29–33. doi: 10.1016/j.cbpa.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Hodge JA, Kawabata TT, Krishnaswami S, et al. The mechanism of action of tofacitinib: an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2016;34(2):318–328. [PubMed] [Google Scholar]
- 5.Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57(12):5023–5038. doi: 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]
- 6.Galluzzo M, D’Adamio S, Servoli S, et al. Tofacitinib for the treatment of psoriasis. Exp Opin Pharmacother. 2016;17(10):1421–1433. doi: 10.1080/14656566.2016.1195812. [DOI] [PubMed] [Google Scholar]
- 7.Leonard WJ, O’Shea JJ. Jaks and STATs: biological implications. Ann Rev Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- 8.Walker JG, Ahern MJ, Coleman M, et al. Characterisation of a dendritic cell subset in synovial tissue which strongly expresses Jak/STAT transcription factors from patients with rheumatoid arthritis. Ann Rhem Dis. 2007;66(8):992–999. doi: 10.1136/ard.2006.060822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Isomaki P, Junttila I, Vidqvist KL, et al. The activity of JAK–STAT pathways in rheumatoid arthritis: constitutive activation of STAT3 correlates with interleukin 6 levels. Rheumatology. 2015;54(6):1103–1113. doi: 10.1093/rheumatology/keu430. [DOI] [PubMed] [Google Scholar]
- 10.Hammaren HM, Ungureanu D, Grisouard J, et al. ATP binding to the pseudokinase domain of JAK2 is critical for pathogenic activation. Proc Natl Acad Sci USA. 2015;112(15):4642–4647. doi: 10.1073/pnas.1423201112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ungureanu D, Wu J, Pekkala T, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol. 2011;18(9):971–976. doi: 10.1038/nsmb.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Min X, Ungureanu D, Maxwell S, et al. Structural and functional characterization of the JH2 pseudokinase domain of JAK family tyrosine kinase 2 (TYK2) J Biol Chem. 2015;290(45):27261–27270. doi: 10.1074/jbc.M115.672048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babon JJ, Liau NP, Kershaw NJ. JAK1 Takes a FERM hold of type II cytokine receptors. Structure. 2016;24(6):840–842. doi: 10.1016/j.str.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Ferrao R, Wallweber HJ, Ho H, et al. The structural basis for class II cytokine receptor recognition by JAK1. Structure. 2016;24(6):897–905. doi: 10.1016/j.str.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallweber HJ, Tam C, Franke Y, et al. Structural basis of recognition of interferon-alpha receptor by tyrosine kinase 2. Nat Struct Mol Biol. 2014;21(5):443–448. doi: 10.1038/nsmb.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vijayakrishnan L, Venkataramanan R, Gulati P. Treating inflammation with the Janus kinase inhibitor CP-690,550. Trends Pharmacol Sci. 2011;32(1):25–34. doi: 10.1016/j.tips.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Lim CP, Cao X. Structure, function, and regulation of STAT proteins. Mol Biosyst. 2006;2(11):536–550. doi: 10.1039/b606246f. [DOI] [PubMed] [Google Scholar]
- 18.Babon JJ, Lucet IS, Murphy JM, et al. The molecular regulation of Janus kinase (JAK) activation. Biochem J. 2014;462(1):1–13. doi: 10.1042/BJ20140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai PS, Rosa DA, Magdy Ali A, et al. A STAT inhibitor patent review: progress since 2011. Exp Opin Ther Pat. 2015;25(12):1397–1421. doi: 10.1517/13543776.2015.1086749. [DOI] [PubMed] [Google Scholar]
- 20.Liongue C, Taznin T, Ward AC. Signaling via the CytoR/JAK/STAT/SOCS pathway: emergence during evolution. Mol Immunol. 2016;71:166–175. doi: 10.1016/j.molimm.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Sen M, Thomas SM, Kim S, et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2(8):694–705. doi: 10.1158/2159-8290.CD-12-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liongue C, Ward AC. Evolution of the JAK–STAT pathway. JAK–STAT. 2013;2(1):e22756. doi: 10.4161/jkst.22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brooks AJ, Dai W, O’Mara ML, et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. 2014;344(6185):1249783. doi: 10.1126/science.1249783. [DOI] [PubMed] [Google Scholar]
- 24.Mavers M, Ruderman EM, Perlman H. Intracellular signal pathways: potential for therapies. Curr Rheumatol Rep. 2009;11(5):378–385. doi: 10.1007/s11926-009-0054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz DM, Bonelli M, Gadina M, O’Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. 2016;12(1):25–36. doi: 10.1038/nrrheum.2015.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mao X, Ren Z, Parker GN, et al. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol Cell. 2005;17(6):761–771. doi: 10.1016/j.molcel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 27.O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368(2):161–170. doi: 10.1056/NEJMra1202117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hofmann SR, Ettinger R, Zhou YJ, et al. Cytokines and their role in lymphoid development, differentiation and homeostasis. Curr Opin Allergy Clin Immunol. 2002;2(6):495–506. doi: 10.1097/00130832-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol. 2015;194(1):21–27. doi: 10.4049/jimmunol.1401867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morinobu A, Gadina M, Strober W, et al. STAT4 serine phosphorylation is critical for IL-12-induced IFN-gamma production but not for cell proliferation. Proc Natl Acad Sci USA. 2002;99(19):12281–12286. doi: 10.1073/pnas.182618999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Decker T. Emancipation from transcriptional latency: unphosphorylated STAT5 as guardian of hematopoietic differentiation. EMBO J. 2016;35(6):555–557. doi: 10.15252/embj.201693974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohr A, Chatain N, Domoszlai T, et al. Dynamics and non-canonical aspects of JAK/STAT signalling. Eur J Cell Biol. 2012;91(6–7):524–532. doi: 10.1016/j.ejcb.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 33.O’Shea JJ, Schwartz DM, Villarino AV, et al. The JAK–STAT pathway: impact on human disease and therapeutic intervention. Ann Rev Med. 2015;66:311–328. doi: 10.1146/annurev-med-051113-024537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basquiera AL, Soria NW, Ryser R, et al. Clinical significance of V617F mutation of the JAK2 gene in patients with chronic myeloproliferative disorders. Hematology. 2009;14(6):323–330. doi: 10.1179/102453309X12473408860226. [DOI] [PubMed] [Google Scholar]
- 35.Levine RL. JAK-mutant myeloproliferative neoplasms. Curr Top Microbiol Immunol. 2012;355:119–133. doi: 10.1007/82_2011_170. [DOI] [PubMed] [Google Scholar]
- 36.Jeong EG, Kim MS, Nam HK, et al. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 2008;14(12):3716–3721. doi: 10.1158/1078-0432.CCR-07-4839. [DOI] [PubMed] [Google Scholar]
- 37.Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2009;106(23):9414–9418. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elliott NE, Cleveland SM, Grann V, et al. FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood. 2011;118(14):3911–3921. doi: 10.1182/blood-2010-12-319467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodig SJ, Meraz MA, White JM, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93(3):373–383. doi: 10.1016/S0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 40.Macchi P, Villa A, Giliani S, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID) Nature. 1995;377(6544):65–68. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- 41.Russell SM, Tayebi N, Nakajima H, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. 1995;270(5237):797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- 42.Sohn SJ, Barrett K, Van Abbema A, et al. A restricted role for TYK2 catalytic activity in human cytokine responses revealed by novel TYK2-selective inhibitors. J Immunol. 2013;191(5):2205–2216. doi: 10.4049/jimmunol.1202859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36(4):515–528. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dupuis S, Dargemont C, Fieschi C, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293(5528):300–303. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- 45.Dupuis S, Jouanguy E, Al-Hajjar S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33(3):388–391. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 46.Uzel G, Sampaio EP, Lawrence MG, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol. 2013;131(6):1611–1623. doi: 10.1016/j.jaci.2012.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hambleton S, Goodbourn S, Young DF, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci USA. 2013;110(8):3053–3058. doi: 10.1073/pnas.1220098110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dasgupta A, Chen KH, Tian L, Archer SL. Gone fission: an asymptomatic STAT2 mutation elongates mitochondria and causes human disease following viral infection. Brain. 2015;138(Pt 10):2802–2806. doi: 10.1093/brain/awv237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong Z, Wen Z, Darnell JE., Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264(5155):95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 50.Abroun S, Saki N, Ahmadvand M, et al. STATs: an old story, yet mesmerizing. Cell J. 2015;17(3):395–411. doi: 10.22074/cellj.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conti HR, Whibley N, Coleman BM, et al. Signaling through IL-17C/IL-17RE is dispensable for immunity to systemic, oral and cutaneous candidiasis. PloS One. 2015;10(4):e0122807. doi: 10.1371/journal.pone.0122807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolk K, Kunz S, Witte E, et al. IL-22 increases the innate immunity of tissues. Immunity. 2004;21(2):241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 53.Freeman AF, Holland SM. The hyper-IgE syndromes. Immunol Allergy Clin North Am. 2008;28(2):277–91, viii. doi:10.1016/j.iac.2008.01.005. [DOI] [PMC free article] [PubMed]
- 54.Flanagan SE, Haapaniemi E, Russell MA, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Gen. 2014;46(8):812–814. doi: 10.1038/ng.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milner JD, Vogel TP, Forbes L, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. 2015;125(4):591–599. doi: 10.1182/blood-2014-09-602763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koskela HL, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905–1913. doi: 10.1056/NEJMoa1114885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wong AL, Soo RA, Tan DS, et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann Oncol. 2015;26(5):998–1005. doi: 10.1093/annonc/mdv026. [DOI] [PubMed] [Google Scholar]
- 58.Cohen AC, Nadeau KC, et al. Cutting edge: decreased accumulation and regulatory function of CD4 + CD25(high) T cells in human STAT5b deficiency. J Immunol. 2006;177(5):2770–2774. doi: 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- 59.Kofoed EM, Hwa V, Little B, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349(12):1139–1147. doi: 10.1056/NEJMoa022926. [DOI] [PubMed] [Google Scholar]
- 60.Sigurdsson S, Nordmark G, Goring HH, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Gen. 2005;76(3):528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Gen. 2010;42(12):1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu K, Hou S, Jiang Z, et al. JAK2 and STAT3 polymorphisms in a Han Chinese population with Behcet’s disease. Invest Ophthalmol Vis Sci. 2012;53(1):538–541. doi: 10.1167/iovs.11-8440. [DOI] [PubMed] [Google Scholar]
- 63.Ellinghaus D, Ellinghaus E, Nair RP, et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am J Hum Gen. 2012;90(4):636–647. doi: 10.1016/j.ajhg.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Remmers EF, Plenge RM, Lee AT, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357(10):977–986. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duetsch G, Illig T, Loesgen S, et al. STAT6 as an asthma candidate gene: polymorphism-screening, association and haplotype analysis in a Caucasian sib-pair study. Hum Mol Gen. 2002;11(6):613–621. doi: 10.1093/hmg/11.6.613. [DOI] [PubMed] [Google Scholar]
- 66.Cacalano NA, Migone TS, Bazan F, et al. Autosomal SCID caused by a point mutation in the N-terminus of Jak3: mapping of the Jak3-receptor interaction domain. EMBO J. 1999;18(6):1549–1558. doi: 10.1093/emboj/18.6.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pfizer receives positive CHMP opinion in europe for XELJANZ® (tofacitinib citrate) for the treatment of moderate to severe active rheumatoid arthritis receives positive CHMP opinion in Europe for XELJANZ® (tofacitinib citrate) for the treatment of moderate to severe active rheumatoid arthritis pfizer receives positive CHMP opinion in Europe for XELJANZ® (tofacitinib citrate) for the treatment of moderate to severe active rheumatoid arthritis receives positive CHMP opinion in Europe for xeljanz (tofacitinib citrate) for the treatment of moderate to severe active rheuamtoid arthritis. http://www.pfizer.com/news/press-release/press-releasedetail/pfizer_receives_positive_chmp_opinion_in_europe_for_xeljanz_tofacitinib_citrate_for_the_treatment_of_moderate_to_severe_active_rheumatoid_arthritis. . Accessed 10 Feb 2017.
- 68.Changelian PS, Flanagan ME, Ball DJ, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302(5646):875–878. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]
- 69.Zerbini CA, Lomonte AB. Tofacitinib for the treatment of rheumatoid arthritis. Exp Rev Clin Immunol. 2012;8(4):319–331. doi: 10.1586/eci.12.19. [DOI] [PubMed] [Google Scholar]
- 70.Dowty ME, Lin J, Ryder TF, et al. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a Janus kinase inhibitor, in humans. Drug Metab Dispos. 2014;42(4):759–773. doi: 10.1124/dmd.113.054940. [DOI] [PubMed] [Google Scholar]
- 71.Gupta P, Wang R, Kaplan I, et al. A phase 1 study to estimate the the effect of ketoconazole on the pharmacokinetics of tofacitinib (CP-690,550) in healthy volunteers [abstract no. P1-64] Clin Pharmacol Ther. 2012;91(Suppl. 1):S31. [Google Scholar]
- 72.Chow V, Ni G, LaBadie R, Chan G. An open label study to estimate the effect of fluconazole on the pharmacokinetics of CP-690,550 in healthy adult subjects (Abstract) Clin Pharmacol Ther. 2008;83(Suppl 36):PI–93. [Google Scholar]
- 73.Xeljanz® [package insert]. New York: Pfizer Labs; 2016.
- 74.Lambda M, Wang R, Kaplan I, Salageanu J, Tarabar S, Krishnaswami S. The effect of rifampin on the pharmacokinetics of tofacitinib (CP-690,550) in healthy volunteers (Abstract no. PI-73) Clin Pharmacol Ther. 2012;91(Suppl. 1):S35. [Google Scholar]
- 75.Gupta P, Alvey C, Wang R, et al. Lack of effect of tofacitinib (CP-690,550) on the pharmacokinetics of the CYP3A4 substrate midazolam in healthy volunteers: confirmation of in vitro data. Br J Clin Pharmacol. 2012;74(1):109–115. doi: 10.1111/j.1365-2125.2012.04168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lamba M, Wang R, Fletcher T, Alvey C, Kushner J, Stock TC. Extended-release once-daily formulation of tofacitinib: evaluation of pharmacokinetics compared with immediate-release tofacitinib and impact of food. J Clin Pharmacol. 2016;56(11):1362–1371. doi: 10.1002/jcph.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ports WC, Khan S, Lan S, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. 2013;169(1):137–145. doi: 10.1111/bjd.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kremer JM, Bloom BJ, Breedveld FC, et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60(7):1895–1905. doi: 10.1002/art.24567. [DOI] [PubMed] [Google Scholar]
- 79.Tanaka Y, Suzuki M, Nakamura H, et al. Tofacitinib study I: phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res. 2011;63(8):1150–1158. doi: 10.1002/acr.20494. [DOI] [PubMed] [Google Scholar]
- 80.Kremer JM, Cohen S, Wilkinson BE, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64(4):970–981. doi: 10.1002/art.33419. [DOI] [PubMed] [Google Scholar]
- 81.Coombs JH, Bloom BJ, Breedveld FC, et al. Improved pain, physical functioning and health status in patients with rheumatoid arthritis treated with CP-690,550, an orally active Janus kinase (JAK) inhibitor: results from a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2010;69(2):413–416. doi: 10.1136/ard.2009.108159. [DOI] [PubMed] [Google Scholar]
- 82.Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367(6):495–507. doi: 10.1056/NEJMoa1109071. [DOI] [PubMed] [Google Scholar]
- 83.Fleischmann R, Cutolo M, Genovese MC, et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 2012;64(3):617–629. doi: 10.1002/art.33383. [DOI] [PubMed] [Google Scholar]
- 84.Burmester GR, Blanco R, Charles-Schoeman C, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381(9865):451–460. doi: 10.1016/S0140-6736(12)61424-X. [DOI] [PubMed] [Google Scholar]
- 85.van Vollenhoven RF, Fleischmann R, Cohen S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367(6):508–519. doi: 10.1056/NEJMoa1112072. [DOI] [PubMed] [Google Scholar]
- 86.Kremer J, Li ZG, Hall S, et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159(4):253–261. doi: 10.7326/0003-4819-159-4-201308200-00006. [DOI] [PubMed] [Google Scholar]
- 87.van der Heijde D, Tanaka Y, Fleischmann R, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013;65(3):559–570. doi: 10.1002/art.37816. [DOI] [PubMed] [Google Scholar]
- 88.Lee EB, Fleischmann R, Hall S, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370(25):2377–2386. doi: 10.1056/NEJMoa1310476. [DOI] [PubMed] [Google Scholar]
- 89.Strand V, van Vollenhoven RF, Lee EB, Fleischmann R, Zwillich SH, Gruben D, et al. Tofacitinib or adalimumab versus placebo: patient-reported outcomes from a phase 3 study of active rheumatoid arthritis. Rheumatology. 2016;55(6):1031–41. doi: 10.1093/rheumatology/kev442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ostergaard M, Edmonds J, McQueen F, et al. An introduction to the EULAR-OMERACT rheumatoid arthritis MRI reference image atlas. Ann Rheum Dis. 2005;64(Suppl. 1):i3–i7. doi: 10.1136/ard.2004.031773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Conaghan PG, Ostergaard M, Bowes MA, Wu C, Fuerst T, van der Heijde D, et al. Comparing the effects of tofacitinib, methotrexate and the combination, on bone marrow oedema, synovitis and bone erosion in methotrexate-naive, early active rheumatoid arthritis: results of an exploratory randomised MRI study incorporating semiquantitative and quantitative techniques. Ann Rheum Dis. 2016;75(6):1024–33. doi: 10.1136/annrheumdis-2015-208267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wollenhaupt J, Silverfield J, Lee EB, et al. Safety and efficacy of tofacitinib, an oral janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, longterm extension studies. J Rheumatol. 2014;41(5):837–852. doi: 10.3899/jrheum.130683. [DOI] [PubMed] [Google Scholar]
- 93.Genovese MC, van Vollenhoven RF, Wilkinson B, et al. Switching from adalimumab to tofacitinib in the treatment of patients with rheumatoid arthritis. Arthritis Res Ther. 2016;18:145. doi: 10.1186/s13075-016-1049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coates LC, FitzGerald O, Helliwell PS, Paul C. Psoriasis, psoriatic arthritis, and rheumatoid arthritis: is all inflammation the same? Semin Arthritis Rheum. 2016. doi:10.1016/j.semarthrit.2016.05.012. [DOI] [PubMed]
- 95.Boy MG, Wang C, Wilkinson BE, et al. Double-blind, placebo-controlled, dose-escalation study to evaluate the pharmacologic effect of CP-690,550 in patients with psoriasis. J Invest Dermatol. 2009;129(9):2299–2302. doi: 10.1038/jid.2009.25. [DOI] [PubMed] [Google Scholar]
- 96.Papp KA, Menter A, Strober B, et al. Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: a phase 2b randomized placebo-controlled dose-ranging study. Br J Dermatol. 2012;167(3):668–677. doi: 10.1111/j.1365-2133.2012.11168.x. [DOI] [PubMed] [Google Scholar]
- 97.Papp KA, Menter MA, Abe M, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173(4):949–961. doi: 10.1111/bjd.14018. [DOI] [PubMed] [Google Scholar]
- 98.Papp KA, Krueger JG, Feldman SR, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: long-term efficacy and safety results from 2 randomized phase-III studies and 1 open-label long-term extension study. J Am Acad Dermatol. 2016. doi:10.1016/j.jaad.2016.01.013. [DOI] [PubMed]
- 99.Bachelez H, van de Kerkhof PC, Strohal R, et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: a phase 3 randomised non-inferiority trial. Lancet. 2015;386(9993):552–561. doi: 10.1016/S0140-6736(14)62113-9. [DOI] [PubMed] [Google Scholar]
- 100.Caso F, Del Puente A, Peluso R, et al. Emerging drugs for psoriatic arthritis. Exp Opin Emerg Drugs. 2016;21(1):69–79. doi: 10.1517/14728214.2016.1146679. [DOI] [PubMed] [Google Scholar]
- 101.Danehy S. Pfizer announces positive top-line results from the first phase 3 trial of investigational tofacitinib in adults with psoriatic arthritis. 2016. http://www.businesswire.com/news/home/20160405005842/en/Pfizer-Announces-Positive-Top-Line-Results-Phase-3. Accessed 1 May 2016.
- 102.Asahina A, Etoh T, Igarashi A, et al. Oral tofacitinib efficacy, safety and tolerability in Japanese patients with moderate to severe plaque psoriasis and psoriatic arthritis: a randomized, double-blind, phase 3 study. J Dermatol. 2016. doi:10.1111/1346-8138.13258. [DOI] [PMC free article] [PubMed]
- 103.Braun J, Kiltz U, Heldmann F, Baraliakos X. Emerging drugs for the treatment of axial and peripheral spondyloarthritis. Exp Opin Emerg Drugs. 2015;20(1):1–14. doi: 10.1517/14728214.2015.993378. [DOI] [PubMed] [Google Scholar]
- 104.Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329–342. doi: 10.1038/nri3661. [DOI] [PubMed] [Google Scholar]
- 105.Akiho H, Yokoyama A, Abe S, et al. Promising biological therapies for ulcerative colitis: a review of the literature. World J Gastrointest Pathophysiol. 2015;6(4):219–227. doi: 10.4291/wjgp.v6.i4.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367(7):616–624. doi: 10.1056/NEJMoa1112168. [DOI] [PubMed] [Google Scholar]
- 107.Panes J, Su C, Bushmakin AG, Cappelleri JC, et al. Randomized trial of tofacitinib in active ulcerative colitis: analysis of efficacy based on patient-reported outcomes. BMC Gastroenterol. 2015;15:14. doi: 10.1186/s12876-015-0239-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pfizer (PFE)’s oral tofacitinib meets primary and key secondary endpoints in two late-stage trials. 2016. http://www.biospace.com/News/pfizers-oral-tofacitinib-meets-primary-and-key/412814. Accessed 18 Mar 2016.
- 109.Nielsen OH, Seidelin JB, Ainsworth M, Coskun M. Will novel oral formulations change the management of inflammatory bowel disease? Exp Opin Invest Drugs. 2016. doi:10.1517/13543784.2016.1165204. [DOI] [PubMed]
- 110.Sandborn WJ, Ghosh S, Panes J, et al. A phase 2 study of tofacitinib, an oral Janus kinase inhibitor, in patients with Crohn’s disease. Clin Gastroenterol Hepatol. 2014;12(9):1485–n93 e2. doi:10.1016/j.cgh.2014.01.029. [DOI] [PubMed]
- 111.Panés J, Sandborn WJ, Schreiber S, et al., editors. OP021 Efficacy and safety of oral tofacitinib for induction therapy in patients with moderate-to-severe Crohns disease: results of a phase 2b randomised placebo-controlled trial. 11th Congress of European Crohns and Colitis Organization, 2016, Amsterdam.
- 112.D’Haens G, Higgins PDR, Colombel J-F, et al., editors. OP022 efficacy and safety of oral tofacitinib for maintenance therapy in patients with moderate-to-severe Crohns disease: results of a phase 2b randomised placebo-controlled trial. 11th Congress of European Crohn’s and Colitis Organization, 2016, Amsterdam.
- 113.Quintas-Cardama A, Kantarjian H, Cortes J, Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov. 2011;10(2):127–140. doi: 10.1038/nrd3264. [DOI] [PubMed] [Google Scholar]
- 114.Deisseroth A, Kaminskas E, Grillo J, et al. U.S. Food and Drug Administration approval: ruxolitinib for the treatment of patients with intermediate and high-risk myelofibrosis. Clin Cancer Res. 2012;18(12):3212–3217. doi: 10.1158/1078-0432.CCR-12-0653. [DOI] [PubMed] [Google Scholar]
- 115.Srdan Verstovsek M, Passamonti F, Rambaldi A, et al., editors. Long-term results from a phase II open-label study of ruxolitinib in patients with essential thrombocythemia refractory to or intolerant of hydroxyurea. 56th American Society of Hematology Annual Meeting and Exposition, 2014, San Francisco (CA).
- 116.FDA grants breakthrough therapy designation for Incyte’s ruxolitinib (Jakafi®) in acute graft-versus-host disease (GVHD). 2016. http://www.businesswire.com/news/home/20160623005540/en/FDA-Grants-Breakthrough-Therapy-Designation-Incyte%E2%80%99s-Ruxolitinib. Accessed 1 July 2016.
- 117.Shi JG, Chen X, Emm T, et al. The effect of CYP3A4 inhibition or induction on the pharmacokinetics and pharmacodynamics of orally administered ruxolitinib (INCB018424 phosphate) in healthy volunteers. J Clin Pharmacol. 2012;52(6):809–818. doi: 10.1177/0091270011405663. [DOI] [PubMed] [Google Scholar]
- 118.Shi JG, Fraczkiewicz G, Williams WV, Yeleswaram S. Predicting drug-drug interactions involving multiple mechanisms using physiologically based pharmacokinetic modeling: a case study with ruxolitinib. Clin Pharmacol Ther. 2015;97(2):177–185. doi: 10.1002/cpt.30. [DOI] [PubMed] [Google Scholar]
- 119.MacFarlane LA, Todd DJ. Kinase inhibitors: the next generation of therapies in the treatment of rheumatoid arthritis. Int J Rheum Dis. 2014;17(4):359–368. doi: 10.1111/1756-185X.12293. [DOI] [PubMed] [Google Scholar]
- 120.Wenzel J, van Holt N, Maier J, et al. JAK-1/2 inhibitor ruxolitinib controls a case of chilblain lupus erythematosus. J Invest Dermatol. 2016. doi:10.1016/j.jid.2016.02.015. [DOI] [PubMed]
- 121.Hornung T, Janzen V, Heidgen FJ, et al. Remission of recalcitrant dermatomyositis treated with ruxolitinib. N Engl J Med. 2014;371(26):2537–2538. doi: 10.1056/NEJMc1412997. [DOI] [PubMed] [Google Scholar]
- 122.Xing L, Dai Z, Jabbari A, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nature Med. 2014;20(9):1043–1049. doi: 10.1038/nm.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Klaeschen AS, Wenzel J. Upcoming therapeutic targets in cutaneous lupus erythematous. Exp Rev Clin Pharmacol. 2016:1–12. doi:10.1586/17512433.2016.1145543. [DOI] [PubMed]
- 124.Punwani N, Scherle P, Flores R, et al. Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J Am Acad Dermatol. 2012;67(4):658–664. doi: 10.1016/j.jaad.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 125.Craiglow BG, Tavares D, King BA. Topical Ruxolitinib for the Treatment of Alopecia Universalis. JAMA Dermatol. 2016;152(4):490–1. doi: 10.1001/jamadermatol.2015.4445. [DOI] [PubMed] [Google Scholar]
- 126.Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Exp Opin Invest Drugs. 2014;23(8):1067–1077. doi: 10.1517/13543784.2014.918604. [DOI] [PubMed] [Google Scholar]
- 127.Shi JG, Chen X, Lee F, et al. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J Clin Pharmacol. 2014;54(12):1354–1361. doi: 10.1002/jcph.354. [DOI] [PubMed] [Google Scholar]
- 128.Keystone EC, Taylor PC, Drescher E, et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann Rhem Dis. 2015;74(2):333–340. doi: 10.1136/annrheumdis-2014-206478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tanaka Y, Emoto K, Cai Z, et al. Efficacy and safety of baricitinib in Japanese patients with active rheumatoid arthritis receiving background methotrexate therapy: a 12-week, double-blind, randomized placebo-controlled study. J Rheumatol. 2016;43(3):504–511. doi: 10.3899/jrheum.150613. [DOI] [PubMed] [Google Scholar]
- 130.van Vollenhoven RF. Small molecular compounds in development for rheumatoid arthritis. Curr Opin Rheumatol. 2013;25(3):391–397. doi: 10.1097/BOR.0b013e32835fd828. [DOI] [PubMed] [Google Scholar]
- 131.Fleischmann R TT, Schlichting DE, Macias WL, et al. Baricitinib, methotrexate, or baricitinib plus methotrexate in patients with early rheumatoid arthritis who had received limited or no treatment with disease-modifying anti-rheumatic drugs (DMARDs): phase 3 trial results [abstract]. Arthritis Rheumatol. 2015;67(Suppl. 10).
- 132.Genovese MC, Kremer J, Zamani O, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med. 2016;374(13):1243–1252. doi: 10.1056/NEJMoa1507247. [DOI] [PubMed] [Google Scholar]
- 133.Dougados M, van der Heijde D, Chen Y-C, et al. LB0001 baricitinib, an oral Janus kinase (JAK)1/JAK2 inhibitor, in patients with active rheumatoid arthritis (RA) and an inadequate response to CDMARD therapy: results of the phase 3 RA-Build Study. Ann Rheum Dis. 2015;74(Suppl. 2):79. [Google Scholar]
- 134.Taylor PCKE, van der Heijde D, Tanaka Y, et al. Baricitinib versus placebo or adalimumab in patients with active rheumatoid arthritis (RA) and an inadequate response to background methotrexate therapy: results of a phase 3 study [abstract] Arthritis Rheumatol. 2015;67(Suppl. 10):2015. [Google Scholar]
- 135.Walsh N. Swift response seen for baricitinib in RA: response by week four predicts subsequent low disease activity, remission. 2016.
- 136.Emery P, Gaich CL, DeLozier AM, et al. Patient-reported outcomes from a phase 3 study of baricitinib in patients with rheumatoid arthritis with inadequate response to conventional synthetic disease-modifying antirheumatic drugs. 2015 ACR/ARHP Annual Meeting, 29 Sep 2015, San Francisco (CA). Arthritis Rheumatol. 2015.
- 137.Papp K, Menter MA, Raman M, et al. A randomized phase 2b trial of baricitinib, an oral JAK1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol. 2016. doi:10.1111/bjd.14403. [DOI] [PubMed]
- 138.Montealegre AR, Brogan P, Berkun Y, et al. Preliminary response to Janus kinase inhibition with baricitinib in chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperatures (CANDLE). 8th International Congress of Familial Mediterranean Fever and Systemic Autoinflammatory Diseases. Pediatric Rheumatol. 2015.
- 139.Jabbari A, Dai Z, Xing L, et al. Reversal of alopecia areata following treatment with the JAK1/2 inhibitor baricitinib. EBioMedicine. 2015;2(4):351–355. doi: 10.1016/j.ebiom.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Saridomichelakis MN, Olivry T. An update on the treatment of canine atopic dermatitis. Veterinary J. 2016;207:29–37. doi: 10.1016/j.tvjl.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 141.Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. 2015;73(3):395–399. doi: 10.1016/j.jaad.2015.06.045. [DOI] [PubMed] [Google Scholar]
- 142.Strand V, Ahadieh S, French J, et al. Systematic review and meta-analysis of serious infections with tofacitinib and biologic disease-modifying antirheumatic drug treatment in rheumatoid arthritis clinical trials. Arthritis Res Ther. 2015;17:362. doi: 10.1186/s13075-015-0880-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Winthrop KL, Yamanaka H, Valdez H, et al. Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014;66(10):2675–2684. doi: 10.1002/art.38745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Curtis JR, Xie F, Yun H, et al. Real-world comparative risks of herpes virus infections in tofacitinib and biologic-treated patients with rheumatoid arthritis. Ann Rheum Dis. 2016. doi:10.1136/annrheumdis-2016-209131. [DOI] [PMC free article] [PubMed]
- 145.Busque S, Leventhal J, Brennan DC, et al. Calcineurin-inhibitor-free immunosuppression based on the JAK inhibitor CP-690,550: a pilot study in de novo kidney allograft recipients. Am J Transplant. 2009;9(8):1936–1945. doi: 10.1111/j.1600-6143.2009.02720.x. [DOI] [PubMed] [Google Scholar]
- 146.Vincenti F, Tedesco Silva H, Busque S, et al. Randomized phase 2b trial of tofacitinib (CP-690,550) in de novo kidney transplant patients: efficacy, renal function and safety at 1 year. Am J Transplant. 2012;12(9):2446–2456. doi: 10.1111/j.1600-6143.2012.04127.x. [DOI] [PubMed] [Google Scholar]
- 147.Wathes R, Moule S, Milojkovic D. Progressive multifocal leukoencephalopathy associated with ruxolitinib. N Engl J Med. 2013;369(2):197–198. doi: 10.1056/NEJMc1302135. [DOI] [PubMed] [Google Scholar]
- 148.Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases: elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16–25. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vincenti F, Silva HT, Busque S, et al. Evaluation of the effect of tofacitinib exposure on outcomes in kidney transplant patients. Am J Transplant. 2015;15(6):1644–1653. doi: 10.1111/ajt.13181. [DOI] [PubMed] [Google Scholar]
- 150.Charles-Schoeman C, Wicker P, Gonzalez-Gay MA, et al. Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral Janus kinase inhibitor. Semin Arthritis Rheum. 2016. doi:10.1016/j.semarthrit.2016.05.014. [DOI] [PubMed]
- 151.Scheller J, Garbers C, Rose-John S. Interleukin-6: from basic biology to selective blockade of pro-inflammatory activities. Semin Immunol. 2014;26(1):2–12. doi: 10.1016/j.smim.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 152.Wu JJ, Strober BE, Hansen PR, et al. Effects of tofacitinib on cardiovascular risk factors and cardiovascular outcomes based on phase III and long-term extension data in patients with plaque psoriasis. J Am Acad Dermatol. 2016;75(5):897–905. doi: 10.1016/j.jaad.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 153.Kume K, Amano K, Yamada S, et al. Tofacitinib improves arterial stiffness despite up-regulating serum cholesterol with chronic cardiovascular disease in methotrexate-resistant active rheumatoid arthritis patients: a cohort study. 2014 ACR/ARHP Annual Meeting, Boston (MA), 2014.
- 154.Charles-Schoeman C, Fleischmann R, Davignon J, et al. Potential mechanisms leading to the abnormal lipid profile in patients with rheumatoid arthritis versus healthy volunteers and reversal by tofacitinib. Arthritis Rheumatol. 2015;67(3):616–625. doi: 10.1002/art.38974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kremer JM, Kivitz AJ, Simon-Campos JA, et al. Evaluation of the effect of tofacitinib on measured glomerular filtration rate in patients with active rheumatoid arthritis: results from a randomised controlled trial. Arthritis Res Ther. 2015;17:95. doi: 10.1186/s13075-015-0612-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mahajan S, Hogan JK, Shlyakhter D, et al. VX-509 (decernotinib) is a potent and selective janus kinase 3 inhibitor that attenuates inflammation in animal models of autoimmune disease. J Pharmacol Exp Ther. 2015;353(2):405–414. doi: 10.1124/jpet.114.221176. [DOI] [PubMed] [Google Scholar]
- 157.Gadina M, Schwartz DM, O’Shea JJ. Editorial: decernotinib. A next-generation Jakinib. Arthritis Rheumatol. 2016;68(1):31–34. doi: 10.1002/art.39463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Huang JYJ, Yogaratnam J, Shen J. The effect of deuteration of VX-509 (Decernotinib) on drug–drug interactions (DDI) with midazolam. Ann Rheum Dis. 2015;74(Suppl. 2):740. [Google Scholar]
- 159.Rendic S, Di Carlo FJ. Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev. 1997;29(1–2):413–580. doi: 10.3109/03602539709037591. [DOI] [PubMed] [Google Scholar]
- 160.Genovese MC, van Vollenhoven RF, Pacheco-Tena C, et al. VX-509 (decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol. 2016;68(1):46–55. doi: 10.1002/art.39473. [DOI] [PubMed] [Google Scholar]
- 161.Hirota T, Ieiri I. Drug-drug interactions that interfere with statin metabolism. Exp Opin Drug Metab Toxicol. 2015;11(9):1435–1447. doi: 10.1517/17425255.2015.1056149. [DOI] [PubMed] [Google Scholar]
- 162.Fleischmann RM, Damjanov NS, Kivitz AJ, et al. A randomized, double-blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheum. 2015;67(2):334–343. doi: 10.1002/art.38949. [DOI] [PubMed] [Google Scholar]
- 163.Genovese MC, Yang F, Ostergaard M, Kinnman N. Efficacy of VX-509 (decernotinib) in combination with a disease-modifying antirheumatic drug in patients with rheumatoid arthritis: clinical and MRI findings. Ann Rheum Dis. 2016;75(11):1979–1983. doi: 10.1136/annrheumdis-2015-208901. [DOI] [PubMed] [Google Scholar]
- 164.Namour F, Desrivot J, Van der Aa A, et al. Clinical confirmation that the selective JAK1 inhibitor filgotinib (GLPG0634) has a low liability for drug-drug interactions. Drug Metab Lett. 2016;10(1):38–48. doi: 10.2174/1872312810666151223103353. [DOI] [PubMed] [Google Scholar]
- 165.Namour F, Diderichsen PM, Cox E, et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIB dose selection. Clin Pharmacokin. 2015;54(8):859–874. doi: 10.1007/s40262-015-0240-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chantal Tasset PH, Annegret Van der Aa, Meuleners L, et al., editors. The JAK1-selective inhibitor GLPG0634 is safe and rapidly reduces disease activity in patients with moderate to severe rheumatoid arthritis; results of a 4-week dose ranging study. American College of Rheumatology Meeting, 2013.
- 167.Kavanaugh A, Ponce L, Cseuz R, Reshetko O, et al. Filgotinib (GLPG0634), an oral JAK1 selective inhibitor is effective as monotherapy in patients with active rheumatoid arthritis: results from a phase 2B dose ranging study [abstract]. Arthritis Rheuml. 2015;67(Suppl. 10).
- 168.Westhovens R, Alten R, Pavlova D, Enríquez-Sosa F, Mazur M, Greenwald M, Van der Aa A, Vanhoutte F, Tasset C, Harrison P. Filgotinib (GLPG0634), an oral JAK1 selective inhibitor is effective in combination with methotrexate in patients with active rheumatoid arthritis: results from a phase 2B dose ranging study [abstract]. Arthritis Rheumatol. 2015;67(Suppl 10).
- 169.Efficacy and safety of GLPG0634 in subjects with active Crohn’s disease. 2016.
- 170.Vermeire S, Schreiber S, Petryka R, et al. Filgotinib (GLPG0634), an oral JAK1 selective inhibitor, induces clinical remission in patients with moderate-to-severe Crohn’s disease: results from the phase 2 FITZROY study interim analysis. Gastroenterology. 2016;150(4 Suppl. 1):S1267. doi: 10.1016/S0016-5085(16)34280-9. [DOI] [Google Scholar]
- 171.Galapagos. GLPG0778/555. Immuno and Inflammatory. http://www.glpg.com/glpg-0778-555. 2016.
- 172.Voss J, Graff C, Schwartz A, et al. THU0127 pharmacodynamics of a novel JAK1 selective inhibitor in rat arthritis and anemia models and in healthy human subjects. EULAR 06.12.2014, Paris, France. Ann Rheum Dis. 2014;222.
- 173.Kremer JM, Emery P, Camp HS, et al. A phase 2b study of ABT-494, a selective JAK1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti-TNF therapy. Arthritis Rheum. 2016. doi:10.1002/art.39801. [DOI] [PMC free article] [PubMed]
- 174.Genovese MC, Smolen JS, Weinblatt ME, Burmester GR, Meerwein S, Camp HS, et al. A randomized phase 2b study of ABT-494, a selective JAK1 inhibitor in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 2016;75(6):1024–33. doi: 10.1002/art.39808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Takeuchi T, Tanaka Y, Iwasaki M, Ishikura H, Saeki S, Kaneko Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann Rheum Dis. 2016;75(6):1057–64. doi: 10.1136/annrheumdis-2015-208279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Oda K, Cao YJ, Sawamoto T, et al. Human mass balance, metabolite profile and identification of metabolic enzymes of [(1)(4)C]ASP015K, a novel oral Janus kinase inhibitor. Xenobiotica. 2015;45(10):887–902. doi: 10.3109/00498254.2015.1026864. [DOI] [PubMed] [Google Scholar]
- 177.Yamazaki S, Inami M, Ito M, et al. ASP015K: a novel JAK inhibitor demonstrated potent efficacy in adjuvant-induced arthritis model in rats. Arthritis Rheum. 2012;64(Suppl. 10):2084. [Google Scholar]
- 178.Papp K, Pariser D, Catlin M, et al. A phase 2a randomized, double-blind, placebo-controlled, sequential dose-escalation study to evaluate the efficacy and safety of ASP015K, a novel Janus kinase inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol. 2015;173(3):767–776. doi: 10.1111/bjd.13745. [DOI] [PubMed] [Google Scholar]
- 179.Ludbrook VJ, Hicks KJ, Hanrott KE, Patel JS, Binks MH, Wyres MR, et al. Investigation of selective JAK1 inhibitor GSK2586184 for the treatment of psoriasis in a randomized placebo-controlled phase 2a study. Br J Dermatol. 2016;174(5):985–95. doi: 10.1111/bjd.14399. [DOI] [PubMed] [Google Scholar]
- 180.van Vollenhoven RF, Layton M, Kahl L, et al. DRESS syndrome and reversible liver function abnormalities in patients with systemic lupus erythematosus treated with the highly selective JAK-1 inhibitor GSK2586184. Lupus. 2015;24(6):648–649. doi: 10.1177/0961203315573347. [DOI] [PubMed] [Google Scholar]
- 181.Kahl L, Patel J, Layton M, Binks M, Hicks K, Leon G, et al. Safety, tolerability, efficacy and pharmacodynamics of the selective JAK1 inhibitor GSK2586184 in patients with systemic lupus erythematosus. Lupus. 2016;25(13):1420–1430. doi: 10.1177/0961203316640910. [DOI] [PubMed] [Google Scholar]
- 182.Bissonnette R, Luchi M, Fidelus-Gort R, et al. A randomized, double-blind, placebo-controlled, dose-escalation study of the safety and efficacy of INCB039110, an oral janus kinase 1 inhibitor, in patients with stable, chronic plaque psoriasis. J Dermatol Treat. 2016:1–7. doi:10.3109/09546634.2015.1115819. [DOI] [PubMed]
- 183.Yiu ZZ, Warren RB. Novel oral therapies for psoriasis and psoriatic arthritis. Am J Clin Dermatol. 2016;17(3):191–200. doi: 10.1007/s40257-016-0179-3. [DOI] [PubMed] [Google Scholar]
- 184.Luchi M, Fidelus-Gort R, Douglas D, et al. A randomized, dose-ranging, placebo-controlled, 84-day study Of INCB039110, a selective Janus kinase-1 inhibitor, in patients with active rheumatoid arthritis [abstract] Arthritis Rheum. 2013;65(Suppl. 10):1797. [Google Scholar]
- 185.Park JS, Kwok SK, Lim MA, et al. STA-21, a promising STAT-3 inhibitor that reciprocally regulates Th17 and Treg cells, inhibits osteoclastogenesis in mice and humans and alleviates autoimmune inflammation in an experimental model of rheumatoid arthritis. Arthritis Rheum. 2014;66(4):918–929. doi: 10.1002/art.38305. [DOI] [PubMed] [Google Scholar]
- 186.Nowell MA, Richards PJ, Fielding CA, et al. Regulation of pre-B cell colony-enhancing factor by STAT-3-dependent interleukin-6 trans-signaling: implications in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2006;54(7):2084–2095. doi: 10.1002/art.21942. [DOI] [PubMed] [Google Scholar]
- 187.Shim SH, Sung MW, Park SW, Heo DS. Absence of STAT1 disturbs the anticancer effect induced by STAT3 inhibition in head and neck carcinoma cell lines. Int J Mol Med. 2009;23(6):805–810. doi: 10.3892/ijmm_00000196. [DOI] [PubMed] [Google Scholar]
- 188.Yesylevskyy SO, Ramseyer C, Pudlo M, et al. Selective inhibition of STAT3 with respect to STAT1: insights from molecular dynamics and ensemble docking simulations. J Chem Info Model. 2016;56(8):1588–1596. doi: 10.1021/acs.jcim.6b00198. [DOI] [PubMed] [Google Scholar]
- 189.Costa-Pereira AP, Tininini S, Strobl B, et al. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc Natl Acad Sci USA. 2002;99(12):8043–8047. doi: 10.1073/pnas.122236099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Okusaka T, Ueno H, Ikeda M, et al. Phase 1 and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor, in patients with advanced hepatocellular carcinoma. Hepatol Res. 2015;45(13):1283–1291. doi: 10.1111/hepr.12504. [DOI] [PubMed] [Google Scholar]
- 191.Ogura M, Uchida T, Terui Y, et al. Phase I study of OPB-51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015;106(7):896–901. doi: 10.1111/cas.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mandal PK, Morlacchi P, Knight JM, et al. Targeting the Src homology 2 (SH2) domain of signal transducer and activator of transcription 6 (STAT6) with cell-permeable, phosphatase-stable phosphopeptide mimics potently inhibits Tyr641 phosphorylation and transcriptional activity. J Med Chem. 2015;58(22):8970–8984. doi: 10.1021/acs.jmedchem.5b01321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Koo MY, Park J, Lim JM, et al. Selective inhibition of the function of tyrosine-phosphorylated STAT3 with a phosphorylation site-specific intrabody. Proc Natl Acad Sci USA. 2014;111(17):6269–6274. doi: 10.1073/pnas.1316815111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, et al. Tofacitinib ameliorates murinelupus and its associated vascular dysfunction. Arthritis Rheum. 2016;69(1):148–160. doi: 10.1002/art.39818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jabbari A, Nguyen N, Cerise JE, et al. Treatment of an alopecia areata patient with tofacitinib results in regrowth of hair and changes in serum and skin biomarkers. Exp Dermatol. 2016;25(8):642–643. doi: 10.1111/exd.13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Harris JE, Rashighi M, Nguyen N, et al. Rapid skin repigmentation on oral ruxolitinib in a patient with coexistent vitiligo and alopecia areata (AA) J Am Acad Dermatol. 2016;74(2):370–371. doi: 10.1016/j.jaad.2015.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Harel S, Higgins CA, Cerise JE, et al. Pharmacologic inhibition of JAK–STAT signaling promotes hair growth. Sci Adv. 2015;1(9):e1500973. doi: 10.1126/sciadv.1500973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.O’Shea JJ, Kanno Y, Chan AC. In search of magic bullets: the golden age of immunotherapeutics. Cell. 2014;157(1):227–240. doi: 10.1016/j.cell.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Telliez JB, Dowty ME, Wang L, Jussif J, Lin T, Li L, et al. Discovery of a JAK3-selective inhibitor: functional differentiation of JAK3-selective inhibition over pan-JAK or JAK1-selective inhibition. ACS Chem Biol. 2016;11(12):3442–51. doi:10.1021/acschembio.6b00677. [DOI] [PubMed]
- 200.Wiegman CH, Adcock IM, Rothaul A, Main M, Morgan F. The Selective Pan-Janus Kinase (jak) Inhibitor Vr588 demonstrates potent anti-inflammatory activity in a murine chronic house dust mite (hdm) model of asthma (Abstract) J Aerosol Med Pulmonary Drug Delivery. 2015;28(3):A22–A23. [Google Scholar]
- 201.Wu H, Yan S, Chen J, Luo X, Li P, Jia X, et al. JAK1-STAT3 blockade by JAK inhibitor SHR0302 attenuates inflammatory responses of adjuvant-induced arthritis rats and decreases Th17 and total B cells. Joint Bone Spine. 2016;83(5):525–32. doi: 10.1016/j.jbspin.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 202.SHR 0302. Jiangsu Hengrui Medicine Co. http://adisinsight.springer.com/drugs/800042343. Accessed 20 Feb 2017.
- 203.JTE 052. Originator: Japan Tobacco. http://adisinsight.springer.com/drugs/800035951. Accessed 20 Feb 2017.