

Abstract
Twenty-nine patients with thalassemia and a median age of 6 years (range 1.1–33 years) were given a BMT from an alternative donor. Six of the 29 donors were HLA-phenotypically identical and two were mismatched relatives, 13 were mismatched siblings and eight were mismatched parents. Six patients received no antigen (relatives), 15 patients one antigen, five patients two antigen and three patients three antigen disparate grafts. Twenty-three patients were in class 2 or class 3, whereas six patients were in class 1. Thirteen patients were given BUCY, nine patients BUCY plus ALG, six patients BUCY plus TBI or TLI and one patient BUCY with prior cytoreductive-immunosuppressive treatment as conditioning. As GVHD prophylaxis four patients received MTX, 22 CsA + MTX + methylprednisolone (MP) and three patients CsA + MP. Thirteen of 29 patients (44.8%) had sustained engraftment. The probability of graft failure or rejection was 55%. There were no significant differences between antigen disparities and graft failure. The incidence of grade II–IV acute GVHD was 47.3% and chronic GVHD was 37.5%. The incidence of acute GVHD was higher in patients receiving one or two antigen disparate in the GVHD direction grafts (vs no antigen) (P EQ 0.04; odds ratio 10.8; 95% CI 1.5–115). The probability of overall and event-free survival was 65% and 21%, respectively, with median follow-up of 7.5 years (range 0.6–17 years) for surviving patients. The degree of HLA disparity between patient and donor did not have a significant effect on survival. The incidence of nonhematologic toxicity was low. Transplant-related mortality was 34%. GVHD (acute or chronic) was a major contributing cause of death (50%) followed by infections (30%). We conclude that at present, due to high graft failure and GVHD rates, BMT from alternative donors should be restricted to patients who have poor life expectancies because they cannot receive adequate conventional treatment or because of alloimmunization to minor blood antigens. Bone Marrow Transplantation (2000) 25, 815–821.
Keywords: bone marrow transplantation, phenotypically identical related donor, mismatched related donor, graft failure, GVHD
Main
Currently, bone marrow transplantation (BMT) from an HLA-identical related donor has been established as the only curative therapy for thalassemia, with long-term event-free survival in 53–94% of patients according to the class of risk.1,2,3,4 One major limitation of BMT is the lack of suitable donors. Since only 30–35% of potential transplant candidates have HLA-identical siblings, attention has turned to alternative donor transplants including those using a partially mismatched related donor5,6,7,8 or phenotypically matched unrelated donor.9,10 In general, the results of BMT from unrelated donors are inferior to those using matched sibling donors due to an increased incidence and severity of GVHD, rejection, and infections.9,10,11 In a non-malignant BMT setting such as aplastic anemia, the results of unrelated BMT for adults are generally unsatisfactory, although improved survival in younger patients has been reported.12 There are few case reports of unrelated BMT for thalassemia.13 In addition, the search for an unrelated donor is costly, time-consuming and has been attained for less than half of the patients who seek an alternative donor. In contrast to the lengthy unrelated donor search, for more than 90% of patients an HLA-mismatched related donor is readily available.5 Most reports of BMT from partially mismatched related donors are restricted to malignancies. Historically, transplantation of related bone marrow genetically disparate at one or more HLA loci for patients with malignancies or aplastic anemia has been associated with an increased risk of both graft failure and severe GVHD,14,15,16 although the results of recent studies in malignant17 and non-malignant18 settings are encouraging.
During the last two decades conventional therapy has improved the prognosis of thalassemia. However, despite such improvement it still remains a progressive disease with therapy-related complications (such as hepatitis, liver fibrosis, cardiac disease) progressing with time. Conventional treatment may postpone but not eliminate these complications with the associated morbidity and mortality. In contrast to this, BMT can prevent or delay progression of the aforementioned complications. Approximately 70% of thalassemic patients lack a related matched donor.19 For those patients who lack HLA-identical sibling donors, alternative sources of bone marrow must be sought. It is well known that thalassemia is more frequent in many developing societies where the mortality from this disease remains extremely high and few patients survive to adulthood because it is not possible to give conventional treatment due to inadequate transfusion services and cost of chelating treatment. In such circumstances an alternative BMT from a related donor may be the only salvage therapy for at least some groups of patients. In a preliminary study we reported some results of alternative BMT in patients with thalassemia.20 In the present retrospective study we examined survival, rejection and transplant-related complications after BMT from an alternative donor in a larger number of patients with thalassemia. These results represent the largest single-institution series of patients who have received an alternative BMT for thalassemia.
Patients and methods
Patients
From July 1982 to December 1998, 29 patients with thalassemia received a BMT from an alternative donor. Six of the 29 donors were HLA-phenotypically identical and two were mismatched relatives (cousin four, aunt two, uncle one, grandmother one), 13 were mismatched siblings and eight were mismatched parents. Characteristics of the patients and donors are shown in Table 1. There were 14 males and 15 females with a median age of 6 years (range 1.1–33 years). All but one patient was less than 16 years old.
Table 1.
Patient and donor characteristics before BMT
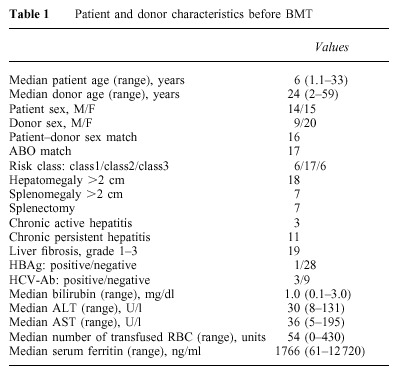
Patients were categorized into three groups according to three risk factors (hepatomegaly greater than 2 cm, portal fibrosis of any degree, and inadequate chelation with desferrioxamine). Class 1 patients had no risk factors, class 2 patients had two of these factors and class 3 patients had all three factors.2 Six patients were in class 1, 17 patients in class 2, and six patients were in class 3. Three of the six class 1 patients had never been transfused because of religious reasons (these three patients were 1.1, 1.3 and 1.4 years old and their Hb level remained between 64 and 68 g/l), while all class 2 and 3 patients had received inadequate conventional therapy mainly because it was not possible to give adequate treatment in their own country or because of patient non-compliance with chelating therapy. The distribution of patients according to geographical regions was as follows: Italy 11 patients, Middle East six patients, India three patients, Iran three patients, other European countries three patients, Argentina, Azerbaijan and Pakistan one patient from each country. Patients were referred to our center for alternative BMT on their physicians’ advice. After a detailed explanation, informed consent was obtained from parents and, where appropriate, patients.
Graft failure was defined as primary and secondary failure. Primary graft failure is characterized by the absence of allogeneic recovery by day >21 with or without autologous reconstitution. Secondary graft failure was defined as early and late failure (losing the graft after transient engraftment within 60 days and after 60 days, respectively) with thalassemia recurrence or aplastic bone marrow. Evaluation of graft status was performed by globin-chain synthesis and by in situ Y chromosome hybridization in sex-mismatched or by analysis of variable tandem repeat (VNTR) poly- morphism in sex-matched pairs as described earlier.4
Histocompatibility studies
Donor selection was based on HLA A, B and DR histocompatibility. All donor–patient pairs had serological HLA typing. Typing for class I HLA-A, B and C antigens was performed using a standard two-stage complement-dependent microcytotoxicity assay and for class II HLA-DR and DQ antigens using nylon wool-purified B cells in a microcytotoxic assay. For 10 patients HLA-DR and DQ polymorphysms had also been determined using molecular genetic analysis with sequence-specific oligonucleotides. Anti-donor lymphocytotoxic antibodies were assayed in an extended incubation microcytotoxicity crossmatch with negative results in all patients. Bidirectional mixed lymphocyte cultures (MLC) were tested in all patients. Reactivity was expressed as stimulation index and relative response index. The patients, all mismatched at class II HLA, were MLC reactive. HLA disparity between recipient–donor pairs is shown in Table 2. Three patients disparate for one antigen were homozygous at the mismatched locus and hence were a one locus mismatch with respect to rejection direction and no locus mismatch with respect to GVHD direction. All the remaining patients and donors were heterozygous at the mismatched locus, and therefore were disparate both in the direction of GVHD and in the direction of rejection. In two patients who received their BMT in 1982 typing of HLA class II antigens was incomplete. These patients had a positive MLC and were classified as class II HLA incompatible.
Table 2.
HLA disparity

Transplant procedure
Patients received different conditioning regimens. Two patients received BUCY in association with TBI, 13 patients BUCY only, nine patients BUCY plus anti-lymphocyte globulin (ALG) (Pressimmune; Behringwerke, Marburg, Germany or Lymphoglobuline; Merrieux, France), four patients BUCY plus total lymphoid irradiation (TLI) and one patient received a new conditioning regimen including cytoreductive-immunosuppressive and hypertransfusion-intensive chelation therapies (from day −45 to day −12 hydroxyurea 30 mg/kg/day, azathioprine 3 mg/kg/day along with hypertransfusions and continuous i.v. desferrioxamine 40 mg/kg/day and from day −17 to day −13 fludarabine (Fludara; Schering AG, Berlin, Germany, 20 mg/m2/day) to reduce the hyperplastic bone marrow and increase immunosuppression before BUCY started on day −9 (Table 3). As GVHD prophylaxis, four patients received MTX only, 22 patients CsA + MTX + methylprednisolone (MP) and three patients CsA + MP. Acute and chronic GVHD were staged by standard criteria.21 Patients received the marrow infusion 36 h after the last dose of CY on day −0. The median marrow cell dose infused was 4.9 × 108/kg (range 1.9–10 × 108/kg).
Table 3.
Characteristics of BMT and engraftment

Patients were maintained in strict isolation in single rooms with positive pressure HEPA-filtered air and were given prophylactic broad-spectrum antibiotics and amphotericin B until the granulocyte level exceeded 0.5 × 109/l. Patients also received acyclovir as herpes virus prophylaxis and trimethoprim/sulfamethoxazole for prophylaxis of Pneumocystis carini. All blood products administered were irradiated to 30 Gy.
Statistical analysis
The endpoints were overall survival, event-free survival and graft failure or rejection. The probabilities of survival, event-free survival and rejection were estimated using the method of Kaplan and Meier.22 Associations between survival, graft failure and the pre-transplant variables (patient or donor age, sex mismatch, pre-transplant transfusions, serum ferritin, liver function tests, chronic hepatitis, liver fibrosis, risk classes, spleen size, HLA disparity, infused marrow cell dose, dose of cyclophosphamide, dose of busulfan, ALG, TBI/TLI, year of transplant) were tested in univariate analyses using the Pearson chi-squared statistics. Variables significant at the P < 0.1 level were assessed by logistic linear regression analyses using the Statistix statistical package.23 This package was also used for engraftment characteristics. The results were analyzed as of 31 July 1999.
Results
Engraftment
All 29 patients were evaluable for engraftment. Nineteen of 29 patients (65.5%) showed allogeneic engraftment and six of them subsequently lost their grafts with autologous reconstitution. Thus, 13 patients (44.8%) had sustained engraftment. The median times to granulocyte count of >0.5 × 109/l and platelet count of >20 × 109/l were 19 days (range 11–46) and 18 days (range 7–63), respectively (Table 3). Two patients with persistent aplasia received a second BMT from the same donor after conditioning with CY + ALG + TLI or Orthoclone (OKT3) without signs of engraftment. Among six patients who received HLA-phenotypically identical bone marrow, three had sustained engraftment. The degree of HLA disparity did not influence sustained engraftment (Table 4). There was no correlation between conditioning regimen and sustained engraftment (Table 5).
Table 4.
Engraftment and survival according to HLA disparity

Table 5.
Engraftment and survival according to conditioning regimens

Transplant-related complications
None of the patients experienced veno-occlusive disease. Two patients developed moderate and one severe mucositis with gastrointestinal hemorrhage. Two patients had moderate to severe hemorrhagic cystitis. Overall incidence of infectious complications was high (62%). There were no significant differences between antigen disparities and the incidence of infectious complications (data not shown). Among patients with infectious complications 14 developed bacteremia with Gram-positive or Gram-negative species (three of these patients also had fungal infections), three Candida fungaemia and one CMV interstitial pneumonia.
Transplant-related mortality
Overall, 10 patients (34%) died, seven during the first 100 days after BMT. Neither the degree of HLA disparity (Table 4) nor the conditioning regimen (Table 5) influenced death. Causes of death included GVHD (acute or chronic) in five patients, sepsis with or without hemorrhage in two patients, interstitial CMV pneumonia in one patient, ARDS in one patient and cardiac disease in one patient which occurred 4 years after BMT while he was receiving conventional treatment for graft failure with autologous reconstitution.
GVHD
Nineteen patients were evaluable for acute GVHD (aGVHD). Nine of these patients (47.3%) developed grade II–IV and six of them (31.5%) grade III–IV acute GVHD. The incidence of grade II–IV aGVHD in patients with no, one and two antigen disparities in the GVHD direction was 14%, 64% and 50%, respectively (P = 0.05 for no antigen vs one or two; by Fisher's exact test). None of the patients with three antigen disparities was evaluable for aGVHD. A logistic regression analysis confirmed that the incidence of aGVHD was higher in patients receiving one or two antigen disparate graft in the GVHD direction (vs no antigen) (P = 0.04; odds ratio 10.8; 95% CI 1.5–115). The risk of acute GVHD was similar whether there was MLC reactivity or not. Three of eight patients evaluable for chronic GVHD (37.5%) developed moderate (two patients) or severe (one patient) disease. Two of these patients died with chronic GVHD and one surviving patient has a Karnofsky performance score of 90%.
Graft failure or rejection
Overall, 16 patients (55%) had graft failure or rejection. Of these 16 patients, 10 (62.5%) had primary graft failure with thalassemia recurrence (eight patients) or persistent aplasia (two patients) and six (37.5%) developed secondary graft failure with thalassemia recurrence. The median time to graft failure was 21 days (range 12–273). Graft failure or rejection was not affected by the overall degree of HLA disparity between donor and patient (Table 4), HLA disparity in the rejection direction or by different conditioning regimens (Table 5). Other variables not influential on graft failure on univariate and logistic linear regression analysis were: patient or donor age, sex mismatch, class of risk, pre-transplant transfusions, serum ferritin, liver function tests, chronic hepatitis, liver fibrosis, spleen size, years from transplant, infused marrow cell dose, dose of cyclophosphamide, dose of busulfan and the use of TBI/TLI or ALG in the conditioning regimen.
Survival
Of 29 patients, 19 are surviving (65%) at a median follow-up of 7.5 years (range 0.6–17 years). The probability of overall and event-free survival was 65% and 21%, respectively (Figure 1). Thirteen patients with thalassemia recurrence continue to receive conventional treatment. The degree of HLA disparity between patient and donor did not have a significant effect on survival. None of the remaining variables listed in the statistical section was influential on survival on univariate or logistic linear regression analysis.
Figure 1.
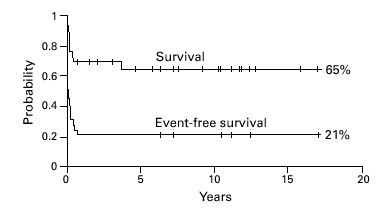
Overall and event-free survival for 29 patients receiving BMT for thalassemia from an alternative donor.
Discussion
At present the decision to perform a BMT from an alternative donor for thalassemia remains an ethical dilemma. Although conventional treatment has dramatically improved survival and quality of life of patients with thalassemia, therapy-related complications such as hepatitis or other infections, liver fibrosis and cardiac disease progress with time and shorten the life of these patients. In certain circumstances where adequate conventional treatment is not available and the search for an unrelated donor is impossible, BMT from an alternative related donor may become the only available approach for some patients. Most of our patients did not receive adequate treatment because this was impossible to give in their countries, due to religious reasons or to patient non-compliance with desferrioxamine. Obviously, the vast majority of these patients belonged to class 2 or class 3 and had associated hepatitis or liver fibrosis. In such circumstances, even a low number of cured patients may, however, justify alternative BMT for those patients with thalassemia whose survival expectancies are poor while they receive inadequate conventional treatment. Nonhematologic moderate to severe toxicity was not very common and involved the gastrointestinal (10%) and renal (6.8%) systems. GVHD (acute or chronic) was a major contributing cause of death (50%) followed by infections (30%) in this study.
Despite the combined GVHD prophylaxis given to most of our patients, the incidence of acute GVHD was high in our series. We found that patients receiving one or two antigen grafts mismatched in the GVHD direction have a high risk of developing aGVHD as compared to the patients receiving grafts zero antigen mismatched in the GVHD direction.
Our data show that the rate of graft failure or rejection after BMT from an alternative donor is high. In contrast to these data, Henslee-Downey et al5 and Soiffer et al17 observed relatively few rejections (12–13%) in patients with malignancies after BMT from mismatched related donors using irradiation containing regimens. One possible explanation of this high failure rate is that unlike hematological malignancies patients with non-malignant diseases, such as aplastic anemia and thalassemia have an increased risk of graft failure even after HLA-identical sibling BMT probably as a result of absence of prior immunosuppressive treatment, compared with patients with hematological malignancies. It has been suggested that the incidence of graft failure increases with the increasing degree of HLA disparity.5,24,25 We could not confirm these data probably because of the small number of patients. It is of interest to note that only two patients with graft failure developed marrow aplasia, while all other patients had full thalassemia recurrence.
Although the probability of overall survival was 65%, event-free survival was lower in our patients (21%). In the present series, HLA disparity did not significantly influence survival. Surprisingly, the results of transplant in patients receiving HLA-phenotypically identical marrow were not better than those of the patients receiving one or two antigen disparate grafts.
There were two main obstacles to successful alternative BMT in this study. The first obstacle was the higher graft failure despite increased immunosuppression in the conditioning regimens given to most patients. This study involved various conditioning regimens, which may have affected the incidence of graft rejection. However, the data we obtained showed that conditioning regimen had no significant influence on rejection. On the other hand, a high rejection rate demonstrates that the conditioning regimens used probably did not provide sufficient immunologic ablation to achieve successful engraftment in the majority of our patients. Over the last few years, investigations into new approaches to immunomodulation after related mismatched transplantation have achieved high engraftment rates and control of acute GVHD. Indeed, the results of recent studies using conditioning regimens including TBI with or without additional TLI in association with T cell depletion for BMT from related mismatch donors are encouraging in terms of sustained engraftment and control of GVHD.5,17 Undertaking of BMT early in the course of the disease before the patient develops therapy-related complications may be one of the keys to reducing graft failure and transplant-related mortality after BMT from alternative donors.
The second obstacle was the high incidence of acute GVHD and the associated morbidity and mortality. T cell depletion of marrow reduces the incidence and severity of acute GVHD,26,27 but may contribute to an increased risk of graft failure.28 However, T cell depletion of specific populations of CD6+ and CD3+ cells with a relatively low incidence of graft failure and GVHD after BMT from HLA non-identical related donors appears promising.17,29 Taking into consideration the fact that thalassemic patients have an increased incidence of graft failure even after HLA-identical sibling BMT we decided to avoid T cell depletion and used combined immunosuppression after BMT to control GVHD. Our data showed that this type of immunosuppression did not decrease either the incidence or the severity of acute GVHD in these patients. Therefore, decreasing the incidence of GVHD remains the major challenge after mismatched related transplants.
This study shows that BMT from an alternative donor can cure some groups of patients with thalassemia. Outcome may be improved by adopting new conditioning and GVHD prophylaxis regimens that decrease the rate of graft failure and the incidence of severe aGVHD. At present, due to high graft failure and GVHD rates this type of transplant should be restricted to patients who have poor life expectancies as a result of not receiving adequate conventional treatment or because of alloimmunization to minor blood antigens.
Acknowledgements
The authors thank Ms Law Aileen for linguistic assistance and all the members of our BMT Team for their exemplary care of these patients. This work has been supported by the Berloni Foundation against Thalassemia, Pesaro and by the Italian Association against Leukemia Pesaro Section.
References
- 1.Thomas ED, Bukner CD, Sanders JE. Marrow transplantation for thalassemia. Lancet. 1982;2:227–228. doi: 10.1016/S0140-6736(82)90319-1. [DOI] [PubMed] [Google Scholar]
- 2.Lucarelli G, Galimberti M, Polchi P. Bone marrow transplantation in patients with thalassemia. New Engl J Med. 1990;332:417–421. doi: 10.1056/NEJM199002153220701. [DOI] [PubMed] [Google Scholar]
- 3.Lucarelli G, Clift RA, Galimberti M. Marrow transplantation for patients with thalassemia. Results in class 3 patients. Blood. 1996;80:2082–2088. [PubMed] [Google Scholar]
- 4.Lucarelli G, Clift RA, Galimberti M. Bone marrow transplantation in adult thalassemic patients. Blood. 1999;4:1164–1167. [PubMed] [Google Scholar]
- 5.Henslee-Downey PJ, Abhyankar SH, Parrish RS. Use of partially mismatched related donors extends access to allogeneic marrow transplant. Blood. 1997;10:3864–3872. [PubMed] [Google Scholar]
- 6.Cain Y, Takaue Y, Watanabe A. Partially mismatched pediatric transplants with allogeneic CD34+ blood cells from a related donor. Blood. 1998;9:3123–3130. [PubMed] [Google Scholar]
- 7.Polchi P, Lucarelli G, Galimberti M. Haploidentical bone marrow transplantation from mother to child with advanced leukemia. Bone Marrow Transplant. 1995;16:529–535. [PubMed] [Google Scholar]
- 8.Aversa F, Tabilio A, Velardi A. Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. New Engl J Med. 1998;339:1186–1193. doi: 10.1056/NEJM199810223391702. [DOI] [PubMed] [Google Scholar]
- 9.Balduzzi A, Gooley T, Anasetti C. Unrelated donor marrow transplantation in children. Blood. 1995;8:3247–3256. [PubMed] [Google Scholar]
- 10.Kernan NA, Bartsch G, Ash RC. Analysis of 462 transplantations from unrelated donors facilitated by the National Marrow Donor Program. New Engl J Med. 1993;328:593–602. doi: 10.1056/NEJM199303043280901. [DOI] [PubMed] [Google Scholar]
- 11.Marks DI, Cullis JO, Ward KN. Allogeneic bone marrow transplantation for chronic myeloid leukemia using sibling and volunteer donors. A comparison of complications in the first 2 years. Ann Intern Med. 1993;119:207–214. doi: 10.7326/0003-4819-119-3-199308010-00005. [DOI] [PubMed] [Google Scholar]
- 12.Margolis D, Camitta B, Pietryga D. Unrelated donor bone marrow transplantation to treat severe aplastic anemia in children and young adults. Br J Haematol. 1996;94:65–72. doi: 10.1046/j.1365-2141.1996.d01-1772.x. [DOI] [PubMed] [Google Scholar]
- 13.La Nasa G, Vacca A, Pizzati A. Role of HLA extended haplotypes in unrelated bone marrow. Bone Marrow Transplant. 1993;12:186–189. [Google Scholar]
- 14.Anasetti C, Beatty PG, Storb R. Effect of HLA compatibility on graft-versus-host disease, relapse and survival after bone marrow transplantation for patients with leukemia or lymphoma. Hum Immunol. 1990;29:79–91. doi: 10.1016/0198-8859(90)90071-V. [DOI] [PubMed] [Google Scholar]
- 15.Beatty PG, Clift RA, Mickelson EM. Marrow transplantation from related donors other than HLA-identical siblings. New Engl J Med. 1985;313:765–771. doi: 10.1056/NEJM198509263131301. [DOI] [PubMed] [Google Scholar]
- 16.Beatty PG, Di Bartolomeo P, Storb R. Treatment of aplastic anemia with marrow grafts from related donors other than HLA genotypically-matched siblings. Clin Transplant. 1987;1:117–122. [Google Scholar]
- 17.Soiffer RJ, Mauch P, Fairclough D. CD6+ T cell depleted allogeneic bone marrow transplantation from genotypically HLA non-identical related donors. Marrow Transplant Rev. 1998;1:9–13. [PubMed] [Google Scholar]
- 18.Tzeng CH, Chen PM, Fan S. CY/TBI 800 as a pretransplant regimen for allogeneic bone marrow transplantation for severe aplastic anemia using HLA-haploidentical family donors. Bone Marrow Transplant. 1996;18:273–277. [PubMed] [Google Scholar]
- 19.Delfini C, Polchi P, Izzi T. Bone marrow donors other than genotypically identical siblings for patients with thalassemia. Exp Hematol. 1985;13:197–200. [PubMed] [Google Scholar]
- 20.Polchi P, Galimberti M, Lucarelli G. HLA-mismatched bone marrow transplantation in thalassemia. Bone Marrow Transplant. 1993;12:67–69. [PubMed] [Google Scholar]
- 21.Thomas ED, Storb R, Clift RA. Bone marrow transplantation. New Engl J Med. 1975;292:895–902. doi: 10.1056/NEJM197504242921706. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan EL, Meier P. Non parametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. doi: 10.1080/01621459.1958.10501452. [DOI] [Google Scholar]
- 23.Siegel J (ed) Statistix. Version 4.0. User's Manual. 1992. [Google Scholar]
- 24.Lamb LS, Gee AP, Parrish RS. Acute rejection of marrow grafts in patients transplanted from a partially mismatched related donor: clinical and immunologic characteristics. Bone Marrow Transplant. 1996;17:1021–1027. [PubMed] [Google Scholar]
- 25.Camitta B, Ash R, Menitove K. Bone marrow transplantation for children with severe aplastic anemia: use of donors other than HLA-identical siblings. Blood. 1989;74:1852–1857. [PubMed] [Google Scholar]
- 26.Wagner JE, Donnenberg AD, Noga SJ. Lymphocyte depletion of donor bone marrow by counterflow centrifugation elutriation: results of a phase I clinical trial. Blood. 1988;72:1168–1176. [PubMed] [Google Scholar]
- 27.Young JW, Papadopoulos EB, Cunningham I. T-cell depleted allogeneic bone marrow transplantation in adults with acute non-lymphocytic leukemia in first remission. Blood. 1992;79:3380–3387. [PubMed] [Google Scholar]
- 28.Kernan NA. Graft Versus Host Disease: Immunology, Pathophysiology and Treatment. 1990. Graft failure following transplantation of T cell depleted marrow. In: Deeg JH, Burakoff SJ, Ferra J, Atkinson K (eds) pp. 557–564. [Google Scholar]
- 29.Munn RK, Henslee-Downey PJ, Romond EH. Treatment of leukemia with partially matched related bone marrow transplantation. Bone Marrow Transplant. 1997;19:421–427. doi: 10.1038/sj.bmt.1700681. [DOI] [PubMed] [Google Scholar]