

Abstract
The respiratory mucosa is responsible for gas exchange and is therefore, of necessity, exposed to airborne pathogens, allergens, and foreign particles. It has evolved a multi-faceted, physical and immune defense system to ensure that in the majority of instances, potentially injurious invaders are repelled. Inflammation, predominantly mediated by effector cells of the granulocyte lineage including neutrophils and eosinophils, is a form of immune defense. Where inflammation proves unable to remove an inciting stimulus, chronic inflammatory disease may supervene because of the potential for tissue damage conferred by the presence of large numbers of frustrated, activated granulocytes. Successful recovery from inflammatory disease and resolution of inflammation rely on the clearance of these cells. Ideally, they should undergo apoptosis prior to phagocytosis by macrophage, dendritic, or epithelial cells. The outcome of inflammation can have serious sequelae for the integrity of the respiratory mucosa leading to disease. Therapeutic strategies to drive resolution of inflammation may be directed at the induction of granulocyte apoptosis and the enhancement of granulocyte clearance.
Supplementary information
The online version of this article (doi:10.1038/mi.2008.31) contains supplementary material, which is available to authorized users.
Respiratory System and Granulocyte-Mediated Mucosal Inflammation
The respiratory mucosa is the largest body surface area to be exposed to, and require defense from, the external environment. It is not surprising, therefore, that a robust, vigorous, and rapidly responsive immune defense system has evolved over millennia of exposure to diverse and increasingly sophisticated microbes. In addition, air-borne irritant particulate matter such as allergens, biopersistent fibers such as asbestos and combustion-derived nanoparticles (e.g., diesel exhaust particles) must be overcome to maintain a high level of function. The architecture of the respiratory system provides a first layer of defense against air-borne microorganisms and particulate matter, effectively blocking the lower airways from anything greater than 5 μm in diameter. Inhaled air is warmed and filtered through the nasal hair and nasopharyngeal passages before negotiating the larynx (which ordinarily prevents any intrusion from matter destined for the gastrointestinal system and allows phonation) down the large tubular trachea and into the upper bronchi. The initially large bronchial airways repeatedly bifurcate until they reach an extremely narrow gauge and form alveoli at which point gas exchange with the capillary network that lines the alveolar wall becomes possible. Pseudostratified columnar-ciliated epithelium predominates in the trachea and bronchi forming the mucociliary escalator. Any intruding microorganism or misplaced particulate matter slipping out of the air stream in the upper airways is trapped in the muco-ciliary escalator and forced up and out by the regular beating motion of the respiratory cilia.
Should these initial defenses be breached then an intruder must encounter an uninviting but nonspecific chemical milieu including lysozyme, endogenous antimicrobial agents, adverse pH, IgA, and surfactants.1 Should this fail then a more goal-directed system is brought on line. The respiratory mucosa has developed a sensitive and specific recognition strategy that allows it to identify microbe-specific protein patterns such as lipopolysaccharide, lipotechoic acid, formylated peptides, flagellin, and non-methylated DNA. Once detected, these galvanize resident alveolar macrophages and initiate a variety of proinflammatory pathways that instigate the classical elements of inflammation (calor (heat), rubor (redness), dolor (pain), tumor (swelling), and loss of function as described by Celsus and (debatably) Virchow)2 driven by increased vascular permeability leading to a proteinaceous infiltrate and leucocyte recruitment. The Toll-like receptors (TLRs) mediate most of this form of recognition and though they each have a specific role, TLRs can function in unison to expand their powers of recognition.3 Resident alveolar macrophages deal with the majority of insults that trigger this alert system but if they are overwhelmed recruited leucocytes including granulocytes lend assistance.
The invading microbes are rendered highly visual to recruited granulocytes by comprehensive opsonization mediated by both complement-dependent and -independent means. They are then ingested (phagocytosed) by neutrophils or, if they should prove resistant to this because of size or learned subversion, are subjected to a chemical onslaught (exocytosis) as neutrophils forcibly externalize toxic granule substances such as lactoferrin and myeloperoxidase as well as reactive oxygen species (ROS).4, 5 A subset of neutrophils (and other leucocytes potentially6) employs web-like neutrophil extracellular traps to ensnare and kill resistant organisms but must themselves die in the process.7 In beneficial neutrophil-dominant inflammation, the organisms or foreign particles are detected and phago-cytosed by neutrophils, which then undergo an organized, non-provocative programmed cell death (apoptosis) that promotes their own recognition and removal by macrophages or dendritic cells. At this point, the interface between the innate and adaptive immune systems occurs as macrophages, which migrate to the lympho-reticular system following ingestion of apoptotic neutrophils, act as antigen-presenting cells allowing the lymphocyte population to complete the resolution and remembrance process. The next time this particular organism is encountered a preprepared, specific response should be available to ensure it has less opportunity to make an impact.8
The lungs are the arena for another type of granulocyte-driven inflammatory response. Eosinophils are present in larger numbers within the lungs of asthma sufferers and are recruited in greater numbers in response to sensitizing allergens such as pollen, house dust mite, and animal dander. This response has no obvious beneficial effects and it is still unclear why it should occur. The eosinophil is a useful and active defender against parasitic infection and has an armament specific to that end. It has been noted that as parasitic infections have been largely eradicated in Western society, the incidence of this abnormal eosinophil response (termed allergy or atopy) has increased and it seems that eosinophilic inflammation occurs almost as an outlet for redundancy.1
Inflammation does not always resolve neatly and, unfortunately, for diverse reasons, pulmonary inflammatory disease is one of the biggest drains on health resources in this and many other countries (respiratory disease cost in the United Kingdom was £6.6 billion in 2004).9, 10 Non-resolving or chronic inflammation is established when an acute inflammatory response fails to counter an instigating stimulus. In pulmonary infection, this may occur because a microorganism can subvert host defense by surviving either within inflammatory cells (Mycobacterium tuberculosis) or inside a protective micro-environment (Pseudomonas aeruginosa).11, 12 Persistent inflammation may occur because of a recurrent stimulus like tobacco smoke (chronic obstructive pulmonary disease (COPD)), allergens (asthma), or long (>20 μm) biopersistent fibers (asbestosis).13 Inflammation is also chronic where the host immune system malfunctions and becomes under- or overactive or misdirected as occurs in autoimmune disease (rheumatoid arthritis, systemic sclerosis), immune deficiency syndromes (chronic granulomatous disease, severe combined immunodeficiency syndrome), cystic fibrosis (CF), or adult respiratory distress syndrome/acute lung injury (ARDS/ALI). In some cases, the etiology is unknown and subject to debate (idiopathic pulmonary fibrosis (IPF)). Regardless of the cause, it is apparent that the over-recruitment, ineffective clearance, and hence accumulation and misdirected or frustrated activation of neutrophils and eosinophils lead to tissue damage and an inciting feedback loop of inflammation that perpetuates chronicity13 (Figure 1).
Figure 1.
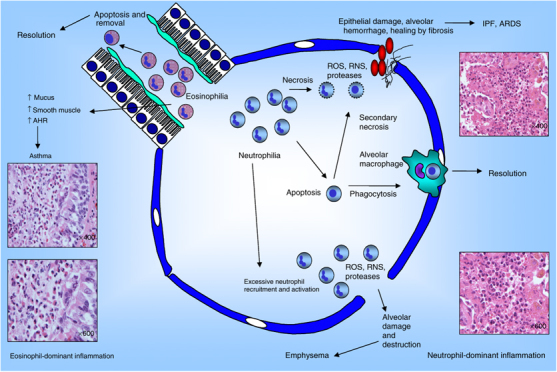
Diagram showing both resolution and failure of resolution of eosinophil- and neutrophil-dominant inflammatory processes. Neutrophilia at the respiratory mucosa is resolved by apoptosis of infiltrating neutrophils and phagocytic clearance by macrophages. It fails to resolve where neutrophils are in great excess or are not efficiently cleared and undergo secondary necrosis following apoptosis. This leads to alveolar damage and destruction followed by fibrotic healing. Histology on the right-hand side of the diagram shows neutrophil-dominant inflammation. Eosinophil-dominant inflammation is resolved by the same mechanisms and fails to resolve for the same reasons. The effects of eosinophil-dominant inflammation in the asthmatic airway are depicted and an example of the histology demonstrated on the left-hand side of the diagram. Histology was kindly provided by William Wallace (Pathology Department, Edinburgh Royal Infirmary).
It is evident that granulocytes play an important role in the pathogenesis of pulmonary inflammation but other cell types such as macrophages, dendritic cells, mast cells, and lymphocytes are equally and potentially more influential in subsets of inflammatory respiratory disease. In this review, our focus will be on granulocyte research as expert reviewers have addressed the role of other cell types across the spectrum of respiratory disease and the interplay of different cell types within single diseases. Our aim will be to address current understanding of granulocyte biology in relation to respiratory inflammatory diseases and prospects for driving resolution of inflammation at the respiratory mucosa to develop effective treatments for lung-based disease.
Granulocytes
Pluripotent hematopoietic stem cells in the bone marrow gene-rate the common myeloid progenitor cell. This cell differen-tiates into a common granulocyte progenitor cell, which when given appropriate stimulation can produce any granulocyte lineage. Potential granulocyte lineages include neutrophils, eosinophils, and basophils. Basophils are the least well studied and least numerous of the granulocytes, and their physiological function seems to overlap somewhat with that of eosinophils. They are probably meant to counter parasites but their relevance to Western society is in the production of numerous medi-ators necessary for the incitement of the asthmatic response. Neutrophils and eosinophils are discussed in greater detail below. Granulocytes are terminally differentiated and having therefore exited the cell cycle, they remain in the G0 phase of growth for the duration of their lives. However, a recent report suggests that postmitotic neutrophils are capable of radical pheno-typic switching to take on major characteristics of macrophages when stimulated with growth factors, whereas another study shows that mouse neutrophils can be switched to a dendritic cell phenotype following in vitro incubation with colony-stimulating factor-1.14, 15 Extravasated granulocytes recruited to the tissues ought to die by apoptosis whilst non-migratory, circulating granulocytes are removed by the combined efforts of the bone marrow, liver, and spleen. There is renewed interest in granulocyte life cycle and differentiation as pharmaco-logical inhibitors of the cell-cycle machinery have been shown to promote granulocyte apoptosis.16, 17
Neutrophils
Neutrophils are 12–15 μm in diameter and account for 70% of the circulating leucocyte population, which corresponds to approximately 2.5–7.5×109/l neutrophils. They survive in the circulation for approximately 7–10 h but if compelled by inflammatory circumstance or chemical persuasion can extend their life span up to 48 h and beyond. This population turnover requires efficient production by the bone marrow and prompt clearance by that same organ with help from the spleen and liver. Neutrophils possess at least four different types of granules termed: primary (azurophilic), secondary (specific), gelatinase, and secretory; each carries a specific arsenal of toxic chemicals that the neutrophil uses against non-host elements. The neutro-phil is a key effector cell at the front line of immune defense and is efficacious (and lifesaving) in the majority of instances because of its versatility. Neutrophils prove to be effective phagocytes because on accomplishment of this task they are programmed to die a prompt, quiet and importantly, contained, apoptotic death. This limits the time available to internalized microorganisms and prevents hijacking of cellular controls to enable subversion (as is thought to occur when M. tuberculosis is internalized by macrophages18). This timely death also signals larger scale phagocytes such as macrophages, dendritic cells, and epithelial cells to initiate a phagocytic response. If a neutro-phil is unable to phagocytose an invader then it will disgorge granule contents into the surrounding environment causing tissue damage and amplifying the inflammatory response. This should not be construed as a purely detrimental response as it serves to block potential routes of entry for invading orga-nisms as tissue damage causes capillary shutdown and collapses lymphatics, sends a strong signal of imperiled defense, and allows time for an alternative immune strategy to be adopted.8
Neutrophils may also employ extracellular traps to ensnare and kill resistant organisms. Neutrophil extracellular trap formation involves the extrusion of fine chromatin and granule-coated tendrils that are microbicidal and fungicidal. This effort proves fatal to the neutrophil.19 Finally, neutrophils are not just blunt effectors but also smooth operators responsible for negotiating the recruitment and education of other arms of the immune system. It is becoming appreciated that there is a false division between the innate and adaptive immune response systems and that a more realistic model involves a continual interplay between constituents of these systems. Neutrophils not only alert antigen-presenting cells and lymphocytes to danger but regulate their response to it and in return receive counter regulation. Appropriate resolution of an inflammatory immune response is finely balanced.5, 8
It is not surprising, given the constant exposure of the vast respiratory mucosa to threat, the fine balance of the immune response and the pivotal inflammatory role played by neutro-phils, that neutrophil-dominant inflammation has been implicated in the pathophysiology of numerous inflammatory respiratory diseases including pneumonia, COPD, IPF, CF, and ARDS.
Eosinophils
Eosinophils are approximately 12–17 μm in size and, under normal circumstances, account for less than 5% of the circulating leucocyte population. They may survive for up to 12 h in the circulation but have the ability to extend their longevity to over a week if required. Not only are they bigger than neutrophils but they also wear more flamboyant colors when stained by the Romanowsky method (methylene blue and eosin), which accounts for their name. Eosinophils may also be distinguished by their production of Charcot–Leyden crystals (manufactured from lysophospholipase, an eosinophil-derived enzyme), which are often visible in their cytoplasm. Eosinophils, like neutrophils, are supplied with numerous granules though their constituents differ including major basic protein, eosinophil cationic protein, eosinophil peroxidase, and eosinophil-derived neurotoxin. This array is certainly capable of tissue damage and probably evolved to combat helminthic infection. Increasingly, new roles for eosinophils are being identified and it seems likely that they have a role in combating viral infection as eosinophil cationic protein and eosinophil-derived neurotoxin have been shown to degrade single-stranded RNA viruses. They are said to play an early role in innate immunity by production of important cytokines such as interleukin (IL)-4 though this is probably only physiologically important in the gastrointestinal tract. They can also modulate adaptive immunity by specific activation of T cells.20
Eosinophils are implicated in a different spectrum of disease from neutrophils, of which the most common variety is allergic/atopic disease, which includes asthma, allergic rhinitis, and eczema. In other countries, parasitic infection is still common including schistosomiasis, dranunculoriasis, ascariasis, filariasis, and hookworm. Eosinophil numbers may be increased in various cancers as well as in rare conditions such as Churg–Strauss syndrome, aspergillosis, and eosinophilic pneumonia. In eosinophil-dominant disease, it has been consistently demonstrated that eosinophil excess due to excessive recruitment, apoptosis avoidance, and failed clearance has a correlation with disease pathology.
Granulocyte Apoptosis
Neutrophils and to a lesser extent eosinophils are short-lived cells, a feature that may help to limit their potential for causing damage by ensuring that they cannot be subverted by pathogens. They are explosively reactive cells and it is therefore surprising that their death is a model of contained self-restraint. Apoptosis is a physiological marvel that allows cells with an incendiary cargo (granules) to package it efficiently and safely (in plasma membrane) so that the cellular environment is protected. In addition, the cell nucleus condenses and chromatin is chopped up and re-organized into packages termed nucleosomes. Throughout this process, the cell membrane is retained intact though glycoprotein and phospholipid signals are displayed to attract macrophages and facilitate interaction and uptake (efferocytosis). Fluorescently-tagged Annexin-V is often used to label phosphatidylserine residues, which are characteristically flipped to the outer membrane of apoptotic cells thereby enabling flow cytometric identification of apoptosis. Another member of this family, Annexin-1, has been shown to induce granulocyte apoptosis but is also released by both neutrophils and macrophages to enhance phagocytosis of apoptotic cells.21, 22 The whole process is amplified by many orders of magnitude during inflammation whether it be neutrophil or eosinophil dominant. Unfortunately, apoptotic cells cannot stay that way forever, and if they are not cleared by phagocytes then they undergo secondary necrosis and all the good work is undone. Large-scale granulocyte recruitment must necessarily be followed by large-scale granulocyte apoptosis and clearance by macrophages. It is extremely important that this is taken into account when plotting to drive granulocyte apoptosis to promote resolution of inflammation.4, 23
Neutrophil and eosinophil apoptosis are similar but not identical processes. It is generally believed, though not universally,24 that there are two pathways by which apoptosis proceeds, both of which are ultimately dependent on the caspase family (Figure 2). The intrinsic pathway occurs when the cell faces withdrawal of growth/survival factors, genotoxic stress, or ultraviolet irradiation. This pathway relies on proapoptotic members of the Bcl-2 family, which escape regulation by their antiapoptotic counterparts and translocate to the mitochondria facilitating liberation of cytochrome c. The various components of the apoptosome (Apaf-1, cytochrome c etc.) then assemble to cleave the inactive zymogen, procaspase-9, to active caspase-9, which inexorably commits the cell to caspase-3-mediated apoptosis. The extrinsic pathway proceeds through external cell-membrane death receptors such as the tumor necrosis factor receptor (TNFR), the Fas receptor (FasR), and TNF-related apoptosis-inducing ligand receptor (TRAILR). Ligand activation of these receptors promotes clustering of receptors and association with their internal adaptor proteins (TNFR-associated death domain protein and Fas-associated death domain protein) in the lipid-raft. Multiple procaspase-8 molecules assemble at the adaptor proteins (formation of the death-inducing signaling complex), and their physical approximation generates an autocatalytic reaction initiating the caspase cascade. The death receptor CD137 has also been implicated in neutrophil and eosinophil apoptosis but it is currently unclear what its physiological role is.25 In neutrophils the importance of caspase action in death receptor-mediated apoptosis has previously been a matter of debate but evidence of caspase-independent apoptosis has been effectively countered by work demonstrating that culture conditions and concentrations of caspase inhibitor had not been optimally utilized in these studies.26
Figure 2.
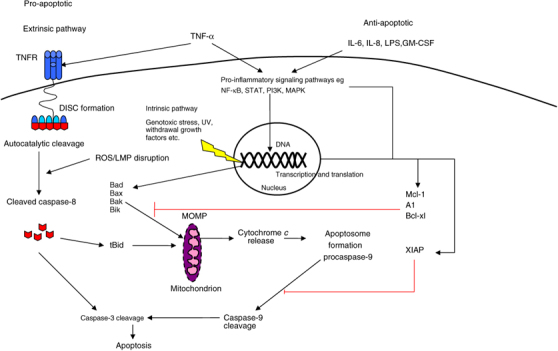
Neutrophil apoptosis. The intrinsic and extrinsic pathways are shown along with some examples of pro and antiapoptotic signaling. Proinflammatory signaling pathways may directly influence transcription of survival proteins e.g., NF-κB promotes transcription of XIAP or increase protein stability e.g., ERK (of the MAPK family) maintains XIAP levels. TNF-α may have both early proapoptotic action and late antiapoptotic effects. TNFR is the TNF receptor. DISC refers to the death-inducing signaling complex, which is composed of TNFR-associated death domain protein (TRADD), Fas-associated death domain protein (FADD), and procaspase-8. MOMP is mitochondrial outer membrane permeabilization. ERK, extracellular signal-regulated kinase; LMP, Lysosomal membrane permeabilisation; NF-κB, nuclear factor-κB; TNF-α, tumor necrosis factor-α; TNFR, tumor necrosis factor receptor.
To further complicate the story, it is apparent that there is a degree of cross talk between the intrinsic and extrinsic pathways. Caspase-8 may alternatively cleave Bid, which can translocate to the mitochondrial membrane and permeabilize it allowing apoptosis to proceed through intrinsic pathway apoptotic machinery.23, 25
Eosinophil apoptosis most likely occurs along approximately the same lines; however, controversy still reigns with regard to the caspase family. It has previously been stated that caspase-3, -8, and -9 have no demonstrable role in eosinophil apoptosis. Subsequently, it has been suggested that perhaps caspase-9 plays a role and that therefore the mitochondrial pathway is of importance. This work was confirmed by observations with regard to the effects of known eosinophil apoptosis-inducing agents such as glucocorticoids, which appear to mediate their effects through mitochondrial permeabilization, with caspase activation merely a downstream effect. In contrast, Fas ligation has been shown to promote eosinophil apoptosis in a caspase-3- and -8-dependent manner, and although mitochondrial integrity was disrupted it proved non-essential for apoptotic progression.21, 27, 28, 29 and 30
Support for the predominance of mitochondria-driven granu-locyte apoptosis is provided by the increasing evidence that survival proteins are key determinants of neutrophil longevity. The antiapoptotic Bcl-2 family member, Mcl-1, is present in both neutrophils and eosinophils, but other family members show contrasting expression. Neutrophils express A1, eosinophils express Bcl-xL but Bcl-2 has not been shown to be constitutively present in either cell at demonstrable levels. Interestingly, however, in eosinophils, Bcl-2 expression can be stimulated by IL-5.31 These survival proteins mediate their effects by marshaling proapoptotic Bcl-2 family counterparts away from the mitochondrial membrane. Proapoptotic Bcl-2 family members are capable of causing permeabilization of the outer mitochondrial membrane (MOMP) when numbers predominate over those of their chaperones. Bax is perhaps the best-characterized member of this family and appears to play a pivotal role in neutrophil apoptosis. In eosinophils, however, Bax fails to be downregulated by antiapoptotic survival factors weakening its position as a key player in apoptosis.32 The role of Mcl-1 in neutrophil apoptosis has received a great deal of attention and it appears that this protein is an essential component of neutrophil viability; a contention that is supported by the neutropenic pheno-type of the Mcl-1 knockout mouse in comparison with the increased apoptotic phenotype of the A1 knockout mouse.33, 34
Modulation of granulocyte apoptosis
Granulocyte longevity is necessarily highly regulated and conse-quently highly variable from a matter of hours up to several days for neutrophils and extending to 2 weeks for eosinophils. Persistent markers of bacterial infection such as lipopolysaccharide, pathogen-derived molecules that act as ligands for TLR-2, -4 and -9, and inflammatory cytokines such as IL-6, IL-8, granulocyte macrophage-colony stimulating factor, and TNF can delay neutrophil apoptosis whereas resolving infection indicated by successful neutrophil phagocytosis of bacteria, removal of bacterial products, and downregulation of inflammatory cytokines will promote apoptosis.3, 35
Granulocyte longevity is also extremely dependent on intra-cellular signaling pathways. Perhaps the most important of which is that controlled by the pivotal transcription factor of inflammatory cytokines, nuclear factor (NF)-κB. NF-κB can be activated by lipopolysaccharide, and is known to control the survival proteins XIAP (an inhibitor of caspase-3, -8, and -9) and Bcl-xl thus enhancing neutrophil and eosinophil longevity by tangible means. Pharmacological inhibitors of NF-κB such as gliotoxin promote neutrophil apoptosis and, in combination with TNF-α, cause dramatically enhanced apoptosis. This feature may partially explain why TNF-α has opposing effects on neutrophils at early and late time points.36, 37 Other important signaling pathways include the phosphoinositide 3-kinase pathway, which can be stimulated by granulocyte macrophage-colony stimulating factor to promote longevity by upregulation of Mcl-1 in neutrophils and which has an emerging role in the maintenance of eosinophilic inflammation.38, 39 Similarly, the extracellular signal-regulated kinase pathway is likely to be important for survival factor-mediated antiapoptotic effects at sites of inflammation. However, direct inhibition of either of these pathways will not promote granulocyte apoptosis per se.4, 40
Perhaps the most interesting and clinically relevant modulation of granulocyte apoptosis is that achieved with the use of glucocorticosteroids. These drugs extend neutrophil life span but promote eosinophil apoptosis. This effect seems to correlate with their ability to upregulate Mcl-1 in neutrophils but downregulate the same protein in eosinophils.41 Dexamethasone has also been shown to promote macrophage phagocytosis of apoptotic neutrophils.42 The impact of putative inflammation-resolving agents on clearance of apoptotic neutrophils is of paramount importance as prompt removal is essential to avoid secondary necrosis and loss of toxic contents. This is perhaps why the discovery of a new class of endogenous lipid-derived proresolution agents has been so exciting. The lipoxin family produced by neutro-phils and macrophages limits the recruitment of neutrophils to inflammatory sites and enhances macrophage phagocytosis of apoptotic neutrophils. This occurs as part of a natural brake on progression of inflammation and there is hope that enhancement of this pathway may provide a novel therapeutic strategy to counter inflammatory disease, a hypothesis that has already shown promise in several animal models.43 Another exciting prospect for therapeutic modulation of granulocyte apoptosis has emerged with the discovery of active cell-cycle machinery in neutrophils, which can be inhibited to promote apoptosis. Cyclin-dependent kinase inhibitor drugs promote neutro-phil apoptosis and drive resolution of inflammation in animal models. Given that these drugs are already in use for the treatment of cancer, it is possible that they could make the transition from bench to bedside for the management of inflammatory disease in the near future.17, 44
Alveolar Macrophages and Phagocytosis of Apoptotic Granulocytes
In normal healthy lungs, the predominant alveolar leucocyte is the macrophage, a cell that becomes resident following differentiation from a bone marrow-produced circulating precursor monocyte. Alveolar macrophages occupy a unique position at the interface between inhaled air (and hence the external environment) and the circulation (in the form of the alveolar capillary network, which is closely juxtaposed to the alveolus to promote efficient gaseous exchange) and because key macrophage functions include chemotaxis, phagocytosis, and cytotoxi-city they are lynch pins of immune defense. Importantly, during pulmonary infective or inflammatory disease, a key role in resolution of inflammation falls to both alveolar and recruited circulating macrophages. These cells are responsible for the removal of apoptotic neutrophils and to some extent eosinophils (though epithelial cells may be equally important for eosinophil removal) by efferocytosis. In order for an apoptotic cell to be recognized by a macrophage it must display specific signals. The most obvious change in the apoptotic granulocyte's plasma membrane is the externalization of phosphatidylserine residues but this in itself is not sufficient to expedite phagocytosis. The search for a macrophage-docking receptor on granulocytes has been exhaustive but inconclusive and currently a combination of various glycoproteins and phospholipids is of postulated importance. These putative receptors include the recently identified Tim,5, 45 stabilin-2,46 and BAI1.47 The process of efferocytosis, literally meaning “burying the dead”, is a proresolution strategy in itself. Macrophages that consume apoptotic neutrophils switch to a resolution phenotype that allows them to secrete TGF-β and IL-1048 as opposed to proinflammatory cytokines such as IL-6, IL-8, and TNF-α. This is in contrast to macrophages that have consumed necrotic neutrophils where the opposite is true and inflammation is actively propagated. In addition, the proresolution phenotype includes the production and enhanced responsiveness to lipoxins, protectins, and resolvins. These mediators enhance macrophage phagocytosis and promote proresolution cytokine production.43
Dysregulation of Granulocyte Apoptosis/phagocytosis and Inflammatory Diseases in The Respiratory Tract
There has been a tendency, certainly in clinical circles, to question the validity of inflammation-based hypotheses for various diseases based on the efficacy or lack of efficacy of gluco-corticosteroid medications. Given the complexity of the inflammatory response and the relative bluntness of this therapeutic tool, it is overly simplistic to make such assumptions. Steroid medications have certainly been a paradigmatic therapy in the treatment of inflammatory disease but they do not and will not drive resolution of inflammation in all settings. With increased understanding of the mechanisms of the inflammatory response and a new focus on its resolution, it is hoped that novel incisive or pleiotropic therapy combinations may be developed to address the inflammatory lung diseases discussed below.
COPD
In most cases of COPD (α-1-antitrypsin-deficient patients are a notable exception), the respiratory mucosa is damaged by repeated exposure to inhaled toxic chemicals leading to chronic inflammation, reduced immunity, and susceptibility to respiratory infections. COPD is a prevalent, largely smoking-related (though there is an increasingly recognized occupational contribution) disease in this country (in others it is related to the burning of bio-fuels), which presents with increasing breathlessness and a productive cough. It is an obstructive airways disease but unlike asthma this obstruction is irreversible with inhaled therapies. The mainstay of current treatment is with inhaled or oral corticosteroids and antibiotics when exacerbations are judged infective. (For interested readers, the refs.1, 23, 49, 50 and 51 give an overview of COPD pathophysiology.)
Neutrophils are likely to play an important role in this condition and they are found in increased numbers throughout the respiratory tract. The highest concentrations of neutrophils are found in sputum and bronchoalveolar lavage fluid (BAL) (which is perhaps representative of rapid airway-directed migration) but numbers of neutrophils are also increased in lung parenchyma and airway smooth muscle. Neutrophilic inflammation is characteristic of COPD exacerbations and there is a correlation between the resting burden of lung-based neutrophils and seve-rity of disease phenotype. Neutrophilic inflammation appears to be driven by the irritant force as smoking drives alveolar macrophages and epithelial cells to express increased levels of IL-8, a potent neutrophil chemoattractant. IL-8 also stimulates neutrophils to release myeloperoxidase whereas TNF-α and leukotriene B4 (also produced by epithelial cells, mast cells, and T lymphocytes) cause neutrophil activation (degranulation, reactive oxygen species production). Meanwhile, macrophage phagocytic function is impaired by cigarette smoking and cannot keep pace with the increased neutrophil burden.52 Enhanced neutrophil recruitment and activity combined with decreased macrophage phagocytosis weigh the scales heavily toward neutrophil-mediated tissue damage. COPD lungs are subjected to high levels of toxic neutrophil products including reactive oxygen species, elastase, and proteinases. These toxic substances overload the capacity of native antiproteinases and antioxidants to neutralize them, which leads to damaged epithelial cilia and decreased mucociliary clearance. Damage to the respiratory mucosa means that alveolar cells are replaced by goblet cells that increase mucous production. Furthermore, airways are progressively remodeled by the reparative process so that they become thicker, less-efficient conductors of air. There is some controversy with regard to the longevity of neutrophils isolated from the airways of COPD patients. Studies have shown both enhanced longevity and no enhancement of longevity whereas peripheral blood neutrophils appear to have an increased life span in keeping with systemic inflammation.
Chronic obstructive pulmonary disease is notoriously resis-tant to glucocorticoid therapy perhaps because these drugs promote neutrophil longevity by upregulating the survival protein Mcl-1.41 It is also known that smoking promotes dysfunction of histone deacetylase–2, an enzyme that is usually recruited by glucocorticoid receptors to switch off the transcription of pro-inflammatory genes. The small benefits attributable to steroid therapy in COPD may stem from enhancement of macrophage phagocytosis of apoptotic cells. Of other therapies currently in use, theophylline (a nonspecific adenosine antagonist and phosphodiesterase inhibitor) is known to restore histone deacetylase-2 function, reversing corticosteroid resistance and reducing IL-8 concentrations and sputum neutrophilia. Long-acting β2 agonists (originally employed to relax smooth muscle in peripheral airways) have now been shown to inhibit neutro-philic inflammation as measured by sputum or BAL analysis, and they are reported to drive neutrophil apoptosis though the circumstances of this are difficult to ascertain. It is clear that potential therapies must consider targeting neutrophil recruitment as well as striving to drive neutrophil apoptosis and removal.49, 50, 53 and 54
Asthma
In asthma, the respiratory mucosa is hypersensitive to exposures that are comfortably processed under normal conditions and a characteristic inflammatory response ensues. It is part of a spectrum of atopic or allergic disorders, where an enhanced sensitivity to particular antigens or environmental conditions results in a disease flare. Exacerbations of asthma are characterized by narrowing of the airways (bronchoconstriction), which leads to shortness of breath, wheezing, and cough. (For interested readers, these refs.1, 55, 56, 57, 58, 59 and 60 give an overview of asthma pathophysiology.)
Recent work has suggested that there may be many subtle mucosal abnormalities that contribute to the pathogenesis of asthma. For example, it appears that exacerbations of asthma caused by rhinovirus may be facilitated by a defect in interferon λ induction, which increases susceptibility to invasive disease whereas viral exacerbations or damage caused by chronic inflammatory disease facilitate epidermal growth factor receptor signaling (a wound repair response), which enhances neutrophil function and chemotaxis.61, 62
The allergy arm of this disease is mediated by IgE, which is produced by B cells in response to an initial exposure to a given antigen. IgE attaches to mast cells and basophils priming them to release histamine, leukotrienes, and ILs at subsequent exposures. Traditionally, asthma is an eosinophil-dominant disease and eosinophils are heavily recruited by the same TH2 cell type signaling (IL-4, IL-5, IL-6) responsible for the production of IgE. This process was thought to be under the direct control of IL-5 but as anti-IL-5 therapy has proved ineffective, it is suspected that there is a greater redundancy in the system. Anti-IL-5 therapy failed to significantly reduce eosinophil numbers in the asthmatic airway but did decrease deposition of extra-cellular matrix proteins suggesting a role for Il-5 in airway remodeling.63, 64 Interestingly, an IL-4 analog (pitrakinra), which prevents binding of IL-4 and IL-13 to IL-4α receptor complex, attenuated the late-phase response (bronchoconstriction measured by reduction in Forced Expiratory Volume in 1 s) to allergen challenge. It is as yet unclear whether this success was due to inhibition of IL-4 alone, IL-13 alone, or due to combined inhibition.65 In addition, a role for TH17 cells has been postulated particularly in forms of asthma where neutrophil recruitment contributes to pathogenesis.66
Regardless of the super-intending mechanism, it is clear that a heavy burden of apoptosis-resistant eosinophils concomitantly resistant to macrophage phagocytosis, yet capable of disgorging their formidable armament onto the respi-ratory mucosa, have significant potential to cause tissue damage and airway remodeling. Thankfully, asthma is usually responsive to glucocorticoid therapy partly because steroids are effective at promoting eosinophil apoptosis.67 This apoptosis may, in direct contrast to experience in neutrophils, be mediated by downregulation of Mcl-1. Rarely, asthma may be steroid resistant and there are different theories as to how resistance might arise.
First, it is clear that smokers with asthma respond poorly to steroids because of the defective action of histone deacetylase-2 (discussed earlier with regard to COPD patients). Second, it is apparent that a subset of asthmatics as well as end-stage or severe asthmatics manifests a neutrophil-dominant inflammation for reasons that are unknown. It has been postulated that these patients may represent a different disease or disease phenotype.68 Third, it is known that airway epithelial damage results in smooth muscle hypertrophy causing significant luminal narrowing without a requirement for a significant bronchoconstrictor stimulus and finally, it is postulated that resis-tant asthmatics might express gene polymorphisms encoding defective caspase machinery.60
Regardless of efficacy, the side effect profile (adrenal suppression, osteoporosis, peptic ulcer disease) of higher dose steroid therapy in the subset of resistant patients makes it less than ideal as they potentially require prolonged or maintenance treatment. It should be emphasized, however, that maintenance therapy with routine doses of inhaled steroids has established symptom control and reduced mortality in the majority of patients without these side effects becoming a problem. Anti-IgE therapy is showing promise in a subset of patients, and increased eosino-phil apoptosis and downregulation of inflammatory cytokines have been demonstrated in peripheral blood samples taken from asthmatics treated with omalizumab (a recombinant humanized monoclonal antibody that selectively binds IgE). Future developments will probably include anti-IL-4, anti-IL-13, and potentially anti-Th17-based therapies, now that there is a rationale for their development. However, it is not always best practice to attempt a one-hit, magic-bullet cure especially in inflammatory disease where there is considerable redundancy in the cytokine system. If an agent was developed that could promote eosinophil apoptosis without detriment to macrophage phagocytosis and with a negligible side effect profile, it would be an extremely useful addition to the pharmacopoeia.
Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis is a condition characterized by the devastation of the respiratory mucosa though its pathophysiology is complex. There is a great deal of debate about the relative importance of inflammation vs. aberrant wound repair in the pathogenesis of IPF. There is not sufficient space to address that debate in this article, suffice to say that there is enough evidence to justify an argument in support of a significant neutrophil-derived component to the disease model. (For interested readers, these references give an overview of the pathophysiology of IPF and a flavor of the debate.1, 69, 70, 71, 72 and 73)
The argument is that the initial and subsequent “exacerbatory” insults are acute and inflammatory even if the characteristic phenotype is conveyed by a disordered “healing” process. Healing by fibrosis results in significant loss of lung architecture and vital respiratory mucosa to scarring. Neutrophils are known to be present in BAL from IPF patients and are also significantly increased within lung tissue. In addition, there is evidence of the toxic chemicals produced by neutrophils including myeloperoxidase, elastase, collagenases, and proteases. The importance of this chemical insult is supported by the elastase knockout mouse, which is significantly protected against lung injury and also fails to upregulate TGF-β.74 A recent clinical paper has shown that significant neutrophilia within the BAL fluid of IPF patients correlates with increased mortality.75 It has been argued that the terminal scarring process of IPF occurs because of a significant loss of lung architecture and that where lung architecture is preserved there is potential for reversal of the remodeling process and resolution.72 It has also been suggested that the fibrotic response may in a sense be driven by a persistent but ineffective proresolution of inflammation phenotype. This hypothesis is drawn from the observation that TGF-β is a key proresolution molecule but is also intimately involved in fibrogenesis. In a disease model where non-resolving low-level inflammation results in sustained TGF-β production, it is possible to conceive that a fibrotic healing phenotype might evolve. Therapeutic strategies that drive neutrophils toward apoptosis will certainly have something to add to treatment of this disease if they can prevent or ameliorate the inflammatory insult (whether it be acute, chronic, or relapsing) that must be responsible for such extreme distortion of lung architecture.69, 70
ARDS/ALI
Adult respiratory distress syndrome/acute lung injury is the pulmonary component of the multi-organ dysfunction syndrome (MODS) and represents a global failing of lung function in response to a specific, nonspecific, or unidentifiable stimulus. Effectively, these conditions are the result of an aggressive mucosal inflammatory immune response. (The refs.1, 76, 77, 78, 79 and 80 cover the wider pathophysiology of ARDS/ALI.).
Characterized by the pathological reaction termed diffuse alveolar damage, ARDS/ALI seems to be a neutrophil dominant disease (interestingly, diffuse alveolar damage is also seen in the terminal stages of IPF). Analysis of BAL fluid from patients with early-stage ARDS demonstrates increased numbers of activated neutrophils and their numbers correlate with severity of lung injury. Indeed, persistence of BAL neutrophilia at day 7 is associated with increased mortality. The pulmonary circulation contains a large sequestered neutrophil population termed the “marginated” pool that does not normally circulate but is loosely adhered to the vessel walls. This population can be mobilized into the circulation by steroid therapy or by exercise. Neutrophils are subject to slow transit through the pulmonary microvasculature where blood vessel diameters are smaller than their own. The ability of neutrophils to progress depends on their considerable properties of distensibility. In ARDS/ALI, neutrophils are recruited early in large numbers, and an alteration in their rheological properties (they become less deformable) means that they struggle to maneuver through vessels whereas cytokine excess encourages their adhesion to vessel walls and subsequent translocation into the lung parenchyma and airways. The influx of huge numbers of neutrophils and proteinaceous inflammatory edema fluid from permeabilized vessels impairs gas exchange and makes adequate ventilation extremely difficult.
Neutrophil influx is driven by alveolar macrophage production of IL-8, and excessive levels of this cytokine have been found to be predictive of progression to ARDS in susceptible patients.81 There is no effective, specific therapy for this condition and yet some patients survive with supportive therapy alone, implying that successful resolution is possible and can be mediated by physiological mechanisms. Neutrophils isolated from the BAL of ARDS patients have enhanced longevity, are activated, and may cause insurmountable damage to the respiratory mucosa leading to healing by fibrosis and permanent scarring with loss of lung architecture and function. In experimental models where neutrophils are depleted, ALI caused by endotoxin is attenuated. In other studies, enhancement of neutrophil apoptosis also reduced inflammatory parameters and tissue damage. The picture is complicated by apparent ARDS/ALI in neutropenic human patients. It is also clear that given the extravagant neutrophil influx associated with this condition, it would likely require a dual strategy of enhancing both neutrophil apoptosis and neutrophil clearance to ensure that secondary necrosis (and tissue damage) is prevented by prompt removal.76, 82
Pneumonia
Pneumonia is the term given to an infection within the lower respiratory tract that is sufficiently significant to result in visible changes on a chest radiograph.83 There are many potential causative organisms including viruses and bacteria. Invading pathogens that overcome immediate host defenses are nonetheless recognized as foreign by their non-host constituents such as lipopolysaccharide or formylated peptides in their outer membranes. TLRs, CD14, and G-protein coupled receptors (e.g., FPR) found on macrophages and other respiratory mucosal cells are responsible for this detection and subsequently stimulate an acute inflammatory reaction by the production of cytokines such as TNF-α, IL-1, IL-6, and IL-8. This proinflammatory milieu prolongs neutrophil life span and promotes neutrophil activation to allow effective microbial phagocytosis and killing.3, 35 and 84 If the inflammatory reaction is successful in containing the pathogens and they are cleared by phagocytosis or killed, then the inflammatory reaction remains localized to a single lobe or segment of the lung. The upregulation of anti-inflammatory, proresolution molecules should ensure containment of inflammation. Neutrophil phagocytosis of bacteria and the induction of proresolution cytokines such as IL-10 and TGF-β promote neutrophil apoptosis and removal by macrophages.85
The alternative is non-resolution with enhanced neutrophil longevity and local tissue damage (resulting in abscess, empyema, or bronchiectasis) and/or loss of containment resulting in global lung inflammation characterized by ARDS or spread through the circulation to other organs (sepsis) with the potential for MODS. Streptococcal pneumonia (the pathogen responsible is Streptococcus pneumoniae) is often cited as a paradigm of resolving inflammation as despite an acute inflammatory response characterized by a massive neutrophil influx (giving the pathological appearance referred to as “red hepatization”) it is possible for the lung to achieve complete recovery.13 There is, however, a sub-population of severe pneumonia that behaves aggressively or is not contained by the patient's immune system and antibiotic therapy resulting in admission to intensive care and potentially MODS, sepsis, terminal decline, and death.
Antibiotics are central to the treatment of pneumonia but they are not always sufficient and it is possible that addressing acute inflammation associated with pneumonia may prove a successful adjuvant therapy. In this setting, it is unclear whether the ideal strategy is to dampen down the inflammatory response or to augment it in the hope that a supra-physiological immune reaction will prove successful in eradicating the organism responsible. Glucocorticoid therapy has been used as an adjuvant to antibiotics in this kind of patient with varying success rates in different trials.86 The theory is that steroids should dampen inflammation through downregulation of NF-κB activation and consequently the inflammatory cytokines under its direct transcriptional control. A recent study that utilized a physiological steroid dose (supra-physiological doses had been employed in previous studies) has shown a significant reduction in length of hospital stay and mortality in Intensive Care Unit patients with severe community-acquired pneumonia.87 It is possible therefore that in a subgroup of patients with pneumonia, strategies to aid resolution of inflammation (in combination with antibiotic therapy) may be warranted.
Cystic fibrosis
Cystic fibrosis is the commonest inherited disease in Caucasian populations affecting one in 2,500 births. It seems surprising that a defect in salt transport (the genetic abnormality affects the cystic fibrosis transmembrane regulator gene (CFTR), which encodes a chloride channel) should be responsible for such widespread organ pathology including pancreatic insufficiency, bronchiectasis, liver dysfunction, infertility, and gut defects. Mortality (on average at age 34 in Western society) is most frequently from respiratory failure caused by aggressive bronchiectasis.
There are competing hypotheses as to the root cause of the repeated infective exacerbations that lead to bronchiectasis in CF. It is postulated that a relative dehydration of the respiratory mucosa means that the mucociliary escalator is compromised by viscid mucus and that altered mucosal pH results in malfunction of host antimicrobial peptides. There may also be a failure of normal bacterial internalization processes and it has been suggested that an intrinsic proinflammatory phenotype is conferred by CFTR malfunction. The proinflammatory phenotype theory postulates that an overburdened endosomal system encumbered by the processing of faulty CFTR signals distress, which leads to activation of NF-κB. Production of IL-8 is stimulated resulting in enhanced neutrophil recruitment. Regardless, the net result is a failure of mucosal immunity, and a succession of bacterial infections occur (initially, Staphylococcus aureus and/or Haemophilus influenzae but subsequently and devastatingly P. aeruginosa, Burkholderia cepacia, and Stenotrophomonas maltophilia). With bacterial colonization, innate defense is roused and a significant neutrophil influx occurs. This is usually thwarted, initially by microbial resistance and subsequently by distorted lung architecture. This results in an insurmountable accumulation of inflammatory cells and toxic damage from chemicals eluted by their necrotic carcasses. Elastase, reactive oxygen species, and myeloperoxidase cause further lung damage, and the clearance of inflammatory cells is so poor that the lungs literally become clogged with inflammatory cell DNA. Eventually, there is a terminal paucity of functional gas-exchange equipment, and respiratory failure and death supervene if lung transplant is not possible.88
Recent work on P. aeruginosa has provided an interesting insight into the mechanisms by which this organism thwarts immune defense. It was known that P. aeruginosa could effectively hide from the immune system in a protective and impenetrable bio-film. A more subtle effect mediated by pseudomonas produced pyocyanin has now been recognized. This chemical promotes neutrophil death but inhibits macrophage clearance resulting in neutrophil death by necrosis. This leads to the spillage of cathepsin G and elastase, which can cleave the important chemokine receptor CXCR1 on other neutrophils. This receptor would usually allow IL-8-mediated enhancement of neutrophil killing prowess, which enabling neutrophils to kill pseudomonas. In CF patients, neutrophils have little CXCR1 for the above reason and are consequently effectively crippled as well as out-maneuvered. Even more detrimental, fragments of this receptor stimulate the TLR system resulting in enhanced neutrophil recruitment and prolongation of the inflammatory response.89
Anti-inflammatory therapy in the form of non-steroidal anti-inflammatory drugs has been used to mild benefit in CF but it appears that the early introduction of a potent anti-inflammatory proresolution therapeutic strategy might be of enhanced benefit. Neutrophils in CF patients are dysfunctional and ineffective at restraining or removing typical CF pathogens, so driving their apoptosis and removal might be of more benefit than allowing them to remain in the hope that they are contri-buting to defense against these microbes.88
Animal Models of Granulocyte-Mediated Pulmonary Inflammatory Disease
There are many methods available for the production of rele-vant animal models of inflammatory disease. These include direct chemical/other injury, constitutive and inducible transgenics, viral vector delivery of relevant genes, adoptive cell transfer, and direct infection strategies. The advantage of the animal model is that it reproduces the complexity of the wider immune/inflammatory response on an organ or system-wide basis as opposed to cell-line strategies that can only examine individual cellular responses. A selection of key rodent models and their major advantages/disadvantages are shown in Table 1. A more comprehensive discussion of this field is not possible due to space limitations but there are excellent reviews available in this area.90, 91, 92, 93, 94 and 95 Clearly, reliable animal models are central to the development of efficacious and safe pharma-ceutical agents that drive granulocyte apoptosis and clearance to enhance resolution of inflammation.
Table 1. Selected rodent models of inflammatory lung disease.
| Disease modelled | Technique | Strategy/basis for technique | Phenotype/cytokine expression | Advantage | Disadvantage | Outcome | References |
|---|---|---|---|---|---|---|---|
| IPF | Bleomycin lung injury | Known pulmonary toxi-city causing fibrosis in human cancer patients | T-cell-independent, CCL2 and 12 required, inflammatory cell recruitment, TGF-β | Well-known, characterized, quick (14–28 days), multiple routes of administration | Disease resolves in mice but not in humans. Variable response between mouse strains. | Dependent on time point (inflammation vs. fibrosis) | 94, 96 and 97 |
| FITC | Direct chemical injury | T-cell-independent, inflammatory cell and fibrocyte recruitment, IL-13 | Visualization as FITC deposition denoted by green immuno-fluorescence, persistent | Variable efficacy of dose | Lymphocyte-independent pulmonary fibrosis by day 21 | 93, 94 | |
| Irradiation | Radiation injury | Monocyte/lymphocyte-derived lymphotactin, RANTES, CCL-2 and 7, CXCL-10, TGF-β | Different susceptibility of mouse strains allows genetic study | Slow (24 weeks) | Model of radiation fibrosis | 93, 94 | |
| Silica | Resistant, fibrogenic particles administered intratracheally | Inflammatory cell recruitment, IL-1, TNF-α, IL-10, TH2 | Persistent | Specialized aerosolization equipment required (non-essential), lengthy (60 days) | Inflammatory injury followed by fibrosis after min 30 days | 94 | |
| Transgenic TGF-α | TGF-α increased in IPF patients’ BAL | TGF-α overexpression. Fibrosis without inflammation | Incisive single-cytokine system | Not representative of complexity of actual disease state | Pulmonary fibrosis at 4 days | 98 | |
| Adenovirus delivery of GMCSF, TNF, TGF-β, IL-1b | Overexpression of important cytokines | Various | Incisive single-cytokine system | As before, vigorous immune response to virus, epithelium trophic | Cytokine dependent | 99, 100, 101 and 102 | |
| IPF and asthma | Transgenic IL-13 | IL-13 elevated in IPF patients and asthmatics | IL-13, CCR1,-2,-5,-10, TGF-β,IL-11, MMP-1,VEGF | More complex-cytokine pattern | As above plus TH2 phenotype | Eosinophil-rich inflammation followed by fibrotic foci long term | 103 |
| Asthma | Ovalbumin/HDM/cockroach/ragweed sensitization | Allergen sensitization | TH2, IgE, eosinophilia, airway hyper-responsiveness | Models TH2 inflammatory response, quick | High-dose, infrequent exposure as opposed to low-dose frequent allergen exposure in human disease, eosinophils less likely to degranulate. Effective mouse therapies do not necessarily translate (e.g., anti-Il-5) | Eosinophilic inflammation | 92, 104 |
| COPD | Inhalation of smoke. Chronic smoke exposure | Smoking causative of COPD | Mild COPD (Gold 1,2) | Simple design. Relevant to etiology of disease in humans | Time-consuming, humans tend to have more advanced disease at presentation | Mild COPD model | 105 |
| Neutrophil elastase KO mouse | Elastase a key neutrophil product | Smoke damage resistant | Incisive | Simplistic | 59% protection from emphysema | 74 | |
| Variety of transgenic KO mice plus smoke exposure (MMP1,9,12,TNFR 1+2) | Relevance of MMPs in COPD development | Various | Examine importance of a single chemokine to COPD and smoke-related inflammation | Difficult technique requiring expertise. | KO dependent | 106, 107 | |
| α1-AT “Pallid mouse” | α1-AT deficiency predisposes to emphysema in humans | CD4+ cells significantly increased in tissue | Has human corollary in α1-AT deficiency phenotype | Small minority human COPD due to α1-AT deficiency | Panlobular emphysema | 106, 107 | |
| Itgb6 null mice | Alteration in TGF-β responsiveness | TGF-β deplete, MMP12 overactivity, age-dependent emphysema | Chronic progressive model | Complex | Age-dependent emphysema | 106, 107 | |
| CF | CFTR gene knockouts (various, approx.11 models) | CF single-gene disease | Failed mucociliary clearance, inflammatory cell recruitment, parenchymal interstitial thickening, pseudomonal susceptibility | Multiple phenotypes generated by different CFTR mutations | Phenotypes not directly applicable to human genotypes | Various | 91 |
| ALI/ARDS | Hyperoxia | Exposure to 95% O2 | TNF, IFN-γ, ROS,IL-12, IL-18 | Quick | O2 chamber required, | Hyperoxic lung injury | 108 |
| LPS IT | Sepsis associated with ARDS | ROS, NF-κB, IL-6, IL-8 | Widely used, well-characterized | Overly simplistic | Model of sepsis-related ARDS/ALI | 109 | |
| Hemorrhage/resus lung injury | Venesection to shock +/−resus | CREB, ROS, NF-κB, IL-6, IL-8 | Models clinical events | Technically difficult | Model of traumatic ARDS/ALI | 110, 111 | |
| Infective | Respiratory reovirus 1/L induction of diffuse alveolar damage | Overlapping phases of exudation including hyaline membranes, regeneration, and healing via resolution and or repair with fibrosis. | Fibro-reparative phase modeled as well as initial insult | Technically difficult | Neutrophilic inflammation | 112 |
Abbreviations: ARDS/ALI, adult respiratory distress syndrome/acute lung injury; CF, cystic fibrosis; COPD, chronic obstructive pulmonary disease; IPF, idiopathic pulmonary fibrosis.
Conclusion
The respiratory mucosa is frequently threatened with microbial invasion because it is responsible for gas exchange. Consequently, it has developed a sophisticated, sensitive surveillance system and a rapid-response immune policing arm to ensure that it usually prevails in a given interaction. In inflammatory disease, this system is thwarted or dysfunctional with devastating results. The respiratory mucosal immune and inflammatory response can be volatile and damaging for lung function and architecture. To understand respiratory inflammatory disease, it is essential to fully appreciate the biology of archetypal inflammatory cells such as granulocytes. To drive granulocyte apoptosis and enhance granulocyte clearance is a research priority that will hopefully enable amelioration or cure of prevalent (and obscure) respiratory inflammatory conditions.
Acknowledgements
We thank the Wellcome Trust (WT082181) and the Medical Research Council (G0601481) for their support of the work that has enabled us to contribute to this review, and our friends and colleagues within the field whose work constitutes the bulk of the information provided.
PowerPoint slides
References
- 1.Seaton A, Seaton D, Leitch AG. Crofton and Douglas's respiratory diseases. Blackwell Science; 2000. [Google Scholar]
- 2.Rather LJ. Disturbance of function (functio laesa): the legendary fifth cardinal sign of inflammation, added by Galen to the four cardinal signs of celsus. Bull NY Acad. Med. 1971;47,:303–322. [PMC free article] [PubMed] [Google Scholar]
- 3.Sabroe I, Whyte MK. Toll-like receptor (TLR)-based networks regulate neutrophilic inflammation in respiratory disease. Biochem. Soc. Trans. 2007;35,:1492–1495. doi: 10.1042/BST0351492. [DOI] [PubMed] [Google Scholar]
- 4.Rossi AG, Hallett JM, Sawatzky DA, Teixeira MM, Haslett C. Modulation of granulocyte apoptosis can influence the resolution of inflammation. Biochem. Soc. Trans. 2007;35,:288–291. doi: 10.1042/BST0350288. [DOI] [PubMed] [Google Scholar]
- 5.Serhan CN. Resolution of inflammation: state of the art, definitions and terms. FASEB. J. 2007;21,:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.von Kockritz-Blickwede M. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. 2008;111,:3070–3080. doi: 10.1182/blood-2007-07-104018. [DOI] [PubMed] [Google Scholar]
- 7.Brinkmann V. Neutrophil extracellular traps kill bacteria. Science. 2004;303,:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 8.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 2006;6,:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 9.Holgate ST. The future of lung research in the UK. Thorax. 2007;62,:1028–1032. doi: 10.1136/thx.2007.088971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.British Thoracic Society. Health Select Committee inquiry into Health InequalitiesSubmission from the British Thoracic Society–08.01.08 (2008).
- 11.Lachmann PJ. Microbial subversion of the immune response. Proc. Natl. Acad. Sci. USA. 2002;99,:8461–8462. doi: 10.1073/pnas.132284499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prince LR. Subversion of a lysosomal pathway regulating neutrophil apoptosis by a major bacterial toxin, pyocyanin. J. Immunol. 2008;180,:3502–3511. doi: 10.4049/jimmunol.180.5.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am. J. Respir. Crit. Care. Med. 1999;160,:S5–11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- 14.Sasmono RT. Mouse neutrophilic granulocytes express mRNA encoding the macrophage colony-stimulating factor receptor (CSF-1R) as well as many other macrophage-specific transcripts and can transdifferentiate into macrophages in vitro in response to CSF-1. J. Leukoc. Biol. 2007;82,:111–123. doi: 10.1189/jlb.1206713. [DOI] [PubMed] [Google Scholar]
- 15.Araki H. Reprogramming of human postmitotic neutrophils into macrophages by growth factors. Blood. 2004;103,:2973–2980. doi: 10.1182/blood-2003-08-2742. [DOI] [PubMed] [Google Scholar]
- 16.Opferman JT. Life and death during hematopoietic differentiation. Curr. Opin. Immunol. 2007;19,:497–502. doi: 10.1016/j.coi.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Rossi AG. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 2006;12,:1056–1064. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- 18.Sundaramurthy V, Pieters J. Interactions of pathogenic mycobacteria with host macrophages. Microbes Infect. 2007;9,:1671–1679. doi: 10.1016/j.micinf.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 19.Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007;5,:577–582. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- 20.Rothenberg ME, Hogan SP. The eosinophil. Annu. Rev. Immunol. 2006;24,:147–174. doi: 10.1146/annurev.immunol.24.021605.090720. [DOI] [PubMed] [Google Scholar]
- 21.Perretti M, Solito E. Annexin 1 and neutrophil apoptosis. Biochem. Soc. Trans. 2004;32,:507–510. doi: 10.1042/bst0320507. [DOI] [PubMed] [Google Scholar]
- 22.Scannell M. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 2007;178,:4595–4605. doi: 10.4049/jimmunol.178.7.4595. [DOI] [PubMed] [Google Scholar]
- 23.Bianchi SM, Dockrell DH, Renshaw SA, Sabroe I, Whyte MK. Granulocyte apoptosis in the pathogenesis and resolution of lung disease. Clin. Sci. 2006;110,:293–304. doi: 10.1042/CS20050178. [DOI] [PubMed] [Google Scholar]
- 24.Maianski NA, Roos D, Kuijpers TW. Tumor necrosis factor alpha induces a caspase-independent death pathway in human neutrophils. Blood. 2003;101,:1987–1995. doi: 10.1182/blood-2002-02-0522. [DOI] [PubMed] [Google Scholar]
- 25.Simon HU. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol. Rev. 2003;193,:101–110. doi: 10.1034/j.1600-065X.2003.00038.x. [DOI] [PubMed] [Google Scholar]
- 26.Cowburn AS, White JF, Deighton J, Walmsley SR, Chilvers ER. z-VAD-fmk augmentation of TNF alpha-stimulated neutrophil apoptosis is compound specific and does not involve the generation of reactive oxygen species. Blood. 2005;105,:2970–2972. doi: 10.1182/blood-2004-07-2870. [DOI] [PubMed] [Google Scholar]
- 27.Simon HU. Regulation of eosinophil and neutrophil apoptosis--similarities and differences. Immunol. Rev. 2001;179,:156–162. doi: 10.1034/j.1600-065X.2001.790115.x. [DOI] [PubMed] [Google Scholar]
- 28.Simon HU. Molecules involved in the regulation of eosinophil apoptosis. Chem. Immunol. Allergy. 2006;91,:49–58. doi: 10.1159/000090229. [DOI] [PubMed] [Google Scholar]
- 29.Simon H, Alam R. Regulation of eosinophil apoptosis: transduction of survival and death signals. Int. Arch. Allergy Immunol. 1999;118,:7–14. doi: 10.1159/000024025. [DOI] [PubMed] [Google Scholar]
- 30.Daigle I, Simon HU. Critical role for caspases 3 and 8 in neutrophil but not eosinophil apoptosis. Int. Arch. Allergy Immunol. 2001;126,:147–156. doi: 10.1159/000049506. [DOI] [PubMed] [Google Scholar]
- 31.Ochiai K, Kagami M, Matsumura R, Tomioka H. IL-5 but not interferon-gamma (IFN-gamma) inhibits eosinophil apoptosis by up-regulation of bcl-2 expression. Clin. Exp. Immunol. 1997;107,:198–204. doi: 10.1046/j.1365-2249.1997.d01-884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weinmann P, Gaehtgens P, Walzog B. Bcl-Xl- and Bax-alpha-mediated regulation of apoptosis of human neutrophils via caspase-3. Blood. 1999;93,:3106–3115. [PubMed] [Google Scholar]
- 33.Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood. 2007;109,:1620–1626. doi: 10.1182/blood-2006-03-013771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamasaki A. Accelerated neutrophil apoptosis in mice lacking A1-a, a subtype of the bcl-2-related A1 gene. J. Exp. Med. 1998;188,:1985–1992. doi: 10.1084/jem.188.11.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabroe I, Dower SK, Whyte MK. The role of Toll-like receptors in the regulation of neutrophil migration, activation, apoptosis. Clin. Infect. Dis. 2005;41(Suppl 7):S421–S426. doi: 10.1086/431992. [DOI] [PubMed] [Google Scholar]
- 36.Ward C. NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J. Biol. Chem. 1999;274,:4309–4318. doi: 10.1074/jbc.274.7.4309. [DOI] [PubMed] [Google Scholar]
- 37.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-kappaB in the resolution of inflammation. Nat. Med. 2001;7,:1291–1297. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 38.Pinho V. Phosphoinositide-3 kinases critically regulate the recruitment and survival of eosinophils in vivo: importance for the resolution of allergic inflammation. J. Leukoc. Biol. 2005;77,:800–810. doi: 10.1189/jlb.0704386. [DOI] [PubMed] [Google Scholar]
- 39.Pinho V. Tissue- and stimulus-dependent role of phosphatidylinositol 3-kinase isoforms for neutrophil recruitment induced by chemoattractants in vivo. J. Immunol. 2007;179,:7891–7898. doi: 10.4049/jimmunol.179.11.7891. [DOI] [PubMed] [Google Scholar]
- 40.Savill J, Haslett C. Granulocyte clearance by apoptosis in the resolution of inflammation. Semin. Cell. Biol. 1995;6,:385–393. doi: 10.1016/S1043-4682(05)80009-1. [DOI] [PubMed] [Google Scholar]
- 41.Sivertson KL, Seeds MC, Long DL, Peachman KK, Bass DA. The differential effect of dexamethasone on granulocyte apoptosis involves stabilization of Mcl-1L in neutrophils but not in eosinophils. Cell. Immunol. 2007;246,:34–45. doi: 10.1016/j.cellimm.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heasman SJ, Giles KM, Ward C, Rossi AG, Haslett C, Dransfield I. Glucocorticoid-mediated regulation of granulocyte apoptosis and macrophage phagocytosis of apoptotic cells: implications for the resolution of inflammation. J. Endocrinol. 2003;178,:29–36. doi: 10.1677/joe.0.1780029. [DOI] [PubMed] [Google Scholar]
- 43.Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 2007;25,:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 44.Hallett JM, Leitch AE, Riley NA, Duffin R, Haslett C, Rossi AG. Novel pharmacological strategies for driving inflammatory cell apoptosis and enhancing the resolution of inflammation. Trends Pharmacol. Sci. 2008;29,:250–257. doi: 10.1016/j.tips.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi N. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27,:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park SY. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15,:192–201. doi: 10.1038/sj.cdd.4402242. [DOI] [PubMed] [Google Scholar]
- 47.Park D. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450,:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 48.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, PAF. J. Clin. Invest. 1998;101,:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barnes, P.J. Alveolar macrophages in chronic obstructive pulmonary disease (COPD). Cell. Mol. Biol. 50, Online Pub:OL627-OL637 (2004). [PubMed]
- 50.Barnes PJ. New molecular targets for the treatment of neutrophilic diseases. J. Allergy Clin. Immunol. 2007;119,:1055–1062. doi: 10.1016/j.jaci.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 51.MacNee W. Pathogenesis of chronic obstructive pulmonary disease. Clin. Chest Med. 2007;28,:479–513. doi: 10.1016/j.ccm.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 52.Kirkham PA, Spooner G, Rahman I, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation products. Biochem. Biophys. Res. Commun. 2004;318,:32–37. doi: 10.1016/j.bbrc.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 53.Barnes PJ, Ito K, Adcock IM. Corticosteroid resistance in chronic obstructive pulmonary disease: inactivation of histone deacetylase. Lancet. 2004;363,:731–733. doi: 10.1016/S0140-6736(04)15650-X. [DOI] [PubMed] [Google Scholar]
- 54.Quint JK, Wedzicha JA. The neutrophil in chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2007;119,:1065–1071. doi: 10.1016/j.jaci.2006.12.640. [DOI] [PubMed] [Google Scholar]
- 55.Barnes PJ. Pathophysiology of asthma. Br. J. Clin. Pharmacol. 1996;42,:3–10. doi: 10.1046/j.1365-2125.1996.03721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Busse WW, Banks-Schlegel S, Wenzel SE. Pathophysiology of severe asthma. J. Allergy Clini. Immunol. 2000;106,:1033–1042. doi: 10.1067/mai.2000.111307. [DOI] [PubMed] [Google Scholar]
- 57.Hanania NA. Targeting airway inflammation in asthma: current and future therapies. Chest. 2008;133,:989–998. doi: 10.1378/chest.07-0829. [DOI] [PubMed] [Google Scholar]
- 58.Holgate ST. Understanding the pathophysiology of severe asthma to generate new therapeutic opportunities. J. Allergy Clin. Immunol. 2006;117,:496–506. doi: 10.1016/j.jaci.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 59.Maddox L, Schwartz DA. The pathophysiology of asthma. Annu. Rev. Med. 2002;53,:477–498. doi: 10.1146/annurev.med.53.082901.103921. [DOI] [PubMed] [Google Scholar]
- 60.Walsh GM, Sexton DW, Blaylock MG. Corticosteroids, eosinophils and bronchial epithelial cells: new insights into the resolution of inflammation in asthma. J. Endocrinol. 2003;178,:37–43. doi: 10.1677/joe.0.1780037. [DOI] [PubMed] [Google Scholar]
- 61.Contoli M. Role of deficient type III interferon-[lambda] production in asthma exacerbations. Nat. Med. 2006;12,:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 62.Uddin M, Seumois G, Lau LC, Rytila P, Davies DE, Djukanovic R. Enhancement of neutrophil function by the bronchial epithelium stimulated by epidermal growth factor. Eur. Respir. J. 2008;31,:714–724. doi: 10.1183/09031936.00144307. [DOI] [PubMed] [Google Scholar]
- 63.Flood-Page P. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J. Clin. Invest. 2003;112,:1029–1036. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. Eosinophil's role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am. J. Respir. Crit. Care. Med. 2003;167,:199–204. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- 65.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370,:1422–1431. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- 66.Bullens DM. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir. Res. 2006;7,:135–13. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meagher LC, Cousin JM, Seckl JR, Haslett C. Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J. Immunol. 1996;156,:4422–4428. [PubMed] [Google Scholar]
- 68.Wenzel SE. A different disease, many diseases or mild asthma gone bad? Challenges of severe asthma. Eur. Respir. J. 2003;22,:397–398. doi: 10.1183/09031936.03.00027403. [DOI] [PubMed] [Google Scholar]
- 69.Collard HR. Acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care. Med. 2007;176,:636–643. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Du Bois R, King TE., Jr Challenges in pulmonary fibrosis x 5: the NSIP/UIP debate. Thorax. 2007;62,:1008–1012. doi: 10.1136/thx.2004.031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hunninghake GW, Schwarz MI. State of the Art. Does current knowledge explain the pathogenesis of idiopathic pulmonary fibrosis?: a perspective. Proc. Am. Thorac. Soc. 2007;4,:449–452. doi: 10.1513/pats.200702-036MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wallace WA, Fitch PM, Simpson AJ, Howie SE. Inflammation-associated remodelling and fibrosis in the lung - a process and an end point. Int. J. Exp. Pathol. 2007;88,:103–110. doi: 10.1111/j.1365-2613.2006.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Strieter RM. What differentiates normal lung repair and fibrosis? Inflammation, resolution of repair, fibrosis. Proc. Am. Thorac. Soc. 2008;5,:305–310. doi: 10.1513/pats.200710-160DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chua F. Mice lacking neutrophil elastase are resistant to bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 2007;170,:65–74. doi: 10.2353/ajpath.2007.060352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kinder BW, Brown KK, Schwarz MI, Ix JH, Kervitsky A, King TE., Jr Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest. 2008;133,:226–232. doi: 10.1378/chest.07-1948. [DOI] [PubMed] [Google Scholar]
- 76.Abraham E. Neutrophils and acute lung injury. Crit. Care. Med. 2003;31,:S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 77.Bellingan GJ. The pulmonary physician in critical care * 6: The pathogenesis of ALI/ARDS. Thorax. 2002;57,:540–546. doi: 10.1136/thorax.57.6.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leaver SK, Evans TW. Acute respiratory distress syndrome. BMJ. 2007;335,:389–394. doi: 10.1136/bmj.39293.624699.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martin TR. Interactions between mechanical and biological processes in acute lung injury. Proc. Am. Thorac. Soc. 2008;5,:291–296. doi: 10.1513/pats.200801-005DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am. J. Respir. Cell. Mol. Biol. 2005;33,:319–327. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Donnelly SC. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet. 1993;341,:643–647. doi: 10.1016/0140-6736(93)90416-E. [DOI] [PubMed] [Google Scholar]
- 82.Oeckler RA, Hubmayr RD. Ventilator-associated lung injury: a search for better therapeutic targets. Eur. Respir. J. 2007;30,:1216–1226. doi: 10.1183/09031936.00104907. [DOI] [PubMed] [Google Scholar]
- 83.British Thoracic Society BTS Guidelines for the Management of Community Acquired Pneumonia in Adults. Thorax. 2001;56(Suppl 4):IV1–I64. doi: 10.1136/thorax.56.suppl_4.iv1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Droemann D. Decreased apoptosis and increased activation of alveolar neutrophils in bacterial pneumonia. Chest. 2000;117,:1679–1684. doi: 10.1378/chest.117.6.1679. [DOI] [PubMed] [Google Scholar]
- 85.Watson RW, Redmond HP, Wang JH, Condron C, Bouchier-Hayes D. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J. Immunol. 1996;156,:3986–3992. [PubMed] [Google Scholar]
- 86.Cazzola M, Matera MG, Pezzuto G. Inflammation--a new therapeutic target in pneumonia. Respiration. 2005;72,:117–126. doi: 10.1159/000084039. [DOI] [PubMed] [Google Scholar]
- 87.Confalonieri M. Hydrocortisone infusion for severe community-acquired pneumonia: a preliminary randomized study. Am. J. Respir. Crit. Care. Med. 2005;171,:242–248. doi: 10.1164/rccm.200406-808OC. [DOI] [PubMed] [Google Scholar]
- 88.Elizur A, Cannon CL, Ferkol TW. Airway inflammation in cystic fibrosis. Chest. 2008;133,:489–495. doi: 10.1378/chest.07-1631. [DOI] [PubMed] [Google Scholar]
- 89.Sabroe I, Whyte MK. Incapacitating the immune system in cystic fibrosis. Nat. Med. 2007;13,:1417–1418. doi: 10.1038/nm1207-1417. [DOI] [PubMed] [Google Scholar]
- 90.Brodmerkel CM, Vaddi K. Transgenic animals in inflammatory disease models. Curr. Opin. Biotechnol. 2003;14,:652–658. doi: 10.1016/j.copbio.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 91.Guilbault C, Saeed Z, Downey GP, Radzioch D. Cystic fibrosis mouse models. Am. J. Respir. Cell. Mol. Biol. 2007;36,:1–7. doi: 10.1165/rcmb.2006-0184TR. [DOI] [PubMed] [Google Scholar]
- 92.Kips JC. Murine models of asthma. Eur. Respir. J. 2003;22,:374–382. doi: 10.1183/09031936.03.00026403. [DOI] [PubMed] [Google Scholar]
- 93.Moeller A. Models of pulmonary fibrosis. Drug Discov. Today Dis. Models. 2006;3,:243–249. doi: 10.1016/j.ddmod.2006.09.006. [DOI] [Google Scholar]
- 94.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;294,:L152–L160. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- 95.Renshaw SA, Loynes CA, Elworthy S, Ingham PW, Whyte MK. Modeling inflammation in the zebrafish: how a fish can help us understand lung disease. Exp. Lung Res. 2007;33,:549–554. doi: 10.1080/01902140701756778. [DOI] [PubMed] [Google Scholar]
- 96.Harrison JH, Jr, Lazo JS. High dose continuous infusion of bleomycin in mice: a new model for drug-induced pulmonary fibrosis. J. Pharmacol. Exp. Ther. 1987;243,:1185–1194. [PubMed] [Google Scholar]
- 97.Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell. Biol. 2008;40,:362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2004;286,:L741–L749. doi: 10.1152/ajplung.00208.2003. [DOI] [PubMed] [Google Scholar]
- 99.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta 1 induces prolonged severe fibrosis in rat lung. J. Clin. Investig. 1997;100,:768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xing Z. Gene transfer for cytokine functional studies in the lung: the multifunctional role of GM-CSF in pulmonary inflammation. J. Leukoc. Biol. 1996;59,:481–488. doi: 10.1002/jlb.59.4.481. [DOI] [PubMed] [Google Scholar]
- 101.Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1 beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Investig. 2001;107,:1529–1536. doi: 10.1172/JCI12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miyazaki Y. Expression of a tumor-necrosis-factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis - a mouse model of progressive pulmonary fibrosis. J. Clin. Investig. 1995;96,:250–259. doi: 10.1172/JCI118029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Elias JA. Transgenic modeling of interleukin-13 in the lung. Chest. 2003;123,:339S–345S. doi: 10.1378/chest.123.3_suppl.339S. [DOI] [PubMed] [Google Scholar]
- 104.Zosky GR, Sly PD. Animal models of asthma. Clin. Exp. Allergy. 2007;37,:973–988. doi: 10.1111/j.1365-2222.2007.02740.x. [DOI] [PubMed] [Google Scholar]
- 105.Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2008;294,:L612–L631. doi: 10.1152/ajplung.00390.2007. [DOI] [PubMed] [Google Scholar]
- 106.Fujita M, Nakanishi Y. The pathogenesis of COPD: lessons learned from in vivo animal models. Med. Sci. Monit. 2007;13,:RA19–RA24. [PubMed] [Google Scholar]
- 107.Shapiro SD. Transgenic and gene-targeted mice as models for chronic obstructive pulmonary disease. Eur. Respir. J. 2007;29,:375–378. doi: 10.1183/09031936.00087606. [DOI] [PubMed] [Google Scholar]
- 108.Dos Santos CC. Hyperoxic acute lung injury and ventilator-induced/associated lung injury: new insights into intracellular signaling pathways. Crit. Care. 2007;11,:126–12. doi: 10.1186/cc5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kabir K. Characterization of a murine model of endotoxin-induced acute lung injury. Shock. 2002;17,:300–303. doi: 10.1097/00024382-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 110.Moine P, Shenkar R, Kaneko D, Le Tulzo Y, Abraham E. Systemic blood loss affects NF-kappa B regulatory mechanisms in the lungs. Am. J. Physiol. Lung. Cell. Mol. Physiol. 1997;273,:L185–L192. doi: 10.1152/ajplung.1997.273.1.L185. [DOI] [PubMed] [Google Scholar]
- 111.Bahrami S. Small-volume fluid resuscitation with hypertonic saline prevents inflammation but not mortality in a rat model of hemorrhagic shock. Shock. 2006;25,:283–289. doi: 10.1097/01.shk.0000208808.03148.ea. [DOI] [PubMed] [Google Scholar]
- 112.London L, Majeski EI, Paintlia MK, Harley RA, London SD. Respiratory reovirus 1/L induction of diffuse alveolar damage: a model of acute respiratory distress syndrome. Exp. Mol. Pathol. 2002;72,:24–36. doi: 10.1006/exmp.2001.2414. [DOI] [PubMed] [Google Scholar]