

Abstract
The development of new mechanisms of resistance among pathogens, the occurrence and transmission of genes responsible for antibiotic insensitivity, as well as cancer diseases have been a serious clinical problem around the world for over 50 years. Therefore, intense searching of new leading structures and active substances, which may be used as new drugs, especially against strain resistant to all available therapeutics, is very important. Dihydrofolate reductase (DHFR) has attracted a lot of attention as a molecular target for bacterial resistance over several decades, resulting in a number of useful agents. Trimethoprim (TMP), (2,4-diamino-5-(3′,4′,5′-trimethoxybenzyl)pyrimidine) is the well-known dihydrofolate reductase inhibitor and one of the standard antibiotics used in urinary tract infections (UTIs). This review highlights advances in design, synthesis, and biological evaluations in structural modifications of TMP as DHFR inhibitors. In addition, this report presents the differences in the active site of human and pathogen DHFR. Moreover, an excellent review of DHFR inhibition and their relevance to antimicrobial and parasitic chemotherapy was presented.
Subject terms: Drug development, Antibiotics, Antibiotics, Drug development, Antibiotics
Introduction
Infections caused by multidrug-resistant (MDR) bacteria are significant global healthcare problem. Thus, understanding the mechanisms of drug resistance is one of the most important trends underway research both in the treatment of infectious diseases, as well as work on treatment methods of cancer [1, 2]. The molecular target connecting these equally important directions of research is dihydrofolate reductase (DHFR), an enzyme that catalyzes the nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reduction of dihydrofolate to tetrahydrofolate (7,8-DHF to 5,6,7,8-THF) in microbial and eukaryotes cells [3]. The crucial role of DHFR is related to biosynthesis pathways of the thymidylate and purines, as well as several other amino acids like glycine, methionine, serine, and N-formyl-methionyl tRNA [4–6]. Inhibition of folate metabolizing enzymes leads to an imbalance in the pathways involved in active synthesizing thymidylate, disrupts DNA replication, and eventually causes cell death [7]. Therefore, DHFR is a very good example of a well-established molecular target of new active compounds, which could be approved as antibacterial drugs and therapeutic agents against variety of fatal disorders e.g., cancer [8, 9]. Several classes of compounds have been explored for their potential antifolate activity, among the most outstanding are diaminopyrimidine [10, 11], diaminoquinazolin [12], diaminopteridine [13], and also diaminotriazines [14]. Inhibitors of DHFR characterize widespread application in the treatment of cancer and rheumatoid arthritis (methotrexate, MTX), bacterial DHFR enzyme (trimethoprim, TMP), and Plasmodium falciparum (pf)DHFR (pyrimethamine, PYR). However, the most successful inhibitor against bacterial DHFR is TMP that belongs to the diaminopyrimidine class of compounds [15]. This review summarizes the field of structural modifications of TMP analogues and other nonclassical antifolates as a wide group of DHFR inhibitors. Moreover, we present first and most recent attempts to develop a potent and selective structure targeting infections caused by the bacterial, protozoan, fungal infections, and malaria disease and several of other directions e.g., leishmaniasis, African trypanosomiasis, Chagas’ disease, cancer, rheumatoid disease, and perspectives.
TMP-clinical usage and main mechanisms of resistance
TMP, (2,4-diamino-5-(3′,4′,5′-trimethoxybenzyl)-pyrimidine) is the well-known DHFR inhibitor and widely prescribed antimicrobial agent (Fig. 1). This synthetic antimicrobial drug was fully described by Roth et al. [16] and has been used clinically since the 1960s [17]. TMP belongs to a group of antibacterial agents called diaminopyrimidines [5, 18]. During the process of DHFR–TMP binding, the protonated aminopyrimidine group of TMP interacts with the carboxylate group of the enzyme, via a pair of N–H…O hydrogen bonds, generating a hydrogen-bonded ring motif [19]. Matthews et al. [20] based X-ray crystallography studies confirmed, that TMP binds differently at the active sites of chicken (vertebrate enzyme) and Escherichia coli (ecDHFR). Analysis of these data permits identification of differences that are important in accounting for TMP’s selectivity. A key finding of these results was that residues on opposite sides of the active-site cleft in chicken DHFR are about 1.5–2.0 A further apart than are structurally equivalent residues in the E. coli enzyme. The X-ray structural results strongly suggest that loss of a potential hydrogen bond between the 4-amino group of TMP and the backbone carbonyl of Val115 in enzyme is the major factor responsible for this drug to be more potent inhibition of bacterial DHFR [20].
Fig. 1.

Structures of classical and nonclassical DHFR inhibitors
TMP administrated alone inhibits the growth of a wide range of bacteria [21]. First clinically usage of TMP was reported in 1962 in the treatment of Proteus septicemia with sulfonamides with polymyxin and it is still considered the first-line drug of choice, active in vitro against most aerobic Gram-negative and Gram-positive bacteria [22–25]. This drug is used extensively in various parts of the world alone or in combination with sulfamethoxazole (SMX) for the treatment of respiratory, enteric, or skin ailments [24, 26, 27]. Navarro-Martinez et al. [28] reported that TMP also inhibited Candida albicans DHFR (caDHFR) by competing with 7,8-dihydrofolate although the value of inhibitory constant for C. albicans enzyme is 7000-fold greater than E. coli enzyme.
TMP is the gold standard of treatment of UTIs and uncomplicated UTIs in women, which is caused mainly by E. coli, but also by Proteus mirabilis, Klebsiella spp., other Enterobacteriaceae spp. and Staphylococcus saprophyticus (5% each) [29–31]. In addition, it is also active in vitro against Enterococci spp., however, its role as a therapeutic option for enterococcal infections is controversial [32]. Clinical reports also present TMP activity against certain types of malaria caused by P. falciparum [33].
The combination of TMP with SMX, applied as cotrimoxazole (STX) has proven synergistic activity against a wide range of respiratory, intestinal, urinary tract pathogens and as to be both inexpensive and effective treatment against Pneumocystis carinii [34]. TMP alone has only weak activity against the DHFR of P. carinii [35, 36]. TMP–SMX combination is used for the treatment of UTI, although increasing resistance trends among uropathogens, primarily E. coli, complicate the choice of antibiotics in the outpatient management of UTI and among a large number of microorganisms, including P. carinii [25, 37, 38]. Furthermore, Stuck et al. [39] reported that resistance for standard combination TMP–SMX in the treatment of UTI cause choice of quinolones in 78,5% of cases. In turn, a large number of studies have described the results of in vitro antifungal activity of TMP–SMX combination against clinical isolates, such as Aspergillus fumigatus, Cryptococcus neoformans [31]. Afeltra et al. [40] showed that SMX is active in vitro against A. fumigatus and therefore might help to prevent invasive aspergillosis in AIDS patients receiving SXT prophylaxis. In successive studies the same team of researchers evaluate the in vitro activities of seven different sulfa compounds and pentamidine (PNT) against Aspergillus isolates comprising six different species in two different media. As reported, SMX, sulfadiazine, and PNT were active in vitro however more studies with different media and animal models and clinical studies are necessary to elucidate the potential of these drugs for the treatment of Aspergillus infections [41]. In contrast, Hanafy et al. [42] reported that SMX showed moderate activity against both strains of A. fumigatus and C. neoformans var. grubii (minimum inhibitory concentration (MIC) values were 32–128 μg ml−1). In this report, sulfaphenazole showed the highest activity (MIC values for A. fumigatus and C. neoformans var. grubii were respectively, 64 μg ml−1 and 4–8 μg ml−1). In other report, Ajayi et al. [43] presented the successful treatment of group of children with orbital histoplasmosis caused by Histoplasma capsulatum var. duboisii (one unsuccessfully treated with amphotericin-B) with a combination of TMP and sulphamethoxazole (Septrin) and surgical drainage. Treatment was devoid of side effects and all patients improved markedly during therapy with resolution of the lesions, and with no recurrence of infection in three patients. These findings suggested that Septrin could be a safe alternative to amphotericin-B for the treatment of African histoplasmosis [43]. As an ingredient in cotrimoxazole, TMP is used extensively for the prophylaxis and/or therapy of potentially life-threatening opportunistic infections in patients with AIDS. What is notable, a major drawback to the use of TMP–sulfa combinations in immunocompromised patients is the occurrence of hypersensitivity reactions to the sulfa drug that can be severe enough to require discontinuation of treatment, when the side effects cannot be managed with steroids [44, 45].
The main mechanisms of resistance to TMP includes the production of additional plasmids-, transposons-, cassettes-mediated drug-resistant DHFR [23, 46], impermeability (Klebsiella pneumoniae, Serratia marcescens) [47, 48], alternative metabolic pathways and overproduction of resistant chromosomal DHFR enzyme [49–53].
Podnecky et al. [54] showed that the molecular mechanisms governing TMP resistance to be due to BpeEF- OprC efflux pump expression in Burkholderia pseudomallei strains. It was noticed that in all of the Australian and Thai isolates assessed, TMP resistance was attributed to expression of BpeEF- OprC efflux pump but not changes in the DHFR target, indicating that efflux is the predominant TMP resistance mechanism in B. pseudomallei [54]. Naturally insensitive DHFR enzymes are found among, for instance, Bacteroides species, Clostridium species, Neisseria species, and Moraxella catarrhalis [55].
Resistance to TMP is caused by modifications in the target enzyme DHFR (dfr) encoded by dfr-genes. So far 30 dfr-genes usually associated with integrons are described [56]. Integrons are integrated in transposons predominantly located on plasmids and can insert, excise, and express mobile gene cassettes, often antibiotic resistance genes. This results in an efficient horizontal spread of antibiotic resistance between bacteria [57, 58]. Overproduction of chromosomal DHFR caused by promoter mutation has reportedly occurred in E. coli. A single amino acid substitution in the dhfr gene and altered chromosomally encoded DHFR has been considered responsible for resistance to TMP [59]. It was determined that a single point mutation in the DHFR enzymes, be it transposon, plasmid, or chromosomally encoded is responsible for the reduced susceptibility of Staphylococci to TMP. The single amino acid substitution Phe to Tyr in the Staphylococcus aureus strain B71 (F98Y) resulted in a 64-fold increase in the MIC of TMP for the strain [60]. This mutation resulted in the loss of a single hydrogen bond between the 4-amino group of TMP and the enzyme. The decrease in affinity was identified as the mechanism of resistance at the protein level [61, 62]. In turn, resistance of TMP among Streptococcus pneumoniae is determined by mutations to the DHFR gene caused by six amino acid changes (Glu20-Asp, Pro70-Ser, Gln81-His, Asp92-Ala, Ile100-Leu, and Leu135-Phe) [63]. In strains of TMP-resistant Haemophilus influenzae, changes in both promoter and coding regions of the dhfr genes have been found [64]. In case of Gram-positive bacteria it has been detected several horizontally transmissible dfr-genes, e.g., dfrG have been identified in one of the emm1-2.2 clinical isolates Streptococcus pyogenes [65, 66]. Moreover DHFR encoded by dfrA was found in S. aureus, Staphylococcus epidermidis, Staphylococcus hominis, and Staphylococcus haemolyticus [67–69]. Currently more than 25 variants of dfrA gene resistance have been reported [70, 71]. Tang et al. [72] found and reported in K. pneumoniae, in a single putative multireplicon plasmid pHS0091147, the coexistence of blaKPC-2 and dfrA25, which carried five resistance genes (blaKPC-2, blaCTA-M-14, blaTEM-1, sul1, and dfrA25). In turn, Bosse et al. [73] reported on the presence of the mobilize plasmids dfrA14 in the Pasteurellaceae spp. In addition, widely reported PCR assays confirmed, that mobilize plasmids conferred TMP resistance in Actinobacillus pleuropneumoniae TMP-insensitive Staphylococci spp. and Gram-negative bacteria [62]. The most frequent mechanisms responsible for acquired resistance to TMP alone or with combination with SMX have been described [53]. The wide spectrum of TMP action used in monotherapy and in combination with SMX confirms its great importance as an antibiotic. On the other hand, constantly increasing resistance is a negative prognosis. This fact is the reason for searching for new, more selective active compounds, obtained by different structures modifications. TMP as a drug used for decades is an excellent target, amplified by the broad-spectrum activity and inhibiting of the key enzyme DHFR for prokaryotic and eukaryotic organisms. Below we present various modifications of nonclassical analogues, both first and recent attempts to improve TMP activity.
Nonclassical antifolates and close TMP analogues
One of the classifications of DHFR inhibitors, based on chemical structures, includes classical and nonclassical antifolates [74]. The classical inhibitors are folate analogues, which have a heterocyclic ring bound to the aryl group and glutamate tail. The literature provides a few examples of these antifolates as MTX and pralatrexate (Fig. 1). On the other hand, nonclassical antifolates, used as a means of ensuring bacterial selectivity against mammalian cells, have a lipophilic side chain and have been obtained in order to overcome the resistance to classic antifolates [15]. These compounds are more lipophilous than the classical antifolates and enter cells by passive diffusion [75]. Figure 1 shows selected examples of nonclassical inhibitors including TMP, PYR, trimetrexate, and pyritrexim, PYR is the most widely used DHFR inhibitor, closed analogue of TMP, which was introduced to therapy over 60 years ago [16, 76]. This nonclassical inhibitor presented high affinity to plasmodial and human DHFR (hDHFR). It is still used (in combination with sulfonamides or dapsone) to the treatment of malaria and also in case of toxoplasmosis [77–79]. However, treatment of PYR has been associated with a number of dose-limiting toxicities e.g., myelosuppresion [76].
In order to search for new modifications, Welsch et al. [80] examined the chemical structures of DHFR inhibitors with well-documented activity and conserved binding features using co-crystal structures of inhibitors containing diamino-1,3,5-triazine or 2,4-diaminopirimidine rings. In addition, diaphorase-coupled assay was used to determine the DHFR IC50 activity values [81]. A higher selectivity was observed for Toxoplasma gondii (tgDHFR) meta-bihenyl analog TRC-19 (Fig. 2) obtained by the introduction of a second acrylic ring. Enzymatic assay for tgDHFR indicated TRC-19 as a promising compound with an I50 value of 9 nM because it was 89-fold selective than PYR (I50 = 230 nM) [80].
Fig. 2.

Structures of selected antifolates and TMP analogues
Based on rational drugs design, Hopper et al. [82] identified several high and potent arylpiperazine-based tgDHFR inhibitors. Incorporation of electron donating 2-methoxy group resulted in 2-methoxypyrimidine analog, compound 3 (Fig. 2), which had 16-fold better selectivity for tgDHFR and tgDHFR I50 = 1.6 nM) compared with PYR.
Cycloguanil (Fig. 1) is a 2,4-diaminotriazine antimalarial drug which, like PYR, inhibits DHFR plasmodial and is an active proguanil metabolite, approved for prophylaxis and infectious treatment by P. falciparum or vivax [83]. Tonelli et al. [84] obtained and investigated a series of 1-aryl-4,6-diamino-1,2-dihydrotriazines analogues of cycloguanil as new, promising inhibitors of influenza A and B virus.
Brodimoprim (Fig. 1) is a TMP analog originated in Roche, which was used alone in case of respiratory tract infections, but has been withdrawn from treatment [85]. This agent presented similar antibacterial, toxicological properties to TMP and was out-licensed. It has a long elimination half-life of 32–35 h [85].
After the successful marketing of TMP as cotrimoxazole, projects to find new inhibitors of DHFR useful as antibacterial agents, were intensively pursued in Burroughs Wellcome and Roche for years, but did not result in the development of any new agent beyond phase I (except brodimoprim) [86]. Understanding the known mechanism of resistance to TMP led to the design of iclaprim (Ro 48–2622, AR-100), a new inhibitor with improved affinity towards the S. aureus DHFR (Fig. 1) [87]. This novel 2,4-diaminopyrimidine exhibits potent, rapid bactericidal activity against major Gram-positive pathogens, including methicillin-susceptible S. aureus, MRSA, and F98Y TMP-resistant strains. Iclaprim binds and inhibits bacterial DHFR in a similar manner to TMP [60, 65]. Oefner et al. [88] based enzyme binding and X-ray crystallographic studies elucidated the binding mode of iclaprim. It was reported that the increased hydrophobic interactions between iclaprim and DHFR account for increased affinity and, unlike TMP, enable iclaprim to inhibit even the resistant enzyme with nanomolar affinity, thus overcoming the mechanism of TMP resistance [88]. Moreover, this analog presented activity in case of methicillin-resistant S. aureus (MRSA) strains, penicillin-susceptible pneumococci, β-hemolytic streptococci, enterococci, as well as Legionella, Listeria and Chlamydia spp, but showed moderate activity against some Enterobacteriaceae, H. influenzae, and M. catarrhalis [89–93]. In addition, Ro 48–2622 (iclaprim) characterized a good distribution in tissue. There was confirmed, that this agent was safe, well tolerated, specifically, and selectively inhibited bacterial DHFR in a similar manner to TMP. However, in the case of Ro 48–2622, a lack of affinity to the hDHFR enzyme was observed in more than five orders of magnitude of higher concentrations [92, 93].
Phthalazine derivative Ro-64–5781 (Fig. 2) showed high activity against TMP-resistant strains and Gram-positive bacteria e.g., Staphylococci. Unfortunately, due to low solubility and high protein binding amounting to 98.3%, it was disqualified for further research [94].
Another class of agents, where 2,4-diamino-pyrimidines were replaced by dihydrofthalazine or tetrahydroisoquinoline molecules, also were synthesized. The most attractive compound B (Fig. 2) was active against the DHFR from TMP-resistant S. pneumonia [94].
Rashid et al. [95] synthesized variations of hydroxy trimethoprim by incorporating benzylidene moieties to a 4-NH2 group of compounds 3–5 (Fig. 3). The analogues 3a–h, except 3c, showed very poor antimicrobial activity. In contrast, the potency of the benzyloxy derivatives 4a–e was noticed. Biological evaluations provided that demethylation of the methoxy group at 4-position resulted in comparable activity to TMP. Activities of compound 4b, as well as compounds 5c and 5d against S. aureus and E. coli, were excellent. However replacement of methoxy group by chlorine or methyl (5c and 5d) not improved significantly activity against S. aureus. The structure-activity relationship (SAR) also revealed that benzyloxy derivatives had excellent in vitro antibacterial properties against S. aureus and E. coli strains. Among the obtained new compounds 4a–d exhibited the most significant activity [95].
Fig. 3.
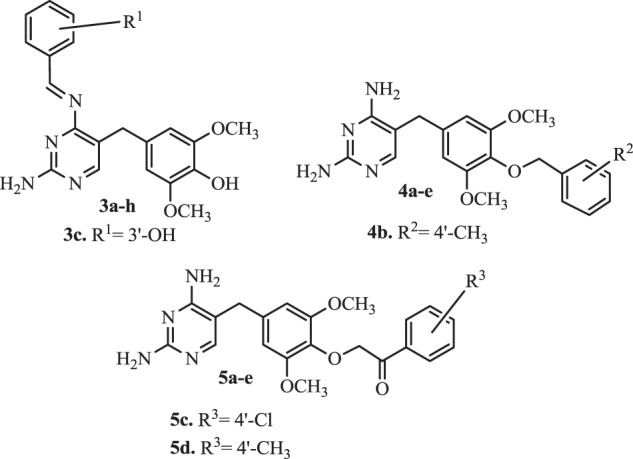
Rashid’s hydroxyl TMP analogues
In turn, Morgan et al. [96] revealed that the series of 2,4-pyrimidines substituted at 4th position by a nucleus with NH linker diminishes the antimalarial activity. As observed, the incorporation of cyclohexylamine instead of a substituted aniline results in a significant increase in activity. Preliminary SAR suggests that associated potential hydrophobic interactions may require flexibility of the substrate. Increased substitution of the cyclohexyl group also results in an increase in activity [97]. The idea of increasing flexibility in new inhibitors was taken from observation of a novel molecule WR99210 (Fig. 2) and some of its elements were incorporated in TMP core to make its congeners [98]. These molecules have sufficient flexibility to withstand this steric repulsion to counteract the resistance, provided better binding to the active site of bacterial DHFR compare with phenyl pyrimidines. Such flexible antifolates may be effective in PYR-resistant P. falciparum [96].
Tarnchompoo et al. [99] made efforts to design a series of 14 novel 4-benzyloxy derivatives of benzyldiaminopyrimidine. The most active TMP analogues A and B (Fig. 2) presented particularly antimalarial activities, strong in vitro antibacterial activity and formed hydrogen bond interactions with Asp27, Leu5, and Phe92 in the active site of DHFR enzyme.
Next new 15 molecules (Fig. 2) were obtained by Shaikh et al. [100]. Series of compounds were tested in vitro against P. falciparum (pfDHFR) specific inhibition, both wild and tetramutant strains. The potency of these molecules was evaluated by in vitro enzyme specific assay. It was concluded, that derivatives with an alkyl substitution on the phenyl ring (S1–10) were more potent against the wild rather than mutant type strain. However the molecules with -O- as the linker (S1–2, S1–3 and S1–4) had low activity against both mutant and wild strains. On the other hand, the introduction of NH as a linker led to potentiate activity on the wild strain. Compound S3–1 without substitution on the phenyl ring and linked through NH showed 40.3% inhibition against wild-type plasmodium strains [100].
First attempts to obtain TMP derivatives
The important achievements in the area of synthesis and biological evaluations analogues of TMP were reported by Roth et al. [101–111] in the series of publications. In the first part of studies, Roth et al. [101] reported that the 4′-hydroxy 1 analog of TMP was active against ecDHFR in vitro as a pattern (Table 1), however, methyl moiety of the 4′-methoxy substituent (analog 2) appears to contribute slightly in increasing the binding to ecDHFR [101]. Removal of the 6-alkyl substituent had the rather remarkable effect on considerably depressing in vitro antimalarial activity and markedly increasing antibacterial activity. The 6-methyl substituent of the compound 2 did not increase antimalarial activity, as evidenced the Plasmodium berghei enzyme data [101]. A 6-hydroxy derivative 3 had no appreciable antibacterial activity [101]. This compound is not ionized at physiological pH (dissociation constant (pKa) at 20 °C values ~3.5 and10.7), in contrast 4′-hydroxy 1, which has a pKa of 7.1 [102].
Table 1.
Various modification of TMP structure
 | |||||
|---|---|---|---|---|---|
| No. | 2′ | 3′ | 4′ | 5′ | 6′ |
| 1 A | H | OCH3 | OH | OCH3 | H |
| 2 A | H | OCH3 | OCH3 | OCH3 | CH3 |
| 3 A | H | OCH3 | OCH3 | OCH3 | OH |
| 4 B | H | OCH3 | OCH3 | OCH3 | H |
| 5 B | H | OCH3 | OH | OCH3 | H |
| 6 A | H | OCH3 | OCHCOOH | OCH3 | H |
| 7 A | H | OCH3 | O(CH2)5COOH | OCH3 | H |
| 8 A | H | OCH3 | H | OCH3 | H |
| 9 A | H | OCH3 | OCH3(CH2)2Cl | OCH3 | H |
| 10 A | H | OCH3 | OCH3CH=CH2 | OCH3 | H |
| 11 A | H | OCH3 | CH | OCH3 | H |
| 12 A | H | OCH3 | C2H5 | OCH3 | H |
| 13 A | H | OCH3 | OH | OCH3 | H |
| 14 A | H | OCH3 | C4H9−n | OCH3 | H |
| 15 A | OCH3 | OCH3 | OCH3 | H | H |
| 16 A | H | OCH3 | OCH3 | H | H |
| 17 A | H | c-OCH2CH(CH3)O | c-OCH2CH(CH3)O | CH3 | H |
| 18 A | c-OCH(CH3)CH2 | c-OCH(CH3)CH2 | H | CH3 | H |
| 19 A | H | CH2CH3 | CH2CH3 | CH2CH3 | H |
| 20 A | H | CH2CH3 | CH2CH3 | H | H |
| 21 A | H | Pr−i | OH | Pr−i | H |
| 22 A | H | OCH3 | OH | C2H5 | H |
| 23 A | H | C2H5 | OH | Pr−i | H |
| 24 A | H | Pr−n | OH | Pr−n | H |
Ruckman et al. [103] noticed that the 3-deazatrimethoprim analog 4 (I50 = 140 µM; pKa = 9.03 ± 0.02) (Table 1) was about 300-fold less active as an inhibitor of ecDHFR than TMP (pKa = 7.1) [102], however 4′-demethyl analog 5 (I50 = 37 µM) was about four times more active than 4. It was reported that the considerable loss of activity was affected by replacement of the N-3 atom of TMP by carbon, but is not related to the increased basicity of analog 4. The derivative 5 appeared more active than 4, but it was about one-half less active as TMP [92]. Later studies of TMP analogues included incorporating the para substituent in the benzene ring (Table 1). All of these compounds were identified as very potent in vitro inhibitors of ecDHFR and the compounds all have I50ecDHFR values between 0.5 and 2.0 µM. Allyloxy derivative 9 (I50ecDHFR = 0.52 µM) and γ-chloropropoxy 10 (I50ecDHFR = 0.5 µM) had activities fairly close to that of TMP (I50ecDHFR = 0.5–0.7 µM). Compound 6 (I50ecDHFR = 37.0 µM) with an anionic carboxyl functionality, indicated significantly lower activity because of hydrophobic bonding between inhibitor and protein, and also interfered by hydration sheath surrounding the ionized center. The carboxymethyl derivative 7 (I50ecDHFR = 6.2 µM) did not cause these disturbances because the charge center was far enough away from the ring. The diversity in side chains resulted in similar activity as TMP [104]. Kompis et al. [105] provided additional supporting data for these conclusions. Their 4′-carboxy and 4′-(aminomethyl) derivatives, for example, both of which have their charged centers within two atoms of the benzene ring, were found to be considerably less active than compound 7. Significantly, the differences in activity were apparently related rather to the size of the substituent than to the polarity or lipophilicity [105]. Derivative 8 (I50ecDHFR = 5.6 µM) is significantly less active than the others (one-tenth that of TMP) [104].
Compound 9 appeared to be the most attractive and also very active as an inhibitor of ecDHFR. Furthermore, compounds 9 and 10 were selected for in vivo antibacterial studies in mice and pharmacokinetic investigations in dogs. This report proved that TMP and two selected analogues were rapidly absorbed but extensively metabolized (10% of the dose excreted in the urine as the intact drug in 24 h). In addition, TMP had longer plasma t1/2 than any of the analogues and also greatest bioavailability. It was suggested that 4′ether chain of TMP with three types of substitutions has apparently created greater opportunity for metabolism, causing more rapid elimination and decreased bioavailability. Compounds 9 and 10 could be interesting for human and veterinary medicines because of their antibacterial activity and physicochemical properties [104]. It was reported that the aliphatic ether derivatives had less activity against rat liver DHFR (rlDHFR) than compounds lacking the ether chain (8, 11, 13; I50rlDHFR = 9000; 12000; 9200 µM respectively). This effect was caused by the out of plane α-carbon, which interfered with binding to the mammalian DHFR, in contrast to bacterial enzyme [104]. Compound 12 (I50rlDHFR = 12000 µM) involving the 4′-ethyl group provided evidence for the same trend. However, it will be noted that structure 14 (I50rlDHFR = 58000 µM), which contained the more lipophilic substituent was in vitro active against the Gram-positive organisms [104].
In the successive report, Stuart et al. [106] presented an evaluation of the various methoxy substitution effects (Table 1). The 2,3,4-trimethoxy analog of TMP (15) was compared with compounds: 16 bearing the 3,4-dimethoxy groups and ortho-substituted isomeric dihydrobenzofuran derivatives 17 and 18. It was reported that compound 15 (values of 50% inhibnecDHFR = 92; 50% inhibnrlDHFR = 5800) was 6-fold more in vitro active than TMP (values of 50% inhibnecDHFR = 0.7; DHFR50% inhibnrlDHFR = 34000) against the rat liver enzyme but considerably less active than analog 16 (values of 50% inhibnecDHFR = 0.6; 50% inhibnrlDHFR = 8600) against ecDHFR. It was also found, that the lack of a 5- or 3-substituent of 16 would be the cause of low activity against ecDHFR [106]. In another report, Roth et al. [107] analyzed terms of inhibitory in vitro activities against four DHFR isoenzymes: ecDHFR, Neisseria gonorrhoeae (ngDHFR), P. berghei (pbDHFR), and rlDHFR. Derivatives with n-propyl or isopropyl substitutes at the 3- and 5-positions were more active than TMP against ngDHFR. It was found, that TMP derivatives selectivity were lower than the standard compound. These studies revealed, that the activities against rlDHFR were at least 2 orders of magnitude less than against enzymes from bacterial strains. In turn, the profiles of activity were different for ecDHFR but remarkably similar for rlDHFR, ngDHFR, and pbDHFR in all three series. In addition the selectivity for this enzyme was dependent on the nature of the 4-substituent, high polar in case 4-amino analogues and low for 4-hydroxy compounds, possibly as a result of solvation differences [107]. With complex substituents, the environment of each atom in the active site must be taken into account to adequately explain SARs e.g., methylene moiety will be largely in contact with solvent, but the solvated hydroxyl function, particularly if in the 5- (down) position, will require desolvation to interact favorably with the hydrophobic side chains of the enzyme, which costs energy [107].
In addition, 3,4,5-triethyl analog 19 (I50ecDHFR = 0.3 µM; I50rlDHFR = 3000 µM; I50ngDHFR = 22 µM; I50pgDHFR = 0.75 µM) (Table 1) and TMP (I50ecDHFR = 0.7 µM; I50rlDHFR = 3700 µM; I50ngDHFR = 45 µM; I50pgDHFR = 12 µM) characterized very close activity in case ecDHFR. It was suggested that the shape of these substituents is the most important feature. The selectivity study found that TMP was tenfold more active inhibitor to ecDHFR than 3,4-dimethoxy analog 16 (I50ecDHFR = 7.0 µM; I50rlDHFR = 7100 µM; I50ngDHFR = 52.6 µM; I50pgDHFR = n.d). In contrast, TMP was 3–4 times more active on the vertebrate isozyme than derivative with 3,4-diethyl group 20 (I50ecDHFR = 2.0 µM; I50rlDHFR = 5200 µM; I50ngDHFR = 35.6 µM; I50pgDHFR = 3.6) [107]. Roth et al. [108] suggested, that a comparison between the binding properties of TMP and its triethyl analog, as well as the disubstituted congeners, made clear the fact, that the methoxy groups of TMP were partially responsible for its selectivity for bacterial, as opposed to vertebrate DHFR.
In the next report, Roth et al. [109] made efforts to obtain lipophilic TMP derivatives with 3,5-dialkyl-4-hydroxyl substituents in the benzene ring. Biological studies revealed, that the synthesized derivatives showed a higher level of in vitro activity in relation to N. gonorrhoeae than the standard. The 3,5-diisopropyl-4-hydroxy analog 21 (I50ngDHFR = 2.9 µM; MIC = 9.2 μg ml−1) (Table 1) was chosen as a strong candidate for an antigonoccocal agent. In vitro antibacterial activity against ten selected organisms in relation to TMP (I50ngDHFR = 45 µM; MIC = 240 μg ml−1) data showed, that the tested compounds in serial dilutions were carried out at levels of 0.1, 0.3, 1, 3, etc., and differences of ±1 dilution were significantly less active than TMP, particularly against Gram-negative organisms. However, it was shown that the most promising compounds among this group against ecDHFR were derivatives 23 and 24 [109]. TMP in combination with sulfonamide has been found effective in the treatment of gonorrhea, but relatively high doses were required. The compound 21 presented 25-fold greater antigonococcal activity compared with TMP. It was suggested also that compound 21 might serve as a therapeutic agent for gonorrhoea, without sulfa synergy combination. These experiences proved, that conjugation with enzyme might well occur with the straight, not branched chain of 3,5-dialkyl-4-hydroxybenzyl derivatives. The phenolic derivatives, which presented lower selectivity to bacterial DHFR were prime targets for the study because of their considerably greater activity than their 4-methoxy counterparts [109]. Further extended pharmacokinetic and metabolic evaluations in dogs indicated, that compound 21 was not extensively conjugated by metabolic enzymes, and one of the methyl groups was metabolized and 3-isopropyl-4-hydroxy-5-(carboxyethyl)benzyl derivative was excreted. Based on these results compound 21 was disqualified to further development [109].
A wide range of studies by Roth et al. indicates the searching of new TMP derivatives based on the study of activity-structure relationship. It was confirmed that the character of the substituent and the position of substitution in the aromatic ring is important for selectivity and potential activity, mainly against Gram-negative bacteria. As presented, some modifications indicated an increase in affinity of analogues to the enzyme DHFR of vertebrates.
TMP analogues containing quinoline ring
Further research focused on the design and synthesis of quinoline ring analogues [110]. Derivatives 25–32 (Fig. 4) with 4-methyl-8-methoxy substitution demonstrated excellent in vitro inhibiting properties of bacterial DHFR. It was observed that 8-ethyl derivative 26 showed in vitro the best inhibitory properties expressed as I50 values (I50ecDHFR = 0.24; I50rlDHFR = 4400; I50ngDHFR = 90) and also about 3–6 fold greater than 25 (I50ecDHFR = 0.68; I50rlDHFR = 25000; I50ngDHFR = 89) against the ecDHFR, rlDHFR, and N. gonorrhoeae (ngDHFR) enzymes [110]. In vitro antibacterial activities of compounds 25 and 26, expressed as a ratio when compared with TMP activity. Serial dilutions of 1–3–10–30…etc. were carried out, and the MICs were divided by that of TMP. These compounds were more active against the Gram-positive organisms (S. aureus and Streptococcus faecalis) than TMP, where (MICCompound/MICTMP ratio for 25 and 26 were 1;03 and 0,03;0,03). The compounds 31 and 32, the 4-methyl-8-methoxy and 2,4-dimethyl-8-methoxy analogues, have the same activity as standard in case S. aureus and MICCompound/MICTMP ratio were equal 1 [110].
Fig. 4.

Structures of selected TMP analogues containing quinoline ring
In addition, the high activity against Proteus species e.g., Proteus vulgaris and P. mirabilis is to be noted in particular. The 4-methyl derivative 30 (MICCompound/MICTMP ratio in case P. vulgaris and P. mirabilis were equal 1) and its 8-methoxy-congener 28 (MICCompound/MICTMP ratio in case P. vulgaris and P. mirabilis were equal 3) also had similar activity [110]. Furthermore, the unsubstituted compound 27 also presented excellent properties in activity (MICCompound/MICTMP ratio in case P. vulgaris and P. mirabilis were equal 10 and 3 respectively) [110]. Based on stereo view of TMP in ternary complex with active site of E. coli DHFR and NADPH, Ruckman et al. [110] suggested that avoidance of bulky side chains (for example, 29), as well as simplicity of aromatic substitution, aid to effective penetration through the bacterial cell wall. Moreover, it was observed, that the specificity for bacterial DHFR was considerably increased in the tetrahydroquinoline series compared with its aromatic analogues [110]. These conclusions were observed in previous report of Painter’s team, where was attempting to adjust log P values of side chains to aid passage into cells, shapes, and bulk are considered as well [111]. Davis et al. [112] designed compounds 33–36 (Fig. 4) with basic or methoxy substituents at the 2- or 4-positions of the quinoline ring, expecting the highest level of antibacterial selectivity. It was found that all of the 6-quinolyl-1-methyl derivatives exhibited highly in vitro inhibitory properties against ecDHFR, caused by the presence of a 1-substituent, as in the quinoline ring of compound 33 (I50ecDHFR = 0.75 µM). This investigation revealed, that the most interesting compounds were 34 and 35 (I50ecDHFR = 0.75 and 0.77 µM respectively), where both of them had basic substitutes at the 2-position of the quinoline ring [112]. Activity of these compounds was also highly specific against bacterial DHFR, compare with a vertebrate counterpart. Protonation on the nitrogen N-1 of the quinoline ring is a possible cause of this specificity. Appropriately, substitution at the 4- and 8-position in the quinoline ring could be compared with the 3- and 5-substituents of benzylpyrimidines, which probably occupy similar positions in space when bound to DHFR [112]. Compound 36 showed excellent in vitro inhibitory properties (I50ecDHFR = 0.3 µM) but presented poor selectivity towards ecDHFR. On the other side, it was noticed, that the diethylamino analog was neither very active nor very selective. Very likely, the decreased activity against bacterial DHFR was due to the bulk of the diethylamino moiety [112].
The further report suggested that the gem-dimethyl substituents of the dihydroquinoline derivatives were responsible for contributing to potent bacterial DHFR inhibition, as well as the high selectivity. Johnson et al. [113] obtained twelve 2,4-diamino-5-[(1,2-dihydro-6-quinolyl)methyl]pyrimidines containing gem-dimethyl or fluoroethyl substituents at the 2-position of the dihydroquinoline ring. Quantitation of affinity differences for some of the more active ecDHFR inhibitors (I50 < 0.2 µM) was made on the basis of apparent K¡ determination [113]. Compound 37 (I50 < 0.2 µM) has a 13-fold greater affinity than TMP. The most important derivatives of this group, 37 and 38 (Fig. 4) showed better properties than TMP in relation to Gram-positive organisms as showed relative in vitro antibacterial activities [113]. Moreover, values of MICCompound/MICTMP for compound 37 in case S. pyogenes and S. aureus were 0.03 and 0.001, respectively. In addition, derivative 37 (app KiE.coli = 0.10 nM) presented highly active antibacterial profile and one order of magnitude higher selectivity for the ecDHFR compared with the control. The compound 38 (e.g. I50E.coli = 0.9 µM) showed activity in vitro against Gram-negative organisms but less than TMP [113]. Further studies presented a synthesis of a novel series of 3,4-dimetoxy-5-alkenyl derivatives of TMP. The 3,4-dimethoxy-alkenyl, 5-alkyl and 5-alkoxy analogues presented high broad-spectrum antibacterial activity, however, the pharmacokinetic studies of four selected compounds in dogs and rats and in vivo abdominal sepsis model in rats showed no advantages over TMP [114].
Compared with 1,2,3,4-tetrahydroquinoline analog 39 (Fig. 4), the addition of 1-methyl substituent, as in the case of the derivative 40, resulted in an increase in selectivity and, at the same time, the effect of weakening the binding effect with the enzyme [115]. On the basis of in vitro screening of antibacterial activity, it has been confirmed, that high affinity of some compounds to ecDHFR does not translate into adequate inhibitory activity, it was concluded that compound 40 was too similar to TMP to provide any significant advantage as a broad-spectrum antibacterial; furthermore, its activity against E. coli in vitro was not compelling [114]. On the basis of the efficacy study and in vivo tests, it was concluded that this series of compounds had no potential for further research in the search of various bacterial DHFR inhibitors [115].
TMP analogues as active agents against toxoplasmosis, Mycobacterium avium and pneumocystis
Infections caused by opportunistic pathogens are a major problem in the clinical treatment of patients with AIDS and other types of immunodeficiency syndromes. T. gondii (tg), P. carinii (pc), Mycobacterium avium (ma) belong to a group of the known organisms, that may cause significant morbidity and mortality in patients within the above-mentioned disorders [116]. Both TMP, and TMP–SMX are used in treatment and prophylaxis infection causes of Pneumocistis jirovecii [117]. However presence mutations of the TMP/SMX resistant have been documented [118–120]. Also with Mycobacterium tuberculosis, especially multiresistant strains, public health risks are emerging in many countries and new drugs are urgently needed [94]. In fact, no DHFR inhibitors are currently used for tuberculosis treatment. One of used drug, Epiroprim (Ro 11-8958) (Fig. 5) has recently been evaluated against various M. tuberculosis and found to exhibit weak activity [121]. In turn, Queener et al. [122], as well as Then et al. [123], based on potency and selectivity tests toward purified recombinant pcDHFR, have suggested, that Ro 11-8958 was the strongest inhibitor among the series of their TMP analogues. Some of the new inhibitors, synthesized as potential antipneumocystis or antitoxoplasma agents by Roskowsky et al. [124, 125], potently in vitro inhibited the maDHFR. Moreover, encouraging results, with respect to both potency and selectivity, were obtained for 2´,5´-disubstituted benzylpyrimidines. Some of these examples are presented below.
Fig. 5.

Structures of selected TMP analogues against Toxoplasmosis, Mycobacterium avium, and Pneumocystis carinii DHFR enzyme
Rosowsky’s analogues: 2,4-diamino-5-[2-methoxy-5-alkoxybenzyl]pyrimidines (41a–e) and 2,4-diamino-5-[2-methoxy-5-(ω-carboxyalkyloxy)benzyl]pyrimidines (41f–k) (Fig. 5), with the general structure as 41, were synthesized and evaluated for the ability to inhibit partially purified enzymes: pc-, tg-, ma-, and rlDHFR [124, 125]. Eleven new derivatives, in which the 2,4-diaminopyrimidine group of TMP was retained, were investigated. The pattern of substitution in the benzyl moiety was modified on the pattern of TMP (i.e., 3′,4′,5′trimethoxy) or PTX (i.e., 2′,5′-dimethoxy). Compound 41g, 2,4-diamino-5-[(2-methoxy-4-carboxy-butyloxy)benzyl]pyrimidine, inhibited the pcDHFR enzyme (IC50 of 0.049 µM) 140 times stronger than TMP (IC50 of 12 µM) and approximation to PTX (IC50 of 0.031 µM). In addition, its SI (defined as the ratio IC50(rat liver)/IC50(P. carinii)), was >104-fold higher than PTX and eightfold higher than TMP [125]. Moreover, analog 41c was the most potent of the alkyl derivatives against pc-, tg-, and rlDHFR, where SIpc = 1.9; SItg = 2.1; SIma = 12. In contrast values for TMP SIpc = 11; SItg = 44; SIma = 680. It was observed an increase in potency of molecules towards pc-, tg-, and maDHFR dependent from the length of the 5′-O-alkyl group, where the number n = 4–6. The activity of the 5′-O-(ω-carboxyalkyl) derivatives decreased, where the length of the group was greater than n = 4 [124, 125].
In earlier report Kuyper et al. [126] presented one of the first examples of rational structure-based design of DHFR inhibitors. The synthesis of new DHFR inhibitors combining the high selectivity of TMP and the high potency of piritrexim. Series of novel 2,4-diamino-6-(2′,5′-disubstitutedbenzyl)-pyrido[2,3-d]-pyrimidines with O-(ω-carboxyalkyl) or ω-carboxy-1-alkynyl groups on the benzyl moiety and three N-(2,4-diaminopteridin-6-yl)methyl)-2′-(ω-carboxy-1-alkynyl)-dibenz-[b,f]-azepines were also synthesized [126]. The highest affinity for ecDHFR was observed with the O-(5- carboxypentyl) analog 42 (Fig. 5). Inhibition assays against ecDHFR revealed a progressive decrease in the Ki values from 2.6 to 0.035 nM as the number of CH2 groups was increased from 1 to 3, followed by stabilization of the Ki in the 0.025–0.066 nM range as this number was increased from 3 to 6 [126, 127].
Forsch et al. [128] synthesized and tested the 2,4-diamino-5-[3,4-dimethoxy-5-(5-carboxy-1-pentynyl)benzylpyrimidine 43 and 2,4-diamino-5-[3,4-dimethoxy-5-(4-carboxy-phenylethynyl)benzylpyrimidine 44 (Fig. 5), in which the 3′,4′,5′-trisubstituted pattern of TMP was retained. These analogues contained a COOH group linked to the benzyl ring via a spacer. The SI for each compound was calculated by dividing its 50% inhibitory concentration (IC50) against rat DHFR by its IC50 against pc, tg, or maDHFR Analogues 45 (SIma = 69) [5-(5-carboxy-1-pentynyl) moiety] and 46 (SIma = 150) [5-(4-carboxy-phenylethynyl) moiety] (Fig. 5) were the most interesting. It was reported, that both such substitution and 3,4,5-trisubstituted benzyl ring were more conducive to binding and selectivity of maDHFR. Compounds 43 (SIpc = 6.3; SIma = 24) and 44 (SIpc = 73; SIma = 210) presented the highest species selectivity for pcDHFR and maDHFR, respectively [128].
Roskowsky et al. [129] based on the previous report [126], obtained seven new 2,4-diamino-5-[2′-methoxy-5′-(substituted benzyl)]pyrimidines. Group of compounds with a carboxyphenyl or carboxyphenoxy group in the side chain were tested for in vitro ability to inhibit pc, tg, and maDHFR and their potencies (IC50 values) and selectivities (SI values as IC50(rat liver DHFR)/IC50(P. avium, T. gondii, or M. avium DHFR)). Biological evaluations disclosed, that the 5′-(3-carboxyphenyl)ethynyl analog 47 (IC50pc = 23; IC50tg = 5.5; SIpc = 28; SItg = 120 µM) (Fig. 5) was 520 times stronger against pcDHFR and 510 times less active against tgDHFR compared with TMP (IC50pc = 1200; IC50tg = 2800; SIpc = 14; SItg = 65 µM) [126, 129].
Compound 48, 2,4-diamino-[2′-methoxy-5′-(4-carboxy-phenyl)ethynylbenzyl]pyrimidine, (IC50ma = 3.7 µM; SIma = 2200) (Fig. 5) was the most selective inhibitor of maDHFR and displayed 2200-fold more selectivity than TMP (IC50ma = 180000 µM; SIma = 610). Analog 47 (IC50ma = 1.5 µM; SIma = 430) was slightly more potent than 48 against the maDHFR but was less selective. It was noticed, that the compounds with bulkier 5′-substituents containing an extra phenyl ring presented less efficient cell penetration, which provided lower activity toward maDHFR [126, 129].
Compounds bearing adamantane subunit
The analogues with the adamantane ring constituted another group of TMP derivatives. Such type of compounds might characterize higher blood–brain barrier penetration, accumulation in lipids, and also better taken up by cells. On the other hand the most important clinically use of these structures e.g. 1-aminoadamantane is antiviral treatment [130]. Orzeszko et al. [131, 132] reported in their previous investigation, that combination pyrimidine ring with the adamant moiety provides compounds of potent antimicrobial properties.
It was found, that (adamant-1-yl)-pyrimidines 49–51 (Fig. 6) presented significant antimicrobial activity (MIC were about 50 µg ml−1) against Bacillus stearothermophilus and S. aureus [131, 132]. The synthesis of these new compounds was already described [133–135]. The further investigation confirmed, that the novel 5-adamantan-1-ylmethyl-pyrimidines poses antibacterial and antifungal properties, however only 5-adamantan-1-ylmethyl-pirymidine-2,4-diamine 49 inhibited the growth B. stearothermophilus, Bacillus subtilis, and Enterococcus faecalis. Moreover, the combination of this derivative with SMX led to an increase in effective inhibition e.g., E. coli ATTCC 295922 [136].
Fig. 6.
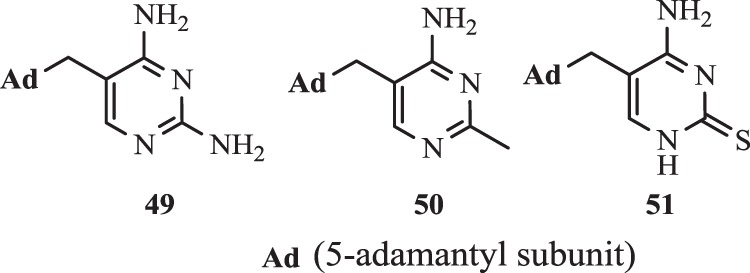
Structures of compounds 49–51
New generation of propargyl-linked inhibitors (PLA)—overview of achievements
Pelphrey et al. [137] designed and synthesized analogues 52a–52i (Fig. 7) with a propargyl-based linker between a substituted 2,4-diaminopyrimidine and a trimethoxyphenyl ring. It was found, that the propargyl linker increases the distance between the pyrimidine and acrylic rings, which allows for optimal match of the arylic ring in the hydrophobic pocket of the enzyme [137].
Fig. 7.

Structures of propargyl-linked inhibitors—A
Liu et al. [138] inspired by previous methodology (Pelphrey et al. [137]), identified two compounds with methyl groups at the meta (53) and para (54) (Fig. 7) positions on the second ring. Significantly, these compounds were extremely in vitro potent and also exhibited very strong selectivity for the DHFR from Candida glabrata (cgDHFR) IC50DHFRcg = 0.55 and 0.6 nM and enzyme selectivity DHFRh/cg = 1364 and 2350 respectively for analog 53 and 54 [138].
In next report, Liu et al. [139] described the incorporation of biphenyl moieties in the hydrophobic domain, what led to obtaining another series of compounds, which showed over 2300-fold more selectivity and very high potency against cgDHFR. In addition, they had significant MIC values (1.5 μg ml−1) compared with standard antifungal agents [139].
Kuper et al. [140] validated hypothesis, that 5′-(ω-carboxyalkyloxy) moieties would improve selectivity to pathogenic DHFR by enhanced ionic interactions with the positively charged residues of enzyme’s active site. Based on these conclusions, Cody et al. [141] designed and synthesized 2,4-diamino-5-[3,4-di-methoxy-5′(5-carboxy-1-pentynyl)]-benzylpyrimidine (PY1011) and 2,4-diamino-5[2-methoxy-5-(4-carboxy-butyloxy)-benzyl]-pyrimidine (PY957) (Fig. 7). DHFR enzyme inhibition (IC50) and SI (rl/pc) relative to rat liver DHFR (rl) data revealed that PY1011(IC50pc = 1.0 nM; rl/pc = 5000) was 5000-fold more selective towards pcDHFR over rat liver DHFR [141]. In addition, compound PY957(IC50pc = 0.049 nM; rl/pc = 80) has 80-fold selectivity for pcDHFR in comparison to TMP(IC50pc = 13000 nM; rl/pc = 14). Probably 5′-(ω-carboxyalkyloxy) side chain of these compounds forms ionic interactions with Arg in the substrate binding pocket of DHFR [141].
Birdsall et al. [142] and Morgan et al. [143] obtained a series of related to TMP analogues with a 4′-Br substituent (brodimoprim), that had 5′-O substitutes. These studies revealed, that the 4,6-dicarboxylate analog showed a 1000-fold tighter binding to Lactobacillus casei DHFR (lcDHFR) than brodimoprim [142, 143].
Lombardo et al. [144] evaluated the antibacterial activity of the series of 16 propargyl-linked antifolates (PLAs), including TMP. It was reported, that ten compounds showed the antibacterial activity against K. pneumoniae. The most potent activity presented derivatives 55–60 (Fig. 7) and 61–64 (Fig. 8) with MIC values at ≤1 μg ml−1. Importantly, compounds 59 (IC50ec = 0.033 ± 0.002 µM) and 60 (IC50ec = 0.183 ± 0.045 µM) showed activity against E. coli [144]. Specifically of interest to this study here, a prevalent gene (dfrA1) coding for an insensitive DHFR confers 190- or 1000-fold resistance to TMP for K. pneumoniae and E. coli, respectively [144]. The SARs and crystal structures will be critical in driving the design of broadly active inhibitors against wild-type and resistant DHFR. These studies confirmed, that two potent PLAs: 65 and 66 were active against wild-type resistant DHFR from Enterobacteriaceae and dfrA1 protein. A number of positive interactions that are unique to the PLAs explain much of the potency observed against the dfrA1 enzyme relative to TMP [144]. Although the substitution of Gln28 is detrimental to TMP binding, the carboxamide forms a hydrogen bond with the pyridyl nitrogen of both compounds 65 and 66 [144].
Fig. 8.

Structures of propargyl-linked inhibitors—B
Beierlein et al. [145] synthesized a group of 2′,5′-dimethoxyphenylpyrimidine analogues of TMP 65–68 (Fig. 8). It was shown, that these compounds proved excellent potency and selectivity against Bacillus anthracis (baDHFR). The best compound 67 in this series makes several key interactions with the active site of baDHFR e.g., additional hydrogen bond with the backbone carbonyl of Val7, the carbonyl oxygen of Met6 forms a hydrogen bond with the 4-amino group [145]. In addition, there are several van der Waals interactions involving the pyrimidine ring and Ala8, Val32, Met6, and Val7. The ethyl group at the C6 position makes favorable lipophilic contacts with Leu21. The analog 67, presenting an 88-fold increase in potency against baDHFR(IC50ba = 0.89 µM) relative to TMP(IC50ba = 71 nM) [145]. In addition selectivity ratio (h/ba) were 1.4 and 1.7, respectively. Improvements in potency and selectivity, while maintaining good drug like properties, should lead to a corresponding increase in antibacterial activity [145].
Hajian et al. [146] reported the activity of series of PLA against the M. tuberculosis. Several of the evaluated PLA potently inhibited the growth of M. tuberculosis with MIC values less than 1 μg ml−1. In addition, this study revealed, that the PLA-COOH compounds maintain their high level of activity against MDR and extensively drug resistant strains [146].
S. aureus belongs to a group of pathogen considered as a major cause of hospital-acquired infections, most frequently associated with the bloodstream, skin and soft tissue, ventilator-assisted pneumonia and catheters. Nowadays, the increasing frequency of infections caused by MRSA is a major therapeutic problem [147]. The occurrence of a common F98Y mutation shared by resistant S. aureus mutants and plasmid-encoded DHFRs provided rationale that new antifolates that are effective against this mutant could expand coverage for these resistant enzymes [148]. Therefore optimizing the PLAs to overcome the central F98Y resistance mechanism would lead to robust inhibitors capable of targeting multiple enzymes that possess this substitution [149]. Several research groups have focused on the development of next generation PLAs that maintain activity against many of the important pathogenic bacterial DHFR enzymes while expanding coverage to include both mutant and naturally TMP-insensitive DHFR enzymes that give rise to TMP resistance within S. aureus and other Gram-positive strains. Bellow, we presented several significant studies in this field.
Frey et al. [150] have developed a novel series of DHFR inhibitors based on TMP scaffold, which included an extended linker between substituted phenyl rings and 2,4-diaminopyrimidine. The main goal of incorporation of the propargyl linker was increasing hydrophobic interactions with DHFR. Compounds 69–73 and 74–76 (Fig. 8) were potent both against the wild-type and mutant S. aureus enzymes and inhibit the wild-type enzyme with IC50 values in the nanomolar range and lose only two- to fourfold potency when evaluated with the TMP-resistant F98Y enzyme [150]. Moreover, it was observed, that an alternate conformation of the cofactor NADPH determined the reduction of the hydrophobic contacts between the residues in the active site of DHFR and the inhibitors. This was probably the cause why background resistance mutation F98Y was observed for these ligands [150]. Other studies for the novel class of PLA as DHFR inhibitors were conducted for both Gram-positive and Gram-negative bacteria. These studies have shown that PLA acted similarly to TMP passively diffused through membranes, strongly inhibited DHFR enzyme and often inhibited the growth of pathogenic cells with submicromole MIC values. The compounds similar to UCP1021 (Fig. 9) had high MIC values against K. pneumoniae and also Gram-positive pathogens like MRSA and S. pyogenes [150].
Fig. 9.

Structures of propargyl-linked inhibitors—C
Viswanathan et al. [151] obtained three new biphenyl derivatives 77–79 and series of pyridyl inhibitors 80–84, as well as compounds containing alternative heterocycles functionalities 85–87 (Fig. 8). The second series of novel PLA containing a pyridyl substituent delivered a group of compounds with activity against both MRSA and S. pyogenes, at MIC values below 0.1 µg ml−1, with minimal cytotoxicity against mammalian cells [151]. Significantly, the incorporation of the additional basic heterocyclic ring not only improved the solubility of the compounds but also dramatically lowered cytotoxicity to mammalian [151, 152].
Scocchera et al. [153] developed a new eight relative to earlier PLA hybrid antifolates with high enzyme affinity and increased antibacterial activity against MRSA and E. coli. In addition, these studies revealed, that a carboxylate moiety can be incorporated into the PLA without compromising the ability to gain access to the target enzyme—DHFR. Scocchera’s team, based on high-resolution crystal structures of the compounds 88 and 89 (Fig. 8) conjugates with S. aureus DHFR, reported, that their affinity in the active site was enhanced by water-mediated contacts between the carboxylate and Arg57 [153].
In another report, Reeve et al. [154] reported the first identification of two plasmid-borne genes found in clinical S. aureus isolates from United States hospitals, as well as identify the molecular mechanisms of resistance to several common antibiotics. The clinical impact of these genes, dfrG and dfrK, is significant as the resulting proteins confer high levels of TMP resistance and render this commonly used antibiotic useless. A series of charged propargyl-linked antifolates are presented that directly target the acquired resistance elements and potently inhibit the resistant enzymes and bacteria. Several of PLA (UCP1039, UCP1164, UCP1172, UCP1173, and UCP1175) presented highly potent activity against TMP-resistant strains [154]. Furthermore, it was noticed, that compounds, which contained a pyridyl C-ring UCP1039 and series possessing an ionized carboxylic acid on the distal C-ring (UCP1164, UCP1172, UCP1173, and UCP1175) showed the most potent activity. Overall, the PLAs were most potent against strains carrying dfrG and dfrK with MIC values as low as 0.1563 mg ml−1, twofold lower than the MIC for TMP against wild-type S. aureus [154]. Compound UCP1173 showed the most potent activity against strains possessing dfrA with MIC values of 1.25 and 2.5 mg ml−1. Interestingly, compound UCP1172 is the antipodal point of UCP1173 but does not significantly inhibit dfrA-possessing strains [154].
In successive report, Reeve et al. [155] have designed, synthesized, and evaluated a series of another 12 PLAs. These compounds were generally much more potent than TMP against TMP resistant DHFR from S. aureus. Analysis of several resistant clinical isolates shows that the mutation F98Y is highly prevalent, especially in combination with secondary mutations, H149R or H30N [156]. Importantly, the presence of a carboxylate moiety (91–94) (Fig. 9) exhibited good potency (Ki values less than 16 nM)against the difficult double mutant enzymes e single mutant enzymes F98Y/H30N. The analogue 90 bound to NADPH and S. aureus mutations, as well as saDHFR (F98Y), saDHFR (H30N), and saDHFR (F98Y/H30N) show that the mutations specifically diminish the binding of the 2- and 4-amino groups on the pyrimidine ring [155]. The carboxylate moiety forms water-mediated hydrogen bonds with an active site arginine, compensating for some of the lost pyrimidine interactions and restoring affinity to the mutant enzymes [155]. It was the noticed that the –COOH moiety formed water-mediated hydrogen bonds with an active site Arg, improved affinity to mutant enzymes and also compensated some of the lost pyrimidine interactions. The PLA–COOH compounds led to maintain potency for wild-type enzymes overcome the resistance caused by mutations in enzymes and pathogens [155].
In next study, Keshipeddy et al. [157] have demonstrated a series of enantiomerically pure PLA that displays low nanomolar inhibition of the wild-type DHFR and potent activity against a range of MRSA strains (MIC values 0.04–0.72 μg ml−1). It was confirmed, that the stereogenicity of the propargyl center is critical for activity. Among ten of enantiomerically pure PLA, Keshipeddy et al. [157] have identified, ten enantiomerically pure PLA, enantiomer S-27 (Fig. 9) was strong both at enzymatic and cellular level against the wild-type and mutant F98Y DHFR. Unexpectedly, crystal structures of a pair of individual enantiomers in the wild-type DHFR revealed that the single change in configuration of the stereocenter drove the selection of an alternative NADPH cofactor, with the minor α-anomer appearing with R-27 [157].
Antibiotic resistance is a naturally evolving phenomenon, it is critical to map compound design to contemporary resistance profiles found in clinical strains of bacteria to properly target the prevailing molecular mechanisms during lead optimization. Therefore, medicinal chemistry efforts are often successful at overcoming these resistance mechanisms through rational modification of the antibiotic structure, leading to successive generations of agents with improved activity. Often, these efforts require innovations in synthetic chemistry to efficiently access significant new derivatives.
Other targets
DHFR is a ubiquitous enzyme and exists in a wide range of organisms, but is a drug target, that has not been thoroughly investigated in Leishmania and Trypanosomes [158, 159]. One of the urgent directions for new drugs design is the treatment of parasitic diseases caused by protozoa, such as leishmaniasis, African trypanosomiasis and Chagas’ disease. Molecular modeling suggests, that there are significant differences in the structures of the actives sites of the leishmanial and trypanosomal enzymes compared with the human enzyme, which should make it possible to selectively DHFR inhibitors [160]. In preliminary studies, Sirawaraporn et al. [161] prepared an extensive series of substituted 5-benzyl-2,4-diaminopyrimidines (with general structure 95) (Fig. 10). They evaluated these against the enzymes from Leishmania major (a causative organism of leishmaniasis), Trypanosoma cruzi (the causative organism of Chagas’disease) and Trypanosoma brucei (the causative organism of African trypanosomiasis) compared the hDHFR. Modeling studies suggested, that there may be a hydrophobic pocket adjacent to the 6-position of the pyrimidine ring. These studies for the compounds 99–100 with substituents at this position (Fig. 10) indicated, that an ethyl substituent may increase the inhibitory activity and selectivity [161]. Based on these conclusions, Chowdhury et al. [162] described the design of a set of 6-substituted pyrimidines, band the preparation and evaluation of new compounds 101–105 (Fig. 10). The compounds were examined against the recombinant DHFR from L. major, T. cruzi, T. brucei, and hDHFR [162].
Fig. 10.

Structures of TMP analogues directed at new targets
Biological assays revealed, that, both the heptyloxy and octyloxy compounds showed a small increase in activity and selectivity towards L. major DHFR on the addition of a 6-substitutent, however, the effect seemed largest with the ethyl substituent (100 and 102) [162]. In the case of the T. cruzi enzyme, there was a marginal effect on activity on addition of the ethyl group (100 and 102) but a large loss in activity and selectivity with larger substituents (benzyl 101 and propyl 103). Compound 105 showed surprising activity, but showed no selectivity [162].
DHFR enzyme as anticancer target
Folate metabolism has long been recognized as an important and attractive target for chemotherapy because of its crucial role in the biosynthesis of nucleic acid precursors [163]. DHFR inhibition has long been recognized as a target for the development of therapeutic agents against certain cancers as well as bacterial, parasitic infections, and chronic inflammatory diseases [164]. DHFR is an essential enzyme which catalyzes the reduction of dihydrofolate acid (DHF) to tetrahydrofolic acid (THF) using reduced NADPH as a cofactor and plays a major role in the biosynthesis nucleic acids. Therefore, DHFR couples with thymidylate synthase (TS), which catalyzes the reductive methylation of deoxyuridine monophosphate in deoxythymidine monophosphate using N5 -N10-methylenetetrahydrofolate (5,10-Methylene THF) as a cofactor [165–168]. Despite the extensive targeting of DHFR in pathogenic organisms [153, 169–171], hDHFR is also targeted by anticancer agents [172–175]. MTX targets hDHFR and is a well-known anticancer drug for the treatment of leukemia, breast cancer, lung cancer, osteosarcoma, and lymphoma [176]. MTX is a strong competitor of DHF with regard to binding hDHFR, as it causes elevated accumulation of DHF when it binds hDHFR, which in turn induces the feedback inhibition of TS. Over the last 30 years, many research projects have focused on the search for new compounds active against this enzyme, most often derivatives of MTX, which is confirmed by extensive literature in this field [176, 177]. However, there are only a few reports that would confirm the biological activity of TMP analogues targeting to anticancer properties. Singh et al. [178] modified antibacterial agent TMP to compounds 106 and 107 (Fig. 10) with promising anticancer applications. These two compounds had significant tumor growth inhibitory activities over 60 human tumor cell lines and exhibited appreciable interactions with DHFR [178]. Algul et al. [179] have developed a new nonclassical series of propargyl-linked DHFR inhibitors. It was observed, that interactions propargyl-linked inhibitors with Leu 22, Thr 56, Ser 59, Ile 60 could potently inhibit hDHFR, opposite to weak inhibition of hDHFR by TMP. Therefore the novel potent propargyl-linked antifolates could provide structures, that may be excellent anticancer drugs?. Based on SARs, Algul et al. [179] reported, that hydrophobic substitutions at C6 and the propargylic position impart increased anticancer potency. Significantly, propargyl-linked compound 108 (Fig. 10) exhibited 3500-fold greater potency than TMP. In addition, this propargyl linker was valuable in extending the trimethoxyphenyl ring closer to critical hydrophobic residues, including Leu 22, Thr 56, Ser 59, and Ile 60 [179]. The new direction of research is using amide bond as a linker in the TMP derivative molecule. So far, there have been no reports that would present the synthesis and study of biological activity of TMP analogues with the proposed modification of the methylene bridge replacement by an amide bond. Designed and obtained by us TMP derivatives, with general structures 107a–c and 108a–c (Fig. 10), had an amide bond, fragment of the structure, which makes the molecule similar to netropsin, representative of the group of minor groove binding agents [180, 181]. These compounds are well-known for their strong affinity for DNA. They bind within the minor groove of B-DNA at sites consisting of four or five consecutive AT base pairs [182]. So new compounds could bind to DNA structures and also have an increased affinity to the human enzyme DHFR, so thus show anticancer effects [180, 181]. Recent trends in medical chemistry suggest the developing of multitargeting and multifunctional compounds, in addition it is a worldwide medical research strategy [183]. The term “designed multiple ligands” was coined by Morphy and Rankovic to describe the abovementioned compounds [184]. The mode of designed multiple ligands could offer several potential advantages as increase of therapeutic efficacy or decrease development of cancer drug resistance [185, 186]. In another attempt, the quinazoline analogues were synthesized to have closed resemblance with MTX and inhibited the mammalian DHFR in cancerous cells [187]. The application of these quinazoline analogues was also recommended for combined inhibition of epidermal growth factor receptor and hDHFR. Besides its use in cancer therapy, hDHFR is also known as an important target for the treatment of rheumatoid arthritis [1, 188]. Potential inhibitors of hDHFR (109–111) (Fig. 10), were identified recently by Rana et al. [189] using 3D-QSAR pharmacophore modeling. 3D-QSAR pharmacophore modeling has become an increasingly acceptable approach for probing novel lead candidates in computational drug designing as fundamental platforms to develop effective chemotherapeutics in cancer and rheumatoid medications [189].
Conclusions and perspectives
DHFR identified in 1958 as a target for TMP synthetic antimicrobial agents, has been considered to be a universal enzyme present in various organisms. This enzyme is actively studied as a target for many new compounds. Following the successful marketing of TMP and cotrimoxazole, intensive research on improved TMP and the search for new DHFR inhibitors was carried out. Search for an improved TMP continued within the Roche group in the 70s and 80s; the nature of desired improvement was, however, not well defined. The focus was on broad-spectrum agents. Activity better than TMP against Gram-negative bacteria was, however, never reached. However, successful discovery of the Hitchings’s group about the structural variation of diaminopyrimidines and related structures led to obtaining highly selective and potent agents and many useful drugs, as TMP. Nowadays, TMP seems to be the optimal benchmark for the development of antimicrobials with a broad spectrum of action against Gram-negative bacteria e.g., E. coli, Staphylococci, parasites as P. falciparum, several protozoa as Toxoplasma and fungi e.g., P. carinii. The evaluation of antifolates as inhibitors of the DHRF enzyme has been and still is the subject of work of many scientific teams. Based on the above overview it could be seen, that one of the significant strategies to obtain new potential compounds is combine potency of known drugs e.g., TMP, MTX, or PTX. Importantly, molecular docking, dynamic molecular simulations, and pharmacokinetic studies help to identify promising directions for new projects. Recently, Shah et al. [190] synthesized and evaluated several of new 6-substituted pyrrolo[2,3-d]pyrimidines as DHFR inhibitors using hDHFR X-ray crystal structures and P. jirovecii homology models were used to design and predict potent and selective pjDHFR inhibitors. In turn, Ouyang et al. [191] synthesized potential antitubercular derivatives of TMP, on the pattern of a docked compound and M. tuberculosis DHFR complex. These evaluations provided to obtain three series of compounds containing a hydrophobic side chain with affinity to the enzyme pocket. In another report, Thakkar et al. [192] described design and synthesis of new benzothiazole analogues as novel antimalarial and antimicrobial analogues, however important stage of rational drugs design was in silico docking study and pharmacokinetic evaluations. These approach provided to further development of lead compounds.
On the other side, MDR bacteria, caused by gene mutation, that encode DHFR enzyme, anticancer, and rheumatoid arthritis diseases, still encourage many kinds of research to develop new molecules. TMP, and other nonclassical antifolates are excellent templates to design new active, both antibacterial and anticancer drugs. The above literature review presents an extensive range of research literature on the first and most recent achievements on DHFR inhibitors and underlines new directions in developing and modeling DHFR inhibitors, that can be useful to design new structures.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Askari BS, Krajinovic M. Dihydrofolate reductase gene variations in susceptibility to disease and treatment outcomes. Curr Genom. 2010;11:578–83. doi: 10.2174/138920210793360925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schnell JR, Dyson HJ, Wright PE. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu Rev Biophys Biomol Struct. 2004;33:119–40. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- 3.Schweitzer B, Dicker AP, Bertino JR. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990;4:2441–52. doi: 10.1096/fasebj.4.8.2185970. [DOI] [PubMed] [Google Scholar]
- 4.Blakley RL. Eukaryotic dihydrofolate reductase. Adv Enzymol Relat Areas Mol Biol. 1995;70:23–102. doi: 10.1002/9780470123164.ch2. [DOI] [PubMed] [Google Scholar]
- 5.Heaslet H, Harris M, Fahnoe K, et al. Structural comparison of chromosomal and exogenous dihydrofolate reductase from Staphylococcus aureus in complex with the potent inhibitor trimethoprim. Proteins. 2009;76:706–17. doi: 10.1002/prot.22383. [DOI] [PubMed] [Google Scholar]
- 6.Hong W, Wang Y, Chang Z. The identification of novel Mycobacterium tuberculosis DHFR inhibitors and the investigation of their binding preferences by using molecular modelling. Sci Rep. 2015;16:15328. doi: 10.1038/srep15328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rashid N, Thapliyal C, Chaudhuri P. Dihydrofolate reductase as a versatile drug target in healthcare. JPP. 2016;7:247–57. [Google Scholar]
- 8.Bhosle A, Chandra N. Structural analysis of dihydrofolate reductases enables rationalization of antifolate binding affinities and suggests repurposing possibilities. FEBS J. 2016;283:1139–67. doi: 10.1111/febs.13662. [DOI] [PubMed] [Google Scholar]
- 9.Selassie CD, Li RL, Poe MR, Hansch C. Optimization of hydrophobic and hydrophilic substituent interactions of 2,4-diamino-5-(substituted-benzyl)pyrimidines with dihydrofolate reductase. J Med Chem. 1991;34:46–54. doi: 10.1021/jm00105a008. [DOI] [PubMed] [Google Scholar]
- 10.Nammalwar B, Bourne CR, Wakeham N. Modified 2,4‐diaminopyrimidine‐based dihydrofolate reductase inhibitors as potential drug scaffolds against Bacillus anthracis. Bioorg Med Chem. 2015;23:203–11. doi: 10.1016/j.bmc.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson RG, Rosowsky A. Dicyclic and tricyclic diaminopyrimidine derivatives as potent inhibitors ofCryptosporidiumparvumdihydrofolate reductase: structure‐activity and structure‐selectivity correlations. Antimicrob Agents Chemother. 2001;45:3293–3303. doi: 10.1128/AAC.45.12.3293-3303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srinivasan B, Skolnick J. Insights into the slow‐onset tight‐binding inhibition of Escherichia colidihydrofolatereductase: detailed mechanistic characterization of pyrrolo [3,2‐f] quinazoline‐1,3‐diamine and its derivatives as novel tight‐binding inhibitors. FEBS J. 2015;282:1922–1938. doi: 10.1111/febs.13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson HC, Biggadike K, McKilligin E, et al. 6,7‐disubstituted 2,4‐diaminopteridines: novel inhibitors of Pneumocystis carinii and Toxoplasma gondiidihydrofolate reductase. Antimicrob Agents Chemother. 1996;40:1371–5. doi: 10.1128/aac.40.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinivasan B, Tonddast‐Navaei S, Skolnick J. Ligand binding studies, preliminary structure‐activity relationship and detailed mechanistic characterization of 1‐phenyl‐6,6‐dimethyl‐1,3,5‐triazine‐2,4‐diamine derivatives as inhibitors of Escherichia coli dihydrofolate reductase. Eur J Med Chem. 2015;103:600–614. doi: 10.1016/j.ejmech.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srinivasan B, Tonddast-Navaei S, Roy A, Zhou H, Skolnick J. Chemical space of Escherichia coli dihydrofolate reductase inhibitors: New approaches for discovering novel drugs for old bugs. Med Res Rev. 2019;39:684–705. doi: 10.1002/med.21538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roth B, Falco EA, Hitchings GH, Bushby SRM. 5-benzyl-2,4-diaminopyrimidines as antibacterial agents.I. Synthesis and Antibacterial Activity in vitro. J Med Pharm Chem. 1962;5:1103–23. doi: 10.1021/jm01241a004. [DOI] [PubMed] [Google Scholar]
- 17.Skold O. Resistance to trimethoprim and sulfonamides. Vet Res. 2011;32:261–73. doi: 10.1051/vetres:2001123. [DOI] [PubMed] [Google Scholar]
- 18.Burchall JJ. The development of the diaminopyrimidines. J Antimicrob Chemother. 1979;5:3–14. doi: 10.1093/jac/5.supplement_b.3. [DOI] [PubMed] [Google Scholar]
- 19.Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs—antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzym Inhib Med Chem. 2014;29:379–87. doi: 10.3109/14756366.2013.787422. [DOI] [PubMed] [Google Scholar]
- 20.Matthews DA, Bolins JT, Burridge JM, Filmann DJ, Volzll KW, Kraut J. Dihydrofolate reductase. the stereochemistry of inhibitor selectivity. J Biol Chem. 1985;260:392–9. [PubMed] [Google Scholar]
- 21.Darrell JH, Garrod LP, Waterworth PM. Trimethoprim: laboratory and clinical studies. J Clin Pathol. 1968;21:202–9. doi: 10.1136/jcp.21.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasanen A, Sundquist H. Trimethoprim alone in the treatment of urinary tract infections: eight years of experience in Finland. Rev Infect Dis. 1982;4:358–65. doi: 10.1093/clinids/4.2.358. [DOI] [PubMed] [Google Scholar]
- 23.Lacey RW. Do sulphonamide-trimethoprim combinations select less resistance to trimethoprim than the use of trimethoprim alone? J Med Microbiol. 1982;15:403–27. doi: 10.1099/00222615-15-4-403. [DOI] [PubMed] [Google Scholar]
- 24.Huovinen P, Sundström L, Swedberg G, Sköld O. Trimethoprim and sulfonamide resistance. Antimicrob Agents Chemother. 1995;39:279–89. doi: 10.1128/aac.39.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noall EW, Sewards HF, Waterworth PM. Successful treatment of a case of Proteus septicaemia. Br Med J. 1962;2:1101–2. doi: 10.1136/bmj.2.5312.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bushby SR. Trimethoprim-sulfamethoxazole: in vitro microbiological aspects. J Infect Dis. 1973;128:442–62. doi: 10.1093/infdis/128.supplement_3.s442. [DOI] [PubMed] [Google Scholar]
- 27.Taur Y, Smith MA. Adherence to the infectious diseases society of America guidelines in the treatment of uncomplicated urinary tract infection. Clin Infect Dis. 2007;44:769–74. doi: 10.1086/511866. [DOI] [PubMed] [Google Scholar]
- 28.Navarro-Martínez MD, Cabezas-Herrera J, Rodríguez-López JN. Antifolates as antimycotics? a connection between the folic acid cycle and the ergosterol biosynthesis pathway in Candida albicans. Int J Antimicrob Agents. 2006;28:560–7. doi: 10.1016/j.ijantimicag.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 29.Kahlmeter G. An international survey of the antimicrobial susceptibility of pathogens from uncomplicated urinary tract infections: the ECO.SENS Project. J Antimicrob Chemother. 2003;51:69–76. doi: 10.1093/jac/dkg028. [DOI] [PubMed] [Google Scholar]
- 30.Kahlmeter G, Poulsen HO. Antimicrobial susceptibility of Escherichia coli from community-acquired urinary tract infections in Europe: the ECO.SENS study revisited. Int J Antimicrob Agents. 2012;39:45–51. doi: 10.1016/j.ijantimicag.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 31.Naber KG, Schito G, Botto H, Palou J, Mazzei T. Surveillance study in Europe and Brazil on clinical aspects and Antimicrobial Resistance Epidemiology in Females with Cystitis (ARESC): implications for empiric therapy. Eur Urol. 2008;54:1164–75. doi: 10.1016/j.eururo.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Crider SR, Colby SD. Susceptibility of enterococci to trimethoprim and trimethoprim-sulfamethoxazole. Antimicrob Agents Chemother. 1985;27:71–5. doi: 10.1128/aac.27.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamilton-Miller JM. Reversal of activity of trimethoprim against gram-positive cocci by thymidine, thymine and ‘folates. J Antimicrob Chemother. 1998;22:35–9. doi: 10.1093/jac/22.1.35. [DOI] [PubMed] [Google Scholar]
- 34.Maskell JP, Sefton AM, Hall LMC. Multiple mutations modulate the function of dihydrofolate reductase in trimethoprim-resistant Streptococcus pneumoniae. Antimicrob Agents Chemother. 2001;45:1104–8. doi: 10.1128/AAC.45.4.1104-1108.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salter AJ. Trimethoprim-sulfamethoxazole: an assessment of more than 12 years of use. Rev Infect Dis. 1982;4:196–236. doi: 10.1093/clinids/4.2.196. [DOI] [PubMed] [Google Scholar]
- 36.Allegra CJ, Kovacs JA, Drake JC, Swan JC, Chabner BA, Masur H. Activity of antifolates against Pneumocystis carinii dihydrofolate reductase and identification of a potent new agent. J Exp Med. 1987;165:926–31. doi: 10.1084/jem.165.3.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hooton TM, Besser R, Foxman B, Fritsche TR, Nicolle LE. Acute uncomplicated cystitis in an era of increasing antibiotic resistance: a proposed approach to empirical therapy. Clin Infect Dis. 2004;39:75–80. doi: 10.1086/422145. [DOI] [PubMed] [Google Scholar]
- 38.Wormser GP, Keusch GT. Drugs five years later: trimethoprim-sulfamethoxazole in the United States. Ann Intern Med. 1979;91:420–9. doi: 10.7326/0003-4819-91-3-420. [DOI] [PubMed] [Google Scholar]
- 39.Stuck AK, Täuber MG, Schabel M, Lehmann T, Suter H, Mühlemann K. Determinants of quinolone versus trimethoprim-sulfamethoxazole use for outpatient urinary tract infection. Antimicrob Agents Chemother. 2012;56:1359–63. doi: 10.1128/AAC.05321-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afeltra J, Meis JF, Mouton JW, Verweij PE. Prevention of invasive aspergillosis in AIDS by sulfamethoxazole. AIDS. 2001;15:1067–8. doi: 10.1097/00002030-200105250-00020. [DOI] [PubMed] [Google Scholar]
- 41.Afeltra J, Meis JF, Vitale RG, Mouton JW, Verweij PE. In vitro activities of pentamidine, pyrimethamine, trimethoprim, and sulfonamides against Aspergillus species. Antimicrob Agents Chemother. 2002;46:2029–31. doi: 10.1128/AAC.46.6.2029-2031.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanafy A, Uno J, Mitani H, Kang Y, Mikami Y. In-vitro antifungal activities of sulfa drugs against clinical isolates of Aspergillus and Cryptococcus species. Nihon Ishinkin Gakkai Zasshi. 2007;48:47–50. doi: 10.3314/jjmm.48.47. [DOI] [PubMed] [Google Scholar]
- 43.Ajayi BG, Osuntokun B, Olurin O, Kale OO, Junaid TA. Orbital histoplasmosis due to Histoplasma capsulatum var. duboisii: successful treatment with Septrin. J Trop Med Hyg. 1986;89:179–87. [PubMed] [Google Scholar]
- 44.Egere JU, Gugnani HC, Okoro AN, Suseelan AV. African histoplasmosis in Eastern Nigeria: report of two culturally proven cases treated with spectrin and amphotericin B. J Trop Med Hyg. 1978;81:225–9. [PubMed] [Google Scholar]
- 45.Brilhante RS, Fechine MA, Cordeiro RA, et al. In vitro effect of sulfamethoxazole-trimethoprim against Histoplasma capsulatum var. capsulatum. Antimicrob Agents Chemother. 2010;54:3978–9. doi: 10.1128/AAC.00793-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amyes SGB, Towner KJ. Trimethoprim resistance: epidemiology and molecular aspects. J Med Microbiol. 1990;31:1–19. doi: 10.1099/00222615-31-1-1. [DOI] [PubMed] [Google Scholar]
- 47.Smith HW. Mutants of Klebsiella pneumoniae resistant to several antibiotics. Nature. 1976;259:307–8. doi: 10.1038/259307a0. [DOI] [PubMed] [Google Scholar]
- 48.Traub WH, Kleber I. Selected and spontaneous variants of Serratia marcescens with combined resistance against chloramphenicol, nalidixic acid, and trimethoprim. Chemotherapy. 1977;23:436–51. doi: 10.1159/000222013. [DOI] [PubMed] [Google Scholar]
- 49.Flensburg J, Skold O. Massive overproduction of dihydrofolate reductase in bacteria as a response to the use of trimethoprim. Eur J Biochem. 1987;162:473–6. doi: 10.1111/j.1432-1033.1987.tb10664.x. [DOI] [PubMed] [Google Scholar]
- 50.Goldstein FW, Papadopoulou B, Acar JF. The changing pattern of trimethoprim resistance in Paris, with a review of the worldwide experience. Rev Infect Dis. 1986;8:725–37. doi: 10.1093/clinids/8.5.725. [DOI] [PubMed] [Google Scholar]
- 51.Hamilton-Miller J.M.T. Antimicrobial Drug Resistance. 1984. Resistance to Antibacterial Agents Acting on Antifolate Metabolism; pp. 173–190. [Google Scholar]
- 52.Smith DR, Calvo JM. Nucleotide sequence of dihydrofolate reductase genes from trimethoprim-resistant mutants of Escherichia coli. Mol Gen Genet. 1982;187:72–8. doi: 10.1007/BF00384386. [DOI] [PubMed] [Google Scholar]
- 53.Then RL. Mechanisms of resistance to trimethoprim, the sulfonamides, and trimethoprim-sulfamethoxazole. Rev Infect Dis. 1982;4:261–9. doi: 10.1093/clinids/4.2.261. [DOI] [PubMed] [Google Scholar]
- 54.Podnecky NL, Wuthiekanun V, Peacock SJ, Schweizer HP. The BpeEF-OprC efflux pump is responsible for widespread trimethoprim resistance in clinical and environmental Burkholderia pseudomallei isolates. Antimicrob Agents Chemother. 2013;57:4381–6. doi: 10.1128/AAC.00660-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.White PA, McIver CJ, Rawlinson WD. Integrons and gene cassettes in the enterobacteriaceae. Antimicrob Agents Chemother. 2001;45:2658–61. doi: 10.1128/AAC.45.9.2658-2661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grape M, Farra A, Kronvall G, Sundstrom L. Integrons and gene cassettes in clinical isolates of co-trimoxazole-resistant Gram-negative bacteria. Clin Microbiol Infect. 2005;11:185–92. doi: 10.1111/j.1469-0691.2004.01059.x. [DOI] [PubMed] [Google Scholar]
- 57.Blahna MT, Zalewski CA, Reuer J, Kahlmeter G, Foxman B, Marrs CF. The role of horizontal gene transfer in the spread of trimethoprim-sulfamethoxazole resistance among uropathogenic Escherichia coli in Europe and Canada. J Antimicrob Chemother. 2006;57:666–72. doi: 10.1093/jac/dkl020. [DOI] [PubMed] [Google Scholar]
- 58.Dionisio F, Matic I, Radman M, Rodrigues OR, Taddei F. Plasmids spread very fast in heterogeneous bacterial communities. Genetics. 2002;162:1525–32. doi: 10.1093/genetics/162.4.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huovinen P. Trimethoprim resistance. Antimicrob Agents Chemother. 1987;31:1451–6. doi: 10.1128/aac.31.10.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dale GE, Broger C, D’Arcy A, et al. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J Mol Biol. 1997;266:23–30. doi: 10.1006/jmbi.1996.0770. [DOI] [PubMed] [Google Scholar]
- 61.Pikis A, Donkersloot JA, Rodriquez WJ, Keith JM. A conservative amino acid mutation in the chromosome-encoded dihydrofolate reductase confers trimethoprim resistance in Streptococcus pneumoniae. J Infect Dis. 1998;178:700–6. doi: 10.1086/515371. [DOI] [PubMed] [Google Scholar]
- 62.Eliopoulos GM, Huovinen P. Resistance to trimethoprim-sulfamethoxazole. Clin Infect Dis. 2001;32:1608–14. doi: 10.1086/320532. [DOI] [PubMed] [Google Scholar]
- 63.Adrian PV, Klugman KP. Mutations in the dihydrofolate reductase gene of trimethoprim-resistant isolates of Streptococcus pneumoniae. Antimicrob Agents Chemother. 1997;41:2406–13. doi: 10.1128/aac.41.11.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Groot R, Sluijter M, de Bruyn A, et al. Genetic characterization of trimethoprim resistance in Haemophilus influenzae. Antimicrob Agents Chemother. 1996;40:2131–6. doi: 10.1128/aac.40.9.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dale GE, Broger C, Hartman PG, et al. Characterization of the gene for the chromosomal dihydrofolate reductase (DHFR) of Staphylococcus epidermidis ATCC 14990: the origin of the trimethoprim-resistant S1 DHFR from Staphylococcus aureus? J Bacteriol. 1995;177:2965–70. doi: 10.1128/jb.177.11.2965-2970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bergmann R, van der Linden M, Chhatwal GS, Nitsche-Schmitz DP. Factors that cause trimethoprim resistance in Streptococcus pyogenes. Antimicrob Agents Chemother. 2014;58:2281–8. doi: 10.1128/AAC.02282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bergmann R, Sagar V, Nitsche-Schmitz DP, Chhatwal GS. First detection of trimethoprim resistance determinant dfrG in Streptococcus pyogenes clinical isolates in India. Antimicrob Agents Chemother. 2012;56:5424–5. doi: 10.1128/AAC.01284-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dale GE, Langen H, Page MG, Then RL, Stuber D. Cloning and characterization of a novel, plasmid-encoded trimethoprim-resistant dihydrofolate reductase from Staphylococcus haemolyticus MUR313. Antimicrob Agents Chemother. 1995;39:1920–4. doi: 10.1128/aac.39.9.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rouch DA, Messerotti LG, Loo LS, Jackson CA, Skurray LA. Trimethoprim resistance transposon Tn4003 from Staphylococcus aureus encodes genes for a dihydrofolate reductase and thymidylate synthetase flanked by three copies of IS257. Mol Microbiol. 1989;3:161–75. doi: 10.1111/j.1365-2958.1989.tb01805.x. [DOI] [PubMed] [Google Scholar]
- 70.Coque TM, Singh KW, Weinstock GM, Murray BE. Characterization of dihydrofolate reductase genes from trimethoprim-susceptible and trimethoprim-resistant strains of Enterococcus faecalis. Antimicrob Agents Chemother. 1999;43:141–7. doi: 10.1128/aac.43.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee JC, Oh JY, Cho JW, et al. The prevalence of trimethoprim-resistance-conferring dihydrofolate reductase genes in urinary isolates of Escherichia coli in Korea. J Antimicrob Chemother. 2001;47:599–604. doi: 10.1093/jac/47.5.599. [DOI] [PubMed] [Google Scholar]
- 72.Tang Y, Shen P, Liang W, Jin J, Jiang X. A putative multi-replicon plasmid co-harboring beta-lactamase genes blaKPC-2, blaCTX-M-14 and blaTEM-1 and trimethoprim resistance gene dfrA25 from a Klebsiella pneumoniae sequence type (ST) 11 strain in China. PLoS One. 2017;12:e0171339. doi: 10.1371/journal.pone.0171339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bossé JT, Li Y, Walker S, et al. Langford Identification of dfrA14 in two distinct plasmids conferring trimethoprim resistance in Actinobacilluspleuropneumoniae. J Antimicrob Chemother. 2015;70:2217–22. doi: 10.1093/jac/dkv121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sekiguchi J, Tharavichitkul P, Miyoshi-Akiyama T. al. Cloning and characterization of a novel trimethoprim-resistant dihydrofolate reductase from a nosocomial isolate of Staphylococcus aureus CM.S2 (IMCJ1454) Antimicrob Agents Chemother. 2005;49:3948–51. doi: 10.1128/AAC.49.9.3948-3951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang M, Yang J, Yuan M, Xue L, Li H, Tian C, et al. Synthesis and antiproliferative activity of a series of novel 6-substituted pyrido[3,2-d]pyrimidines as potential nonclassical lipophilic antifolates targeting dihydrofolate reductase. Eur J Med Chem. 2017;128:88–97. doi: 10.1016/j.ejmech.2017.01.033. [DOI] [PubMed] [Google Scholar]
- 76.Gangjee A, Mavandadi F, Kisliuk RL, Queener SF. Synthesis of classical and nonclassical 2-amino-4-oxo-6-methyl-5-substituted pyrrolo[2,3-d]pyrimidine antifolate inhibitors of thimidale synthase. J Med Chem. 1999;42:2272–9. doi: 10.1021/jm980586o. [DOI] [PubMed] [Google Scholar]
- 77.Burchall JJ, Hitchings GH. Inhibitor binding analysis of dihydrofolate reductases from various species. Inhibitor binding analysis of dihydrofolate reductases from various species. Mol Pharm. 1965;1:126–36. [PubMed] [Google Scholar]
- 78.McKeage K, Scott L. Atovaquone/Proguanil: A revives of its use for the prophylaxis of Plasmodium falciparum malaria. Drugs. 2003;63:597–623. doi: 10.2165/00003495-200363060-00006. [DOI] [PubMed] [Google Scholar]
- 79.Elsheikha HM. Congenital toxoplasmosis: priorities for further health promotion action. Public Health. 2008;122:335–53. doi: 10.1016/j.puhe.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 80.Welsch ME, Zhou J, Gao Y, Yan Y, Porter G, Agnihotri G, et al. Discovery of potent and selective leads against toxoplasma gondii dihydrofolate reductase via structure-based design. ACS Med Chem Lett. 2016;7:1124–9. doi: 10.1021/acsmedchemlett.6b00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sharma H, Landau MJ, Vargo MA, Spasov KA, Anderson KS. First threedimensional structure of Toxoplasma gondii thymidylate synthase-dihydrofolate reductase: insights for catalysis, interdomain interactions, and substrate channeling. Biochemistry. 2013;52:7305–17. doi: 10.1021/bi400576t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hopper AT, Brockman A, Wise A, Gould J, Barks J, Radke JB, et al. Discovery of selective toxoplasma gondii dihydrofolate reductase inhibitors for the treatment of toxoplasmosis. J Med Chem. 2019;62:1562–76. doi: 10.1021/acs.jmedchem.8b01754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.https://www.amazon.com/Antibiotic-Chemotherapy-Book-Expert-Consult-ebook/dp/B0055XI3GA- (ACCESS: FEBRUARY, 2019).
- 84.Tonelli M, Naesens L, Gazzarrini S, Santucci M, Cichero E, Tasso B. Host dihydrofolate reductase (DHFR)-directed cycloguanil analogues endowed with activity against influenza virus and respiratory syncytial virus. Eur J Med Chem. 2017;135:467–78. doi: 10.1016/j.ejmech.2017.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Braunsteiner AR, Finsinger F. Brodimoprim: therapeutic efficacy and safety in the treatment of bacterial infections. J Chemother. 1993;5:507–11. [PubMed] [Google Scholar]
- 86.Peppard WJ, Schuenke CD. Iclaprim, a diaminopyrimidine dihydrofolate reductase inhibitor for the potential treatment of antibiotic-resistant staphylococcal infections. Curr Opin Investig Drugs. 2018;9:210–25. [PubMed] [Google Scholar]
- 87.Schneider P, Hawser S, Islam K. Iclaprim, a novel diaminopyrimidine with potent activity on trimethoprim sensitive and resistant bacteria. Bioorg Med Chem Lett. 2003;13:4217–21. doi: 10.1016/j.bmcl.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 88.Oefner C, Bandera M, Haldimann A, Laue H, Schulz H, Mukhija S, et al. Increased hydrophobic interactions of iclaprim with Staphylococcus aureus dihydrofolate reductase are responsible for the increase in affinity and antibacterial activity. J Antimicrob Chemother. 2009;63:687–98. doi: 10.1093/jac/dkp024. [DOI] [PubMed] [Google Scholar]
- 89.Sincak CA, Schmidt JM. Iclaprim, a novel diaminopyrimidine for the treatment of resistant Gram-positive infections. Ann Pharmacother. 2009;43:1107–14. doi: 10.1345/aph.1L167. [DOI] [PubMed] [Google Scholar]
- 90.Andrews J, Honeybourne D, Ashby J, et al. Concentrations in plasma, epithelial lining fluid, alveolar macrophages and bronchial mucosa after a single intravenous dose of 1.6 mg/kg of iclaprim (AR-100) in healthy men. J Antimicrob Chemother. 2007;60:677–80. doi: 10.1093/jac/dkm242. [DOI] [PubMed] [Google Scholar]
- 91.Laue H, Valensise T, Seguin A, Lociuro S, Islam K, Hawser S. In the vitro bactericidal activity of iclaprim in human plasma. Antimicrob Agents Chemother. 2009;53:4542–4. doi: 10.1128/AAC.00766-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Peppard WJ, Schuenke CDIclaprim. a diaminopyrimidinedihydrofolate reductase inhibitor for the potential treatment of antibiotic-resistant staphylococcal infections. Curr Opin Investig Drugs. 2008;9:210–25. [PubMed] [Google Scholar]
- 93.Wyss PC, Gerber P, Hartman PG, et al. Novel dihydrofolate reductase inhibitors. structure-based versus diversity-based library design and high- throughput synthesis and screening. J Med Chem. 2003;46:2304–12. doi: 10.1021/jm020495y. [DOI] [PubMed] [Google Scholar]
- 94.Then RL. Antimicrobial dihydrofolate reductase inhibitors—achievements and future options: review. Rev J Chemother. 2004;16:3–12. doi: 10.1179/joc.2004.16.1.3. [DOI] [PubMed] [Google Scholar]
- 95.Rashid U, Ahmad W, Hassan FS, et al. Design, synthesis, antibacterial, activity and docking study of some new trimethoprim derivatives. Bioorg Med Chem Lett. 2016;26:5749–53. doi: 10.1016/j.bmcl.2016.10.051. [DOI] [PubMed] [Google Scholar]
- 96.Morgan J, Haritakul R, Keller PA. Antimalarial activity of 2, 4-diaminopyrimidines. Lett Drug Des Discov. 2008;5:277–80. [Google Scholar]
- 97.Warhurst D. Antimalarial drug discovery: development of inhibitors of dihydrofolate reductase active in drug resistance. Drug Discov Today. 1998;3:538–46. [Google Scholar]
- 98.Canfield CJ, Milhous WK, Ager AL, Rossan RN, Sweeney TR, Lewis NJ, et al. Ps-15: A potent, orally active antimalarial from a new class of folic acid antagonists. Am J Trop Med Hyg. 1993;49:121–6. doi: 10.4269/ajtmh.1993.49.121. [DOI] [PubMed] [Google Scholar]
- 99.Tarnchompoo B, Sirichaiwat C, Phupong W, et al. Development of 2,4-diaminopyrimidines as antimalarials based on inhibition of the S108N and C59R+S108N mutants of dihydrofolate reductase from pyrimethamine-resistant Plasmodium falciparum. J Med Chem. 2002;45:1244–52. doi: 10.1021/jm010131q. [DOI] [PubMed] [Google Scholar]
- 100.Shaikh MS, Rana J, Gaikwad D, et al. Antifolate agents against wild and mutant strains of Plasmodium falciparum. Indian J Pharm Sci. 2014;76:116–24. [PMC free article] [PubMed] [Google Scholar]
- 101.Roth B, Strelitz JZ, Rauckman BS. 2,4-Diamino-5-benzylpyrimidines and analogues as antibacterial agents. 2. C-Alkylation of pyrimidines with Mannich bases and application to the synthesis of trimethoprim and analogues. J Med Chem. 1980;23:379–84. doi: 10.1021/jm00178a007. [DOI] [PubMed] [Google Scholar]
- 102.Roth B, Strelitz JZ. Protonation of 2, 4-diaminopyrimidines. I. Dissociation constants and substituent effects. J Org Chem. 1969;34:821. [Google Scholar]
- 103.Rauckman BS, Roth B. 2,4-Diamino-5-benzylpyrimidines and analogues as antibacterial agents. 3. C-Benzylation of aminopyridines with phenolic Mannich bases. Synthesis of 1- and 3-deaza analogues of trimethoprim. J Med Chem. 1980;23:384–91. doi: 10.1021/jm00178a008. [DOI] [PubMed] [Google Scholar]
- 104.Roth B, Aig E, Rauckman BS, et al. 2, 4-Diamino-5-benzylpyrimidines and analogues as antibacterial agents. 5. 3’,5’-Dimethoxy-4’-substituted-benzyl analogues of trimethoprim. J Med Chem. 1981;24:933–41. doi: 10.1021/jm00140a005. [DOI] [PubMed] [Google Scholar]
- 105.Kompis I, Then R, Boehni E, Rey-Bellet G, Zanetti G, Montavon M. Synthesis and antimicrobial activity of C(4’)-substituted analogs of trimethoprim. Eur J Med Chem. 1980;15:17. [Google Scholar]
- 106.Stuart A, Paterson T, Roth B, Aig E. 2,4-diamino-5-benzylpyrimidines and analogues as antibacterial agents. 6. One-Step synthesis of new trimethoprim derivatives and activity analysis by molecular modeling. J Med Chem. 1983;26:667–73. doi: 10.1021/jm00359a009. [DOI] [PubMed] [Google Scholar]
- 107.Roth B, Rauckman BS, Ferone R, Baccanari DP, Champness JN, Hyde RM. 2,4-Diamino-5-benzylpyrimidines as antibacterial agents. 7. Analysis of the effect of 3,5-dialkyl substituent size and shape on binding to four different dihydrofolate reductase enzymes. J Med Chem. 1987;30:348–56. doi: 10.1021/jm00385a017. [DOI] [PubMed] [Google Scholar]
- 108.Roth B, Aig E. 2,4-Diamino-5-benzylpyrimidines as antibacterial agents. 8. The 3,4,5-triethyl isostere of trimethoprim. A Study Specificity. J Med Chem. 1987;30:1998–2004. doi: 10.1021/jm00394a012. [DOI] [PubMed] [Google Scholar]
- 109.Roth B, Baccanari DP, Sigel CW, et al. 2,4-Diamino-5-benzylpyrimidines and analogues as antibacterial agents. 9. Lipophilic trimethoprim analogues as antigonococcal agents. J Med Chem. 1988;31:122–9. doi: 10.1021/jm00396a018. [DOI] [PubMed] [Google Scholar]
- 110.Rauckman BS, Tidwell MY, Johnson JV, Roth B. 2,4-Diamino-5-benzylpyrimidines and analogues as antibacterial agents. 10. 2,4-Diamino-5-(6-quinolylmethyl)- and -[(tetrahydro-6-quinolyl) methyl]pyrimidine derivatives. Further specificity studies. J Med Chem. 1989;32:1927–35. doi: 10.1021/jm00128a040. [DOI] [PubMed] [Google Scholar]
- 111.Painter GR, Grunwald R, Roth B. Interaction of the antifolate antibiotic trimethoprim with phosphatidylcholine membranes: a 13C and 31P nuclear magnetic resonance study. Mol Pharm. 1988;33:551. [PubMed] [Google Scholar]
- 112.Davis SE, Rauckman BS, Chan JH, Roth B. 2,4-Diamino-5-benzylpyrimidines and analogues as antibacterial agents. 11. Quinolylmethyl analogues with basic substituents conveying specificity. J Med Chem. 1989;32:1936–42. doi: 10.1021/jm00128a041. [DOI] [PubMed] [Google Scholar]
- 113.Johnson JV, Rauchman BS, Baccanari DP, Roth B. 2,4-Diamino-5-benzylpyrimidines and analogues as antibacterial agents. 12. 1,2-Dihydroquinolylmethyl analogues with high activity and specificity for bacterial dihydrofolate reductase. J Med Chem. 1989;32:1942–9. doi: 10.1021/jm00128a042. [DOI] [PubMed] [Google Scholar]
- 114.Roth Barbara, Tidwell Mary Y., Ferone Robert, Baccanari David P., Sigel Carl W., DeAngelis Diane, Elwell Lynn P. 2,4-Diamino-5-benzylpyrimidines as antibacterial agents. 13. Some alkenyl derivatives with high in vitro activity against anaerobic organisms. Journal of Medicinal Chemistry. 1989;32(8):1949–1958. doi: 10.1021/jm00128a043. [DOI] [PubMed] [Google Scholar]
- 115.Roth B, Cheng CC. Recent progress in the medicinal chemistry of 2,4-diaminopyrimidines. Prog Med Chem. 1982;19:269–331. doi: 10.1016/s0079-6468(08)70332-1. [DOI] [PubMed] [Google Scholar]
- 116.Masur H. Problems in the management of opportunistic infections in patients infected with human immunodeficiency virus. J Infect Dis. 1990;161:858–64. doi: 10.1093/infdis/161.5.858. [DOI] [PubMed] [Google Scholar]
- 117.Masur H, Brooks JT, Benson CA, Holmes KK, Pau AK, Kaplan JE. Prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: updated Guidelines from the Centers for Disease Control and Prevention, National Institutes of Health, and HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis. 2014;58:1308–11. doi: 10.1093/cid/ciu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yoon C, Subramanian A, Chi A, et al. Dihydropteroate synthase mutations in pneumocystis pneumonia: impact of applying different definitions of prophylaxis, mortality endpoints and mutant in a single Cohort. Med Mycol. 2013;51:568–75. doi: 10.3109/13693786.2013.770604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang L, Crothers K, Atzori C, et al. Dihydropteroate synthase gene mutations in pneumocystis and sulfa resistance. Emerg Infect Dis. 2004;10:1721–8. doi: 10.3201/eid1010.030994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ponce CA, Chabe M, George C, et al. High prevalence of pneumocystis jirovecii dihydropteroate synthase gene mutations in patients with a first episode of Pneumocystis pneumonia in Santiago, Chile, and clinical response to trimethoprim-sulfamethoxazole therapy. Antimicrob Agents Chemother. 2017;61:e01290–16. doi: 10.1128/AAC.01290-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dosso M, Ouattara L, Cherif AM, Bouzid SA, Haller L, Fernex M. Experimental in vitro efficacy study on the interaction of epiroprim plus isoniazid against Mycobacterium tuberculosis. Chemotherapy. 2001;47:123–7. doi: 10.1159/000048511. [DOI] [PubMed] [Google Scholar]
- 122.Queener SF. New drug developments for opportunistic infections in immunocompromised patients: Pneumocystis carinii. J Med Chem. 1995;38:4739–59. doi: 10.1021/jm00024a001. [DOI] [PubMed] [Google Scholar]
- 123.Then R. L., Hartman P. G., Kompis I., Santi D. Advances in Experimental Medicine and Biology. Boston, MA: Springer US; 1993. Selective Inhibition of Dihydrofolate Reductase from Problem Human Pathogens; pp. 533–536. [DOI] [PubMed] [Google Scholar]
- 124.Rosowsky A, Forsch RA, Queener SF. Inhibition of Pneumocystis carinii, Toxoplasma gondii, and Mycobacterium avium dihydrofolate reductases by 2,4-diamino-5-[2-methoxy-5-(omega-carboxyalkoxy)benzyl]pyrimidines: marked improvement in potency relative to trimethoprim and species selectivity relative to piritrexim. J Med Chem. 2002;45:233–41. doi: 10.1021/jm010407u. [DOI] [PubMed] [Google Scholar]
- 125.Rosowsky A, Chen H, Fu H, Queener SF. Synthesis of new 2,4-diaminopyrido[2,3-d] pyrimidine and 2,4-diaminopyrrolo[2,3-d]pyrimidine inhibitors of Pneumocystis carinii, Toxoplasma gondii, and Mycobacterium avium dihydrofolate reductase. Bioorg Med Chem. 2003;11:59–67. doi: 10.1016/s0968-0896(02)00325-5. [DOI] [PubMed] [Google Scholar]
- 126.Kuyper LF, Roth B, Baccanari DP, Ferone R, Beddell CR, Champness JN, et al. Receptor-based design of dihydrofolate reductase inhibitors: comparison of crystallographically determined enzyme-binding with enzyme affinity in a series of carboxy-substituted trimethoprim analogues. J Med Chem. 1985;28:303–11. doi: 10.1021/jm00381a008. [DOI] [PubMed] [Google Scholar]
- 127.Chan DCM, Fu H, Forsch RA, Queener SF. Design, synthesis, and antifolate activity of new analogues of piritrexim and other diaminopyrimidine dihydrofolate reductase inhibitors with ω-carboxyalkoxy or ω-carboxy-1-alkynyl substitution in the side chain. J Med Chem. 2005;48:4420–31. doi: 10.1021/jm0581718. [DOI] [PubMed] [Google Scholar]
- 128.Forsch RA, Queener SF, Rosowsky A. Preliminary in vitro studies on two potent, water-soluble trimethoprim analogues with exceptional species selectivity against dihydrofolate reductase from Pneumocystis carinii and Mycobacterium avium. Bioorg Med Chem Lett. 2004;14:1811–5. doi: 10.1016/j.bmcl.2003.12.103. [DOI] [PubMed] [Google Scholar]
- 129.Rosowsky A, Forsch RA, Queener SF. Further studies on 2,4-diamino-5-(2′,5′-disubstituted benzyl)pyrimidines as potent and selective inhibitors of dihydrofolate reductases from three major opportunistic pathogens of AIDS. J Med Chem. 2003;46:1726–36. doi: 10.1021/jm020466n. [DOI] [PubMed] [Google Scholar]
- 130.Davies WL, Grunert RR, Haff RF, et al. Antiviral activity of 1-adamantanamine (amantadine) Science. 1964;144:862–3. doi: 10.1126/science.144.3620.862. [DOI] [PubMed] [Google Scholar]
- 131.Orzeszko B, Kazimierczuk Z, Maurin JK, et al. Novel adamantylated pyrimidines and their preliminary biological evaluations. Farmaco. 2004;59:929–37. doi: 10.1016/j.farmac.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 132.Orzeszko B, Laudy AE, Starościak BJ, et al. Synthesis and antibacterial activity of adamantyl substituted pyrimidines. Acta Pol Pharm. 2004;61:455–60. [PubMed] [Google Scholar]
- 133.Farooq O, Marcelli M, Prakash GKS, Olah GA. Electrophilic reactions at single bonds. 22. Superacid-catalyzed electrophilic formylation of adamantane with carbon monoxide competing with Koch-Haaf carboxylation. J Am Chem Soc. 1988;110:864–7. [Google Scholar]
- 134.Kraus GA, Siclovan TM. Bridgehead intermediates in organic synthesis. A reproducible synthesis of adamantane-containing compounds. J Org Chem. 1994;59:922. [Google Scholar]
- 135.Mancuso AJ, Swern D. Activated dimethyl sulfoxide: useful reagents for synthesis. Synthesis. 1981;3:165–85. [Google Scholar]
- 136.Orzeszko B, Fedoryński M, Laudy AE, Starościak BJ, Orzeszko A. Synthesis and antibacterial activity of 5-adamantan-1-yl-methyl analogues of trimethoprim. Acta Pol Pharm. 2006;63:374–7. [PubMed] [Google Scholar]
- 137.Pelphrey PM, Popov VM, Joska TM, et al. Highly efficient ligands for dihydrofolate reductase from Cryptosporidium hominis and Toxoplasma gondii inspired by structural analysis. J Med Chem. 2007;50:940–50. doi: 10.1021/jm061027h. [DOI] [PubMed] [Google Scholar]
- 138.Liu J, Bolstad D, Smith A, et al. Structure-guided development of efficacious antifungal agents targeting Candida glabrata dihydrofolate reductase. Chem Biol. 2008;15:990–6. doi: 10.1016/j.chembiol.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu J, Bolstad D, Smith A, et al. Probing the active site of Candida glabrata dihydrofolate reductase with high-resolution crystal structures and the synthesis of new inhibitors. Chem Biol Drug Des. 2009;73:62–74. doi: 10.1111/j.1747-0285.2008.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kuyper LF, Roth B, Baccanari DP, et al. Receptor-based design of dihydrofolate reductase inhibitors: comparison of crystallographically determined enzyme binding with enzyme affinity in a series of carboxy-substituted trimethoprim analogues. J Med Chem. 1985;28:303–11. doi: 10.1021/jm00381a008. [DOI] [PubMed] [Google Scholar]
- 141.Cody V, Pace J, Chisum K, Rosowsky A. New insights into DHFR interactions: analysis of Pneumocystis carinii and mouse DHFR complexes with NADPH and two highly potent trimethoprim derivatives. Proteins. 2006;65:959–69. doi: 10.1002/prot.21131. [DOI] [PubMed] [Google Scholar]
- 142.Birdsall B, Feeney J, Pascual C, et al. A 1H NMR study of the interactions and conformations of rationally designed brodimoprim analogues in complexes with Lactobacillus casei dihydrofolate reductase. J Med Chem. 1984;27:1672–6. doi: 10.1021/jm00378a025. [DOI] [PubMed] [Google Scholar]
- 143.Morgan WD, Birdsall B, Polshalov VL, Sali D, Kompis I, Feeney L. Solution structure of a brodimoprim analogue in its complex with Lactobacillus casei dihydrofolate reductase. Biochemistry. 1995;34:11690–702. doi: 10.1021/bi00037a006. [DOI] [PubMed] [Google Scholar]
- 144.Lombardo MN, G-Dayanandan N, Wright DL, Anderson AC. Crystal structures of trimethoprim-resistant DfrA1 rationalize potent inhibition by propargyl-linked antifolates. ACS Infect Dis. 2016;2:149–56. doi: 10.1021/acsinfecdis.5b00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Beierlein JM, Frey KM, Bolstad DB, et al. Synthetic and crystallographic studies of a new inhibitor series targeting Bacillus anthracis dihydrofolate reductase. J Med Chem. 2008;51:7532–40. doi: 10.1021/jm800776a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hajian B, Scocchera E, Keshipeddy S, et al. Propargyl-linked antifolates are potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. PLoS One. 2016;11:e0161740. doi: 10.1371/journal.pone.0161740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bush K, Leal J, Fathima S, et al. The molecular epidemiology of incident methicillin-resistant Staphylococcus aureus cases among hospitalized patients in Alberta, Canada: a retrospective cohort study. Antimicrob Resist Infect Control. 2015;14:35. doi: 10.1186/s13756-015-0076-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Frey KM, Lombardo MN, Wright DL, Anderson AC. Towards the understanding of resistance mechanisms in clinically isolated trimethoprim-resistant, methicillin-resistant Staphylococcus aureus dihydrofolate reductase. J Struct Biol. 2010;170:93–7. doi: 10.1016/j.jsb.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Frey KM, Georgiev I, Donald BR, Anderson AC. Predicting resistance mutations using protein design algorithms. Proc Natl Acad Sci USA. 2010;107:13707–12. doi: 10.1073/pnas.1002162107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Frey KM, Liu J, Lombardo MN, Bolstad DB, Wright DL, Anderson AC. Crystal structures of wild-type and mutant methicillin-resistant Staphylococcus aureus dihydrofolate reductase reveal an alternate conformation of NADPH that may be linked to trimethoprim resistance. J Mol Biol. 2009;387:1298–308. doi: 10.1016/j.jmb.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Viswanathan K, Frey KM, Scocchera EW, et al. Toward new therapeutics for skin and soft tissue infections: propargyl-linked antifolates are potent inhibitors of MRSA and Streptococcus pyogenes. PLoS One. 2012;7:e29434. doi: 10.1371/journal.pone.0029434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lamb KM, Lombardo MN, Alverson J, Priestley ND, Wright DL, Anderson AC. Crystal structures of Klebsiella pneumoniae dihydrofolate reductase bound to propargyl-linked antifolates reveal features for potency and selectivity. Antimicrob Agents Chemother. 2014;58:7484–91. doi: 10.1128/AAC.03555-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Scocchera E, Reeve SM, Keshipeddy S, et al. Charged nonclassical antifolates with activity against Gram-positive and Gram-negative pathogens. ACS Med Chem Lett. 2016;7:692–6. doi: 10.1021/acsmedchemlett.6b00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Reeve SM, Scocchera EW, G-Dayanadan N, et al. MRSA Isolates from United States hospitals carry dfrG and dfrK resistance genes and succumb to propargyl-linked antifolates. Cell Chem Biol. 2016;23:1458–67. doi: 10.1016/j.chembiol.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Reeve SM, Scocchera E, Ferreira JJ, et al. Charged propargyl-linked antifolates reveal mechanisms of antifolate resistance and inhibit trimethoprim-resistant MRSA strains possessing clinically relevant mutations. J Med Chem. 2016;59:6493–500. doi: 10.1021/acs.jmedchem.6b00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dale G, Broger C, D’Arcy A, Hartman P, DeHoogt R, Jolidon S, et al. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J Mol Biol. 1997;266:23–30. doi: 10.1006/jmbi.1996.0770. [DOI] [PubMed] [Google Scholar]
- 157.Keshipeddy S, Reeve SM, Anderson AC, Wright DL. Nonracemic antifolates stereoselectively recruit alternate cofactors and overcome resistance in S. aureus. J Am Chem Soc. 2015;137:8983–90. doi: 10.1021/jacs.5b01442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tobias AM, Toska D, Lange K, et al. Expression, purification, and inhibition profile of dihydrofolate reductase from the filarial nematode Wuchereria bancrofti. PLoS One. 2018;13:e0197173. doi: 10.1371/journal.pone.0197173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gilbert IH. Inhibitors of dihydrofolate reductase in Leishmania and trypanosomes. Biochim Biophys Acta. 2002;62126:249–57. doi: 10.1016/s0925-4439(02)00088-1. [DOI] [PubMed] [Google Scholar]
- 160.Chowdhury SF, Guerrero RH, Brun R, Ruiz-Perez LM, et al. Synthesis and testing of 5-benzyl-2,4-diaminopyrimidines as potential inhibitors of Leishmanial and Trypanosomal dihydrofolate reductase. J Enzym Inhib. 2002;17:293–302. doi: 10.1080/1475636021000059083. [DOI] [PubMed] [Google Scholar]
- 161.Sirawaraporn W, Sertsrivanich R, Booth RG, Hansch C, et al. Mol Biochem Parasitol. 1988;31:79. doi: 10.1016/0166-6851(88)90147-8. [DOI] [PubMed] [Google Scholar]
- 162.Chan DCM, Anderson AC. Towards species-specific antifolates. Curr Med Chem. 2006;13:377–98. doi: 10.2174/092986706775527938. [DOI] [PubMed] [Google Scholar]
- 163.Borst P, Ouellette M. New mechanisms of drug resistance in parasitic protozoa. Annu Rev Microbiol. 1995;49:427–60. doi: 10.1146/annurev.mi.49.100195.002235. [DOI] [PubMed] [Google Scholar]
- 164.Foye WO, Lemke TL, Williams DA. Principles of Medicinal Chemistry. 4th ed. Media, PA: Williams and Wilkins; 2005. [Google Scholar]
- 165.El-Gazzar YI, Georgey HH, El-Messery SM, et al. Synthesis, biological evaluation and molecular modeling study of new (1,2,4-triazole or 1,3,4-thiadiazole)-methylthio-derivatives of quinazolin-4(3H)-one as DHFR inhibitors. Bioorg Chem. 2017;72:282–92. doi: 10.1016/j.bioorg.2017.04.019. [DOI] [PubMed] [Google Scholar]
- 166.Berman EM, Werbel LM. The renewed potential for folate antagonists in contemporary cancer chemotherapy. J Med Chem. 1991;34:479–85. doi: 10.1021/jm00106a001. [DOI] [PubMed] [Google Scholar]
- 167.Kisliuk RL. Folate Biochemistry in Relation to Antifolate Selectivity. In: Jackman AL, editor. Antifolate drugs in cancer therapy. cancer drug discovery and development. Totowa, NJ: Humana Press; 1999. pp. 13–36. [Google Scholar]
- 168.Zhang Q, Nguyen T, Mcmichael M, et al. New small-molecule inhibitors of dihydrofolate reductase inhibit Streptococcus mutans. Int J Antimicrob Agents. 2015;46:174–82. doi: 10.1016/j.ijantimicag.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yuthavong Y, Tarnchompoo B, Vilaivan T, et al. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. PNAS. 2012;109:16823–8. doi: 10.1073/pnas.1204556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Then RL. Antimicrobial dihydrofolate reductase inhibitors-achievements and future options. Rev J Chemother. 2004;16:3–12. doi: 10.1179/joc.2004.16.1.3. [DOI] [PubMed] [Google Scholar]
- 171.Snapka RM, Ge S, Trask J, et al. Unbalanced growth in mouse cells with amplified dhfr genes. Cell Prolif. 1997;30:385–99. doi: 10.1046/j.1365-2184.1997.00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kubbies M, Stockinger H. Cell cycle-dependent DHFR and t-PA production in cotransfected, MTX-amplified CHO cells revealed by dual-laser flow cytometry. Exp Cell Res. 1990;188:267–71. doi: 10.1016/0014-4827(90)90169-b. [DOI] [PubMed] [Google Scholar]
- 173.Neradil J, Pavlasova G, Veselska R. New mechanisms for an old drug; DHFR- and non-DHFR-mediated effects of methotrexate in cancer cells. Klin Onkol. 2012;25:87–92. [PubMed] [Google Scholar]
- 174.Rodríguez M, Coma S, Noé V. Development and effects of immunoliposomes carrying an antisense oligonucleotide against DHFR RNA and directed toward human breast cancer cells overexpressing HER2. Antisense Nucleic Acid Drug Dev. 2002;12:311–25. doi: 10.1089/108729002761381294. [DOI] [PubMed] [Google Scholar]
- 175.Giletti A, Esperon P. Genetic markers in methotrexate treatments. Pharmacogn J. 2018;18:689–703. doi: 10.1038/s41397-018-0047-z. [DOI] [PubMed] [Google Scholar]
- 176.Gangjee A, Li W, Kisliuk R, et al. Design, synthesis, and X-ray crystal structure of classical and nonclassical 2-amino-4-oxo-5-substituted-6-ethylthieno[2,3-d]pyrimidines as dual thymidylate synthase and dihydrofolate reductase inhibitors and as potential antitumor agents. J Med Chem. 2009;52:4892–902. doi: 10.1021/jm900490a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wright DL, Anderson AC. Antifolate agents: a patent review (2006–10) Expert Opin Ther Pat. 2011;21:1293–308. doi: 10.1517/13543776.2011.587804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Singh P, Kaur M, Sachdeva S. Mechanism inspired the development of rationally designed dihydrofolate reductase inhibitors as anticancer agents. J Med Chem. 2012;55:6381–90. doi: 10.1021/jm300644g. [DOI] [PubMed] [Google Scholar]
- 179.Algul O, Paulsen JL, Anderson AC. 2,4-Diamino-5-(2’-arylpropargyl)pyrimidine derivatives as new nonclassical antifolates for human dihydrofolate reductase inhibition. J Mol Graph Model. 2011;29:608–13. doi: 10.1016/j.jmgm.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wróbel A, Drozdowska D Synthesis and some biological properties of new trimethoprim analogues [abstract P290]. In: Proceedings of the 7th edition of the EFMC International Symposium on Advances in Synthetic and Medicinal Chemistry (EFMC-ASMC'17). Vienna (Austria) 2017.
- 181.Wróbel A, Drozdowska D Solid-phase synthesis of thirteen trimethoprim analogues as DNA-binding agents. [abstract P56]. In: 25th Young Research Fellows Meeting. France (Orléans) 2018.
- 182.Bailly C, Chaires JB, Sequence-specific DNA. minor groove binders. Design and synthesis of netropsin and distamycin analouges. Bioconjugate Chem. 1999;9:513–38. doi: 10.1021/bc980008m. [DOI] [PubMed] [Google Scholar]
- 183.Viegas-Junior C, Danuello A, da Silva BV, Barreiro EJ, Fraga CA. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr Med Chem. 2007;14:1829–52. doi: 10.2174/092986707781058805. [DOI] [PubMed] [Google Scholar]
- 184.Morphy R, Rankovic Z. Designed multiple ligands. an emerging drug discovery paradigm. J Med Chem. 2005;48:6523–43. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 185.Espinoza-Fonseca LM. The benefits of the multi-target approach in drug design and discovery. Bioorg Med Chem. 2006;14:896–7. doi: 10.1016/j.bmc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 186.O’Boyle NM, Meegan MJ. Designed multiple ligands for cancer therapy. Curr Med Chem. 2011;18:4722–37. doi: 10.2174/092986711797535344. [DOI] [PubMed] [Google Scholar]
- 187.Rao AS, Road K. A study on dihydrofolate reductase and its inhibitors: a review. Int J. 2013;4:2535–47. [Google Scholar]
- 188.Lin JT, Mbewe B, Taylor SM, et al. Increased prevalence of dhfr and dhps mutants at delivery in Malawian pregnant women receiving intermittent preventive treatment for malaria. Trop Med Int Health. 2013;18:175–8. doi: 10.1111/tmi.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rana RM, Rampogu S, Zeb A, et al. In silico study probes potential inhibitors of human dihydrofolate reductase for cancer therapeutics. J Clin Med. 2019;8:233. doi: 10.3390/jcm8020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shah K, Lin X, Queener SF, et al. Targeting species-specific amino acid residues: design, synthesis and biological evaluation of 6-substituted pyrrolo[2,3-d]pyrimidines as dihydrofolate reductase inhibitors and potential anti-opportunistic infection agents. Bioorg Med Chem. 2018;26:2640–50. doi: 10.1016/j.bmc.2018.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ouyang Y, Yang H, Zhang P, et al. Synthesis of 2,4-diaminopyrimidine core-based derivatives and biological evaluation of their anti-tubercular actives. Molecules. 2017;22:1592–619. doi: 10.3390/molecules22101592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Thakkar SS, Thakor P, Ray A. Benzothiazole analogues: Synthesis, characterization, MO calculations with PMG6 and DFT, in silico studies and in vitro antimalarial as DHFR inhibitors and antimicrobial actives. Bioorg Med Chem. 2017;25:5396–406. doi: 10.1016/j.bmc.2017.07.057. [DOI] [PubMed] [Google Scholar]