

Abstract
Defining the immune physiology of culture-adapted Mesenchymal stromal Cells (MSCs) derived from distinct tissue compartments informs their potential utility as pharmaceuticals. Here we have investigated the comparative immune plasticity of MSCs and Hepatic Stellate Cells (HeSCs) isolated from human and murine bone marrow and liver respectively. Although both bone marrow MSCs (BM-MSCs) and HeSCs share mesenchymal phenotype and overall molecular genetic responses to inflammatory cues, HeSCs differ from BM-MSCs in a meaningful manner. We show that culture-adapted HeSCs express substantially higher levels of HGF (hepatocyte Growth factor), MMP-1 (Matrix metalloproteinase-1) and CCL2 (chemokine (C-C motif) ligand 2 than BM-MSCs. Both human BM-MSCs and HeSCs inhibit T cell proliferation by a shared Indoleamine, 2,3 Dioxygenase (IDO)-dependent mechanism. However, HeSCs are distinct from BM-MSCs by their significantly higher expression of IL-8, CCL11 and GMCSF when co-cultured with activated PBMCs. We have investigated MSCs and HeSCs derived from murine systems to describe interspecies comparability. Murine BM-MSCs inhibit T cell proliferation through inducible nitric oxide synthase (iNOS) but not IDO. However, Murine HeSCs inhibit T cell proliferation through a mechanism distinct from either IDO or iNOS. Altogether, these results suggest that although culture-adapted BM-MSCs and HeSCs display a similar phenotype, their secretome and immune plasticity are in part distinct likely mirroring their tissular origins. In addition, the discordance in immune biology between mouse and human sourced HeSC and BM-MSCs speaks to the importance of comparative biology when interrogating rodent systems for human translational insights.
Keywords: Mesenchymal Stromal cells, Hepatic Stellate Cells, Secretome, Transcriptome, Immune suppression
Graphical Abstract
Introduction
Bone marrow-derived Mesenchymal Stromal Cells (BM-MSCs) are under intense clinical investigation as a cellular pharmaceutical due to their anti-inflammatory and regenerative properties[1]. Culture adapted cells meeting the International Society for Cell Therapy (ISCT) definition of MSCs can also be derived from an array of non-marrow tissue sources that deploy comparable in vitro features of suppression of T-cell proliferation or regenerative functionalities[2, 3]. Though culture-adapted MSCs from distinct tissue sources share phenotype and functionalities, there are meaningful cell biological differences which likely reflect imprinting from the tissue of origin[4–6]. The comparative analysis of functional features of MSCs from distinct tissue sources may well provide important insights on the biology of the endogenous progenitors from which they arise as well as how best to exploit these properties with translational intent as a cell pharmaceutical. The Hepatic stellate cell (HeSC) is the primary hepatic non-parenchymal mesenchymal progenitor of the liver. HeSCs are the storage of retenyl esters in normal liver and play an important role in the immune tolerance[7, 8]. Conversely, HeSC are also responsible for dense matrix production and deposition following chronic liver injury[7]. It has been demonstrated that Human HeSCs display characteristics shared with BM-MSCs[9–11]. In addition, of the participation of liver HeSCs and exogenous MSCs recruited from outside of the liver in hepatic injury and fibrosis are entirely not clear[12]. Yet, several clinical trials aim to utilize BM-MSCs to treat liver fibrosis[13, 14]. To address this issue, we characterized the phenotype and function of HeSCs and BM-MSCs of human and murine origin to understand their cellular physiology so to inform our understanding of tissue source imprinting on their immune plasticity.
Results and Discussion
The ISCT has provided consensus minimal criteria guidelines to define MSCs[2]. There are no significant differences in the phenotypical markers (CD45−CD105+CD44+CD73+CD90+) expressed by human bone marrow-derived MSCs and liver-derived HeSCs (n=3 donors each) (Fig.1A). MSCs are known to express adhesion molecules as part of their anchorage-dependent growth[15]. Since HeSCs are located in the perivascular space of Disse in between hepatic sinusoidal endothelium and hepatocytes, and interact with the lymphoid, myeloid populations, we have investigated their expression profile[8]. Both BM-MSCs and HeSCs express identical adhesive marker profile (Fig.1B), suggesting their similarities in physical interaction with microenvironment. Adipogenic induction of human BM-MSCs and HeSCs demonstrate that both BM-MSCs and HeSCs exhibit oil droplet accumulation, demonstrating their shared mesenchymal plasticity, however BM-MSCs exhibit more robust induction than HeSCs. In addition, BMSCs exhibit strong differentiation in to chondrocytes and osteocytes compared to HeSCs (Fig.1C). We investigated the comparative mRNA expression of 40 genes, relevant to MSCs’ immunobiology and regenerative properties. Although both BM-MSCs and HeSCs display nearly identical mRNA expression patterns, CCL2, MMP-1, and HGF are highly expressed on HeSCs compared to BM-MSCs (Fig.1D). The T-cell immunosuppressive potential of MSCs is largely dependent upon its response to IFNγ. Therefore MSCs’ responsiveness to IFNγ has been proposed as a surrogate measure of their in vivo suppressive potential[3]. Comparative IFNγ phenotypic responsiveness of BM-MSCs and HeSCs are identical since IFNγ upregulates MHC class I, class II and B7-H1 but not costimulatory molecules B7-1 and B7-2 in both populations (Fig.1E). We also analyzed the expression of 11 genes that are known to respond to IFNγ by MSCs. BM-MSCs and HeSCs upregulate all of these genes at comparable levels (Fig.1F). We analyzed the modulation of HeSC specific CCL2, MMP-1, and HGF expression by IFNγ, TNFα, LPS, TGFβ and IFNα. Although CCL2, MMP-1 and HGF are highly expressed by resting HeSCs, no differences were observed between activated BM-MSCs and HeSCs in response to inflammatory stimuli (Fig.1G).
Figure 1. Phenotype and molecular genetic responses of human MSCs and HeSCs.
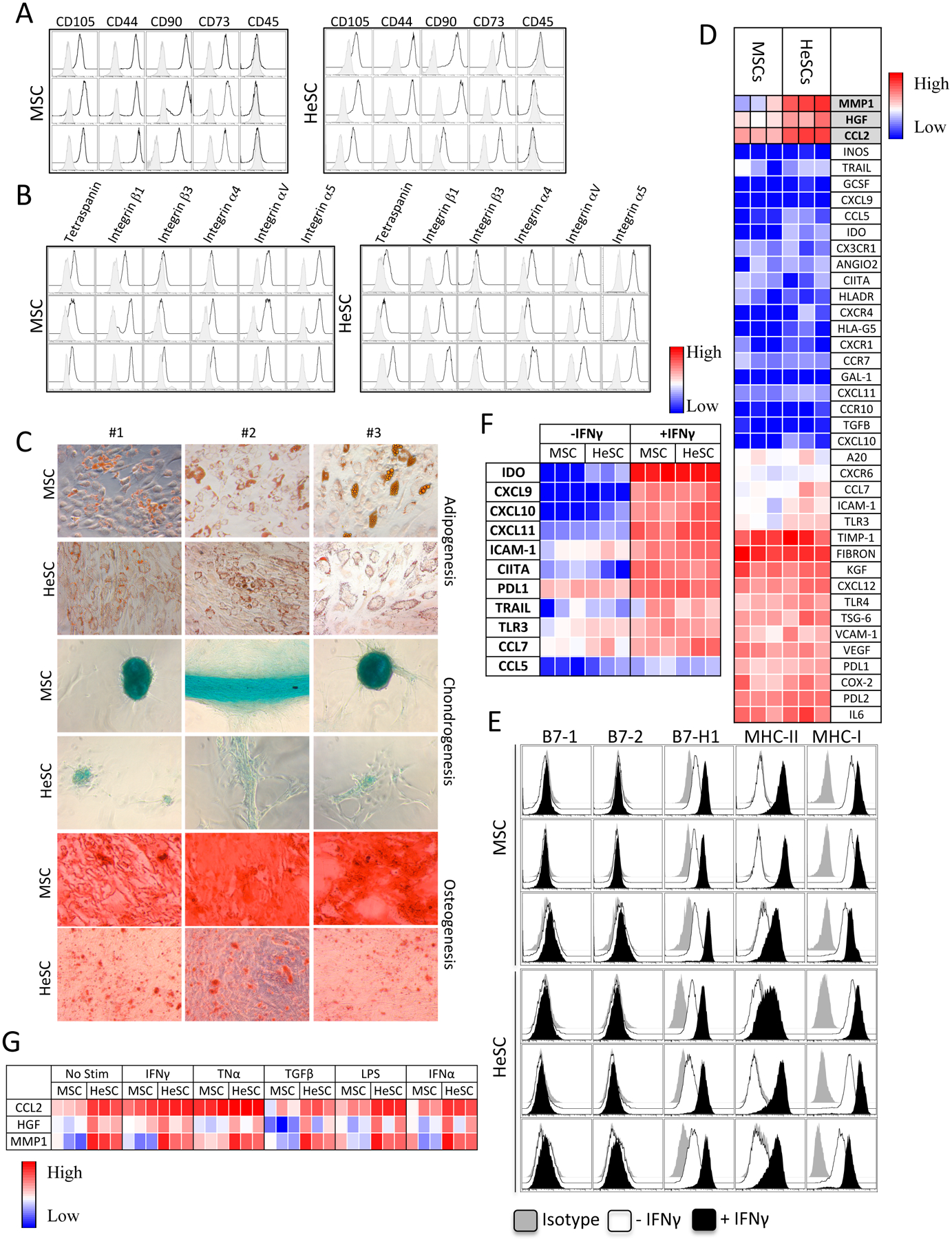
Human BM-MSCs and primary liver derived HeSCs (n=3 donors each) were analyzed for (A) Phenotypical markers (CD105+CD44+CD90+CD73+CD45−) and (B) adhesive molecule profile, Tetraspanin (CD63), Integrin β1 (CD29), Integrin β3 (CD61), Integrin α4 (CD49d), Integrin alpha V (CD51), Integrin alpha 5 (CD49e). Open and grey histograms represent marker and isotype controls respectively. (C) BM-MSCs/HeSCs were subjected to adipogenesis, chondrogenesis and osteogenesis induction for a period of 15–20 days and stained with Oil Red O, Alcian Blue and Alizarin Red S respectively.. (D) Human BM-MSCs/HeSCs (n=3 donors each) were analyzed for the expression of 40 genes through Fluidigm nanoscale quantitative PCR. Heat map represents the relative CT values that are normalized to endogenous GAPDH control. (E) BM-MSCs/HeSCs were stimulated with IFNγ for 48 hours and analyzed for the expression of HLA-ABC, HLA-DR, B7–1, B7–2 and B7H1 by flow cytometry. Open and dark histograms represent – or + IFNγ respectively. Grey histograms at each plot represent the isotype control. (F) BM-MSC or HeSCs (n=3 donors each) were stimulated with 20 ng/mL IFNγ for 48 hours, and total cDNA were generated from RNA. Transcriptional profiles of 11 genes were investigated in Fluidigm nanoscale quantitative PCR. Heat map represents the relative CT values that are normalized to endogenous GAPDH control. (G) BM-MSC or HeSCs (n=3 donors each) were stimulated with 20–25ng/ml IFNγ, TNFα, LPS, TGFβ and IFNα for 48 hours. Expression levels of CCL2, HGF and MMP-1 were assessed by Fluidigm nanoscale quantitative PCR. Heat map represents the relative CT values that are normalized to endogenous GAPDH control.
BM-MSC and HeSCs inhibit allogeneic T cell proliferation at comparable levels (Fig.2A,B). Human BM-MSC T-cell immunosuppressive mechanisms are largely dependent on IFNγ induced Indoleamine, 2,3 Dioxygenase (IDO)[16]. Both human BM-MSCs and HeSCs upregulate IDO upon IFNγ stimulation (Fig.2C,D). Blocking of IDO catalytic activity with 1-Methyl Tryptophan (1-MT) negates the suppressive effect of both human MSCs and HeSCs on T-cell proliferation (Fig.2E,F,G). Thus IDO plays a central role in T-cell immunosuppressive properties of both human BM-MSCs and HeSCs. We explored the role of inducible Nitric Oxide Synthase (iNOS) on human MSC and HeSC’s immunosuppressive properties. Although IFNγ upregulates iNOS moderately on human MSCs and HeSCs, blocking of its activity with L-NMMA do not reverses their inhibitory potential on T cell proliferation (Fig.2H,I,J). These results demonstrate the role of IDO but not iNOS on the T-cell suppressive effect of human MSCs and HeSCs.
Figure 2. Immunosuppressive mechanism of human BM-MSCs and HeSCs.
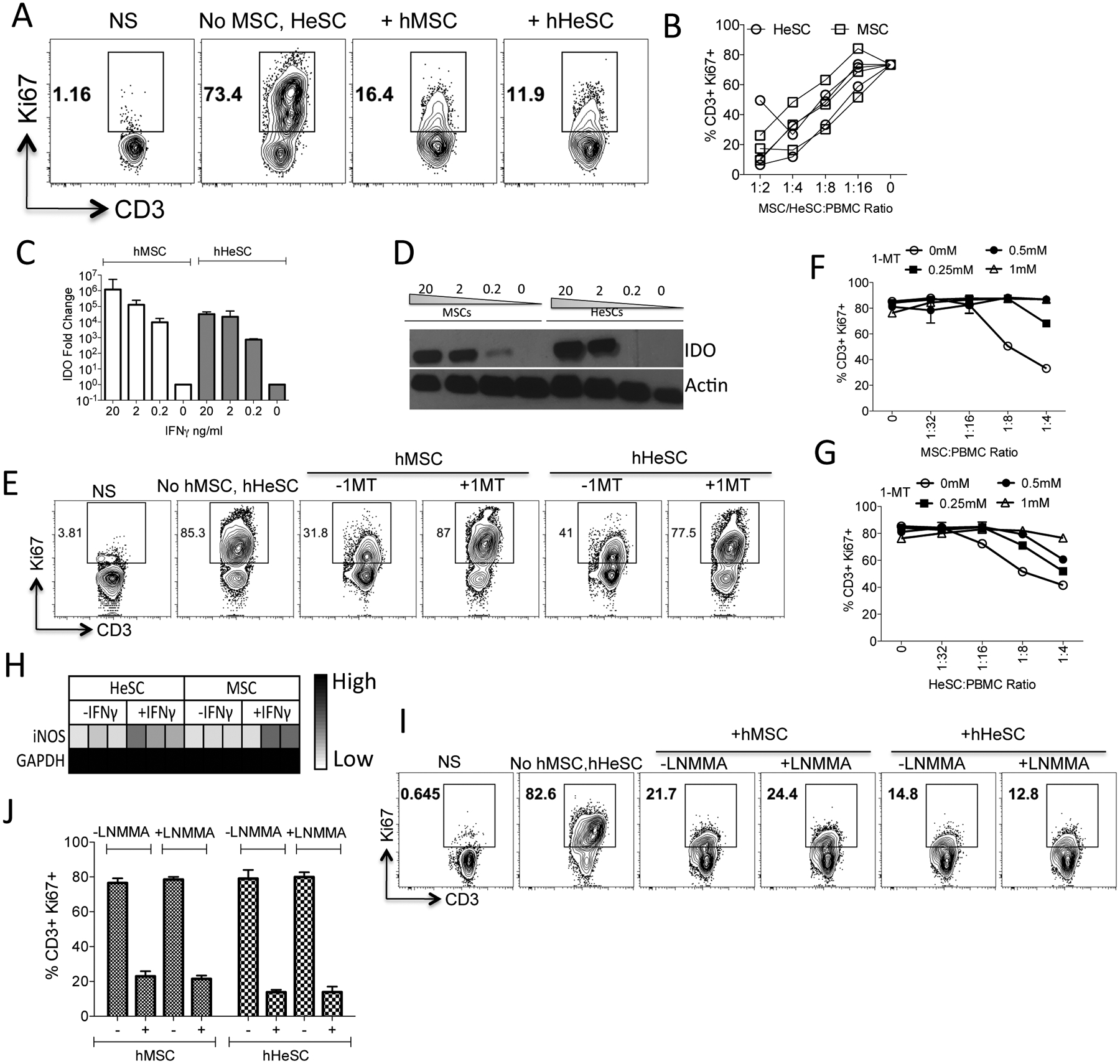
Human MSCs or HeSCs (N=3 donors) were cultured with Staphylococcus Enterotoxin B (SEB) activated PBMCs. Four days post culture, T-cell proliferation was measured by Ki67 intracellular staining. (A) Representative Flow cytometery plot and (B) cumulative dose dependent effect of BM-MSCs or HeSCs effect on T-cell proliferation (%CD3+Ki67+) is shown. (C) Human BM-MSCs or HeSCs were stimulated with indicated concentrations of IFNγ for 48 h. Expression levels of IDO mRNA were quantitated by quantitative SYBR Green real-time PCR. GAPDH mRNA levels were used as internal controls. (D) Western blot analysis of human BM-MSCs and HeSCs for IDO expression at protein levels. Actin was used as an internal control. Human BM-MSCs or HeSCs were cocultured with SEB activated PBMCs in the presence and absence of IDO blocker 1-MT. Four days after, T-cell proliferation was measured by flow cytometry. (E) Representative flow cytometry plot and dose dependent effect of 1-MT on T cell proliferation in a differential ratio of (F) BM-MSCs or (G) HeSCs with PBMCs is shown with mean and standard deviation. (H) MSCs or HeSCs were stimulated with IFNγ and the expression levels of iNOS were determined through quantitative PCR. Human BM-MSCs or HeSCs were cocultured with SEB activated PBMCs in the presence and absence of 1mM iNOS blocker L-NMMA. Four days after, T-cell proliferation was measured by flow cytometry. (I) Representative flow cytometry plot and (J) cumulative from independent cocultures is shown with mean and standard deviation. Similar results were obtained in at least two experiments with another donor.
In order to define the secretome responses of MSCs and HeSCs, we analyzed 28 biologically relevant cytokines and chemokines released in the supernatant of the human BM-MSCs/HeSCs as well as BM-MSCs/HeSCs co-cultured with Staphylococcal Enterotoxin B (SEB) activated human Peripheral Blood Mononuclear Cells (PBMCs). Three major pattern of cytokine modulation that are common to MSCs and HeSCs were observed. They are (1) Downregulated Cytokines (2) Upregulated Cytokines (3) Unmodulated Cytokines. IFNγ, TNFα, CCL4, IL2R, IL-12, IL-13, CCL3, IL-10 were downregulated in both BM-MSC and HeSC cocultures with PBMCs compared to PBMC cultures alone (Fig.3A). None of the downregulated cytokines were produced in MSC/HeSC cultures without PBMCs. Thus the down regulation was due to the direct effect of MSCs/HeSCs on the inhibition of SEB activated T cell proliferation which reflects down regulated responses of PBMCs upon interaction with MSCs/HeSCs. GCSF, VEGF, IL-7, CXCL9, CXCL10, IFNα, FGF-basic and CCL2 were upregulated by MSC/HeSC and PBMC cocultures (Fig.3B). Of these, VEGF, IL-7, IFNα, and FGF-basic are constitutively secreted by MSCs/HeSCs in the absence of PBMC interaction (Fig.3B). However, GCSF, CXCL9, and CXCL10 are de novo upregulated by both MSCs and HeSCs upon interaction with PBMCs (Fig.3B). But CXCL9, CXCL10 and GCSF were produced substantially only on MSC/HeSC coculture with PBMCs but not on their individual cultures. Thus these three cytokines were considered as de novo induced cytokines of MSC/HeSC interaction with PBMCs. To identify the source of this induction, we cultured MSCs/HeSCs with PBMCs in a two-chamber transwell system. We have evaluated the RNA levels of MSCs/HeSCs cultured with and without activated PBMCs. Our results have demonstrated that CXCL9, CXCL0 and GCSF were substantially upregulated on MSCs and HeSCs upon interaction with activated PBMCs (Fig.S1). In addition, there are a number of leukines and morphogens, including IL-2, IL-4, CCL5, IL-17, IL1RA, IL-15, EGF, IL-1beta that are not significantly modulated by MSCs/HeSCs (Fig.3C). HGF, CCL2, and IL-8 were secreted at high levels constitutively by HeSCs compared to BM-MSCs in the absence of activated PBMCs (Fig.3D). We also confirmed that HeSCs constitutively secrete higher levels of MMP-1 compared to MSCs (Fig. S2). Although CCL11 is also secreted at high levels by resting HeSCs, PBMC interaction significantly blunts its expression (Fig.3E). In contrast, GMCSF is substantially upregulated in the cocultures of HeSCs and activated PBMCs in comparison to BM-MSCs (Fig.3F). A two-chamber transwell system analysis demonstrates that activated PBMCs up regulate GMCSF mRNA specifically by HeSCs, implying that HeSCs are the source of GMCSF production (Fig.3G). Altogether, although MSCs and HeSCs share both resting and PBMC-activated secretome profile, there are significant differences (HGF, MMP-1, CCL2, IL-8, CCL11, and GM-CSF) in their immune plasticity response (Table S1). These observations are congruent with the hepatic origin of HeSC and the important roles that HGF, MMP-1, CCL2 and IL-8 have been found to play in hepatic injury and regenerative response in which endogenous HeSC likely participate. HGF and MMP-1 have been shown to play a major role in the liver regeneration and attenuation of fibrosis [17, 18]. In contrast IL-8 and CCL2 play an important role in the accumulation of macrophage in the liver to promote fibrosis [19, 20]. CCL11 play a major role on the recruitment of leukocytes into the injured liver[21]. Downreguation of CCL11 in HeSCs upon its interaction with PBMCs suggests that HeSCs are responsive to leukocytes in regulating their infiltration. In contrast, GMCSF is upregulated on HeSCs upon its interaction with activated PBMCs. Considering that liver is susceptible to fibrosis when subjected to chronic inflammation, it may well be that HeSC sourced GM-CSF leads to autocrine endogenous HeSC activation as well as neutrophil and myeloid cell activation in a manner which promotes fibrosis [22, 23]. Altogether HeSCs constitutively secrete and modulate the expression of unique cytokines and chemokines which have combined significance for liver regeneration and fibrosis.
Figure 3. Secretome signature responses of human BM-MSCs and HeSCs.
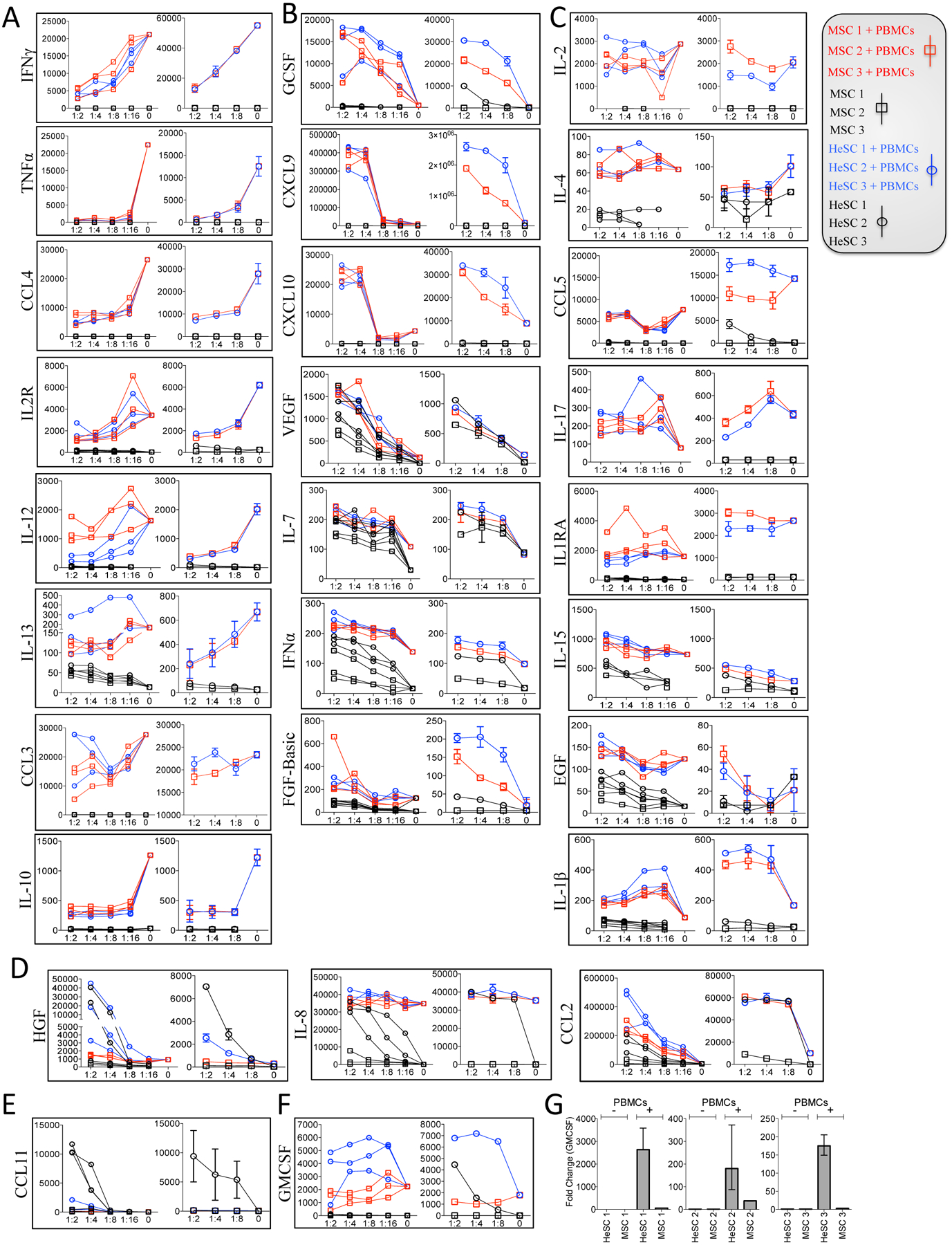
The supernatant of SEB-activated PBMCs co-cultured with BM-MSCs (Red-Square) or HeSCs (Blue-Circle) at the indicated ratio was collected on day 4. BM-MSC (Black-Square) or HeSC (Black-Circle) cultures without PBMCs were collected at the indicated ratios which indicates the constitutive secretion of cytokines/chemokines by MSCs/HeSCs. Quantitative levels of 28 cytokines were assayed through luminex xMAP (multi-analyte profiling) technology. Cytokine responses of three MSC/HeSC donors cultured with PBMC from a single donor (Left) and one BM-MSC/HeSC donor cultured with second distinct PBMC donor (Right) was shown in each box. X-axis represents BM-MSC/HeSC and PBMC ratio along with BM-MSCs/HeSCs without PBMCs. Y axis represents cytokine concentration in Picogram/milliliter. Downregulated, upregulated and unmodulated cytokines that are common to MSC and HeSC cocultures with/without SEB activated PBMCs are shown in Figure 3A, 3B and 3C. (A) Down regulation and (B) Up regulation of cytokines upon BM-MSC or HeSC addition in activated PBMC cultures are shown (C) Low/no modulation of cytokines upon BM-MSC or HeSC addition is also shown. Distinct cytokine secretion pattern of MSCs and HeSCs with and without SEB activated PBMCs is shown in Figure 3D, 3E and 3F. (D) Significant secretion of IL-8, HGF and CCL2 by resting HeSCs (Black Circle) and HeSC coculture with activated PBMCs (Blue Circle). (E) Unique secretion of CCL11 by resting HeSCs, which is down regulated upon cocultured with activated PBMCs. (F) De novo upregulation of GMCSF in HeSC cocultures with SEB activated PBMCs. (G) BM-MSCs or HeSCs were cultured in the presence and absence of SEB-activated PBMCs in a 2-chamber transwell plate. Three days after culture, BM-MSCs or HeSCs were harvested, and the expression levels of GMCSF mRNA were measured through real-time quantitative PCR. Expression levels were expressed after endogenous GAPDH normalization. Cumulative results from three independent donors are shown.
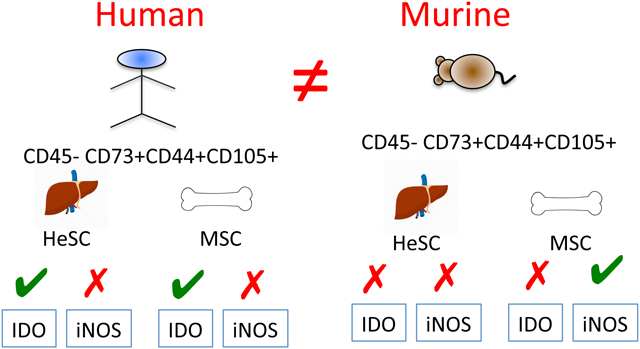
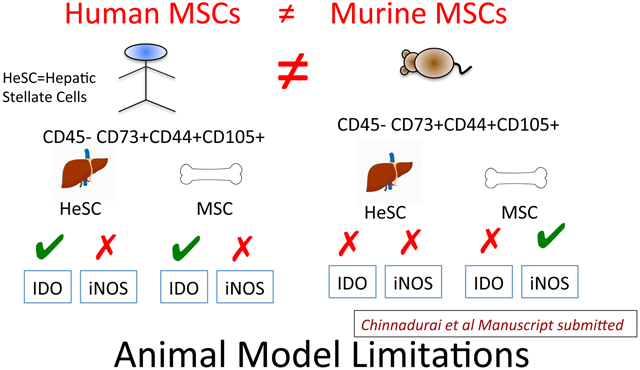
Several in vivo animal model studies were done in the past to study the biology and translational application/significance of MSCs and HeSCs. However it was unclear about their translational relevance to human. Species-specific differences in the immunobiology of human and murine MSC are well described [24]. In order to define if meaningful species-specific differences exist between mouse and human HeSCs and BM-MSCs, we investigated the immunosuppressive mechanisms of murine-derived BM-MSCs and liver-derived HeSCs. Cultured primary murine HeSCs expressed markers of MSC identity such as CD45-CD105+CD44+CD73+ (Fig.4A). Although CD73 expression on cultured HeSCs was reported already [25], we detected their expression in flow cytometry only after permeabilization. IFNγ stimulation upregulated IDO and iNOS mRNA expression on both murine MSCs and HeSCs (Fig.4B,C). We also confirmed that IFNγ induced iNOS expression by Western Blot analysis (Fig.4D). Blocking of iNOS activity through the iNOS blocker L-NMMA reversed the immunosuppressive properties of murine MSCs on CD8+ and CD4+ T cell proliferation (Fig.4E,F). However, 1-MT had no effect on murine BM-MSCs immunosuppressive properties (Fig.4E,F). In contrast, neither 1-MT or L-NMMA block the immunosuppressive properties of murine HeSCs on CD8+ and CD4+ T cell proliferation (Fig.4G,H). These results suggest that T-cell immunosuppressive mechanisms of murine BM-MSCs and HeSCs are meaningfully distinct.
Figure 4. Immunosuppressive mechanism of murine BM-MSCs and HeSCs.
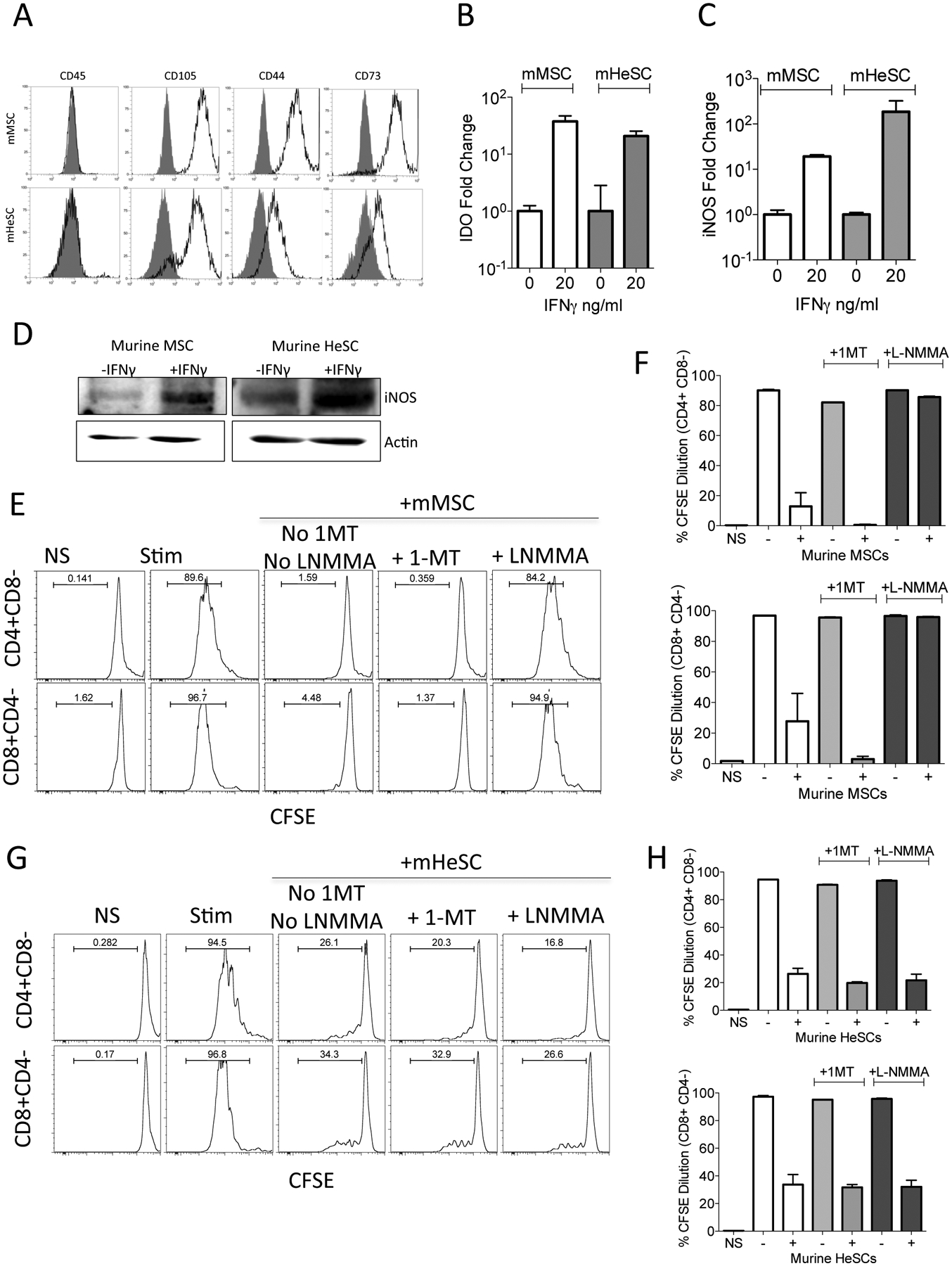
Murine HeSCs were isolated from C57BL/B6 mice and were cultured for 10–14 days. Murine BM-MSCs were isolated from bone marrow flush of culture expanded until CD45+cell populations disappear. (A) HeSCs or BM-MSCs were stained for CD45, CD73, CD105, CD44 and acquired through flow cytometer. Murine BM-MSCs and HeSCs were stimulated with 25ng/ml murine IFNγ for 48 hours. Relative mRNA levels of (B) IDO and (C) iNOS were determined through quantitative real-time PCR. (D) Western blot analysis of murine BM-MSCs and HeSCs were performed to analyze iNOS protein. Actin was used as an internal control. (E, F) Murine BM-MSCs or (G, H) Murine HeSCs were cocultured with αCD3αCD28 activated CFSE labeled CD3+ purified T cells from the spleens of C57BL/B6 mice with and without 1mM 1-MT and L-NMMA respectively. Three days later T cell proliferation was measured by CFSE dilution. (E, G) Representative FACS plots and (F, H) cumulative of CD4+ and CD8+ T populations from three independent cocultures are shown with mean and standard deviation. Similar results were obtained in two independent experiments.
Conclusion:
Our study confirms that human BM-MSCs and HeSCs inhibit T cell suppression through IDO mechanism suggesting its dominant role in the immune physiology of human mesenchymal cell types from liver and bone marrow. However, distinct from BM-MSC, we have shown that culture adapted HeSC express substantially different immune plasticity. The difference in the immune plasticity account for the tissue specific origin and functions of HeSCs and MSCs from liver and bone marrow respectively.
Lastly, the IDO-independent immunosuppressive pathways of BM-MSCs and HeSCs of murine origin suggest interspecies biological distinctive biology that needs to be taken into account in pre-clinical animal modelling.
Supplementary Material
Acknowledgements:
This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases award R01DK109508 to JG. This project is supported in part by a grant from the When Everyone Survives Foundation to R.C. Some of the work reported was conducted while Frank A. Anania was employed by Emory University. The opinions expressed are those of Frank A. Anania and do not reflect the view of the US Food and Drug Administration, the Department of Health and Human Services, or the United States Government. RD is supported by a Post-Doctoral fellowship (ID-17POST33670196) from the American Heart Association.
References
- 1.Galipeau J, Sensebe L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell stem cell. 2018;22:824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dominici M, Le Blanc K, Mueller I et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. [DOI] [PubMed] [Google Scholar]
- 3.Krampera M, Galipeau J, Shi Y et al. Immunological characterization of multipotent mesenchymal stromal cells--The International Society for Cellular Therapy (ISCT) working proposal. Cytotherapy. 2013;15:1054–1061. [DOI] [PubMed] [Google Scholar]
- 4.Billing AM, Ben Hamidane H, Dib SS et al. Comprehensive transcriptomic and proteomic characterization of human mesenchymal stem cells reveals source specific cellular markers. Scientific reports. 2016;6:21507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guimaraes-Camboa N, Cattaneo P, Sun Y et al. Pericytes of Multiple Organs Do Not Behave as Mesenchymal Stem Cells In Vivo. Cell stem cell. 2017;20:345–359 e345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu M, Shaw G, Murphy M et al. Induced Pluripotent Stem Cell-Derived Mesenchymal Stromal Cells Are Functionally and Genetically Different From Bone Marrow Derived-Mesenchymal Stromal Cells. Stem cells. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koyama Y, Brenner DA. Liver inflammation and fibrosis. The Journal of clinical investigation. 2017;127:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nature reviews Gastroenterology & hepatology. 2017;14:397–411. [DOI] [PubMed] [Google Scholar]
- 9.Castilho-Fernandes A, de Almeida DC, Fontes AM et al. Human hepatic stellate cell line (LX-2) exhibits characteristics of bone marrow-derived mesenchymal stem cells. Experimental and molecular pathology. 2011;91:664–672. [DOI] [PubMed] [Google Scholar]
- 10.Raicevic G, Najar M, Najimi M et al. Influence of inflammation on the immunological profile of adult-derived human liver mesenchymal stromal cells and stellate cells. Cytotherapy. 2015;17:174–185. [DOI] [PubMed] [Google Scholar]
- 11.Covas DT, Panepucci RA, Fontes AM et al. Multipotent mesenchymal stromal cells obtained from diverse human tissues share functional properties and gene-expression profile with CD146+ perivascular cells and fibroblasts. Experimental hematology. 2008;36:642–654. [DOI] [PubMed] [Google Scholar]
- 12.Kisseleva T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology. 2017;65:1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alfaifi M, Eom YW, Newsome PN et al. Mesenchymal stromal cell therapy for liver diseases. Journal of hepatology. 2018;68:1272–1285. [DOI] [PubMed] [Google Scholar]
- 14.Haldar D, Henderson NC, Hirschfield G et al. Mesenchymal stromal cells and liver fibrosis: a complicated relationship. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2016;30:3905–3928. [DOI] [PubMed] [Google Scholar]
- 15.Chinnadurai R, Garcia MA, Sakurai Y et al. Actin cytoskeletal disruption following cryopreservation alters the biodistribution of human mesenchymal stromal cells in vivo. Stem cell reports. 2014;3:60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chinnadurai R, Rajan D, Qayed M et al. Potency Analysis of Mesenchymal Stromal Cells Using a Combinatorial Assay Matrix Approach. Cell reports. 2018;22:2504–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakamura T, Sakai K, Nakamura T et al. Hepatocyte growth factor twenty years on: Much more than a growth factor. Journal of gastroenterology and hepatology. 2011;26 Suppl 1:188–202. [DOI] [PubMed] [Google Scholar]
- 18.Iimuro Y, Nishio T, Morimoto T et al. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology. 2003;124:445–458. [DOI] [PubMed] [Google Scholar]
- 19.Zimmermann HW, Seidler S, Gassler N et al. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PloS one. 2011;6:e21381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. 2014;147:577–594 e571. [DOI] [PubMed] [Google Scholar]
- 21.Jaruga B, Hong F, Sun R et al. Crucial role of IL-4/STAT6 in T cell-mediated hepatitis: up-regulating eotaxins and IL-5 and recruiting leukocytes. Journal of immunology. 2003;171:3233–3244. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Z, Xu MJ, Cai Y et al. Neutrophil-Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. Cellular and molecular gastroenterology and hepatology. 2018;5:399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pillarisetty VG, Miller G, Shah AB et al. GM-CSF expands dendritic cells and their progenitors in mouse liver. Hepatology. 2003;37:641–652. [DOI] [PubMed] [Google Scholar]
- 24.Ren G, Su J, Zhang L et al. Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem cells. 2009;27:1954–1962. [DOI] [PubMed] [Google Scholar]
- 25.Fausther M, Sheung N, Saiman Y et al. Activated hepatic stellate cells upregulate transcription of ecto-5’-nucleotidase/CD73 via specific SP1 and SMAD promoter elements. American journal of physiology Gastrointestinal and liver physiology. 2012;303:G904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.