

Abstract
Store-operated calcium entry (SOCE) modulates cytosolic calcium in multiple cells. Endoplasmic reticulum (ER) localized STIM1 and plasma membrane (PM)-localized ORAI1 are two main components of SOCE. STIM1:ORAI1 association requires STIM1 oligomerization, its re-distribution to ER-PM junctions, and puncta formation. However, little is known about the negative regulation of these steps to prevent calcium overload. Here, we identified Tmem178 as a negative modulator of STIM1 puncta formation in myeloid cells. Using site-directed mutagenesis, co-immunoprecipitation assays and FRET imaging, we determined that Tmem178:STIM1 association occurs via their transmembrane motifs. Mutants that increase Tmem178:STIM1 association reduce STIM1 puncta formation, SOCE activation, impair inflammatory cytokine production in macrophages and osteoclastogenesis. Mutants that reduce Tmem178:STIM1 association reverse these effects. Furthermore, exposure to plasma from arthritic patients decreases Tmem178 expression, enhances SOCE activation and cytoplasmic calcium. In conclusion, Tmem178 modulates the rate-limiting step of STIM1 puncta formation and therefore controls SOCE in inflammatory conditions.
Keywords: SOCE, Tmem178, STIM1, Macrophage activation, Osteoclastogenesis
1. Introduction
Cytosolic calcium is an important second messenger that modulates numerous signaling pathways and therefore participates in multiple cellular functions, such as adhesion, migration, differentiation, secretion, and others [1,2]. Dysregulated calcium signaling has been involved in autoimmune and inflammatory diseases, including rheumatoid arthritis [3–5]. Unlike other second messengers, cytosolic calcium is not synthesized by the cell, but its levels are dynamically regulated by its release from intracellular calcium storage sites, such as the endoplasmic reticulum (ER), and influxes from the extracellular calcium pool [6]. Cytosolic calcium fluxes exhibit numerous patterns, in terms of duration and amplitude [1,7], thus making this second messenger a versatile signal for activating numerous downstream events.
Store-operated calcium entry (SOCE) has been identified as one of the major mechanisms for regulation of cytosolic calcium levels. SOCE is activated upon release of calcium from the ER to refill the depleted ER calcium stores by allowing extracellular calcium entry [2]. Plasma membrane (PM) localized ORAI1 [8–11] and ER localized STIM1 [12–14] have been identified as the two main components of SOCE. In vitro experiments to induce ER calcium depletion with either stimulation of ER calcium release by activation of the PLC/IP3 cascade with ATP, or suppression of ER calcium reuptake by blockade of SERCA2 with thapsigargin (TG) demonstrated that STIM1 undergoes oligomerization and redistribution to ER-PM junctions to form puncta structures and bind to ORAI1. Formation of the STIM1:ORAI1 complex allows extracellular calcium entry and promotes ER calcium refilling [15–18]. In turn, the increased calcium concentration in the ER provides a negative feedback signal through the binding of calcium to the N-terminus of STIM1, followed by STIM1 de-oligomerization, reduced STIM1 puncta and inactivation of SOCE [19,20].
Mutations in either ORAI1 or STIM1 have been identified in humans. Loss of function or null mutations result in severe combined immunodeficiency (SCID) like disease with chronic infections, autoimmunity, muscular hypotonia and defects in tooth development [5]. Mice with complete or conditional deletion of Stiml and/or Orail in hematopoietic cells show defects in T cells [21,22], neutrophils [23] and osteoclasts [24–26], which are multinucleated cells derived from the fusion of bone marrow macrophages. In contrast, gain of function mutations in the patients or in transgenic animal models result in York platelet and Stormorken syndromes, characterized by bleeding disorders with thrombocytopenia, short stature and skeletal muscle weakness [5]. Because SOCE activation is important for multiple cellular responses, this modality of calcium entry must be tightly controlled to prevent cytotoxicity due to excessive influx of calcium [27].
In the past few years, a number of proteins have been reported to modulate SOCE via their association with STIM1 or ORAI1 [28]. The majority of the proteins that have been identified to interact with STIM1 are positive regulators of SOCE and facilitate STIM1 oligomerization, ER-PM translocation, puncta formation, and/or STIM1:ORAI1 association [29–38]. Few negative regulators of SOCE interacting with STIM1 have been described [39–41], although in the majority of the cases the mechanism remains elusive. EB1 was shown to restrict STIM1 translocation to ER-PM junctions, however, EB1 deficiency exhibited no [42] or minor [41] increase of SOCE. SARAF was shown to accelerate STIM1 de-oligomerization after ER calcium refilling to prevent calcium overload [39]. These reports suggest that negative regulators of STIM1 activation and/or localization might exist for fine-tune regulation of SOCE-mediated calcium influx.
We recently identified Tmem178 as a negative regulator of osteoclast formation in mice and humans by regulating the calcium/NFATc1 axis [43]. We also discovered that Tmem178−/− mice developed more severe cytokine storm syndrome symptoms due to hyper-activation of macrophage pro-inflammatory responses following TLR9 ligand stimulation or viral infection [44]. Initial studies in HEK293T cells overexpressing Tmem178 indicated that Tmem178 affects ER calcium release, however the effects were very mild suggesting that Tmem178 might modulate calcium levels in myeloid cells via other modalities. We now demonstrate that Tmem178 negatively regulates cytosolic calcium levels in macrophages and osteoclasts primarily by limiting STIM1 localization to ER-PM junctions and puncta formation and thus by modulating SOCE activation. Using site-directed mutagenesis, co-immunoprecipitation assays and FRET imaging, we further determined that Tmem178 associate with STIM1 via the transmembrane residues L212 and M216 in Tmem178 and G225 in STIM1. Our data provides important insights into fine-tune regulation of calcium fluxes via the Tmem178:STIM1 complex in myeloid cells during inflammatory conditions.
2. Materials and methods
2.1. Chemicals
Fura-2 AM and thapsigargin (TG) were purchased from Fisher Scientific (NH, USA). Adenosine 5’-triphosphate (ATP) and all other reagents were obtained from Sigma (MO, USA).
2.2. Cells and mice
HEK293T cells were purchased from ATCC and cultured in Dulbecco’s Modified Eagle Medium (DMEM, GIBCO, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (GIBCO, NY, USA), penicillin (100 IU/ml), streptomycin (100 μg/ml) (GIBCO, NY, USA), and 1 mM sodium pyruvate (GIBCO, NY, USA). PLAT-E cells were a kind gift from Dr. T Kitamura and cultured in the same medium as HEK293T cells. Mycoplasma contamination was tested and it was negative in these two cell lines. To generate BMMs, bone marrow cells were flushed by centrifugation from femurs and tibias of 6–8 week old C57BL/6 mice and cultured in petri dishes for 3 days with α−10 medium [α-minimum essential medium (α-MEM, Sigma, MO, USA) containing 10% heat-inactivated fetal bovine serum (GIBCO, NY, USA), penicillin (100 IU/ml) plus streptomycin (100 μg/ml) (GIBCO, NY, USA), 2 mM glutamine (Corning, NY, USA)] and 10% CMG14–12 cell conditioned medium as a source of M-CSF (100 μg/ml). For osteoclastogenesis, BMMs were plated in 96-well plates at a concentration of 5000 cells/ well and cultured in α-MEM + 10% heat-inactivated fetal bovine serum in the presence of 100 ng/ml RANKL and 1% CMG 14–12 cell conditioned medium for 5 days. Medium was changed daily. Mature osteoclasts were fixed with 4% formaldehyde (Polysciences, PA, USA) and enumerated after TRAP staining (Sigma, MO, USA). All experiments were approved by the Washington University School of Medicine animal care and use committee. Mice were housed in cages and were fed with food and water ad libitum, with a 12 h light and 12 h dark cycle. Tmem178 KO mice were described previously [43].
2.3. Single-cell Ca2+ measurements
2 × 105 WT or Tmem178−/− BMMs were plated in 29 mm glass bottom dishes. Adherent cells were incubated for 30 min in the dark with 2 μM Fura-2 diluted in hanks’ balanced salt solution (HBSS) plus 2 mM CaCl2, 1 mM MgSO4 and 10 ng/ml M-CSF (Biolegend, CA, USA). Next, cells were washed twice with Ca2+ free HBSS containing 1 mM MgSO4, and maintained in Ca2+ free HBSS buffer plus 10 ng/ml M-CSF (Biolegend, CA, USA). The stained cells were treated with 100 μM ATP, 100 μM histamine, or 1 μM TG followed by addition of 2 mM CaCl2. For the oATP pretreated experiment, the cells were treated with 100 μM oATP when incubated with Fura-2. The stained cells were then further maintained in 100 μM oATP during the measurement of calcium fluxes. An Olympus IX-71 inverted microscope with a Lamda-LS illuminator, Fura-2 (340/380) filter set, a 20 × 0.3 NA. objective lens, and a Photometrics Coolsnap HQ2 CCD camera was used to capture images at a frequency of 1 image pair every 2 s. Relative fluorescence ratio at wavelengths of 340 nM and 380 nM (F340/F380) was utilized for the assessment of cytoplasmic calcium level. The calcium fluxes were quantified by the area under the curve, as indicated. An average of 40 cells per field was recorded and analyzed.
2.4. Mutagenesis
Tmem178ΔNT (deletion of TM1 plus N-terminal loop, 618–974 bp) and Tmem178ΔCT (deletion of the C-terminal region, 81–914 bp) were amplified from full length human Tmem178 cDNA and inserted into the pMX-blasticidin retroviral vector using the BamH1/Xho1 restriction sites. All Tmem178 mutants were constructed by overlapping PCR and contain the HA-tag in their C-terminal end.
STIM1ΔNT (deletion of N terminal region, 1208–2944 bp) and STIM1ΔCT (deletion of C-terminal region of STIM1, 569–1270 bp) were amplified and ligated into the pMX-blasticidin retroviral vector via BamH1 and Xho1 restriction sites. STIM1ΔNT&SOAR (deletion of the N-terminal region and SOAR region, 1208–1591 bp + 1913–2944 bp)was constructed via overlapping PCR. STIM1 ACT contains the myc-tag after the signal peptide and all other STIM1 mutants contain the myc-tag in their C-terminal end.
2.5. Lentivirus and retrovirus generation and cell transduction
HEK293T cells were plated in 6 cm dishes and transfected with 1 μg shRNA targeting Stim1 (targeting sequence CCCTTCCTTTCTTTGCAA TAT in the 3UTR region) or with shRNA Ctrl (targeting a scramble sequence) using the puromycin-resistant PLKo.1 lentiviral vector, in the presence of 1 ng packaging plasmid (Delta 8.2), 0.125 μg envelope plasmid (VSVg) and polyjet transfection reagent (Signagen, MD, USA). 24 h later, medium was replaced and cells were incubated for additional 24 h. BMMs were infected by adding 50% lentiviral supernatant diluted in α−10 medium containing 10% CMG 14–12 and 8 μg/ml polybrene for 24 h. Infected cells were selected for 24 h in medium containing 2 μg/ ml puromycin.
PLAT-E cells were plated in 6 cm dishes and transfected with 2 μg of Tmem178 or STIM1 constructs in pMX-blasticidin retroviral vectors to generate retrovirus used to infect BMMs. Medium was replaced 24 h later and the supernatant containing the retrovirus was collected after an additional incubation of 24 h. BMMs were infected with 50% supernatant containing indicated retrovirus in α−10 medium containing 10% CMG 14–12 as source of M-CSF and 4 ng/ml polybrene for 24 h. Infected cells were then selected with 1 ng/ml blasticidin for 48 h before being used for indicated experiments.
HEK293T cells were cultured in 6 cm dishes and transfected with 2 μg of full length STIM1 or STIM1 mutants and 3 μg full length Tmem178 or Tmem178 mutants using polyjet transfection reagent (Signagen, MD, USA) and cultured for 36–48 h prior to be used for indicated experiments.
2.6. Co-immunoprecipitation and Western blot
HEK293T cells were lysed in TNE buffer [10 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40 and 10% (vol/vol) glycerol]. For IP experiments, 400 μg protein lysates were incubated with 1 μg anti-HA antibody (Biolegend, 901501, CA, USA) or 1 ng anti-Myc antibody (Santa Cruz, sc-40, CA, USA) overnight at 4 °C. 10 μl protein A/G beads (Thermo Fisher Scientific, NJ, USA) were then added for 3 h at 4 °C, followed by centrifugation. Precipitated beads were lysed with 1 × SDS loading buffer and subjected to Western blot by using specific antibodies for HA (Cell signal Technology, 3724, 1:1000, MA, USA) and Myc (Biolegend, 906301, 1:1000, CA, USA).
For NFATc1 Western blot, BMMs cultured with M-CSF and RANKL for 3 days to generate pre-OCs were lysed in TNE buffer and 40 ng lysates were utilized for Western blot analysis using the following antibodies for NFATc1 (Santa Cruz, sc-7294, 1:500, CA, USA) and actin (Sigma, A5441, 1:5000, MO, USA).
2.7. Immunofluorescence
HEK293T cells (5 × 104) expressing indicated STIM1 or Tmem178 mutants were plated on glass coverslips pre-coated with poly-lysine, fixed in 4% formaldehyde for 20 min and permeabilized with 0.1% Triton-X-100 in phosphate-buffered saline (PBS) for 6 min. Cells were then washed with PBS twice and blocked in 0.2% BSA at room temperature (RT) for 30 min prior to overnight incubation at 4 °C with the anti-HA antibody (Cell signal Technology, 3724, 1:1600, MA, USA), anti-Myc antibody (Biolegend, 906301, 1:1000, CA, USA), or the anti calnexin antibody (Santa Cruz, Sc-6465, 1:25, CA, USA). Cells were gently washed and secondary antibodies (Thermo Fisher Scientific, A11058 and A21206, 1:1000, NJ, USA) were added at RT for 1 h. Coverslips were mounted using VECTASHIELD anti-fade mounting medium with DAPI (Vector Laboratories, H-1200, Burlingame, CA, USA). Fluorescent signals were captured by using a Nikon Eclipse 80i microscope and a Nikon DS-Qi1MC camera (Nikon, CO, USA).
2.8. Confocal microscopy & FRET imaging
Confocal microscopy, including the imaging required for FRET, was performed on an inverted Nikon A1Rsi laser scanning confocal microscope using a 40 × 1.4 NA oil-immersion objective lens (Nikon Instruments Inc., NY, USA). 445 and 514 nm lasers were used for CFP and YFP excitation respectively, with only the 445 nm laser being used for FRET event detection. CFP and YFP fluorescent signals were collected individually by two separate gallium arsenide phosphide photomultiplier tubes (GaAsP PMTs) using bandpass filters, 465–505 nm for CFP and 518–558 nm for YFP. Throughout the data acquisition process, samples were maintained at 37 °C with 5% CO2, controlled by a Tokai Hit stage-top incubation system (Shizuoka, Japan). The Nikon PerfectFocus system was engaged full-time throughout the imaging so as to correct for any real-time fluctuations in z-axis focal position. Acquisition was performed using Nikon NIS-Elements software (Nikon Instruments Inc., NY, USA.)
For fluorescence resonance energy transfer (FRET) measurements, CFP-Tmem178 and STIM1-YFP, or ORAI1-CFP and STIM1-YFP constructs were co-expressed in HEK293T cells. 29 mm glass bottom dishes (Cellvis, D29–14-1.5-N, CA, USA) pre-coated with poly-L-lysine were utilized for both cell culture and construct transduction. 36 h after transfection, cells were washed twice with 1 mM MgSO4 in HBSS, followed by a 1 h balancing process in HBSS containing 1 mM MgSO4 and 1mM CaCl2. After the balancing, cells were washed twice with HBSS containing 1 mM MgSO4 and were taken for imaging. 1 μM TG or 100 μM ATP were added to the media for 1 min after data acquisition began. Time-lapse images were captured every 15 s for a 5 min period. The final datasets were analyzed using the FRET module in the Nikon NIS-Elements software, and included the bleed-through correction based on a previously published method [45]. The average FRET efficiency was calculated from the corrected FRET value obtained from inside a given region of interest (ROI), and used for statistical analysis. In particular, four channels were set for the FRET analysis, including Dd, Da, Ad and Aa. Dd is the channel in which the wavelength of excitation and emission are both for CFP. Da is the channel in which the wavelength of excitation is for CFP while the wavelength of emission is for YFP. Ad is the channel in which the wavelength of excitation is for YFP while the wavelength of emission is for CFP. Aa is the channel in which the wavelength of excitation and emission are both for YFP. FRET efficiency was calculated using the following formula: FRET efficiency = (Da-Dd*Da/Dd-Aa*Da/Aa)/Dd*100, and presented as percentage [FRET efficiency (%)]. Data are shown as mean ± s. e.m.
2.9. STIM1 puncta analysis
To analyze the spatial organization of STIM1-YFP, images were first collected following the same data acquisition settings for FRET imaging and were subsequently processed through an intensity-based segmentation methodology for selecting regions containing STIM1-YFP positive signal. Measurements of puncta, including number of puncta, size (as measured in μm2), and integrated fluorescence intensity, were calculated. All analyses were performed in Nikon NIS-Elements software (Nikon, NY, USA).
2.10. Molecular modeling and docking
STIM1 and Tmem178 molecular modeling was performed using the software FR-t5-M [46] and I-TASSER [47]. Based on Co-IP results and the software SPR [48] for the prediction of protein-protein interfaces, STIM1ACT and full length Tmem178 were docked into a complex by the software ZDock v3.0.2 [49]. The model with the highest confidence among the top ten was selected by using METop program [46]. The selected Tmem178-STIM1 model was further adjusted and exhibited bythe software PyMol.
2.11. Semi-quantitative RT-PCR
WT or Tmem178−/− BMMs expressing indicated constructs were plated in 24-well plates and stimulated with 100 ng/ml LPS for 4 h. The stimulated cells were lysed in TRIZOL (Invitrogen, CA, USA) and total RNA was isolated and processed for cDNA by using the high capacity cDNA Reverse Transcription Kit (Applied Biosystems, CA, USA). Realtime PCR was performed with SYBR Green PCR Master Mix (Applied Biosystems, UK) in 7300 Real Time PCR System (Thermo Fisher Scientific, MA, USA). Rel mRNA expr was calculated by 2[ − (Ct target gene−ct Gapdh], where Ct represents the threshold cycle for each transcript, and Gapdh is the reference. The sequences of the specific primers are:
Il-6, Forward: TTCTCTGGGAAATCGTGGAAA.
Reverse: TGCAAGTGCATCATCGTTGTT.
Il-1β, Forward: GCTTCCTTGTGCAAGTGTCTGA.
Reverse: TCAAAAGGTGGCATTTCACAGT.
Tnfα, Forward: CTGTAGCCCACGTCGTAGC.
Reverse: TTGAGATCCATGCCGTTG.
Stim1, Forward: GGCGTGGAAGTCATCAGAAGT.
Reverse: TCAGTACAGTCCCTGTCATGG.
Orai1 , Forward: GATCGGCCAGAGTTACTCCG.
Reverse: TGGGTAGTCATGGTCTGTGTC.
Tmem178, Forward: ATGACAGGGATATTTTGCACCAT.
Reverse: CCGGTTCAAGTCATAGGAGACACT.
Gapdh, Forward: ACCCAGAAGACTGTGGATGG.
Reverse: TTCAGCTCAGGGATGACCTT.
2.12. LPS-induced sepsis
6–8 weeks old WT and Tmem178−/− C57BL/6 male mice (WT n = 12; Tmem178−/− n = 17) were injected with 25 mg/kg Lipopolysaccharides (LPS) from Escherichia coli 0111:B4 (Sigma). Blood was collected by submandibular bleed 2 h after LPS administration, and serum was harvested by centrifugation at 8000g for 5 min at 4° to analyze TNFa levels by ELISA (BD Biosciences).
2.13. Statistical analysis
Data are represented as mean ± SD or mean ± SEM as indicated. Statistical significance was analyzed by two-tailed one type Student’s t-test or in experiment with multiple comparisons by the one-way or two-way ANOVA followed by Bonferroni post-tests, as indicated. *P < 0.05, **P < 0.01, ***P < 0.001.
3. Results
3.1. Tmem178 negatively regulates SOCE
We recently reported that loss of Tmem178 leads to higher levels of basal intracellular calcium during osteoclastogenesis [43]. Due to its confined expression in bone marrow monocytes/macrophages (BMMs) and osteoclasts, but not T cells [44], we hypothesized that Tmem178 is a specific modulator of intracellular calcium in myeloid cells. To determine how Tmem178 modulates calcium levels in the primary cells, we stimulated WT and Tmem178 deficient BMMs and OC precursors (pre-OCs) with 100 nM ATP or 1 μM thapsigargin (TG), to induce ER calcium depletion, followed by the addition of 2 mM extracellular calcium, to activate SOCE. Remarkably, we observed that calcium levels are significantly higher in Tmem178−/− BMMs compared with WT cells under all conditions (Fig. 1A–D). Similar results were observed in WT and Tmem178−/− pre-OCs (Fig. 1E and F). Quantitative analysis of the area under the curve starting at the time of stimulation with TG and ATP or 2 mM calcium reveals higher differences in SOCE activation rather than ER calcium release between the WT and Tmem178−/− BMMs (Fig. 1B and D) or pre-OCs (Fig. 1F).
Fig. 1.
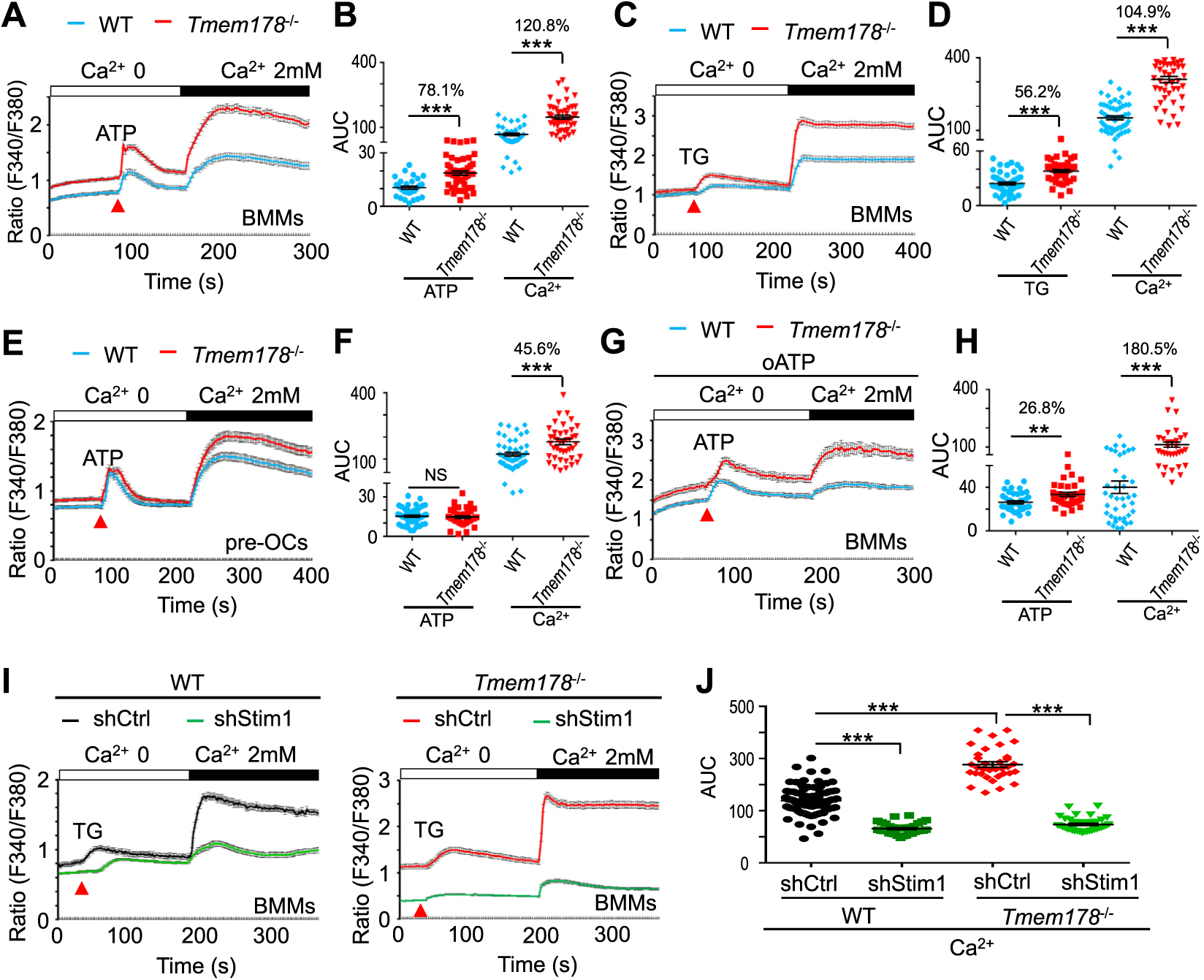
Tmem178 is a negative regulator of SOCE in macrophages and osteoclasts. (A–D) Calcium traces (A, C) and the quantification of the area under the curve (AUC) (B, D) in WT BMMs (n=38) and Tmem178−/− BMMs (n=51) treated with ATP (100 μM) or stimulated with TG (1 μM) followed by addition of 2mM calcium (WT, n=55; Tmem178−/−, n=48). Data shown are representative of 4–6 independent experiments. (E, F) Calcium fluxes and AUC in WT (n=43) and Tmem178−/− (n=52) pre-OCs stimulated with ATP and 2mM calcium. (G, H) Calcium fluxes and AUC in WT (n=44) and Tmem178−/− (n=37) pre-OCs pretreated with 100mM oATP and stimulated with ATP and 2mM calcium. (I, J) Calcium fluxes and AUC following stimulation with TG and 2mM calcium in WT BMMs (Left) or Tmem178−/− BMMs (Right) infected with shRNA control (WT, n=84; Tmem178−/−, n=45) or shRNA Stim1 (WT, n=37; Tmem178−/−, n=74). Data shown are representative of four independent experiments and indicated number of cells is from an individual experiment. Data are presented as mean ± SEM. NS, not significant; **P < 0.01, ***P < 0.001.
Because ATP induces SOCE via activation of P2Y receptors, but can also modulate calcium levels independent of SOCE through the ionotropic P2X receptors, we monitored calcium fluxes in cells pretreated with oxidized ATP (oATP), an antagonist of P2X receptors. Tmem178−/− BMMs still exhibit significantly higher calcium levels than WT cells (Fig. 1G and H), suggesting that Tmem178 mainly modulates ATP/P2Y mediated SOCE activation.
The effects of Tmem178 on SOCE activation are not due to differences in Stim1 and Orai1 mRNA levels between WT and Tmem178−/− cells (Supplemental Fig. 1A). Moreover, knock-down of Stim1 by sh-RNA (Supplemental Fig. 1B) strongly reduces intracellular calcium levels in both WT and Tmem178−/− BMMs compared to the shRNA scramble control (Ctrl) groups (Fig. 1I–J). These results indicate that Tmem178 modulates calcium fluxes via SOCE activation in primary macrophages and pre-OCs.
3.2. Tmem178/STIM1 association is dependent on ER-calcium
We previously reported that ectopic expression of Tmem178 in HEK293T cells reduced ER calcium release but did not alter TG-induced SOCE compared to empty vector (EV) expressing cells [43]. However, at that time we only compared the peak calcium levels reached after stimulation with extracellular calcium and did not measure calcium fluxes overtime. To better evaluate the effects of ectopic expression of Tmem178 on SOCE activation, we measured ATP- and TG-induced SOCE in HEK293T expressing full length Tmem178 or in EV controls. Interestingly, while ER-calcium release is not negatively impacted by the expression of Tmem178, ATP-induced SOCE is significantly reduced in Tmem178 expressing cells (Fig. 2A and B). Under these conditions, co-IP experiments show that Tmem178 is associated with STIM1 (Fig. 2C, quantification in Supplemental Fig. 2A). Importantly, the formation of the STIM1/ORAI1 complex in response to ATP stimulation is reduced in cells expressing Tmem178 compared to EV controls (Fig. 2C; quantification in Supplemental Figs. 2A and 2B). In contrast, minor differences in calcium fluxes are observed between HEK293 expressing Tmem178 or EV following TG stimulation (Supplemental Figs. 2C and 2D). This result is supported by the observation that the binding of Tmem178 to STIM1 is reduced following TG treatment and the STIM1/ORAI1 complex is forming regardless of Tmem178 expression (Fig. 2C and quantification in Supplemental Figs. 2A and 2B).
Fig. 2.
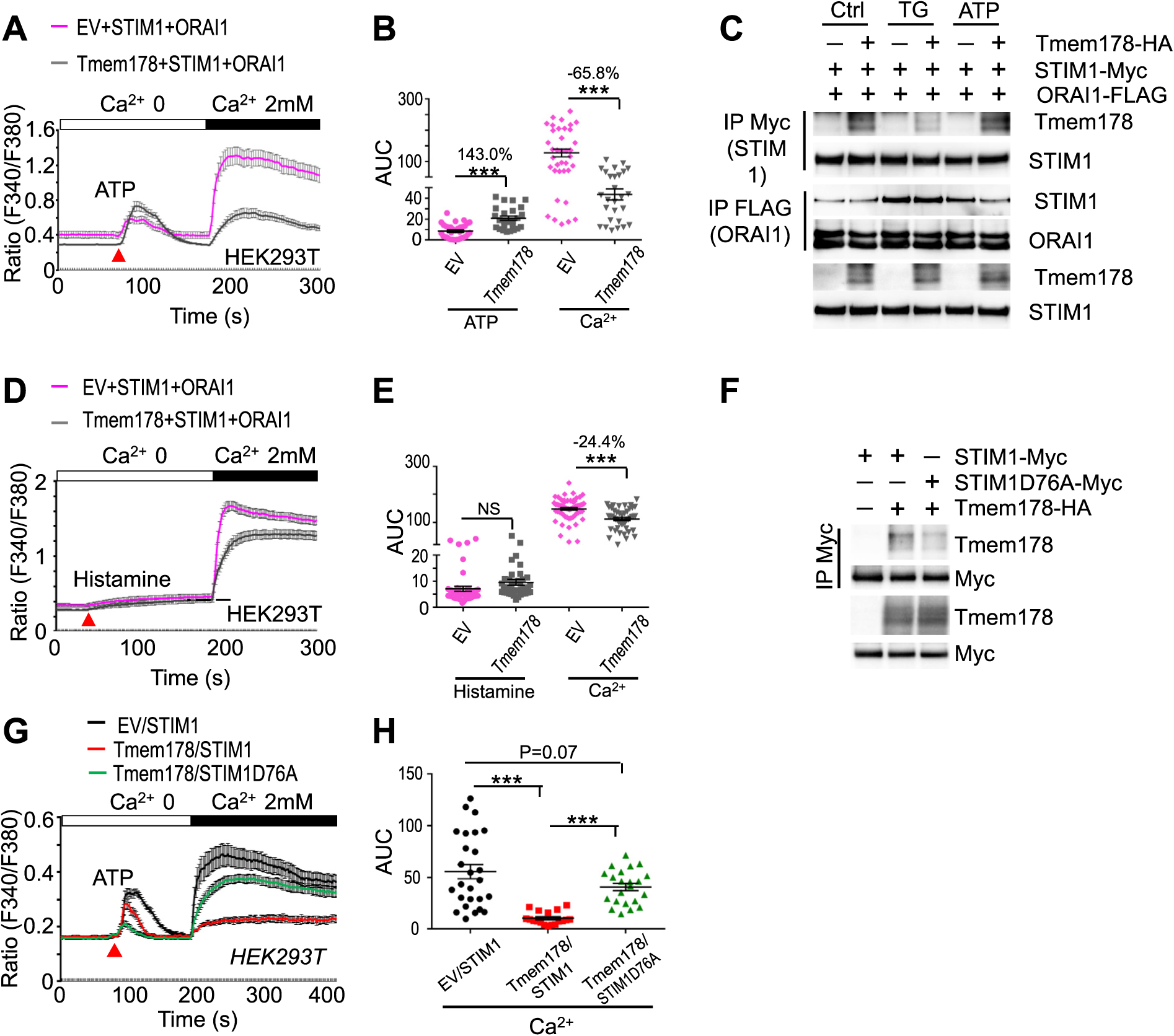
Tmem178: STIM1 association is dependent on ER-calcium content. (A, B) Calcium fluxes and the area under the curve (AUC) in HEK 293 T cells transfected with STIM1, ORAI1 and Tmem178 (n = 28) or empty vector (EV) (n = 37) and treated with ATP and 2 mM calcium. Data shown are representative of 5 independent experiments. (C) HEK293 cells expressing Tmem178-HA, STIM1-Myc and ORAI1-FLAG cultured in calcium-free medium and stimulated with 1 μMTG or 100 μM ATP for 5 min. Cell lysates were subjected to Co-IP with anti-Myc or anti-FLAG antibodies followed by western blotting to detect the association between STIM1 and Tmem178, or STIM1 and ORAI1. STIM1 and Tmem178 levels in total cell lysates are shown at the bottom. (D, E) Calcium fluxes and AUC following stimulation with histamine (100 μM) and addition of 2mM calcium in HEK 293 T cells transfected with STIM1, ORAI1 and Tmem178 (n = 47) or EV (n = 62). (F) Co-IPs showing association between the HA-tagged Tmem178 and STIM1D76A-myc in HEK293T cells. (G, H) ATP-induced calcium fluxes followed by addition of 2 mM extracellular calcium in HEK 293 T cells transfected with EV (n = 27) or full-length Tmem178 (n = 19), together with STIM1 and ORAI1; or full-length Tmem178 (n = 22), together with STIM1D76A and ORAI1. Calcium traces from indicated number of cells are from a represenative experiment. Data are presented as mean ± SEM. NS, not significant; ***P < 0.001.
Since ATP induces a transient ER calcium response compared to the irreversible effects of TG, we wondered whether the negative regulation of SOCE in HEK293T cells ectopically expressing Tmem178 might be dependent on the amplitude of ER calcium release. To address this possibility, we stimulated the cells with ionomycin, which empties the ER calcium stores, or with histamine, which only induces a partial ER calcium release, followed by the addition of extracellular calcium to activate SOCE. Confirming our hypothesis, ectopic expression of Tmem178 reduces histamine-induced SOCE (Fig. 2D and E) but is not sufficient to affect ionomycin-induced SOCE (Supplemental Figs. 2E and 2F).
To further test whether binding of STIM1 to ER calcium is required for the Tmem178: STIM1 association, we performed co-IP experiments in HEK293T cells expressing wildtype Tmem178 and the STIM1D76A mutant which lacks the calcium binding site. We observed that Tmem178 does not bind to STIM1D76A in basal conditions (Fig. 2F) and expression of Tmem178 does not decrease SOCE activation in cells expressing STIM1D76A compared to wildtype STIM1 (Fig. 2G and H). All together these results indicate that the ability of Tmem178 to negatively regulate SOCE is dependent on the ER-calcium content.
3.3. The N- and C-terminal regions of STIM1 are not required for the binding to Tmem178
Next we wanted to identify the domains involved in the Tmem178:STIM1 interaction. STIM1 is a single transmembrane protein located in the ER with the N-terminal domain facing the ER lumen and the C-terminal domain towards the cytosol [2]. First, we generated Myc-tagged STIM1ΔNT (deletion of the N terminal region) and STIM1ΔCT (deletion of the C-terminal region) mutants (schemes in Fig. 3A and Supplemental Fig. 3B), and assessed their association with HA-tagged Tmem178 by Co-IP in HEK293T cells in basal conditions. Interestingly, STIM1ΔNT hardly interacts with Tmem178 while STIM1 ACT strongly binds to Tmem178 compared to full length STIM1 (Fig. 3B, quantified in Supplemental Fig. 3A; Supplemental Fig. 3B). Previous studies demonstrated that STIM1 binds to ORAI1 via its C-terminal domain [50–52] (red box in Fig. 3A). To determine whether the reduced interaction between STIM1ΔNT and Tmem178 is due to the constitutive binding of STIM1ΔNT to ORAI1, we generated a STIM1ANT mutant that lacks the SOAR region, the domain of STIM1 required for binding to ORAI1 [50,53,54] (red box in Fig. 3A). Co-IP assay confirmed these previous findings (Supplemental Fig. 3C). Interestingly, we observed that the association between STIM1ANT&SOAR and Tmem178 is recovered (Fig. 3B, lane 4 and quantified in Supplemental Fig. 3A). Importantly, all of the STIM1 mutants are localized in the ER (Supplemental Fig. 4A). These results suggest that 1) the N- and C-terminal regions of STIM1 are not involved in the binding to Tmem178 and 2) Tmem178 and ORAI1 compete for binding to STIM1.
Fig. 3.
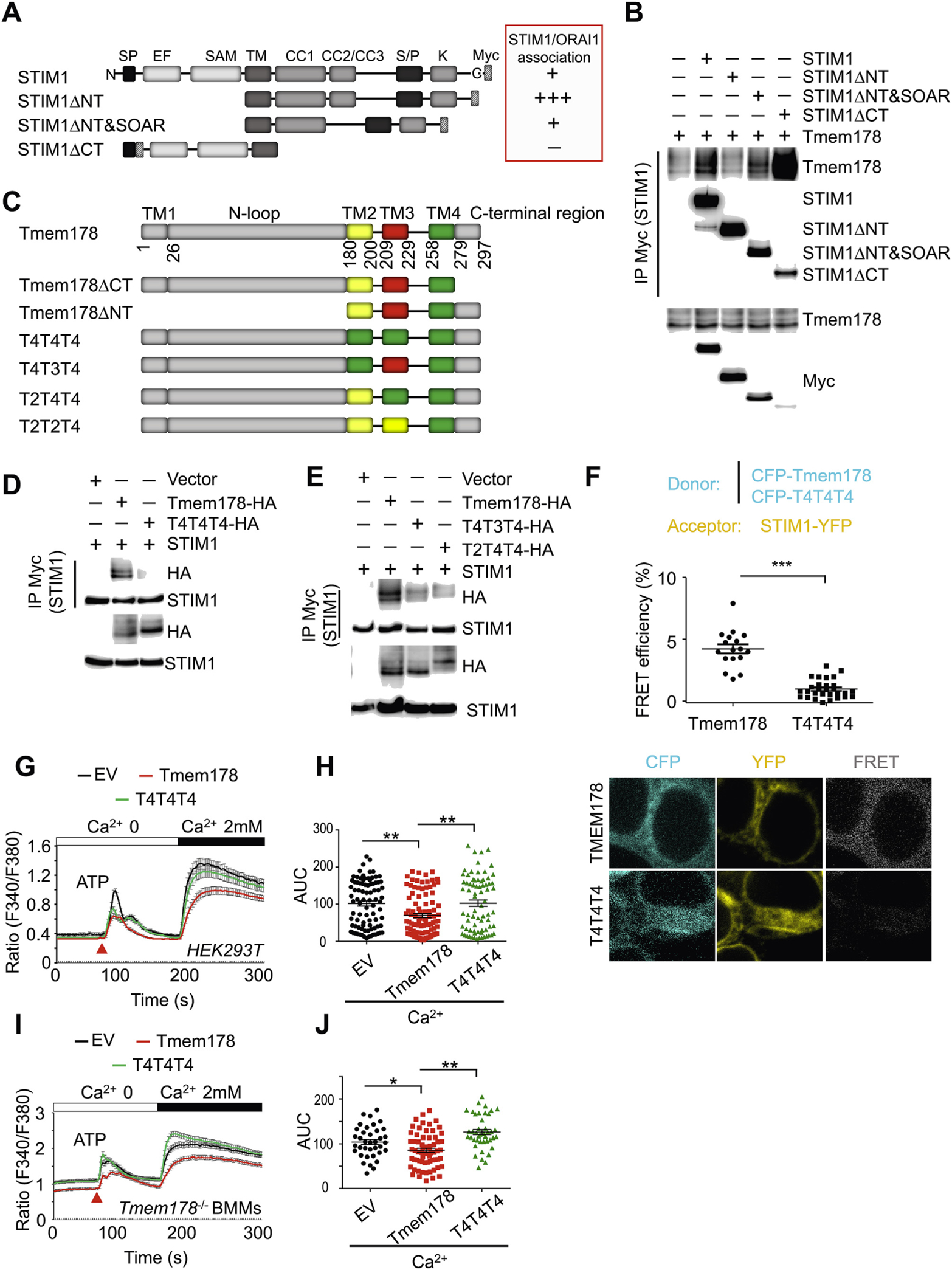
STIM1 and Tmem178 associate via their transmembrane regions. (A) Schematic diagram of indicated STIM1 mutants. Based on published data, the binding of each STIM1 mutant with ORAI1 is shown in the red box as + + +, +, and − (from the strongest to the weakest). (B) Co-IP and Western blot showing association between Tmem178-HA and indicated Myc-tagged STIM1 mutants in HEK293T cells. (C) Schematic diagram ofTmem178 mutants. (D, E) Co-IPs followed by Western blot showing association between the HA-tagged Tmem178 mutants and STIM1-Myc in HEK293T cells. (F) Quantification of % FRET efficiency (top) and representative images (bottom) between STIM1-YFP and CFP- Tmem178 (n = 17), or STIM1-YFP and CFP-T4T4T4 (n = 29) in HEK293T cells. One of 3 independent experiments is shown. (G, H) Calcium traces and AUC in HEK 293 T cells expressing STIM1, ORAI1 and EV (n = 88), full-length Tmem178 (n = 98) or Tmem178- T4T4T4 mutant (n = 82) stimulated with ATP and 2 mM calcium. (I, J) Calcium traces and AUC in Tmem178−/− BMMs infected with EV (n = 37), full-length Tmem178 (n = 70) or Tmem178-T4T4T4 mutant (n = 42) stimulated with ATP and 2mM calcium. Calcium traces from indicated number of cells are from a representative experiment. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.3. Tmem178 transmembrane regions 2 and 3 are required for the binding with STIM1
Based on the above findings, the transmembrane (TM) region of STIM1 is the most likely region mediating the association with Tmem178. In fact, we confirmed that neither the C- nor the N-terminal region of Tmem178, which encompasses the first TM motif, are required for the binding with full length STIM1. Since deletion of the sole TM domain of STIM1 will affect the protein cellular localization, and Tmem178 is predicted to be a four transmembrane protein, as schematically shown in Fig. 3C, we turned our focus on which TM domain of Tmem178 is required for binding to STIM1. In order to avoid possible conformational changes by deleting any of the TM motifs of Tmem178, we generated TM swapping domain mutants. Compared to TM4, the TM2 and TM3 regions of Tmem178 share some similarities. Hence, we generated the following four mutants (scheme in Fig. 3C): T4T4T4 (replacement of TM2 and TM3 with TM4), T4T3T4 (replacement of TM2 with TM4), and T2T4T4 (replacement of TM3 with TM4). We confirmed ER localization of all the swapping mutants, similar to wild type Tmem178 (Supplemental Fig. 4B). Intriguingly, the T4T4T4 mutant exhibits reduced binding capacity to STIM1 (Fig. 3D; quantified in Supplemental Fig. 4C). Although to a less extent, reduced binding to STIM1 is also observed in the T3T4T3 and T2T4T4 mutants (Fig. 3E; quantified in Supplemental Fig. 4D).
To further confirm these findings, we used FRET microscopy. First we expressed STIM1-YFP together with CFP-Tmem178 or Tmem178-CFP in HEK293T cells and confirmed FRET signal only in cells expressing the CFP-tagged Tmem178 at the N-terminus (Fig. 3F top and not shown). Nex, we tested the FRET signal between STIM1-YFP and CFP-T4T4T4 mutant and observed that the FRET efficiency, defined as the proportion of the donor molecules that have transferred excitation state energy to the acceptor molecules, is largely reduced (Fig. 3F bottom).
To further determine the biological significance of these results, we analyzed calcium levels following ATP-induced SOCE activation in HEK293T cells expressing wild type Tmem178, the T4T4T4 mutant or EV as control. Cells expressing the T4T4T4 mutant or EV show increased calcium levels compared to cells expressing wild type Tmem178 (Fig. 3G and H). Similar results were observed in Tmem178−/− BMMs expressing the T4T4T4 mutant versus wild type Tmem178 (Fig. 3I and J). All together, these results suggest that the TM2 and the TM3 regions of Tmem178 are required for binding to STIM1 and that the association between Tmem178 and STIM1 negatively regulates SOCE.
3.5. STIM1 G225 modulates the interaction with L212 and M216 in Tmem178
To identify the specific amino acids mediating the association between Tmem178 and STIM1, we performed molecular modeling and docking of STIM1 and Tmem178. Based on the strong association from the co-IP results in Fig. 3B, we utilized STIM1ΔCT and full length Tmem178 for the docking study. Several amino acids are predicted to modulate the association between these two proteins, with six amino acids (F214, V218, V221, G225, W228 and I232) in the TM region of STIM1 and four (V209, L212, M216 and F220) in the TM3 region of Tmem178 (Fig. 4A). Based on the docking analysis, we used alanine scanning mutagenesis to generate six STIM1ΔCT mutants, in which each predicted amino acid was mutated to alanine, and co-expressed each mutant with full length Tmem178 in HEK293T cells. All of the STIM1ΔCT mutants except STIM1ΔCTG225A retain binding to Tmem178. However, contrary to our expectation, STIM1CTG225A shows stronger binding to Tmem178 than STIM1ACT (Fig. 4B; quantified in Supplemental Fig. 5A). We confirmed STIM1ΔG225A mutant (Fig. 4C; quantified in Supplemental Fig. 5B). One possible reason for this finding is that the mutant G225A contains a relatively long side chain, which might facilitate the interaction between the two TM regions (Supplemental Fig. 5C). To test this possibility, we used the tryptophan scanning approach since the tryptophan contains a very large natural side chain that could maximize the probability for docking affinity between two proteins [55]. Surprisingly, however, we repeatedly found that the STIM1G225W mutant has reduced association with Tmem178 (Fig. 4D, lane 4 and quantified in Supplemental Fig. 5D).
Fig. 4.
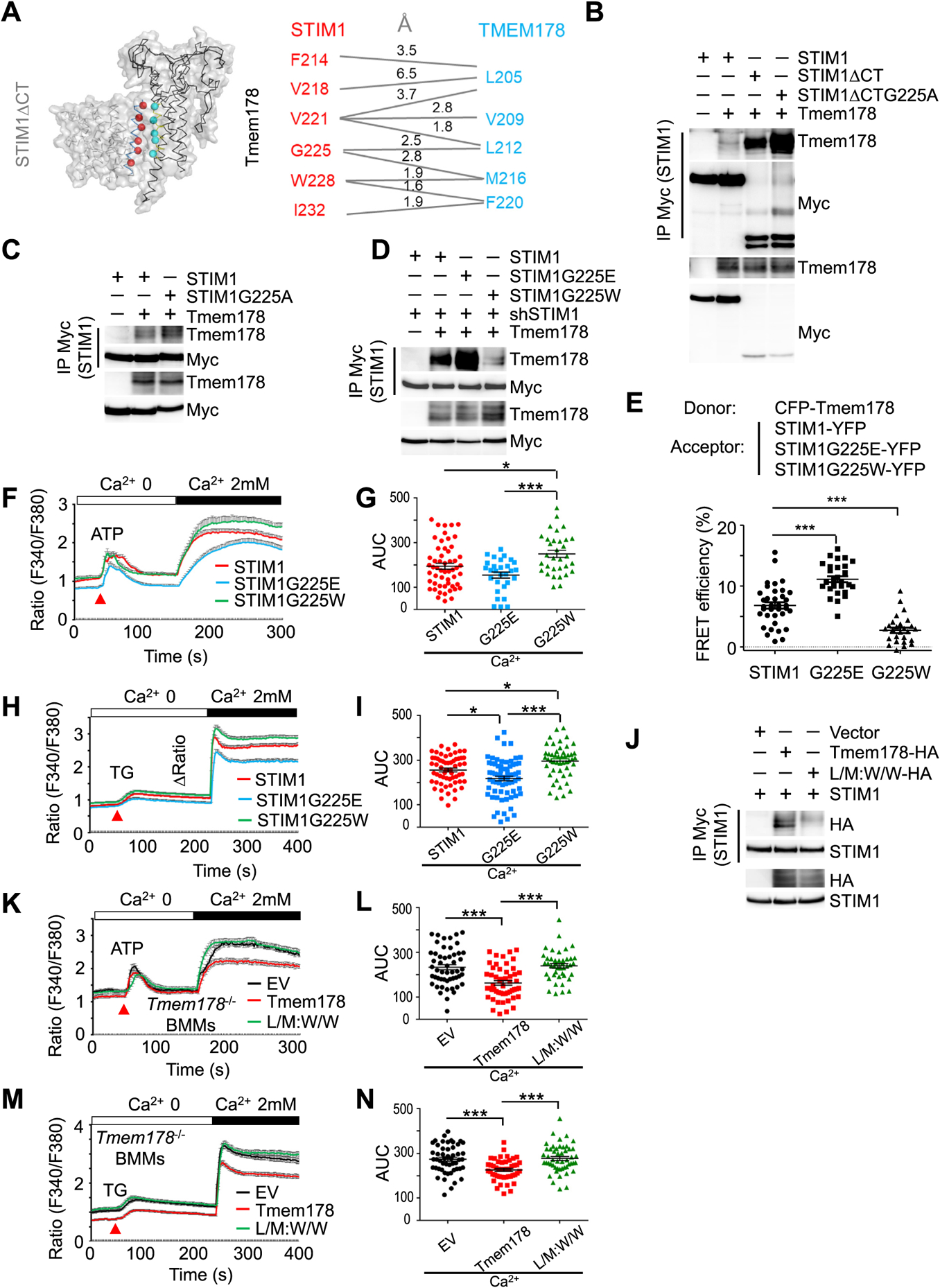
STIM1 G225 modulates the interaction with L212 and M216 in Tmem178. (A) Molecular modeling and docking of STIM1ACT and full length Tmem178 with predicted amino acids involved in the association. (B-D) Co-IP and Western blot showing association between Myc-tagged STIM1 or indicated mutants and Tmem178-HA in HEK293T cells. (E) Quantification of % of FRET efficiency signals between CFP-Tmem178 and STIM1-YFP (n = 17), STIM1G225E -YFP (n = 12), or STIM1G225W- YFP (n = 12) in HEK293T cells. (F, G) ATP-induced calcium fluxes in shRNA-Stim1 infected BMMs expressing either wild type STIM1 (n = 54), STIM1G225E (n = 29) or STIM1G225W (n = 32). (H, I) TG-induced calcium fluxes in shRNA-Stim1 infected BMMs expressing either wild type STIM1 (n = 62), STIM1G225E (n = 70) or STIM1G225W (n = 49). (J) Co-IP and Western blot showing association between Myc-tagged STIM1 and HA-tagged Tmem178 or the Tmem178 double mutant L212W&M216W. (M, N) Calcium fluxes in ATP- or TG-stimulated Tmem178−/− BMMs infected with EV, Tmem178, or Tmem178 L/M:W/ W mutant. An average of 50 cells per condition were analyzed. Data are shown as mean ± SEM. One way ANOVA was used in (E). *P < 0.05, ***P < 0.001. Data shown are representative of 3–4 independent experiments. Calcium traces from indicated number of cells are from a representative experiment.
The hydrophile scanning approach has been reported to provide additional insights on interactions identified by the alanine scanning approach [56]. Therefore, we generated STIM1G225E (Supplemental Fig. 5C) and found increased association with Tmem178 (Fig. 4D, lane 3 and quantified in Supplemental Fig. 5D). We further confirmed these findings by FRET imaging. We observed increased FRET efficiency between CFP-Tmem178 and STIM1G225E-YFP compared to STIM1-YFP, and reduced signal with STIM1G225W-YFP (Fig. 4E). Importantly, we confirmed the ER localization of STIM1G225E and STIM1G225W, and their equal ability to bind to ORAI1 (Supplemental Figs. 5E and 5F).
Next, we measured calcium fluxes in Stim1 deficient BMMs (Supplemental Fig. 5G) expressing wild type STIM1, STIM1G225E or STIM1G225W. Quantification of the area under the curve of the calcium traces following ATP-induced SOCE shows lower calcium levels in cells expressing the STIM1G225E mutant, which exhibits strong association with Tmem178, and higher calcium in cells expressing the STIM1G225W mutant, which weakly binds to Tmem178 (Fig. 4F and G). Similar results are observed following TG-induced SOCE activation (Fig. 4H and I).
The docking analysis also predicted that the G225 site in the TM domain of STIM1 associates with the L212 and M216 sites in the TM3 region of Tmem178. Therefore, we hypothesized that mutations in Tmem178 L212 and M216 could affect the association with STIM1 and consequently calcium levels. The Tmem178-L212W&M216W double mutant (L/M:W/W) is localized normally as shown in Supplemental Fig. 5H, but the association between L/M:W/W and STIM1 is drastically reduced when compared to wild type Tmem178 (Fig. 4J, quantified in Supplemental Fig. 5I). As a result, intracellular calcium levels following ATP-induced SOCE (Fig. 4K and L) and TG-induced SOCE (Fig. 4M and 4N) are higher in the Tmem178−/− BMMs expressing the Tmem178 L/ M:W/W mutant compared to wild type Tmem178. Taken together, these results indicate that the G225 residue in the TM region of STIM1 and L212 and M216 in the TM3 region of Tmem178 modulate the association between STIM1 and Tmem178 and regulate the activation of SOCE.
3.6. Tmem178 limits STIM1 puncta formation
To associate with ORAI1 and activate SOCE, STIM1 must undergo oligomerization and re-distribution to ER-PM junctions through formation of puncta structures [57]. To determine whether Tmem178 limits STIM1 puncta formation, first we analyzed TG-treated WT and Tmem178−/− BMMs expressing STIM1-YFP. Confocal microscopy shows that both TG-treated WT and Tmem178−/− cells form puncta structures compared to untreated cells (Fig. 5A). Tmem178−/− BMMs have increased numbers of puncta per μm2 compared to WT (Fig. 5B). Moreover, each punctum exhibits significantly higher intensity (Fig. 5C) and larger size (Fig. 5D).
Fig. 5.
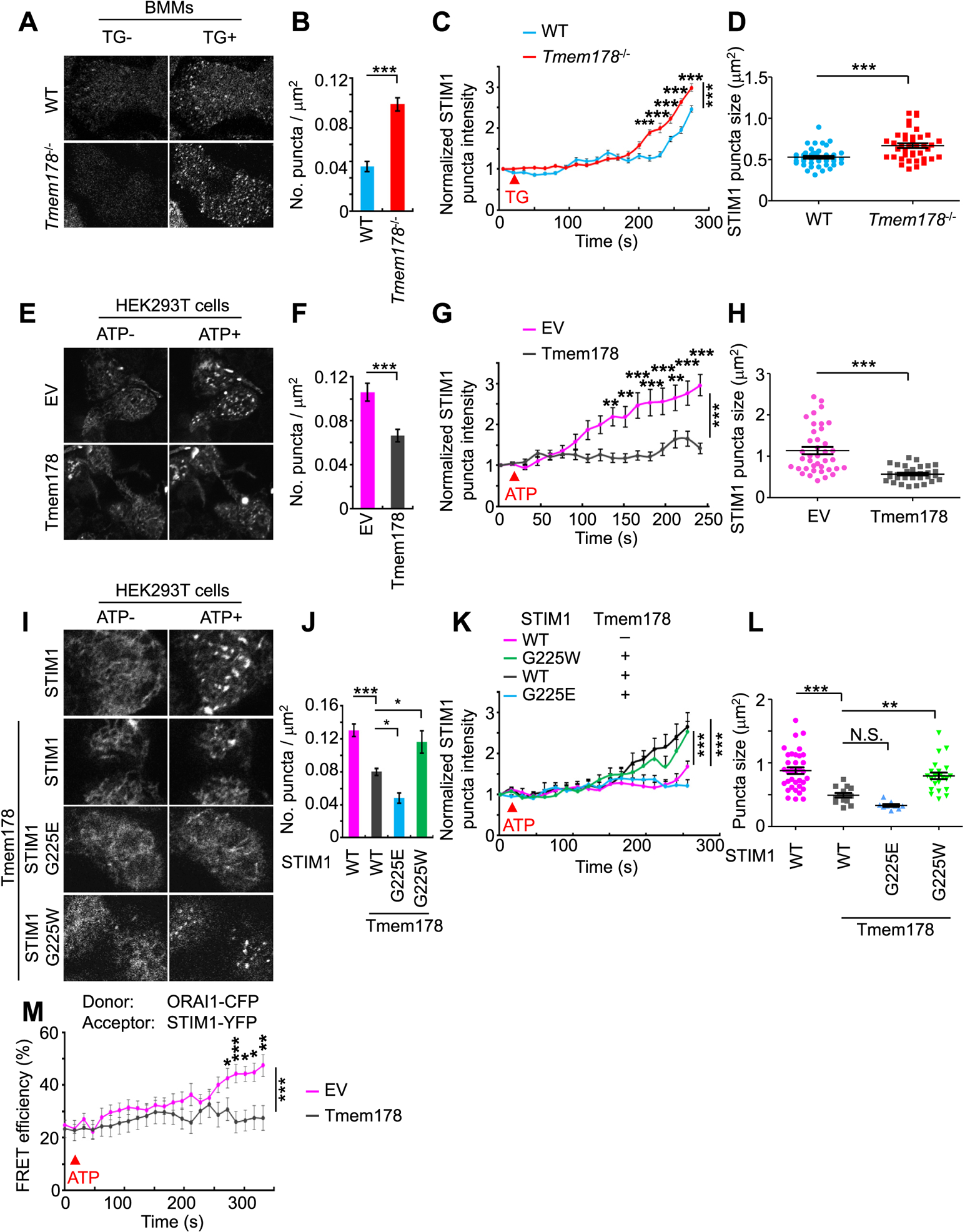
Tmem178 binding to STIM1 limits STIM1 puncta formation and the association with ORAI1. (A) Representative images of STIM1 puncta in WT (Top, n=43) and Tmem178−/− BMMs (Bottom, n=42) transduced with STIM1-YFP. Images were acquired before and after TG stimulation. (B–D) Quantification of the number of puncta per μm2, puncta intensity (normalized to baseline signal intensity) and puncta size. (E) Representative images of STIM1 puncta formation in HEK293T cells co-transfected with STIM1-YFP and empty vector EV (Top, n=39) or Tmem178 (Bottom, n=33) before and after ATP stimulation. (F - H) Data are quantified as in (B–D). (I) Representative images of STIM1 puncta formation following ATP stimulation in shRNA STIM1 HEK293T cells expressing STIM1-YFP alone (n=33), or CFP-Tmem178 together with STIM1-YFP (n=14), STIM1G225E-YFP (n=9), or STIM1G225W-YFP (n=21). (J–L) Data are quantified as in (B–D). (M) Time course showing ATP-induced % FRET efficiency signals in HEK293T cells between ORAI1-CFP and STIM1-CFP in the presence of Tmem178 (pink, n=25) or EV (gray, n=21). One out of 3 independent experiments is shown. Data are represented as mean ± SEM. Student’s t-test was performed in B, D, F and H. One way ANOVA was performed in J and L. Two way ANOVA was performed in C, G, K and M. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Next, we analyzed the STIM1 puncta formation in HEK293T cells expressing EV or Tmem178, before and after ATP stimulation. Ectopic expression of Tmem178 largely diminishes ATP-induced puncta formation compared to EV cells (Fig. 5E). The numbers of puncta (Fig. 5F), the intensity (Fig. 5G) and the size (Fig. 5H) of each punctum are significantly reduced in cells expressing Tmem178.
Finally, we monitored the puncta formation in ATP treated HEK293T cells co-expressing Tmem178 together with wild type STIM1, STIM1G225E or STIM1G225W. Consistent with the above results, ectopic expression of Tmem178 reduces STIM1 puncta formation (Fig. 5I and J). Furthermore, puncta formation is slightly but significantly reduced in cells expressing Tmem178 and STIM1G225E (mutant which strongly binds to Tmem178), whereas is increased in cells co-expressing Tmem178 and STIM1G225W (mutant which fails to bind to Tmem178) (Fig. 5I and J). Taken together, these results indicate that the association between Tmem178 and STIM1 reduces puncta formation.
STIM1 puncta formation leads to the association between STIM1 and ORAI1 and therefore promotes SOCE. Next, we investigated whether the reduced puncta formation by Tmem178 impairs the STIM1:ORAI1 association. We performed FRET imaging using ORAI1-CFP as donor and STIM1-YFP as acceptor in HEK293T cells expressing EV or Tmem178. We found that Tmem178 significantly reduces ATP-induced STIM1:ORAI1 association (Fig. 5M), which is consistent with the co-IP analysis shown in Fig. 2C, lane 5 and 6. Taken together, these results suggest that the association between Tmem178 and STIM1 reduces puncta formation, thereby affecting the binding of STIM1 with ORAI1 and limiting SOCE.
3.7. Loss of Tmem178:STIM1 association enhances macrophage responses and osteoclastogenesis
Next, we addressed the physiological relevance of the Tmem178:STIM1 association. We previously reported that the expression of Tmem178 is significantly reduced in peripheral blood mononuclear cells (PBMCs) treated with plasma from patients with systemic juvenile idiopathic arthritis (sJIA) [43], leading to the hypothesis that these cells have increased intracellular calcium levels. We obtained plasma from 5 age matched healthy donors and 6 sJIA patients. Demographic information on the healthy donors and the sJIA patients can be found in Table 1 and Table 2. Except for one patient, all the other sJIA patients have joint damage and were treated with anti-TNF (Etanercept) alone, or in combination with NSAID, MTX, and prednisone (Table 2). Consistent with our previous report [43], we found reduced expression of Tmem178 in sJIA plasma treated macrophages compared to healthy plasma treated cells (Fig. 6A), regardless of the medication received. We also found that the basal cytosolic calcium levels in macrophages exposed to sJIA plasma are higher compared to cells exposed to healthy plasma, both in basal conditions or after treatment with TG and 2 mM calcium (Fig. 6B–D). Importantly, exposure of Tmem178−/− macrophages to sJIA plasma does not lead to any further increase in SOCE activation compared to either Tmem178−/− treated with healthy plasma or WT cells treated with sJIA plasma (Fig. 6C and D). This finding indicates that loss of Tmem178 in WT cells exposed to sJIA plasma drives the increase in cytosolic calcium.
Table 1.
Demographic and clinical characteristics of the healthy donor plasma samples indicating the gender, ethnicity and age in years at time of collection.
| Sample | Gender | Ethnicity | Age at sample collection (years) |
|---|---|---|---|
| 142A | M | White | 8.6 |
| 151A | F | Hispanic/White | 10.2 |
| 162A | F | Hispanic/White | 7.9 |
| 164A | M | Asian | 15.2 |
| 338A | M | White | 8.9 |
Table 2.
Demographic and clinical characteristics of the sJIA patient plasma samples indicating the gender, ethnicity, age in years at disease onset and at time of collection, presence of joint damage evaluated radiographically and treatment at time of collection
| Sample | Gender | Ethnicity | Age at disease onset (years) | Age at sample collection (years) | Joint damage | Treatment at time of sample collection |
|---|---|---|---|---|---|---|
| 2L | M | Hispanic/White | 8.8 | 17.6 | Yes | Anti-TNF (Etanercept) |
| 5G | M | Hispanic/White | 12.2 | 16.3 | Yes | NSAID, MTX, Anti-TNF (Etanercept) |
| 10 N | F | Asian | 2.7 | 7.9 | Yes | NSAID, Anti-TNF(Etanercept) |
| 14G | M | White | 3.2 | 8.9 | Yes | NSAID, MTX, Anti-TNF(Etanercept), prednisone 1.03mg/kg/day |
| 21J | F | Hispanic/White | 1.7 | 9.5 | Yes | NSAID, MTX, Anti-TNF(Etanercept), prednisone 0.42mg/kg/day |
| 123M | M | White | 11.8 | 15.9 | No | None |
Fig. 6.
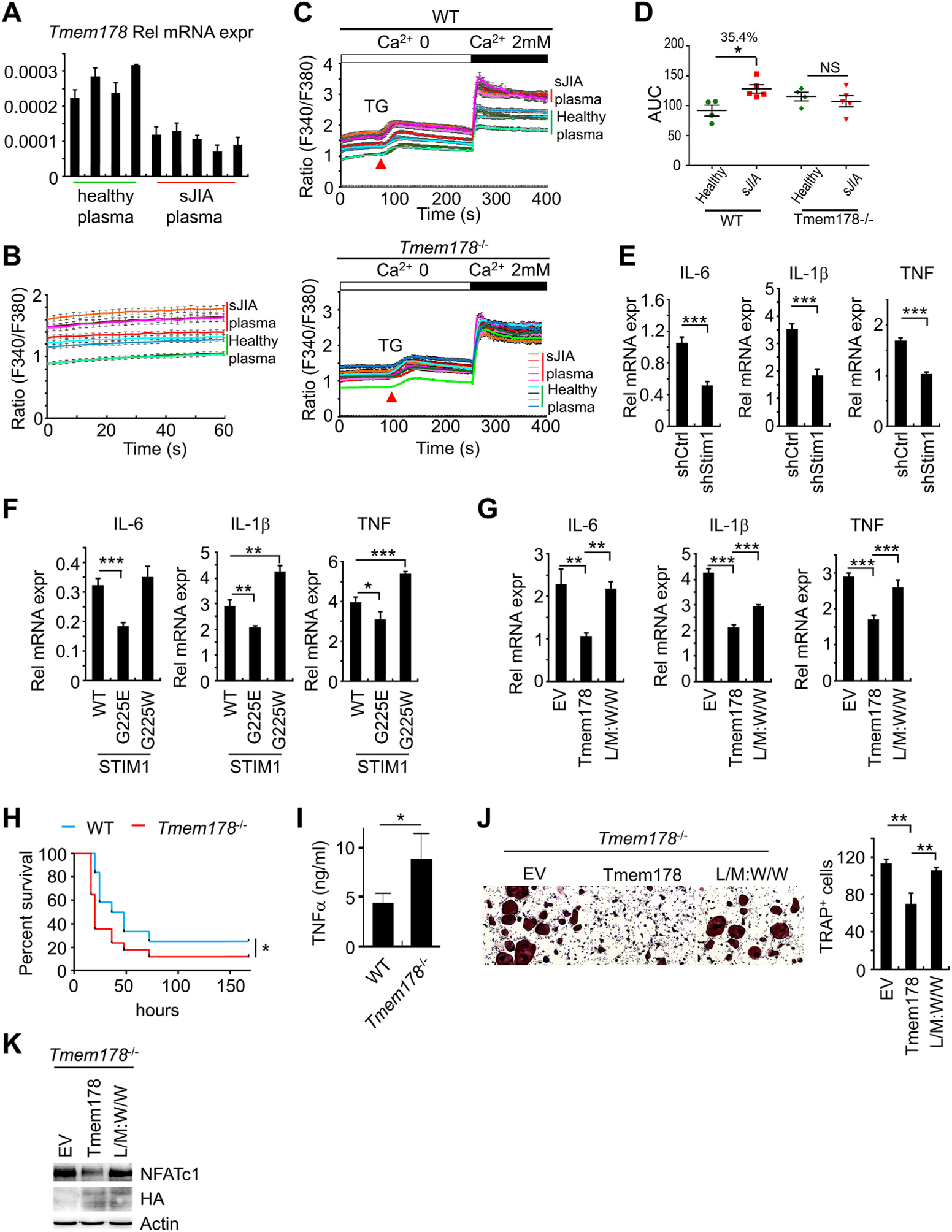
The association between Tmem178 and STIM1 restrains macrophage activation and osteoclastogenesis. (A) Tmem178 mRNA levels in macrophages exposed to 10% healthy plasma or sJIA plasma for 24 h. (B-D) Calcium traces and area under the curve (AUC) in macrophages treated with 10% healthy plasma (N = 4) or sJIA plasma (N = 5) for 24 h in basal conditions (B) or following stimulation with TG and 2 mM calcium in WT (top) or Tmem178−/− (bottom) BMMs (traces from 50 to 70 cells per experiment are shown) (C, D). (E) mRNA levels of inflammatory cytokines in shCtrl or shRNA-Stim1 BMMs stimulated with 100 ng/ml LPS for 4h. (F) RT- PCR as in (E) in shRNA Stim1 BMMs expressing wild type STIM1, STIM1G225E or STIM1G225W. (G) RT-PCR as in (E) in Tmem178−/− BMMs expressing EV, wild type Tmem178 or L/M: W/W mutant. (H) WT (blue line, n = 12) or Tmem178−/− (red line, n = 17) mice were injected (i.p.) with 25 mg/kg LPS. The Kaplan-Meier survival analysis was performed, and Log-rank test was used to determine the statistical significance. (I) Serum of WT or Tmem178−/− mice were collected after 2 h challenge with LPS. TNFa levels were measured by ELISA. (J) Representative images (Left) and quantification (Right) of TRAP stained Tmem178−/− osteoclasts expressing wild type Tmem178 or L/M: W/W mutant. (K) Western blot for NFATc1 in Tmem178−/− pre-OCs expressing HA-tagged Tmem178 or L/M: W/W mutant. Actin and HA western blots are used as loading controls. In B - D, data are represented as mean ± SEM, in all other panels as mean ± SD. Student’s t-test was performed in E and I. One way ANOVA was performed in F, G and J. *P < 0.05_, **P < 0.01, ***P < 0.001. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Monocytes and macrophages from patients with sJIA produce high levels of inflammatory cytokines, including IL-6, IL-1p and TNFa [58]. We also recently reported that Tmem178−/− mice develop more severe cytokine storm syndrome symptoms due to hyper activation of macrophage pro-inflammatory responses [44]. Thus, we hypothesized that Tmem178 negatively regulates cytokine expression in macrophages via SOCE. To address this possibility, we used three complementary approaches. First, we knocked-down Stim1 in BMMs by using sh-RNA Stim1 and observed significantly reduced expression of Il-6, Il-1j5 and Tnfa transcripts in response to LPS stimulation compared with scrambled shRNA control cells (Fig. 6E and Supplemental Fig. 5G). Next, we infected Stim1 deficient BMMs with wild type STIM1, STIM1G225E or STIM1G225W, and measured the expression of inflammatory cytokines after LPS stimulation. Cells expressing STIM1G225E (which strongly binds to Tmem178) show reduced inflammatory cytokine transcripts compared to wild type STIM1, while ectopic expression of STIM1G225W (which cannot bind to Tmem178) enhances their levels (Fig. 6F). Finally, we infected Tmem178−/− BMMs with wild type Tmem178 or the Tmem178 L/M:W/W mutant, which does not bind to STIM1. Ectopic expression of wild type Tmem178 reduces inflammatory cytokine levels while ectopic expression of the L/M:W/W mutant increases their transcripts to levels comparable to EV expressing cells (Fig. 6G). These results suggest that Tmem178 negatively regulates the expression of inflammatory cytokines in macrophages mainly via its association with STIM1. Most importantly, to assess the significance of these in vitro observations, we evaluated the role of Tmem178 during an inflammatory response in vivo using the endotoxin model of sepsis. Strikingly, Tmem178−/− mice rapidly succumb to systemic LPS, with the majority of knock-out mice dying within 16 h of challenge (Fig. 6H). This mortality is associated with a significant increase in TNFa levels detected in the serum 2 h post-LPS administration (Fig. 6I). All together, these results suggest that Tmem178 negatively regulates the expression of inflammatory cytokines in macrophages mainly via its association with STIM1.
Bone erosion due to excessive osteoclast activation is also observed in sJIA patients [59]. We previously reported that loss of Tmem178 augmented osteoclast differentiation in peripheral blood mononuclear cells (PBMCs) exposed to sJIA plasma [43]. Therefore, we asked whether the association between Tmem178 and STIM1 is also required to negatively regulate osteoclastogenesis. Tmem178−/− BMMs expressing EV, wilde type Tmem178 or the L/M:W/W mutant lacking binding to STIM1 were stimulated with RANKL and M-CSF to induce osteoclast differentiation. We found that cells expressing wild type Tmem178 have reduced osteoclastogenesis compared to EV (Fig. 6J). In contrast, cells expressing the L/M:W/W mutant that does not associate well with STIM1 show significantly increased osteoclastogenesis (Fig. 6J). Consistently, NFATc1, a calcium-dependent transcription factor and the master regulator of osteoclastogenesis, is reduced in cells expressing wild type Tmem178, but increased in cells expressing L/M:W/W (Fig. 6K). Taken together, these results suggest that Tmem178 negatively regulates osteoclastogenesis via binding to STIM1.
4. Discussion
SOCE activation is a process tightly controlled to assure proper regulation of intracellular calcium fluxes and mutations in its two components STIM1 and ORAI1 have been implicated in immune disfunctions, autoimmunity, susceptibility to chronic infections, just to name a few [5]. Recent studies have identified a number of proteins interacting with STIM1 that modulate SOCE activation [29–36,38–41]. Most of these proteins are positive regulators of SOCE and facilitate STIM1 oligomerization, its proper localization to ER-PM junctions, puncta formation, and/or the association with ORAI1. However, SOCE must also be properly inactivated to impede calcium overfilling. Although the primary negative regulator of SOCE is ER calcium itself, very few proteins that either act as an ER anchor for STIM1 or facilitate the STIM1 de-oligomerization process have been described [12,39,41]. Here, we identified Tmem178 as a negative modulator of STIM1 puncta formation. Genetic deletion of Tmem178 and inflammation-induced downregulation of Tmem178 enhance STIM1 puncta formation, increase SOCE-mediated calcium influx and induce pro-inflammatory cytokine expression in macrophages and osteoclastogenesis. Tmem178 is specifically expressed in monocytes/macrophages and osteoclasts, but not T cells [44], therefore our results provide new insights into fine-tune regulation of calcium levels in myeloid cells during inflammatory conditions.
We have previously reported that loss of Tmem178 enhances basal calcium levels in the osteoclasts leading to enhanced NFATc1 and osteoclastogenesis. At the time, the mechanism by which Tmem178 regulated calcium levels in the primary cells was not investigated. Studies in HEK293T cells overexpressing the SOCE components STIM1 and ORA1 along with Tmem178 showed a slight reduction in ER calcium release following TG treatment when compared to EV controls, but no differences in peak calcium levels following addition of extracellular calcium. In this study, we more carefully analyzed calcium fluxes in primary BMMs and pre-OCs and in the overexpression system using different modalities to induce ER-store depletion and thus to activate SOCE. In primary BMMs lacking Tmem178 we observe significant effects on ER-calcium store depletion and SOCE, following either ATP or TG treatment. Quantitative analysis shows that the changes in calcium levels following SOCE activation are more robust than the effects on ER calcium release. In the overexpression system, ectopic expression of Tmem178 can significantly reduce SOCE only when the cells are treated with histamine or ATP. In contrast, Tmem178 has minimal to no effects in limiting SOCE following TG or ionomycin. A similar observation was found in HEK293T cells overexpressing SARAF and showing reduced ATP- but not TG-mediated SOCE. This phenomenon could be ascribed to the robust activation of STIM1 and association with ORAI1 that cannot be limited by ectopic expression of Tmem178 following TG or ionomycin. Such finding also suggests that Tmem178 inhibitory effects on SOCE are dependent on ER-calcium content. Supporting the notion that Tmem178:STIM1 association is dependent on ER calcium, we found that Tmem178 does not interact with the STIM1 mutant lacking the ER calcium binding site and under these conditions Tmem178 does not reduce SOCE activation.STIM1 oligomerization is a process dynamically regulated by the formation of puncta structures [12,60]. Recently, two STIM1 partners, STIMATE and SARAF, have been reported to modulate STIM1 oligomerization and de-oligomerization, respectively. STIMATE facilitates STIM1 oligomerization [32] and thus is a positive regulator of SOCE, whereas SARAF enhances STIM1 de-oligomerization and therefore downregulates SOCE activation to impede ER calcium overfilling [39]. We find that Tmem178 can also interact with STIM1. However, differently from STIMATE and SARAF, Tmem178 modulates the rate-limiting step of STIM1 puncta formation. By using FRET imaging and confocal microscopy we find that Tmem178 binds to STIM1 in the ER, and reduces the number and size of STIM1 puncta structures following ATP or TG-mediated ER calcium depletion. By contrast, puncta formation is increased in cells lacking Tmem178. Both SARAF and Tmem178 likely affect STIM1 conformation, yet they associate via different motifs. SARAF associates with the C-terminal region of STIM1[61], whereas the Tmem178:STIM1 association occurs via their TM regions. It remains to be determined whether binding of STIM1 to Tmem178 facilitates STIM1 association with SARAF.
A novel aspect of the Tmem178:STIM1 interaction is that it occurs via their TM domains. Most of the studies on STIM1 have focused on the roles of the N- and C-terminal regions in regulating SOCE activation. The importance of the TM region of STIM1 has been only recently identified through a tryptophan scanning approach [53]. This approach identified that the C227W mutant constitutively associates with ORAI1 and activates SOCE. This study also reported that the triple mutant G223/225/226 W reduces SOCE activation [53]. However, a specific role for these sites was not identified. Our data demonstrate that the STIM1 G225 site is critical for the regulation of SOCE through its interaction with the L212 &M216 in the TM3 region of Tmem178. Furthermore, the G225 site exhibits dual functions based on what this amino acid is mutated to: G225W enhances SOCE, whereas G225E attenuates it. We think that this dual function is ascribed to the different ability of the two mutants to associate with Tmem178. G225W exhibits reduced association with Tmem178, possibly due to steric hindrance, whereas G225E enhances the association, suggesting that hydrophobic interactions might be required for binding to Tmem178.
Our data suggest that Tmem178 and ORAI1 compete for binding to STIM1. STIM1ACT, which lacks the ORAI1 binding domain, exhibits strong association with Tmem178, while STIM1ANT, which has increased binding to ORAI1, poorly associates with Tmem178. Similarly, STIM1D76A, which constitutively associates with ORAI [5], exhibits reduced association with Tmem178. Interestingly, deletion of SOAR, the ORAI1 binding motif [50,53,54], in the STIM1ANT mutant, restores the association with Tmem178. Physiologically, the association between STIM1 and ORAI1 is enhanced after calcium store-depletion [62], whereas the interaction between Tmem178 and STIM1 is reduced under these conditions (Fig. 2A). Thus, it is plausible that, at least in myeloid cells, SOCE could be modulated by the ratio of Tmem178 and ORAI1. For example, SOCE activation could be reduced in cells with high Tmem178/ORAI1 ratio and vice versa. While ORAI1 levels are stable, at least in macrophages and osteoclasts, we found that Tmem178 levels are significantly reduced in these cells under inflammatory conditions. Tmem178 expression is downregulated in macrophages treated with plasma from sJIA patients with erosive disease, regardless of what type of therapies the patients are receiving. Consistently, SOCE is enhanced in these conditions. Future studies are needed to determine whether downregulation of Tmem178 expression is a feature of patients with joint damage and what circulating factors are responsible for modulation of Tmem178 expression.
A large body of evidence has demonstrated the involvement of SOCE activation in T cell functions [63]. Fewer studies have implicated regulation of calcium fluxes through SOCE in macrophages. Although Vaeth et al. reported that calcium signaling, but not SOCE, is required for inflammatory cytokine production in macrophages [64], other groups found that SOCE deficiency reduces cytokine expression in pathological conditions [65,66]. We recently reported that chelation of calcium dampens inflammatory cytokine expression in WT and Tmem178−/− macrophages [44]. However, whether increased SOCE activation enhances inflammatory responses in macrophages and/or their differentiation into osteoclasts has not been investigated. Here we provide evidence that loss of Tmem178 increases SOCE, enhances inflammatory cytokine production in macrophages and rises the number of osteoclasts. In vivo, Tmem178−/− mice are more susceptible to LPS-induced sepsis, produce higher levels of pro-inflammatory cytokines in mouse models of cytokine storm syndrome, a lethal complication of sJIA [44], and present with exuberant bone erosion under inflammatory conditions [43]. Based on these results, we propose a model in which Tmem178 acts as an ER anchor for STIM1 to reduce SOCE and constrain macrophage and osteoclast activation. When Tmem178 levels are downregulated (inflammatory conditions), STIM1 loses its anchor in the ER, forms puncta structures near ER-PM junctions to bind to ORAI1 and potentiate SOCE activation, leading to increased inflammatory cytokine production and contributing to disease progression. Our work unveils a previously poorly understood mechanism for fine-tuned regulation of calcium levels regulating macrophage inflammatory responses and bone erosion via the Tmem178: STIM1 complex.
5. Conclusions
Proper regulation of calcium fluxes is critical for optimal macrophage inflammatory responses and osteoclastogenesis. Aberrant regulation of SOCE has been linked to immune cell dysfunctions and autoimmunity. Here we report a novel regulator of SOCE, Tmem178, which specifically regulates SOCE in myeloid cells. Tmem178 associates with STIM1 in the ER, acting as an anchor for STIM1 to limit STIM1 puncta formation, the binding to ORAI1 and SOCE activation. As a result, Tmem178: STIM1 association reduces pro-inflammatory macrophage responses and osteoclastogenesis. Decreased Tmem178 levels in monocytes/macrophages exposed to sJIA plasma leads to higher intracellular calcium concentration and increased inflammatory cytokine production, highlighting the importance of regulation of calcium homeostasis in myeloid cells in pathological conditions.
Supplementary Material
Acknowledgements
We thank Deborah Veis for constructive discussions. This work was supported by NIH Grants R01 AR053628 and AR066551 (to R.F.), Shriners Hospital Grant 85100 (to R.F.), NIH Grants R01GM112003 and R21GM126532 (to Y.Z.), the Welch Foundation BE-1913 (to Y.Z.), the American Cancer Society RSG-16-215-01 TBE (to Y.Z.), National Key R &D Program of China 2018YFC0910500 (to W.D.), Shanghai Municipal Science and Technology Major Project 2017SHZDZX01 (to W.D.), National Natural Science Foundation of China 81672736 (to W.D.), Shanghai Municipal Commission of Science and Technology 14DZ2252000 (to W.D.), Shanghai Sailing Program 16YF1408600 (to W.D.). Calcium imaging was performed in the Hope Center and the Center for Investigation of Membrane Excitability Diseases (CIMED) Live Cell Imaging Facility at WUSM. Confocal and FRET imaging (Nikon A1Rsi) and analysis were performed through the use of Washington University Center for Cellular Imaging (WUCCI) supported by Washington University School of Medicine, The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015-505) and the Foundation for Barnes-Jewish Hospital (3770).
Footnotes
Declare of interests
The authors declare that they have no conflicts of interest with the contents of this article.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jaut.2019.04.015.
References
- [1].Berridge MJ, Bootman MD, Roderick HL, Calcium signalling: dynamics, homeostasis and remodelling, Nat. Rev. Mol. Cell Biol 4 (2003) 517–529. [DOI] [PubMed] [Google Scholar]
- [2].Soboloff J, Rothberg BS, Madesh M, Gill DL, STIM proteins: dynamic calcium signal transducers, Nat. Rev. Mol. Cell Biol 13 (2012) 549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Berridge MJ, The inositol trisphosphate/calcium signaling pathway in health and disease, Physiol. Rev 96 (2016) 1261–1296. [DOI] [PubMed] [Google Scholar]
- [4].Feske S, Calcium signalling in lymphocyte activation and disease, Nat. Rev. Immunol 7 (2007) 690–702. [DOI] [PubMed] [Google Scholar]
- [5].Lacruz RS, Feske S, Diseases caused by mutations in ORAI1 and STIM1, Ann. N. Y. Acad. Sci 1356 (2015) 45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bagur R, Hajnoczky G, Intracellular Ca(2 + ) sensing: its role in calcium homeostasis and signaling, Mol. Cell 66 (2017) 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cheng H, Lederer WJ, Calcium sparks, Physiol. Rev 88 (2008) 1491–1545. [DOI] [PubMed] [Google Scholar]
- [8].Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. , A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function, Nature 441 (2006) 179–185. [DOI] [PubMed] [Google Scholar]
- [9].Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, et al. , CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry, Science 312 (2006) 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, et al. , Genomewide RNAi screen of Ca(2 + ) influx identifies genes that regulate Ca(2 + ) releaseactivated Ca(2 + ) channel activity, Proc. Natl. Acad. Sci. U.S.A 103 (2006) 9357–9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, Neems DS, et al. , Biochemical and functional characterization of Orai proteins, J. Biol. Chem 282 (2007) 16232–16243. [DOI] [PubMed] [Google Scholar]
- [12].Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr.et al. , STIM is a Ca2+ sensor essential for Ca2 + -store-depletion-triggered Ca2+ influx, Curr. Biol 15 (2005) 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. , STIM1, an essential and conserved component of store-operated Ca2+ channel function, J. Cell Biol 169 (2005) 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, et al. , STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane, Nature 437 (2005) 902–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sabala P, Amler E, Baranska J, Intracellular Ca2+ signals induced by ATP and thapsigargin in glioma C6 cells. Calcium pools sensitive to inositol 1,4,5-trisphosphate and thapsigargin, Neurochem. Int 31 (1997) 55–64. [DOI] [PubMed] [Google Scholar]
- [16].Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS, Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation, Nature 454 (2008) 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhou Y, Meraner P, Kwon HT, Machnes D, Oh-hora M, Zimmer J, et al. , STIM1 gates the store-operated calcium channel ORAI1 in vitro, Nat. Struct. Mol. Biol 17 (2010) 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhou Y, Srinivasan P, Razavi S, Seymour S, Meraner P, Gudlur A, et al. , Initial activation of STIM1, the regulator of store-operated calcium entry, Nat. Struct. Mol. Biol 20 (2013) 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shen WW, Frieden M, Demaurex N, Local cytosolic Ca2+ elevations are required for stromal interaction molecule 1 (STIM1) de-oligomerization and termination of store-operated Ca2+ entry, J. Biol. Chem 286 (2011) 36448–36459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lewis RS, Store-operated calcium channels: new perspectives on mechanism and function, Cold Spring Harb. Perspect. Biol 3 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vaeth M, Eckstein M, Shaw PJ, Kozhaya L, Yang J, Berberich-Siebelt F, et al. , Store-operated Ca(2 + ) entry in follicular T cells controls humoral immune responses and autoimmunity, Immunity 44 (2016) 1350–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vaeth M, Maus M, Klein-Hessling S, Freinkman E, Yang J, Eckstein M, et al. , Store-operated Ca(2 + ) entry controls clonal expansion of T cells through metabolic reprogramming, Immunity 47 (2017) 664–679 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Clemens RA, Chong J, Grimes D, Hu Y, Lowell CA, STIM1 and STIM2 cooperatively regulate mouse neutrophil store-operated calcium entry and cytokine production, Blood 130 (2017) 1565–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kajiya H, Okamoto F, Nemoto T, Kimachi K, Toh-Goto K, Nakayana S, et al. , RANKL-induced TRPV2 expression regulates osteoclastogenesis via calcium oscillations, Cell Calcium 48 (2010) 260–269. [DOI] [PubMed] [Google Scholar]
- [25].Hwang SY, Putney JW, Orai1-mediated calcium entry plays a critical role in osteoclast differentiation and function by regulating activation of the transcription factor NFATc1, FASEB J. 26 (2012) 1484–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hwang SY, Foley J, Numaga-Tomita T, Petranka JG, Bird GS, Putney JW Jr., Deletion of Orai1 alters expression of multiple genes during osteoclast and osteoblast maturation, Cell Calcium 52 (2012) 488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kass GE, Orrenius S, Calcium signaling and cytotoxicity, Environ. Health Perspect 107 (Suppl 1) (1999) 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lopez JJ, Albarran L, Gomez LJ, Smani T, Salido GM, Rosado JA, Molecular modulators of store-operated calcium entry, Biochim. Biophys. Acta 1863 (2016) 2037–2043. [DOI] [PubMed] [Google Scholar]
- [29].Shambharkar PB, Bittinger M, Latario B, Xiong Z, Bandyopadhyay S, Davis V, et al. , TMEM203 is a novel regulator of intracellular calcium homeostasis and is required for spermatogenesis, PLoS One 10 (2015) e0127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Quintana A, Rajanikanth V, Farber-Katz S, Gudlur A, Zhang C, Jing J, et al. , TMEM110 regulates the maintenance and remodeling of mammalian ER-plasma membrane junctions competent for STIM-ORAI signaling, Proc. Natl. Acad. Sci. U.S.A 112 (2015) E7083–E7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Krapivinsky G, Krapivinsky L, Stotz SC, Manasian Y, Clapham DE, POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters, Proc. Natl. Acad. Sci. U.S.A 108 (2011) 19234–19239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jing J, He L, Sun A, Quintana A, Ding Y, Ma G, et al. , Proteomic mapping of ER-PM junctions identifies STIMATE as a regulator of Ca(2)( + ) influx, Nat. Cell Biol 17 (2015) 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen YJ, Chang CL, Lee WR, Liou J, RASSF4 controls SOCE and ER-PM junctions through regulation ofPI(4,5)P2, J. Cell Biol 216 (2017) 2011–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Srikanth S, Jung HJ, Kim KD, Souda P, Whitelegge J, Gwack Y, A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells, Nat. Cell Biol 12 (2010) 436–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Srikanth S, Jew M, Kim KD, Yee MK, Abramson J, Gwack Y, Junctate is a Ca2 + -sensing structural component of Orai1 and stromal interaction molecule 1 (STIM1), Proc. Natl. Acad. Sci. U.S.A 109 (2012) 8682–8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Miao Y, Miner C, Zhang L, Hanson PI, Dani A, Vig M, An essential and NSF independent role for alpha-SNAP in store-operated calcium entry, eLife 2 (2013) e00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sharma S, Quintana A, Findlay GM, Mettlen M, Baust B, Jain M, et al. , An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry, Nature 499 (2013) 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Maschalidi S, Nunes-Hasler P, Nascimento CR, Sallent I, Lannoy V, Garfa-Traore M, et al. , UNC93B1 interacts with the calcium sensor STIM1 for efficient antigen cross-presentation in dendritic cells, Nat. Commun 8 (2017) 1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Palty R, Raveh A, Kaminsky I, Meller R, Reuveny E, SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling, Cell 149 (2012) 425–438. [DOI] [PubMed] [Google Scholar]
- [40].Srivats S, Balasuriya D, Pasche M, Vistal G, Edwardson JM, Taylor CW, et al. , Sigma 1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai1, J. Cell Biol 213 (2016) 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chang CL, Chen YJ, Quintanilla CG, Hsieh TS, Liou J, EB1 binding restricts STIM1 translocation to ER-PMjunctions and regulates store-operated Ca(2 + ) entry, J. Cell Biol 217 (2018;:−58) 2047–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Grigoriev I, Gouveia SM, van der Vaart B, Demmers J, Smyth JT, Honnappa S, et al. , STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER, Curr. Biol 18 (2008) 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Decker CE, Yang Z, Rimer R, Park-Min KH, Macaubas C, Mellins ED, et al. , Tmem178 acts in a novel negative feedback loop targeting NFATc1 to regulate bone mass, Proc. Natl. Acad. Sci. U.S.A 112 (2015) 15654–15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mahajan S, Decker CE, Yang Z, Veis D, Mellins ED, Faccio R, Plcgamma2/ Tmem178 dependent pathway in myeloid cells modulates the pathogenesis of cytokine storm syndrome, J. Autoimmun (2019. March 15), 10.1016/j.jaut.2019.02.005 pii: S0896–8411(18)30697–8 [Epub ahead ofprint]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Youvan DC, Silva CM, Bylina EJ, Coleman WJ, Dilworth MR, Yang MM, Calibration of fluorescence resonance energy transfer in microscopy using genetically engineered GFP derivatives on nickel chelating beads, Biotechnology 3 (1997) 1–18. [Google Scholar]
- [46].Dai W, Song T, Wang X, Jin X, Deng L, Wu A, et al. , Improvement in lowhomology template-based modeling by employing a model evaluation method with focus on topology, PLoS One 9 (2014) e89935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y, The I-TASSER Suite: protein structure and function prediction, Nat. Methods 12 (2015) 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dai W, Wu A, Ma L, Li YX, Jiang T, Li YY, A novel index of protein-protein interface propensity improves interface residue recognition, BMC Syst. Biol 10 (2016) 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pierce BG, Hourai Y, Weng Z, Accelerating protein docking in ZDOCK using an advanced 3D convolution library, PLoS One 6 (2011) e24657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, et al. , STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1, Cell 136 (2009) 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ma G, Zheng S, Ke Y, Zhou L, He L, Huang Y, et al. , Molecular determinants for STIM1 activation during store-operated Ca2+ entry, Curr. Mol. Med 17 (2017) 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Covington ED, Wu MM, Lewis RS, Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1, Mol. Biol. Cell 21 (2010) 1897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ma G, Wei M, He L, Liu C, Wu B, Zhang SL, et al. , Inside-out Ca(2 +) signalling prompted by STIM1 conformational switch, Nat. Commun 6 (2015) 7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S, SOAR and the polybasic STIM1 domains gate and regulate Orai channels, Nat. Cell Biol 11 (2009) 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bass RB, Miller AS, Gloor SL, Falke JJ, The PICM chemical scanning method for identifying domain-domain and protein-protein interfaces: applications to the core signaling complex of E. coli chemotaxis, Methods Enzymol. 423 (2007) 3–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Boersma MD, Sadowsky JD, Tomita YA, Gellman SH, Hydrophile scanning as a complement to alanine scanning for exploring and manipulating protein-protein recognition: application to the Bim BH3 domain, Protein Sci. 17 (2008) 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gudlur A, Zhou Y, Hogan PG, STIM-ORAI interactions that control the CRAC channel, Curr. Top. Membr 71 (2013) 33–58. [DOI] [PubMed] [Google Scholar]
- [58].Mellins ED, Macaubas C, Grom AA, Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions, Nat. Rev. Rheumatol 7 (2011) 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Woo P, Systemic juvenile idiopathic arthritis: diagnosis, management, and outcome, Nat. Clin. Pract. Rheumatol 2 (2006) 28–34. [DOI] [PubMed] [Google Scholar]
- [60].Wu MM, Buchanan J, Luik RM, Lewis RS, Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane, J. Cell Biol 174 (2006) 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jha A, Ahuja M, Maleth J, Moreno CM, Yuan JP, Kim MS, et al. , The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function, J. Cell Biol 202 (2013) 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cahalan MD, STIMulating store-operated Ca(2 +) entry, Nat. Cell Biol 11 (2009) 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hogan PG, Lewis RS, Rao A, Molecular basis of calcium signaling in lymphocytes: STIM and ORAI, Annu. Rev. Immunol 28 (2010) 491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Vaeth M, Zee I, Concepcion AR, Maus M, Shaw P, Portal-Celhay C, et al. , Ca2 + signaling but not store-operated Ca2+ entry is required for the function of macrophages and dendritic cells, J. Immunol 195 (2015) 1202–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Velmurugan GV, Huang H, Sun H, Candela J, Jaiswal MK, Beaman KD, et al. , Depletion of H2S during obesity enhances store-operated Ca2+ entry in adipose tissue macrophages to increase cytokine production, Sci. Signal 8 (2015) ra128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liang SJ, Zeng DY, Mai XY, Shang JY, Wu QQ, Yuan JN, et al. , Inhibition of Orai1 store-operated calcium channel prevents foam cell formation and atherosclerosis, Arterioscler. Thromb. Vasc. Biol 36 (2016) 618–628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.