

Abstract
The development of colorectal cancer (CRC) is a multistep process initiated by a benign polyp that has the potential to evolve into in situ carcinoma through the interactions between environmental and genetic factors. CRC incidence rates are constantly increased for young adult patients presenting an advanced tumor stage. The majority of CRCs arise from colonic adenomas originating from aberrant cell proliferation of colon epithelium. Endoscopic polypectomy represents a tool for early detection and removal of polyps, although the occurrence of cancers after negative colonoscopy shows a significant incidence. It has long been recognized that the aberrant regulation of Wingless/It (Wnt)/β-Catenin signaling in the pathogenesis of colorectal cancer is supported by its critical role in the differentiation of stem cells in intestinal crypts and in the maintenance of intestinal homeostasis. For this review, we will focus on the development of adenomatous polyps through the interplay between renewal signaling in the colon epithelium and reactive oxygen species (ROS) production. The current knowledge of molecular pathology allows us to deepen the relationships between oxidative stress and other risk factors as lifestyle, microbiota, and predisposition. We underline that the chronic inflammation and ROS production in the colon epithelium can impair the Wnt/β-catenin and/or base excision repair (BER) pathways and predispose to polyp development. In fact, the coexistence of oxidative DNA damage and errors in DNA polymerase can foster C>T transitions in various types of cancer and adenomas, leading to a hypermutated phenotype of tumor cells. Moreover, the function of Adenomatous Polyposis Coli (APC) protein in regulating DNA repair is very important as therapeutic implication making DNA damaging chemotherapeutic agents more effective in CRC cells that tend to accumulate mutations. Additional studies will determine whether approaches based on Wnt inhibition would provide long-term therapeutic value in CRC, but it is clear that APC disruption plays a central role in driving and maintaining tumorigenesis.
1. Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide. In fact, CRC is the third most commonly diagnosed cancer in males and the second in females, with 1.8 million new cases and almost 861,000 deaths in 2018 according to the World Health Organization [1]. The risk of developing CRC is influenced by environmental and genetic factors. The incidence and mortality rates of CRC vary widely worldwide. Current literature suggests that incidence rates are constantly increased for patients diagnosed under the age of 50, who often present with symptomatic diagnosis and with a more advanced tumor stage [2]. Up to 35% of CRCs are thought to be due to heritable factors, but currently, only 5% to 10% of CRCs are attributable to high-risk mutations in known CRC susceptibility genes.
The development of CRC is considered to be a multistep process, initiated by the development of a benign polyp that has the potential to evolve into an in situ carcinoma by the accumulation of additional somatic mutations [3]. The majority of CRCs arise from colonic adenomas, originating from aberrant cell proliferation of colon epithelium.
Adenoma prevalence is about 40% in Western populations, ranging from 50 to 75 years, and is prevalently male associated [4]. Screening and surveillance programs are useful to identify precursor lesions and to prevent the death from CRC. Endoscopic polypectomy represents a tool for early detection and removal of polyps. The occurrence of cancers after negative colonoscopy shows a significant incidence since adenomas could be missed during colonoscopy or biological changes could occur in tumor growth rates [5].
Many risk factors for polyp development have been correlated to lifestyles and can play a potential role in gut mucosal inflammation consequent to dysbiosis [6, 7]. Altered microbiome composition is associated with signal activation involving mitochondria, or/and altered redox homeostasis, proinflammatory cytokines induction, and stimulation of the immune system [8]. It is well known that chronic inflammation and reactive oxygen species (ROS) production in the colon epithelium can impair the Wingless/It (Wnt)/β-catenin [9] and/or base excision repair (BER) pathways [10, 11], enhance a cascade of molecular reactions in cells, and alter the metabolic state of tissues and predispose to polyp development.
Adenomas represent the main precursors of CRC in high-risk family groups with a history of polyposis syndrome and in the wide-ranging population. Indeed, genetic events like gain or loss of functions on molecules, necessary for functional homeostasis of the intestinal cells, can lead to the polyp development [12].
For this review, we will focus on the development of adenomatous polyps through oxidative stress and signaling in the colon epithelium interactions. The current knowledge of molecular pathology will be highlighted with particular regard to the relationships between oxidative stress and other risk factors, like lifestyle, microbiota, and predisposition. In fact, only the understanding of the complex interrelations between these factors and the body's response and immune defense will allow correct prevention of early events in colon carcinogenesis.
2. Epidemiology and Risk Factors of Intestinal Polyps
Although the development of polyps is strongly correlated with the development of CRC, their malignant potential differs among different subtypes [13].
At least three subtypes of polyps can be distinguished on the basis of histology and the underlying molecular pathway: tubular/villous adenomas, hyperplastic polyps, and sessile/traditional serrated adenomas. Tubular/villous adenomas are characterized by an adenomatous histotype, whereas both hyperplastic polyps and sessile/traditional serrated adenomas have a serrated histotype [14]. The prevalence of hyperplastic polyps is higher than that of tubular/villous adenomas and sessile/traditional serrated adenomas [13]. Although an increased risk for malignant transformation has been described for hyperplastic polyps, their tumorigenic potential is considered to be lower than that of tubular/villous adenomas and sessile/traditional serrated adenomas [13]. In addition, the risk of developing cancer is strongly associated with the number and size of previously encountered polyps [15]. Therefore, the development of multiple colonic polyps with malignant potential will result in an increased lifetime risk of developing CRC. Owing to the malignant potential of tubular/villous adenomas, patients diagnosed with adenomatous polyposis, i.e., the constitutive development of multiple colorectal adenomas, are at increased risk of developing CRC.
The different histological types of polyps show a variable anatomic distribution in the large bowel. Even though the different types of polyps may be disseminated in all the large bowel, adenomas and hyperplastic polyps are prevalently located in distal colon [16–18], and sessile serrated polyps are often found in proximal colon and they seem to be the precursor of up to 30% of CRC [19–23]. In a study conducted in an Italian population, serrated lesions were recognized in the proximal colon in 38% of cases, while 40.7% were in the left colon, and 14.3% in the rectum [24]. Various risk factors, unmodifiable and/or modifiable, may influence polyp onset. The unmodifiable factors are represented by age, gender, and ethnicity while the modifiable ones, including unhealthy diet, tobacco smoking, excessive alcohol consumption, physical inactivity, and obesity, have been independently associated with the risk of polyps and altered composition of the intestinal microbiota [25] (Table 1).
Table 1.
Possible risk factors associated with colorectal adenoma onset.
| Risk factors | Effects on adenoma onset | References |
|---|---|---|
| Age | Possible onset before 50 years | [26, 27] |
| Major incidence after 50 years | [28] | |
| Peak at 70 years | [16] | |
|
| ||
| Gender | Higher incidence in males than in females | [16, 29] |
|
| ||
| Ethnicity | High incidence in Western and African Americans populations | [4, 30, 31] |
|
| ||
| Persistent organic pollutants | ||
| Organochlorine pesticides | Wnt and Hedgehog/Gli1 pathway activation. Increased ROS | [32] |
| Dichlorodiphenyldichloroethylene | ||
| Polychlorinated biphenyls | Predisposition to gut inflammation | [33] |
| Dioxin-like PBS126 | ||
|
| ||
| Heavy metals | ||
| Arsenic | Altered gut-associated immunity and microbiome | [34] |
| Cadmium | Intestinal inflammation, modified microbiome | [35] |
|
| ||
| Antibiotics | Changes in gut microbiota | [36] |
|
| ||
| Food additives | Changes in gut microbiota | [36] |
|
| ||
| Diet | ||
| Excessive processed red meat consumption | Gut dysbiosis by HCA and PAH production. Heme iron associated with aldehyde generation | [37–40] |
| High saturated fat intake | Intestinal inflammation | [41–43] |
|
| ||
| Lifestyles | ||
| Stress | Increased stress hormones, altered rate of cell growth | [44, 45] |
| Lack of physical exercise | Overweight and obesity | [46] |
|
| ||
| Obesity | High TNF-α, IGF-1, and adiponectin | [47] |
|
| ||
| Cigarette smoking | Oxidative stress, chronic inflammation, genetic/epigenetic alterations by BaP/HCA generation | [25, 48–50] |
|
| ||
| Heavy alcohol drinking | Production of acetaldehyde | [51] |
|
| ||
| Gut microbiota alterations | ||
| Fusobacterium nucleatum | Colon cells adhesion by FadA, β-catenin activation | [52] |
| Escherichia coli | Inflammation and DNA breaks by CDT and colibactin | [51] |
| Bacteroides fragilis | Wnt pathway activation by BFT. ROS production | [53] |
| Enterococcus faecalis | Superoxide production | [54] |
| Helicobacter pylori | Increased serrated polyps | [55] |
Bacteroides fragilis toxin (BFT); cytolethal distending toxin (CDT); heterocyclic amines (HCAs); polycyclic aromatic hydrocarbons (PAHs); benzo[a]pyrene (BaP).
2.1. Unmodifiable Factors
The prevalence rate of polyps increases with age, and their major incidence occurs after 50 years [28], with a peak at 70 years [16]. Colorectal polyps are uncommon before age 40, but some reports indicate a rising incidence of them before 50 years [56, 57] and even among young patients from 20 to 39 years of age, with a prevalent detection of serrated lesions [26, 27]. Unlike most studies that affirm that adenomas are more common in males than in females [16, 29], Klein et al. highlight the prevalence of adenomas in the left colon in women compared with men [18], whereas Brettahuer et al. did not observe any significant sex differences [30]. In contrast, sessile serrated polyps were commonly detected in females in the proximal colon [22]. Several studies demonstrated a significant association between ethnicity and risk to develop polyps. The prevalence of colorectal polyps may vary across countries and racial/ethnic groups. High incidence of colorectal polyps is found in Western countries and ranges from about 30% to 50% of the population [4, 30]. Solakoğlu et al. identified a higher percentage (81%) of adenomas in patients through a screening colonoscopy based retrospective study conducted in Turkey [57]. African Americans show a higher incidence of more aggressive colorectal polyps than Caucasians and prevalently located in the proximal colon [58]. In particular, blacks have an increased prevalence of precursor lesions >9 mm as compared to white individuals [31] and show a higher number of polyps at a young age compared to Hispanic Americans [59], with whom they share an increased risk of adenomas than whites at an older age [60]. Katsidzira et al. conducted a study that demonstrated racial differences in the incidence of polyps in Zimbabwe: a less occurrence of polyps among black Africans (5%) and more frequent onset of adenomatous polyps in Caucasians (8%) and Asians (9%), whereas colorectal cancer was more diffused among black Africans [61]. These geographic differences might be supported by diversities in lifestyles and exposition to different environmental factors. Indeed, there is a large body of evidence that correlates modifiable aspects as dietary habits, or less modifiable aspects as pollutant exposure, with modifications of the structure of the gut microbiota, inducing carcinogenesis of the colorectum.
2.2. Environmental Factors
Environmental influences alone or in association with lifestyle may be responsible for the development of colorectal polyps. An increased risk of polyps in the large intestine is associated with chronic low-dose exposure to persistent organic pollutants, such as organochlorine pesticides (OCPs) and polychlorinated biphenyls (PCBs). Lee et al. reported a strong association of colorectal polyps and cancer with the OCP β-hexachlorocyclohexane, as well as low-chlorinated PCBs [48]. The OCP p, p′-dichlorodiphenyldichloroethylene (p,p′-DDE), considered the main metabolite of dichlorodiphenyltrichloroethane (DDT), could induce the activation of Wnt/β-catenin and Hedgehog/Gli1 (Hh) pathways, resulting in the overexpression of c-Myc and cyclin D1 and adenocarcinoma cells proliferation. p,p′-DDE increases ROS levels in colorectal cancer cells by NADPH oxidase (NOX) activation and reduces the expression of antioxidant enzymes [32]. PCBs are also considered potent activators of oxidative stress by increasing the activity of NOX through phosphorylation of p47phox [62].
PCBs are responsible for altered gut barrier functions and permeability by modifying the expression of tight junction proteins in the intestinal mucosa [62]. These evidences suggest that a similar mechanism could predispose to the gut inflammation state and formation of colorectal polyps. Moreover, xenobiotics, as persistent organic pollutants, can interact with gut microbiota modifying its structure and inducing carcinogenesis of colorectum [63]. OCPs are absorbed in the small intestine and accumulate mainly in the adipose tissue, from which they join the bloodstream [64], whereas PCBs are mainly removed from the body in feces [48]. Dioxin-like PBS126 exposure significantly alters the gut microbiota equilibrium, predisposing to intestinal inflammation [33].
Heavy metal contact consequent to food uptake or occupational exposure is considered responsible for gut microbiota dysbiosis. Metabolites of arsenic alter gut bacterial species, the production of butyrate, and the gut-associated immune system [34]. Cadmium exposure determines an inflammatory response and tight junction alterations in the intestinal barrier that increase gut permeability and favour the Bacteroidetes in the microbiome [35]. Antibiotics, nanoparticles as environmental pollution, and additives used for food preservation influence and alter the composition and function of the gut microbiota, predisposing to the onset of diseases [36].
2.3. Modifiable Factors
In recent decades, extensive epidemiological and experimental researches have highlighted the key role of the diet in preventing the CRC onset. Dietary behaviours influence the composition of gut microbiota. Indeed, bacterial metabolism of bile acids, heme iron, and complex carbohydrates is crucial for barrier function and immune homeostasis and inflammation. The illness has been ascribed to the excessive consumption of red and processed meat [65], which is associated with an elevated risk of adenomas in the descending and sigmoid colon [37]. Voskuil et al. in a case-control study suggest an association between habitual methods of meat preparation and adenoma formation [66]. In fact, the daily intake of red meat cooked at high temperature, with production of polycyclic aromatic hydrocarbons such as benzo(a)pyrene, or saturated fat consumption, low fiber diet [67, 68], heterocyclic amines produced during cooking of red meat [38], and N-nitroso compounds can induce genotoxic effects [69]. In particular, the risk for single adenomas was found to be associated with the heterocyclic amines 2-amino-3,8-dimethylimidazo[4,5]quinoxaline (MeIQx) and 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) and benzo[a]pyrene [37]. The exposure to benzo[a]pyrene generates intestinal inflammation and also gut microbiota modifications [39]. Heterocyclic amines introduced by diet are metabolized and converted to mutagens and DNA adducts by microbe intestinal flora. Indeed, diet prevalently based on red meat and animal fats promotes dysbiosis with a selection of bacterial species that alter bile acid metabolism, with proinflammatory and prooncogenic effects [40]. A crucial role in the colorectal adenoma risk is also referred to a diet rich in heme iron contained in red meat that leads to modification gut barrier homeostasis and microbiota associated with the selection of bacteria linked to inflammation and colorectal adenoma [41]. Heme iron induces the oxidation of dietary polyunsaturated fatty acids and the consequent production of unsaturated aldehydes, such as malondialdehyde and 4-hydroxynonenal (HNE), which are cytotoxic [42]. Colon cells harbouring Adenomatous Polyposis Coli (APC) mutations in rats are resistant to apoptosis induced by HNE, suggesting that lipoperoxidation by heme iron confers to these cells a selective survival advantage [70]. Luminal lipoperoxidation is associated with high levels of mucosal inflammation markers, such as the myeloperoxidase activity with ROS production, increased gene expression of interleukin 6 (IL-6) and transforming growth factor β (TGF-β), and enhanced cellular permeability due to reduced expression of the junctional adhesion molecules [41].
The toxicity of the nitrosylated heme iron seems to act for site-specific etiology of colon adenoma. In fact, an increased risk is related to nonnitrosylated heme iron uptake with advanced distal adenoma, as well as nitrosylated heme iron with proximal adenoma [42].
Resistant starch stimulates gut bacterial fermentation to produce short-chain fatty acids (SCFAs) into the colon, which are known to modulate immune responses in the intestine [71]. SCFAs act as a gut barrier by reduction of oxygen concentrations and induction of hypoxia-induced factor (HIF). Nevertheless, butyrate one of SCFAs shows a “paradox” behaviour since it prevents intestinal polyp formation or modulates the expression of genes to elude the aberrant cell colon proliferation through the activation of apoptosis or upregulation of detoxifying enzymes [72]. Botma et al. observed an increased risk of colorectal adenomas associated with a “snack” dietary pattern in a prospective cohort study involving patients affected with Lynch syndrome [73]. High-fat diet intake can increase the expression of macrophage markers and inflammatory mediators in the adipose tissue, which are associated with an increased number of large polyps [43]. Moreover, increased consumption of fat is associated with high production of hydrogen sulphide, inducing inflammation and cell proliferation, by sulphate-reducing bacteria [51, 54].
Another risk factor for colorectal adenoma development is the lack of physical activity [44]. Several biological mechanisms have been proposed to explain the impact and the protective effect of the physical activity on the less likely development of colorectal adenomas. Active lifestyle confers health benefits since it increases the intestinal motility and the immune system functions, decreases the systemic inflammation, reduces the insulin resistance and the obesity, and increases the activity of free radical scavenger in the antioxidative systems [74, 75].
A study highlighted the association of total stressful life events and colon polyps in African Americans, supporting the evidence that stress hormones alter the rate of cell growth and proliferation [45]. A study based on a multiethnic colorectal screening [46] observed that individuals with an active lifestyle showed a significantly lower prevalence of adenoma risk, compared to counterparts who had reduced physical activity and demonstrated the development of adenomas in distal colon prevalently. Consistent with these findings, weight loss associated with physical exercise has been found to reduce biomarkers of oxidative stress, such as oxidized low-density protein (LDL), in postmenopausal women [76]. Significant inverse association of physical activity and colon adenoma suggests the importance of its role in colon cancer prevention, particularly in males [77]. During adolescence, physical activity can reduce the risk of colorectal adenoma later in life [78].
In obese subjects, the increased body mass index ≥30 (BMI) or overweight (25 ≤ BMI ≤ 29.9) is considered a risk factor for colonic adenomas [79]. Botma et al. found a significant association between BMI and increased risk of adenomas in men, in a prospective cohort study conducted on carriers of DNA mismatch repair (MMR) gene mutations [80].
A recent case-control study involving African Americans showed an increased risk of colorectal adenoma onset associated with high circulating levels of TNF-α, IGF-1, and the metabolic biomarker adiponectin, which is secreted from the abdominal fat tissue and can induce cell proliferation [47]. Numerous studies analyzed the relationship among the metabolic syndrome associated-diseases, such as obesity, hypertension, and diabetes mellitus, and the development of colorectal adenomas, as well as the association of high triglyceride/high-density lipoprotein cholesterol ratio with serrated polyps [81, 82].
In recent years, there has been a growing interest in the understanding of the relationships between the hepatic manifestation of the metabolic syndrome, represented by nonalcoholic fatty liver disease (NAFLD), and the development of colorectal polyps [83, 84]. NAFLD is a significant risk factor for adenomatous and hyperplastic polyps in males as compared with females [85, 86], although Li et al. had demonstrated that NAFLD is a greater factor for the adenomatous polyp development in women than in men [87]. Moreover, an association among NAFLD, alcohol consumption, and colorectal adenomas has been identified [88] but has not still been well investigated. Randomized controlled studies on the interactions among physical activities, dietary factors, modulation of immune responses, and the microbiota will be crucial to advance the customized guidelines in the prevention of colorectal adenomas.
Smoking and alcohol consumption increased the risk of adenomas. The association between smoking and adenomas has been pointed out in a prospective cohort study [49]. Cigarette smoking components, such as benzo[a]pyrene and heterocyclic amines, represent a significant modifiable risk factor for polyps since they induce chronic inflammation consequent to oxidative stress and genetic/epigenetic alterations. In particular, tobacco use is associated with the development of serrated polyps [48, 50] and with the development of large and flat colorectal polyps [89]. A study of Fu et al. described a strong association between cigarette smoking and synchronous hyperplastic polyps and adenomas, due to a stronger relationship of cigarette smoking with hyperplastic polyps than with adenomas [25].
Alcohol is metabolized by bacteria expressing alcohol dehydrogenase that converts ethanol to acetaldehyde, which is a carcinogenic factor. Moreover, alcohol interferes with folate metabolism that is involved in DNA methylation [51]. Diergaarde et al. correlated an increased risk of adenomas with alcohol assumption and smoking and also a decreased risk of adenomas with a diet enriched with fruit and fibers in a Dutch case-control study involving individuals affected with hereditary nonpolyposis colorectal cancer (HNPCC) [90]. The association was not significantly confirmed by Winkels et al. [49].
3. Gut Microbiota
The colonic mucosa is in contact with the intestinal bacteria and their metabolic products. Normal microbiota is composed of obligate anaerobic bacteria. The gut microbiota plays a pivotal role in physiological homeostasis of the intestine by the modulation of immune responses, enhancement of epithelial barrier function, and stimulation of cell proliferation. Alterations in the gut microbiome (dysbiosis) drive the signals between mitochondria and epithelial mucosal cells and induce inflammasome signaling through the activation of immune cells until they change the epithelial barrier function [8]. In the past few years, several studies have definitively shown that gut microbes exert distinct impacts on DNA damage, DNA methylation, chromatin structure, and noncoding RNA expression in colon epithelial cells [91]. Some genes and pathways that are altered by gut microbes are also related to CRC development, particularly those involved in cell proliferation and Wnt signaling [91]. Dysbiosis is the consequence of diet factors or potentially harmful microorganisms which can induce inflammatory processes. Among them, Fusobacterium nucleatum (Fn), an intermediate and driver microbe, attaches to the colon epithelial cells by its adhesion molecule FadA that interacts with E-Cadherin, inhibiting its oncosuppressive activity [52]. E-Cadherin is known to maintain the integrity of mucosa at cell-cell junctions of colon epithelial cells, whereas the binding with FadA increases the endothelial permeability and allows activating β-catenin and uncontrolled cell proliferation, predisposing the host to the development of adenomas, induces oxidative stress, and stimulates the immune system [51]. Altered functions of the intestinal barrier allow other bacteria, called passengers, to cross the colon epithelium. Indeed, dysregulation of the barrier is associated with the increased production of IL-23 [92] and the induction of proinflammatory cytokines, including IL-17 that generates a proinflammatory microenvironment recruiting tumor-infiltrating immune cells (i.e., tumor-infiltrating lymphocytes, TILs) [6, 7, 51]. In addition, McCoy and coauthors observed a significant positive link between IL-10 and TNF-α gene expression and Fn in colorectal adenomas, suggesting their potential role in gut mucosal inflammation [93]. Proinflammatory cytokines can also induce DNA methyltransferases (DNMT) with the silencing of tumor suppressor genes. Inflammation associated with Fn activates the Wnt/β–catenin pathway through the production of chemical mediators as cyclooxygenase-2 (COX-2) and Prostaglandin E2, producing a tumor microenvironment and finally promoting CRC progression. Indeed, COX-2 generates reactive aldehydes that modify proteins, induce damage DNA, and can also activate RAS and PI3K signaling pathways. This scenario is also associated with ROS production by inflammatory cells.
Escherichia coli (Ec) is a gut commensal and its oncogenic potential is linked with the ability of some strains to produce toxins such as cytolethal distending toxin and colibactin, which promote inflammation and are involved in colon carcinogenesis through DNA breaks and mutations [51, 54]. Colibactin is a genotoxin encoded by the multienzymatic machinery tumor-promoting polyketide synthase (pks) islands that increase cell proliferation and are characterized by double-stranded DNA breaks and impaired DNA repair [53, 54].
Enterotoxigenic Bacteroides fragilis (Bf) secretes the Bacteroides fragilis toxin (BFT) and is associated with inflammatory Th17 cells. It activates spermine oxidase (SPO) with ROS production and DNA damage [54]. BFT binds to a specific colon cell receptor and activates Wnt and NF-kB signaling pathways. This determines an increase in cell proliferation, the production of proinflammatory mediators, and DNA damage [53]. Among other bacteria, Enterococcus faecalis (Ef) produces superoxide that induces DNA mutations, while Helicobacter pylori (Hp) infection is associated with an increased risk of serrated polyps [55].
Signs of dysbiosis occur early in the colorectal adenoma-carcinoma sequence so that the mucosal microbiota shows distinct structural modifications during the different steps of colorectal carcinogenesis [94]. Adenomas were found to be rich in Escherichia coli, Pseudomonas veronii, and members of the Enterobacteriaceae, while B. fragilis increases during adenoma-to-carcinoma progression [94].
Differences in gut bacteria composition were detected in patients with various histological types of polyps. Fn, Ef, Streptococcus bovis (Sb), enterotoxigenic Bf, and lower numbers of Lactobacillus spp., Roseburia spp., and Bifidobacterium spp. were detected in the colon of patients with tubular adenoma and villous/tubulovillous polyps compared to healthy subjects and patients with hyperplastic polyps or sessile serrated polyps [95]. These findings suggest that microbiota affects the development of adenomatous polyps, but not sessile serrated adenomas, which are located in specific areas of colorectum [96].
Microenvironment homeostasis and differential expression of Wnt signaling components are influenced by bacterial colonization [97]. Furthermore, functional interactions were suggested by the association between loss-of-function mutations in tumor pathway genes (including Wnt) and displacements in the abundance of specific sets of bacterial taxa [98]. In the intestinal epithelium, the expression of antimicrobial peptides regulates defense against infection and homeostasis thanks also to the cholinergic nervous system that controls antimicrobial gene expression. The release of acetylcholine from neurons induced by infections can stimulate muscarinic signaling in the epithelium, inducing downstream the expression of the canonical Wnt signal which, by determining the expression of type C lectin and lysozyme, integrates the host defense [99].
In animal models of colon carcinogenesis, the use of nonsteroidal anti-inflammatory drugs (NSAIDs) can inhibit antigen-presenting cells and control intestinal inflammation and intestinal immune homeostasis via the canonical signal Wnt [100]. In fact, commensal-polarized macrophages induce gene mutation, chromosomal instability, and endogenous transformation through microbiome-induced bystander effects (MIBE) that activate Wnt/β-catenin signaling [101]. The administration of NSAIDs may contribute to the downregulation of the canonical Wnt/β-catenin pathways acting as PPAR agonists, which can promote cell cycle arrest, cell differentiation, and apoptosis and reduce inflammation, oxidative stress, proliferation, invasion, and cell migration [102].
In addition, the type I interferons (IFNs) produced in the gut under the influence of microbiota play an important role in controlling the proliferation and function of the intestinal epithelium in the context of β-catenin activation [103]. The mechanism of bacterial production of ROS in phagocytes in response to ligand binding with formyl peptide receptors (FPR) and subsequent activation of NADPH oxidase 2 (Nox2) was well defined while the response to microbial signals from Nox1 has not been fully investigated in epithelial cells [104].
ROS enzymatically generated may modulate many signal transduction pathways inducing transient oxidation of sensitive thiol groups in sensory proteins. Examples of redox-sensitive proteins include tyrosine phosphatases that act as MAPK pathway regulators, focal kinase adhesion, and components involved in NF-kB activation [104].
Changes in gut commensal bacteria environment can confer resistance to/or promote infection by pathogenic bacteria and activate inflammation signaling by toll-like and interleukin-1 receptors (TLR and IL-1R) [105]. This is further evidenced by the observations that tumor suppression activity has been demonstrated by some negative TLR and IL-1R signaling regulators and commensal bacteria in the gastrointestinal tract. Modulators of innate immunity can act as a bridge between the inflammatory signaling TLR/IL-1R and the oncogenic RAS signaling pathway, which represents the first necessary path to the onset of colon cancer [106, 107].
4. Interplay between Colon Epithelium Renewing and Oxidative Stress
The colon epithelium is constantly renewed and arises from only a few intestinal stem cells residing at the crypt base. The epithelial cell layer is derived and is progressively differentiated from these amplifying cells until the top of the villi [108]. The stem cell niche is a microenvironment required to maintain the “staminality” of a stem cell proper lineage ratios; it also supports the absorptive, secretory, and barrier functions [109].
The maintenance and regeneration of epithelial organs take place through the simultaneous production of proliferative and differentiation signals. Deregulation of these signals in intestinal epithelium leads to the genesis of small lesions called aberrant crypt foci, whose expansion causes the adenoma that can progress to in situ carcinoma and then to invasive adenocarcinoma [110]. Molecular studies have long defined that these stages on the development of colon cancer are driven by a progressive accumulation of somatic/genetic alterations which confers an advantage in uncontrolled growth [111]. Besides the mechanistic studies focused on the intrinsic alterations of tumor cells, in recent years, the involvement of the activation of the local rather than peripheral immune response has been considered as a fundamental component of the prevention of neoplastic transformation of the epithelium of the colon. The theory of immune-surveillance, postulated for several decades ago, hypothesized that a normal cell that acquires oncogenic mutations can be recognized as foreign and eliminated by the immune system [112, 113]. Actually, different findings support this theory, since cancer progression resulted in changes in the composition of tumor-infiltrating cells in the suppressive immune microenvironment [114].
4.1. Self-Renewal Dysregulation of the Intestinal Epithelium
The rapid self-renewal of the intestinal epithelium and the differentiation gradient in the crypt-villus structure is controlled by the integration of multiple and redundant cell signaling pathways, such as PI3K/AKT, ERK1/2, β–Catenin/GSK3, SMAD, NICD, and JAK/STAT1, which are activated via mechanisms of hormesis by paracrine ligands released from all cells of the tissue community. Finally, the levels of the first and second messengers elicit the effects on the intestinal epithelium renewal and differentiation [115]. In the colon mucosa, the control of the balance between differentiation and renewal is mainly guided by Hedgehog (Hh), bone morphogenetic protein (BMP), Notch, Hippo, and Wnt signaling pathways [108, 116–120].
The Hedgehog signaling acts as a prodifferentiative force in the development and normal homeostasis of the intestinal epithelium. The expression signatures suggest that stromal Hh signaling activity exerts a prodifferentiative influence on intestinal epithelial stem cells, mediated at least in part by the modulation of BMP signaling factors secreted by stromal cells that negatively regulate self-renewal of Lgr5+ intestinal stem cells, and constrains the expansion of intestinal epithelium, therefore attenuating colorectal cancer formation [121]. In the adult intestinal homeostasis, a reduction of Hh signaling increases Wnt activity and intestinal stem cell compartment [122–124].
The Hippo kinase cascade pathway promotes the cytoplasmic localization of YAP/TAZ, which restricts cell proliferation and induces apoptosis. The enterocyte self-renewal and crypt regeneration are triggered by YAP through Wnt/β-catenin signaling. This mechanism also stimulates epithelial cell proliferation following epithelial damage and may facilitate the promotion of associated cancer development to colitis through signals of chronic inflammation and excessive tissue regeneration [125].
Differentiation and proliferation of the epithelium in the intestine are redox-sensitive and regulated by NADPH oxidases [126, 127]. These physiological processes must be highly controlled and balanced. The ROS produced by enzymes of the NADPH oxidase family behave as second messengers for cellular signaling [128].
In the gut, NADPH oxidase 1 (NOX1) is the major expressed NADPH oxidase and, together with p47phox (alias Neutrophil cytosol factor 1) and NADPH oxidase organizer 1 (NOXO1), mediates ROS formation that facilitates the proliferation of colon epithelial cells [129]. NOXO1 acts as a mediator of constant redox-dependent signaling in the differentiation, proliferation, or cell survival. In epithelial cells, the absence of NOXO1 promotes proliferation and reduction of apoptosis supporting malignant transformation and tumor development [129]. NOXO1 is also needed to maintain the activity of the Notch signaling pathway by enabling the activity of a disintegrin and metalloproteinases (ADAM) [130].
Notch also plays an important role in the renewal of the colon epithelium. It is conserved in the cell-cell communication pathway and mediates cell fate decisions during development and in adult tissues [131]. Upon ligand binding, Notch receptors undergo two successive proteolytic cleavages: an ectodomain cleavage followed by intramembrane proteolysis mediated by γ-secretase. This process releases the Notch intracellular domain (NICD), which translocates to the nucleus and binds CSL (an acronym for CBF-1/RBPJ-κ in Homo sapiens/Mus musculus respectively, Suppressor of Hairless in Drosophila melanogaster, Lag-1 in Caenorhabditis elegans) to activate its target genes like Hes-1 and Hes-5 which contain CSL binding sites [131–133]. The control of Notch receptors proteolysis, throughout the gastrointestinal tract, also regulates the intestinal injury/regenerative responses and drives intestinal inflammation and colon cancer initiation [129, 134].
The involvement of Wnt dysregulation in colorectal tumorigenesis is supported by its critical role in the differentiation of stem cells in intestinal crypts and in the maintenance of intestinal homeostasis [135]. Wnt signal transduction is based on the autocrine and paracrine interaction of secreted Wnt glycoproteins, rich in cysteine, essential for intestinal morphogenesis and for the maintenance of architecture and homeostasis in the adult intestinal epithelium [120, 136].
Furthermore, these pathways are necessary for the function of immune cells [137] of both innate and adaptive responses; in particular, they are necessary for the development of T lymphocytes [138]. For these reasons, it is not at all abstruse that the Wnt molecules seem to be also involved in immune diseases such as thyroiditis and psoriasis [139].
The aberrant regulation of Wnt/β-catenin signaling in the pathogenesis of colorectal cancer progression has long been recognized [140, 141], and it has been observed in 100% of CRCs [142]. Wnt proteins are about 40 kDa in size and contain many conserved cysteines [143]. They exhibit lipid modifications necessary for more efficient signaling and for both their secretion and ability to bind to Frizzled receptors. The changes by palmitoleic acid, a monounsaturated fat, linked to conserved serines seem to play an important role in the lipids for signaling [120]. The role of the lipids is also reflected by the requirement for Porcupine (Porc), a multipass transmembrane O-acyltransferase in the endoplasmic reticulum (ER), which is essential for Wnt palmitoylation and maturation, and is active only in Wnt-producing cells [120].
Importantly, the Wnt pathways transduce signals into many signaling cascades and protein phosphorylation amplifies the signal by modifying multiple substrate molecules. The signaling is activated by binding a Wnt protein ligand to a receptor of the Frizzled family, which sends the biological signal to the Dishevelled protein inside the cell. Three Wnt signaling pathways have been characterized: the β-catenin/canonical pathway, the noncanonical planar cell polarity (PCP) pathway, and the noncanonical Wnt/Ca2+ pathway. The noncanonical Wnt pathways control the activity of small GTPases altering the mechanism of the Actin and the cytoskeletal rearrangement; in this way, the Wnt molecules can stimulate JNK activity (N-terminal kinase c-Jun) or promote adhesion and cellular movement by calcium activating CaMK II and calmodulin [144]. In the absence of Wnt ligands, the cytoplasmic destructive complex of Adenomatous Polyposis Coli/Axin (APC/Axin) regulates the exit of the canonical Wnt pathway by controlling the stability of β-catenin in the cytoplasm, where it binds and phosphorylates the β-catenin through two constitutively active serine-threonine kinases (CK1a and GSK3a/b). This continuous elimination of β-catenin prevents it from reaching the nucleus and the Wnt target genes are then repressed by the DNA-bound T-cell factor/lymphoid enhancer factor (TCF/LEF) proteins [120]. The mutations in the molecules that are part of the Wnt/β-catenin pathway (in particular, the truncating mutations in the APC gene) lead to the formation of constitutive nuclear TCF/β-catenin complexes and to the uncontrolled transcription of target genes. This decompensation of Wnt signaling is present in at least 80% of colorectal carcinomas [145]. The APC protein, associated with the microtubule cytoskeleton, has an important effect on the structure and differentiation of intestinal epithelial cells. Thus, APC loss of functions in intestinal cells can lead to the polyp development [146, 147]. Recently, a novel role for APC mutated in reducing the action of the immune system has been suggested, preventing the control of intestinal inflammation. In particular, a deficiency in the APC protein reduces nuclear transcription factor NFAT, thereby preventing T-reg lymphocyte activation and then a failure in the control of local inflammation in the intestine [9]. Recent studies have shown that chronic inflammation and ROS production can activate the Wnt/β-catenin pathways, but the mechanisms involved remain unclear.
4.2. Oxidative Stress
Oxidative damage has been suggested to promote tumor initiation and progression by increasing mutation rates and activating oncogenic pathways [148]. On the other hand, also proinflammatory cytokines trigger oxidative stress that increases mucosal permeability and compromise the regenerative potential of the intestinal epithelium [149].
The most studied source of oxidative stress is attributable to the ROS formation. In fact, the intrinsic and extrinsic environmental stress factors, such as bacterial toxins or lipid overload, can induce ROS production through the activation of phagocytes and resident cells. This process involved the ROS production through microsomes, peroxisomes, and impairment of mitochondrial metabolism-related pathways [150, 151]. For some time, it has been proposed that inflammation is implicated in the onset of chronic degenerative diseases and tumors. This can be explained, at least in part, by overproduction and/or lack of H2O2 degradation.
Mitochondrial processes play an important role in tumor initiation and progression through alteration in glucose metabolism, production of ROS, and compromise of intrinsic apoptotic function [152]. Genetic and epigenetic alterations of Krebs cycle enzymes favour the shift of cancer cells from oxidative phosphorylation to anaerobic glycolysis. The process of maintaining redox homeostasis is driven by genome-wide transcriptional clustering with mitochondrial retrograde signaling and coupled with the glucose metabolic pathway and cell division cycle. Abnormalities of Krebs cycle enzymes cause ectopic production of Krebs cycle intermediates (oncometabolites) such as 2-hydroxyglutarate and citrate. These oncometabolites, which are important driving forces of cancer pathogenesis and progression, can stabilize hypoxia-inducible factor 1 (HIF1) and nuclear factor-like 2 (Nrf2), inhibit p53 and prolyl hydroxylase 3 (PDH3) activities, and regulate DNA/histone methylation, which in turn activate cell growth signaling. They also stimulate increased glutaminolysis, glycolysis, and production of ROS. Genetic alterations in Krebs cycle enzymes are also involved in increased fatty acid β-oxidations and induction of epithelial-mesenchymal transition (EMT) [153]. The central route for oxidative metabolism is the tricarboxylic acid (TCA) cycle responsible for the production of NADH and FADH2, which fuel the mitochondrial electron transport chain to generate ATP and source of metabolic intermediates required for anabolic reactions important for proliferating cells, which require the precursors for the synthesis of lipids, proteins, and nucleic acids [154].
The average of the ATP concentration over the cell cycle is higher and the pHi is globally more acidic in normal proliferating cells. The NAD+/NADH and NADP+/NADPH redox ratios are, respectively, five and ten times higher in cancer cells compared to the normal cell population as the effect of aerobic glycolysis or Warburg effect in cancer cells [155]. In normal proliferating cells, the Warburg effect is an example of homeostasis of redox status by transiently shifting metabolic flux from OXPHOS to glycolysis to avoid ROS generation during DNA synthesis and protect genome integrity [128]. In contrast, in tumor cells, the Warburg effect determines an alteration of the redox state derived from the glucose metabolic path reprogrammed by the OXPHOS dysfunction; this supports glycolysis and the excessive loss of ROS responsible for cancer progression [128]. For these reasons, the Warburg effect should be downregulated in the precancerous phase, while it should be used in the antitumor response since the response to oxidative stress can improve the action of targeted molecular agents. This implies that the antitumor cellular response induced by oxidative stress in the postcancerous phase should not be downregulated when cancer cells are still present [156].
Inflammation is associated with the production of ROS and oxidative damage of macromolecules such as 7,8-dihydro-8-oxoguanine (8-oxoG) in DNA. Most of these small base modifications are repaired by BER pathway by the pivotal role of the OGG1 and MUTYH glycosylases. OGG1 binds the 8-oxoG base with high affinity and then the complex interacts with canonical RAS family GTPases to catalyze the replacement of GDP with GTP, thus serving as a guanine nuclear exchange factor. OGG1-mediated activation of RAS leads to the phosphorylation of the mitogen-activated kinases MEK1,2/ERK1,2 and the increase of the downstream gene expression [10, 11]. A recent eminent study shows that OGG1 inhibition is able to alleviate inflammatory conditions in vivo [157]. MUTYH is a base excision repair glycosylase that removes adenine opposite 8-oxoguanine [158]. MUTYH has evolved from an OG:A mispair glycosylase to a multifunctional scaffold for rapid DNA damage response to a wide variety of DNA damaging signaling including PARP activation, ATR signaling, and SIRT6 activity. MUTYH inhibits the repair of alkyl-DNA damage and cyclopyrimidine dimers interaction with mismatch repair [159]. Many of the MAP variants encompass amino acid changes that occur at positions surrounding the two-metal cofactor-binding sites of MUTYH. One of these cofactors, found in nearly all MUTYH orthologs, is a [4Fe-4S]2+ cluster coordinated by four Cys residues located in the N-terminal catalytic domain [159] that may be a redox-sensitive target [160]. In the model of ulcerative colitis, Mutyh plays a major role in maintaining intestinal integrity by affecting the inflammatory response. Adenomas from Mutyh-/- mice had a greater infiltrate of Foxp3+ T regulatory cells, granulocytes, macrophages, myeloid-derived suppressor cells, and strong expression of TGF-β-latency-associated peptide and IL6. Then, MUTYH loss is associated with an increase in CRC risk involving immunosuppression and altered inflammatory response [161].
5. Major Colorectal Adenomatous Polyposis Syndromes
The major colorectal adenomatous polyposis syndromes that predispose to the development of CRC are divided into two groups based on predisposition: autosomal recessive and autosomal dominant disorders. The known autosomal recessive adenomatous polyposis syndromes are MUTYH-associated polyposis (MAP) and NTHL1 associated tumor syndrome. MUTYH and NTHL1 are DNA glycosylase genes of the BER. The first has a key role in the repair of oxidative DNA damage; the second, together with OGG1, does not contribute significantly to autosomal recessive polyposis [162]. BER is a single-strand DNA repair mechanism used by cells to maintain genomic integrity. This pathway is involved in the correction of events caused by oxidative damage, alkylation and deamination, and defects in its components give high-penetrant predisposition to develop polyposis [162]. For this reason, in these forms of polyposis, the greatest damage caused by oxidative stress occurs.
The MAP is the second most common high-penetrant Mendelian cancer syndrome associated with adenomatous polyposis [163]. Tumor sequencing has identified a specific mutation signature associated with germline MUTYH mutations (base excision repair defects), evidenced by an increase of somatic G:C to T:A base pair transversion [164]. This somatic signature might aid the invariant classification of rare MUTYH variants. Two variants c.536A>G, p.Tyr179Cys and c.1187G>A, p.Gly396Asp account for 70% to 80% of pathogenic MUTYH mutations in Europeans; homozygous p.Tyr179Cys mutations have a more aggressive phenotype than homozygous p.Gly396Asp or compound heterozygous p.Gly396Asp/p.Tyr179-Cys mutations [164, 165].
In the NTHL1-associated tumor syndrome, an association between base excision repair defects and specific somatic mutation signature in adenomas is found [162]. NTHL1, a novel recessive polyposis and CRC-predisposing gene, is the second DNA glycosylase gene of the BER pathway with a high-penetrant predisposition to develop polyposis. At first, p.Gln90 nonsense mutation was detected in the NTHL1 gene, and then the other eight different pathogenic variants, all of which are nonsense or frameshift mutations, have been detected in patients with NTHL1-associated polyposis [162, 166, 167]. Patients develop multiple adenomatous polyps and CRC between the ages of 40 and 65 years, with a strongly resembling MAP phenotype [162, 166]. There are differences in the somatic mutation spectrum associated with the inactivation of the two DNA glycosylases. For NTHL1, somatic nonsense mutations involving C:G>T:A transitions were detected. The reason for this difference lies in the substrate specificity of the two enzymes. In the BER pathway, the 8-oxoguanine produced after oxidative damage is recognized and excised by the DNA glycosylase OGG1; then MUTYH removes the adenine base incorporated opposite to 8-oxoguanine [168]. Conversely, NTHL1 specifically targets oxidized pyrimidines and products of cytosine oxidation, which are strongly mutagenic due to their ability to mispair with adenine, causing C>T transitions [168]. Phenotypic characterization of additional families will increase the knowledge of tumor spectrum and cancer risk in association with NTHL1-associated tumor syndrome. Mutations in the NTHL1 gene are extremely rare in the population and this syndrome is at least fivefold less frequent than MAP [167].
The polyposis-associated syndromes with autosomal dominant predisposition are familial adenomatous polyposis (FAP) and polymerase proofreading-associated polyposis (PPAP).
FAP accounts for approximately 1% of CRC and is the most common high-penetrant Mendelian syndrome that predisposes to adenomatous polyposis [169]. FAP is the major hereditary predisposition event leading to CRC development and is caused by truncating mutations in the APC gene [140, 170]. APC is essential for the development and homeostasis and its inactivation facilitates tumorigenesis. Heterozygotes develop multiple colonic polyps due to the loss of heterozygosity that for unclear reasons favours the growth of colonocytes in humans. Virtually, all patients with FAP will develop colorectal cancer unless the colon is removed. Somatic truncation mutations are also found in more than 80% of sporadic colorectal cancers and loss of heterozygosity (LOH) of chromosome 5q is found in 30–40% of CRC cases [171, 172]. Oncogenic APC mutations cluster in the mutation cluster region (MCR) [173]. APC is best known as a scaffold protein in the β-catenin destruction complex in the Wnt pathway, and its activity is antagonized by canonical Wnt signaling. Mutations in APC disrupt the degradation complex, deleting the Axin interaction domain that confers the turnover of β-catenin. This causes the stabilization of β-catenin and constitutive activation of the canonical Wnt pathway. Also, the loss of APC leads to the accumulation of nuclear β-catenin which activates the targets of the canonical Wnt pathway, the transcription factors T-cell factor (TCF), and lymphoid enhancer factor (LEF) [174]. APC alterations are an initiating event for sporadic CRC except for those carrying a CpG island methylator phenotype (CIMP) or hypermutable microsatellite instability (MSI) due to a defect in the MMR genes [175]. Patients with a familial risk of FAP have shown lower levels of ROS in the whole blood than patients with sporadic CRC [176]. This suggests that oxidative stress may play a crucial role in sporadic CRCs while its action could be less pivotal when the APC gene is mutated and CRC has an earlier onset. APC contributes to adenoma formation but some of its roles remain to elucidate. Inactivation of APC contributes to cancer development through processes besides the Wnt signaling. Conversely, recent evidences support the hypothesis of a new potential role of gain of function of APC truncations in colon cancer initiation and progression in addition to the loss of function [12]. Intracellular and/or oxidative microenvironment could promote the production of APC protein. This hypothesis is supported by the demonstration, in a recent paper, that in HT29 CRC cell line full-length APC requires mitochondrial respiratory ROS production to stimulate apoptosis [177]. But to better understand this paradox, first, it is necessary to examine the three models regarding APC. The two-hit hypothesis states that one of the copies of the gene is inactivated by a truncated mutation and the other by similar mutation or LOH [178]. The “three-hit hypothesis” was subsequently proposed, which states that mutant APC proteins retain some functions; thus, the third hit could affect the residual part of the gene with copy number gains or deletions [179]. The hypothesis of the functional implications of β-catenin shuttling is poorly understood. It might be possible according to the theory called “just-right nuclear export activity” that the loss of the central nuclear export signals—adjacent to the MCR region—has reduced nuclear export activity that compromises APC tumor-suppressing function [180, 181].
A novel function of APC is also regulating DNA repair modulating BER [182]. APC has also been found to shuttle into the nucleus, where it inhibits the assembly of base excision repair [183, 184] directly binding to APE1 endonuclease. The DNA repair inhibitory (DRI) domain of APC is located in the N-terminal region and is retained in APC mutants, allowing CRC cells to accumulate genetic alterations and to be more susceptible to DNA damaging chemotherapeutic agents [12].
In this context, the APC role in apoptosis supports tumor suppressor function. In those cells with a high extent of damaged DNA, the APC level increases and blocks BER leading to apoptosis. But the overall role of APC in carcinogenesis remains paradoxal [182]. Thus, this function of APC to unbalance BER may favour chromosome instability (CIN) and then carcinogenesis (Figure 1). In this scenario, since APC does not help the repair of DNA damage, for example, from chemotherapeutic agents that are therefore more effective in a CRC cell with an APC mutation than with wild type APC, indeed with increased APC levels, the CRC cell, thus damaged, undergoes apoptosis.
Figure 1.
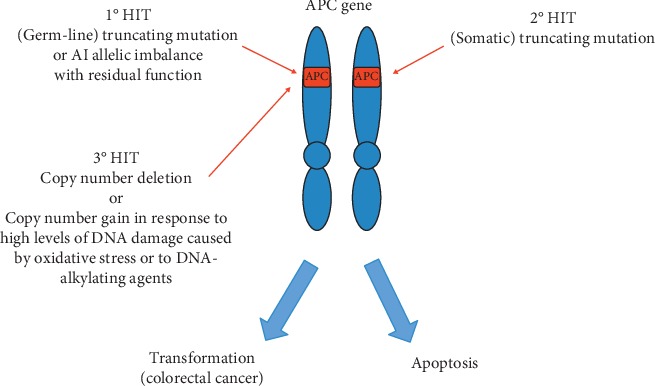
Hypothetical model of interaction between APC and oxidative stress in carcinogenesis. The figure shows a hypothetical model of inactivation of the APC gene that takes into account the influence of oxidative stress. In this model, the known “three-hit hypothesis” is further updated stating that mutant APC protein retains some functions. Thus, the third hit could affect the residual part of the gene with copy number gains or deletions. We now propose APC copy number gain in response to high levels of DNA damage caused by oxidative stress or by DNA-alkylating agents.
Another mechanism of inactivation is a decrease in transcript expression, as previously described [185, 186], in which APC showed a reduced germline expression regardless of the presence of the mutation.
Of the ∼20% of sporadic CRCs that have intact APC gene, many contain mutations in the N-terminal phosphorylation sites in β-catenin, sites that mediate its proteosomal degradation. This could represent a compelling genetic argument that APC suppresses intestinal neoplasia through the inhibition of the canonical Wnt/β-catenin pathway.
PPAP is an autosomal dominant syndrome caused by monoallelic germline mutations in the exonuclease (proofreading) domains of POLE and POLD1 [187–189]. POLE and POLD1 encode ε and δ polymerases, respectively, and accurate proofreading via their exonuclease domains is required to correct mispaired bases inserted during DNA replication. POLD1 (Polδ) has exonuclease and polymerase activities and is critical in BER and MMR [190]. Both POLE and POLD1 have been associated with an increased risk of endometrial cancer and furthermore POLD1 has been associated with breast and brain tumors in addition to CRC and endometrial cancer [191]. One of the common germline variants in POLE, encoding the pathogenic p.Lys424Val, has been encountered with variable frequencies in different cohorts of individuals with unexplained polyposis and/or early-onset and familial CRC [190, 191]. In PPAP patients, the proofreading activity of POLE or POLD1 is impaired, whereas the polymerase activity is unaffected. As a consequence, these patients accumulate base substitutions during life, which eventually results in the development of hypermutated tumors [190, 191]. The clinical phenotype of PPAP has not yet been precisely established, but the data strongly indicate that PPAP results in a high-penetrant predisposition to develop polyposis, early-onset CRC, and extracolonic tumors, including endometrial, stomach, and duodenal tumors [190, 191].
The study from Haraldsdottir supported the hypothesis of the coexistence of somatic MMR alterations with somatic mutations in DNA polymerase POLE or POLD1 in patients with hypermutated colon and endometrial cancers without germline MMR mutations [192]. It is not yet clear if mutations in the DNA polymerase genes are the initiating events causing MMR gene mutations or vice versa. As the critical role of POLD1 in BER as in MMR, the potential function of oxidative stress in PPAP polyposis should be investigated.
Serrated Polyposis Syndrome (SPS) is another genetic disease of the colon whose inheritance remains unknown: both autosomal recessive and autosomal dominant patterns have been suggested [193]. The serrated lesions, unlike conventional adenomas which are uniformly dysplastic, contain no dysplasia and include the hyperplastic polyps. Approximately half of the cancers in the serrated pathway have microsatellite instability. Sessile serrated polyps, common in the proximal colon, the same location where hypermethylated cancers are more common, have a high prevalence of mutations in the BRAF oncogene and hypermethylation due to epigenetic inactivation of the promoter region of MLH1 gene. Some evidences suggest that the sessile serrated polyp-to-cancer sequence takes 10 to 20 years, the same time frame generally accepted for the conventional adenoma-to-cancer sequence. The serrated neoplasia is characterized by the presence of multiple serrated adenomas including sessile and traditional serrated adenomas [194]. First- and second-degree relatives of individuals with SPS are at increased risk of developing CRC, which occurs on average in subjects aged between 50 and 60 years, but the percentage remains uncertain [195]. The serrated pathway is also characterized by mutations of KRAS which, together with BRAF, are thought to initiate the development of serrated adenomas via activation of the MAP kinase pathway. Activation of Wnt signaling in the progression of this kind of adenoma to carcinoma is less clear and in the past individuals with SPS were tested for APC and MUTYH mutations, founding some missense APC mutations in patients with serrated pathway neoplasia. Other affected individuals were found to carry a mutation in SMAD4, BMPR1A, and PTEN [196–198]. To date, the genetic basis of SPS remains to be determined. Recently, a germline mutation in RING finger protein 43 (RNF43) was found to segregate in a family with SPS phenotype [199]. RNF43 encodes an E3 ubiquitin ligase that negatively regulates Wnt signaling [200]. Somatic RNF43 mutations have been identified in up to 18% of CRCs with molecular features of SPS [201, 202].
6. Conclusion and Therapeutic Implications
The molecular biology of early carcinogenesis is controlled by genomic susceptibility, metabolic reprogramming, and microenvironment.
The development of adenomatous polyps passes through the interaction between oxidative stress and pathways involved in the colon epithelium renewal and immune defense. Recent studies have shown that chronic inflammation and oxidative stress can activate the Wnt/β-catenin pathways, but the mechanisms involved remain unclear. The most studied source of oxidative stress is ROS whose production is induced through the activation of phagocytes and resident cells after stimulation by environmental stress factors, such as bacterial toxins or lipid overload. Mitochondria may play an important role in tumor initiation and progression through metabolic processes and ROS production.
Inflammation is also associated with oxidative damage of macromolecules such as 7,8-dihydro-8-oxoguanine (8-oxoG) in DNA. Most of these small base modifications are repaired by the BER. The administration of NSAIDs may contribute to the downregulation of the canonical Wnt/β-catenin pathways acting as PPAR agonists. In fact, PPAR agonists can reduce inflammation, oxidative stress, and proliferation promoting cell cycle arrest, cell differentiation, and apoptosis.
As regards ROS, data on their role in the pathogenesis of colorectal cancer are accumulating. Patients with advanced cancer of the colon, pancreas, and breast showed extensive granulocyte activation with the release of ROS, which could be an important factor in the process of carcinogenesis.
In the control of intestinal inflammation, a new role for mutated APC has been suggested in reducing the action of the immune system. In particular, a deficiency in the APC protein reduces nuclear transcription factor NFAT, thereby preventing T-reg lymphocyte activation and then a failure in the control of local inflammation in the intestine.
Given the multiple roles of APC, new therapeutic opportunities could be addressed. APC restoration leading to tumor regression has been observed. In this model, the reacquisition of the self-renewal and multilineage differentiation capability and reestablishment of the normal crypt-villus homeostasis can restore homeostasis in the intestinal crypt. Moreover, the function of APC protein in regulating DNA repair is very important as a therapeutic implication making DNA damaging chemotherapeutic agents more effective in CRC cells that tend to accumulate mutations. Additional studies will determine whether approaches based on Wnt inhibition would provide long-term therapeutic value in CRC, but it is clear that APC disruption plays a central role in driving and maintaining tumorigenesis.
The findings that high levels of ROS were found in the blood of patients with sporadic CRC and in the normal-appearing rectal mucosa of patients with history of CRC, compared to patients with familial risk of FAP, not only confirm the crucial role of oxidative stress in CRC but also suggest a minor role of ROS when APC expression is completely lost, as in patients with FAP.
Also, microbiota may influence microenvironmental homeostasis and differential expression of Wnt signaling components. The association between loss-of-function mutations in genes of different pathways, including Wnt, and shifts in the abundances of specific sets of bacterial taxa are suggestive of potential functional interactions. Differences in lifestyles and exposition to environmental factors may induce microbiota changes and immune response modulation in the colon epithelial microenvironment on the light of homeostatic adaptation to the oxidative stress, thus inducing adenoma onset in specific colorectal areas. The therapeutic approach should take into account the composition of the microbiota and the levels of oxidative stress.
So in this review ultimately we conclude that oxidative stress may be important in inducing colorectal adenomas through the interaction with Wnt signaling and DNA damage response. A deeper insight into the pathophysiological mechanisms influencing the crosstalk among Wnt signaling and/or microbiota dysregulation, oxidative stress, and DNA damage could contribute to better elucidate the causes responsible for CRC onset, with the aim of providing new clinical approaches for the prevention and develop innovative therapeutic strategies for this tumor.
Acknowledgments
This study was supported by the Italian Ministry of Instruction, University and Research (GMA, MCC).
Abbreviations
- 8-oxoG:
7,8-dihydro-8-oxoguanine
- ADAM:
A disintegrin and metalloproteinases
- APC:
Adenomatous polyposis Coli
- APE:
Apurinic/Apyrimidinic Endonuclease
- ATR:
Serine/threonine-protein kinase
- BER:
Base excision repair
- BMI:
Body mass index
- BMP:
Bone morphogenetic protein
- BMPR1A:
Bone morphogenetic protein receptor, type IA
- BRAF:
B-Raf proto-oncogene, serine/threonine kinase
- CIN:
Chromosome instability
- CIMP:
CpG island methylator phenotype
- COX-2:
Cyclooxygenase 2
- CaMK II:
Ca 2+ /calmodulin-dependent protein kinase II
- CRC:
Colorectal cancer
- DNMT:
DNA methyltransferases
- ERK:
Extracellular signal-regulated kinase
- FAD/FADH:
Flavin adenine dinucleotide/semiquinone
- FPR:
Formyl peptide receptors
- GSK3:
Glycogen synthase kinase 3
- Hg:
Hedgehog
- HNE:
4-hydroxynonenal
- HIF:
Hypoxia-induced factor
- IGF-1:
Insulin-like growth factor
- IFNs:
Type I interferons
- IL-1R:
Interleukin-1 receptor
- IL-6:
Interleukin 6
- JAK:
Janus kinase
- JNK:
c-Jun N-terminal kinase
- LDL:
Low-density protein
- MAP:
MUTYH-associated polyposis
- MAPK:
Mitogen-activated protein kinase
- MIBE:
Microbiome-induced bystander effects
- MHL1:
MutL homolog 1
- MMR:
DNA mismatch repair
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- NAFLD:
Nonalcoholic fatty liver disease
- NER:
Nucleotide excision repair
- NFAT:
Nuclear factor of activated T-cells
- NICD:
Notch intracellular domain
- NSAIDs:
Nonsteroidal anti-inflammatory drugs
- NF-kB:
Nuclear factor-kB
- NOX:
NADPH oxidase
- NOXO1:
NADPH oxidase organizer 1
- NRF2:
Nuclear factor erythroid 2-related factor 2
- NTHL1:
Nth like DNA glycosylase 1
- KRAS:
Kirsten RAt Sarcoma
- OCPs:
Organochlorine pesticides
- OGG1:
8-oxoguanine DNA glycosylase-1
- OXPHOS:
Oxidative phosphorylation
- PPAP:
Polymerase proofreading-associated polyposis
- PARP:
Poly (ADP-ribose) polymerase
- PDH3:
Prolyl hydroxylase 3
- PI3K:
Phosphoinositide-3-kinase–protein kinase
- PTEN:
Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase
- PCBs:
Polychlorinated biphenyls
- POLD1:
Polymerase delta 1
- POLE:
DNA polymerase epsilon catalytic subunit A
- PPAR:
Peroxisome proliferator-activated receptors
- ROS:
Reactive oxygen species
- RFP43:
Ring Finger Protein 43
- SCFAs:
Short-chain fatty acids
- SIRT6:
Sirtuin 6
- SMAD:
Small mother against decapentaplegic
- STAT:
Signal transducers and activators of transcription
- TCA:
Tricarboxylic acid
- TAZ:
Transcriptional coactivator with PDZ-binding motif
- TILs:
Tumor-infiltrating lymphocytes
- TNF:
Tumor necrosis factor
- TCF/LEF:
T-cell factor/lymphoid enhancer factor
- TGF-β:
Transforming growth factor-beta
- TLR:
Toll-like receptors
- WNT:
Wingless/It
- YAP:
Yes-associated protein.
Conflicts of Interest
The authors declare that they have no conflicts of interest regarding the publication of this paper.
References
- 1.Bray F., Ferlay J., Soerjomataram I., et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Connell L. C., Mota J. M., Braghiroli M. I., Hoff P. M. The rising incidence of younger patients with colorectal cancer: questions about screening, biology, and treatment. Current Treatment Options in Oncology. 2017;18(4):p. 23. doi: 10.1007/s11864-017-0463-3. [DOI] [PubMed] [Google Scholar]
- 3.Haggar F. A., Preen D. B., Pereira G., Holman C. D., Einarsdottir K. Cancer incidence and mortality trends in Australian adolescents and young adults. BMC Cancer. 2012;12:1982–2007. doi: 10.1186/1471-2407-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonnington S. N., Rutter M. D. Surveillance of colonic polyps: are we getting it right? World Journal of Gastroenterology. 2016;22(6):1925–1934. doi: 10.3748/wjg.v22.i6.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim N. H., Jung Y. S., Jeong W. S., et al. Miss rate of colorectal neoplastic polyps and risk factors for missed polyps in consecutive colonoscopies. Intestinal Research. 2017;15(3):411–418. doi: 10.5217/ir.2017.15.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui G., Yuan A., Goll R., Florholmen J. IL-17A in the tumor microenvironment of the human colorectal adenoma-carcinoma sequence. Scandinavian Journal of Gastroenterology. 2012;47(11):1304–1312. doi: 10.3109/00365521.2012.725089. [DOI] [PubMed] [Google Scholar]
- 7.Su X., Ye J., Hsueh E. C., et al. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. The Journal of Immunology. 2010;184(3):1630–1641. doi: 10.4049/jimmunol.0902813. [DOI] [PubMed] [Google Scholar]
- 8.Jackson D. N., Theiss A. L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes. 2019;26:1–20. doi: 10.1080/19490976.2019.1592421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agüera-González S., Burton O. T., Vázquez-Chávez E., et al. Adenomatous polyposis coli defines treg differentiation and anti-inflammatory function through microtubule-mediated NFAT localization. Cell Reports. 2017;21(1):181–194. doi: 10.1016/j.celrep.2017.09.020. [DOI] [PubMed] [Google Scholar]
- 10.Boldogh I., Hajas G., Aguilera-Aguirre L., et al. Activation of ras signaling pathway by 8-oxoguanineDNA glycosylase bound to its excision product, 8-oxoguanine. The Journal of Biological Chemistry. 2012;287(25):20769–20773. doi: 10.1074/jbc.C112.364620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.German P., Szaniszlo P., Hajas G., et al. Activation of cellular signaling by 8-oxoguanine DNA glycosylase-1-initiated DNA base excision repair. DNA Repair (AMST) 2013;12(10):856–863. doi: 10.1016/j.dnarep.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L., Shay J. W. Multiple roles of APC and its therapeutic implications in colorectal cancer. Journal of the National Cancer Institute. 2017;109(8) doi: 10.1093/jnci/djw332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lieberman D. A., Rex D. K., Winawer S. J., et al. Guidelines for colonoscopy surveillance after screening and polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2012;143(3):844–857. doi: 10.1053/j.gastro.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Lucci-Cordisco E., Risio M., Venesio T., Genuardi M. The growing complexity of the intestinal polyposis syndromes. American Journal of Medical Genetics. Part A. 2013;161(11):2777–2787. doi: 10.1002/ajmg.a.36253. [DOI] [PubMed] [Google Scholar]
- 15.Martínez M. E., Baron J. A., Lieberman D. A., et al. A pooled analysis of advanced colorectal neoplasia diagnoses after colonoscopic polypectomy. Gastroenterology. 2009;136(3):832–841. doi: 10.1053/j.gastro.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corley D. A., Jensen C. D., Marks A. R., et al. Variation of adenoma prevalence by age, sex, race, and colon location in a large population: implications for screening and quality programs. Clinical Gastroenterology and Hepatology. 2013;11(2):172–180. doi: 10.1016/j.cgh.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pommergaard H. C., Burcharth J., Rosenberg J., Raskov H. The association between location, age and advanced colorectal adenoma characteristics: a propensity-matched analysis. Scandinavian Journal of Gastroenterology. 2017;52(1):1–4. doi: 10.1080/00365521.2016.1218929. [DOI] [PubMed] [Google Scholar]
- 18.Klein J. L., Okcu M., Preisegger K. H., Hammer H. F. Distribution, size and shape of colorectal adenomas as determined by a colonoscopist with a high lesion detection rate: influence of age, sex and colonoscopy indication. United European Gastroenterology Journal. 2016;4(3):438–448. doi: 10.1177/2050640615610266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J. F., Tang S. J., Lash R. H., Wu R., Yang Q. Anatomic distribution of sessile serrated adenoma/polyp with and without cytologic dysplasia. Archives of Pathology & Laboratory Medicine. 2015;139(3):388–393. doi: 10.5858/arpa.2013-0523-oa. [DOI] [PubMed] [Google Scholar]
- 20.IJspeert J. E., van Doorn S. C., van der Brug Y. M., et al. The proximal serrated polyp detection rate is an easy-to-measure proxy for the detection rate of clinically relevant serrated polyps. Gastrointestinal Endoscopy. 2015;82(5):870–877. doi: 10.1016/j.gie.2015.02.044. [DOI] [PubMed] [Google Scholar]
- 21.Kim K. H., Kim K. O., Jung Y., et al. Clinical and endoscopic characteristics of sessile serrated adenomas/polyps with dysplasia/adenocarcinoma in a Korean population: a Korean Association for the Study of Intestinal Diseases (KASID) multicenter study. Scientific Reports. 2019;9(1):p. 3946. doi: 10.1038/s41598-019-40559-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kahi C. J., Hewett D. G., Norton D. L., Eckert G. J., Rex D. K. Prevalence and variable detection of proximal colon serrated polyps during screening colonoscopy. Clinical Gastroenterology and Hepatology. 2011;9(1):42–46. doi: 10.1016/j.cgh.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 23.Leggett B., Whitehall V. Role of serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138(6):2088–2100. doi: 10.1053/j.gastro.2009.12.066. [DOI] [PubMed] [Google Scholar]
- 24.Rotondano G., Bianco M. A., Cipolletta L., Marmo R. Prevalence and characteristics of serrated lesions of the colorectum in Italy: a multicentre prospective cohort study. Digestive and Liver Disease. 2015;47(6):512–517. doi: 10.1016/j.dld.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Fu Z., Shrubsole M. J., Smalley W. E., et al. Lifestyle factors and their combined impact on the risk of colorectal polyps. American Journal of Epidemiology. 2012;176(9):766–776. doi: 10.1093/aje/kws157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim H. Y., Kim S. M., Seo J. H., et al. Age-specific prevalence of serrated lesions and their subtypes by screening colonoscopy: a retrospective study. BMC Gastroenterology. 2014;14(1):p. 82. doi: 10.1186/1471-230x-14-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jung Y. S., Kim N. H., Park J. H., Park D. I., Sohn C. I. Appropriate surveillance interval after colonoscopic polypectomy in patients younger than 50 years. Journal of Korean Medical Science. 2019;34(12):p. e101. doi: 10.3346/jkms.2019.34.e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hussein Kamareddine M., Ghosn Y., Karam K., et al. Adenoma detection before and after the age of 50: a retrospective analysis of Lebanese outpatients. BMJ Open Gastroenterology. 2018;5(1) doi: 10.1136/bmjgast-2018-000253.e000253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lieberman D. A., Holub J. L., Moravec M. D., et al. Prevalence of colon polyps detected by colonoscopy screening in asymptomatic black and white patients. Journal of the American Medical Association. 2008;300(12):1417–1422. doi: 10.1001/jama.300.12.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bretthauer M., Kaminski M. F., Løberg M., et al. Population-based colonoscopy screening for colorectal cancer: a randomized clinical trial. JAMA Internal Medicine. 2016;176(7):894–902. doi: 10.1001/jamainternmed.2016.0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lieberman D. A., Williams J. L., Holub J. L., et al. Race, ethnicity, and sex affect risk for polyps >9 mm in average-risk individuals. Gastroenterology. 2014;147(2):351–358. doi: 10.1053/j.gastro.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song L., Liu J., Jin X., et al. p, p′-Dichlorodiphenyldichloroethylene induces colorectal adenocarcinoma cell proliferation through oxidative stress. PLoS One. 2014;9(11) doi: 10.1371/journal.pone.0112700.e112700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petriello M. C., Hoffman J. B., Vsevolozhskaya O., Morris A. J., Hennig B. Dioxin-like PCB 126 increases intestinal inflammation and disrupts gut microbiota and metabolic homeostasis. Environmental Pollution. 2018;242:1022–1032. doi: 10.1016/j.envpol.2018.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gokulan K., Arnold M. G., Jensen J., et al. Exposure to arsenite in CD-1 mice during juvenile and adult stages: effects on intestinal microbiota and gut-associated immune status. mBio. 2018;9(4) doi: 10.1128/mbio.01418-18.e01418–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tinkov A. A., Gritsenko V. A., Skalnaya M. G., et al. Gut as a target for cadmium toxicity. Environmental Pollution. 2018;235:429–434. doi: 10.1016/j.envpol.2017.12.114. [DOI] [PubMed] [Google Scholar]
- 36.Jin Y., Wu S., Zeng Z., Fu Z. Effects of environmental pollutants on gut microbiota. Environmental Pollution. 2017;222:1–9. doi: 10.1016/j.envpol.2016.11.045. [DOI] [PubMed] [Google Scholar]
- 37.Sinha R., Peters U., Cross A. J., et al. Meat, meat cooking methods and preservation, and risk for colorectal adenoma. Cancer Research. 2005;65(17):8034–8041. doi: 10.1158/0008-5472.can-04-3429. [DOI] [PubMed] [Google Scholar]
- 38.Jamin E. L., Riu A., Douki T., et al. Combined genotoxic effects of a polycyclic aromatic hydrocarbon (B(a)P) and an heterocyclic amine (PhIP) in relation to colorectal carcinogenesis. PLoS One. 2013;8(3) doi: 10.1371/journal.pone.0058591.e58591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ribière C., Peyret P., Parisot N., et al. Oral exposure to environmental pollutant benzo[a]pyrene impacts the intestinal epithelium and induces gut microbial shifts in murine model. Scientific Reports. 2016;6(1) doi: 10.1038/srep31027.31027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jia W., Xie G., Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nature Reviews. Gastroenterology & Hepatology. 2018;15(2):111–128. doi: 10.1038/nrgastro.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin O. C. B., Olier M., Ellero-Simatos S., et al. Haem iron reshapes colonic luminal environment: impact on mucosal homeostasis and microbiome through aldehyde formation. Microbiome. 2019;7(1):p. 72. doi: 10.1186/s40168-019-0685-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bastide N., Morois S., Cadeau C., et al. Heme iron intake, dietary antioxidant capacity, and risk of colorectal adenomas in a large cohort study of French women. Cancer Epidemiology, Biomarkers & Prevention. 2016;25(4):640–647. doi: 10.1158/1055-9965.epi-15-0724. [DOI] [PubMed] [Google Scholar]
- 43.Day S. D., Enos R. T., McClellan J. L., et al. Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine. 2013;64(1):454–462. doi: 10.1016/j.cyto.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dowswell G., Ryan A., Taylor A., et al. Designing an intervention to help people with colorectal adenomas reduce their intake of red and processed meat and increase their levels of physical activity: a qualitative study. BMC Cancer. 2012;12(1):p. 255. doi: 10.1186/1471-2407-12-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ashktorab H., Hassanzadeh Namin H., Taylor T., et al. Role of life events in the presence of colon polyps among African Americans. BMC Gastroenterology. 2013;13(1):p. 101. doi: 10.1186/1471-230x-13-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanchez N. F., Stierman B., Saab S., et al. Physical activity reduces risk for colon polyps in a multiethnic colorectal cancer screening population. Biomed Central Research Notes. 2012;5(1):p. 312. doi: 10.1186/1756-0500-5-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ashktorab H., Soleimani A., Nichols A., et al. Adiponectin, leptin, IGF-1, and tumor necrosis factor alpha as potential serum biomarkers for non-invasive diagnosis of colorectal adenoma in African Americans. Frontiers in Endocrinology. 2018;9:p. 77. doi: 10.3389/fendo.2018.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee Y. M., Kim S. A., Choi G. S., et al. Association of colorectal polyps and cancer with low-dose persistent organic pollutants: a case-control study. PLoS One. 2018;13(12) doi: 10.1371/journal.pone.0208546.e0208546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winkels M., Botma A., Van Duijnhoven F. J., et al. Smoking increases the risk for colorectal adenomas in patients with lynch syndrome. Gastroenterology. 2012;142(2):241–247. doi: 10.1053/j.gastro.2011.10.033. [DOI] [PubMed] [Google Scholar]
- 50.Buchanan D. D., Sweet K., Drini M., et al. Risk factors for colorectal cancer in patients with multiple serrated polyps: a cross-sectional case series from genetics clinics. PLoS One. 2010;5(7) doi: 10.1371/journal.pone.0011636.e11636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Irrazábal T., Belcheva A., Girardin S. E., Martin A., Philpott D. J. The multifaceted role of the intestinal microbiota in colon cancer. Molecular Cell. 2014;54(2):309–320. doi: 10.1016/j.molcel.2014.03.039. [DOI] [PubMed] [Google Scholar]
- 52.Rubinstein M. R., Wang X., Liu W., et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesion. Cell Host & Microbe. 2013;14(2):195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun J., Kato I. Gut microbiota, inflammation and colorectal cancer. Genes & Diseases. 2016;3(2):130–143. doi: 10.1016/j.gendis.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tilg H., Adolph T. E., Gerner R. R., Moschen A. R. The intestinal microbiota in colorectal cancer. Cancer Cell. 2018;33(6):954–964. doi: 10.1016/j.ccell.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 55.Kumar A., Kim M., Lukin D. J. Helicobacter pylori is associated with increased risk of serrated colonic polyps: analysis of serrated polyp risk factors. Indian Journal of Gastroenterology. 2018;37(3):235–242. doi: 10.1007/s12664-018-0855-8. [DOI] [PubMed] [Google Scholar]
- 56.Karsenti D., Tharsis G., Burtin P., et al. Adenoma and advanced neoplasia detection rates increase from 45 years of age. World Journal of Gastroenterology. 2019;25(4):447–456. doi: 10.3748/wjg.v25.i4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Solakoğlu T., Atalay R., Köseoğlu H., et al. Analysis of 2222 colorectal polyps in 896 patients: a tertiary referreal hospital study. The Turkish Journal of Gastroenterology. 2014;25(2):175–179. doi: 10.5152/tjg.2014.5059. [DOI] [PubMed] [Google Scholar]
- 58.Jackson C. S., Vega K. J. Higher prevalence of proximal colon polyps and villous histology in African-Americans undergoing colonoscopy at a single equal access center. Journal of Gastrointestinal Oncology. 2015;6(6):638–643. doi: 10.3978/j.issn.2078-6891.2015.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ashktorab H., Paydar M., Namin H. H., et al. Prevalence of colorectal neoplasia among young African Americans and Hispanic Americans. Digestive Diseases and Sciences. 2014;59(2):446–450. doi: 10.1007/s10620-013-2898-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lebwohl B., Capiak K., Neugut A. I., Kastrinos F. Risk of colorectal adenomas and advanced neoplasia in Hispanic, black and white patients undergoing screening colonoscopy. Alimentary Pharmacology and Therapeutics. 2012;35(12):1467–1473. doi: 10.1111/j.1365-2036.2012.05119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katsidzira L., Gangaidzo I. T., Mapingure M. P., Matenga J. A. Retrospective study of colorectal cancer in Zimbabwe: colonoscopic and clinical correlates. World Journal of Gastroenterology. 2015;21(8):2374–2380. doi: 10.3748/wjg.v21.i8.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi Y. J., Seelbach M. J., Pu H., et al. Polychlorinated biphenyls disrupt intestinal integrity via NADPH oxidase-induced alterations of tight junction protein expression. Environmental Health Perspectives. 2010;118(7):976–981. doi: 10.1289/ehp.0901751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Atashgahi S., Shetty S. A., Smidt H., de Vos W. M. Flux, impact, and fate of halogenated xenobiotic compounds in the gut. Frontiers in Physiology. 2018;9:p. 888. doi: 10.3389/fphys.2018.00888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mrema E. J., Rubino F. M., Brambilla G., et al. Persistent organochlorinated pesticides and mechanisms of their toxicity. Toxicology. 2013;307:74–88. doi: 10.1016/j.tox.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 65.Song M., Garrett W. S., Chan A. T. Nutrients, foods, and colorectal cancer prevention. Gastroenterology. 2015;148(6):1244–1260. doi: 10.1053/j.gastro.2014.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Voskuil D. W., Kampman E., Grubben M. J., et al. Meat consumption and meat preparation in relation to colorectal adenomas among sporadic and HNPCC family patients in The Netherlands. European Journal of Cancer. 2002;38(17):2300–2308. doi: 10.1016/s0959-8049(02)00455-0. [DOI] [PubMed] [Google Scholar]
- 67.Harris D. L., Washington M. K., Hood D. B., Roberts L. J., Ramesh A. Dietary fat-influenced development of colon neoplasia in APC Min mice exposed to benzo(a)pyrene. Toxicologic Pathology. 2009;37(7):938–946. doi: 10.1177/0192623309351722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chadolias D., Zissimopoulos A., Nena E., et al. Association of occupational exposures and work characteristics with the occurrence of gastrointestinal disorders. Hippokratia. 2017;21(2):74–79. [PMC free article] [PubMed] [Google Scholar]
- 69.Hemeryck L. Y., Van Hecke T., Vossen E., De Smet S., Vanhaecke L. DNA adductomics to study the genotoxic effects of red meat consumption with and without added animal fat in rats. Food Chemistry. 2017;230:378–387. doi: 10.1016/j.foodchem.2017.02.129. [DOI] [PubMed] [Google Scholar]
- 70.Pierre F., Tache S., Guéraud F., et al. APC mutation induces resistance of colonic cells to lipoperoxide-triggered apoptosis induced by faecal water from haem-fed rats. Carcinogenesis. 2007;28(2):321–327. doi: 10.1093/carcin/bgl127. [DOI] [PubMed] [Google Scholar]
- 71.Maslowski K. M., Vieira A. T., Ng A., et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Canani R. B., Costanzo M. D., Leone L., et al. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World Journal of Gastroenterology. 2011;17(12):1519–1528. doi: 10.3748/wjg.v17.i12.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Botma A., Vasen H. F., van Duijnhoven F. J., et al. Dietary patterns and colorectal adenomas in Lynch syndrome: the GEOLynch cohort study. Cancer. 2013;119(3):512–521. doi: 10.1002/cncr.27726. [DOI] [PubMed] [Google Scholar]
- 74.Simioni C., Zauli G., Martelli A. M., et al. Oxidative stress: role of physical exercise and antioxidant nutraceuticals in adulthood and aging. Oncotarget. 2018;9(24):17181–17198. doi: 10.18632/oncotarget.24729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perše M. Oxidative stress in the pathogenesis of colorectal cancer: cause or consequence? BioMed Research International. 2013;2013:9. doi: 10.1155/2013/725710.725710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Duggan C., Tapsoba J. D., Wang C. Y., et al. Dietary weight loss, exercise, and oxidative stress in postmenopausal women: a randomized controlled trial. Cancer Prevention Research. 2016;11(9):835–843. doi: 10.1158/1940-6207.capr-16-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brenner D. R., Shaw E., Yannitsos D. H., et al. The association between recreational physical activity, sedentary time, and colorectal polyps in a population screened for colorectal cancer. Cancer Epidemiology. 2018;53:12–20. doi: 10.1016/j.canep.2017.12.017. [DOI] [PubMed] [Google Scholar]
- 78.Rezende L. F. M., Lee D. H., Keum N., et al. Physical activity during adolescence and risk of colorectal adenoma later in life: results from the nurses’ health study II. British Journal of Cancer. 2019;121(1):86–94. doi: 10.1038/s41416-019-0454-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Omata F., Deshpande G. A., Ohde S., Mine T., Fukui T. The association between obesity and colorectal adenoma: systematic review and meta-analysis. Scandinavian Journal of Gastroenterology. 2013;48(2):136–146. doi: 10.3109/00365521.2012.737364. [DOI] [PubMed] [Google Scholar]
- 80.Botma A., Nagengast F. M., Braem M. G., et al. Body mass index increases risk of colorectal adenomas in men with Lynch syndrome: the GEOLynch cohort study. Journal of Clinical Oncology. 2010;28(28):4346–4353. doi: 10.1200/jco.2010.28.0453. [DOI] [PubMed] [Google Scholar]
- 81.Shapero T. F., Chen G. I., Devlin T., et al. Obesity increases prevalence of colonic adenomas at screening colonoscopy: a Canadian community-based study. Canadian Journal of Gastroenterology & Hepatology. 2017;2017:8. doi: 10.1155/2017/8750967.8750967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fliss-Isakov N., Zelber-Sagi S., Webb M., et al. Distinct metabolic profiles are associated with colorectal adenomas and serrated polyps. Obesity (Silver Spring) 2017;25(Suppl 2):S72–S80. doi: 10.1002/oby.22001. [DOI] [PubMed] [Google Scholar]
- 83.Duvnjak M., Stojsavljević S., Jukić L. V., Duvnjak L. S. Risk factors for colorectal adenoma-acknowledging the burden of NAFLD. Journal of Clinical and Translational Hepatology. 2019;2(7):97–98. doi: 10.14218/jcth.2019.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen J., Bian D., Zang S., et al. The association between nonalcoholic fatty liver disease and risk of colorectal adenoma and cancer incident and recurrence: a meta-analysis of observational studies. Expert Review of Gastroenterology & Hepatology. 2019;13(4):385–395. doi: 10.1080/17474124.2019.1580143. [DOI] [PubMed] [Google Scholar]
- 85.Chen Q. F., Zhou X. D., Sun Y. J., et al. Sex-influenced association of non-alcoholic fatty liver disease with colorectal adenomatous and hyperplastic polyps. World Journal of Gastroenterology. 2017;23(28):5206–5215. doi: 10.3748/wjg.v23.i28.5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stadlmayr A., Aigner E., Steger B., et al. Nonalcoholic fatty liver disease: an independent risk factor for colorectal neoplasia. Journal of Internal Medicine. 2011;270(1):41–49. doi: 10.1111/j.1365-2796.2011.02377.x. [DOI] [PubMed] [Google Scholar]
- 87.Li Y., Liu S., Gao Y., et al. Association between NAFLD and risk of colorectal adenoma in Chinese han population. Journal of Clinical and Translational hepatology. 2019;7(2):99–105. doi: 10.14218/JCTH.2019.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bhatt B. D., Lukose T., Siegel A. B., Brown R. S., Jr., Verna E. C. Increased risk of colorectal polyps in patients with non-alcoholic fatty liver disease undergoing liver transplant evaluation. Journal of Gastrointestinal Oncology. 2015;6(5):459–468. doi: 10.3978/j.issn.2078-6891.2015.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhan T., Hahn F., Hielscher T., et al. Multiple behavioral factors are associated with occurrence of large, flat colorectal polyps. International Journal of Colorectal Disease. 2017;32(4):575–582. doi: 10.1007/s00384-016-2717-0. [DOI] [PubMed] [Google Scholar]
- 90.Diergaarde B., Braam H., Vasen H. F., et al. Environmental factors and colorectal tumor risk in individuals with hereditary nonpolyposis colorectal cancer. Clinical Gastroenterology and Hepatology. 2007;5(6):736–742. doi: 10.1016/j.cgh.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 91.Allen J., Sears C. L. Impact of the gut microbiome on the genome and epigenome of colon epithelial cells: contributions to colorectal cancer development. Genome Medicine. 2019;11(11):1–18. doi: 10.1186/s13073-019-0621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grivennikov S. I., Wang K., Mucida D., et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491(7423):254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McCoy A. N., Araújo-Pérez F., Azcárate-Peril A., et al. Fusobacterium is associated with colorectal adenomas. PLoS One. 2013;8(1) doi: 10.1371/journal.pone.0053653.e53653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakatsu G., Li X., Zhou H., et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nature Communications. 2015;6(1):p. 8727. doi: 10.1038/ncomms9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weng M. T., Chiu Y. T., Wei P. Y., et al. Microbiota and gastrointestinal cancer. Journal of the Formosan Medical Association. 2019;118(Suppl 1):S32–S41. doi: 10.1016/j.jfma.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 96.Rezasoltani S., Asadzadeh Aghdaei H., Dabiri H., et al. The association between fecal microbiota and different types of colorectal polyp as precursors of colorectal cancer. Microbial Pathogenesis. 2018;124:244–249. doi: 10.1016/j.micpath.2018.08.035. [DOI] [PubMed] [Google Scholar]
- 97.Neumann P. A., Koch S., Hilgarth R. S., et al. Gut commensal bacteria and regional Wnt gene expression in the proximal versus distal colon. American Journal of Pathology. 2014;184(3):592–599. doi: 10.1016/j.ajpath.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burns M. B., Montassier E., Abrahante J., et al. Colorectal cancer mutational profiles correlate with defined microbial communities in the tumor microenvironment. PLoS Genetics. 2018;14(6) doi: 10.1371/journal.pgen.1007376.e1007376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Labed S. A., Wani K. A., Jagadeesan S., et al. Intestinal epithelial Wnt signaling mediates acetylcholine-triggered host defense against infection. Immunity. 2018;48(5):963–978. doi: 10.1016/j.immuni.2018.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Swafford D., Shanmugam A., Ranganathan P., et al. Canonical Wnt signaling in CD11c+ APCs regulates microbiota-induced inflammation and immune cell homeostasis in the colon. Journal of Immunology. 2018;200(9):3259–3268. doi: 10.4049/jimmunol.1701086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang X., Yang Y., Huycke M. M. Commensal-infected macrophages induce dedifferentiation and reprogramming of epithelial cells during colorectal carcinogenesis. Oncotarget. 2017;8(60):102176–102190. doi: 10.18632/oncotarget.22250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vallée A., Lecarpentier Y., Vallée J. N. Targeting the canonical WNT/β-Catenin pathway in cancer treatment using non-steroidal anti-inflammatory drugs. Cells. 2019;8(7):p. E726. doi: 10.3390/cells8070726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Katlinskaya Y. V., Katlinski K. V., Lasri A., et al. Type I interferons control proliferation and function of the intestinal epithelium. Molecular Cell Biology. 2016;36(7):1124–1135. doi: 10.1128/MCB.00988-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jones R. M., Neish A. S. Redox signaling mediated by the gut microbiota. Free Radical Biology and Medicine. 2017;105:41–47. doi: 10.1016/j.freeradbiomed.2016.10.495. [DOI] [PubMed] [Google Scholar]
- 105.Pickard J. M., Zeng M. Y., Caruso R., Núñez G. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunological Reviews. 2017;279(1):70–89. doi: 10.1111/imr.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Klimesova K., Kverka M., Zakostelska Z., et al. Altered gut microbiota promotes colitis-associated cancer in IL-1 receptor-associated kinase M-deficient mice. Inflammatory Bowel Diseases. 2013;19(6):1266–1277. doi: 10.1097/MIB.0b013e318281330a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li T. T., Ogino S., Qian Z. R. Toll-like receptor signaling in colorectal cancer: carcinogenesis to cancer therapy. World Journal Gastroenterology. 2014;20(47):17699–17708. doi: 10.3748/wjg.v20.i47.17699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nature Reviews Molecular Cell Biology. 2014;15:19–33. doi: 10.1038/nrm3721. [DOI] [PubMed] [Google Scholar]
- 109.Lander A. D., Kimble J., Clevers H., et al. What does the concept of the stem cell niche really mean today? BMC Biology. 2012;10:p. 19. doi: 10.1186/1741-7007-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vries R. G., Huch M., Clevers H. Stem cells and cancer of the stomach and intestine. Molecular Oncology. 2010;4(5):373–384. doi: 10.1016/j.molonc.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kinzler K. W., Vogelstein B. The Genetic Basis of Human Cancer. New York, NY, USA: McGraw-Hill; 1998. [Google Scholar]
- 112.Schreiber R. D., Old L. J., Smyth M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 113.Finn O. J. Cancer immunology. The New England Journal of Medicine. 2008;358:2704–2715. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 114.Galon J., Costes A., Sanchez-Cabo F., et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 115.Medema J. P., Vermeulen L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature. 2011;474:318–326. doi: 10.1038/nature10212. [DOI] [PubMed] [Google Scholar]
- 116.Qi Z., Li Y. H., Zhao B., et al. BMP restricts stemness of intestinal Lgr5(+) stem cells by directly suppressing their signature genes. Nature Communication. 2017;8:p. 13824. doi: 10.1038/ncomms13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.López-Arribillaga E., Rodilla V., Pellegrinet L., et al. Bmi1 regulates murine intestinal stem cell proliferation and self-renewal downstream of notch. Development. 2015;142(1):41–50. doi: 10.1242/dev.107714. [DOI] [PubMed] [Google Scholar]
- 118.Guo Z., Ohlstein B. Stem cell regulation. Bidirectional notch signaling regulates Drosophila intestinal stem cell multipotency. Science. 2015;350(6263) doi: 10.1126/science.aab0988.aab0988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gregorieff A., Liu Y., Inanlou M. R., Khomchuk Y., Wrana J. L. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature. 2015;526(7575):715–718. doi: 10.1038/nature15382. [DOI] [PubMed] [Google Scholar]
- 120.Clevers H., Loh K. M., Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014;346(6205) doi: 10.1126/science.1248012.1248012 [DOI] [PubMed] [Google Scholar]
- 121.Shyer A. E., Huycke T. R., Lee C., Mahadevan L., Tabin C. J. Bending gradients: how the intestinal stem cell gets its home. Cell. 2015;161(3):569–580. doi: 10.1016/j.cell.2015.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Büller N. V., Rosekrans S. L., Metcalfe C., et al. Stromal Indian hedgehog signaling is required for intestinal adenoma formation in mice. Gastroenterology. 2015;148(1):170–180. doi: 10.1053/j.gastro.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 123.Kosinski C., Stange D. E., Xu C., et al. Indian hedgehog regulates intestinal stem cell fate through epithelial-mesenchymal interactions during development. Gastroenterology. 2010;139(3):893–903. doi: 10.1053/j.gastro.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.van Dop W. A., Heijmans J., Büller N. V., et al. Loss of Indian Hedgehog activates multiple aspects of a wound healing response in the mouse intestine. Gastroenterology. 2010;139(5):1665–1676. doi: 10.1053/j.gastro.2010.07.045. [DOI] [PubMed] [Google Scholar]
- 125.Deng F., Peng L., Li Z., et al. YAP triggers the Wnt/β-catenin signalling pathway and promotes enterocyte self-renewal, regeneration and tumorigenesis after DSS-induced injury. Cell Death and Disease. 2018;9(2):p. 153. doi: 10.1038/s41419-017-0244-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bhattacharyya A., Chattopadhyay R., Mitra S., Crowe S. E. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiological Review. 2014;94(2):329–354. doi: 10.1152/physrev.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pérez S., Taléns-Visconti R., Rius-Pérez S., Finamor I., Sastre J. Redox signaling in the gastrointestinal tract. Free Radical Biology and Medicine. 2017;104:75–103. doi: 10.1016/j.freeradbiomed.2016.12.048. [DOI] [PubMed] [Google Scholar]
- 128.Zhang S., Yang C., Yang Z., et al. Homeostasis of redox status derived from glucose metabolic pathway could be the key to understanding the Warburg effect. American Journal of Cancer Research. 2015;5(4):1265–1280. [PMC free article] [PubMed] [Google Scholar]
- 129.Moll F., Walter M., Rezende F., et al. NoxO1 controls proliferation of colon epithelial cells. Frontiers in Immunology. 2018;9:p. 973. doi: 10.3389/fimmu.2018.00973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brandes R. P., Harenkamp S., Schürmann C., et al. The cytosolic NADPH oxidase subunit NoxO1 promotes an endothelial stalk cell phenotype. Arteriosclerosis, Thrombosis and Vascular Biology. 2016;36(8):846–854. doi: 10.1161/ATVBAHA.116.307132. [DOI] [PubMed] [Google Scholar]
- 131.Kopan R., Ilagan M. X. The canonical notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chillakuri C. R., Sheppard D., Lea S. M., Handford P. A. Notch receptor–ligand binding and activation: insights from molecular studies. Seminars in Cell & Developmental Biology. 2012;23(4):421–428. doi: 10.1016/j.semcdb.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Goto N., Ueo T., Fukuda A., et al. Distinct roles of HES1 in normal stem cells and tumor stem-like cells of the intestine. Cancer Research. 2017;77(13):3442–3454. doi: 10.1158/0008-5472.CAN-16-3192. [DOI] [PubMed] [Google Scholar]
- 134.Dempsey P. J. Role of ADAM10 in intestinal crypt homeostasis and tumori-genesis. Biochimica et Biophysica Acta. 2017;1864(11):2228–2239. doi: 10.1016/j.bbamcr.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sebio A., Kahn M., Lenz H. J. The potential of targeting Wnt/β-catenin in colon cancer. Expert Opinion on Therapeutic Targets. 2014;18(6):611–615. doi: 10.1517/14728222.2014.906580. [DOI] [PubMed] [Google Scholar]
- 136.Karimaian A., Bannazadeh Baghi H., Yousefi B. The crosstalk between Wnt/β-catenin signaling pathway with DNA damage response and oxidative stress: implications in cancer therapy. DNA Repair (AMST) 2017;51:14–19. doi: 10.1016/j.dnarep.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 137.Suryawanshi A., Tadagavadi R. K., Swafford D., Manicassamy S. Modulation of inflammatory responses by Wnt/β-catenin signaling in dendritic cells: a novel immunotherapy target for Autoimmunity and cancer. Frontiers in Immunology. 2016;7:p. 460. doi: 10.3389/fimmu.2016.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.van Loosdregt J., Coffer P. J. The role of WNT signaling in mature T cells: T cell factor is coming home. Journal of Immunology. 2018;201(8):2193–2200. doi: 10.4049/jimmunol.1800633. [DOI] [PubMed] [Google Scholar]
- 139.Gudjonsson J. E., Johnston A., Stoll S. W., et al. Evidence for altered Wnt signaling in psoriatic skin. Journal of Investigative Dermatology. 2010;130(7):1849–1859. doi: 10.1038/jid.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kinzler K. W., Nilbert M. C., Su L. K., et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253(5020):661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 141.Nishisho I., Nakamura Y., Miyoshi Y., et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253(5020):665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 142.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tanaka K., Kitagawa Y., Kadowaki T. Drosophila segment polarity gene product porcupine stimulates the posttranslational N-glycosylation of wingless in the endoplasmic reticulum. Journal of Chemical Biology. 2002;277(15):12816–12823. doi: 10.1074/jbc.M200187200. [DOI] [PubMed] [Google Scholar]
- 144.Katoh M. WNT/PCP signaling pathway and human cancer. Oncology Report. 2005;14(6):1583–1588. [PubMed] [Google Scholar]
- 145.Schneikert J., Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut. 2007;56(3):417–425. doi: 10.1136/gut.2006.093310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Samowitz W. S., Slattery M. L., Sweeney C., et al. APC mutations and other genetic and epigenetic changes in colon cancer. Molecular Cancer Research. 2007;5(2):165–170. doi: 10.1158/1541-7786.MCR-06-0398. [DOI] [PubMed] [Google Scholar]
- 147.Fodde R., Kuipers J., Rosenberg C., et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nature Cell Biology. 2001;3(4):433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 148.Moloney J. N., Cotter T. G. ROS signalling in the biology of cancer. Seminars in Cell & Developmental Biology. 2018;80:50–64. doi: 10.1016/j.semcdb.2017.05.023. [DOI] [PubMed] [Google Scholar]
- 149.Ahmad R., Sorrell M. F., Batra S. K., Dhawan P., Singh A. B. Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal Immunology. 2017;10(2):307–317. doi: 10.1038/mi.2016.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pessayre D. Role of mitochondria in non-alcoholic fatty liver disease. Journal of Gastroenterology and Hepatology. 2007;22(Suppl. 1):S20–S27. doi: 10.1111/j.1440-1746.2006.04640.x. [DOI] [PubMed] [Google Scholar]
- 151.Fransen M., Lismont C., Walton P. The peroxisome-mitochondria connection: how and why? International Journal of Molecular Sciences. 2017;18(6):p. E1126. doi: 10.3390/ijms18061126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fogg V. C., Lanning N. J., Mackeigan J. P. Mitochondria in cancer: at the crossroads of life and death. Chinese Journal of Cancer. 2011;30(8):526–539. doi: 10.5732/cjc.011.10018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sajnani K., Islam F., Smith R. A., Gopalan V., Lam A. K. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie. 2017;135:164–172. doi: 10.1016/j.biochi.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 154.Desideri E., Vegliante R., Ciriolo M. R. Mitochondrial dysfunctions in cancer: genetic defects and oncogenic signaling impinging on TCA cycle activity. Cancer Letters. 2015;356(2):217–223. doi: 10.1016/j.canlet.2014.02.023. [DOI] [PubMed] [Google Scholar]
- 155.Moreira J. D., Hamraz M., Abolhassani M., et al. The redox status of cancer cells supports mechanisms behind the Warburg effect. Metabolites. 2016;6(4):p. E33. doi: 10.3390/metabo6040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Takaki A., Kawano S., Uchida D., et al. Paradoxical roles of oxidative stress response in the digestive system before and after carcinogenesis. Cancers (Basel) 2019;11(2):p. E213. doi: 10.3390/cancers11020213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Visnes T., Cázares-Körner A., Hao W., et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science. 2018;362(6416):834–839. doi: 10.1126/science.aar8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kantor M., Sobrado J., Patel S., Eiseler S., Ochner C. Hereditary colorectal tumors: a literature review on MUTYH-associated polyposis. Gastroenterology Research and Practice. 2017;2017:4. doi: 10.1155/2017/8693182.8693182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Raetz A. G., David S. S. When you’re strange: unusual features of the MUTYH glycosylase and implications in cancer. DNA Repair (AMST) 2019;80:16–25. doi: 10.1016/j.dnarep.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tamir S., Paddock M. L., Darash-Yahana-Baram M., et al. Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochimica et biophysicaacta. 2015;1853(6):1294–1315. doi: 10.1016/j.bbamcr.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 161.Grasso F., Di Meo S., De Luca G., et al. The MUTYH base excision repair gene protects against inflammation-associated colorectal carcinogenesis. Oncotarget. 2015;6(23):19671–19684. doi: 10.18632/oncotarget.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Weren R. D., Ligtenberg M. J., Kets C. M., et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nature Genetics. 2015;47(6):668–671. doi: 10.1038/ng.3287. [DOI] [PubMed] [Google Scholar]
- 163.Aretz S., Genuardi M., Hes F. J. Clinical utility gene card for: MUTYH-associated polyposis (MAP), autosomal recessive colorectal adenomatous polyposis, multiple colorectal adenomas, multiple adenomatous polyps (MAP)-update 2012. European Journal of Human Genetics. 2013;21(1) doi: 10.1038/ejhg.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Al-Tassan N., Chmiel N. H., Maynard J., et al. Inherited variants of MYH associated with somatic G: C ⟶ T: a mutations in colorectal tumors. Nature Genetics. 2002;30(2):227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 165.Nielsen M., Joerink-van de Beld M. C., Jones N., et al. Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH-associated polyposis. Gastroenterology. 2009;136(2):471–476. doi: 10.1053/j.gastro.2008.10.056. [DOI] [PubMed] [Google Scholar]
- 166.Rivera B., Castellsagué E., Bah I., van Kempen L. C., Foulkes W. D. Biallelic NTHL1 mutations in a woman with multiple primary tumors. New England Journal of Medicine. 2015;373(20):1985–1986. doi: 10.1056/NEJMc1506878. [DOI] [PubMed] [Google Scholar]
- 167.Weren R. D., Ligtenberg M. J., Geurts van Kessel A., De Voer R. M., Hoogerbrugge N., Kuiper R. P. NTHL1 and MUTYH polyposis syndromes: two sides of the same coin? Journal of Pathology. 2018;244(2):135–142. doi: 10.1002/path.5002. [DOI] [PubMed] [Google Scholar]
- 168.David S. S., O’Shea V. L., Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447(7147):941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mulvihill J. J. The frequency of hereditary large bowel cancer. Progress in Clinical and Biological Research. 1983;115:61–75. [PubMed] [Google Scholar]
- 170.Groden J., Thliveris A., Samowitz W., et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66(3):589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 171.Fearnhead N. S., Britton M. P., Bodmer W. F. The ABC of APC. Human Molecular Genetics. 2001;10(7):721–733. doi: 10.1093/hmg/10.7.721. [DOI] [PubMed] [Google Scholar]
- 172.Powell S. M., Zilz N., Beazer-Barclay Y., et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359(6392):235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 173.Miyoshi Y., Nagase H., Ando H., et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Human Molecular Genetics. 1992;1(4):229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 174.Morin P. J., Sparks A. B., Korinek V., et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275(5307):1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 175.Worthley D. L., Leggett B. A. Colorectal cancer: molecular features and clinical opportunities. Clinical Biochemist Reviews. 2010;31(2):31–38. [PMC free article] [PubMed] [Google Scholar]
- 176.van der Logt E. M., Roelofs H. M., Wobbes T., Nagengast F. M., Peters W. H. High oxygen radical production in patients with sporadic colorectal cancer. Free Radical Biology and Medicine. 2005;39(2):182–187. doi: 10.1016/j.freeradbiomed.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 177.Cristofaro M., Contursi A., D’Amore S., et al. Adenomatous polyposis coli (APC)-induced apoptosis of HT29 colorectal cancer cells depends on mitochondrial oxidative metabolism. Biochimica et Biophysica Acta. 2015;1852(9):1719–1728. doi: 10.1016/j.bbadis.2015.05.009. [DOI] [PubMed] [Google Scholar]
- 178.Knudson A. Alfred Knudson and his two-hit hypothesis. Lancet Oncology. 2001;2(10):642–645. doi: 10.1016/s1470-2045(01)00524-1. [DOI] [PubMed] [Google Scholar]
- 179.Segditsas S., Rowan A. J., Howarth K., et al. APC and the three-hit hypothesis. Oncogene. 2009;28(1):146–155. doi: 10.1038/onc.2008.361. [DOI] [PubMed] [Google Scholar]
- 180.Henderson B. R., Fagotto F. The ins and outs of APC and beta-catenin nuclear transport. Embo Reports. 2002;3(9):834–839. doi: 10.1093/embo-reports/kvf181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rosin-Arbesfeld R., Townsley F., Bienz M. The APC tumour suppressor has a nuclear export function. Nature. 2000;406(6788):1009–1012. doi: 10.1038/35023016. [DOI] [PubMed] [Google Scholar]
- 182.Jaiswal A. S., Narayan N. A novel function of adenomatous polyposis coli (APC) in regulating DNA repair. Cancer Letter. 2008;271(2):272–280. doi: 10.1016/j.canlet.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Neufeld K. L., Zhang F., Cullen B. R., White R. L. APC-mediated downregulation of beta-catenin activity involves nuclear sequestration and nuclear export. EMBO Reports. 2000;1(6):519–523. doi: 10.1093/embo-reports/kvd117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Henderson B. R. Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nature Cell Biology. 2000;2(9):653–656. doi: 10.1038/35023605. [DOI] [PubMed] [Google Scholar]
- 185.Curia M. C., De Iure S., De Lellis L., et al. Increased variance in germline allele-specific expression of APC associates with colorectal cancer. Gastroenterology. 2012;142(1):71–77. doi: 10.1053/j.gastro.2011.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Aceto G. M., Fantini F., De Iure S., et al. Correlation between mutations and mRNA expression of APC and MUTYH genes: new insight into hereditary colorectal polyposis predisposition. Journal of Experimental and Clinical Cancer Research. 2015;34(131) doi: 10.1186/s13046-015-0244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Palles C., Cazier J. B., Howarth K. M., et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature Genetics. 2013;45(2):136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Heitzer E., Tomlinson I. Replicative DNA polymerase mutations in cancer. Current Opinion in Genetics and Development. 2014;24:107–113. doi: 10.1016/j.gde.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rayner E., van Gool I. C., Palles C. A panoply of errors: polymerase proofreading domain mutations in cancer. Nature Reviews Cancer. 2016;16(2):71–81. doi: 10.1038/nrc.2015.12. [DOI] [PubMed] [Google Scholar]
- 190.Longley M. J., Pierce A. J., Modrich P. DNA polymerase delta is required for human mismatch repair in vitro. The Journal of Biological Chemistry. 1997;272(16) doi: 10.1074/jbc.272.16.10917. [DOI] [PubMed] [Google Scholar]
- 191.Bellido F., Pineda M., Aiza G. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genetics in Medicine. 2016;18(4):325–332. doi: 10.1038/gim.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Haraldsdottir S., Hampel H., Tomsic J., et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014;147(6):1308–1316. doi: 10.1053/j.gastro.2014.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Guarinos C., Sánchez-Fortún C., Rodríguez-Soler M., et al. Serrated polyposis syndrome: molecular, pathological and clinical aspects. World Journal of Gastroenterology. 2012;18(20):2452–2461. doi: 10.3748/wjg.v18.i20.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Snover D. C., Ahnen D. J., Burt R. W., Odze R. D. Serrated polyps of the colon and rectum and serrated (“hyperplastic”) polyposis. In: Bosman S. T., Carneiro F., Hruban R. H., et al., editors. WHO Classification of Tumours of the Digestive System. Berlin, Germany: Springer-Verlag; 2010. [Google Scholar]
- 195.Win K., Walters R. J., Buchanan D. D., et al. Cancer risks for relatives of patients with serrated polyposis. American Journal of Gastroenterology. 2012;107(5):770–778. doi: 10.1038/ajg.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mongin C., Coulet F., Lefevre J. H., et al. Unexplained polyposis: a challenge for geneticists, pathologists and gastroenterologists. Clinical Genetics. 2012;81(1):38–46. doi: 10.1111/j.1399-0004.2011.01676.x. [DOI] [PubMed] [Google Scholar]
- 197.Cheah P. Y., Wong Y. H., Chau Y. P. Germline bone morphogenesis protein receptor 1A mutation causes colorectal tumorigenesis in hereditary mixed polyposis syndrome. American Journal of Gastroenterology. 2009;104(12):3027–3033. doi: 10.1038/ajg.2009.542. [DOI] [PubMed] [Google Scholar]
- 198.Heald B., Mester J., Rybicki L., Orloff M. S., Burke C. A., Eng C. Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology. 2010;139(6):1927–1933. doi: 10.1053/j.gastro.2010.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yan H. H., Lai J. C., Ho S. L., et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut. 2017;66(9):1645–1656. doi: 10.1136/gutjnl-2016-311849. [DOI] [PubMed] [Google Scholar]
- 200.de Lau W., Peng W. C., Gros P., Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes & Development. 2014;28(4):305–316. doi: 10.1101/gad.235473.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Giannakis M., Hodis E., Jasmine Mu X., et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nature Genetics. 2014;46(12):1264–1266. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Leggett B., Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138(6):2088–2100. doi: 10.1053/j.gastro.2009.12.066. [DOI] [PubMed] [Google Scholar]