

Abstract
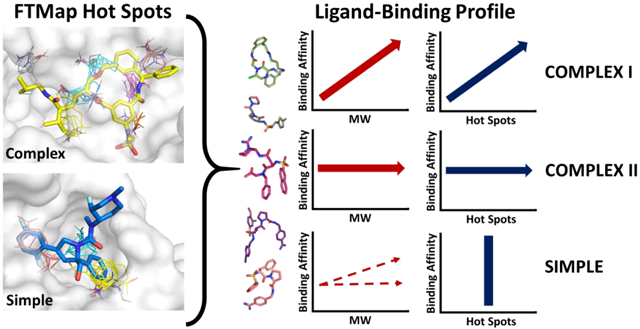
Beyond Rule-of-5 (bRo5) compounds are increasingly used in drug discovery. Here we analyze 37 target proteins that have bRo5 drugs or clinical candidates. Targets can benefit from bRo5 drugs if they have “Complex” hot spot structure with four or more hots spots, including some strong ones. Complex I targets show positive correlation between binding affinity and molecular weight. These targets are conventionally druggable, but reaching additional hot spots enables improved pharmaceutical properties. Complex II targets, mostly protein kinases, also have strong hot spots but show no correlation between affinity and ligand molecular weight, and the primary motivation for creating larger drugs is to increase selectivity. Each target considered as Complex III has some specific reason for requiring bRo5 drugs. Finally, targets with “Simple” hot spot structure, i.e., three or fewer weak hot spots, must use larger compounds that interact with surfaces beyond the hot spot region to achieve acceptable affinity.
Keywords: drug discovery, binding hot spot, drug target protein, druggability, kinase inhibitors
Graphical Abstract
INTRODUCTION
Lipinski’s Rule of five (Ro5) provides simple conditions to determine if a chemical compound with a certain pharmacological or biological activity has properties consistent with being an orally active drug.1 The aim of this rule has been to rationalize compound design (in particular compound library design) so as not to make molecules that are too apolar, floppy, and large, and thus have a lower chance of exhibiting oral bioavailability and other desirable pharmaceutical properties. While the Ro5 has served as a very useful guideline for developing orally bioavailable small-molecule drugs, and its importance is widely recognized, many compounds that violate the Ro5 are good drug candidates, and it is increasingly accepted that drug development efforts should not be restricted to the Ro5 chemical space.2–4 In particular, Doak et al.3 provided a comprehensive overview and discussion of close to 500 drugs and clinical candidates with MW ranging from 500–2000 Da, including discussions of structure-property trends. More recently, DeGoey et al.4 discussed learnings from AbbVie drug projects in beyond the Ro5 space. The increased interest in “beyond Rule of five” (bRo5) compounds is driven by at least four observations. First, some natural products lie outside the Ro5 space but have fairly good oral availability, encouraging the development of bRo5 drug candidates, especially synthetic macrocycles.5–9 Second, an increasing number of bRo5 compounds are in clinical trials and are becoming approved by the FDA.4, 10–12 For example, over 30% of approved kinase inhibitors are bRo5 compounds.12 Third, increasing attention is being given to small-molecules that disrupt protein–protein interactions (PPIs),13 and around 50% of such compounds discussed in the scientific literature are bRo5.14–18 Fourth, although oral bioavailability is an important criterion for most successful drugs, many therapeutics used in today’s medical practice are administered parenterally.19 Thus, discovery efforts toward parenteral drugs may be useful to provide treatment options for indications with high unmet medical need, or to validate a potential ‘first-in-class’ target before intensive effort on oral therapeutics is put in place.19 In fact, many proteins have large binding sites, and the use of larger and more complex compounds can increase selectivity.20
Recognizing the need for improved understanding of drugs and clinical candidates in the bRo5 space, Doak et al.21 analyzed the interactions between a set of such compounds and their targets. The underlying assumption of their analysis was that these targets have special characteristics, such as possessing binding sites that are “large, highly lipophilic, highly polar, flexible, flat, or featureless”, and thus contain few opportunities for the binding of conventional Ro5-compliant ligands21. Accordingly, they selected 48 bRo5 drugs and their targets, and analyzed the interactions within each specific ligand-receptor pair, focusing on pocket size and shape, buried ligand surface area, ligand interface, ligand and target interface non-polar atom ratios, hydrogen bond donor and acceptor interactions, affinity, and ligand efficiencies.
While the results by Doak et al.21 already provide important guidance for the design of bioactive drugs for difficult targets, here we describe a substantially expanded analysis of these bRo5 targets and their available ligands. We studied only globular proteins, and after removing the DNA/RNA binding and membrane proteins from their set we focused on 37 targets. First, we identified the binding hot spots of each protein target using FTMap22. Next, to study how ligands interact with these proteins, we extended the analysis beyond the specific protein-ligand pair Doak et al.21 selected, to include all compounds, whether Ro5 or bRo5, that bind the target proteins and have structures available in the Protein Data Bank23 (PDB) and a known ligand binding affinity.24–26 Finally, we analyzed the binding site hot spots and ligand binding profiles of each target and classified them into four groups with different mechanisms explaining how bRo5 drugs increase the likelihood of success. In addition to the structure-based study of ligand binding to hot spots we also show results from a ligand-based analysis of the chemical tractability of the targets and the physicochemical properties of all their ligands collected from ChEMBL,27 to eliminate bias from our structure-based ligand dataset.
BINDING SITE ANALYSIS OF bRo5 PROTEIN TARGETS
Analysis of the 37 target proteins was focused on the binding hot spots of the protein-ligand structure. Hot spots are small regions on the protein surface that contribute a disproportionate amount to the ligand-binding free energy, and hence ligands generally overlap with one or more hot spots.28–31 For determining the hot spots we used the protein mapping program FTMap.22 FTMap (http://ftmap.bu.edu/) distributes small organic probe molecules of different sizes, shapes, and polarities on the surface of the protein to be studied, finds the most favorable positions for each probe type, clusters the probes, and ranks the clusters on the basis of their average energy. Regions that bind several different probe clusters are called consensus sites (CSs) and are the predicted binding hot spots. The hot spots predicted by FTMap have been shown to agree with the hot spots determined by a variety of experimental methods.32–38 Moreover, the strength of the hot spot, defined as the number of probe clusters included, indicates a relative energetic importance of the hot spot in the context of binding small molecules.32–38 Thus, the hot spots on each protein are ranked on the basis of the number of probe clusters they contain. Following the notation established in the FTMap server (http://ftmap.bu.edu), the hot spots are numbered starting from 0 for the strongest; the number of probe clusters in each hot spot is shown in parenthesis after the hot spot rank. For example, a hot spot labeled 1(20) indicates that it is the second strongest hotspot (second to hot spot 0) and that it has a strength of 20 probe clusters. Moreover, a binding site of 0(20), 1(10), and 4(5) indicates that it includes three hotspots: 0, 1, and 4, each with 20, 10, and 5 probe clusters, respectively. Hot spots with fewer than five probe clusters were disregarded. We note that in all figures showing FTMap results, each probe cluster is represented by a single probe at the center of cluster.
We have previously studied the hot spot properties on proteins that bind Ro5 drugs and demonstrated that a hot spot of at least 16 probe clusters is required for druggability.39 Furthermore, traditional Ro5 druggable targets have one or more additional “secondary” hot spots (with three or more probe clusters) in close proximity to the strong “primary” hot spot.39 FTMap studies have also been performed for targets that bind macrocycles5 and inhibitors of protein-protein interactions.40 Based on these latter studies, we expected that bRo5 ligands always would be needed for targets with hot spots that are too far from each other or are too weak, in both cases necessitating larger ligands.5,39,40 As will be shown, some of the targets studied by Doak et al.21 indeed exhibit these properties. However, we found that the majority (22 out of 37) of the selected proteins that are targets for bRo5 drugs and clinical candidates can also bind small compounds (MW < 500 Da) with high affinity. In some cases, the smaller compounds can have even higher affinity than the larger ones. This observation raises the following questions: Which proteins require or benefit from bRo5 ligands, and can such targets be prospectively identified on the basis of the ligand-free protein structure?
Among the 37 protein targets mapped here, we identified two distinct structures of hot spot ensembles in the binding sites. We will refer to these hot spot structures as “complex” and “simple” (Figure 1). A complex hot spot structure defines a binding site that consists of 4 or more hot spots, whereas the simple hot spot ensemble has three or fewer hot spots. Based on the number of hot spots alone, we have classified the 37 protein targets as complex or simple (Table 1). 24 targets have a complex hot spot structure (mean number of hot spots = 5.63) and the remaining 13 have simple hot spot structure (mean number of hot spots = 2.15), resulting in significant difference (p < 0.001 by the Wilcoxon-Mann-Whitney test, Figure S1). Furthermore, the total number of probe clusters in all hot spots in the binding site can also be used to help determine whether a given hot spot ensemble should be classified as complex or simple. The mean number of probe clusters in complex hot spot structures is 68.88, whereas in simple hot spot structures it is 29.88, and this difference is also significant (p < 0.001 by the Wilcoxon-Mann-Whitney test, Figure S1). The consequence of complex versus simple hot spot structures will be discussed later in the context of the protein-ligand binding profiles.
Figure 1.
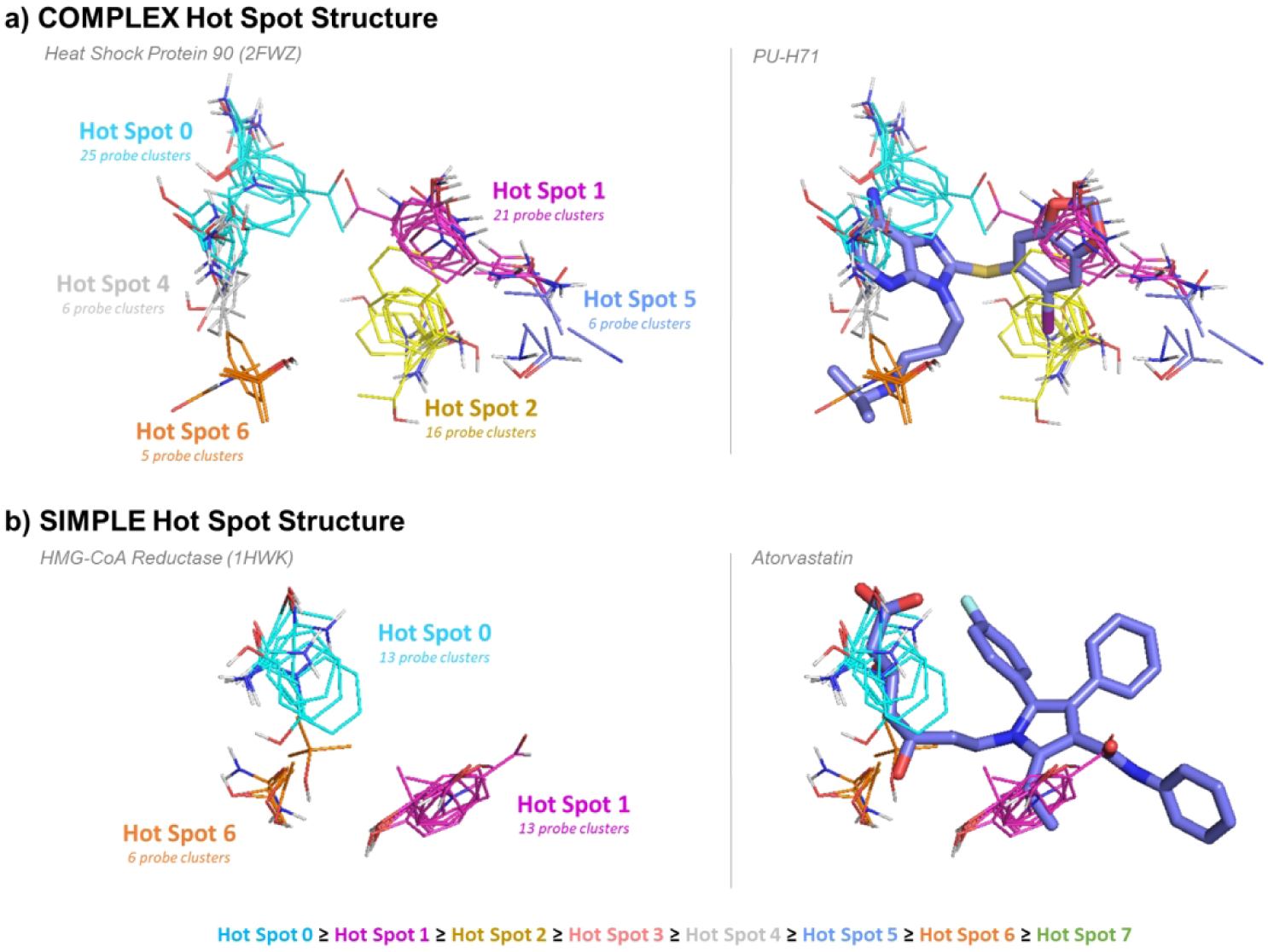
Complex versus simple binding site hot spot structure, determined by FTMap. The hot spots are labeled according to rank (starting at 0) and colored according to the standard FTMap output (http://ftmap.bu.edu). The color of each hot spot ranked 0 though 7 is shown explicitly at the bottom of the figure. a) A complex hot spot structure consists of four or more hot spots. Heat Shock Protein 90 (PDB: 2FWZ) has the hot spots 0(25), 1(21), 2(16), 4(6), 5(6), and 6(5). The hot spots are labeled on the left, and the compound PU-H71 is overlaid on the right. b) A simple hot spot structure has three or fewer hot spots. HMG-CoA Reductase (PDB: 1HWK) has the hot spots 0(13), 1(13), and 6(6). The hot spots are labeled on the left, and Atorvastatin is overlaid on the right.
Table 1.
Structures mapped and additional ligand-bound structures considered
| Target Name | PDB ID | Chains | Drug Class | Hot Spot Structure | Na | MW < 500 | MW > 500 | Hot Spots utilized by ligands | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Nb | Nhac | Nb | Nhac | |||||||
| HIV-1 Protease / Saquinavir | 3OXC | A B | eRo5 | Complex I | 327 | 12 | 6 | 315 | 272 | 0(21), 1 (18), 2(18), 3(13), 4(11), 5(8), 6(5) |
| Heat Shock Protein 90 / PU-1471 | 2FWZ | A | eRo5 | Complex I | 155 | 147 | 58 | 8 | 6 | 0 (25), 1 (21), 2 (16), 4 (6), 5 (6), 6 (5) |
| Thrombin / Argatroban | 1DWC | H | bRo5 | Complex I | 150 | 109 | 65 | 41 | 30 | 0(26), 2(14), 5(6), 6(6), 7(5) |
| MAP Kinase P38 / PF03715455 | 2YIS | A | bRo5 | Complex I | 144 | 117 | 66 | 27 | 22 | 0(26), 1 (21), 2(10), 3(7) |
| Bromodomain BRD4 / Fedratinib | 4OGJ | A B | eRo5 | Complex I | 113 | 103 | 19 | 10 | 4 | 0(21), 1 (18), 2(11), 3(8), 4(8), 5(7), 6(5), 7(5) |
| Renin / Aliskiren | 2V0Z | C | eRo5 | Complex I | 62 | 36 | 20 | 26 | 24 | 0 (24), 1 (15), 2 (12), 3 (8), 4 (8), 5 (7), 6 (7) |
| PPAR-γ / AMG-131 | 3FUR | A | eRo5 | Complex I | 52 | 40 | 10 | 12 | 11 | 2 (9), 4 (8), 5 (8), 6 (7), 8 (6) |
| MAP Kinase Kinase / Cobimetinib | 4AN2 | A | eRo5 | Complex I | 25 | 18 | 11 | 7 | 7 | 0(18), 1 (17), 2(14), 3(13), 4(13) |
| E3 ubiquitin-protein ligase XIAP / Birinapant | 4KMP | A B | bRo5 | Complex I | 20 | 13 | 2 | 7 | 4 | 0(18), 1 (16), 2(16), 3(11), 4(11), 5(7) |
| Epidermal Growth Factor Receptor / Lapatinib | 1XKK | A | eRo5 | Complex II | 59 | 47 | 38 | 12 | 10 | 0 (16), 1 (15), 2 (12), 3 (9), 5 (7), 6 (7) |
| Hepatocyte Growth Factor Receptor / BMS-777607 | 3F82 | A | eRo5 | Complex II | 54 | 36 | 32 | 18 | 16 | 0 (14), 1 (12), 2 (11), 3 (10), 4 (9), 5 (8), 7 (6) |
| Anaplastic Lymphoma Kinase / Ceritinib | 4MKC | A | eRo5 | Complex II | 32 | 23 | 21 | 9 | 7 | 0 (24), 3 (8), 5 (6), 6 (6), 7 (5) |
| Tyrosine Protein Kinase ABL1 / Nilotinib | 3CS9 | A | eRo5 | Complex II | 28 | 18 | 16 | 10 | 9 | 0(20), 2(15), 3(14), 4(13) |
| VEGFR-2 / Nintedanib | 3C7Q | A | eRo5 | Complex II | 24 | 16 | 11 | 8 | 7 | 0 (20), 1 (17), 2 (10), 3 (9), 4 (8), 6 (8), 7 (6) |
| Polo-like Kinase 1 / Volasertib | 3FC2 | A | eRo5 | Complex II | 11 | 4 | 3 | 7 | 6 | 0(28), 1 (19), 2(12), 3(7), 7(5) |
| Glucocorticoid Receptor / Mometasone furoate | 4P6W | A | eRo5 | Complex II | 9 | 6 | 6 | 3 | 3 | 0 (25), 1 (21), 2 (13), 3 (7), 4 (7), 5 (6) |
| Cyclophilin A / Cyclosporine A | 1CWA | A | bRo5 | Complex III | 8 | 5 | 0 | 3 | 3 | 0(27), 1 (16), 4(11), 5(6) |
| Bcl-2 / Navitoclax | 4LVT | A | bRo5 | Complex III | 7 | 0 | 0 | 7 | 7 | 0(22), 2(17), 4(10), 5(5) |
| DOT1-like Histone H3 Methyltransferase / EPZ - 5676 | 4HRA | A | eRo5 | Complex III | 5 | 2 | 0 | 3 | 3 | 1 (17), 2(12), 3(11), 4(6) |
| Lanosterol 14-alpha Demethylase / Itraconazole | 5EQB | A | bRo5 | Complex III | 3 | 2 | 0 | 1 | 1 | 0(23), 1 (17), 2(10), 4(7), 6(5) |
| Isoleucyl-tRNA Synthetase / Pseudomonic Acid A | 1QU2* | A | eRo5 | Complex III | 2 | 0 | 0 | 2 | 1 | 0 (14), 1 (14), 2 (14), 3 (11), 4 (8), 7 (5) |
| FK506 Binding Protein / Rapamycin | 4DRI | A B | bRo5 | Complex III | 2 | 0 | 0 | 2 | 2 | 0 (27), 1 (20), 2 (16), 3 (13), 4 (6), 5 (5) |
| Toll-like Receptor 4 / Eritoran | 2Z65 | C | bRo5 | Complex III | 1 | 0 | 0 | 1 | 1 | 0 (22), 1 (20), 2 (13), 3 (12), 4 (10), 5 (8), 6 (5), 7 (5) |
| FGFR 1 / BGJ-398 | 3TT0 | B | eRo5 | Complex III | 1 | 0 | 0 | 1 | 1 | 0(18), 1 (17), 3(12), 5(6), 6(5) |
| Phosphoinositide-3 Kinase / Pictilisib (GDC-0941) | 3DBS | A | eRo5 | Simple | 65 | 53 | 36 | 12 | 10 | 4 (8) |
| Soluble Acetylcholine Receptor / Tubocurarine | 3PMZ | D E | eRo5 | Simple | 36 | 27 | 10 | 9 | 8 | 0 (19), 3 (13), 5 (9) |
| Protein Farnesyltransferase / Lonafanib | 1O5M | A B | eRo5 | Simple | 34 | 17 | 15 | 17 | 13 | 0 (19), 2 (15) |
| Kinesin Eg5 / Ispinesib | 4A5Y | A | eRo5 | Simple | 28 | 24 | 12 | 4 | 3 | 0 (17), 2 (17), 3 (12) |
| HMG-CoA Reductase / Atorvastatin | 1HWK | A B | eRo5 | Simple | 19 | 6 | 5 | 13 | 13 | 0 (13), 1 (13), 6 (6) |
| Hepatitis C Virus NS5b Subunit / Beclabuvir | 4NLD | A | eRo5 | Simple | 8 | 4 | 3 | 4 | 4 | 2 (10) |
| Hepatitis C Virus NS34A Protease / Simeprevir | 3KEE | A E | bRo5 | Simple | 5 | 0 | 0 | 5 | 5 | 1 (16), 3 (10) |
| DNA-directed RNA-polymerase / Rifampicin | 4KMU* | C | bRo5 | Simple | 3 | 0 | 0 | 3 | 3 | 0 (17) |
| Tubulin-Alpha Chain / Paclitaxel | 1JFF | B | bRo5 | Simple | 2 | 0 | 0 | 2 | 1 | 2 (16), 5 (8) |
| Integrin Aplha-IIB / Eptifibatide | 2VDN* | B | bRo5 | Simple | 2 | 1 | 0 | 1 | 0 | 2 (17) |
| Smoothened Homolog / Taladegib | 4JKV* | A | eRo5 | Simple | 1 | 0 | 0 | 1 | 1 | 0 (20), 1 (19), 3 (12) |
| Na-K ATPase / Ouabain | 3A3Y | A | bRo5 | Simple | 1 | 0 | 0 | 1 | 1 | 1 (17), 2 (12), 3(8) |
| α-Amylase / 3-Acarbose | 1PPI | A | bRo5 | Simple | 1 | 0 | 0 | 1 | 0 | 0 (18), 1 (16), 3 (11) |
Indicates domain splitting was performed prior to FTMap (see Methods)
N number of structures with bound ligand and known binding affinity;
N number of ligand-bound structures
NHA number of high affinity structures with bound ligand for the <500 Da and >500 Da cases
EXTENDED LIGAND DATA SET FOR bRo5 PROTEIN TARGETS
While for each target Doak et al.21 described only one particular bound drug or clinical candidate, to study how the protein can bind different ligands we analyzed all ligand-bound structures with at least 90% sequence identity that were available in the PDB23 and had binding affinity information24–26. The number of additional ligand-bound structures varied from 0 to 327 per protein, resulting in an extended dataset that includes 1499 ligand-protein complexes with structural data (Table 1). All ligands with MW > 500 Da were labeled either “extended Ro5” (eRo5) or “beyond Ro5” (bRo5) as defined by Doak et al.21 (see Methods). The eRo5 category describes drugs that violate the Ro5 MW threshold of 500 Da, but maintain MW < 700 Da and ClogP < 7.5, and otherwise satisfy all the Ro5 criteria. The bRo5 class describes ligands that have either very large MW > 700 Da, or violate any of the Ro5 criteria beyond the eRo5 limits. The original set selected by Doak et al.21 included 26 targets with a bound eRo5 ligand and 22 targets with a bRo5 ligand. However, we restricted consideration to globular proteins, and hence our set of targets contains 23 targets with eRo5 and 14 targets with bRo5 ligands. While this set is skewed towards eRo5 compounds, we think that it is still important to explore why drugs with MW > 500 Da are frequently more successful in clinical trials than smaller candidates that are also available for most of these targets.
All ligands have experimentally determined binding affinity (BA) values in the form of KD, KI, or IC50. These were converted to pBA values defined as pBA = −log KD, −log KI, or −log IC50, depending on which measure was reported. Ligands with pBA>7 were considered high affinity. We note that the decision to restrict consideration to compounds with known binding affinity was adopted to exclude co-factors, fragments, and crystallization additives that frequently occur in X-ray crystal structures. The compounds in our extended set are restricted to those for which experimental crystal structures have been reported bound to the target protein, and so they clearly do not represent all ligands that bind to the 37 targets. Information gained from considering other known ligands with reported affinity values but no crystal structures are examined in the ligand-based analysis at the end of our study.
TARGETS WITH COMPLEX HOT SPOT STRUCTURE
As discussed, the hot spot structure of the ligand binding site of a target is defined as complex if it includes 4 or more hot spots. Among the 37 proteins considered, 24 satisfy this condition. In addition, in most such targets the hot spot ensemble is strong, i.e., the ensemble includes at least one hot spot with 20 or more probe clusters (Figure 1, Table 1). These complex targets were further classified based on the ligand-binding profile, where data is available, and primarily on the correlation between binding affinity and MW (Figure S1, Table S1). For targets classified as Complex I we observe a positive correlation between binding affinity and molecular weight of the ligands (Figure S2), whereas no such correlation exists for Complex II targets (Figure S4). This classification scheme is outlined in Figure 2, and is further discussed in detail below. However, eight of the 24 targets have too few ligands with structure and binding data (see Table 1). For each target in this group, defined as Complex III, the motivation for developing eRo5/bRo5 drugs or drug candidates will be individually discussed.
Figure 2.
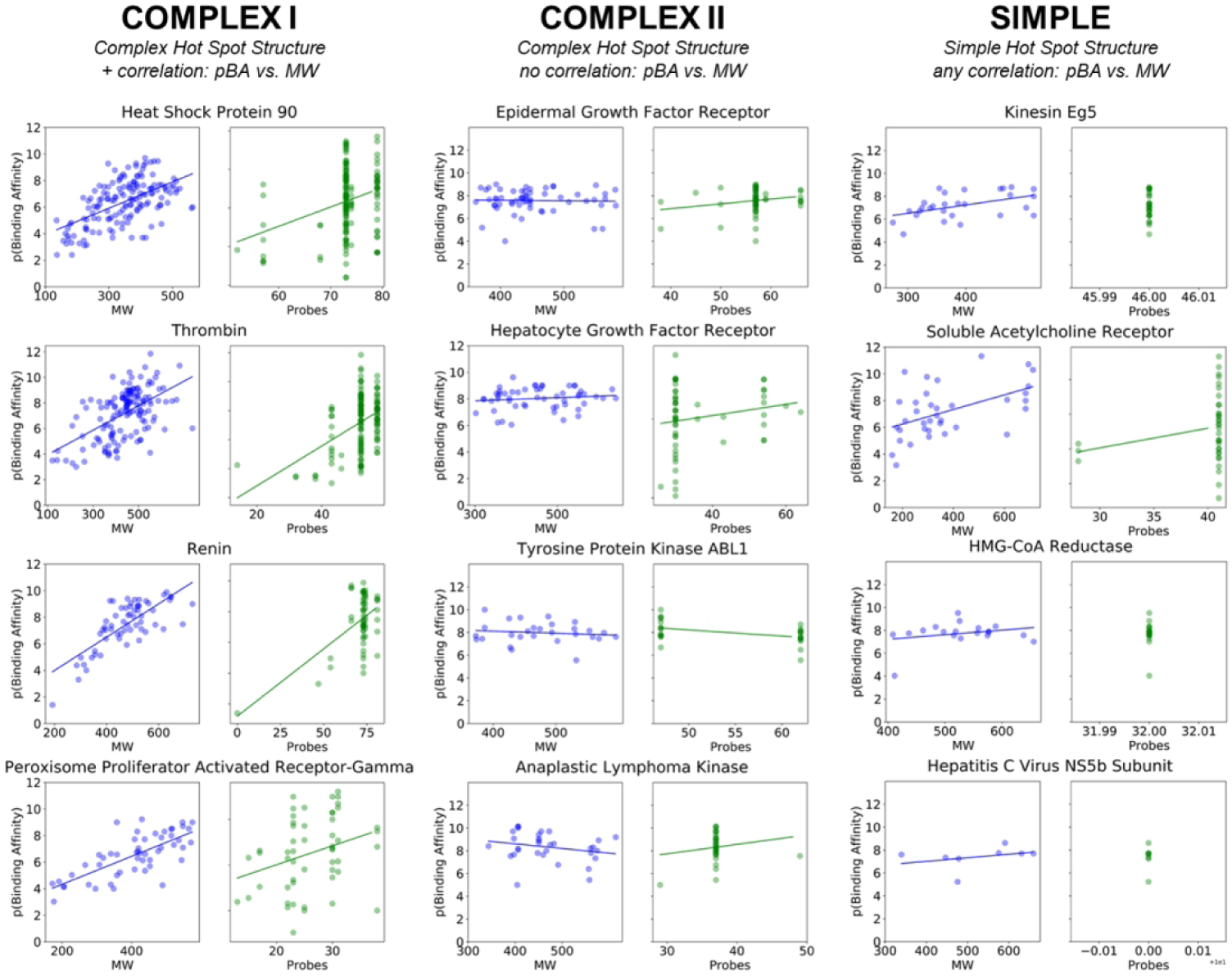
Examples of characteristic structure-based ligand profiles for Complex I, Complex II, and Simple protein targets which bind eRo5/bRo5 drugs and clinical candidates. Examples of Complex I targets shown are the heat shock protein 90, thrombin, renin, and PPAR-γ. Examples of Complex II targets shown are EGFR, HGFR, ABL1, and ALK. Examples of targets with simple hot spot structure are kinesin Eg5, soluble acetylcholine receptor, HMG-CoA reductase, and HCV NS5b. Only ligands that bind to the above proteins with known structure and binding affinity are shown (see Methods). Plots with ligands in blue (left) show the pBA [pBA = −log(KD or KI or IC50)] versus molecular weight, and green (right) show pBA versus total number of probe clusters.
Complex I Targets.
The nine targets in this group have multiple, strong hot spots at the binding site, and individual ligands interact with different combinations of the hot spots, ranging from a subset to all hot spots. These complex binding sites are very capable of binding small ligands (MW < 500 Da) with high affinity (pBA > 7) (Table 1). However, the successful drugs and clinical candidates for these targets are large and classified as eRo5/bRo5. To understand why these larger candidates are more successful, we consulted the ligand binding profiles for each Type I target, and observed that these targets exhibit a positive correlation between pBA and molecular weight, and between pBA and number of probe clusters reached (p < 0.05, Table S1, Figure S2). Thus, as ligands increase in molecular weight, they reach more hot spots and gain binding affinity (Figure 2, Figure S2). Furthermore, as will be discussed, we notice that the approved drugs or promising clinical candidates frequently tend to lie at the higher end of the molecular weight spectrum and occupy all or almost all hot spots, in spite of the potential disadvantages in terms of compound developability of moving into the bRo5 range. In spite of these general characteristics, each target may have some specific properties motivating development of eRo5/bRo5 drugs as discussed below.
Thrombin.
According to FTMap, thrombin has five binding hot spots 0(26), 2(14), 5(6), 6(6), and 7(5) in the inhibitor binding site, totaling 57 probe clusters, and exhibits the characteristic Complex I positive correlations between pBA and MW (p < 0.001) (Table S1 and Figure S2). Overall, 144 ligands were identified to bind thrombin with known structure and affinity. Of the 89 that bound with high affinity, 59 have MW < 500 Da and 30 have MW > 500 Da. Argatroban, the bRo5 ligand discussed by Doak et al.21 is the only FDA approved non-peptidic direct thrombin inhibitor. It reaches all five hot spots and has a molecular weight just above 500 Da (1DWC:MIT, MW = 509.64 Da, KI = 39 nM, pBA = 7.41). Ligands exist that are much smaller, such as GR157368 (1QHR:157, MW = 226.31 Da, IC50 = 130 nM, pBA = 6.89), but these bind only a subset of the hot spots and display somewhat lower affinity (Figure 3a). Two inhibitors that are quite large, but with MW slightly less than Argatroban, Inogatran (1K21:IGN, MW = 438.6 Da, KI = 4.2 nM, pBA = 8.38) and Ximelagatran (4BAH:MEL, MW = 473.6 Da, KI = 2.01 nM, pBA = 8.7), bind to four of the five available hot spots, 0(26), 2(14), 5(6), and 6(6). While these two inhibitors had sufficiently high affinity, they were withdrawn from the clinic due to liver toxicity.41, 42 In fact, many low MW thrombin inhibitors have problems, including poor selectivity, inherent toxicity, high-plasma protein binding, poor metabolic stability, rapid elimination from the blood, low anticoagulant activity, and poor oral bioavailability.43 Thus, it appears that inhibitors that reach all hot spots of thrombin can have superior pharmaceutical properties in spite of extending into the bRo5 range. This is possibly because the large amount of binding energy that is potentially made available by exploiting all hot spots gives latitude to compromise the binding complementarity in certain locations on the compound, allowing stronger binding groups that bring pharmaceutical liabilities to be replaced with slightly weaker binding but more structurally benign functionality, while still achieving sufficient overall binding affinity. In addition, the more complex structure of the drugs is likely to reduce the chances of off-target binding.
Figure 3.
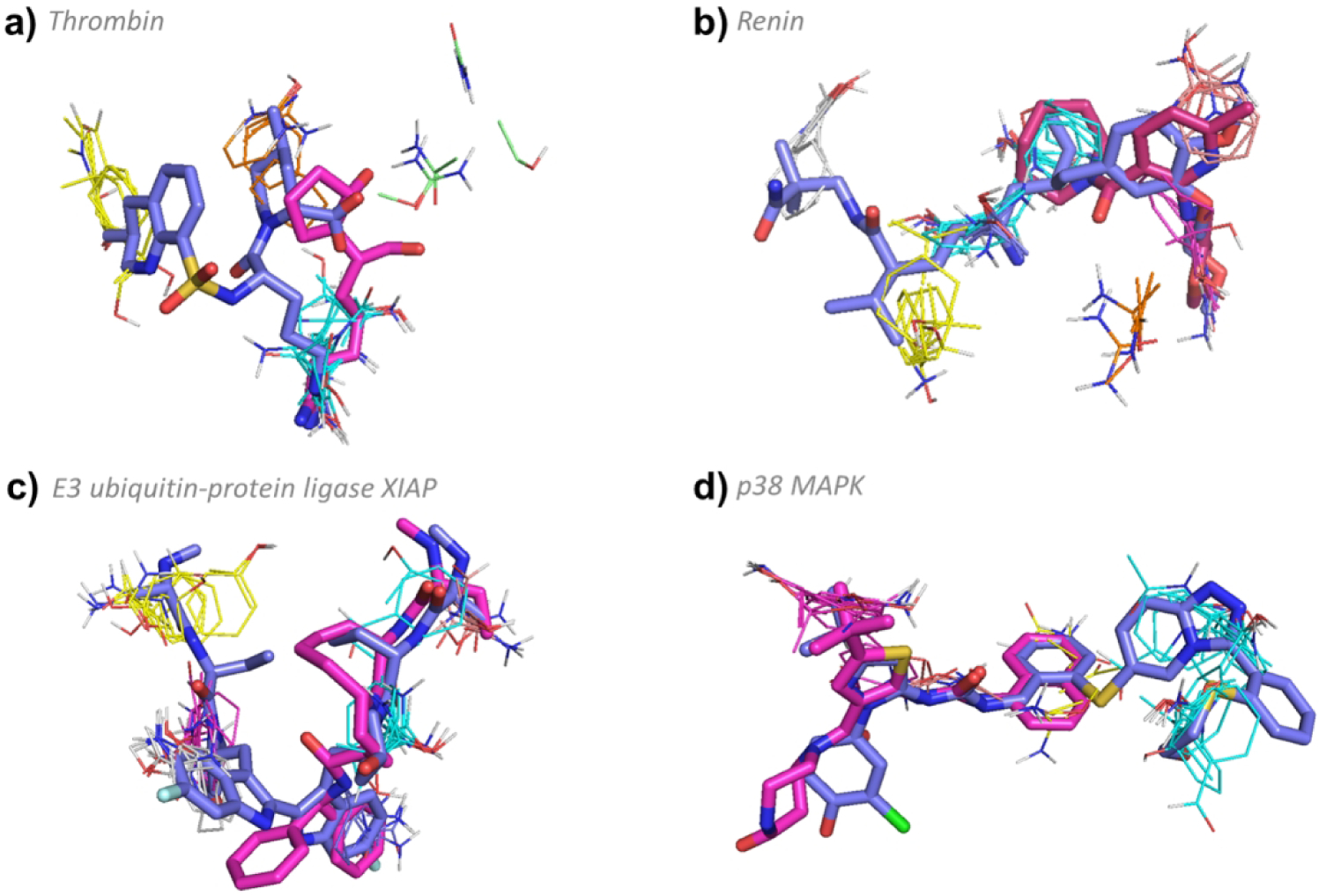
FTMap determined hot spot structure of Complex I targets with example ligands bound. Blue ligands represent the eRo5/bRo5 drug/clinical candidate identified by Doak et al., whereas pink ligands represent smaller compounds, and white ligands (where applicable) show compounds larger than the Doak et al. identified ligand. FTMap hot spots are colored according to standard output, shown in Figure 1. a) Hot spot structure of thrombin (1DWC). Argatroban (1DWC:MIT, MW = 509.64 Da, KI = 39 nM, pBA = 7.41) and GR157368 (1QHR:157, MW = 226.31 Da, IC50 = 130 nM, pBA = 6.89) are shown in blue and pink, respectively. b) Hot spot structure of renin (2V0Z). Aliskiren (2V0Z:C41, MW = 551.76 Da, KI = 0.6nM, pBA = 9.22) and N-piperidin-3-ylpyrimidine-5-carboxamide (5SZ9:74Y, MW = 313.39 Da, IC50 = 38 μM, pBA = 4.42) are shown in blue and pink, respectively. c) Hot spot structure of E3 ubiquitin-protein ligase XIAP (4KMP). Birinapant (4KMP:GT6, MW = 806.94 Da, KD = 45 nM, pBA = 7.35) and (2JK7:BI6, MW = 486.61 Da, KI = 67 nM, pBA = 7.17) are shown in blue and pink, respectively. d) Hot spot structure of MAP kinase p38 (2YIS). PF03715455 (2YIS:YIS MW = 700.27 Da, IC50 = 1.7 nM, pBA = 8.77) and (3P7B:P7B, MW = 464.58 Da, IC50 = 18 nM, pBA = 7.74) are shown in blue and pink, respectively.
Renin.
Renin is another typical Type I protein. 62 ligands were identified to bind renin with known structure and affinity, including high affinity ligands both with MW below and above 500 Da. Overall, there is a positive correlation between pBA and MW (p < 0.001 (Table S1, Figure S2). The bRo5 inhibitor Aliskiren (2V0Z:C41, MW = 551.76 Da, KI = 0.6 nM, pBA = 9.22), discussed by Doak et al.21 reaches six of the seven hot spots, 0(24), 1(15), 2(12), 3(8), 4(8), and 5(7), overlapping with a total of 74 probe clusters (Figure 3b). Many peptidomimetic inhibitors achieve high affinity by binding to various subsets of these hot spots. For example, remikiren (3D91:REM, MW = 630.84, KD = 131 pM, pBA = 9.88) binds to one fewer hot spot than Aliskiren, as it does not reach hot spot 4(8). However, peptide-like compounds such as remikiren, enalkiren and zanikiren are poorly absorbed and rapidly metabolized, and hence were not successful in clinical development44, 45. In fact, Aliskiren is the only FDA approved direct renin inhibitor available in the United States, and while it does not have the highest affinity, it does extend into a deep and narrow pocket to reach hot spot 1(15), and has somewhat better bioavailability than the peptidomimetics.44, 45 Reaching all hot spots, including 1(15), in the inhibitor binding pocket seems to be one of the keys to the success of Aliskiren, perhaps again because the extra binding energy thereby achieved provides scope to optimize regions of the inhibitor for improved pharmaceutical properties.
E3 ubiquitin-protein ligase XIAP is a member of the “inhibitor of apoptosis protein” (IAP) family and is endogenously regulated by second mitochondrial activator of caspases (SMAC) via its binding to the BIR domain of XIAP. The bRo5 compound birinapant (4KMP:GT6, MW = 806.94 Da, KD = 45 nM, pBA = 7.35), considered by Doak et al.21, is a bivalent SMAC-mimetic compound currently in clinical trials for the treatment of cancer (Figure 3c). In order to understand the binding of bivalent inhibitors we have mapped the XIAP dimer (4KMP). Birinapant reaches all FTMap identified hot spots: 0 (18), 1 (16), 2 (16), 3 (11), 4 (11), and 5 (7), thus stabilizing the dimer. However, most of the 20 compounds that bind to XIAP with known structure and affinity are monovalent SMAC-mimetic inhibitors and only reach the hot spots on one of the XIAP proteins. One such example is BI6 (2JK7:BI6, MW = 486.61 Da, KI = 67 nM, pBA = 7.17), which binds to hot spots 0(18), 3(11), and 5(7) with relatively high affinity (Figure 3c). Overall, there is a positive correlation between MW and pBA for the 20 XIAP inhibitors identified, but high affinity is achieved with both small (MW < 500 Da) and eRo5/bRo5 compounds. All XIAP inhibitors, including birinapant, exhibit moderate selectivity and also bind to other members of the IAP family of proteins.46 It may not be necessary to bind all hot spots in XIAP to inhibit its activity, however, birinapant appears to be the most successful clinical candidate at this time. More generally, the monovalent inhibitors are approximately 100–1000 times less potent than the corresponding bivalent compounds at the cellular level.47
P38 mitogen activated protein kinase (p38 MAPK) has a complex binding site consisting of four hot spots in the DFG-out conformation (2YIS), and shows a strong positive correlation between ligand pBA and MW (p < 0.001) (Figure S1). As will be discussed, this property makes the two MAP kinases (p38 MAPK and MEK1) considered here unique among the other kinases in Table 1, since the latter exhibit no correlation between pBA and MW. In fact, the inhibitors of p38 MAPK and MEK1 are type III kinase inhibitors that bind to an allosteric site that is adjacent to the ATP-binding pocket, and the mode of binding is very different from those of the type I and type II inhibitors that bind to ATP binding site in other kinases.48 The bRo5 inhibitor selected by Doak et al.21, PF-03715455 (2YIS:YIS MW = 700.27 Da, IC50 = 1.7 nM, pBA = 8.77) is a type III kinase inhibitor that binds to hot spots 1(21), 2(10), and 3(7) in the allosteric site, and also reaches 0(26) near the ATP site (Figure 3d). Another type III p38 MAPK inhibitor, BIRB-796 (1KV2:B96 MW = 527.66 Da, KD = 0.1 nM, pBA = 10) also binds to all four hot spots, and achieves even higher affinity. While large inhibitors that bind to all four hot spots generally achieve the highest affinity, smaller inhibitors can bind a subset of the hot spots and still achieve fairly high affinity. For example, (3P7B:P7B, MW = 464.58 Da, IC50 = 18 nM, pBA = 7.74) is a type III inhibitor that binds only the allosteric site in the DFG-out conformation (Figure 3d). Other kinase inhibitors that only bind near the ATP binding site can also achieve a range of affinities, such as (3HVC:GG5 MW = 239.25, KI = 600 nM, pBA = 6.22) and neflamapimod (3HP5:52P MW = 436.26 Da, KI = 0.8 nM, pBA = 9.09). While some high affinity inhibitors mentioned here (BIRB-796, neflamapimod) have advanced to clinical trials for inflammatory diseases, their progress has been hampered by adverse findings such as skin disorders, infection, and a lack of sustained efficacy.49 The development of the bRo5 inhibitor PF-03715455, considered by Doak et al.,21 was also discontinued50, but for business reasons rather than for safety and/or efficacy concerns.51 Thus, while p38 MAPK remains a challenging drug target, it appears that eRo5/bRo5 ligands which bind all hot spots, including those in the hydrophobic allosteric binding pocket, achieve the highest affinity and likely have improved selectivity as well.
HIV-1 aspartic protease.
This enzyme represents a special case of Complex I targets. We found 327 compounds that bind HIV-1 protease with known structure and affinity. Of the compounds that bind with high affinity, 97.8% of ligands are large, having MW > 500 Da, and bind to all seven hot spots in the complex binding site. This is explained by recognizing that the binding site resides at a very flexible region of the dimer (Figure 4a).52 Specifically, FTMap of the protease (3OXC) reveals seven hot spots: 0(21), 1(18), 2(18), 3(13), 4(11), 5(8), and 6(5), and it appears that reaching all hot spots is beneficial for the stability of the flexible dimer.52 We identified only six ligands which bind with high affinity that have MW < 500 Da. One example is XK216 (1HWR:216, MW = 406.52, KI = 4.6 nM, pBA = 8.34), an experimental cyclic urea inhibitor that binds all hot spots incredibly efficiently (Figure 4b). Larger ligands, such as the FDA-approved Saquinavir (3EL4:ROC, MW = 670.84 Da, KD = 67.4 nM, pBA = 7.17), do not necessarily have higher affinity, but do expand further into the binding site (Figure 4b). Based on its hot spot structure we consider HIV-1 protease to be in the Complex I group of targets. In fact, due to the flexibility of the dimer, small inhibitors that do not reach all hot spots would certainly have low binding affinity, resulting in positive correlation between affinity and MW. However, because all biomedically relevant HIV-1 protease inhibitors are large and bind to all hot spots, such a correlation cannot be directly observed (p > 0.05, see Figure 4c and Table S1).
Figure 4.
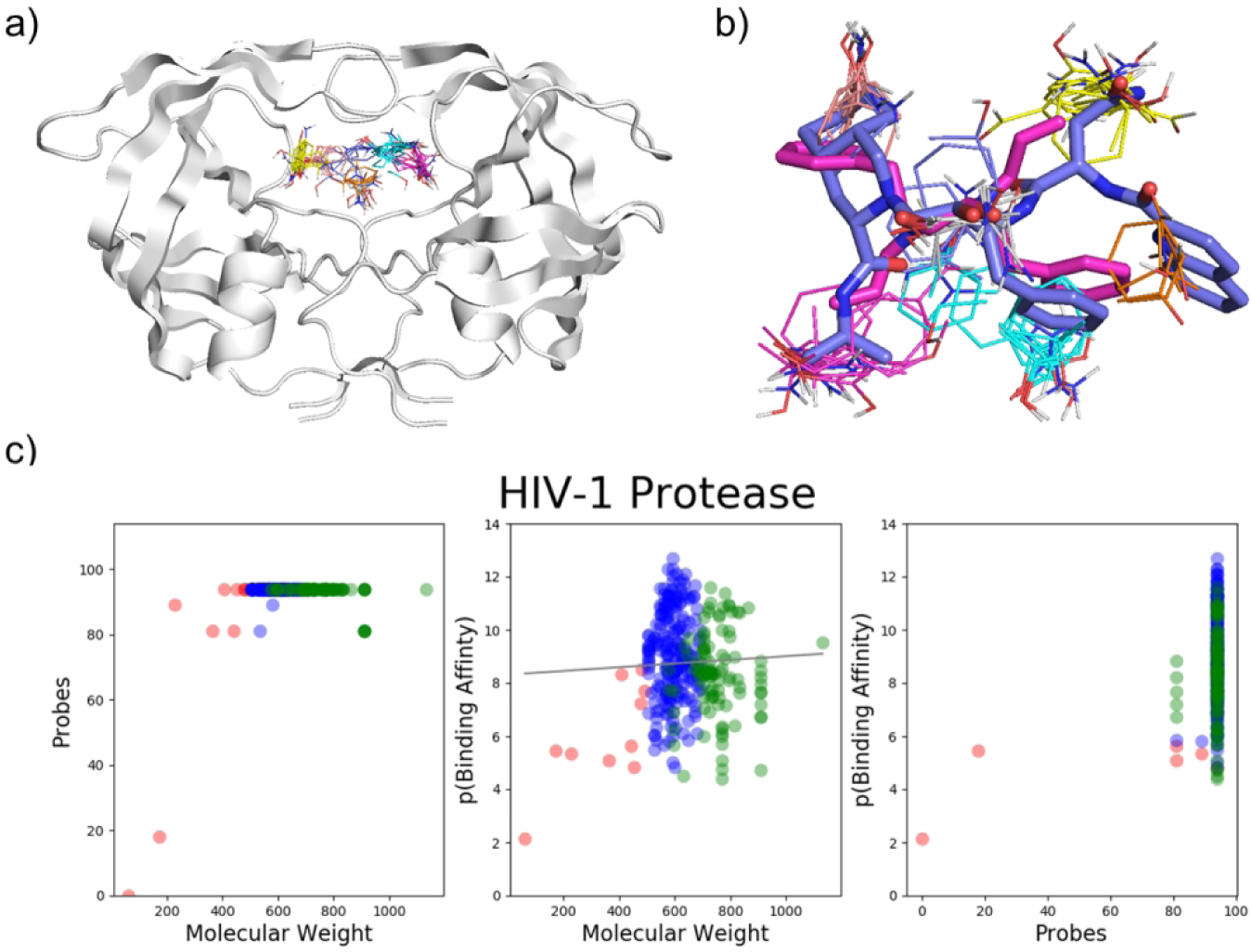
HIV-1 Protease binding hot spots. a) FTMap hot spots of HIV-1 Protease (3OXC): 0(21), 1(18), 2(18), 3(13), 2(111) 5(8), 6(5). b) Hot spot structure of HIV-1 Protease (3OXC). Saquinavir (3EL4:ROC, MW = 670.84 Da, KD = 67.4 nM, pBA = 7.17) and XK216 (1HWR:216, MW = 406.52, KI = 4.6 nM, pBA = 8.34) are shown in blue and pink, respectively. FTMap hot spots are colored according to standard output, shown in Figure 1. c) Three plots showing data on ligands that bind HIV-1 protease with known structure (90% or greater similarity) and binding affinity.
Heat shock protein 90 (Hsp90).
Current inhibitors of Hsp90 target the ATP-binding pocket in the N-terminal domain,53 which includes six hot spots: 0(25), 1(21), 2(16), 4(6), 5(6), and 6(5). The ligands that bind this pocket show a positive correlation between pBA and MW (p < 0.001, Table S1), characteristic to Complex I targets. Most inhibitors have been developed from geldanamycin (1YET: GDM, MW = 560.64 Da, KD = 1.2 μM, pBA = 5.92), a macrocyclic natural product that binds to most of the hot spots (Figure S3). Geldanamycin is an effective Hsp90 inhibitor but cannot be used in vivo due to hepatotoxicity.54 The inhibitor PU-H71 (2FWZ: H71, MW=512.37 Da, IC50=50 nM, pBA = 7.30), considered by Doak et al.21 is a purine based analog, which is slightly smaller than geldanamycin, and reaches five hot spots: 0(25), 1(21), 2(16), 4(6), and 6(5) (Figure S3).55 PU-H71 has demonstrated marked specificity for malignant cells, displaying minimal toxicity to normal tissue. Although PU-H71 and many other inhibitors are in active oncology trials, there are currently no approved Hsp90 targeted drugs.55
Mitogen-activated protein kinase kinase (MEK1).
MEK1 has an allosteric binding site adjacent to the ATP binding pocket, similar to p38 MAPK, but can be accessed in the DFG-in conformation. All 25 ligands identified to bind MEK1 with known structure and affinity bind the DFG-in conformation, and among them 17 bind the allosteric pocket, which has three hot spots: 0(18), 1(17), and 2(14), while eight compounds primarily bind the ATP binding site: 3(13) and 4(13). Cobimetinib (4AN2:EUI, MW = 531.31 Da, IC50 = 0.9 nM, pBA = 9.05), the inhibitor considered by Doak et al.,21 is one of only three FDA approved Type III kinase inhibitors, and it binds to all three hot spots in the allosteric binding site. Some smaller inhibitors such as (4ANB:YQY, MW = 477.22 Da, IC50 = 6.6 nM, pBA = 8.8) bind with less, but still high affinity via two hot spots in the allosteric site, 1(17) and 2(14), but have not been approved for clinical use. Two other inhibitors, Trametinib (MW = 615.39 Da) and Binimetinib (MW = 441.23 Da), have been approved by the FDA for the treatment of certain cancers, but there is no co-crystallized structure to determine how the drugs associate with the binding hot spots of MEK1. Notably, MEK1 and p38 MAPK differ from the other protein kinases, discussed as complex II targets, because they have an allosteric site adjacent to the ATP binding site, which enables inhibitors to bind with very high affinity, shown by the strong positive correlation between the pBA and MW of ligands (p < 0.001, Table S1).
Bromodomain-containing protein 4 (BRD4).
The bromodomain family has emerged as a target for cancer therapy. BRD4 is able to bind traditional Ro5 compounds, dual kinase-bromodomain inhibitors, and bivalent inhibitors. Due to the presence of bivalent inhibitors, the hot spots in Table 1 were obtained by considering BRD4 as a dimer. FTMap of the complex reveals three hot spots on each monomer, chain A: 0(21), 3(8), 6(5) and chain B: 1(18), 2(11), 5(7), and two hot spots in the interface of the dimer: 4(8) and 7(5). Taken together, the hot spot structure of BRD4 is incredibly complex. However, the binding cavity of a single chain in BRD4 on its own provides sufficient hot spots for Ro5 drugs such as the cell-permeable small molecule JQ1 (3MXF:JQ1, MW = 458.00 Da, KD = 49 nM, pBA = 7.31), which exhibits high potency and specificity towards BRD4 (Figure S3).56 A number of eRo5 compounds have been identified to bind BRD4 as well, including the dual BRD4-kinase inhibitor fedratinib (4OGJ: 2TA, MW = 524.68 Da, KD = 164 nM, pBA = 6.79) considered by Doak et al.21 (Figure S3). Most eRo5 bromodomain inhibitors are clinical kinase inhibitors that also inhibit bromodomains with therapeutically relevant potencies.57 An example of bivalent inhibitors is (5KHM: XNH, MW = 479.57 Da, KI = 5 nM, pBA = 8.3), which binds two hot spots on each monomer (Figure S3). Overall, there is a positive correlation between affinity and MW of compounds that bind BRD4 (p < 0.001), as with all targets classified as complex I. Bivalent inhibitors are able to achieve higher potency and specificity than both the dual BRD4-kinase inhibitors and traditional Ro5 compounds such as JQ1.58
Peroxisome proliferator-activated receptor γ (PPARγ).
The activity of PPARγ can be modulated with agonists, partial agonists, and antagonists. The protein is flexible, and the binding of the ligands affect the position of the activating helix (H12), which regulates the binding of cofactor proteins.59 The impact of the ligand on the shape of the binding site is shown by the difference between mapping a ligand-bound structure and a ligand-free structure. FTMap of the structure with bound INT131 (3FUR:Z12, MW = 514.21 Da, KI = 10 nM, pBA = 8), a selective partial agonist considered by Doak et al.,21 identifies a very large binding site with five weak hot spots 2(9), 4(8), 5(8), 6(7), and 8(6), while the two strongest hot spots, 0(15) and 1(12), are located at H12, in the cofactor binding site (Table 1, Figure S3). In contrast, FTMap of the ligand-free PPARγ structure (2HWR) finds 10 hot spots: 0(23), 1(9), 2(9), 3(9), 4(9), 5(7), 6(6), 7(6), 8(6), and 9(6) in the binding site, emphasizing that the protein has a complex hot spot structure (Table 2, Figure S3). The best known PPARγ ligands are the thiazolidinedione (TZD) class of full agonists that include rosiglitazone (1ZGY:BRL, MW = 357.43 Da, KI = 1.0 nM, pBA = 9) and pioglitazone (2XKW:P1B, MW = 356.44 Da, KI = 1320 nM, pBA = 5.88).60 The TZDs have been clinically validated as anti-diabetic agents, but their use has been associated with serious side effects, including weight gain, peripheral edema, hepatotoxicity, increased risk of congestive heart failure, and bone fracture. Although the maximal efficacies of the partial agonist, INT131, and full agonist, rosiglitazone, were similar with respect to improvements in glucose tolerance, INT131 had less effect on heart and lung weights, weight gain, hemodilution, and plasma volume. Thus, INT131 appears to selectively modulate PPARγ responses, showing antidiabetic efficacy while exhibiting an improved hemodynamic and cardiovascular adverse effect profile compared to the full agonists.
Table 2.
Hots spots based on the mapping of bound and unbound target structures
| Target Name | Hot Spot Structure | PDB ID | Chai n | Bound | PDB ID | Chai n | Unbound |
|---|---|---|---|---|---|---|---|
| FTMap Associated Hotspots | FTMap Associated Hotspots | ||||||
| Thrombin | Complex I | 1DWC | H | 0(26), 2(14), 5(6), 6(6), 7 (5) | 1SGI | E | 0(28), 1 (16), 2(15), 4(11) |
| MAP Kinase P38 | Complex I | 2YIS | A | 0(26), 1 (21), 2 (10), 3 (7) | 4E5B | A | 0(15), 1 (13), 2(12), 5(10) |
| Renin | Complex I | 2V0Z | C | 0 (24), 1 (15), 2 (12), 3 (8), 4 (8), 5 (7), 6 (7) | 1BBS | A | 0 (23), 1 (19), 2 (15), 3 (9), 4 (8), 5 (8), 6 (7) |
| PPAR-γ | Complex I | 3FUR | A | 2 (9), 4 (8), 5 (8), 6 (7), 8 (6) | 2HW R | B | 0 (23), 1 (9), 2 (9), 3 (9), 4 (9), 5 (7), 6 (6), 7 (6), 8 (6), 9 (6) |
| Epidermal Growth Factor Receptor | Complex II | 1XKK | A | 0 (16), 1 (15), 2 (12), 3 (9), 5 (7), 6 (7) | 4TKS | A | 0(20), 1 (20), 2 (17), 3 (14), 4 (5) |
| Hepatocyte Growth Factor Receptor | Complex II | 3F82 | A | 0 (14), 1 (12), 2 (11), 3 (10), 4 (9), 5 (8), 7 (6) | 1R1W | A | 0 (16), 2 (13), 3 (10), 5 (8), 6 (7), 7 (5), 9 (5) |
| Anaplastic Lymphoma Kinase | Complex II | 4MKC | A | 0 (24), 3 (8), 5 (6), 6 (6), 7 (5) | 4FNX | A | 0 (23), 1 (20) |
| Tyrosine Protein Kinase ABL1 | Complex II | 3CS9 | A | 0(20), 2(15), 3(14), 4(13) | 2HZ4 | C | 0(26), 1 (19), 2(11) |
| VEGFR- 2 | Complex II | 3C7Q | A | 0 (20), 1 (17), 2 (10), 3 (9), 4 (8), 6 (8), 7 (6) | 1VR2 | A | 0 (26), 1 (14), 2 (13), 3 (12), 4 (12), 5 (7), 6 (6) |
| Polo-like Kinase 1 | Complex II | 3FC2 | A | 0(28), 1 (19), 2(12), 3(7), 7(5) | 2V5Q | A | 0(25), 1 (15), 3(11), 4 (9) |
| Cyclophilin A | Complex III | 1CWA | A | 0(27), 1 (16), 4(11), 5 (6) | 5KV0 | A | 0 (26), 2 (11), 3 (11), 6 (5) |
| Bcl-2 | Complex III | 4LVT | A | 0(22), 2 (17), 4 (10), 5 (5) | 1GJH | A | 0 (20), 1 (14), 4 (12) |
| Phosphoinositide-3 Kinase | Simple | 3DBS | A | 4 (8) | 1E8Y | A | 0 (16) |
| Protein Farnesyltransferase | Simple | 1O5M | A B | 0 (19), 2 (15) | 1FT1 | B | 1 (17), 3 (16) |
| Kinesin Eg5 | Simple | 4A5Y | A | 0 (17), 2 (17), 3 (12) | 4A28 | A | 1 (19) |
| Hepatitis C Virus NS5b Subunit | Simple | 4NLD | A | 2 (10) | 2ZKU | B | - |
| Hepatitis C Virus NS34A Protease | Simple | 3KEE | A E | 1 (16), 3(10) | 1DXP | A | - |
| Tubulin-Alpha Chain | Simple | 1JFF | B | 2 (16), 5 (8) | 2XRP | G | 2 (17), 4 (10) |
| Integrin Alpha-11B | Simple | 2VDN* | B | 2 (17) | 2VDL | B | 3 (13) |
| Na-K ATPase | Simple | 3A3Y | A | 1 (17), 2(12), 3 (8) | 5AW8 | A | 0(10), 6(5), 11 (5) |
| α-Amylase | Simple | 1PPI | A | 0 (18), 1 (16), 3 (11) | 1KXV | A | 1 (15), 2(14), 3(12) |
Indicates domain splitting was performed prior to FTMap (see Methods)
The ligand binding profiles for all Complex I targets are shown in Figure S2. Overall, Complex I targets can bind small ligands with high affinity, but additional affinity can be gained by adopting eRo5/bRo5 ligands that reach to additional hot spots. While the motivation for employing larger compounds is sometimes to improve potency, in many cases it appears instead to be for the purpose of gaining additional binding energy that can be traded off for improved pharmaceutical properties.
Complex II Targets.
The Complex II group is comprised of the seven proteins that have complex hot spot structures and show no correlation between affinity and MW (p > 0.05), which distinguishes their ligand binding profiles from Complex I targets (Figure 2, Table S1, Figure S4). For these targets, high affinity binding is usually achieved at low ligand molecular weights (i.e. MW < 400 Da), however larger ligands have nonetheless been explored. Six of the seven Complex II targets are kinases. As mentioned previously, over 30% of FDA approved kinase inhibitors have MW > 500 Da, placing them in the eRo5/bRo5 category.12 Typically, small kinase inhibitors bind at the ATP binding site, which is present in the DFG-in conformation of the protein, whereas larger inhibitors often reach into the so-called “back pocket” present only in the DFG-out conformation. The lack of increase in binding affinity with increased molecular weight of the ligands for Complex II targets indicates that the reason for using bRo5 ligands is not for improved affinity. Indeed, it is well recognized that the reason for developing eRo5/bRo5 inhibitors for kinases is most often to improve selectivity.61 Since this statement applies to all kinases considered here in the Complex II group, we discuss only three examples in detail, as well as the only non-kinase member of this group, the glucocorticoid receptor.
Tyrosine protein kinase ABL1.
Inhibitors of ABL1 variants represent first-line therapy for most patients with chronic myelogenous leukemia (CML). We identified numerous ABL1 inhibitors, with MW values ranging from 372 Da to 595 Da, that bind either the DFG-in or DFG-out conformation. FTMap analysis of the DFG-out conformation (3CS9) shows four hot spots, two near the ATP binding site 0(20), and 4(13), and two in the DFG back pocket 2(15) and 3(14). In contrast, mapping the DFG-in conformation (4TWP) reveals five hot spots:0(19), 2(11), 3(9), 4(7), and 6(5), all located near the ATP binding site. Thus, in the DFG-in conformation the kinase has a higher density of hot spots than in the DFG-out conformation (Figure 5a). Small ligands, such as the experimental inhibitor PPY-A (2Z60:P3Y, MW=372.42, KI = 20.0 nM, pBA = 7.70) can bind the DFG-in conformation with high affinity (Figure 6a), whereas larger drugs such as Nilotinib (3CS9:NIL MW = 529.52 Da, KD = 4.9 nM, pBA = 8.31), usually bind the DFG-out conformation (Figure 6a). Nilotinib utilizes all four hot spots in the DFG-out conformation by extending into the DFG-out pocket, but it achieves only slightly higher binding affinity than smaller inhibitors. Nilotinib is structurally related to imatinib, the first FDA approved Type II kinase inhibitor. The ABL1 inhibitors dasatinib, bosutinib, and ponatinib, which were approved for the treatment of imatinib resistant or intolerant CML, also bind to all four hot spots in the DFG-out conformation and are bRo5 compounds. Thus, for ABL1, bRo5 compounds appear to be most successful, yet all have comparable affinity to the smaller inhibitors (pBA versus MW p-value > 0.05) (Table S1). This may be attributed to the overall shift in hot spots from the ATP site in the DFG-in conformation to the back pocket in the DFG-out conformation (Figure 5a).
Figure 5.
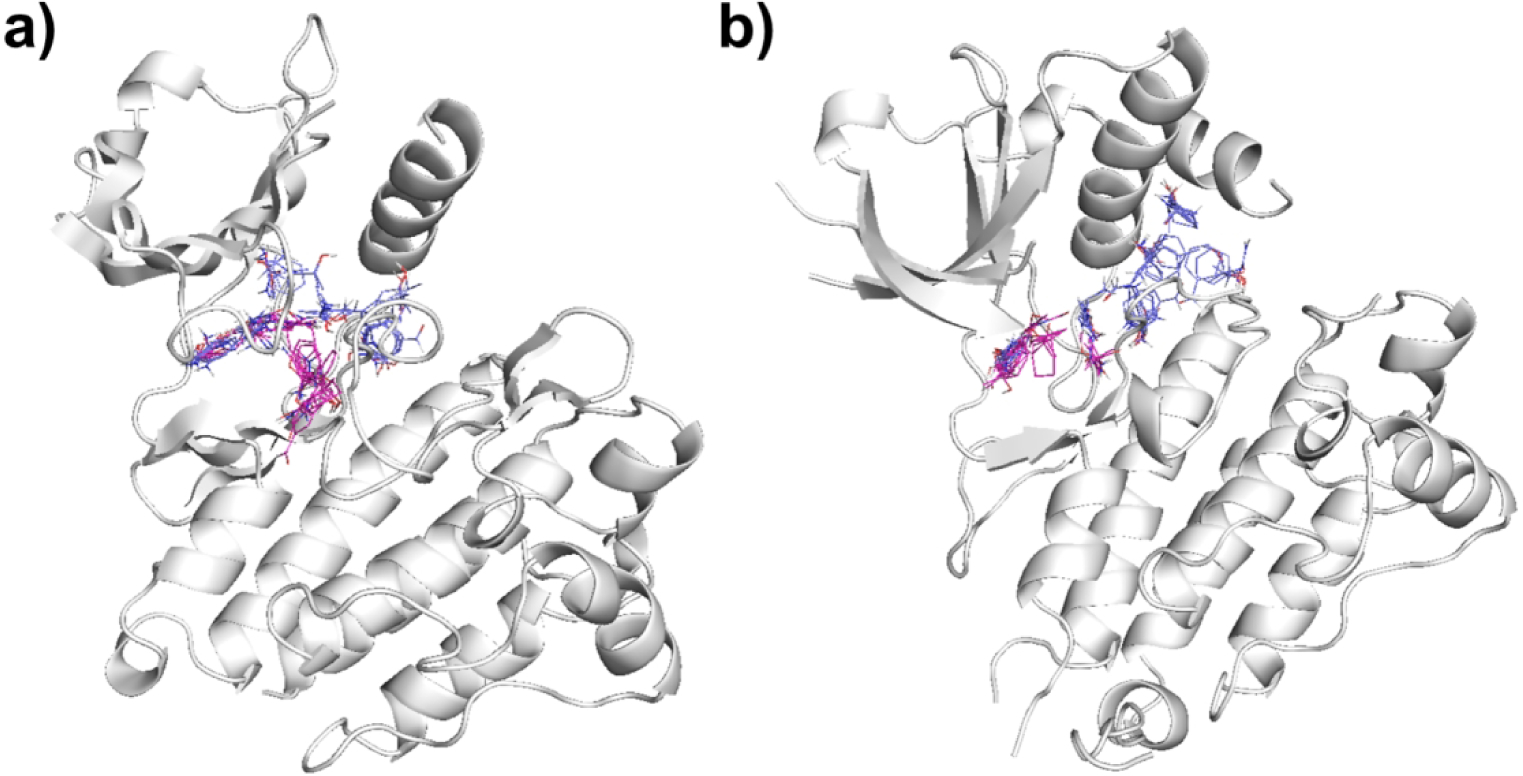
FTMap hot spot structure of kinase DFG-in versus DFG-out conformations. Hot spots in pink represent the FTMap results for the DFG-in conformation, whereas blue represent DFG-out hot spots. The protein cartoon shows the DFG-out conformation. a) Hot spots of tyrosine protein kinase ABL1 in DFG-in (4TWP) and DFG-out (3CS9) conformations. b) Anaplastic Lymphoma Kinase in DFG-in (4MKC) and DFG-out (5IUH) conformations.
Figure 6.
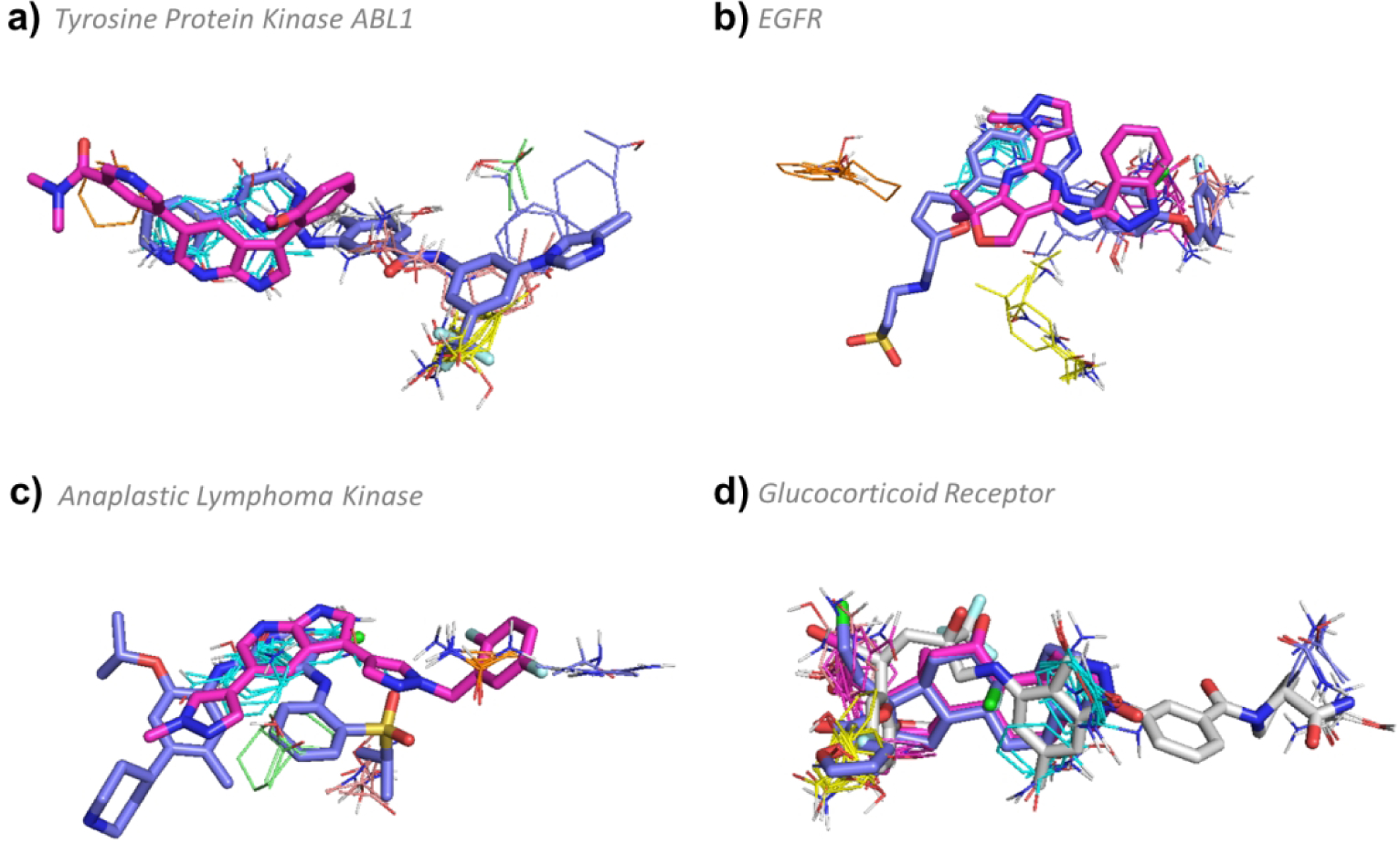
FTMap determined hot spot structure of Complex II targets with example ligands bound. Pink ligands represent smaller compounds, whereas blue ligands represent the eRo5/bRo5 drug/clinical candidate identified by Doak et. al. White ligands (where applicable) show alternative compounds. FTMap hot spots are colored according to standard output, which is described in the supplementary information. a) Hot spot structure of tyrosine protein kinase ABL1 (3CS9). Nilotinib (3CS9:NIL MW = 529.52 Da, KD = 4.9 nM, pBA = 8.31) and (2Z60:P3Y, MW=372.42, KI = 20.0 nM, pBA = 7.70) are shown in blue and pink, respectively. b) Hot spot structure of epidermal growth factor receptor (1XKK). Lapatinib (1XKK:FMM MW = 581.06 Da, KI = 3 nM, pBA = 8.52) and (5EDR:5N4, MW = 361.4 Da, KI = 34.3 nM, pBA = 7.46) are shown in blue and pink, respectively. c) Hot spot structure of anaplastic lymphoma kinase (4MKC). Certinib (4MKC:4MK, MW = 558.13 Da, KI = 3.7 μM, pBA = 5.43) and 7-azaindole based inhibitor (4JOA:3DK, MW = 390.39 Da, IC50 = 29 nM, pBA = 7.54) are shown in blue and pink, respectively. d) Hot spot structure of glucocorticoid receptor (4P6W). Mometasone furoate (4P6W:MOF, MW = 521.43 Da, KI = 0.7 nM, pBA = 9.15), (1P93:DEX MW = 392.46 Da, KD = 19 nM, pBA = 7.72), and (3K23:JZN, MW = 655.68 Da, IC50 = 6.3 nM, pBA = 8.2) are shown in blue, pink, and white, respectively.
Epidermal growth factor receptor (EGFR).
In the active, or DFG-in, conformation, the binding site of EGFR is complex and includes 6 hot spots, 0(16), 1(15), 2(12), 3(9), 5(7), and 6(7). Of the 59 compounds identified for EGFR, 55 bind the DFG-in conformation and 4 bind the DFG-out conformation. All structurally characterized ligands that bind in the DFG-in conformation associate with the same trio of hot spots 0(16), 1(15), 5(7) in the ATP site, and most extend in different directions to also engage with various combinations of the remaining hot spots, or to regions between hot spots. For example, the smallest ligand identified (5EDR:5N4, MW = 361.4 Da, KI = 34.3 nM, pBA = 7.46) binds only the core trio of hot spots (Figure 6b). The FDA approved drug Lapatinib (1XKK:FMM MW = 581.06 Da, KI = 3 nM, pBA = 8.52) also binds this core trio in the DFG-in conformation, but additionally extends to hot spot 3(9) and toward Arg 803 on the other side of the ATP binding site (Figure 6b). The four compounds that bind the DFG-out conformation (FTMap: 5HG5) only bind the strong hot spot 0(25) in the ATP site, and extend outwards toward to the surface of the kinase. Thus, no additional hot spots are reached in these DFG-out binding compounds and overall, there is no correlation between affinity and MW (p > 0.05; see Figure 2, Table S1). Because high affinity can be accomplished with very small ligands, we propose that the motivation for creating larger compounds for EGFR is to improve selectivity rather than affinity.
Anaplastic lymphoma kinase (ALK).
The clinical candidate Certinib (4MKC:4MK, MW = 558.13 Da, KI = 3.7 μM, pBA = 5.43) binds to the hot spots 0(24), 3(8), and 7(5) in the DFG-in conformation of ALK (Figure 6c). A smaller compound, a 7-azaindole based inhibitor (4JOA:3DK, MW = 390.39 Da, IC50 = 29 nM, pBA = 7.54), also binds to the DFG-in conformation, but a slight conformational change enables the compound to extend deeper into the binding crevice towards hot spots 5(6) and 6(6), achieving fairly high affinity with a low molecular weight (Figure 6c). Larger Inhibitors such as the pyrazolylamine derivative (5IUH:34Y, MW = 556.66 Da, IC50 = 402 nM, pBA = 6.4) bind ALK in the DFG-out conformation, which opens a large back pocket for binding. FTMap analysis of ALK in the DFG-out conformation (5IUH) reveals five new hot spots: 0(19), 1(11), 2(11), 3(10), and 5(7) concentrated around the DFG-out back pocket, and retains one hot spot 4(8) in the ATP binding site (Figure 5b). Overall, the ligands that bind ALK do not show a positive correlation between pBA and MW (p > 0.05), consistent with other kinases (Figure 2, Table S1). The shift in the locations and strengths of the binding hot spots on ALK between the DFG-in and DFG-out conformations is similar, but more exaggerated, than seen for ABL1 (Figure 5). Thus, a potential reason why a ligand that reaches the hot spots in the DFG-out pocket yields similar affinity to smaller ligands that only are concentrated around ATP binding site may be the weakened ATP binding site in the DFG-out conformation.
The kinases described above show that larger inhibitors generally do not result in improved binding affinity. Similar patterns exist for the additional kinases in our study, namely the hepatocyte growth factor receptor (HGFR), the vascular endothelial growth factor receptor 2 (VEGFR-2), and polo-like kinase 1 (PLK1). The ligand binding profiles are provided in Figure S4, and corresponding statistics are provided in Table S1. Based on the observation that kinases can typically bind small ligands with high affinity and do not gain substantial additional affinity by adopting bRo5 ligands, we conclude that the reason for employing larger compounds is likely not for the purpose of improving potency. Instead, the motivation is to achieve greater selectivity, which is accomplished by occupying regions of the binding site that are unique to the protein of interest. Although the fibroblast growth factor receptor (FGFR) is a typical kinase, it has only one ligand with known structure and affinity, and hence will be discussed in the Complex III group. As mentioned previously, the inhibitors of MAP kinase P38 and MAP kinase kinase considered by Doak et al.21 are Type III kinase inhibitors, and bind very differently from the Type I and Type II inhibitors discussed for the other kinases here. In addition to kinases, we discuss the glucocorticoid receptor here, because based on its binding profile it belongs to the Complex II group.
Glucocorticoid receptor.
Glucocorticoid receptor (4P6W) has a complex hot spot structure with six hot spots, 0(25), 1(21), 2(13), 3(7), 4(7) and 5(6). We do not observe a correlation between pBA and MW (p > 0.05), and thus the binding profile of the glucocorticoid receptor is similar to those of kinases (Figure S4). All ligands identified bind three core hot spots 0(25), 1(21), and 3(7), while larger ligands also extend to the more distant secondary hot spots 4(7) and 5(6) in one direction and 2(13) in another direction. For example, the small FDA-approved agonist of the glucocorticoid receptor, dexamethasone (1P93:DEX MW = 392.46 Da, KD = 19 nM, pBA = 7.72), binds only to the three core hot spots (Figure 6d). Dexamethasone is a typical corticosteroid with many side effects, including fluid retention, increased appetite, mood changes, skin rash, bruising or discoloration. The larger FDA-approved drug mometasone furoate (4P6W:MOF, MW = 521.43 Da, KI = 0.7 nM, pBA = 9.15), which is used as a topical corticosteroid in ointments and nasal spray, extends beyond the three hot spots to overlap with hot spot 2(13) (Figure 6d). It has a glucocorticoid receptor binding affinity 22 times stronger than dexamethasone and higher than many other corticosteroids as well.62 A much larger compound (3K23:JZN, MW = 655.68 Da, IC50 = 6.3 nM, pBA = 8.2), which also extends towards hot spot 2(13), similar to mometasone furoate, and towards hot spots 4(7) and 5(6), however, does not gain more affinity (Figure 6d).
Targets in the Complex III Group.
Eight proteins with complex hot spot structures have been co-crystallized with fewer than 9 ligands of known binding affinity, and hence we were unable to confidently construct ligand-binding profiles. However, the small ligand-binding profiles are shown in Figure S5, where possible. Although we believe that some of these targets behave similarly to Complex I targets while others to Complex II, without substantial binding profiles they cannot be added to these groups, and hence are defined to form the Complex III group. In addition, some of the targets can benefit from eRo5/bRo5 drugs or drug candidates for very specific reasons, and hence each of the nine targets is individually discussed here, without any attempt of trying to find an overarching motivation.
Cyclophilin A.
Cyclophilin A has a complex hot spot structure, including four strong hot spots: 0(27), 1(16), 4(11), and 5(6). The drug considered by Doak et. al.21 is cyclosporine A (1CWA:PRD_000142, MW = 1,202.61 Da, KD = 36.8 nM, pBA = 7.43) which is a macrocycle that reaches all four hot spots, and extends outward; the complex has been shown to inhibit calcineurin, acting as an immunosuppressant (Figure 7a).63, 64 Smaller compounds, such as (3RDD:EA4, MW = 251.28 Da, KI = 16800 nM, pBA = 4.77), bind only three of the hot spots on cyclophilin A and have very weak affinity. In addition, such compounds do not competitively inhibit calcineurin and hence have no biological significance (Figure 7a). Reaching all four hot spots creates a span of 19 Å (between 0(27) and 5(6)), and requires a very long molecule. However, the main reason for the high MW is that the cyclophilin-bound cyclosporine directly contributes to the binding of calcineurin, and this function requires additional atoms expanding the macrocycle.63
Figure 7.
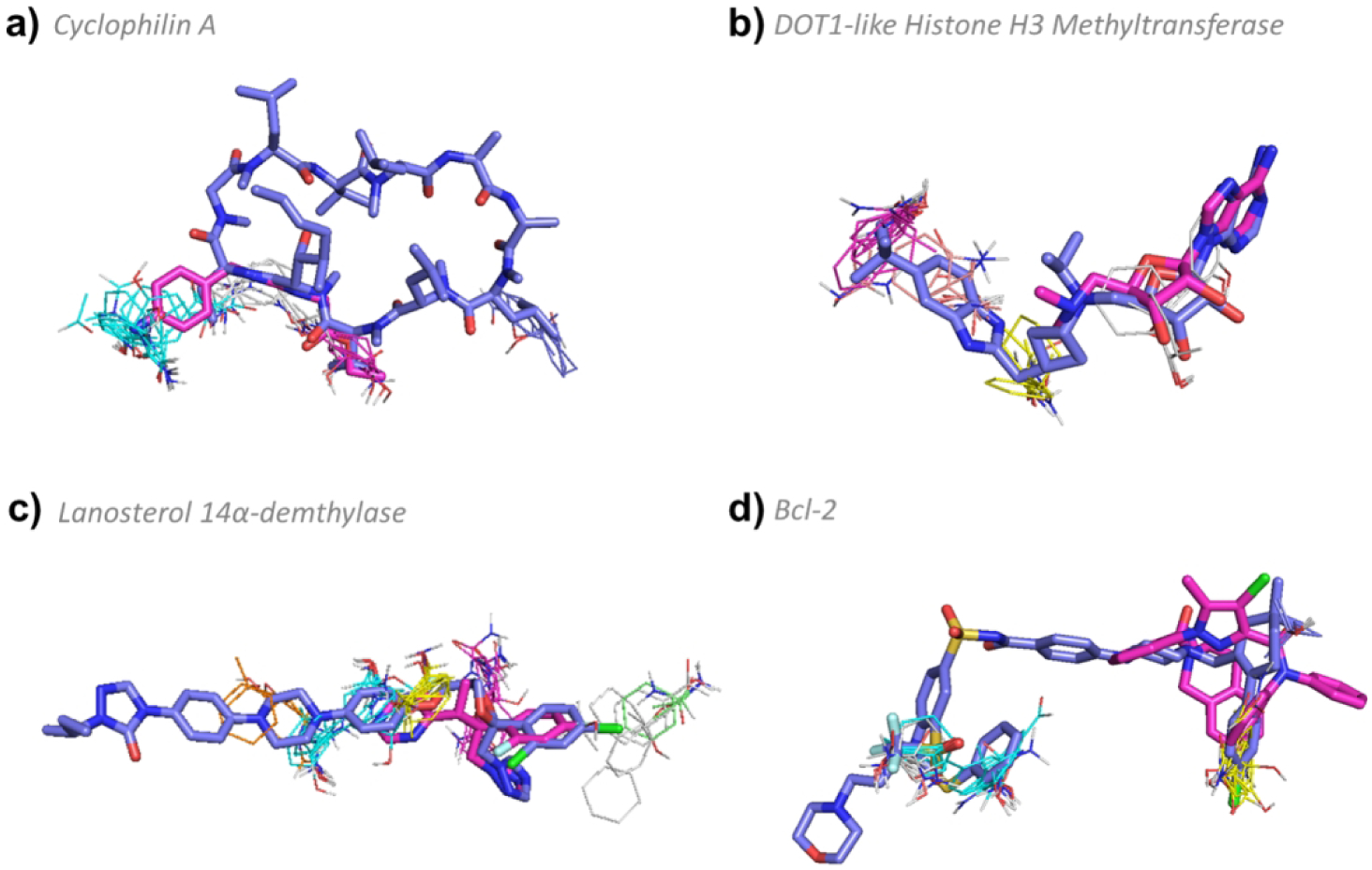
FTMap determined hot spot structure of Complex III targets with example ligands bound. Pink ligands represent smaller compounds, whereas blue ligands represent the eRo5/bRo5 drug/clinical candidate identified by Doak et al. White ligands (where applicable) show alternative compounds. FTMap hot spots are colored according to standard output as shown in Figure 1. a) Hot spot structure of cyclophilin A (1CWA). Cyclosporine A (1CWA:PRD_000142, MW = 1,202.61 Da, KD = 36.8 nM, pBA = 7.43) and (3RDD:EA4, MW = 251.28 Da, KI = 16800 nM, pBA = 4.77) are shown in blue and pink, respectively. b) Hot spot structure of DOT1-like histone H3 methyltransferase (4HRA). EPZ-5676 (4HRA:EP6 MW = 562.71 Da, KI = 0.08 nM, pBA = 10.1) and (4EK9:EP4, MW = 294.31 Da, KI = 9 μM, pBA = 5.04) are shown in blue and pink, respectively. c) Hot spot structure of lanosterol 14α-demethylase (5EQB). Itraconazole (5EQB:1YN, MW = 705.63 Da, IC50 = 19.4 nM, pBA = 7.71) and voriconazole (5HS1:VOR, MW = 349.31 Da, KI = 174 μM, pBA = 3.76) are shown in blue and pink, respectively. d) Hot spot structure of Bcl-2 (4LVT). Navitoclax (4LVT:1XJ, MW = 974.61 Da, KI = 0.044 nM, pBA = 10.36) and (2W3L:DRO, MW = 576.09 Da, IC50 = 30 nM, pBA = 7.52) are shown in blue and pink, respectively.
DOT1-like histone H3 methyltransferase.
This protein binds a clinical candidate, EPZ-5676 (4HRA:EP6 MW = 562.71 Da, KI = 0.08 nM, pBA = 10.1), with very high affinity at hot spots 1(17), 2(12), 3(11), and 4(6) (Figure 7b). While the primary hot spot of 17 probe clusters is slightly weaker than in typical complex targets, the hot spot structure is still relatively complex, with four hot spots. The smallest ligand, EPZ000004 (4EK9:EP4, MW = 294.31 Da, KI = 9 μM, pBA = 5.04), only binds hot spots 2(12) and 4(6), and has very low affinity (Figure 7b). A slightly larger ligand, S-adenosyl-L-homocysteine (3QOX:SAH, MW = 384.4 Da, KI = 0.27 μM, pBA = 6.57), binds to three hotspots, 2(12), 3(11) and 4(6), with higher affinity. However, the three highest affinity inhibitors, including EPZ–5676 which is currently in clinical trials for advanced leukemia,65 bind to all four hot spots (46 total probe clusters), and all have MW > 500 Da. EPZ–5676 has both the highest MW and affinity. Thus, for this protein it appears that a bRo5 compound that binds all hot spots is required to achieve high affinity binding.
Lanosterol 14α-demethylase.
Lanosterol 14α-demethylase (5EQB) has a large, complex binding site consisting of five hot spots: 0(23), 1(17) 2(10), 4(7), and 6(5) located inside a large cavity, with an additional three hot spots buried even deeper inside the cavity. The bRo5 drug identified by Doak et al.,21 is itraconazole (5EQB:1YN, MW = 705.63 Da, IC50 = 19.4 nM, pBA = 7.71), a high affinity antifungal medication66 that binds to all five hot spots in the binding site (Figure 7c). The distance between 4(7) and 6(5) is over 18.5 Å, too large to bridge by a Ro5 ligand. Of the three ligands found to bind lanosterol 14α-demethylase with known structure and affinity, itraconazole is the largest, protruding out of the cavity, and has the highest binding affinity.
However, there exist smaller inhibitors such as fluconazole (4WMZ:TPF, MW = 306.27 Da, KI = 30 μM, pBA = 4.52) that binds only to 2(10) and 4(7), and voriconazole (5HS1:VOR, MW = 349.31 Da, KI = 174 μM, pBA = 3.76) that binds to 0(23), 2(10), and 6(5) (Figure 7c). All three are azole compounds, with the heterocyclic nitrogen atom binding to the heme iron atom and a halogenated phenyl ring interacting with a hydrophobic pocket of the enzyme binding site. Itraconazole has a broader spectrum of activity than fluconazole, and is better for antifungal prophylaxis67, but is less well tolerated68. These differences are limited, and it is not clear whether itraconazole has advantages over the smaller compounds, or simply was the first lanosterol 14α-demethylase inhibitor developed.
Bcl-2.
The unique hot spot structure of Bcl-2 (4LVT) has two groups of hot spots 0(22), 4(10) and 2(17), 5(5) separated from one another on the surface of Bcl-2 by approximately 16 Å, requiring a relatively large ligand in order to bind all hot spots. The FDA-approved Navitoclax (4LVT:1XJ, MW = 974.61 Da, KI = 0.044 nM, pBA = 10.36) binds all hot spots and achieves very high affinity (Figure 7d). All but one of the compounds identified to bind Bcl-2 are very large, bridge the gap between the two sets of hot spots, and have high affinity (pBA > 7). We only identified one smaller inhibitor (2W3L:DRO, MW = 576.09 Da, IC50 = 30 nM, pBA = 7.52) which binds half of the hot spots: 2(17) and 5(5), but the MW > 500 Da and affinity is reduced relative to Navitoclax (Figure 7d).
Isoleucyl-tRNA Synthetase.
Aminoacyl-tRNA synthetase is an enzyme that catalyzes the binding of an amino acid onto tRNA via ATP. The binding site of the isoleucyl-tRNA synthetase is narrow crevice that accommodates both isoleucine and ATP,69 and contains six relatively weak hot spots 0(14), 1(14), 2(14), 3(11), 4(8), and 7(5). The inhibitor pseudomonic acid A (1QU2:MRC, MW = 500.6 Da, KD = 0.14 nM, pBA = 9.85), considered by Doak et al.21 binds to all six hot spots (Figure S5). The maximum distance between hot spots is over 18 Å, thus a relatively long ligand is required to bind to all hot spots. In addition, the 9-hydroxynonanoic acid moiety of the pseudomonic acid A extends beyond the hot spot ensemble to a hydrophobic surface region, resulting in a molecule that is over 20 Å long. Nevertheless, it is almost within the 500 Da range.
FK506 Binding Protein.
The FK506-binding protein 12 (FKBP12) binds the immunosuppressant drug rapamycin (4DRI:RAP, MW = 914.17 Da, KI = 1.0 nM, pBA = 9), which works by inducing inhibitory protein complexes with the kinase mTOR.70 The hot spots between FKBP12 and mTOR are 0(27), 1(20), 2(16), 3(13), 4(6), and 5(5), and rapamycin interacts with all of them (Figure S5). In principle, 0(27) and 4(6) alone may support a Ro5 ligand of FKBP12 with moderate binding affinity. However, the main biological function of rapamycin is to interact with both with FKBP12 and the FRB fragment of mTOR, so additional moieties are needed for interaction with both proteins, and rapamycin is able to achieve that as a fairly large macrocyclic compound.
Toll-like Receptor 4.
Toll-like receptor 4 (TLR4) detects lipopolysaccharides found in most Gram-negative bacteria, and hence plays a fundamental role in pathogen recognition and activation of innate immunity. Eritoran is a TLR4 antagonist (2Z65:E55, MW = 1313.7 Da, IC50 = 1.5 nM, pBA = 8.82).71 The natural substrates of TLR4 are large lipopolysaccharides that bind to four major hot spots 0(22), 1(20), 2(13), 3(12) in a deep cavity. Eritoran, a lipid derivative, interacts with these hots spots (and additional four hot spots) to compete with the substrate, resulting in a very large molecule with four long “arms” reaching into the hot spots (Figure S5). Thus, in this case, it appears a large compound may be needed to compete with the natural substrate.
Fibroblast growth factor receptor 1 (FGFR1).
Small molecular inhibitors target the ATP site of the kinase domain of FGFR1, which includes the hot spots 0(18), 1(17), 3(12), 5(6), and 6(5). A number of Ro5 kinase inhibitors such as dovitinib (5A46: 38O, MW = 392.43 Da, no binding affinity data) strongly overlap with the hot spot 0(18), and lightly overlap with the hot spots 1(17), 3(12), and 5(6) located deeper in the ATP binding site. All such compounds showed toxicities related to VEGFR inhibition, such as hypertension, cardiovascular events, and proteinuria.72 In contrast, the compound studied by Doak et al.,21 BGJ398 (3TT0:07J, MW = 560.5 Da, KI = 13 nM, pBA = 7.89) associates with all hot spots, including a strong overlap with 1(17), 3(12), and 5(6) deep in the pocket, and is a selective fibroblast growth factor receptor (FGFR) inhibitor that does not bind to VEGFR.73 Similar to other kinases, the use of an eRo5 compound substantially improves selectivity, but there are not enough compounds with structure and affinity data to generate a ligand binding profile.
TARGETS WITH SIMPLE HOT SPOT STRUCTURE
Targets with “simple” hot spot structures have three or fewer hot spots in the ligand binding site. The total number of probes in the binding site is generally less than 50 probe clusters (Figure 1) and the primary hot spot is weak, with less than 20 probe clusters, which places the targets at or below the threshold for druggability, based on our previous study.39 These weak binding sites are often located in a deep cavity, narrow canyon, or are otherwise isolated from additional hot spots. Generally, all ligands that bind these proteins must contact all hot spots in the ensemble, and then reach outward for increased binding affinity, and possibly increased selectivity, by interacting with the surrounding surface regions, which do not have additional hot spots. As a result, some proteins with simple host spot structure show a positive correlation between pBA versus MW while others do not (Figure 2, Figure S6, Table S1).
Kinesin Eg5.
The binding site of kinesin Eg5 consists of three hot spots, 0(17), 2(17) and 3(12), that lie close together in a fairly deep pocket. The primary hot spot has sufficient strength, at 17 probe clusters, to render the binding site weak but druggable.39 As a result of kinesin Eg5’s weak and compact hot spot ensemble, every ligand with known structure and affinity binds to all three hot spots in the binding site. Among these compounds, MW ranges from 274 to 517 Da, and a positive correlation exists between pBA and MW (p < 0.01, see Table S1 and Figure 2). The smallest compound, (S)-enastron (2X7C:KZ9, MW = 274.34 Da, IC50 = 2 μM, pBA = 5.7) lies within 3 Å of all three hot spots in the cavity, but does not strongly overlap with the hot spots and has low affinity (Figure 8a). However, the clinical candidate ispinesib (4A5Y:G7X, MW = 517.06 Da, KI = 2.3 nM, pBA = 8.64), which has the largest molecular weight of all drugs identified for kinesin Eg5, has stronger overlap with the hot spots, extends deeper into the binding pocket, and interacts with the walls of the cavity, resulting in a much higher binding affinity (Figure 8a). While we found only 28 kinesin Eg5 inhibitors with known structure and affinity, over 100 different inhibitors have been described in the scientific literature and there have been at least 35 Phase 1 or Phase 2 clinical trials initiated.74 Some of the compounds brought to clinical trials have MW < 500 Da, but ispinesib shows promise as being one of the most advanced drug candidates.75
Figure 8.
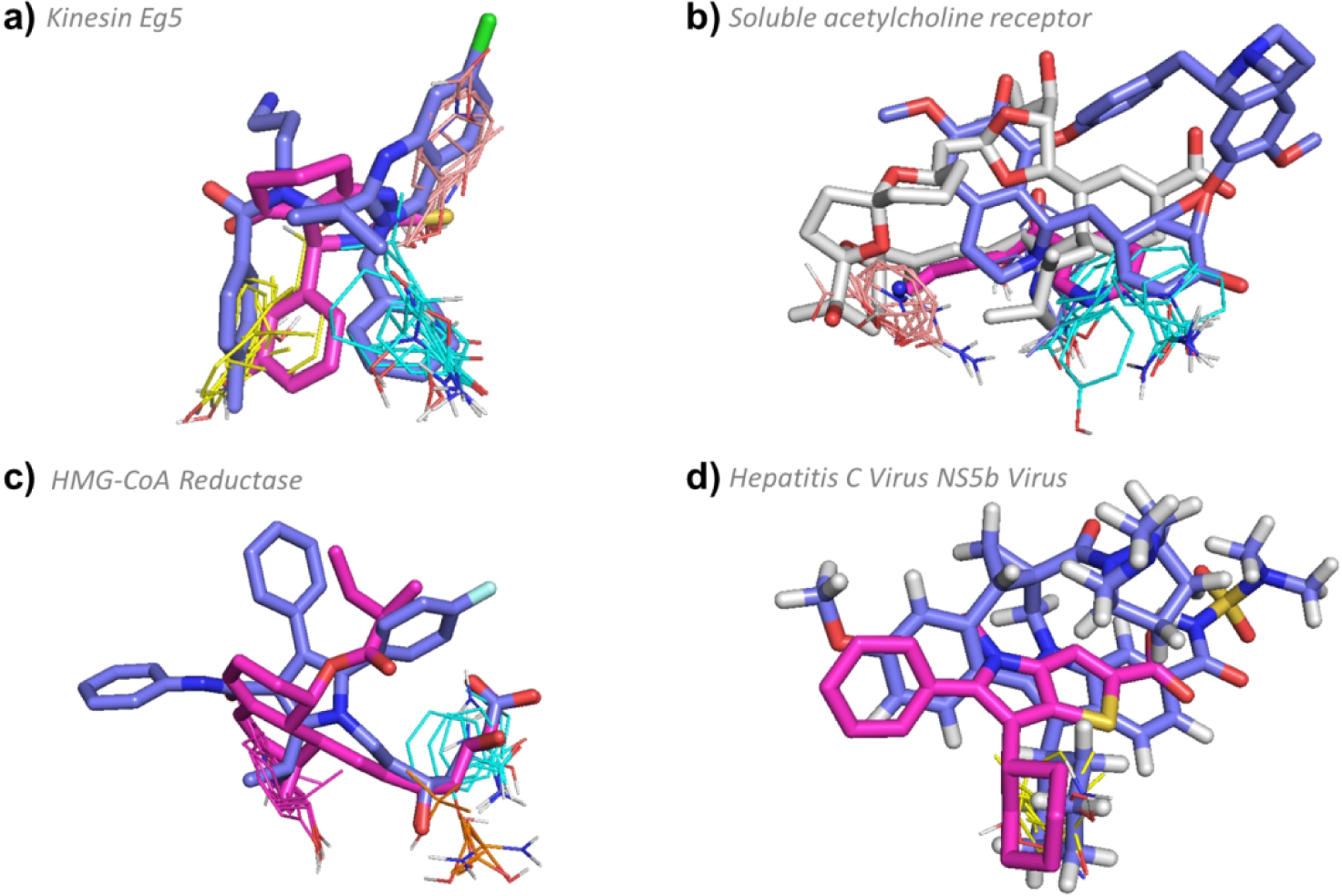
FTMap determined hot spot structure of Type III targets with example ligands bound. Pink ligands represent smaller compounds, whereas blue ligands represent the eRo5/bRo5 drug/clinical candidate identified by Doak et al. White ligands (where applicable) show compounds larger than the Doak et al., identified ligand. FTMap hot spots are colored according to standard output as shown in Figure 1. a) Hot spot structure of kinesin Eg5 (4A5Y). Ispinesib (4A5Y:G7X, MW = 517.06 Da, KI = 2.3 nM, pBA = 8.64), and (S)-enastron (2X7C:KZ9, MW = 274.34 Da, IC50 = 2 μM, pBA = 5.7) are shown in blue and pink, respectively. b) Hot spot structure of soluble acetylcholine receptor (3PMZ). Tubocurarine (3PMZ:TUB MW = 609.73 Da, KI = 3481.0 nM, pBA = 5.46), (2WNL:AN5, MW = 178.23 Da, KI = 120 μM, pBA = 6), and pinnatoxin A (4XHE:40P, MW = 711.92 Da, KD < 0.05 nM, pBA = 10.3) are shown in blue, pink, and white, respectively. c) Hot spot structure of HMG-CoA reductase (1HWK). Atorvastatin (1HWK:117, MW = 558.64 Da, KI = 6.2 nM, pBA = 8.21) and mevastatin (1HW8:114, MW = 408.53 Da, IC50 = 23 nM, pBA = 7.64) are shown in blue and pink, respectively . d) Hot spot structure of Hepatitis C Virus NS5b Subunit (4NLD). Beclabuvir (4NLD:2N7, MW = 659.84 Da, IC50 = 0.02 μM, pBA = 7.7) and (2WCX:VGC, MW = 339.45 Da, IC50 = 0.025 μM, pBA = 7.6) are shown in blue and pink, respectively.
Soluble acetylcholine receptor.
Similar to kinesin Eg5, most ligands that bind the soluble acetylcholine receptor bind to all three hots spots in the site: 0(19), 3(13), and 5(9), totaling 41 probe clusters (Figure 2). The site is located in a deep inter-chain crevice, and the larger ligands form significant interactions with the sides of the crevice, increasing the binding affinity, as shown by the positive correlation between pBA versus MW (p < 0.01, see Table S1 and Figure 2). One of the smallest compounds to bind (2WNL:AN5, MW = 178.23 Da, KI = 120 μM, pBA = 6) only barely touches the three hot spots, whereas one of the largest compounds, the macrocyclic neurotoxin pinnatoxin A (4XHE:40P, MW = 711.92 Da, KD < 0.05 nM, pBA = 10.3) extends deeper into the crevice and interacts significantly with the walls of the protein, resulting in a much higher binding affinity (Figure 8b). The FDA-approved inhibitor tubocurarine (3PMZ:TUB MW = 609.73 Da, KI = 3.48 μM, pBA = 5.46) is a macrocycle that also extends into the crevice and interacts with the walls of the protein, but with much weaker affinity (Figure 8b).
HMG-CoA reductase.
As was the case with kinesin Eg5 and soluble acetylcholine receptor, all ligands of HMG-CoA reductase, the target of statins,76 bind to the same three hot spots, 0(13), 1(13), and 6(6). The MW of the 19 HMG-CoA reductase ligands in the PDB ranges from 408 Da to 655 Da, and there is no correlation between pBA and MW (p > 0.05, see Table S1 and Figure 2). The best known FDA-approved statin, Atorvastatin (1HWK:117, MW = 558.64 Da, KI = 6.2 nM, pBA = 8.21), interacts with the cavity walls and has acceptable binding affinity (Figure 8c), similar to the other approved statins that have MWs between 420 Da and 580 Da. The smallest known statin, an experimental compound called Mevastatin (1HW8:114, MW = 408.53 Da, IC50 = 23 nM, pBA = 7.64) also binds to the three hot spots and extends up the cavity wall, resulting in sufficient binding affinity (Figure 8c). However, the side effects of Mevastatin include those of other statins, such as myalgias, abdominal pain, and nausea, and this compound additionally has a higher chance of giving more severe side effects related to myotoxicity and hepatotoxicity.77 The increased toxicity is most likely associated with reduced selectivity, and Mevastatin was never marketed as a drug. We note that the inhibitor binding site of HMG-CoA reductase is very large and includes two additional hot spots, 4(9) and 5(7), that are far from the three main hot spots and are not utilized by any of the ligands (Figure S7). It is plausible, based on a structural analysis, that a very large ligand might extend to reach the additional hot spots at the other end of the cavity. However, since the additional hot spots are weak, doing so would likely result in at best a modest improvement in affinity, leaving little scope to trade excess affinity for improved pharmaceutical properties.
Hepatitis C Virus subunits.
The Hepatitis C Virus (HCV) NS5b RNA Polymerase has only one very weak hot spot, 2(10), located in a shallow cavity on the surface of the protein. There are no other hot spots nearby and all eight ligands identified that bind to HCV NS5b with known structure and affinity overlap with this one hot spot (Figure S6). The ligands’ MW ranges from 339 to 659 Da and extend around the hot spot in multiple directions, but overall there is no increase in pBA with MW (p-value > 0.05; Table S1, Figure S6). Small compounds, such as (2WCX:VGC, MW = 339.45 Da, IC50 = 25 nM, pBA = 7.6), do not reach into the surrounding area to the same extent as larger ligands such as Beclabuvir (4NLD:2N7, MW = 659.84 Da, IC50 = 20 nM, pBA = 7.7), which is currently in phase II clinical trials (Figure 8d). Sofosbuvir (MW = 529.45 Da) has been approved as a drug, but there is no structure of the Sofosbuvir-HCV NS5B complex in the PDB. Similar to HCV NS5b RNA Polymerase, HCV NS3/4a protease also presents a simple hot spot structure of 1(16) and 3(10), and drugs extend significantly outside of the shallow binding pocket, yet there is no correlation between pBA and MW (p > 0.05; Table S1, Figure S6). Notably, all five drugs identified to bind the NS3/4a subunit have MW > 740 Da and high affinity (pBA > 8.4; Table 1, Figure S6). In the case of these two targets, therefore, the motivation for using bRo5 ligands is not for greater affinity, but likely to achieve greater selectivity by interacting with regions of the protein beyond the very simple hot spot ensemble.
Protein Farnesyltransferase.
Farnesyltransferase post-translationally modifies proteins by adding an isoprenoid lipid called a farnesyl group to the -SH of the cysteine near the end of target proteins to form a thioether linkage. This process causes farnesylated proteins to become membrane-associated due to the hydrophobic nature of the farnesyl group. Farnesyl transferase inhibitors inhibit farnesylation of a wide range of target proteins, including Ras. It is thought that these agents block Ras activation through inhibition of the enzyme farnesyl transferase, ultimately resulting in cell growth arrest. Lonafarnib (1O5M:336, MW = 638.8 Da, KI = 8.3 nM, pBA = 8.08)78 considered by Doak et al.,21 is a farnesyltransferase inhibitor that has been investigated in a human clinical trial as a treatment for progeria. The hot spots in the binding site are 0(19) and 2(15), but 0(19) overlaps with the location of farnesyl diphosphate. The latter also interacts with most ligands, including Lonafarnib. The current version of FTMap does not enable mapping in the presence of farnesyl diphosphate, and hence may not provide valid information on the hot spots. However, we note that Ro5 farnesyltransferase inhibitors have also been developed. For example, tipifarnib (4LNG:JAN, MW = 489.4 Da, no binding affinity data) was submitted to FDA. Although the application was rejected, the drug is still under development.
Phosphoinositide-3 Kinase.
Phosphoinositide 3-kinases (PI3Ks) are a family of enzymes involved in cellular functions such as cell growth, proliferation, differentiation, motility, survival and intracellular trafficking, which in turn are involved in cancer. Several inhibitors of PI3Ks have been reported in the literature, such as wortmannin (1E7U:KWT, MW = 428.4 Da, KI = 4.2 nM, pBA = 8.38).79 While these compounds have been widely used to elucidate the functional role of PI3K, their toxicity and lack of selectivity have limited their therapeutic potential. A number of pharmaceutical companies have thus developed PI3K isoform-specific inhibitors that are FDA approved, including the PIK3CD inhibitor, idelalisib (4XE0:40L, MW = 415.42 Da, IC50 = 19 nM, pBA = 7.72), and the dual PIK3CA and PIK3CD inhibitor, copanlisib (5G2N:6E2, MW =480.52 Da, no binding affinity data). In contrast, pictilisib, a pan PI3K inhibitor considered by Doak et al.,21 is still in clinical trials. All inhibitors have substantial toxicity issues, and it is not yet clear whether pictilisib (3DBS:GD9, MW = 513.63 Da, KI = 3.0 nM, pBA = 8.52) will provide advantages.80 PI3K is a large protein that substantially differs from protein kinases. Mapping finds only the hot spot 4(8) at the ligand binding site, while the stronger hot spots are placed deeper in the protein. However, mapping the unbound structure (1E8Y) places 0(16) at the inhibitor binding site, but overall, the site is weak, and needs fairly large molecules for strong binding.
Table 1 lists six additional simple targets that have three or fewer ligands with known structure and affinity, and thus do not provide data for the construction of meaningful ligand binding profiles. In summary, the motivation for adopting bRo5 compounds for targets with simple hot spot structure can be to achieve improved affinity, selectivity, or perhaps other pharmaceutical properties, by contacting additional regions of the protein around the relatively small and weak hot spot ensemble. Thus, while for Complex I and Complex II targets the use of bRo5 compounds was driven mainly by the need to improve selectivity or other pharmaceutical properties, for targets with simple hot spot structure bRo5 ligands may be a means to compensate for borderline or poor druggability. Importantly, simple and complex targets can easily be distinguished based on their binding site hot spot structure, as identified using FTMap.
CONSERVATION OF HOT SPOTS: BOUND VERSUS UNBOUND PROTEIN STRUCTURES
The structural analysis performed above was completed by mapping the proteins from the protein-ligand complexes selected by Doak et. al.21 However, the number, strength, and arrangement of binding hot spots are properties of the target protein itself, and hence can be determined by the analysis of ligand-free protein structure. Here, the FTMap hot spot calculations were repeated with the unbound structures corresponding to the drug-target complexes selected by Doak et al.21 whenever such were available. Using a threshold of 95% sequence similarity, we identified unbound structures for 21 of the 37 protein targets and applied FTMap. Since ligand binding may promote pocket formation by induced fit or conformational selection, we expected to find weaker hot spots in the ligand-free structures, and a few cases confirmed this expectation (Table 2). However, apart from a slight decrease in the number of probe clusters and minor shifts in hot spot locations, our results show remarkable conservation of major hot spots in ligand binding sites across the unbound and bound structures (Table 2).
Unbound structures were found for 12 proteins that have complex hot spot structures in the bound form. The “complex” classification was conserved in the unbound form for 9 of these proteins: MAPK p38, thrombin, PPAR-γ, renin, cyclophilin A, EGFR, HGFR, VEGFR-2, and polo-like kinase 1. The “complex” classification of the hot spot structure was lost in three of the 12 targets, however, due to a drop in the number of hot spots below four. For ABL1 and ALK the number of hot spots in the binding site dropped from four to three and two hot spots, respectively, yet the primary hot spot remained strong, with a strength of 26 and 23 probe clusters, respectively. As a result, these targets are still distinct from those designated as having a “simple” hot spot structure because the primary hot spot is so strong, and most simple hot spot structures have only a weak (< 20 probe clusters) primary hot spot (Table 2). In the case of Bcl-2, the number of hot spots dropped from 4 in the bound structure to 2 in the unbound structure, and the strength of the primary hot spot dropped from 22 probe clusters to 20 (Table 2). However, the geometry of the hot spots for Bcl-2 was conserved across the unbound and bound structures, as both show two distinct hot spot regions separated from each other by about 11 Å.
The nine targets with a “simple” hot spot structure that had also crystallized in the unbound form also showed good conservation of hot spots between the bound and unbound structures. Specifically, seven of these targets, PI3K, protein farnesyltransferase, kinesin Eg5, tubulin-alpha chain, integrin alpha 11-B, Na-K ATPase, and α-amylase show very close conservation of hot spots, keeping one to three hot spots in total with the strongest hot spot below 20 probe clusters (Table 2). However, the already weak hot spots for the two hepatitis C virus proteins, NS3/4a and NS5b, were lost in the unbound structure. Thus, the weak sites in these proteins remain weak or become even weaker in the unbound conformation, and their “simple” binding site classification is retained. Overall, while the agreement between FTMap results for unbound and bound structures is not perfect, in most cases the binding site structure is sufficiently conserved, and is useful in determining if and how a target may benefit from bRo5 ligands.
LIGAND-BASED ANALYSIS
The structure-based analysis suggests that most of the targets studied from Doak et al.21 can bind both Ro5 and eRo5/bRo5 compounds with high affinity, however some of the larger compounds tend to be more successful in clinical trials for the reasons outlined above. A number of targets (11 of 37) have very limited structure-based ligand profiles however, with fewer than five ligands identified, and thus analysis of their ligand binding profiles was not possible. To check whether these targets generally follow the same pattern of binding both Ro5 and eRo5/bRo5 ligands and are considered druggable, we pulled all ligands for each target from ChEMBL,27 including those that do not have a structure deposited in the PDB. The results of our ChEMBL ligand-based analysis are shown in Table 3; only targets with more than 40 high affinity ligands were considered. As the result of this condition, 8 of the 37 targets were excluded. Generally, the number of ligands in each ChEMBL profile greatly increased relative to the structure based profile, especially for the targets that had fewer than five ligands in the structure-based analysis, namely FK506 binding protein and FGFR1 (Complex III), and hepatitis C virus NS34A protease, integrin alpha-IIB, smoothened homolog, and Na-K ATPase (Simple).
Table 3.
Results of the ChEMBL-based ligand analysis
| Target name | Hot Spot Structure | Total No. of ChEMBL ligands | Liganablea | Tractableb | Ro5 compliance | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LIGexp | LE/LLE | Ro5_3c | Ro5_4d | MW>500 | ||||||||
| No. | % | No. | % | No. | % | No. | % | No. | % | |||
| HIV-1 Protease | Complex I | 5638 | 3561 | 63.16 | 75 | 2.11 | 1922 | 53.97 | 183 | 5.14 | 3166 | 88.91 |
| Heat Shock Protein 90 | Complex I | 841 | 335 | 39.83 | 92 | 27.46 | 326 | 97.31 | 247 | 73.73 | 69 | 20.6 |
| Thrombin | Complex I | 5921 | 2297 | 38.79 | 1230 | 53.55 | 2051 | 89.29 | 1057 | 46.02 | 1012 | 44.06 |
| MAP Kinase P38 | Complex I | 4772 | 2548 | 53.39 | 408 | 16.01 | 2186 | 85.79 | 1509 | 59.22 | 506 | 19.86 |
| Bromodomain BRD4 | Complex I | 1088 | 222 | 20.40 | 17 | 7.66 | 220 | 99.1 | 149 | 67.12 | 29 | 13.06 |
| Renin | Complex I | 4038 | 2813 | 69.66 | 392 | 13.94 | 1665 | 59.19 | 276 | 9.81 | 2413 | 85.78 |
| PPAR-γ | Complex I | 2134 | 917 | 42.97 | 1 | 0.11 | 405 | 44.17 | 108 | 11.78 | 531 | 57.91 |
| MAP Kinase Kinase | Complex I | 867 | 461 | 53.17 | 150 | 32.54 | 416 | 90.24 | 263 | 57.05 | 176 | 38.18 |
| E3 ubiquitin-protein ligase XIAP | Complex I | 1168 | 308 | 26.37 | 14 | 4.55 | 174 | 56.49 | 63 | 20.45 | 242 | 78.57 |
| Epidermal Growth Factor Receptor | Complex II | 7047 | 2959 | 41.99 | 504 | 17.03 | 2673 | 90.33 | 1963 | 66.34 | 801 | 27.07 |
| Hepatocyte Growth Factor Receptor | Complex II | 3864 | 2382 | 61.65 | 755 | 31.7 | 2042 | 85.73 | 1316 | 55.25 | 967 | 40.6 |
| Anaplastic Lymphoma Kinase | Complex II | 1688 | 1020 | 60.43 | 151 | 14.8 | 835 | 81.86 | 392 | 38.43 | 584 | 57.25 |
| Tyrosine Protein Kinase ABL1 | Complex II | 2032 | 1084 | 53.35 | 401 | 36.99 | 963 | 88.84 | 678 | 62.55 | 296 | 27.31 |
| VEGFR- 2 | Complex II | 8031 | 3894 | 48.49 | 688 | 17.67 | 3585 | 92.06 | 2597 | 66.69 | 839 | 21.55 |
| Polo-like Kinase 1 | Complex II | 737 | 281 | 38.13 | 34 | 12.1 | 254 | 90.39 | 161 | 57.3 | 105 | 37.37 |
| Glucocorticoid Receptor | Complex II | 3510 | 2584 | 73.62 | 221 | 8.55 | 2146 | 83.05 | 930 | 35.99 | 752 | 29.1 |
| Cyclophilin A | Complex III | 231 | 74 | 32.03 | 1 | 1.35 | 12 | 16.22 | 10 | 13.51 | 59 | 79.73 |
| Bcl-2 | Complex III | 865 | 427 | 49.36 | 3 | 0.7 | 109 | 25.53 | 41 | 9.6 | 361 | 84.54 |
| DOT1-like Histone H3 Me-transferase | Complex III | 100 | 42 | 42.00 | 28 | 66.67 | 40 | 95.24 | 5 | 11.9 | 34 | 80.95 |
| FK506 Binding Protein | Complex III | 491 | 156 | 31.77 | 4 | 2.56 | 62 | 39.74 | 32 | 20.51 | 110 | 70.51 |
| FGFR1 | Complex III | 1605 | 661 | 41.18 | 87 | 13.16 | 624 | 94.4 | 369 | 55.82 | 270 | 40.85 |
| Phosphoinositide-3 Kinase | Simple | 1912 | 704 | 36.82 | 315 | 44.74 | 686 | 97.44 | 515 | 73.15 | 161 | 22.87 |
| Protein Farnesyltransferase | Simple | 2411 | 1204 | 49.94 | 373 | 30.98 | 1070 | 88.87 | 570 | 47.34 | 543 | 45.1 |
| Kinesin Eg5 | Simple | 1060 | 396 | 37.36 | 105 | 26.52 | 362 | 91.41 | 287 | 72.47 | 55 | 13.89 |
| HMG-CoA Reductase | Simple | 287 | 199 | 69.34 | 26 | 13.07 | 154 | 77.39 | 91 | 45.73 | 64 | 32.16 |
| Hepatitis C Virus NS5b Subunit | Simple | 1267 | 302 | 23.84 | 57 | 18.87 | 227 | 75.17 | 128 | 42.38 | 128 | 42.38 |
| Hepatitis C Virus NS34A Protease | Simple | 498 | 254 | 51.00 | 1 | 0.39 | 212 | 83.46 | 1 | 0.39 | 249 | 98.03 |
| Integrin Alpha-IIB | Simple | 2436 | 1027 | 42.16 | 613 | 59.69 | 779 | 75.85 | 545 | 53.07 | 415 | 40.41 |
| Smoothened Homolog | Simple | 710 | 340 | 47.89 | 59 | 17.35 | 316 | 92.94 | 261 | 76.76 | 39 | 11.47 |
| Na-K ATPase | Simple | 223 | 55 | 24.66 | 11 | 20 | 47 | 85.45 | 22 | 40 | 24 | 43.64 |
Liganable - number, percent of compounds with pKI > 7
Tractable - number, percent of compounds with LE > 0.3 and LLE > 5, percent based on total ligandable
Ro5_3 - number of ligands meeting 3 of the 4 Ro5 criteria;
Ro5_4 - number of ligands meeting 4 of the 4 Ro5 criteria, percents based on total ligandable
Overall, the ChEMBL ligand analysis confirms that the targets selected by Doak et al.21 bind both Ro5 and eRo5/bRo5 compliant ligands. Across all targets, ligands were composed of an average of ~43% Ro5 compliant ligands, and ~ 46% eRo5/bRo5 ligands (i.e. MW > 500 Da), see Table 3. Furthermore, we assessed the ligandability of each target with the recently defined LIGexp3 index,81 which is defined as the proportion of compounds tested against a target with a pKi > 7, our “high affinity” threshold discussed previously. Our results confirm that on average, about half (~45%) of ligands bind the target with high affinity. Lastly, we assessed the chemical tractability of each target by calculating the percentage of each target’s compounds that have a ligand efficiency greater than 0.3 and lipophilic ligand efficiency greater than 5, where LE = pBA/Number of Heavy Atoms and LLE = pBA-clogP (i.e. LE > 0.3 and LLE > 5).82 We note that this concept is used to assess the tractability of a target for drug discovery, and is not related to synthetic tractability – i.e. the ease of synthesis of particular chemotypes.83 While some targets show high ligand efficiency, the majority actually have fairly low ligand efficiency, which is not surprising since these targets favor eRo5/bRo5 ligands.
There are a few targets in our set with ligands that are especially skewed towards eRo5/bRo5. For example, Bcl-2 has only 7 ligands in the structure-based ligand profiles and all have MW > 500 Da and high affinity. The ChEMBL-based ligand profile confirms that Bcl-2 most likely requires a bRo5 drug, as the percentage of Ro5 compliant ligands is very low (9.6%), the percentage of eRo5/bRo5 ligands is very high (84.5% of ligands has MW > 500 Da), and tractability is very low (LE > 0.3 and LLE > 5 for 0.7% of ligands), see Table 3. Very similar profiles exist for HIV-1 protease and E3 ubiquitin-protein ligase XIAP (Complex I), cyclophilin A (Complex III) and hepatitis C virus NS34a (Simple), thus confirming that the limited structure-based ligand profiles are representative of the greater ChEMBL-based ligand profile. We note that PPAR-γ presents somewhat of an exception as the chemical tractability appears very low (0.1%) because ligands generally satisfy the LE > 0.3 criteria, but notoriously fail the LLE > 5 criteria because the binding site is highly lipophilic.
It was shown that Complex II targets, comprising mostly kinases, have strong hot spots resulting in a highly druggable active site, with the motivation for using bRo5 ligands being improved selectivity rather than potency or druglikeness. For the Complex II targets that are kinases, the results of the ligand based analysis are in full agreement with these findings. More than 10% of the ligands of each kinase fulfil the LE/LLE criteria (Table 3). At least 85% and 55% of the ligands of these kinases (except for those targeting ALK) fulfil the Ro5_3 and Ro5_4 criteria. ALK ligands, however, are typically larger and more lipophilic than other kinase ligands in this set. The only non-kinase target in this class considered here is glucocorticoid receptor, which has the lowest LE/LLE compliance at 8.5%. Most of the ligands of the glucocorticoid receptor fulfil the LE criterion; however, they notoriously fail the LLE criterion. This is in line with our previous study classifying nuclear hormone receptors as challenging targets for druglike molecules.55 Notably, glucocorticoid receptor has the highest ligandability of all type II targets (0.74).
CONCLUSIONS
The goal of this study has been to understand why some targets benefit from drugs which violate the traditional Ro5 criteria. To investigate this question, we started with a set of 37 proteins which have eRo5/bRo5 drugs or clinical candidates from Doak et. al21. Each of the drugs and clinical candidates in this set has MW > 500 Da, thus we focused on the MW criteria of the Ro5, but also calculated all other Ro5 properties. Initial analysis of these targets and all ligands that bind to them revealed that, in addition to eRo5/bRo5 compounds, 22 targets can also bind multiple small compounds (MW < 500 Da) with high affinity. Thus, in these cases the need for ligands outside the Ro5 is not driven by poor druggability per se. Structural analysis of the targets revealed that the targets of eRo5/bRo5 drugs have one of two distinct hot spot structures, referred to as “complex” and “simple”, and both differ from the hot spots structures seen in most proteins that bind FDA approved Ro5 drugs.39 Analysis of how small and large ligands engage with the hot spot ensemble at the binding site shed light on why eRo5/bRo5 ligands are often more successful than their smaller alternatives even when these also have high affinity. Specifically, from the 37 proteins studied here, different mechanisms by which expanding to bRo5 improved drug properties emerged. Among the targets with complex hot spot structure, the proteins within the Complex I group have similar motivation for eRo5/bRo5 ligand development, as supported by the binding profiles. We also find a common motivation among the targets classified as Complex II, but not for the targets in the Complex III group that are discussed individually. However, improving binding is a common motivation for targets with simple hot spot structure.
Complex I targets have a “complex” hot spot structure, and most bind both small compounds (MW < 500 Da) and bRo5 compounds with high affinity, yet the bRo5 compounds are generally more successful. In these targets we observe that there is an increase in binding affinity with increased molecular weight. However, small ligands with fairly high affinity generally exist, and so it does not seem that enhanced binding affinity is the main driver for why bRo5 ligands have been more successful in clinical trials. As shown for thrombin, many smaller drug candidates had problems due to inherent toxicity, high-plasma protein binding, poor metabolic stability, rapid elimination from the blood, and low in vivo activity, while the bRo5 compound Argatroban has improved properties. These improved pharmaceutical properties are presumably not inherent in the use of larger compounds; indeed, the opposite might be expected. But good properties were evidently not achieved with Ro5-compliant compounds. A potential explanation is that in moving to the bRo5 range, the extra binding energy gained by engaging more hot spots provides scope for pharmaceutically problematic functionality on ligand to be substituted with more benign structures that individually bind less well, while retaining sufficient overall binding affinity for the compound as a whole. In addition, the higher complexity of the compounds may reduce the likelihood of off-target binding. We emphasize that these observations are based on only a small set of targets, and it is possible that other targets with complex hot spot structures do have pharmaceutically optimized Ro5 ligands. However, in previous work we have mapped a substantial number of proteins that have only Ro5 drugs, and in most cases we have found at most three hot spots, albeit strong ones, in the ligand binding site.39 This finding supports the notion that binding sites with more than three strong enough hot spots can provide means to overcome poor pharmaceutical behavior by trading off extra binding energy for property improvements in the context of a bRo5 compound.
Complex II targets also have complex hot spot structure, but they benefit from bRo5 compounds for a different reason than Complex I targets, since binding affinity does not improve with ligands of increased molecular weight. All but one Complex II targets are protein kinases, with small ligands (i.e. MW < 400 Da) that bind with high affinity, and we conclude that the primary motivation for expanding to eRo5/bRo5 ligands for Complex II targets is to obtain improved selectivity rather than higher binding affinity. In addition, we have discussed eight targets that have a complex hot spot structure, but too few ligands to see whether or not a correlation exists between MW and binding affinity. In spite of only five ligands, a positive correlation suggests that DOT1-like histone H3 methyltransferase could be classified as Complex I, whereas FGFR1 is a typical kinase and behaves like a Complex II target. Nevertheless, in view of the limited data, we placed these proteins in a separate group called Complex III, together with six more targets that benefit from eRo5/bRo5 drugs for various reasons.
In contrast to the targets that have complex binding hot spot structures with many strong hot spots, targets with simple hot spot structures only contain three or fewer relatively weak hot spots, in many cases rendering the target only barely druggable.39 Almost all ligands which bind such targets utilize all hot spots, and then expand beyond to interact with additional surface regions of the protein, likely to increase binding affinity and/or selectivity. The binding sites of proteins with Simple hot spot structure are frequently located in deep pockets or narrow crevices, and hence modest increases in MW can significantly increase the protein-ligand interface area and improve binding affinity, particularly if the interacting surfaces are hydrophobic. As a result, for some proteins with simple hot spot structure the binding affinity of the ligands increases with increasing molecular weight, while in other cases the binding affinity is not affected.
Since only a small fraction of ligands of the target proteins considered here have been co-crystallized with the proteins, we also studied all ligands available in the ChEMBL27 database. Results confirmed general agreement between the ligand profiles, indicating that the limited structure-based ligand profiles were representatives of the true ligand profiles based on all ligands without co-crystalized structures. Specifically, for targets that appeared druggable with both small and large ligands, we often identified a substantial number of small (MW < 500 Da) ligands with high affinity (pBA > 7) in the general ligand dataset. In contrast, for many targets with no co-crystallized high affinity Ro5 ligands, the fraction of large ligands (MW > 500 Da) was very high in the general ligand dataset.
Importantly, the hot spots of most proteins can be found, without major differences, by mapping either unbound or bound protein structures. Thus, once the X-ray structure of a protein is available, one can map the unbound structure or a structure co-crystallized with any ligand to determine the hot spot structure of the binding site. The hot spots structures will then indicate if it may be useful to consider bRo5 ligands for the target in question. In particular, our results reveal the somewhat counterintuitive finding that bRo5 ligands can be beneficial for targeting complex binding sites on highly druggable proteins, as well as proteins with simple binding sites that are only borderline druggable. Moreover, we highlight that bRo5 drugs can show benefits through a number of different mechanisms and not just increased binding affinity, including improved selectivity along with other improved pharmaceutical properties such as reduced toxicity. Thus, the data encourage the consideration of bRo5 ligands in drug discovery against chemically tractable targets, in cases where achieving selectivity or good pharmaceutical properties is problematic, in addition to being an approach for low druggability targets.
METHODS
FTMap of protein structures.
The FTMap server (http://ftmap.bu.edu)22 was used to identify the binding hot spots for the 37 holo structures listed in Table 1, and the 21 apo structures in Table 2. Domain splitting was performed by Protein Domain Parser84 on four of the holo structures prior to FTMap hot spot identification: DNA-directed RNA-polymerase (4KMU), Integrin Alpha-IIB (2VDN), Smoothened Homolog (4JKV), Isoleucyl-tRNA Synthetase (1QU2).
Data set for structure-based ligand analysis.
The set of ligands which bind each target with known structure and binding affinity were identified by the following procedure. For any given target, all structures in the PDB which have 90% or greater sequence similarity were identified. These structures were aligned to the protein target of interest using the PyMOL™,85 align function, and the het atoms in the PDB file were isolated and analyzed to extract only the ligands which bind within 15 Å of the drug / clinical candidate of interest. The ligands were further filtered by eliminating any ligands which do not have known binding affinity in either the PDB Bind Database24, Binding MOAD Database25, or The Binding Database26. We accepted binding affinity data in the form of KD, KI, and IC50, however, where multiple binding affinity data is available for one ligand, we set priority as: KD > KI > IC50. Moreover, if multiple affinities of the preferable measurement exist, the highest affinity was selected. Furthermore, when two or more PDB IDs presented the same ligand (i.e. same ligand name in the PDB) and had the same binding affinity, we arbitrarily selected one representative and discarded the duplicate ligands. Notably, all but five protein-ligand complexes were solved with x-ray crystallography (resolution: mean = 2.08Å and standard deviation=0.45Å); the distribution is plotted in Figure S8.
Binding site identification by FTMap.
The binding hot spots of each target represent the available hot spots for drug development in the binding region of the drug / clinical candidate of interest. The most intuitive definition of the binding site is simply a collection of all hot spots utilized by the ligands identified in the PDB. This definition is superb for targets with many similar ligands identified. However, for targets where very few ligands are identified, a more robust rule for the binding site is necessary. Based on the targets with many ligands, the ligand-based binding site generally corresponds to all hot spots within ~8 A of the drug / clinical candidate of interest. Manual filtering is necessary to ensure all hot spots are in the same cavity (i.e. a hot spot around the other side of the protein is not actually within 8 A) Therefore, in the absence of ligands, the binding site for any given target can be estimated as all hot spots within 8 A of any atom of the drug / clinical candidate of interest. The binding site listed for each target in Table 1 consists of all the hot spots utilized by the ligands identified with known structure and affinity via the procedure above.
Hot spot – ligand proximity analysis.
For each target protein, the aligned ligands were assessed for their proximity to each of the hot spots identified by FTMap. If any atom in the ligand was within 3 Å of the high density probe center of the hot spot, it was considered an associated hot spot and the number of probe clusters in the hot spot was added to the total number of probe clusters associated. Only FTMap hot spots with five or more probe clusters were considered, for simplicity.
ERo5/bRo5 property calculation.
Physiochemical descriptors for the extended and beyond Ro5 classification were calculated using the open-source cheminformatics software RDKit (https://www.rdkit.org/). Specifically, the molecular weight (MW), number of hydrogen bond donors (HBD), number of hydrogen bond acceptors (HBA), calculated octanol-water partition coefficient (CLogP), topological polar surface area (TPSA), and the number of rotatable bonds (NRotB) were calculated from the SMILES string given for each ligand in the PDB. The eRo5 and bRo5 definitions by Doak et al.21 were used to classify the ligands. For reference, ligands that satisfy all of the following were classified eRo5: MW 500–700 Da, ClogP ≤ 7.5, HBD ≤ 5. HBA ≤ 10, TPSA ≤ 200, NRotB ≤ 20. Ligands that have MW > 500 Da and satisfy one of the following were classified as bRo5: MW > 700 Da, ClogP > 7.5 HBD > 5, HBA > 10, PSA > 200, NRotB > 20, in agreement with the classification used by Doak and colleagues.21
Data set for ligand-based analysis.
As the final step of our study, we examined the properties of all ligands with pBA > 7, which bind each of the 39 proteins. Ligands of the selected 39 protein targets were collected from ChEMBL.27 The database has been searched by the target and gene name and ligands with definite target activity were downloaded. Here we considered both Ki and IC50 values for this analysis and converted the affinities to pBA values. According to the activity threshold used in the target based analysis we considered only ligands with pBA > 7, however, the total number of measured ligands is also shown in Table 3. For statistically meaningful results the ligand based analysis was restricted to targets having at least 40 qualified ligands in ChEMBL. Eight targets (soluble acetylcholine receptor, Lanosterol 14-alpha demethylase, DNA-directed RNA-polymerase, tubulin-alpha Chain, toll-like receptor 4, alpha-amylase, isoleucyl-tRNA synthetase and glutamate-gated chloride channel) have less than 40 qualified ligands and therefore were not considered.
Supplementary Material
ACKNOWLEDGEMENTS
This investigation was supported by grant R35-GM118078 from the National Institute of General Medical Sciences. G.M.K. is supported by the National Brain Research Program (2017-1.2.1-NKP-2017-00002) of the National Research, Development and Innovation Office, Hungary.
ABBREVIATIONS USED
- Ro5
Rule of five
- eRo5
extended Rule of Five
- bRo5
beyond Rule of Five
- MW
Molecular Weight
- FDA
Food and Drug Association
- PPI
Protein-Protein Interaction
- PDB
Protein Data Bank
- CSs
Consensus Sites
- BA
Binding Affinity
- pBA
negative logarithm of binding affinity in form of KD, KI, or IC50,
- NRotB
number of rotatable bonds
- TPSA
topological polar surface area
Biographies
Megan Egbert is a graduate student, pursuing a Ph.D. in Biomedical Engineering at Boston University. Megan obtained a Bachelor of Science in Engineering in Chemical Engineering and a minor in Biochemistry at the University of Michigan, Ann Arbor. Following her Bachelor’s degree, Megan worked at Intel Corporation as a process engineer. Her current research is focused on extracting information from the binding site of proteins and modeling the binding of protein-small molecule interactions, specifically focusing on beyond rule of 5 drug – protein interactions.
Adrian Whitty received a B.Sc. in Chemistry from King’s College London in 1985, followed by M.S. and Ph.D. degrees in Chemistry from the University of Illinois at Chicago. After completing postdoctoral work with William P. Jencks FRS at Brandeis University, he joined the biopharmaceutical company Biogen, where he worked in the Department of Drug Discovery, rising to the position of Director of Physical Biochemistry. In 2008 he moved to Boston University, where he is an Associate Professor in the Department of Chemistry, and in the Department of Pharmacology and Experimental Therapeutics. His research interests include protein-protein and protein-ligand binding, especially as related to the discovery of small molecule inhibitors of protein-protein interactions, and to the quantitative, mechanistic analysis of growth factor receptor activation and signaling.
György M. Keserű obtained his Ph.D. at Budapest, Hungary and joined Sanofi heading a chemistry research lab. He moved to Gedeon Richter in 1999 as the Head of Computer-aided Drug Discovery. He earned D.Sc. from the Hungarian Academy of Science in 2003 and he was invited for a research professorship at the Budapest University of Technology and Economics. Since 2007 he was appointed as the Head of Discovery Chemistry at Gedeon Richter. He contributed to the discovery of the antipsychotic Vraylar® (cariprazine). He served as a director general of the Research Centre for Natural Sciences (RCNS) at the Hungarian Academy of Sciences. From 2015 he is heading the Medicinal Chemistry Research Group at RCNS. His research interests include medicinal chemistry and drug design.
Sandor Vajda received an M.S. in Applied Mathematics and Ph.D. in chemistry in Budapest, Hungary. He held research positions in the Department of Engineering, University of Warwick, England, and the Department of Chemistry, Princeton University. He was a faculty member in Hungary and at the Mount Sinai School of Medicine, New York, NY. Currently he is Professor of Biomedical Engineering and Chemistry at Boston University. He also directs the Biomolecular Engineering Research Center at BU. Dr. Vajda has been active in method development for modeling of biological macromolecules, with emphasis on molecular interactions and drug design. In 2004 he and his graduate students founded SolMap Pharmaceuticals, a start-up company focusing on fragment-based drug design. SolMap was acquired by Forma Therapeutics, Cambridge, MA, in 2008.
Footnotes
Supporting information
The Supporting Information is available free of charge on the ACS Publications website. Contents include graphical representation and statistical analysis of structure-based ligand binding profiles for all targets with more than five ligands; figures for supporting data are mentioned in the text (PDF).
REFERENCES
- (1).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv. Rev 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- (2).Shultz MD Two decades under the influence of the rule of five and the changing properties of approved oral drugs. J. Med. Chem 2018, 62, 1701–1714. [DOI] [PubMed] [Google Scholar]
- (3).Doak BC; Over B; Giordanetto F; Kihlberg J Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol 2014, 21, 1115–1142. [DOI] [PubMed] [Google Scholar]
- (4).DeGoey DA; Chen HJ; Cox PB; Wendt MD Beyond the rule of 5: Lessons learned from abbvie’s drugs and compound collection. J. Med. Chem 2018, 61, 2636–2651. [DOI] [PubMed] [Google Scholar]
- (5).Villar EA; Beglov D; Chennamadhavuni S; Porco JA; Kozakov D; Vajda S; Whitty A How proteins bind macrocycles. Nat. Chem. Biol 2014, 10, 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Whitty A; Zhong M; Viarengo L; Beglov D; Hall DR; Vajda S Quantifying the chameleonic properties of macrocycles and other high-molecular-weight drugs. Drug Discov. Today 2016, 21, 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Driggers EM; Hale SP; Lee J; Terrett NK The exploration of macrocycles for drug discovery--an underexploited structural class. Nat. Rev. Drug. Discov 2008, 7, 608–624. [DOI] [PubMed] [Google Scholar]
- (8).Mallinson J; Collins I Macrocycles in new drug discovery. Future Med. Chem 2012, 4, 1409–1438. [DOI] [PubMed] [Google Scholar]
- (9).Marsault E; Peterson ML Macrocycles are great cycles: Applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem 2011, 54, 1961–2004. [DOI] [PubMed] [Google Scholar]
- (10).Doak BC; Kihlberg J Drug discovery beyond the rule of 5 - opportunities and challenges. Expert Opin. Drug Discov 2017, 12, 115–119. [DOI] [PubMed] [Google Scholar]
- (11).Santos GB; Ganesan A; Emery FS Oral administration of peptide-based drugs: Beyond Lipinski’s rule. ChemMedChem 2016, 11, 2245–2251. [DOI] [PubMed] [Google Scholar]
- (12).Wu P; Nielsen TE; Clausen MH Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [DOI] [PubMed] [Google Scholar]
- (13).Arkin MR; Tang Y; Wells JA Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem Biol 2014, 21, 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Overington JP; Al-Lazikani B; Hopkins AL Opinion - how many drug targets are there? Nat. Rev. Drug Disc 2006, 5, 993–996. [DOI] [PubMed] [Google Scholar]
- (15).Basse MJ; Betzi S; Morelli X; Roche P 2P2IDB V2: Update of a structural database dedicated to orthosteric modulation of protein-protein interactions. Database (Oxford) 2016, DOI: 10.1093/database/baw007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fry DC Small-molecule inhibitors of protein-protein interactions: How to mimic a protein partner. Curr. Pharm. Des 2012, 18, 4679–4684. [DOI] [PubMed] [Google Scholar]
- (17).Wells JA; McClendon CL Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- (18).Arkin MR; Wells JA Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Disc 2004, 3, 301–317. [DOI] [PubMed] [Google Scholar]
- (19).Zhang MQ; Wilkinson B Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol 2007, 18, 478–488. [DOI] [PubMed] [Google Scholar]
- (20).Tonddast-Navaei S; Srinivasan B; Skolnick J On the importance of composite protein multiple ligand interactions in protein pockets. J. Comput. Chem 2017, 38, 1252–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Doak BC; Zheng J; Dobritzsch D; Kihlberg J How beyond rule of 5 drugs and clinical candidates bind to their targets. J. Med. Chem 2016, 59, 2312–2327. [DOI] [PubMed] [Google Scholar]
- (22).Kozakov D; Grove LE; Hall DR; Bohnuud T; Mottarella SE; Luo L; Xia B; Beglov D; Vajda S The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc 2015, 10, 733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang R; Fang X; Lu Y; Wang S The PDBbind database: Collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem 2004, 47, 2977–2980. [DOI] [PubMed] [Google Scholar]
- (25).Hu L; Benson ML; Smith RD; Lerner MG; Carlson HA Binding MOAD (mother of all databases). Proteins 2005, 60, 333–340. [DOI] [PubMed] [Google Scholar]
- (26).Liu T; Lin Y; Wen X; Jorissen RN; Gilson MK BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2006, 35, D198–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gaulton A; Bellis LJ; Bento AP; Chambers J; Davies M; Hersey A; Light Y; McGlinchey S; Michalovich D; Al-Lazikani B; Overington JP ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).DeLano WL Unraveling hot spots in binding interfaces: Progress and challenges. Curr. Opin. Struct. Biol 2002, 12, 14–20. [DOI] [PubMed] [Google Scholar]
- (29).Ciulli A; Williams G; Smith AG; Blundell TL; Abell C Probing hot spots at protein-ligand binding sites: A fragment-based approach using biophysical methods. J. Med. Chem 2006, 49, 4992–5000. [DOI] [PubMed] [Google Scholar]
- (30).Metz A; Pfleger C; Kopitz H; Pfeiffer-Marek S; Baringhaus KH; Gohlke H Hot spots and transient pockets: Predicting the determinants of small-molecule binding to a protein-protein interface. J. Chem. Inf. Model 2012, 52, 120–133. [DOI] [PubMed] [Google Scholar]
- (31).Hall DR; Kozakov D; Whitty A; Vajda S Lessons from hot spot analysis for fragment-based drug discovery. Trends Pharmacol. Sci 2015, 36, 724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dennis S; Kortvelyesi T; Vajda S Computational mapping identifies the binding sites of organic solvents on proteins. Proc. Natl. Acad. Sci. U S A 2002, 99, 4290–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Landon MR; Lancia DR Jr.; Yu J; Thiel SC; Vajda S Identification of hot spots within druggable binding regions by computational solvent mapping of proteins. J. Med. Chem 2007, 50, 1231–1240. [DOI] [PubMed] [Google Scholar]
- (34).Landon MR; Lieberman RL; Hoang QQ; Ju S; Caaveiro JM; Orwig SD; Kozakov D; Brenke R; Chuang GY; Beglov D; Vajda S; Petsko GA; Ringe D Detection of ligand binding hot spots on protein surfaces via fragment-based methods: Application to DJ-1 and glucocerebrosidase. J. Comput. Aid. Mol. Des 2009, 23, 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chuang GY; Kozakov D; Brenke R; Beglov D; Guarnieri F; Vajda S Binding hot spots and amantadine orientation in the influenza a virus M2 proton channel. Biophys. J 2009, 97, 2846–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Buhrman G; O’Connor C; Zerbe B; Kearney BM; Napoleon R; Kovrigina EA; Vajda S; Kozakov D; Kovrigin EL; Mattos C Analysis of binding site hot spots on the surface of Ras GTPase. J. Mol. Biol 2011, 413, 773–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zerbe BS; Hall DR; Vajda S; Whitty A; Kozakov D Relationship between hot spot residues and ligand binding hot spots in protein-protein interfaces. J. Chem. Inf. Model 2012, 52, 2236–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Golden MS; Cote SM; Sayeg M; Zerbe BS; Villar EA; Beglov D; Sazinsky SL; Georgiadis RM; Vajda S; Kozakov D; Whitty A Comprehensive experimental and computational analysis of binding energy hot spots at the NF-κB essential modulator/IKKβ protein-protein interface. J. Am. Chem. Soc 2013, 135, 6242–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kozakov D; Hall DR; Napoleon RL; Yueh C; Whitty A; Vajda S New frontiers in druggability. J. Med. Chem 2015, 58, 9063–9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kozakov D; Hall DR; Chuang GY; Cencic R; Brenke R; Grove LE; Beglov D; Pelletier J; Whitty A; Vajda S Structural conservation of druggable hot spots in protein-protein interfaces. Proc. Natl. Acad. Sci. U S A 2011, 108, 13528–13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cully M Milestone 9: Ximelagatran sets the stage for NOACs. Nat. Rev. Cardiol 2017, DOI: 10.1038/nrcardio.2017.179. [DOI] [PubMed] [Google Scholar]
- (42).Keisu M; Andersson TB Drug-induced liver injury in humans: The case of ximelagatran. Handb. Exp. Pharmacol 2010, 407–418. [DOI] [PubMed] [Google Scholar]
- (43).Riester D; Wirsching F; Salinas G; Keller M; Gebinoga M; Kamphausen S; Merkwirth C; Goetz R; Wiesenfeldt M; Sturzebecher J; Bode W; Friedrich R; Thurk M; Schwienhorst A Thrombin inhibitors identified by computer-assisted multiparameter design. Proc. Natl. Acad. Sci. U S A 2005, 102, 8597–8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Pool JL Direct renin inhibition: Focus on aliskiren. J. Manag. Care Pharm 2007, 13, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sepehrdad R; Frishman WH; Stier CT Jr.; Sica DA Direct inhibition of renin as a cardiovascular pharmacotherapy: Focus on aliskiren. Cardiol. Rev 2007, 15, 242–256. [DOI] [PubMed] [Google Scholar]
- (46).Lukacs C; Belunis C; Crowther R; Danho W; Gao L; Goggin B; Janson CA; Li S; Remiszewski S; Schutt A; Thakur MK; Singh SK; Swaminathan S; Pandey R; Tyagi R; Gosu R; Kamath AV; Kuglstatter A The structure of XIAP BIR2: Understanding the selectivity of the BIR domains. Acta Crystallogr. D. Biol. Crystallogr 2013, 69, 1717–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cong H; Xu L; Wu Y; Qu Z; Bian T; Zhang W; Xing C; Zhuang C Inhibitor of apoptosis protein (IAP) antagonists in anticancer agent discovery: Current status and perspectives. J. Med. Chem 2019, DOI: 10.1021/acs.jmedchem.8b01668. [DOI] [PubMed] [Google Scholar]
- (48).Pargellis C; Tong L; Churchill L; Cirillo PF; Gilmore T; Graham AG; Grob PM; Hickey ER; Moss N; Pav S Inhibition of p38 map kinase by utilizing a novel allosteric binding site. Nat. Struct. Mol. Biol 2002, 9, 268–272. [DOI] [PubMed] [Google Scholar]
- (49).Millan DS; Bunnage ME; Burrows JL; Butcher KJ; Dodd PG; Evans TJ; Fairman DA; Hughes SJ; Kilty IC; Lemaitre A; Lewthwaite RA; Mahnke A; Mathias JP; Philip J; Smith RT; Stefaniak MH; Yeadon M; Phillips C Design and synthesis of inhaled p38 inhibitors for the treatment of chronic obstructive pulmonary disease. J. Med. Chem 2011, 54, 7797–7814. [DOI] [PubMed] [Google Scholar]
- (50).Norman P Investigational p38 inhibitors for the treatment of chronic obstructive pulmonary disease. Expert Opin. Investig. Drugs 2015, 24, 383–392. [DOI] [PubMed] [Google Scholar]
- (51).Janin J; Henrick K; Moult J; Eyck LT; Sternberg MJ; Vajda S; Vakser I; Wodak SJ CAPRI: A critical assessment of predicted interactions. Proteins 2003, 52, 2–9. [DOI] [PubMed] [Google Scholar]
- (52).Ishima R; Freedberg DI; Wang YX; Louis JM; Torchia DA Flap opening and dimer-interface flexibility in the free and inhibitor-bound HIV protease, and their implications for function. Structure 1999, 7, 1047–1055. [DOI] [PubMed] [Google Scholar]
- (53).Seo YH Small molecule inhibitors to disrupt protein-protein interactions of heat shock protein 90 chaperone machinery. J. Cancer Prev 2015, 20, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Stebbins CE; Russo AA; Schneider C; Rosen N; Hartl FU; Pavletich NP Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 1997, 89, 239–250. [DOI] [PubMed] [Google Scholar]
- (55).Trendowski M Pu-h71: An improvement on nature’s solutions to oncogenic Hsp90 addiction. Pharmacol. Res 2015, 99, 202–216. [DOI] [PubMed] [Google Scholar]
- (56).Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE Selective inhibition of bet bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ciceri P; Muller S; O’Mahony A; Fedorov O; Filippakopoulos P; Hunt JP; Lasater EA; Pallares G; Picaud S; Wells C; Martin S; Wodicka LM; Shah NP; Treiber DK; Knapp S Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol 2014, 10, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Tanaka M; Roberts JM; Seo HS; Souza A; Paulk J; Scott TG; DeAngelo SL; Dhe-Paganon S; Bradner JE Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol 2016, 12, 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Frkic RL; Marshall AC; Blayo AL; Pukala TL; Kamenecka TM; Griffin PR; Bruning JB PPARγ in complex with an antagonist and inverse agonist: A tumble and trap mechanism of the activation helix. iScience 2018, 5, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Taygerly JP; McGee LR; Rubenstein SM; Houze JB; Cushing TD; Li Y; Motani A; Chen JL; Frankmoelle W; Ye G; Learned MR; Jaen J; Miao S; Timmermans PB; Thoolen M; Kearney P; Flygare J; Beckmann H; Weiszmann J; Lindstrom M; Walker N; Liu J; Biermann D; Wang Z; Hagiwara A; Iida T; Aramaki H; Kitao Y; Shinkai H; Furukawa N; Nishiu J; Nakamura M Discovery of int131: A selective PPARγ modulator that enhances insulin sensitivity. Bioorg. Med. Chem 2013, 21, 979–992. [DOI] [PubMed] [Google Scholar]
- (61).Badrinarayan P; Sastry GN Rational approaches towards lead optimization of kinase inhibitors: The issue of specificity. Curr. Pharm. Des 2013, 19, 4714–4738. [DOI] [PubMed] [Google Scholar]
- (62).Hubner M; Hochhaus G; Derendorf H Comparative pharmacology, bioavailability, pharmacokinetics, and pharmacodynamics of inhaled glucocorticosteroids. Immunol. Allergy Clin. North Am 2005, 25, 469–488. [DOI] [PubMed] [Google Scholar]
- (63).Jin L; Harrison SC Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc. Natl. Acad. Sci. U S A 2002, 99, 13522–13526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Liu J; Farmer JD Jr; Lane WS; Friedman J; Weissman I; Schreiber SL Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [DOI] [PubMed] [Google Scholar]
- (65).Daigle SR; Olhava EJ; Therkelsen CA; Basavapathruni A; Jin L; Boriack-Sjodin PA; Allain CJ; Klaus CR; Raimondi A; Scott MP; Waters NJ; Chesworth R; Moyer MP; Copeland RA; Richon VM; Pollock RM Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Monk BC; Tomasiak TM; Keniya MV; Huschmann FU; Tyndall JD; O’Connell JD 3rd; Cannon RD; McDonald JG; Rodriguez A; Finer-Moore JS; Stroud RM Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc. Natl. Acad. Sci. U S A 2014, 111, 3865–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Winston DJ; Maziarz RT; Chandrasekar PH; Lazarus HM; Goldman M; Blumer JL; Leitz GJ; Territo MC Intravenous and oral itraconazole versus intravenous and oral fluconazole for long-term antifungal prophylaxis in allogeneic hematopoietic stem-cell transplant recipients. A multicenter, randomized trial. Ann. Intern. Med 2003, 138, 705–713. [DOI] [PubMed] [Google Scholar]
- (68).Martin MV The use of fluconazole and itraconazole in the treatment of candida albicans infections: A review. J. Antimicrob. Chemother 1999, 44, 429–437. [DOI] [PubMed] [Google Scholar]
- (69).Hurdle JG; O’Neill AJ; Chopra I Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother 2005, 49, 4821–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Marz AM; Fabian AK; Kozany C; Bracher A; Hausch F Large FK506-binding proteins shape the pharmacology of rapamycin. Mol. Cell. Biol 2013, 33, 1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Opal SM; Laterre PF; Francois B; LaRosa SP; Angus DC; Mira JP; Wittebole X; Dugernier T; Perrotin D; Tidswell M; Jauregui L; Krell K; Pachl J; Takahashi T; Peckelsen C; Cordasco E; Chang CS; Oeyen S; Aikawa N; Maruyama T; Schein R; Kalil AC; Van Nuffelen M; Lynn M; Rossignol DP; Gogate J; Roberts MB; Wheeler JL; Vincent JL; Group AS Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The access randomized trial. JAMA 2013, 309, 1154–1162. [DOI] [PubMed] [Google Scholar]
- (72).Dieci MV; Arnedos M; Andre F; Soria JC Fibroblast growth factor receptor inhibitors as a cancer treatment: From a biologic rationale to medical perspectives. Cancer Discov. 2013, 3, 264–279. [DOI] [PubMed] [Google Scholar]
- (73).Guagnano V; Furet P; Spanka C; Bordas V; Le Douget M; Stamm C; Brueggen J; Jensen MR; Schnell C; Schmid H; Wartmann M; Berghausen J; Drueckes P; Zimmerlin A; Bussiere D; Murray J; Graus Porta D Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamin o]-pyrimidin-4-yl}−1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem 2011, 54, 7066–7083. [DOI] [PubMed] [Google Scholar]
- (74).Olziersky AM; Labidi-Galy SI Clinical development of anti-mitotic drugs in cancer. Adv. Exp. Med. Biol 2017, 1002, 125–152. [DOI] [PubMed] [Google Scholar]
- (75).Purcell JW; Davis J; Reddy M; Martin S; Samayoa K; Vo H; Thomsen K; Bean P; Kuo WL; Ziyad S; Billig J; Feiler HS; Gray JW; Wood KW; Cases S Activity of the kinesin spindle protein inhibitor ispinesib (SB-715992) in models of breast cancer. Clin. Cancer Res 2010, 16, 566–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Istvan ES; Deisenhofer J Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [DOI] [PubMed] [Google Scholar]
- (77).Fouty BW; Rodman DM Mevastatin can cause G1 arrest and induce apoptosis in pulmonary artery smooth muscle cells through a p27KIP1-independent pathway. Circ. Res 2003, 92, 501–509. [DOI] [PubMed] [Google Scholar]
- (78).Strickland CL; Weber PC; Windsor WT; Wu Z; Le HV; Albanese MM; Alvarez CS; Cesarz D; del Rosario J; Deskus J; Mallams AK; Njoroge FG; Piwinski JJ; Remiszewski S; Rossman RR; Taveras AG; Vibulbhan B; Doll RJ; Girijavallabhan VM; Ganguly AK Tricyclic farnesyl protein transferase inhibitors: Crystallographic and calorimetric studies of structure-activity relationships. J. Med. Chem 1999, 42, 2125–2135. [DOI] [PubMed] [Google Scholar]
- (79).Walker EH; Pacold ME; Perisic O; Stephens L; Hawkins PT; Wymann MP; Williams RL Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [DOI] [PubMed] [Google Scholar]
- (80).Folkes AJ; Ahmadi K; Alderton WK; Alix S; Baker SJ; Box G; Chuckowree IS; Clarke PA; Depledge P; Eccles SA; Friedman LS; Hayes A; Hancox TC; Kugendradas A; Lensun L; Moore P; Olivero AG; Pang J; Patel S; Pergl-Wilson GH; Raynaud FI; Robson A; Saghir N; Salphati L; Sohal S; Ultsch MH; Valenti M; Wallweber HJ; Wan NC; Wiesmann C; Workman P; Zhyvoloup A; Zvelebil MJ; Shuttleworth SJ The identification of 2-(1h-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem 2008, 51, 5522–5532. [DOI] [PubMed] [Google Scholar]
- (81).Vukovic S; Huggins DJ Quantitative metrics for drug-target ligandability. Drug Discov. Today 2018, 23, 1258–1266. [DOI] [PubMed] [Google Scholar]
- (82).Hopkins AL; Keseru GM; Leeson PD; Rees DC; Reynolds CH The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov 2014, 13, 105–121. [DOI] [PubMed] [Google Scholar]
- (83).Wehling M Assessing the translatability of drug projects: What needs to be scored to predict success? Nat. Rev. Drug Discov 2009, 8, 541–546. [DOI] [PubMed] [Google Scholar]
- (84).Alexandrov N; Shindyalov I PDP: Protein domain parser. Bioinformatics 2003, 19, 429–430. [DOI] [PubMed] [Google Scholar]
- (85).Schrodinger LLC. The PyMOL molecular graphics system, version 1.8 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.