

Abstract
Mechanisms that govern the shift from joint homeostasis to osteoarthritis (OA) remain unknown. Here, we identify a pathway used for joint development and homeostasis, and its role in OA. Using a combination of transgenic, pharmacological, and surgical conditions in mouse and human tissues, we found that TGF-β signaling promotes joint homeostasis through regulation of the IL-36 family. We identified IL-36 receptor antagonist (IL-36 in mice and IL-36RN in humans) as a potential disease-modifying OA drug. Specifically, OA development was associated with IL-36α up-regulation and IL-36Ra down-regulation in mice with tissue-specific postnatally induced ablation of Tgfbr2, mice treated with a TGF-β signaling inhibitor, mice with posttraumatic OA, and aging mice with naturally occurring OA. In human cartilage, OA severity was associated with decreased TGFBR2 and IL-36RN, whereas IL-36α increased. Functionally, intra-articular treatment with IL-36Ra attenuated OA development in mice, and IL-36RN reduced MMP13 in human OA chondrocytes. These findings highlight the relevance of TGFBR2–IL-36 interplay in joint homeostasis and IL-36RN as a potential therapeutic agent for OA.
INTRODUCTION
Osteoarthritis (OA) is a debilitating disease expected to affect 67 million people in the United States by 2030 (1). In addition to cartilage damage, patients with OA develop osteophytosis, subchondral bone sclerosis, and synovitis. OA causes multiple symptoms including joint tenderness, joint stiffness, and pain. Current management of OA is limited by the lack of therapeutic interventions that alter the rate of progression of the disease. Although preventing the enzymatic cartilage catabolism of early to mid-stage OA has therapeutic potential, the ideal targets—the upstream regulatory mediators of these enzymes—have yet to be identified (2–5). Given that the potent anabolic mechanisms used during early joint development may also be used in adult homeostasis, they provide a potential means of overcoming OA-related cartilage catabolism. Contemporary organ disease models of OA suggest that changes in subchondral bone sclerosis, osteophytosis, articular cartilage destruction, and synovitis are not independent; rather, these tissues are structurally distinct, but functionally interdependent, entities (6,7). Although proinflammatory cytokines have been historically associated with advanced OA, they now appear to also have regulatory roles in early stages of OA (8). Specifically, interleukin-1β (IL-1 β) (9–12) and tumor necrosis factor–α (TNF-α) (10,13,14) subfamilies appear to regulate matrix metalloproteases (MMPs) such as MMP13 and A disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS5) during cartilage degradation.
The TGF-β cytokine superfamily has garnered attention with respect to joint development and OA. Previously, we reported that mice with prenatally inactivated TGF-β type 2 receptor (Tgfbr2) in osteochondral progenitors (Tgfbr2Prx1KO) do not undergo joint formation (15). Furthermore, we applied evolutionary pathway analysis to RNA-based microarray data obtained from laser capture microdissection of developing mouse joint cells, which showed that a TGFBR2-dependent signaling mechanism that tightly regulated cytokine expression is required for proper joint formation (16).
Recent reports suggest that proper TGF-β signaling is also required to maintain adulthood cartilage homeostasis. Not only is disrupted TGF-β signaling evident during OA progression (17), but TGF-β signaling deficiencies induced in normal joints can initiate OA-like phenotypes in mice as well (18–20). Corresponding interventions have produced promising results: Manipulating TGF-β signaling in OA joints appears to have potential for opposing cartilage catabolism induced by IL-1 and TNF-α (21,22). Moreover, both systemically delivered TGF-β and subchondral mesenchyme-localized TGF-β signaling deficiencies are capable of attenuating surgically induced OA progression (23,24). Overall, these results suggest that joints require proper TGF-β signaling during both development and maintenance. Because TGF-β signaling has multifaceted niche-dependent roles within joints, systemic actions outside the joint, and an as-yet incomplete dossier of transcriptional regulation targets, interventions that directly affect TGF-β signaling have a high risk for causing multiorgan side effects. Therefore, developing a clinically effective TGF-β–derived disease-modifying OA drug (DMOAD) requires the elucidation of relevant local and downstream signaling mechanisms.
The aim of the present study was to identify and characterize TGFBR2 downstream cytokine signaling networks with high potential for translational OA therapy. Specifically, we found that IL-36α expression was greatly up-regulated in Tgfbr2Prx1KO embryos at the time and location of failed joint segmentation compared to controls.
IL-36α belongs to one of several IL-1 cytokine subfamilies (25,26). This IL-36 subfamily includes three activating ligands (IL-36α, IL-36β, and IL-36γ), a receptor antagonist (named IL-36Ra in mice and IL-36RN in humans), and their common subfamily-specific receptor (IL-36R) (27). Previous reports suggest that IL-36 subfamily members play critical roles in inflammatory diseases (28–32). In synovial fibroblasts and articular chondrocytes, IL-36β stimulated expression of inflammatory mediators. However, in animal models for rheumatoid arthritis, the IL-36R–blocking antibody did not affect the phenotype (33–35). More recently, Conde et al. (36) have reported that chondrocytes from patients with OA have an increased expression of IL-36α compared to chondrocytes from healthy individuals.
In this study, we investigated the role of the TGFBR2/IL-36α axis in human and mouse osteoarthritic joints and in isolated chondrocytes. We found that inactivation of TGF-β signaling led to OA; inhibition of IL-36α signaling activity by IL-36Ra attenuated these pathological changes and reduced articular cartilage degeneration in different animal models of OA. In human cartilage, the severity of OA in osteoarthritic joints was associated with a decrease of TGFBR2 and IL-36RN, and attenuation of TGF-β signaling in primary human chondrocytes led to an increase of catabolic factors that was attenuated by IL-36RN.
RESULTS
Tgfbr2Prx1KO mice exhibit uncavitated joints associated with IL-36α and IL-36R overexpression
Compared to control Tgfbr2flox/flox mice, which exhibited knee joint cavities containing cartilage, menisci, and supporting ligaments, knees from P0 (postnatal day 0) Tgfbr2Prx1KO mice lacking Tgfbr2 in osteochondral progenitors had an uncavitated and disorganized cluster of cells and abnormal condyle morphology (Fig. 1, A and B). We then took advantage of our previous microarray studies that compared gene expression profiles of messenger RNAs (mRNAs) obtained by laser capture microscopy (LCM) from interzone cells of Tgfbr2Prx1KO to Tgfbr2flox/flox interzone cells, and by further analyses, we found that mRNA expression of IL-36α in Tgfbr2Prx1KO mice was 40-fold up-regulated compared to control (Fig. 1C). The unique receptor for IL-36α, named IL-36R, was also up-regulated more than fourfold in Tgfbr2Prx1KO (Fig. 1C). IL-36β, IL-36γ, and IL-36Ra were only slightly up-regulated (two- to fourfold) in Tgfbr2Prx1KO mice. These results, which suggest a link between TGF-β and IL-36α signaling in joint development, were confirmed by reverse transcription polymerase chain reaction (RT-PCR) for IL-36α and IL-36R (Fig. 1D) and IHC throughout embryonic joint development (E14.5 and P0; Fig. 1, E and F) for IL-36α.
Fig. 1. Ablation of Tgfbr2 in Tgfbr2Prx1KO causes defects in joint development and up-regulation of IL-36α expression.
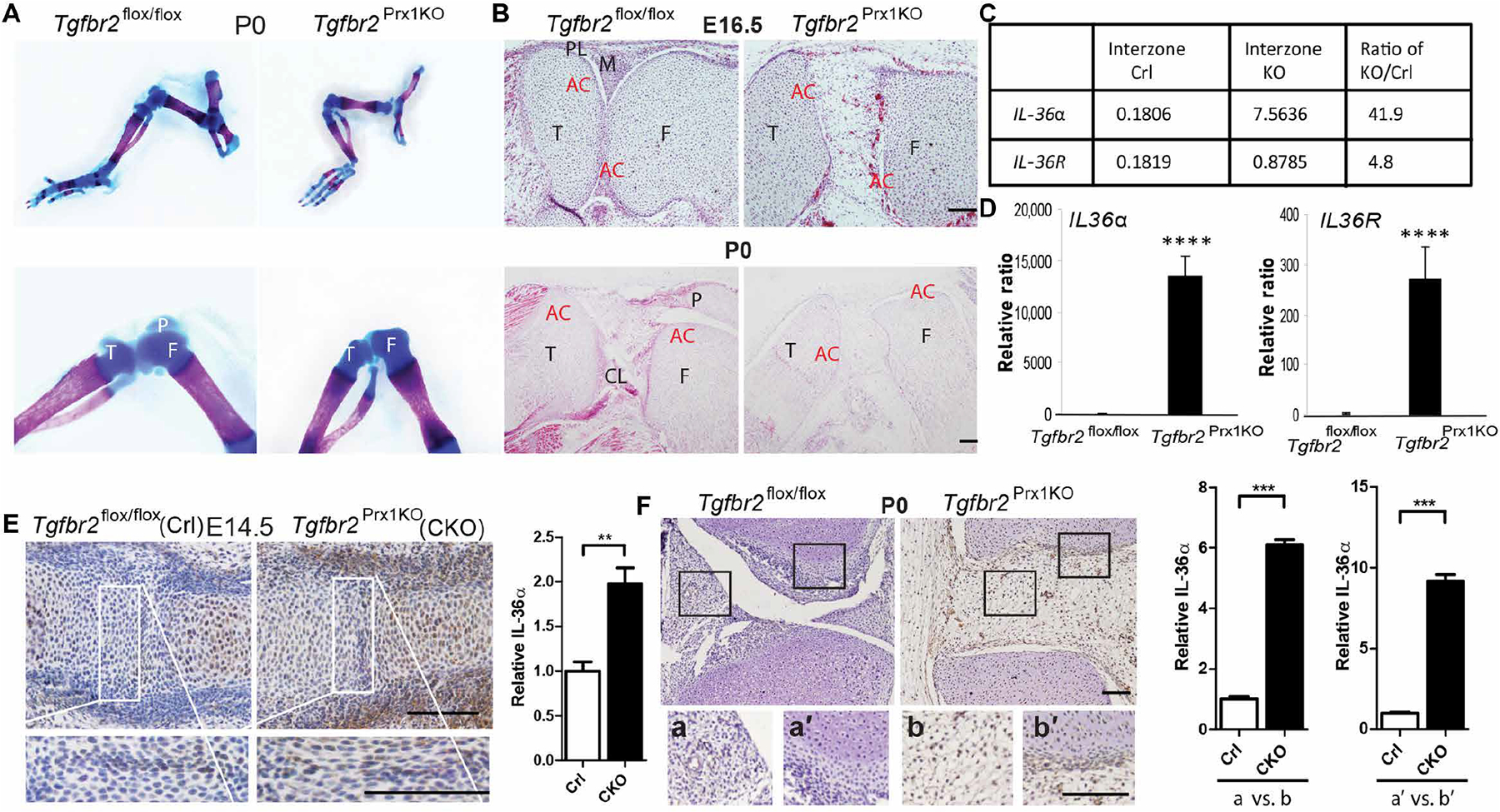
(A) Whole-mount alcian blue and alizarin red staining for P0 hind limbs of Tgfbr2flox/flox and Tgfbr2Prx1KO mice at low (top) and high (bottom) magnification. (B) Sectional hematoxylin and eosin staining of knees from E16.5 and P0 hind limbs. The tibia (T), femur (F), meniscus (M), patellar (P), patellar ligament (PL), cruciate ligament (CL), and articular cartilage (AC) are labeled. n = 3 mice per group. (C) Microarray analysis (16) of the complementary DNAs from LCM in the interzone cells of both Tgfbr2flox/flox (control, Crl) and Tgfbr2Prx1KO knockout (KO) at E14.5 (embryonic day 14.5). Left (control) and middle (KO) columns represent normalized signal data; the right column represents KO/control ratio of normalized signal data. (D) Quantitative RT-PCR (qRT-PCR) analysis of LCM samples showing expression of IL-36α in the Tgfbr2flox/flox interzone, compared to the Tgfbr2Prx1KO presumptive interzone. All n = 3 replicates per group are shown. Data are means ± SD. ****P < 0.0001 (unpaired Student’s t test). Limb buds were dissected from E14.5 (E) or P0 (F) Tgfbr2flox/flox and Tgfbr2Prx1KO embryos, and then embedded in paraffin. Sections including interphalangeal (E) or knee (F) joints were subjected to immunohistochemistry (IHC) analysis using anti-IL-36α antibody [distal side: right for (E) and top for (F)], respectively. n ≥ 4 embryos per group with n ≥ 6 IHC sections analyzed per embryo. Quantification of IHC is shown in the right panels (n = 3 embryos per group). White/black frames indicate comparable interzone and knee joint regions shown in high magnification in the bottom panels (a, a′, b, and b′). Data are means ± SEM. **P < 0.01 and ***P < 0.001 (unpaired Student’s t test). Scale bars, 100 μm.
Tgfbr2−/− mice spontaneously develop an OA-like phenotype and have increased damage from posttraumatic OA
We then explored whether TGFBR2 signaling was also critical for postnatal joint homeostasis. To that end, we used a Prx1-CreER tamoxifen-inducible system (system’s recombination efficiency reported in the Supplemental Materials and fig. S1) to generate Tgfbr2−/− mice, where Tgfbr2 is deleted from osteochondral progenitors upon tamoxifen exposure.
By P6, Tgfbr2−/− mice showed a decrease in cellularity of the superficial layer of the articular cartilage and discernable signs of chondrocyte hypertrophy that become more evident at P31, indicated by increased percentages of hypertrophic chondrocytes (fig. S2, A to D). Long-term Tgfbr2−/− mice developed a spontaneous knee OA phenotype that progressed to complete loss of articular cartilage by postnatal month 9 (P9M) to P12M (Fig. 2A and fig. S3A). Specifically, by P3M, Tgfbr2−/− mice showed proteoglycan loss, hypertrophic chondrocytes, and cells with endoplasmic reticular ballooning. By P6M, proteoglycan loss, fibrillation, and moderately deep lesions were noted. By P9M to P12M, full-thickness cartilage lesions, osteophytes, subchondral sclerosis, and synovitis were observed (Fig. 2A). At P3M to P6M, IHC analyses showed that the articular cartilage of mutant mice had increased expressions for Collagen X and MMP13 (fig. S3, B to D). After the cartilage had been completely degraded (P14M, fig. S4A), we found increased expressions for MMP13, the subchondral bone, and synovium (fig. S4B).
Fig. 2. Postnatal ablation of Tgfbr2 in Tgfbr2−/− mutants leads to OA development, mobility-related behavioral changes, and accelerated posttraumatic OA.
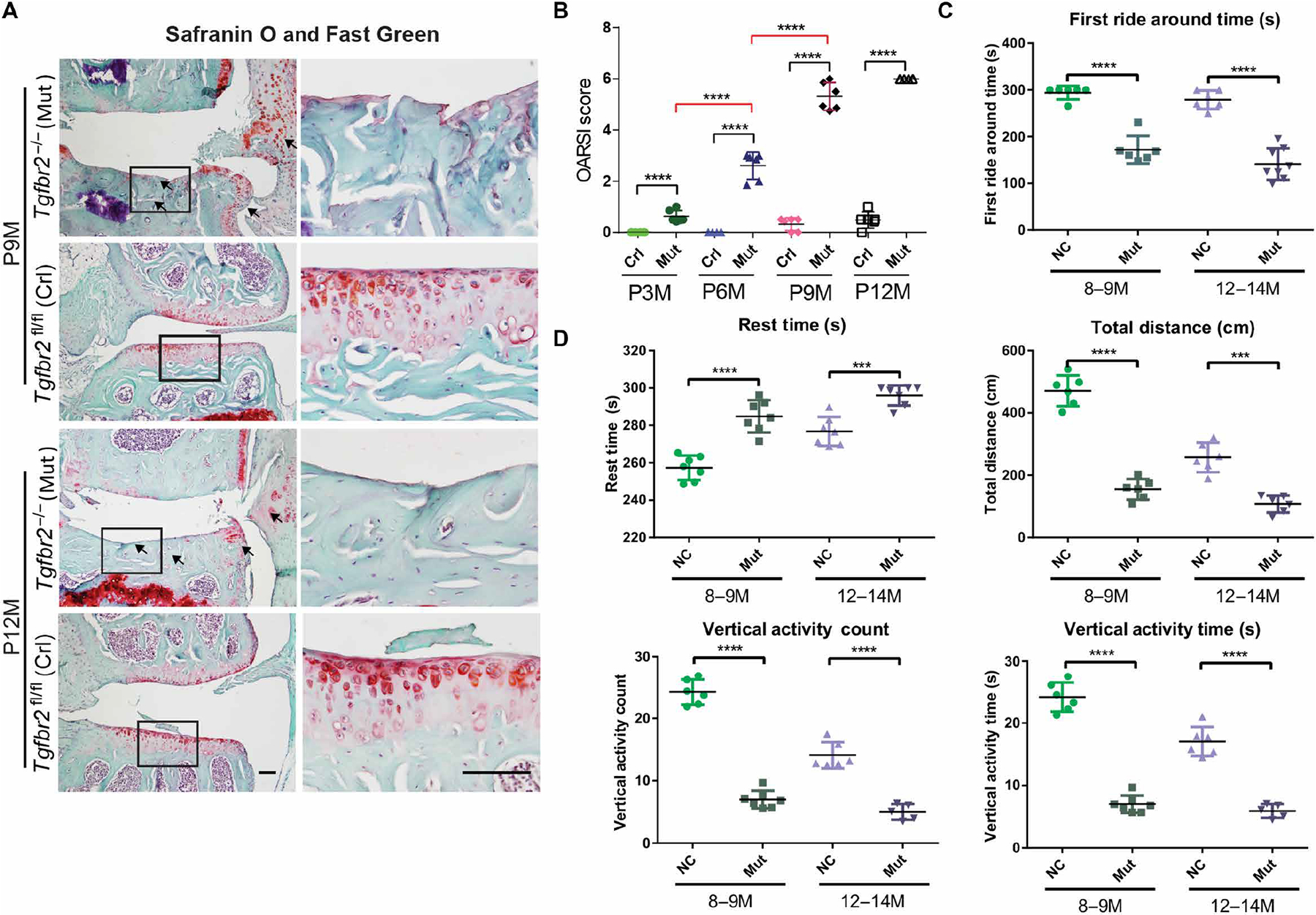
(A) Coronal Safranin O– and Fast Green–stained sections of murine medial tibial plateaus. Black frames indicate regions shown in high magnification (right column). Black arrows indicate degraded articular cartilage, subchondral bone sclerosis, and synovial reaction. (B) OARSI score analysis in 3-, 6-, 9-, and 12-month-old Tgfbr2−/− (Mut) mice treated with tamoxifen and in Tgfbr2fl/fl (control, Crl) mice. (C) Rotarod measurements of first ride around time and (D) timed open-field measurements of distance traveled, rest time, rearing event count, and mean rearing event duration. n ≥ 5 mice per group. Error bars denote SEM. ***P < 0.001 and ****P < 0.0001 [one-way analysis of variance (ANOVA) and Tukey’s multiple comparison post test]. Scale bars, 100 μm.
Structurally, we graded the Safranin O and Fast Green sections using the Osteoarthritis Research Society International (OARSI) scoring system for rodents with scores that range from 0 to a maximal score of 6 (37). We found that OARSI scores were clearly higher in mutants than in controls (tamoxifen-treated Tgfbr2flox/flox) at all time points (Fig. 2B). Furthermore, in mutants, OARSI scores increased significantly over time, again indicating the progressive nature of OA when Tgfbr2 is inactivated (P < 0.0001). Functionally, we evaluated the relative knee joint mobility using behavioral analyses performed at P9M and P12M. Using rotarod analysis (Fig. 2C), we found that Tgfbr2−/− mice were unable to stay atop the rotating apparatus as long as their control counterparts. Using open-field analysis (Fig. 2D), we found that in the same amount of time, mutant mice voluntarily traveled shorter distances, had longer rest times, and engaged in less rearing activity than controls. These results indicate that mutants might have painful, less functional joints. To evaluate a role for TGFBR2 signaling in other forms of OA, we induced posttraumatic OA via the DMM (destabilization of the medial meniscus) procedure in Tgfbr2−/− and control mice (fig. S5). Both Tgfbr2−/− and Tgfbr2fl/fl DMM-operated mice exhibited significantly higher cartilage degeneration with higher OARSI scores, as early as 4 weeks after DMM, than their sham counterparts (fig. S5, A and B). Intra-articular (i.a.) injections of TGF-α1 had no effect on the increased susceptibility of Tgfbr2−/− mice to developing posttraumatic OA, confirming the efficiency of the inducible ablation of TGFBR2 in affecting cell responsiveness to TGF-β. I.a. injections of TGF-β1 increased Safranin O staining in Tgfbr2fl/fl control mice subjected to DMM, although OARSI scores were not statistically different between treated and untreated groups (fig. S5, A and B).
Up-regulated IL-36α and down-regulated IL-36Ra are observed in both Tgfbr2−/− mice and wild-type mice treated with a TGF-β signaling inhibitor
Next, we determined whether our prenatal findings, indicating an interplay between TGFBR2 and IL-36α/IL-36R, extended to postnatal joint homeostasis and OA progression. At P3M, before macrostructural cartilage damage had occurred (Fig. 3A), IHC analyses showed that in mutants, IL-36α/R was up-regulated, whereas IL-36Ra was down-regulated compared to controls (Fig. 3, B to D). We found that in P3M Tgfbr2−/− mice, both IL-36α and IL-36R were up-regulated in the articular cartilage and superficial layers of the menisci and synovia (Fig. 3, B and C). This expression pattern persisted at late stages of OA (fig. S6A). We also found that IL-36Ra was down-regulated in the articular cartilage (Fig. 3D) but up-regulated in the synovia (fig. S6B) of Tgfbr2−/− mice.
Fig. 3. Genetic ablation of Tgfbr2 and pharmacological inhibition of TGF-β signaling induces up-regulation of IL-36α and IL-36R; IL-36Ra is down-regulated.
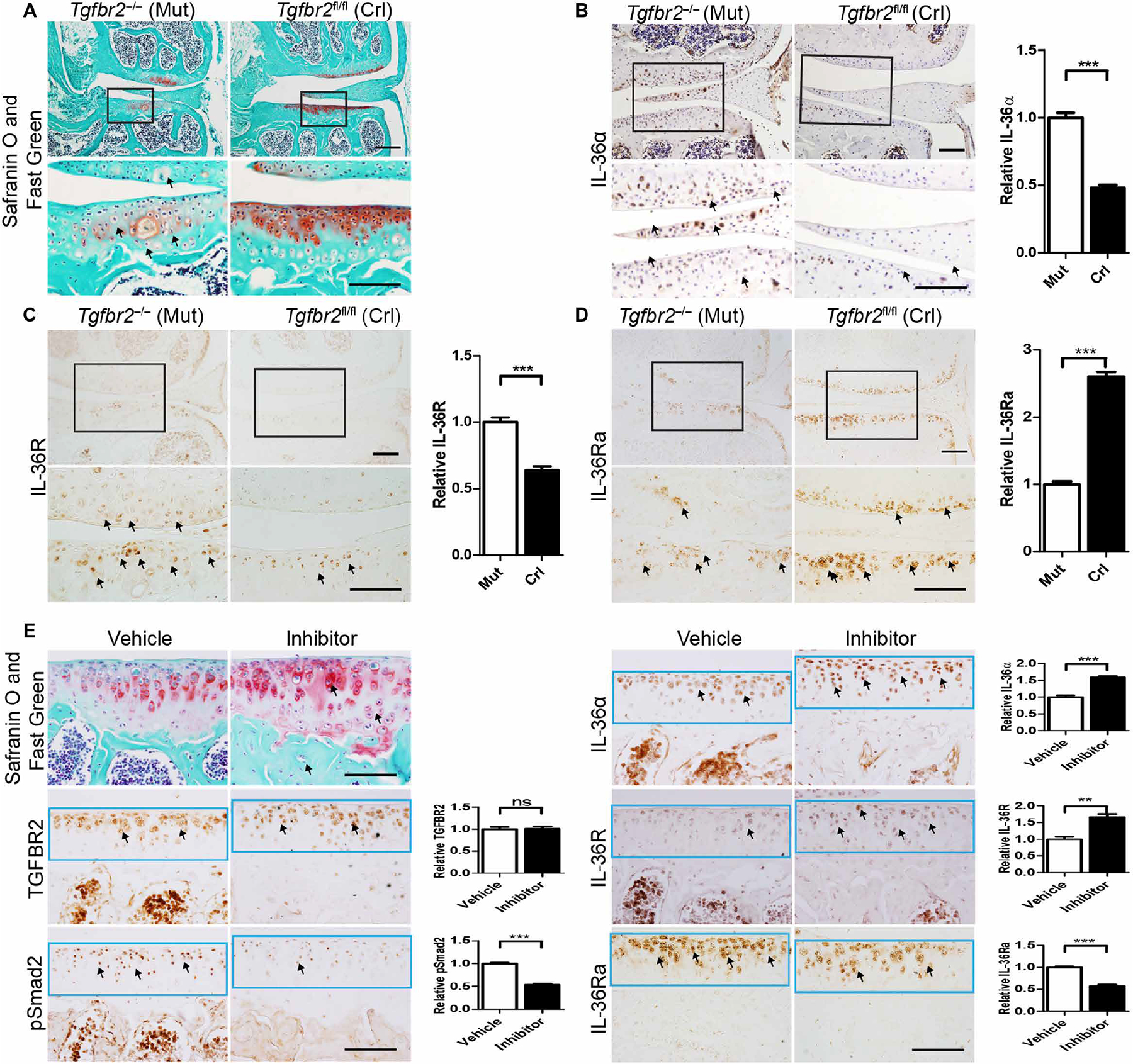
Sections from Tgfbr2−/− (Mut) mice and Tgfbr2fl/fl (Crl) 3-month-old mice were subjected to the following: (A) Safranin O and Fast Green staining of articular cartilage in coronal sections of the tibia medial compartment. Black frames indicate regions shown in higher magnification (bottom row); black arrows indicate hypertrophic chondrocytes. IHC analyses for (B) IL-36α, (C) IL-36R, and (D) IL-36Ra, respectively, where black arrows indicate positive cells. (E) Daily intraperitoneal injections of SB-505124 (5 mg/kg body weight) or an equivalent volume of vehicle (controls) were administered to mice from P21 to P50. Top left: Safranin O and Fast Green staining of articular cartilage in coronal sections of the tibia medial compartment from mice treated with vehicle or SB-505124 for 30 days and analyzed 1 day after the last injection. Bottom, middle, and top right: IHC for TGFBR2, pSmad2, IL-36α, IL-36R, and IL-36Ra. Quantification of IHC density in the region of interest (ROI) of the articular cartilage (black or light blue frame) shown in the right panels of each group image, relative to either mutant [mut; (B), (C), and (D)] or vehicle (E), which were given a value of 1. Black arrows indicate positive cells. n ≥ 4 mice per group. Data are means ± SEM. ns, nonsignificant. **P < 0.01 and ***P < 0.001 (unpaired Student’s t test). Scale bars, 100 μm.
We examined the relationship between the IL-36α system and TGF-β signaling using a pharmacological approach by treating wildtype mice with an inhibitor of TGF-β signaling, SB-505124 (38). Mice that received SB-505124 showed an enhanced chondrocyte hypertrophic phenotype (with a ratio of hypertrophic chondrocytes of 25.22 ± 0.67% in SB-505124–treated mice versus 16.13 ± 0.68% in vehicle-treated mice; P < 0.005, n = 4) that was associated with increased IL-36α and IL-36R but decreased IL-36Ra expression (Fig. 3E). Mice treated with SB-505124 via intraperitoneal injection from P21 to P50 did not show any noticeable side effects. As expected, we found unchanged TGFBR2 expression but a decrease in phosphorylated Smad2, indicating the effectiveness of SB-505124 in interfering with SMAD signaling (Fig. 3E). Together, these results suggest that inhibition of TGF-β signaling by either genetic (Tgfbr2−/− mice) or pharmacological (SB-505124) means induces an OA-like phenotype that occurs with a concomitant increase in IL-36α/R and a decrease in IL-36Ra expression.
In wild-type mice, IL-36Ra attenuates posttraumatic OA and IL-36α exacerbates the phenotype
We then evaluated whether the TGFBR2/IL-36α axis was involved in the DMM-induced posttraumatic OA in wild-type mice (Fig. 4A). We found that, after DMM surgery, TGFBR2 expression decreased within the regions of increased mechanical loading (i.e., weight-bearing region of the medial tibial plateau) as early as 3 days after surgery, and this expression pattern was associated with a decrease in IL-36Ra and increases in IL-36α, IL-36R, and Mmp13 within the same regions. This inverse expression pattern between TGFBR2–IL-36Ra and IL-36α–IL-36R was consistent 5 days after DMM (Fig. 4B). We found an increase in TGFBR2 expression in the interfacing region between the articular cartilage and the synovium (fig. S7, A and B), a region that we had previously reported to show slowly proliferating, multipotent, TGFBR2-expressing cells (39). Two and 12 weeks after DMM surgery, within the formed arthritic lesions expression of TGFBR2, IL-36Ra, IL-36α, IL-36R, and Mmp13 became almost undetectable (fig. S8). These results suggest that dampened TGF-β signaling might induce a hyperactivation of the IL-36α signaling to trigger the onset, but not maintenance, of OA. Our data suggested that, in articular cartilage, the abnormal mechanical loading caused by DMM leads to down-regulation of TGFBR2 that triggers an early down-regulation of IL-36Ra and up-regulation of IL-36α/IL-36R to induce expression of catabolic factors like Mmp13 causing OA progression.
Fig. 4. DMM induces posttraumatic OA and changes in expression of TGFBR, IL-36α, IL-36Ra, IL-36R, and MMP13.
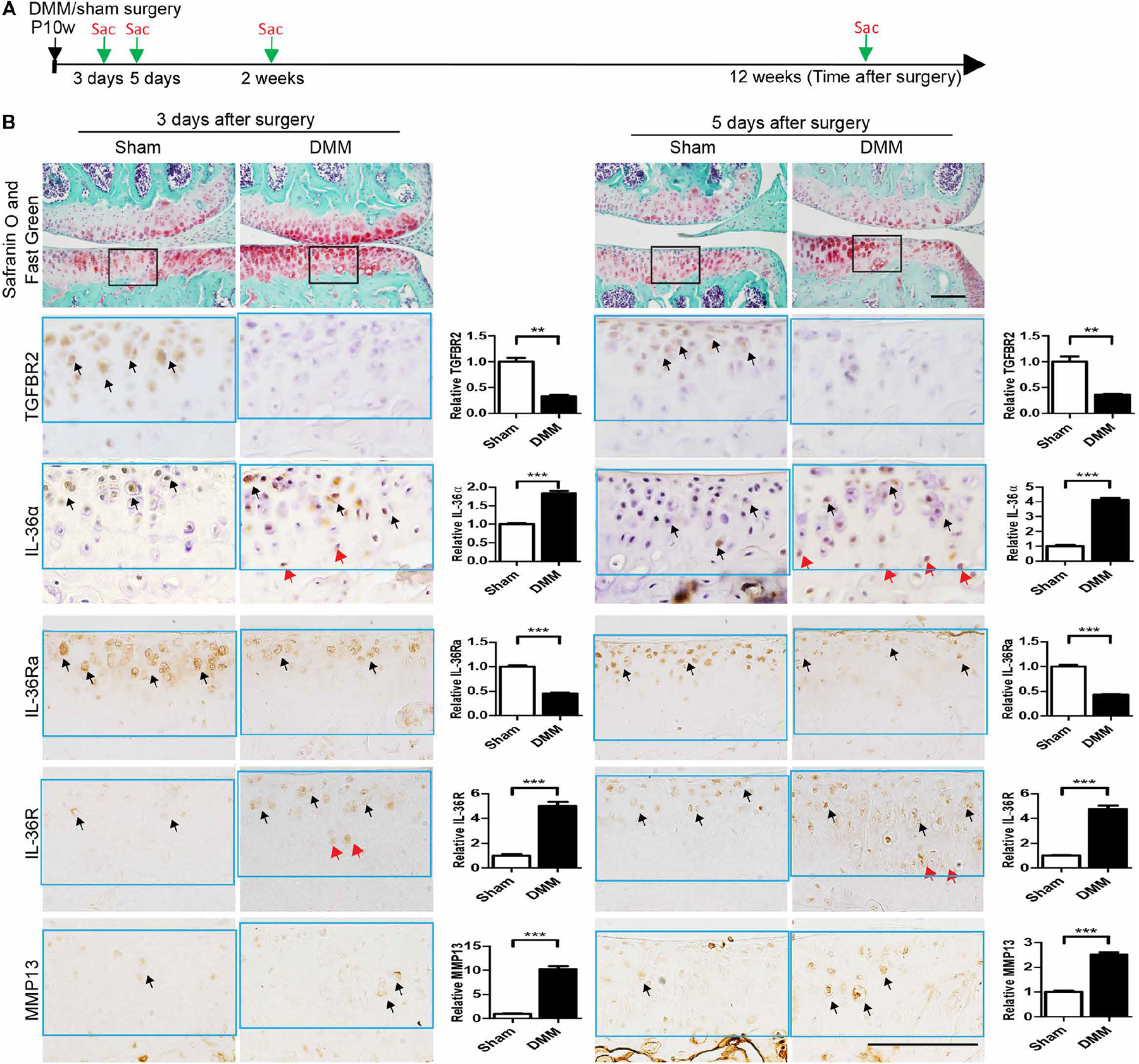
(A) C57BL/6 mice subjected to DMM or sham surgery at postnatal week 10 (P10w) were euthanized 3 and 5 days and 2 and 12 weeks after surgery, respectively, and evaluated histologically. Later time points are shown in fig. S6. n = 3 mice per group. (B) Safranin O and Fast Green staining (top) of articular cartilage in coronal sections of the tibia medial compartment of the knee. IHC (brown stain, bottom) for TGFBR2, IL-36α, IL-36Ra, IL-36R, and MMP13 was performed on adjacent sections. Black frames on Safranin O and Fast Green sections indicate the cartilage loading regions shown in higher magnification for IHC experiments (bottom panels). Black arrows indicate IHC-positive cells in articular cartilage. Red arrows indicate IHC-positive cells in the deep zone of the articular cartilage. Quantification of IHC density in the ROI (light blue frame) shown in the right panels of each group image, relative to sham control, which was given a value of 1. Data are means ± SEM. **P < 0.01 and ***P < 0.001 (unpaired Student’s t test). n = 3 mice per group. Scale bars, 100 μm.
Pharmacological inhibition of IL-36α/R attenuates OA progression, whereas IL-36α signaling hyperactivation exacerbates OA phenotypes
To evaluate the potential of IL-36α/R signaling as a target for pharmacological OA therapy, we performed i.a. injections of IL-36Ra or phosphate-buffered saline (PBS) (as control vehicle) in Tgfbr2−/−, as well as in DMM-induced OA mice, and their respective controls (Fig. 5A). To determine the appropriate dose regimen, IL-36Ra was injected into the knee joint of Tgfbr2−/− mice and controls (3 months old), respectively, at doses of 10, 50, or 250 ng per injection daily for 3 days. The length of treatment was based on the observation that in DMM-induced OA, IL-36α signaling is increased at 3 days after surgery but such an increase is not maintained throughout OA progression (Fig. 4), indicating that IL-36α might be needed to trigger but not to sustain the progression of the disease. One month after injections, no significant effects were noted in Tgfbr2−/− mice that received IL-36α/R at either a dose of 10 or 50 ng per injection (fig. S9). In Tgfbr2−/− mice that received IL-36Ra at a dose of 250 ng per injection, 1 month after injection, we noted attenuation of the OA progression. This was indicated by a decrease in chondrocyte hypertrophy, as well as by less fibrillation, reduced matrix destruction, and decreased expression of collagen 10 and Mmp13 (Fig. 5B). The positive effects of IL-36Ra were detectable 3 months after the injection as indicated by the lower OARSI scores (Fig. 5C).
Fig. 5. Intra-articular injection of IL-36Ra attenuates the OA-like phenotype caused by either ablation of Tgfbr2 or DMM surgery; IL-36α worsens the OA-like phenotypes.
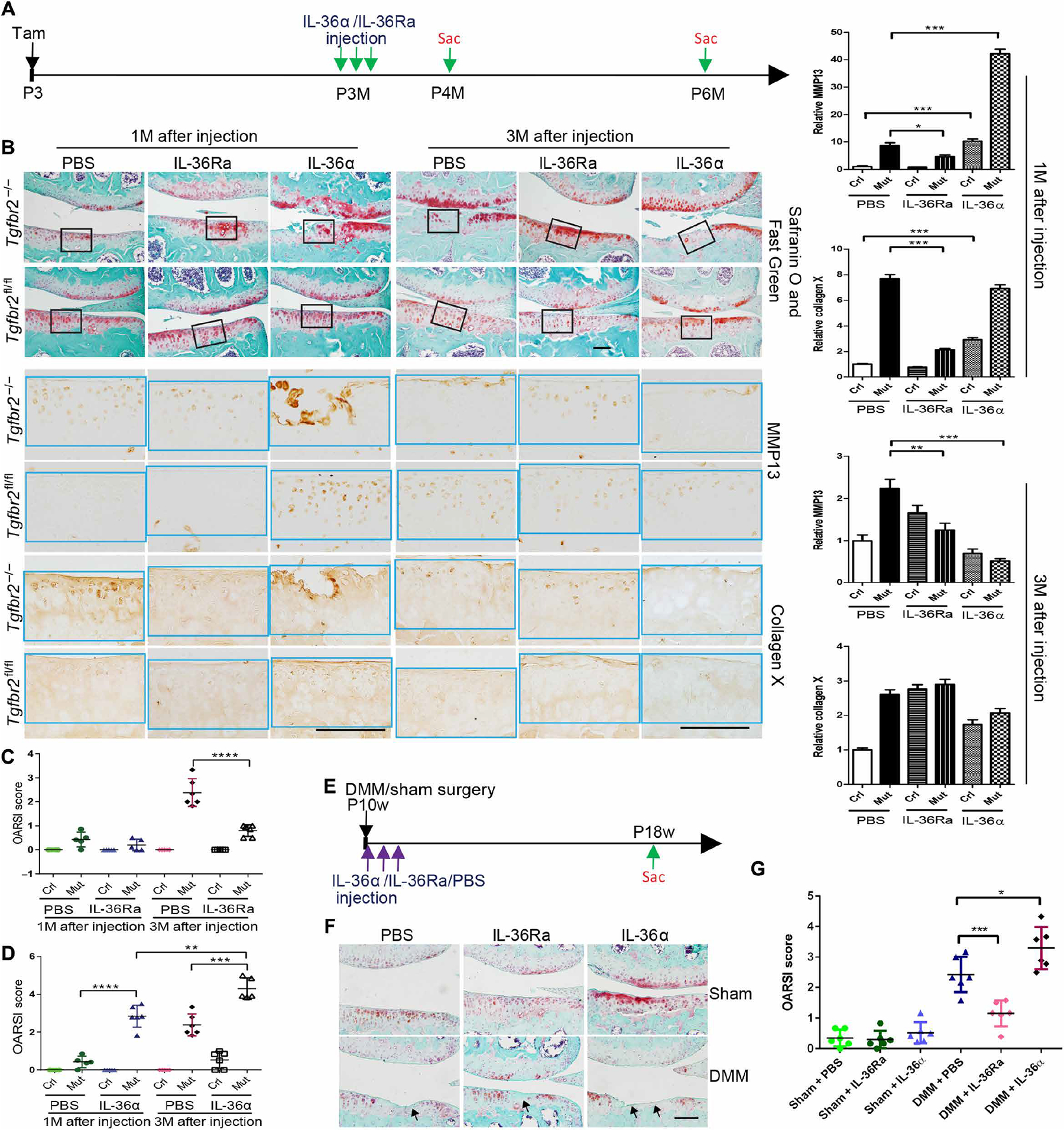
(A) Tgfbr2−/− (Mut) mice and Tgfbr2fl/fl (Crl) mice injected with tamoxifen (Tam) at P3 were intra-articularly injected daily for 3 days with either IL-36α (250 ng per injection) or IL-36Ra (250 ng per injection) or vehicle at P3M. Mice were euthanized at either P4M or P6M and evaluated histologically. n ≥ 5 animals per group. (B) Safranin O and Fast Green staining (top) of articular cartilage in coronal sections of the tibia medial compartment of the knee. IHC (brown stain, bottom) for Mmp13 and collagen 10 was performed on adjacent sections. Black frames on Safranin O and Fast Green sections indicate the regions shown in high magnification for IHC experiments (bottom). Quantification of IHC density in the ROI (light blue frame) shown in the right panels of each group image, relative to Crl, which was given a value of 1. Data are means ± SEM. **P < 0.01 and ***P < 0.001 (unpaired Student’s t test). n = 5 mice per group. Scale bars, 100 μm. (C and D) OARSI score analysis 1 and 3 months after injection. PBS-injected groups are from the same experiment and were used as the control group for both of the IL-36Ra–injected groups (C) and for the IL-36α–injected group (D). n ≥ 5 per group. **P < 0.01, ***P < 0.001, and ****P < 0.0001 (one-way ANOVA and Tukey’s multiple comparison post test). (E) C57BL/6 mice subjected to DMM or sham surgeries at P10w were intra-articularly injected daily for 3 days with either IL-36α (250 ng) or IL-36Ra (250 ng) or vehicle. The first injection was performed 4 hours after surgery. Mice were euthanized 8 weeks after surgery and evaluated histologically. (F) Safranin O and Fast Green staining (top) of articular cartilage in coronal sections of the tibia medial compartment of the knee. (G) OARSI score analysis. n = 6 per group. *P < 0.05 and ***P < 0.001 (one-way ANOVA and Tukey’s multiple comparison post test).
On the other hand, i.a. injections of IL-36α worsened OA of Tgfbr2−/− mice at both 1 and 3 months after injection (Fig. 5, B and D). IL-36α injections given to control mice induced a mild OA-like phenotype (Fig. 5, B and D). To further determine the role of IL-36α signaling in DMM-induced posttraumatic OA, we performed i.a. injections of either IL-36Ra, IL-36α, or PBS in either mice subjected to DMM or sham mice, daily for 3 days starting after surgery. Animals were euthanized 8 weeks later (Fig. 5E). Injection of IL-36Ra partially attenuated the OA process as indicated by the decreased degeneration (>52%) of the articular cartilage by OARSI scores (Fig. 5, F and G), whereas IL-36 α’s injection exacerbated DMM surgery and worsened OA (Fig. 5, F and G). Together, our studies show that IL-36Ra can have therapeutic effects in OA, especially in the presence of an upstream down-regulation of TGF-β signaling (either genetically or DMM-induced).
Expression pattern of TGFBR2 and IL-36 family in articular cartilage along age-related joint pathology progression
Previous studies have reported that in human cartilage, TGFBR2 signaling decreases with age (40). To evaluate the potential role of TGFBR2/IL-36 signaling in age-related spontaneous OA, we examined the expression patterns of TGFBR2/pSmad2 and IL-36α/R/Ra from 2 to 23 months of age in the knee cartilage of wild-type mice (Fig. 6). First, we found evidence of gradually developed OA-like phenotype over time especially after 16 to 17 months (Fig. 6). Furthermore, we found decreased TGFBR2/pSmad2 expression over time. For IL-36α/R, expression increased to peak amounts at P12M to P13M and then gradually decreased when IL-36Ra decreased. These results in aging mice are consistent with the findings observed in Tgfbr2−/− mice, SB-505124–treated mice, and DMM-subjected mice, where a decrease in TGFBR2 corresponds to a derangement of the IL-36α/R/Ra system and is related to OA development.
Fig. 6. Age-dependent expression patterns of IL-36α/IL-36R/IL-36Ra, TGFBR2, and pSmad2 in knee articular cartilage of wild-type mice.
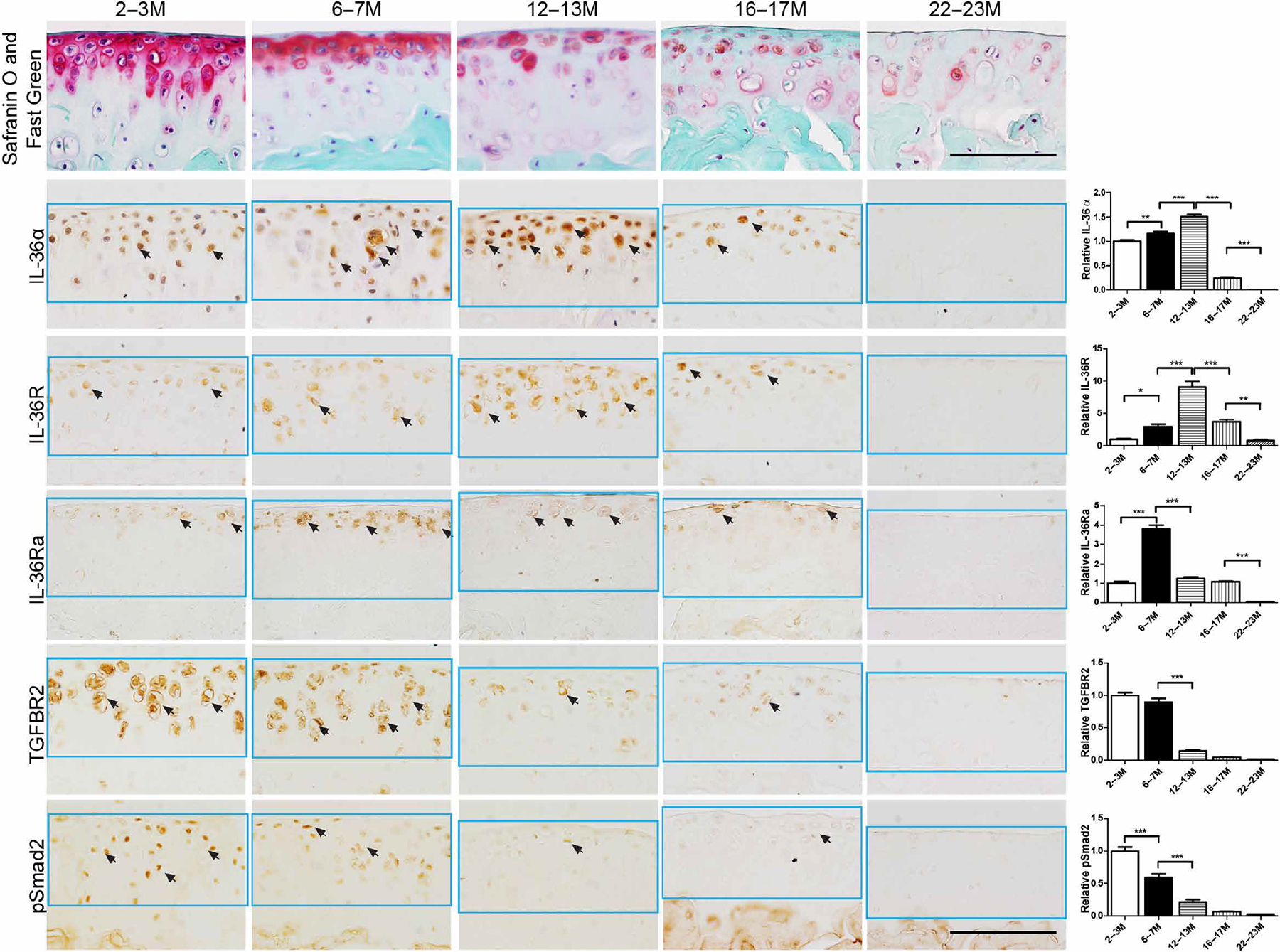
Top row: Representative sagittal Safranin O– and Fast Green–stained sections of femoral cartilage taken from the medial compartment. Bottom rows: IHC for IL-36α, IL-36R, IL-36Ra, TGFBR2, and phosphorylated Smad2 (pSmad2). Sections are adjacent to those stained for Safranin O and Fast Green (top row) and positive cells are stained brown. Black arrows indicate positive cells. Knees were harvested at 2 to 3M, 6 to 7M, 12 to 13M, 16 to 17M, and 22 to 23M of age. Results were consistent in n = 4 mice per group. Scale bars, 100 μm.
IL-36α/IL-36R expression is increased in pathologic human cartilage
To build upon our preclinical relationships regarding TGFBR2 expression, IL-36α/R expression, and OA development, we examined specimens from human organ donors with or without morphological/histological OA-like degenerative changes, as well as specimens from subjects with end-stage OA cartilage obtained perioperatively during total joint arthroplasty. Joints, obtained from organ donors with no previous history of joint disease, were graded for morphologic OA-like changes based on Collins grading and histologic OA-like appearance based on the OARSI Osteoarthritis Cartilage Histopathology Assessment System (OOCHAS) grading system (41–43). The articular cartilage was then shaved from the condyle, digested, and prepared for RT-PCR analysis (Fig. 7, A to E).
Fig. 7. Analysis of IL-36α, IL-36R, IL-36RN, Mmp13, and Tgfbr2 in histological and end-stage OA.
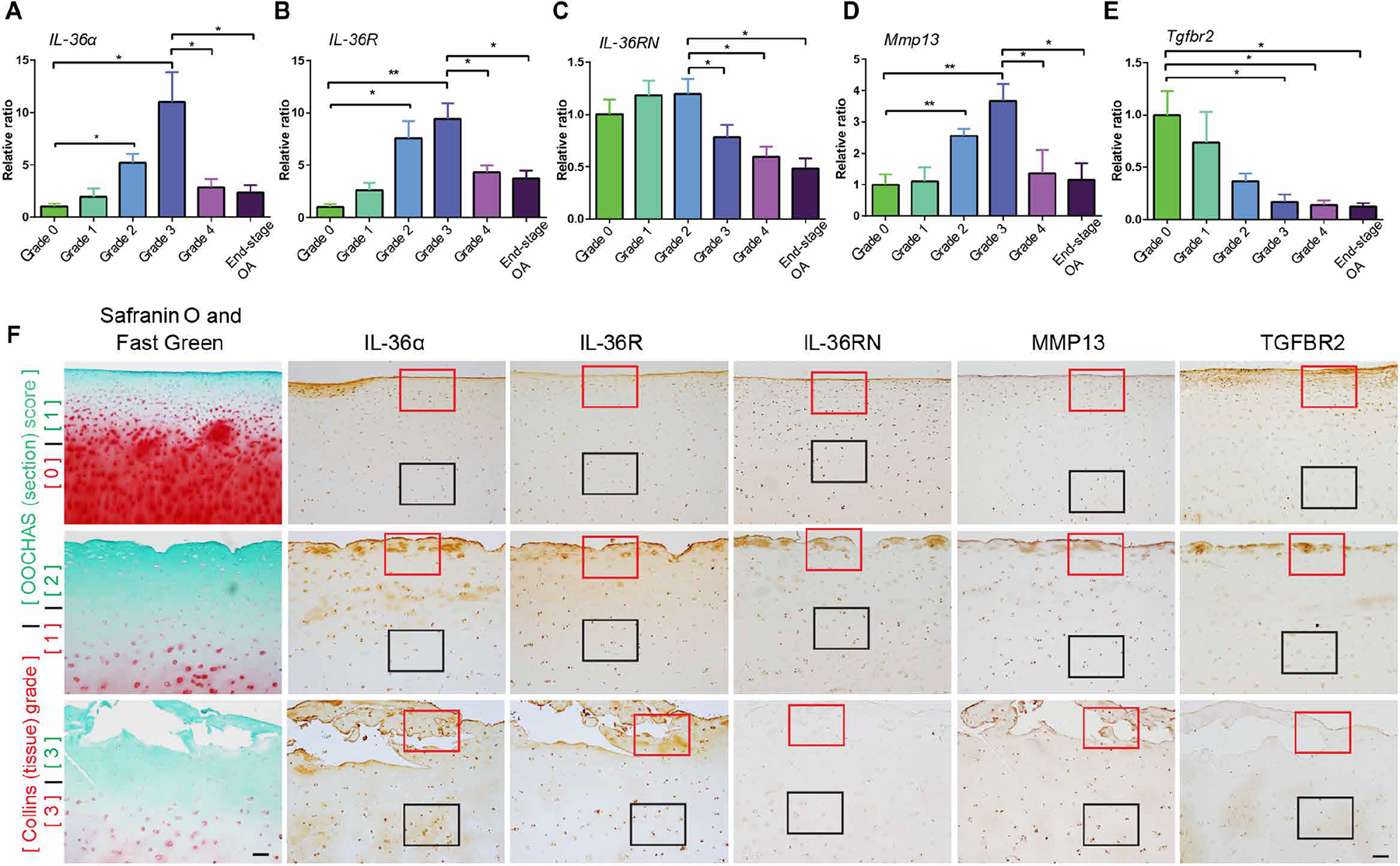
(A to E) RT-PCR results for IL-36α, IL-36R, IL-36RN, Mmp13, and Tgfbr2. Tissues were collected from human tissue donors graded with the Collins method and from patients with end-stage OA. n = 4 donors per group. Data are means ± SEM. *P < 0.05 and **P < 0.01 (one-way ANOVA and Tukey’s multiple comparison post test). (F) Histologically sectioned samples from donor cartilage specimens spanning a range of macroscopic morphological (Collins grades) and histologic (OOCHAS scores) cartilage degradation states. Intact donor condyles were assigned morphological grades (Collins grades, 0 to 3 labeled in red) and then histologically processed. Sections were Safranin O– and Fast Green–stained and scored using the OOCHAS histopathology system (OOCHAS scores of 1 to 3 labeled in green). Adjacent sections were subjected to IHC of IL-36α, IL-36R, IL-36Ra, Mmp13, and TGFBR2, respectively. Red and black frame squares indicate representative and comparable regions in the superficial zone and the middle zone. n = 4 per group. Scale bars, 100 μm.
To better understand the spatiotemporal expression patterns of target gene expression during the OA-like degenerative process, we performed IHC analysis for IL-36α, IL-36R, IL-36RN, MMP13, and TGFBR2 on histological sections spanning a progressive range of cartilage degeneration states (Fig. 7F). Specifically, before IHC, sampled sections were stained for Safranin O and Fast Green, and scored using the OOCHAS histopathology system. We then identified the subpopulation of scored sections encompassing this progressive cartilage degradation range (OOCHAS scores of 1 to 3) and performed IHC on adjacent sections.
We found gradually increasing expression of IL-36α, IL-36R, and Mmp13 at both mRNA (Collins grades 0 to 3; Fig. 7, A to E) and protein (OOCHAS grades 1 to 3; Fig. 7F) levels. We found a decrease in IL-36RN (human form of IL-36Ra) expression at both mRNA (Collins grade 3; Fig. 7C) and protein levels (OOCHAS grade 3; Fig. 7F). Higher-magnification results of ROIs in Fig. 7F are shown in fig. S10. This pattern correlated with the severity of cartilage degeneration, with a drastic decrease noted at Collins grade 3. In end-stage OA specimens, clusters of cells highly expressing IL-36RN and TGFBR2 were found in the upper cartilage layer within the vertical fissures of OA damage (OOCHAS grade 3) according to histological characteristics (fig. S10). From grades 0 to 3, TGFBR2 protein (by IHC) and Tgfbr2 mRNA (by RT-PCR) gradually decreased, and this pattern paralleled increases in IL-36α, IL-36R, and Mmp13 proteins and mRNAs, whereas IL-36RN decreased by grade 3, presumably indicating that TGFBR2 signaling may be an upstream mediator of the changes in IL-36α/IL-36R/IL-36RN and Mmp13 expression. The articular cartilage of joints affected by severe OA, either from subjects with end-stage disease that underwent arthroplasty or from donors that showed severe histological degenerative changes (Collins grade 4), exhibited substantial decreases in proteins (figs. S10 and S11) and mRNA for all targets (Fig. 7, A to E). Similar to our preclinical aging results, this pattern is likely indicative of an extremely pathological OA environment in which chondrocytes become metabolically silenced and/or dramatically decreased in number.
Further RT-PCR analyses of anabolic factors (Sox9, collagen 2, and aggrecan) and catabolic factors (Adamts4, Adamts5, and Mmp3) were performed. We found that OA severity (Collins grade from grade 0 to grade 4) was associated with a decrease of collagen 2 and an increase in the mRNA expression of Adamts4, Adamts5, and Mmp3 (fig. S12). Decreased mRNA of Sox9 and aggrecan were associated with more severe OA (Collins grades 3 and 4).
In human chondrocytes, inhibition of canonical TGF-β signaling induces a dose-dependent increase of IL-36α, IL-36R, and Mmp13 and decrease of IL-36RN
We used primary cultures of isolated human healthy chondrocytes to evaluate the effect of blocking TGFBR2 signaling on IL-36/IL-36R expression and the effect of IL-36RN treatment on Mmp13 expression. First, SB-505124 treatment induced a dose-dependent increase of IL-36α and IL-36R, but a decrease of IL-36RN as evaluated by RT-PCR (Fig. 8, A to C). SB-505124 treatment also induced a dose-dependent increase of Mmp13, as evaluated by RT-PCR and Western immunoblotting analyses (Fig. 8, D and E). These effects were partially rescued by TGF-β1 treatment. The inhibition of TGF-β signaling by SB-505124 was again confirmed by a decrease in pSmad2, and the TGF-β1 rescue effect was confirmed by an increase in pSmad2 (Fig. 8E). Second, IL-36α treatment induced a dose-dependent increase in Mmp13 expression (Fig. 8, E and F). IL-1β was used as a positive control (44). Western immunoblot analyses showed that treatment with either IL-36α or IL-1β was sufficient to increase pERK1/2 (phosphorylated extracellular signal–regulated kinase 1/2), pP65, pP38, and pJNK1/2 (phosphorylated c-Jun N-terminal kinase 1/2) (fig. S13). The results suggest that IL-36α regulates MMP13 expression via the nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways. In addition, IL-36α treatment induced a dose-dependent increase in Adamts4 and Mmp3 expression, but a dose-dependent decrease in Sox9, collagen 2, and aggrecan expression, suggesting that IL-36α plays an essential role for chondrocyte homeostasis as a catabolic factor (fig. S14). A low concentration (2 ng/ml) of IL-36α increased Sox9 expression (fig. S14) and a dose-dependent decrease of Adamts5. Third, in human articular chondrocytes obtained from samples of end-stage OA, IL-36RN induced a dose-dependent decrease in Mmp13 expression at both protein and mRNA levels (Fig. 8, G to I).
Fig. 8. Interplay between TGFBR2 signaling, IL-36α/IL-36R/IL-36RN signaling, and Mmp13 expression in isolated human articular chondrocytes.
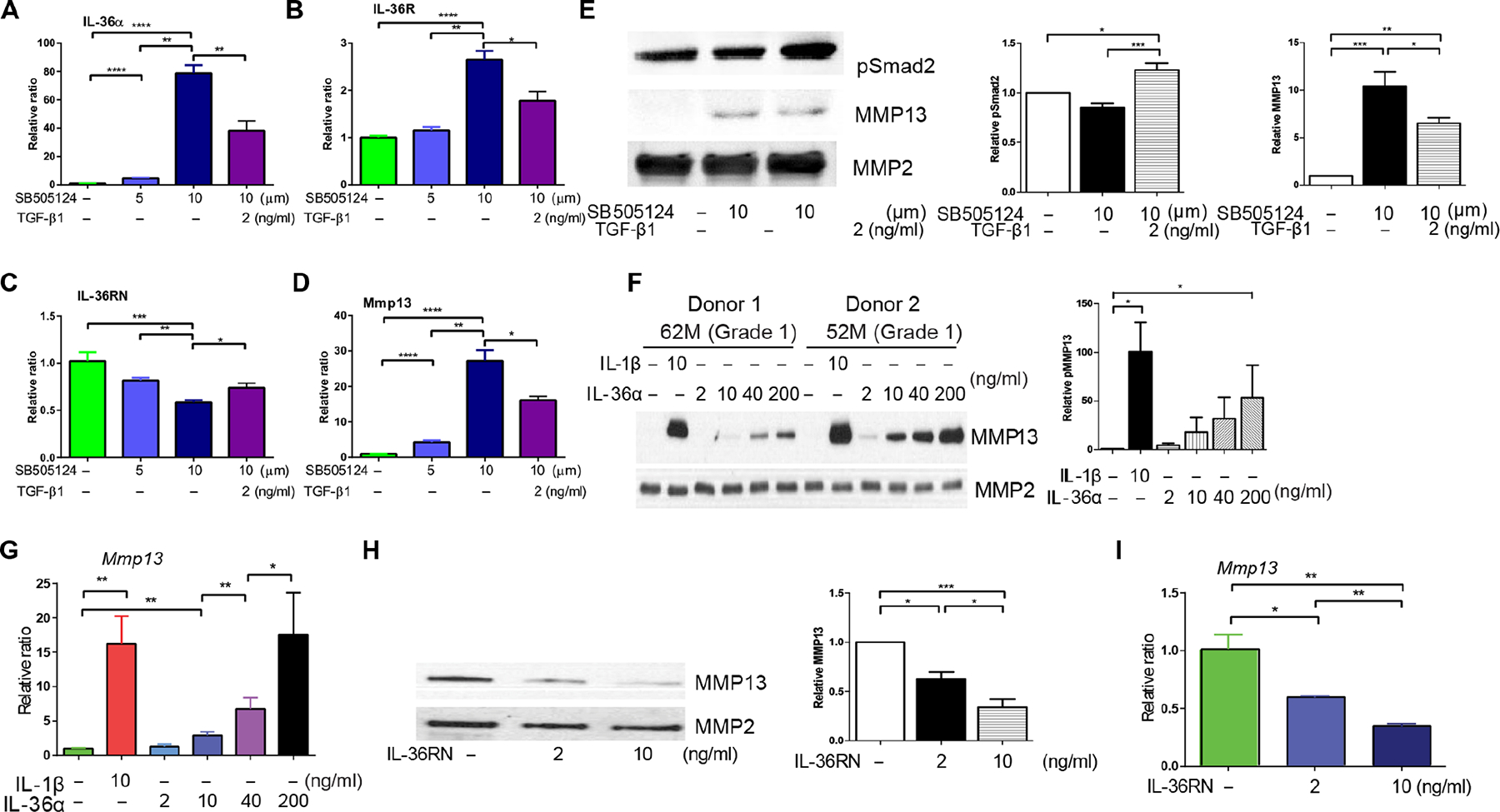
(A to E) Human chondrocytes isolated from donors with mild cartilage degradation (Collins grade 1) were treated with SB-505124 (5 to 10 μM) with and without TGF-β1 (2 ng/ml) for 24 hours, and extracted mRNAs were subjected to qRT-PCR. The data reflect n = 3 independent experiments done using cells from n = 3 different tissue donors. In addition, in (E), total cell lysates were immunoblotted for phosphorylated Smad2; conditioned medium was immunoblotted for MMP13. Total MMP2 was used as a loading control. Blots are representative experiments done using cells from n = 3 different tissue donors. (F and G) Human chondrocytes from donors with mild cartilage degeneration (Collins grade 1) were treated for 24 hours with either IL-36α (2 to 200 ng/ml) or IL-1β (10 ng/ml, positive control). Conditioned medium was immunoblotted for MMP13. Total MMP2 was used as a loading control. Blots are representative experiments done using cells from n = 4 different tissue donors. mRNA extracted from cells in parallel experiments was subjected to qRT-PCR (n = 4 replicates per group). (H and I) Human chondrocytes from donors with end-stage OA (Collins grade 4) were treated for 24 hours with IL-36RN (2 to 10 ng/ml). Conditioned medium was immunoblotted for MMP13. Total MMP2 was used as a loading control. Blots are representative experiments done using cells from n = 3 end-stage OA joints. mRNA extracted from cells in parallel experiments was subjected to qRT-PCR (n = 3 replicates per group). *P < 0.05, **P < 0.01, and ****P < 0.0001 (one-way ANOVA and Tukey’s multiple comparison post test). Quantification of Western blots (relative protein expression normalized by internal control protein MMP2) shown in the right panels of each group image. Data are means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 (one-way ANOVA and Tukey’s multiple comparison post test). n = 3 replicates per group.
DISCUSSION
TGF-β signaling has been widely reported to have a central role in synovial joint development and homeostasis—leading to the notion that manipulating TGF-β signaling may represent a promising means to therapeutically combat clinical OA (15, 19, 23, 45, 46). However, because TGF-β level intervention is expected to induce severe negative multiorgan side effects, development of a clinically effective TGF-β–derived DMOAD requires that we understand the relevant local and downstream signaling mechanisms in the joint. In this study, we identified IL-36α and IL-36Ra/N as critical components of a highly conserved mechanism by which TGFBR2 signaling regulates joint development and joint homeostasis. We presented the feasibility of using IL-36RN as a therapy for OA.
To evaluate the feasibility of developing a clinically effective TGFBR2/IL-36α/IL-36Ra/N-derived DMOAD, we investigated the extent to which this motif was relevant to adult joint homeostasis and clinical OA. We found that this three-part motif was conserved in different murine and human systems: When one component (namely, TGFBR2) of the system was altered, the other two were affected accordingly.
First, postnatal Tgfbr2 inactivation in joint progenitor cells resulted in spontaneous OA development that was concomitant with increased expression of IL-36α. This OA phenotype is consistent with the phenotype of Col2-CreER::Tgfbr2flox/flox mutant mice (19). The protective role of TGFBR2 signaling observed in these progenitor (Prx1-CreER) and cartilage (Col2-CreER) cell populations is opposite to the effect reported in Nestin-CreER mesenchymal stromal cells (23). These apparently contradictory findings may indicate a multifaceted and niche-specific role for TGFBR2 signaling in joint homeostasis and OA progression and between the bone marrow and joint environments. Thus, it is possible that the local prohomeostatic effect of TGFBR2 signaling within the joint is insufficient to protect it from opposing marrow-derived systemic changes in TGF-β signaling.
Furthermore, pharmacological TGFBRI inhibition via SB-505124 produced an enhanced chondrocyte hypertrophy phenotype in wildtype mice and a dose-dependent increase in Mmp13 expression in healthy primary human chondrocytes—both with a corresponding increase in IL-36α. These results suggest that proper TGF-β signaling is required to maintain homeostatic concentrations of IL-36α/R and Mmp13. Although results observed in TGFBR2-deficient mice and SB-505124–treated mice are similar, we cannot exclude the possibility that SB-505124’s effects are due to the inhibition of ALK family members eliciting TGF-β/TGFBR2-independent signaling and that effects observed in TGFBR2-deficient mice are due to noncanonical SMAD-independent signaling. Direct treatment with IL-36α produced an OA-like phenotype in wild-type mice and a dose-dependent increase in Mmp13 expression in normal primary human chondrocytes. In agreement with our results, Conde et al. (36) have recently reported that OA chondrocytes express more IL-36α than healthy chondrocytes, and IL-36α induced expression of MMP13, NOS2 (nitric oxide synthase 2), and COX-2 (cyclooxygenase 2) while activating NF-κB and p38 MAPK pathways. TGF-β signaling has been reported to be inactivated in spontaneous and instability-induced OA due to reduction in TGF-α expression and Smad2 phosphorylation (47, 48). Consistent with these reports, we found a decreased expression of TGFBR2 in the arthritic lesions induced by DMM. Further consistent with our findings are reports showing that TGFBR2 signaling in cartilage decreases with age in humans (40). We previously reported the characterization of joint TGFBR2-expressing progenitors cells with slow proliferative and multidifferentiating traits and their localization within specific niches, such as the synovial-articular cartilage region (39). The increased number of TGFBR2-expressing cells after DMM in the synovial-articular cartilage region might be due to a protective response from these cells and may account for the more severe DMM-induced OA phenotype noted in Tgfbr2−/− mice compared to controls. Furthermore, pharmacological inhibition of IL-36α/R with IL-36Ra attenuated the posttraumatic OA progression in wild-type mice, suggesting potential therapeutic applications of IL-36RN in human posttraumatic OA.
Last, mice with spontaneous age-related OA exhibited decreased TGFBR2 expression and increased IL-36α expression in articular cartilage, indicating that a TGFBR2–IL-36 interplay subsists in physiological conditions (aging) in which the TGFBR2 system has not been experimentally manipulated. Increased IL-36Ra expression in the middle zone of articular cartilage at P6M to P7M might suggest a potential endogenous chondroprotective effect by blocking IL-36R signaling, while at late ages (P12M to P13M), a decrease of IL-36Ra corresponds to the peak expression for IL-36α and IL-36R.
In joints from human donors, we found that the pattern of decreased TGFBR2 and increased IL-36α not only was present but also became more dramatic as a function of the degree of cartilage degeneration (among samples with different Collins grades) and proximity to the lesion site (within a given sample). These IHC findings were confirmed by RT-PCR analysis on isolated primary chondrocytes from donors with high-grade degenerative changes.
Having established the consistency and clinical relevance of this TGFBR2–IL-36 motif, we then investigated the feasibility of using IL-36α inhibition as a clinically effective DMOAD. We hypothesized that the excess IL-36α activity could be blocked pharmacologically using its receptor antagonist IL-36Ra/N. Our experiments produced consistent results across mouse and human studies: In mice, i.a. IL-36Ra treatment was sufficient to arrest the spontaneous OA progression of TGFBR2-deficient mice in both the short and long term, and mitigated the DMM-induced OA in wild-type mice. In primary human chondrocytes isolated from OA joints, IL-36RN treatment reduced Mmp13 expression in a dose-dependent manner. Together, our results provide a strong body of evidence in support of using IL-36RN as a future therapy for OA. Furthermore, the highly conserved nature of the TGF-β–IL-36 system between mice and humans implies that preclinical studies can be used to rapidly accelerate our understanding of how TGF-β signaling regulates IL-36α and, subsequently, how IL-36 signaling is involved in catabolic OA processes. The presence of this pathway in an injury-induced OA model reinforces the concept of a general, high-level regulatory role for the TGFBR2/IL-36 axis in OA pathogenesis and implies that IL-36RN treatment is also a potential therapy for patients that develop OA due to known joint injury (49).
Future studies are needed to evaluate IL-36RN as a potential DMOAD in the context of OA as an organ disease affecting other joint tissues (beside articular cartilage) and to confirm that IL-36RN’s potent effect within the joint occurs without the negative systemic effects. Future investigations are also warranted to determine whether the effects seen after TGFBR2 ablation may not be exclusively IL-36 dependent, as indicated by the fact that IL-36Ra attenuated but did not totally rescue the spontaneous OA observed in Tgfbr2−/− mice. In our study, we used i.a. injection (versus systemic) as a route for IL-36Ra/RN administration to avoid potential systemic effects, and therefore, it is more suitable for clinical use. We recognize that intra-articularly–injected drugs can be rapidly cleared. However, in patients with OA that fail systemic drug treatment, long-term beneficial effects have been reported after i.a. injections of other drugs, although results are still debated (50–54). In future studies, we will evaluate systemic versus i.a. effects of IL-36Ra/RN. We speculate that the sustained beneficial effects by the short course of i.a. administration of IL-36Ra is related to the likelihood that IL-36α signaling is needed to initiate the OA process but not in OA progression. This is supported by the high expression pattern of IL-36α/IL-36R at the early stage of the disease (3 days after DMM) and the almost disappearance at later stages (3 months after DMM). Further evidence for a predominantly early role of the TGFBR2/IL-36 signaling axis in OA development is provided by the observation that in TGFBR2-deficient mice, substantial IL-36α up-regulation was observed before the cartilage started to degrade. Similarly, although we observed a substantial and grade-dependent increase in IL-36α expression in donor-derived human tissues, these donors had no reported medical history of joint disease. Thus, early IL-36RN treatment has promise as a clinical DMOAD: It provides a potential means to restore joint homeostasis before any irreversible damage has occurred and to halt the OA process when it is just started. Because this window of opportunity might be passed by the time a patient presents with joint pain, there is a rationale for pursuing future studies aimed at evaluating IL-36α and IL-36RN as potential early biomarkers for OA in subjects at high risk to develop OA (i.e., postinjury). In summary, we have identified and characterized a TGFBR2/IL-36 signaling axis in joint homeostasis and joint degeneration in OA and demonstrated the viability of pharmacologically manipulating this axis by IL-36Ra/RN to attenuate the OA process.
MATERIALS AND METHODS
Study design
The aim of this study was to explore the role of the TGFBR2/IL-36 axis in joint development, joint homeostasis maintenance, and the preclinical use of IL-36R antagonists as potential therapy for OA. Microarray analyses and immunohistological staining implicated IL-36α in articular cartilage development and OA. Ablation of Tgfbr2 induced OA-like phenotypes but rescued by i.a. injection of IL-36Ra, which was analyzed using histological staining and the OARSI histological scoring system. Inversely related expression patterns between TGFBR2 and IL-36α were further confirmed in DMM-induced and aging-induced OA by histological staining, immunohistological analyses, or RT-PCR. We used only male mice for DMM surgery, because tamoxifen is widely reported to have effects in females, including in cartilage, and male mice are much more susceptible to the development of OA. An investigator who was blinded to the study design performed the histological analyses. Using OA grading based on the OARSI murine scoring system, coded slides were assigned randomly to blind the relevance to the experiment. After grading, histology scores were decoded and assigned to their experimental group. A statistician was consulted before the study to determine the minimum number of animals that would be required for a specific study on the basis of power analysis using relevant published data. Experiments had been done with three to six animals/donors per group as indicated in the figure legends. Human subjects were randomly chosen, with male and female subjects represented equally. Primary data are reported in data file S1.
Human subjects
Two types of primary human joint specimens were collected. First, we collected fresh human femoral condyle and tibial plateau specimens (n = 28) from subjects receiving total knee arthroplasty at the University of North Carolina Hospitals (Chapel Hill, NC, USA) for a diagnosis of end-stage OA. Subjects with a previous history of joint trauma were not considered for enrollment. The study was granted an exemption by the University of North Carolina Institutional Review Board for these discarded “end-stage OA” specimens. In addition, human joint specimens (n = 70) were obtained from cadaveric donors with no previous documented history of joint disease by way of the National Disease Research Interchange (Philadelphia, PA, USA) and Gift of Hope Organ and Tissue Donor Network (Itasca, IL, USA). These donor specimens were derived from both knee (n = 66) and ankle (n = 4) joints, and cartilage samples were isolated as previously reported (55). Sectioned joints were morphologically graded for OA-like cartilage degeneration using the Collins method (42).
Paraffin embedding, histological sectioning, and IHC were performed as above on both end-stage OA (n = 21) and donor cartilage specimens (n = 36). The OOCHAS histopathology grading system was applied to adjacent Safranin O and Fast Green-stained sections to provide a microscopic counterpart to the macroscopic Collins grades (41, 43). mRNA was extracted from shaved cartilage samples of both end-stage OA (n = 4) and donor (n = 30) knee joints and used for RT-PCR. Primary human chondrocytes were isolated from the remaining end-stage OA (n = 3) and donor ankle cartilage (n = 4) specimens; cells were cultured for in vitro assays.
Statistical analysis
Data are presented as the means ± SEM or ±SD. Statistical software (GraphPad Prism) was used to evaluate the results of experiments. Differences among multiple groups were assessed by one-way ANOVA followed by Tukey’s multiple comparison post test for all possible comparisons. Comparison of two groups was performed using an unpaired Student’s t test. Significance was set at P < 0.05.
Supplementary Material
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/11/491/eaan2585/DC1
Materials and Methods
Fig. S1. Early postnatal Prx1-mediated ablation of Tgfbr2 generates long-term TGFBR2 deficits in joint cell populations.
Fig. S2. In mice with TGFBR2-deficient joint cell populations, evidence of abnormal cartilage morphology appears within days of Tgfbr2 ablation and persists through early postnatal development.
Fig. S3. Postnatal ablation of Tgfbr2 leads to an OA-like cartilage phenotype at P3M and P6M.
Fig. S4. Postnatal ablation of Tgfbr2 leads to a severe OA-like phenotype in multiple joint tissues that is concomitant with up-regulation of MMP13 at P14M.
Fig. S5. Postnatal ablation of Tgfbr2 in Tgfbr2−/− mutants leads to accelerated posttraumatic OA.
Fig. S6. Postnatal ablation of Tgfbr2 leads to up-regulation of IL-36α
Fig. S7. DMM surgery induces an expression change of TGFBR2 in synovium region interfacing with the articular cartilage.
Fig. S8. DMM surgeries induce expression changes of TGFBR2, IL-36 α, IL-36r, and IL-36Ra in articular cartilage.
Fig. S9. Low dose of i.a. injection of IL-36Ra and IL-36 α has no effect on the articular phenotype of Tgfbr2−/− mice.
Fig. S10. Analysis of IL-36 α, IL-36R, IL-36RN, MMP13, and TGFBR2 in histological and end-stage OA.
Fig. S11. Histologically prepared sections of end-stage OA cartilage specimens.
Fig. S12. mRNA expression of Sox9, collagen 2, aggrecan, Adamts4, Adamts5, and Mmp3 in histological and end-stage OA.
Fig. S13. Human chondrocytes treated with IL-36 α exhibit a dose-dependent activation of MAPK and NF-κB signaling pathways.
Fig. S14. RT-PCR results of Sox9, collagen 2, aggrecan, Adamts4, Adamts5, and Mmp3 in IL-36 α–treated human articular chondrocytes.
Data file S1. Primary data.
Acknowledgments:
We are grateful to S. Murakami (Case Western Reserve University) for providing the Prx1-CreER transgenic mice. We acknowledge the support of J. Temple in assisting in some of the survival surgeries, in collecting the human specimens from joint arthroplasty subjects, and in proofreading the manuscript. We acknowledge the support of D. J. Del Gaizo and C. W. Olcott (University of North Carolina Hospitals) in procuring end-stage OA specimens. We acknowledge the technical support of Y. Zhao (Department of Medicine, University of North Carolina) in isolating primary human chondrocytes from donor cartilage and Western blotting. We would like to acknowledge the Gift of Hope Organ and Tissue Donor Network, the donor families, and A. Margulis for collecting donor tissues.
Funding: Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award number 1R01AR057042-02 (to A.S.), by the National Institute on Aging (R01 AG044034 to R.L.), by Rush Ciba-Geigy Endowed Chair (to S.C.), and by Rush Woman’s Board Endowed Chair (to A.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the
REFERENCES AND NOTES
- 1.Hootman JM, Helmick CG, Projections of US prevalence of arthritis and associated activity limitations. Arthritis Rheum. 54, 226–229 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma H-L, Flannery CR, Peluso D, Kanki K, Yang Z, Majumdar MK, Morris EA, Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 434, 644–648 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Neuhold LA, Killar L, Zhao W, Sung M-LA, Warner L, Kulik J, Turner J, Wu W, Billinghurst C, Meijers T, Poole AR, Babij P, DeGennaro LJ, Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Invest 107, 35–44 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saito T, Fukai A, Mabuchi A, Ikeda T, Yano F, Ohba S, Nishida N, Akune T, Yoshimura N, Nakagawa T, Nakamura K, Tokunaga K, Chung U.-i., Kawaguchi H, Transcriptional regulation of endochondral ossification by HIF-2α during skeletal growth and osteoarthritis development. Nat. Med 16, 678–686 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Yang S, Kim J, Ryu J-H, Oh H, Chun C-H, Kim BJ, Min BH, Chun J-S, Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med 16, 687–693 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Loeser RF, Goldring SR, Scanzello CR, Goldring MB, Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 64, 1697–1707 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suri S, Walsh DA, Osteochondral alterations in osteoarthritis. Bone 51, 204–211 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Felson DT, Clinical practice. Osteoarthritis of the knee. N. Engl. J. Med 354, 841–848 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Chadjichristos C, Ghayor C, Kypriotou M, Martin G, Renard E, Ala-Kokko L, Suske G, de Crombrugghe B, Pujol JP, Galera P, Sp1 and Sp3 transcription factors mediate interleukin-1 beta down-regulation of human type II collagen gene expression in articular chondrocytes. J. Biol. Chem 278, 39762–39772 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Saklatvala J, Tumour necrosis factor α stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature 322, 547–549 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shakibaei M, Schulze-Tanzil G, John T, Mobasheri A, Curcumin protects human chondrocytes from IL-l1beta-induced inhibition of collagen type II and beta1-integrin expression and activation of caspase-3: An immunomorphological study. Ann. Anat 187, 487–497 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Stöve J, Huch K, Günther KP, Scharf HP, Interleukin-1beta induces different gene expression of stromelysin, aggrecan and tumor-necrosis-factor-stimulated gene 6 in human osteoarthritic chondrocytes in vitro. Pathobiology 68, 144–149 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Guerne PA, Carson DA, Lotz M, IL-6 production by human articular chondrocytes. Modulation of its synthesis by cytokines, growth factors, and hormones in vitro. J. Immunol 144, 499–505 (1990). [PubMed] [Google Scholar]
- 14.Reboul P, Pelletier JP, Tardif G, Cloutier JM, Martel-Pelletier J, The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes. A role in osteoarthritis. J. Clin. Invest 97, 2011–2019 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spagnoli A, O’Rear L, Chandler RL, Granero-Molto F, Mortlock DP, Gorska AE, Weis JA, Longobardi L, Chytil A, Shimer K, Moses HL, TGF-β signaling is essential for joint morphogenesis. J. Cell Biol 177, 1105–1117 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Longobardi L, Li T, Myers TJ, O’Rear L, Ozkan H, Li Y, Contaldo C, Spagnoli A, TGF-β type II receptor/MCP-5 axis: At the crossroad between joint and growth plate development. Dev. Cell 23, 71–81 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finnson KW, Chi Y, Bou-Gharios G, Leask A, Philip A, TGF-b signaling in cartilage homeostasis and osteoarthritis. Front. Biosci 4, 251–268 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Yang X, Chen L, Xu X, Li C, Huang C, Deng C-X, TGF-β/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol 153, 35–46 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen J, Li J, Wang B, Jin H, Wang M, Zhang Y, Yang Y, Im H-J, O’Keefe R, Chen D, Deletion of the transforming growth factor α receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 65, 3107–3119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q, Tan QY, Xu W, Qi HB, Chen D, Zhou S, Ni ZH, Kuang L, Guo JY, Huang JL, Wang XX, Wang ZQ, Su N, Chen L, Chen B, Jiang WL, Gao Y, Chen HG, Du XL, Xie YL, Chen L, Cartilage-specific deletion of Alk5 gene results in a progressive osteoarthritis-like phenotype in mice. Osteoarthritis Cartilage 25, 1868–1879 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blaney Davidson EN, Remst DFG, Vitters EL, van Beuningen HM, Blom AB, Goumans M-J, van den Berg WB, van der Kraan PM, Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol 182, 7937–7945 (2009). [DOI] [PubMed] [Google Scholar]
- 22.van der Kraan PM, Blaney Davidson EN, Blom A, van den Berg WB, TGF-β signaling in chondrocyte terminal differentiation and osteoarthritis: Modulation and integration of signaling pathways through receptor-Smads. Osteoarthritis Cartilage 17, 1539–1545 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Zhen G, Wen C, Jia X, Li Y, Crane JL, Mears SC, Askin FB, Frassica FJ, Chang W, Yao J, Carrino JA, Cosgarea A, Artemov D, Chen Q, Zhao Z, Zhou X, Riley L, Sponseller P, Wan M, Lu WW, Cao X, Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med 19, 704–712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie L, Tintani F, Wang X, Li F, Zhen G, Qiu T, Wan M, Crane J, Chen Q, Cao X, Systemic neutralization of TGF-β attenuates osteoarthritis. Ann. N. Y. Acad. Sci 1376, 53–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Debets R, Timans JC, Homey B, Zurawski S, Sana TR, Lo S, Wagner J, Edwards G, Clifford T, Menon S, Bazan JF, Kastelein RA, Two novel IL-1 family members, IL-1 delta and IL-1 epsilon, function as an antagonist and agonist of NF-kappa B activation through the orphan IL-1 receptor-related protein 2. J. Immunol 167, 1440–1446 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA, IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Dinarello C, Arend W, Sims J, Smith D, Blumberg H, O’Neill L, Goldbach-Mansky R, Pizarro T, Hoffman H, Bufler P, Nold M, Ghezzi P, Mantovani A, Garlanda C, Boraschi D, Rubartelli A, Netea M, van der Meer J, Joosten L, Mandrup-Poulsen T, Donath M, Lewis E, Pfeilschifter J, Martin M, Kracht M, Muehl H, Novick D, Lukic M, Conti B, Solinger A, Kelk P, van de Veerdonk F, Gabel C, IL-1 family nomenclature. Nat. Immunol 11, 973(2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Towne JE, Sims JE, IL-36 in psoriasis. Curr. Opin. Pharmacol 12, 486–490 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Chustz RT, Nagarkar DR, Poposki JA, Favoreto S Jr., Avila PC, Schleimer RP, Kato A, Regulation and function of the IL-1 family cytokine IL-1F9 in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol 45, 145–153 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramadas RA, Ewart SL, Medoff BD, LeVine AM, Interleukin-1 family member 9 stimulates chemokine production and neutrophil influx in mouse lungs. Am. J. Respir. Cell Mol. Biol 44, 134–145 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frey S, Derer A, Messbacher M-E, Baeten DLP, Bugatti S, Montecucco C, Schett G, Hueber AJ, The novel cytokine interleukin-36α is expressed in psoriatic and rheumatoid arthritis synovium. Ann. Rheum. Dis 72, 1569–1574 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Scheibe K, Kersten C, Schmied A, Vieth M, Primbs T, Carlé B, Knieling F, Claussen J, Klimowicz AC, Zheng J, Baum P, Meyer S, Schürmann S, Friedrich O, Waldner MJ, Rath T, Wirtz S, Kollias G, Ekici AB, Atreya R, Raymond EL, Mbow ML, Neurath MF, Neufert C, Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology 156, 1082–1097.e11 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Magne D, Palmer G, Barton JL, Mezin F, Talabot-Ayer D, Bas S, Duffy T, Noger M, Guerne P-A, Nicklin MJH, Gabay C, The new IL-1 family member IL-1F8 stimulates production of inflammatory mediators by synovial fibroblasts and articular chondrocytes. Arthritis Res. Ther 8, R80(2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derer A, Groetsch B, Harre U, Böhm C, Towne J, Schett G, Frey S, Hueber AJ, Blockade of IL-36 receptor signaling does not prevent from TNF-induced arthritis. PLOS ONE 9, e101954(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamacchia C, Palmer G, Rodriguez E, Martin P, Vigne S, Seemayer CA, Talabot-Ayer D, Towne JE, Gabay C, The severity of experimental arthritis is independent of IL-36 receptor signaling. Arthritis Res. Ther 15, R38(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conde J, Scotece M, Abella V, Lois A, Lopez V, Garcia-Caballero T, Pino J, Gómez-Reino JJ, Gómez R, Lago F, Gualillo O, IL-36α: A novel cytokine involved in the catabolic and inflammatory response in chondrocytes. Sci. Rep 5, 16674(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glasson SS, Chambers MG, Van Den Berg WB, Little CB, The OARSI histopathology initiative—Recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 18 (Suppl 3), S17–S23 (2010). [DOI] [PubMed] [Google Scholar]
- 38.DaCosta Byfield S, Major C, Laping NJ, Roberts AB, SB-505124 is a selective inhibitor of transforming growth factor-β type I receptors ALK4, ALK5, and ALK7. Mol. Pharmacol 65, 744–752 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Li T, Longobardi L, Myers TJ, Temple JD, Chandler RL, Ozkan H, Contaldo C, Spagnoli A, Joint TGF-β type II receptor-expressing cells: Ontogeny and characterization as joint progenitors. Stem Cells Dev. 22, 1342–1359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Kraan PM, Blaney Davidson EN, van den Berg WB, A role for age-related changes in TGFβ signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res. Ther 12, 201(2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Custers RJ, Creemers LB, Verbout AJ, van Rijen MH, Dhert WJ, Saris DB, Reliability, reproducibility and variability of the traditional Histologic/Histochemical Grading System vs the new OARSI Osteoarthritis Cartilage Histopathology Assessment System. Osteoarthritis Cartilage 15, 1241–1248 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Kuettner KE, Cole AA, Cartilage degeneration in different human joints. Osteoarthritis Cartilage 13, 93–103 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, Salter D, van den Berg WB, Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthritis Cartilage 14, 13–29 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Mengshol JA, Vincenti MP, Brinckerhoff CE, IL-1 induces collagenase-3 (MMP-13) promoter activity in stably transfected chondrocytic cells: Requirement for Runx-2 and activation by p38 MAPK and JNK pathways. Nucleic Acids Res. 29, 4361–4372 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen CG, Thuillier D, Chin EN, Alliston T, Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheum. 64, 3278–3289 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Serra R, Johnson M, Filvaroff EH, LaBorde J, Sheehan DM, Derynck R, Moses HL, Expression of a truncated, kinase-defective TGF-β type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J. Cell Biol 139, 541–552 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blaney Davidson EN, Vitters EL, van der Kraan PM, van den Berg WB, Expression of transforming growth factor-β (TGFβ) and the TGFβ signalling molecule SMAD-2P in spontaneous and instability-induced osteoarthritis: Role in cartilage degradation, chondrogenesis and osteophyte formation. Ann. Rheum. Dis 65, 1414–1421 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blaney Davidson EN, Scharstuhl A, Vitters EL, van der Kraan PM, van den Berg WB, Reduced transforming growth factor-β signaling in cartilage of old mice: Role in impaired repair capacity. Arthritis Res. Ther 7, R1338–R1347 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson DD, Chubinskaya S, Guilak F, Martin JA, Oegema TR, Olson SA, Buckwalter JA, Post-traumatic osteoarthritis: Improved understanding and opportunities for early intervention. J. Orthop. Res 29, 802–809 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Camurcu Y, Sofu H, Ucpunar H, Kockara N, Cobden A, Duman S, Single-dose intra-articular corticosteroid injection prior to platelet-rich plasma injection resulted in better clinical outcomes in patients with knee osteoarthritis: A pilot study. J. Back Musculoskelet. Rehabil 31, 603–610 (2018). [DOI] [PubMed] [Google Scholar]
- 51.McAlindon TE, LaValley MP, Harvey WF, Price LL, Driban JB, Zhang M, Ward RJ, Effect of intra-articular triamcinolone vs saline on knee cartilage volume and pain in patients with knee osteoarthritis: A randomized clinical trial. JAMA 317, 1967–1975 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takamura J, Seo T, Strand V, A single intra-articular injection of Gel-200 for treatment of symptomatic osteoarthritis of the knee is more effective than phosphate buffered saline at 6 months: A subgroup analysis of a multicenter, Randomized Controlled Trial. Cartilage, 1947603518768015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taniguchi Y, Yoshioka T, Kanamori A, Aoto K, Sugaya H, Yamazaki M, Intra-articular platelet-rich plasma (PRP) injections for treating knee pain associated with osteoarthritis of the knee in the Japanese population: A phase I and IIa clinical trial. Nagoya J. Med. Sci 80, 39–51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian K, Cheng H, Zhang J, Chen K, Intra-articular injection of methylprednisolone for reducing pain in knee osteoarthritis: A systematic review and meta-analysis. Medicine (Baltimore) 97, e0240(2018). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Chubinskaya S, Merrihew C, Cs-Szabo G, Mollenhauer J, McCartney J, Rueger DC, Kuettner KE, Human articular chondrocytes express osteogenic protein-1. J. Histochem. Cytochem 48, 239–250 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Kawanami A, Matsushita T, Chan YY, Murakami S, Mice expressing GFP and CreER in osteochondro progenitor cells in the periosteum. Biochem. Biophys. Res. Commun 386, 477–482 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Glasson SS, Blanchet TJ, Morris EA, The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 15, 1061–1069 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, Ladunga I, Ulitsky-Lazareva B, Muruganujan A, Rabkin S, Vandergriff JA, Doremieux O, PANTHER: A browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 31, 334–341 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McLeod MJ, Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology 22, 299–301 (1980). [DOI] [PubMed] [Google Scholar]
- 60.Long DL, Loeser RF, p38γ mitogen-activated protein kinase suppresses chondrocyte production of MMP-13 in response to catabolic stimulation. Osteoarthritis Cartilage 18, 1203–1210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greene MA, Loeser RF, Function of the chondrocyte PI-3 kinase-Akt signaling pathway is stimulus dependent. Osteoarthritis Cartilage 23, 949–956 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/11/491/eaan2585/DC1
Materials and Methods
Fig. S1. Early postnatal Prx1-mediated ablation of Tgfbr2 generates long-term TGFBR2 deficits in joint cell populations.
Fig. S2. In mice with TGFBR2-deficient joint cell populations, evidence of abnormal cartilage morphology appears within days of Tgfbr2 ablation and persists through early postnatal development.
Fig. S3. Postnatal ablation of Tgfbr2 leads to an OA-like cartilage phenotype at P3M and P6M.
Fig. S4. Postnatal ablation of Tgfbr2 leads to a severe OA-like phenotype in multiple joint tissues that is concomitant with up-regulation of MMP13 at P14M.
Fig. S5. Postnatal ablation of Tgfbr2 in Tgfbr2−/− mutants leads to accelerated posttraumatic OA.
Fig. S6. Postnatal ablation of Tgfbr2 leads to up-regulation of IL-36α
Fig. S7. DMM surgery induces an expression change of TGFBR2 in synovium region interfacing with the articular cartilage.
Fig. S8. DMM surgeries induce expression changes of TGFBR2, IL-36 α, IL-36r, and IL-36Ra in articular cartilage.
Fig. S9. Low dose of i.a. injection of IL-36Ra and IL-36 α has no effect on the articular phenotype of Tgfbr2−/− mice.
Fig. S10. Analysis of IL-36 α, IL-36R, IL-36RN, MMP13, and TGFBR2 in histological and end-stage OA.
Fig. S11. Histologically prepared sections of end-stage OA cartilage specimens.
Fig. S12. mRNA expression of Sox9, collagen 2, aggrecan, Adamts4, Adamts5, and Mmp3 in histological and end-stage OA.
Fig. S13. Human chondrocytes treated with IL-36 α exhibit a dose-dependent activation of MAPK and NF-κB signaling pathways.
Fig. S14. RT-PCR results of Sox9, collagen 2, aggrecan, Adamts4, Adamts5, and Mmp3 in IL-36 α–treated human articular chondrocytes.
Data file S1. Primary data.