

Abstract
Adenovirus infection has a tremendous impact on the cellular silencing machinery. Adenoviruses express high amounts of non-coding virus associated (VA) RNAs able to saturate key factors of the RNA interference (RNAi) processing pathway, such as Exportin 5 and Dicer. Furthermore, a proportion of VA RNAs is cleaved by Dicer into viral microRNAs (mivaRNAs) that can saturate Argonaute, an essential protein for miRNA function. Thus, processing and function of cellular miRNAs is blocked in adenoviral-infected cells. However, viral miRNAs actively target the expression of cellular genes involved in relevant functions such as cell proliferation, DNA repair or RNA regulation. Interestingly, the cellular silencing machinery is active at early times post-infection and can be used to control the adenovirus cell cycle. This is relevant for therapeutic purposes against adenoviral infections or when recombinant adenoviruses are used as vectors for gene therapy. Manipulation of the viral genome allows the use of adenoviral vectors to express therapeutic miRNAs or to be silenced by the RNAi machinery leading to safer vectors with a specific tropism. This article is part of a "Special Issue entitled:MicroRNAs in viral gene regulation".
Keywords: Adenovirus, miRNAs, VA RNA, TIA-1, RNAi
Research highlights
► The silencing machinery is active at early times post adenoviral infection. ► Later, Adenoviruses have evolved to both use and block the RNA silencing machinery. ► Adenovirus blocks RNAi by expression of VA RNA, able to saturate Exp5, Dicer and Ago. ► VA RNAs are processed to viral miRNAs able to control the expression of target genes. ► This should be considered in the design of adenoviral vectors for gene therapy.
1. Introduction
RNA interference (RNAi) is a posttranscriptional mechanism of gene silencing highly conserved in eukaryotes. RNAi controls gene expression by interaction of complementary sequences between a target mRNA and a single stranded molecule of RNA: small interference (siRNA) or micro-RNA (miRNA). siRNA binds the target mRNA with a perfect complementarity. Instead, miRNA shows a partial matching, generally, with the 3′UTR of target mRNA. The interaction between the small RNA and the target results in gene silencing when the siRNA or the miRNA is bound to the RNA-induced silencing complex RISC. Thus, RISC is guided to the target mRNA by the small RNA. Binding to the target with perfect complementarity induces RISC-mediated cleavage of the target RNA. miRNA guiding of RISC induces gene silencing by an as yet not fully understood mechanism that leads to translational repression and deadenylation and degradation of mRNA targets [1].
miRNA precursors are transcribed by RNA polymerase II as long primary transcripts named pri-miRNAs. Pri-miRNAs can contain one or more immature miRNAs. The pri-miRNA is processed by a nuclear RNase III called Drosha generating imperfectly pairing stem–loop molecules of approximately 70 nucleotides known as pre-miRNAs. Pre-miRNAs are transferred to the cytosol by exportin 5 (Exp5) where another RNase III, Dicer, generates a mature double-stranded miRNA of approximately 22 nucleotides. Following Dicer cleavage, the resulting RNA duplex is loaded into RISC. Argonaute (Ago), one of the proteins of the complex, separates the two strands of the double-stranded RNA. The guide strand remains in RISC as a mature miRNA, whereas the other strand (the passenger strand) is degraded [2], [3], [4], [5]. Then, the RISC-miRNA complex is prepared for regulation of target mRNAs.
miRNA regulation of gene expression contributes to essential processes such as differentiation, apoptosis or proliferation [6]. The expression levels of one or a combination of several miRNAs can induce, or serve as a signature for, a specific medical condition, such as cancer, obesity, diabetes, or neurodegenerative disease [7], [8], [9]. Cellular signalling and cellular stresses can upregulate or downregulate the expression of several miRNAs triggering new cell behaviour [10], [11]. Alteration of the cellular miRNA content seems particularly evident when cells are infected with a virus (reviewed recently in [12], [13], [14], [15], [16]). Viral infections alter the miRNA profile of infected cells by several means. The infection per se can change expression levels of several cellular miRNAs [17], [18], [19], [20], [21], [22]. This may be induced by the virus or may result from the cellular response to the viral infection. Furthermore, several viruses have been shown to express viral miRNAs that can control the levels of viral proteins or can decrease the expression of cellular factors with antiviral properties [12], [13], [14], [15], [16]. Some viruses have been studied in more detail to understand their impact on miRNA expression and functionality. Here, we focus on adenoviruses, which serve as a good example to demonstrate the variety of strategies that may be used by a virus to alter the miRNA machinery. Expression of cellular miRNAs is altered upon adenoviral infection. Moreover, adenoviruses express several viral miRNAs. Finally, the virus blocks some of the key players of the silencing machinery, resulting in the inhibition of cellular miRNA processing and function.
2. Adenovirus
Adenoviruses have been studied for more than 50 years [23]. Investigation on viral function has contributed to the understanding of important aspects of cellular physiology. In addition, recombinant versions of adenovirus have been extensively used as vectors for gene therapy. In fact, these agents are the most frequently used viral vectors in clinical trials.
An icosahedral capsid with protruding fiber proteins stores the linear double-stranded genome of adenovirus. The adenoviral genome has approximately 35–36 kilobases which contain early and late genes and inverted terminal repeats at both ends (Fig. 1A). Fiber-mediated binding to the cell surface starts the viral infection, which can be divided into an early and a late phase [24]. In the early phase of viral infection, the viral genome reaches the nucleus of the host cell and early viral genes are transcribed and translated. The function of early proteins is devoted to blocking the host antiviral response and apoptosis, activating cell-cycle to facilitate viral replication and inducing the expression of late viral genes. E1a proteins are the first to be expressed upon viral infection and execute several of these tasks. E1a activates transcription and blocks retinoblastoma (Rb) leading to activation of E2F and its target genes causing uncontrolled growth. However, E1a has been shown to have both oncogenic and tumor-suppressive properties [25]. In cells expressing E1a, a balance between these two activities could regulate the properties of cellular transformation which could be beneficial for the viral cell cycle. Since adenovirus infects mainly differentiated non-dividing epithelial tissues, E1a-mediated promotion of cell proliferation could help for epithelial-to-mesenchymal cell transformation and viral DNA replication, while tumor-suppressive properties of E1a could promote mesenchymal-to-epithelial transformation, thereby providing a cellular environment that could facilitate further steps in virus multiplication (reviewed in [25]).
Fig. 1.

A. Schematic of adenovirus genome. The alternative splicing of E1a pre-mRNA is shown. U-rich stretches are indicated. Arrows represent direction of transcription. MLP is the major late promoter. E genes are early and L genes are late. B. Structure of adenovirus type 2 VA I RNA. The structure is divided into three regions associated to the indicated functions. The arrows indicate Dicer cleavage sites for mivaRNAI-137 and mivaRNAI-138.
E1a-mediated activation of E2F allows expression of E2 early viral genes, required for replication [24]. Once the viral genome has replicated, expression of the delayed early genes IVa2 and IX marks initiation of the late phase of the viral cycle. Expression of late transcripts leads to production of capsid proteins such as penton, hexon or fiber, and other factors required for viral packaging and release. All these proteins, a total of 18, derive from a single pre-mRNA that is processed by alternative splicing to different mRNAs. In fact, processing of most of the viral pre-mRNAs from both the early and the late phase requires proper alternative splicing and polyadenylation. Furthermore, alternative splicing of some of the pre-mRNAs differs throughout the viral cycle (Fig. 1A). For instance, E1a pre-mRNA is preferentially spliced to a 13s mRNA during the early phase, in contrast to a 9s mRNA in the late phase. Differences in alternative splicing are also observed between the initiation versus end of the late phase [26]. Several viral and cellular factors have been involved in the regulation of the alternative splicing of adenoviral pre-mRNAs. A well-described example is cellular SF2/ASF and SRp30 proteins, whose inactivation by dephosphorylation leads to a difference in alternative splicing that results in production of viral 52-55K or IIIa protein at early or late time points, respectively [26].
Cellular splicing factors are modified during the viral infection so that adequate alternative splicing of viral transcripts can occur. However, these changes have a negative impact on the alternative splicing of cellular pre-mRNAs. In fact, several cellular pathways are affected during the adenovirus cell-cycle. DNA repair enzymes are inactivated to avoid the concatemerization of the adenoviral genome [27]. The interferon (IFN) pathway is blocked to impede the host antiviral response by affecting several key molecules [28]. Furthermore, cellular mRNA transport to the cytoplasm is repressed while transport of adenoviral RNA is not [29]. In the cytoplasm, translation of cellular mRNAs is also inhibited, so adenoviral mRNAs are preferentially translated [30]. Finally, several essential proteins of the RNAi silencing pathway are blocked or saturated during adenoviral infection. Since some mammalian viruses are inhibited by host-derived siRNA or miRNA [31], this RNAi-blocking strategy could enhance infection. Like adenovirus, other mammalian viruses have evolved ways to block the RNAi pathway which include expression of proteins such as primate foamy virus-1 Tas [31], HIV Tat [32], influenza virus NS1 [33], [34], vaccinia virus E3L [34], HCV core [35], ebola virus VP35, VP30 and VP40 [36], [37], HTLV Rex [38] or coronavirus 7a accessory protein [39]. Moreover, certain viral RNAs also have the ability to inhibit the silencing pathway. A good example is TAR RNA, expressed by HIV-1 virus, which binds and sequesters the host-derived TAR RNA binding protein (TRBP), a necessary cofactor for Dicer function [40]. Another example is adenovirus virus-associated (VA) RNAs [41], [42] reviewed here.
3. Adenoviral-mediated inhibition of RNAi: VA RNAs
VA RNAs are short non-coding RNAs transcribed by cellular RNA polymerase III from the adenoviral genome. Adenovirus can express one or two VA RNAs. VAI is found in all adenovirus serotypes, whereas VAII is detected only in some of them. The length of different VA RNAs is similar, VAI is 157–160 nucleotides long while VAII is 158–163 nucleotides long [43]. The nucleotide sequences between VAI and VAII RNAs of a particular virus are significantly different from one another. The same diversity can be observed when analyzing VAI or VAII among different serotypes, especially if they belong to different groups (A to F) [44], [45]. Comparison of adenovirus serotypes has shown conserved regions in the 5′ end and near the 3′ terminus. It has been shown that these sequences are required for VA RNA transcription initiation and termination. Besides these homologies, highly conserved sequences up to 4 nucleotides are present throughout the VA gene that seem to be required for maintenance of the VA RNA secondary structure [44], [45].
3.1. VA RNA structure
Secondary structure models for VA RNA have been based on extensive mutagenesis and structure probing studies together with comparative sequence analysis [44], [45], [46], [47], [48], [49], [50]. The results of these analyses clearly show that VA RNAs fold into a stem–loop structure (Fig. 1B). The structure is fairly well conserved in all VA RNAs with slight differences among different groups. VA secondary structure can be divided into three regions: the terminal stem (including the paired 5′ and 3′ ends), the panhandle apical stem and a more structured central domain [46], [49]. These domains are associated with functional activities.
The terminal and apical stems are highly conserved structures. Mutations in one strand of the stem are compensated for by complementary changes in the other strand [51]. This is also the case for a pair of highly conserved based-paired tetranucleotides GGGU/ACCC located in the central domain [44], [45] This suggests that for VA functionality, the nucleotide sequence is less important than the specific structure. However, since some single point mutations in the central domain reduce VA activity, specific sequences may also have a role in VA functionality [52]. Recently, it has been described that VAI apical stem can adopt two different conformations with different functional activities that co-exist in the infected cell [53].
3.2. VA RNA function
Most of the studies regarding VA RNA structure and functionality have used human adenovirus serotypes 2 and 5 as a model. Inactivation of VAI in adenovirus 2 causes a 10-20-fold decrease in viral growth. When both VA RNAs are deleted, viral growth decreases 60-fold [51], [54], [55], [56], [57]. Deletion of VAII alone has little impact on viral growth [54], suggesting that VAII might exert non-essential functions and compensate in part for the lack of VAI in VAI-mutated adenovirus.
3.2.1. VAI and PKR
Early observations showed that protein synthesis is decreased in cells infected with adenovirus lacking VAI as compared to wild type adenovirus, suggesting an active role of VAI in promoting translation [54]. This effect was associated with the phosphorylation of the α-subunit of the eukaryotic translation initiation factor 2 (eIF2α) [58], [59]. When eIF2α is phosphorylated, it has increased affinity for the eIF-2 subunit β, which prevents recharging of the initiation complex with GTP and results in translational inhibition. Later, protein kinase R (PKR) was identified as the kinase responsible for eIF2α phosphorylation and the subsequent arrest in translation [56], [60]. Induced by interferon, PKR is a serine-threonine kinase that becomes activated by dsRNA, a common intermediate of viral replication, and acts as a major component of the cellular anti-viral pathway. It is through the inhibition of PKR that VAI eliminates the block in the translational machinery and thus allows the efficient production of viral proteins. As previously described, VA RNA structure can be divided into three major domains: apical stem, central domain and terminal domain (Fig. 1B). The apical stem–loop is required for binding to PKR and the central domain is involved in the inhibition of PKR activation [28], [46], [47], [48], [49], [61]. The terminal stem is not directly involved in PKR inhibition, but is essential for binding to Exp 5, which mediates VA transport from the nucleus to the cytoplasm, where PKR is localized [62].
3.2.2. VA RNA as a silencing suppressor
Since the silencing suppressor function of VA RNAs is intimately related to VA RNA biogenesis, a detailed description is provided here. VA RNAs are transcribed by RNA polymerase III in the nucleus of infected cells [45]. VAI synthesis starts during the early phase of infection and rapidly accelerates during late phase. As a result, VAI molecules accumulate to very high level (108 molecules/cell) [63], [64]. VAII is also synthesized at high levels in the late phase but accumulates at lower concentration than VAI (107 molecules/cell). This lower expression is not surprising since VAI promoter is stronger than VAII promoter. In fact VAI and VAII are expressed to similar levels when both VA genes are placed under the same promoter [41]. On the other hand, VAII can increase its concentration when VAI is not transcribed suggesting transcriptional competition between both RNAs [55]. VA RNA transcription initiation is under the control of two internal regions named box A and box B. Transcriptional termination occurs in an A-rich region that is copied into a terminal U-tail on the viral transcripts [65]. These regions are highly conserved across several serotypes.
VA RNAs have a stem–loop structure similar to that of pre-miRNAs and are subjected to a similar processing. Both pre-miRNAs and VA RNAs are exported to the cytosol by interaction with Exp5 (Fig. 2 ). Specifically, Exp5 interacts with the terminal stem of VAI, and binding requires VA double-stranded stem (> 14 nucleotides), a base-paired 5′ end and a 3–8-nucleotide protruding 3´end [62]. Binding of VAI to Exp5 is very efficient and in fact, VAI expressed from a plasmid is able to bind and saturate Exp5 [42]. This saturation leads to the inhibition of the transport to the cytoplasm of pre-miRNAs and siRNA precursors called shRNAs. Thus, RNAi is blocked upon VAI expression. Silencing inhibition can be partially rescued by over-expression of Exp5, indicating that VAI inhibition is partly due to competition for Exp5 [42]. It is unclear whether VAII expression can also saturate Exp5 or whether saturation of Exp5 also occurs in the context of adenoviral infection. It is probable that VA RNA also blocks Exp 5 in infected cells because expression levels of VAI are higher in infected than in transfected cells. Furthermore, silencing is also blocked in adenoviral-infected cells [41].
Fig. 2.
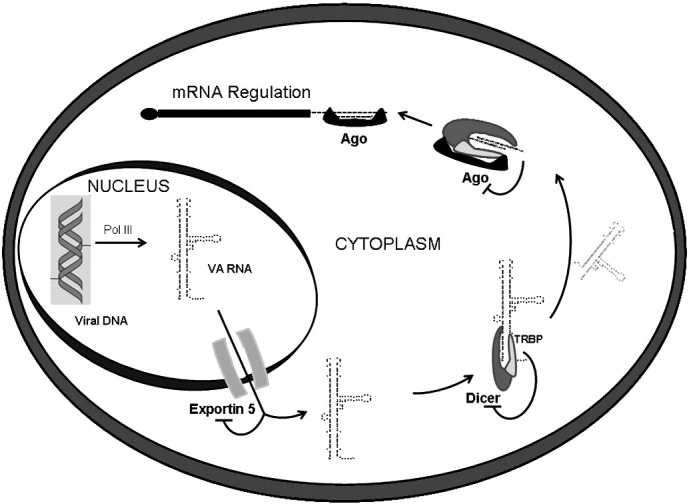
Schematic of mivaRNA biogenesis. VA RNA is transcribed from the viral genome by cellular polymerase (pol) III and is transported to the cytoplasm by Exp5. There, VA RNA binds Dicer and is processed to mivaRNAs able to bind Ago and regulate the expression of target genes. VA RNA is expressed to high levels and saturates Exp5, Dicer and Ago.
There are additional molecular mechanisms that mediate silencing inhibition in the presence of VA RNAs. VA RNAs can bind and block Dicer activity both in vivo and in vitro [42]. This has been clearly shown in infected cells or in cells transfected with plasmids that express either VAI or VAII RNAs. In agreement with this, extracts obtained from cells infected with wild-type adenovirus are not competent for Dicer activity in vitro, whereas normal activity is observed in extracts from cells infected with a mutant that does not express VA RNAs. Deletion of VAII alone leads to Dicer-inactive extracts, while extracts obtained with viruses deleted in VAI show partial Dicer activity. Interestingly, Dicer activity is restored when an excess of dsRNA is used for the reaction. These data suggest that in infected cells VAI, and to a lesser extent VAII, can act as RNA decoys that bind Dicer and sequester it, preventing Dicer from binding to cellular pre-miRNA substrates.
VA RNAs also induce saturation of Ago as an additional mechanism to block RNAi. Ago saturation may require high amounts of VA RNAs, as transfection of a plasmid that express VAI does not lead to inhibition of siRNA-mediated silencing, indicating that Ago is not saturated under these circumstances [42]. However, extracts obtained at 16 hours post-infection, when VA RNAs can be detected in the cytoplasm, showed a reduction in RISC activity in vitro [41]. Ago saturation is not due to a direct effect of VA RNAs. Instead, Ago becomes saturated due to the high level of viral-derived miRNAs generated by Dicer processing of VA RNAs.
4. Viral miRNAs produced by adenovirus
As mentioned above, VA RNAs are able to bind and saturate Dicer. Dicer is an RNase capable of processing stem–loop dsRNAs such as shRNAs or pre-miRNAs, into siRNAs or miRNAs, respectively. Dicer recognizes free ends and cuts the duplex at a distance of approximately 22 nucleotides [66]. Therefore, it was not unexpected to find that VA RNAs are processed by Dicer into smaller dsRNAs named mivaRNAs [41], [67], [68], [69]. VA RNA processing has been described in vitro but also in vivo, in cells that express VA RNAs after transfection or adenoviral infection. Both VAI and VAII are processed, but the VAII processed fragment is more difficult to detect, perhaps due to the lower expression levels of VAII RNA [41]. Only the terminal stem of VA RNA is processed while the apical stem is not affected. The efficiency of Dicer cleavage is mainly determined from the 3′ end of the stem, but the length of the stem and the interaction with the loop also influence Dicer activity. VA structure has a long stem and a large bulge that could negatively affect Dicer processing of VA RNAs. In fact, VA RNA processing by Dicer is not very efficient, since only 2-5% of the total VAI RNA is cleaved [67]. As a result, most of the VAI in the cell is full-length and only a minor amount is processed into mivaRNA. The larger piece that remains after Dicer processing is not detectable, probably because it is unstable. Given that there are 108 molecules of VAI per infected cell, Dicer processing generates over 106 molecules of mivaRNAs/cell. Not only are mivaRNAs abundant, but they are also functional. They can bind the RISC complex and inhibit the expression of luciferase reporter genes with complementary sequences in vivo and in vitro [41], [67]. This is observed in cells transfected with a plasmid that expresses the VA RNAs or in adenovirus-infected cells [67]. mivaRNAs can also decrease the expression of reporter genes whose targets lack perfect complementarity with the mivaRNA, so they can function as miRNAs [68]. Similar to adenoviral VA RNAs, HIV-1 TAR RNA is also inefficiently processed by Dicer to yield viral miRNAs that inhibit the expression of genes with complementary sequences [70], [71], [72], [73]. TAR-derived miRNAs target cellular genes to protect the infected cell from apoptosis [73]. In HIV-1 infected cells, small RNAs have also been described that contain sequences from HIV-1 NEF or Rev Response Element (RRE), or sequences complementary to the primer binding site [71]. The latter small RNA has been shown to bind RISC, control the expression of reporter constructs and modulate HIV replication.
Similar to the miRNAs derived from HIV-1 genome, the sequence of the mivaRNAs has been deciphered using several technologies: primer extension combined with Northern-blot analysis, analysis of target cleavage products, S1 mapping, and sequencing of small RNAs [41], [67], [68], [69]. The latter technique has generated unexpected results. According to sequencing results, the most abundant mivaRNA sequences derive from the 3′ end of VAI and VAII and they were named after their starting position. mivaRI-137 and mivaRI-138 are processed from VAI, and mivaRII-138 is processed from VAII [69]. mivaRNAs were also detected from the 5′ end of VAI but they accumulate to much lower levels than those of the 3′ end. Furthermore, mivaRNAs from the 5′ end are incorporated into RISC with lower efficiencies, but they form RISC complexes with good stability [74]. Cloning and sequencing of cytoplasmic small RNAs in infected cells showed that the amount of small RNAs derived from VAII was twice the amount of those derived from VAI [69]. Since the amount of VAII is 1/20 lower than that of VAI, this result suggests that VAII could be preferentially processed by Dicer. Cloning and sequencing of Ago2 bound miRNAs in adenovirus-infected cells showed that the largest proportion derive from VAII RNA [69]. 60% of the small RNAs bound to Ago2 derived from VAII RNA while only 20% derived from VAI. This suggests that VAII mivaRNAs could be preferentially bound to Ago2. Furthermore, only 10% of the RNAs bound to Ago2 are cellular miRNAs in adenovirus-infected cells, compared to more than 50% in mock infected cells. These results suggest that Ago2, and probably the other cellular Agos, are saturated with viral mivaRNAs. This could explain reduced RISC efficiency in infected cells. Thus, the reduction in RISC effectiveness is not due to a specific inhibition of the enzymatic complex, but likely due to a saturation of RISC with mivaRNAs. By sheer numbers, mivaRNAs would outcompete cellular miRNAs, leading to a reduction in miRNA incorporation into RISC. In summary, inhibition of the RNAi pathway by adenoviral VAs can be defined as the result of a progressive saturation of critical enzymes of the RNAi machinery, including Ago2, Exp5, and Dicer, which induces a competitive displacement of cellular RNAi precursors.
5. Viral miRNAs target cellular genes involved in essential processes
An obvious question is whether the synthesis of adenoviral mivaRNAs is simply a consequence of the VA molecule binding to RNAi pathway enzymes to cause pathway inhibition by saturation, or whether adenovirus could also exploit the RNAi pathway to generate regulatory mivaRNAs that are actively important for viral viability. There are several results supporting the second option. As described earlier, mivaRNAs are functional inhibitors of the expression of genes containing a complementary sequence [41], [67], [68]. Moreover, mivaRNAII is found associated with polyribosomes, suggesting a possible role in the regulation of gene expression at a translational level [69]. Finally, specific inhibition of mivaRNAs with 2-o-methyl (2ome)-modified oligonucleotides affects viral growth [67]. As 2ome oligonucleotides should not affect Ago saturation but miRNA interaction with a target, this result indicates that mivaRNA binding to a target is important for adenovirus viability.
Several strategies have been followed to identify mivaRNA targets. Bioinformatic analysis of the viral genome revealed no potential targets in the adenovirus genome [67], excluding regulation of viral gene expression by their own small RNAs as observed in other viruses such as SV40, Epstein–Barr virus, or Herpes Simplex virus type I [14]. In fact, the nucleotide sequence complementary to the target binding seed sequence of major mivaRNAs is underrepresented in the adenovirus genome, suggesting that the virus has evolved to avoid expression control by mivaRNAs. To identify potential cellular targets of mivaRNAs, genomic and bioinformatic studies were performed [75]. It is known that miRNAs induce mRNA decay, leading to reduced accumulation of target mRNAs. Therefore, the expression pattern of cells expressing VAI and VAII was analyzed by microarray. This analysis allowed the identification of 462 downregulated mRNAs and 637 upregulated mRNAs after VA RNA expression. Genes activated by NFκβ, including the inflammatory cytokines interleukin-6 (IL6) and IL1, were particularly abundant among upregulated mRNAs. This may result from VA RNA activation of cytoplasmic dsRNA-sensor RIG-I, a well-known IFN inducer [76], [77]. Functional analysis of downregulated genes indicated that 10% were related to RNA metabolism, splicing or translation or encoded RNA-binding proteins. The reason for this is unclear. We speculate that expression of proteins that process or bind cellular RNA could be detrimental for adenoviral RNA and VA RNA could downregulate the expression of genes that encode for RNA binding proteins by an unknown mechanism.
In order to identify cellular mRNAs targeted by mivaRNAs, downregulated genes were scanned searching for sequences complementary to the seed sequence of abundant mivaRNAs: mivaRI-137, mivaRI-138 and mivaRII-138 [75]. It is important to notice that only one nucleotide difference in Dicer processing yields mivaRI-137 and mivaRI-138 which contain different seed sequences and consequently can bind different targets. Out of the 462 genes downregulated by expression of VA RNAs, 305 contained in their 3′ UTR a putative target of the abundant mivaRNAs. Fifty of these were studied in more detail. They were not chosen randomly: 22 had good target sites for mivaRI-138, 4 for mivaRI-137, 12 for mivaRII-138 and 12 were relevant genes of known function. Adequate quantitative RT-PCR amplification was obtained for 48 of these genes, and their mRNA levels were studied in cells expressing VA RNAs after plasmid transfection or at different time-points after viral infection. Out of the 48 characterized genes, 30 of them were downregulated in infected or transfected VA RNA expressing cells. If the numbers could be scaled up linearly, out of 305 downregulated genes in the array, as many as 190 cellular genes could be targeted by abundant adenoviral miRNAs. Inhibitions were normally higher in infected cells than in cells transfected with plasmids that express VA RNAs, VAI RNA or mivaRI-138. This could reflect the fact that mivaRNAs levels in infected cells, being higher, saturate the RISC machinery so the complex could be more efficient in silencing mivaRNA target genes. Alternatively, other mechanisms related to viral infection could potentiate the efficacy of the RISC complex. In fact, eight genes were downregulated in VA-expressing transfected cells but not in cells infected with wild-type adenovirus, indicating that they could escape mivaRNA control during infection. Out of the 38 genes regulated by VA RNA, all were decreased by expression of VAI RNA and 31 respond to mivaRI-138 [75]. This was unexpected as Ago is bound preferentially to mivaRII-138 [69]. Also, since overexpression of adenoviral mivaRNAs should saturate RISC, increased levels of RISC-controlled genes might have been expected. However, there was no upregulation of well-characterized miRNA targets. It may be that under our experimental conditions viral miRNA production was not high enough to saturate RISC or did not lead to production of high levels of mivaRII-138.
Adenoviral miRNA-controlled genes can be classified according to their cellular functions [75]. Seven genes are associated to cell signalling (ADCY9, ARHGEF7, MAP3K3, DUSP3, PPP1R3C, PTP4A1 and TGFbR3), nine are related to cell growth and apoptosis (PTP4A1, TGFBR3, LY6K, CCND1, BNIP3L, BNIP3, TIA-1, ETS1 and CDK8), nine are implicated in DNA transcription or DNA repair (ETS1, CDK8, RBPSUH, PHF20, TH1L, PHC2, POLS, EXO1 and UBE2N), and finally, five are involved in RNA metabolism (DAZAP2, MDN1, CUGBP1, QKI and TIA1). It is unknown whether these genes are direct targets of mivaRNAs, with the sole exception being TIA-1, which has been characterized in more detail. mivaRI-138 has been shown to bind directly to the 3′UTR of TIA-1 and decrease TIA-1 mRNA and protein expression.
Several hypotheses could explain an antiviral role for TIA-1. TIA-1 is a well characterized RNA-binding protein that regulates the expression of proapoptotic molecules at the RNA level [78], [79], [80]. Thus, adenovirus infection could proceed favourably with decreased TIA-1 and, therefore, in a less apoptotic environment. Furthermore, TIA-1 is essential for the formation of cytoplasmic stress granules [81] associated with several cellular stresses, including viral infections. Stress granules would act as mRNA hijackers that induce translational arrest. A decrease in TIA-1 levels would inhibit, at least in part, the formation of stress granules, and would overcome the translational arrest that may block viral growth [82], [83], [84]. Finally, TIA-1 binds to U-rich sequences in mRNAs to increase splicing or to block translation [78], [79], [85]. Most adenoviral transcripts have U-rich stretches that could allow TIA-1 binding. E1a pre-mRNA has several U-rich sequences located in regions that can be alternatively spliced (Fig. 1A). In fact, overexpression of TIA-1 changes the alternative splicing of E1a and leads to increased E1a levels (Aparicio et al., unpublished results). Thus, a negative loop of regulation may be in action. At early times of infection TIA-1 levels are generally high and could help to increase E1a expression. Then, E1a would activate transcription of VA RNAs that are processed to mivaRNAs. Subsequently, mivaRI-138 would downregulate TIA-1 leading to decreased E1a levels and to a different alternative splicing of E1a mRNAs at late times post-infection. Thus mivaRI-138 downregulation of TIA-1 could contribute to the shift between the early and the late phases of adenovirus infection.
Similar to TIA-1, expression of other mivaRNA target genes could affect the adenoviral cell cycle. In fact, some of the processes potentially targeted by mivaRNAs such as apoptosis or DNA repair are inhibited in adenovirus-infected cells. However, we have evaluated adenoviral replication in cells overexpressing several mivaRNA target genes without observing a relevant decrease in adenoviral titers in any of the cases tested (unpublished observations). However, we cannot exclude that several of these genes need to be controlled at the same time to have a positive impact on adenoviral growth. Alternatively, regulation of mivaRNA targets may be more relevant in nature than in the favourable conditions for viral growth used in the laboratory. Also, miRNA regulation is not rapid and may be more important in persistent than in lytic infections. In fact, an association has been suggested between VA RNA expression and the ability of adenovirus to establish persistent infections [69].
6. Cellular miRNAs altered by adenoviral infection
Given the negative impact of VA RNA expression on the RNA machinery, adenoviral infection should lead to a decrease in all cellular miRNAs. However, miRNA expression profiling performed in Hep2 cells infected with adenovirus type 3 for 72 hours showed that 44 miRNAs have increased expression and only 36 miRNAs showed decreased expression compared to non-infected controls [86]. This result was not contradictory to previous observations (see above), as it was demonstrated by studying 5 of these miRNAs in more detail. All of these were upregulated at 6 hours post-adenoviral infection, prior to VA RNA accumulation, but all decreased at 72 hours post infection, probably due to VA RNA expression. However, the extent of the decrease was different. Some miRNAs reached levels lower than those in control cells while others did not. Further experiments will be required to validate these results with other adenoviral serotypes and to determine whether the higher accumulation of cellular miRNAs at early times post-infection is relevant for viral viability.
Interestingly, miR-27b and miR-125b were downregulated at late times of adenoviral infection. miR-27b was also found to be downregulated in cells infected with Herpesvirus saimiri and cytomegalovirus [87], [88]. miR-125b was reduced after human papillomavirus infection (HPV) and exogenous expression of miR-125b markedly inhibited HPV DNA synthesis [89]. Thus, miR-27b and miR-125b could have a general antiviral role.
Expression of miR-27a was also downregulated in infected cells. Indeed, not only miR-27a but also miR-520h, miR-7b and miR-197 were downregulated upon expression of E1a in cancer cells [90]. Since overexpression of miR-520h led to invasion and metastasis of cancer cells, one may speculate that E1a downregulation of miR-520h is linked to the tumor suppressing activities of E1a [90].
7. Adenoviral vectors and miRNAs: Looking towards the RNAi pathway for better gene therapy
Adenoviruses have been widely used as vectors for gene therapy. Adenoviruses present several advantages for use as therapeutic vectors: well-established methods for manipulation and propagation, episomal location in infected cells, capacity to accommodate large transgenes and ability to infect non-dividing cells [91]. Transduction with adenoviral vectors allows efficient expression of therapeutic genes in different organs, especially in the liver and to a lesser extent in the lung and brain. Initial vectors, the so-called first-generation adenoviral vectors, lacked E1 protein and induced high immune responses, caused, in part, by type I IFN activation of VA RNAs [76], [77]. Therefore, first-generation adenoviral vectors allow only short-term expression of the transgene. In order to overcome this inconvenience, more recent strategies include the use of “gutless” or helper-dependent (HD) vectors. These vectors lack all viral genes thus avoiding cellular immune responses, have a high capacity for accommodation of transgene elements and maintain long-term expression of their genetic material. Conditionally replicating adenoviruses (CRAd) have also been developed to preferentially destroy tumor cells. Among CRAd oncolytic adenovirus, dl1520/ ONYX-015 contains deletions in the E1b gene and Ad-D24 or ONYX-411 lack a portion of E1a gene which restrict viral growth in non-tumor cells. These replication-competent adenoviruses can also accommodate therapeutic transgenes. A clever strategy to increase the oncolytic potential of adenovirus is the use of VA RNA-deleted adenoviruses. Such viruses can selectively grow in cells with inactive PKR, such as tumor cells with activated Ras or Epstein–Barr virus (EBV) positive tumors in which PKR is blocked by expression of EBV RNAs similar to VA RNAs [92], [93], [94]. Finally, adenoviral vectors have also been used as vaccines [95]. Adenoviral-based vaccines expressing VA RNAs may exploit VA activation of type I IFN to act as an adjuvant and boost the immune response against the desired antigen [76], [77]. This function of VA RNA is independent of VA RNA influence on the RNAi machinery. However, VA RNAs could affect the RNAi machinery when they are expressed from adenoviral vectors.
CRAd and first generation adenoviral vectors contain VA genes, and as stated previously, VA RNA expression should saturate the RNAi machinery, leading to the loss of regulation exerted by cellular miRNAs. This should have toxic consequences in the transduced cell. The saturation effect should be even more prominent if the vector is overexpressing shRNA as a transgene. In fact, overexpression of shRNAs from adenoassociated viral vectors leads to lethality in mice by saturation of the cellular RNAi machinery [96]. However, shRNAs delivered from first-generation adenoviral vectors efficiently downregulated the expression of the target Abcc2 gene in mice without detectable impact on the accumulation or functionality of cellular miRNAs [97]. The lack of toxicity in this case could be explained by two reasons. First, cellular miRNAs are very stable, and only a long-term saturation of the silencing machinery by overexpression of shRNAs or by VA RNAs could disrupt miRNA-mediated regulation. However, first generation adenoviral vectors only express transgenes for short-term [97]. Second, from first-generation adenoviral vectors, VA RNA levels do not reach saturating levels since these vectors lack E1a, which increases transcription of VA RNAs and allows replication of the viral genome, providing new DNA copies for VA RNA expression.
Even if VA RNAs expressed from adenoviral vectors do not appear to interfere with the processing and functionality of therapeutic shRNA, the removal of VA genes would eliminate the possibility of a sequestering of RNAi machinery and this might increase shRNA potency. Elimination of VA sequences together with all other viral genes results in HD vectors, able to express transgenes over long periods of time. In one example, shRNA expression from HD vectors did not saturate the RNAi pathway, but did cause activation of the interferon response [98]. This may be of interest as an adjuvant for the treatment of viral infections such as hepatitis B or hepatitis C virus (HBV or HCV). There are other examples of HD vectors expressing functional shRNAs without detectable unwanted secondary effects, such as those targeting type I collagen for cartilage formation, SREBP-1 for type 2 diabetes, HBV, or huntingtin for Huntington disease [99], [100], [101], [102]. Several oncolytic vectors expressing shRNAs have also been successful at silencing oncogenic genes such as K-Ras, angiogenic factors VEGF or IL-8, antiapoptotic genes Apollon, Survivin, XIAP or Bcl2, proliferation inducers such as Ki67 or telomerases such as hTert [103]. Unfortunately in these reports, there is no information about the functionality of the RNAi machinery in cells infected with these vectors.
Interestingly, the RNAi pathway has been used to modulate viral tropism in oncolytic vectors. An oncolytic adenovirus was designed with p53-dependent expression of miRNAs that target adenoviral genes [104]. When the vector transduced p53-negative tumors, the virus could replicate and exert its oncolytic abilities. Oncolytic adenoviruses may be toxic since they also reach healthy tissues, especially the liver. However, with the modified oncolytic adenovirus, p53 expression in the liver or other organs activated production of miRNAs that target adenoviral genes, therefore suppressing viral replication and infection. Thus, the vector turns into a liver-safe oncolytic virus. A second approach to modulating viral tropism and decreasing the toxicity of oncolytic vectors was to exploit their regulation by cellular miRNAs. Oncolytic viruses were constructed with miR-122 binding sites in the 3´UTR of E1a [105], [106]. In the liver, where high levels of miR-122 are expressed, adenoviral replication was abrogated. However, the oncolytic potential was retained in tumor cells. In summary, miRNA-specific control of wild type virus provides a new and attractive strategy for designing safer gene therapy vectors.
8. Concluding remarks
We are beginning to understand and to better exploit the relationship between adenoviral infection and the RNAi machinery. Adenoviruses have evolved to simultaneously both use and block this process. Expression and processing of high amounts of VA RNAs and mivaRNAs may lead to saturation of the pathway and, therefore, to the blocking of cellular miRNA control. At the same time, key enzymes of the RNAi machinery are loaded with non-coding viral RNAs, so they can efficiently control the expression of viral miRNA target genes. The main question that should still be addressed is: what is the benefit for the virus of such a delicate equilibrium? Several authors have shown that the silencing machinery may have an antiviral role. Theoretically, replication intermediates of RNA viruses or abundant secondary structures of viral RNAs could be cleaved by Dicer to generate antiviral miRNAs. In fact, blocking Dicer function with silencing suppressors or specific shRNAs results in increased adenoviral titers ([107], Carnero et al., unpublished results). However, the antiviral role of RNAi has not been clearly demonstrated for animal viruses. Dicer processing of viral sequences to generate antiviral miRNAs has never been shown in viral-infected animal cells. Future experiments should also address the functional relevance of mivaRNAs. Viability should be studied in adenoviral versions that produce functional VA RNAs processed to mivaRNAs that are mutated in the seed sequence. Viral viability should also be studied in cells that overexpress or downregulate mivaRNA target genes in different environments. Animal models could be used to perform these experiments as it has been recently shown that Syrian hamsters are partially permissive for adenoviral replication, which is impeded in murine cells [108], [109]. Finally, special attention should be paid to the putative role of cellular miRNAs in viral infection. Taken together, all of these approaches should lead to the identification and validation of proteins and miRNAs with anti-adenoviral properties, which may also have relevance to other viruses of therapeutic interest. Moreover, these studies should allow the development of recombinant adenoviruses with increased efficacy for gene therapy applications.
Acknowledgments
We would like to thank Cristian Smerdou and Ruben Hernández-Alcoceba for critically reading of the manuscript and Paul Miller for English editorial work. The work in Fortes laboratory was supported by MICINN (BIO2006-13225 and 2009/09295), through the “UTE project CIMA” and by the project RNAREG (CSD2009-00080), funded by the Ministry of Science and Innovation under the programme CONSOLIDER INGENIO 2010.
Footnotes
This article is part of a "Special Issue entitled:MicroRNAs in viral gene regulation".
References
- 1.Fabian M.R., Sonenberg N., Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 2.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Tomari Y., Zamore P.D. Perspective: machines for RNAi. Genes Dev. 2005;19:517–529. doi: 10.1101/gad.1284105. [DOI] [PubMed] [Google Scholar]
- 4.Kim V.N. MicroRNA biogenesis: coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 5.Kim V.N., Han J., Siomi M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 6.Medina P.P., Slack F.J. microRNAs and cancer: an overview. Cell Cycle. 2008;7:2485–2492. doi: 10.4161/cc.7.16.6453. [DOI] [PubMed] [Google Scholar]
- 7.Dykxhoorn D.M., Lieberman J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu. Rev. Med. 2005;56:401–423. doi: 10.1146/annurev.med.56.082103.104606. [DOI] [PubMed] [Google Scholar]
- 8.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 9.Zhang C. Novel functions for small RNA molecules. Curr. Opin. Mol. Ther. 2009;11:641–651. [PMC free article] [PubMed] [Google Scholar]
- 10.Leung A.K., Sharp P.A. MicroRNA functions in stress responses. Mol. Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newman M.A., Hammond S.M. Emerging paradigms of regulated microRNA processing. Genes Dev. 2010;24:1086–1092. doi: 10.1101/gad.1919710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cullen B.R. Five questions about viruses and microRNAs. PLoS Pathog. 2010;6:e1000787. doi: 10.1371/journal.ppat.1000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skalsky R.L., Cullen B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010;64:123–141. doi: 10.1146/annurev.micro.112408.134243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Umbach J.L., Cullen B.R. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009;23:1151–1164. doi: 10.1101/gad.1793309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cullen B.R. Viral and cellular messenger RNA targets of viral microRNAs. Nature. 2009;457:421–425. doi: 10.1038/nature07757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottwein E., Cullen B.R. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe. 2008;3:375–387. doi: 10.1016/j.chom.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Randall G., Panis M., Cooper J.D., Tellinghuisen T.L., Sukhodolets K.E. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L.M., Hu Z.B., Zhou Z.X., Chen X., Liu F.Y. Serum microRNA profiles serve as novel biomarkers for HBV infection and diagnosis of HBV-positive hepatocarcinoma. Cancer Res. 2010;70:9798–9807. doi: 10.1158/0008-5472.CAN-10-1001. [DOI] [PubMed] [Google Scholar]
- 19.Cameron J.E., Fewell C., Yin Q., McBride J., Wang X. Epstein–Barr virus growth/latency III program alters cellular microRNA expression. Virology. 2008;382:257–266. doi: 10.1016/j.virol.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeung M.L., Bennasser Y., Myers T.G., Jiang G., Benkirane M. Changes in microRNA expression profiles in HIV-1-transfected human cells. Retrovirology. 2005;2:81. doi: 10.1186/1742-4690-2-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y., Wang F., Xu J., Ye F., Shen Y. Progressive miRNA expression profiles in cervical carcinogenesis and identification of HPV-related target genes for miR-29. J. Pathol. 2011 doi: 10.1002/path.2873. [DOI] [PubMed] [Google Scholar]
- 22.Bouzar A.B., Willems L. How HTLV-1 may subvert miRNAs for persistence and transformation. Retrovirology. 2008;5:101. doi: 10.1186/1742-4690-5-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ginsberg H.S. The life and times of adenoviruses. Adv. Virus Res. 1999;54:1–13. doi: 10.1016/s0065-3527(08)60363-2. [DOI] [PubMed] [Google Scholar]
- 24.Shenk T. Adenoviridae: The viruses and their replication; Philadelphia: 1996. Raven publishers editor. [Google Scholar]
- 25.Chinnadurai G. Opposing oncogenic activities of small DNA tumor virus transforming proteins. Trends Microbiol. 2011;19:174–183. doi: 10.1016/j.tim.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Estmer Nilsson C., Petersen-Mahrt S., Durot C., Shtrichman R., Krainer A.R. The adenovirus E4-ORF4 splicing enhancer protein interacts with a subset of phosphorylated SR proteins. EMBO J. 2001;20:864–871. doi: 10.1093/emboj/20.4.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stracker T.H., Carson C.T., Weitzman M.D. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 28.Ghadge G.D., Swaminathan S., Katze M.G., Thimmapaya B. Binding of the adenovirus VAI RNA to the interferon-induced 68-kDa protein kinase correlates with function. Proc. Natl. Acad. Sci. U.S.A. 1991;88:7140–7144. doi: 10.1073/pnas.88.16.7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pilder S., Moore M., Logan J., Shenk T. The adenovirus E1B-55K transforming polypeptide modulates transport or cytoplasmic stabilization of viral and host cell mRNAs. Mol. Cell. Biol. 1986;6:470–476. doi: 10.1128/mcb.6.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xi Q., Cuesta R., Schneider R.J. Tethering of eIF4G to adenoviral mRNAs by viral 100k protein drives ribosome shunting. Genes Dev. 2004;18:1997–2009. doi: 10.1101/gad.1212504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lecellier C.H., Dunoyer P., Arar K., Lehmann-Che J., Eyquem S. A cellular microRNA mediates antiviral defense in human cells. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- 32.Bennasser Y., Le S.Y., Benkirane M., Jeang K.T. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity. 2005;22:607–619. doi: 10.1016/j.immuni.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Bucher E., Hemmes H., de Haan P., Goldbach R., Prins M. The influenza A virus NS1 protein binds small interfering RNAs and suppresses RNA silencing in plants. J. Gen. Virol. 2004;85:983–991. doi: 10.1099/vir.0.19734-0. [DOI] [PubMed] [Google Scholar]
- 34.Li W.X., Li H., Lu R., Li F., Dus M. Interferon antagonist proteins of influenza and vaccinia viruses are suppressors of RNA silencing. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1350–1355. doi: 10.1073/pnas.0308308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y., Kato N., Jazag A., Dharel N., Otsuka M. Hepatitis C virus core protein is a potent inhibitor of RNA silencing-based antiviral response. Gastroenterology. 2006;130:883–892. doi: 10.1053/j.gastro.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 36.Haasnoot J., de Vries W., Geutjes E.J., Prins M., de Haan P. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 2007;3:e86. doi: 10.1371/journal.ppat.0030086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fabozzi G., Nabel C.S., Dolan M.A., Sullivan N.J. Ebolavirus proteins suppress the effects of small interfering RNA by direct interaction with the mammalian RNA interference pathway. J. Virol. 2011;85:2512–2523. doi: 10.1128/JVI.01160-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abe M., Suzuki H., Nishitsuji H., Shida H., Takaku H. Interaction of human T-cell lymphotropic virus type I Rex protein with Dicer suppresses RNAi silencing. FEBS Lett. 2010;584:4313–4318. doi: 10.1016/j.febslet.2010.09.031. [DOI] [PubMed] [Google Scholar]
- 39.Karjee S., Minhas A., Sood V., Ponia S.S., Banerjea A.C. The 7a accessory protein of severe acute respiratory syndrome coronavirus acts as an RNA silencing suppressor. J. Virol. 2010;84:10395–10401. doi: 10.1128/JVI.00748-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennasser Y., Yeung M.L., Jeang K.T. HIV-1 TAR RNA subverts RNA interference in transfected cells through sequestration of TAR RNA-binding protein, TRBP. J. Biol. Chem. 2006;281:27674–27678. doi: 10.1074/jbc.C600072200. [DOI] [PubMed] [Google Scholar]
- 41.Andersson M.G., Haasnoot P.C., Xu N., Berenjian S., Berkhout B. Suppression of RNA interference by adenovirus virus-associated RNA. J. Virol. 2005;79:9556–9565. doi: 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu S., Cullen B.R. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 2004;78:12868–12876. doi: 10.1128/JVI.78.23.12868-12876.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akusjarvi G., Mathews M.B., Andersson P., Vennstrom B., Pettersson U. Structure of genes for virus-associated RNAI and RNAII of adenovirus type 2. Proc. Natl. Acad. Sci. U.S.A. 1980;77:2424–2428. doi: 10.1073/pnas.77.5.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma Y., Mathews M.B. Comparative analysis of the structure and function of adenovirus virus-associated RNAs. J. Virol. 1993;67:6605–6617. doi: 10.1128/jvi.67.11.6605-6617.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma Y., Mathews M.B. Structure, function, and evolution of adenovirus-associated RNA: a phylogenetic approach. J. Virol. 1996;70:5083–5099. doi: 10.1128/jvi.70.8.5083-5099.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mellits K.H., Mathews M.B. Effects of mutations in stem and loop regions on the structure and function of adenovirus VA RNAI. EMBO J. 1988;7:2849–2859. doi: 10.1002/j.1460-2075.1988.tb03141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mellits K.H., Pe'ery T., Mathews M.B. Role of the apical stem in maintaining the structure and function of adenovirus virus-associated RNA. J. Virol. 1992;66:2369–2377. doi: 10.1128/jvi.66.4.2369-2377.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clarke P.A., Pe'ery T., Ma Y., Mathews M.B. Structural features of adenovirus 2 virus-associated RNA required for binding to the protein kinase DAI. Nucleic Acids Res. 1994;22:4364–4374. doi: 10.1093/nar/22.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furtado M.R., Subramanian S., Bhat R.A., Fowlkes D.M., Safer B. Functional dissection of adenovirus VAI RNA. J. Virol. 1989;63:3423–3434. doi: 10.1128/jvi.63.8.3423-3434.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pe'ery T., Mellits K.H., Mathews M.B. Mutational analysis of the central domain of adenovirus virus-associated RNA mandates a revision of the proposed secondary structure. J. Virol. 1993;67:3534–3543. doi: 10.1128/jvi.67.6.3534-3543.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhat R.A., Domer P.H., Thimmappaya B. Structural requirements of adenovirus VAI RNA for its translation enhancement function. Mol. Cell. Biol. 1985;5:187–196. doi: 10.1128/mcb.5.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rahman A., Malhotra P., Dhar R., Kewalramani T., Thimmapaya B. Effect of single-base substitutions in the central domain of virus-associated RNA I on its function. J. Virol. 1995;69:4299–4307. doi: 10.1128/jvi.69.7.4299-4307.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wahid A.M., Coventry V.K., Conn G.L. The PKR-binding domain of adenovirus VA RNAI exists as a mixture of two functionally non-equivalent structures. Nucleic Acids Res. 2009;37:5830–5837. doi: 10.1093/nar/gkp595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thimmappaya B., Weinberger C., Schneider R.J., Shenk T. Adenovirus VAI RNA is required for efficient translation of viral mRNAs at late times after infection. Cell. 1982;31:543–551. doi: 10.1016/0092-8674(82)90310-5. [DOI] [PubMed] [Google Scholar]
- 55.Bhat R.A., Thimmappaya B. Adenovirus mutants with DNA sequence perturbations in the intragenic promoter of VAI RNA gene allow the enhanced transcription of VAII RNA gene in HeLa cells. Nucleic Acids Res. 1984;12:7377–7388. doi: 10.1093/nar/12.19.7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kitajewski J., Schneider R.J., Safer B., Munemitsu S.M., Samuel C.E. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell. 1986;45:195–200. doi: 10.1016/0092-8674(86)90383-1. [DOI] [PubMed] [Google Scholar]
- 57.Bhat R.A., Thimmappaya B. Construction and analysis of additional adenovirus substitution mutants confirm the complementation of VAI RNA function by two small RNAs encoded by Epstein–Barr virus. J. Virol. 1985;56:750–756. doi: 10.1128/jvi.56.3.750-756.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levin D.H., Petryshyn R., London I.M. Characterization of double-stranded-RNA-activated kinase that phosphorylates alpha subunit of eukaryotic initiation factor 2 (eIF-2 alpha) in reticulocyte lysates. Proc. Natl. Acad. Sci. U.S.A. 1980;77:832–836. doi: 10.1073/pnas.77.2.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petryshyn R., Levin D.H., London I.M. Double-stranded RNA-dependent eIF-2alpha protein kinase. Methods Enzymol. 1983;99:346–362. doi: 10.1016/0076-6879(83)99070-5. [DOI] [PubMed] [Google Scholar]
- 60.O'Malley R.P., Mariano T.M., Siekierka J., Mathews M.B. A mechanism for the control of protein synthesis by adenovirus VA RNAI. Cell. 1986;44:391–400. doi: 10.1016/0092-8674(86)90460-5. [DOI] [PubMed] [Google Scholar]
- 61.McKenna S.A., Kim I., Liu C.W., Puglisi J.D. Uncoupling of RNA binding and PKR kinase activation by viral inhibitor RNAs. J. Mol. Biol. 2006;358:1270–1285. doi: 10.1016/j.jmb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 62.Gwizdek C., Ossareh-Nazari B., Brownawell A.M., Doglio A., Bertrand E. Exportin-5 mediates nuclear export of minihelix-containing RNAs. J. Biol. Chem. 2003;278:5505–5508. doi: 10.1074/jbc.C200668200. [DOI] [PubMed] [Google Scholar]
- 63.Reich P.R., Forget B.G., Weissman S.M. RNA of low molecular weight in KB cells infected with adenovirus type 2. J. Mol. Biol. 1966;17:428–439. doi: 10.1016/s0022-2836(66)80153-5. [DOI] [PubMed] [Google Scholar]
- 64.Soderlund H., Pettersson U., Vennstrom B., Philipson L., Mathews M.B. A new species of virus-coded low molecular weight RNA from cells infected with adenovirus type 2. Cell. 1976;7:585–593. doi: 10.1016/0092-8674(76)90209-9. [DOI] [PubMed] [Google Scholar]
- 65.Fowlkes D.M., Shenk T. Transcriptional control regions of the adenovirus VAI RNA gene. Cell. 1980;22:405–413. doi: 10.1016/0092-8674(80)90351-7. [DOI] [PubMed] [Google Scholar]
- 66.Zhang H., Kolb F.A., Jaskiewicz L., Westhof E., Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118:57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 67.Aparicio O., Razquin N., Zaratiegui M., Narvaiza I., Fortes P. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J. Virol. 2006;80:1376–1384. doi: 10.1128/JVI.80.3.1376-1384.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sano M., Kato Y., Taira K. Sequence-specific interference by small RNAs derived from adenovirus VAI RNA. FEBS Lett. 2006;580:1553–1564. doi: 10.1016/j.febslet.2006.01.085. [DOI] [PubMed] [Google Scholar]
- 69.Xu N., Segerman B., Zhou X., Akusjarvi G. Adenovirus virus-associated RNAII-derived small RNAs are efficiently incorporated into the rna-induced silencing complex and associate with polyribosomes. J. Virol. 2007;81:10540–10549. doi: 10.1128/JVI.00885-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ouellet D.L., Plante I., Landry P., Barat C., Janelle M.E. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucleic Acids Res. 2008;36:2353–2365. doi: 10.1093/nar/gkn076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yeung M.L., Bennasser Y., Watashi K., Le S.Y., Houzet L. Pyrosequencing of small non-coding RNAs in HIV-1 infected cells: evidence for the processing of a viral-cellular double-stranded RNA hybrid. Nucleic Acids Res. 2009;37:6575–6586. doi: 10.1093/nar/gkp707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klase Z., Kale P., Winograd R., Gupta M.V., Heydarian M. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol. Biol. 2007;8:63. doi: 10.1186/1471-2199-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Klase Z., Winograd R., Davis J., Carpio L., Hildreth R. HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression. Retrovirology. 2009;6:18. doi: 10.1186/1742-4690-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu N., Gkountela S., Saeed K., Akusjarvi G. The 5'-end heterogeneity of adenovirus virus-associated RNAI contributes to the asymmetric guide strand incorporation into the RNA-induced silencing complex. Nucleic Acids Res. 2009;37:6950–6959. doi: 10.1093/nar/gkp764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aparicio O., Carnero E., Abad X., Razquin N., Guruceaga E. Adenovirus VA RNA-derived miRNAs target cellular genes involved in cell growth, gene expression and DNA repair. Nucleic Acids Res. 2010;38:750–763. doi: 10.1093/nar/gkp1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Minamitani T., Iwakiri D., Takada K. Adenovirus virus-associated RNAs induce type I interferon expression through a RIG-I-mediated pathway. J. Virol. 2011;85:4035–4040. doi: 10.1128/JVI.02160-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamaguchi T., Kawabata K., Kouyama E., Ishii K.J., Katayama K. Induction of type I interferon by adenovirus-encoded small RNAs. Proc. Natl. Acad. Sci. U.S.A. 2010;107:17286–17291. doi: 10.1073/pnas.1009823107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Forch P., Puig O., Kedersha N., Martinez C., Granneman S. The apoptosis-promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol. Cell. 2000;6:1089–1098. doi: 10.1016/s1097-2765(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 79.Forch P., Puig O., Martinez C., Seraphin B., Valcarcel J. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5' splice sites. EMBO J. 2002;21:6882–6892. doi: 10.1093/emboj/cdf668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Forch P., Valcarcel J. Molecular mechanisms of gene expression regulation by the apoptosis-promoting protein TIA-1. Apoptosis. 2001;6:463–468. doi: 10.1023/a:1012441824719. [DOI] [PubMed] [Google Scholar]
- 81.Gilks N., Kedersha N., Ayodele M., Shen L., Stoecklin G. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell. 2004;15:5383–5398. doi: 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McInerney G.M., Kedersha N.L., Kaufman R.J., Anderson P., Liljestrom P. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell. 2005;16:3753–3763. doi: 10.1091/mbc.E05-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Emara M.M., Brinton M.A. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. U.S.A. 2007;104:9041–9046. doi: 10.1073/pnas.0703348104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.White J.P., Cardenas A.M., Marissen W.E., Lloyd R.E. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe. 2007;2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 85.Le Guiner C., Lejeune F., Galiana D., Kister L., Breathnach R. TIA-1 and TIAR activate splicing of alternative exons with weak 5' splice sites followed by a U-rich stretch on their own pre-mRNAs. J. Biol. Chem. 2001;276:40638–40646. doi: 10.1074/jbc.M105642200. [DOI] [PubMed] [Google Scholar]
- 86.Qi Y., Tu J., Cui L., Guo X., Shi Z. High-throughput sequencing of microRNAs in adenovirus type 3 infected human laryngeal epithelial cells. J. Biomed. Biotechnol. 2010;2010:915980. doi: 10.1155/2010/915980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cazalla D., Yario T., Steitz J.A. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science. 2010;328:1563–1566. doi: 10.1126/science.1187197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Buck A.H., Perot J., Chisholm M.A., Kumar D.S., Tuddenham L. Post-transcriptional regulation of miR-27 in murine cytomegalovirus infection. RNA. 2010;16:307–315. doi: 10.1261/rna.1819210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nuovo G.J., Wu X., Volinia S., Yan F., di Leva G. Strong inverse correlation between microRNA-125b and human papillomavirus DNA in productive infection. Diagn. Mol. Pathol. 2010;19:135–143. doi: 10.1097/PDM.0b013e3181c4daaa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Su J.L., Chen P.B., Chen Y.H., Chen S.C., Chang Y.W. Downregulation of microRNA miR-520h by E1A contributes to anticancer activity. Cancer Res. 2010;70:5096–5108. doi: 10.1158/0008-5472.CAN-09-4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Campos S.K., Barry M.A. Current advances and future challenges in Adenoviral vector biology and targeting. Curr. Gene Ther. 2007;7:189–204. doi: 10.2174/156652307780859062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cascallo M., Gros A., Bayo N., Serrano T., Capella G. Deletion of VAI and VAII RNA genes in the design of oncolytic adenoviruses. Hum. Gene Ther. 2006;17:929–940. doi: 10.1089/hum.2006.17.929. [DOI] [PubMed] [Google Scholar]
- 93.Cascallo M., Capella G., Mazo A., Alemany R. Ras-dependent oncolysis with an adenovirus VAI mutant. Cancer Res. 2003;63:5544–5550. [PubMed] [Google Scholar]
- 94.Wang Y., Xue S.A., Hallden G., Francis J., Yuan M. Virus-associated RNA I-deleted adenovirus, a potential oncolytic agent targeting EBV-associated tumors. Cancer Res. 2005;65:1523–1531. doi: 10.1158/0008-5472.CAN-04-3113. [DOI] [PubMed] [Google Scholar]
- 95.Lasaro M.O., Ertl H.C. New insights on adenovirus as vaccine vectors. Mol. Ther. 2009;17:1333–1339. doi: 10.1038/mt.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grimm D., Streetz K.L., Jopling C.L., Storm T.A., Pandey K. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 97.Narvaiza I., Aparicio O., Vera M., Razquin N., Bortolanza S. Effect of adenovirus-mediated RNA interference on endogenous microRNAs in a mouse model of multidrug resistance protein 2 gene silencing. J. Virol. 2006;80:12236–12247. doi: 10.1128/JVI.01205-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Witting S.R., Brown M., Saxena R., Nabinger S., Morral N. Helper-dependent adenovirus-mediated short hairpin RNA expression in the liver activates the interferon response. J. Biol. Chem. 2008;283:2120–2128. doi: 10.1074/jbc.M704178200. [DOI] [PubMed] [Google Scholar]
- 99.Zhang F., Yao Y., Hao J., Zhou R., Liu C. A dual-functioning adenoviral vector encoding both transforming growth factor-beta3 and shRNA silencing type I collagen: construction and controlled release for chondrogenesis. J. Control. Release. 2010;142:70–77. doi: 10.1016/j.jconrel.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 100.Ruiz R., Witting S.R., Saxena R., Morral N. Robust hepatic gene silencing for functional studies using helper-dependent adenoviral vectors. Hum. Gene Ther. 2009;20:87–94. doi: 10.1089/hum.2008.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rauschhuber C., Xu H., Salazar F.H., Marion P.L., Ehrhardt A. Exploring gene-deleted adenoviral vectors for delivery of short hairpin RNAs and reduction of hepatitis B virus infection in mice. J. Gene Med. 2008;10:878–889. doi: 10.1002/jgm.1207. [DOI] [PubMed] [Google Scholar]
- 102.Huang B., Schiefer J., Sass C., Landwehrmeyer G.B., Kosinski C.M. High-capacity adenoviral vector-mediated reduction of huntingtin aggregate load in vitro and in vivo. Hum. Gene Ther. 2007;18:303–311. doi: 10.1089/hum.2006.160. [DOI] [PubMed] [Google Scholar]
- 103.Pei D.S., Di J.H., Chen F.F., Zheng J.N. Oncolytic-adenovirus-expressed RNA interference for cancer therapy. Expert Opin. Biol. Ther. 2010;10:1331–1341. doi: 10.1517/14712598.2010.512002. [DOI] [PubMed] [Google Scholar]
- 104.Gurlevik E., Woller N., Schache P., Malek N.P., Wirth T.C. p53-dependent antiviral RNA-interference facilitates tumor-selective viral replication. Nucleic Acids Res. 2009;37:e84. doi: 10.1093/nar/gkp374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ylosmaki E., Hakkarainen T., Hemminki A., Visakorpi T., Andino R. Generation of a conditionally replicating adenovirus based on targeted destruction of E1A mRNA by a cell type-specific MicroRNA. J. Virol. 2008;82:11009–11015. doi: 10.1128/JVI.01608-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cawood R., Chen H.H., Carroll F., Bazan-Peregrino M., van Rooijen N. Use of tissue-specific microRNA to control pathology of wild-type adenovirus without attenuation of its ability to kill cancer cells. PLoS Pathog. 2009;5:e1000440. doi: 10.1371/journal.ppat.1000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.de Vries W., Haasnoot J., van der Velden J., van Montfort T., Zorgdrager F. Increased virus replication in mammalian cells by blocking intracellular innate defense responses. Gene Ther. 2008;15:545–552. doi: 10.1038/gt.2008.12. [DOI] [PubMed] [Google Scholar]
- 108.Thomas M.A., Spencer J.F., La Regina M.C., Dhar D., Tollefson A.E. Syrian hamster as a permissive immunocompetent animal model for the study of oncolytic adenovirus vectors. Cancer Res. 2006;66:1270–1276. doi: 10.1158/0008-5472.CAN-05-3497. [DOI] [PubMed] [Google Scholar]
- 109.Bortolanza S., Alzuguren P., Bunuales M., Qian C., Prieto J. Human adenovirus replicates in immunocompetent models of pancreatic cancer in Syrian hamsters. Hum. Gene Ther. 2007;18:681–690. doi: 10.1089/hum.2007.017. [DOI] [PubMed] [Google Scholar]