

Abstract
Viruses do not carry their own protein biosynthesis machinery and the translation of viral proteins therefore requires that the virus usurps the machinery of the host cell. To allow optimal translation of viral proteins at the expense of cellular proteins, virus families have evolved a variety of methods to repress the host translation machinery, while allowing effective viral protein synthesis. Many viruses use noncanonical mechanisms that permit translation of their own RNAs under these conditions. Viruses have also developed mechanisms to evade host innate immune responses that would repress translation under conditions of viral infection, in particular PKR activation in response to double-stranded RNA (dsRNA). Importantly, the study of viral translation mechanisms has enormously enhanced our understanding of many aspects of the cellular protein biosynthesis pathway and its components. A number of unusual mechanisms of translation initiation that were first discovered in viruses have since been observed in cellular mRNAs, and it has become apparent that a diverse range of translation mechanisms operates in eukaryotes, allowing subtle regulation of this essential process.
Keywords: Virus, Translation, Protein synthesis, PKR, PERK, Upstream open reading frame, IRES, Frameshifting, 4EBP, Reinitiation
I. Introduction
Viruses are obligate intracellular parasites and therefore depend on host cells for their replication. Viruses have evolved a number of ways in which to modify the translation apparatus of the host cell to ensure preferential translation of virus mRNAs, and use alternative and novel mechanisms to initiate translation of viral mRNAs under such conditions. The study of viral translation mechanisms has been a rich source of information about cellular protein synthesis and its regulation.
Viruses are diverse in the ways they interact with the host and replicate. For example, their genomes can be made up of DNA or RNA, and within the RNA viruses the genomes may be positive-sense RNA, negative-sense RNA, or dsRNA. Production of mRNA transcripts may take place in the nucleus and therefore the virus can make use of host cell enzymes. Alternatively, this process may occur in the cytoplasm, in which case the virus must encode, or bring with it, its own transcriptional system. Viral mRNAs also differ in their structure; some viral mRNAs are capped and polyadenylated, and these viruses often adopt novel ways in which to ensure the viral mRNAs are preferentially translated over cellular mRNAs, either through modification of the host cell translational machinery and/or adoption of novel translational mechanisms. There are also a large number of viruses that do not produce capped mRNAs and have evolved novel strategies to direct initiation of protein synthesis.
No virus has been discovered that encodes its own translation system, and in fact this is one of the main distinguishing features between a virus and a living cell. However, the recent discovery of the “giant” virus, Acanthamoeba polyphaga mimivirus1, 2 has challenged this long-held belief. tRNAs have been discovered in a number of such giant viruses and this new mimivirus has also been shown to encode a number of translation factors and four aminoacyl-tRNA synthetases. The virus encodes homologs of eukaryotic initiation factor (eIF)4E, eIF4A, and eIF1 and also possesses a homolog of the release factor eRF1. Although the virus therefore encodes proteins involved in all stages of translation, it does not encode any ribosomal components. It has been speculated2 that these viral components probably represent the remains of a more complex translational system that has been gradually lost over time, rather than an acquisition of cellular components.
In summary, to enable efficient synthesis of viral proteins a virus needs to be able to do one or more of the following: (1) Modify the host translation machinery to favor the translation of viral rather than host encoded mRNAs. Many viral RNAs are uncapped and/or contain highly structured 5′-untranslated regions (UTRs) that would inhibit the scanning ribosome, so the viral RNA would compete poorly with host encoded mRNAs for the translation machinery. (2) Use novel mechanisms that allow the selective synthesis of viral proteins. (3) Circumvent the host defense mechanisms that function to inhibit translation following viral infection.
A detailed molecular understanding of these three processes has the potential to provide unique insights into viral replication strategies and therefore highlight potential new targets for antiviral therapies.
II. Viral Modification of Host Translation Machinery
A. Cap-Dependent Translation
Translation in mammalian cells is a multistep, highly regulated process (see chapter by Fraser, this volume). It can be considered as three phases; initiation, elongation, and termination. All three phases are regulated, although initiation is thought to be the rate-limiting step for the whole process. For initiation to occur the eIF4F complex, comprised of the cap-binding protein eIF4E, the helicase eIF4A, and the bridging protein eIF4G, binds to the mRNA. The 40S ribosomal subunit is recruited via an eIF4G–eIF3–40S interaction together with the ternary complex, which contains eIF2, GTP, and initiator met-tRNAi. The resulting complex is known as the 48S complex. The scanning model of translation initiation predicts that this complex then scans along the mRNA until the start codon is reached.3
The poly(A)-binding protein (PABP) interacts with both the polyA tail and the N-terminal half of eIF4G to circularize the mRNA (4, 5; Fig. 1 ). An interaction between eIF4B, which binds to eIF4A, and PABP further stabilizes this circularization.6 The rate of translation initiation in mammalian cells is also controlled by sequence elements within the 5′- and 3′-UTRs of mRNAs which regulate this process by providing sites for interaction of regulatory proteins and RNAs. These include upstream open reading frames (uORFs), microRNA (miRNA) target sites, and polyadenylation elements.7, 8 Transcription of mRNAs from mammalian DNA virus genomes such as herpesviruses and adenoviruses occur in the nucleus and results in the production of capped viral mRNAs. These viruses need to establish conditions to permit selective translation of viral mRNAs over cellular transcripts. It is therefore desirable to stimulate cap-dependent translation pathways and at the same time inhibit or downregulate the translation of host mRNAs, which otherwise would be similarly stimulated, to provide the virus with a selective advantage.
Fig. 1.
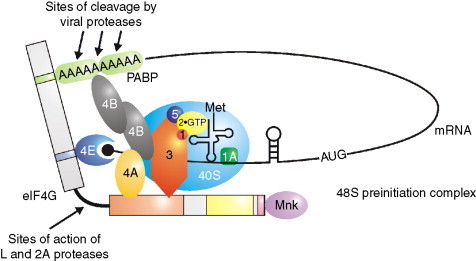
The scanning model of translation initiation. The scanning model of translation initiation predicts that the 48S preinitiation complex moves along the mRNA in a 5′–3′ direction until it encounters an AUG codon that is in a good context. Many viruses produce proteases that cleave protein components of this complex to inhibit cap-dependent scanning. Thus the cleavage of eIF4G dissociates the ribosome binding ability of the complex from cap recognition. The cleavage of PABP by viral proteases will prevent the interaction of the 5′ and 3′ ends of the mRNA and also reduce the stability of the complex.
B. Viral Regulation of 4E-BP1
A major mechanism of regulation of cap-dependent translation is mediated by the eIF4E-binding proteins (4E-BPs), which regulate the formation of the eIF4F complex (Fig. 2 ) (discussed in chapter by Fraser, this volume). There are three 4E-BPs in mammals, all of which act by binding to eIF4E and inhibiting its interaction with eIF4G, leading to an inhibition of cap-dependent translation initiation. Hyperphosphorylation of 4E-BP1 releases eIF4E and allows it to interact with eIF4G in the eIF4F complex. This hyperphosphorylation is stimulated by growth factors via the mammalian target of rapamycin (mTOR) signaling pathway, and is a target of regulation for a number of viruses. How such regulation contributes to viral replication is described below.
Fig. 2.
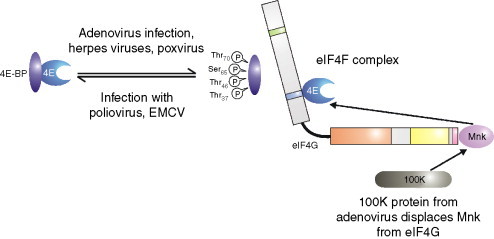
Regulation of eIF4E by 4E-BPs. The availability of eIF4E (the cap-binding protein) for eIF4F complex formation (which also contains eIF4G (the bridging protein)) and eIF4A (a deadbox helicase), is controlled by interaction with its binding partners the 4E-BPs which bind to and sequester eIF4E. The interaction of eIF4E with 4E-BP is regulated by phosphorylation and viral infection controls, either positively or negatively, the phosphorylation status of this protein. When bound to eIF4G, eIF4E can be phosphorylated by Mnk1 and it has been suggested that this may increase the affinity of eIF4E for the cap. The 100 K protein from adenovirus displaces Mnk1 from eIF4G and so prevents the phosphorylation of eIF4E.
1. Herpesviruses
The herpesviruses are a large family of dsDNA viruses and are responsible for a number of different diseases of vertebrates, such as cold sores caused by herpes simplex virus-1 (HSV-1) and chicken pox caused by varicella zoster virus (VZV) in humans. Viruses of the herpesvirus family have adapted to colonize a variety of terminally differentiated cells9 in which translation rates are generally low. These viruses establish latent infections during which a restricted subset of viral mRNAs is expressed at a low level, so that the virus evades the host immune system. The virus replication may undergo periodic reactivation to a productive lytic infectious cycle, accompanied by major changes in viral gene expression. This switch from latent to lytic infection requires the induction of protein synthesis, and 4E-BP modification appears to play an essential role in this process.10
The switch from latent to lytic infection in HSV-1-infected sensory neurons is accompanied by induction of 4E-BP1 phosphorylation. Inactivation of 4E-BP1 in HSV-1-infected cells is increased further by proteasome-dependent degradation, and a decrease in the level of this protein accompanies the increase in phosphorylation.11 The viral ICP0 protein, an important master regulator of lytic reactivation, is required for both these processes. 4E-BP1 phosphorylation during HSV-1 infection is sensitive to the effects of rapamycin, suggesting that virus-induced signaling through mTOR (see chapter by Blenis and Mahoney, this volume) is required for inactivation of this protein.11
Direct regulation of the mTOR pathway occurs in cells infected with Epstein–Barr virus (EBV). The protein product of the LMP-2A gene activates mTOR via PI3 kinase/Akt signaling12 and it has been suggested that the resulting phosphorylation and inactivation of 4E-BP1 may be required to increase translation in the transformed cells.10
For other viruses in this family, such as human cytomegalovirus (hCMV), it would appear that additional mechanisms are also required, as rapamycin is not sufficient to abolish the 4E-BP1 phosphorylation induced in the virus infection.13, 14 Infection of B cells with Kaposi's sarcoma associated herpesvirus (KSHV) also stimulates hyperphosphorylation of 4E-BP1, although complete release of eIF4E from 4E-BP1 is not observed in this case.15
2. Poxviruses
The best known poxviruses are smallpox virus and the smallpox vaccine virus, vaccinia virus. Recently, the effects of poxvirus infection on the eIF4F complex have been studied for the first time. Poxviruses replicate in the cytoplasm of infected cells and manufacture capped viral mRNAs using a viral methyltransferase complex16 and therefore must effectively compete with host mRNAs for the eIF4F complex. It has now been shown that in vaccinia virus-infected cells 4E-BP1 is inactivated through its hyperphosphorylation.17 In addition, the overall amount of 4E-BP1 decreases following vaccinia virus infection, so the virus is able to inactivate this protein through two different mechanisms.
3. Adenoviruses
Adenoviruses (AdV) are also DNA viruses and are widespread in humans and birds. These viruses have been shown to increase the phosphorylation of 4E-BP1, allowing eIF4E to bind to eIF4G and stimulate formation of the eIF4F complex.18 This suggests that the regulation of 4E-BP1 by multiple mechanisms is a common mechanism of regulation of translation initiation used by most DNA viruses.
4. Picornaviruses
The picornaviruses are a large family of positive-sense RNA viruses including important animal and human pathogens such as foot-and mouth-disease virus (FMDV) and poliovirus (PV). The picornaviruses have uncapped mRNAs that are translated by the cap-independent mechanism of internal ribosome entry. Encephalomyocarditis virus (EMCV) and, to a lesser extent, PV, affect 4E-BP activity by increasing the ability of this protein to bind to and sequester eIF4E in order to silence cap-dependent translation of host mRNAs and permit selective translation of EMCV and PV mRNAs.19 Upon infection with EMCV, 4E-BP1 becomes dephosphorylated, and this coincides with the shutoff of protein synthesis that occurs. Dephosphorylation of 4E-BP1 in PV-infected cells lags behind the shutoff of cellular protein synthesis, and it appears that in this situation protein synthesis inhibition is initiated by the cleavage of eIF4G. Further evidence of a role for dephosphorylation of 4E-BP1 in inhibition of protein synthesis during EMCV and PV infections was demonstrated by addition of rapamycin, an inhibitor of 4E-BP phosphorylation, to virus-infected cells. This results in enhanced synthesis of EMCV and PV viral proteins.20
5. Rhabdoviruses
Rhabdoviruses are negative-stranded RNA viruses. Inactivation of 4E-BP1 by dephosphorylation and downregulation of host protein synthesis is also observed in cells infected with vesicular stomatitis virus (VSV), despite the fact that VSV mRNAs are capped.21 However, multiple other factors control the translation of viral mRNA including a relocalization of certain hnRNPs from the nucleus to the cytoplasm.22
C. Other Regulation of eIF4F Assembly
1. Herpesviruses
In addition to their regulation of 4E-BP1, some herpesviruses stimulate the assembly of eIF4F complexes in cells directly using a range of mechanisms.11, 13, 14 For example, it has been shown that the ICP6 protein produced in HSV-1 lytic infection is required for increased eIF4F complex formation and interacts directly with eIF4G, suggesting that it has a chaperone function.10 hCMV infection leads to an increase in the abundance of eIF4E, eIF4G, and PABP, and to enhanced eIF4F assembly.14 Reactivation from latency in KSHV-infected cells also leads to a stimulation of eIF4F assembly. Interestingly, no corresponding increase in the level of PABP association with the eIF4F complex was observed, and PABP was seen to redistribute from the cytoplasm to the nucleus. This is perhaps surprising given the role of PABP in stimulating translation.5 It was suggested that alternative eIF4F complexes lacking PABP could selectively promote the synthesis of viral, but not host, proteins, so that KSHV-encoded mRNAs would compete more effectively for host translation machinery in infected cells.15
2. Poxviruses
Poxvirus infection results in the reorganization of discrete cytoplasmic regions into replication factories. The poxvirus vaccinia virus induces the redistribution of eIF4E, eIF4G, and PABP to these replication compartments. It is not fully understood how redistribution of initiation factors occurs although it has been proposed that this may be important in selectively promoting translation of viral mRNAs.17
D. Regulation of eIF4E Phosphorylation
eIF4E is phosphorylated on residue serine 209 by the MAP-kinase signal-integrating kinases Mnk1 and Mnk2 (reviewed in Ref. 23 and the chapter by Blenis and Mahoney, this volume). The Mnks bind to eIF4G, bringing the kinase into close proximity to eIF4E.24, 25, 26 The exact role of eIF4E phosphorylation in translational regulation is still unresolved, and although Mnk1 and Mnk2 are essential for constitutive and inducible phosphorylation of eIF4E they are not required for cell growth or development.27 However, it is thought that phosphorylation of eIF4E leads to stimulation of cap-dependent translation28 and this is associated with tumorigenesis.29 Changes in the phosphorylation state of eIF4E are often seen in virus-infected cells and these have been shown to affect virus replication.
1. Adenovirus
Adenovirus infection results in changes in eIF4E phosphorylation that are important for virus replication. Adenovirus mRNAs are capped, but they are selectively translated during late viral infection. During this stage, the first protein to be synthesized is the 100 K protein, encoded by the L4 transcription unit, and this is produced in very large amounts.30 The 100 K protein then binds to the carboxyl-terminus of eIF4G at, or near, the site that is normally occupied by the eIF4E kinase Mnk1. By competing with Mnk1 for binding, the 100 K protein acts as a direct inhibitor of Mnk1 and displaces this protein from eIF4G.31, 32 The removal of Mnk1 from eIF4G results in the dephosphorylation of eIF4E, which is thought to be associated with the inhibition of cellular cap-dependent translation, although the precise mechanism by which this occurs is not understood. Late AdV mRNAs are capped but are still translated when cellular protein synthesis is inhibited as they possess a common 5′-noncoding region (5′-NCR) known as the tripartite leader. The tripartite leader allows the late mRNAs to be selectively translated by an alternate mechanism of initiation known as ribosome shunting. During late infection, the 100 K protein enhances the binding of eIF4G and eIF4A to the tripartite leader complex at the 5′ end of the adenovirus mRNA.33 The activity of the 100 K protein is stimulated by tyrosine phosphorylation,33 and this phosphorylation event is necessary to promote viral translation following shunting and does not affect the binding of this protein to eIF4G.
Similar decreases in eIF4E phosphorylation are observed in VSV and influenza virus infections.21, 34 In contrast, HSV-1 induces the phosphorylation of eIF4E, which promotes its association with eIF4G and may enhance cap-dependent (and therefore viral) protein synthesis, although it is not fully understood how selective translation of viral protein synthesis is achieved (reviewed in Ref. 21).
E. Cleavage of eIF4G
eIF4G is the central component of the eIF4F cap-binding complex and is frequently targeted during virus infection. There are a number of functional homologs of eIF4G including eIF4GI and II.
1. Picornaviruses
Many picornavirus infections induce a rapid inhibition of host cell translation. In the case of the entero and rhinoviruses and FMDV, this shutoff is associated with the cleavage of eIF4G. The entero- and rhinovirus 2A proteases cleave eIF4GI such that the protein is separated into an N-terminal one-third, containing the eIF4E-binding site, and a C-terminal two-thirds, to which eIF3 and eIF4A bind.35, 36, 37 The bridging function of eIF4GI between the cap-binding activity of eIF4E and the helicase and 40S recruitment roles of eIF4A and eIF3 is therefore lost.
The FMDV L-protease similarly cleaves eIF4GI at a site close to, but distinct from, the 2A protease cleavage site.38 A secondary cleavage event is mediated by a second FMDV protease, 3C, in a species-specific manner.39 Both cleavage events result in separation of the eIF4E-binding domain from the C-terminal portion of the protein, similar to the effects of the entero rhinovirus 2A protease (Fig. 1).
The effects of eIF4GI cleavage on host translation are more complex than was originally thought. Experiments conducted in the presence of inhibitors of viral RNA synthesis indicated that, although eIF4GI is still cleaved under these conditions, host translational shutoff is minimal.40 It was subsequently shown that 2A protease also cleaves eIF4GII. Cleavage of both proteins is required for the virus to inhibit host translation.41 In the case of PV, eIF4GII is cleaved with slower kinetics than eIF4GI, and eIF4GII cleavage is therefore the rate-limiting step for induction of host translational shutoff.41 FMDV L-protease also cleaves both eIF4GI and II, but with similar kinetics for each protein.38
The significance of eIF4GII cleavage is not certain, as it is much less abundant than eIF4GI in cells and is no more active in supporting translation initiation. Moreover, the central domain of eIF4G, which lacks the eIF4E-binding domain, can support translation initiation on capped mRNAs. This eIF4G p100 domain is fourfold less effective than intact eIF4F in mediating translation initiation on capped mRNAs, but is more active than intact eIF4F for initiation on PV RNA.42 It is likely that, when viral RNA synthesis increases the pool of PV RNA in the cell, the p100 fragment of eIF4G is redirected to PV RNA at the expense of host translation. Other effects of picornavirus infection, such as PABP cleavage, may also be involved in mediating the inhibition of host translation that occurs during picornavirus infection.
Picornavirus translation is directed by internal ribosome entry sites (IRESs) within the 5′-UTRs of the viral RNAs. The central one-third of eIF4G, containing the eIF3 and one eIF4A-binding domain, is sufficient to support translation initiation from these IRESs.43 This allows picornavirus RNAs to compete effectively for the host translation machinery following infection, although the situation appears to be more complicated than this (see Section III). An exception to this is hepatitis A virus (HAV), which does require full-length eIF4G and eIF4E for translation initiation, and hence does not cleave eIF4G or induce shutoff.42
2. Caliciviruses
The caliciviruses are an important family of viruses, being the main cause of outbreaks of viral gastroenteritis in man (noroviruses) and the causative agents of a number of animal diseases. Infection with feline calicivirus (FCV) induces cleavage of eIF4GI and II, somewhat closer to the N-terminus than the picornavirus 2A protease cleavage site.44 This cleavage occurs late in infection and correlates with host translational shutdown. Despite this induction of cleavage, FCV requires intact eIF4G to mediate translation of viral mRNAs. It may be that in this case, cleavage of eIF4GI results in a cleavage product that retains the eIF4E-binding site, but removes the PABP-binding site. This would make sense as FCV mRNA translation requires eIF4E,45 although the PABP requirement is currently unknown. However, translation of mRNA from the related calicivirus murine norovirus (MNV) is insensitive to FMDV L-protease treatment and it therefore seems that intact eIF4G is not required for translation of MNV mRNAs.46 It appears that even within the same family of viruses, different requirements for specific initiation factors in viral translation exist.
3. Retroviruses
Retroviruses have RNA genomes that undergo reverse transcription in infected cells to give a full-length dsDNA copy, which then integrates into the host genome. Human immunodeficiency virus (HIV) is a lentivirus responsible for acquired immunodeficiency syndrome (AIDS). Proteases encoded by HIV-1 and -2 cleave eIF4GI, but not eIF4GII, in infected cells and in vitro. 47, 48 Unlike the picornavirus proteases, this cleavage occurs at multiple sites and results in inhibition of HIV IRES-driven translation, in addition to host translation.47 Several other retrovirus proteases cleave eIF4GI and eIF4GII at sites in a similar location when the protease is expressed in cells or introduced into cell-free systems.49 The significance of this result is not clear, as most retroviruses other than HIV do not inhibit host translation.
F. Targeting PABP
It is now well accepted that PABP plays a central role in stimulation of translation. By binding to poly(A) tails on capped mRNAs, PABP can mediate the circularization of mRNAs by simultaneously binding to eIF4G at the 5′ end of the mRNA, thus promoting the recycling of ribosomes; this has been termed the “closed loop model.”4, 5 A number of viruses have been shown to target PABP as a mechanism of inhibiting host cell translation.
1. Picornaviruses
It has been shown that infection of cells with the picornaviruses PV and coxsackie virus B3 (CVB3) results in the cleavage of PABP.50, 51 Furthermore, it was demonstrated that the viral 2A proteases directly cleaved PABP between M487-G488.50, 51 PABP contains four RNA recognition motifs (RRMs) that participate in eIF4G- and RNA-binding and a conserved C-terminal domain that interacts with other factors such as eIF4B.52, 53 Cleavage of PABP by the picornavirus 2A proteases separates the RRMs from the C-terminus and results in inhibition of protein synthesis, although PABP cleavage does not fully correlate with shutoff. In PV-infected cells PABP is cleaved by the viral 3C protease at different sites to the 2A proteases.54 Recent work has provided new information on the role of PABP cleavage in picornavirus infections. It is known that PABP also stimulates picornavirus IRES-directed translation through its interaction with poly(A) tails and eIF4G.55, 56
One question that has been the focus of interest for picornavirologists for many years is the mechanism of switching from translation to replication of the viral RNAs. As these two processes are occurring in opposite directions on the same RNA, it is believed that something must stall translation to allow replication to occur. It has also been shown recently that HAV 3C protease cleaves PABP in vivo and in vitro. 57 The resulting N-terminal cleavage product binds to the HAV 5′-UTR (to the pY1 region upstream of the IRES) and suppresses translation of the HAV mRNA. HAV does not induce host cell shutoff and infection does not result in cleavage of eIF4G. A model has been proposed in which PABP binds to the poly(A) tail on the HAV RNA early in infection and stimulates translation. Once enough viral proteins have accumulated, the 3C protease cleaves PABP and the N-terminal cleavage product binds to the pY1 region of the 5′-UTR of HAV RNA, inhibiting translation. The RNA is then cleared of ribosomes to allow replication to occur in the opposite direction.57
This model is also likely to apply to other picornaviruses, as it has now been shown that PABP cleavage by PV 3C protease also inhibits translation directed by the PV IRES, both on RNAs with and without poly(A) tails.58 It was also demonstrated that expression of a PABP that is resistant to cleavage by 3C protease within cells resulted in reduced production of viral RNA and reduced virus production. This suggests that PABP cleavage may be important in promoting the switch from translation to replication in picornavirus infections.
2. Caliciviruses
It is not only in picornavirus infections that PABP cleavage is seen. The caliciviruses norovirus (NV) and FCV also induce PABP cleavage54 and this results in inhibition of translation of polyadenylated RNAs in vitro. The 3C-like protease is responsible for this cleavage. PABP cleavage does not occur until relatively late in FCV infection. As calicivirus RNAs are also polyadenylated it is possible that PABP also stimulates translation of viral RNAs and that PABP cleavage would inhibit viral translation, but this has not yet been demonstrated. In line with the model described above, it is tempting to speculate that PABP cleavage in calicivirus infections may also modulate the switch from translation to replication of the viral RNAs.
3. Rubella Virus
A different mechanism of targeting PABP that does not involve its cleavage has recently been described in rubella virus infection. The rubella virus capsid protein binds to PABP and there is an increase in PABP levels during infection.59 Addition of the rubella virus capsid protein to in vitro translation reactions inhibited translation of viral RNAs, but this inhibition could be rescued by the addition of PABP. An inhibition of host protein synthesis was also observed, although this was not complete. The authors suggested that the binding of the capsid protein to PABP may mediate the switch between translation and packaging of the new genomes, in a similar manner to the model described for picornaviruses and caliciviruses above.
4. Rotaviruses
Rotaviruses belong to the Reoviridae family and have segmented dsRNA genomes. Rotaviruses cause gastroenteritis and infect many animal species; in humans, they are responsible for severe diarrheal disease in infants which is a cause of high mortality in the developing world. These viruses replicate in the cytoplasm of infected cells where capped, nonpolyadenylated viral mRNAs are made.60 The rotavirus nonstructural protein NSP3 binds to the N-terminal region of eIF4GI, both in vitro and in infected cells and also binds to the 3′-terminal sequence common to all rotavirus mRNAs, similar to PABP binding to cellular mRNAs.61 Rotavirus-infected cells also undergo inhibition of cellular protein synthesis and this has been attributed to the novel action of the NSP3 protein. The NSP3 protein specifically evicts PABP from the eIF4F complex by competing for binding to eIF4GI. In spite of this similar function, there is no sequence homology between PABP and NSP3. It is therefore believed that NSP3 binding to a consensus sequence in the 3′ end of rotavirus mRNAs recruits the eIF4F complex to the viral mRNAs via NSP3 binding to eIF4GI, effectively circularizing the viral mRNAs. It has been proposed that NSP3 functions in a similar way to PABP binding to polyadenylated eukaryotic mRNAs. However, more recent data have questioned this model as it has been demonstrated that NSP3 (and its interaction with eIF4GI) is not required for rotavirus mRNA translation.62 In this study, RNAi-induced silencing of NSP3 in infected cells had no effect on viral protein production (except NSP3) or virus replication, although it did result in a less severe shutoff of host cell protein synthesis. In fact, viral progeny production was enhanced in the NSP3-silenced cells. Similarly, these authors suggested that eIF4GI is also not required for viral protein synthesis, as silencing of this factor also had no effect on virus production. A new model on the role of NSP3 was put forward that proposes that binding of NSP3 to the viral mRNAs either protects them from degradation or prevents binding of the virus polymerase, thereby ensuring they are utilized for translation.62 The exact role remains to be determined.
5. Bunyaviruses
Bunyaviruses possess tripartite, negative-sense RNA genomes and are responsible for a febrile illness in humans that is mosquito-borne. Bunyavirus mRNAs, like rotavirus mRNAs, are capped but not polyadenylated. Infection of cells with these viruses induces shutoff of host cell protein synthesis, possibly through the inhibition of transcription. It has been shown recently that a translational enhancer element (TEE) within the 3′-UTR of Bunyamwera virus S segment mRNA is able to substitute for a poly(A) tail.63 A similar element has been shown to exist in Dengue virus mRNA.64 Translation of the bunyavirus mRNAs requires eIF4GI but does not require PABP, although the exact role of eIF4GI is currently unknown. Furthermore, bunyavirus infection of cells in culture resulted in a redistribution of PABP localization from the cytoplasm to the nucleus, and it was suggested that this may contribute to the inhibition of translation of host (polyadenylated) mRNAs. The viral N protein binds to PABP in the cytoplasm but it is not yet known if this protein is directly involved in the nuclear relocalization.
G. Cap-Independent Translation
An alternative mechanism of translation initiation that is used in mammalian cells is termed internal ribosome entry. In this case, a complex, highly structured RNA element (an internal ribosome entry site or IRES) is formed in the 5′-UTR of the mRNA and the ribosome is recruited via the IRES to an AUG start codon that may be a considerable distance from the 5′ end of the mRNA.48 Accessory proteins termed IRES trans-acting factors (ITAFs) are usually required by cellular IRESs and the data suggest that these act as RNA chaperones that permit the IRES to attain the correct structure to recruit the 40S ribosomal subunit.48 In general, IRES-mediated translation is used under conditions of pathophysiological cell stress which include genotoxic shock, temperature shock, hypoxia, and viral infection.48
Members of several different families of RNA viruses are able to bypass the canonical, cap-dependent, translation initiation process by employing this strategy of internal initiation of protein synthesis. The translation initiation factors required, and mechanisms used, by different viral IRESs vary considerably.
1. Picornaviruses
The picornavirus RNAs are not capped, but are covalently linked to a small peptide known as VPg at the 5′ terminus; this peptide is rapidly lost once the virus enters the cell, leaving an uncapped RNA. Picornavirus 5′-UTRs tend to be long and structured, with many upstream AUG codons that are not used for translation initiation. This suggested that a cap-dependent scanning mechanism of translation initiation was unlikely to be utilized, and led to the discovery of the first IRESs in the 5′-UTRs of EMCV65 and PV RNA.66 These were identified by construction of dicistronic reporter RNAs, in which the viral 5′-UTR was placed between two cistrons and was able to promote translation of the downstream cistron.
IRESs were subsequently identified in many different picornavirus RNAs and divided into two major categories, within which there are common secondary structural and mechanistic features. Type I IRESs are found in entero- and rhinoviruses, such as PV, and recruit ribosomes to an AUG codon at the 3′ end of the IRES. A ribosomal scanning process then transports the 40S subunit and associated factors to the next AUG codon further downstream, where translation initiation occurs. Type II IRESs, located in cardio- and aphthovirus mRNAs (e.g., EMCV), are similar in length to type I IRESs, at about 450 nucleotides (nt). They also recruit the 40S ribosomal subunit directly to an AUG codon at the 3′ end of the IRES, but in this case this AUG is the initiation codon67 (Fig. 3 ). The FMDV IRES is structurally related to the type II IRESs, but only a minority of translation initiation occurs at the site of ribosome recruitment. The remaining ribosomes initiate translation at the next AUG downstream following a scanning process, so this IRES uses a hybrid of type I and type II mechanisms.
Fig. 3.
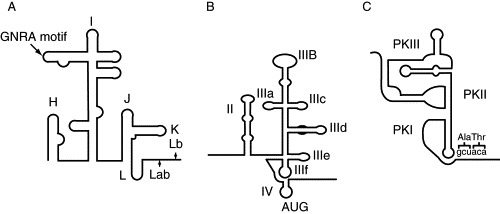
IRES secondary structures. (A) Secondary structure model of the FMDV IRES, a type II picornaviral IRES. The FMDV IRES directs translation initiation from both AUG Lab and AUG Lb sites. Most picornaviral IRESs use a single initiation codon, analogous to FMDV AUG10 for type II and AUG11 for type I IRESs. (B) A structural model of the hepatitis C virus (HCV) IRES. The basal part of domain III is involved in 40S ribosomal subunit binding, and the apical loops of this domain in binding to eIF3. (C). The cricket paralysis virus (CrPV) IRES adopts a triple pseudoknot structure. PKI mimics a tRNA in the ribosomal P site, allowing initiation to occur at a GCU codon in the A site.
Both type I and type II IRESs require the entire canonical translation initiation machinery, with the exception of the cap-binding protein eIF4E, and the eIF4E-binding domain of eIF4G. In vitro reconstruction of initiation complexes using purified and recombinant initiation factors and 40S subunits indicated that eIF4G binds directly to the J-K domain of type II IRESs, that this binding is stimulated by eIF4A, and that this induces a conformational change in the region surrounding the initiation codon that is likely to promote 43S complex recruitment.68 Recently, similar experiments on the type I PV IRES have indicated that an analogous mechanism is used to recruit eIF4G-4A to domain V of the IRES and to induce structural changes at the 3′ border of the IRES.69 Other structural features of both classes of IRES are also required for initiation, and therefore eIF4G/4A recruitment alone is not sufficient for IRES activity.
In addition to their requirement for components of the canonical translation initiation machinery, many picornavirus IRESs need to recruit noncanonical IRES ITAFs to achieve optimal activity. A number of ITAFs that interact with specific IRESs have been identified, although in some cases the physiological role of these factors is questionable. Well-characterized examples include the polypyrimidine tract-binding protein (PTB), which was first shown to interact with the human rhinovirus (HRV),70 and was subsequently found to be required for efficient PV and EMCV translation in cells.71 A role for the autoantigen La in stimulation of PV IRES activity has also been demonstrated in vitro and in cell culture.72 ITAFs are thought to act by modulating the secondary structure of the IRES such that canonical initiation factors are more effectively recruited, and such a role was demonstrated for PTB and ITAF45 binding to the FMDV IRES73 and more recently in cells.74
Picornavirus infection is frequently associated with rapid shutoff of host translation, providing a rationale for the use of IRESs to maintain viral translation under these conditions. The enterovirus 2A and FMDV L-proteases, for example, cleave eIF4GI and II such that the N-terminal eIF4E-binding domain is separated from the remainder of the protein. Although picornavirus IRESs show unaffected or even enhanced activity in the presence of 2A protease, some of this stimulation is thought to be independent of inhibition of host translation or expression of the C-terminal fragment of eIF4G.75 Stimulation of IRES activity can still occur when 2A protease with a mutant active site is expressed, or when eIF4G is resistant to 2A cleavage.76 The effects of this protease on IRES activity are therefore more complex than a simple competition between full-length and truncated eIF4G mediating host and viral translation, and it is probable that other factors are regulated by the protease and have an effect on picornavirus IRESs.
Two further categories of picornavirus IRES have also been identified. The HAV IRES forms a minor class of its own, and requires the full canonical initiation machinery including eIF4E and full-length eIF4G, although the viral RNA is not capped.42 It has been suggested, although not yet experimentally proven, that the requirement for eIF4E may be due to its conformational effects on eIF4G.42 A fourth, and very distinct, class of picornavirus IRES elements was recently identified in porcine teschovirus-1 (PTV-1),77 and subsequently in several other picornaviruses such as avian encephalomyelitis virus.78 These IRESs are distinct from other picornavirus IRESs in their initiation factor requirements and mechanism of action, and instead are very similar to the HCV and pestivirus IRESs. The PTV-1 and AEV IRES elements show sequence and structural homology to the hepatitis C virus (HCV) IRES and act similarly to directly recruit the 48S complex in the absence of the eIF4 factors. The PTV-1 IRES has also been shown to interact directly with the 40S ribosomal subunit and eIF3.79 This suggests that exchange of genetic information between picornaviruses and flaviviruses has occurred at some point.
2. HCV and Pestiviruses
The mechanism of initiation used by the PTV-1 IRES and its relatives was initially identified in HCV and pestivirus RNAs.80, 81 HCV belongs to a subgroup of the Flaviviridae family and is a major cause of human disease, causing blood-borne hepatitis that can result in the development of hepatocellular carcinoma. The flavi- and pestiviruses are positive-sense RNA viruses with uncapped structured 5′-UTRs that direct synthesis of the viral polyprotein. The HCV and pestivirus 5′-UTRs are somewhat shorter than those of the picornaviruses, and almost the entire UTR, approximately 330 nt, is required for IRES activity. HCV and related IRESs can bind directly to the 40S subunit (in the absence of any initiation factors) such that the start codon is positioned close to the ribosomal P site. The IRESs then bind directly to eIF3 and require this factor and the ternary eIF2/GTP/Met-tRNAi complex to form correctly positioned 48S* complexes. The IRES can then recruit the 60S subunit and assemble functional 80S ribosomes.82
These IRESs do not require any components of the eIF4F complex, and it was recently demonstrated that at high magnesium concentrations the HCV IRES is able to initiate translation independently of the eIF2/GTP/Met-tRNAi ternary complex.83 eIF2- and eIF5-independent HCV IRES activity was mediated by eIF5B in a manner analogous to prokaryotic translation initiation.84 This ability to function in an eIF2-independent manner displays an intriguing similarity to the dicistrovirus intergenic region (IGR) IRESs and is likely to allow the HCV IRES to function under conditions of cell stress that induce eIF2α phosphorylation.
The minimal factor requirements for HCV IRES activity have allowed close study in vitro and it has been possible to use cryo-electron microscopy (cryo-EM) to determine the tertiary structure of the IRES in complex with the 40S ribosomal subunit.85 The HCV IRES was shown to induce conformational changes in the 40S subunit, some of which are very similar to those induced by eIF1 and eIF1A binding to the 40S subunit. This suggests that the HCV IRES may function in a similar manner to these factors to promote initiation.86 A pathway for recruitment of HCV IRES RNA to the 40S subunit has been determined using directed hydroxyl radical probing, providing a further indication of similar, but distinct, effects of the HCV IRES and eIF1/1A binding to the 40S subunit.87
The secondary structure of the HCV and pestivirus IRESs has been clearly defined (Fig. 3). Domain III is necessary for 40S binding and for subsequent eIF3 recruitment, but domain II is not required to recruit these components. However, deletion of domain II results in severely impaired IRES activity, and this region of the IRES was shown to be important for 80S formation.88 Domain II folds independently of the remainder of the IRES, and is responsible for the conformational changes induced in the 40S subunit by HCV IRES binding.85 An analysis of the role of this domain in subunit joining indicated that it promotes eIF5-induced GTP hydrolysis and release of eIF2/GDP.89
Despite its ability to initiate translation by binding directly to the ribosome, there is some evidence for a role for several different ITAFs in HCV IRES activity. Several proteins have been shown to bind to the HCV IRES, but the most convincing evidence is for a role for the autoantigen La. This factor interacts directly with the HCV IRES at a site close to the start codon, and stimulates 40S binding in vitro. Depletion of La in cultured cells led to a loss of IRES activity.72
3. Dicistrovirus IGR IRESs
The most recent class of IRES to be identified was found in the IGR of the cricket paralysis virus (CrPV) genome and other members of the Dicistroviridae family90, 91 of insect-infecting viruses that belong to the picornavirus superfamily. These viruses have a naturally discistronic genome and IRESs have been discovered in both the 5′-UTR and the IGR. The CrPV IGR IRES is approximately 200 nt long, and adopts a high degree of secondary structure, with three pseudoknots (PKI–III) that are involved in different aspects of the IRES activity.92 The CrPV IGR IRES and its relatives recruit the 40S and 60S ribosomal subunits independently of any host initiation factors or Met-tRNAi. The IGR IRES binds to the 40S subunit via the PKII–PKIII domains, and is positioned correctly for elongation by PKI, which occupies the P site of the ribosome. A GCU codon is positioned in the A site and recruits its cognate tRNA such that the first amino acid is alanine91 (Fig. 3).
The mechanism of initiation by the CrPV IGR IRES has been analyzed in detail by in vitro reconstitution experiments. The IRES mediates peptide synthesis following incubation with the 40S and 60S ribosomal subunits, the elongation factors eEF1A and eEF2, and aminoacylated tRNAs.93, 94 The first translocation step on the IRES occurs without peptide bond formation. The IRES therefore mimics both the initiator and elongator tRNAs by its interactions with the ribosomal P site.
The structure of the IGR IRES in complex with 40S and 80S ribosomes has been revealed by cryo-EM and crystallography.95, 96 These structures demonstrate the ability of the IRES to mimic a tRNA in the ribosomal P site, and to form contacts with the A, P, and E sites. This binding pattern is very different to that of the HCV IRES, which interacts predominantly with the solvent side of the 40S subunit, although there is some overlap in the E site.85 Despite this difference in binding, the CrPV and HCV IRESs induce similar conformational changes in the 40S subunit, suggesting that these changes may be intrinsic to translation initiation.
The ability of the CrPV IGR IRES to initiate translation in the absence of initiation factors allows it to function effectively under conditions in which host translation is inhibited. The IRES is relatively inactive in wild-type yeast, but is activated by eIF2α phosphorylation, or by depletion of various other canonical initiation factors.97, 98 This implies that the IRES competes with host mRNAs for ribosomes, and is able to do so effectively under conditions of cell stress.
The IRES found in the 5′-UTR of the dicistroviruses is very different in both structure and mode of action to that of the IGR IRES. It has been demonstrated that the 5′-UTR of the dicistrovirus Rhopalosiphum padi virus (RhPV) is mostly unstructured and requires only a minimal set of factors for function, eIF2, eIF3, and eIF1, although the addition of eIF4F did stimulate 48S complex formation on the IRES.99 It may be that this simplified mode of action is responsible for the ability of the IRES to direct translation initiation in mammalian, insect and plant systems.100
4. HIV
Two IRESs have been identified in HIV-1 RNA. The first to be identified is located in the coding region of the gag gene and directs synthesis of an N-terminally truncated Gag protein.101 The second IRES is located in the 5′-UTR and directs protein synthesis during the G2/M phase of the cell cycle.102 An unusual form of internal ribosome entry has been described in HIV-2. HIV-2 viral particles contain Gag p57 (the translation of which initiates at the first codon, AUG1), and two shorter isoforms of p50 (which initiates at AUG2) and p44 (which initiates at AUG3). An IRES that directs translation of all three isoforms of the Gag protein is located in a highly structured region which is downstream of the authentic AUG1 start codon and spans to AUG3.103 This IRES is therefore required to deliver the preinitiation complex upstream to produce Gag p57 polyprotein from the first AUG codon.103
H. miRNAs
miRNAs have recently emerged as major regulators of gene expression (discussed in detail in the chapter by Sarnow, this volume). Metazoan miRNAs function predominantly by binding via imperfect complementarity to sites in the 3′-UTR of mRNAs and repressing gene expression, both at the level of translation and by mRNA degradation. This important mode of gene regulation has been found to affect the life cycles of a number of viruses in a variety of different ways (Fig. 4 ).
Fig. 4.
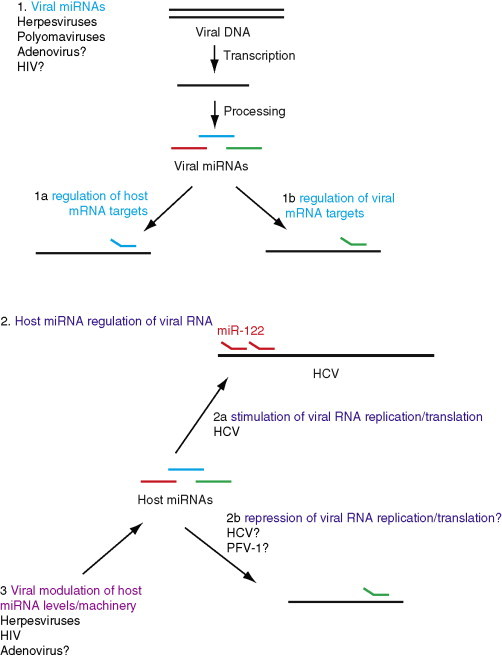
Viral interactions with the miRNA pathway. (1) Some viral families, in particular the herpesviruses, express their own miRNAs using the host miRNA processing pathway. Viral miRNAs may regulate host or viral mRNA targets and may be important for viral latency. (2) A positive role for a host miRNA in viral infection has been demonstrated for miR-122 binding to hepatitis C virus RNA. Negative regulation of viral gene expression by host miRNAs has been suggested by some studies, although its relevance in viral infection is not yet clear. (3) Several viruses modulate levels of host miRNAs.
1. Viral miRNAs
The first observation that viruses utilize the miRNA pathway came from the herpesviruses. These large DNA viruses encode their own miRNAs within their genomes and express these miRNAs during both latent and lytic infection.
Cloning and sequencing of small RNAs derived from B cells infected with the γ-herpesvirus EBV led to the identification of the first five viral miRNAs.104 miRNAs expressed by a number of other α, β, and γ-herpesviruses that infect a range of species have subsequently been identified by cloning and computational methods,105 implying that this is a general mechanism used by this viral family. Conservation of sequence or genomic location between miRNAs derived from different viruses was generally not observed.105
miRNAs are also expressed by the small DNA virus Simian virus 40 (SV40) and several other polyomaviruses.106, 107, 108 Two miRNAs, derived from opposite strands of a single pre-miRNA, are expressed by these viruses. A number of small RNAs derived from adenovirus VA RNAI, and predominantly from VA RNAII, were identified and shown to associate with the RNAi-induced silencing complex (RISC),109 although a functional role has not yet been ascribed to these small RNAs.
It is likely that viral miRNA expression is limited to DNA viruses, as Drosha and Dicer-dependent processing of miRNA precursors results in the destruction of the parent RNA, and would therefore be undesirable for a virus with an RNA genome. Cloning of small RNAs from cells infected with HCV, yellow fever virus (YFV), or HIV-1 did not yield any miRNAs derived from these RNAs and viruses.105 miRNAs derived from both strands of the HIV-1 TAR RNA have since been detected in infected cell lines and primary cells.110 However, HIV-1-derived miRNAs were undetectable in another recent study, so it is questionable whether any expression that does occur is at a sufficiently high level to have functional consequences.111
Viral miRNAs are similar to those of the host, as they are derived from longer hairpin transcripts expressed by RNA pol II or III and undergo processing to yield mature, cytoplasmic miRNAs. Identification of novel viral miRNAs is therefore amenable to computational analysis.105, 112 EBV is now known to encode at least 23 miRNAs,112 located in two genomic clusters, within introns of the BART and BHRF1 genes. miRNAs derived from each cluster show differential expression patterns across different stages of viral latency.113 The temporal regulation of herpesviral miRNA expression in general is not yet well understood, as most viral miRNAs have been cloned exclusively from cells at a particular stage of the viral life cycle. However, HSV-1 miRNAs show distinct expression profiles, with four miRNAs expressed in latency, one during productive replication, and one throughout infection.114 SV40 miRNAs are only expressed in late infection.107
2. Regulation of Viral Targets by Viral miRNAs
The first identified target for a viral miRNA was the SV40 large T antigen (TAg). The viral miR-S1 is expressed antisense to the TAg mRNA and acts in an siRNA-like manner to cleave and degrade the TAg transcript.107 This mechanism appears to function to mediate downregulation of antigen levels late in the viral life cycle and thus allows the virus to evade a cytotoxic T cell response.107 The location and function of this miRNA appear to be conserved in other polyomaviruses.106, 115
Regulation of a viral transcript by an antisense viral miRNA has also been observed or predicted for some herpesvirus miRNAs. miR-H2-3p, which is expressed during latent HSV-1 infection, is antisense to the viral ICP0 transcript. Surprisingly, despite its exact complementarity to its target, miR-H2-3p does not significantly affect ICP0 mRNA levels, but reduces the expression of the encoded protein at the level of translation.114 This regulation is likely to be important for establishment and maintenance of viral latency, as the ICP0 protein is important for productive replication and is thought to be involved in reactivation from latency.114
A search for imperfect targets for viral miRNAs within viral 3′-UTRs yielded a number of attractive candidates, suggesting that viruses also use this mechanism to regulate their own gene expression. This was confirmed experimentally for hCMV miR-UL112-1, which downregulates expression of the major immediate early gene IE1 by binding to its 3′-UTR.116 IE1 protein is important for the switch from latent to lytic infection, as are some of the other predicted viral targets of herpesvirus miRNAs, so viral miRNAs may play a general role in regulation of herpesvirus latency.
3. Regulation of Cellular Targets by Viral miRNAs
An important function of herpesvirus miRNAs appears to be in the regulation of host gene expression. Various host mRNA targets of herpesvirus miRNAs have been detected, and the viral miRNAs appear to function in a similar manner to cellular miRNAs, by binding to the 3′-UTR with imperfect complementarity and negatively regulating translation and RNA stability. EBV miR-BHRF1-3 downregulates the chemokine CXCL-11, a T cell attractant, and may thus allow infected cells to escape the T cell response,117 whereas miR-BART-5 targets the proapoptotic factor PUMA and protects infected cells from apoptosis.118 Several KSHV miRNAs repress expression of BCLAF1, a protein involved in apoptosis (248), and others downregulate THBS1, a multifunctional protein which has a role in recruitment of monocytes and T cells to sites of infection.119 Despite a lack of sequence homology, miRNAs from several different herpesviruses bind to adjacent sites in the 3′-UTR of MICB mRNA and inhibit expression of MICB protein, which is involved in immune surveillance.120 The herpesvirus miRNA targets identified to date appear to allow these viruses to regulate the host immune response or apoptosis, and thus are likely to be important in allowing effective infection.
A particularly intriguing viral miRNA is KSHV miR-K11, which has an identical seed sequence to cellular miR-155, and has been shown to regulate many of the same targets.121 miR-155 levels are upregulated in cells infected with EBV,122 and a similar viral orthologue is encoded by another oncolytic herpesvirus, Marek's disease virus type 1 (MDV-1).123 miR-155 overexpression is linked to various cancers, so these results suggest that viral miRNA expression, or virus-induced modulation of host miRNA expression, may contribute to oncogenesis.
4. Modulation of the Host miRNA Machinery by Viral Infection
Viral infection can affect both the cellular miRNA machinery as a whole, and the expression of individual miRNAs. In addition to its effects on miR-155 expression, discussed above, EBV modulates the levels of multiple host miRNAs, with different effects in latent and lytic infections.124, 125 hCMV also regulates the expression of specific host miRNAs, some of which affect viral replication.126 HIV-1-dependent downregulation of the miR-17-92 cluster had a positive effect on viral replication, mediated by the Tat cofactor PCAF.127
Inhibition of the miRNA and RNAi pathways by viruses is well established as a mechanism of evasion of the host immune response in lower eukaryotes, but few examples exist in mammalian systems. Adenovirus infection results in a global inhibition of host miRNA expression and activity. Expression of VA RNAI led to inhibition of processing of ectopically expressed pre-miR30, and to inhibition of RNAi induced by short hairpin RNA (shRNA) precursors that require Dicer cleavage for activity. This activity was mediated by VA RNAI competition with pre-miRNAs and shRNAs for binding to both exportin-5, required for pre-miRNA export from the nucleus, and to the cytoplasmic processing enzyme Dicer.128 VA-derived small RNAs also compete with endogenous miRNAs for incorporation into the RISC.109 VA RNAI is expressed at very high levels in infected cells, so it is possible that its interference with the miRNA and RNAi pathways may be a consequence of this expression and have little functional relevance for the virus. Although some studies have suggested repression of the miRNA pathway by retroviral transcription factors,129, 130 a recent analysis did not identify any such activity.111
5. Regulation of Viruses by Host miRNAs
A direct effect of a cellular miRNA on virus replication has been observed in the case of the liver-specific miR-122. miR-122 binds to the 5′-UTR of HCV RNA and is required to maintain viral RNA abundance.131 The mechanism by which this occurs has not yet been resolved. No effects of miR-122 on HCV translation were initially observed, suggesting that the miRNA acts at the level of viral RNA replication,131 whereas translational stimulation mediated by miR-122 binding to the HCV 5′-UTR was observed in a subsequent study.132 It is possible that multiple mechanisms may operate. It is not yet known whether this mechanism is unique to HCV, or whether other viruses may use similar strategies.
Various miRNAs, including several induced by interferon stimulation, have been shown to negatively regulate HCV,133 and miRNA-dependent repression of other viruses, including primate foamy virus 1 (PFV-1) has also been observed.130 However, the extent to which these miRNAs regulate virus replication in naturally infected tissues remains unclear. The strong requirement for conservation of complementary sequences to an miRNA seed for regulation to occur, coupled with rapid viral evolution, would suggest that any miRNA that has a detrimental effect on virus replication would quickly be evaded. Tissue specificity is an important feature of the expression of many cellular miRNAs. It is possible that viruses may have evolved to selectively avoid targeting by host miRNAs that are expressed in the tissue they infect, and therefore results obtained outside normal target cells should be interpreted with caution.
Attempts to target viruses by introduction of artificial-binding sites for tissue-specific host miRNAs were effective in restricting viral tropism, but some viral escape mutants evolved.134, 135
In conclusion, the interplay between viruses and the miRNA pathway is complex and varied, and has many important consequences for viral infection.
III. Novel Mechanisms that Permit the Synthesis of Viral Proteins
Viruses have evolved a number of unconventional mechanisms that allow the translation of distal ORFs, contributing to the complexity of gene expression from compact viral genomes. These include (i) leaky scanning, where the AUG of the 5′-most ORF is poorly recognized and the ribosomes scan and initiate at a downstream ORF, (ii) reinitiation, where a posttermination complex remains associated with the RNA and reinitiates at a downstream ORF, and (iii) frameshifting. Although few of these mechanisms of translation initiation have been described in host mRNAs thus far, it is interesting to speculate that, as in the case of IRESs, viruses could use or adapt a preexisting system to initiate synthesis of viral proteins.
A. Presence of a Cap Analogue on Virus mRNAs
A number of RNA viruses have a protein known as VPg covalently linked to the 5′ end of the viral RNA. In picornaviruses, the VPg protein is small and is removed from the RNA following viral entry, such that an uncapped viral RNA serves as a substrate for translation, which occurs by internal initiation. Many of these novel mechanisms have been attributed to the presence of RNA structures within the virus genome that are involved in translation initiation, however, recent work has described the presence of a proteinaceous “cap-substitute” on calicivirus RNAs that directs translation of the viral mRNAs. The VPg on calicivirus RNAs is much larger than its picornaviral counterpart and has a different function, as it is retained on the RNA and serves as a cap substitute that directs translation of the viral mRNAs.
The calicivirus VPg protein binds to eIF4E at a site distinct from both the cap and 4E-BP1-binding sites. In the case of FCV, the VPg:eIF4E interaction is required for translation initiation (at least in vitro) on the viral mRNA, but in the case of MNV this interaction is not required for efficient translation, although the presence of VPg on the mRNA is necessary. It has also been reported that human norovirus VPg binds to eIF3, suggesting that VPg has multiple interactions with key components of the translational apparatus.136 The interaction of calicivirus VPg with eIF4E is unique amongst mammalian RNA viruses but a similar interaction occurs on plant potyvirus mRNAs (reviewed in Ref. 137). In this case, plants that do not express eIF(iso)4E are resistant to infection with turnip mosaic virus. Potyvirus VPg is thought to bind eIF4E in competition with the cap structure.138 Hence, even though the principles of VPg-directed translation are common, the specific mechanisms by which the VPg proteins interact with eIF4E are distinct.
B. Stealing Caps from Host mRNAs
Influenza virus is a major human health problem with worldwide prevalence. Influenza virus is well known for causing pandemic outbreaks and is a zoonotic disease, infecting pigs, avian species and horses, as well as humans. Influenza A viruses belong to the Orthomyxoviridae family and have a single-stranded, negative-sense RNA genome which is made up of eight segments. In influenza virus-infected cells there is a dramatic inhibition of host cell translation while the viral mRNAs are selectively translated.139 It is well established that influenza virus mRNAs acquire their 5′ caps through a process of “cap snatching” or “cap stealing” during which the virus polymerase complex “snatches” the 5′ 10–12 nt from host nuclear mRNAs which then prime the synthesis of viral mRNAs. The viral mRNAs therefore contain host cell sequences at their 5′ ends, which are followed by conserved viral sequences that are known to bind the viral polymerase. It is believed that polymerase binding to these conserved sequences prevents the snatching of caps from the viral mRNAs140 and contributes to the selective translation of viral mRNAs during infection; the cellular mRNAs being degraded once decapped. The polymerase itself is a complex of three subunits, PA, PB1, and PB2. The PB1 subunit is involved in binding to the caps on cellular mRNAs that are then thought to be cleaved by the endonuclease activity of the polymerase. The crystal structure of the PA subunit has recently been solved and the endonuclease active site was shown to reside in the amino-terminal end of the protein.141, 142
As described above, influenza virus infection results in changes to the eIF4F complex as eIF4E is dephophorylated and eIF4G is hyperphosphoryated.34 A study to understand the components of the eIF4F complex that are required for translation of viral mRNAs has recently demonstrated that influenza virus translation has no requirement for eIF4E, as viral mRNAs are translated in cells depleted of eIF4E and in cells treated with rapamycin.143 The authors suggested that the polymerase is able to substitute for eIF4E by binding to the conserved sequences in the 5′-UTR of the viral mRNAs and recruiting initiation factors. This seems to be another example of how some capped viral mRNAs display a reduced requirement for eIF4E in a similar manner to the adenoviruses.
C. Substitution of the Entire eIF4F Complex with a Viral Protein
Whereas influenza virus substitutes the eIF4E component of the eIF4F cap-binding complex with a viral protein, a recent report has demonstrated the unique ability of hantaviruses to replace the entire eIF4F complex with just one viral protein.144 The hantaviruses are rodent-borne viruses of the Bunyaviridae family and the genome is made up of three negative sense, single-stranded RNA molecules. Hantaviruses include Sin Nombre virus and Hantaan virus, viruses associated with high-mortality rates and symptoms including hantavirus pulmonary syndrome and haemorrhagic fever, respectively.
The nucleocapsid (N) protein of Sin Nombre virus has been shown to uniquely possess activities that mimic all three components of the eIF4F complex, eIF4E (as N binds to the 5′ end of capped mRNAs), eIF4G (as N recruits the 43S complex to the 5′ cap), and eIF4A (as N replaces the RNA helicase). It was shown that N enhances translation of capped mRNAs as a whole, but preferentially stimulates translation of capped mRNAs that contained 44 nt of 5′ noncoding sequence from the virus. Following inhibition of translation by the addition of a picornavirus 2A protease to RRL such that eIF4G is cleaved, N rescues translation of capped mRNAs. Finally, it was shown that N increased the rate of recruitment of the 43S complex to mRNAs.145 In summary, N protein increases the translation of both viral and cellular mRNAs, although the enhancement is greater on viral mRNAs. Hantaviruses do not induce shutoff of cellular protein synthesis but the binding of N to viral mRNAs is likely to permit their preferential translation over host mRNAs.
D. Frameshifting
There are many examples of important mammalian virus pathogens including retroviruses (e.g., HIV-1) and coronaviruses (SARS) that employ frameshifting during translation. In most of the systems examined to date, frameshifting is required for the expression of viral replicases. In retroviruses, frameshifting is necessary for the synthesis of Gag–Pol and Gag–Pro–Pol polyproteins and the production of reverse transcriptases, whereas for the majority of other viruses it is essential to permit the synthesis of RNA-dependent RNA polymerases.146
Ribosomal frameshifting is a process that alters the triplet decoding of the mRNA by the elongating ribosome. A specific signal in the mRNA causes the ribosome to change reading frame from the 0 to the − 1 frame and translation then continues in the new frame.146 In eukaryotes frameshift signals require two elements, a heptanucleotide “slippery sequence,” where the ribosome changes reading frame, and a stimulatory element that is located a few nucleotides downstream in the form of an RNA pseudoknot147 or a stem-loop.148 A spacer region of 6–8 nt between the slippery sequence and the stimulatory RNA element is also required, and frameshifting efficiency is dependent upon the length of this sequence (Fig. 5 ; 147, 149). Several models for how frameshifting occurs have been proposed (reviewed in 146, 150) and the model that is most consistent with experimental data suggests that ribosomal pausing at the stimulatory RNA element increases the time that the ribosome is held over the slippery sequence and this permits the tRNA to realign in the − 1 frame.151, 152
Fig. 5.
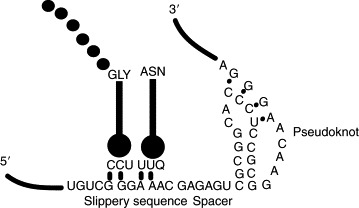
Basic mechanism of action of frameshifting. Two elements are required in the viral RNA for frameshifting to take place; a heptanucleotide slippery sequence and a structured downstream stimulatory element which is present in the form of either a RNA pseudoknot or a hairpin-stem. Ribosome pausing, due to the presence of the RNA structure, over the slippery sequence permits the tRNA to realign in the − 1 frame.
1. Coronaviruses
Coronaviruses are a family of animal viruses with a large positive stranded RNA genome. In this family of viruses the replicase gene is composed of two partially overlapping ORFs, 1a and 1b, and the fused polyprotein 1a/1b (pp1ab) is synthesized by programmed − 1 ribosomal frameshifting.147, 153 The first characterized frameshift signal described for pp1a/pp1b in coronaviruses was in avian infectious bronchitis virus (IBV; Ref. 147); subsequently, very similar mechanisms were shown to achieve this mode of translation elongation in other coronaviruses. It was shown by mutational analysis that the slippery sequence in IBV is UUUAAAC154 and that the downstream frameshift stimulatory element 6–7 nt away from this sequence is a hairpin (H)-type mRNA pseudoknot (Ref. 147; Fig. 6 ).
Fig. 6.
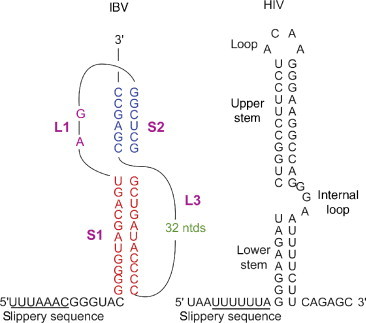
Structures of the stimulatory sequences that are found in IBV and HIV. The structure of the stimulatory sequence found in IBV is an RNA pseudoknot whereas the structured element in HIV forms a complex stem-loop.
To carry out detailed structural analysis of ribosomes paused on frameshift sequences, a variant of the frameshift sequence of IBV was generated in which the slippery sequence was changed to CGAGGCA and ribosomes, stalled in the act of decoding the frameshift signal, were purified and subjected to cryo-EM.152 This allowed images to be generated of 80S ribosomes stalled over the 1a/1b IBV pseudoknot frameshift signal and important mechanistic details of the frameshift process have been derived from these reconstructions.152 Importantly, these data indicate that translocation is the point in the elongation cycle at which frameshifting occurs. The RNA pseudoknot interacts with the ribosome in close association with the putative 80S helicase at the entrance to the mRNA channel as well as with the 18S rRNA helix 16, rpS9, rpS2, and the ubiquitous eukaryotic ribosomal regulatory protein RACK1. Eukaryotic elongation factor 2 (eEF2) was shown to be trapped in the A site of the ribosome and this would prevent binding of tRNA to this site until the frameshift has been completed. In the presence of the pseudoknot the P site tRNA is distorted and bent toward the A site that contains eEF2. Taken together these data allowed the following three step model for frameshifting to be proposed.152
-
(i)
The helicase at the entrance of the mRNA tunnel on the elongating ribosome is unable to unwind the pseudoknot structure in the mRNA, pausing the ribosome.
-
(ii)
The blockage imposed by the pseudoknot partially inhibits the movement of the tRNA, bound to the mRNA by the codon–anticodon pairing, during translocation. The tRNA is unable to return to the A site due to the presence of eEF2, and the resulting strain on the tRNA causes it to bend in a (+) sense direction such that it now moves to the roof of the P site.
-
(iii)
Due to the strain of these opposing forces the codon–anticodon interactions breaks. The tRNA then relaxes and moves in the (−) sense 5′ direction and repairs with the mRNA in the − 1 position.152
This elegant model agrees well with earlier data and supports a number of other models that have been proposed.146, 150, 155
Severe acute respiratory syndrome (SARS) in humans is caused by a novel coronavirus156 and the frameshift element in this virus is highly related to those described previously although with some interesting differences. In SARS-CoV the site of frameshifting signal that allows the production of pp1ab is also a U_UUA_AAC stretch and this is found 12 bases upstream of the 1a stop codon. The frameshift is very efficient; using reporter-based systems, frameshift frequencies of between 14% and 27% were measured in vitro and in vivo. 157, 158 As with other coronaviruses there is a downstream pseudoknot, and disruption of base pairing in this element substantially reduces the efficiency of frameshifting.157, 158 However, there are notable differences in this pseudoknot when compared to other coronaviruses. The pseudoknot conforms to the H type structure found in IBV, but there is extensive base pairing in loop 3.157 Most of loop 3 can be deleted without great effect. However, there appears to be an essential conformation that needs to be maintained to achieve maximum frameshifting efficiency.146, 159
2. HIV and Related Lentiviruses
Frameshifting is essential to HIV replication and related lentiviruses (e.g., SIV, HIV-2) as it controls both the expression of Gag–Pol polyproteins and importantly the precise ratio of the Gag:Gag–Pol polyproteins.160 Even small variations in the Gag and Gag:Pol ratio can have adverse effects on the virus in terms of infectivity.161 The site of frameshifting for HIV is a U_UUU-UUA stretch located within the gag/pol overlap 200 nt upstream of the gag termination codon.151, 162 The downstream stimulatory element found at the gag–pol junction is a simple but very stable RNA stem-loop.148, 163, 164, 165, 166 For HIV-1 the NMR data suggest that the stimulatory RNA is comprised of a two stem structure (Fig. 6). There is an 11 bp helical stem and a highly ordered hairpin loop, the top of which contains an ACAA tetraloop (Fig. 6). A less stable stem is also present which is separated from the upper loop.148, 166 It has been suggested that the lower stem acts as a “positioning element” that permits the stem-loop to induce the ribosomal pausing required to perturb ribosome translocation.166
3. Frameshift Sequences as Putative Drug Targets
In the longer term a full understanding of frameshift regions is likely to be of considerable importance in the development of antiviral drugs. For example, HIV has an absolute requirement for a − 1 ribosome frameshift during translation and this would provide an attractive new target to interfere with the viral lifecycle. It may be feasible to disrupt this process using small molecules, peptides, or oligonucleotides.167
E. Reinitiation
Eukaryotic translation initiation generally occurs close to the 5′ end of the mRNA. In some mRNAs, the 5′-proximal AUG is followed by a short uORF, and if this is fewer than 30 codons a significant percentage of the ribosomes that have completed uORF translation may resume scanning (as 40S subunits) and reinitiate translation at a downstream AUG codon.168 Since not all ribosomes resume scanning after termination, the presence of the uORF results in a decrease in translation of the major ORF.168 The uORF must be translated rapidly for reinitiation to occur,169, 170, 171 suggesting that rescanning occurs only if some of the initiation factors that promoted initiation at the uORF AUG remain ribosome-associated during uORF translation. Although the mechanism of reinitiation is not fully understood, it has been shown that in mammalian systems efficient reinitiation following uORF translation only occurs if the complete eIF4F complex, or eIF4A, 4B, and the central domain of eIF4G participated in the original initiation event.172
Some viral mRNAs are able to mediate translation reinitiation following translation of a long ORF, and special mechanisms have been developed to promote this event.
1. Caliciviruses
The best studied case of translation reinitiation occurring following translation of a long ORF is found in caliciviruses. The subgenomic mRNA encoding the structural proteins of FCV is bicistronic with two overlapping cistrons. The first ORF of the RNA encodes the viral major capsid protein (VP1) and the second cistron encodes the minor capsid protein VP2. The two uORFs overlap by 4 nt in FCV (A UGA), one in norovirus (UAA UG) and eight in rabbit hemorrhagic disease virus (AUGUCUGA).173, 174 The expression of the downstream ORF requires a termination–reinitiation event which is different from those identified in mammalian systems studied to date since it is independent of eIF4G or the eIF4F complex. Instead reinitiation requires an interaction of eIF3 and the 40 S ribosomal subunit with a sequence element that is present in the 3′-terminal 70 nt of the upstream ORF, denoted the termination upstream ribosomal-binding site (TURBS).175, 176 Two short sequence motifs present in TURBS are required for reinitiation, the first of which is a pentameric UGGGA sequence that is complementary to the apical loop of helix 26 in the mammalian 18S rRNA. Evidence for a direct interaction between FCV mRNA and 18S rRNA was obtained using a yeast model system where mutations were introduced into both RNAs.128 Thus when the yeast 18S rRNA was mutated such that it was adapted to the FCV sequence or vice versa there was a dramatic increase in the translation of the downstream frame.128 This UGGA motif is conserved in caliciviruses.175 The second motif is not conserved among caliciviruses but is located at an equivalent position in the TURBS of FCV and RHDV,175 and it is the secondary structure of these sequences, and not the primary sequence, that is important for function.176 It has been proposed that the binding of posttermination eIF3/40S complexes to TURBS retains them in a position suitable for reinitiation once they have acquired the eIF2/GTP/Met tRNAi ternary complex.175, 176 In agreement with this hypothesis it has been shown that eIF3 is involved in termination and recycling of ribosomal complexes, thus providing in part an explanation for the interaction of eIF3 with the 40S ribosomal subunit (Ref. 177 and discussed in the chapter by Fraser, this volume).
2. Influenza B Virus
In influenza B virus the genes encoding the M1 (matrix protein 1) and the BM2 proteins, both of which are important for virus viability,178 are located on segment 7 of the viral genome.179 The termination codon of M1 overlaps the start codon of BM2 (UAA UG) and the BM2 polypeptide is expressed by termination-dependent reinitiation.179, 180 The mRNA sequence requirements for reinitiation are similar to those identified for the caliciviruses and are dependent on a 45 nt stretch of RNA that is immediately upstream of the UAAUG pentanucleotide and includes the UGGGA motif. The process of reinitiation and termination is thought to involve the interaction of a stem-loop structure in this region that has complementarity with the apical loop of helix 26 in 18S rRNA.181
3. Pneumovirinae
There are two genera of pneumovirinae: the pneumoviruses, including human respiratory syncytial virus (RSV) and the metapneumoviruses including avian pneumovirus (APV) and human MPV (hMPV). All these viruses cause acute respiratory infections in their hosts. The pneumovirinae direct the synthesis of eight mRNA transcripts, encoding nine primary translation products and the M2 transcripts all contain two uORFs, M2-1 and M2-2, which are overlapping.182 Expression of the RSV M2-2 ORF occurs via an unusual coupled translation event in which termination of translation of the M2-1 ORF is required before translation of M2-2 can be initiated.183, 184, 185 A number of regions in the RSV M2-1 ORF have been shown to play a role in coupled translation. The most important region is not between the overlapping cistrons, but is located 150 nts upstream in the M2-1 ORF and contains stable structural elements.184, 185 A similar mechanism is used to initiate the translation of M2-2 transcripts of APV and hMPV, although there are differences in the efficiencies of the process, which appear to be due to lack of stimulatory sequences in the M2-1 ORF.185 The reinitiation mechanism is likely to be quite different from that found in influenza BM2 and calicivirus subgenomic mRNAs, but may well involve the same principle of capturing some of the posttermination 40S subunits and restraining them in a position suitable for reinitiation.
F. Leaky Scanning and Shunting
Although translation initiation generally occurs at the proximal 5′ AUG codon that is reached after scanning of the 5′-UTR, alternative sites of translation initiation also exist which contribute to the complexity of viral genomes. It is known that the sequences flanking the AUG codon contribute to the efficiency of translation initiation. A purine, particularly adenosine, at − 3, and guanosine at + 4, when the A of AUG is designated + 1, gives the greatest enhancement to the initiation event. In many viral mRNAs the first AUG codon is in a suboptimal context, so the scanning ribosome may migrate past this first AUG codon and initiate at an alternative downstream AUG. This process is termed “leaky scanning.” The process of shunting is occasionally observed following leaky scanning and examples of such a mechanism are provided by studies of adenovirus, sendai virus, and avian reoviruses.
1. Adenoviruses
Adenoviruses contain a double-stranded linear DNA genome and the timing of the expression of mRNA from this genome is dependent upon the stage of infection. During early stages of infection there is a production of proteins for viral DNA replication whereas in late infection this switches to proteins that are required for the assembly of viral capsids.186 Most late adenovirus transcription is initiated from the major late promoter (MLP). mRNAs derived from this promoter all possess a common 5′-NCR of 212 nt in length called the tripartite leader which arises from splicing of three small exons.187, 188 There is an inhibition of host protein synthesis following adenovirus infection. The continued selective translation of viral mRNAs under these conditions is due to the presence of the tripartite leader which is able to direct ribosome shunting.189, 190 Shunting involves the loading of the 40S subunit at the 5′ end of the capped tripartite leader-containing mRNAs, followed by linear scanning over a short distance and then a direct translocation of the 40S subunits via “shunting elements” to the start codon.190 Within the tripartite leader, the elements that are required for translation initiation during late viral infection are an unstructured 5′ end to the mRNA189 and hairpin structures that have complementarity to the 18S rRNA at the 3′ end.190
2. Reoviruses
The S1 mRNAs transcribed by the fusogenic avian (ARV) and Nelson Bat (NBV) reoviruses encode three unrelated proteins from sequential partially overlapping ORFs. The 5′-ORF encodes the p10 fusion-associated small transmembrane protein that is responsible for the syncytium-inducing phenotype of these reoviruses.191 The second ORF encodes a nonstructural nucleocytoplasmic protein (p17) that has no known function.192 The terminal ORF encodes the sigma C protein. This is similar to the mammalian reovirus sigma 1 protein that has a function in cell adhesion.193 Analysis of the start codons of these three ORFs revealed that the first AUG codon is in a suboptimal context, whereas the sequence surrounding the AUG of the second uORF provides a good context for translation initiation with a highly conserved A at the − 3 position. The start codon of sigma C is in a strong context but it is a considerable distance from the 5′-terminal cap structure.194 By altering the sequences surround the AUGs of the uORFs that encoded the p10 and p17 proteins to optimal contexts it was shown that the coordinated expression of these proteins occurred by leaky scanning.194 However, the translation initiation of sigma C was shown to be independent of leaking scanning, reinitation, and internal ribosome entry and it was proposed that sigma C translation is mediated by an atypical shunting mechanism.194
3. Sendai Virus
Sendai virus belongs to the parainfluenza virus family, is a pathogen of mice, and has a negative-sense RNA genome. In Sendai Virus P/C mRNA there are five start codons that are located 81–201 nt from the 5′ end of the mRNA. A sixth start codon is located more than 1500 nt from the 5′ end, and together these generate eight protein products, as alternative C-termini are also used.195 Initiation from the first three start codons, which are ACG, AUG, and AUG, can be explained by the leaky scanning model. The sequence that surrounds the first unusual ACG codon is in an otherwise optimal context (GCCACG GAT) with a purine at + 4 and − 3.196 If the ACG sequence is mutated to AUG there is increased expression of the encoded C′ protein, but the expression of the P and C proteins initiated from the second and third start codons is ablated, presumably because there is no leaky scanning. The expression of the proteins from the downstream start codons is not affected under these circumstances, suggesting that the initiation of these proteins is independent of scanning.197, 198 It was shown that in vitro translation of these downstream Y proteins was initiated by ribosomal shunting,195 but in vivo this occurred by both shunting and proteolytic processing of the C′ and/or C proteins.199
G. Stop-Go Reprogramming
All proteins encoded by picornaviruses are present in a long single ORF which must be “cleaved” to give the functional polypeptides. The FMDV 2A peptide is encoded between sequences that specify capsid and replicative functions of the virus and this is the major site for the processing of this polyprotein.200 It has been shown that the 2A region of FMDV (and other picornaviruses) mediates “cleavage” of its own C-terminus to release it from the 2B region. Interestingly, the 2A peptide is active when placed between reporter proteins and the cleavage reaction is not therefore dependent on viral (protease) sequences outside of this region.200 Instead the data suggest that the 2A peptide is able to interact with the ribosome and direct translational recoding.201 A detailed analysis of this recoding reaction shows that the ribosomes pause over the final amino acid of the 2A peptide. The recruitment of release factors to this peptide catalyzes termination and releases the peptide, the sequence of which includes the penultimate amino acid encoded by this region (glycine), while the codon of the terminal amino acid (proline) remains in the A site of the ribosome.201 These 2A like peptides have been termed CHYSEL (cis-acting hydrolase element) peptides and their ability to act independently of any proteolytic activity in the “host” cell has lead to a number of biotechnological uses.201
IV. Mechanisms to Overcome Host-Mediated Translational Shut Down Caused by Phosphorylation of eIF2
A. Inhibition of PKR
In mammalian cells there are four proteins that control the formation of ternary complex that is required to bring the initiator tRNAimet to the ribosome by inducing phosphorylation of eIF2α (Fig. 7 ). These are the GCN2, PERK, HRI, and PKR kinases. These proteins are activated by different external stimuli, generally under conditions of cell stress, and the particular kinase that is activated is dependent upon the stress induced. PKR activation represents one of the major host responses to viral infection. Exposure of cells to interferon stimulates the production of PKR202 which then is activated in response to the presence of dsRNA in cells. This often occurs as a result of viral infection by RNA viruses, either because the virus has dsRNA elements within its genome, or because viral replication induces the temporary formation of dsRNA intermediates which could originate from bidirectional transcription. The interaction of PKR with dsRNA initiates dimerization, autophosphorylation, and the subsequent activation of this kinase (Fig. 8 ). Active PKR dimers then phosphorylate the alpha subunit of eIF2 at serine 51 and this has the net effect of preventing further ternary complex formation (Fig. 3) such that the translation of host and viral RNAs is inhibited.203, 204, 205
Fig. 7.
Regulation of ternary complex formation. Ternary complex is comprised of eIF2, GTP, and tRNAimet. The activity of eIF2 is regulated by kinases that phosphorylate this protein at Ser 51 on the alpha subunit. Both PKR and PERK are activated by viral infection and these phosphorylate and inactivate eIF2 which in turn inhibits protein synthesis.
Fig. 8.
Mechanism of action of PKR. Double stranded (ds) viral RNA is bound by PKR causing dimerisation, autophophorylation, and activation of this protein. The activated enzyme then phosphorylates the alpha subunit of eIF2 which will inhibit ternary complex formation.
1. Inhibition of dsRNA-Dependent Protein Kinase (PKR) Activity
Viruses have developed a number of mechanisms to counteract the effects of PKR and there are viral proteins that inhibit PKR activation, sequester dsRNA, inhibit PKR dimerisation, activate antagonist phosphatases, or degrade PKR (Fig. 9 ).
Fig. 9.
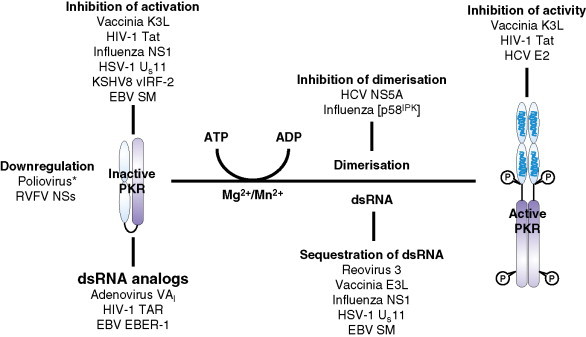
Mechanisms of inactivation of PKR by viral proteins. Viruses have developed a wide range of mechanisms to prevent activation of PKR. These include the production of viral proteins that are able to prevent PKR activation by inhibiting PKR dimerisation and dsRNA-binding proteins that sequester dsRNA.
Many viruses employ mechanisms to inhibit PKR dimerization and thereby prevent PKR activation in the presence of dsRNA. For example, the KSHV-8(HHV-8) protein vIRF-2 binds directly to PKR and prevents activation by inhibiting the autophosphorylation of the protein.206
HCV encodes two PKR inhibitors, the E2 envelope and the NS5A protein. The cytosolic form of the E2 protein is unglycosylated, and has been shown to directly bind to and inhibit PKR function.207 Similarly, NS5A interacts with PKR and inactivates its kinase function.208, 209 This interaction occurs via the interferon sensitivity determining region (ISDR) and there is a correlation between a negative response to interferon (IFN) of some HCV-chronically infected patients and mutations in a region of NS5A, although the relevance of the NS5A/PKR interaction in this regard is still unclear.210, 211, 212 Interestingly, it has also been shown that the NS5A/PKR interaction may be involved in the development of liver carcinoma.213, 214, 215
2. Inactivation of PKR by Functional Competitors of eIF2α
HIV-1 TAR is a highly conserved stable RNA stem-loop that interacts with the viral Tat protein to regulate viral transcription.216 Although low levels of TAR bind and activate PKR,217 high levels of TAR RNA inhibit PKR activity.218, 219 In addition, Tat directly contributes to the downregulation of PKR219 as this protein is a substrate for PKR and acts as a competitor of eIF2α.220, 221 The phosphorylation of Tat by PKR leads to more robust viral transcription since this phosphorylation increases the interaction of Tat with TAR.222
The vaccinia virus K3L gene encodes an 88 amino acid protein that is 30% identical to the N-terminus of eIF2α.223 The K3L protein acts as a competitive inhibitor of PKR and a pentapeptide motif is important for this effect. K3L is produced early in vaccina virus replication, suggesting that this serves to ensure continued translation once dsRNA is produced by the virus.224
EBV produces two small noncoding RNAs EBER-1 and EBER-2. The EBER RNAs play an important role in downregulating PKR activity and preventing virus-induced apoptosis.12 EBER-1 inhibits the protein kinase activity of PKR directly in vitro by competing with dsRNA activators for binding to the enzyme.225 Moreover, expression of EBER-1 in EBV negative cells protects against IFN-induced apoptosis.226 Similarly, the adenovirus VAI RNA which is synthesized in large quantities following viral infection binds to PKR.227 This RNA blocks the activation of PKR in the presence of dsRNA and as a consequence both the autophosphorylation of PKR and the subsequent phosphorylation of eIF2a are inhibited.134
3. Sequestration of dsRNA
Many viruses synthesize dsRNA-binding proteins that prevent activation of PKR by interacting with and sequestering any free dsRNA molecules.228, 229 The first such dsRNA-binding protein identified in this regard was reovirus sigma 3.230 The reovirus sigma 3 protein does not have catalytic activity and it was shown that inhibition of PKR by this protein could be overcome by the addition of excess dsRNA,229 suggesting that the protein acts as a competitive inhibitor by directly binding to dsRNA.
Influenza NS1 protein is a multifunctional protein involved in both protein–protein and protein–RNA interactions.231 This protein dimerizes upon binding to poly(A), and NS1–RNA complexes block PKR activation.231 Similarly, the E3L protein from vaccinia virus contains a dsRNA-binding domain which binds to and sequesters dsRNAs produced during virus infection and so inhibits PKR activation.232
Both EBV and HSV-1 encode proteins, SM protein and Us 11, respectively, that bind to dsRNA and interact directly with PKR.233, 234 Both of these proteins contain an RXP domain comprised of multiple copies of the amino acid sequence RXP. This domain has been demonstrated to be a dsRNA recognition motif234 and is involved in the interaction of these proteins with PKR.233, 234, 235
4. Degradation of PKR
In PV-infected cells PKR is activated by the presence of dsRNA, resulting in an increase in the phosphorylation of eIF2α that leads to a decrease in protein synthesis rates. To circumvent this, PV infection also initiates a series of events that lead to the degradation of PKR.236 The detailed mechanism of this degradation is not fully understood, although it has been suggested that cellular rather than viral proteases are required.237
Finally, it has been shown recently that the NSs protein of rift valley fever virus (RVFV) induces specific degradation of PKR.238 Following infection with RVFV there is a dramatic decrease in PKR protein levels with little change in the levels of the mRNA. In the presence of proteasome inhibitors there is a decrease in RVFV growth and an increase in PKR, strongly suggesting that the degradation is mediated via the proteasome pathway. Since other related viruses do not have this activity it was suggested that the extraordinary pathogenicity of RVFV is due to the acquired PKR degradation function of NSs.238
5. Additional Mechanisms to Overcome PKR Activation
Although many viruses have devised novel ways in which to circumvent the effects of PKR, it is interesting to note that viruses belonging to the Dicistroviridae family which infect insects use a unique mechanism to initiate their translation which is independent of ternary complex.91, 92, 93 In this case viral protein synthesis is stimulated when eIF2α is phosphorylated.92
An alternative mechanism is adopted by the alphavirus sindbis virus (SV) where activation of PKR by viral infection causes almost complete phosphorylation of eIF2α. However, under these conditions there is still efficient translation of SV 26S mRNA. Downstream of the initiation codon on the subgenomic RNA there is a stable stem-loop, that is able to stall ribosomes on the correct site to initiate translation of SV 26S mRNA.81 The data suggest that eIF2A delivers the Met-tRNAi to the stalled 40S ribosome, bypassing the requirement for a functional eIF2.239
B. PERK Regulation
PERK is a type I transmembrane protein that attenuates protein synthesis during endoplasmic reticulum (ER) stress (see chapter by Kedersha and Anderson, this volume), including viral infection. Virus infection has a profound effect on the ER and this can result in activation of the unfolded protein response (UPR) leading to PERK-mediated eIF2α phosphorylation240, 241, 242, 243, 244 and transient translational arrest (reviewed in Ref. 245). Viruses have developed a unique set of mechanisms to overcome the effects of PERK activation and alternatively use the UPR to selectively enhance the synthesis of viral proteins.
The herpesvirus human cytomegalovirus (HCMV) is a widespread opportunistic pathogen that can cause disease and death of newborn infants or individuals that are immunocompromised. Infection of cells with HCMV results in the production of large amounts of viral glycoproteins and an increase of Ca2+ release from the ER to the cytosol. This induces cell stress, which benefits the virus, as chaperone induction by the UPR extends the protein folding capacity of the ER.246, 247 However, virus infection also induces the phosphorylation of PERK and the translational upregulation of the transcription factor ATF4, yet the virus prevents the activation of the ATF4-dependent pathway that would lead to cell apoptosis.246, 247 This is mediated by viral protein pUL38 which promotes PERK activation and ATF4 production but suppresses the persistent c-Jun N-terminal kinase (JNK) phoshorylation and ER-stress-induced cell death.244 It has also been shown that the LMP1 protein from EBV works in a similar manner.240 Again this protein induces the phosphorylation of PERK and this translationally upregulates the production of ATF4. ATF4 then trans-activates the promoter of LMP1 leading to increased transcription and expression of this protein and enhanced proliferation of the infected B cells.240
In HSV-1 infected cells PERK is inactivated and not affected by the acute ER stress that occurs as a result of the viral infection.241 This is mediated by the viral glycoprotein (B) (gB) of HSV-1 that specifically associates with the luminal domain of PERK and suppresses its activation.241 Interestingly, gB appears also to directly regulate viral protein accumulation in a PERK-dependent manner.241 Similarly the HCV envelope protein E2, an ER-bound protein, has been shown to directly bind to PERK and inhibit its activation. Mammalian cells that stably express E2 are resistant to the effects of ER-stress inducers, and E2 can relieve the translational repression induced by PERK.243
V. General Conclusions
As viruses do not possess their own translational machinery they are completely dependent on the host cell for this critical step in their replication cycle. It is clear that viruses have adopted a number of elegant mechanisms to inhibit host cell translation, or at least modify translation factors, in order to effectively compete with cellular mRNAs (Fig. 10). Viruses, especially RNA viruses with their high RNA replication error rates, are the best example we have of natural selection (as opposed to directed selection in agricultural and laboratory practice) operating in real time before our very eyes. The most “successful” viral strains or species are, by definition, those which are most abundant in the environment, and by this criterion H1N1 swine flu is proving a more successful virus than SARS was, notwithstanding the much higher mortality rate (almost 10%) in SARS infection. Of course, such success may be short lived. For example, we expect swine flu to peak in the next couple of years and then decline, while other virus strains or species may be considered more successful because they maintain a more enduring high abundance, even though they never reach such extreme peaks.
Fig. 10.
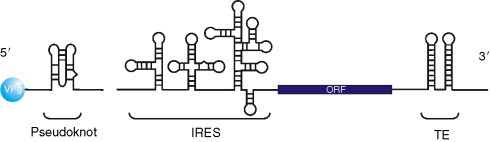
Alternative mechanisms of translation initiation on viral RNAs. The caliciviruses possess a 5′ proteinaceous cap substitute VPg. Some viruses such as coronaviruses and HIV possess frameshift sequences within the mRNAs; these can be located in the 5′-UTR, coding region or 3′-UTR of the mRNAs. Internal initiation of translation on picornavirus RNA is mediated by IRESs within the 5′-UTR. Other viruses have translational enhancers within the 3′-UTR that may substitute for a poly(A) tail.
The drivers behind virus evolution seem relatively straightforward: (i) efficient transmission between host organisms, (ii) efficient redirection of host cell mechanisms, especially translation, toward viral multiplication at the expense of host cell functions, and (iii) an ability to evade the host cell defense mechanisms, both extracellular (e.g., immune system) and intracellular. Against these, there is evolutionary pressure on the host to elaborate antiviral defense mechanisms, which in turn puts pressure on the viruses to develop mechanisms of negating or bypassing these host cell defenses. Although this picture is almost certainly oversimplified, there is little doubt that virus evolution is relatively simple as compared with higher eukaryote whole organism evolution.
Viruses have contributed enormously to our understanding of eukaryotic protein synthesis mechanisms, and mRNA structure and function. This is in part due to the fact that, irrespective of whether the viral genome is DNA or RNA, positive or negative strand, double or single stranded, all viruses are dependent on the cellular protein synthesis machinery, whereas they vary in their dependence on other facets of host-cell gene expression. The contribution of virus research to our understanding of mRNA structure and function, and the mechanism of protein biosynthesis is highlighted by the following list of discoveries (in approximate chronological order) all made through exploiting viruses: eubacterial translation initiation sites (RNA bacteriophages), poly(A) tails (vaccinia), 5′ caps (reovirus), PKR (PV), translation of only the first cistron of polycistronic mRNAs (tobacco mosaic virus), leaky termination (plant RNA viruses), the scanning ribosome mechanism (reovirus), eIF4G (PV), IRESs (picornaviruses), programmed-1 frame-shifting (retroviruses and coronaviruses), ribosome shunting (adenovirus), and reinitiation after translation of a long ORF (pneumoviruses, caliciviruses, influenza B viruses). Moreover, this list has not closed because there are unusual viral RNA translation mechanisms that remain to be fully elucidated, for example, the extraordinary translational stop–restart mechanism promoted by the FMDV 2A peptide. Some of the earlier discoveries in the above list exploited the fact that in the era before recombinant DNA techniques and transcription vectors had been developed, viruses were often the best source of a single mRNA species (e.g., PV, tobacco mosaic virus) or a very limited number of mRNAs (reovirus), with the added advantage of being able to synthesize defined radiolabeled mRNAs in some cases (e.g., reovirus). Viruses also provide excellent tools with which to study cellular protein synthesis or novel viral mechanisms of initiation. For example, the picornavirus proteases that cleave eIF4G are widely used to study cap-independent mechanisms of initiation, and a range of viral IRES elements that require different factors for function are routinely used to dissect translational control processes, such as miRNA-mediated regulation of gene expression. However, the more recent discoveries are due to the fact that viruses, particularly positive-strand RNA viruses with small genomes, have evolved to exploit the extremes of behavior (or what might be thought the weaknesses) in the host cell translation machinery (e.g., programmed frameshifting, leaky termination, shunting, reinitiation after a long ORF). Some of these viral “tricks” (e.g., IRESs, and perhaps shunting) have evolved to enable efficient viral protein synthesis to proceed despite the virus-induced shutdown of host cell protein synthesis, and, certainly in the case of IRESs, the discovery has proved highly relevant to mRNA translation in the uninfected host cell.
Many other viral “tricks” have evolved to allow more than one protein to be expressed from a single RNA: frameshifting and leaky termination give two proteins with the same N-termini, while reinitiation after a long ORF gives two unrelated proteins as an alternative to relying entirely on proteolytic processing of a polyprotein by virus-encoded proteases, or synthesis of monocistronic subgenomic mRNA(s). It is not yet clear whether these events also occur in the translation of some cellular mRNAs. If they do, they certainly concern only a very limited number of cellular mRNAs, and without the clues provided by the viral examples as to the sequence and structure of the required RNA motifs, the task of identifying such cellular mRNAs would be like trying to find the proverbial needle in a field of haystacks. Even if they do not occur in any cellular mRNA, the viral mechanisms are always instructive, because they provide insights into issues such as why does reinitiation after a long ORF not normally occur with the vast majority of cellular mRNAs. Moreover, if the viral mRNA motifs directing leaky termination, frameshifting, or reinitiation after a long ORF, are absent from all essential cellular mRNAs, they provide potential targets for the development of novel antiviral agents targeted against the intracellular phase of the infectious process (although the high mutation rate of RNA viruses may well pose problems with this approach). Thus, while the “tricks” are essential for viral multiplication, they are also a potential Achilles heel.
Viruses not only interfere with host translational processes to improve competition for factors, etc., but they must also respond to a number of host antiviral “attacks”—this means that viruses have evolved ways in which to manipulate the host to ensure their replication is not inhibited. Viral countermeasures against host attack, and the delicate balance between the host and virus, are only beginning to be understood and it is likely that we will learn much more about this in the future.
There is no doubt that a greater understanding of how viruses regulate host translation, and identification of viral strategies, will aid in the development of novel antiviral therapies; the diversity and ingenuity of viruses is also likely to further surprise us.
Acknowledgments
We thank Dr. Mark Coldwell (University of Southampton UK) for very kindly drawing many of the figures, Prof. M. Mathews (New Jersey Medical School) for the PKR figure, Dr. I. Brierley (University of Cambridge) for his advice and also providing the frameshifting figures, and Dr. K. Spriggs (University of Nottingham) for reading the manuscript. Catherine Jopling is a BBSRC David Phillips Fellow and Anne Willis holds a BBSRC Professorial Fellowship.
References
- 1.La Scola B., Audic S., Robert C., Jungang L., de Lamballerie X., Drancourt M. A giant virus in amoebae. Science. 2003;299:2033. doi: 10.1126/science.1081867. [DOI] [PubMed] [Google Scholar]
- 2.Raoult D., Audic S., Robert C., Abergel C., Renesto P., Ogata H. The 1.2-megabase genome sequence of mimivirus. Science. 2004;306:1344–1350. doi: 10.1126/science.1101485. [DOI] [PubMed] [Google Scholar]
- 3.Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lloyd R.E. Translational control by viral proteinases. Virus Res. 2006;119:76–88. doi: 10.1016/j.virusres.2005.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wendel H.G., Silva R.L.A., Malina A., Mills J.R., Zhu H., Ueda T. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21:3232–3237. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bushell M., Wood W., Carpenter G., Pain V.M., Morley S.J., Clemens M.J. Disruption of the interaction of mammalian protein synthesis eukaryotic initiation factor 4B with the poly(A)-binding protein by caspase- and viral protease-mediated cleavages. J Biol Chem. 2001;276:23922–23928. doi: 10.1074/jbc.M100384200. [DOI] [PubMed] [Google Scholar]
- 7.Gebauer F., Hentze M.W. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5:827–835. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson R.J., Standart N. How do microRNAs regulate gene expression? Sci STKE. 2007;2007:re1. doi: 10.1126/stke.3672007re1. [DOI] [PubMed] [Google Scholar]
- 9.Damania B. Oncogenic gamma-herpesviruses: comparison of viral proteins involved in tumorigenesis. Nat Rev Microbiol. 2004;2:656–668. doi: 10.1038/nrmicro958. [DOI] [PubMed] [Google Scholar]
- 10.Mohr I. Phosphorylation and dephosphorylation events that regulate viral mRNA translation. Virus Res. 2006;119:89–99. doi: 10.1016/j.virusres.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Walsh D., Perez C., Notary J., Mohr I. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J Virol. 2005;79:8057–8064. doi: 10.1128/JVI.79.13.8057-8064.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clemens M.J. Epstein-Barr virus: inhibition of apoptosis as a mechanism of cell transformation. Int J Biochem Cell Biol. 2006;38:164–169. doi: 10.1016/j.biocel.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 13.Kudchodkar S.B., Yu Y.J., Maguire T.G., Alwine J.C. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J Virol. 2004;78:11030–11039. doi: 10.1128/JVI.78.20.11030-11039.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang F.Z., Weber F., Croce C., Liu C.G., Liao X., Pellett P.E. Human cytomegalovirus infection alters the expression of cellular microRNA species that affect its replication. J Virol. 2008;82:9065–9074. doi: 10.1128/JVI.00961-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arias C., Walsh D., Harbell J., Wilson A.C., Mohr I. Activation of host translational control pathways by a viral developmental switch. PLoS Pathog. 2009;5:e1000334. doi: 10.1371/journal.ppat.1000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin S.A., Moss B. Modification of RNA by messenger-RNA guanylyltransferase and messenger-RNA (guanine-7-)methyltransferase from vaccinia virions. J Biol Chem. 1975;250:9330–9335. [PubMed] [Google Scholar]
- 17.Walsh D., Mohr I. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 2004;18:660–672. doi: 10.1101/gad.1185304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Shea C.C., Choi S., McCormick F., Stokoe D. Adenovirus overrides cellular checkpoints for protein translation. Cell Cycle. 2005;4:883–888. doi: 10.4161/cc.4.7.1791. [DOI] [PubMed] [Google Scholar]
- 19.Gingras A.C., Svitkin Y., Belsham G.J., Pause A., Sonenberg N. Activation of the translational suppressor 4E-BP1 following infection with encephalomyocarditis virus and poliovirus. Proc Natl Acad Sci USA. 1996;A93:5578–5583. doi: 10.1073/pnas.93.11.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beretta L., Svitkin Y.V., Sonenberg N. Rapamycin stimulates viral protein synthesis and augments the shutoff of host protein synthesis upon picornavirus infection. J Virol. 1996;70:8993–8996. doi: 10.1128/jvi.70.12.8993-8996.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Connor J.H., Lyles D.S. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J Virol. 2002;76:10177–10187. doi: 10.1128/JVI.76.20.10177-10187.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kneller E.L.P., Connor J.H., Lyles D.S. hnRNPs relocalize to the cytoplasm following infection with vesicular stomatitis virus. J Virol. 2009;83:770–780. doi: 10.1128/JVI.01279-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gingras A.C., Raught B., Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 24.Fukunaga R., Hunter T. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 1997;16:1921–1933. doi: 10.1093/emboj/16.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waskiewicz A.J., Flynn A., Proud C.G., Cooper J.A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16:1909–1920. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wells S.E., Hillner P.E., Vale R.D., Sachs A.B. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell. 1998;2:135–140. doi: 10.1016/s1097-2765(00)80122-7. [DOI] [PubMed] [Google Scholar]
- 27.Umbach J.L., Kramer M.F., Jurak I., Karnowski H.W., Coen D.M., Cullen B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheper G.C., Proud C.G. Does phosphorylation of the cap-binding protein eIF4E play a role in translation initiation? Eur J Biochem. 2002;269:5350–5359. doi: 10.1046/j.1432-1033.2002.03291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willcocks M.M., Carter M.J., Roberts L.O. Cleavage of eukaryotic initiation factor eIF4G and inhibition of host-cell protein synthesis during feline calicivirus infection. J Gen Virol. 2004;85:1125–1130. doi: 10.1099/vir.0.19564-0. [DOI] [PubMed] [Google Scholar]
- 30.Riley D., Flint S.J. RNA-binding properties of a translational activator, the adenovirus L4 100-kilodalton protein. J Virol. 1993;67:3586–3595. doi: 10.1128/jvi.67.6.3586-3595.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuesta R., Xi Q.R., Schneider R.J. Adenovirus-specific translation by displacement of kinase Mnk1 from cap-initiation complex elF4F. EMBO J. 2000;19:3465–3474. doi: 10.1093/emboj/19.13.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y., Yao Y., Xu H., Lambeth L., Smith L.P., Kgosana L. A functional microRNA-155 ortholog encoded by the oncogenic Marek's disease virus. J Virol. 2009;83:489–492. doi: 10.1128/JVI.01166-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia T., O'Hara A., Araujo I., Barreto J., Carvalho E., Sapucaia J.B. EBV microRNAs in primary lymphomas and targeting of CXCL-11 by ebv-mir-BHRF1-3. Cancer Res. 2008;68:1436–1442. doi: 10.1158/0008-5472.CAN-07-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feigenblum D., Schneider R.J. Modification of eukaryotic initiation-factor 4F during infection by influenza-virus. J Virol. 1993;67:3027–3035. doi: 10.1128/jvi.67.6.3027-3035.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Etchison D., Milburn S.C., Edery I., Sonenberg N., Hershey J.W. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J Biol Chem. 1982;257:14806–14810. [PubMed] [Google Scholar]
- 36.Lamphear B.J., Kirchweger R., Skern T., Rhoads R.E. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap-dependent and cap-independent translational initiation. J Biol Chem. 1995;270:21975–21983. doi: 10.1074/jbc.270.37.21975. [DOI] [PubMed] [Google Scholar]
- 37.Lamphear B.J., Yan R., Yang F., Waters D., Liebig H.D., Klump H. Mapping the cleavage site in protein synthesis initiation factor eIF-4 gamma of the 2A proteases from human Coxsackievirus and rhinovirus. J Biol Chem. 1993;268:19200–19203. [PubMed] [Google Scholar]
- 38.Gradi A., Foeger N., Strong R., Svitkin Y.V., Sonenberg N., Skern T. Cleavage of eukaryotic translation initiation factor 4GII within foot-and-mouth disease virus-infected cells: identification of the L-protease cleavage site in vitro. J Virol. 2004;78:3271–3278. doi: 10.1128/JVI.78.7.3271-3278.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sullivan C.S., Grundhoff A.T., Tevethia S., Pipas J.M., Ganem D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature. 2005;435:682–686. doi: 10.1038/nature03576. [DOI] [PubMed] [Google Scholar]
- 40.Perez L., Carrasco L. Lack of direct correlation between p220 cleavage and the shut-off of host translation after poliovirus infection. Virology. 1992;189:178–186. doi: 10.1016/0042-6822(92)90693-j. [DOI] [PubMed] [Google Scholar]
- 41.Gradi A., Svitkin Y.V., Imataka H., Sonenberg N. Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc Natl Acad Sci USA. 1998;A95:11089–11094. doi: 10.1073/pnas.95.19.11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ali I.K., McKendrick L., Morley S.J., Jackson R.J. Activity of the hepatitis A virus IRES requires association between the cap-binding translation initiation factor (eIF4E) and eIF4G. J Virol. 2001;75:7854–7863. doi: 10.1128/JVI.75.17.7854-7863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pestova T.V., Shatsky I.N., Hellen C.U. Functional dissection of eukaryotic initiation factor 4F: the 4A subunit and the central domain of the 4G subunit are sufficient to mediate internal entry of 43S preinitiation complexes. Mol Cell Biol. 1996;16:6870–6878. doi: 10.1128/mcb.16.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson J.E., Pestova T.V., Hellen C.U., Sarnow P. Initiation of protein synthesis from the A site of the ribosome. Cell. 2000;102:511–520. doi: 10.1016/s0092-8674(00)00055-6. [DOI] [PubMed] [Google Scholar]
- 45.Goodfellow I., Chaudhry Y., Gioldasi I., Gerondopoulos A., Natoni A., Labrie L. Calicivirus translation initiation requires an interaction between VPg and eIF4E. EMBO Rep. 2005;6:968–972. doi: 10.1038/sj.embor.7400510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chaudhry Y., Nayak A., Bordeleau M.E., Tanaka J., Pelletier J., Belsham G.J. Caliciviruses differ in their functional requirements for eIF4F components. J Biol Chem. 2006;281:25315–25325. doi: 10.1074/jbc.M602230200. [DOI] [PubMed] [Google Scholar]
- 47.Ohlmann T., Prevot D., Decimo D., Roux F., Garin J., Morley S.J. In vitro cleavage of eIF4GI but not eIF4GII by HIV-1 protease and its effects on translation in the rabbit reticulocyte lysate system. J Mol Biol. 2002;318:9–20. doi: 10.1016/S0022-2836(02)00070-0. [DOI] [PubMed] [Google Scholar]
- 48.Ventoso I., Sanz M.A., Molina S., Berlanga J.J., Carrasco L., Esteban M. Translational resistance of late alphavirus mRNA to eIF2 alpha phosphorylation: a strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 2006;20:87–100. doi: 10.1101/gad.357006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alvarez E., Menendez-Arias L., Carrasco L. The eukaryotic translation initiation factor 4GI is cleaved by different retroviral proteases. J Virol. 2003;77:12392–12400. doi: 10.1128/JVI.77.23.12392-12400.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joachims M., Van Breugel P.C., Lloyd R.E. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J Virol. 1999;73:718–727. doi: 10.1128/jvi.73.1.718-727.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kerekatte V., Keiper B.D., Badorff C., Cai A.L., Knowlton K.U., Rhoads R.E. Cleavage of poly(A)-binding protein by coxsackievirus 2A protease in vitro and in vivo: another mechanism for host protein synthesis shutoff ? J Virol. 1999;73:709–717. doi: 10.1128/jvi.73.1.709-717.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mangus D.A., Evans M.C., Jacobson A. Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol. 2003;4:14. doi: 10.1186/gb-2003-4-7-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mullin C., Duning K., Barnekow A., Richter D., Kremerskothen J., Mohr E. Interaction of rat poly(A)-binding protein with poly(A)- and non-poly(A) sequences is preferentially mediated by RNA recognition motifs 3 + 4. FEBS Lett. 2004;576:437–441. doi: 10.1016/j.febslet.2004.09.054. [DOI] [PubMed] [Google Scholar]
- 54.Kuyumcu-Martinez M., Belliot G., Sosnovtsev S.V., Chang K.O., Green K.Y., Lloyd R.E. Calicivirus 3C-like proteinase inhibits cellular translation by cleavage of poly(A)-binding protein. J Virol. 2004;78:8172–8182. doi: 10.1128/JVI.78.15.8172-8182.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bergamini G., Preiss T., Hentze M.W. Picornavirus IRESes and the poly(A) tail jointly promote cap-independent translation in a mammalian cell-free system. RNA. 2000;6:1781–1790. doi: 10.1017/s1355838200001679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Michel Y.M., Borman A.M., Paulous S., Kean K.M. Eukaryotic initiation factor 4G-poly(A) binding protein interaction is required for poly(A) tail-mediated stimulation of picornavirus internal ribosome entry segment-driven translation but not for X-mediated stimulation of hepatitis C virus translation. Mol Cell Biol. 2001;21:4097–4109. doi: 10.1128/MCB.21.13.4097-4109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y., Feigenblum D., Schneider R.J. A late adenovirus factor induces eIF-4e dephosphorylation and inhibition of cell protein-synthesis. J Virol. 1994;68:7040–7050. doi: 10.1128/jvi.68.11.7040-7050.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonderoff J.M., Larey J.L., Lloyd R.E. Cleavage of poly(A)-binding protein by poliovirus 3C proteinase inhibits viral internal ribosome entry site-mediated translation. J Virol. 2008;82:9389–9399. doi: 10.1128/JVI.00006-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ilkow C.S., Mancinelli V., Beatch M.D., Hobman T.C. Rubella virus capsid protein interacts with poly(A)-binding protein and inhibits translation. J Virol. 2008;82:4284–4294. doi: 10.1128/JVI.02732-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Imai M., Akatani K., Ikegami N., Furuichi Y. Capped and conserved terminal structures in human rotavirus genome double-stranded-RNA segments. J Virol. 1983;47:125–136. doi: 10.1128/jvi.47.1.125-136.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Piron M., Vende P., Cohen J., Poncet D. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J. 1998;17:5811–5821. doi: 10.1093/emboj/17.19.5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montero H., Arias C.F., Lopez S. Rotavirus nonstructural protein NSP3 is not required for viral protein synthesis. J Virol. 2006;80:9031–9038. doi: 10.1128/JVI.00437-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blakqori G., van Knippenberg I., Elliott R.M. Bunyamwera Orthobunyavirus S-segment untranslated regions mediate poly(A) tail-independent translation. J Virol. 2009;83:3637–3646. doi: 10.1128/JVI.02201-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiu W.W., Kinney R.M., Dreher T.W. Control of translation by the 5′- and 3′-terminal regions of the dengue virus genome. J Virol. 2005;79:8303–8315. doi: 10.1128/JVI.79.13.8303-8315.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jang S.K., Krausslich H.G., Nicklin M.J., Duke G.M., Palmenberg A.C., Wimmer E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol. 1988;62:2636–2643. doi: 10.1128/jvi.62.8.2636-2643.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pelletier J., Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334:320–325. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- 67.Jackson R.J., Kaminski A. Internal initiation of translation in eukaryotes: the picornavirus paradigm and beyond. RNA. 1995;1:985–1000. [PMC free article] [PubMed] [Google Scholar]
- 68.Kolupaeva V.G., Lomakin I.B., Pestova T.V., Hellen C.U. Eukaryotic initiation factors 4G and 4A mediate conformational changes downstream of the initiation codon of the encephalomyocarditis virus internal ribosomal entry site. Mol Cell Biol. 2003;23:687–698. doi: 10.1128/MCB.23.2.687-698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Breyne S., Yu Y.P., Unbehaun A., Pestova T.V., Hellen C.U.T. Direct functional interaction of initiation factor eIF4G with type 1 internal ribosomal entry sites. Proc Natl Acad Sci USA. 2009;A106:9197–9202. doi: 10.1073/pnas.0900153106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hunt S.L., Jackson R.J. Polypyrimidine-tract binding protein (PTB) is necessary, but not sufficient, for efficient internal initiation of translation of human rhinovirus-2 RNA. RNA. 1999;5:344–359. doi: 10.1017/s1355838299981414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Florez P.M., Sessions O.M., Wagner E.J., Gromeier M., Garcia-Blanco M.A. The polypyrimidine tract binding protein is required for efficient picornavirus gene expression and propagation. J Virol. 2005;79:6172–6179. doi: 10.1128/JVI.79.10.6172-6179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Costa-Mattioli M., Svitkin Y., Sonenberg N. La autoantigen is necessary for optimal function of the poliovirus and hepatitis C virus internal ribosome entry site in vivo and in vitro. Mol Cell Biol. 2004;24:6861–6870. doi: 10.1128/MCB.24.15.6861-6870.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pilipenko E.V., Pestova T.V., Kolupaeva V.G., Khitrina E.V., Poperechnaya A.N., Agol V.I. A cell cycle-dependent protein serves as a template-specific translation initiation factor. Genes Dev. 2000;14:2028–2045. [PMC free article] [PubMed] [Google Scholar]
- 74.Monie T.P., Perrin A.J., Birtley J.R., Sweeney T.R., Karakasiliotis I., Chaudhry Y. Structural insights into the transcriptional and translational roles of Ebp1. EMBO J. 2007;26:3936–3944. doi: 10.1038/sj.emboj.7601817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roberts L.O., Seamons R.A., Belsham G.J. Recognition of picornavirus internal ribosome entry sites within cells; influence of cellular and viral proteins. RNA. 1998;4:520–529. doi: 10.1017/s1355838298971989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dobrikova E.Y., Grisham R.N., Kaiser C., Lin J., Gromeier M. Competitive translation efficiency at the picornavirus type 1 internal ribosome entry site facilitated by viral cis and trans factors. J Virol. 2006;80:3310–3321. doi: 10.1128/JVI.80.7.3310-3321.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaku Y., Chard L.S., Inoue T., Belsham G.J. Unique characteristics of a picornavirus internal ribosome entry site from the porcine teschovirus-1 talfan. J Virol. 2002;76:11721–11728. doi: 10.1128/JVI.76.22.11721-11728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bakhshesh M., Groppelli E., Willcocks M.A., Royall E., Belsham G.J., Roberts L.O. The picornavirus avian encephalomyelitis virus possesses a hepatitis C virus-like internal ribosome entry site element. J Virol. 2008;82:1993–2003. doi: 10.1128/JVI.01957-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pisarev A.V., Chard L.S., Kaku Y., Johns H.L., Shatsky I.N., Belsham G.J. Functional and structural similarities between the internal ribosome entry sites of hepatitis C virus and porcine teschovirus, a picornavirus. J Virol. 2004;78:4487–4497. doi: 10.1128/JVI.78.9.4487-4497.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brown E.A., Zhang H., Ping L.H., Lemon S.M. Secondary structure of the 5′ nontranslated regions of hepatitis C virus and pestivirus genomic RNAs. Nucleic Acids Res. 1992;20:5041–5045. doi: 10.1093/nar/20.19.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ueda T., Watanabe-Fukunaga R., Fukuyama H., Nagata S., Fukunaga R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol. 2004;24:6539–6549. doi: 10.1128/MCB.24.15.6539-6549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pestova T.V., Shatsky I.N., Fletcher S.P., Jackson R.J., Hellen C.U. A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev. 1998;12:67–83. doi: 10.1101/gad.12.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lancaster A.M., Jan E., Sarnow P. Initiation factor-independent translation mediated by the hepatitis C virus internal ribosome entry site. RNA. 2006;12:894–902. doi: 10.1261/rna.2342306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thompson S.R., Gulyas K.D., Sarnow P. Internal initiation in Saccharomyces cerevisiae mediated by an initiator tRNA/eIF2-independent internal ribosome entry site element. Proc Natl Acad Sci USA. 2001;98:12972–12977. doi: 10.1073/pnas.241286698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spahn C.M., Kieft J.S., Grassucci R.A., Penczek P.A., Zhou K., Doudna J.A. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science. 2001;291:1959–1962. doi: 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]
- 86.Passmore L.A., Schmeing T.M., Maag D., Applefield D.J., Acker M.G., Algire M.A. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol Cell. 2007;26:41–50. doi: 10.1016/j.molcel.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 87.Fraser C.S., Hershey J.W., Doudna J.A. The pathway of hepatitis C virus mRNA recruitment to the human ribosome. Nat Struct Mol Biol. 2009;16:397–404. doi: 10.1038/nsmb.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Otto G.A., Puglisi J.D. The pathway of HCV IRES-mediated translation initiation. Cell. 2004;119:369–380. doi: 10.1016/j.cell.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 89.Locker N., Easton L.E., Lukavsky P.J. HCV and CSFV IRES domain II mediate eIF2 release during 80S ribosome assembly. EMBO J. 2007;26:795–805. doi: 10.1038/sj.emboj.7601549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sasaki J., Nakashima N. Translation initiation at the CUU codon is mediated by the internal ribosome entry site of an insect picorna-like virus in vitro. J Virol. 1999;73:1219–1226. doi: 10.1128/jvi.73.2.1219-1226.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Woolaway K.E., Lazaridis K., Belsham G.J., Carter M.J., Roberts L.O. The 5′ untranslated region of Rhopalosiphum padi virus contains an internal ribosome entry site which functions efficiently in mammalian, plant, and insect translation systems. J Virol. 2001;75:10244–10249. doi: 10.1128/JVI.75.21.10244-10249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jan E., Sarnow P. Factorless ribosome assembly on the internal ribosome entry site of cricket paralysis virus. J Mol Biol. 2002;324:889–902. doi: 10.1016/s0022-2836(02)01099-9. [DOI] [PubMed] [Google Scholar]
- 93.Jan E., Kinzy T.G., Sarnow P. Divergent tRNA-like element supports initiation, elongation, and termination of protein biosynthesis. Proc Natl Acad Sci USA. 2003;100:15410–15415. doi: 10.1073/pnas.2535183100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pestova T.V., Hellen C.U. Translation elongation after assembly of ribosomes on the Cricket paralysis virus internal ribosomal entry site without initiation factors or initiator tRNA. Genes Dev. 2003;17:181–186. doi: 10.1101/gad.1040803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Costantino D.A., Pfingsten J.S., Rambo R.P., Kieft J.S. tRNA-mRNA mimicry drives translation initiation from a viral IRES. Nat Struct Mol Biol. 2008;15:57–64. doi: 10.1038/nsmb1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Spahn C.M., Jan E., Mulder A., Grassucci R.A., Sarnow P., Frank J. Cryo-EM visualization of a viral internal ribosome entry site bound to human ribosomes: the IRES functions as an RNA-based translation factor. Cell. 2004;118:465–475. doi: 10.1016/j.cell.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 97.Deniz N., Lenarcic E.M., Landry D.M., Thompson S.R. Translation initiation factors are not required for Dicistroviridae IRES function in vivo. RNA. 2009;15:932–946. doi: 10.1261/rna.1315109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Triboulet R., Mari B., Lin Y.L., Chable-Bessia C., Bennasser Y., Lebrigand K. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science. 2007;315:1579–1582. doi: 10.1126/science.1136319. [DOI] [PubMed] [Google Scholar]
- 99.Terenin I.M., Dmitriev S.E., Andreev D.E., Shatsky I.N. Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nat Struct Mol Biol. 2008;15:836–841. doi: 10.1038/nsmb.1445. [DOI] [PubMed] [Google Scholar]
- 100.Xi Q.R., Cuesta R., Schneider R.J. Tethering of eIF4G to adenoviral mRNAs by viral 100k protein drives ribosome shunting. Genes Dev. 2004;18:1997–2009. doi: 10.1101/gad.1212504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Buck C.B., Shen X., Egan M.A., Pierson T.C., Walker C.M., Siliciano R.F. The human immunodeficiency virus type 1 gag gene encodes an internal ribosome entry site. J Virol. 2001;75:181–191. doi: 10.1128/JVI.75.1.181-191.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brasey A., Lopez-Lastra M., Ohlmann T., Beerens N., Berkhout B., Darlix J.L. The leader of human immunodeficiency virus type 1 genomic RNA harbors an internal ribosome entry segment that is active during the G2/M phase of the cell cycle. J Virol. 2003;77:3939–3949. doi: 10.1128/JVI.77.7.3939-3949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Herbreteau C.H., Weill L., Decimo D., Prevot D., Darlix J.L., Sargueil B. HIV-2 genomic RNA contains a novel type of IRES located downstream of its initiation codon. Nat Struct Mol Biol. 2005;12:1001–1007. doi: 10.1038/nsmb1011. [DOI] [PubMed] [Google Scholar]
- 104.Pfeffer S., Zavolan M., Grasser F.A., Chien M., Russo J.J., Ju J. Identification of virus-encoded microRNAs. Science. 2004;304:734–736. doi: 10.1126/science.1096781. [DOI] [PubMed] [Google Scholar]
- 105.Pfeffer S., Sewer A., Lagos-Quintana M., Sheridan R., Sander C., Grasser F.A. Identification of microRNAs of the herpesvirus family. Nat Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 106.Seo G.J., Fink L.H., O'Hara B., Atwood W.J., Sullivan C.S. Evolutionarily conserved function of a viral microRNA. J Virol. 2008;82:9823–9828. doi: 10.1128/JVI.01144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sullivan C.S., Sung C.K., Pack C.D., Grundhoff A., Lukacher A.E., Benjamin T.L. Murine Polyomavirus encodes a microRNA that cleaves early RNA transcripts but is not essential for experimental infection. Virology. 2009;387:157–167. doi: 10.1016/j.virol.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Takyar S., Hickerson R.P., Noller H.F. MRNA helicase activity of the ribosome. Cell. 2005;120:49–58. doi: 10.1016/j.cell.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 109.Xuan B.Q., Qian Z.K., Torigoi E., Yu D. Human cytomegalovirus protein pul38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J Virol. 2009;83:3463–3474. doi: 10.1128/JVI.02307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ouellet D.L., Plante I., Landry P., Barat C., Janelle M.E., Flamand L. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucleic Acids Res. 2008;36:2353–2365. doi: 10.1093/nar/gkn076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lin J., Cullen B.R. Analysis of the interaction of primate retroviruses with the human RNA interference machinery. J Virol. 2007;81:12218–12226. doi: 10.1128/JVI.01390-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grundhoff A., Sullivan C.S., Ganem D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA. 2006;12:733–750. doi: 10.1261/rna.2326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cai X., Schafer A., Lu S., Bilello J.P., Desrosiers R.C., Edwards R. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. 2006;2:e23. doi: 10.1371/journal.ppat.0020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ventoso I., Blanco R., Perales C., Carrasco L. HIV-1 protease cleaves eukaryotic initiation factor 4G and inhibits cap-dependent translation. Proc Natl Acad Sci USA. 2001;98:12966–12971. doi: 10.1073/pnas.231343498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Seo G.J., Chen C.J., Sullivan C.S. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology. 2009;383:183–187. doi: 10.1016/j.virol.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 116.Murphy E., Vanicek J., Robins H., Shenk T., Levine A.J. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc Natl Acad Sci USA. 2008;105:5453–5458. doi: 10.1073/pnas.0711910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xu N., Segerman B., Zhou X., Akusjarvi G. Adenovirus virus-associated RNAII-derived small RNAs are efficiently incorporated into the RNA-induced silencing complex and associate with polyribosomes. J Virol. 2007;81:10540–10549. doi: 10.1128/JVI.00885-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Choy E.Y., Siu K.L., Kok K.H., Lung R.W., Tsang C.M., To K.F. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J Exp Med. 2008;205:2551–2560. doi: 10.1084/jem.20072581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Samols M.A., Skalsky R.L., Maldonado A.M., Riva A., Lopez M.C., Baker H.V. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007;3:e65. doi: 10.1371/journal.ppat.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nachmani D., Stern-Ginossar N., Sarid R., Mandelboim O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe. 2009;5:376–385. doi: 10.1016/j.chom.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 121.Gottwein E., Mukherjee N., Sachse C., Frenzel C., Majoros W.H., Chi J.T. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450:1096–1099. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ylosmaki E., Hakkarainen T., Hemminki A., Visakorpi T., Andino R., Saksela K. Generation of a conditionally replicating adenovirus based on targeted destruction of E1A mRNA by a cell type-specific MicroRNA. J Virol. 2008;82:11009–11015. doi: 10.1128/JVI.01608-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ziegelbauer J.M., Sullivan C.S., Ganem D. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat Genet. 2009;41:130–134. doi: 10.1038/ng.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cameron J.E., Fewell C., Yin Q., McBride J., Wang X., Lin Z. Epstein-Barr virus growth/latency III program alters cellular microRNA expression. Virology. 2008;382:257–266. doi: 10.1016/j.virol.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Godshalk S.E., Bhaduri-McIntosh S., Slack F.J. Epstein-Barr virus-mediated dysregulation of human microRNA expression. Cell Cycle. 2008;7:3595–3600. doi: 10.4161/cc.7.22.7120. [DOI] [PubMed] [Google Scholar]
- 126.Wang X.M., Flynn A., Waskiewicz A.J., Webb B.L.J., Vries R.G., Baines I.A. The phosphorylation of eukaryotic initiation factor eIF4E in response to phorbol esters, cell stresses, and cytokines is mediated by distinct MAP kinase pathways. J Biol Chem. 1998;273:9373–9377. doi: 10.1074/jbc.273.16.9373. [DOI] [PubMed] [Google Scholar]
- 127.Tsukiyama-Kohara K., Iizuka N., Kohara M., Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J Virol. 1992;66:1476–1483. doi: 10.1128/jvi.66.3.1476-1483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lu S., Cullen B.R. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J Virol. 2004;78:12868–12876. doi: 10.1128/JVI.78.23.12868-12876.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bennasser Y., Le S.Y., Benkirane M., Jeang K.T. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity. 2005;22:607–619. doi: 10.1016/j.immuni.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 130.Lecellier C.H., Dunoyer P., Arar K., Lehmann-Che J., Eyquem S., Himber C. A cellular microRNA mediates antiviral defense in human cells. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- 131.Jopling C.L., Yi M., Lancaster A.M., Lemon S.M., Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 132.Henke J.I., Goergen D., Zheng J., Song Y., Schuttler C.G., Fehr C. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008;27:3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pedersen I.M., Cheng G., Wieland S., Volinia S., Croce C.M., Chisari F.V. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kelly E.J., Hadac E.M., Greiner S., Russell S.J. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat Med. 2008;14:1278–1283. doi: 10.1038/nm.1776. [DOI] [PubMed] [Google Scholar]
- 135.Yuan P.W., Bartlam M., Lou Z.Y., Chen S.D., Zhou J., He X.J. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature. 2009;458:909–913. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- 136.Daughenbaugh K.F., Fraser C.S., Hershey J.W.B., Hardy M.E. The genome-linked protein VPg of the Norwalk virus binds eIF3, suggesting its role in translation initiation complex recruitment. EMBO J. 2003;22:2852–2859. doi: 10.1093/emboj/cdg251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dreher T.W., Miller W.A. Translational control in positive strand RNA plant viruses. Virology. 2006;344:185–197. doi: 10.1016/j.virol.2005.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Leonard S., Plante D., Wittmann S., Daigneault N., Fortin M.G., Laliberte J.F. Complex formation between potyvirus VPg and translation eukaryotic initiation factor 4E correlates with virus infectivity. J Virol. 2000;74:7730–7737. doi: 10.1128/jvi.74.17.7730-7737.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Garfinkel M.S., Katze M.G. How does influenza virus regulate gene expression at the level of mRNA translation? Let us count the ways. Gene Expr. 1993;3:109–118. [PMC free article] [PubMed] [Google Scholar]
- 140.Shih S.R., Krug R.M. Surprising function of the three influenza viral polymerase proteins: selective protection of viral mRNAs against the cap-snatching reaction catalyzed by the same polymerase proteins. Virology. 1996;226:430–435. doi: 10.1006/viro.1996.0673. [DOI] [PubMed] [Google Scholar]
- 141.Dias A., Bouvier D., Crepin T., McCarthy A.A., Hart D.J., Baudin F. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature. 2009;458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 142.Yue Z.Y., Shatkin A.J. Double-stranded RNA-dependent protein kinase (PKR) is regulated by reovirus structural proteins. Virology. 1997;234:364–371. doi: 10.1006/viro.1997.8664. [DOI] [PubMed] [Google Scholar]
- 143.Burgui I., Yanguez E., Sonenberg N., Nieto A. Influenza virus mRNA translation revisited: is the eIF4E cap-binding factor required for viral mRNA translation? J Virol. 2007;81:12427–12438. doi: 10.1128/JVI.01105-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mir M.A., Panganiban A.T. A protein that replaces the entire cellular eIF4F complex. EMBO J. 2008;27:3129–3139. doi: 10.1038/emboj.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mir M.A., Panganiban A.T. Characterization of the RNA chaperone activity of hantavirus nucleocapsid protein. J Virol. 2006;80:6276–6285. doi: 10.1128/JVI.00147-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brierley I., Dos Ramos F.J. Programmed ribosomal frameshifting in HIV-1 and the SARS-CoV. Virus Res. 2006;119:29–42. doi: 10.1016/j.virusres.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Brierley I., Digard P., Inglis S.C. Characterization of an efficient coronavirus ribosomal frameshifting signal—requirement for an RNA pseudoknot. Cell. 1989;57:537–547. doi: 10.1016/0092-8674(89)90124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gaudin C., Mazauric M.H., Traikia M., Guittet E., Yoshizawa S., Fourmy D. Structure of the RNA signal essential for translational frameshifting in HIV-1. J Mol Biol. 2005;349:1024–1035. doi: 10.1016/j.jmb.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 149.Kollmus H., Honigman A., Panet A., Hauser H. The sequences of and distance between 2 cis-acting signals determine the efficiency of ribosomal frameshifting in human-immunodeficiency-virus type-1 and human T-cell leukemia-virus type-II in-vivo. J Virol. 1994;68:6087–6091. doi: 10.1128/jvi.68.9.6087-6091.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Giedroc D.P., Cornish P.V. Frameshifting RNA pseudoknots: structure and mechanism. Virus Res. 2009;139:193–208. doi: 10.1016/j.virusres.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jacks T., Madhani H.D., Masiarz F.R., Varmus H.E. Signals for ribosomal frameshifting in the rous-sarcoma virus gag-pol region. Cell. 1988;55:447–458. doi: 10.1016/0092-8674(88)90031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Namy O., Moran S.J., Stuart D.I., Gilbert R.J.C., Brierley I. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature. 2006;441:244–247. doi: 10.1038/nature04735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Brierley I., Rolley N.J., Jenner A.J., Inglis S.C. Mutational analysis of the RNA pseudoknot component of a coronavirus ribosomal frameshifting signal. J Mol Biol. 1991;220:889–902. doi: 10.1016/0022-2836(91)90361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Brierley I., Jenner A.J., Inglis S.C. Mutational analysis of the slippery-sequence component of a coronavirus ribosomal frameshifting signal. J Mol Biol. 1992;227:463–479. doi: 10.1016/0022-2836(92)90901-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Terenin I.M., Dmitriev S.E., Andreev D.E., Royall E., Belsham G.J., Roberts L.O. A cross-kingdom internal ribosome entry site reveals a simplified mode of internal ribosome entry. Mol Cell Biol. 2005;25:7879–7888. doi: 10.1128/MCB.25.17.7879-7888.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Drosten C. Kuala Lumpur; Malaysia: 2003. Presented at the 6th Asia Pacific Congress of Medical Virology. [PubMed] [Google Scholar]
- 157.Baranov P.V., Henderson C.M., Anderson C.B., Gesteland R.F., Atkins J.F., Howard M.T. Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology. 2005;332:498–510. doi: 10.1016/j.virol.2004.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Plant E.P., Perez-Alvarado G.C., Jacobs J.L., Mukhopadhyay B., Hennig M., Dinman J.D. A three-stemmed mRNA pseudoknot in the SARS coronavirus frameshift signal. PLoS Biol. 2005;3:e172. doi: 10.1371/journal.pbio.0030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Brierley I., Gilbert R.J.C., Pennell S. RNA pseudoknots and the regulation of protein synthesis. Biochem Soc Trans. 2008;36:684–689. doi: 10.1042/BST0360684. [DOI] [PubMed] [Google Scholar]
- 160.Cen S., Niu M.J., Saadatmand J., Guo F., Huang Y., Nabel G.J. Incorporation of Pol into human immunodeficiency virus type 1 Gag virus-like particles occurs independently of the upstream Gag domain in Gag-Pol. J Virol. 2004;78:1042–1049. doi: 10.1128/JVI.78.2.1042-1049.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shehu-Xhilaga M., Crowe S.M., Mak J. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J Virol. 2001;75:1834–1841. doi: 10.1128/JVI.75.4.1834-1841.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Biswas P., Jiang X., Pacchia A.L., Dougherty J.P., Peltz S.W. The human immunodeficiency virus type 1 ribosomal frameshifting site is an invariant sequence determinant and an important target for antiviral therapy. J Virol. 2004;78:2082–2087. doi: 10.1128/JVI.78.4.2082-2087.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Marcheschi R.J., Staple D.W., Butcher S.E. Programmed ribosomal frameshifting is SIV is induced by a highly structured RNA stem-loop. J Mol Biol. 2007;373:652–663. doi: 10.1016/j.jmb.2007.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Staple D.W., Butcher S.E. Pseudoknots: RNA structures with diverse functions. PLoS Biol. 2005;3:e213. doi: 10.1371/journal.pbio.0030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Staple D.W., Butcher S.E. Solution structure and thermodynamic investigation of the HIV-1 frameshift inducing element. J Mol Biol. 2005;349:1011–1023. doi: 10.1016/j.jmb.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 166.Stojdl D.F., Abraham N., Knowles S., Marius R., Brasey A., Lichty B.D. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J Virol. 2000;74:9580–9585. doi: 10.1128/jvi.74.20.9580-9585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gareiss P.C., Miller B.L. Ribosomal frameshifting: an emerging drug target for HIV. Curr Opin Investig Drugs. 2009;10:121–128. [PubMed] [Google Scholar]
- 168.Kozak M. Effects of intercistronic length on the efficiency of reinitiation by eukaryotic ribosomes. Mol Cell Biol. 1987;7:3438–3445. doi: 10.1128/mcb.7.10.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kontos H., Napthine S., Brierley I. Ribosomal pausing at a frameshifter RNA pseudoknot is sensitive to reading phase but shows little correlation with frameshift efficiency. Mol Cell Biol. 2001;21:8657–8670. doi: 10.1128/MCB.21.24.8657-8670.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kozak M. Constraints on reinitiation of translation in mammals. Nucleic Acids Res. 2001;29:5226–5232. doi: 10.1093/nar/29.24.5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Somogyi P., Jenner A.J., Brierley I., Inglis S.C. Ribosomal pausing during translation of an RNA pseudoknot. Mol Cell Biol. 1993;13:6931–6940. doi: 10.1128/mcb.13.11.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Poyry T.A.A., Kaminski A., Jackson R.J. What determines whether mammalian ribosomes resume scanning after translation of a short upstream open reading frame? Genes Dev. 2004;18:62–75. doi: 10.1101/gad.276504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Meyers G. Characterization of the sequence element directing translation reinitiation in RNA of the calicivirus, rabbit hemorrhagic disease virus. J Virol. 2007;81:9623–9632. doi: 10.1128/JVI.00771-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Meyers G. Translation of the minor capsid protein of a calicivirus is initiated by a novel termination-dependent reinitiation mechanism. J Biol Chem. 2003;278:34051–34060. doi: 10.1074/jbc.M304874200. [DOI] [PubMed] [Google Scholar]
- 175.Luttermann C., Meyers G. The importance of inter- and intramolecular base pairing for translation reinitiation on a eukaryotic bicistronic mRNA. Genes Dev. 2009;23:331–344. doi: 10.1101/gad.507609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Poyry T.A.A., Kaminski A., Connell E.J., Fraser C.S., Jackson R.J. The mechanism of an exceptional case of reinitiation after translation of a long ORF reveals why such events do not generally occur in mammalian mRNA translation. Genes Dev. 2007;21:3149–3162. doi: 10.1101/gad.439507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Pisarev A.V., Hellen C.U., Pestova T.V. Recycling of eukaryotic posttermination ribosomal complexes. Cell. 2007;131:286–299. doi: 10.1016/j.cell.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jackson D., Zurcher T., Barclay W. Reduced incorporation of the influenza B virus BM2 protein in virus particles decreases infectivity. Virology. 2004;322:276–285. doi: 10.1016/j.virol.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 179.Horvath C.M., Williams M.A., Lamb R.A. Eukaryotic coupled translation of tandem cistrons—identification of the influenza-b virus bm2 polypeptide. EMBO J. 1990;9:2639–2647. doi: 10.1002/j.1460-2075.1990.tb07446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Powell M.L., Napthine S., Jackson R.J., Brierley I., Brown T.D.K. Characterization of the termination–reinitiation strategy employed in the expression of influenza B virus BM2 protein. RNA. 2008;14:2394–2406. doi: 10.1261/rna.1231008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Powell M.L., Brown T.D.K., Brierley I. Translational termination-re-initiation in viral systems. Biochem Soc Trans. 2008;36:717–722. doi: 10.1042/BST0360717. [DOI] [PubMed] [Google Scholar]
- 182.Easton A.J., Domachowske J.B., Rosenberg H.F. Animal pneumoviruses: molecular genetics and pathogenesis. Clin Microbiol Rev. 2004;17:390–412. doi: 10.1128/CMR.17.2.390-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ahmadian G., Randhawa J.S., Easton A.J. Expression of the ORF-2 protein of the human respiratory syncytial virus M2 gene is initiated by a ribosomal termination-dependent reinitiation mechanism. EMBO J. 2000;19:2681–2689. doi: 10.1093/emboj/19.11.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gould P.S., Easton A.J. Coupled translation of the respiratory syncytial virus M2 open reading frames requires upstream sequences. J Biol Chem. 2005;280:21972–21980. doi: 10.1074/jbc.M502276200. [DOI] [PubMed] [Google Scholar]
- 185.Gould P.S., Easton A.J. Coupled translation of the second open reading frame of M2 mRNA is sequence dependent and differs significantly within the subfamily Pneumovirinae. J Virol. 2007;81:8488–8496. doi: 10.1128/JVI.00457-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Russell W.C. Adenoviruses: update on structure and function. J Gen Virol. 2009;90:1–20. doi: 10.1099/vir.0.003087-0. [DOI] [PubMed] [Google Scholar]
- 187.Berget S.M., Moore C., Sharp P.A. Spliced segments at 5′ terminus of adenovirus 2 late messenger-RNA. Proc Natl Acad Sci USA. 1977;A74:3171–3175. doi: 10.1073/pnas.74.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Moore C.L., Skolnikdavid H., Sharp P.A. Analysis of RNA cleavage at the adenovirus-2 l3 polyadenylation site. EMBO J. 1986;5:1929–1938. doi: 10.1002/j.1460-2075.1986.tb04446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yueh A., Schneider R.J. Translation by ribosome shunting on adenovirus and hsp70 mRNAs facilitated by complementarity to 18S rRNA. Genes Dev. 2000;14:414–421. [PMC free article] [PubMed] [Google Scholar]
- 190.Zhang B., Seitz S., Kusov Y., Zell R., Gauss-Muller V. RNA interaction and cleavage of poly(C)-binding protein 2 by hepatitis A virus protease. Biochem Biophys Res Commun. 2007;364:725–730. doi: 10.1016/j.bbrc.2007.09.133. [DOI] [PubMed] [Google Scholar]
- 191.Shmulevitz M., Marcato P., Lee P.W.K. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene. 2005;24:7720–7728. doi: 10.1038/sj.onc.1209041. [DOI] [PubMed] [Google Scholar]
- 192.Costas C., Martinez-Costas J., Bodelon G., Benavente J. The second open reading frame of the avian reovirus S1 gene encodes a transcription-dependent and CRM1-independent nucleocytoplasmic shuttling protein. J Virol. 2005;79:2141–2150. doi: 10.1128/JVI.79.4.2141-2150.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Grande A., Rodriguez E., Costas C., Everitt E., Benavente J. Oligomerization and cell-binding properties of the avian reovirus cell-attachment protein sigma C. Virology. 2000;274:367–377. doi: 10.1006/viro.2000.0473. [DOI] [PubMed] [Google Scholar]
- 194.Racine T., Barry C., Roy K., Dawe S.J., Shmulevitz M., Duncan R. Leaky scanning and scanning-independent ribosome migration on the tricistronic S1 mRNA of avian reovirus*. J Biol Chem. 2007;282:25613–25622. doi: 10.1074/jbc.M703708200. [DOI] [PubMed] [Google Scholar]
- 195.Latorre P., Kolakofsky D., Curran J. Sendai virus Y proteins are initiated by a ribosomal shunt. Mol Cell Biol. 1998;18:5021–5031. doi: 10.1128/mcb.18.9.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Curran J., Kolakofsky D. Replication of paramyxoviruses. Adv Virus Res. 1999;54:403–422. doi: 10.1016/s0065-3527(08)60373-5. [DOI] [PubMed] [Google Scholar]
- 197.Curran J., Kolakofsky D. Scanning independent ribosomal initiation of the sendai virus-Y proteins in vitro and in vivo. EMBO J. 1989;8:521–526. doi: 10.1002/j.1460-2075.1989.tb03406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Curran J., Kolakofsky D. Scanning independent ribosomal initiation of the Sendai virus X-protein. EMBO J. 1988;7:2869–2874. doi: 10.1002/j.1460-2075.1988.tb03143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.de Breyne S., Stalder R., Curran J. Intracellular processing of the Sendai virus C′ protein leads to the generation of a Y protein module: structure-functional implications. FEBS Lett. 2005;579:5685–5690. doi: 10.1016/j.febslet.2005.09.052. [DOI] [PubMed] [Google Scholar]
- 200.Doronina V.A., de Felipe P., Wu C., Sharma P., Sachs M.S., Ryan M.D. Dissection of a co-translational nascent chain separation event. Biochem Soc Trans. 2008;36:712–716. doi: 10.1042/BST0360712. [DOI] [PubMed] [Google Scholar]
- 201.Doronina V.A., Wu C., de Felipe P., Sachs M.S., Ryan M.D., Brown J.D. Site-specific release of nascent chains from ribosomes at a sense codon. Mol Cell Biol. 2008;28:4227–4239. doi: 10.1128/MCB.00421-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Meurs E., Chong K.L., Galabru J., Thomas N.S.B., Kerr I.M., Williams B.R.G. Molecular cloning and characterisation of the human double-stranded RNA-dependent protein kinase induced by interferon. Cell. 1990;62:379–390. doi: 10.1016/0092-8674(90)90374-n. [DOI] [PubMed] [Google Scholar]
- 203.Balachandran S., Roberts P.C., Brown L.E., Truong H., Pattnaik A.K., Archer D.R. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity. 2000;13:129–141. doi: 10.1016/s1074-7613(00)00014-5. [DOI] [PubMed] [Google Scholar]
- 204.Galabru J., Hovanessian A. Autophosphorylation of the protein-kinase dependent on double-stranded-RNA. J Biol Chem. 1987;262:15538–15544. [PubMed] [Google Scholar]
- 205.Strong R., Belsham G.J. Sequential modification of translation initiation factor elF4GI by two different foot-and-mouth disease virus proteases within infected baby hamster kidney cells: identification of the 3C(pro) cleavage site. J Gen Virol. 2004;85:2953–2962. doi: 10.1099/vir.0.80254-0. [DOI] [PubMed] [Google Scholar]
- 206.Burysek L., Pitha P.M. Latently expressed human herpesvirus 8-encoded interferon regulatory factor 2 inhibits double-stranded RNA-activated protein kinase. J Virol. 2001;75:2345–2352. doi: 10.1128/JVI.75.5.2345-2352.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Pavio N., Taylor D.R., Lai M.M.C. Detection of a novel unglycosylated form of hepatitis C virus E2 envelope protein that is located in the cytosol and interacts with PKR. J Virol. 2002;76:1265–1272. doi: 10.1128/JVI.76.3.1265-1272.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Gale M., Tan S.L., Wambach M., Katze M.G. Interaction of the interferon-induced PKR protein kinase with inhibitory proteins p58(IPK) and vaccinia virus K3L is mediated by unique domains: implications for kinase regulation. Mol Cell Biol. 1996;16:4172–4181. doi: 10.1128/mcb.16.8.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gale M.J., Korth M.J., Tang N.M., Tan S.L., Hopkins D.A., Dever T.E. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 210.Enomoto N., Sakuma I., Asahina Y., Kurosaki M., Murakami T., Yamamoto C. Comparison of full-length sequences of interferon-sensitive and resistant hepatitis-c virus 1b—sensitivity to interferon is conferred by amino-acid substitutions in the NS5a region. J Clin Invest. 1995;96:224–230. doi: 10.1172/JCI118025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Enomoto N., Sakuma I., Asahina Y., Kurosaki M., Murakami T., Yamamoto C. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N Engl J Med. 1996;334:77–81. doi: 10.1056/NEJM199601113340203. [DOI] [PubMed] [Google Scholar]
- 212.Kmieciak D., Kruszyna L., Migdalski P., Lacinski M., Juszczyk J., Trzeciak W.H. Mutations within protein kinase R-binding domain of NS5A protein of hepatitis C virus (HCV) and specificity of HCV antibodies in pretreatment sera of HCV-chronically infected patients and their effect on the result of treatment. Jap J Infect Dis. 2006;59:92–99. [PubMed] [Google Scholar]
- 213.De Mitri M.S., Cassini R., Bagaglio S., Morsica G., Andreone P., Marino N. Evolution of hepatitis C virus non-structural 5A gene in the progression of liver disease to hepatocellular carcinoma. Liver Int. 2007;27:1126–1133. doi: 10.1111/j.1478-3231.2007.01537.x. [DOI] [PubMed] [Google Scholar]
- 214.De Mitri M.S., Morsica G., Cassini R., Bagaglio S., Zoli M., Alberti A. Prevalence of wild-type in NS5A-PKR protein kinase binding domain in HCV-related hepatocellular carcinoma. J Hepatol. 2002;36:116–122. doi: 10.1016/s0168-8278(01)00235-5. [DOI] [PubMed] [Google Scholar]
- 215.Gimenez-Barcons M., Wang C.F., Chen M., Sanchez-Tapias J.M., Saiz J.C., Gale M. The oncogenic potential of hepatitis C virus NS5A sequence variants is associated with PKR regulation. J Interferon Cytokine Res. 2005;25:152–164. doi: 10.1089/jir.2005.25.152. [DOI] [PubMed] [Google Scholar]
- 216.Muesing M.A., Smith D.H., Capon D.J. Regulation of messenger-RNA accumulation by a human-immunodeficiency-virus transactivator protein. Cell. 1987;48:691–701. doi: 10.1016/0092-8674(87)90247-9. [DOI] [PubMed] [Google Scholar]
- 217.Roy S., Agy M., Hovanessian A.G., Sonenberg N., Katze M.G. The integrity of the stem structure of human-immunodeficiency-virus type-1 tat-responsive sequence RNA is required for interaction with the interferon-induced 68,000-MR protein-kinase. J Virol. 1991;65:632–640. doi: 10.1128/jvi.65.2.632-640.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Gunnery S., Rice A.P., Robertson H.D., Mathews M.B. Tat-responsive region RNA of human immunodeficiency virus-1 can prevent activation of the double-stranded-RNA-activated protein-kinase. Proc Natl Acad Sci USA. 1990;A87:8687–8691. doi: 10.1073/pnas.87.22.8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Roy S., Katze M.G., Parkin N.T., Edery I., Hovanessian A.G., Sonenberg N. Control of the interferon-induced 68-kilodalton protein-kinase by the HIV-1 tat gene-product. Science. 1990;247:1216–1219. doi: 10.1126/science.2180064. [DOI] [PubMed] [Google Scholar]
- 220.Brand S.R., Kobayashi R., Mathews M.B. The Tat protein of human immunodeficiency virus type 1 is a substrate and inhibitor of the interferon-induced, virally activated protein kinase, PKR. J Biol Chem. 1997;272:8388–8395. doi: 10.1074/jbc.272.13.8388. [DOI] [PubMed] [Google Scholar]
- 221.McMillan N.A.J., Chun R.F., Siderovski D.P., Galabru J., Toone W.M., Samuel C.E. Hiv-1 Tat directly interacts with the interferon-induced, double-stranded RNA-dependent kinase, PKR. Virology. 1995;213:413–424. doi: 10.1006/viro.1995.0014. [DOI] [PubMed] [Google Scholar]
- 222.Endo-Munoz L., Warby T., Harrich D., McMillan N.A.J. Phosphorylation of HIV Tat by PKR increases interaction with TAR RNA and enhances transcription. Virol J. 2005;2:17. doi: 10.1186/1743-422X-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Carroll K., Elroystein O., Moss B., Jagus R. Recombinant vaccinia virus k3l gene-product prevents activation of double-stranded RNA-dependent, initiation factor-2-alpha-specific protein-kinase. J Biol Chem. 1993;268:12837–12842. [PubMed] [Google Scholar]
- 224.Beattie E., Paoletti E., Tartaglia J. Distinct patterns of IFN sensitivity observed in cells infected with vaccinia K3l(-) and E3l(-) mutant viruses. Virology. 1995;210:254–263. doi: 10.1006/viro.1995.1342. [DOI] [PubMed] [Google Scholar]
- 225.Sharp T.V., Schwemmle M., Jeffrey I., Laing K., Mellor H., Proud C.G. Comparative-analysis of the regulation of the interferon-inducible protein-kinase PKR by Epstein-Barr-virus RNAs Eber-1 and Eber-2 and adenovirus VA(I) RNA. Nucleic Acids Res. 1993;21:4483–4490. doi: 10.1093/nar/21.19.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Nanbo A., Yoshiyama H. Epstein-Barr virus-encoded poly(A)(-) RNA confers resistance to apoptosis mediated through Fas by blocking the PKR pathway in human epithelial intestine 407 cells. J Virol. 2005;79:12280–12285. doi: 10.1128/JVI.79.19.12280-12285.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Katze M.G., De Corato D., Safer B., Galabru J., Hovanessian A. Adenovirus VAI RNA complexes with the 68,000 Mr protein kinase to regulate its autophosphorylation and activity. EMBO J. 1987;6:689–697. doi: 10.1002/j.1460-2075.1987.tb04809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Davies M.V., Chang H.W., Jacobs B.L., Kaufman R.J. The E3l and K3l vaccinia virus gene-products stimulate translation through inhibition of the double-stranded RNA-dependent protein-kinase by different mechanisms. J Virol. 1993;67:1688–1692. doi: 10.1128/jvi.67.3.1688-1692.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Yueh A., Schneider R.J. Selective translation initiation by ribosome jumping in adenovirus-infected and heat-shocked cells. Genes Dev. 1996;10:1557–1567. doi: 10.1101/gad.10.12.1557. [DOI] [PubMed] [Google Scholar]
- 230.Langland J.O., Cameron J.M., Heck M.C., Jancovich J.K., Jacobs B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006;119:100–110. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 231.Lu Y., Wambach M., Katze M.G., Krug R.M. Binding of the influenza-virus ns1 protein to double-stranded-RNA inhibits the activation of the protein-kinase that phosphorylates the eIF-2 translation initiation-factor. Virology. 1995;214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- 232.Shors T., Kibler K.V., Perkins K.B., Seidler-Wulff R., Banaszak M.P., Jacobs B.L. Complementation of vaccinia virus deleted of the E3L gene by mutants of E3L. Virology. 1997;239:269–276. doi: 10.1006/viro.1997.8881. [DOI] [PubMed] [Google Scholar]
- 233.Khoo D., Perez C., Mohr I. Characterization of RNA determinants recognized by the arginine- and proline-rich region of Us11, a herpes simplex virus type 1-encoded double-stranded RNA binding protein that prevents PKR activation. J Virol. 2002;76:11971–11981. doi: 10.1128/JVI.76.23.11971-11981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Poppers J., Mulvey M., Perez C., Khoo D., Mohr I. Identification of a lytic-cycle Epstein-Barr virus gene product that can regulate PKR activation. J Virol. 2003;77:228–236. doi: 10.1128/JVI.77.1.228-236.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Cassady K.A., Gross M. The herpes simplex virus type 1 U(s)11 protein interacts with protein kinase R in infected cells and requires a 30-amino-acid sequence adjacent to a kinase substrate domain. J Virol. 2002;76:2029–2035. doi: 10.1128/jvi.76.5.2029-2035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Black T.L., Safer B., Hovanessian A., Katze M.G. The cellular 68,000-MR protein-kinase is highly autophosphorylated and activated yet significantly degraded during poliovirus infection—implications for translational regulation. J Virol. 1989;63:2244–2251. doi: 10.1128/jvi.63.5.2244-2251.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Black T.L., Barber G.N., Katze M.G. Degradation of the interferon-induced 68,000-mr protein-kinase by poliovirus requires RNA. J Virol. 1993;67:791–800. doi: 10.1128/jvi.67.2.791-800.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Habjan M., Pichlmair A., Elliott R.M., Overby A.K., Glatter T., Gstaiger M. NSs protein of rift valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J Virol. 2009;83:4365–4375. doi: 10.1128/JVI.02148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Walsh D., Arias C., Perez C., Halladin D., Escandon M., Ueda T. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol Cell Biol. 2008;28:2648–2658. doi: 10.1128/MCB.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Lee D.Y., Sugden B. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to chive its own synthesis. Blood. 2008;111:2280–2289. doi: 10.1182/blood-2007-07-100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Mulvey M., Arias C., Mohr I. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol. 2007;81:3377–3390. doi: 10.1128/JVI.02191-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Mulvey M., Arias C., Mohr I. Resistance of mRNA translation to acute endoplasmic reticulum stress-inducing agents in herpes simplex virus type 1-infected cells requires multiple virus-encoded functions. J Virol. 2006;80:7354–7363. doi: 10.1128/JVI.00479-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Pavio N., Romano P.R., Graczyk T.M., Feinstone S.M., Taylor D.R. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2 alpha kinase PERK. J Virol. 2003;77:3578–3585. doi: 10.1128/JVI.77.6.3578-3585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Yin Q., McBride J., Fewell C., Lacey M., Wang X., Lin Z. MicroRNA-155 is an Epstein-Barr virus-induced gene that modulates Epstein-Barr virus-regulated gene expression pathways. J Virol. 2008;82:5295–5306. doi: 10.1128/JVI.02380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Harding H.P., Calfon M., Urano F., Novoa I., Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- 246.Isler J.A., Maguire T.G., Alwine J.C. Production of infectious human cytomegalovirus virions is inhibited by drugs that disrupt calcium homeostasis in the endoplasmic reticulum. J Virol. 2005;79:15388–15397. doi: 10.1128/JVI.79.24.15388-15397.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Isler J.A., Skalet A.H., Alwine J.C. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol. 2005;79:6890–6899. doi: 10.1128/JVI.79.11.6890-6899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]