

Highlights
► Robust cell culture, mouse, and nonhuman primate models of SARS-CoV infection have been developed for the study of innate immune pathogenesis. ► Host mechanisms of innate immune sensing of SARS-CoV are unknown, but there is evidence for the involvement of RLRs and TLRs. ► Aberrant proinflammatory cytokine and Interferon Stimulated Gene responses are associated with phenotypes of severe SARS-CoV disease. ► SARS-CoV proteins that modulate innate immune responses antagonize the Interferon response and avoid detection by host sensing mechanisms.
Abstract
SARS-CoV is a pathogenic coronavirus that emerged from a zoonotic reservoir, leading to global dissemination of the virus. The association SARS-CoV with aberrant cytokine, chemokine, and Interferon Stimulated Gene (ISG) responses in patients provided evidence that SARS-CoV pathogenesis is at least partially controlled by innate immune signaling. Utilizing models for SARS-CoV infection, key components of innate immune signaling pathways have been identified as protective factors against SARS-CoV disease, including STAT1 and MyD88. Gene transcription signatures unique to SARS-CoV disease states have been identified, but host factors that regulate exacerbated disease phenotypes still remain largely undetermined. SARS-CoV encodes several proteins that modulate innate immune signaling through the antagonism of the induction of Interferon and by avoidance of ISG effector functions.
SARS-CoV: the first viral pandemic of the new millenium
In 2002 the first viral pandemic of the millennium emerged from the Guangdong province in Southern China. Severe Acute Respiratory Syndrome (SARS) presented as initial ‘flu-like’ symptoms (cough, sore throat, and fever) that could progress to atypical pneumonia in patients with severe SARS disease [1, 2]. A rapid response from scientists identified a novel coronavirus as the causative agent of SARS, named SARS-Coronavirus (SARS-CoV, Figure 1a) and angiotensin converting enzyme 2 (ACE2) as the viral receptor [3]. Despite identification of the virus, the disease spread from China to other Southeast Asia countries, becoming a global threat with significant outbreaks reported in Singapore, Hong Kong, Taiwan, and Canada [4]. At the end of the epidemic, 774 of the 8096 confirmed cases resulted in death (a mortality rate of 9.6%) [5]. By July of 2003 the virus was controlled by public health measures, but no vaccines or antivirals are currently approved for the treatment of SARS-CoV should the virus re-emerge [6, 7].
Figure 1.
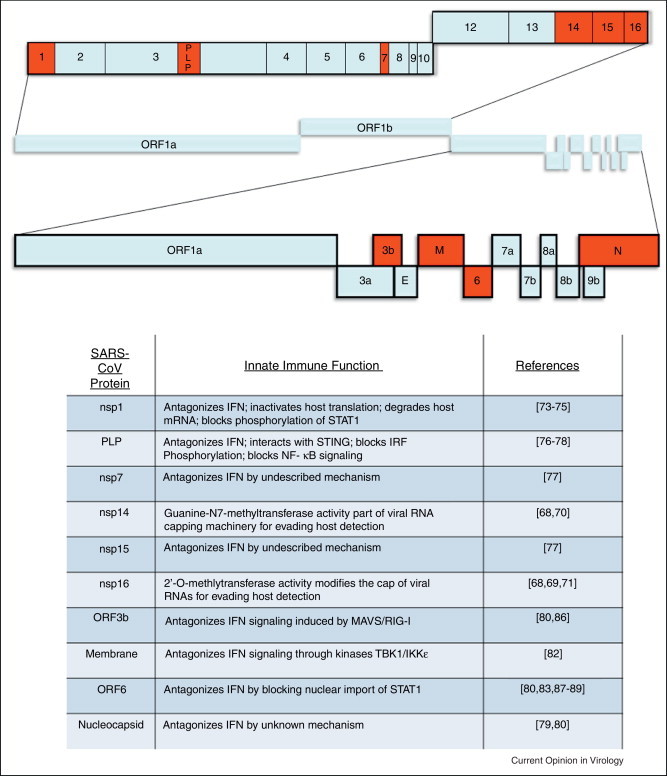
The SARS-CoV genome and functions of SARS-CoV innate immune antagonists. (a) The typical coronavirus genome size is quite large in comparison to many other positive-sense RNA viruses; within the SARS-CoV genome of 29.7 kB at least ten genes with potential functions that modulate innate immunity have been characterized (highlighted here in red). Like other members of the viral family Coronaviridae, SARS-CoV has a positive-sense, single-stranded RNA genome that is amenable to manipulation using reverse genetic techniques [90]. In SARS-CoV the first open reading frame (ORF) encodes the 16 nonstructural proteins that make up the viral replicase, while the ensuing ORFs encode four structural proteins that compose the virion, as well as eight accessory proteins. The SARS-CoV accessory proteins share no homology to the accessory proteins of other human coronaviruses, and while dispensable for replication in vitro, encode functions that probably impact viral pathogenesis in vivo [91]. While SARS-CoV was a novel virus not previously recognized before the 2002 outbreak, other coronaviruses have been associated with disease in humans. Coronaviruses known to infect humans include HCoV-HKU1, HCoV-OC43, HCoV-229E, and HCoV-NL63, which also cause respiratory infections but are generally much less severe than SARS [92]. (b) Transcription and subsequent signaling of Interferon is vital for activating the antiviral response in host cells. Because of this, many viruses (including SARS-CoV) encode proteins that antagonize the IFN response to viral infection. Of the SARS-CoV viral proteins listed here, eight have been identified as Interferon antagonists and two have been implicated in the viral RNA capping machinery.
SARS disease in patients with poor outcome was marked by the progression to Acute Respiratory Distress Syndrome (ARDS): approximately 25% of SARS cases were diagnosed with ARDS and the ARDS-associated mortality rate exceeded 50%[8]. Elderly SARS patients had a poor prognosis with mortality rates of 50% in patients over 65 year of age [9]. In SARS patients with ARDS, the acute phase characterized by pulmonary edema, severe hypoxia, and the accumulation of inflammatory cells in the lungs could progress to ARDS late phase fibrosis, organizing pneumonia, systemic inflammation responses, and multiple organ failure [10, 11]. Consistent with ARDS progression, the primary targets of SARS-CoV infection are ciliated cells of the airway epithelium and alveolar Type II pneumocytes [12, 13]. ARDS is also associated with the induction of inflammatory cytokines including IL-1, IL-6, IL-8, CXCL-10, and TNFα, many of which were highly expressed in the lungs of SARS patients [14, 15]. In many viral infections the antiviral cytokine Interferon (IFN) acts not only to control viral infections, but also to program the adaptive immune response to promote viral clearance [16]. However, in patients with severe SARS disease, aberrant IFN, Interferon Stimulated Genes (ISGs), and cytokine responses were observed compared to healthy individuals providing evidence that SARS is an innate immune regulated disease [17, 18].
Elucidation of innate immune pathogenesis mechanisms through models of SARS-CoV infection
Initial models of SARS-CoV innate immune pathogenesis were viral infection of cell lines including Vero E6, Caco-2, and Huh-7 cells, as well as PBMCs; however, these systems may not yield relevant biological information consistent with SARS-CoV infection of pneumocytes, because they are not derived from lung tissues [19, 20, 21]. Human Airway Epithelial Cultures (HAEs) are primary cell lines of pseudostratified mucocilliary epithelium that replicate the morphological and physiological characteristics of human airways. HAEs can be infected with SARS-CoV, are derived directly from normal lung tissues, and contain the relevant epithelial cell types within human airways for SARS-CoV infection, but HAEs are difficult to procure and are highly heterogeneous [13, 22]. Recently, the 2B4 cell line derived from a clonally selected Calu-3 cell population with high expression of ACE2 (the SARS-CoV receptor) was developed that forms differentiated pseudostratified columnar epithelia highly permissible to SARS-CoV infection. SARS-CoV infection of 2B4 cells provides data on innate immune responses within a biologically relevant and easily replicated in vitro system [23••].
Small animal models of SARS-CoV infection have benefits into the elucidation of innate immune pathogenesis beyond cell culture systems due to their ability to model the interaction of lung epithelium and immune cell types within an infected organism. While hamsters and ferrets have been considered for use as small animal models of SARS-CoV infection, a robust mouse model has been more vigorously pursued because of the relative ease of genetic manipulation of the host, as well as greater availability of immunological reagents [24, 25, 26]. SARS-CoV epidemic isolates replicate in young mice but do not cause clinical disease, limiting the use of these models for pathogenesis studies [27, 28]. SARS-CoV infected aged mice (12 months) exhibit minor clinical illness, but do not address pathogenic mechanisms associated with SARS disease in senescent or non-senescent populations [29, 30, 31]. Infections using the mouse coronavirus MHV-1 have also been proposed as models for SARS-CoV infection [32]. Recently, mouse adapted SARS coronaviruses (MA-SARS-CoV) have been developed by serial passage through the lungs of mice yielding several different MA-SARS-CoV strains [33, 34]. Infection of 6-10 week old mice with SARS-CoV adapted by 15 serial passages (MA15-SARS-CoV) causes morbidity and mortality, viral replication in the lungs, and lung pathology associated with mild SARS disease [33, 34]. In addition, MA-SARS-CoV infections of aged mice exhibit exacerbated SARS disease that mimics the age-dependent and ARDS phenotypes seen in humans [35, 36•]. Currently, studies are underway to determine the response of recombinant inbred lines of mice (known as the Collaborative Cross) to MA15-SARS-CoV infection, utilizing Genome Wide Associate Studies to map quantitative trait loci that contribute to in vivo phenotypes (e.g. weight loss or lung pathology) [37•]. These studies offer an unbiased approach to determining the contributions of many different genes to the complex trait of SARS-CoV disease, and could identify novel host factors involved in SARS-CoV pathogenesis.
The use of primate models of SARS-CoV infection is typically limited due to ethical concerns and expense. However, infection of nonhuman primates with SARS-CoV is a model more relevant to humans for testing of drug treatments and vaccines. SARS-CoV replicates in the lungs of primate species, including African green monkeys, cynomolgus macaques, and rhesus macaques [38]. Infection of cynomolgus macaques with SARS-CoV replicates aspects of the human disease, including lung pathology of diffuse alveolar damage (DAD) found in humans [39]. Additionally, a comparison of SARS-CoV infection of young adult cynomolgus macaques to aged cynomolgus macaques found age-dependent susceptibility to SARS disease resembling the same trend in humans [40••]. More recently, it has been shown that SARS-CoV causes increased severity of disease in African green monkeys compared to cynomolgus macaques, and that the increased lung injury is probably associated with differential innate immune signaling [41•].
Host antiviral innate immune detection and response to SARS-CoV infection
Innate immune signaling is the earliest differentiation of pathogens from cellular molecules that alerts host cells to the presence of invading viruses. Pattern Recognition Receptors (PRRs), such as the RIG-I-Like Receptors (RLRs, Figure 2a) and Toll-Like Receptors (TLRs, Figure 3a and b) recognize Pathogen Associated Molecular Patterns (PAMPs) from viral components or replication intermediates, resulting in signaling cascades that initiate an antiviral state in cells as a result of infection [42, 43]. PRRs are distributed on plasma membranes, endosomal membranes, and within the cytosol of host cells to ensure maximal detection of viral PAMPs including nucleic acid motifs, carbohydrate moieties, glycoproteins, lipoproteins or other small molecules present within the viral life cycle, but absent from normal cellular components.
Figure 2.
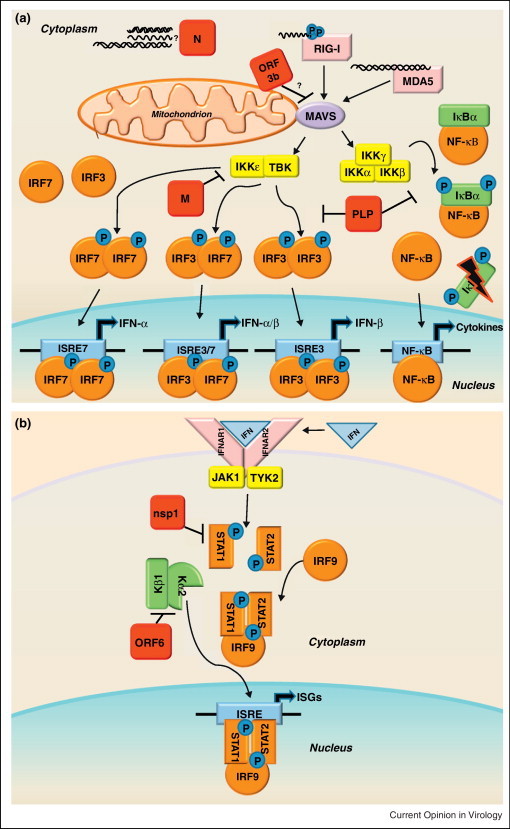
(a) RLR family of innate immune receptors induce Type I interferon. The family of RIG-I Like Receptors (RLRs) contains three cytosolic RNA helicases that recognize non-self RNA species resulting from viral replication [93]. The two signaling sensors within the RLR family are retinoic acid-inducible gene I (RIG-I) and melanoma differentiation associated factor 5 (MDA5). The third RLR, laboratory of genetics and physiology 2 (LGP2, not shown), facilitates recognition of viral PAMPs by RIG-I and MDA5, but is dispensable for their signaling [94]. RIG-I recognizes primarily 5′ppp-RNA molecules with secondary motifs of dsRNA or ssRNA of short length [95, 96]. MDA5 recognizes longer dsRNA motifs than RIG-I [97]. Following binding of viral RNAs, RIG-I and MDA5 interact with the mitochondrial membrane bound adaptor molecule MAVS (mitochondrial antiviral signaling protein, also referred to as IPS-1, VISA, or CARDIF) to transduce the signal via complexes of kinases: the IKKɛ/TBK1 complex and the IKKα/IKKβ/IKKγ complex. The IKKɛ/TBK1 kinases phosphorylate the transcription factors IRF3 and IRF7, which then form homodimers or heterodimers. Upon dimerization, the transcription factors enter the nucleus to initiate transcription of Type I IFNs (IFN-α and IFN-β). While IRF3 is nearly ubiquitously expressed in cells, IRF7 is an ISG typically expressed at low levels, so it is thought that IRF3 mediates transcription of the majority of early IFN expression. The IKKα/IKKβ/IKKγ kinases phosphorylate IκBα, targeting this repressor protein of NF-κB for degradation. Activation of NF-κB leads to transcription of proinflammatory cytokines, and NF-kB mediated transcription has also been linked to the pathogenesis of ARDS [46]. SARS-CoV encodes proteins that antagonize RLR family signaling, shown here in red.
(b) Interferon signals through the JAK-STAT pathway to induce interferon stimulated genes. The secretion of IFN-α and IFN-β molecules from an infected cell leads to an autocrine and paracrine signaling through the IFNαβ Receptor (composed of the IFNAR1 and IFNAR2 subunits) resulting in the activation of the JAK-STAT pathway. The JAK/TYK2 kinases phosphorylate the transcription factors STAT1 and STAT2, which form heterodimers complexed with IRF9. The STAT complex translocates to the nucleus leading to the transcription of Interferon Stimulated Genes (ISGs) that establish an antiviral state in the cell. Because neighboring cells can receive IFN stimulation before infection, it is a crucial pathway to preventing viral spread in the host. SARS-CoV also encodes proteins that antagonize the JAK-STAT pathway, shown here in red. Mice deficient in STAT1 showed an increased susceptibility to SARS-CoV infection [98]. Although there were no differences in mice deficient in IFN receptors, STAT1−/− mice showed increased weight loss, viral titer, and lung pathology compared to wild type over the course of MA15-SARS-CoV infection, demonstrating that STAT has important IFN independent role in SARS-CoV infection [80]. Severe lung pathology in STAT1−/− mice infected with MA15-SARS-CoV was associated with the infiltration of immune cells and fibrotic lung response. The STAT1−/− dependent prolonged expression of inflammatory cytokines (IL-1, IL-6, IL-10, IL-12, and TNFα) and chemokines (CCL2, CCL3, CCL4, CCL7, and CCL20), could be a transcriptional regime responsible for fibrotic phenotypes within the lungs. Additionally, ISG responses were significantly lower in STAT1−/− mice compared to wild type or IFNAR−/− mice, leading to the conclusion that STAT1 dependent, IFNAR1 independent ISG expression was protective in these mice [80]. It remains unclear how STAT1 controls ISGs independent of IFNAR expression, or which ISGs have important potential roles in SARS-CoV pathogenesis.
Figure 3.
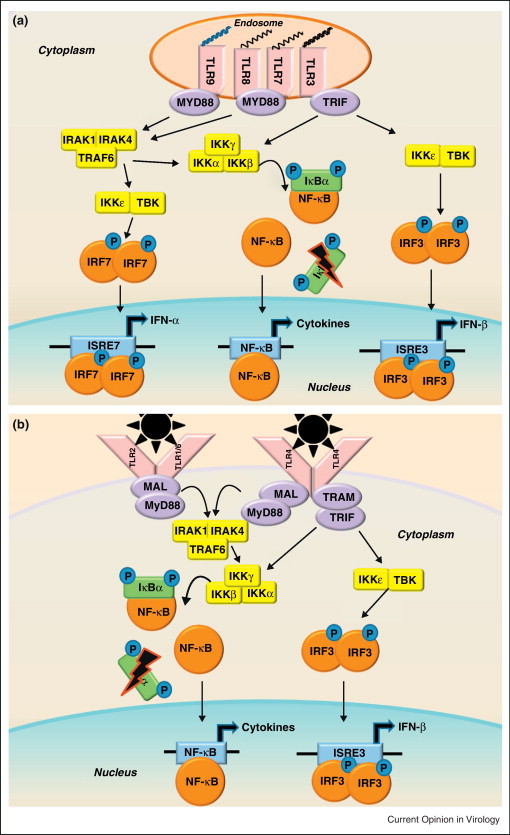
Pathogen associated molecular pattern sensing by Toll-Like Receptors. (a) In the endosomal compartment, TLRs recognize viral nucleic acid PAMPs: TLR3 recognizes dsRNAs, TLR7/8 recognizes ssRNAs, and TLR9 recognizes CpG DNA motifs. (b) On the surface of cells, TLR2 and TLR4 are known to recognize viral glycoproteins [47, 99]. TLR2/6 heterodimers help to activate the innate immune response to RSV, though the viral PAMP recognized has not been determined [100]. TLR1/2 heterodimers have been shown to recognize viral glycoproteins, though their potential role in respiratory virus infection has not been determined [99]. While there are many TLRs that recognize viral PAMPs, they signal through common adaptor molecules, including MyD88, MAL, TRAM, and TRIF. The TLR adaptor molecules signal through the IKKɛ/TBK1 complex and the IKKα/IKKβ/IKKγ complex similarly to RLRs, but can also recruit an IRAK-1/IRAK4/TRAF6 complex capable of activating the transcription factors IRF3, IRF7, and NF-κB. Activation of these transcription factors leads to the transcription of Type I IFNs and proinflammatory cytokines. Due to the considerable crosstalk between TLR and RLR signaling, it is difficult to discriminate between transcriptional products generated by the two sensor families, but it is likely that both play an important role in the innate immune response to SARS-CoV infection.
RIG-I like receptor signaling
The RIG-I Like Receptors are cytoplasmic sensors that detect viral RNA PAMPs in a wide range of cell types (Figure 2a). The RLRs RIG-I and MDA5 are ISGs that are transcribed during SARS-CoV infection in vitro [23••]. MHV, another coronavirus, is recognized by MDA5 in brain macrophages and microglial cells, and by RIG-I and MDA5 in oligodendrocyte cells [44, 45•]. Although it is not known whether SARS-CoV is recognized by RLRs, MHV and SARS-CoV are likely to have similar replication intermediates (putative RLR ligands), so it is likely that SARS-CoV could be detected by the same sensors. RLR signaling leads to the activation of several transcription factors: IRF3, IRF7, and NF-κB. IRF3and IRF7 initiate transcription of Type I IFNs (IFN-α and IFN-β), important for an antiviral response. NF-κB mediated transcription of proinflammatory cytokines has been linked to the pathogenesis of ARDS [46]. In vitro SARS-CoV infections have demonstrated that the expression of NF-κB generated transcripts, such as IL-6 and IL-8, happens as early as 12 h post infection, while IRF3/IRF7 transcription of Type I IFNs is delayed until 48 h post infection [23••]. Similarly, in the macaque model of age-dependent SARS-CoV pathogenesis NF-κB induced genes are more highly expressed in aged macaques that have significantly increased lung injury compared to young adult macaques where higher expression of IFNs was observed [40••]. While the correlation of severe SARS-CoV disease with different transcriptional regimes is promising, the key to finding determinants of increased SARS-CoV pathogenesis may be how innate immune sensing mechanisms initiate transcription at critical junctures during infection and which types of innate immune sensing are protective.
Toll-Like Receptor signaling
The Toll-Like Receptor family of membrane bound sensors also recognizes viral PAMPs, although no TLR has been directly implicated in the recognition of SARS-CoV. On the surface of cells, TLR1, TLR2, TLR4, and TLR6 have been implicated in the recognition of PAMPs from other viruses (Figure 3a), while the endosomal receptors TLR3, TLR7, TLR8, and TLR9 detect viral nucleic acid PAMPs (Figure 3b). TLR4 recognizes viral glycoproteins of RSV, is expressed on the surface of lung epithelium, is a potential entry co-factor for respiratory viruses, and has been identified as a protective host factor against MHV-1 in a respiratory model of SARS disease [47, 48, 49]. Transcription of TLRs increased in mice following infection with MA15-SARS-CoV and in human dendritic cells infected with SARS-CoV [50••, 51]. Additionally, the activation of TLR3 has protective effects in a mouse model of SARS-CoV infection [52]. While there are many TLRs that recognize viral PAMPs, they signal through the common adaptor molecule MyD88, with the exception of TLR3 that uses the adaptor TRIF (Figure 3a and b). Infection of MyD88−/− mice established a protective role for TLR adaptors in MA15-SARS-CoV infection: while wild-type mice experienced transient weight loss, from which they recovered after 7 days, MyD88−/− mice lost significantly more weight, and all of the MyD88−/− mice died by day 6 post-infection [53]. Additionally, higher viral loads, severe lung pathology and differences in cytokines and chemokines were observed in MyD88−/− mice compared to wild-type mice [53]. Currently, studies are in progress to determine the roles of other TLR adaptor proteins (TRIF, MAL, and TRAM) in SARS-CoV infections, as well as what TLR(s) or TLR ligand(s) contribute to the protective role of MyD88.
Innate immune signaling effector molecules and SARS-CoV pathogenesis
Following detection of virus by the host cells, the production of cytokines, chemokines, and ISGs continues the innate immune response to viral infection by mediating inflammation and cellular antiviral processes. The importance of these effector responses to the prognosis of SARS patients is underscored by the observation that IFN (Type I and Type II IFN), chemokine (CXCL10 and CCL2), and ISG (CIG5, MXA, IFITM1, IFIT3) hyperimmune responses persisted in patients who succumbed to SARS, indicating that differences in expression patterns of innate immune effector molecules may be a determinant of SARS disease outcome [18].
Interferon
Interferons are potent cytokines of critical importance in controlling viral infections and priming adaptive immune responses [54]. Several studies of antiviral treatments tested against SARS-CoV replication show administration of Type I IFN inhibits SARS-CoV growth in cell culture as well as viral replication in cynomolgus macaques and mouse models [39, 40••, 55, 56, 57, 58, 59]. Despite the potential importance of IFNs in controlling SARS-CoV replication, infection of mice deficient in Type I, II, or III IFN receptors showed minimal phenotypic difference in weight loss, viral titer, lung pathology, and mortality from wild-type mice in the MA15-SARS-CoV model [60•]. However, mice deficient in STAT1, a critical transcription factor for IFN signaling (Figure 2b), were significantly more susceptible to MA15-SARS-CoV infection than wild-type or IFNAR1−/− mice [60•]. Transcriptional analysis from these studies showed that ISGs were induced even in the absence of IFNAR1, demonstrating that there may be compensatory mechanisms through other innate immune signaling to protect against severe SARS-CoV disease in the absence of IFN [50••]. In the SARS-CoV infection model, mice deficient in Type I, Type III, or Type I and Type III IFN receptors had slightly higher levels of viral replication in the lungs [61]. In 2B4 cells expression of Type III IFN was detected 24 h earlier than Type I IFN transcripts, demonstrating a delay in Type I IFN signaling and a potentially protective role of Type III IFN following SARS-CoV infection [23••]. While IFNs continue to be an attractive potential antiviral strategy if SARS were to re-emerge, their role as a protective component of the innate immune response during SARS-CoV infection still needs additional investigation, particularly into protective innate immune mechanisms that occur in the absence of IFN signaling.
Cytokines, Chemokines, and ISGs
Proinflammatory cytokines and chemokines (many of which are ISGs) may be part of a necessary initial immune response to pathogens, but exacerbated expression of these factors is associated with immunopathology and ARDS [46, 62]. In vitro studies found that SARS-CoV infection initiates a proinflammatory cytokine response at 24 h post infection, but that IFNs and ISGs are delayed in expression until 48 h post infection [23••]. Although the consequences of the timing of these signals are not yet understood, SARS-CoV infection of susceptible aged mice leads to elevated levels of proinflammatory cytokines with ARDS association, including TNFα, IL-6, and IL-1β [30, 31]. Chemokine receptors CCR1, CCR2, and CCR5 have protective roles during MA15-SARS-CoV infection in the mouse model as well as SARS-CoV infection of human DCs, indicating the importance of cell recruitment in controlling SARS-CoV infections [51, 53]. Transcriptional profiles of ISGs associated with increased SARS-CoV disease have been described in several model systems, but the consequences of ISG signaling responses to SARS-CoV infection has not been characterized [23••, 31, 40••, 41•, 50••]. Although it is known that antiviral ISGs of the IFITM family restrict SARS-CoV entry into host cells and several ISGs such as MxA, OAS1, RNaseL, PKR, IFIT, Viperin, and TRIM5α have defined functions in the context of other viral infections, these are only a subset of this large family of molecules, most of which have antiviral properties that are not yet well understood [63••, 64]. Additional studies to determine the crucial ISGs that control SARS-CoV infection or contribute to SARS disease could be exploited for the development of antiviral therapies. Extant reagents to study the overexpression of ISGs singly and in combination could determine which ISGs are effective at initiating an antiviral state against SARS-CoV infection [65••].
Modulation of innate immune response by SARS-CoV: evasion of innate immune detection
Evasion of innate immune responses to SARS-CoV infection requires avoidance of detection by cellular PRRs. During the SARS-CoV replication cycle, the segregation of viral dsRNA intermediates in the interior of Double Membrane Vesicles (DMVs) may potentially shield viral PAMPS from recognition by cytosolic PRRs [66, 67]. It is unknown whether small viral ssRNA or dsRNA degradation products are also sequestered within DMVs or can be sensed by PRRs. The lack of a 5′cap distinguishes viral mRNAs from other eukaryotic mRNAs, and many viruses (including SARS-CoV) have evolved mechanisms to mimic host capping machinery. In vitro capping of SARS-CoV RNAs requires nsp14 and an nsp16/nsp10 complex [68•, 69]. The guanine-N7-methyltransferase activity of SARS-CoV nsp14 is the initial step to building an RNA cap that is structurally similar to the RNA cap used by the host, making it more difficult for the host to discriminate viral non-self RNAs from self mRNAs [68•, 70]. Additionally, nsp16 of SARS-CoV has been identified as a 2′-O-methlytransferase capable of modifying the cap of viral RNAs, which seems to be of particular import in evading recognition by host PRRs such as MDA5, as well as host ISGs such as IFIT family members IFIT1 and IFIT2 [71, 72••].
Strategy of antagonism of innate immune molecules by SARS-CoV: block IFN
To counter innate immune signaling, SARS-CoV encodes eight proteins that antagonize the IFN response to prevent activation of antiviral effectors in host cells (Figures 1b and 2a, b).
Nonstructural proteins
The first nonstructural protein SARS-CoV nsp1 antagonizes Type I IFN by three mechanisms: inactivation of host translational machinery, degradation of host mRNAs, and inhibition of phosphorylation of STAT1 [73, 74, 75•]. While nsp1 mediates host mRNA degradation, SARS-CoV mRNAs are not susceptible to the cleavage or subsequent degradation [75•]. Part of the third nonstructural protein, SARS-CoV PLP is a papain like protease that antagonizes IFN by blocking phosphorylation IRF3 [76]. PLP prevents IRF3 phosphorylation in cell culture but not with purified components of the signaling pathway, indicating that a direct interaction between PLP and IRF3 does not take place [77]. More recently, findings that PLP interacts with STING resulted in a proposed mechanism of IFN antagonism by disruption of the signaling complex that leads to phosphorylation of IRF3 [78]. PLP also disrupts NF-κB signaling in addition to IRF3 signaling, possibly by a similar mechanism [77]. In addition to SARS-CoV nsp1 and PLP, SARS-CoV nsp7 and nsp15 have both been identified as potential IFN antagonists, but are not well described [77]. The majority of the SARS-CoV nonstructural proteins are required for replication, including those that have been identified as IFN antagonists. Their essential functions in viral replication may be due at least partly to their innate immune modulatory functions.
Structural proteins
In addition to functioning as components of the SARS-CoV virion two structural proteins antagonize IFN signaling. The Nucleocapsid (N) protein of SARS Co-V is capable of blocking Type I IFN when induced by Sendai virus or polyI:C, but not upstream signaling components such as RIG-I, MDA5, MAVS, IKKɛ, TBK1 or TRIF, indicating that N exerts its effects before these signaling mediators [79•, 80]. Additional studies of MHV Nucleocapsid identified IFN antagonism activity through RNaseL mediated host translation shutoff, but this has not yet been shown with SARS-CoV N [81]. The SARS-CoV Membrane (M) protein blocks transcription of IFN-β when stimulated by dsRNA as well as components of the RIG-I signaling pathway including RIG-I, MAVS, IKKɛ, and TBK1, but not the transcription factor IRF3, suggesting that the block in signaling is prior to IRF3 initiation of transcription [82]. SARS-CoV M also co-immunoprecipitated with RIG-I, IKKɛ, and TBK1, suggesting that SARS-CoV M interacts with a complex formed by these proteins as a mechanism for disrupting IFN-β transcription. SARS-CoV M was not identified as an IFN antagonist by the Venezuelan Equine Encephalitis Virus Replicon or Newcastle Disease Virus-GFP screens, demonstrating the need for multiple approaches to identify all of the IFN antagonist proteins within the SARS-CoV genome [80, 83]. Structural components of the SARS-CoV virion acting as antagonists of IFN may be important for blocking innate immune responses immediately upon introduction of the virion into the cell; however, the temporal nature of antagonism of IFN signaling by SARS-CoV is not yet well understood.
Accessory proteins
SARS-CoV encodes eight accessory proteins that share no homology with proteins from other human coronaviruses and are dispensable for viral replication [84, 85]. SARS-CoV ORF3b protein was identified as an antagonist of Type I IFN capable of inhibiting RIG-I and MAVS mediated induction of IFN-β by the transcription factors IRF3 and NF-κB [80, 86]. However, ORF3b does not inhibit TNFα mediated activation of NF-κB transcription, leading to speculation that the disruption of NF-κB signaling is specific for induction by the RLRs [86]. SARS-CoV ORF3b temporally distributes to the mitochondrial outer membrane, indicating that the mechanism of IFN antagonism may involve MAVS, also located on the mitochondria [86]. SARS-CoV ORF6 protein antagonizes IFN by inhibiting signaling of the JAK-STAT pathway downstream of IFNAR by blocking nuclear translocation of the transcription factor STAT1 [80, 83, 87]. ORF6 binds to karyopherin-α2, and tethers karyopherin-β1 on internal membranes, disrupting formation of the complex of proteins associated with the nuclear import of STAT1 [87]. The C-terminus of ORF6 interferes with proteins with NLS-signals, disrupting the classical nuclear import pathway [88, 89]. The disruption of nuclear transport is specific to a nuclear import pathway, indicating that there are potentially many other transcription factors that modulate innate immunity that could be affected by SARS-CoV ORF6.
Many of the SARS-CoV gene products that modulate IFN signaling have been identified by overexpression in cell culture using individual viral components, a system that may not accurately reflect innate immune signaling that occurs during SARS-CoV infection in vivo. Additional studies are needed to elucidate IFN antagonism by these proteins in the context of infection, particularly because SARS-CoV proteins can form large complexes during viral infection, and the role of these complexes in potentially modulating innate immune responses is not yet known. Because of the complicated replication scheme utilized by coronaviruses like SARS-CoV, some viral proteins may be temporally expressed at different levels during viral infection or compartmentalized in different areas of the cell, which are factors that still need to be investigated in the context of how viral proteins affect innate immune signaling.
SARS-CoV pathogenesis: innate immune factors still at large
SARS-CoV is a highly pathogenic respiratory virus where the mechanisms of severe disease are largely mediated by innate immune pathways. Excellent models exist for studying SARS-CoV pathogenesis that replicate findings from the SARS outbreak in humans: cell lines for studying in vitro responses in human lung epithelial cells, mouse models of fibrosis and GWAS mapping of traits, as well as primate models of comparative species infection and age-dependent phenotypes. Due to the development of these models, SARS-CoV is uniquely suited for a systems biology based platform to compare respiratory virus infection in multiple relevant model systems as an unbiased approach to identify novel host modulators of innate immunity in the context of viral infections. In addition, SARS-CoV encodes many proteins that antagonize the host's Interferon response, but questions remain about the effects of these antagonists of viral pathogenesis during SARS-CoV infection in vivo. Of the currently well-described innate immune signaling pathways, there is evidence to support that RLR and TLR sensors detect and respond to SARS-CoV infection, but no mechanism or SARS-CoV ligand for these receptors has been determined. Unique gene transcription signatures associated with defined temporal expression of proinflammatory cytokines and ISGs in models of severe SARS-CoV disease have been described, but few of these genes have been evaluated for their role in SARS pathogenesis or the host antiviral response to SARS-CoV, which could help identify novel immunomodulatory therapies in the event of SARS-CoV re-emergence. In future studies, SARS-CoV could be particularly useful as a comparative model for Influenzavirus or RSV infection to evaluate common targets for antiviral strategies as well as unique mechanisms of innate immune pathogenesis across multiple virus families with similar tropisms.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgement
This work was supported by the National Institutes of Health (NIH) grant 1R01AI075297-02.
References
- 1.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.-R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A.M. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New England Journal of Medicine. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W. A novel coronavirus associated with severe acute respiratory syndrome. New England Journal of Medicine. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hui D.S.C., Chan M.C.H., Wu A.K., Ng P.C. Severe acute respiratory syndrome (SARS): epidemiology and clinical features. Postgraduate Medical Journal. 2004;80:373–381. doi: 10.1136/pgmj.2004.020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization: Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003 on World Wide Web URL: http://www.who.int/csr/sars/country/table2004_04_21/en/index.html.
- 6.Stockman L.J., Bellamy R., Garner P. SARS: systematic review of treatment effects. PLoS Medicine. 2006;3:e343. doi: 10.1371/journal.pmed.0030343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolles M., Deming D., Long K., Agnihothram S., Whitmore A., Ferris M., Funkhouser W., Gralinski L., Totura A., Heise M. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. Journal of Virology. 2011;85:12201–12215. doi: 10.1128/JVI.06048-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lew T.W.K., Kwek T.-K., Tai D., Earnest A., Loo S., Singh K., Kwan K.M., Chan Y., Yim C.F., Bek S.L. Acute respiratory distress syndrome in critically ill patients with severe acute respiratory syndrome. The Journal of the American Medical Association. 2003;290:374–380. doi: 10.1001/jama.290.3.374. [DOI] [PubMed] [Google Scholar]
- 9.World Health Organization: SARS case fatality ratio, incubation period on World Wide Web URL: http://www.who.int/csr/sarsarchive/2003_05_07a/en/.
- 10.Tsushima K., King L.S., Aggarwal N.R., Gorordo A.D., D’Alessio F.R., Kubo K. Acute lung injury review. Internal Medicine. 2009;48:621–630. doi: 10.2169/internalmedicine.48.1741. [DOI] [PubMed] [Google Scholar]
- 11.Hwang D.M., Chamberlain D.W., Poutanen S.M., Low D.E., Asa S.L., Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Modern Pathology. 2004;18:1–10. doi: 10.1038/modpathol.3800247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chow K.-C., Hsiao C.-H., Lin T.-Y., Chen C.-L., Chiou S.-H. Detection of severe acute respiratory syndrome associated coronavirus in pneumocytes of the lung. American Journal of Clinical Pathology. 2004;121:574–580. doi: 10.1309/C0EDU0RAQBTXBHCE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sims A.C., Baric R.S., Yount B., Burkett S.E., Collins P.L., Pickles R.J. Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: role of ciliated cells in viral spread in the conducting airways of the lungs. Journal of Virology. 2005;79:15511–15524. doi: 10.1128/JVI.79.24.15511-15524.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong S.L., Chui P., Lim B., Salto-Tellez M. Elucidating the molecular physiopathology of acute respiratory distress syndrome in severe acute respiratory syndrome patients. Virus Research. 2009;145:260–269. doi: 10.1016/j.virusres.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baas T., Taubenberger J.K., Chong P.Y., Chui P., Katze M.G. SARS-CoV virus-host interactions and comparative etiologies of acute respiratory distress syndrome as determined by transcriptional and cytokine profiling of formalin-fixed paraffin-embedded tissues. Journal of Interferon & Cytokine Research. 2006;26:309–317. doi: 10.1089/jir.2006.26.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cervantes-Barragan L., Kalinke U., Zust R., Konig M., Reizis B., Lopez-Macias C., Thiel V., Ludewig B. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. The Journal of Immunology. 2009;182:1099–1106. doi: 10.4049/jimmunol.182.2.1099. [DOI] [PubMed] [Google Scholar]
- 17.Wong C.K., Lam C.W.K., Wu A.K.L., Ip W.K., Lee N.L.S., Chan I.H.S., Lit L.C.W., Hui D.S.C., Chan M.H.M., Chung S.S.C. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clinical & Experimental Immunology. 2004;136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cameron M.J., Ran L., Xu L., Danesh A., Bermejo-Martin J.F., Cameron C.M., Muller M.P., Gold W.L., Richardson S.E., Poutanen S.M. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. Journal of Virology. 2007;81:8692–8706. doi: 10.1128/JVI.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leong W.F., Tan H.C., Ooi E.E., Koh D.R., Chow V.T.K. Microarray and real-time RT-PCR analyses of differential human gene expression patterns induced by severe acute respiratory syndrome (SARS) coronavirus infection of Vero cells. Microbes and Infection. 2005;7:248–259. doi: 10.1016/j.micinf.2004.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dosch S.F, Mahajan S.D., Collins A.R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-κB pathway in human monocyte macrophages in vitro. Virus Research. 2009;142:19–27. doi: 10.1016/j.virusres.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L., Wo J., Shao J., Zhu H., Wu N., Li M., Yao H., Hu M., Dennin R.H. SARS-coronavirus replicates in mononuclear cells of peripheral blood (PBMCs) from SARS patients. Journal of Clinical Virology: The Official Publication of the Pan American Society for Clinical Virology. 2003;28:239–244. doi: 10.1016/S1386-6532(03)00195-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sims A.C., Burkett S.E., Yount B., Pickles R.J. SARS-CoV replication and pathogenesis in an in vitro model of the human conducting airway epithelium. Virus Research. 2008;133:33–44. doi: 10.1016/j.virusres.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23••.Yoshikawa T., Hill T.E., Yoshikawa N., Popov V.L., Galindo C.L., Garner H.R., Peters C.J., Tseng C.-T. Dynamic innate immune responses of human bronchial epithelial cells to severe acute respiratory syndrome-associated coronavirus infection. PLoS ONE. 2010;5:e8729. doi: 10.1371/journal.pone.0008729. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes for the first time innate immune responses in the 2B4 cell culture model of SARS-CoV infection, including transcription of Interferons, proinflammatory cytokines, and Interferon stimulated genes (ISGs).
- 24.Martina B.E.E., Haagmans B.L., Kuiken T., Fouchier R.A.M., Rimmelzwaan G.F., van Amerongen G., Peiris J.S.M., Lim W., Osterhaus A.D.M.E. Virology: SARS virus infection of cats and ferrets. Nature. 2003;425 doi: 10.1038/425915a. 915–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Brand J.M.A., Haagmans B.L., Leijten L., van Riel D., Martina B.E.E., Osterhaus A.D.M.E., Kuiken T. Pathology of experimental SARS coronavirus infection in cats and ferrets. Veterinary Pathology Online. 2008;45:551–562. doi: 10.1354/vp.45-4-551. [DOI] [PubMed] [Google Scholar]
- 26.Roberts A., Vogel L., Guarner J., Hayes N., Murphy B., Zaki S., Subbarao K. Severe acute respiratory syndrome coronavirus infection of golden syrian hamsters. Journal of Virology. 2005;79:503–511. doi: 10.1128/JVI.79.1.503-511.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subbarao K., McAuliffe J., Vogel L., Fahle G., Fischer S., Tatti K., Packard M., Shieh W.-J., Zaki S., Murphy B. Prior infection and passive transfer of neutralizing antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. Journal of Virology. 2004;78:3572–3577. doi: 10.1128/JVI.78.7.3572-3577.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glass W.G., Subbarao K., Murphy B., Murphy P.M. Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV) pulmonary infection of mice. The Journal of Immunology. 2004;173:4030–4039. doi: 10.4049/jimmunol.173.6.4030. [DOI] [PubMed] [Google Scholar]
- 29.Roberts A., Paddock C., Vogel L., Butler E., Zaki S., Subbarao K. Aged BALB/c mice as a model for increased severity of severe acute respiratory syndrome in elderly humans. Journal of Virology. 2005;79:5833–5838. doi: 10.1128/JVI.79.9.5833-5838.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baas T., Roberts A., Teal T.H., Vogel L., Chen J., Tumpey T.M., Katze M.G., Subbarao K. Genomic analysis reveals age-dependent innate immune responses to severe acute respiratory syndrome coronavirus. Journal of Virology. 2008;82:9465–9476. doi: 10.1128/JVI.00489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rockx B., Baas T., Zornetzer G.A., Haagmans B., Sheahan T., Frieman M., Dyer M.D., Teal T.H., Proll S., van den Brand J. Early upregulation of acute respiratory distress syndrome-associated cytokines promotes lethal disease in an aged-mouse model of severe acute respiratory syndrome coronavirus infection. Journal of Virology. 2009;83:7062–7074. doi: 10.1128/JVI.00127-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Albuquerque N., Baig E., Ma X., Zhang J., He W., Rowe A., Habal M., Liu M., Shalev I., Downey G.P. Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. Journal of Virology. 2006;80:10382–10394. doi: 10.1128/JVI.00747-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Day C.W., Baric R., Cai S.X., Frieman M., Kumaki Y., Morrey J.D., Smee D.F., Barnard D.L. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology. 2009;395:210–222. doi: 10.1016/j.virol.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts A., Deming D., Paddock C.D., Cheng A., Yount B., Vogel L., Herman B.D., Sheahan T., Heise M., Genrich G.L. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathogens. 2007;3:e5. doi: 10.1371/journal.ppat.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheahan T., Whitmore A., Long K., Ferris M., Rockx B., Funkhouser W., Donaldson E., Gralinski L., Collier M., Heise M. Successful vaccination strategies that protect aged mice from lethal challenge from influenza virus and heterologous severe acute respiratory syndrome coronavirus. Journal of Virology. 2011;85:217–230. doi: 10.1128/JVI.01805-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36•.Frieman M., Yount B., Agnihothram S., Page C., Donaldson E., Roberts A., Vogel L., Woodruff B., Scorpio D., Subbarao K. Molecular determinants of severe acute respiratory syndrome coronavirus pathogenesis and virulence in young and aged mouse models of human disease. Journal of Virology. 2012;86:884–897. doi: 10.1128/JVI.05957-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the pathogenesis of mouse adapted SARS-CoV infection in young and old mice, as well as the contributions of amino acid changes in mouse adapted SARS-CoV to virulence.
- 37•.Aylor D.L., Valdar W., Foulds-Mathes W., Buus R.J., Verdugo R.A., Baric R.S., Ferris M.T., Frelinger J.A., Heise M., Frieman M.B. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Research. 2011;21:1213–1222. doi: 10.1101/gr.111310.110. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the approach of using the Collaborative Cross, a recombinant inbred panel of mouse strains, to define contributions of quantitative trait loci to disease phenotypes, including SARS-CoV disease.
- 38.McAuliffe J., Vogel L., Roberts A., Fahle G., Fischer S., Shieh W.-J., Butler E., Zaki S., St. Claire M., Murphy B. Replication of SARS coronavirus administered into the respiratory tract of African Green, rhesus and cynomolgus monkeys. Virology. 2004;330:8–15. doi: 10.1016/j.virol.2004.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haagmans B.L., Kuiken T., Martina B.E., Fouchier R.A.M., Rimmelzwaan G.F., van Amerongen G., van Riel D., de Jong T., Itamura S., Chan K.-H. Pegylated interferon-α protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nature Medicine. 2004;10:290–293. doi: 10.1038/nm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40••.Smits S.L., de Lang A., van den Brand J.M.A., Leijten L.M., van Ijcken W.F., Eijkemans M.J.C., van Amerongen G., Kuiken T., Andeweg A.C., Osterhaus A.D.M.E. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathogens. 2010;6:e1000756. doi: 10.1371/journal.ppat.1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the differences in pathogenesis of SARS-CoV disease in young adult and aged macaques, including differences in the transcription of innate immune genes that contribute to ARDS.
- 41•.Smits S.L., van den Brand J.M.A., de Lang A., Leijten L.M.E., van IJcken W.F., van Amerongen G., Osterhaus A.D.M.E., Andeweg A.C., Haagmans B.L. Distinct severe acute respiratory syndrome coronavirus-induced acute lung injury pathways in two different nonhuman primate species. Journal of Virology. 2011;85:4234–4245. doi: 10.1128/JVI.02395-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the increased susceptibility to SARS-CoV infections in African green monkeys compared to cynomolgus macaques, including the contributions of proinflammatory cytokines and chemokines to increased SARS-CoV disease.
- 42.Rathinam V.A.K., Fitzgerald K.A. Cytosolic surveillance and antiviral immunity. Current Opinion in Virology. 2011;1:455–462. doi: 10.1016/j.coviro.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arpaia N., Barton G.M. Toll-like receptors: key players in antiviral immunity. Current Opinion in Virology. 2011;1:447–454. doi: 10.1016/j.coviro.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roth-Cross J.K., Bender S.J., Weiss S.R. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces Type I interferon in brain macrophages/microglia. Journal of Virology. 2008;82:9829–9838. doi: 10.1128/JVI.01199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45•.Li J., Liu Y., Zhang X. Murine coronavirus induces Type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. Journal of Virology. 2010;84:6472–6482. doi: 10.1128/JVI.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the detection of the MHV by the cytosolic PRRs MDA5 and RIG-I, and the contributions of IRF3 and IF-κB mediated signaling to the induction of Interferons in the context of Coronavirus infection.
- 46.Galani V., Tatsaki E., Bai M., Kitsoulis P., Lekka M., Nakos G., Kanavaros P. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): an up-to-date cell-specific review. Pathology – Research and Practice. 2010;206:145–150. doi: 10.1016/j.prp.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 47.Kurt-Jones E.A., Popova L., Kwinn L., Haynes L.M., Jones L.P., Tripp R.A., Walsh E.E., Freeman M.W., Golenbock D.T., Anderson L.J. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nature Immunology. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 48.Marchant D., Singhera G.K., Utokaparch S., Hackett T.L., Boyd J.H., Luo Z., Si X., Dorscheid D.R., McManus B.M., Hegele R.G. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. Journal of Virology. 2010;84:11359–11373. doi: 10.1128/JVI.00804-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khanolkar A., Hartwig S.M., Haag B.A., Meyerholz D.K., Harty J.T., Varga S.M. Toll-like receptor 4 deficiency increases disease and mortality after mouse hepatitis virus Type 1 infection of susceptible C3H mice. Journal of Virology. 2009;83:8946–8956. doi: 10.1128/JVI.01857-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50••.Zornetzer G.A., Frieman M.B., Rosenzweig E., Korth M.J., Page C., Baric R.S., Katze M.G. Transcriptomic analysis reveals a mechanism for a prefibrotic phenotype in STAT1 knockout mice during severe acute respiratory syndrome coronavirus infection. Journal of Virology. 2010;84:11297–11309. doi: 10.1128/JVI.01130-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the differences in transcriptional responses of innate immune genes in wild type, IFNAR1−/−, and STAT1−/− mice, including differences in ISG trancription that potentially contribute to fibrotic phenotypes of SARS-CoV disease.
- 51.Law H., Cheung C., Sia S., Chan Y., Peiris J.M., Lau Y. Toll-like receptors, chemokine receptors and death receptor ligands responses in SARS coronavirus infected human monocyte derived dendritic cells. BMC Immunology. 2009;10:35. doi: 10.1186/1471-2172-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao J., Zhao J., Van Rooijen N., Perlman S. Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS-CoV-infected mice. PLoS Pathogens. 2009;5:e1000636. doi: 10.1371/journal.ppat.1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheahan T., Morrison T.E., Funkhouser W., Uematsu S., Akira S., Baric R.S., Heise M.T. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathogens. 2008;4:e1000240. doi: 10.1371/journal.ppat.1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le Bon A., Tough D.F. Links between innate and adaptive immunity via type I interferon. Current Opinion in Immunology. 2002;14:432–436. doi: 10.1016/s0952-7915(02)00354-0. [DOI] [PubMed] [Google Scholar]
- 55.Dahl H., Linde A., Strannegard O. In vitro inhibition of SARS virus replication by human interferons. Scandinavian Journal of Infectious Diseases. 2004;36:829–831. doi: 10.1080/00365540410021144. [DOI] [PubMed] [Google Scholar]
- 56.Stroher U., DiCaro A., Li Y., Strong J.E., Aoki F., Plummer F., Jones S.M., Feldmann H. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon-alpha. Journal of Infectious Diseases. 2004;189:1164–1167. doi: 10.1086/382597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sainz B., Jr., Mossel E.C., Peters C.J., Garry R.F. Interferon-beta and interferon-gamma synergistically inhibit the replication of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) Virology. 2004;329:11–17. doi: 10.1016/j.virol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barnard D.L., Day C.W., Bailey K., Heiner M., Montgomery R., Lauridsen L., Chan P.K., Sidwell R.W. Evaluation of immunomodulators, interferons and known in vitro SARS-coV inhibitors for inhibition of SARS-coV replication in BALB/c mice. Antiviral Chemistry & Chemotherapy. 2006;17:275–284. doi: 10.1177/095632020601700505. [DOI] [PubMed] [Google Scholar]
- 59.Kumaki Y., Ennis J., Rahbar R., Turner J.D., Wandersee M.K., Smith A.J., Bailey K.W., Vest Z.G., Madsen J.R., Li J.K.K. Single-dose intranasal administration with mDEF201 (adenovirus vectored mouse interferon-alpha) confers protection from mortality in a lethal SARS-CoV BALB/c mouse model. Antiviral Research. 2011;89:75–82. doi: 10.1016/j.antiviral.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60•.Frieman M.B., Chen J., Morrison T.E., Whitmore A., Funkhouser W., Ward J.M., Lamirande E.W., Roberts A., Heise M., Subbarao K. SARS-CoV pathogenesis is regulated by a STAT1 dependent but a Type I, II and III interferon receptor independent mechanism. PLoS Pathogens. 2010;6:e1000849. doi: 10.1371/journal.ppat.1000849. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the contribution of STAT1 to a protective immune response to SARS-CoV disease, while also demonstrating minimal differences in SARS-CoV disease phenotypes in the absence of Interferon receptors.
- 61.Mordstein M., Neugebauer E., Ditt V., Jessen B., Rieger T., Falcone V., Sorgeloos F., Ehl S., Mayer D., Kochs G. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. Journal of Virology. 2010;84:5670–5677. doi: 10.1128/JVI.00272-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pugin J., Ricou B., Steinberg K.P., Suter P.M., Martin T.R. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1. American Journal of Respiratory and Critical Care Medicine. 1996;153:1850–1856. doi: 10.1164/ajrccm.153.6.8665045. [DOI] [PubMed] [Google Scholar]
- 63••.Huang I.C., Bailey C.C., Weyer J.L., Radoshitzky S.R., Becker M.M., Chiang J.J., Brass A.L., Ahmed A.A., Chi X., Dong L. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathogens. 2011;7:e1001258. doi: 10.1371/journal.ppat.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes IFITM protein family mediated restriction of entry and replication of SARS-CoV, elucidating an antiviral function of ISGs.
- 64.Schoggins J.W., Rice C.M. Interferon-stimulated genes and their antiviral effector functions. Current Opinion in Virology. 2011;1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65••.Schoggins J.W., Wilson S.J., Panis M., Murphy M.Y., Jones C.T., Bieniasz P., Rice C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes an overexpression screen to determine the contribution of Interferon stimulated genes to the antiviral response against a panel of important human and animal viruses.
- 66.Snijder E.J., van der Meer Y., Zevenhoven-Dobbe J., Onderwater J.J.M., van der Meulen J., Koerten H.K., Mommaas A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. Journal of Virology. 2006;80:5927–5940. doi: 10.1128/JVI.02501-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knoops K., Kikkert M., van den Worm S.H.E., Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J., Mommaas A.M., Snijder E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biology. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68•.Bouvet Ml, Debarnot C., Imbert I., Selisko B., Snijder E.J., Canard B., Decroly E. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathogens. 2010;6:e1000863. doi: 10.1371/journal.ppat.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the capping mechanisms of SARS-CoV nonstructural proteins, including the requirement for nsp14, nsp10, and nsp16 in the formation of SARS-CoV viral RNA caps in vitro.
- 69.Chen Y., Su C., Ke M., Jin X., Xu L., Zhang Z., Wu A., Sun Y., Yang Z., Tien P. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathogens. 2011;7:e1002294. doi: 10.1371/journal.ppat.1002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen Y., Cai H., Pan Ja, Xiang N., Tien P., Ahola T., Guo D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3484–3489. doi: 10.1073/pnas.0808790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Daffis S., Szretter K.J., Schriewer J., Li J., Youn S., Errett J., Lin T.-Y., Schneller S., Zust R., Dong H. 2[prime]-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72••.Zust R., Cervantes-Barragan L., Habjan M., Maier R., Neuman B.W., Ziebuhr J., Szretter K.J., Baker S.C., Barchet W., Diamond M.S. Ribose 2[prime]-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nature Immunology. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the importance of viral RNA cap modifications by Coronaviruses in evading detection by the innate immune sensor MDA5 and avoiding the induction of Interferons.
- 73.Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. Journal of Virology. 2007;81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kamitani W., Huang C., Narayanan K., Lokugamage K.G., Makino S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nature Structural & Molecular Biology. 2009;16:1134–1140. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75•.Huang C., Lokugamage K.G., Rozovics J.M., Narayanan K., Semler B.L., Makino S. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathogens. 2011;7:e1002433. doi: 10.1371/journal.ppat.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the escape of SARS-CoV viral RNAs from nsp1 dependent RNA cleavage, whereas host mRNAs are more susceptible to RNA degradation.
- 76.Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C.J., Tseng C.-T.K., Baker S.C. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. Journal of Biological Chemistry. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-κB signaling. Journal of Virology. 2009;83:6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun L., Xing Y., Chen X., Zheng Y., Yang Y., Nichols D.B., Clementz M.A., Banach B.S., Li K., Baker S.C. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS ONE. 2012;7:e30802. doi: 10.1371/journal.pone.0030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79•.Lu X., Pan Ja, Tao J., Guo D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes. 2011;42:37–45. doi: 10.1007/s11262-010-0544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the Interferon antagonist activity of the SARS-CoV nucleocapsid protein, which occurs upstream of the cellular PRRs RIG-I and MDA5.
- 80.Kopecky-Bromberg S.A., Martinez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. Journal of Virology. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ye Y., Hauns K., Langland J.O., Jacobs B.L., Hogue B.G. Mouse hepatitis coronavirus A59 nucleocapsid protein is a Type I interferon antagonist. Journal of Virology. 2007;81:2554–2563. doi: 10.1128/JVI.01634-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Siu K.-L., Kok K.-H., Ng M.-H.J., Poon V.K.M., Yuen K.-Y., Zheng B.-J., Jin D.-Y. Severe acute respiratory syndrome coronavirus M protein inhibits Type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKɛ complex. Journal of Biological Chemistry. 2009;284:16202–16209. doi: 10.1074/jbc.M109.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-{kappa}B signaling. Journal of Virology. 2009;83:6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Narayanan K., Huang C., Makino S. SARS coronavirus accessory proteins. Virus Research. 2008;133:113–121. doi: 10.1016/j.virusres.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yount B., Roberts R.S., Sims A.C., Deming D., Frieman M.B., Sparks J., Denison M.R., Davis N., Baric R.S. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. Journal of Virology. 2005;79:14909–14922. doi: 10.1128/JVI.79.23.14909-14922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Freundt E.C., Yu L., Park E., Lenardo M.J., Xu X.-N. Molecular determinants for subcellular localization of the severe acute respiratory syndrome coronavirus open reading frame 3b protein. Journal of Virology. 2009;83:6631–6640. doi: 10.1128/JVI.00367-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Frieman M., Yount B., Heise M., Kopecky-Bromberg S.A., Palese P., Baric R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/golgi membrane. Journal of Virology. 2007;81:9812–9824. doi: 10.1128/JVI.01012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hussain S., Perlman S., Gallagher T.M. Severe acute respiratory syndrome coronavirus protein 6 accelerates murine hepatitis virus infections by more than one mechanism. Journal of Virology. 2008;82:7212–7222. doi: 10.1128/JVI.02406-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hussain S., Gallagher T. SARS-coronavirus protein 6 conformations required to impede protein import into the nucleus. Virus Research. 2010;153:299–304. doi: 10.1016/j.virusres.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yount B., Curtis K.M., Fritz E.A., Hensley L.E., Jahrling P.B., Prentice E., Denison M.R., Geisbert T.W., Baric R.S. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12995–13000. doi: 10.1073/pnas.1735582100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Frieman M., Yount B., Sims A., Deming D., Morrison T., Sparks J., Denison M., Heise M., Baric R., Perlman S. The nidoviruses: SARS coronavirus accessory ORFs encode luxury functions. Advances in Experimental Medicine and Biology. 2006;581:149–152. doi: 10.1007/978-0-387-33012-9_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wevers B.A., van der Hoek L. Recently discovered human coronaviruses. Clinics in Laboratory Medicine. 2009;29:715–724. doi: 10.1016/j.cll.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yoneyama M., Kikuchi M., Matsumoto K., Imaizumi T., Miyagishi M., Taira K., Foy E., Loo Y.-M., Gale M., Akira S. Shared and unique functions of the DExD/H-Box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. The Journal of Immunology. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 94.Satoh T., Kato H., Kumagai Y., Yoneyama M., Sato S., Matsushita K., Tsujimura T., Fujita T., Akira S., Takeuchi O. LGP2 is a positive regulator of RIG-I and MDA5-mediated antiviral responses. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1512–1517. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F., Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 96.Baum A., Sachidanandam R., Garcia-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2011;107:16303–16308. doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kato H., Takeuchi O., Mikamo-Satoh E., Hirai R., Kawai T., Matsushita K., Hiiragi A., Dermody T.S., Fujita T., Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation associated gene 5. The Journal of Experimental Medicine. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hogan R.J., Gao G., Rowe T., Bell P., Flieder D., Paragas J., Kobinger G.P., Wivel N.A., Crystal R.G., Boyer J. Resolution of primary severe acute respiratory syndrome-associated coronavirus infection requires Stat1. Journal of Virology. 2004;78:11416–11421. doi: 10.1128/JVI.78.20.11416-11421.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boehme K.W., Guerrero M., Compton T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. The Journal of Immunology. 2006;177:7094–7102. doi: 10.4049/jimmunol.177.10.7094. [DOI] [PubMed] [Google Scholar]
- 100.Murawski M.R., Bowen G.N., Cerny A.M., Anderson L.J., Haynes L.M., Tripp R.A., Kurt-Jones E.A., Finberg R.W. Respiratory syncytial virus activates innate immunity through toll-like receptor 2. Journal of Virology. 2009;83:1492–1500. doi: 10.1128/JVI.00671-08. [DOI] [PMC free article] [PubMed] [Google Scholar]