

Highlights
► Virus entry is an indispensable first step for cell infection. ► Viruses may escape or hijack innate immune receptors for cell entry. ► Anti-viral innate immunity can be inhibited by entering virion-incorporated proteins. ► Viruses might hijack autophagy early post entry for promoting their replication.
Abstract
Entry into a cell submits viruses to detection by pattern recognition receptors (PRRs) leading to an early innate anti-viral response. Several viruses evolved strategies to avoid or subvert PRR recognition at the step of virus entry to promote infection. Whereas viruses mostly escape from soluble PRR detection, endocytic/phagocytic PRRs, such as the mannose receptor or DC-SIGN, are commonly used for virus entry. Moreover, virion-incorporated proteins may also offer viruses a way to dampen anti-viral innate immunity upon virus entry, and entering viruses might usurp autophagy to improve their own infectivity.
Introduction
Viruses can enter cells by many routes which reflect the evolutionary interface between host and viruses, for which anti-viral innate immune evasion might have been positively selected to improve cell entry [1, 2]. Besides the host innate immune defence ensured by hundred of antimicrobial peptides (AMP) which can be rapidly mobilized to neutralize viruses [3], innate pathogen recognition is mediated by dozens of soluble, membrane bound or cytosolic germ-line encoded pattern recognition receptors (PRRs) which detect conserved pathogen associated molecular patterns (PAMPs) [4]. Soluble serum PRRs include collectins, ficolins and pentraxins that may opsonise viruses leading to their complement-dependent destruction. Membrane-bound endocytic/phagocytic PRRs, including the mannose receptor (MR), scavenger receptors (SR), and dendritic cell-specific ICAM grabbing non-integrin (DC-SIGN) may also directly recognize viruses to mediate virus uptake.
Moreover, several membrane bound and cytosolic-distributed PRRs have for major functions to transduce intracellular signals to elicit innate immune responses. Toll-like receptors (TLRs) are membrane-expressed signaling PRRs: while TLR1/2/4/5/6/10 are distributed on the cell surface, TR3/7/8/9 are located within endosomal compartments [5]. Detection of cytosolic-located PAMPs can be achieved by specific PRRs which include the retinoic acid-inducible gene I (RIG-I)-like receptors (RLR), RIG-I and MDA5 [4]. Following recognition of viral PAMPs, these PRRs transduce intracellular signals to activate nuclear factor-kappa B (NF-κB) and/or type I IFN (IFN-I) regulatory transcription factors (IRF)3 and/or IRF7, leading to proinflammatory cytokine and IFN-I expression by the infected cells. Newly synthesized IFN-I is secreted and binds to IFN-I receptor (IFNAR) inducing the expression of hundreds of IFN stimulating genes (ISGs) with direct anti-viral effect [4].
Overall, innate PRRs are a line of defence that viruses have to escape to establish a successful cellular infection. However, very little is known about how viruses might modulate innate immunity in virus entry. In this review we focused on mechanisms used by viruses to modulate innate immunity in order to both facilitate their entry into a cell and to counteract immediate cell autonomous antiviral responses postvirus entry.
Innate immunity modulation for enhancing virus entry
Evasion and subversion of soluble PRR
Mannose-binding lectin (MBL) is a serum lectin of the collectin family that plays an important role in innate immunity [6]. MBL binds to carbohydrates on the surface of a wide range of pathogens and activates the lectin pathway of complement. However, the degree of glycosylation of viral glycoproteins is a general factor in determining sensitivity to MBL recognition (Figure 1 ). For example, the sensitivity to MBL detection of seasonal H1N1 strains of influenza virus (highly sensitive) and A (H1N1) pandemic viruses (poorly sensitive), is depending on the extent of glycosylation of their respective hemagglutinin (HA). The loss of a single N-linked glycan from the HA of influenza virus is associated with resistance to MBL detection and increased virulence [7•]. MBL can also bind to high-mannose glycans on human immunodeficiency virus (HIV)-1 envelope glycoprotein gp120 [8]; the level of glycosylation of gp120 is variable, depending on infected cell [9] what might allow prevention of MBL binding.
Figure 1.
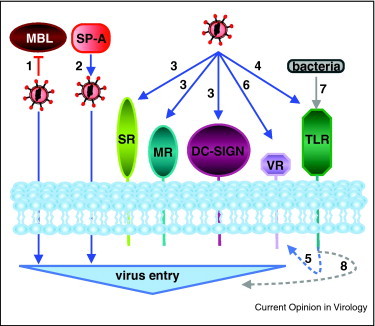
General viral strategies to manipulate innate PRR to enhance virus entry. Viruses may express glycosylated surface proteins which do not allow collectin binding, such as MBL, and subsequent complement-mediated destruction (1). Alternatively, viruses may benefit from SP-A recognition to improve cell entry, through unknown mechanism (2). Viruses may directly bind several different endocytic/phagocytic PRRs (SR, MR, DC-SIGN) to usurp intracellular routes for efficient entry (3). Virus binding to TLR (4) may also promote specific virus receptor (VR) expression (5) to enhance further virus entry (6). Finally, co-infecting bacteria may contribute to virus entry via TLR signaling (7) through still undefined process (8).
A high number of virus-mediated diseases are associated with MBL gene polymorphisms, clearly suggesting that avoiding MBL detection is important for viruses. For instance, hepatitis B and C virus persistence and disease progression was linked to MBL polymorphisms. Recently, MBL was shown to interact with hepatitis C virus (HCV) E1/E2 envelope glycoproteins leading to inhibition of virus entry [10]. These new results provide a molecular explanation for the role of MBL in HCV disease.
Finally, another way to block MBL-dependent virus neutralization was recently reported for human astroviruses (HAstVs); the coat protein of HAstVs binds to MBL and inhibits the mannan-mediated activation of the lectin pathway of complement [11•].
Besides avoiding or inhibiting collectin functions, viruses may hijack collectins to facilitate infection (Figure 1). The surfactant protein (SP-A) is an innate immune factor of the lung, amniotic fluid and vaginal tract. SP-A binds to high-mannose carbohydrate residues of several human cytomegalovirus (CMV) glycoproteins. SP-A binding to CMV stimulates virus entry in permissive lung rat cells [12]. Whether this is true for human cells is not known. Interestingly, SP-A binds to HIV gp120 mannose carbohydrate what enhance the uptake of viral particles by dendritic cells (DCs) [13].
Subversion of endocytic/phagocytic PRR
Phagocytosis is an innate defence system of specialized phagocytes, i.e. macrophages, neutrophiles and DC, first described more than 120 years ago by Ilya Ilitch Metchnikov (Nobel prize in medicine 1908) [14]. It is only recently that this process appeared as a mechanism promoting virus entry. Adenovirus targeted to the Fcγ receptor 1 of hematopoietic cells gives rise not only to adenovirus aggregates which are phagocytosed, but also to single particles which enter into the cells by endocytosis [15]. Interestingly, both phagocytosis and endocytosis of adenoviruses were shown to cooperate in order to optimize viral gene delivery. Thus, phagocytosis might facilitate entry of aggregated viruses, but not the one of single particles. However, virus phagocytosis was also reported by Clement et al. who have shown that plasma membrane protrusions of fibroblastic cells are formed around entering herpes simplex virus (HSV)-1 that further enter cells in phagocytosis-like particles [16]. A recent study has shown that the additional binding to the αVβ3-integrin routes HSV-1 to an acidic vesicular compartment [17••]. In regard to the role of integrins in phagocytosis [18], it might be determined whether the αVβ3-integrin routes HSV-1 through phagosome-like vesicles. Finally, the giant mimivirus was also shown to use phagocytosis to infect macrophages [19]. Although the big size of mimiviruses may explain why they evolved to subvert phagocytosis, this cellular process might be usurped by other viruses.
As phagocytosis, macropinocytosis is an endocytic mechanism with a role in immune defence (although it is primarily used for the non-selective internalization of fluid and membrane). First reports have shown that macropinocytosis is an infectious entry route for adenovirus serotype 3 and vaccinia virus [20, 21]. Recently, it was described that influenza A virus, well described to use a clathrin-mediated endocytosis pathway to enter into a cell, can enter host cells through an alternative pathway with the molecular characteristics of macropinocytosis, in serum-rich conditions [22].
Subversion of mannose receptor
MR (CD206) is a type-I transmembrane glycoprotein which binds mannose, fucose or N-acetylglucosamine sugar residues on the surface of a broad array of pathogens and mediates endocytosis and phagocytosis. MR was previously reported to be associated with efficient macrophage entry of highly HA glycosylated influenza A virus strains when compared with less glycosylated strains [23]. Indeed, MR was recently reported to directly bind influenza A virus glycans on HA through its carbohydrate recognition domain what promote virus entry, and similar observations were done for another endocytic PRR, the macrophage galactose-type lectin (MGL) (Figure 1) [24•]. MR also contributes to virus binding and entry of HBV [25], dengue virus [26] and HIV-1 [27] although for the latter MR-dependent entry is not associated with productive HIV-1 infection in macrophages.
Subversion of the scavenger receptors
SR represents a large family of transmembrane PRR involved in endocytosis/phagocytosis that recognizes several different PAMPs [28]. SR-BI, a class B SR, which binds a variety of lipoproteins (high-density lipoproteins (HDL), low-density lipoproteins (LDL)), is utilized by HCV to gain entry into hepatocytes (Figure 1). SR-BI binds the envelope E2 glycoprotein of HCV [29]. However, HCV may be associated with the serum factors LDL and HDL. These associations shield the virus from neutralizing antibodies. However, LDL/HDL on HCV could bind SR-BI and contribute to viral entry [30••]. HDL improves HCV entry by accelerating SR-BI-mediated endocytosis, and HCV and HDL binding to SR-BI, as well as the lipid transfer activities of SR-BI, are required for SR-BI involvement in HCV entry [31, 32]. Interestingly, the SR-BI-dependent recognition of HCV by dendritic cells leads to cross-presentation of viral antigens what might contribute to elicit an host CD8+ T cell anti-HCV response [33].
CD163, another SR contributes to porcine and respiratory syndrome virus entry possibly through direct interaction between CD163 and viral proteins [34] suggesting that SR binding might be a widely used pathway to promote viral entry.
Subversion of DC-SIGN
DC-SIGN is a type II transmembrane protein expressed on DC, endothelium and macrophage subpopulations. DC-SIGN is an evolutionary positively selected pathogen receptor that binds to high mannose carbohydrates of a range of microorganisms and contributes to facilitate host cell entry of several different viruses including HIV-1 (Figure 1) [1], HCV [35], influenza A viruses [36] and coronavirus [37]. In this latter case, it was shown that a single N-linked glycosylation on the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates MBL binding what interfere with coronavirus interaction with DC-SIGN on type II alveolar cells and endothelial cells. Such competition for virus binding may play a major role in viral spread and pathogenicity [38•].
Evasion and subversion of signaling PRR
The high variability of components exposed on the surface of viruses might contribute to limit broad recognition by surface expressed TLRs. In contrast, endocytosed viruses are exposed to vesicular TLRs which detect viral genomic PAMPs [4]. Accordingly, evolutionary genetic studies indicate that intracellular TLR have evolved under stronger positive selective pressure than plasma membrane-expressed TLRs suggesting that host defence mechanisms have to face a variety of highly mutating viruses and adapt to offer a proper anti-viral response [39]. Nevertheless, the list of viruses recognized by or signaling from cell surface TLR (TLR2 and TLR4), is growing: wild-type measles virus (MeV) [40], the respiratory syncytial virus (RSV) [41], CMV [42], HSV-1 [43], mouse mammary tumor virus (MMTV) [44], Epstein-bar virus (EBV) [45], lymphocytic choriomeningitis virus (LCMV) [46], mouse hepatitis virus (MHV-68) [47], vaccinia virus (VV) [48], and human rhinovirus (HRV)6 [49]. Whereas, contribution of TLR recognition in virus-induced proinflammatory and/or IFN-I anti-viral responses was clearly reported [50••], contribution to cell entry remains largely unappreciated.
However, viruses might benefit from stimulating TLRs (Figure 1). Wild-type HA MeV activates TLR2 as a means to upregulate the expression of CD150, its viral entry receptor [40]. Similarly, MMTV binding to TLR4 on DC leads to an increased expression of its own entry receptor, CD71 [44]. However in both cases, it is not known whether the enhanced expression of the virus entry receptor further promtes virus entry.
Viruses can also indirectly use cell surface bound TLR to improve entry and/or infectivity (Figure 1). For example, gonococci coinfection enhances both HIV-1 entry and replication, through a TLR2-dependent signaling mechanism [51]. Whether other viruses may benefit from PRRs activation by coinfecting pathogens has to be investigated. However, commensal bacteria stimulating TLR4 were shown to offer resistance to HIV-1 infection [52]. How virus infections may benefit from pathogenic bacteria co-infections while not from non-pathogenic bacteria remains to be fully understood.
Virus entry-dependent anti-viral innate immunity modulation
Inhibition of IFN-I
Subsequently to entry, the ability of viruses to counteract cellular antiviral innate response mostly involves non-structural proteins, which are not incorporated in entering virions. Few examples are however known of immediate antiviral modulation possibly imposed soon postentry by virion-incorporated proteins (Figure 2 ).
Figure 2.
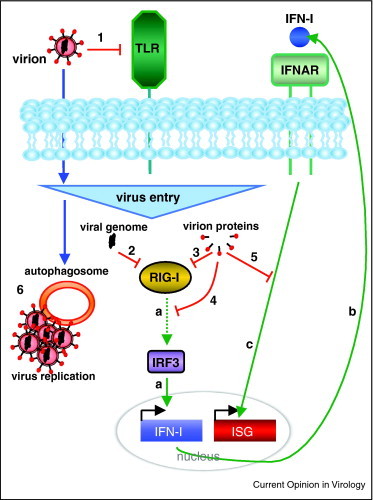
General viral strategies to avoid, inhibit or usurp innate antiviral responses immediately postentry. Variability of surface expressed viral proteins limits their detection by plasma membrane TLR (1). Within the cytosol, viral genomic PAMPs can be recognized par intracytosolic PRR, such as RIG-I. RIG-I induces IFN-I synthesis via the transcription factor IRF3 (a). Secreted IFN-I activates then its own receptor IFNAR (b) which transduces signals leading to antiviral ISG effectors production (c). Postvirus entry, certain viral genomes may escape from RIG-I detection either directly (2) or via inhibitory virion-incorporated proteins (3). IRF3 activation may also be inhibited by virion-incorporated proteins (4), to prevent IFN-I production. Moreover, virion-incorporated proteins could inhibit IFNAR-dependent signals to prevent antiviral ISG production (5). Independently, autophagy induction upon virus entry might be hijacked to facilitate subsequent virus replication (6).
Inside the cytoplasm, viruses can be recognized by several PRRs, including two DExD/H box RNA helicases RIG-I and MDA5, ultimately driving IFN-I production [53]. Only examples of direct modulation of RIG-I by virus-incorporated proteins were yet reported (Figure 2). RIG-I detects unique 5′ triphosphate on virus genomic RNA and triggers IFN-I induction [54]. RNA viruses elicit IFN-I production upon RIG-I recognition soon after virus entry and delivery of their genome inside the cytosol. However, it was shown that genomic RNAs of several emergent double-stranded (ds)RNA viruses evade RIG-I detection because of the cleavage of triphosphates at the RNA 5′ end, by a viral function [55]. Beyond, virion-incorporated proteins such as ebola-VP35 can also prevent RIG-I-mediated detection by competing for RNA genome interaction [56]. Another mechanism for direct RIG-I modulation was recently described with the protease of HIV-1 which promotes the lysosomal degradation of RIG-I [57]. Moreover, immunoglobulin-dependent dengue virus entry into immunoglobulin Fc region receptor (FcR)-bearing cells promotes activation of hydroxyacetone kinase and autophagy-related genes (ATG)5-ATG12, which disrupt the RIG-I and MDA-5 signaling cascade and prevent IFN-I production [58].
To prevent IFN-I induction, IRF3, a downstream effector of several cytosolic PRRs, is a very common putative target of virion proteins (Figure 2). The immediate-early (IE)62 protein is an abundant tegument protein of the alphaherpesvirus varicella-zoster virus (VZV). IE62 promotes inactivation of IRF3 by preventing its phosphorylation by TANK-binding kinase (TBK)1 [59•]. Moreover, although IE62 is not able to interact with TBK1 or IRF3, unproductive TBK1-IRF3 complexes are maintained, limiting the redistribution of trapped-TBK1 to activate other IRF3 substrates. Another example is the phosphoprotein P of rabies virus that binds to the ribonucleoprotein of free virions, which prevent TBK1 dependent phosphorylation of IRF3 [60]. Similarly, ebola virus VP35 also inactivates IRF3 [61] and the nucleoprotein of LCMV dampens IFN-I induction by preventing IRF3 activation [62].
Interestingly, viral strategies evolved to inhibit IFN-I induction might also depend on host-incorporated proteins within free infectious virions. Although most of the host-incorporated proteins contribute to virus infectivity, it is unknown whether they contribute to modulate early innate immune events. Interestingly, the propyl isomerase (PIN)1 integrated into HIV-1 viral particles [63] and required for uncoating of the virus [64], regulates ubiquitination and proteasome-dependent degradation of IRF3, what might prevent IFN-I induction [65]. Moreover, PIN1 regulates the expression of the cytidine deaminase APOBEC3G an innate restriction factor that inhibits HIV-1 replication [66].
Finally, IFNAR-depending signals might also be the target of virions proteins (Figure 2). Thus the VZV-IE63 protein can prevent IFN-I response by inhibiting the phosphorylation of the eukaryotic initiation factor 2 (eIF-2α), a downstream event of the IFNAR signaling pathway which inhibits translation [67]. Moreover, the phosphoprotein P of MeV inhibits IFNAR-dependent signalisation by binding to and preventing the activation of STAT1, a downstream intermediate of this receptor [68].
Autophagy subversion
Autophagy is a catabolic lysosomal mechanism which plays a crucial role as an innate defence mechanism by promoting virus or virus-derived component degradation and delivery of virus RNA to TLR-containing endosomes leading to IFN-I induction [69, 70]. However, several viruses evolved strategies to avoid or usurp autophagy to their own benefit [71]. Although the manipulation of autophagy upon virus entry to improve infectivity was not yet reported, several studies reported autophagy induction upon virus receptor engagement (Figure 2). Vaccine MeV entry induces autophagy through the direct engagement of the regulatory complement activation receptor CD46 [72••], a receptor also for human herpes virus 6 (HHV6) and several serotypes of adenoviruses [73, 74]. Although CD46-mediated autophagy induction does not enhance MeV entry [72••], whether MeV benefits from autophagy induction to replicate is not yet known. TLR3, TLR4 and TLR7, PRRs that recognize virus genome PAMPs, were also reported to induce autophagy upon ligand binding [75], what might be hijacked by some viruses to promote their own replication. Moreover, it is also shown that autophagy-associated proteins can be recruited to nascent phagosomes and accelerate phagosome maturation [76]. Moreover, clathrin-associated plasma membrane contributes to the formation of autophagosomes [77••]. Since phagocytosis and clathrin-dependent endocytosis are both involved in virus entry, their links with autophagy proteins or with the entire autophagy process, respectively, might benefit for virus entry/replication. Further studies are required to determine whether viruses induce/use autophagy or autophagy-associated proteins upon entry to enhance entry and infectivity.
Conclusion
Different evolutionary selected strategies that promote viral entry into cells through subversion of innate immunity were described. Viruses might hijack innate cell surface PRR to enter a cell. Viruses may also evade anti-viral innate response by avoiding PRR recognition and/or inhibiting PRR downstream signaling intermediates. A recent global genomic analysis on the HIV/host interface highlighted that most genetic variability that could account for differences in susceptibility to disease occurs in genes coding for cellular membrane proteins of the host as well as in the viral envelope genes suggesting that the genetic variability of cell surface expressed proteins might be a defence mechanism for virus binding/entry prevention [1]. Moreover, a recent bottom-up approach based on both literature-curation and literature-omics data integration to analyze the interactions between viral proteins and host proteins of the IFN-I response highlighted that viruses target significantly transcription factors, signaling intermediates and membranous receptors [78•]. These global results display the potential of the studies that remain to be done to fully depict innate immune manipulation in virus entry.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgments
We are thankful to Dr. Isabel P. Grégoire for critical reading of the manuscript. Our work is supported by INSERM, UCBLyon-1, ANR-08-JCJC-0064-01 and Cluster 10 infectiologie Rhône-Alpes.
References
- 1.Bozek K., Lengauer T. Positive selection of HIV host factors and the evolution of lentivirus genes. BMC Evol Biol. 2010;10:186. doi: 10.1186/1471-2148-10-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mercer J., Helenius A. Virus entry by macropinocytosis. Nat Cell Biol. 2009;11:510–520. doi: 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- 3.Ding J., Chou Y.Y., Chang T.L. Defensins in viral infections. J Innate Immun. 2009;1:413–420. doi: 10.1159/000226256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennan K., Bowie A.G. Activation of host pattern recognition receptors by viruses. Curr Opin Microbiol. 2010;13:503–507. doi: 10.1016/j.mib.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 5.Kumar H., Kawai T., Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 6.Ip W.K., Takahashi K., Ezekowitz R.A., Stuart L.M. Mannose-binding lectin and innate immunity. Immunol Rev. 2009;230:9–21. doi: 10.1111/j.1600-065X.2009.00789.x. [DOI] [PubMed] [Google Scholar]
- 7•.Job E.R., Deng Y.M., Tate M.D., Bottazzi B., Crouch E.C., Dean M.M., Mantovani A., Brooks A.G., Reading P.C. Pandemic H1N1 influenza A viruses are resistant to the antiviral activities of innate immune proteins of the collectin and pentraxin superfamilies. J Immunol. 2010;185:4284–4291. doi: 10.4049/jimmunol.1001613. [DOI] [PubMed] [Google Scholar]; This paper shows that the degree of glycosylation in the globular head of the HA correlates with the ability of H1N1 viruses to infect human respiratory epithelial cells and to inhibit MBL and SP-D binding.
- 8.Hart M.L., Saifuddin M., Uemura K., Bremer E.G., Hooker B., Kawasaki T., Spear G.T. High mannose glycans and sialic acid on gp120 regulate binding of mannose-binding lectin (MBL) to HIV type 1. AIDS Res Hum Retroviruses. 2002;18:1311–1317. doi: 10.1089/088922202320886352. [DOI] [PubMed] [Google Scholar]
- 9.Raska M., Takahashi K., Czernekova L., Zachova K., Hall S., Moldoveanu Z., Elliott M.C., Wilson L., Brown R., Jancova D. Glycosylation patterns of HIV-1 gp120 depend on the type of expressing cells and affect antibody recognition. J Biol Chem. 2010;285:20860–20869. doi: 10.1074/jbc.M109.085472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown K.S., Keogh M.J., Owsianka A.M., Adair R., Patel A.H., Arnold J.N., Ball J.K., Sim R.B., Tarr A.W., Hickling T.P. Specific interaction of hepatitis C virus glycoproteins with mannan binding lectin inhibits virus entry. Protein Cell. 2010;1:664–674. doi: 10.1007/s13238-010-0088-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11•.Hair P.S., Gronemus J.Q., Crawford K.B., Salvi V.P., Cunnion K.M., Thielens N.M., Arlaud G.J., Rawal N., Krishna N.K. Human astrovirus coat protein binds C1q and MBL and inhibits the classical and lectin pathways of complement activation. Mol Immunol. 2010;47:792–798. doi: 10.1016/j.molimm.2009.10.006. [DOI] [PubMed] [Google Scholar]; This paper describes a novel mechanism evolved by human astroviruses to inhibit MBL detection-depending innate immune response through direct inhibition of MBL-mediated activation of the lectin pathway of complement.
- 12.Weyer C., Sabat R., Wissel H., Kruger D.H., Stevens P.A., Prosch S. Surfactant protein A binding to cytomegalovirus proteins enhances virus entry into rat lung cells. Am J Respir Cell Mol Biol. 2000;23:71–78. doi: 10.1165/ajrcmb.23.1.3859. [DOI] [PubMed] [Google Scholar]
- 13.Gaiha G.D., Dong T., Palaniyar N., Mitchell D.A., Reid K.B., Clark H.W. Surfactant protein A binds to HIV and inhibits direct infection of CD4+ cells, but enhances dendritic cell-mediated viral transfer. J Immunol. 2008;181:601–609. doi: 10.4049/jimmunol.181.1.601. [DOI] [PubMed] [Google Scholar]
- 14.Stuart L.M., Ezekowitz R.A. Phagocytosis and comparative innate immunity: learning on the fly. Nat Rev Immunol. 2008;8:131–141. doi: 10.1038/nri2240. [DOI] [PubMed] [Google Scholar]
- 15.Meier O., Gastaldelli M., Boucke K., Hemmi S., Greber U.F. Early steps of clathrin-mediated endocytosis involved in phagosomal escape of Fcgamma receptor-targeted adenovirus. J Virol. 2005;79:2604–2613. doi: 10.1128/JVI.79.4.2604-2613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clement C., Tiwari V., Scanlan P.M., Valyi-Nagy T., Yue B.Y., Shukla D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J Cell Biol. 2006;174:1009–1021. doi: 10.1083/jcb.200509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17••.Gianni T., Gatta V., Campadelli-Fiume G. {alpha}V{beta}3-integrin routes herpes simplex virus to an entry pathway dependent on cholesterol-rich lipid rafts, dynamin2. Proc Natl Acad Sci U S A. 2010;107:22260–22265. doi: 10.1073/pnas.1014923108. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using cells expressing or not the alphaVbeta3-integrin, this paper shows that the binding of HSV-1 to this integrin routes specifically viruses to acidic compartments, through a dynamin2-dependent pathway, known to be involved in the phagocytosis internalization process.
- 18.Dupuy A.G., Caron E Integrin-dependent phagocytosis: spreading from microadhesion to new concepts. J Cell Sci. 2008;121:1773–1783. doi: 10.1242/jcs.018036. [DOI] [PubMed] [Google Scholar]
- 19.Ghigo E., Kartenbeck J., Lien P., Pelkmans L., Capo C., Mege J.L., Raoult D. Ameobal pathogen mimivirus infects macrophages through phagocytosis. PLoS Pathog. 2008;4:e1000087. doi: 10.1371/journal.ppat.1000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amstutz B., Gastaldelli M., Kalin S., Imelli N., Boucke K., Wandeler E., Mercer J., Hemmi S., Greber U.F. Subversion of CtBP1-controlled macropinocytosis by human adenovirus serotype 3. EMBO J. 2008;27:956–969. doi: 10.1038/emboj.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercer J., Helenius A Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 22.de Vries E., Tscherne D.M., Wienholts M.J., Cobos-Jimenez V., Scholte F., Garcia-Sastre A., Rottier P.J., de Haan C.A. Dissection of the influenza a virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011;7:e1001329. doi: 10.1371/journal.ppat.1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reading P.C., Miller J.L., Anders E.M. Involvement of the mannose receptor in infection of macrophages by influenza virus. J Virol. 2000;74:5190–5197. doi: 10.1128/jvi.74.11.5190-5197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24•.Upham J.P., Pickett D., Irimura T., Anders E.M., Reading P.C. Macrophage receptors for influenza A virus: role of the macrophage galactose-type lectin and mannose receptor in viral entry. J Virol. 2010;84:3730–3737. doi: 10.1128/JVI.02148-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that influenza virus interacts with the MR and the macrophage galactose-type lectin (MGL). Whereas these interactions are independent on sialic acid, influenza virus also bound to the sialic acid on the MR. Interestingly, influenza virus strain which infect macrophages poorly was not recognized by the MR and MGL.
- 25.Op den Brouw M.L., Binda R.S., Geijtenbeek T.B., Janssen H.L., Woltman A.M. The mannose receptor acts as hepatitis B virus surface antigen receptor mediating interaction with intrahepatic dendritic cells. Virology. 2009;393:84–90. doi: 10.1016/j.virol.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 26.Miller J.L., de Wet B.J., Martinez-Pomares L., Radcliffe C.M., Dwek R.A., Rudd P.M., Gordon S. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008;4:e17. doi: 10.1371/journal.ppat.0040017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trujillo J.R., Rogers R., Molina R.M., Dangond F., McLane M.F., Essex M., Brain J.D. Noninfectious entry of HIV-1 into peripheral and brain macrophages mediated by the mannose receptor. Proc Natl Acad Sci U S A. 2007;104:5097–5102. doi: 10.1073/pnas.0611263104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Areschoug T., Gordon S. Scavenger receptors: role in innate immunity and microbial pathogenesis. Cell Microbiol. 2009;11:1160–1169. doi: 10.1111/j.1462-5822.2009.01326.x. [DOI] [PubMed] [Google Scholar]
- 29.Scarselli E., Ansuini H., Cerino R., Roccasecca R.M., Acali S., Filocamo G., Traboni C., Nicosia A., Cortese R., Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30••.Dreux M., Pietschmann T., Granier C., Voisset C., Ricard-Blum S., Mangeot P.E., Keck Z., Foung S., Vu-Dac N., Dubuisson J. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J Biol Chem. 2006;281:18285–18295. doi: 10.1074/jbc.M602706200. [DOI] [PubMed] [Google Scholar]; This paper shows that HDL accelerates cell entry of HCV pseudo-particles and that the lipid transfer function of SR-BI is involved in the potency of SR-BI to promote HCV entry. However, no interaction between HDL and HCV pseudo-particles was detected.
- 31.Eyre N.S., Drummer H.E., Beard M.R. The SR-BI partner PDZK1 facilitates hepatitis C virus entry. PLoS Pathog. 2010:6. doi: 10.1371/journal.ppat.1001130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dreux M., Dao Thi V.L., Fresquet J., Guerin M., Julia Z., Verney G., Durantel D., Zoulim F., Lavillette D., Cosset F.L. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog. 2009;5:e1000310. doi: 10.1371/journal.ppat.1000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barth H., Schnober E.K., Neumann-Haefelin C., Thumann C., Zeisel M.B., Diepolder H.M., Hu Z., Liang T.J., Blum H.E., Thimme R. Scavenger receptor class B is required for hepatitis C virus uptake and cross-presentation by human dendritic cells. J Virol. 2008;82:3466–3479. doi: 10.1128/JVI.02478-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Gorp H., Van Breedam W., Van Doorsselaere J., Delputte P.L., Nauwynck H.J. Identification of the CD163 protein domains involved in infection of the porcine reproductive and respiratory syndrome virus. J Virol. 2010;84:3101–3105. doi: 10.1128/JVI.02093-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pohlmann S., Zhang J., Baribaud F., Chen Z., Leslie G.J., Lin G., Granelli-Piperno A., Doms R.W., Rice C.M., McKeating J.A. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J Virol. 2003;77:4070–4080. doi: 10.1128/JVI.77.7.4070-4080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Londrigan S.L., Turville S.G., Tate M.D., Deng Y.M., Brooks A.G., Reading P.C. N-Linked glycosylation facilitates sialic acid-independent attachment and entry of Influenza A viruses into cells expressing DC-SIGN or L-SIGN. J Virol. 2011;85:2990–3000. doi: 10.1128/JVI.01705-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han D.P., Lohani M., Cho M.W. Specific asparagine-linked glycosylation sites are critical for DC-SIGN- and L-SIGN-mediated severe acute respiratory syndrome coronavirus entry. J Virol. 2007;81:12029–12039. doi: 10.1128/JVI.00315-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38•.Zhou Y., Lu K., Pfefferle S., Bertram S., Glowacka I., Drosten C., Pohlmann S., Simmons G. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J Virol. 2010;84:8753–8764. doi: 10.1128/JVI.00554-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports that severe acute respiratory syndrome-coronavirus spike glycoprotein binds to MBL what inhibits virus infectivity through prevention of binding of the virus to its receptor DC-SIGN. A single N-linked glycosylation site is responsible for this interaction.
- 39.Casanova J.L., Abel L., Quintana-Murci L. Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu Rev Immunol. 2011;29:447–491. doi: 10.1146/annurev-immunol-030409-101335. [DOI] [PubMed] [Google Scholar]
- 40.Bieback K., Lien E., Klagge I.M., Avota E., Schneider-Schaulies J., Duprex W.P., Wagner H., Kirschning C.J., Ter Meulen V., Schneider-Schaulies S. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol. 2002;76:8729–8736. doi: 10.1128/JVI.76.17.8729-8736.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murawski M.R., Bowen G.N., Cerny A.M., Anderson L.J., Haynes L.M., Tripp R.A., Kurt-Jones E.A., Finberg R.W. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J Virol. 2009;83:1492–1500. doi: 10.1128/JVI.00671-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szomolanyi-Tsuda E., Liang X., Welsh R.M., Kurt-Jones E.A., Finberg R.W. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J Virol. 2006;80:4286–4291. doi: 10.1128/JVI.80.9.4286-4291.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurt-Jones E.A., Chan M., Zhou S., Wang J., Reed G., Bronson R., Arnold M.M., Knipe D.M., Finberg R.W. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A. 2004;101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burzyn D., Rassa J.C., Kim D., Nepomnaschy I., Ross S.R., Piazzon I. Toll-like receptor 4-dependent activation of dendritic cells by a retrovirus. J Virol. 2004;78:576–584. doi: 10.1128/JVI.78.2.576-584.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaudreault E., Fiola S., Olivier M., Gosselin J. Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J Virol. 2007;81:8016–8024. doi: 10.1128/JVI.00403-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou S., Halle A., Kurt-Jones E.A., Cerny A.M., Porpiglia E., Rogers M., Golenbock D.T., Finberg R.W. Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J Neuroimmunol. 2008;194:70–82. doi: 10.1016/j.jneuroim.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michaud F., Coulombe F., Gaudreault E., Kriz J., Gosselin J. Involvement of TLR2 in recognition of acute gammaherpesvirus-68 infection. PLoS ONE. 2010;5:e13742. doi: 10.1371/journal.pone.0013742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu J., Martinez J., Huang X., Yang Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood. 2007;109:619–625. doi: 10.1182/blood-2006-06-027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Triantafilou K., Vakakis E., Richer E.A., Evans G.L., Villiers J.P., Triantafilou M. Human Rhinovirus recognition in non-immune cells is mediated by Toll like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune responseNew article. Virulence. 2011;2:22–29. doi: 10.4161/viru.2.1.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50••.Barbalat R., Lau L., Locksley R.M., Barton G.M. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. 2009;10:1200–1207. doi: 10.1038/ni.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that non-nucleic structures of viruses can induce type I IFN induction. It reports that vaccinia virus ligands can specifically induce type I IFN induction via recognition by TLR2, via a process requiring receptor internalization in inflammatory monocytes.
- 51.Zhang J., Li G., Bafica A., Pantelic M., Zhang P., Broxmeyer H., Liu Y., Wetzler L., He J.J., Chen T. Neisseria gonorrhoeae enhances infection of dendritic cells by HIV type 1. J Immunol. 2005;174:7995–8002. doi: 10.4049/jimmunol.174.12.7995. [DOI] [PubMed] [Google Scholar]
- 52.Ahmed N., Hayashi T., Hasegawa A., Furukawa H., Okamura N., Chida T., Masuda T., Kannagi M. Suppression of human immunodeficiency virus type 1 replication in macrophages by commensal bacteria preferentially stimulating Toll-like receptor 4. J Gen Virol. 2010;91:2804–2813. doi: 10.1099/vir.0.022442-0. [DOI] [PubMed] [Google Scholar]
- 53.Wilkins C., Gale M., Jr. Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.K., Schlee M. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 55.Habjan M., Andersson I., Klingstrom J., Schumann M., Martin A., Zimmermann P., Wagner V., Pichlmair A., Schneider U., Muhlberger E. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE. 2008;3:e2032. doi: 10.1371/journal.pone.0002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cardenas W.B., Loo Y.M., Gale M., Jr., Hartman A.L., Kimberlin C.R., Martinez-Sobrido L., Saphire E.O., Basler C.F. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol. 2006;80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Solis M., Nakhaei P., Jalalirad M., Lacoste J., Douville R., Arguello M., Zhao T., Laughrea M., Wainberg M.A., Hiscott J. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J Virol. 2011;85:1224–1236. doi: 10.1128/JVI.01635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ubol S., Phuklia W., Kalayanarooj S., Modhiran N. Mechanisms of immune evasion induced by a complex of dengue virus and preexisting enhancing antibodies. J Infect Dis. 2010;201:923–935. doi: 10.1086/651018. [DOI] [PubMed] [Google Scholar]
- 59•.Sen N., Sommer M., Che X., White K., Ruyechan W.T., Arvin A.M. Varicella-zoster virus immediate-early protein 62 blocks interferon regulatory factor 3 (IRF3) phosphorylation at key serine residues: a novel mechanism of IRF3 inhibition among herpesviruses. J Virol. 2010;84:9240–9253. doi: 10.1128/JVI.01147-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports that VZV inhibits IRF3 function independently on virus replication. IE62, an abundant virion-associated protein is shown to be sufficient to prevent IRF3 activation, while maintaining TBK1-IRF3 complex formation.
- 60.Brzozka K., Finke S., Conzelmann K.K. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J Virol. 2005;79:7673–7681. doi: 10.1128/JVI.79.12.7673-7681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Basler C.F., Mikulasova A., Martinez-Sobrido L., Paragas J., Muhlberger E., Bray M., Klenk H.D., Palese P., Garcia-Sastre A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol. 2003;77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martinez-Sobrido L., Zuniga E.I., Rosario D., Garcia-Sastre A., de la Torre J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol. 2006;80:9192–9199. doi: 10.1128/JVI.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ott D.E., Coren L.V., Johnson D.G., Kane B.P., Sowder R.C., II, Kim Y.D., Fisher R.J., Zhou X.Z., Lu K.P., Henderson L.E. Actin-binding cellular proteins inside human immunodeficiency virus type 1. Virology. 2000;266:42–51. doi: 10.1006/viro.1999.0075. [DOI] [PubMed] [Google Scholar]
- 64.Misumi S., Inoue M., Dochi T., Kishimoto N., Hasegawa N., Takamune N., Shoji S. Uncoating of human immunodeficiency virus type 1 requires prolyl isomerase Pin1. J Biol Chem. 2010;285:25185–25195. doi: 10.1074/jbc.M110.114256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saitoh T., Tun-Kyi A., Ryo A., Yamamoto M., Finn G., Fujita T., Akira S., Yamamoto N., Lu K.P., Yamaoka S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol. 2006;7:598–605. doi: 10.1038/ni1347. [DOI] [PubMed] [Google Scholar]
- 66.Watashi K., Khan M., Yedavalli V.R., Yeung M.L., Strebel K., Jeang K.T. Human immunodeficiency virus type 1 replication and regulation of APOBEC3G by peptidyl prolyl isomerase Pin1. J Virol. 2008;82:9928–9936. doi: 10.1128/JVI.01017-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ambagala A.P., Cohen J.I. Varicella-Zoster virus IE63, a major viral latency protein, is required to inhibit the alpha interferon-induced antiviral response. J Virol. 2007;81:7844–7851. doi: 10.1128/JVI.00325-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Devaux P., von Messling V., Songsungthong W., Springfeld C., Cattaneo R. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology. 2007;360:72–83. doi: 10.1016/j.virol.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 69.Lee H.K., Iwasaki A. Autophagy and antiviral immunity. Curr Opin Immunol. 2008;20:23–29. doi: 10.1016/j.coi.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Orvedahl A., MacPherson S., Sumpter R., Jr., Talloczy Z., Zou Z., Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dreux M., Chisari F.V. Viruses and the autophagy machinery. Cell Cycle. 2010;9:1295–1307. doi: 10.4161/cc.9.7.11109. [DOI] [PubMed] [Google Scholar]
- 72••.Joubert P.E., Meiffren G., Gregoire I.P., Pontini G., Richetta C., Flacher M., Azocar O., Vidalain P.O., Vidal M., Lotteau V. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe. 2009;6:354–366. doi: 10.1016/j.chom.2009.09.006. [DOI] [PubMed] [Google Scholar]; This paper identifies a direct molecular connection between the autophagy machinery and the cellular receptor CD46-Cyt-1, a receptor for several viruses including MeV. Moreover, MeV binding to CD46-Cyt-1 induces the rapid formation of autophagosomes upon virus entry, through this molecular pathway.
- 73.Fleischli C., Sirena D., Lesage G., Havenga M.J., Cattaneo R., Greber U.F., Hemmi S. Species B adenovirus serotypes 3, 7, 11 and 35 share similar binding sites on the membrane cofactor protein CD46 receptor. J Gen Virol. 2007;88:2925–2934. doi: 10.1099/vir.0.83142-0. [DOI] [PubMed] [Google Scholar]
- 74.Santoro F., Kennedy P.E., Locatelli G., Malnati M.S., Berger E.A., Lusso P. CD46 is a cellular receptor for human herpesvirus 6. Cell. 1999;99:817–827. doi: 10.1016/s0092-8674(00)81678-5. [DOI] [PubMed] [Google Scholar]
- 75.Delgado M.A., Elmaoued R.A., Davis A.S., Kyei G., Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sanjuan M.A., Dillon C.P., Tait S.W., Moshiach S., Dorsey F., Connell S., Komatsu M., Tanaka K., Cleveland J.L., Withoff S., Green D.R. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 77••.Ravikumar B., Moreau K., Jahreiss L., Puri C., Rubinsztein D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports that the internalization of clathrin-coated vesicles provide membrane reservoir for the formation of autophagosomes, through a mechanisms involving the interaction of the heavy chain of clathrin with the autophagy-related potein ATL16L1. Thus, plasma membrane can be at the origine of autophagosome formation.
- 78•.Navratil V., de Chassey B., Meyniel L., Pradezynski F., Andre P., Rabourdin-Combe C., Lotteau V. System-level comparison of protein-protein interactions between viruses and the human type I interferon system network. J Proteome Res. 2010;9:3527–3536. doi: 10.1021/pr100326j. [DOI] [PubMed] [Google Scholar]; This paper evaluates protein-protein interactions between 62 viral proteins belonging to 34 viruses of 13 different families and 70 host proteins of the type I IFN response and highlights that viruses significantly target more than 50% of the proteins of the IFN-I response; viruses target transcription factors (>70% of targeted proteins), signaling intermediates (>50%) and TLR receptors (25%).