

Abstract
Most clinical applications of human multipotent mesenchymal stromal cells (MSCs) for cell therapy, tissue engineering, regenerative medicine, and treatment of immune and inflammatory diseases require a phase of isolation and ex vivo expansion allowing a clinically meaningful cell number to be reached. Conditions used for cell isolation and expansion should meet strict quality and safety requirements. This is particularly true for the growth medium used for MSC isolation and expansion. Basal growth media used for MSC expansion are supplemented with multiple nutrients and growth factors. Fetal bovine serum (FBS) has long been the gold standard medium supplement for laboratory-scale MSC culture. However, FBS has a poorly characterized composition and poses risk factors, as it may be a source of xenogenic antigens and zoonotic infections. FBS has therefore become undesirable as a growth medium supplement for isolating and expanding MSCs for human therapy protocols. In recent years, human blood materials, and most particularly lysates and releasates of platelet concentrates have emerged as efficient medium supplements for isolating and expanding MSCs from various origins. This review analyzes the advantages and limits of using human platelet materials as medium supplements for MSC isolation and expansion. We present the modes of production of allogeneic and autologous platelet concentrates, measures taken to ensure optimal pathogen safety profiles, and methods of preparing PLs for MSC expansion. We also discuss the supply of such blood preparations. Produced under optimal conditions of standardization and safety, human platelet materials can become the future ‘gold standard’ supplement for ex vivo production of MSCs for translational medicine and cell therapy applications.
Introduction
Stem cell (SC)-based therapy, using multipotent mesenchymal stromal cells (MSCs) derived from adult tissues of mesodermal origin, has great potential in various clinical applications in cell therapy, tissue engineering, regenerative medicine, and in treating immune and inflammatory diseases [1], [2], [3], [4], [5]. Current clinical applications using MSCs include treatments for bone diseases, repair of cartilage, control of graft versus host disease, facilitation of engraftment of bone marrow transplants, and treatment of myocardial infarction [6]. Researchers and clinicians alike hope that SC therapy will prove beneficial in other therapeutic fields such as treating musculoskeletal damage, cancer, and neurological disorders.
One technical impairment to the practical use of MSC-based cell therapy in regenerative medicine and immunosuppression, is the low levels at which these cells are present in tissues such as bone marrow, umbilical cord blood, adipose tissues, dental pulp, etc. Thus, for many clinical indications, MSCs have to be isolated and expanded ex vivo to reach a clinically meaningful cell count needed for therapeutic use and differentiation under a selective environment, into various cell lineages like osteoblasts, chondrocytes, myoblasts, hepatocytes, neuronal cells, and vascular smooth muscle cells.
To be acceptable for clinical applications, this phase of ex vivo expansion should be achieved under safe culture conditions meeting the requirements of good manufacturing practices (GMPs) and good tissue culture practices. A key compliance factor is the growth medium used for culture [7], [8], [9], [10]. Successful MSC isolation and culture protocols require special media containing an enriched substrate of proteins, bioactive molecules, growth factors and various nutrients. Until now, fetal bovine serum (FBS), also often referred to as fetal calf serum, has been used to supplement the basal culture medium needed for successful ex vivo expansion of MSCs, and still represents the gold standard as a medium supplement in most cell culture settings.
Drawbacks of FBS
The regulatory approval of cell therapy clinical trials based on MSCs grown in FBS-supplemented media has been impeded due to two well-identified drawbacks of FBS. First, despite attempts to clear bovine proteins after MSC expansion, residual bovine components may induce immune reactions in MSC recipients, in particular, immunogenic xeno-carbohydrate structures may be internalized by these cells during culture, possibly explaining MSC transplantation failure [11], [12]. Indirect evidence of such side-effects comes from rodent experimental studies where strong immunogenic reactions to β2-microglobulin originating from the FBS-based culture medium were observed [13]. Similarly, inoculation of human peptide-pulsed dendritic cells grown in FBS-supplemented medium into patients with malignant melanomas resulted in the generation of immunoglobulin G (IgG) and IgM antibodies to bovine proteins and the occurrence of anaphylactic reactions [14]. Likewise, generation of antibodies to FBS components and development of arthus-like reactions were observed in 12 human immunodeficiency virus (HIV)-1 infected patients, who received multiple infusions of lymphocytes from their HIV-1-uninfected identical twin siblings cultured in medium supplemented with FBS. This took place despite thorough washing of the cells before infusion [15].
A second regulatory and safety concern is the risk of zoonotic infections associated with the use of FBS. The recent occurrence of natural transmissions of zoonotic diseases illustrates possible exchanges of humans with natural reservoirs of biologic agents found in animals [16], and the inherent risks of emerging diseases [17]. Examples of such infections originating from animal pathogens include HIV, and Ebola, Hanta, Lassa, and Nipah viruses and other paramyxoviruses, the equine morbilli virus, West Nile virus (WNV), the severe acute respiratory syndrome (SARS) coronavirus [18], [19], and, more recently, the H7N9 flu virus [20] and the new Middle East respiratory syndrome coronavirus (MERS-CoV) [21]. Infectious risks from bovine sources include the transmission of viruses, such as bovine viral diarrhea virus and bovine parvovirus [22], and of prions responsible for bovine spongiform encephalopathy (BSE, or mad cow disease) in cows and its human equivalent, variant Creutzfeldt-Jakob disease (vCJD) [23]. The World Health Organization (WHO) and several regulatory authorities are recommending restrictions on, or even banning, the use of materials from bovine origin, particularly for therapeutic applications, including cell therapy protocols [24], [25].
Additional drawbacks of FBS include (a) the well-recognized inconsistency in quality that requires testing samples from several lots before identifying a suitable batch for a given MSC source, and (b) growing ethical concerns about the suffering imposed on animals by production methods of serum from calves [26], [27].
Animal serum-free media: searching for FBS substitutes
Chemically-defined media
Attempts to develop ‘chemically defined’ medium supplements using nutritional sources of exclusively non-animal origin or recombinant growth factor combinations, such as platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), transforming growth factor (TGF)-β and epithelium growth factor (EGF) [28], have so far not turned out to be practically feasible for expanding MSCs, in contrast to what has been achieved for well-established stable cell lines to produce recombinant therapeutic proteins [29], [30]. The difficulty with using chemically-defined media for primary cell cultures may be associated with their inherent variability and specificity. In addition, it is anticipated that chemically defined media with a specific mix of growth factors will have to be developed to adjust to the sources of the SCs (e.g. bone marrow, adipose tissues, cord blood, dental pulp, etc.) or even possibly to the donor. As a single growth factor cannot be expected to readily provide the diverse physiological functions exhibited by such a complex protein and nutrient supplement as FBS, the limited number of licensed growth factors complying with therapeutic applications complicates the development of chemically-defined media. The use of human blood materials, possibly complemented with a mix of recombinant growth factors [31] appears to be the most pragmatic and safest approach available so far to substitute for FBS for expanding clinical-grade SCs [8], [32].
Animal serum-free media
Initially, the use of human blood platelet-derived materials, such as human serum, as a source of essential nutrients, attachment factors and growth factors for cell growth was reported to be somewhat controversial [7]. The scientific rationale behind such use is the presence of a wide range of growth factors entrapped in platelets [33]. Platelets or thrombocytes, with a concentration range of 150,000–400,000/μL, are irregularly shaped non-nucleated bodies, released into the blood by the fragmentation of megakaryocytes. In vivo, platelets play a major role in hemostasis, assisting the formation of blood clots and blood vessels and in wound healing after injury [34]. Under physiological conditions, growth factors are released by platelet activation and this release can be reproduced in vitro to prepare growth factor-rich fluids. One of the first clear demonstrations of the positive effect of growth factor-rich human blood materials on ex vivo expansion of bone marrow-derived (BM)-MSCs was, to the best of our knowledge, published in 2003 [35]. Those authors used a platelet gel releasate obtained by in vitro thrombin activation of platelet-rich plasma (PRP). They showed that BM-MSCs expanded in a medium containing up to 10% human platelet releasate retained their ability to express markers of chondrogenic and osteogenic differentiation after several passages. Two years later, it was clearly demonstrated that growth medium supplemented with platelet concentrate lysate, as the sole source of nutrients, could be used to expand human BM-MSC. The lysate was made from apheresis platelet concentrates that were frozen at −80°C and subsequently thawed to induce platelet lysis and release their growth factors into the plasma compartment. The platelet concentrate lysate was centrifuged at 900 × g to eliminate cell debris. Mean respective contents of PDGF-AB, TGF-β1, basic (b)-FGF, and insulin-like growth factor (IGF-1) were approximately 31, 51, 92 and 107 ng/mL, significantly more than those in FBS. Medium supplemented with 5% platelet lysate (PL) promoted BM-MSC expansion, decreased the time required to reach confluence, and increased the colony forming unit-fibroblast (CFU-F) size, compared to medium containing 10% FBS. The immunophenotype of expanded MSCs was not modified for at least four passages, and MSCs maintained their ability to differentiate into osteogenic, chondrogenic, and adipogenic lineages and exert immunosuppressive activity [36]. Since those pioneering works, many additional studies were conducted, which generally confirmed those observations. Some groups compared several types of blood materials including plasma, serum, and PLs in an attempt to identify the most suitable for a given type of MSC [37]. A recent review indicated that PLs (or activated platelet releasates) are superior or at least similar to FBS for expanding BM-MSC and adipose tissue-derived (AT)-SCs. A 5% content of platelet materials is often sufficient to replace 10% FBS, although 10% may provide optimal results for other MSCs [38].
Human blood fractions for preparing PLs
As described above, there are several modes of preparing human blood materials for applications in ex vivo SC expansion. The preparation method can exert significant impacts on the platelet, leucocyte, and plasma protein content [39] in a platelet concentrate, and ultimately the concentration and type of growth factors released. It is therefore important to document the way platelet concentrates or other blood fractions are prepared as these production variables may influence SC expansion and regenerative capacity [38] even though these still need to be more fully explored. We examine below the different modes of preparing blood components and platelet growth factor-rich fractions.
Human serum can be prepared for use as growth medium supplement by letting whole blood, collected without anticoagulant, clot overnight at 4°C [37], [40]. The serum is recovered by centrifugation at approximately 2000 × g for 15 min and then aliquoted and kept frozen at −30°C until use. However, human serum is not usually available from blood establishments, which is a drawback for routine applications.
Platelet concentrates are most often used to prepare PLs for SC expansion. Fig. 1 summarizes the main production modes of PL materials for SC expansion and regenerative medicine as recently reviewed [41]. Autologous platelet concentrates are obtained using various types of medical devices that differ in the methodology for platelet separation and result in products with variable platelet and leucocyte contents. As such, they produce variable ranges and contents of growth factors [42]. Allogeneic platelet concentrates prepared by blood establishments are likely to be a major source of platelet concentrates for SC expansion in the future. Three basic types of allogeneic platelet concentrates can be prepared by blood establishments and can be used (usually after 5 days of storage at 22 ± 2°C, when they are no longer suitable for transfusion) as starting materials to prepare medium supplements for MSC expansion. During processing, they may or may not be subjected to a leucoreduction step, usually by dedicated filtration, to reduce the content of white blood cells (WBCs), and may exclusively be formulated in plasma or in a combination of plasma and additive solution. Their production should comply with principles of GMPs that ensure that they meet established characteristics for transfusion use.
Fig. 1.
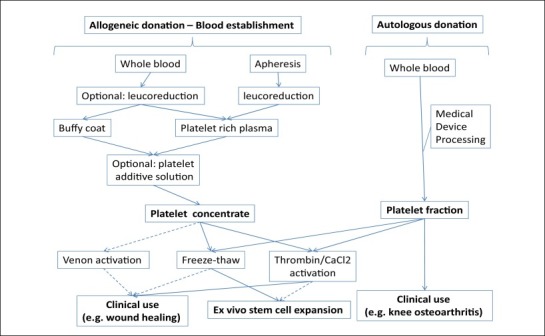
Mode of preparation of allogeneic and autologous platelet concentrates and PLs for use as a medium supplement for stem cell expansion and for regenerative medicine.
Whole blood-derived platelet concentrates are obtained as by-products of red blood cell (RBC) preparations and are known as ‘random-donor’ platelets; they are processed from PRP or from the buffy coat (BC). Typically, 450 mL of whole blood is collected in 63 mL of an anticoagulant solution made of citrate and cell nutrients, such as glucose and adenine, in a polyvinyl chloride bag. The donation procedure should take less than 12–15 min to limit the risks of activation of blood coagulation and platelets, respectively. The donation can be kept for up to 24 h at 22 ± 2°C. The blood is not cooled down to 2–6°C if platelet concentrates are prepared, as cool temperatures lead to loss of platelet viability and generation of platelet and leucocyte microaggregates.
The first centrifugation step of whole blood influences the characteristics of the platelet concentrates. Precise conditions of g-forces, acceleration, time, and deceleration should therefore be applied [39], as they impact separation of the various blood components and determine the additional processing conditions needed to obtain platelet concentrates of a defined composition. In the PRP method, the whole-blood donation is subjected to light spinning centrifugation (typically 1000 × g) for about 10 min at 21–22°C under conditions validated to segregate RBCs from the upper half containing a platelet and plasma mixture, called PRP. Platelets are then concentrated by heavy spinning centrifugation at about 3000 × g for 5 min at 21–22°C, with validated acceleration and deceleration curves. The platelet concentrate bag is left stationary at room temperature for approximately 1 h and then the concentrate is resuspended in 50–70 mL of plasma [39]. Platelets derived from whole blood are mostly produced by the PRP method in the US.
In the BC procedure, anticoagulated whole blood is subjected to hard spinning centrifugation at approximately 3000 × g for 5 min at 21–22°C with validated acceleration and deceleration curves to separate ‘cell-free’ plasma in the top layer, a middle layer called BC (that contains roughly 90% of the platelets, 70% of WBCs, and 10% of RBCs) and an RBC bottom layer [39]. The BC layer is transferred to a satellite bag. About 20–30 mL of plasma is returned to the BC layer and gently mixed before again being subjected to light spinning centrifugation at approximately 1000 × g for 6 min at 21–22°C, with validated acceleration and deceleration curves. The PRP supernatant is then placed in a platelet storage bag and stored at 22 ± 2°C. In newer procedures, part of the plasma can be replaced by a platelet additive solution. The BC method is favored in Europe as leucocyte depletion by BC removal reduces the frequency of febrile transfusion reactions, and facilitates a high level of leucodepletion when combined with leucocyte filtration. Therefore most whole blood-derived allogeneic platelets used for MSC expansion in Europe are likely derived from BCs. Platelet concentrates from whole blood require the pooling of platelets from four to six donations to obtain an adult therapeutic dose equivalent to that of a single apheresis platelet procedure.
Automated cell separators can be used to prepare apheresis platelet concentrates in a closed system. A dedicated extracorporeal procedure and collection machine using intermittent or continuous centrifugation are used to separate and collect platelets. Various cell separators are available for platelet collection and have different collection principles [43]. Whole blood is anticoagulated at the point of withdrawal in a controlled manner and at a 9:1–11:1 blood/anticoagulant ratio. Other components are usually returned to the donor, unless also collected. The entire procedure takes at least 1.5 h. Such platelet concentrates have a larger volume (typically 200–300 mL), and the total platelet content is about 6–8 fold that of a platelet concentrate from a single whole-blood donor.
Quality criteria of platelet concentrate composition
Several variables in platelet concentrate production methods can influence the platelet, leucocyte and protein contents, and therefore potentially impacting MSC expansion. For instance, apheresis platelet concentrates usually have significantly lower WBC contamination than standard BC-derived or PRP-derived platelet concentrates [44]. In addition, platelets undergo physiological and functional changes upon storage which may affect therapeutic benefits to the recipient. Whether such changes affect SC expansion has not been evaluated. Procedures to prepare platelet concentrates may include a leucoreduction step using different types of leucoreduction filters [45], [46]. When leucocytes are present in platelet concentrates, lysates contain higher amounts of cytokines that can influence MSC growth. In addition, until recently, platelet concentrates were suspended in 100% plasma. However, residual plasma may mitigate transfusion-related adverse reactions in recipients when used intravenously. Therefore, platelet storage additive solutions containing sodium/potassium chloride, citrate, phosphate, and mannitol may be used to substitute for part, usually 2/3, of the plasma volume [47], [48]. This affects the protein content and reduces the presence of plasma nutrients in the PLs. However, (gamma-irradiated) BCs pooled in a T-sol additive solution and subjected to freeze–thaw cycles, centrifuged, and filtered could be used for expanding BM-MSCs [49]. Furthermore, plasma-poor platelet concentrates in the additive solution, subjected to pathogen reduction, were found suitable for stimulation of BM-MSC growth and maintenance of their properties [50]. Further studies are deemed necessary to confirm absence of impact of complete plasma removal on ex vivo expansion of MSCs from bone marrow or other sources. It should be noted that some research institutes utilize platelet concentrates from blood group O donors with possible dilution with blood group AB plasma [40], [51].
Table 1 summarizes the most significant characteristics of allogeneic platelet concentrates available from blood establishments using current production procedures. All therapeutic platelet units should have a platelet count exceeding 2 × 1011. Limits in the acceptable residual leucocyte content (of <109 to <106) depend upon whether the platelet concentrate is stored in 100% plasma or a combination of plasma (30–40%) and additive solution (60–70%), and whether a leucoreduction step is used.
Table 1.
Preparation methods of platelet concentrates for transfusion: impacts on protein, platelet, and leucocyte contents per unit
| Preparation method |
Suspended in |
Leuco-reduction |
Counts/unit |
||
|---|---|---|---|---|---|
| Plasma | Additive solution | Platelet | Leukocytes | ||
| Whole blood (buffy coat or PRP method) | 100% | >2 × 1011 | <109 | ||
| 100% | Yes | <106 | |||
| 30–40% | 60–70% | 0.3 × 109 | |||
| 30–40% | 60–70% | Yes | <106 | ||
| Apheresis | 100% | <0.3 × 109 | |||
| 100% | Yes | <106 | |||
| 30–40% | 60–70% | <0.3 × 109 | |||
| 30–40% | 60–70% | Yes | <106 | ||
PRP, platelet-rich plasma.
Platelet concentrates are stored at 22 ± 2°C under continuous gentle agitation in a bag made of cell-compatible plastic with enhanced oxygen permeability and diffusion of carbon dioxide. The pH should remain above pH 6.4. Temperatures below +20 °C may induce platelet activation and release platelet storage components into the plasma. Proper storage conditions limit the risk of activation and maintain an unaltered hemostatic state for intravenous infusion. Agitation enables good mixing and gas exchange. Folding of the bags and foaming should be avoided. Platelet components are transported to hospitals, with no agitation, in an insulated container to ensure maintenance of an optimal storage temperature. Platelets used for transfusion can be stored for up to 5–7 days, after which time they can no longer be used for transfusions and are discarded. Expired platelet concentrates can be used as source material to prepare growth factor preparations for SC expansion. Such products may be frozen at −20°C or colder until being processed into PLs, which facilitate the logistics of preparation. A recent study has confirmed that human PLs can be produced from expired platelet concentrates and are suitable, in all aspects evaluated, as growth supplement for ex vivo expansion of BM-MSCs, as no difference between PLs from fresh and expired platelet concentrates were observed [52].
Each blood donation is subjected to ABO/RhD grouping to ensure compatibility for the recipient of blood components. Serum from AB-blood-group-type donors (AB serum) and platelets from group-O donors are reportedly used by some research groups for SC expansion [40]. Also, lysates of platelet concentrate prepared from BCs collected from blood-group-O-type donors pooled in AB plasma were used as an MSC growth culture supplement [37], [51].
In countries with a mature regulatory system, blood establishments responsible for producing blood components, including platelet fractions, should have a strict quality system in place based on principles of GMP and comply with local regulations [53], [54] to ensure the quality and consistency in meeting specifications. Anticoagulants, any additive solutions, and collection and storage bags should be licensed. Preparation equipment should be validated and premises maintained under clean and hygienic conditions and monitored. Preparation of platelet concentrates and pooling should be done using aseptic procedures, and either multiple bag configurations or sterile connecting devices used following a validated procedure for closed-system processing. Although variability linked to individual donors cannot be eliminated, such requirements ensure a relative consistency in the characteristics of the various types of allogeneic platelet concentrates, particularly platelet and leucocyte counts, used to prepare PLs for SC expansion, which should contribute to higher reproducibility.
Control of infectious risks from allogeneic PLs
Blood-borne pathogens
Table 2 shows the most relevant blood-borne pathogens and indicates the relative risks of their transmission by various classes of blood products. Further information can be found in various reviews and international guidelines [55], [56], [57]. Risks of transmission of plasma-borne viruses represent, in principle, the most serious infectious concerns with the use of PLs for cell therapy and regenerative medicine. Among those, the enveloped viruses HIV, hepatitis B virus (HBV), and HCV are the most pathogenic, although there are increasing concerns from risks posed by emerging agents like WNV, and dengue (DENV) and Chikungunya (CHIKV) viruses [57], [58], as well as the non-enveloped HAV, parvovirus B19 (B19V), and HEV [57], [59], [60]. Microbial filtration of PLs at 0.2 μm prior to the addition to culture medium should ensure removal of bacteria and parasites. In addition, a freeze–thaw step, as used to produce some PLs, should destroy parasites.
Table 2.
Blood-borne pathogens: types and evidence of transmission by blood products
| Pathogen | Family | Genomea | Size (nm) |
Evidence of transmission |
||
|---|---|---|---|---|---|---|
| Whole blood, red blood cells, platelets | Plasma for transfusion | Industrial plasma products | ||||
|
Enveloped viruses | ||||||
| Human immunodeficiency virus I and II | Retro | ss-RNA | 80–100 | + | + | +b |
| Hepatitis B virus | Hepadna | ds-DNA | 40–48 | + | + | +b |
| Hepatitis C virus | Toga | ss-RNA | 40–50 | + | + | +b |
| Hepatitis delta virus | Delta | ss-RNA | 36 | + | + | +b |
| Human T cell leukemia I and II virus | Retro | ss-RNA | 80–100 | + | −c | − |
| Epstein Barr virus | Herpes | ds-DNA | 120–220 | + | −c | − |
| Cytomegalovirus | Herpes | ds-DNA | 180–200 | + | −c | − |
| Human herpes 8 virus | Herpes | ds-DNA | 120–200 | ? | − | − |
| TT virus | Circo | ss-DNA | 30–50 | + | + | +b |
| Hepatitis G virus | Flavi | ss-RNA | 40–60 | + | + | +b |
| West Nile virus | Flavi | ss-RNA | 40–60 | + | + | −d |
| Dengue virus | Flavi | ss-RNA | 40–60 | + | + | −d |
|
Chikungunya virus |
Alpha |
ss-RNA |
60–70 |
+ |
+ |
−d |
| Non-enveloped viruses | ||||||
| Hepatitis A virus | Picorna | ss-RNA | 27–32 | + | + | +b |
| Hepatitis E virus | Calici | ss-RNA | 35–39 | + | + | +b |
|
Human erythro B19 virus |
Parvo |
ss-DNA |
18–26 |
+ |
+ |
+b |
| Bacteria | ||||||
|
Spirochetes |
+ |
− |
− |
|||
| Parasites | ||||||
| Babesia microti | + | − | − | |||
| Plasmodium (malaria) | + | − | − | |||
| Leishmania (Leishmaniosis) | + | − | − | |||
|
Trypanosoma cruzi(Chagas disease) |
+ |
− |
− |
|||
| Unconventional transmissible spongiform encephalopathy agent | ||||||
| Creutzfeldt-Jakob disease agent | − | − | − | |||
| Variant Creutzfeldt-Jakob disease agent | + | ? | −?e | |||
ss, single-stranded; ds, double-stranded.
Most cases of transmissions took place prior to implementation of robust viral inactivation and removal procedures.
Intracellular viruses destroyed by freeze–thaw or removed by filtration.
Lack of evidence of transmission is likely due to implementation of robust viral inactivation and removal treatments.
Investigative studies using spiked TSE agents suggesting the existence of efficient removal steps during plasma fractionation, but these data cannot necessarily be extrapolated to the endogenous form of the TSE agent in human blood (2007).
So far, there are four recorded cases of transmission of a new vCJD disease agent by cellular blood components collected in the UK [61], [62]. There is yet no evidence of transmission of vCJD by other blood products, including pooled plasma products at this point [63]. Currently, the vCJD safety of human blood components, including PLs, relies on epidemiological population control and leucoreduction of blood components [64], [65]. Prion-removal filtration, at least of some blood components [66] and blood donor testing for vCJD-positive donations [67] are being developed.
Safety nets: donor screening and donation testing
Allogeneic platelet concentrates used for the ex vivo expansion of clinical-grade MSC should comply with quality and safety regulatory requirements established by national regulatory authorities for the production of blood components for transfusion [54]. In countries with advanced blood-collection services and mature regulatory systems, well-established safety nets are in place to prepare blood components. Safety measures include (a) epidemiological control of the population from whom blood is collected, (b) screening of donors, and (c) testing of blood donations for known viral markers. In addition, a strict traceability system should be in place, with the potential to perform back-tracing if a donor is retrospectively found to have a risk factor [54]. Each blood donation should be individually controlled using approved tests for at least HIV1/HIV2 antibodies, hepatitis B surface antigen (HBsAg), and HCV antibodies. Only non-reactive donations can be used for therapeutic applications. Specific epidemiological situations may require testing for other agents or markers, such as anti-human T-cell leukemia virus (HTLV) I/II and WNV [54]. Nucleic acid testing (NAT) of individual donations or mini-pools of HCV, HBV, HIV, HAV, WNV (where appropriate) and/or B19V is performed in some countries to eliminate window-period donations. With modern procedures for donor screening and donation viral testing in place, the residual risk of transmission is estimated at 1 per 2.3 million donations for HIV, 1 per 1.8 million for HCV [68], [69], and approximately 1 per 300,000 donations for HBV [70]. The constant exposure of the blood supply to emerging viruses such as WNV, DENV, and CHIKV which might not be tested for yet [57], [71] could be regarded as justifying implementation of viral-inactivation treatments of blood components [55] including platelet concentrates for SC expansion.
Viral inactivation of individual platelet concentrates
In a limited number of countries, mostly in Europe, two technologies for viral inactivation of platelet concentrates for transfusion have been licensed [72]. Both target the binding and alteration of nucleic acids by a photoactivation process. One procedure (Intercept®) uses 150 μM of a synthetic psoralen (amotosalen hydrochloride or S-59) [73], [74]. This small, soluble planar molecule passes through cell membranes and capsids of viruses and reversibly intercalates into helical regions of nucleic acids. Upon illumination with ultraviolet A (UVA; at 300–400 nm), covalent cross links to pyrimidines in RNA and DNA are generated, which block replication and transcription of viral nucleic acids [75]. Both random-donor and apheresis platelets suspended in plasma with or without additive solutions can be processed with this system. Residual amotosalen and free photoproducts generated during illumination are reduced by exposure to a compound-adsorption device. After treatment, the platelet concentrate is transferred to a storage container. The second procedure (Mirasol®) combines a riboflavin (vitamin B2) photosensitizer, a naturally occurring compound, and UV (265–370 nm) illumination. The riboflavin planar molecule intercalates between the bases of the DNA or RNA. Upon UV illumination, riboflavin generates reactive oxygen species that oxidize guanine in nucleic acids and causes strand breakage of genomic material of viruses precluding their proliferation [76]. Riboflavin and the photo-byproducts formed (lumichrome) are generally regarded as non-toxic and therefore not eliminated [77], [78]. Most studies were performed with platelet concentrates stored in plasma [79], but more recently, treatment has been applied to BC platelet concentrates (BCPCs) [80] or apheresis platelet concentrates [81] prepared with a platelet additive solution. A third viral inactivation technology of platelet concentrates for transfusion is in development. It uses UVC illumination without the addition of a photosensitizer [82]. In vitro data indicate that viral inactivation of platelet concentrates by photoactivation methods leads to some degree of platelet activation [81]. Whether these alterations significantly influence the use of virally inactivated platelet concentrates for SC expansion is not known. Psoralen/UV-treated human serum was found to be appropriate for in vitro production of clinical-grade islets of Langerhans (for treating type I diabetes), MSCs for treating graft versus host disease (GvHD) grade III or IV and T cells (for immune therapy against malignant melanoma) [83], [84]. Recently, plasma-poor platelet in an additive solution were subjected to pathogen reduction by psoralen/UV treatment, followed by 3 cycles of freezing/thawing, and centrifugation to remove membranes, and were found to stimulate MSC growth in clinical-scale cultures [50]. In the absence of viral inactivation treatments, quarantine of PLs until donors’ retesting for viral markers can be an option [49].
Viral inactivation of pooled platelet concentrates
In recent years, the preparation of PLs from pools of at least 15 and up to 30–50 donations has been a trend to increase product batch-to-batch consistency. However, pooling increases the need for implementation of dedicated viral-inactivation treatment(s) when PLs are used for clinical-grade SC expansion. A solvent/detergent (S/D) treatment developed for inactivating lipid-enveloped viruses (e.g., HIV, HBV, and HCV) in plasma [85] was adapted for pooled PL supplements for ex vivo SC expansion and other uses in regenerative medicine [86]. The treatment contributes to an extensive release of platelet growth factors [86]. Pooling allows the preparation of a standardized product [87] usable as a supplement for growth medium for ex vivo expansion of AT-MSCs. Proliferation rates of AT-MSCs are similar to or better than those achieved using FBS supplements; expanded MSCs preserve their typical immunophenotype for at least seven passages, and exhibit an unaltered differentiation capacity into osteogenic, chondrogenic, and adipogenic lineages [88]. S/D treatments have been used for over 20 years by the plasma product industry with excellent efficacy, safety, and robustness for inactivating lipid-enveloped viruses [59], [89]. However, S/D treatment has no impact on non-enveloped viruses, potentially requiring combination with another viral-reduction step.
Bacterial contamination
Due to the production mode and storage at room temperature, platelet concentrates have a 0.1–0.6% risk of contamination by adventitious bacteria [90], [91] mostly as a result of improper donor arm cleaning at the time of venipuncture [92], [91]. Improved donor screening, skin disinfection, and bacterial screening [93] can reduce such risks [94]. Bacterial screening can be implemented as part of the qualification of platelet units intended for use in preparing PLs for SC expansion. The two pathogen inactivation techniques of single platelet donations described before also inactivate a wide range of bacteria [75] and can therefore be useful in that regard. S/D treatment was also shown not to alter the capacity of the complement system of plasma to inactivate gram-negative bacteria and also some gram-positive bacteria [95].
Preparation and use of PLs for SC expansion
In principle, platelets can be activated either by adhesion to molecules that are exposed on injured endothelium, such as von Willebrand factor (vWF), collagen, fibronectin, and laminin, or by physiologic agonists such as thrombin, ADP, collagen, thromboxane A2, epinephrine, and platelet-activating factors [96]. However, in practice, a limited number of methods of preparation of PLs from allogeneic or autologous platelet concentrates (Fig. 1) have been developed to prepare supplements for SC expansion. In most procedures, PLs are obtained by freeze/thaw cycles or by thrombin activation [38]. PLs are mostly produced from autologous or allogeneic whole blood, or from allogeneic apheresis platelet concentrates. To the best of our knowledge, there is limited application using autologous apheresis platelet concentrates due to technical and regulatory restrictions in the use of apheresis systems. Preparation methods used for allogeneic platelet concentrates can however be adapted to concentrates of autologous origin.
Autologous whole blood-derived PLs
Autologous platelet materials can only be used for clinical applications [97] where only small population of MSCs are needed [7], [10]. The possibility of using relatively low (≤5%) PL concentrations can expand applications of autologous PLs. Generally, autologous PLs are prepared by activation with human thrombin or by the addition of calcium salts to counterbalance the anticoagulant added for blood collection. Thrombin and calcium ions activate platelets and convert fibrinogen into fibrin, resulting in the formation of a platelet gel and a liquid PL. Batroxobin, a venom toxin from Bothrops atrox, can also be used to convert fibrinogen to fibrin without immediate activation of platelets, a feature that is believed to delay the release of growth factors [98]. Autologous or single-donor PLs can also be prepared by a freeze–thaw process (−80/37°C), with centrifugation at 2600 × g for 30 min and storage at −80 °C until use [99]. Data showed that donor recipient-matched or autologous PLs are feasible for therapeutic use in cooperation with MSC products, and that plasma proteins are required for SC expansion [99]. PLs from younger donors appear to enhance MSC expansion and lead to more-pronounced osteogenic differentiation [100].
Allogeneic whole blood-derived platelet concentrates
Several studies described the preparation of lysates from allogeneic platelet concentrates prepared from whole blood [49], [101]. In one procedure [51], whole blood (450 ± 45 mL) is centrifuged at 4250 × g for 13 min at 22°C to isolate the BC fraction. Four BC units (blood group O) and 1 unit of plasma (blood group AB) are pooled. Such blood group selection is meant to avoid exposure of MSCs to ABO blood group antigens and isoagglutinins [51]. The units are then centrifuged at 341 × g for 6 min at 22°C to obtain PRP which is transferred to a storage bag after inline filtration to deplete WBCs. To prepare the lysate, the PRP is frozen at −30°C. After thawing at 37°C, a minimum of 10 units of lysed human PRP (representing 40 BCs) and 10 plasma units is pooled to reduce the impact of individual donor variations. This mixture is stored at −30°C until use. After being thawed, the lysate is centrifuged at 4000 × g for 15 min at 4°C to reduce the presence of platelet membrane fragments which may represent a risk for alloimmunization against MHC class I and human platelet antigens [51].
Alternative methods to prepare PLs have been described. Whole-blood donations of four AB-blood-group-type donors are collected to prepare BC-derived platelet concentrates by centrifugation of whole blood [102] and the concentrate is then suspended in AB plasma [40]. Several protocols to obtain PLs were compared. One method uses shock freezing twice in liquid nitrogen followed by 30 min of centrifugation at 3000 × g, as also done before [36]. The supernatant is then sterile-filtered through 0.2-μm-pore filters and stored as aliquots at −80 °C. Alternatively, platelets are activated using either (a) human thrombin at a final concentration of 1 unit/mL, (b) 5 μM ADP/5 μM epinephrine, or (c) 1 mM thrombin receptor-activated peptide (TRAP)-6 for 45 min at room temperature with gentle agitation. In these procedures, calcium is not used for platelet activation to prevent increased osteogenic differentiation. Lysates are then centrifuged to remove the clot, sterile-filtered, and frozen. After thawing, the lysate is centrifuged at 1500 × g for 5 min as new fibrin clots may form [40]. One procedure uses leucoreduced BC-derived allogeneic platelets from whole blood (450 ± 45 mL) that are frozen at −30°C and thawed at 37°C. Such lysates obtained from 40 BCs are pooled, aliquoted, and stored at −30°C. Immediately before use, the aliquots are thawed and centrifuged at room temperature for 15 min at 4000 × g to remove cell debris and reduce the formation of aggregates during cell culturing [101]. These lysates can be used as medium supplements to expand and support the immunomodulatory capacity of BM-MSCs [103].
Recently, platelet concentrates were prepared from whole blood (109 platelets/mL) within 24 h after blood collection, washed, subjected to three cycles of freezing/thawing at −80 and 37°C, centrifuged at 900 × g for 30 min, and stored at −80°C. These lysates can stimulate dental-pulp SCs when used with 5% PL, but not 10% PLs [104]. Lysates from whole blood-derived PLT concentrates (at approximately 9.1 × 108/mL) which are subjected to four cycles of freezing/thawing and centrifugation at 3300 × g for 30 min were effective for AT-MSC expansion, with maintenance of the immunophenotype and proliferative and differentiation capacities up to passage 10 [105].
Production of PLs from gamma irradiated BC-derived platelets was also reported. The PLs are frozen and stored at −30 to −45°C for up to 18 months, and were stable for up to 14 days based on their capacity to expand BM-MSC [49].
Allogeneic apheresis platelet concentrates
In one procedure, platelets collected by apheresis from 10 healthy volunteers (5 × 1011 PLTs) are frozen at −80°C soon after collection and subsequently thawed at 37°C. Heparin is added to avoid gel formation, and lysates are centrifuged three times at 900 × g for 30 min to eliminate platelet bodies. The lysates are pooled for use in generating and expanding MSCs from bone marrow [106]. The morphology, phenotype, and differentiation capacity of MSCs were similar whether expanded in PLs or FBS. However, the clonogenic efficiency and proliferative capacity of MSCs expanded in PL appeared superior, while those expanded in FBS were more suitable for preventing/treating alloreactivity-related immune complications [106].
Other procedures obtain lysates by freezing at −80°C, followed by thawing and centrifugation at 1400 × g. These lysates were found to accelerate BM-MSC proliferation and enhance MSC osteogenic differentiation [107]. Apheresis platelets (∼2.5 × 1011/unit) frozen/thawed at −80/37°C, centrifuged at 900 × g for 30 min, filtered at 0.22 μm, and stored at −20°C were found, when used at 7.5% in growth medium, to be similar to 10% FBS at expanding MSCs, while the morphology and phenotype were the same.
A procedure for preparing PLs using sonication was recently described. Apheresis platelet concentrates samples from five or six donors were pooled in ethylene vinyl acetate (EVA) at a ratio between the bag surface and platelet concentrate volume of <0.24. Ultrasound was applied for 30 min at a frequency of 20 kHz, a condition found to release 74% of PDGF-AB. After centrifugation at 1600 × g for 15 min at room temperature the supernatant was filtered and stored at −20°C. This PL enhanced proliferation rates of BM-MSC which differentiated into adipogenic, osteogenic and chondrogenic lineages. These BM-MSCs maintained their immunosuppressive activity, and cell karyotypes showed no genetic alterations [108].
Advantages of human blood products
There are several advantages of using human blood products for SC expansion. First, due to their human origin, these products are devoid of immunological risks specifically associated with the use of xenoprotein-based products. Second, the infrastructure for blood collection and quality/safety testing is well established, at least in industrialized countries with an advanced blood-collection system and good national regulatory supervision. In countries like the US, Germany, Switzerland, France, and others, blood products are regulated as pharmaceutical/medicinal products and are produced following the principles of GMP, which contributes to optimized product consistency, viral safety, and traceability. Recent WHO guidelines encourage the concept of GMP implementation in blood establishments at the global level [54] which should increase the availability of quality source material.
Impacts of platelet growth factors on MSC expansion and differentiation potentials
PLs (PL) are used for tissue stem/progenitor cell expansion, differentiation into various lineages in vitro, and for clinical applications that favor expansion of MSCs as an FBS substitute for regenerative medicine from bench to bed site. Several studies determined the protein composition of PLs, in particular the growth factor content [41], [109]. The total plasma protein content was close to 40–60 g/L but the composition differed, depending upon the use of a platelet-stabilizing solution to at least partially replace the plasma. In addition, the contents of fibrinogen, other coagulation factors, and fibronectin were higher in PLs obtained by a freeze–thaw procedure, compared to those produced by activation with thrombin or calcium chloride. High fibrinogen levels more than likely require the addition of anticoagulant heparin at 0.6–2 IU/mL (final concentration in the culture medium) or more during cell culture, to avoid clotting due to conversion of fibrinogen into fibrin. Higher heparin concentration may impair cellular proliferation [110]. Alternatively, serum-converted PL can be prepared by calcium activation to convert fibrinogen into insoluble fibrin before being added to the medium [111].
As mentioned above, platelet activation and freeze–thawing both induce the release of a complex blend of growth factors, including PDGF-AA, -AB and -BB, TGF-β1 and -β2, EGF, VEGF, b-FGF, BDNF, and HGF. A variety of other cytokines, including G-CSF, GM-CSF, interleukin (IL)-1α, IL-7, IL-8, MIP-1α, MIP-1β, and interferon (IFN)-γ, are present at pg/mL levels. PL also contains high concentrations of chemokines (CCL5/RANTES and CXCL1/2/3) and, in particular, attachment factors (fibronectin and vitronectin), cell adhesion molecules, coagulation factors, mitogens, protease inhibitors, proteoglycans, serotonin, and other potent biomolecules [33], [112]. The rich content in growth factors has been used to produce PL as a substitute for FBS in the culture expansion of MSCs derived from various human tissues [4], [37], [40], [113] and under conditions where these cells maintain their ability to differentiate [36], [51], [114], [115]. To enhance consistency, pools of platelet concentrates, or PRP were developed with viral inactivation treatment using an S/D treatment to improve the viral safety [86]. The resulting growth factor-rich PL was demonstrated to allow the ex vivo expansion of AT-MSCs [88]. Table 3 provides an indication of ranges of growth factor content in PLs and PRP.
Table 3.
Approximate contents of growth factors in human platelet lysates [41]
| Growth factor | Content (ng/mL) |
|---|---|
| Platelet-derived growth factor (PDGF)-AB | ∼50–300 |
| PDGF-BB | ∼10–30 |
| PDGF-AA | ∼1–10 |
| Tumor growth factor (TGF)-β1 | ∼50–300 |
| TGF-β2 | ∼0.5 |
| Brain-derived neurotrophic factor (BDNF) | ∼10–50 |
| Vascular endothelial growth factor (VEGF) | ∼5–20 |
| Endothelial growth factor (EGF) | ∼0.5–20 |
| Insulin-like growth factor (IGF)-1 | ∼50–200 |
The cytokines identified in platelets play important roles in cell proliferation, chemotaxis, cell differentiation, regeneration, and angiogenesis [116]. A particular advantage of PRP is that these native cytokines are all present in normal physiological ratios. A wide array of bioactive growth factors are found in the lysate of platelet cells and in platelet alpha granules [117]. Positive dose–response relationships was found between platelet concentrations and proliferation of human MSCs, proliferation of fibroblasts, and production of type I collagen [118].
Sanchez-Gonzalez et al. reviewed some of the more extensively studied platelet growth factors, that is, peptidic growth factors in platelet alpha granules [119], [120], [121] and PRP [40], [120], [122], [123], [124], [125], and many more, both discovered and undiscovered. PLs contain these potent factors involved in recruiting tissue stem/progenitor cells to the site of injury and interacting with them, thus stimulating differentiation and angiogenesis, which are critical for MSC culture and tissue therapeutic healing [126], [127], [128], [129], [130], [131], [132]. By two-dimensional difference gel electrophoresis (2D-DIGE), MALDI-TOF analyses and complementary Western blotting, protein qualitative and quantitative differences could be observed in various types of platelet materials, also revealing that fibrinogen and apolipoprotein A1 differentially affected AT- and BM-MSC proliferation, showing a role in ex vivo expansion of MSC. The study also illustrated the role that proteomic studies can have in identifying impacts of known compositions of growth factors on MSC expansion [133].
Platelets growth factors: a family of cytokines for MSC survival
Successful use of MSCs in regenerative repair requires the survival of transplanted cells. The ability of various growth factors like FGF, EFG and, in particular, VEGF to promote survival has been studied. MSCs treated with VEGF in vitro and MSCs carrying the VEGF gene in vivo increase the survival of stromal cells. Rodent hearts that had undergone myocardial infarction and had been injected with MSCs along with VEGF showed a higher number of MSCs at the injection site [134]. The surge in MSC survival was attributed to an increase in Akt signaling causing a reduction in infarct size, less fibrosis, increased vascularity, and thicker ventricular walls [135], [136]. PDGFs are potent mitogens for connective tissue cells and exert their function by interacting with related receptor tyrosine kinases, promoting cellular proliferation and inhibiting apoptosis. IGF mediates many of the growth-promoting effects of growth hormone.
Platelet cytokines for MSC proliferation and differentiation
TGF-β is known to influence cells from the chondrogenic lineage in vivo and promotes initial stages of mesenchymal condensation, prechondrocyte proliferation, production of the extracellular matrix (ECM), and deposition of cartilage-specific molecules, while inhibiting terminal differentiation [137], [138], [139]. When applied to MSCs in vitro to study chondrocyte regeneration, cells showed increased proliferation and a bias towards a chondrogenic lineage [137], [140]. While all three isoforms (types 1–3) induce proliferation of MSCs and chondrocyte formation, TGF-β3 exhibits the most pronounced effect on chondrogenesis and consistently increased proliferation of MSCs [141], [142], making it a prime factor for inducing chondrogenesis from implanted MSCs. Similarly, bone morphogenic protein (BMP)-2 to -7 growth factors, which belong to the TGF-β superfamily, are known to affect bone formation. While BMP-2, -4, -6, and -7 induce MSCs to form osteoblasts, BMP-2 has the greatest impact on differentiation [143]. MSCs overexpressing BMP-2 and implanted with the ECM protein, collagen I, as in a hydrogel system, increased proliferation of MSC differentiation into bone; this model was used to study cranial closure in swine [144], [145]. Another member of the same family, BMP-3, increased MSC proliferation 3-fold [146]. Since all these factors affect bone formation at different rates and some have a greater effect on proliferation, synergistic pairs of these growth factors can be used at optimal doses and at specific points during the bone-regeneration process. One such search for synergistic pairs led to combination treatment of MSCs with TGFβ3 and BMP-2, and chondrogenic differentiation was found to be enhanced [146], [147].
MSC proliferation and survival are regulated by the signaling of various cytokines
MSCs are self-renewing cells that have the capacity to differentiate into adipocytes, chondrocytes, myocytes, and osteocytes. MSC expansion and/or differentiation towards a specific cell lineage can be stimulated by numbers of growth/differentiation factors like FGF, EGF, IGF, PDGF, VEGF, Wnt, and TGF-β. Certain basic growth factors like FGF and EGF promotes MSC expansion without inducing differentiation, while others in addition to their proliferative and pro-survival effects also induce differentiation of MSCs towards a specific lineage. For example, TGF-β 3 can increase proliferation of MSCs while inducing chondrogenesis. Similarly, BMP-3 increases MSC proliferation while inducing osteogenesis.
TGF-β signaling occurs when TGF-β, or factors from its family, binds a type II serine-threonine kinase receptor recruiting another such transmembrane protein (receptor I). Receptor I phosphorylates the primary intracellular downstream molecules, e.g. Sma and Mad related proteins (SMADS), causing their translocation into nuclei and specific gene transcription. Receptor I can be ALK-1, -2, -3, or -6 that signals SMAD-1, SMAD 5, and SMAD 8, or can be ALK-4, -5, or -7 that signals SMAD 2 and SMAD 3. Signaling via SMAD 1, SMAD 5, or SMAD 8 is required for MSC chondrocyte differentiation, while signaling through SMAD 2 or SMAD 3 blocks chondrocyte differentiation [148]. TGF-β and members of this growth factor family can also signal via the mitogen-activated protein kinase (MAPK), Rho GTPase and phosphoinositide-3 kinase (PI3K) pathways in MSCs [149]. The effect of BMP-2 on proliferation and osteogenic differentiation of MSCs was shown to occur via sustained signaling of the MAPK, extracellular signal-regulated kinase (ERK) [150]. The mitogenic effects of BMP-3, on the other hand, were found to be mediated by TGF-β/activin signaling and not by any of the MAPK signaling pathways, with ALK-4, SMAD 2, and SMAD 3 being key players involved in MSC proliferation and differentiation [146].
MSCs pretreated with TGF-α exhibited increased survival. This improvement was also attributed to VEGF signaling, although direct signaling through the EGF receptor (EGFR) cannot be discounted. TGF-α increases VEGF production via the p38 MAPK pathway and enhances recovery [151]. Therefore, in ischemic cardiac tissues, MSCs supplanted with VEGF have so far been the best choice for increased survival, leading to improved vascularity in ischemic cardiac tissues and isolated islets. Several other platelet growth factors were proven to increase MSC survival. MSC transplantation with BDNF and nerve growth factor (NGF) into rodents after traumatic brain injury produced significantly higher numbers of engrafted cells compared to MSCs transplanted with no growth factor [152]. PDGF-BB was found to reduce the loss of cells by apoptosis by 46% as seen between days 5 and week 3 in rats following an acute myocardial infarction [153]. Contrary to its limited effect on MSC expansion, HGF caused a slight increase in MSC survival. PI3K signaling was implicated in this increase [154]. Another major growth factor studied for its effects on MSC survival is EGF. Since initial studies showed that EGF in a soluble state does not cause differentiation of MSCs but enhances expansion, it was hypothesized that soluble EGF would similarly enhance survival of MSCs subjected to pro-death cytokines such as FasL and TRAIL in vitro. Contrary to what was expected, soluble EGF did not protect MSCs, but increased cell death in the presence of FasL [155].
Human PL presents a pool of bioactive molecules for cell growth in culture. The PDGF and TGF-β families of cytokines that affect cell morphogenesis, proliferation, and differentiation were found to be abundant in PLs with the potential to influence MSC culture. The selection of specific growth factors derived from PLs can be used to expand MSCs or differentiate MSCs towards a specific lineage. PDGF-AB/BB, TGF-β1 and bFGF are essential stimuli for the proliferation of MSCs. The three factors bFGF, PDGF-BB and TGF-β1 seem to be necessary for optimal proliferation of MSC; however, these three factors are insufficient on their own, even when using cell culture multiwell plates coated with human fibronectin and human placental collagen I–III. Therefore, in addition to the essential components, PDGF-BB, TGF-β1, bFGF, and other constituents of PLs are important for their full biologic activity [49].
Supply of human platelets
Platelet concentrates are obtained as a by-product of RBC concentrates made from whole blood, or they are prepared by apheresis. A recent survey by the WHO indicated that there were approximately 92 million whole-blood donations globally in 2008 [156], the majority being collected in countries with a high or medium development index. Although not all blood donations are currently used to prepare platelet concentrates, these figures provide some idea of the volume of platelet concentrates potentially available for use for SC expansion based on a mean volume of 30 mL of platelet concentrate per donation. It is also now established that PLs produced from expired platelet concentrates are suitable as supplements for SC expansion [157].
Conclusions
There is now substantial and well-established scientific evidence that PLs and releasates can be used to replace FBS for ex vivo expansion of SCs. Allogeneic materials present the advantage of being available worldwide as a side-product of the preparation of RBC concentrates that are by far the major driver of whole-blood collection in the world. In addition, apheresis procedures can be implemented for the dedicated collection of platelet concentrates if there is a need. Principles of GMP (donor screening, viral testing, traceability, production monitoring, etc.) that are in place for collecting blood components used for traditional transfusion or further manufacture [158] needs constitute a professional framework that can definitely support the quality and safety requirements of allogeneic platelet concentrates as source materials for preparing growth factor-rich PLs for therapeutic-scale SC expansion and regenerative medicine. In addition, the possibility of performing autologous platelet collection represents an alternative source of PLs when a small expansion culture volume is acceptable to reach clinically meaningful MSC numbers. One can expect that further improvements in the ex vivo expansion of MSCs and their clinical applications will allow the development of fully autologous treatment procedures. We believe that human PLs made from well-controlled platelet concentrate sources and production procedures have the potential to become the next clinical-grade gold standard as supplements for the isolation and ex vivo expansion of SCs.
Acknowledgement
This study was supported by a grant from the National Science Council of Taiwan 102-2320-B-038-002 and NSC 101-2314-B038-039-MY2.
References
- 1.Barry F.P., Murphy J.M. Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol. 2004;36:568–584. doi: 10.1016/j.biocel.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Deans R.J., Moseley A.B. Mesenchymal stem cells: biology and potential clinical uses. Exp Hematol. 2000;28:875–884. doi: 10.1016/s0301-472x(00)00482-3. [DOI] [PubMed] [Google Scholar]
- 3.Kassem M., Abdallah B.M. Human bone-marrow-derived mesenchymal stem cells: biological characteristics and potential role in therapy of degenerative diseases. Cell Tissue Res. 2008;331:157–163. doi: 10.1007/s00441-007-0509-0. [DOI] [PubMed] [Google Scholar]
- 4.Le Blanc K., Ringden O. Immunomodulation by mesenchymal stem cells and clinical experience. J Internal Med. 2007;262:509–525. doi: 10.1111/j.1365-2796.2007.01844.x. [DOI] [PubMed] [Google Scholar]
- 5.Pittenger M.F., Mackay A.M., Beck S.C., Jaiswal R.K., Douglas R., Mosca J.D. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 6.Granero-Molto F., Weis J.A., Longobardi L., Spagnoli A. Role of mesenchymal stem cells in regenerative medicine: application to bone and cartilage repair. Expert Opin Biol Ther. 2008;8:255–268. doi: 10.1517/14712598.8.3.255. [DOI] [PubMed] [Google Scholar]
- 7.Berger M.G., Veyrat-Masson R., Rapatel C., Descamps S., Chassagne J., Boiret-Dupre N. Cell culture medium composition and translational adult bone marrow-derived stem cell research. Stem Cells. 2006;24:2888–2890. doi: 10.1634/stemcells.2006-0387. [DOI] [PubMed] [Google Scholar]
- 8.Dimarakis I., Levicar N. Cell culture medium composition and translational adult bone marrow-derived stem cell research. Stem Cells. 2006;24:1407–1408. doi: 10.1634/stemcells.2005-0577. [DOI] [PubMed] [Google Scholar]
- 9.Perez-Ilzarbe M., Diez-Campelo M., Aranda P., Tabera S., Lopez T., del Canizo C. Comparison of ex vivo expansion culture conditions of mesenchymal stem cells for human cell therapy. Transfusion. 2009;49:1901–1910. doi: 10.1111/j.1537-2995.2009.02226.x. [DOI] [PubMed] [Google Scholar]
- 10.Sotiropoulou P.A., Perez S.A., Salagianni M., Baxevanis C.N., Papamichail M. Cell culture medium composition and translational adult bone marrow-derived stem cell research. Stem Cells. 2006;24:1409–1410. doi: 10.1634/stemcells.2005-0654. [DOI] [PubMed] [Google Scholar]
- 11.Heiskanen A., Satomaa T., Tiitinen S., Laitinen A., Mannelin S., Impola U. N-glycolylneuraminic acid xenoantigen contamination of human embryonic and mesenchymal stem cells is substantially reversible. Stem Cells. 2007;25:197–202. doi: 10.1634/stemcells.2006-0444. [DOI] [PubMed] [Google Scholar]
- 12.Spees J.L., Gregory C.A., Singh H., Tucker H.A., Peister A., Lynch P.J. Internalized antigens must be removed to prepare hypoimmunogenic mesenchymal stem cells for cell and gene therapy. Mol Ther. 2004;9:747–756. doi: 10.1016/j.ymthe.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 13.Kievits F., Boerenkamp W.J., Ivanyi P. H-2-dependent binding of xenogeneic beta 2-microglobulin from culture media. J Immunol. 1988;140:4253–4255. [PubMed] [Google Scholar]
- 14.Mackensen A., Drager R., Schlesier M., Mertelsmann R., Lindemann A. Presence of IgE antibodies to bovine serum albumin in a patient developing anaphylaxis after vaccination with human peptide-pulsed dendritic cells. Cancer Immunol Immunother. 2000;49:152–156. doi: 10.1007/s002620050614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selvaggi T.A., Walker R.E., Fleisher T.A. Development of antibodies to fetal calf serum with arthus-like reactions in human immunodeficiency virus-infected patients given syngeneic lymphocyte infusions. Blood. 1997;89:776–779. [PubMed] [Google Scholar]
- 16.Ivan A., Indrei L.L. Emergence of transmissible disorders, a continuous process – a new type of viral meningoencephalitis. Rev Med Chir Soc Med Nat Iasi. 2000;104:51–55. [PubMed] [Google Scholar]
- 17.Kaaden O.R., Eichhorn W., Essbauer S. Recent developments in the epidemiology of virus diseases. J Vet Med B: Infect Dis Vet Public Health. 2002;49:3–6. doi: 10.1046/j.1439-0450.2002.00530.x. [DOI] [PubMed] [Google Scholar]
- 18.Abbott A., Cyranoski D. Biologists seek to head off future sources of infection. Nature. 2003;423:3. doi: 10.1038/423003b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng S.K. Possible role of an animal vector in the SARS outbreak at Amoy Gardens. Lancet. 2003;362:570–572. doi: 10.1016/S0140-6736(03)14121-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao R., Cao B., Hu Y., Feng Z., Wang D., Hu W. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 21.Anonymous Abu Dhabi 5 camel connection to new coronavirus? Science. 2013;340 127–127. [Google Scholar]
- 22.Robertson J.S. Bovine serum – regulatory issues. Dev Biol (Basel) 2006;123:269–272. discussion 291–308. [PubMed] [Google Scholar]
- 23.Hill A.F., Desbruslais M., Joiner S., Sidle K.C.L., Gowland I., Collinge J. The same prion strain causes vCJD and BSE. Nature. 1997;389:448–450. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 24.CHMP . European Medicine Agency; 2003. Note for guidance on the use of bovine serum in the manufacture of human biological medicinal products. CPMP/BWP/1793/02. October. http://www.emea.eu.int. [Google Scholar]
- 25.WHO, 2006. WHO guidelines on tissue infectivity distribution in transmissible spongiform encephalopathies. http://www.who.int/bloodproducts.
- 26.Gstraunthaler G. Alternatives to the use of fetal bovine serum: serum-free cell culture. Altex Altern Tierexp. 2003;20:275–281. [PubMed] [Google Scholar]
- 27.van der Valk J., Mellor D., Brands R., Fischer R., Gruber F., Gstraunthaler G. The humane collection of fetal bovine serum and possibilities for serum-free cell and tissue culture. Toxicol In Vitro. 2004;18:1–12. doi: 10.1016/j.tiv.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Kuznetsov S.A., Friedenstein A.J., Robey P.G. Factors required for bone marrow stromal fibroblast colony formation in vitro. Br J Haematol. 1997;97:561–570. doi: 10.1046/j.1365-2141.1997.902904.x. [DOI] [PubMed] [Google Scholar]
- 29.Burnouf T. Recombinant plasma proteins. Vox Sang. 2011;100:68–83. doi: 10.1111/j.1423-0410.2010.01384.x. [DOI] [PubMed] [Google Scholar]
- 30.Grillberger L., Kreil T.R., Nasr S., Reiter M. Emerging trends in plasma-free manufacturing of recombinant protein therapeutics expressed in mammalian cells. Biotechnol J. 2009;4:186–201. doi: 10.1002/biot.200800241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fekete N., Rojewski M.T., Lotfi R., Schrezenmeier H. Essential components for ex vivo proliferation of mesenchymal stromal cells. Tissue Eng Part C Methods. 2014;20:129–139. doi: 10.1089/ten.TEC.2013.0061. [DOI] [PubMed] [Google Scholar]
- 32.Bieback K. Platelet lysate as replacement for fetal bovine serum in mesenchymal stromal cell cultures. Transf Med Hemotherapy. 2013;40:326–335. doi: 10.1159/000354061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blair P., Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 2009;23:177–189. doi: 10.1016/j.blre.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kisucka J., Butterfield C.E., Duda D.G., Eichenberger S.C., Saffaripour S., Ware J. Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc Natl Acad Sci U S A. 2006;103:855–860. doi: 10.1073/pnas.0510412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lucarelli E., Beccheroni A., Donati D., Sangiorgi L., Cenacchi A., Del Vento A.M. Platelet-derived growth factors enhance proliferation of human stromal stem cells. Biomaterials. 2003;24:3095–3100. doi: 10.1016/s0142-9612(03)00114-5. [DOI] [PubMed] [Google Scholar]
- 36.Doucet C., Ernou I., Zhang Y., Llense J.R., Begot L., Holy X. Platelet lysates promote mesenchymal stem cell expansion: a safety substitute for animal serum in cell-based therapy applications. J Cell Physiol. 2005;205:228–236. doi: 10.1002/jcp.20391. [DOI] [PubMed] [Google Scholar]
- 37.Bieback K., Hecker A., Kocaomer A., Lannert H., Schallmoser K., Strunk D. Human alternatives to fetal bovine serum for the expansion of mesenchymal stromal cells from bone marrow. Stem Cells. 2009;27:2331–2341. doi: 10.1002/stem.139. [DOI] [PubMed] [Google Scholar]
- 38.Pawitan J.A. Platelet rich plasma in xeno-free stem cell culture: the impact of platelet count and processing method. Curr Stem Cell Res Ther. 2012;7:329–335. doi: 10.2174/157488812802481508. [DOI] [PubMed] [Google Scholar]
- 39.Anonymous . 10th edition. Council of Europe Publishing; 2011. Guide to the preparation, use and quality assurance of blood components. [Google Scholar]
- 40.Kocaoemer A., Kern S., Kluter H., Bieback K. Human AB serum and thrombin-activated platelet-rich plasma are suitable alternatives to fetal calf serum for the expansion of mesenchymal stem cells from adipose tissue. Stem Cells. 2007;25:1270–1278. doi: 10.1634/stemcells.2006-0627. [DOI] [PubMed] [Google Scholar]
- 41.Burnouf T., Goubran H.A., Chen T.M., Ou K.L., El-Ekiaby M., Radosevic M. Blood-derived biomaterials and platelet growth factors in regenerative medicine. Blood Rev. 2013;27:77–89. doi: 10.1016/j.blre.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Mazzucco L., Balbo V., Cattana E., Guaschino R., Borzini P. Not every PRP-gel is born equal. Evaluation of growth factor availability for tissues through four PRP-gel preparations: fibrinet, RegenPRP-Kit, Plateltex and one manual procedure. Vox Sang. 2009;97:110–118. doi: 10.1111/j.1423-0410.2009.01188.x. [DOI] [PubMed] [Google Scholar]
- 43.Burgstaler E.A. Blood component collection by apheresis. J Clin Apheresis. 2006;21:142–151. doi: 10.1002/jca.20043. [DOI] [PubMed] [Google Scholar]
- 44.Singh R.P., Marwaha N., Malhotra P., Dash S. Quality assessment of platelet concentrates prepared by platelet rich plasma-platelet concentrate, buffy coat poor-platelet concentrate (BC-PC) and apheresis-PC methods. Asian J Transfus Sci. 2009;3:86–94. doi: 10.4103/0973-6247.53882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pietersz R.N.I., van der Meer P.F., Steneker I., Hinloopen B., Dekker W.J.A., van Zanten A.P. Preparation of leukodepleted platelet concentrates from pooled buffy coats: prestorage filtration with Autostop (TM) BC. Vox Sang. 1999;76:231–236. doi: 10.1159/000031057. [DOI] [PubMed] [Google Scholar]
- 46.van der Meer P., Pietersz R., Reesink H. Leucoreduced platelet concentrates in additive solution: an evaluation of filters and storage containers. Vox Sang. 2001;81:102–107. doi: 10.1046/j.1423-0410.2001.00084.x. [DOI] [PubMed] [Google Scholar]
- 47.Azuma H., Hirayama J., Akino M., Ikeda H. Platelet additive solution – electrolytes. Transf Apher Sci. 2011;44:277–281. doi: 10.1016/j.transci.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 48.Eriksson L., Hogman C.F. Platelet concentrates in an additive solution prepared from pooled buffy coats. 1. In vitro studies. Vox Sang. 1990;59:140–145. doi: 10.1111/j.1423-0410.1990.tb00848.x. [DOI] [PubMed] [Google Scholar]
- 49.Fekete N., Gadelorge M., Furst D., Maurer C., Dausend J., Fleury-Cappellesso S. Platelet lysate from whole blood-derived pooled platelet concentrates and apheresis-derived platelet concentrates for the isolation and expansion of human bone marrow mesenchymal stromal cells: production process, content and identification of active components. Cytotherapy. 2012;14:540–554. doi: 10.3109/14653249.2012.655420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iudicone P., Fioravanti D., Bonanno G., Miceli M., Lavorino C., Totta P. Pathogen-free, plasma-poor platelet lysate and expansion of human mesenchymal stem cells. J Transl Med. 2014;12:28. doi: 10.1186/1479-5876-12-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schallmoser K., Bartmann C., Rohde E., Reinisch A., Kashofer K., Stadelmeyer E. Human platelet lysate can replace fetal bovine serum for clinical-scale expansion of functional mesenchymal stromal cells. Transfusion. 2007;47:1436–1446. doi: 10.1111/j.1537-2995.2007.01220.x. [DOI] [PubMed] [Google Scholar]
- 52.Jonsdottir-Buch S.M., Lieder R., Sigurjonsson O.E. Platelet lysates produced from expired platelet concentrates support growth and osteogenic differentiation of mesenchymal stem cells. PLoS One. 2013;8:e68984. doi: 10.1371/journal.pone.0068984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Commission E. European Commission; 2003. Commission Directive 2003/94/EC of 8 October 2003 laying down the principles and guidelines of good manufacturing practice in respect of medicinal products doe human use and investigational medicinal products for human use. 14.10.2003. (Union, O. j. o. t. E. ed.) pp. 22–26. [Google Scholar]
- 54.WHO Guidelines on good manufacturing practices for blood establishments. Annex 4. WHO Tech Rep Series. 2011;961:148–214. [Google Scholar]
- 55.Allain J.P., Bianco C., Blajchman M.A., Brecher M.E., Busch M., Leiby D. Protecting the blood supply from emerging pathogens: the role of pathogen inactivation. Transfus Med Rev. 2005;19:110–126. doi: 10.1016/j.tmrv.2004.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Busch M.P., Kleinman S.H., Nemo G.J. Current and emerging infectious risks of blood transfusions. JAMA. 2003;289:959–962. doi: 10.1001/jama.289.8.959. [DOI] [PubMed] [Google Scholar]
- 57.Dodd R.Y. Emerging pathogens and their implications for the blood supply and transfusion transmitted infections. Br J Haematol. 2012;159:135–142. doi: 10.1111/bjh.12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lanteri M.C., Busch M.P. Dengue in the context of safe blood and global epidemiology: to screen or not to screen? Transfusion. 2012;52:1634–1639. doi: 10.1111/j.1537-2995.2012.03747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burnouf T., Radosevich M. Reducing the risk of infection from plasma products: specific preventative strategies. Blood Rev. 2000;14:94–110. doi: 10.1054/blre.2000.0129. [DOI] [PubMed] [Google Scholar]
- 60.Prowse C., Ludlam C.A., Yap P.L. Human parvovirus B19 and blood products. Vox Sang. 1997;72:1–10. doi: 10.1046/j.1423-0410.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- 61.Lefrere J.-J., Hewitt P. From mad cows to sensible blood transfusion: the risk of prion transmission by labile blood components in the United Kingdom and in France. Transfusion. 2009;49:797–812. doi: 10.1111/j.1537-2995.2008.02044.x. [DOI] [PubMed] [Google Scholar]
- 62.Llewelyn C.A., Hewitt P.E., Knight R.S.G., Amar K., Cousens S., Mackenzie J. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–421. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- 63.Burnouf T., Padilla A. Current strategies to prevent transmission of prions by human plasma derivatives. Transf Clin Biol. 2006;13:320–328. doi: 10.1016/j.tracli.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 64.Gregori L., McCombie N., Palmer D., Birch P., Sowemimo-Coker S.O., Giulivi A. Effectiveness of leucoreduction for removal of infectivity of transmissible spongiform encephalopathies from blood. Lancet. 2004;364:529–531. doi: 10.1016/S0140-6736(04)16812-8. [DOI] [PubMed] [Google Scholar]
- 65.Vamvakas E.C. Universal white blood cell reduction in Europe: has transmission of variant Creutzfeldt-Jakob disease been prevented? Transf Med Rev. 2011;25:133–144. doi: 10.1016/j.tmrv.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 66.Cancelas J.A., Rugg N., Pratt P.G., Worsham D.N., Pehta J.C., Banks K. Infusion of P-Capt prion-filtered red blood cell products demonstrate acceptable in vivo viability and no evidence of neoantigen formation. Transfusion. 2011;51:2228–2236. doi: 10.1111/j.1537-2995.2011.03133.x. [DOI] [PubMed] [Google Scholar]
- 67.Edgeworth J.A., Farmer M., Sicilia A., Tavares P., Beck J., Campbell T. Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet. 2011;377:487–493. doi: 10.1016/S0140-6736(10)62308-2. [DOI] [PubMed] [Google Scholar]
- 68.Busch M.P., Glynn S.A., Stramer S.L., Strong D.M., Caglioti S., Wright D.J. A new strategy for estimating risks of transfusion-transmitted viral infections based on rates of detection of recently infected donors. Transfusion. 2005;45:254–264. doi: 10.1111/j.1537-2995.2004.04215.x. [DOI] [PubMed] [Google Scholar]
- 69.Dodd R.Y. Current risk for transfusion transmitted infections. Curr Opin Hematol. 2007;14:671–676. doi: 10.1097/MOH.0b013e3282e38e8a. [DOI] [PubMed] [Google Scholar]
- 70.Zou S.M., Stramer S.L., Dodd R.Y. Donor testing and risk: current prevalence, incidence, and residual risk of transfusion-transmissible agents in US allogeneic donations. Transf Med Rev. 2012;26:119–128. doi: 10.1016/j.tmrv.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 71.de Mendoza C., Altisent C., Aznar J.A., Batlle J., Soriano V. Emerging viral infections – a potential threat for blood supply in the 21st century. Aids Rev. 2012;14:279–289. [PubMed] [Google Scholar]
- 72.Reesink H.W., Panzer S., McQuilten Z.K., Wood E.M., Marks C., Wendel S. Pathogen inactivation of platelet concentrates. Vox Sang. 2010;99:85–95. doi: 10.1111/j.1423-0410.2010.01319.x. [DOI] [PubMed] [Google Scholar]
- 73.Lin L., Cook D.N., Wiesehahn G.P., Alfonso R., Behrman B., Cimino G.D. Photochemical inactivation of viruses and bacteria in platelet concentrates by use of a novel psoralen and long-wavelength ultraviolet light. Transfusion. 1997;37:423–435. doi: 10.1046/j.1537-2995.1997.37497265344.x. [DOI] [PubMed] [Google Scholar]
- 74.Lin L., Hanson C.V., Alter H.J., Jauvin V., Bernard K.A., Murthy K.K. Inactivation of viruses in platelet concentrates by photochemical treatment with amotosalen and long-wavelength ultraviolet light. Transfusion. 2005;45:580–590. doi: 10.1111/j.0041-1132.2005.04316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klein H.G. Pathogen inactivation technology: cleansing the blood supply. J Intern Med. 2005;257:224–237. doi: 10.1111/j.1365-2796.2005.01451.x. [DOI] [PubMed] [Google Scholar]
- 76.Ruane P.H., Edrich R., Gampp D., Keil S.D., Leonard R.L., Goodrich R.P. Photochemical inactivation of selected viruses and bacteria in platelet concentrates using riboflavin and light. Transfusion. 2004;44:877–885. doi: 10.1111/j.1537-2995.2004.03355.x. [DOI] [PubMed] [Google Scholar]
- 77.Corbin F., 3rd Pathogen inactivation of blood components: current status and introduction of an approach using riboflavin as a photosensitizer. Int J Hematol. 2002;76(Suppl 2):253–257. doi: 10.1007/BF03165125. [DOI] [PubMed] [Google Scholar]
- 78.Goodrich R.P. The use of riboflavin for the inactivation of pathogens in blood products. Vox Sang. 2000;78(Suppl 2):211–215. [PubMed] [Google Scholar]
- 79.AuBuchon J.P., Herschel L., Roger J., Taylor H., Whitley P., Li J.Z. Efficacy of apheresis platelets treated with riboflavin and ultraviolet light for pathogen reduction. Transfusion. 2005;45:1335–1341. doi: 10.1111/j.1537-2995.2005.00202.x. [DOI] [PubMed] [Google Scholar]
- 80.Castrillo A., Cardoso M., Rouse L. Treatment of buffy coat platelets in platelet additive solution with the mirasol (R) pathogen reduction technology system. Transf Med Hemother. 2013;40:44–48. doi: 10.1159/000345679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cookson P., Thomas S., Marschner S., Goodrich R., Cardigan R. In vitro quality of single-donor platelets treated with riboflavin and ultraviolet light and stored in platelet storage medium for up to 8 days. Transfusion. 2012;52:983–994. doi: 10.1111/j.1537-2995.2011.03388.x. [DOI] [PubMed] [Google Scholar]
- 82.Mohr H., Steil L., Gravemann U., Thiele T., Hammer E., Greinacher A. A novel approach to pathogen reduction in platelet concentrates using short-wave ultraviolet light. Transfusion. 2009;49:2612–2624. doi: 10.1111/j.1537-2995.2009.02334.x. [DOI] [PubMed] [Google Scholar]
- 83.Stahle M., Carlsson B., Le Blanc K., Korsgren O., Knutson F. Photochemical pathogen inactivation of human serum enables its large-scale application in clinical cell transplantation. Vox Sang. 2010;98:e364–e365. doi: 10.1111/j.1423-0410.2009.01257.x. [DOI] [PubMed] [Google Scholar]
- 84.Stahle M.U., Brandhorst D., Korsgren O., Knutson F. Pathogen inactivation of human serum facilitates its clinical use for islet cell culture and subsequent transplantation. Cell Transpl. 2011;20:775–781. doi: 10.3727/096368910X539056. [DOI] [PubMed] [Google Scholar]
- 85.Horowitz B., Bonomo R., Prince A.M., Chin S.N., Brotman B., Shulman R.W. Solvent/detergent-treated plasma: a virus-inactivated substitute for fresh frozen plasma. Blood. 1992;79:826–831. [PubMed] [Google Scholar]
- 86.Burnouf T., Tseng Y.H., Kuo Y.P., Su C.Y. Solvent/detergent treatment of platelet concentrates enhances the release of growth factors. Transfusion. 2008;48:1090–1098. doi: 10.1111/j.1537-2995.2008.01691.x. [DOI] [PubMed] [Google Scholar]
- 87.Su C.Y., Kuo Y.P., Lin Y.C., Huang C.T., Tseng Y.H., Burnouf T. A virally inactivated functional growth factor preparation from human platelet concentrates. Vox Sang. 2009;97:119–128. doi: 10.1111/j.1423-0410.2009.01180.x. [DOI] [PubMed] [Google Scholar]
- 88.Shih D.T., Chen J.C., Chen W.Y., Kuo Y.P., Su C.Y., Burnouf T. Expansion of adipose tissue mesenchymal stromal progenitors in serum-free medium supplemented with virally inactivated allogeneic human platelet lysate. Transfusion. 2011;51:770–778. doi: 10.1111/j.1537-2995.2010.02915.x. [DOI] [PubMed] [Google Scholar]
- 89.Dichtelmuller H.O., Biesert L., Fabbrizzi F., Gajardo R., Groner A., von Hoegen I. Robustness of solvent/detergent treatment of plasma derivatives: a data collection from Plasma Protein Therapeutics Association member companies. Transfusion. 2009;49:1931–1943. doi: 10.1111/j.1537-2995.2009.02222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Blajchman M.A., Beckers E.A., Dickmeiss E., Lin L., Moore G., Muylle L. Bacterial detection of platelets: current problems and possible resolutions. Transf Med Rev. 2005;19:259–272. doi: 10.1016/j.tmrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 91.de Korte D., Marcelis J.H., Soeterboek A.M. Determination of the degree of bacterial contamination of whole-blood collections using an automated microbe-detection system. Transfusion. 2001;41:815–818. doi: 10.1046/j.1537-2995.2001.41060815.x. [DOI] [PubMed] [Google Scholar]
- 92.Blajchman M.A. Bacterial contamination of cellular blood components: risks, sources and control. Vox Sang. 2004;87(Suppl1):98L 103. doi: 10.1111/j.1741-6892.2004.00441.x. [DOI] [PubMed] [Google Scholar]
- 93.Montag T. Strategies of bacteria screening in cellular blood components. Clin Chem Lab Med. 2008;46:926–932. doi: 10.1515/CCLM.2008.176. [DOI] [PubMed] [Google Scholar]
- 94.Jenkins C., Ramirez-Arcos S., Goldman M., Devine D.V. Bacterial contamination in platelets: incremental improvements drive down but do not eliminate risk. Transfusion. 2011;51:2555–2565. doi: 10.1111/j.1537-2995.2011.03187.x. [DOI] [PubMed] [Google Scholar]
- 95.Chou M.L., Wu Y.W., Su C.Y., Lee L.W., Burnouf T. Impact of solvent/detergent treatment of plasma on transfusion-relevant bacteria. Vox Sang. 2012;102:277–284. doi: 10.1111/j.1423-0410.2011.01560.x. [DOI] [PubMed] [Google Scholar]
- 96.Mininkova A.I. Investigation of platelets by the flow cytofluorometric technique (a review of literature). Part 2. Klinicheskaia Laboratornaia Diagnostika. 2011:25–30. [PubMed] [Google Scholar]
- 97.Centeno C.J., Schultz J.R., Cheever M., Freeman M., Faulkner S., Robinson B. Safety and complications reporting update on the re-implantation of culture-expanded mesenchymal stem cells using autologous platelet lysate technique. Curr Stem Cell Res Therapy. 2011;6:368–378. doi: 10.2174/157488811797904371. [DOI] [PubMed] [Google Scholar]
- 98.Mazzucco L., Balbo V., Cattana E., Borzini P. Platelet-rich plasma and platelet gel preparation using Plateltex(R) Vox Sang. 2008;94:202–208. doi: 10.1111/j.1423-0410.2007.01027.x. [DOI] [PubMed] [Google Scholar]
- 99.Horn P., Bokermann G., Cholewa D., Bork S., Walenda T., Koch C. Impact of individual platelet lysates on isolation and growth of human mesenchymal stromal cells. Cytotherapy. 2010;12:888–898. doi: 10.3109/14653249.2010.501788. [DOI] [PubMed] [Google Scholar]
- 100.Lohmann M., Walenda G., Hemeda H., Joussen S., Drescher W., Jockenhoevel S. Donor age of human platelet lysate affects proliferation and differentiation of mesenchymal stem cells. PLoS One. 2012:7. doi: 10.1371/journal.pone.0037839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schallmoser K., Strunk D. Generation of a pool of human platelet lysate and efficient use in cell culture. Methods Mol Biol. 2013;946:349–362. doi: 10.1007/978-1-62703-128-8_22. [DOI] [PubMed] [Google Scholar]
- 102.Janetzko K., Kluter H., van Waeg G., Eichler H. Fully automated processing of buffy-coat-derived pooled platelet concentrates. Transfusion. 2004;44:1052–1058. doi: 10.1111/j.1537-2995.2004.03296.x. [DOI] [PubMed] [Google Scholar]
- 103.Flemming A., Schallmoser K., Strunk D., Stolk M., Volk H.D., Seifert M. Immunomodulative efficacy of bone marrow-derived mesenchymal stem cells cultured in human platelet lysate. J Clin Immunol. 2011;31:1143–1156. doi: 10.1007/s10875-011-9581-z. [DOI] [PubMed] [Google Scholar]
- 104.Chen B., Sun H.H., Wang H.G., Kong H., Chen F.M., Yu Q. The effects of human platelet lysate on dental pulp stem cells derived from impacted human third molars. Biomaterials. 2012;33:5023–5035. doi: 10.1016/j.biomaterials.2012.03.057. [DOI] [PubMed] [Google Scholar]
- 105.Blande I.S., Bassaneze V., Lavini-Ramos C., Fae K.C., Kalil J., Miyakawa A.A. Adipose tissue mesenchymal stem cell expansion in animal serum-free medium supplemented with autologous human platelet lysate. Transfusion. 2009;49:2680–2685. doi: 10.1111/j.1537-2995.2009.02346.x. [DOI] [PubMed] [Google Scholar]
- 106.Bernardo M.E., Avanzini M.A., Perotti C., Cometa A.M., Moretta A., Lenta E. Optimization of in vitro expansion of human multipotent mesenchymal stromal cells for cell-therapy approaches: further insights in the search for a fetal calf serum substitute. J Cell Physiol. 2007;211:121–130. doi: 10.1002/jcp.20911. [DOI] [PubMed] [Google Scholar]
- 107.Chevallier N., Anagnostou F., Zilber S., Bodivit G., Maurin S., Barrault A. Osteoblastic differentiation of human mesenchymal stem cells with platelet lysate. Biomaterials. 2010;31:270–278. doi: 10.1016/j.biomaterials.2009.09.043. [DOI] [PubMed] [Google Scholar]
- 108.Bernardi M., Albiero E., Alghisi A., Chieregato K., Lievore C., Madeo D. Production of human platelet lysate by use of ultrasound for ex vivo expansion of human bone marrow-derived mesenchymal stromal cells. Cytotherapy. 2013;15:920–929. doi: 10.1016/j.jcyt.2013.01.219. [DOI] [PubMed] [Google Scholar]
- 109.Burnouf T., Lee C.Y., Luo C.W., Kuo Y.P., Chou M.L., Wu Y.W. Human blood-derived fibrin releasates: composition and use for the culture of cell lines and human primary cells. Biologicals. 2012;40:21–30. doi: 10.1016/j.biologicals.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 110.Hemeda H., Kalz J., Walenda G., Lohmann M., Wagner W. Heparin concentration is critical for cell culture with human platelet lysate. Cytotherapy. 2013;15:1174–1181. doi: 10.1016/j.jcyt.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 111.Mojica-Henshaw M.P., Jacobson P., Morris J., Kelley L., Pierce J., Boyer M. Serum-converted platelet lysate can substitute for fetal bovine serum in human mesenchymal stromal cell cultures. Cytotherapy. 2013;15:1458–1468. doi: 10.1016/j.jcyt.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 112.Borzini P., Mazzucco L. Platelet gels and releasates. Curr Opin Hematol. 2005;12:473–479. doi: 10.1097/01.moh.0000177831.70657.e8. [DOI] [PubMed] [Google Scholar]
- 113.Mizuno N., Shiba H., Ozeki Y., Mouri Y., Niitani M., Inui T. Human autologous serum obtained using a completely closed bag system as a substitute for foetal calf serum in human mesenchymal stem cell cultures. Cell Biol Int. 2006;30:521–524. doi: 10.1016/j.cellbi.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 114.Capelli C., Domenghini M., Borleri G., Bellavita P., Poma R., Carobbio A. Human platelet lysate allows expansion and clinical grade production of mesenchymal stromal cells from small samples of bone marrow aspirates or marrow filter washouts. Bone Marrow Transpl. 2007;40:785–791. doi: 10.1038/sj.bmt.1705798. [DOI] [PubMed] [Google Scholar]
- 115.Salvade A., Della Mina P., Gaddi D., Gatto F., Villa A., Bigoni M. Characterization of platelet lysate cultured mesenchymal stromal cells and their potential use in tissue-engineered osteogenic devices for the treatment of bone defects. Tissue Eng Part C Methods. 2010;16:201–214. doi: 10.1089/ten.TEC.2008.0572. [DOI] [PubMed] [Google Scholar]
- 116.Werner S., Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83:835–870. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 117.Bennett N.T., Schultz G.S. Growth-factors and wound healing – biochemical properties of growth factors and their receptors. Am J Surg. 1993;165:728–737. doi: 10.1016/s0002-9610(05)80797-4. [DOI] [PubMed] [Google Scholar]
- 118.Liu Y.W., Kalen A., Risto O., Wahlstrom O. Fibroblast proliferation due to exposure to a platelet concentrate in vitro is pH dependent. Wound Repair Regeneration. 2002;10:336–340. doi: 10.1046/j.1524-475x.2002.10510.x. [DOI] [PubMed] [Google Scholar]
- 119.Anitua E., Andia I., Ardanza B., Nurden P., Nurden A.T. Autologous platelets as a source of proteins for healing and tissue regeneration. Thromb Haemost. 2004;91:4–15. doi: 10.1160/TH03-07-0440. [DOI] [PubMed] [Google Scholar]
- 120.Sanchez-Gonzalez D.J., Mendez-Bolaina E., Trejo-Bahena N.I. Platelet-rich plasma peptides: key for regeneration. Int J Peptides. 2012;2012 doi: 10.1155/2012/532519. 532519–532519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Siljander P.R.M. Platelet-derived microparticles – an updated perspective. Thromb Res. 2011;127:S30–S33. doi: 10.1016/S0049-3848(10)70152-3. [DOI] [PubMed] [Google Scholar]
- 122.Borzini P., Mazzucco L. Platelet-rich plasma (PRP) and platelet derivatives for topical therapy. What is true from the biologic view point? ISBT Sci Series. 2007;2:272–281. [Google Scholar]
- 123.Cole B.J., Seroyer S.T., Filardo G., Bajaj S., Fortier L.A. Platelet-rich plasma: where are we now and where are we going? Sports Health. 2010;2:203–210. doi: 10.1177/1941738110366385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Eppley B.L., Woodell J.E., Higgins J. Platelet quantification and growth factor analysis from platelet-rich plasma: implications for wound healing. Plastic Reconstruct Surg. 2004;114:1502–1508. doi: 10.1097/01.prs.0000138251.07040.51. [DOI] [PubMed] [Google Scholar]
- 125.Frechette J.P., Martineau I., Gagnon G. Platelet-rich plasmas: growth factor content and roles in wound healing. J Dental Res. 2005;84:434–439. doi: 10.1177/154405910508400507. [DOI] [PubMed] [Google Scholar]
- 126.Anitua E., Sanchez M., Orive G., Andia I. The potential impact of the preparation rich in growth factors (PRGF) in different medical fields. Biomaterials. 2007;28:4551–4560. doi: 10.1016/j.biomaterials.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 127.Celotti F., Colciago A., Negri-Cesi P., Pravettoni A., Zaninetti R., Sacchi M.C. Effect of platelet-rich plasma on migration and proliferation of SaOS-2 osteoblasts: role of platelet-derived growth factor and transforming growth factor-beta. Wound Repair Regen. 2006;14:195–202. doi: 10.1111/j.1743-6109.2006.00110.x. [DOI] [PubMed] [Google Scholar]
- 128.Driver V.R., Hanft J., Fylling C.P., Beriou J.M., Autologel Diabetic Foot Ulcer Study G. A prospective, randomized, controlled trial of autologous platelet-rich plasma gel for the treatment of diabetic foot ulcers. Ostomy/wound Manage. 2006;52:68–70. 72, 74 passim. [PubMed] [Google Scholar]
- 129.Knighton D.R., Ciresi K., Fiegel V.D., Schumerth S., Butler E., Cerra F. Stimulation of repair in chronic, nonhealing, cutaneous ulcers using platelet-derived wound healing formula. Surg Gynecol Obstet. 1990;170:56–60. [PubMed] [Google Scholar]
- 130.Knighton D.R., Ciresi K.F., Fiegel V.D., Austin L.L., Butler E.L. Classification and treatment of chronic nonhealing wounds. Successful treatment with autologous platelet-derived wound healing factors (PDWHF) Anna Surg. 1986;204:322–330. doi: 10.1097/00000658-198609000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McAleer J.P., Sharma S., Kaplan E.M., Persich G. Use of autologous platelet concentrate in a nonhealing lower extremity wound. Adv Skin Wound Care. 2006;19:354–363. doi: 10.1097/00129334-200609000-00010. [DOI] [PubMed] [Google Scholar]
- 132.O’Connell S.M., Impeduglia T., Hessler K., Wang X.J., Carroll R.J., Dardik H. Autologous platelet-rich fibrin matrix as cell therapy in the healing of chronic lower-extremity ulcers. Wound Repair Regen. 2008;16:749–756. doi: 10.1111/j.1524-475X.2008.00426.x. [DOI] [PubMed] [Google Scholar]
- 133.Kinzebach S., Dietz L., Kluter H., Thierse H.J., Bieback K. Functional and differential proteomic analyses to identify platelet derived factors affecting ex vivo expansion of mesenchymal stromal cells. BMC Cell Biol. 2013;14:48. doi: 10.1186/1471-2121-14-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pons J., Huang Y., Arakawa-Hoyt J., Washko D., Takagawa J., Ye J. VEGF improves survival of mesenchymal stem cells in infarcted hearts. Biochem Biophys Res Commun. 2008;376:419–422. doi: 10.1016/j.bbrc.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 135.Tang J., Wang J., Zheng F., Kong X., Guo L., Yang J. Combination of chemokine and angiogenic factor genes and mesenchymal stem cells could enhance angiogenesis and improve cardiac function after acute myocardial infarction in rats. Mol Cell Biochem. 2010;339:107–118. doi: 10.1007/s11010-009-0374-0. [DOI] [PubMed] [Google Scholar]
- 136.Wang X.H., Hu Q.S., Mansoor A., Lee J., Wang Z.L., Lee T. Bioenergetic and functional consequences of stem cell-based VEGF delivery in pressure-overloaded swine hearts. Am J Physiol-Heart Circ Physiol. 2006;290:H1393–H1405. doi: 10.1152/ajpheart.00871.2005. [DOI] [PubMed] [Google Scholar]
- 137.Bonewald L.F., Dallas S.L. Role of active and latent transforming growth factor beta in bone formation. J Cell Biochem. 1994;55:350–357. doi: 10.1002/jcb.240550312. [DOI] [PubMed] [Google Scholar]
- 138.Canalis E., McCarthy T.L., Centrella M. Effects of platelet-derived growth factor on bone formation in vitro. J Cell Physiol. 1989;140:530–537. doi: 10.1002/jcp.1041400319. [DOI] [PubMed] [Google Scholar]
- 139.van der Kraan P.M., Davidson E.N.B., Blom A., van den Berg W.B. TGF-beta signaling in chondrocyte terminal differentiation and osteoarthritis. Modulation and integration of signaling pathways through receptor-Smads. Osteoarthritis Cartilage. 2009;17:1539–1545. doi: 10.1016/j.joca.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 140.Longobardi L., O’Rear L., Aakula S., Johnstone B., Shimer K., Chytil A. Effect of IGF-1 in the chondrogenesis of bone marrow mesenchymal stem cells in the presence or absence of TGF-beta signaling. J Bone Miner Res. 2006;21:626–636. doi: 10.1359/jbmr.051213. [DOI] [PubMed] [Google Scholar]
- 141.Ogawa T., Akazawa T., Tabata Y. In vitro proliferation and chondrogenic differentiation of rat bone marrow stem cells cultured with gelatin hydrogel microspheres for TGF-beta 1 release. J Biomater Sci-Polym Ed. 2010;21:609–621. doi: 10.1163/156856209X434638. [DOI] [PubMed] [Google Scholar]
- 142.Weiss S., Hennig T., Bock R., Steck E., Richter W. Impact of growth factors and PTHrP on early and late chondrogenic differentiation of human mesenchymal stem cells. J Cell Physiol. 2010;223:84–93. doi: 10.1002/jcp.22013. [DOI] [PubMed] [Google Scholar]
- 143.Luu H.H., Song W.-X., Luo X., Manning D., Luo J., Deng Z.-L. Distinct roles of bone morphogenetic proteins in osteogenic differentiation of mesenchymal stem cells. J Orthopaedic Res. 2007;25:665–677. doi: 10.1002/jor.20359. [DOI] [PubMed] [Google Scholar]
- 144.Chang S.C.N., Chung H.-Y., Tai C.-L., Chen P.K.T., Lin T.-M., Jeng L.-B. Repair of large cranial defects by hBMP-2 expressing bone marrow stromal cells: comparison between alginate and collagen type I systems. J Biomed Mater Res Part A. 2010;94A:433–441. doi: 10.1002/jbm.a.32685. [DOI] [PubMed] [Google Scholar]
- 145.Lou J.R., Xu F., Merkel K., Manske P. Gene therapy: adenovirus-mediated human bone morphogenetic protein-2 gene transfer induces mesenchymal progenitor cell proliferation and differentiation in vitro and bone formation in vivo. J Orthopaedic Res. 1999;17:43–50. doi: 10.1002/jor.1100170108. [DOI] [PubMed] [Google Scholar]
- 146.Stewart A., Guan H., Yang K. BMP-3 promotes mesenchymal stem cell proliferation through the TGF-beta/activin signaling pathway. J Cell Physiol. 2010;223:658–666. doi: 10.1002/jcp.22064. [DOI] [PubMed] [Google Scholar]
- 147.Simmons C.A., Alsberg E., Hsiong S., Kim W.J., Mooney D.J. Dual growth factor delivery and controlled scaffold degradation enhance in vivo bone formation by transplanted bone marrow stromal cells. Bone. 2004;35:562–569. doi: 10.1016/j.bone.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 148.Valcourt U., Gouttenoire J., Moustakas A., Herbage D., Mallein-Gerin F. Functions of transforming growth factor-beta family type I receptors and smad proteins in the hypertrophic maturation and osteoblastic differentiation of chondrocytes. J Biol Chem. 2002;277:33545–33558. doi: 10.1074/jbc.M202086200. [DOI] [PubMed] [Google Scholar]
- 149.Zhang Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lou J., Tu Y., Li S., Manske P.R. Involvement of ERK in BMP-2 induced osteoblastic differentiation of mesenchymal progenitor cell line C3H10T1/2. Biochem Biophys Res Commun. 2000;268:757–762. doi: 10.1006/bbrc.2000.2210. [DOI] [PubMed] [Google Scholar]
- 151.Herrmann J.L., Wang Y., Abarbanell A.M., Weil B.R., Tan J., Meldrum D.R. Preconditioning mesenchymal stem cells with transforming growth factor-alpha improves mesenchymal stem cell-mediated cardioprotection. Shock. 2010;33:24–30. doi: 10.1097/SHK.0b013e3181b7d137. [DOI] [PubMed] [Google Scholar]
- 152.Mahmood A., Lu D.Y., Wang L., Chopp M. Intracerebral transplantation of marrow stromal cells cultured with neurotrophic factors promotes functional recovery in adult rats subjected to traumatic brain injury. J Neurotrauma. 2002;19:1609–1617. doi: 10.1089/089771502762300265. [DOI] [PubMed] [Google Scholar]
- 153.Krausgrill B., Vantler M., Burst V., Raths M., Halbach M., Frank K. Influence of cell treatment with PDGF-BB and reperfusion on cardiac persistence of mononuclear and mesenchymal bone marrow cells after transplantation into acute myocardial infarction in rats. Cell Transpl. 2009;18:847–853. doi: 10.3727/096368909X471134. [DOI] [PubMed] [Google Scholar]
- 154.Forte G., Minieri M., Cossa P., Antenucci D., Sala M., Gnocchi V. Hepatocyte growth factor effects on mesenchymal stem cells: proliferation, migration, and differentiation. Stem Cells. 2006;24:23–33. doi: 10.1634/stemcells.2004-0176. [DOI] [PubMed] [Google Scholar]
- 155.Fan V.H., Au A., Tamama K., Littrell R., Richardson L.B., Wright J.W. Tethered epidermal growth factor provides a survival advantage to mesenchymal stem cells. Stem Cells. 2007;25:1241–1251. doi: 10.1634/stemcells.2006-0320. [DOI] [PubMed] [Google Scholar]
- 156.WHO . World Health Organization; 2011. Blood Safety – Key global fact and figures in 2011. (Organization, W. H. ed.) [Google Scholar]
- 157.Dietz A.B., Padley D.J., Gastineau D.A. Infrastructure development for human cell therapy translation. Clin Pharmacol Ther. 2007;82:320–324. doi: 10.1038/sj.clpt.6100288. [DOI] [PubMed] [Google Scholar]
- 158.WHO WHO Expert Committee on biological standardization – WHO Recommendations for the production, control and regulation of human plasma for fractionation. World Health Organization Tech Rep Series. 2007 1–340, back cover. [Google Scholar]