

Abstract
The discovery of angiotensin-I-converting enzyme 2 (ACE2) and a (pro)renin receptor has renewed interest in the physiology of the renin-angiotensin system (RAS). Through the ACE2/angiotensin-(1–7)/Mas counter-regulatory axis, ACE2 balances the vasoconstrictive, proliferative, fibrotic and proinflammatory effects of the ACE/angiotensin II/AT1 axis. The (pro)renin receptor system shows an angiotensin-dependent function related to increased generation of angiotensin I, and an angiotensin-independent aspect related to intracellular signalling. Activation of ACE2 and inhibition of ACE and renin have been at the core of the RAS regulation. The aim of this review is to discuss the biochemistry and biological functions of ACE, ACE2 and renin within and beyond the RAS, and thus provide a perspective for future bioactives from natural plant and/or food resources related to the three proteases.
Keywords: Angiotensin, ACE, ACE2, Renin, (Pro)renin receptor
Résumé
La découverte de l’enzyme de conversion de l’angiotensine 2 (AC2) et un récepteur à la prorénine est une avancée récente dans la compréhension de la physiologie du système rénine-angiotensine. Au sein de l’axe inhibiteur de l’enzyme de conversion de l’angiotensine 2/angiotensine 1/MAS, l’AC2 contrebalance l’effet vasoconstricteur, prolifératif, fibrosant et pro-inflammatoire de l’axe ACE/angiotensine 2/AT1. Le récepteur à la prorénine a une fonction angiotensine dépendante, liée à l’augmentation de la production d’angiotensine 1, et un aspect indépendant de l’angiotensine, lié aux signaux intracellulaires. L’activation de l’AC2 et l’inhibition de l’ACE de la rénine ont été considérées comme au centre de la régulation du système rénine-angiotensine. L’objet de cette revue générale est de discuter les fonctions biochimiques et biologiques de l’ACE, de l’AC2 et de la rénine au sein et au-delà du système rénine-angiotensine et ainsi de proposer une perspective de développement d’agents actifs extraits de plantes naturelles ou d’alimentation, produits liés à ces trois protéases.
Mots clés: Angiotensine, Inhibiteurs de l’enzyme de conversion, Inhibiteur de l’enzyme de conversion II, Rénine, Récepteur de la prorénine
Background
The renin-angiotensin system (RAS) is not only an endocrine but also a paracrine and an intracrine system [1]. In mammals, the intravascular RAS plays a key role in maintaining blood pressure homeostasis and fluid and salt balance, and the tissue or local RAS is involved in physiological and pathological processes, such as tissue growth and remodelling, development and inflammation [2]. In a classical RAS, the substrate angiotensinogen (AGT), which is released into the circulation from the liver, is degraded by the enzyme renin that originates in the kidney, generating the inactive angiotensin I (Ang I). When this decapeptide comes into contact with angiotensin-I-converting enzyme (ACE) at the endothelial surface of blood vessels, the C-terminal dipeptide is cleaved, giving rise to angiotensin II (Ang II), the main effector molecule of the RAS. Through interactions with specific receptors, particularly its type 1 or AT1 receptor, Ang II stimulates a wide variety of signalling pathways in the heart, blood vessels, kidneys, adipose tissue, pancreas and brain, initiating most of the physiological and pathophysiological effects that have been attributed to the RAS [3]. Due to the function of directly generating the main effector Ang II, ACE – together with the classical axis ACE/Ang II/AT1 – has been at the core of RAS studies since its discovery.
In 2000, a homologue of ACE, known as angiotensin-I-converting enzyme 2 (ACE2), was cloned by two independent research groups [4], [5]. Evidence indicates that ACE2 negatively regulates the activated RAS by degrading Ang II to the heptapeptide Ang-(1–7). Moreover, through the Mas receptor (a G protein-coupled receptor), the resulting Ang-(1–7) counterbalances the cardiovascular effects of Ang II by opposing many AT1 receptor-mediated actions [6]. ACE2 and the axis ACE2/Ang-(1–7)/Mas are becoming the focus of intense research regarding the RAS [1]. The discovery of a (pro)renin receptor ([P]RR) and the introduction of renin inhibitors have also brought (pro)renin back into the spotlight [7]. Far from being a straightforward cascade containing one substrate (AGT), two proteases (renin and ACE), two peptides (Ang I and Ang II) and one receptor (AT1), the RAS currently consists of several axes upstream and downstream of the classical cascade, which include more than two dozen peptidases, nearly a dozen Ang fragments and at least six different receptors [8]. Here, we review three critical proteases (ACE, ACE2 and renin) within and beyond the RAS and thus intend to find new connections between natural plant and/or food resources and the RAS.
Occurrence, gene encoding and structure of ACE
ACE (EC 3.4.15.1) is a monomeric glycoprotein that is distributed in many tissues and biological fluids. There are two isoforms of ACE in humans: somatic ACE (sACE) and germinal ACE (gACE). Somatic ACE is found in many types of endothelial and epithelial cells [9]. Germinal ACE or testicular ACE is present exclusively in germinal cells in the male testis. Although ACE is a type I integral membrane protein, it can also be released as a soluble enzyme into extracellular fluids, such as plasma and seminal and cerebrospinal fluids, following post-translational proteolytic cleavage by a membrane protein sheddase or secretase [10], [11], [12].
Somatic ACE and gACE are encoded by a single gene containing 26 exons. The promoter for sACE is situated in the 5′ flanking region of the first exon, whereas that for gACE is within intron 12, which results in different lengths for the two isoforms. The longer sACE (150–180 kDa) is transcribed from exon 1 to exon 26, excluding exon 13, whereas the shorter gACE (90–110 kDa) is transcribed from exon 13 to exon 26. Exon 13 encodes a unique sequence for the N-terminus of gACE, whereas downstream exons encode a common sequence for both isozymes [13].
Somatic ACE and gACE both consist of a 28-residue hydrophilic C-terminal cytoplasmic domain, a 22-residue hydrophobic transmembrane domain that anchors the protein in the membrane and an N-terminal ectodomain (Fig. 1 ) that is heavily glycosylated with mannose, galactose, fructose, N-acetylneuraminic acid and N-acetylglucosamine [14]. The ectodomain of sACE is further divided into two similar domains (N domain and C domain) encoded by the homologous exons 4–11 and 17–24, respectively, and each domain contains an active His-Glu-X-X-His (HEXXH) sequence [9]. Somatic ACE is the only known metallopeptidase with two homologous active sites [15], which implies that there has been a gene duplication event during evolution [16]. Except for a unique sequence constituting its N-terminus, gACE is identical to the C-terminal half of sACE [17]. Due to cleavage of the membrane-bound residues by ACE secretase, soluble circulating ACE lacks a transmembrane portion and a cytosolic domain [18].
Figure 1.
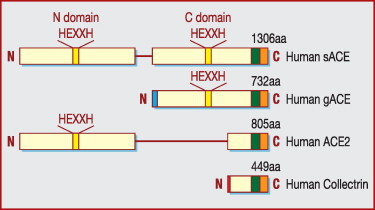
Schematic representation of the primary structures of sACE, gACE, ACE2 and collectrin [6]. ACE: angiotensin-I-converting enzyme; gACE: germinal ACE; HEXXH: His-Glu-X-X-His; sACE: somatic ACE.
The three-dimensional X-ray crystallographic structure of a deglycosylated truncated version of gACE (C domain of sACE), reveals a preponderance of α-helices with a zinc ion and two chloride ions incorporated. A deep narrow channel separates the molecule into two subdomains and the active site is located toward the bottom of this channel. An N-terminal ‘lid’ on the top of molecule appears to allow only small peptide substrates access to the active site cleft. In fact, the structure bears little similarity to that of carboxypeptidase A (M14 family) on which the initial drug development of ACE inhibitors was based. Instead, it resembles rat neurolysin (M3 family) and Pyrococcus furiosus carboxypeptidase (M32 family), despite sharing little sequence similarity with these two proteins [17]. Corradi et al. [18] reported the crystal structure of the N domain of sACE. Similarly, it has an ellipsoid shape with a central groove dividing it into two subdomains, one of which contains the N-terminal region that covers the central binding cavity. But the structure reveals differences in the active site and it contains only one chloride ion, equivalent to chloride II of gACE. The three-dimensional structures of C domains (based on gACE) and N domains provide an opportunity to design domain-selective ACE inhibitors that may exhibit new pharmacological profiles [17], [18].
Catalytic mechanism of ACE
According to the catalytic mechanism and the critical amino acid residue involved, peptidases are classified into four major types: serine, cysteine, aspartic and metallo [15]. ACE is an M2 family metallopeptidase: MA(E), the gluzincins [19]. Two histidine residues of the functional motif HEXXH and a third distant glutamate positioned 23–24 residues further towards the C-terminus are the ligands for the zinc cofactor [20]. An activated water molecule complexed to Zn2+ serves as the nucleophile to attack the carbonyl group of the targeted peptide bond [15].
The activity of ACE is also chloride dependent. Chloride primarily activates the active sites of ACE and enhances the binding of substrates [9]. Each active domain of ACE displays differences in sensitivity to chloride activation [21]. The activity of the C domain of sACE depends highly on chloride ion concentration and is inactive in its absence, whereas the N domain can be completely activated at relatively low concentrations of this anion and is still active in the absence of chloride [22], [23]. Germinal ACE depends on chloride to a lesser extent compared with the C domain of sACE [9]. Cushman and Cheung [24] reported an optimal in vitro ACE activity of rabbit rung acetone extract in the presence of 300 mM NaCl at pH 8.1–8.3.
The two active domains of sACE are also subtly different in substrate specificity. They hydrolyze bradykinin almost equally but the C domain active site can hydrolyze Ang I, substrate P [23] and Hippuryl-His-Leu [25] more efficiently, while the N domain active site preferentially hydrolyzes Ang-(1–7) [26], luteinizing hormone-releasing hormone (LH-RH) [23], the haemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro (AcSDKP) [27] and Alzheimer amyloid β-peptide (Aβ) [28]. Fuchs et al. [29] proved that the C-terminal catalytic domain was the main site of Ang I cleavage in mice. The differentiation of catalytic specificity might be due to very subtle variation in substrate-specific amino acids [22] and chloride-induced conformational alteration of active sites [23].
ACE acts as an exopeptidase to cleave dipeptides from the free C-termini of two typical substrates, Ang I and bradykinin. For certain substrates such as cholecystokinin [30], substrate P [31] and LH-RH [32], which have amidated C-termini, ACE not only displays exopeptidase activity but also acts as an endopeptidase [33]. The most prominent example of endopeptidase activity is ACE hydrolyzing the synthetic Aβ-(1–40) peptide into four fragments: an Aβ-(8–40) peptide and the others corresponding to products of Aβ-(1–7) hydrolysis [34]. Thus, ACE might have a more general impact on the metabolism of biologically active peptides than previously recognized [16]. The two substrates used most often for measuring ACE activity and inhibition in vitro – Hippuryl-His-Leu and N-[3-(2-furyl)acryloyl]-L-phenylalanylglycylglycine (FAPGG) – only have the N-termini blocked and substrates with two termini blocked have been developed [22].
Biological impact of ACE
ACE was originally isolated in 1956 as a ‘hypertensin-converting enzyme’ [35]. In the RAS, ACE cleaves the decapeptide Ang I-(1–10) (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu) into the octapeptide Ang II-(1–8) by removing the C-terminal dipeptide His-Leu. When the RAS is overactive, Ang II exerts its harmful effects primarily via the AT1 receptor, whereas the AT2 receptor may oppose and counterbalance those effects mediated by AT1 receptor to exert protective actions [1]. Ang II is a potent vasoconstrictor, stimulates the release of aldosterone and antidiuretic hormone or vasopressin and increases the retention of sodium and water. These effects act directly in concert to raise blood pressure. A nonapeptide derivative of Ang I, des-Asp1-Ang I-(2–10), prevents infarction-related and non-infarction-related cardiac injuries and disorders. His-Leu can be cleaved from the peptide by ACE to produce Ang III-(2–8) [14], which has 40% of the vasoconstriction activity of Ang II. Ang III exerts its effects, in principle, in a similar manner to Ang II, and may be equally or even more important in mediating the release of vasopressin [1]. ACE also degrades Ang-(1–9) to Ang-(1–7) then further degrades this peptide to the inactive Ang-(1–5) (Fig. 2 ). In addition, ACE (also termed kininase II) inactivates the vasodilators bradykinin-(1–9) (Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg) and kallidin (Lys-bradykinin) in the kallikrein-kinin system, by cleaving the C-terminal dipeptide Phe-Arg. ACE eventually cleaves its primary metabolite bradykinin-(1–7) into the shorter fragment bradykinin-(1–5) [36].
Figure 2.
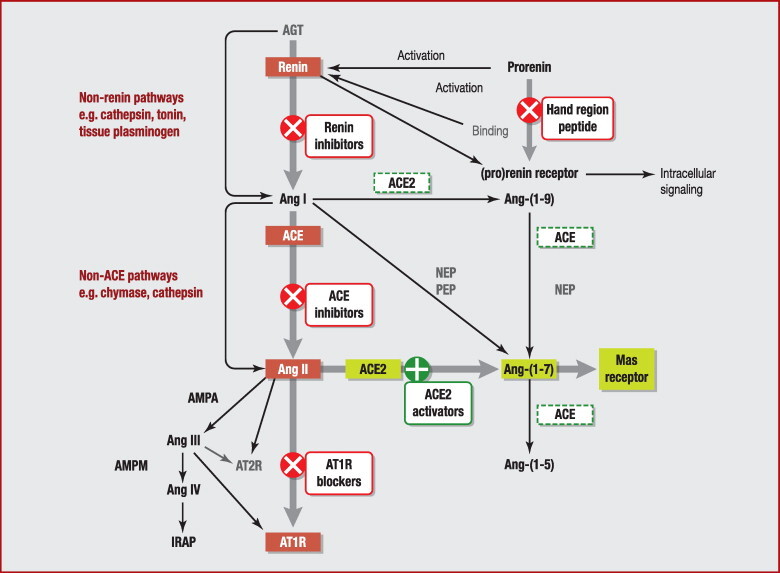
Multiple effector systems of the renin-angiotensin system. ACE: angiotensin-I-converting enzyme; ACE2: angiotensin-I-converting enzyme 2; AGT: angiotensinogen; AMPA: aminopeptidase A; AMPM: aminopeptidase A; Ang I: angiotensin I; Ang II: angiotensin II; AT1R: angiotensin II receptor, type 1; AT2R: angiotensin II receptor, type 2; IRAP: insulin-regulated aminopeptidase; NEP: neutral endopeptidase; PEP: prolyl endopeptidase.
Through Ang II and aldosterone, ACE may also be implicated in the impairment of nitric oxide bioavailability and cell oxidative stress, augmenting the generation of reactive oxygen species and peroxynitrite [37], [38]. With the ability to hydrolyze neuropeptides such as enkephalin [39], [40], substrate P, neurotensin [31] and LH-RH, ACE may be involved in the functioning of the brain and nervous system. ACE may affect the digestive system by hydrolyzing the peptide hormone cholecystokinin and gastrin [30]. The in vivo experiment conducted by Azizi et al. [41] proved that acute ACE inhibition could increase the level of the natural stem cell regulator AcSDKP in plasma. AcSDKP substantially inhibits cell cycle entry of normal haematopoietic stem cells and protects haemopoiesis against damage caused by cycle-active cytotoxic agents [42]. AcSDKP can also inhibit the proliferation of hepatocytes [43] and lymphocytes [44] and stimulate angiogenesis [45] in vivo. The in vivo antifibrotic effect of ACE inhibition is partially mediated by AcSDKP [46]. ACE may also affect susceptibility to Alzheimer's disease by degrading Aβ and preventing the accumulation of amyloid plaques in vivo [47]. In the brains of amyloid precursor protein Swedish mutation transgenic mice, ACE converts Aβ1–42 to Aβ1–40 and degrades Aβ, and chronic inhibition of ACE with captopril enhances predominant Aβ1–42 deposition [48]. However, through the inhibition of brain ACE activity in the Aβ25–35-injected mice, perindopril ameliorates cognitive impairment and may therefore have a beneficial effect on Alzheimer's disease as well as hypertension [49].
ACE inhibitors
Using the assumed mechanistic analogy to other zinc metallopeptidases, plus the knowledge that several snake-venom peptides potentiate the action of bradykinin by inhibiting ACE, efforts were undertaken to develop orally-active peptide analogues for potential use in the treatment of hypertension [9]. The first such compound, captopril or D-3-mercapto-2-methylpropanoyl-L-proline, is an analogue of the Ala-Pro sequence, with sulphydryl as a strong chelating group for the zinc ion. Its adverse effects, which were the same as those caused by mercapto-containing penicillamine, prompted the design of non-sulphydryl ACE inhibitors [50]. The results were two active inhibitors: enalaprilat and lisinopril. They are both essentially tripeptide analogues with a zinc-co-ordinating carboxyl group and a phenylalanine that occupies the S1 groove in the enzyme. Lisinopril is a lysine analogue of enalaprilat but it is hydrophilic, with greater affinity than enalaprilat. The later compounds are all variations of the first three inhibitors, with most of the differences residing in the functionalities that bind the active site zinc and the S2′ pocket. In addition to phosphonates, ketones are also useful as chelators [51].
Currently, there are more than 10 ACE inhibitors marketed that are widely used as first-line therapy for cardiovascular diseases, including hypertension, heart failure, heart attack and left ventricular dysfunction. According to the functional moiety, they are divided into three types: thiol (captopril), carboxylate (benazepril, enalapril, lisinopril, moexipril, perindopril, quinapril, ramipril, trandolapril) or phosphate (fosinopril). Some ACE inhibitors are now administrated clinically as ethyl-ester prodrugs, which have good bioavailability but are inactive in their own right. They are then converted to the active diacid molecules in vivo by esterases.
As a drug class, ACE inhibitors are very effective, have a relatively low incidence of side effects and are well tolerated. A common side effect of ACE inhibitors is a dry cough, which appears in 5–20% of patients and may result in the discontinuation of treatment. Another serious problem is angioedema, which affects 0.1–0.5% of patients and can be life-threatening. The two side effects have generally been attributed to altered concentrations of bradykinin [51]. Use of ACE inhibitors during the second and third trimesters of pregnancy is contraindicated because of their association with an increased risk of foetopathy, a group of conditions that includes oligohydramnios, intrauterine growth retardation, hypocalvaria, renal dysplasia, anuria, renal failure and death. Exposure to ACE inhibitors during the first trimester of pregnancy may place the infant at increased risk for major congenital malformations [52].
The initial drug development of clinical ACE inhibitors was based on the assumption of an active site related to that of carboxypeptidase A but organized to remove a dipeptide rather than a single amino acid from the C-terminus of its substrate. It is now known that sACE has two active sites, neither of which resembles that of carboxypeptidase A, and that these sites are not identical. Clinical ACE inhibitors, however, show little discrimination between these two active sites [9]. Ang I is hydrolyzed predominantly by the C domain of sACE in vivo [29] but bradykinin is hydrolyzed by both active sites [53]; therefore a C domain-selective inhibitor would allow some degradation of bradykinin by the N domain and this degradation could be enough to prevent the accumulation of excess bradykinin that has been observed during attacks of angioedema. That is, the C domain-selective inhibition could possibly result in specialized control of blood pressure with fewer vasodilator-related adverse effects [51]. A structure-activity study has proved that the group substitution involving the phenyl ring and carbon chain at the sulphonyl and propionyl moieties of captopril is essential for better activity towards the C domain of ACE [54]. There is increasing evidence that the N domain of sACE is responsible for the in vivo degradation of the natural haemoregulatory hormone AcSDKP [27], [41], [55]. So, N domain-selective inhibition might open up novel therapeutic areas. Two phosphinic tetrapeptides, RXPA380 and RXP407, have been found to be highly selective inhibitors of the C and N domains of sACE, respectively [51]. The availability of the three-dimensional structures of the C and N domains of sACE makes the structure-based design of active site-specific inhibitors possible [17], [18].
ACE2
ACE2 (EC 3.4.17.-) is also a type I transmembrane glycoprotein and its expression has now been recognized as being ubiquitous. It appears to be susceptible to cleavage that releases the catalytical active ectodomain. A disintegrin and metalloprotease 17 (ADAM17; also known as tumour necrosis factor-alpha cleavage enzyme [TACE]) is a major protease for soluble ACE2 shedding, while phorbol ester, ionomycin, endotoxin and the proinflammatory cytokines interleukin-1β and tumour necrosis factor-alpha can also acutely induce ectodomain release [56]. Calmodulin binding sites have been identified in the cytoplasmic tail of ACE2 and calmodulin inhibitors are proved to increase ACE2 shedding [57]. The cleavage site resides between amino acids 716 and 741 near the predicted transmembrane domain and residue Lys548 in the ectodomain plays an important role in dictating ACE2 ectodomain shedding [58]. ACE2 has 805 amino acids encoded from 18 exons and shares about 42% sequence identity with the N and C domains of sACE [16]. ACE2 also belongs to the M2 family of metalloproteases but consists of a single active site domain that, by sequence comparison, more closely resembles the N domain than the C domain of sACE (Fig. 1) [11]. In addition to the conserved zinc metallopeptidase consensus sequence HEXXH (amino acids 374–378), there is a conserved glutamate residue 24 residues C-terminal to the second histidine of the zinc motif, which serves as the third zinc ligand [16]. The three-dimensional structure of a truncated extracellular region of human ACE2 shows that the active site domain (residues 19–611) can be further divided into two subdomains I and II, which form two sides of a long deep cleft and are connected only at the floor of the active site cleft by a prominent α-helix. The deeply recessed and shielded proteolytic active site of ACE2 is a common structural feature of proteases and can avoid hydrolysis of correctly folded and functional proteins. The zinc is co-ordinated by His374, His378, Glu402 and one water molecule in the subdomain I near the bottom, whereas a chloride ion is co-ordinated by Arg169, Trp477 and Lys481 in the subdomain II [58]. The ACE2 transmembrane C-terminal domain shares 48% sequence identity with collectrin (Fig. 1), a non-catalytic protein shown to have a critical role in amino acid reabsorption in the kidney [59], pancreatic beta cell proliferation and possibly insulin exocytosis [6].
Unlike sACE and gACE, which are primarily dipeptidylcarboxypeptidases, ACE2 functions predominantly as a monocarboxypeptidase, with a substrate preference for hydrolysis between proline and a hydrophobic or basic C-terminal residue [6]. It is like carboxypeptidase A in its action model but is different in active structure because the latter has an HXXE zinc-binding motif with the third ligand (histidine) positioned 108–135 residues further towards the C-terminus [16]. ACE2 efficiently cleaves a single residue phenylalanine from Ang II to generate Ang-(1–7), with about 400-fold higher catalytic efficiency than the conversion of Ang I to Ang-(1–9) by removing the C-terminal leucine residue (Fig. 2). Other substrates with high catalytic efficiency are apelin-13, dynorphin A-(1–13) and des-Arg9-bradykinin [60]. Arg514 of ACE2 has been identified as a residue critical to substrate selectivity [61]. ACE2 activity is also regulated by chloride ions; it has been proposed that chloride binding induces subtle changes in the conformation of the active site, which either facilitate or hinder substrate binding [6]. For the substrate Ang I, ACE2 activity increases with increased [Cl−] and a plateau is reached at approximately 500 mM Cl−. For the substrate Ang II, an increase in ACE2 activity is observed as [Cl−] increases from 0–100 mM but any further increase in [Cl−] decreases ACE2 activity until a plateau is reached at 500 mM Cl−; ACE2 activity at 500 mM Cl− is even lower than that in the absence of Cl− (0 mM). Consequently, an increase in [Cl−] above 100 mM, which is the physiological concentration in human plasma, increases Ang I and decreases Ang II cleavage by ACE2. This has an effect on the localized concentration of Ang II in the kidney, where ACE2 has a high level of expression and extracellular chloride ion levels fluctuate. Thus, in vivo Cl− sensitivity may serve as a homeostatic regulatory mechanism [61]. ACE2 activity is unaffected by inhibitors of ACE (captopril, lisinopril and enalaprilat) or carboxypeptidase A (benzylsuccinate and potato carboxypeptidase inhibitor) [16].
Biological actions of ACE2
The major function of ACE2 is to counter-regulate ACE activity by reducing Ang II bioavailability and increasing Ang-(1–7) formation. As a result, ACE2 plays a crucial role in maintaining the balance between the two axes ACE2/Ang-(1–7)/Mas and ACE/Ang II/AT1 of the RAS; a chronic and sustained imbalance may lead to pathophysiology of the cardiovascular, renal, pulmonary and central nervous systems [62]. Studies have shown that ACE2 overexpression and recombinant ACE2 treatment can attenuate hypertension in animal models [63], [64], while in humans, there is a strong association between ACE2 polymorphisms and hypertension in Han Chinese [65]. In addition to the Ang II system, ACE2 may regulate blood pressure through other peptide systems, such as bradykinin and/or apelin [6]. ACE2 gene delivery [66] and ACE-(1–7) infusion [67] also have beneficial effects on atherosclerosis, whereas ACE2 deficiency accentuates vascular atherosclerosis and inflammation [67] in animal models. Regarding heart function, ACE2 null mice display impaired cardiac contractility [68] and the loss of ACE2 in wild-type mice accelerates adverse ventricular remodelling by potentiation of Ang II effects by means of the AT1 receptors [69]. Ang-(1–7), through interaction with the Mas receptor, can improve atrial tachyarrhythmias [70], myocardial performance, cardiac modelling and survival [3], [6] in rodent heart failure models. In humans, ACE2 gene variants might be involved in modulation of left ventricular mass in men [71] and soluble ACE2 activity is increased in patients with heart failure and correlates with disease severity to exert cardioprotective actions [72]. Deletion of the ACE2 gene in mice leads to the development of glomerulosclerosis and increased albuminuria [73], while treatment with the ACE2 inhibitor MLN4760 can worsen renal damage in streptozotocin-induced diabetic mice [74]. Chronic treatment with Ang-(1–7) improves renal endothelial dysfunction via the Mas receptor in apolipoprotein E-deficient mice, by increasing levels of endogenous nitric oxide [75], whereas genetic deletion of the Mas receptor in mice leads to a reduction in urine volume, sodium retention, microalbuminuria and reduced renal blood flow, which are associated with upregulation of the AT1 receptor and transforming growth factor-beta messenger ribonucleic acid [76]. This evidence indicates the protective role of ACE2/Ang-(1–7)/Mas in renal function. ACE2 has also been shown to regulate cardiovascular functions in brain regions. Overexpression of ACE2 in the rostral ventrolateral medulla reduces high blood pressure and heart rate in spontaneously hypertensive rats (SHRs) [77]. In the nucleus tractus solitarius of SHRs, ACE2 gene transfer improves baroreceptor heart rate reflex [78]. In the mouse subfornical organ, ACE2 overexpression inhibits AT1 receptor expression and prevents Ang II-mediated pressor and drinking responses [79]. In addition, ACE2/Ang-(1–7)/Mas can exert cerebroprotective functions in endothelin-1-induced ischaemic stroke in rodent models [80]. ACE2 exerts a host of actions on the cardiopulmonary system, which include prevention of endothelial dysfunction, reduction in pulmonary oxidative stress, attenuation of vascular impairment, anti-inflammatory effects and anticardiac remodelling effects. All these properties are responsible for the protective role of ACE2 against pulmonary arterial hypertension [81].
Structure-based drug screening has identified two ACE2 activators: a xanthenone (1-[(2-diethylamino)ethyl-amino]-4-(hydroxymethyl)-7-[(4-methylphenyl)sulphonyloxy]-9H-xanthene-9-one; XNT) and resorcinolnaphthalein. XNT hydrogen bonds with ACE2 residues Lys94, Tyr196, Gly205 and His 195, and resorcinolnaphthalein is involved in three hydrogen bonds with residues Gln98, Gln101 and Gly205. XNT and resorcinolnaphthalein modulate ACE2 activity possibly by two mechanisms. Logically, the closed conformation of the enzyme cannot allow the substrate into its active site. In the presence of compound, the free enzyme may be shifted to the open form, effectively increasing the activity coefficient of the enzyme. Alternatively, product release is a rate-limiting step in ACE2 turnover. ACE2 activity may be enhanced in the presence of compound, as the enzyme-product complex empties more quickly and ACE2 becomes available to start another cycle. XNT and resorcinolnaphthalein enhance in vitro ACE2 activity in a dose-dependent manner and show no significant effects on ACE activity. Administration of XNT to SHRs can result in a decrease in blood pressure, improvements in cardiac function and reversal of myocardial, perivascular and renal fibrosis [82]. The protective role of XNT against hypertension-induced cardiac fibrosis is associated with activation of ACE2, increases in Ang-(1–7) and inhibition of extracellular signal-regulated kinases [83]. Furthermore, ACE2 activation by XNT attenuates thrombus formation and reduces platelet attachment to vessels [84]. XNT also prevents pulmonary vascular remodelling and right ventricular hypertrophy and fibrosis in a rat model of monocrotaline-induced pulmonary hypertension [85]. Development of XNT is being pursued to translate the vasoprotective concept of ACE2 into an effective cardiovascular therapy [62]. Diminazine aceturate, a small molecule antiprotozoal agent that shares structural similarity with XNT, has recently been demonstrated to be a potent activator of ACE2 and to decrease mean arterial pressure and myocardial fibrosis in SHRs [86]. Intracerebroventricular infusion of diminazine aceturate, prior to and following endothelin-1-induced middle cerebral artery occlusion, significantly attenuates cerebral infarct size and neurological deficits in a rat model [80]. These data suggest that diminazine aceturate may serve as a lead compound for the discovery of the next generation of drugs for the treatment of cardiovascular disease and hypertension [86].
Interestingly, peptidase-independent actions of ACE2 have also been elucidated. ACE2 has been identified as an essential receptor for the coronavirus (CoV) that causes severe acute respiratory syndrome (SARS). The spike protein of SARS-CoV attaches the virus to its cellular receptor ACE2 with a binding domain on the spike protein mediating the interaction [87]. This ACE2-spike interaction leads to endocytosis of virus particles through internalization with ACE2, induces the fusion of virus with host cells and establishes SARS-CoV infection with the release of viral RNAs into cytoplasm [6]. The spike protein of SARS-CoV also induces TACE-dependent shedding of the ACE2 ectodomain, with the involvement of the ACE2 cytoplasmic domain. Cellular TACE and the ACE2 cytoplasmic tail promote viral entry and infection. These observations indicate that ACE2 shedding and its causative cellular signals may be attributable to SARS-CoV-induced tissue damage [88]. An ACE2 inhibitor, N-(2-aminoethyl)-1 aziridine-ethanamine, has been identified as being effective in blocking SARS-CoV spike protein-mediated cell fusion [89]. The SARS-CoV receptor function of ACE2 is independent of its catalytic activities for Ang II degradation but ACE2-mediated Ang II degradation is still important for lung protection from SARS pathogenesis [6], [90]. So, the consequence of long-term activation of ACE2 must be determined and the effects of ACE2 activators on the immune competence of animals and their vulnerability to SARS-CoV infection must be tested before these molecules are ready for preclinical trials [64]. ACE2 is also the receptor for the human coronavirus NL63 that was discovered in 2004 in the Netherlands and was shown to be globally distributed [91]. Additionally, due to the high homology with collectrin at its transmembrane, ACE2 binds to B0AT1-family amino acid transporters and contributes to the absorption of neutral amino acids in animal intestines [6], [92]. Thus, ACE2 may be involved in multiple biological functions.
Renin, prorenin and the (pro)renin receptor
Renin (EC 3.4.23.15) is a key enzyme of the RAS. Since its discovery 100 years ago, an impressive quantity of information about renin has been compiled. It is generally accepted that the active renin found in the circulation of mammals almost exclusively originates from the kidney. In addition to systemic renin, there are a number of extrarenal tissues that express renin as part of the local or tissue-specific RASs. Within the kidney, renin is predominantly produced by the juxtaglomerular cells. (Pro)renin gene transcription in these cells is controlled through several mechanisms, among which cyclic adenosine monophosphate (cAMP)/protein kinase A/cAMP response element binding protein (stimulatory) and calcium/protein kinase C (inhibitory) cascades are employed by physiological cues, whereas signal transducers, activators of transcription and nuclear factor κB transcription factors (inhibitory) and members of the nuclear receptor superfamily probably become relevant in pathological situations. Prorenin is stored in vesicles, activated to renin and then released upon demand. The release of renin is under the control of the intracellular cAMP (stimulatory), Ca2+ (inhibitory) and cyclic guanosine monophosphate signalling pathways. Meanwhile, a great number of intrarenally generated or systemically acting factors have been identified that control renin secretion directly at the level of renin-producing cells, by activating the cAMP or Ca2+ signalling pathways [93]. Renin is an aspartyl protease with a typical structure made of two lobes. The cleft between the lobes contains the active site, characterized by two catalytic aspartic residues. Renin is a highly specific enzyme and has only one known substrate (AGT) [94].
Prorenin is the ‘inactive’ precursor of renin. In addition to the main organ, the kidney, other tissues, such as the brain, the adrenal gland, the submandibular gland, the glands of the reproductive system and adipose tissue, are also able to secrete prorenin in the surrounding milieu and in plasma. In contrast to renin, prorenin is released constitutively; renin and prorenin levels are usually well correlated. Under some physiopathological circumstances, such as pregnancy and diabetes, prorenin levels exceed by far those of renin [94]. Prorenin has a prosegment of 43 amino acid residues attached to the N-terminus of mature renin; the prosegment folds into an active site cleft of mature renin to prevent catalytic interaction with AGT [95]. Prorenin-renin conversion occurs in the kidney before renin is released from the juxtaglomerular cells. Several enzymes, including proconvertase 1 and cathepsin B, have been proposed to be responsible for this irreversible cleavage of the prosegment from prorenin to form mature renin. A (reversible) non-proteolytic activation of prorenin may also occur. At acidic pH and/or low temperature, prorenin is capable of undergoing a conformational change, involving the unfolding of the prosegment from the enzymatic cleft. This non-proteolytically-activated prorenin is fully enzymatically active [96]. Under physiological conditions, approximately 2% of plasma prorenin is in the open form and can also display enzymatic activity [94].
The traditional assumptions of renin being just an enzyme responsible for the cleavage of AGT and prorenin being just an ‘inactive’ proenzyme were challenged by the cloning of a human (P)RR in 2002 [94]. The (P)RR is a single transmembrane-domain protein of 350 amino acids, with a large unglycosylated and highly hydrophobic N-terminal domain responsible for renin and prorenin binding and a short cytoplasmic tail of about 20 amino acids involved in intracellular signalling [97]. The (P)RR was first identified on cultured human mesangial cells and bound well to renin and prorenin with a K D in the nanomolar range [96]. Binding of prorenin induced a conformational change in the molecule, increasing its enzymatic activity from virtually zero to values similar to those of active renin in solution without proteolytic removal of the prosegment [93], [98]. Two regions in the human prorenin segment, namely T7PFKR10P (a gate that is not accessible by its specific antibodies until it is loosened from the active site cleft) and I11PFLKR15P (a handle, a protruding pentameric segment), have been identified as being crucial for the non-proteolytic activation [99]. Renin bound to the (P)RR displays a 3- to 5-fold increase in the catalytic efficiency of AGT conversion to Ang I compared with free renin [97], [100]. In addition to the Ang-dependent function on the cell surface related to the increased catalytic activity of receptor-bound (pro)renin (renin activity of activated prorenin and increased activity of renin) that leads to the formation of Ang I from AGT, the (P)RR system also has Ang-independent intracellular effects that are not necessarily related to the RAS [7]. Pathological conditions like high blood pressure upregulate the receptor, whereas elevated (pro)renin concentrations downregulate the receptor via translocation of the transcription factor promyelotic zinc finger protein to the nucleus [98]. In the Ang-independent signalling pathways, prorenin-induced activation of mitogen-activated protein kinase p38 and subsequent phosphorylation of heat shock protein 27 result in actin polymerization, while (pro)renin-induced activation of the extracellular signal-regulated kinase 1/2 (p44/p42) signalling cascade leads to the intracellular expression of profibrotic genes, giving rise to the synthesis of transforming growth factor-β, plasminogen activator inhibitor-1, collagen 1 and fibronectin. These effects potentially increase cardiac and renal hypertrophy and fibrosis [97], [101], [102], [103]. The discovery of the (P)RR has confirmed the hypothesis that renin is also a hormone. It has been suggested that blocking the (P)RR may be a new target for renal and cardiac end-organ protection [1]. Additionally, the mannose-6-phosphate/insulin-like growth factor II receptor can bind both renin and prorenin with high affinity and is believed to serve as a clearance receptor for renin/prorenin. An intracellular renin-binding protein has been also cloned and found to be an inhibitor of renin activity but its deletion affected neither blood pressure nor plasma renin [94].
(Pro)renin receptor antagonist and renin inhibitors
After the discovery of the receptor, a (P)RR antagonist was designed, based on the idea that the prosegment contains a handle region that binds to the receptor, allowing prorenin to become catalytically active in a non-proteolytic manner [96]. The antagonist (also known as hand region peptide, HRP), consisting of 10 amino acids (NH2-RILLKKMPSV-COOH), resembles the handle region (I11PLLKK15P) of rat prorenin prosegment and will thus competitively bind to the (P)RR as a decoy peptide and inhibit the receptor-mediated activation of prorenin [95]. Treatment with the HRP in diabetic mice and rats decreased the renal content of Ang I and II and inhibited the development of nephropathy without affecting hyperglycaemia [104], [105]. In stroke-prone SHRs, continuous subcutaneous administration of the HRP inhibited activation of the tissue RAS without affecting the circulating RAS or arterial pressure and significantly attenuated the development and progression of proteinuria, glomerulosclerosis and cardiac fibrosis [106], [107]. Infusion of the HRP in human (P)RR transgenic rats significantly inhibited the development of glomerulosclerosis, proteinuria, mitogen-activated protein kinase activation and transforming growth factor-β expression in the kidneys [108]. Future research should be able to determine to what degree the beneficial in vivo effects of HRP are due to prorenin blockade [98] and are related to interference with renin binding and the RAS [4]. Decoy effects of the HRP, however, were not confirmed by other research groups [109], [110], [111], [112].
ACE inhibitors and AT1 receptor blockers (ARBs) are proven to be effective therapeutic agents in the treatment of CVD. However, both ACE inhibitors and ARBs lead to a substantial compensatory rise in circulating active renin and Ang peptides that may eventually limit their therapeutic potential [113]. Moreover, the increased Ang I seen with ACE inhibitors can subsequently be converted to Ang II by non-ACE pathways, mediated by chymase and chymotrypsin-like Ang-generating enzyme [114]. In addition to the side effects of ACE inhibitors, such as cough and angioedema, a meta-analysis of randomized controlled trials in 2010 suggested that ARBs are associated with a modestly increased risk of new cancer diagnosis, although conclusions about the exact risk of cancer associated with each particular drug have not been drawn [115]. Based on this study, the United States Food and Drug Administration is conducting a review to assess the possible link between the use of ARBs and cancer. Therefore, direct renin inhibition may be an alternative pharmacological approach to RAS inhibition.
Renin is the rate-limiting step of the RAS and the concept of blocking the RAS at its origin by inhibiting renin has existed for at least 50 years [113]. The first-generation renin inhibitors were peptide analogues of AGT and the second-generation compounds were peptidomimetic agents that are dipeptide transition-state analogue inhibitors of the active site. But these renin inhibitors had limited clinical use due to poor metabolic stability and oral bioavailability, short duration of action, weak antihypertensive activity and high cost of synthesis [114]. A combination of molecular modelling and X-ray crystallographic analysis of the active site of renin led to the development of aliskiren, a new non-peptide low-molecular-weight orally-active renin inhibitor. Aliskiren has a high binding affinity for renin and appears to bind to both the hydrophobic S1-/S3-binding pocket and a large, distinct subpocket that extends from the S3-binding site toward the hydrophobic core of the enzyme. Aliskiren is a potent competitive inhibitor of renin but very poorly inhibits related aspartic peptidases. It shows no effects on cytochrome P450 isoenzyme activities and is not bound extensively to blood proteins, therefore having a low potential for drug interactions [113]. Compared with the earlier renin inhibitors, aliskiren is more resistant to intestinal degradation and possesses significantly improved oral bioavailability. The terminal half-life ranges from 23 to 40 hours, which makes it suitable for once-daily dosing. Aliskiren monotherapy displays an antihypertensive efficacy similar if not superior to that of other first-line antihypertensive drugs, with a safety and tolerability profile similar to that of ARBs. Combined with various antihypertensive agents, aliskiren exhibits synergistic effects [114]. From 2007, aliskiren was approved by regulatory bodies in both Europe and the United States, for use alone and with other agents in the treatment of hypertension [113]. Recent studies showed that aliskiren treatment also markedly increased the plasma concentration of (pro)renin in patients and failed to inhibit the non-catalytic effects of (pro)renin [110], [116]. The pharmaceutical company Novartis reported that the addition of aliskiren to standard therapy for patients recovering from a heart attack did not provide the anticipated effect of limiting adverse changes to the heart's left ventricle. In December 2011, Novartis announced the early termination of the Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE) and advised that aliskiren should not be used in combination with ACE inhibitors or ARBs in patients with diabetes. The study was conducted in type 2 diabetic patients at high risk of fatal and non-fatal cardiovascular and renal events; aliskiren 300 mg was given in addition to an ACE inhibitor or an ARB. The overseeing Data Monitoring Committee concluded that study patients were unlikely to benefit from aliskiren and that there was a higher incidence of adverse events related to non-fatal stroke, renal complications, hyperkalaemia and hypotension in this high-risk population after 18–24 months of combined therapy (www.novartis.com). Aliskiren has been shown to be a very safe antihypertensive after more than 20,000 patient-years of data. However, as we have seen in Ongoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial (ONTARGET) that the combination of telmisartan (an ARB) and ramipril (an ACE inhibitor) is associated with more adverse events without an increase in benefit in patients with vascular disease or high-risk diabetes without heart failure [117], the combination of multiple safe drugs in this class may no longer be a promising strategy.
Future directions of bioactives from natural plant and/or food resources for RAS regulation
Over the last decade, a number of food-derived compounds have been shown to have in vitro ACE inhibitory activity. Some of them display significant antihypertensive activity in rats and humans. Among these compounds, food-derived ACE inhibitory peptides have been most widely studied [14], [118]. A quantitative structure-activity relationship study indicated that a potent ACE inhibitory dipeptide should have a large and hydrophobic amino acid at the C-terminus and a non-polar amino acid or possibly a positively charged amino acid at the N-terminus. For tripeptides, the most favourable residues for the C-terminus are aromatic amino acids, while hydrophobic amino acids are preferred for the N-terminus and positively charged amino acids are preferred for the middle position [119]. The in vitro half-maximal inhibitory concentration (IC50) values of food-derived ACE inhibitory peptides are about 1000-fold higher than that of synthetic captopril but they have higher in vivo activities than would be expected from their in vitro activities. It has been suggested that food-derived peptides might act via different antihypertensive mechanisms, possess higher affinities for tissues and be more slowly eliminated than synthetic captopril [118]. Until now, only two lactotripeptides (VPP and IPP) have been successfully commercialized. Data from van Mierlo et al., however, did not support a blood pressure-lowering effect of the two tripeptides in Caucasians [120]. Given the discrepancy between in vivo and in vitro results, further investigation into the in vivo and clinical antihypertensive effects and mechanisms of food-derived ACE inhibitors is necessary. Three dipeptides (IR, KF and EF) in pea protein hydrolate have been identified as inhibiting renin activity in vitro [121]. Oleic acid and linoleic acid isolated from rice and some cereals also have in vitro renin inhibitory activity [122], [123]. A series of studies have shown that saponins from different food/plant sources, primarily from soybean, can inhibit in vitro renin activity [124], [125], [126], [127]; among them, orally administered soybean saponin can lead to a reduction of blood pressure in SHRs but the direct evidence for saponin suppressing plasma renin activity in vivo is still lacking [124]. Experiments are required to further investigate the biochemical and biological properties of these plant/food-derived non-peptides that are related to renin inhibition. Significant conceptual progress made in the last few years leads us to conclude that ACE2 could serve as a new direction for improved therapeutics for cardiovascular disease. Compared with ACE inhibitor therapy, ACE2 is an endogenous regulator of the RAS. Targeting ACE2 would not only produce the vasoprotective/antiproliferative peptide Ang-(1–7) but would also influence the vasoconstrictive/proliferative effects of the ACE/Ang II/AT1 axis. As a part of the vasoprotective/antiproliferative axis, ACE2 can effectively control fibrosis and structural remodelling and is extremely beneficial for pulmonary hypertension. ACE2 activation may provide improved protection and reversal of ischaemia-induced neural damage. Thus, direct activation of ACE2 could result in better outcomes in cardiovascular disease. Additionally, ACE2 is a multifunctional enzyme with many biologically active substrates. The effects of ACE2 on substrates other than Ang I and Ang II may hold relevance for the treatment of cardiovascular disease [62]. XNT analogues exist in many natural plant resources and have shown various physiological functions. Using these resources to search for in vitro ACE2 activators would be a good starting point for developing plant food-derived ACE2 activating agents. Ongoing experiments would characterize these ACE2 activators. Compared with RAS regulatory drugs, these plant/food-derived bioactives appear more natural and safer to the consumer. As part of a daily diet, bioactives from food sources may result in a much lower healthcare cost. Plant/food-derived RAS regulators could be applied in the prevention of hypertension and as initial treatment in mildly hypertensive individuals [118].
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
This work was supported partly by Fundamental Research Funds for the Central Universities (JUSRP211A06) and the Self-Determined Research Program of Jiangnan University (No. 5815210232110800 & No. 5815210372100530).
References
- 1.Fyhrquist F., Saijonmaa O. Renin-angiotensin system revisited. J Intern Med. 2008;264:224–236. doi: 10.1111/j.1365-2796.2008.01981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen G., Contrepas A. Physiology and pharmacology of the (pro)renin receptor. Curr Opin Pharmacol. 2008;8:127–132. doi: 10.1016/j.coph.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg B. An ACE in the hole alternative pathways of the renin angiotensin system and their potential role in cardiac remodeling. J Am Coll Cardiol. 2008;52:755–757. doi: 10.1016/j.jacc.2008.04.059. [DOI] [PubMed] [Google Scholar]
- 4.Donoghue M., Hsieh F., Baronas E., et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 5.Tipnis S.R., Hooper N.M., Hyde R., et al. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 6.Kuba K., Imai Y., Ohto-Nakanishi T., et al. Trilogy of ACE2: a peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther. 2010;128:119–128. doi: 10.1016/j.pharmthera.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen G., Danser A.H. Prorenin and (pro)renin receptor: a review of available data from in vitro studies and experimental models in rodents. Exp Physiol. 2008;93:557–563. doi: 10.1113/expphysiol.2007.040030. [DOI] [PubMed] [Google Scholar]
- 8.Lazartigues E. A map and new directions for the (pro)renin receptor in the brain: focus on “A role of the (pro)renin receptor in neuronal cell differentiation”. Am J Physiol Regul Integr Comp Physiol. 2009;297:R248–R249. doi: 10.1152/ajpregu.00287.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riordan J.F. Angiotensin-I-converting enzyme and its relatives. Genome Biol. 2003;4:225. doi: 10.1186/gb-2003-4-8-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balyasnikova I.V., Karran E.H., Albrecht R.F., 2nd., et al. Epitope-specific antibody-induced cleavage of angiotensin-converting enzyme from the cell surface. Biochem J. 2002;362:585–595. doi: 10.1042/0264-6021:3620585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hooper N.M., Turner A.J. An ACE structure. Nat Struct Biol. 2003;10:155–157. doi: 10.1038/nsb0303-155. [DOI] [PubMed] [Google Scholar]
- 12.Parkin E.T., Turner A.J., Hooper N.M. Secretase-mediated cell surface shedding of the angiotensin-converting enzyme. Protein Pept Lett. 2004;11:423–432. doi: 10.2174/0929866043406544. [DOI] [PubMed] [Google Scholar]
- 13.Hubert C., Houot A.M., Corvol P., et al. Structure of the angiotensin I-converting enzyme gene. Two alternate promoters correspond to evolutionary steps of a duplicated gene. J Biol Chem. 1991;266:15377–15383. [PubMed] [Google Scholar]
- 14.Murray B.A., FitzGerald R.J. Angiotensin converting enzyme inhibitory peptides derived from food proteins: biochemistry, bioactivity and production. Curr Pharm Des. 2007;13:773–791. doi: 10.2174/138161207780363068. [DOI] [PubMed] [Google Scholar]
- 15.Lew R.A. The zinc metallopeptidase family: new faces, new functions. Protein Pept Lett. 2004;11:407–414. doi: 10.2174/0929866043406481. [DOI] [PubMed] [Google Scholar]
- 16.Turner A.J., Hooper N.M. The angiotensin-converting enzyme gene family: genomics and pharmacology. Trends Pharmacol Sci. 2002;23:177–183. doi: 10.1016/s0165-6147(00)01994-5. [DOI] [PubMed] [Google Scholar]
- 17.Natesh R., Schwager S.L., Sturrock E.D., et al. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature. 2003;421:551–554. doi: 10.1038/nature01370. [DOI] [PubMed] [Google Scholar]
- 18.Corradi H.R., Schwager S.L., Nchinda A.T., et al. Crystal structure of the N domain of human somatic angiotensin I-converting enzyme provides a structural basis for domain-specific inhibitor design. J Mol Biol. 2006;357:964–974. doi: 10.1016/j.jmb.2006.01.048. [DOI] [PubMed] [Google Scholar]
- 19.Rawlings N.D., Barrett A.J. Evolutionary families of peptidases. Biochem J. 1993;290(Pt 1):205–218. doi: 10.1042/bj2900205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coates D. The angiotensin converting enzyme (ACE) Int J Biochem Cell Biol. 2003;35:769–773. doi: 10.1016/s1357-2725(02)00309-6. [DOI] [PubMed] [Google Scholar]
- 21.Wei L., Alhenc-Gelas F., Corvol P., et al. The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J Biol Chem. 1991;266:9002–9008. [PubMed] [Google Scholar]
- 22.Araujo M.C., Melo R.L., Cesari M.H., et al. Peptidase specificity characterization of C- and N-terminal catalytic sites of angiotensin I-converting enzyme. Biochemistry. 2000;39:8519–8525. doi: 10.1021/bi9928905. [DOI] [PubMed] [Google Scholar]
- 23.Jaspard E., Wei L., Alhenc-Gelas F. Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J Biol Chem. 1993;268:9496–9503. [PubMed] [Google Scholar]
- 24.Cushman D.W., Cheung H.S. Spectrophotometric assay and properties of the angiotensin-converting enzyme of rabbit lung. Biochem Pharmacol. 1971;20:1637–1648. doi: 10.1016/0006-2952(71)90292-9. [DOI] [PubMed] [Google Scholar]
- 25.Corradi H.R., Chitapi I., Sewell B.T., et al. The structure of testis angiotensin-converting enzyme in complex with the C domain-specific inhibitor RXPA380. Biochemistry. 2007;46:5473–5478. doi: 10.1021/bi700275e. [DOI] [PubMed] [Google Scholar]
- 26.Deddish P.A., Marcic B., Jackman H.L., et al. N-domain-specific substrate and C-domain inhibitors of angiotensin-converting enzyme: angiotensin-(1-7) and keto-ACE. Hypertension. 1998;31:912–917. doi: 10.1161/01.hyp.31.4.912. [DOI] [PubMed] [Google Scholar]
- 27.Rousseau A., Michaud A., Chauvet M.T., et al. The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J Biol Chem. 1995;270:3656–3661. doi: 10.1074/jbc.270.8.3656. [DOI] [PubMed] [Google Scholar]
- 28.Oba R., Igarashi A., Kamata M., et al. The N-terminal active centre of human angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide. Eur J Neurosci. 2005;21:733–740. doi: 10.1111/j.1460-9568.2005.03912.x. [DOI] [PubMed] [Google Scholar]
- 29.Fuchs S., Xiao H.D., Hubert C., et al. Angiotensin-converting enzyme C-terminal catalytic domain is the main site of angiotensin I cleavage in vivo. Hypertension. 2008;51:267–274. doi: 10.1161/HYPERTENSIONAHA.107.097865. [DOI] [PubMed] [Google Scholar]
- 30.Dubreuil P., Fulcrand P., Rodriguez M., et al. Novel activity of angiotensin-converting enzyme. Hydrolysis of cholecystokinin and gastrin analogues with release of the amidated C-terminal dipeptide. Biochem J. 1989;262:125–130. doi: 10.1042/bj2620125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skidgel R.A., Engelbrecht S., Johnson A.R., et al. Hydrolysis of substance p and neurotensin by converting enzyme and neutral endopeptidase. Peptides. 1984;5:769–776. doi: 10.1016/0196-9781(84)90020-2. [DOI] [PubMed] [Google Scholar]
- 32.Skidgel R.A., Erdos E.G. Novel activity of human angiotensin I converting enzyme: release of the NH2- and COOH-terminal tripeptides from the luteinizing hormone-releasing hormone. Proc Natl Acad Sci U S A. 1985;82:1025–1029. doi: 10.1073/pnas.82.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naqvi N., Liu K., Graham R.M., et al. Molecular basis of exopeptidase activity in the C-terminal domain of human angiotensin I-converting enzyme: insights into the origins of its exopeptidase activity. J Biol Chem. 2005;280:6669–6675. doi: 10.1074/jbc.M412638200. [DOI] [PubMed] [Google Scholar]
- 34.Toropygin I.Y., Kugaevskaya E.V., Mirgorodskaya O.A., et al. The N-domain of angiotensin-converting enzyme specifically hydrolyzes the Arg-5-His-6 bond of Alzheimer's Abeta-(1-16) peptide and its isoAsp-7 analogue with different efficiency as evidenced by quantitative matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2008;22:231–239. doi: 10.1002/rcm.3357. [DOI] [PubMed] [Google Scholar]
- 35.Skeggs L.T., Jr., Kahn J.R., Shumway N.P. The preparation and function of the hypertensin-converting enzyme. J Exp Med. 1956;103:295–299. doi: 10.1084/jem.103.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sivieri D.O., Jr., Bispo-da-Silva L.B., Oliveira E.B., et al. Potentiation of bradykinin effect by angiotensin-converting enzyme inhibition does not correlate with angiotensin-converting enzyme activity in the rat mesenteric arteries. Hypertension. 2007;50:110–115. doi: 10.1161/HYPERTENSIONAHA.106.085761. [DOI] [PubMed] [Google Scholar]
- 37.Imanishi T., Ikejima H., Tsujioka H., et al. Addition of eplerenone to an angiotensin-converting enzyme inhibitor effectively improves nitric oxide bioavailability. Hypertension. 2008;51:734–741. doi: 10.1161/HYPERTENSIONAHA.107.104299. [DOI] [PubMed] [Google Scholar]
- 38.Jung H.A., Hyun S.K., Kim H.R., et al. Angiotensin-converting enzyme I inhibitory activity of phlorotannins from Ecklonia stolonifera. Fisheries Sci. 2006;72:1292–1299. [Google Scholar]
- 39.Leung M.K., Le S., Houston S., et al. Degradation of Met-enkephalin by hemolymph peptidases in Mytilus edulis. Cell Mol Neurobiol. 1992;12:367–378. doi: 10.1007/BF00711539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lund L., Bak A., Friis G.J., et al. The enzymatic degradation and transport of leucine-enkephalin and 4-imidazolidinone enkephalin prodrugs at the blood-brain barrier. Int J Pharm. 1998;172:97–101. [Google Scholar]
- 41.Azizi M., Rousseau A., Ezan E., et al. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J Clin Invest. 1996;97:839–844. doi: 10.1172/JCI118484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genevay M.C., Mormont C., Thomas F., et al. The synthetic tetrapeptide AcSDKP protects cells that reconstitute long-term bone marrow stromal cultures from the effects of mafosfamide (Asta Z 7654) Exp Hematol. 1996;24:77–81. [PubMed] [Google Scholar]
- 43.Lombard M.N., Sotty D., Wdzieczak-Bakala J., et al. In vivo effect of the tetrapeptide, N-acetyl-Ser-Asp-Lys-Pro, on the G1-S transition of rat hepatocytes. Cell Tissue Kinet. 1990;23:99–103. doi: 10.1111/j.1365-2184.1990.tb01336.x. [DOI] [PubMed] [Google Scholar]
- 44.Volkov L., Quere P., Coudert F., et al. The tetrapeptide AcSDKP, a negative regulator of cell cycle entry, inhibits the proliferation of human and chicken lymphocytes. Cell Immunol. 1996;168:302–306. doi: 10.1006/cimm.1996.0080. [DOI] [PubMed] [Google Scholar]
- 45.Liu J.M., Lawrence F., Kovacevic M., et al. The tetrapeptide AcSDKP, an inhibitor of primitive hematopoietic cell proliferation, induces angiogenesis in vitro and in vivo. Blood. 2003;101:3014–3020. doi: 10.1182/blood-2002-07-2315. [DOI] [PubMed] [Google Scholar]
- 46.Castoldi G., di Gioia C.R., Bombardi C., et al. Prevention of myocardial fibrosis by N-acetyl-seryl-aspartyl-lysyl-proline in diabetic rats. Clin Sci (Lond) 2010;118:211–220. doi: 10.1042/cs20090234. [DOI] [PubMed] [Google Scholar]
- 47.Hu J., Igarashi A., Kamata M., et al. Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide (A beta); retards A beta aggregation, deposition, fibril formation; and inhibits cytotoxicity. J Biol Chem. 2001;276:47863–54788. doi: 10.1074/jbc.M104068200. [DOI] [PubMed] [Google Scholar]
- 48.Zou K., Yamaguchi H., Akatsu H., et al. Angiotensin-converting enzyme converts amyloid beta-protein 1-42 (Abeta(1-42)) to Abeta(1-40), and its inhibition enhances brain Abeta deposition. J Neurosci. 2007;27:8628–8635. doi: 10.1523/JNEUROSCI.1549-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamada K., Uchida S., Takahashi S., et al. Effect of a centrally active angiotensin-converting enzyme inhibitor, perindopril, on cognitive performance in a mouse model of Alzheimer's disease. Brain Res. 2010;1352:176–186. doi: 10.1016/j.brainres.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 50.Patchett A.A., Harris E., Tristram E.W., et al. A new class of angiotensin-converting enzyme inhibitors. Nature. 1980;288:280–283. doi: 10.1038/288280a0. [DOI] [PubMed] [Google Scholar]
- 51.Acharya K.R., Sturrock E.D., Riordan J.F., et al. Ace revisited: a new target for structure-based drug design. Nat Rev Drug Discov. 2003;2:891–902. doi: 10.1038/nrd1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cooper W.O., Hernandez-Diaz S., Arbogast P.G., et al. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med. 2006;354:2443–2451. doi: 10.1056/NEJMoa055202. [DOI] [PubMed] [Google Scholar]
- 53.Georgiadis D., Beau F., Czarny B., et al. Roles of the two active sites of somatic angiotensin-converting enzyme in the cleavage of angiotensin I and bradykinin: insights from selective inhibitors. Circ Res. 2003;93:148–154. doi: 10.1161/01.RES.0000081593.33848.FC. [DOI] [PubMed] [Google Scholar]
- 54.San Juan A.A., Cho S.J. 3D-QSAR studies on angiotensin-converting enzyme (ACE) inhibitors: a molecular design in hypertensive agents. Bull Korean Chem Soc. 2005;26:952–958. [Google Scholar]
- 55.Junot C., Gonzales M.F., Ezan E., et al. RXP 407, a selective inhibitor of the N-domain of angiotensin I-converting enzyme, blocks in vivo the degradation of hemoregulatory peptide acetyl-Ser-Asp-Lys-Pro with no effect on angiotensin I hydrolysis. J Pharmacol Exp Ther. 2001;297:606–611. [PubMed] [Google Scholar]
- 56.Jia H.P., Look D.C., Tan P., et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2009;297:L84–L96. doi: 10.1152/ajplung.00071.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lambert D.W., Yarski M., Warner F.J., et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2) J Biol Chem. 2005;280:30113–33019. doi: 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Towler P., Staker B., Prasad S.G., et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem. 2004;279:17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verrey F., Singer D., Ramadan T., et al. Kidney amino acid transport. Pflugers Arch. 2009;458:53–60. doi: 10.1007/s00424-009-0638-2. [DOI] [PubMed] [Google Scholar]
- 60.Vickers C., Hales P., Kaushik V., et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 61.Rushworth C.A., Guy J.L., Turner A.J. Residues affecting the chloride regulation and substrate selectivity of the angiotensin-converting enzymes (ACE and ACE2) identified by site-directed mutagenesis. FEBS J. 2008;275:6033–6042. doi: 10.1111/j.1742-4658.2008.06733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferreira A.J., Santos R.A., Bradford C.N., et al. Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension. 2010;55:207–213. doi: 10.1161/HYPERTENSIONAHA.109.140145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rentzsch B., Todiras M., Iliescu R., et al. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension. 2008;52:967–973. doi: 10.1161/HYPERTENSIONAHA.108.114322. [DOI] [PubMed] [Google Scholar]
- 64.Wysocki J., Ye M., Rodriguez E., et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension. 2010;55:90–98. doi: 10.1161/HYPERTENSIONAHA.109.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Niu W., Qi Y., Hou S., et al. Correlation of angiotensin-converting enzyme 2 gene polymorphisms with stage 2 hypertension in Han Chinese. Transl Res. 2007;150:374–380. doi: 10.1016/j.trsl.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 66.Lovren F., Pan Y., Quan A., et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am J Physiol Heart Circ Physiol. 2008;295:H1377–H1384. doi: 10.1152/ajpheart.00331.2008. [DOI] [PubMed] [Google Scholar]
- 67.Thomas M.C., Pickering R.J., Tsorotes D., et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ Res. 2010;107:888–897. doi: 10.1161/CIRCRESAHA.110.219279. [DOI] [PubMed] [Google Scholar]
- 68.Nakamura K., Koibuchi N., Nishimatsu H., et al. Candesartan ameliorates cardiac dysfunction observed in angiotensin-converting enzyme 2-deficient mice. Hypertens Res. 2008;31:1953–1961. doi: 10.1291/hypres.31.1953. [DOI] [PubMed] [Google Scholar]
- 69.Kassiri Z., Zhong J., Guo D., et al. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail. 2009;2:446–455. doi: 10.1161/CIRCHEARTFAILURE.108.840124. [DOI] [PubMed] [Google Scholar]
- 70.Ferreira A.J., Moraes P.L., Foureaux G., et al. The angiotensin-(1-7)/Mas receptor axis is expressed in sinoatrial node cells of rats. J Histochem Cytochem. 2011;59:761–768. doi: 10.1369/0022155411411712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lieb W., Graf J., Gotz A., et al. Association of angiotensin-converting enzyme 2 (ACE2) gene polymorphisms with parameters of left ventricular hypertrophy in men. Results of the MONICA Augsburg echocardiographic substudy. J Mol Med. 2006;84:88–96. doi: 10.1007/s00109-005-0718-5. [DOI] [PubMed] [Google Scholar]
- 72.Epelman S., Tang W.H., Chen S.Y., et al. Detection of soluble angiotensin-converting enzyme 2 in heart failure: insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J Am Coll Cardiol. 2008;52:750–754. doi: 10.1016/j.jacc.2008.02.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oudit G.Y., Herzenberg A.M., Kassiri Z., et al. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am J Pathol. 2006;168:1808–1820. doi: 10.2353/ajpath.2006.051091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Soler M.J., Wysocki J., Ye M., et al. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. 2007;72:614–623. doi: 10.1038/sj.ki.5002373. [DOI] [PubMed] [Google Scholar]
- 75.Stegbauer J., Potthoff S.A., Quack I., et al. Chronic treatment with angiotensin-(1-7) improves renal endothelial dysfunction in apolipoproteinE-deficient mice. Br J Pharmacol. 2011;163:974–983. doi: 10.1111/j.1476-5381.2011.01295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pinheiro S.V., Ferreira A.J., Kitten G.T., et al. Genetic deletion of the angiotensin-(1-7) receptor Mas leads to glomerular hyperfiltration and microalbuminuria. Kidney Int. 2009;75:1184–1193. doi: 10.1038/ki.2009.61. [DOI] [PubMed] [Google Scholar]
- 77.Yamazato M., Yamazato Y., Sun C., et al. Overexpression of angiotensin-converting enzyme 2 in the rostral ventrolateral medulla causes long-term decrease in blood pressure in the spontaneously hypertensive rats. Hypertension. 2007;49:926–931. doi: 10.1161/01.HYP.0000259942.38108.20. [DOI] [PubMed] [Google Scholar]
- 78.Yamazato M., Ferreira A.J., Yamazato Y., et al. Gene transfer of angiotensin-converting enzyme 2 in the nucleus tractus solitarius improves baroreceptor heart rate reflex in spontaneously hypertensive rats. J Renin Angiotensin Aldosterone Syst. 2011;12:456–461. doi: 10.1177/1470320311412809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Feng Y., Yue X., Xia H., et al. Angiotensin-converting enzyme 2 overexpression in the subfornical organ prevents the angiotensin II-mediated pressor and drinking responses and is associated with angiotensin II type 1 receptor downregulation. Circ Res. 2008;102:729–736. doi: 10.1161/CIRCRESAHA.107.169110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mecca A.P., Regenhardt R.W., O’Connor T.E., et al. Cerebroprotection by angiotensin-(1-7) in endothelin-1-induced ischaemic stroke. Exp Physiol. 2011;96:1084–1096. doi: 10.1113/expphysiol.2011.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shenoy V., Qi Y., Katovich M.J., et al. ACE2, a promising therapeutic target for pulmonary hypertension. Curr Opin Pharmacol. 2011;11:150–155. doi: 10.1016/j.coph.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hernandez Prada J.A., Ferreira A.J., Katovich M.J., et al. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension. 2008;51:1312–1317. doi: 10.1161/HYPERTENSIONAHA.107.108944. [DOI] [PubMed] [Google Scholar]
- 83.Ferreira A.J., Shenoy V., Qi Y., et al. Angiotensin-converting enzyme 2 activation protects against hypertension-induced cardiac fibrosis involving extracellular signal-regulated kinases. Exp Physiol. 2011;96:287–294. doi: 10.1113/expphysiol.2010.055277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fraga-Silva R.A., Sorg B.S., Wankhede M., et al. ACE2 activation promotes antithrombotic activity. Mol Med. 2010;16:210–215. doi: 10.2119/molmed.2009.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ferreira A.J., Shenoy V., Yamazato Y., et al. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179:1048–1054. doi: 10.1164/rccm.200811-1678OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gjymishka A., Kulemina L.V., Shenoy V., et al. Diminazene aceturate is an ACE2 activator and a novel antihypertensive drug. FASEB J. 2010;24 1032.3 [abstract] [Google Scholar]
- 87.Li F., Li W., Farzan M., et al. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 88.Haga S., Yamamoto N., Nakai-Murakami C., et al. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci U S A. 2008;105:7809–7814. doi: 10.1073/pnas.0711241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huentelman M.J., Zubcevic J., Hernandez Prada J.A., et al. Structure-based discovery of a novel angiotensin-converting enzyme 2 inhibitor. Hypertension. 2004;44:903–906. doi: 10.1161/01.HYP.0000146120.29648.36. [DOI] [PubMed] [Google Scholar]
- 90.Kuba K., Imai Y., Rao S., et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Glowacka I., Bertram S., Herzog P., et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol. 2010;84:1198–1205. doi: 10.1128/JVI.01248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Camargo S.M., Singer D., Makrides V., et al. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology. 2009;136:872–882. doi: 10.1053/j.gastro.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Castrop H., Hocherl K., Kurtz A., et al. Physiology of kidney renin. Physiol Rev. 2010;90:607–673. doi: 10.1152/physrev.00011.2009. [DOI] [PubMed] [Google Scholar]
- 94.Danser A.H., Deinum J. Renin, prorenin and the putative (pro)renin receptor. Hypertension. 2005;46:1069–1076. doi: 10.1161/01.HYP.0000186329.92187.2e. [DOI] [PubMed] [Google Scholar]
- 95.Ichihara A., Hayashi M., Kaneshiro Y., et al. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. J Clin Invest. 2004;114:1128–1135. doi: 10.1172/JCI21398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jan Danser A.H., Batenburg W.W., van Esch J.H. Prorenin and the (pro)renin receptor--an update. Nephrol Dial Transplant. 2007;22:1288–1292. doi: 10.1093/ndt/gfl846. [DOI] [PubMed] [Google Scholar]
- 97.Nguyen G., Delarue F., Burckle C., et al. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–1427. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Batenburg W.W., Jan Danser A.H. The (pro)renin receptor: a new addition to the renin-angiotensin system? Eur J Pharmacol. 2008;585:320–324. doi: 10.1016/j.ejphar.2008.02.092. [DOI] [PubMed] [Google Scholar]
- 99.Suzuki F., Hayakawa M., Nakagawa T., et al. Human prorenin has “gate and handle” regions for its non-proteolytic activation. J Biol Chem. 2003;278:22217–22222. doi: 10.1074/jbc.M302579200. [DOI] [PubMed] [Google Scholar]
- 100.Nabi A.H., Kageshima A., Uddin M.N., et al. Binding properties of rat prorenin and renin to the recombinant rat renin/prorenin receptor prepared by a baculovirus expression system. Int J Mol Med. 2006;18:483–488. [PubMed] [Google Scholar]
- 101.Huang Y., Noble N.A., Zhang J., et al. Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int. 2007;72:45–52. doi: 10.1038/sj.ki.5002243. [DOI] [PubMed] [Google Scholar]
- 102.Huang Y., Wongamorntham S., Kasting J., et al. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int. 2006;69:105–113. doi: 10.1038/sj.ki.5000011. [DOI] [PubMed] [Google Scholar]
- 103.Saris J.J., Hoen P.A., Garrelds I.M., et al. Prorenin induces intracellular signaling in cardiomyocytes independently of angiotensin II. Hypertension. 2006;48:564–571. doi: 10.1161/01.HYP.0000240064.19301.1b. [DOI] [PubMed] [Google Scholar]
- 104.Ichihara A., Suzuki F., Nakagawa T., et al. Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor-deficient mice. J Am Soc Nephrol. 2006;17:1950–1961. doi: 10.1681/ASN.2006010029. [DOI] [PubMed] [Google Scholar]
- 105.Takahashi H., Ichihara A., Kaneshiro Y., et al. Regression of nephropathy developed in diabetes by (Pro)renin receptor blockade. J Am Soc Nephrol. 2007;18:2054–2061. doi: 10.1681/ASN.2006080820. [DOI] [PubMed] [Google Scholar]
- 106.Ichihara A., Kaneshiro Y., Takemitsu T., et al. Contribution of nonproteolytically activated prorenin in glomeruli to hypertensive renal damage. J Am Soc Nephrol. 2006;17:2495–2503. doi: 10.1681/ASN.2005121278. [DOI] [PubMed] [Google Scholar]
- 107.Ichihara A., Kaneshiro Y., Takemitsu T., et al. Nonproteolytic activation of prorenin contributes to development of cardiac fibrosis in genetic hypertension. Hypertension. 2006;47:894–900. doi: 10.1161/01.HYP.0000215838.48170.0b. [DOI] [PubMed] [Google Scholar]
- 108.Kaneshiro Y., Ichihara A., Sakoda M., et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol. 2007;18:1789–1795. doi: 10.1681/ASN.2006091062. [DOI] [PubMed] [Google Scholar]
- 109.Batenburg W.W., Krop M., Garrelds I.M., et al. Prorenin is the endogenous agonist of the (pro)renin receptor. Binding kinetics of renin and prorenin in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor. J Hypertens. 2007;25:2441–2453. doi: 10.1097/HJH.0b013e3282f05bae. [DOI] [PubMed] [Google Scholar]
- 110.Feldt S., Batenburg W.W., Mazak I., et al. Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension. 2008;51:682–688. doi: 10.1161/HYPERTENSIONAHA.107.101444. [DOI] [PubMed] [Google Scholar]
- 111.Feldt S., Maschke U., Dechend R., et al. The putative (pro)renin receptor blocker HRP fails to prevent (pro)renin signaling. J Am Soc Nephrol. 2008;19:743–748. doi: 10.1681/ASN.2007091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Muller D.N., Klanke B., Feldt S., et al. (Pro)renin receptor peptide inhibitor “handle-region” peptide does not affect hypertensive nephrosclerosis in Goldblatt rats. Hypertension. 2008;51:676–681. doi: 10.1161/HYPERTENSIONAHA.107.101493. [DOI] [PubMed] [Google Scholar]
- 113.Tabassum N. Aliskiren: a new renin inhibitor as anti-hypertensive. J Appl Pharm Sci. 2011;1:30–33. [Google Scholar]
- 114.Fogari R., Zoppi A. New class of agents for treatment of hypertension: focus on direct renin inhibition. Vasc Health Risk Manag. 2010;6:869–882. doi: 10.2147/VHRM.S4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sipahi I., Debanne S.M., Rowland D.Y., et al. Angiotensin-receptor blockade and risk of cancer: meta-analysis of randomised controlled trials. Lancet Oncol. 2010;11:627–636. doi: 10.1016/S1470-2045(10)70106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schefe J.H., Neumann C., Goebel M., et al. Prorenin engages the (pro)renin receptor like renin and both ligand activities are unopposed by aliskiren. J Hypertens. 2008;26:1787–1794. doi: 10.1097/HJH.0b013e3283060f2e. [DOI] [PubMed] [Google Scholar]
- 117.Yusuf S., Teo K.K., Pogue J., et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547–1559. doi: 10.1056/NEJMoa0801317. [DOI] [PubMed] [Google Scholar]
- 118.Guang C., Phillips R.D. Plant food-derived Angiotensin I converting enzyme inhibitory peptides. J Agric Food Chem. 2009;57:5113–5120. doi: 10.1021/jf900494d. [DOI] [PubMed] [Google Scholar]
- 119.Wu J., Aluko R.E., Nakai S. Structural requirements of Angiotensin I-converting enzyme inhibitory peptides: quantitative structure-activity relationship study of di- and tripeptides. J Agric Food Chem. 2006;54:732–738. doi: 10.1021/jf051263l. [DOI] [PubMed] [Google Scholar]
- 120.van Mierlo L.A., Koning M.M., van der Zander K., et al. Lactotripeptides do not lower ambulatory blood pressure in untreated whites: results from 2 controlled multicenter crossover studies. Am J Clin Nutr. 2009;89:617–623. doi: 10.3945/ajcn.2008.26918. [DOI] [PubMed] [Google Scholar]
- 121.Li H., Aluko R.E. Identification and inhibitory properties of multifunctional peptides from PEA protein hydrolysate. J Agric Food Chem. 2010;58:11471–11476. doi: 10.1021/jf102538g. [DOI] [PubMed] [Google Scholar]
- 122.Takahashi S., Tokiwano T., Hata K., et al. The occurrence of renin inhibitor in rice: isolation, identification, and structure-function relationship. Biosci Biotechnol Biochem. 2010;74:1713–1715. doi: 10.1271/bbb.100233. [DOI] [PubMed] [Google Scholar]
- 123.Takahashi S., Tokiwano T., Suzuki N., et al. Renin inhibitory activity in rice and cereals. J Biol Macromol. 2010;10:83–91. [Google Scholar]
- 124.Hiwatashi K., Shirakawa H., Hori K., et al. Reduction of blood pressure by soybean saponins, renin inhibitors from soybean, in spontaneously hypertensive rats. Biosci Biotechnol Biochem. 2010;74:2310–2312. doi: 10.1271/bbb.100328. [DOI] [PubMed] [Google Scholar]
- 125.Takahashi S., Hori K., Hokari M., et al. Inhibition of human renin activity by saponins. Biomed Res. 2010;31:155–159. doi: 10.2220/biomedres.31.155. [DOI] [PubMed] [Google Scholar]
- 126.Takahashi S., Hori K., Kumagai M., et al. Human renin inhibitory activity in legumes. J Biol Macromol. 2007;7:49–54. [Google Scholar]
- 127.Takahashi S., Hori K., Shinbo M., et al. Isolation of human renin inhibitor from soybean: soyasaponin I is the novel human renin inhibitor in soybean. Biosci Biotechnol Biochem. 2008;72:3232–3236. doi: 10.1271/bbb.80495. [DOI] [PubMed] [Google Scholar]