

SUMMARY
We performed a comprehensive assessment of rare inherited variation in autism spectrum disorders (ASD) by analyzing whole-genome sequences (WGS) of 2,308 individuals from families with multiple affected children. We implicate 69 genes in ASD-risk, including 24 passing genome-wide Bonferroni correction and 16 new ASD-risk genes, most supported by rare inherited variants, a substantial extension of previous findings. Biological pathways enriched for genes harboring inherited variants are distinct from pathways previously implicated by de novo variation, representing cytoskeletal organization and ion transport. Nevertheless, the de novo and inherited genes contribute to a common protein-protein interaction network. We also identified SVs affecting non-coding regions, implicating recurrent deletions in the promoters of DLG2 and NR3C2. Loss of nr3c2 function in zebrafish disrupts sleep and social function, overlapping with human ASD-related phenotypes. These data support the utility of studying multiplex families in ASD and are available through the Hartwell Autism Research and Technology portal.
Graphical Abstract
In Brief
Whole genome sequencing from families with multiple ASD-affected children allows identification of rare inherited variants associated with disease and the definition of a syndromic form of disease caused by mutations in NR3C2.
INTRODUCTION
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by early deficits in social communication and interaction, together with restricted and repetitive patterns of behavior, interest, or activity (American Psychiatric Association, 2013). Global prevalence is 1-2% (CDC, 2014) with heritability estimated at 60-90% (Colvert et al., 2015; Gaugler et al., 2014; Geschwind and Flint, 2015; Hoekstra et al., 2007; Klei et al., 2012; Sandin et al., 2014; Skuse et al., 2005).
Considerable progress in gene discovery has come from studies in families with one affected child (simplex families), identifying de novo copy number variants (CNV) (Levy et al., 2011; Marshall et al., 2008; Sanders et al., 2011; Sebat et al., 2007) and de novo frameshift, splice-acceptor, splice-donor, or nonsense variants (collectively referred to as protein-truncating variants (PTVs)) (De Rubeis et al., 2014; Iossifov et al., 2014; Iossifov et al., 2012; O’Roak et al., 2012; Sanders et al., 2012) that increase ASD risk and account for an estimated 3-5% of ASD cases (Constantino et al., 2010; Gaugler et al., 2014; Ozonoff et al., 2011; Sandin et al., 2014; Werling and Geschwind, 2015). Despite these remarkable advances in identifying de novo (germline) mutations in ASD, by definition, de novo mutations account for none of the substantial heritability of ASD.
To date, recurrent CNVs are the primary established form of inherited risk variation for ASD (Glessner et al., 2009; Leppa et al., 2016; Mefford et al., 2008). Exploration of other types of inherited risk variation (SNVs and indels) has been drawn primarily from families containing only one affected child (De Rubeis et al., 2014; Krumm et al., 2015), which are depleted for inherited risk as compared to families with two or more affected children (multiplex families) (Ronemus et al., 2014; Sebat et al., 2007; Virkud et al., 2009). A recent study by the MSSNG consortium was limited to large rare CNVs and de novo protein-coding variation, despite drawing 40% of samples from multiplex ASD families (Yuen et al., 2017). Thus, a majority of ASD-risk genes, especially those contributing to inherited risk, have yet to be identified. Moreover, without broader knowledge of individual genes contributing to heritable risk for ASD, whether rare de novo and inherited risk variants impact the same biological pathways remains an important and yet, unanswered question. Here, we used WGS to identify both de novo and inherited genetic risk factors for ASD in both coding and non-coding regions of the genome, in the largest cohort of multiplex families evaluated to date.
RESULTS
We analyzed high-coverage WGS data from 2,308 individuals in 493 multiplex ASD families from the Autism Genetic Resource Exchange (AGRE) (Methods; Figure 1; Figure S1; Table S1). This cohort, the Hartwell Autism Research and Technology Initiative (iHART), includes 960 affected children and 217 unaffected children for whom both biological parents were sequenced.
Figure 1. Overview of the Analysis Pipeline.
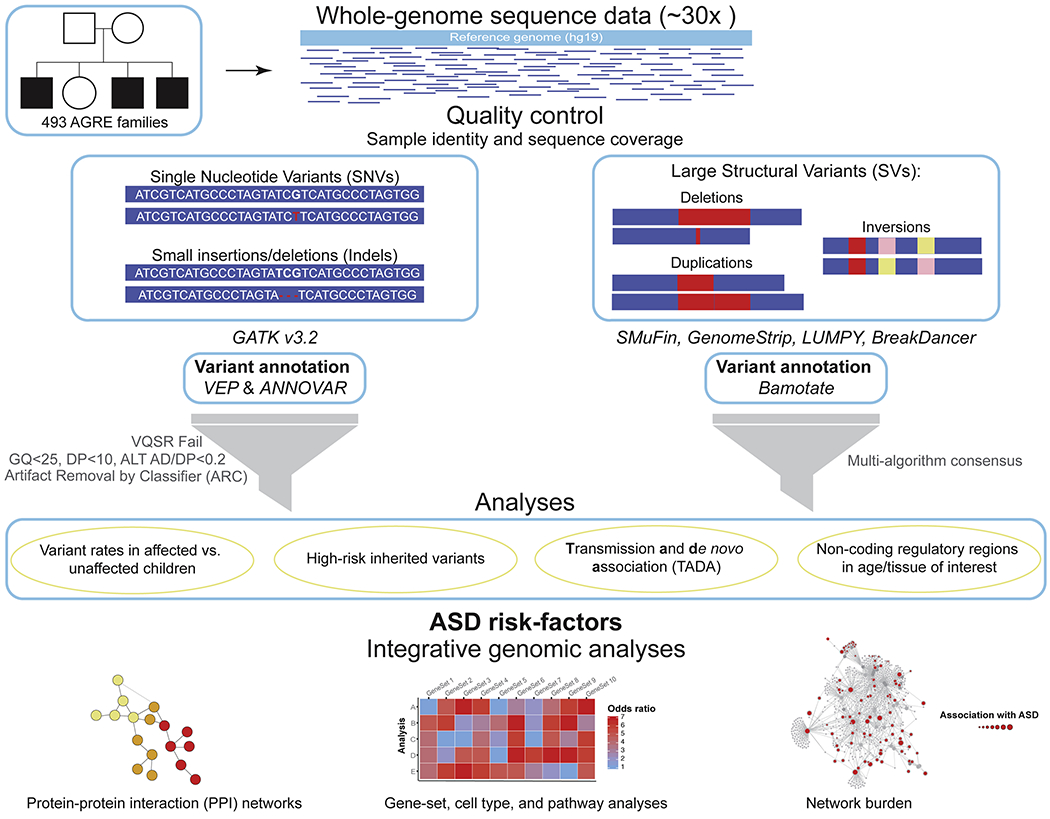
High-coverage whole-genome sequence reads were aligned to the human reference genome (hg19) and quality control checks were applied to insure both sample identity and sequencing coverage (see Figure S1). SNVs and indels were called following GATK’s best practices; annotated using both VEP and ANNOVAR, and then filtered for mildly stringent quality thresholds. All de novo variants were classified by ARC and high-confidence variants were retained (see Figures 3 and S3–S4). Large SVs were identified by four different SV-detection algorithms, three of which used aligned sequence reads and one that performed de novo alignment (SMuFin). Large SVs were annotated using Bamotate and then filtered for high quality variants by using our multi-algorithm consensus pipeline. The resulting variants were then analyzed to identify ASD-risk factors and perform integrative genomic analyses.
Excess of high-risk inherited variants in affected children
Previous studies have shown that siblings discordant for ASD exhibit similar overall mutation rates, but differ in the rates of certain classes of deleterious mutations (e.g., de novo PTV) and in the specific biological processes represented by genes hit with deleterious variants (e.g., chromatin modifiers) (Iossifov et al., 2014; Iossifov et al., 2012; O’Roak et al., 2012; Sanders et al., 2015). Since multiplex ASD families are expected to be enriched for inherited risk variants (Ronemus et al., 2014; Sebat et al., 2007; Virkud et al., 2009), we first assessed the rate of rare inherited variants in affected and unaffected children. We found no excess of rare (allele frequency (AF) ≤0.1%) inherited PTV or missense variants in affected subjects (Figures 2A and S2A–E).
Figure 2. Inherited ASD-risk genes.
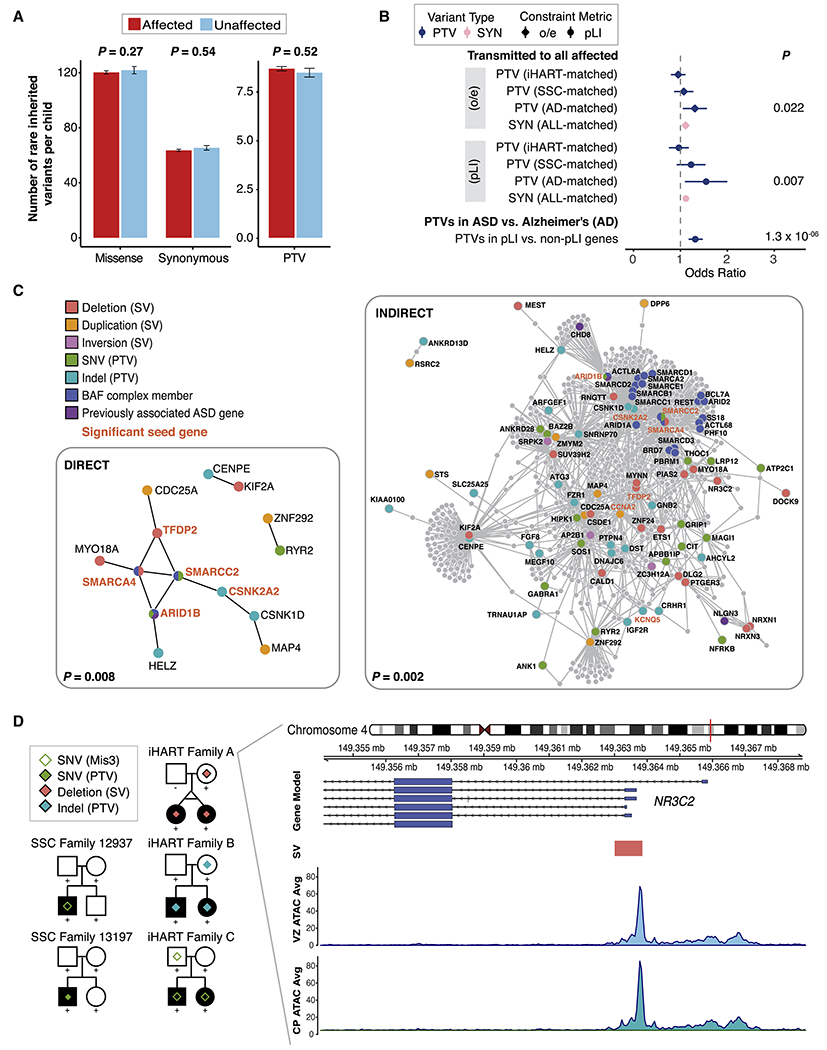
(A) The number of rare inherited coding variants per fully phase-able child is displayed for 960 affected (red) and 217 unaffected (blue) children by variant consequence. Mean ± SE rates are shown. (B) Odds ratios from simulations of high-risk inherited PTV or SYN variants. Results are shown for constrained genes (gnomAD pLI score or gnomAD o/e score) and the cohort used for calculation of the null PTV or SYN rate is displayed (cohort-matched class rate). The odds ratio resulting from a Fisher’s exact test comparing the rate of PTVs in constrained vs. non-constrained genes in the iHART and SSC cohorts to that observed in the Alzheimer’s Disease cohort is also shown. Significant p-values are displayed. Whiskers represent 95% confidence intervals. (C) Direct and indirect PPI networks formed by constrained genes harboring PTVs or SVs (promoter or exon disrupting) transmitted to all affected and no unaffected children in a family. Proteins are colored according to the variant category of the variant identified in the high-risk inherited analysis, and previously known ASD-risk genes (Sanders et al., 2015) are shown in purple. Significant seed genes are bold and orange. P-values from 1,000 permutations. (D) Pedigrees for five ASD families with coding or regulatory NR3C2 variants. Square=male, circle=female, filled shape=individual with ASD, ‘+’=sequenced individual. Both SSC families harbor de novo variants in the proband (a PTV in SSC13197 and a probably damaging missense (Mis3, a “probably damaging” prediction by PolyPhen-2 (Adzhubei et al., 2010)) in SSC12937). iHART families A-C harbor rare inherited variants transmitted to both affected children; including a ~850bp deletion in family A, a PTV in family B, and a Mis3 variant in family C. The NR3C2 promoter-disrupting deletion (orange rectangle, chr4:149363005-149363852) overlaps a functional non-coding regulatory region in developing human brain (chr4:149362706-149367485) (de la Torre-Ubieta et al., 2018). The average ATAC-seq peak read depth from the cortical plate (CP) and ventricular zone (VZ) of developing human brain samples (n=3) are shown below the NR3C2 deletion.
To investigate non-coding regions likely to have the largest association signal (An et al., 2018), we examined whether private (observed in a single family) inherited variants were enriched in the promoter regions of affected vs. unaffected iHART children (Methods). We found no enrichment in affected subjects globally (Methods; P=0.07, quasi-Poisson linear regression), nor when restricting the analysis to promoters of known ASD-risk genes (Methods; P=0.26, quasi-Poisson linear regression). We still found no significant excess of private inherited variants in the promoters of affected subjects when combined with 517 affected and 518 unaffected subjects from the Simons Simplex Cohort (SSC; Methods; All genes P=0.14 and ASD-risk genes P=0.12).
Similarly, we observed no difference in the overall rate of rare inherited SVs nor gene disrupting SVs between affected and unaffected individuals (Figure S2F–M). The absence of substantial rate differences for rare inherited variants is consistent with prior studies, which either found no global signal, or only identified signals in selected candidates (Brandler et al., 2018; De Rubeis et al., 2014; Krumm et al., 2015; Leppa et al., 2016; Werling et al., 2018). Our findings are also consistent with the lower average effect size and reduced penetrance of inherited risk variation relative to de novo pathogenic mutations.
Given the low effect size of inherited risk variants, we further leveraged family structure to identify rare variants transmitted to all affected, but no unaffected children, under the hypothesis that such variants may confer a high disease risk. These high-risk inherited variants were further defined as variants disrupting highly constrained genes (those predicted to be the least tolerant to loss-of-function mutations in the human population; pLI>0.9; Lek et al., 2016; Methods). We identified 98 unique genes harboring these high-risk inherited variants, including 62 PTVs and 40 SVs disrupting a coding exon or promoter. Three genes (NR3C2, NRXN1, and ZMYM2) were disrupted by a PTV in one family and a SV in a second family. To determine if these findings were significant, we performed 1000 permutations under the null, using the observed PTV counts and estimated gene mutation rates (Samocha et al., 2014)(Methods). We observed a striking depletion of PTVs in constrained genes in our cohort (observed=57, expected=255 by permutation; Methods). Indeed, both iHART parents and SSC parents had five times fewer PTVs in constrained genes than expected from previously established de novo rates, while non-constrained genes follow the expected rate (Samocha et al., 2014)(Methods). This finding is consistent with natural selection acting rapidly to eliminate deleterious mutations.
We next updated our simulations to match the empirical ratio of PTVs in highly constrained genes (pLI≥0 .9) versus all genes in three cohorts: the SSC (Werling et al., 2018), this iHART cohort, or an Alzheimer’s Disease (AD) cohort (Bennett et al., 2018)(Methods), the latter selected for comparison because of the lack of ASD comorbidity. We observed a significant enrichment (P<0.05 by permutation, Methods) for high-risk inherited variants disrupting constrained genes in iHART when the PTV ratio was matched to AD (P=0.007), trending enrichment when matched to SSC (P<0.16), and no enrichment when matched to iHART (Figure 2B; Methods). We draw two conclusions from these observations: first, the rare variant burden within constrained genes differs across the iHART, SSC, and AD cohorts: we observe significantly more PTVs in constrained genes in the parents within the ASD cohorts (iHART and SSC) than in the AD cohort (Fisher exact test, P = 1.3x10−6; OR=1.3; 95% confidence interval 1.2-1.5; Figure 2B). Second, we validated the high-risk inherited approach (which identified 98 genes harboring high-risk inherited variants) by observing an excess of PTVs transmitted to all affected and not to unaffected children (transmission disequilibrium) in constrained genes (Figure 2B; P=0.007; Methods). Furthermore, genome-wide PTVs show a trend towards increased PTV transmission to all affected and no unaffected children (P=0.08), suggesting that inherited PTVs – even in non highly-constrained genes – increase ASD liability. Thus, while we find a significant signal for inherited variants in highly constrained genes, larger samples will be needed to reach significance for inherited, lower penetrant variants more broadly.
High-risk inherited coding and non-coding variants form a significant PPI network
Since genes harboring de novo PTVs are enriched in gene networks representing specific biological pathways (Hormozdiari et al., 2015; Krishnan et al., 2016; Parikshak et al., 2013), we reasoned that similar enrichment among genes harboring inherited risk variants would provide orthogonal support for the role of these genes in ASD biology. Indeed, the protein products of the 98 genes harboring high-risk inherited variation form a significant direct protein–protein interaction (PPI) network (P<0.008, Methods, Figure 2C), as well as a significant indirect PPI network (P<0.002) that highlights seven risk genes as significantly connected hubs (corrected seed score P<0.05) (Figure 2C). This PPI network is enriched for members of the BAF (SWI/SNF) complex (two-sided Fisher’s Exact Test, P=0.02, OR=5.9, 95% confidence interval 1.1 – 20.7), including ARID1B, SMARCC2, and SMARCA4, which are involved in chromatin remodeling during cortical neurogenesis and have previously been associated with de novo variation in ASD (Parikshak et al., 2013; Vandeweyer et al., 2014). These data show for the first time that rare inherited and de novo variation impact potentially overlapping molecular processes based on their convergence within a PPI network.
Inherited regulatory deletions disrupt NR3C2 and DLG2
Among the 98 genes harboring high-risk inherited variation, we then focused on NR3C2, which had not been consistently associated with ASD in previous studies (TADA FDR=0.079, De Rubeis et al., 2014; TADA FDR=0.136, Sanders et al., 2015). Our analysis of high-risk inherited variation provides the first evidence of inherited risk in NR3C2, including non-coding structural variation, and further supports NR3C2 as an ASD-risk gene (Figure 2D). The three families with NR3C2 risk variants share striking phenotypic similarities, defining a new syndromic form of ASD characterized by metacarpal hypoplasia, high arched palate, sensory hypersensitivity, and abnormal prosody (Table S1).
A second gene identified by the analysis of high-risk inherited variation was DLG2, which is associated with cognition and learning in mice and humans (Belgard and Geschwind, 2013; Nithianantharajah et al., 2013), but not previously implicated in ASD. We identified three families with the same 2.5Kb deletion in the DLG2 promoter (Figure S2N), which falls in a recently-defined, functional, non-coding regulatory region in developing human brain (de la Torre-Ubieta et al., 2018) (Figure S2N) and likely arose independently because the deletion is found on a different haplotype in each family (Methods, Table S1). No deletions overlap the DLG2 promoter deletion in controls (n=26,565 controls, Methods), suggesting that this region is highly constrained. This rare regulatory mutation is significantly associated with ASD (3 of 484 unrelated affected children versus 0 of 2,889 WGS controls, two-sided Fisher’s Exact Test, P=0.003, OR=Inf, 95% CI=2.47-Inf).
Identification of high-quality de novo variants by machine learning
De novo missense and PTVs have been identified as significant risk factors for ASD in simplex families (De Rubeis et al., 2014; Iossifov et al., 2014; Samocha et al., 2014). However, true de novo mutations may be indistinguishable from data artifacts, especially in WGS data derived from lymphoblastoid cell line (LCL) DNA – which despite wide-spread use in the genetics community, may contain mutations introduced and propagated during cell line transformation that are unrelated to disease biology (Conrad et al., 2011; Genomes Project et al., 2010). We reasoned that removal of LCL-derived artifacts from samples whose biomaterials were limited to LCL DNA (Table S1) would be critical for de novo variant identification in this study – as well as of broad utility for studies using LCLs. Therefore, we developed a supervised random forest model, Artifact Removal by Classifier (ARC), to distinguish true rare de novo variants (RDNVs) from LCL-specific genetic aberrations, as well as artifacts such as sequencing and mapping errors.
We used 76 pairs of monozygotic (MZ) twins with LCL DNA from iHART to train ARC, under the assumption that true de novo variants would be present in both twin pairs, but LCL-derived artifacts would not. ARC incorporates 48 features representing intrinsic genomic properties, (e.g., GC content, de novo hotspots (Michaelson et al., 2012)), sample specific properties (e.g., number of de novo SNVs), signatures of transformation of peripheral B lymphocytes by Epstein-Barr virus (e.g., number of de novo SNVs in immunoglobulin genes), or variant properties (e.g., GATK variant metrics) (Figure 3A). To evaluate ARC, we applied it to WGS from LCL-derived DNA in 17 patients, and compared it to WGS derived from whole blood (WB) in the same patients. The resulting random forest classifier achieved an area under the receiver operating characteristic (ROC) curve of 0.99 and 0.98 in the training and test set, respectively (Figures 3B–C and S3), indicating that ARC very successfully distinguishes true and false de novo variants.
Figure 3. Rare de novo variants in iHART.
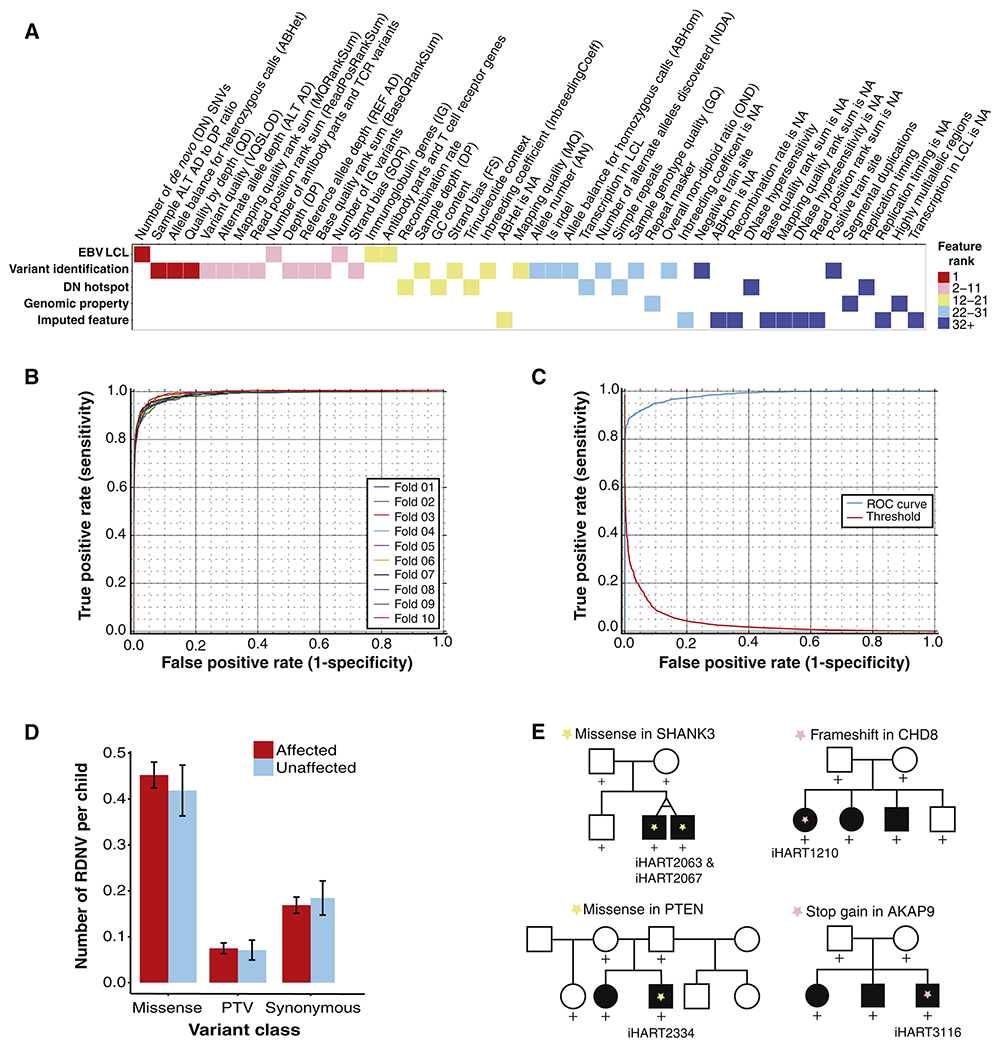
(A) Heat map reflecting the importance ranking for all 48 ARC features, listed on the x-axis in order of rank and sorted by category (signatures of transformation of peripheral B lymphocytes by Epstein-Barr virus (EBV LCL), properties of variant identification, de novo hot spots, intrinsic genomic property, or imputed feature) on the y-axis. (B) ROC curves for 10-fold cross validation for the ARC training set; AUC=0.99. (C) ROC curve for the ARC test set; AUC=0.98. (D) Rate of RDNVs per child is displayed for 575 affected (red) and 141 unaffected (blue) children (716 fully phase-able samples after excluding MZ twins and ARC outliers) by variant consequence. Mean ± SE rates are shown. (E) Pedigrees for iHART families containing RDNVs in previously established ASD-risk genes. Children harboring the RDNV of interest are labeled with their iHART sample ID and a star symbol. The missense variants in SHANK3 and PTEN are predicted to damage the encoded protein (Mis3).
Application of ARC in the 1,177 children for whom both biological parents were also sequenced, successfully eliminated the significantly higher rate of RDNVs in LCL samples (Figures S4A–S4C), and resulted in the expected genome-wide de novo mutation rate (mean=60.1 RDNVs per child; Figure S4B)(Besenbacher et al., 2016; Conrad et al., 2011; Kong et al., 2012; Michaelson et al., 2012; Turner et al., 2016; Yuen et al., 2017). Running ARC similarly corrected mutation rates to reveal that iHART children exhibit the well-known effect of paternal age on de novo mutation rates (increase of 1.46 RDNVs per year of paternal age; Methods; Figure S4D) (Deciphering Developmental Disorders, 2017; Francioli et al., 2015; Goldmann et al., 2016; Michaelson et al., 2012; Study, 2017). These RDNV properties match expectation, confirming that we had high quality RDNVs for downstream analyses.
Evidence for depletion of rare de novo ASD-risk in multiplex families
We hypothesized that the iHART multiplex families would be enriched for inherited risk variants relative to previous studies of simplex families in whom de novo variants primarily contribute to disease risk. Leppa et al. previously found an enrichment of rare de novo CNVs in affected as compared to unaffected children in simplex SSC families, but not in multiplex AGRE families (Leppa et al., 2016). Consistent with that finding, we observed no significant association for de novo missense (P=0.561, quasi-Poisson linear regression), nor PTVs (P=0.873, quasi-Poisson linear regression) in affected individuals in iHART multiplex families (Figure 3D). The rate of rare de novo PTVs in affected children from multiplex families (AffiHART=0.07) was approximately half of that in simplex families (AffKosmicki=0.13) (Iossifov et al., 2014; Kosmicki et al., 2017) and equivalent to the rate in unaffected children (UnaffiHART=0.07) (Table S2). We estimated that our current cohort had >70% power to detect a rate difference for de novo PTVs in affected versus unaffected individuals (Monte Carlo integration; Methods), suggesting a true difference in the underlying architecture of multiplex families, as compared with simplex families. Despite not observing a global excess for damaging RDNVs in affected children, we do identify pathogenic de novo variants in previously established ASD-risk genes (Methods; Figure 3E). Interestingly, we observe these mutations in some, but usually not all, affected family members, in line with a complex etiology where additional risk loci explain ASD in affected siblings, also in agreement previous observations based only on large de novo CNVs (Leppa et al., 2016).
To expand this analysis to non-coding regions, we analyzed promoters, but did not find enrichment for rare de novo promoter variants when looking globally (Methods; P=0.33, quasi-Poisson linear regression), nor when restricting the analysis to promoters of known ASD-risk genes (Methods; P=0.42, quasi-Poisson linear regression). We also increased power by combining our cohort with 517 affected and 518 unaffected children with WGS data from the SSC and still found no evidence for enrichment in promoters (Methods; All genes P=0.25 and ASD-risk genes P=0.31, quasi-Poisson linear regression). These data are accordant with recent results in simplex families (Werling et al., 2018), which suggests that the effect sizes in non-coding regions are on aggregate too small to detect with current sample sizes.
Identification of 16 novel ASD-risk genes enriched for inherited variation
We next used a powerful Bayesian framework, the Transmitted And de novo Association (TADA) test (He et al., 2013) to combine inherited and de novo signals to identify ASD-risk genes (Methods). To further improve power, we combined qualifying variants (Methods) from the iHART cohort with the most recent ASD TADA mega-analysis (Sanders et al., 2015) (Table S3). Our TADA-mega analysis identified 69 genes significantly associated with ASD at FDR<0.1 (Figure 4A, Tables 1 and S3), 16 of which had not previously been identified (Figure 4A, Tables 1 and S3). The 16 novel ASD-risk genes are enriched for genes in which a higher proportion of risk variants are inherited versus de novo (Methods, Figure 4A). For 6 of the 16 novel genes (UIMC1, C16orf13, MLANA, CCSER1, PCM1, FAM98C) and 5 of the 53 previously associated ASD-risk genes (RANBP17, ZNF559, P2RX5, CTTNBP2, CAPN12), ≥70% of the qualifying variants are inherited PTVs (Fisher’s Exact Test, P = 0.015, OR=5.57, 95% CI:1.17-28.35).
Figure 4. 69 ASD-risk genes identified by TADA mega-analysis.
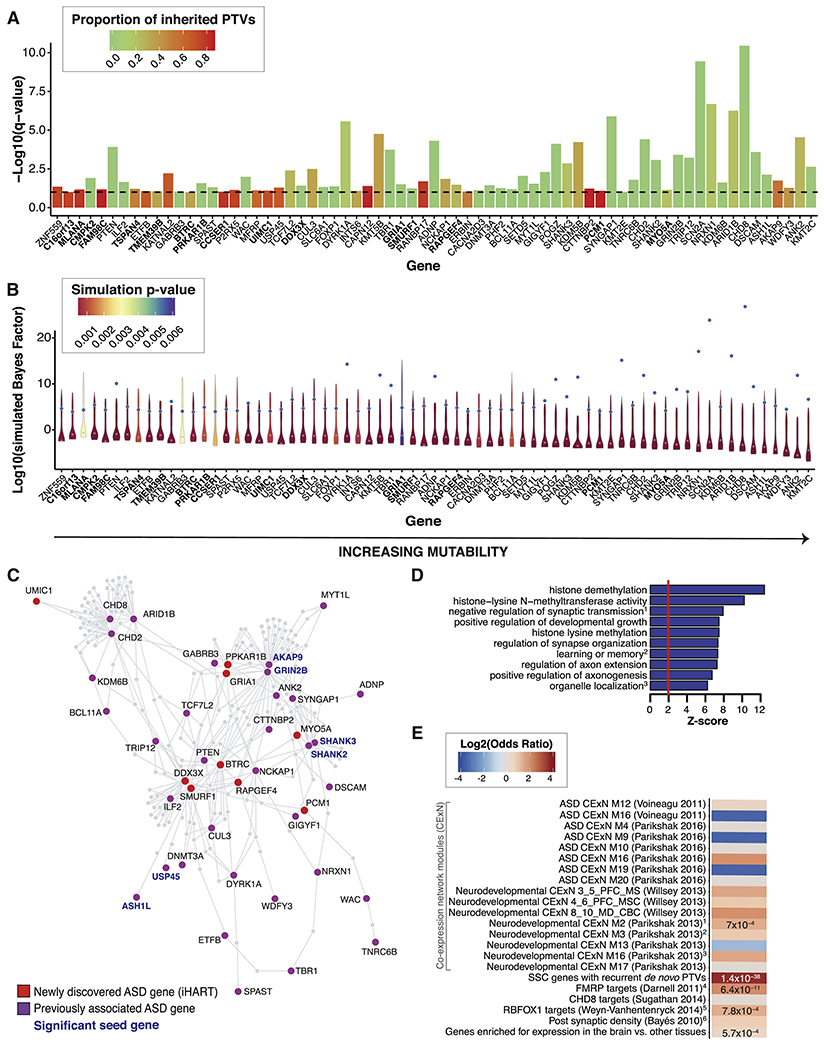
(A,B) The 69 genes identified in the iHART TADA mega-analysis (FDR<0.1) are displayed in order of increasing gene mutability; the 16 novel genes are in bold. (A) The per-gene TADA FDR is displayed as a bar reaching the −log10(q-value). The dashed horizontal line marks the FDR=0.1 threshold. Bars are colored by the proportion of inherited PTVs for each gene (inherited PTVs/(inherited PTVs + de novo PTVs + de novo Mis3 + de novo small deletions)). (B) Violin plots of the simulated Bayes Factors (displayed as log(simulated Bayes Factor), 111 quantiles from the 1.1 million simulations) for each gene. The violin plots are colored by simulation p-value (max p-value=0.006). For each gene, the grey x indicates the median of the simulated Bayes Factors and the blue dot is the Bayes Factor obtained in the iHART TADA-mega analysis. The larger the distance between the median simulated Bayes Factor and the observed TADA-mega analysis Bayes Factor, the lower the probability of having achieved the observed Bayes Factor by chance. (C) Indirect PPI network formed by the 69 ASD-risk genes identified by TADA (FDR<0.1). Proteins encoded by previously known ASD-risk gene (Sanders et al., 2015)) are shown in purple and newly identified ASD-risk genes (iHART TADA-mega analysis) are shown in red. Gene labels for the six significant seed genes are bold and blue. (D) Gene-ontology enrichment for the 69 ASD-risk genes with known biological pathways. Three of the enriched pathways contain one or more of the 16 novel ASD-risk genes (any of the 69 genes in biological pathway are listed, with novel risk genes in bold): (1) negative regulation of synaptic transmission includes ADNP, SLC6A1, and RAPGEF4; (2) learning and memory includes ADNP, GRIA1, NRXN1, PRKAR1B, SLC6A1, and SYNGAP1; and (3) organelle organization includes MYO5A, PCM1, and TCF7L2. (E) Gene-set enrichment results for the 69 ASD-risk genes displayed by the log2(odds ratio), with p-values listed for gene sets surviving multiple test correction (P<0.002); the SSC gene set was included as a positive control. In addition to the gene set “genes enriched for expression in the brain vs. other tissues” which contains almost all of the 16 novel ASD-risk genes, six additional gene-sets contain one or more of the 16 novel ASD-risk genes: (1) TMEM39B and PCM1, (2) CCSER1 and UIMC1, (3) BTRC, PRKAR1B, and MYO5A, (4) RAPGEF4 and MYO5A, (5) BTRC, (6) DDX3X, GRIA1, RAPGEF4, and MYO5A.
Table 1. 69 ASD-risk genes identified in the iHART TADA mega-analysis.
All 69 genes significantly associated with ASD-risk (FDR<0.1) by the iHART TADA mega-analysis are displayed by the number of de novo PTVs identified in the gene. The 16 newly ASD-associated genes are in bold. The 24 underlined genes are the subset of highly-confident genes that reach genome-wide significance after Bonferroni correction.
| dnPTV count | FDR ≤ 0.01 | 0.01 < FDR ≤ 0.05 | 0.05 < FDR ≤ 0.1 |
|---|---|---|---|
| ≥2 | CHD8, SCN2A, ARID1B, SYNGAP1, DYRK1A, CHD2, ANK2, KDM5B, ADNP, POGZ, KMT5B, TBR1, GRIN2B, DSCAM, KMT2C, TCF7L2, TRIP12, ASH1L, CUL3, KATNAL2, GIGYF1 | TNRC6B, WAC, NCKAP1, RANBP17, KDM6B, ILF2, SPAST, FOXP1, AKAP9, CMPK2, DDX3X | WDFY3, PHF2, BCL11A, KMT2E, CACNA2D3* |
| 1 | NRXN1, SHANK2, PTEN, SHANK3, SETD5 | DNMT3A, MYT1L, RAPGEF4, PRKAR1B | MFRP, GABRB3, P2RX5, ETFB, CTTNBP2, INTS6, USP45, ERBIN, TMEM39B, TSPAN4, MLANA, SMURF1, C16orf13, BTRC, CCSER1, FAM98C |
| 0 | - | SLC6A1, ZNF559, CAPN12, GRIA1 | PCM1, MYO5A, UIMC1 |
The CACNA2D3 gene had an FDR<0.1 in this iHART TADA-mega analysis but not the previous mega analysis (Sanders et al., 2015); however, it was previously reported (De Rubeis et al., 2014) and thus is not considered a novel ASD-risk gene.
Since TADA was previously applied to simplex families, the null distribution of the TADA statistic was not known for multiplex families. To ensure that we did not obtain false positives (type I errors) due to family structure alone, we estimated this distribution by simulating Mendelian transmission and de novo mutation across family structures using the observed variant counts (Methods). As expected, genes with the lowest FDR in the TADA-mega analysis showed the largest simulated Bayes factors and lowest p-values (Figures S5A and S5B); the three association statistics consistently reflect ASD-risk association (the smaller the FDR or p-value and the larger the Bayes factor, the stronger the association). All 69 genes with an FDR<0.1 in the TADA-mega analysis obtained a simulated p-value of less than 0.006 (median P=1x10−3). The lowest simulation p-value was for CHD8 (P=9x10−7; Figure 4B), which is a well-established ASD gene. We also leveraged the simulation p-values and applied a stringent Bonferroni correction (P<2.7x10−6) to highlight a high-confidence subset of 24 genes (Methods; Tables 1 and S3). Stringent Bonferroni correction had not been previously utilized to identify genome-wide significant ASD-risk genes. The most comparable approach was applying Fisher’s Exact test to variants found in a large CHD8 resequencing cohort (P=1.01 × 10−5) (Bernier et al., 2014).
The low relative risk estimated for inherited PTVs (De Rubeis et al., 2014) means that genes with primarily inherited risk variants will typically require more ASD carriers than those with primarily de novo risk to reach the same level of association. We identified 119 genes at a relaxed statistical threshold FDR<0.2, 84 of which were previously identified at this threshold (Sanders et al., 2015). For 15 of the 35 genes that had not reached FDR<0.2 in the previous study (Sanders et al., 2015), the majority (≥70%) of qualifying risk variants are inherited PTVs; in contrast, this was only the case for 8 of the 84 genes previously identified (FDR<0.2) (Fisher’s Exact Test, P=7.45x10−5, OR=6.98, 95% CI: 2.39-21.96). Consistently, for these 35 genes, we observe inherited PTV Bayes Factors higher than those obtained in the previous TADA mega-analysis performed in largely simplex families (Sanders et al., 2015) (Kruskal–Wallis test, P=0.0003, Figure S6A). For five of these 35 genes (PCM1, STARD9, GRM6, RHPN1, and SLC10A1) and two of the remaining 84 genes (CTTNBP2 and ZNF559), the largest association signal is from inherited PTVs. Thus these 35 genes are enriched for genes whose association signal is primarily driven by inherited PTVs (Fisher’s Exact Test, P=0.02, OR=6.70, 95% CI: 1.03-73.81) (Methods), further indicating that there is a substantial, previously unrecognized signal from rare inherited variants.
Biological insights from known and novel ASD genes
Gene-set enrichment analyses (Methods) indicated that the set of 69 high-confidence ASD-risk genes identified in the TADA mega-analysis were enriched in a highly co-expressed group of transcriptionally co-regulated genes active during human cerebral cortical neurogenesis (Module M2; Parikshak et al., 2013); FMRP targets (Darnell et al., 2011); RBFOX1 targets (Weyn-Vanhentenryck et al., 2014), and genes enriched for expression in the brain vs. other tissues (Methods; Figure 4E). We also integrated new data from single-cell sequencing of 40,000 cells from human brain (Polioudakis et al., 2018) and previously published single-cell sequencing data (Lake et al., 2018; Nowakowski et al., 2017), which reveals an overall enrichment in mid-gestation and adult glutamatergic projection neurons for both the previously established (Sanders et al., 2015) and 16 newly identified ASD-risk genes (Methods; Figures S6C and S6D).
Many of the 16 new ASD-risk genes from this study fall into biological pathways or gene-sets of interest, including negative regulation of synaptic transmission (RAPGEF4), learning and memory (GRIA1 and PRKAR1B), and cytoskeletal organization (PCM1 and MYO5A) (Figure 4D). Other examples include PRKAR1B, which is in a gene co-expression module comprised of structural synaptic proteins that are highly co-expressed during human cerebral cortical neurogenesis and in which 60 genes harboring RDNVs in ASD probands from early exome sequencing studies are over-represented (Parikshak et al., 2013); and three genes that are found in the post synaptic density of human neocortex (Bayes et al., 2011): GRIA1, RAPGEF4, and DDX3X. RAPGEF4 is also a known FMRP target (Darnell et al., 2011) and was previously suggested as a potential ASD candidate gene, but lacked strong statistical support (Bacchelli et al., 2003). DDX3X was recently reported to account for 1%-3% of unexplained intellectual disability in females (Snijders Blok et al., 2015). Finally, 9 of these 16 new ASD-risk genes form a significant indirect PPI network in concert with previously associated ASD genes (Methods; seed indirect degrees mean permutation P=0.016, and CI degrees mean P=0.024) (Figure 4C).
Pathways harboring primarily de novo variation are dominated by transcriptional and chromatin regulation (De Rubeis et al., 2014). Using gene ontology enrichment analysis, we asked whether inherited ASD-risk variants cluster in distinct biological pathways, and whether those pathways are the same or different from those implicated by de novo variation. Indeed, genes where the majority of the signal is from inherited variants reveal different pathways than those published based on de novo risk, including novel pathways related to ion transport (z = 3.7), cell cycle (z = 4.2), and the microtubule cytoskeleton (z = 5.7) (Figure S6E).
ASD-risk genes form a PPI network with candidate genes harboring high-risk inherited variation
We then asked if the proteins encoded by the 69 ASD-risk genes identified in the TADA mega-analysis (FDR<0.1) interact with the 98 candidate genes harboring high-risk inherited variants. The resulting PPI network formed by these 165 unique genes is significant for all reported network properties (P<0.05, Methods, Figures 5A and S6B). This network reveals interactions between genes with different levels of statistical support, ranging from high-risk inherited candidate genes, established ASD-risk genes, and new ASD-risk genes, which suggests that many of these 98 candidate genes are true ASD-risk genes. This network is preserved even when we limit the PPI analysis to genes emerging from the version of the TADA mega-analysis that excluded de novo variants from the iHART cohort (FDR<0.1; Table S3), with the seed direct and indirect degrees mean both reaching significance (P=0.013 and P=0.0009, respectively). Thus, inherited risk variants critically contribute to this network.
Figure 5. PPI networks formed by ASD-risk genes.
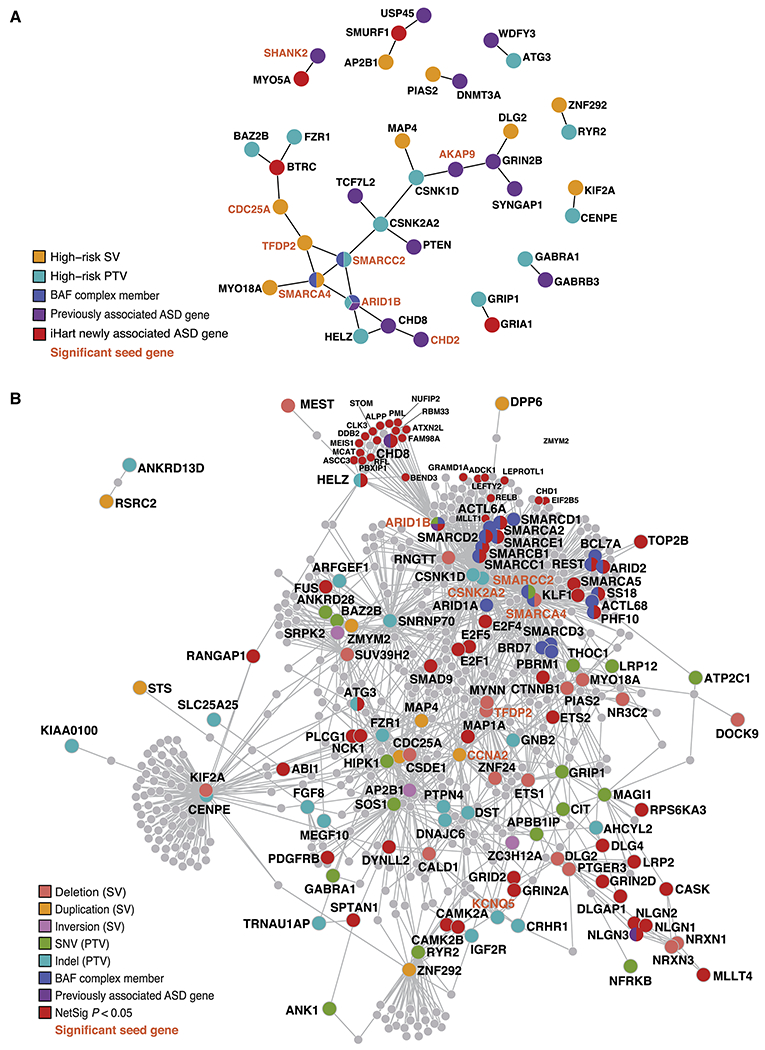
(A,B) Proteins encoded by previously known ASD-risk genes (Sanders et al., 2015) are shown in purple, those belonging to the BAF complex are blue, and those belonging to more than one category are shown with all colors that apply. Gene labels for significant seed genes are bold and orange. (A) Direct PPI network formed by constrained genes harboring high-risk inherited variants (98 genes) and ASD-risk genes identified in the TADA mega-analysis (69 genes, FDR<0.1). The direct PPI network formed by these 165 unique genes is significant for three connectivity metrics: the direct edges count (P=0.036), the seed direct degrees mean (P=0.046), and the CI degrees mean (P=0.005). Proteins encoded by a gene with a high-risk inherited SV are shown in gold, those with PTVs are teal, and those that are a newly identified ASD-risk gene by the iHART TADA mega analysis are shown in red. (B) Indirect PPI networks seeded by genes harboring high-risk inherited variants (98 genes). Proteins are colored according to the variant class identified and NetSig significant genes (P<0.05) are shown in red.
Given that a large number of predicted ASD-risk genes remain unidentified (Ronemus et al., 2014), we applied NetSig to identify high probability candidate genes via integration of PPI and association statistics (Horn et al., 2018). We identified 596 genes that were significantly more directly connected to ASD-risk genes than expected by chance (Figure 5B; Methods; Table S4), 38 of which are enriched in a developmental co-expression module previously shown to contain de novo variants in ASD-probands (module M2, Parikshak et al., 2013, P=0.0003, OR=1.98; 95% confidence interval=1.37-2.81). Interestingly, proteins in the network seeded by 98 high-risk inherited genes interact with NetSig candidates more than expected by chance, both directly (P = 0.02; OR=12.80; 95% confidence interval = 1.07-111.92) and indirectly (P = 4.24x10−16; OR=4.90; 95% confidence interval = 3.45-6.85) (Methods; Figure 5B), providing further evidence that the genes identified by the analysis of high-risk inherited variants are likely to include true ASD-risk genes.
Zebrafish modeling of NR3C2 syndromic ASD
Because previous evidence for NR3C2 was inconsistent (De Rubeis et al., 2014; Sanders et al., 2015), but supported by our analyses, we sought to firmly establish NR3C2 as an ASD-risk gene by in vivo zebrafish modeling. We created a predicted null mutation in the single zebrafish nr3c2 ortholog using CRISPR/Cas9 (Hwang et al., 2013) (Figures S7A and S7B). Homozygous mutant animals are viable, fertile, and morphologically indistinguishable from their wild-type (WT) siblings. We first asked whether nr3c2 mutant zebrafish exhibit abnormal social behaviors by developing and validating (Methods, Figures S7C–H) a modified version of a previously-described social preference assay (Figure 6A) (Dreosti et al., 2015). We found that WT animals display a social preference for conspecifics (Figures S7C and S7F) at three weeks of age or older (data not shown), as reported (Dreosti et al., 2015). We found that on average, nr3c2 +/+ and nr3c2 +/− animals showed a social preference, but nr3c2 −/− animals did not (Figures 6B and 6C). There was no significant difference in the size of nr3c2 −/− animals compared to their nr3c2 +/+ or nr3c2 +/− siblings (Figure S7I), suggesting that the mutant phenotype was not simply due to developmental delay. This result indicates that nr3c2 −/− animals have a social behavioral deficit.
Figure 6. nr3c2 mutant zebrafish exhibit impaired social preference behavior and disrupted sleep at night.
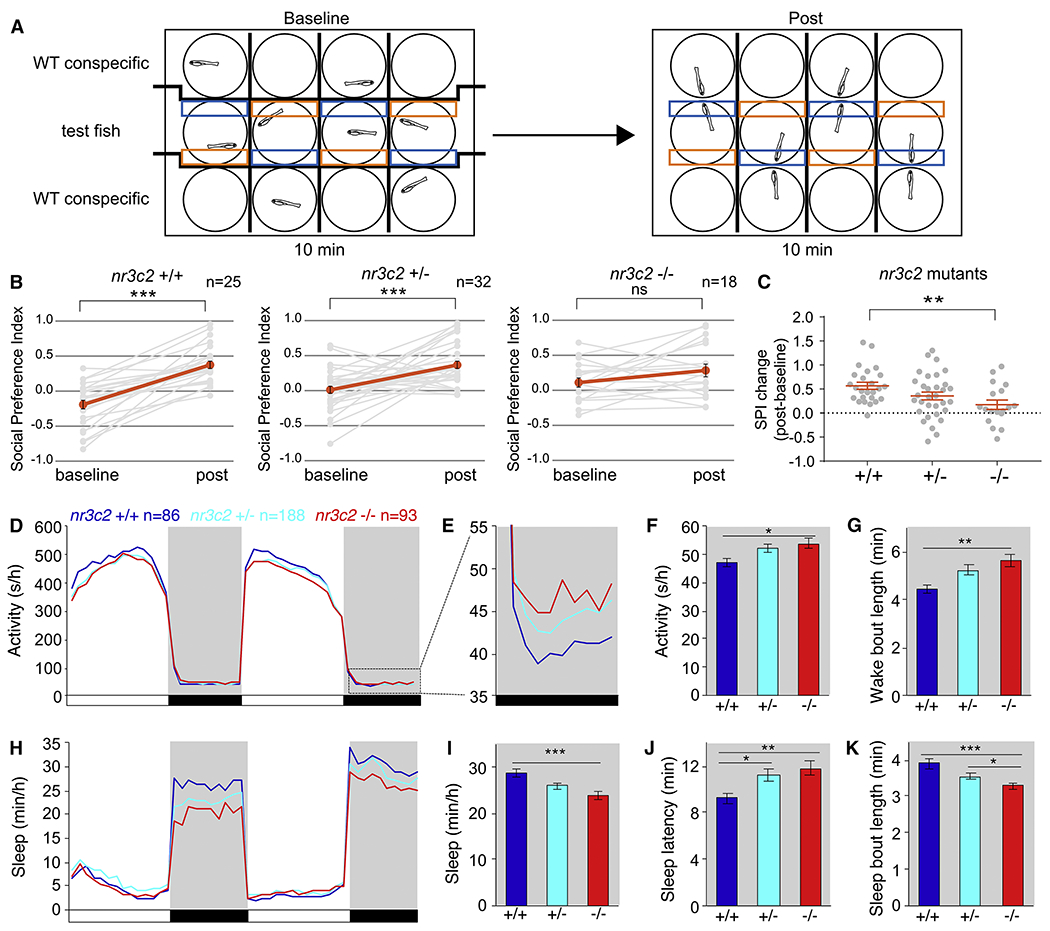
(A) Schematic of social preference behavioral assay. Boxes indicate regions used to quantify time spent by the test fish near (blue) and far (orange) from the conspecific. Thick lines indicate opaque dividers. (B) nr3c2 +/+ and nr3c2 +/− animals on average showed a significant preference for the conspecific but nr3c2 −/− animals did not. (C) The change in social preference index (SPI post – SPI baseline) was significantly smaller for nr3c2 −/− animals compared to their nr3c2 +/+ siblings. Grey data represent individuals. Red data indicate mean ± SEM. (D-K) Compared to their nr3c2 +/+ siblings at night, nr3c2 −/− animals were 14% more active (D-F) and slept 17% less (H,I) due to 27% longer wake bouts (G) and 16% shorter sleep bouts (K). nr3c2 −/− animals also showed a 28% longer sleep latency (time to first sleep bout at night) (J). There was no difference among the three genotypes in the number of sleep bouts at night or in any of these measures during the day (data not shown). Boxed region in (D) is magnified in (E). White and black bars indicate day (14 h) and night (10 h). Grey shading indicates night. Line graphs show mean and bar graphs show mean ± SEM for 5 pooled experiments. n=number of animals. *P<0.05; **P<0.01; ***P<0.001, ns=not significant by paired t test (B), one-way ANOVA with Tukey’s HSD post-hoc test (C), or one-way ANOVA with Holm-Sidak post-hoc test (F,G,I-K). See also Figure S7.
Second, because ASD is often comorbid with disrupted sleep (Maxwell-Horn and Mallow, 2017), we assayed sleep/wake behaviors (Prober et al., 2006) in 5-7 day old nr3c2 mutants. We found that nr3c2 −/− animals were more active and slept less at night compared to their nr3c2 +/− and nr3c2 +/+ siblings (Figures 6D–6F, 6H and 6I). This effect was due to increased sleep latency, longer wake bouts and shorter sleep bouts (Figures 6G, 6J and 6K), indicating defects in both sleep initiation and maintenance, similar to sleep phenotypes observed in individuals with ASD (Ballester et al., 2018; Maxwell-Horn and Malow, 2017). Thus, nr3c2 mutant zebrafish exhibit both social deficits and sleep disturbances, parallel to core and comorbid phenotypes observed in humans with ASD, which is consistent with the genetic evidence implicating NR3C2 as an ASD-risk gene.
DISCUSSION
To date, de novo variants have provided compelling evidence for dozens of ASD-risk genes, but studies in primarily simplex families have yielded little, if any, inherited risk signal. Here, we used WGS to identify over a dozen new genes that are significantly associated with ASD-risk, the majority of which exhibit a contribution from rare inherited mutations. The identification of more than a dozen novel ASD-risk genes was facilitated by studying families ascertained for containing two or more children with ASD, where inherited risk variants are likely to contribute to the observed ASD recurrence (Ronemus et al., 2014; Sebat et al., 2007; Virkud et al., 2009). We provide strong support for 69 ASD-risk genes, 24 of which reach genome-wide significance after Bonferroni correction (Table 1). This extends previous work substantially, as only a few genes had previously passed this threshold. The fact that we did not find differences in the rate of rare inherited variants between affected and unaffected children is consistent with both (1) the known lower effect size of inherited ASD-risk variation (as compared to de novo pathogenic mutations) and (2) the expectation that in multiplex families the unaffected siblings (like their unaffected parents) also carry ASD-risk variation (reduced penetrance), necessitating large sample sizes. Nevertheless, we identified a significant excess of constrained genes harboring inherited PTVs transmitted to all affected children and not transmitted to any unaffected children, and found that these genes converge in a PPI network. This significant PPI network is seeded by known ASD-risk genes, including multiple members of the BAF complex and other chromatin modifiers, and is also enriched for proteins that interact with additional ASD-risk genes, many of which are involved in cortical neurogenesis (Parikshak et al., 2013). Single cell sequencing data reveals that many of these ASD-risk genes are expressed in developing glutamatergic neurons (Figures S6C and S6D), lending further support to the role of ASD-risk genes in neurogenesis.
We employed WGS to enable the detection of non-coding variants and structural variation at high resolution, and identified small non-coding regulatory deletions for both DLG2 and NR3C2. The shared phenotypic features amongst the NR3C2 variant carriers are consistent with a new syndromic form of ASD (Table S1). We were able to infer biological importance of the NR3C2 putative regulatory deletion from its open chromatin state in human developing brain (de la Torre-Ubieta et al., 2018) and phenotypic concordance to the family harboring the coding PTV. We also modeled this syndromic ASD in zebrafish, finding that the mutant animals exhibit both social deficits and sleep disturbances. We also identified a recurrent deletion significantly associated with ASD that disrupts the DLG2 promoter, which further emphasizes the utility of WGS in identifying small functional deletions in non-coding regulatory regions.
More broadly, we found no global enrichment for non-coding variation in promoters – structural variant or otherwise – in affected vs. unaffected children. Consistently, a previously investigation of 53 simplex families found a small enrichment (P=0.03) for private and de novo disruptive variants in fetal brain DNase I hypersensitive sites in probands. However, this signal was limited to DNase I hypersensitive sites within 50Kb of genes that had been previously associated with ASD-risk (Turner et al., 2016). More recent studies are consistent with our lack of enrichment for rare, non-coding variation (Werling et al., 2018). Advances in methods for analyzing the non-coding genome, similar to what has been done to identify functional PTVs (e.g., constraint metrics such as pLI), as well as increased sample sizes, are necessary to improve power for identifying non-coding risk variants.
As previous studies have shown, inherited variation alone does not explain all instances of ASD within multiplex families, consistent with complex genetic contributions that include de novo mutations (Leppa et al., 2016). Given our success in uncovering many ASD-risk genes whose signal is derived at least partially from inherited variation, even modest increases in sample sizes from families with multiple affected children will likely confirm many new genes. Our machine learning classifier, Artifact Removal by Classifier (ARC), will also enable increases in sample sizes when only LCL-derived DNA is available by distinguishing sequencing and cell line artifacts from true de novo variation.
As sample sizes grow, we can confirm whether our observed differences between simplex vs. multiplex families are generalizable, but our data suggest substantial differences in their genetic architecture. Furthermore, with larger cohorts, we may be able to explore additive effects of both common and rare inherited variation and classify risk genes based on inheritance – (1) de novo, (2) inherited, or (3) de novo + inherited – to establish if these distinct gene classes are associated with phenotypic severity and/or specific biological pathways.
One striking finding of our study is that genes where the majority of the autism signal is from inherited variants are in pathways related to ion transport, cell cycle and the microtubule cytoskeleton (Figure S6E). In contrast, genes harboring primarily de novo variation are enriched in pathways related to transcriptional and chromatin regulation. These observations suggest that inherited and de novo variation, the former expected to have smaller effects and reduced penetrance and the latter with larger effects (Kosmicki et al., 2017), may impact distinct biological processes. Nevertheless, the ASD-risk genes identified here contribute to cellular processes that are interconnected at the level of gene co-expression and PPI networks – a pattern of interaction that we hypothesize will be replicated in future studies having more power to assess variants on a broad continuum of effect sizes.
The iHART portal (http://www.ihart.org/home) provides researchers access to these data, facilitating additional analyses of these samples and integration with future cohorts.
STAR ★ METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
The whole-genome sequencing data generated during this study are available from the Hartwell Foundation’s Autism Research and Technology Initiative (iHART) following request and approval of the data use agreement available at http://www.ihart.org. We provide the code for ARC (Artifact Removal by Classifier), our random forest supervised model developed to distinguish true rare de novo variants from LCL-specific genetic aberrations or other types of artifacts such as sequencing and mapping errors, together with a full tutorial at https://github.com/walllab/iHART-ARC. The zebrafish mutant line generated in this study will be deposited to the Zebrafish International Resource Center (ct867, ZFIN ID: ZDB-ALT-190607-1). Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dennis Paul Wall (dpwall@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
ASD multiplex family samples
The UCLA and Stanford IRBs designated this study as “Not human subjects research” and therefore exempt from review; this was due to the study being limited to previously-existing coded data and specimens. Study subjects were carefully selected from the Autism Genetic Resource Exchange (AGRE) (Lajonchere, 2010) and chosen from families including two or more individuals with ASD (those with a “derived affected status” of “autism”, “broad-spectrum”,”nqa”,”asd”, or “spectrum”). Patients with known genetic causes of ASD (15p13 duplication ,15q deletion, 15q duplication, 16p deletion, 16p duplication, 22q duplication, mosaic for deleted Y, mosaic trisomy 12, Trisomy 21 (Down Syndrome), Fragile X) or syndromes with overlapping ASD-features (Gaucher Disease, Marfan’s Syndrome, Sotos Syndrome) were excluded from sequencing. We prioritized ASD-families harboring affected female subjects. We also prioritized monozygotic-twin containing families, in part to facilitate the development of our machine learning model (Artifact Removal by Classifier (ARC)). A complete list of sequenced samples can be found in Table S1.
A total of 2,308 individuals from 493 ASD families from the Autism Genetic Resource Exchange (AGRE) (Table S1) passed quality control. Details for each of these 2,308 samples, including sex, ethnicity, phenotype, and familial relationship, can be found in Table S1. Unless otherwise specified (Methods or Table S1), our analyses included a subset of 1,177 children (960 affected and 217 unaffected children) for whom both biological parents were sequenced.
Purified DNA was obtained from the Rutgers University Cell and DNA Repository (RUCDR; Piscataway, NJ). Where available, DNA from whole blood was used; however, for many samples, only lymphoblastoid cell line (LCL) DNA was available because DNA was not extracted from whole blood at the time of recruitment.
Control cohorts
Throughout this manuscript, we reference several control cohorts used for assessing variant frequencies in samples not ascertained for ASD. These cohorts are described below. The Alzheimer’s Disease cohort was only used for the high-risk inherited simulation analysis. The Genome Aggregation Database (gnomAD) cohort was only used for the analysis of non-coding variants.
Publicly available databases
Unless otherwise specified, the publicly available databases (all annotations provided by ANNOVAR) referenced include: the NHLBI Exome Sequencing Project (ESP, esp6500siv2_all) ( http://evs.gs.washington.edu/EVS/), the Exome Aggregation Consortium (ExAC_ALL annotation from version exac03nonpsych) (Lek et al., 2016), 46 unrelated, whole-genome sequenced (high coverage on the Complete Genomics platform), non-disease samples (http://www.completegenomics.com/public-data/69-genomes/, cg46)(Drmanac et al., 2010) and the 1000 genomes project (1000g2015aug_all) (Auton et al., 2015).
UCLA internal controls
Throughout this manuscript, the use of “UCLA internal controls” refers to a set of 379 unrelated, whole-genome sequenced (30x coverage on Illumina platform, processed by the same bioinformatics pipeline as was used for iHART) samples with a neurodegenerative disorder known as Progressive Supranuclear Palsy (PSP). There is no known etiological overlap or comorbidity between PSP and ASD.
Healthy Non-Phaseable (HNP) samples
Throughout this manuscript, the use of “HNPs” refers to the 922 healthy non-phaseable (no biological parents sequenced) iHART samples. The majority of these samples are parents of affected or unaffected children. Due to the fact that these samples likely harbor genetic ASD-risk variants, these HNPs provide a helpful estimate of allele frequencies but we generally apply more permissive allele frequency filtering to retain inherited risk variants.
Alzheimer’s Disease
The Alzheimer’s Disease (AD) cohort (n=1,173 unrelated samples) was selected as a control group for the high-risk inherited simulation analysis (Bennett et al., 2018). This AD cohort was selected because of the lack of ASD comorbidity and the late-onset of the disease which precludes ASD diagnoses in this cohort.
gnomAD
We used allele frequency estimates from gnomAD (version 2.0.2) (Karczewski et al., 2019) for the analysis of non-coding variants because these data include 15,708 genomes from unrelated individuals which facilitates allele frequency estimation in the non-coding regions of the genome.
Curated Database of Genomic Variants (cDGV)
To assess the population frequency of structural variants in a more precise manner, we manually curated the Database of Genomic Variants (DGV, release date 2015-07-23) (MacDonald et al., 2014). This curation involved removing studies that did not include sample identifications and/or only analyzed targeted genomic regions, as well as SVs detected in non-human samples or individuals diagnosed with intellectual disability (ID) or developmental delay (DD) (ID and DD samples in two studies, Coe et al. 2014 (Coe et al.) and Cooper et al. 2011 (Cooper et al., 2011), were flagged for exclusion by Evan Eichler’s laboratory and their accession numbers were shared with us via personal communication). This resulted in a total of 26,353 unique samples with DGV data. We then removed redundancies in DGV’s SV types by collapsing all SV types in the remaining samples into five different categories: deletions (“deletion” + “loss”), duplications (“duplication” + “gain” + “tandem duplication”), insertions (“insertion” + “mobile element insertion” + “ novel sequence insertion”), inversions, and unknown (“complex” + “gain+loss” + “sequence alteration”). We finally re-calculated the frequency of the different SV categories by continuous genomic intervals, avoiding double-counting SVs (of the same type) identified in the same sample and same region by different studies.
Zebrafish studies
Zebrafish experiments and husbandry followed standard protocols in accordance with Caltech Institutional Animal Care and Use Committee (IACUC) guidelines (animal protocol 1580). Zebrafish behaviors were studied before the onset of sexual differentiation and were performed using siblings with the same genetic background, differing only in nr3c2 genotype, or in treatment with drugs and appropriate vehicle controls. WT and mutant stocks were derived from a TLAB hybrid strain. Animals were raised on a 14:10 hour light:dark cycle, and were housed in petri dishes with 50 animals per dish in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) until 4 days post-fertilization. Animals were then either assayed for sleep/wake behaviors, or were transferred to 0.8 L tanks and fed rotifers (Brachionus plicatilis) twice per day until reaching 2 weeks of age. Animals were then fed brine shrimp (Artemia salina) until 3-4 weeks of age, at which point their social behavior was assayed. Animals were not involved in any previous procedures and were naive to the tests and drugs used. The zebrafish mutant generated in this study will be made available upon request.
METHOD DETAILS
Whole-genome sequencing and data processing
DNA samples were submitted to the New York Genome Center (NYGC) for whole-genome sequencing. DNA samples were examined for quality/quantity, and subsequently genotyped using Illumina Infinium Human Exome-12 v1.2 or Infinium Human Core Exome microarrays (San Diego, CA) according to standard manufacturer protocols. Identity-by-descent estimation and sex checks in PLINK v1.07 (Purcell et al., 2007) were used to validate expected vs. observed family relationships and confirm sample identity based on these genome-wide genotyping data. Contamination was assessed using verifyIDintensity (VII) (Jun et al., 2012); samples exceeding 3% contamination in two or more modes were excluded from sequencing.
Samples passing these array-based identity and quality checks were sequenced at NYGC using the Illumina TruSeq Nano library kits and Illumina’s HiSeq X (San Diego, CA) according to standard manufacturer protocols.
All iHART WGS data were processed through the same bioinformatics pipeline; this pipeline was designed based on GATK’s best practices (DePristo et al., 2011; Van der Auwera et al., 2013). The metadata for each sample are stored in a custom MySQL database where each sample was tracked as it progressed through the sequencing pipeline at the New York Genome Center (NYGC) and bioinformatic processing, and finally the quality assurance metrics were populated based on the resulting processed data. The first step in the pipeline was to align the raw short sequence reads to the human reference genome (human_g1k_v37.fasta). This was accomplished by processing the fastq files with the Burrows-Wheeler Aligner (bwa-mem, version 0.7.8) (Li and Durbin, 2009) to generate BAM files. BAM files were generated in a read-group-aware fashion (properly annotating sequence reads derived from the same flow cell and lane) and thus multiple BAM files were subsequently merged using BamTools (version 2.3.0) (Barnett et al., 2011) to generate a single BAM file per sample. The second step in the pipeline was to mark duplicate reads in the BAM file using the Picard MarkDuplicates tool (version 1.119; http://broadinstitute.github.io/picard/). The third step in the pipeline was to perform local realignment of reads around indels using GATK’s IndelRealigner (version 3.2-2). The fourth step in the pipeline was to genotype each sample, generating a gVCF file. To achieve accuracy at this stage, base quality score recalibration was run using GATK (version 3.2-2) (McKenna et al., 2010). Subsequently, GATK’s HaplotypeCaller (version 3.2-2) was run on each base-recalibrated BAM to identify the variant and non-variant bases in the genome. All four of these steps were performed at the NYGC, resulting in a BAM and a gVCF file for each sample.
The fifth step in the pipeline was to jointly call variants across all AGRE/iHART samples, to generate a VCF file. This was accomplished by combining gVCF files, 200 samples at a time using GATK’s combineGVCFs (version 3.2-2), and then running GATK’s GenotypeGVCFs (version 3.2-2). Step 5 was accomplished by splitting data by chromosome (which increases parallelization) and resulted in one cohort-wide VCF per chromosome. Finally, to help filter out low quality variants within the call set, GATK’s Variant Quality Score Recalibration (VQSR, version 3.2-2) was run to generate well-calibrated quality scores. The final step in the pipeline was to annotate the resulting variant calls (SNVs and indels) in order to generate an annotated VCF file. This was accomplished by annotating with ANNOVAR (version 20160201) (Wang et al., 2010) and then with Variant Effect Predictor (version VEPv83) (McLaren et al., 2016). The resulting VCF contains gene-based, region-based, and filter-based annotations for each identified variant. For all the analyses described in this manuscript, we excluded VQSR failed variants and multi-allelic variants.
Quality control assessment
We performed standard quality control checks on our WGS data to ensure both sequencing/variant quality and sample identity. This included checking relatedness between samples, exclusion of duplicate samples, concordance between genotyping chip and WGS data, concordance between self-declared sex and observed biological sex, exclusion of samples with contamination from other samples, variant quality evaluation with GATK’s VariantEval module (data not shown), and sequencing coverage. A total of 2,308 individuals from 493 ASD families, from the Autism Genetic Resource Exchange (AGRE) passed quality control (Table S1).
Whole-genome sequence coverage
We used SAMtools v1.2 (Li et al., 2009) depth utility to calculate genome-wide (excluding gap regions in the human reference genome, downloaded from the UCSC table browser) per-base sequencing coverage for each sample. In order to reduce memory requirements, the reported depth was truncated at a maximum of 500 reads. Subsequently, we calculated two main summary statistics for each sample using custom scripts: (i) average coverage and (ii) percent of the genome (excluding gap regions) covered at 1X, 10X, 20X, 30X and 40X. On average, 98.97 ± 0.37 % of bases were covered at a depth of ≥10X (Figure S1A–E).
Variant inheritance classifications
Children with only a single parent sequenced are referred to as partially phase-able and children with both parents sequenced are referred to as fully phase-able. For each member of the iHART cohort with at least one parent sequenced (partially or fully phase-able affected or unaffected children), all identified variants were classified based on their observed inheritance. Every variant was categorized into one of eight inheritance types: (i) de novo, (ii) maternally inherited, (iii) paternally inherited, (iv) newly homozygous, (v) newly hemizygous, (vi) missing, (vii) unknown phase, or (viii) uncertain. To perform this classification, we developed a custom script to simultaneously evaluate variant quality and inheritance within each family. Prior to this classification step, all VQSR failed variants and multi-allelic variants were excluded. Additionally, we set permissive quality control thresholds in order to retain sensitivity while removing variants with a high probability of being false positives. Variants were required to have a depth of ≥10x, a genotype quality of ≥25, and a ratio of alternative allele reads/total reads ≥0.2. We assumed that if a variant met these quality thresholds, then the assigned genotype was correct.
While maternally inherited, paternally inherited, and de novo categories are self-explanatory, definitions for the remaining inheritance classifications are more complex. A homozygous variant observed in a child is called a newly homozygous variant if it is heterozygous in both parents. Similarly, a newly hemizygous variant on the X chromosome is defined as a hemizygous variant observed in a male child which is not identified as hemizygous in the corresponding father. A variant was classified as missing (./.) if the variant was called in at least one child in the iHART cohort but did not have sufficient coverage for GATK’s haplotype caller to define a genotype. A variant was classified as unknown phase if a child had an inherited variant and only one biological parent sequenced (unless on a sex chromosome where inheritance can be inferred) or if both parents carry the variant and thus the phase cannot be determined from this site alone. Finally, a variant was classified as uncertain if it could not be classified into another inheritance type; this includes: Mendelian error variants (e.g., heterozygous variants on male sex chromosomes), variants failing the quality control thresholds above (in a child or a parent), or a variant that couldn’t be classified with confidence (e.g., a variant identified in a child but absent in its only sequenced parent could be de novo or inherited). Unless otherwise specified, variants classified as missing, uncertain, or unknown phase were excluded from our analyses.
Detection of large structural variants
We developed a custom pipeline for high-resolution detection of large structural variants (SVs) from whole-genome sequence data (Figure S1F–H). This pipeline combines four different detection algorithms, including: BreakDancer (Chen et al., 2009), LUMPY (Layer et al.), GenomeSTRiP (Handsaker et al.; Handsaker et al.), and Somatic MUtation FINder (SMuFin) (Moncunill et al., 2014) (Methods; Figures 1 and S1F–H).
BreakDancer
We first used the bam2cfg.pl script (part of the BreakDancer v1.1.2 package (Chen et al., 2009) to generate a tab-delimited configuration file per family required to run BreakDancerMax. This configuration file specifies the locations of the BAM files, the desired detection parameters (the upper and lower insert size thresholds to detect SVs) and sample metadata (e.g., read group and sequencing platform); we used default detection parameters. We then ran BreakDancerMax to call SVs per chromosome within families. The resulting output files were combined for all chromosomes and samples and converted into a single VCF file using a custom script (see SV post-detection processing for details about genotyping). We filtered to exclude variants if the identified variant (i) was in a sequence contig, (ii) had a quality score <80, (iii) had <4 supporting reads, or (iv) had a length of <71 base pairs (small indel).
LUMPY
We used SAMtools v1.1 (Li et al., 2009) to extract both the discordant paired-end reads and the split-read alignments per sample, generating two different sorted BAM files required to run LUMPY v0.2.11(Layer et al., 2014). We then ran lumpyexpress to call SVs within families. We merged the resulting VCF files per family (containing raw calls), into a single genotyped VCF file for all the samples in the cohort, using a custom script (see SV post-detection processing for details about genotyping). We filtered to exclude variants if the identified variant (i) was in a sequence contig, (ii) was a small insertion or inversions with a length of <71 base pairs. No filter was applied for small duplications because the min length identified was 74 base pairs.
GenomeSTRiP
We obtained genotyped SV calls generated by the NYGC’s in-house GenomeSTRiP v1.04 standard pipeline (Handsaker et al., 2011; Handsaker et al., 2015). This pipeline consists of three main modules: (i) SVPreprocess: a pre-processing module that was run per sample to generate genome-wide metadata required for next processes; (ii) SVDiscovery: a discovery module, that was run in three large batches to call deletions, producing a VCF file with raw calls detected per batch; and (iii) SVGenotyper: a module run to produce genotyped VCF files per sequencing batch. In total, we received three genotyped VCF files, for sequencing batch one (N=956 samples), two (N=538 samples), and three (N=858 samples). We filtered out variants flagged as “LowQual” and merged the final set of SV calls for downstream analyses.
SMuFin
We adapted Somatic MUtation FINder (SMuFin) (Moncunill et al., 2014), a reference-free approach, for family-based structural variant detection by performing de novo alignment of child reads to the parental reads (Figure S1H), to provide high sensitivity and break point accuracy in the detection of SVs. Families were processed as independent trios and SMuFin was used to directly contrast sequencing reads between the parents and the offspring (Figure S1H). During the detection process, one parental genome is used as the reference genome to identify genetic variants in the children that were absent in that parent and then this process is repeated using the other parental genome as the reference genome. This produced one output file for each parent-offspring comparison run, containing the SVs detected per comparison. We then merged all the SV calls identified in phase-able individuals (i.e., individuals for which at least one biological parent was also sequenced) and classified them according to their inheritance patterns.
SV post-detection processing
We assumed heterozygosity for all SV calls, with two exceptions: (i) SVs identified in sex chromosomes from males, which were annotated as homozygous; and (ii) SVs identified by GenomeSTRiP, whose genotypes were defined by its SVGenotyper module. The inheritance type for all SVs identified in phase-able individuals was classified as: de novo, maternal, paternal, newly homozygous, newly hemizygous, unknown phase, missing, or uncertain – as defined above. For SVs, the missing classification was only applied to BreakDancer calls with a quality score of <80 and/or <4 reads supporting the variant call.
We focused on the analysis of high-confidence SVs, specifically deletions (DELs), duplications (DUPs), and inversions (INVs), by restricting to events identified by at least two detection algorithms and removing SVs that overlapped genomic regions of low complexity (i.e., centromeres, segmental duplications, regions of low mappability, and regions subject to somatic recombination in antibodies and T-cell receptor genes) (Brandler et al., 2016) by more than 50%. We made two exceptions to the rule that at least two detection algorithms must detect an SV. The first exception was to exclude SVs detected by only LUMPY and BreakDancer because this subset of SVs had very low concordance with genotype array data (Table S5). The second exception was to include an SV event if it was called by at least two detection algorithms in one or more family members, but called by only one algorithm in another family member.
Even though WGS theoretically enables high-resolution prediction of breakpoints, the breakpoints called by the detection algorithms can vary due to technical differences between these methods and also between samples (e.g., coverage) despite the fact that they are detecting the same underlying SV event. To adjust for this, SV calls made by different detection algorithms were considered to be the same SV event if they were: (i) called in the same individual, (ii) had a reciprocal overlap of at least 50%, and (iii) shared the same SV type (e.g., DEL) and inheritance pattern. A similar approach was subsequently applied to SVs within a family, where SV events are likely inherited and thus identical; the breakpoints of overlapping SVs (≥50% reciprocal overlap of the same SV type) identified in individuals within the same family, were adjusted to the predicted minimum start and maximum end coordinates predicted (maximum size based on breakpoints) in family members with the SV call.
SVs were defined as rare if they had no more than 50% overlap in (a) regions commonly disrupted by SVs in our Curated Database of Genomic Variants (cDGV; allele frequency ≥0.001) and (b) regions commonly disrupted by the same SV type (allele frequency ≥0.01) in the HNP samples. We also classified SVs as rare if (c) they had a region of ≥500Kb that did not overlap with common SVs in cDGV (allele frequency ≥0.001) or HNP samples (allele frequency ≥0.01).
Finally, in order to facilitate prioritization for likely pathogenic variants, gene-based and region-based annotations were added to the final set of high-confidence SV calls by using custom scripts and the Bamotate annotation tool (Leppa et al., 2016).
Multi-algorithm consensus SV calls
The four algorithms chosen to call SVs use different detection strategies and are suitable for identifying different sizes and types of SVs with varying levels of sensitivity and specificity. Therefore, we ran a multi-algorithm comparison to identify high-quality SVs, identified by at least two methods (as described above). We used BEDTools (Quinlan and Hall, 2010) to intersect SV calls detected by the different algorithms, by performing an all-against-all comparison (Figure S1F and Table S5).
The start and end positions of identical SV events identified for an individual (≥50% reciprocal overlap of the same SV type and inheritance pattern) were reassigned based on the coordinates from the detection algorithm predicted to be more precise in calling breakpoints. By considering the strategy implemented to identify SVs (e.g., split-read methods can detect SVs at single base-pair resolution) for each detection algorithm, we defined the following rank for breakpoint precision accuracy: SMuFin (split-read and de novo assembly method) > LUMPY (split-read and read-pair method, with coordinates assigned within families) > GenomeSTRiP (split-read, read-pair and read-count method, with coordinates assigned within sequencing batch) > BreakDancer (read-pair detection method).
Array-based SV detection is a well-established method with high accuracy for certain SV classes, in particular large deletions (Miller et al., 2010). Thus, to confirm our ranking of algorithms by their SV breakpoint precision, we compared our WGS-based SV calls to SV calls obtained from Illumina genotyping array data (Leppa et al., 2016) on overlapping AGRE samples. Specifically, we identified a high confidence set of heterozygous deletions for which heterozygous deletions were also detected (≥50% reciprocal overlap) in the array data (n=224 SVs). We then used GATK’s VariantEval tool to generate het:hom metrics for SNVs identified within 224 heterozygous deletions.
A heterozygous deletion with accurate break points would include only homozygous SNVs (het:hom ratio of zero). This analysis reavealed no significant differences between these methods (with all of them showing a median het:hom ratio of 0.01), but ranking of mean het:hom ratios was generally consistent with our ranking of algorithms by their SV breakpoint precision: SMuFin (0.028)< LUMPY (0.043) < BreakDancer (0.059) < GenomeSTRiP (0.067).
Joint LUMPY-BreakDancer SV call inspections
Copy Number Variants (CNVs) detected from genotyping array data can be visualized by plotting the B Allele Frequency and Log R Ratio values for array genotyped SNPs within the estimated CNV region and its flanking regions (25% of the length of the CNV on each side); we will refer to this as an “array visualization plot”. Given the low concordance rate between LUMPY and BreakDancer SV calls with other methods (Table S5), we manually inspected array visualization plots generated by using available Illumina genotyping array data (Leppa et al., 2016), for regions with LUMPY-BreakDancer joint SV calls identified in the iHART WGS data.
We randomly selected LUMPY-BreakDancer detected SV events within bins containing events of different sizes/lengths (n=218) and used a custom script to generate array visualization plots for each detected SV region. For each of the 218 SVs, an array visualization plot was generated for the carrier and all corresponding family members. Manual inspection of the array visualization plots was conducted (blinded with respect to the predicted carrier(s) of the LUMPY-BreakDancer SV call), and each SV was categorized as true or false. By treating the array-based true calls as the gold standard, we were able to estimate the validation rate for LUMPY-BreakDancer joint SV calls (Table S5).
Sensitivity to detect rare SVs
A set of rare SVs detected from Illumina genotyping array data (array-SVs) were available for 553 iHART fully phase-able samples (Leppa et al., 2016). We used BEDTools (Quinlan and Hall, 2010) to intersect our set of SV calls (WGS SV calls, DELs and DUPs) with rare SVs identified in genotyping array data (Leppa et al., 2016) in these 553 overlapping samples. We evaluated our sensitivity to detect array-SVs by considering events detected with ≥50% reciprocal overlap by both array and NGS in the same sample – both with and without LUMPY-BreakDancer joint SV calls (Table S5).
Defining rare inherited and private variants
We define rare inherited variants (SNVs and indels) as those with an allele frequency (AF) less than or equal to 0.1% in public databases (1000g, ESP6500, ExACv3.0, cg46), internal controls, and iHART HNP samples and were restricted to those not missing in more than 25% of controls and not flagged as low-confidence by the Genome in a Bottle Consortium (GIAB; Zook et al., 2014). Rare SVs (DELs, DUPs, INVs) were defined as those with an AF<0.001 in cDGV and an AF<0.01 in iHART HNP samples.
We define private variants as variants that are observed in one and only one iHART/AGRE family (AF~0.05%) and are not missing in more than 25% of Ihart HNPs. Additionally, private variants were (i) never observed in any control cohorts (AF=0), (ii) not missing in more than 25% of the PSP control samples, and (iii) not flagged as low-confidence by the GIAB consortium. We only report analyses for iHART private variants in the 1,177 children with both biological parents sequenced (fully phase-able). For non-coding private inherited variants, variants present in gnomAD (version 2.0.2) were also removed.
Non-coding analyses
Definition of non-coding variants
We defined non-coding SNVs and indels as variants that do not occur within a coding transcript, as annotated by VEP. This includes 17 of the 35 VEP consequences: “mature miRNA variant”, “5 prime UTR variant”, “3 prime UTR variant”, “non-coding transcript exon variant“, “intron variant”, “non-coding transcript variant”, “upstream gene variant”, “downstream gene variant”, “TFBS ablation”, “TFBS amplification”, “TF binding site variant”, “regulatory region ablation”, “regulatory region amplification”, “feature elongation”, “regulatory region variant”, “feature truncation”, or “intergenic variant”. If multiple annotations for consequence were present for a single variant, only the first most damaging consequence was considered in order to stringently filter for non-coding variants. Only variants that were not flagged as low-confidence by the GIAB consortium were considered. To increase our accuracy in assessing the allele frequency of these non-coding variants, we also annotated these variants with the Genome Aggregation Database (gnomAD) (version 2.0.2) allele frequencies identified from whole-genome sequencing of over 15K samples not enriched for ASD phenotypes. We defined promoters as 2Kb upstream and 1 Kb downstream from the transcription start site (TSS) by referencing the longest transcript for each gene (ties in transcript length were resolved by selecting the lower Ensembl Transcript ID). The ASD-risk genes used for this analysis are the 69 genes with an FDR<0.1 in the iHART TADA-mega analysis.
Samples included for non-coding analyses
iHART non-coding private variants were identified in the 1,177 children with both biological parents sequenced (fully phase-able) (Naff=960, Nunaff=217). iHART non-coding RDNVs were considered after running ARC to identify high confidence variants and were restricted to those identified in the 716 non-ARC outlier samples (Naff=575, Nunaff=141).
To increase our power for non-coding variants, we obtained data from 519 whole-genome sequenced Simons Simplex Collection (SSC) quads (mother, father, affected child, unaffected child). These data were also generated and processed to a per sample gVCF (GATK version 3.2-2) by NYGC. We then performed joint genotyping, annotation, and quality control using the same pipeline applied to the iHART genomes. After resolving 4 identity crises in these data by quality control, we removed one likely contaminated sample and two samples with unresolvable sex crises. This resulted in 516 quads and 3 trios from the SSC (Naff=517, Nunaff=518). We identified an average of 89 raw RDNVs per child in this cohort. After applying ARC to these data, we obtain an average of 61.83 RDNVs per SSC child which is very similar to the genome-wide expectation and matches the average observed for iHART RDNVs after applying ARC (60.3 RDNVs per child in LCL-derived samples and 59.4 RDNVs per child in WB-derived samples). Given that the SSC cohort is comprised entirely of WB-derived samples, we identified zero ARC outliers (no samples with >90% of their raw RDNVs removed by ARC). The resulting combined iHART + SSC whole-genome cohort includes 1,092 affected and 659 unaffected samples for RDNV analysis and 1,477 affected and 735 unaffected samples for the analysis of private inherited variants.
High-risk inherited variant analysis
To characterize potential high-risk inherited variants, we identified rare damaging variants which were transmitted to all affected individuals in a multiplex family, but not transmitted to unaffected children. High-risk inherited variants were further defined as those which disrupted a gene with a high probability of being loss-of-function (LoF) intolerant (pLI≥0.9, n=3,483 genes) (Lek et al., 2016). Such genes are also referred to as constrained genes because they are under evolutionary constraint – as evidenced by the lack of mutations in such genes in the general human population. Specifically, we considered rare PTVs (AF≤0.001 in public databases and internal controls) or rare SVs (AF≤0.001 in cDGV and AF≤0.01 in HNPs) disrupting an exon or promoter, where the promoter was defined as being 2Kb upstream of the TSS. The families selected for the PTV analysis were restricted to a subset of 346 families with ≥2 genetically distinct (i.e., not a family with just a pair of affected MZ twins) fully phase-able affected children with a variable number of unaffected children. Given the small number of qualifying SVs in these ASD families, all families (n=493) were considered.
We next used protein-protein interaction (PPI) analysis to assess whether the 98 genes harboring high-risk inherited variation showed evidence for biological convergence. To determine if these high-risk inherited variants formed a PPI network, we used the Disease Association Protein-Protein Link Evaluator (DAPPLE) (Rossin et al., 2011) and performed 1,000 permutations (within-degree node-label permutation). Given that PPI databases are incomplete and biased against typically less well-studied neuronal interactions (Parikshak et al., 2015), we frequently also expanded the network to include indirect interactions among the seed genes.
When we combined both high-risk inherited variant classes (PTVs and SVs), we found that the protein products of the 98 genes harboring high-risk inherited variation formed a significant direct PPI network (P < 0.008, 1,000 permutations). The protein products of both of the high-risk inherited variant classes also formed a significant direct PPI network on their own (P < 0.04 for the 61 genes hit by qualifying PTVs and P < 0.02 for the 40 genes hit by qualifying SVs).
Gene set enrichment (Methods) for the 98 genes harboring high-risk inherited variation identified a trend for enrichment for targets of RBFOX1 (Weyn-Vanhentenryck et al., 2014) (P=0.034, uncorrected), which regulates neuronal alternative splicing and previously has been implicated in ASD (Martin et al., 2007; Sebat et al., 2007).
The PPI network formed by the 69 TADA ASD-risk genes and the 98 genes harboring high-risk inherited variants (n=165 unique genes) was significant for all DAPPLE reported network properties (Rossin et al., 2011); this includes: the direct edges count (P=0.036), the seed direct degrees mean (P=0.046), the seed indirect degrees mean (P=0.003), and the CI degrees mean (P=0.005) (Figures 5A and S6B).
Simulations for high-risk inherited variants
We sought to establish how exceptional it was to observe 98 genes harboring high-risk inherited variation. To simplify the simulations, we focused on high-risk inherited PTVs (n=57) identified in the subset of 323 families containing only fully phase-able children (excluding extended families). We also analyzed synonymous variants (SYN) with the same inheritance pattern as high-risk inherited variants (transmitted to all affected and no unaffected children), as a negative control, because this variant class is not expected to confer disease risk. First, we calculated the LCL-artifact rate for SNVs and indels (separately) for each parent in these 323 families. Rare SNVs and indels (AF≤0.1%) on the autosomes, not falling in problematic regions (GIAB regions (Zook et al., 2014), problematic CNV regions (Brandler et al., 2016), or common CNV regions from cDGV), were categorized as being transmitted to at least one vs. never transmitted to any of their offspring. A zero-inflated binomial model was then used to estimate the LCL-artifact rate per parent via maximum-likelihood. The parental LCL-artifact rate for SNVs and indels were highly correlated (Pearson correlation = 0.94); therefore, we used the combined SNV+indel parental LCL-artifact rate. The parental LCL-artifact rates were modest with a median of 0.05 (mean=0.06).
To test for an excess of high-risk inherited PTVs in constrained genes, we performed simulations that permute the location of PTVs (or SYN) across genes and simulate LCL-artifact adjusted Mendelian inheritance. First, we extracted all qualifying variants in the parents (rare AF≤0.1%, PTV (or SYN) – SNVs and indels – in non-GIAB regions). Second, we grouped genes into constraint score bins (spanning the full score range of 0-1) by either gnomAD pLI score or gnomAD o/e score. Third, we computed the per-bin PTV (or SYN) rates in an external cohort (either the SSC (Werling et al., 2018) or the AD cohort (Bennett et al., 2018)). These rates are the empirical ratio of PTVs in highly constrained genes (pLI>0 .9) versus all genes (pLI-PTV)/(PTV) in each cohort: parents from this iHART cohort, parents from the SSC, or samples from the AD cohort. Finally, we counted the observed number of high-risk inherited variants within each constraint score bin, and compared these to 1,000 simulations of a null expectation. Each simulation randomly assigns each PTV to a constraint score bin according to the expected rates computed in one of the cohorts (iHART parents (iHART-matched), SSC parents (SSC-matched), or AD samples (AD-matched)), using a multinomial distribution. Each simulation subsequently simulates transmission to each child assuming a Mendelian transmission of 50% while adjusting for parental LCL artifact rates (for instance, a parent with a 5% LCL rate would have a 5% chance to not transmit a variant to any child, and a 95% to transmit the variant with Mendelian transmission).
For a null expectation based on AD-matched PTV rates, we found that the most constrained genes based on o/e (the lowest o/e bins; o/e (upper confidence interval) < 0.467) are significantly enriched for transmitted-to-all PTVs over the expectation (observed=83, expected=65, P=0.022), and the same holds true for the most constrained genes based on pLI (the highest pLI bins; pLI > 0.889; observed=46, expected=32, P=0.007) (Figure 2B). When matching to the SSC, we find the expectations and p-values are, respectively, 80 (P=0.402) and 39 (P=0.172) (Figure 2B). While we observe both an excess of pLI-PTVs (class imbalance) and an excess of PTVs transmitted to all affected and no unaffected children (transmission disequilibrium) in the bins containing the constrained genes, the number of high-risk inherited variants (PTVs transmitted to all affected and no unaffected children in only constrained genes) were too few (n=57 SNV/indel) to simultaneously test for transmission disequilibrium conditioned on constraint.
We also note that the simulation results were highly sensitive to the estimated parental LCL-artifact rates and the empirical ratio of PTVs in highly constrained genes (pLI≥0 .9) versus all genes (Figure 2B). The deviation of the synonymous variants from the expected odds ratio of 1 is likely due to slightly different LCL-artifact rates for synonymous variants (as opposed to PTVs).
Gene set enrichment
The purpose of gene set enrichment (GSE) analysis is to count the number of genes in common between two sets of genes and determine if there is greater overlap than expected by chance. We use a null model in which the probability that a gene is hit by mutation is proportional to the length of this gene, as previously described in Iossifov et al. Nature 2014 (Iossifov et al., 2014). In this model, we collapse all recurrent hits resulting in a gene being classified as hit (1) or not hit (0) in the sample of interest (e.g., ASD affected children). We then compare the genes targeted by at least one mutation in the sample (T) to a predefined gene set (S), and obtain the overlap (0) from the intersection of T and S. We estimate the probability p(S) that an exonic mutation (and hence the gene) is contained within S by taking the ratio of the sum of the coding lengths of the genes of S and the sum of the coding lengths of all genes.
As described in Iossifov et al. (Iossifov et al., 2014), we then perform a two-sided binomial test of |O| outcomes in |T| opportunities given the probability of success p(S), where ‘‖’ denotes the number of gene members in a set.
For some analyses, only a portion of the genome was considered (e.g., only genes with a pLI≥0.9), and thus all parameters (T, S, and p(S)) were adjusted to remove genes (and gene lengths) not being considered in the count of T. All gene sets (S), were first converted to their HGNC symbol and then matched by HGNC symbol to the target genes (T) before intersecting to obtain O. In the analysis of high-risk inherited variants, the gene set enrichment analysis was adjusted to include only the 3,483 genes which have a pLI≥0.9. In the TADA mega-analysis, the gene set enrichment analysis was adjusted to include only the 18,472 gencodeV19 TADA genes.
We selected 22 gene sets with known or hypothesized biological relevance in the study of ASD. This included four transcriptome co-expression studies: (1) one module downregulated (M12) and one module upregulated (M16) in ASD brain vs. control brain (Voineagu et al., 2011), (2) three modules downregulated (M4, M10, M16) and three modules upregulated (M9, M19, M20) in ASD brain vs. control brain (Parikshak et al., 2016), (3) three neurodevelopmental co-expression networks from multiple human brain regions across human development enriched for genes hit by a single de novo PTV in ASD patients from the Simons Simplex Collection (SSC) (3_5_PFC_MS, 4_6_PFC_MSC, and 8_10_MD_CBC) – after removing the nine high-confidence ASD (hcASD) genes on which these networks were seeded (Willsey et al., 2013), and (4) five neurodevelopmental co-expression modules – constructed agnostic to ASD-risk genes – enriched for ASD-risk genes/variants (M2, M3, M13, M16, and M17) (Parikshak et al., 2013). In addition to these 16 gene sets, we compiled a list of genes with ≥2 SSC probands harboring a de novo PTV (Iossifov et al., 2014); this gene set serves as a positive control. We also included FMRP targets (Darnell et al., 2011), CHD8 targets (Sugathan et al., 2014), RBFOX1 targets (Weyn-Vanhentenryck et al., 2014), and genes encoding proteins identified in the post synaptic density in human neocortex (Bayes et al., 2011). Finally, we used >8,000 samples from various tissues (e.g., brain, heart, liver) in GTEx (Battle et al., 2017) data v6 (dbGap # phs000424.v6.p1) to identify genes enriched for expression in the brain vs. other tissues. These genes had a 2-fold enrichment (FDR<0.05) after regressing out RNA Integrity Number (RIN) and various sequencing covariates (principal component 1 and 2 of sequencing statistics provided by GTEx). Significance should only be considered for gene sets surviving multiple test correction (Bonferroni correction for the 22 gene sets tested or P<0.002).
DLG2 association and haplotype prediction
The 2.5Kb deletion identified in the promoter region of DLG2 is significantly associated with ASD when considering the three independent ASD carriers in the iHART cohort (3 of 484 unrelated, phase-able (at least one parent sequenced), affected children with SVs called) and the lack of any deletions intersecting this DLG2 promoter region deletion amongst 212 unaffected children in this cohort (iHART) and 26,353 cDGV controls (curated DGV) (two-sided Fisher’s Exact Test, P=5.7 x 10−6, OR=Inf, 95% CI=22.7- Inf). In other words, no deletions were found to overlap the DLG2 promoter deletion in public databases nor in any of our controls (n=26,565 controls). However, it is unclear if this SV is detectable by microarray. Since the majority of DGV SV detection was based on microarray, we restricted to only WGS control samples (212 unaffected children in this cohort and 2,677 cDGV controls), and found that this association is significant (two-sided Fisher’s Exact Test, P=0.003, OR=Inf, 95% CI=2.47- Inf).
Given that all carriers of the recurrent, high-risk, inherited SV deletion disrupting the promoter of DLG2 (chr11:85339733-85342186) were of Hispanic or Latino origin, we wanted to eliminate the possibility that this was a rare population-specific event from a common ancestor. When restricting to only Hispanic or Latino (AMR) WGS control samples (98 unaffected children in this cohort and 351 cDGV controls), this association remains significant (two-sided Fisher’s Exact Test, P=0.006, OR=Inf, 95% CI=1.92-Inf). To determine if this SV was always found on the same haplotype, we first extracted all high-confidence SNVs (genotype quality of ≥30 and ≤30% of samples with missing genotypes) in the region surrounding the SV (1Kb upstream and downstream the start and end positions of the SV, respectively). We then ran fastPHASE (Scheet and Stephens, 2006) to estimate the haplotype in this region for all the 2,308 WGS samples included in this study, as well as the corresponding haplotype frequencies (using –F option). Of the 40 possible estimated haplotypes, a different haplotype was found in each of the three families carrying the SV, with haplotype frequencies of 0.469, 0.024 and 0.001 (Table S1).
Artifact Removal by Classifier (ARC)
Artifact Removal by Classifier (ARC) is a random forest supervised model developed to distinguish true rare de novo variants from LCL-specific genetic aberrations or other types of artifacts such as sequencing and mapping errors (https://github.com/walllab/iHART-ARC). To train the model we used rare de novo variants identified in 76 pairs of fully phase-able monozygotic (MZ) twins with WGS data derived from LCL DNA. We performed GATK joint genotyping of variants in MZ twins together with all samples in the iHART cohort and identified the de novo variants as described above. We defined rare de novo variants as de novo variants with a population frequency of zero in the publicly available databases, UCLA internal controls, and HNP samples. In the training set, rare de novo variants identified in both MZ twins were labeled as true variants (positive class), whereas discordant calls were labeled as false variants (negative class). Our final training set consisted of 5,667 positive and 56,018 negative variants.
A random forest classifier with 1,000 decision trees was trained on these positive and negative examples. We used the RandomForestClassifier implementation from the Python scikit-learn package (version 0.18.1). Weights associated with classes were adjusted inversely proportional to the class frequencies by using sklearn ‘balanced’ class weight option to control for class imbalance (many more negative examples than positive). We performed hyperparameter optimization by grid search.
ARC features
Variants in both classes were annotated with 48 features; these features are related to intrinsic genomic properties (e.g., GC content and other properties implicated in de novo hotspots (Michaelson et al., 2012)), sample specific properties (e.g., genome-wide number of de novo SNVs), signatures of transformation of peripheral B lymphocytes by Epstein-Barr virus (e.g., number of de novo SNVs in immunoglobulin genes), or variant properties (e.g., GATK variant metrics). We also annotated whether or not each variant fell into a region flagged as low-confidence (regions for which no high-confidence genotype calls were possible) by the GIAB Consortium (Zook et al., 2014); variants flagged as low-confidence (deemed “GIAB variants”) were retained for calculating sample-level metrics but subsequently removed prior to running the classifier (5,373 positive and 53,622 negative variants remained for use in classifier). For the eleven features which occasionally had missing values, missing values were imputed and a feature “is.X.feature.na” was included to capture this imputation process as an independent feature. All non-missing GATK metrics were taken directly from the VCF, with the exception of ABhet (see ABhet adjustment details below). A complete list of features and the importance (relative importance of each random forest feature was obtained from the RFECV module from scikit-learn) of each feature used in the random forest classifier are shown in Figure 3A.
Evaluation of ARC performance
The performance of the model was first evaluated with the receiver operating characteristic (ROC) curve analysis using a ten-fold cross validation procedure. In the ten-fold cross validation, the entire training set was divided into ten folds such that the ratio of positive to negative examples was constant across folds. We achieved an AUC of >0.98 (Figures S3A and S3B). The ROC and precision recall curves are shown in Figures S3C and S3D.
To assess the generalization error of our model, we additionally performed whole-genome sequencing (~30X) of matched whole blood (WB) and LCL samples from 17 fully phase-able individuals from the iHART cohort (“test set”). These samples were also jointly genotyped with all samples in the iHART cohort so as to preserve variant calling metrics between the training and test set. We followed the same procedure as in the training set to identify and extract rare de novo variants in our WB-LCL matched samples. We assumed that true de novo variants would be those identified in both the WB and LCL sample in a pair (deemed “concordant”). We further assumed that variants detected in only one sample of a pair (deemed “discordant”), would be due to LCL-specific aberrations (if called in LCL) or other sources of errors (if called in WB or LCL). In total, 1,512 concordant rare de novo variants (n=1,291 after excluding GIAB variants) were called in these samples, which we used as positive examples in our test set.
Furthermore, 2,560 discordant rare de novo variants (n=1,898 after excluding GIAB variants) were called in only one sample of a pair (64% of discordant variants found only in LCL, 36% of discordant variants found only in WB) and were used as negative examples. We evaluated a model that was trained using the entire training set on this independent test set and achieved an AUC of 0.98 and an F1 score of 0.89.
To determine a cutoff point for the predicted ARC scores, below which a variant would be considered likely to be an LCL-specific genetic aberration or other type of artifact, we chose a conservative cutoff value. We selected a conservative ARC score threshold (0.4) that achieved a minimum precision and recall rate of 0.92 and 0.80, respectively, in the 10-fold cross validation training set (Figures S3C and S3D); and achieved a precision and recall rate of 0.98 and 0.84, respectively, in the test set (Figure S3H).
ABHet adjustment
ABHet is a variant-level annotation from GATK that aims to estimate if biallelic variants match expected allelic ratios. An ideal heterozygous variant will have a value of close to 0.5 and an ideal homozygous variant will have a value of close to 1.0. ABHet is calculated for a variant based on all samples in the VCF which are not homozygous reference at this site. The ABHet annotation is not currently provided by GATK for indels. Using the ABHet formula below, we manually calculated the ABHet value for all indels.
Additionally, in the training set, we manually adjusted ABHet values by only including the proband and removing his/her twin(s) from the calculation. This corrects for bias introduced by applying the raw GATK metric calculated based on two samples to a single sample because we retain only the proband metrics (and exclude the MZ twin metrics) for shared de novo variants. This systematic bias is particularly apparent when comparing to the sample-level ADDP metric (formula below).
If a variant is present in only one sample in the VCF, then ABHet == 1 − ADDP. In contrast, for shared de novo variants a variant is in two different samples (proband (x) and MZ twin(y)), and ADDPx is rarely equal to ADDPy, and thus ABHetxy != 1 − ADDPx.
Similarly, in the test set we manually adjusted the ABHet values by only including the LCL sample and removing its matched WB sample from the calculation.
Imputing missing values for ARC features
For eleven of the ARC features (Inbreeding coefficient, ABHet, ABHom, Overall non-diploid ratio (OND), Recombination rate, Base quality rank sum, Mapping quality rank sum, DNase hypersensitivity, Read position rank sum, Replication timing, Transcription in LCL), some variants had missing values. In general, we used the mean of all non-missing values to impute the missing values of a feature. However, for GATK’s “OND” feature, missing values were imputed as zero. In order to account for missingness and capture this imputation process in the ARC model, a binary feature “is.X.feature.na” was included for all variants for each of these features, with the exception of the “OND” feature (as OND values were missing for the majority of variants).
For two ARC features, indels were occasionally annotated with multiple values – SimpleRepeats and EncodeCaltechRnaSeqGm12878R2x75. For SimpleRepeats, if multiple values were listed, only the lowest value was retained. For EncodeCaltechRnaSeqGm12878R2x75, only the max value was retained. We chose these features to be most conservative and least likely to bias the classifier. These exceptions are also captured by the “is.indel” feature.
ARC outlier samples
After applying ARC to all 1,377 children (partially or fully phase-able), we identified a subset of outlier samples for which >90% of their raw DN variant calls had an ARC score of less than 0.4. These are samples for which >90% of raw DN variant calls were excluded by ARC (partially phase-able n=2; fully phase-able n=346). These outlier samples were those with the largest number of raw DN variant calls prior to running ARC (biological sequencing source was LCL) and it’s likely that the classifier was unable to confidently distinguish variants in these samples. Unless otherwise mentioned, all ARC outlier samples were excluded from downstream analyses involving de novo variants.
De novo mutation rate vs. paternal age
We evaluated the correlation between DN variant rate in 574 fully phase-able iHART affected children (excluding MZ twins, ARC outliers, and one sample without paternal age information) and paternal age at the child’s birth using a generalized quasi-Poisson linear model, assuming that the counts are distributed as an over-dispersed Poisson distribution (a generalized quasi-Poisson linear model):
Where RC is the rate of DN events per child, FC is the age of the father at the birth of the child and TC is the percent of the child’s genome covered at ≥10X and A and B are whole population parameters, estimated by maximizing the likelihood over all children (as previously described in Iossifov et al. Nature 2014 (Iossifov et al., 2014)). We performed this analysis before and after ARC, considering only DN events not falling in GIAB low-confidence regions.
Given the well-known effect of paternal age on germline mutation, we tested for an effect of paternal age on the number of de novo mutations per affected ASD child and find a robust signal after running ARC (P=3.6 x 10−13), but not prior to application of ARC (Methods; Figure S4D). We observed an increase of 1.46 RDNVs per year of paternal age (95% CI = 1.37-1.55), matching previously published rates (Deciphering Developmental Disorders, 2017; Francioli et al., 2015; Goldmann et al., 2016; Michaelson et al., 2012; Study, 2017).
Rates for rare de novo mutations
When calculating de novo mutation rates, we only considered the 1,177 children with both biological parents sequenced (fully phase-able). Rare de novo variants (absent in all controls) were restricted to those with an ARC score ≥0.4 that were not flagged as low-confidence by the GIAB consortium. We then excluded ARC outlier samples (n=346). Consistent with what is shown in Figure S4B, there was no significant difference in the rate of rare de novo variants based on the biological sequencing source (WB vs. LCL; after ARC and after excluding ARC outliers) when including all MZ twins. However, we observed that shared de novo (TRUE) variants from the LCL MZ twins have slightly inflated ARC scores (median number of de novo variants is 69), as compared to LCL non-MZ twin samples (median number of de novo variants is 57) (Figure S4A). This difference in de novo rates was significant when evaluated using Wilcoxon rank sum test (P=1.28 x 10−12). The inflated ARC scores are likely due to the fact that these LCL MZ twins were used as the ARC training set; therefore, de novo variants from all MZ twin samples were excluded from all de novo rate calculations (n=158, some of which are also ARC outliers n=43). Therefore, all de novo mutation rate calculations were performed using 716 fully phase-able non-MZ twin and non-ARC outlier samples (Naff=575; Nunaff=141).
We observed a mean genome-wide de novo mutation rate of 60.1 RDNVs per child (Figure S4B), which is consistent with previously reported genome-wide de novo mutation rates (mean=64.4; range 54.8-81) (Besenbacher et al., 2016; Conrad et al., 2011; Kong et al., 2012; Michaelson et al., 2012; Turner et al., 2016; Yuen et al., 2017).
Power calculations for RDNVs
Given our observation that children from multiplex families and simplex families have comparable rates of rare de novo synonymous and missense variants in both affected and unaffected children but different rates for rare de novo PTVs (Table S2), we sought to determine if this represented a true difference in the underlying genetic architecture of multiplex ASD families by performing a Monte Carlo integration. This revealed that with the current iHART sample size (Naff=575, Nunaff=141), we had only 51% power to detect an odds ratio greater than or equal to the odds ratio reported in simplex families (OR=0.13/0.07=1.86) (Iossifov et al., 2014; Kosmicki et al., 2017) and that our power to reject the null hypothesis that affected and unaffected children have no difference in the rate of de novo PTVs was 70.8%. We estimate that once we expand our cohort by a factor of 2.5 (Naff=1,438, Nunaff=353), we will have >95% power to detect a rate difference in de novo PTVs if such a difference exists in multiplex ASD families.
Defining pathogenic de novo variants
We defined pathogenic de novo variants (Figure 3E) as missense or PTV variants passing ARC and found in one of the previously established 65 ASD-risk genes (Sanders et al., 2015). Despite finding no global excess of damaging RDNVs in ASD cases in the study, we do identify PTVs and predicted deleterious missense (Mis3) RDNVs in previously established ASD-risk genes, including CHD8, SHANK3, and PTEN (Figure 3E and Table S3). As expected, such pathogenic RDNVs are only found in affected children in our cohort.
TADA mega-analysis
Samples and qualifying variants
We used the Transmitted And De novo Association (TADA) test (He et al., 2013) to combine evidence from rare de novo (DN) or transmitted (inherited) PTVs and de novo Mis3 variants predicted to damage the encoded protein which were identified in ASD cases. Within the 422 iHART families with at least one ASD case and both biological parents sequenced, there were 838 genetically non-identical (only one MZ twin retained) ASD cases available for the TADA analysis. These 838 affected samples, and their biological parents, were treated as independent trios for the TADA analysis. This approach means that siblings were treated as belonging to independent trios, an approach for which we also approximate the null distribution (see details on TADA simulations below). To further increase power for the identification of novel ASD-risk genes, we combined qualifying variants found in ASD cases from the current (iHART) cohort with the most recent TADA mega-analysis (Sanders et al., 2015), which included variants described in the Simons Simplex Collection (SSC) and the Autism Sequencing Consortium (ASC) cohorts, together with small de novo CNV deletions (SmallDel) identified in SSC and Autism Genome Project (AGP) probands (Table S3).
Qualifying variants in the iHART cohort included rare DN/transmitted PTV and rare DN Mis3 variants identified in the 838 affected samples, and not flagged as low-confidence by the GIAB consortium (Zook et al., 2014). Following the allele frequency threshold used in the previous TADA mega-analysis (Sanders et al., 2015), we required transmitted PTVs to have an AF≤0.1% in public databases (1000g, ESP6500, ExACv3.0, cg46), internal controls, and iHART HNP samples. DN variants identified in the iHART cohort were required to be absent in all public databases, internal controls, and HNP samples (AF = 0). High confidence DN variants were obtained by ARC for all non-MZ twin samples. DN variants shared by MZ twins (shared DN variants, used as TRUE examples in the ARC training set) were also included as qualifying variants without filtering on their ARC score. Additionally, for the 185 TADA samples identified as ARC outlier samples, we excluded DN variants in these samples and retained only their inherited PTVs as qualifying variants. We used the PolyPhen-2 (Adzhubei et al., 2010) v2.2.2r395 HDIV predictions from the Whole Human Exome Sequence Space (WHESS dataset) to annotate DN Mis3 variants in the iHART cohort. This method is highly concordant to the method (PolyPhen-2 web application) implemented for the ASC and SSC (Sanders et al., 2015), with our re-annotation resulting in identical Mis3 classifications for 99.8% of the reported DN Mis3 variants in the ASC and SSC cohorts. When multiple qualifying variants in a gene were found in the same sample, only the most damaging variant was retained.
We then tallied qualifying variants from the different cohorts into a gene by variant-type matrix for the TADA analysis, which contained variant counts for a total of 18,472 gencodeV19 genes with HGNC approved gene names (this excludes a subset of genes (193 out of 18,665) from the most recent TADA mega-analysis (Sanders et al., 2015) that could not be easily converted to a single non-redundant HGNC gencodeV19 gene). In particular, counts of DN PTV/Mis3 variants come from 4,689 ASD cases from ASC (De Rubeis et al., 2014), SSC (Iossifov et al., 2014), and iHART (this manuscript), while counts for the transmitted and non-transmitted PTVs are from 3,813 ASD cases and 7,609 controls from the ASC (De Rubeis et al., 2014) and iHART (this manuscript) (Table S3). Finally, counts of DN SmallDel were calculated in 4,687 ASD cases from the SSC (Sanders et al., 2015) and the AGP (Pinto et al., 2014) (Table S3).
Critically, 424 AGRE samples (sample list obtained via personal correspondence with Bernie Devlin, Mark Daly, and Christine Stevens) were included as “cases” in the original ASC TADA analysis (De Rubeis et al., 2014) meaning that all qualifying PTVs identified in these cases were treated as transmitted PTVs because de novo status could not be determined. Given that iHART sequenced 119 of these samples (or the monozygotic twin of one of these samples) and their biological parents, we were able to recover the de novo status for variants identified in these samples (71 samples after excluding ARC outliers) by using the iHART data. Thus, we used iHART data to count qualifying DN PTV and Mis3 variants in these samples and allowed transmitted PTV counts to come from the original study (De Rubeis et al., 2014). To do this in a non-redundant way (without double-counting variants), we looked for all qualifying DN PTVs identified by iHART data in these samples in the ASC VCFs (downloaded from dbGAP (De Rubeis et al., 2014)) and subtracted a transmitted PTV count from the iHART mega-analysis TADA-ready table for each variant found in the ASC VCFs. In three instances, the transmitted PTV count from ASC cases was already zero for the gene harboring the corresponding variant and thus we left it at zero.
TADA parameters
The parameters used for performing the TADA analysis, matched those used in previous TADA mega-analyses (De Rubeis et al., 2014; He et al., 2013; Sanders et al., 2015); including the previously observed aggregate association signals used to estimate relative risk (RR, γ) for each variant class – DN PTV (γ=20), DN SmallDel (γ=15.3), DN Mis3 (γ=4.7), and transmitted PTV (γ=2.3) (use of these parameters facilitated replication of previous findings prior to adding the iHART cohort to perform a mega analysis). PTVs classified as uncertain or missing (as defined previously) in children were excluded. In addition to these RR parameters, we also assumed the fraction of ASD-risk genes (π) to be ≈ 0.05 (1,000 ASD-risk genes divided by a total of 18,472 genes), the PTV frequency parameters (required by TADA to integrate transmitted and non-transmitted PTV variant counts into the model) were ρ=0.1 and v=200 and the gene mutation rates were taken directly from the most recent TADA mega-analysis (Sanders et al., 2015), with PTV and Mis3 gene mutation rates calculated by multiplying the exome mutation rates, originally estimated by Samocha et al. (Samocha et al., 2014), by the fractional constants of 0.074 and 0.32, respectively. This use of gene mutation rates as ground truth (rather than comparing to control samples) facilitates the use of TADA in mega-analyses because differences in sample size and variant detection between studies impact the power of TADA, but are not a potential source of bias.
Novel gene discovery
We applied stringent parameters for declaring a gene as novel – genes had to have an FDR <0.1 in our TADA mega-analysis and lack genome-wide statistical support in all previous studies (Sanders et al., 2015, De Rubeis et al., 2014) with statistical rigor. The CACNA2D3 gene was significantly associated with ASD in the iHART mega-analysis, but not the previous TADA mega-analysis (Sanders et al., 2015); however it was previously reported in De Rubeis et al. (De Rubeis et al., 2014) and thus is not considered as a novel ASD-risk gene. In contrast, MYO5A was reported as a “putative ASD-risk gene” by Yuen et al. (Yuen et al., 2017), however the binomial test they use to obtain an FDR is not applied genome-wide (e.g., they first restrict to genes with ≥2 PTVs in genes with a pLI≥0.9). Furthermore, they apply an FDR threshold of <0.15 (the standard in the field is an FDR<0.1) and they do not provide per gene FDR values. Therefore, we consider MYO5A a novel ASD-risk gene.
Removal of de novo signal
Given that our multiplex ASD familial cohort is expected to be depleted for de novo variation relative to simplex families, we also asked how our novel gene discovery would change if we ignored the contribution of de novo variants in the iHART cohort to the TADA mega-analysis (by assigning a relative risk of one to de novo variants in iHART children). Using this overly conservative approach, a total of 65 ASD-risk genes are identified at an FDR<0.1, including six of the 16 novel genes identified in the iHART TADA mega analysis (C16orf13, CCSER1, MLANA, PCM1, TMEM39B, and TSPAN4) and five additional genes (ASXL3, CDH13, NR3C2, SCN7A, STARD9) Table S3.
Replication of previous TADA-mega analysis
Comparison of the iHART TADA-mega analysis to the previously published findings (Sanders et al., 2015) identified 16 newly-significant (FDR<0.1) ASD-risk genes plus CACNA2D3, which was previously reported as an ASD-risk gene (De Rubeis et al., 2014) (Table 1, Figure S5D). We failed to replicate 13 of the genes previously published with an FDR<0.1 (Sanders et al., 2015) (Figure S5C). The q-values for these 13 genes were borderline significant in iHART (Figure S5C), and their simulation p-values were greater (min p-value=0.01, max p-value=0.06, Figure S5E) than those of the 69 ASD-risk genes we identified in the TADA mega-analysis with high confidence, which include the 16 newly significant genes (min p-value=0.001, max p-value=0.006) (Figures 4B, S5D, and S5F). While some of the genes that failed to replicate in our study may reach genome-wide significance again as sample sizes grow, at this stage our data do not support them.
TADA simulations
The distribution of the TADA statistic (under the null) is known for independent trios (He et al., 2013), but not for multiplex families. Therefore, we estimated the distribution of the null TADA statistic by simulating Mendelian transmission and de novo mutation across the family structures used in our TADA-mega analysis. This simulation was based on the observed qualifying variant counts and family structures from our TADA-mega analysis datasets, which included: (1) iHART multiplex families, (2) ASC and SSC trios and ASC case-control samples, and (3) small deletions from Sanders et al. (Sanders et al., 2015) (Table S3). Simulations under the null model (1.1 million-simulations) were conducted prior to running TADA with the same parameters used for our TADA-mega analysis (see “TADA mega-analysis”).
The occurrence of rare de novo variants (RDNVs) was simulated by randomly shuffling genes carrying the observed qualifying RDNVs across the genome of each sample by redrawing in proportion to the gene-specific mutation rates (derived from Samocha et al. (Samocha et al., 2014)). For example, if an affected sample harbored 8 RDNVs, these RDNVs would be placed in 8 genes in simulation 1, independently in 8 genes in simulation 2, and so on, where the probability of a gene containing an RDNV is proportional to its gene mutation rate. This method was applied to simulate RDNVs in affected children from iHART multiplex families and affected children from ASC and SSC trios.
The occurrence of transmitted (inherited) and non-transmitted variants was simulated by (A) randomly shuffling genes carrying the observed qualifying variants in the parents of a given family, by redrawing in proportion to the gene-specific mutation rates and (B) randomizing the Mendelian inheritance of such variants across all children (affected and unaffected) in the family. Randomization of the Mendelian inheritance simply means that for each simulation a variant can be transmitted to each child, regardless of affected status, with a 50% probability. For example, in Family001, if mom harbored 10 qualifying PTV variants and dad harbored 10 qualifying PTV variants; then in each simulation each of these 20 variants would be randomly placed in a gene according to its gene-specific mutation rate and is either transmitted or not transmitted to each of the children in the family. This method was applied to simulate transmitted and non-transmitted PTVs in the cohorts listed in Table S3.
Finally, we simulated small deletions disrupting 2-7 genes at a time (Sanders et al., 2015). For each observed small deletion containing Ngenes, we selected a contiguous set of Ngenes by redrawing in proportion to the multi-gene mutation rates. Multi-gene mutation rates were calculated by summing single-gene mutation rates of adjacent genes using sliding windows of K genes across the genome. For example, if a small deletion disrupted 5 genes in an affected sample, then for each simulation a contiguous set of 5 genes would be randomly selected for this sample with a probability proportional to the 5-gene-sliding-window multi-gene mutation rates.
The resulting set of simulation-based Bayes factors from TADA were multiplied together. A single p-value for each gene was generated, reflecting how unlikely it is to have observed the Bayes factor obtained in our TADA-mega analysis given the 1.1 million simulation-based Bayes factors observed for this gene (Figures S5A and S5B).
We used the simulation p-values to identify genes reaching genome-wide significance after applying a stringent Bonferroni correction for the total number of genes included in the TADA analysis (0.05/18,472=2.7x10−06). We restricted this to genes obtaining an FDR<0.1 by TADA and a simulation p-value of less than or equal to 2.7x10−06 (Table S3). If we remove the requirement for a TADA-mega analysis FDR<0.1, then a 25th gene, DNAH10, also reaches genome-wide significance implicating that variants in this gene show over transmission to affected children in our multiplex children.
Genes with large inherited PTVs contribution
Given our signal for rare inherited variants, we sought to highlight genes for which a large contribution of the TADA ASD-risk association signal is derived from inherited PTVs. Conservatively, we considered only variants where the inheritance was known (de novo vs. inherited). Therefore, we adjusted the total number of TADA-qualifying variants to ignore PTVs from cases because some of the TADA-mega analysis qualifying variants originate from case-control studies (not iHART) where inheritance is unknown. We applied two methods to identify genes with a large contribution from inherited PTVs. Method 1: The total number of qualifying variants (N) in each TADA gene was defined as NDN.PTV + NDN.SmallDel + NDN.Mis3 + NInherited.PTV; and if NInherited.PTV/N ≥70%, then this gene was considered to have a higher proportion of inherited risk variants. Method 2: Alternatively, we identified genes for which the main driver of the TADA association signal was from inherited PTVs. We defined this class of genes as those where the Bayes Factor from inherited PTVs was greater than the Bayes Factor from all other de novo variant classes (BFinheritedPTV > BFdnPTV & BFinheritedPTV > BFdnSmallDel & BFinheritedPTV >BFdnMis3). For PCM1, the Bayes Factor contribution from inherited PTVs was greater than the Bayes Factor from any class of de novo variants, indicating that the association signal for PCM1 is mainly driven by inherited PTVs.
Single cell RNA-seq
Gene cell type enrichment scores were obtained from an unpublished scRNA-seq dataset of GW17-18 human fetal cortex, a human fetal forebrain scRNA-seq dataset from Nowakowski et al. (Nowakowski et al., 2017), and adult brain scRNA-seq dataset from Lake et al. (Lake et al., 2018) (Figures S6C and S6D). Cell type enrichment lists were grouped into major cell classes (glutamatergic, GABAergic, glial and other support cells). Broadly expressed genes were determined by enrichment in neuronal and glial or other support cell types, or above a mean expression threshold across all cells in the dataset but without cell type specific enrichment. Enrichment log2 odds ratios were calculated using a general linear model (binomial distribution).
Identifying candidate ASD genes with NetSig
In order to identify genes whose encoded proteins directly interact with potential ASD-risk genes more than expected by chance, we ran NetSig (Horn et al., 2018). NetSig requires two input files: (1) a set of genes and their associated q-values (or p-values) and (2) a PPI network. We input the q-values obtained in the iHART TADA mega-analysis and known protein-protein interactions from InWeb v3 (Lage et al., 2007) after converting to HGNC and restricting to genes included in the iHART TADA mega-analysis (12,015 genes); this resulted in a subset of 302,991 known PPIs. Given that iHART SVs were not included in the TADA analysis (Table S3), and therefore do not contribute to the input q-values in this analysis, we explored enrichment for NetSig significant genes using the direct and indirect network seeded by high-risk inherited PTVs (Figure 2C).
Zebrafish experiments
Generation of zebrafish nr3c2 mutant
The zebrafish nr3c2 mutant was generated using CRISPR/Cas9 as described (Hwang et al., 2013) with sgRNA target sequence 5’-GGTGTGTGGTACGAGAGCGG-3’. The mutant contains a 5 bp deletion (open reading frame nucleotides 2120-2124, 5’-CCGCT-3’) that shifts the translational reading frame after amino acid 707 and results in a premature stop codon after amino acid 738, compared to 970 amino acids for the WT protein. The predicted mutant protein lacks the ligand binding domain, and thus should be non-functional. Mutant animals were genotyped using the primers 5’-CTTCCCTGCAGAGCTCAAAG-3’ and 5’-ATAGCCAGCGAACACCACTT-3’, which produce a 164 or 159 bp band for the WT or mutant allele, respectively. nr3c2 heterozygous mutants were outcrossed to the parental TLAB strain for three generations before use in experiments. For each behavioral experiment, nr3c2 +/− animals were in-crossed, generating nr3c2 −/−, −/+ and +/+ sibling progeny. Experiments were performed blind to genotype, and animals were genotyped using PCR after each experiment. Multiple sequence alignments were performed using Megalign Pro (DNASTAR Lasergene).
Pharmacology
MK-801 (M107, Sigma Aldrich) was dissolved in dimethyl sulfoxide (DMSO, 4948-02, Macron Chemicals) as a 100 mM stock solution. Immediately before each experiment, this stock solution was diluted in system water for a final concentration of 20 μM. WT TLAB fish were exposed to either 20 μM MK-801 in 0.02% DMSO or 0.02% DMSO vehicle control for 1 hour prior to behavioral testing. For ethanol experiments, WT TLAB fish were exposed to ethanol (V1016, Koptec) diluted in system water at a final concentration of 0.5% for 1 hour prior to behavioral testing. After each drug treatment, fish were rinsed in fresh system water 3 times before behavioral testing.
Social preference assay
Beginning at 2 weeks of age, and becoming robust at 3 weeks of age, zebrafish show what has been described as social behavior by exhibiting a strong preference to remain in a compartment where they can view conspecifics compared to a compartment where they cannot (Dreosti et al., 2015). This behavior is not simply a result of attraction to a novel or moving object, as it is only elicited by conspecifics of similar size and behavioral patterns (Dreosti et al., 2015; Larsch and Baier, 2018). Based on these observations, we developed a modified version of a previously-described social preference assay (Dreosti et al., 2015). Zebrafish were raised on a 14:10 hour light:dark cycle and were fed rotifers (Brachionus plicatilis) twice per day until reaching 2 weeks of age. Fish were then fed brine shrimp (Artemia salina) until 3-4 weeks of age, at which point their behavior was assayed. The behavioral assay was performed using a flat-bottom 12-well plate containing round wells made of clear plastic (CC7672-7512, CytoOne) and custom-built removable opaque dividers. Single “test” animals, whose behaviors were analyzed, were placed in each of the 4 middle wells of the plate, and a WT conspecific of similar age and size was placed in a well either above or below each middle well. Wells were filled with fresh system water and the plate was placed in a custom-modified, Zebrabox (Viewpoint Life Sciences) that was illuminated with infrared and white LEDs. The 12-well plate was housed in a chamber filled with recirculating water to maintain a constant temperature of 28.5°C. Locomotor activity was monitored using an automated videotracking system (Viewpoint Life Sciences) with a Dinion one-third inch monochrome camera (Dragonfly 2, Point Grey) fitted with a fixed-angle megapixel lens (M5018-MP, Computar) and infrared filter. The tracking mode was used to record the location of each test animal, with the following empirically determined settings: low detection threshold=130; x min size=3; inactivity=5. Animals were given a 5-minute habituation period before the start of data acquisition. During a 10-minute baseline period, opaque dividers were inserted between each well to prevent the animals from seeing each other. The dividers separating each row of wells (but not the dividers separating each column of wells) were then removed, allowing each test animal to view one well containing a conspecific and one empty well. The fish were given another 5-minute habituation period, followed by a 10-minute post-baseline period. For data acquisition, wells containing test fish were divided into two 0.5 cm × 2.2 cm zones, one closest to the well containing a conspecific and one closest to the empty well (indicated as blue and orange boxes in Figure 6A, respectively). The amount of time spent by a test fish in each zone during the baseline and post-baseline periods was recorded.
Social preference of test fish was quantified by calculating the social preference index (SPI) = (time spent in zone near the conspecific − time spent in zone near the empty well)/time spent in both zones. Thus, SPI=1 indicates a fish that spends 100% of its time near a conspecific, SPI=−1 indicates a fish that spends 100% of its time near the empty well, and SPI=0 indicates a fish that spends equal amounts of time near the conspecific and near the empty well. Data analysis and statistical tests were performed using Prism (GraphPad).
To validate the social preference assay, we treated zebrafish with either MK-801, an NMDA receptor antagonist that disrupts rodent (Moy et al., 2013) and zebrafish (Zimmermann et al., 2016) social behaviors, or DMSO vehicle control, for one hour prior to performing the behavioral assay. Animals treated with DMSO on average showed no spatial preference during the baseline period and a strong preference for conspecifics during the post-baseline period (Figure S7C). This behavior was only observed in animals 3 weeks of age or older (data not shown), as previously reported (Dreosti et al., 2015). In contrast, while animals treated with 20 μM MK-801 also on average showed no spatial preference during the baseline period, social preference for conspecifics during the post-baseline period was abolished (Figure S7D). To further validate the assay, we treated zebrafish with ethanol, which has also been shown to reduce preference for conspecifics in 3-week old zebrafish (Dreosti et al., 2015). Similarly, we found that treatment with 0.5% ethanol for 1 hour prior to behavioral testing significantly reduced social preference (Figure S7G). Furthermore, both MK-801 and ethanol treatment significantly suppressed the increase in SPI during the post-baseline period compared to the baseline period (Figures S7E and S7H), indicating reduced social preference. Taken together, these results reproduce observations obtained using a similar assay (Dreosti et al., 2015) and suggest that our assay can identify social interaction defects in zebrafish.
Zebrafish size was quantified by measuring body length from the tip of the mouth through the midline of the body to the end of the tail fin in single frames of video recordings of the social preference assay using ImageJ (Schneider et al., 2012). The social behavioral deficit observed in nr3c2 −/− animals (Figures 6B and 6C) is unlikely to be due to developmental delay because there was no significant difference in the size of nr3c2 −/−, +/− and +/+ siblings when the assay was performed (Figure S7I).
Sleep/wake assay
Sleep/wake analysis was performed as previously described (Prober et al., 2006). Zebrafish were raised on a 14:10 hour light:dark cycle until 4-days post-fertilization, when individual animals were placed into each well of a 96-well plate (7701 −1651, Whatman) containing 650 μL of E3 embryo medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4, pH 7.4). Plates were sealed with an optical adhesive film (4311971, Applied Biosystems) to prevent evaporation. The sealing process introduces air bubbles in some wells, which are discarded from analysis. Animals were blindly assigned a position in the plate and were genotyped by PCR after the behavioral experiment was complete. Locomotor activity was monitored using an automated videotracking system (Viewpoint Life Sciences) with a Dinion one-third inch monochrome camera (Dragonfly 2, Point Grey) fitted with a fixed-angle megapixel lens (M5018-MP, Computar) and infrared filter. The movement of each larva was recorded at 15 Hz using the quantization mode with 1-minute time bins. The 96-well plate and camera were housed inside a custom-modified, Zebrabox (Viewpoint Life Sciences) that was continuously illuminated with infrared LEDs, and illuminated with white LEDs from 9 a.m. to 11 p.m. The 96-well plate was housed in a chamber filled with recirculating water to maintain a constant temperature of 28.5°C. The parameters used for detection were: detection threshold, 15; burst, 29; freeze, 3, which were determined empirically. A movement was defined as a pixel displacement between adjacent video frames preceded and followed by a period of inactivity of at least 67 ms (the limit of temporal resolution). Any one-minute period with no movement was defined as one minute of sleep because this is associated with a significant increase in arousal threshold (Prober et al., 2006). A sleep bout was defined as a continuous string of sleep minutes. Sleep latency was defined as the length of time from lights off at night to the start of the first sleep bout. Data were processed using custom PERL and Matlab (The Mathworks, Inc.) scripts. Statistical tests were performed using Prism (GraphPad).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise noted, statistical calculations were done using R (3.5.1). DAPPLE metrics results for evaluating the significance of PPI networks were all done using 1,000 permutations (within DAPPLE parameter) and P values < 0.05 were considered significant. For gene set enrichment analyses, significance should only be considered for gene sets surviving multiple test correction (Bonferroni correction for the 22 gene sets tested or P<0.002). NetSig genes were considered significant if they obtained a P value <0.05. Unless otherwise specified, enrichment tests (e.g., enrichment of NetSig genes within high-risk inherited PPI networks) was performed by Fisher exact test; we considered P values <0.05 as significant and also report the odds ratio (OR) with its associated 95% confidence interval.
All statistics for Artifact Removal by Classifier (ARC) are described within the Method Details and corresponding figures. The samples included in the training and test set are shown in Table S1. We also re-emphasize that we selected a conservative threshold of ARC score ≥0.4 to consider only RDNVs with extremely high confidence.
Determining rate differences between groups
Unless otherwise specified, rates comparisons between phenotypic groups (affected vs. unaffected) were calculated by taking the number of variants per child and performing a quasi-Poisson linear regression and resulting P values < 0.05 were considered significant. This method enabled us to adjust for both biological sequencing source (WB vs. LCL) and biological sex (male vs. female). Biological sex was not used as a covariate for hemizygous variants because only male children are considered. Unless otherwise noted, rates are displayed as the mean number of variants with error bars representing the standard error. Here we reiterate the sample sizes for each of the rate tests performed: (i) rare inherited coding variants (Naff=960, Nunaff=217), (ii) coding RDNVs (Naff=575, Nunaff=141), (iii) iHART non-coding RDNVs (Naff=575, Nunaff=141), (iv) iHART non-coding inherited variants (Naff=960, Nunaff=217), (v) iHART+SSC non-coding RDNVs (Naff=1092, Nunaff=659), (vi) iHART+SSC non-coding inherited variants (Naff=1477, Nunaff=735).
TADA and TADA simulations
The sample sizes for the TADA-mega analysis are provided in Table S3. Benjamini-Hochberg correction was performed for TADA results and q-values (False Discovery Rate (FDR)) <0.1 were considered significantly associated with ASD. When we apply the field standard FDR<0.1, we identify 69 genome-wide significant genes. The TADA simulations were performed using the same sample sizes (and family structures) as used in the TADA-mega analysis. For the TADA simulations, only genes with a P value < 2.7x10−06 were considered as reaching genome-wide significance because these genes pass the stringent Bonferroni correction for the total number of genes included in the TADA analysis (0.05/18,472=2.7x10−06).
Zebrafish statistics
The Shapiro-Wilk normality test was used to determine whether data in each experiment was normally distributed. Most datasets were normally distributed and were analyzed as mean ± standard error of the mean using parametric statistical tests, except where noted that data was analyzed as median ± 95% confidence interval using non-parametric statistical tests. The specific test used to assess statistical significance in each experiment is described in each Figure Legend. Statistical tests were performed using Prism (GraphPad). Data were considered to be statistically significant if p<0.05.
DATA AND CODE AVAILABILITY
The whole-genome sequencing data generated during this study are available from the Hartwell Foundation’s Autism Research and Technology Initiative (iHART) following request and approval of the data use agreement available at http://www.ihart.org. Access to the whole-genome sequencing data generated in this study will be subject to approval by Autism Speaks and AGRE. Details about the format of the data, access options, and access instructions are included at http://www.ihart.org.
We also freely provide the code for ARC (Artifact Removal by Classifier), our random forest supervised model developed to distinguish true rare de novo variants from LCL-specific genetic aberrations or other types of artifacts such as sequencing and mapping errors, together with a full tutorial at https://github.com/walllab/iHART-ARC.
Interactive genotype/phenotype search engine
To facilitate sharing of iHART data with the broader autism research community and patients, we implemented a set of online data access methods to preview and search genetic variants and phenotypic traits (http://www.ihart.org/home).
Zebrafish data
The zebrafish datasets generated and analyzed in this study, and the code used to generate the data, are available upon request.
Supplementary Material
Table S1. iHART sample information, Related to Figures 1 and 2. Detailed sample information for the 2,308 AGRE samples whole-genome sequenced as part of iHART. Including phenotypic descriptions for NR3C2 families and haplotype predictions for the DLG2 families.
Table S2. RDNV rates in multiplex vs. simplex autism families, Related to Figure 3. Comparison of RDNV rates in affected and unaffected children from ASD simplex vs. multiplex families.
Table S3. TADA results, Related to Figure 4. Results from the TADA mega-analysis, including the sample counts included from each cohort, the qualifying variants identified in iHART/AGRE samples, and the iHART TADA-mega analysis results for all 18,472 genes.
Table S4. NetSig significant genes, Related to Figure 5. The 596 genes that are significantly (P < 0.05) directly connected to potential ASD-risk genes (as defined by their TADA q-value) using NetSig.
Table S5. Structural Variant Method Details, Related to Figure 1. Structural variant method details, including the number of rare SVs identified by each calling algorithm, percentage of SVs also called by at least one other algorithm, sensitivity comparisons to genotyping array data, and LUMPY-BreakDancer joint deletion call validation rate by deletion size.
Figure S1. Whole-genome sequence coverage statistics for 2,308 iHART/AGRE samples and the high-resolution detection of large structural variants (SVs), Related to Figure 1. There were no significant differences in the average fold coverage per sample across the cohort and no differences in the categories of (A) ASD affectation status, (B) sex, or (D) family member type – where family member type was simplified to include Mother, Father, Child (proband, sibling, MZ or DZ twin) and Other (e.g., cousin). (C) The percent of exonic and genomic bases covered at ≥10x in all family members for each of the 422 fully-phaseable iHART families. Exonic regions were defined as those annotated as protein-coding exons in Gencode V19 (>75Mb). Genomic regions were defined as all non-N bases in the reference genome (>2.8Gb). (E) The percentage of genomic bases covered at greater than or equal to 1X, 10X, 20X, 30X, and 40X bases for the 2,308 iHART samples with WGS data. On average, 98.97 ± 0.37 % of bases were covered at a depth of ≥10X. (F) An overview of our custom multi-algorithm consensus SV pipeline for high-resolution detection of large structural variants (SVs) from whole-genome sequence data. The four boxes at the top list the four main algorithms used to call SVs, and the parenthetical describes the detection strategy(s) used by each algorithm: AS, de novo assembly method; SR, split-read method; RP, read-pair method; RC, read-count method. (G) Venn diagrams of structural variants detected by four different algorithms for all and rare (AF < 0.001 in cDGV and AF < 0.01 in iHART HNP samples) SVs (DELs, DUPs and INVs) detected in 1,377 phase-able WGS samples by SMuFin, LUMPY, GenomeSTRiP and BreakDancer, after excluding events with ≥50% overlap with genomic low-complexity regions (Brandler et al., 2016). Additional per-algorithm filters were also applied prior to the generation of this Venn diagram as described in Methods. (H) A schematic overview of the SMuFin detection pipeline. Families are processed as independent trios, where the sequence reads from a child are aligned to the mother’s genome and then the father’s genome, treating the parental genome as the reference genome in both comparisons. Each comparison, or SMuFin execution, results in variants identified in the child by that parent-offspring comparison. All three members of the trio are considered for assigning the corresponding inheritance of variants identified in the child. A variant detected when comparing to both mom and dad is de novo, while a variant detected only when comparing to mom is paternally inherited and a variant detected only when comparing to dad is maternally inherited.
{kind=link}
Figure S2. Additional details on rare inherited PTVs and SVs, Related to Figure 2. (A-D) Rare inherited coding variants by consequence and inheritance. The rate of rare inherited coding variants per fully phase-able child is displayed for 960 affected (red) and 217 unaffected (blue) children by both variant consequence and inheritance, this includes newly hemizygous variants in 563 affected (red) and 100 unaffected (blue) male children. The graph for newly homozygous PTVs (C) was excluded because none were identified in affected or unaffected children. Mean ± SE rates are shown. (E) The rate of private inherited PTVs in 960 affected (red) and 217 unaffected (blue) children iHART children for all genes vs. PTV intolerant genes. We found no excess of inherited private PTVs in mutation intolerant genes (pLI≥0.9) (Lek et al., 2016) in affected subjects (P=0.40, quasi-Poisson linear regression). Mean ± SE rates are shown. (F) The rate of rare inherited SVs per fully phase-able child is displayed for 960 affected (red) and 217 unaffected (blue) children by inheritance type. Mean ± SE rates are shown. (G) The rate of rare inherited SVs per fully phase-able child is displayed for newly hemizygous variants in 563 affected (red) and 100 unaffected (blue) male children. (H-M) The rate of rare inherited SVs per fully phase-able child identified in 960 affected (red) and 217 unaffected (blue) children by inheritance type; this includes newly hemizygous variants in 563 affected (red) and 100 unaffected (blue) male children. Mean ± SE rates are shown. Maternally and paternally inherited SVs by affectation status and gene disruption for deletions (H), duplications (I), and inversions (J). Newly hemizygous SVs by affectation status and gene disruption for deletions (K), duplications (L), and inversions (M). (N) The DLG2 promoter-disrupting 2.5Kb deletion (chr11: 85339733 – 85342186), displayed as an orange rectangle, detected in three independent iHART families. This 2.5Kb deletion is transmitted to all affected members of two different iHART families. This deletion falls in a recently-defined, functional, non-coding regulatory region in developing human brain (chr11:85338026-85340560) (de la Torre-Ubieta et al., 2018); below the deletion we show the average ATAC-seq peak read depth from the cortical plate (CP) and ventricular zone (VZ) of developing human brain samples (n=3).
{kind=link}
Figure S3. ARC performance in the training and test set, Related to Figure 3. The 10-fold cross validation (CV) for the ARC training set (A-D). (A) Receiver Operating Characteristic (ROC) curves for the true positive rate (Sensitivity) is plotted as a function of the false positive rate; (B) Area Under the ROC Curve (ROC AUC) statistics (median=0.992) for each of the 10 folds; (C) Precision rates vs. predicted score cutoffs – the dashed line at the selected score (0.4) highlights that the minimum precision across all 10 folds is >0.9; (D) Recall rates vs. predicted score cutoffs – the dashed line at the selected score (0.4) highlights that the minimum precision across all 10 folds is ~0.8. ARC performance in the test set (E-H). (E) Distribution of all test set variants by ARC score; (F) Distribution of all TRUE test set variants by ARC score – the majority of concordant variants have an ARC score of ≥0.4; (G) Distribution of all FALSE test set variants by ARC score – almost all discordant variants have an ARC score of <0.4; (H) The precision and recall rates vs. predicted ARC score cutoff in the test set – the dashed line at the selected score (0.4) highlights that the precision is >0.95 and the recall is >0.85.
{kind=link}
Figure S4. RDNVs identified in iHART samples before and after ARC, Related to Figure 3. (A) The ARC score distribution for raw de novo variants identified in 1,177 fully phase-able samples – displayed as non-MZ twin samples vs. MZ twin samples. Samples for which DNA was derived from LCL or WB are shown in red and green, respectively. All LCL MZ twins were included in the ARC training set and all WB MZ twins were included in the ARC test set. (B) The number of rare de novo variants identified in LCL (pink) and WB (blue) fully phase-able (non-MZ twin) samples before ARC (N=1,019 samples) and the number of rare de novo variants identified in LCL (pink) and WB (blue) fully phase-able (non-MZ twin) samples after ARC (variants with an ARC score <0.4 are filtered out) and after excluding ARC outlier samples (samples with >90% DNs removed by ARC) (N=716). After ARC, there is no significant difference in the rate of rare de novo variants based on the biological sequencing source (LCLmean=60.3 and WBmean=59.4; LCLmedian=57 and WBmedian=57). The difference in DN rates between the biological sequencing source (LCL vs. WB) was evaluated using Wilcoxon rank sum test. (C) The number of rare de novo coding variants identified per fully phase-able sample displayed as histograms. The coding RDNVs before ARC are from 1,177 fully phase-able samples and after ARC (variants with an ARC score <0.4 are filtered out) and after excluding ARC outlier samples (samples with >90% DNs removed by ARC) (n=831 samples). (D) The correlation between the rate of rare de novo variants and paternal age before and after ARC. This analysis considers 574 fully phase-able ASD children (excluding MZ twins and ARC outliers) for which paternal age was known. The red line is the linear regression line. The graph on the left shows the raw number of rare de novo variants (SNVs and indels) per child by paternal age at the time of the participant’s birth in years. The graph on the right shows the number of rare de novo variants (SNVs and indels) per child after running ARC by paternal age at the time of the participant’s birth in years.
{kind=link}
Figure S5. TADA-mega analysis simulation results, Related to Figure 4. (A) For each of the 18,472 TADA genes, the observed FDR in the iHART TADA-mega analysis is plotted against the simulated p-value. Genes with the smallest FDRs also have small simulated p-values, as expected. (B) The observed Bayes Factor (BF), for genes with a BF>1, in the iHART TADA-mega analysis is plotted against the simulated p-value. Genes with the largest BF also have small simulated p-values, as expected. (C-F) The TADA-mega analysis results from the previous study vs. current iHART study. Genes are sorted by increasing difference in the FDR (q-value) obtained in Sanders et al., 2015 vs. the current iHART TADA-mega analysis. In panels (C) and (D) the per-gene TADA FDR is displayed as the −log10(q-value) (higher dots have a lower FDR) obtained in Sanders et al. (green) and the current iHART study (red) and the horizontal line marks the FDR=0.1 threshold; for (C) the 13 genes with an FDR<0.1 in Sanders et al. that failed replication in iHART (FDR>0.1), and (D) the 16 newly significant genes identified in the iHART mega analysis with an FDR<0.1. Note that the CACNA2D3 gene is significantly associated with ASD in the iHART mega-analysis, but not the previous TADA mega-analysis. However, it was previously reported in De Rubeis et al., 2014 and thus we do not consider it a new risk gene. Below this, in panels (E) and (F), are the per-gene violin plots of Bayes Factors (displayed as log(simulated Bayes Factor)) obtained for each of the 1.1 million TADA simulations. The grey “x” marks the median simulated Bayes Factor, the blue dot indicates the observed Bayes Factor in the iHART TADA mega analysis, and the violin plots are filled according to their simulation p-value; for (E) the 13 genes with an FDR<0.1 in Sanders et al. that failed replication in iHART (FDR>0.1) (max p-value=0.06) and (F) the 16 newly significant genes (plus CACNA2D3) identified in the iHART mega analysis with an FDR<0.1 (max p-value=0.006).
{kind=link}
Figure S6. Biological insights from known and novel ASD-risk genes identified in the TADA-mega analysis, Related to Figure 4. (A) A box plot for the inherited PTV Bayes Factors observed for the 35 genes with an FDR<0.2 in the current iHART TADA mega analysis that had an FDR>0.2 in the et al., 2015 TADA mega analysis (Sanders et al.) (Kruskal–Wallis test, P=0.003). (B) The indirect PPI network formed by the 69 ASD-risk genes and the 98 genes harboring high-risk inherited variants (n=165 unique genes). The resulting indirect PPI was significant for two connectivity metrics – seed indirect degrees mean P=0.003, and CI degrees mean P=0.005. Proteins encoded by a gene with a high-risk inherited PTVs are shown in teal and SVs are shown in gold. Proteins encoded by a previously established ASD-risk gene (Sanders et al., 2015) are shown in purple, newly identified ASD-risk gene (iHART TADA mega analysis) are shown in red, those belonging to the BAF complex are shown in blue, and any protein falling in more than one category is colored with all categorical colors that apply (e.g., ARID1B). The gene label for significant seed genes are bold and blue. (C-D) Enrichment of iHART ASD-risk genes in single cell RNA seq (scRNA-seq) cell type expression signatures. Genes enriched in major cell type classes were obtained from human fetal brain datasets and an adult brain dataset, and the percentage of iHART ASD-risk genes in each cell type class is shown. (C) iHART 69 ASD-risk genes in fetal cell classes (left and center) and adult cell classes (right). (D) iHART 16 novel ASD-risk genes in fetal cell classes (left and center) and adult cell classes (right). Significant log2 odds ratios of neuronal cell type enrichment: Fetal drop-seq glutamatergic, 3; GABAergic 4.7; neuron 4.8. Fetal Nowakowski et al.(Nowakowski et al., 2017) glutamatergic, 1.6; GABAergic 2.4; neuron 5.4. Adult Lake et al. (Lake et al., 2018) Glutamatergic, 2.9; GABAergic 0.65; Neuron 3.4. The Broad expression class was defined as expression in neuronal cell types and glial cell types and Neuron class was defined as expression in glutamatergic and GABAergic cell types. The numbers inside each pie chart indicate the percentage of iHART ASD-risk genes in that cell type. (E) The interaction network for the gene ontology over-represented terms, and associated genes, for the genes enriched in inherited variation (TADA FDR<0.2, proportion of inherited variants ≥70%). We focused on the 23 genes with an FDR<0.2 for which the majority (≥70%) of their qualifying risk variants were inherited PTVs. The z-score is displayed together with each color-coded ontology term (squares) and the genes are color coded by the proportion of qualifying TADA variants that were inherited PTVs (circles).
{kind=link}
Figure S7. NR3C2 protein sequence alignment, zebrafish mutant sequence, and validation of the social preference assay, Related to Figure 6. (A) Multiple sequence alignment for human (Hs), mouse (Mm) and zebrafish (Dr) NR3C2 proteins. Amino acids are colored according to their chemical properties to highlight identical and similar residues. (B) Alignment of WT and mutant zebrafish NR3C2 proteins. Gray shading indicates altered amino acid sequence in the mutant. Blue and red lines indicate DNA binding domain and ligand binding domain, respectively. (C-I) Validation of the social preference assay. (C,D) WT zebrafish treated with DMSO vehicle control showed a significantly higher SPI during the post-baseline period compared to the baseline period, but WT zebrafish treated with 20 μM MK-801 did not. (E) The increase in SPI in the presence of a conspecific was significantly smaller for zebrafish treated with MK-801 compared to controls. (F,G) Both untreated WT zebrafish and WT zebrafish treated with 0.5% ethanol showed a significantly higher SPI during the post-baseline period compared to the baseline period, although the SPI increase was smaller for ethanol-treated animals. (H) The increase in SPI in the presence of a conspecific was significantly smaller for zebrafish treated with 0.5% ethanol compared to controls. (I) There was no significant difference in the body length of nr3c2 +/+, +/− and −/− siblings for the data presented in Figures 6B and 6C. Grey data points and lines represent individual animals. Red lines indicate mean ± SEM (C-H) or median ± 95% confidence interval (I). *P<0.05; **P<0.01; ***P<0.001, ns=not significant by paired t test (C,D,F,G), unpaired t test (E,H), or Kruskal-Wallis test with Dunn’s multiple comparison test (I).
{kind=link}
Highlights.
Identification of rare inherited variants associated with ASD and 16 new ASD risk genes
Inherited risk reveals both new biological pathways and shared PPI with known genes
We develop and validate a machine learning algorithm (ARC) to remove WGS artifacts
NR3C2 mutations define a novel syndromic form of ASD, which we model in zebrafish
ACKNOWLEDGMENTS
We thank Stephanie A. Arteaga, Stephanie N. Kravitz, Cheyenne L. Schloffman, Min Sun, Tor Solli-Nowlan, T.Chang, Hyejung Won, Sasha Sharma, Marlena Duda, Greg Madden Mclnnes, Ravina Jain, Valenti Moncunill, Josep M. Mercader, Montserrat Puiggros, Hailey H. Choi, Anika Gupta, and David Torrents for technical support, and Hannah Hurley and Amina Kinkhabwala for assistance with zebrafish experiments. We acknowledge the Hartwell Foundation for supporting whole genome sequencing and the creation of the iHART database, as well as the Simons Foundation who provided additional support for the genome sequencing. We thank the New York Genome Center for conducting sequencing and initial quality control. We thank the PRACE Research Infrastructure resource MareNostrum Ill based in Spain at the Barcelona Supercomputing Center. We thank A.Gordon, J.Huang, J.Sebat, and D.Antaki for their help in resolving the DLG2 structural variant. We thank J.Sul for helpful discussions and suggesting a machine learning approach. This work has been supported by grants from the NIH (NIMH U24, MH081810, R01MH064547, NS101158, NS070911, NS101665, NS095824, S10OD011939, P30AG10161, R01AG17917, and U01AG61356). We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, E. Wijsman). We appreciate obtaining access to genetic data on SFARI Base. Approved researchers can obtain the SSC population dataset described in this study (https://www.sfari.org/2015/12/11/whole-genome-analysis-of-the-simons-simplex-collection-ssc-2/#chapter-wgs-of-500-additional-ssc-families) by applying at https://base.sfari.org.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nature methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association; (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th edn (Arlington, Virginia, USA: ). [Google Scholar]
- An JY, Lin K, Zhu L, Werling DM, Dong S, Brand H, Wang HZ, Zhao X, Schwartz GB, Collins RL, et al. (2018). Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science (New York, NY) 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchelli E, Blasi F, Biondolillo M, Lamb JA, Bonora E, Barnby G, Parr J, Beyer KS, Klauck SM, Poustka A, et al. (2003). Screening of nine candidate genes for autism on chromosome 2q reveals rare nonsynonymous variants in the cAMP-GEFII gene. Molecular psychiatry 8, 916–924. [DOI] [PubMed] [Google Scholar]
- Ballester P, Martinez MJ, Javaloyes A, Inda MM, Fernandez N, Gazquez P, Aguilar V, Perez A, Hernandez L, Richdale AL, et al. (2018). Sleep Problems in Adults With Autism Spectrum Disorder and Intellectual Disability. Autism research : official journal of the International Society for Autism Research. [DOI] [PubMed] [Google Scholar]
- Barnett DW, Garrison EK, Quinlan AR, Stromberg MP, and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics (Oxford, England) 27, 1691–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A, Brown CD, Engelhardt BE, and Montgomery SB (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayes A, van de Lagemaat LN, Collins MO, Croning MD, Whittle IR, Choudhary JS, and Grant SG (2011). Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nature neuroscience 14, 19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgard TG, and Geschwind DH (2013). Retooling spare parts: gene duplication and cognition. Nature neuroscience 16, 6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, and Schneider JA (2018). Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis 64, S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C, Vulto-van Silfhout AT, et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besenbacher S, Sulem P, Helgason A, Helgason H, Kristjansson H, Jonasdottir A, Jonasdottir A, Magnusson OT, Thorsteinsdottir U, Masson G, et al. (2016). Multinucleotide de novo Mutations in Humans. PLoS genetics 12, e1006315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandler WM, Antaki D, Gujral M, Kleiber ML, Whitney J, Maile MS, Hong O, Chapman TR, Tan S, Tandon P, et al. (2018). Paternally inherited cis-regulatory structural variants are associated with autism. Science (New York, NY) 360, 327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandler WM, Antaki D, Gujral M, Noor A, Rosanio G, Chapman TR, Barrera DJ, Lin GN, Malhotra D, Watts AC, et al. (2016). Frequency and Complexity of De Novo Structural Mutation in Autism. American journal of human genetics 98, 667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC (2014). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. Morbidity and mortality weekly report Surveillance summaries (Washington, DC : 2002) 63, 1–21. [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. (2009). BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nature methods 6, 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LE, et al. (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nature genetics 46, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvert E, Tick B, McEwen F, Stewart C, Curran SR, Woodhouse E, Gillan N, Hallett V, Lietz S, Garnett T, et al. (2015). Heritability of Autism Spectrum Disorder in a UK Population-Based Twin Sample. JAMA psychiatry 72, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. (2011). Variation in genome-wide mutation rates within and between human families. Nature genetics 43, 712–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantino JN, Zhang Y, Frazier T, Abbacchi AM, and Law P (2010). Sibling recurrence and the genetic epidemiology of autism. The American journal of psychiatry 167, 1349–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. (2011). A copy number variation morbidity map of developmental delay. Nature genetics 43, 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, and Geschwind DH (2018). The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172, 289–304. e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders, S. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreosti E, Lopes G, Kampff AR, and Wilson SW (2015). Development of social behavior in young zebrafish. Front Neural Circuits 9, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. (2010). Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science (New York, NY) 327, 78–81. [DOI] [PubMed] [Google Scholar]
- Francioli LC, Polak PP, Koren A, Menelaou A, Chun S, Renkens I, van Duijn CM, Swertz M, Wijmenga C, van Ommen G, et al. (2015). Genome-wide patterns and properties of de novo mutations in humans. Nature genetics 47, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J, et al. (2014). Most genetic risk for autism resides with common variation. Nature genetics 46, 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project, C., Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, and McVean GA (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, and Flint J (2015). Genetics and genomics of psychiatric disease. Science (New York, NY) 349, 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann JM, Wong WS, Pinelli M, Farrah T, Bodian D, Stittrich AB, Glusman G, Vissers LE, Hoischen A, Roach JC, et al. (2016). Parent-of-origin-specific signatures of de novo mutations. Nature genetics 48, 935–939. [DOI] [PubMed] [Google Scholar]
- Grant SG (2016). The molecular evolution of the vertebrate behavioural repertoire. Philosophical transactions of the Royal Society of London Series B, Biological sciences 371, 20150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Korn JM, Nemesh J, and McCarroll SA (2011). Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nature genetics 43, 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, and McCarroll SA (2015). Large multiallelic copy number variations in humans. Nature genetics 47, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, Schellenberg GD, Gibbs RA, Daly MJ, Buxbaum JD, et al. (2013). Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS genetics 9, e1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra RA, Bartels M, Verweij CJ, and Boomsma DI (2007). Heritability of autistic traits in the general population. Archives of pediatrics & adolescent medicine 161, 372–377. [DOI] [PubMed] [Google Scholar]
- Hormozdiari F, Penn O, Borenstein E, and Eichler EE (2015). The discovery of integrated gene networks for autism and related disorders. Genome research 25, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn H, Lawrence MS, Chouinard CR, Shrestha Y, Hu JX, Worstell E, Shea E, Ilic N, Kim E, Kamburov A, et al. (2018). NetSig: network-based discovery from cancer genomes. Nature methods 15, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, and Joung JK (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature biotechnology 31, 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun G, Flickinger M, Hetrick KN, Romm JM, Doheny KF, Abecasis GR, Boehnke M, and Kang HM (2012). Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. American journal of human genetics 91, 839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv. [Google Scholar]
- Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ, Moreno-De-Luca D, Yu TW, Fombonne E, Geschwind D, et al. (2012). Common genetic variants, acting additively, are a major source of risk for autism. Molecular autism 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmicki JA, Samocha KE, Howrigan DP, Sanders SJ, Slowikowski K, Lek M, Karczewski KJ, Cutler DJ, Devlin B, Roeder K, et al. (2017). Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nature genetics 49, 504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A, Zhang R, Yao V, Theesfeld CL, Wong AK, Tadych A, Volfovsky N, Packer A, Lash A, and Troyanskaya OG (2016). Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nature neuroscience 19, 1454–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX, et al. (2015). Excess of rare, inherited truncating mutations in autism. Nature genetics 47, 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage K, Karlberg EO, Storling ZM, Olason PI, Pedersen AG, Rigina O, Hinsby AM, Turner Z, Pociot F, Tommerup N, et al. (2007). A human phenome-interactome network of protein complexes implicated in genetic disorders. Nature biotechnology 25, 309–316. [DOI] [PubMed] [Google Scholar]
- Lajonchere CM (2010). Changing the landscape of autism research: the autism genetic resource exchange. Neuron 68, 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, et al. (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nature biotechnology 36, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsch J, and Baier H (2018). Biological motion as an innate perceptual mechanism driving social affiliation. bioRxiv. [DOI] [PubMed] [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, and Hall IM (2014). LUMPY: a probabilistic framework for structural variant discovery. Genome biology 15, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppa VM, Kravitz SN, Martin CL, Andrieux J, Le Caignec C, Martin-Coignard D, DyBuncio C, Sanders SJ, Lowe JK, Cantor RM, et al. (2016). Rare Inherited and De Novo CNVs Reveal Complex Contributions to ASD Risk in Multiplex Families. American journal of human genetics 99, 540–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, et al. (2011). Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, and Scherer SW (2014). The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic acids research 42, D986–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. (2008). Structural variation of chromosomes in autism spectrum disorder. American journal of human genetics 82, 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CL, Duvall JA, Ilkin Y, Simon JS, Arreaza MG, Wilkes K, Alvarez-Retuerto A, Whichello A, Powell CM, Rao K, et al. (2007). Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 144b, 869–876. [DOI] [PubMed] [Google Scholar]
- Maxwell-Horn A, and Malow BA (2017). Sleep in Autism. Semin Neurol 37, 413–418. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome biology 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, et al. (2008). Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. The New England journal of medicine 359, 1685–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, et al. (2012). Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151, 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, et al. (2010). Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American journal of human genetics 86, 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncunill V, Gonzalez S, Bea S, Andrieux LO, Salaverria I, Royo C, Martinez L, Puiggros M, Segura-Wang M, Stutz AM, et al. (2014). Comprehensive characterization of complex structural variations in cancer by directly comparing genome sequence reads. Nature biotechnology 32, 1106–1112. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nonneman RJ, Shafer GO, Nikolova VD, Riddick NV, Agster KL, Baker LK, and Knapp DJ (2013). Disruption of social approach by MK-801, amphetamine, and fluoxetine in adolescent C57BL/6J mice. Neurotoxicol Teratol 36, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithianantharajah J, Komiyama NH, McKechanie A, Johnstone M, Blackwood DH, St Clair D, Emes RD, van de Lagemaat LN, Saksida LM, Bussey TJ, et al. (2013). Synaptic scaffold evolution generated components of vertebrate cognitive complexity. Nature neuroscience 16, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science (New York, NY) 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozonoff S, Young GS, Carter A, Messinger D, Yirmiya N, Zwaigenbaum L, Bryson S, Carver LJ, Constantino JN, Dobkins K, et al. (2011). Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics 128, e488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Gandal MJ, and Geschwind DH (2015). Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nature reviews Genetics 16, 441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, and Geschwind DH (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta LT, Huang J, et al. (2016). Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. American journal of human genetics 94, 677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polioudakis D, de la Torre-Ubieta L, Langerman J, Elkins AG, Stein JL, Vuong CK, Opland CK, Lu D, Connell W, Ruzzo EK, et al. (2018). A single cell transcriptomic analysis of human neocortical development. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prober DA, Rihel J, Onah AA, Sung RJ, and Schier AF (2006). Hypocretin/orexin overexpression induces an insomnia-like phenotype in zebrafish. J Neurosci 26, 13400–13410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England) 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronemus M, Iossifov I, Levy D, and Wigler M (2014). The role of de novo mutations in the genetics of autism spectrum disorders. Nature reviews Genetics 15, 133–141. [DOI] [PubMed] [Google Scholar]
- Rossin EJ, Lage K, Raychaudhuri S, Xavier RJ, Tatar D, Benita Y, Cotsapas C, and Daly MJ (2011). Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS genetics 7, e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnstrom K, Mallick S, Kirby A, et al. (2014). A framework for the interpretation of de novo mutation in human disease. Nature genetics 46, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, and Reichenberg A (2014). The familial risk of autism. Jama 311, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheet P, and Stephens M (2006). A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. American journal of human genetics 78, 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. (2007). Strong association of de novo copy number mutations with autism. Science (New York, NY) 316, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuse DH, Mandy WP, and Scourfield J (2005). Measuring autistic traits: heritability, reliability and validity of the Social and Communication Disorders Checklist. The British journal of psychiatry : the journal of mental science 187, 568–572. [DOI] [PubMed] [Google Scholar]
- Snijders Blok L, Madsen E, Juusola J, Gilissen C, Baralle D, Reijnders MR, Venselaar H, Helsmoortel C, Cho MT, Hoischen A, et al. (2015). Mutations in DDX3X Are a Common Cause of Unexplained Intellectual Disability with Gender-Specific Effects on Wnt Signaling. American journal of human genetics 97, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Study DDD (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, Ragavendran A, Brand H, Lucente D, Miles J, et al. (2014). CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proceedings of the National Academy of Sciences of the United States of America 111, E4468–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, Raja A, Baker C, Hoekzema K, Stessman HA, et al. (2016). Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. American journal of human genetics 98, 58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current protocols in bioinformatics 43, 11.10.11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeweyer G, Helsmoortel C, Van Dijck A, Vulto-van Silfhout AT, Coe BP, Bernier R, Gerdts J, Rooms L, van den Ende J, Bakshi M, et al. (2014). The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. American journal of medical genetics Part C, Seminars in medical genetics 166c, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virkud YV, Todd RD, Abbacchi AM, Zhang Y, and Constantino JN (2009). Familial aggregation of quantitative autistic traits in multiplex versus simplex autism. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 150b, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, and Geschwind DH (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling DM, Brand H, An JY, Stone MR, Zhu L, Glessner JT, Collins RL, Dong S, Layer RM, Markenscoff-Papadimitriou E, et al. (2018). An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nature genetics 50, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling DM, and Geschwind DH (2015). Recurrence rates provide evidence for sex-differential, familial genetic liability for autism spectrum disorders in multiplex families and twins. Molecular autism 6, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, Zhang Z, Xue C, Herre M, Silver PA, Zhang MQ, et al. (2014). HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell reports 6, 1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RKC, Merico D, Bookman M,J,LH, Thiruvahindrapuram B, Patel RV, Whitney J, Deflaux N, Bingham J, Wang Z, et al. (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nature neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann FF, Gaspary KV, Siebel AM, and Bonan CD (2016). Oxytocin reversed MK-801-induced social interaction and aggression deficits in zebrafish. Behav Brain Res 311, 368–374. [DOI] [PubMed] [Google Scholar]
- Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, and Salit M (2014). Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nature biotechnology 32, 246–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. iHART sample information, Related to Figures 1 and 2. Detailed sample information for the 2,308 AGRE samples whole-genome sequenced as part of iHART. Including phenotypic descriptions for NR3C2 families and haplotype predictions for the DLG2 families.
Table S2. RDNV rates in multiplex vs. simplex autism families, Related to Figure 3. Comparison of RDNV rates in affected and unaffected children from ASD simplex vs. multiplex families.
Table S3. TADA results, Related to Figure 4. Results from the TADA mega-analysis, including the sample counts included from each cohort, the qualifying variants identified in iHART/AGRE samples, and the iHART TADA-mega analysis results for all 18,472 genes.
Table S4. NetSig significant genes, Related to Figure 5. The 596 genes that are significantly (P < 0.05) directly connected to potential ASD-risk genes (as defined by their TADA q-value) using NetSig.
Table S5. Structural Variant Method Details, Related to Figure 1. Structural variant method details, including the number of rare SVs identified by each calling algorithm, percentage of SVs also called by at least one other algorithm, sensitivity comparisons to genotyping array data, and LUMPY-BreakDancer joint deletion call validation rate by deletion size.
Figure S1. Whole-genome sequence coverage statistics for 2,308 iHART/AGRE samples and the high-resolution detection of large structural variants (SVs), Related to Figure 1. There were no significant differences in the average fold coverage per sample across the cohort and no differences in the categories of (A) ASD affectation status, (B) sex, or (D) family member type – where family member type was simplified to include Mother, Father, Child (proband, sibling, MZ or DZ twin) and Other (e.g., cousin). (C) The percent of exonic and genomic bases covered at ≥10x in all family members for each of the 422 fully-phaseable iHART families. Exonic regions were defined as those annotated as protein-coding exons in Gencode V19 (>75Mb). Genomic regions were defined as all non-N bases in the reference genome (>2.8Gb). (E) The percentage of genomic bases covered at greater than or equal to 1X, 10X, 20X, 30X, and 40X bases for the 2,308 iHART samples with WGS data. On average, 98.97 ± 0.37 % of bases were covered at a depth of ≥10X. (F) An overview of our custom multi-algorithm consensus SV pipeline for high-resolution detection of large structural variants (SVs) from whole-genome sequence data. The four boxes at the top list the four main algorithms used to call SVs, and the parenthetical describes the detection strategy(s) used by each algorithm: AS, de novo assembly method; SR, split-read method; RP, read-pair method; RC, read-count method. (G) Venn diagrams of structural variants detected by four different algorithms for all and rare (AF < 0.001 in cDGV and AF < 0.01 in iHART HNP samples) SVs (DELs, DUPs and INVs) detected in 1,377 phase-able WGS samples by SMuFin, LUMPY, GenomeSTRiP and BreakDancer, after excluding events with ≥50% overlap with genomic low-complexity regions (Brandler et al., 2016). Additional per-algorithm filters were also applied prior to the generation of this Venn diagram as described in Methods. (H) A schematic overview of the SMuFin detection pipeline. Families are processed as independent trios, where the sequence reads from a child are aligned to the mother’s genome and then the father’s genome, treating the parental genome as the reference genome in both comparisons. Each comparison, or SMuFin execution, results in variants identified in the child by that parent-offspring comparison. All three members of the trio are considered for assigning the corresponding inheritance of variants identified in the child. A variant detected when comparing to both mom and dad is de novo, while a variant detected only when comparing to mom is paternally inherited and a variant detected only when comparing to dad is maternally inherited.
Figure S2. Additional details on rare inherited PTVs and SVs, Related to Figure 2. (A-D) Rare inherited coding variants by consequence and inheritance. The rate of rare inherited coding variants per fully phase-able child is displayed for 960 affected (red) and 217 unaffected (blue) children by both variant consequence and inheritance, this includes newly hemizygous variants in 563 affected (red) and 100 unaffected (blue) male children. The graph for newly homozygous PTVs (C) was excluded because none were identified in affected or unaffected children. Mean ± SE rates are shown. (E) The rate of private inherited PTVs in 960 affected (red) and 217 unaffected (blue) children iHART children for all genes vs. PTV intolerant genes. We found no excess of inherited private PTVs in mutation intolerant genes (pLI≥0.9) (Lek et al., 2016) in affected subjects (P=0.40, quasi-Poisson linear regression). Mean ± SE rates are shown. (F) The rate of rare inherited SVs per fully phase-able child is displayed for 960 affected (red) and 217 unaffected (blue) children by inheritance type. Mean ± SE rates are shown. (G) The rate of rare inherited SVs per fully phase-able child is displayed for newly hemizygous variants in 563 affected (red) and 100 unaffected (blue) male children. (H-M) The rate of rare inherited SVs per fully phase-able child identified in 960 affected (red) and 217 unaffected (blue) children by inheritance type; this includes newly hemizygous variants in 563 affected (red) and 100 unaffected (blue) male children. Mean ± SE rates are shown. Maternally and paternally inherited SVs by affectation status and gene disruption for deletions (H), duplications (I), and inversions (J). Newly hemizygous SVs by affectation status and gene disruption for deletions (K), duplications (L), and inversions (M). (N) The DLG2 promoter-disrupting 2.5Kb deletion (chr11: 85339733 – 85342186), displayed as an orange rectangle, detected in three independent iHART families. This 2.5Kb deletion is transmitted to all affected members of two different iHART families. This deletion falls in a recently-defined, functional, non-coding regulatory region in developing human brain (chr11:85338026-85340560) (de la Torre-Ubieta et al., 2018); below the deletion we show the average ATAC-seq peak read depth from the cortical plate (CP) and ventricular zone (VZ) of developing human brain samples (n=3).
Figure S3. ARC performance in the training and test set, Related to Figure 3. The 10-fold cross validation (CV) for the ARC training set (A-D). (A) Receiver Operating Characteristic (ROC) curves for the true positive rate (Sensitivity) is plotted as a function of the false positive rate; (B) Area Under the ROC Curve (ROC AUC) statistics (median=0.992) for each of the 10 folds; (C) Precision rates vs. predicted score cutoffs – the dashed line at the selected score (0.4) highlights that the minimum precision across all 10 folds is >0.9; (D) Recall rates vs. predicted score cutoffs – the dashed line at the selected score (0.4) highlights that the minimum precision across all 10 folds is ~0.8. ARC performance in the test set (E-H). (E) Distribution of all test set variants by ARC score; (F) Distribution of all TRUE test set variants by ARC score – the majority of concordant variants have an ARC score of ≥0.4; (G) Distribution of all FALSE test set variants by ARC score – almost all discordant variants have an ARC score of <0.4; (H) The precision and recall rates vs. predicted ARC score cutoff in the test set – the dashed line at the selected score (0.4) highlights that the precision is >0.95 and the recall is >0.85.
Figure S4. RDNVs identified in iHART samples before and after ARC, Related to Figure 3. (A) The ARC score distribution for raw de novo variants identified in 1,177 fully phase-able samples – displayed as non-MZ twin samples vs. MZ twin samples. Samples for which DNA was derived from LCL or WB are shown in red and green, respectively. All LCL MZ twins were included in the ARC training set and all WB MZ twins were included in the ARC test set. (B) The number of rare de novo variants identified in LCL (pink) and WB (blue) fully phase-able (non-MZ twin) samples before ARC (N=1,019 samples) and the number of rare de novo variants identified in LCL (pink) and WB (blue) fully phase-able (non-MZ twin) samples after ARC (variants with an ARC score <0.4 are filtered out) and after excluding ARC outlier samples (samples with >90% DNs removed by ARC) (N=716). After ARC, there is no significant difference in the rate of rare de novo variants based on the biological sequencing source (LCLmean=60.3 and WBmean=59.4; LCLmedian=57 and WBmedian=57). The difference in DN rates between the biological sequencing source (LCL vs. WB) was evaluated using Wilcoxon rank sum test. (C) The number of rare de novo coding variants identified per fully phase-able sample displayed as histograms. The coding RDNVs before ARC are from 1,177 fully phase-able samples and after ARC (variants with an ARC score <0.4 are filtered out) and after excluding ARC outlier samples (samples with >90% DNs removed by ARC) (n=831 samples). (D) The correlation between the rate of rare de novo variants and paternal age before and after ARC. This analysis considers 574 fully phase-able ASD children (excluding MZ twins and ARC outliers) for which paternal age was known. The red line is the linear regression line. The graph on the left shows the raw number of rare de novo variants (SNVs and indels) per child by paternal age at the time of the participant’s birth in years. The graph on the right shows the number of rare de novo variants (SNVs and indels) per child after running ARC by paternal age at the time of the participant’s birth in years.
Figure S5. TADA-mega analysis simulation results, Related to Figure 4. (A) For each of the 18,472 TADA genes, the observed FDR in the iHART TADA-mega analysis is plotted against the simulated p-value. Genes with the smallest FDRs also have small simulated p-values, as expected. (B) The observed Bayes Factor (BF), for genes with a BF>1, in the iHART TADA-mega analysis is plotted against the simulated p-value. Genes with the largest BF also have small simulated p-values, as expected. (C-F) The TADA-mega analysis results from the previous study vs. current iHART study. Genes are sorted by increasing difference in the FDR (q-value) obtained in Sanders et al., 2015 vs. the current iHART TADA-mega analysis. In panels (C) and (D) the per-gene TADA FDR is displayed as the −log10(q-value) (higher dots have a lower FDR) obtained in Sanders et al. (green) and the current iHART study (red) and the horizontal line marks the FDR=0.1 threshold; for (C) the 13 genes with an FDR<0.1 in Sanders et al. that failed replication in iHART (FDR>0.1), and (D) the 16 newly significant genes identified in the iHART mega analysis with an FDR<0.1. Note that the CACNA2D3 gene is significantly associated with ASD in the iHART mega-analysis, but not the previous TADA mega-analysis. However, it was previously reported in De Rubeis et al., 2014 and thus we do not consider it a new risk gene. Below this, in panels (E) and (F), are the per-gene violin plots of Bayes Factors (displayed as log(simulated Bayes Factor)) obtained for each of the 1.1 million TADA simulations. The grey “x” marks the median simulated Bayes Factor, the blue dot indicates the observed Bayes Factor in the iHART TADA mega analysis, and the violin plots are filled according to their simulation p-value; for (E) the 13 genes with an FDR<0.1 in Sanders et al. that failed replication in iHART (FDR>0.1) (max p-value=0.06) and (F) the 16 newly significant genes (plus CACNA2D3) identified in the iHART mega analysis with an FDR<0.1 (max p-value=0.006).
Figure S6. Biological insights from known and novel ASD-risk genes identified in the TADA-mega analysis, Related to Figure 4. (A) A box plot for the inherited PTV Bayes Factors observed for the 35 genes with an FDR<0.2 in the current iHART TADA mega analysis that had an FDR>0.2 in the et al., 2015 TADA mega analysis (Sanders et al.) (Kruskal–Wallis test, P=0.003). (B) The indirect PPI network formed by the 69 ASD-risk genes and the 98 genes harboring high-risk inherited variants (n=165 unique genes). The resulting indirect PPI was significant for two connectivity metrics – seed indirect degrees mean P=0.003, and CI degrees mean P=0.005. Proteins encoded by a gene with a high-risk inherited PTVs are shown in teal and SVs are shown in gold. Proteins encoded by a previously established ASD-risk gene (Sanders et al., 2015) are shown in purple, newly identified ASD-risk gene (iHART TADA mega analysis) are shown in red, those belonging to the BAF complex are shown in blue, and any protein falling in more than one category is colored with all categorical colors that apply (e.g., ARID1B). The gene label for significant seed genes are bold and blue. (C-D) Enrichment of iHART ASD-risk genes in single cell RNA seq (scRNA-seq) cell type expression signatures. Genes enriched in major cell type classes were obtained from human fetal brain datasets and an adult brain dataset, and the percentage of iHART ASD-risk genes in each cell type class is shown. (C) iHART 69 ASD-risk genes in fetal cell classes (left and center) and adult cell classes (right). (D) iHART 16 novel ASD-risk genes in fetal cell classes (left and center) and adult cell classes (right). Significant log2 odds ratios of neuronal cell type enrichment: Fetal drop-seq glutamatergic, 3; GABAergic 4.7; neuron 4.8. Fetal Nowakowski et al.(Nowakowski et al., 2017) glutamatergic, 1.6; GABAergic 2.4; neuron 5.4. Adult Lake et al. (Lake et al., 2018) Glutamatergic, 2.9; GABAergic 0.65; Neuron 3.4. The Broad expression class was defined as expression in neuronal cell types and glial cell types and Neuron class was defined as expression in glutamatergic and GABAergic cell types. The numbers inside each pie chart indicate the percentage of iHART ASD-risk genes in that cell type. (E) The interaction network for the gene ontology over-represented terms, and associated genes, for the genes enriched in inherited variation (TADA FDR<0.2, proportion of inherited variants ≥70%). We focused on the 23 genes with an FDR<0.2 for which the majority (≥70%) of their qualifying risk variants were inherited PTVs. The z-score is displayed together with each color-coded ontology term (squares) and the genes are color coded by the proportion of qualifying TADA variants that were inherited PTVs (circles).
Figure S7. NR3C2 protein sequence alignment, zebrafish mutant sequence, and validation of the social preference assay, Related to Figure 6. (A) Multiple sequence alignment for human (Hs), mouse (Mm) and zebrafish (Dr) NR3C2 proteins. Amino acids are colored according to their chemical properties to highlight identical and similar residues. (B) Alignment of WT and mutant zebrafish NR3C2 proteins. Gray shading indicates altered amino acid sequence in the mutant. Blue and red lines indicate DNA binding domain and ligand binding domain, respectively. (C-I) Validation of the social preference assay. (C,D) WT zebrafish treated with DMSO vehicle control showed a significantly higher SPI during the post-baseline period compared to the baseline period, but WT zebrafish treated with 20 μM MK-801 did not. (E) The increase in SPI in the presence of a conspecific was significantly smaller for zebrafish treated with MK-801 compared to controls. (F,G) Both untreated WT zebrafish and WT zebrafish treated with 0.5% ethanol showed a significantly higher SPI during the post-baseline period compared to the baseline period, although the SPI increase was smaller for ethanol-treated animals. (H) The increase in SPI in the presence of a conspecific was significantly smaller for zebrafish treated with 0.5% ethanol compared to controls. (I) There was no significant difference in the body length of nr3c2 +/+, +/− and −/− siblings for the data presented in Figures 6B and 6C. Grey data points and lines represent individual animals. Red lines indicate mean ± SEM (C-H) or median ± 95% confidence interval (I). *P<0.05; **P<0.01; ***P<0.001, ns=not significant by paired t test (C,D,F,G), unpaired t test (E,H), or Kruskal-Wallis test with Dunn’s multiple comparison test (I).
Data Availability Statement
The whole-genome sequencing data generated during this study are available from the Hartwell Foundation’s Autism Research and Technology Initiative (iHART) following request and approval of the data use agreement available at http://www.ihart.org. Access to the whole-genome sequencing data generated in this study will be subject to approval by Autism Speaks and AGRE. Details about the format of the data, access options, and access instructions are included at http://www.ihart.org.
We also freely provide the code for ARC (Artifact Removal by Classifier), our random forest supervised model developed to distinguish true rare de novo variants from LCL-specific genetic aberrations or other types of artifacts such as sequencing and mapping errors, together with a full tutorial at https://github.com/walllab/iHART-ARC.
Interactive genotype/phenotype search engine
To facilitate sharing of iHART data with the broader autism research community and patients, we implemented a set of online data access methods to preview and search genetic variants and phenotypic traits (http://www.ihart.org/home).
Zebrafish data
The zebrafish datasets generated and analyzed in this study, and the code used to generate the data, are available upon request.