

Abstract
The mTOR pathway integrates a diverse set of environmental cues, such as growth factor signals and nutritional status, to direct eukaryotic cell growth. Over the past two and a half decades, mapping of the mTOR signalling landscape has revealed that mTOR controls biomass accumulation and metabolism by modulating key cellular processes, including protein synthesis and autophagy. Given the pathway’s central role in maintaining cellular and physiological homeostasis, dysregulation of mTOR signalling has been implicated in metabolic disorders, neurodegeneration, cancer and ageing. In this Review, we highlight recent advances in our understanding of the complex regulation of the mTOR pathway and discuss its function in the context of physiology, human disease and pharmacological intervention.
Introduction
In 1964, a team of pharmaceutical prospectors from Ayerst Research Laboratories struck microbial gold in a soil sample from the island of Rapa Nui (Easter Island). From a Streptomyces hygroscopicus soil bacterium, Sehgal and colleagues isolated a novel macrolide with potent antifungal activity, which they named ‘rapamycin’ in deference to its place of origin1. Subsequent studies of rapamycin elaborated on its immunosuppressive, antitumour and neuroprotective properties, generating significant clinical excitement2,3,4. Nonetheless, its mechanism of action remained elusive for more than 20 years until a series of breakthroughs in the early 1990s cracked open both the mystery of rapamycin and one of the most important signalling networks in biology.
In 1990, Schreiber and colleagues demonstrated that rapamycin acts in part by binding the prolyl-isomerase FKBP12 to form a gain-of-function complex that broadly inhibits cell growth and proliferation5,6. However, the full mechanism of action of rapamycin was only elucidated in 1994, when three groups used biochemical affinity purification of the FKBP12–rapamycin complex to identify a large kinase as the mechanistic (originally ‘mammalian’) target of rapamycin (mTOR) in mammals7,8,9. This discovery also revealed homology between mTOR and the yeast TOR/DRR proteins, which had previously emerged as rapamycin targets in genetic screens for rapamycin resistance10,11,12,13.
As intimated by the profound effects of rapamycin treatment, we now know that the mTOR protein kinase lies at the nexus of many major signalling pathways and plays a key part in organizing the cellular and organismal physiology of all eukaryotes. In the two and a half decades since its discovery, mTOR has emerged as the central node in a network that controls cell growth. As such, it integrates information about the availability of energy and nutrients to coordinate the synthesis or breakdown of new cellular components. Dysregulation of this fundamental signalling pathway disrupts cellular homeostasis and may fuel the overgrowth of cancers and the pathologies associated with ageing and metabolic disease.
In this Review, we analyse the signalling landscape of the mTOR pathway, from the inputs that regulate mTOR activation to the downstream effectors that enact its pro-growth programmes. In particular, we highlight how the intimate association between mTOR and the lysosome can facilitate rapid mobilization of nutrients upon stress or starvation. We then discuss how the mTOR pathway responds to metabolic signals in diverse organisms, cell types and tissues. Finally, drawing on recent advances in our understanding of mTOR pathway structure and function, we examine pharmacological approaches that target the pathway and evaluate their therapeutic potential in the treatment of metabolic disease, neurodegeneration, cancer and ageing.
Architecture of mTORC1 and mTORC2
mTOR is a 289-kDa serine/threonine protein kinase in the PI3K-related protein kinases (PIKK) family14. In mammals, it constitutes the catalytic subunit of two distinct complexes known as mTOR complex 1 (mTORC1) and mTORC2. These complexes are distinguished by their accessory proteins and their differential sensitivity to rapamycin, as well as by their unique substrates and functions (Fig. 1a).
Fig. 1: Structure and function of mTORC1 and mTORC2.
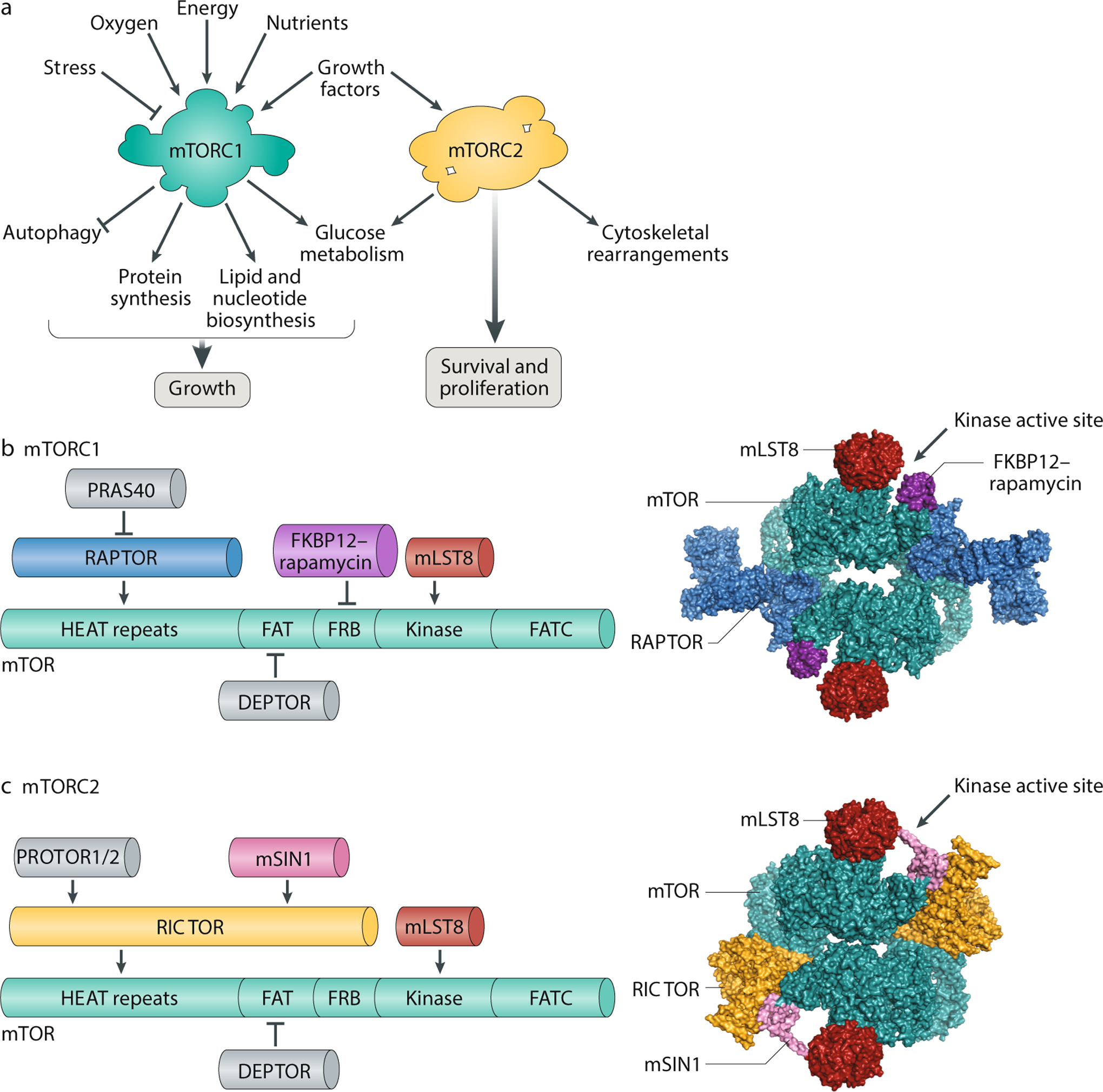
A. mTOR complex 1 (mTORC1) and mTORC2 have distinct signalling roles in the cell. mTORC1 integrates information about nutritional abundance and environmental status to tune the balance of anabolism and catabolism in the cell, while mTORC2 governs cytoskeletal behaviour and activates several pro-survival pathways. Unlike mTORC1, which is acutely inhibited by rapamycin, mTORC2 responds only to chronic rapamycin treatment. B. Components of mTORC1 (left). The domain structure of the mTOR kinase (green) is annotated with binding sites for the other mTORC1 subunits. The N-terminus of mTOR contains clusters of huntingtin, elongation factor 3, a subunit of protein phosphatase 2A and TOR1 (HEAT) repeats, followed by a FRAP, ATM and TRRAP (FAT) domain; the FKBP12–rapamycin binding (FRB) domain; the catalytic kinase domain; and the C-terminal FATC domain. mTOR binds mammalian lethal with SEC13 protein 8 (mLST8), a core component of the complex, and DEP-domain-containing mTOR-interacting protein (DEPTOR), an endogenous inhibitor of mTORC1 activity. Regulatory-associated protein of mTOR (Raptor), the defining subunit of mTORC1, binds mTOR with its own HEAT repeats and is required for lysosomal localization of the complex. Raptor also recruits proline-rich AKT substrate 40 kDa (PRAS40), an insulin-regulated inhibitor of mTORC1 activity. A 5.9-Å reconstruction of mTORC1 (without PRAS40 and DEPTOR) complexed with FKBP12–rapamycin is shown as a surface representation (Protein Database (PDB) ID: 5FLC) (right). C. Components of mTORC2 (left). The mTOR kinase (green) is annotated with the binding sites for the other constituent subunits of mTORC2. These subunits include mLST8, DEPTOR and RICTOR, the defining component of mTORC2. As a scaffolding protein, RICTOR recruits protein associated with rictor 1 or 2 (PROTOR1/2) to the complex, along with MAPK-interacting protein (mSIN1), which contains a pleckstrin homology domain. A 4.9-Å reconstruction of mTORC2 (without DEPTOR and PROTOR) is shown as a surface representation (PDB: 5ZCS) (right).
mTORC1 is nucleated by three core components: mTOR, mammalian lethal with SEC13 protein 8 (mLST8, also known as GβL)15 and its unique defining subunit, the scaffold protein regulatory-associated protein of mTOR (RAPTOR)16,17 (Fig. 1b). While structural data suggest that mLST8 may stabilize the kinase domain of mTOR18, ablation of this protein does not affect phosphorylation of known mTORC1 substrates in vivo19. Meanwhile, RAPTOR is essential for proper subcellular localization of mTORC1 and can recruit substrates of mTORC1 by binding the TOR signalling motifs that are present on several canonical mTOR substrates20,21. In addition, RAPTOR forms a scaffold for the mTORC1 accessory factor proline-rich AKT substrate 40 kDa (PRAS40)22,23, which acts as an endogenous inhibitor of mTORC1 activity alongside DEP-domain-containing mTOR-interacting protein (DEPTOR)24.
In the past decade, structural studies have shed new light on the assembly and catalysis of mTORC1. Cryo-electron microscopy and crystallographic analyses have revealed that mTORC1 dimerizes to form a megaDalton ‘lozenge’, with dimerization occurring along the mTOR HEAT repeats and the mTOR–RAPTOR interface25,26. In isolation, this complex is relatively inactive; a recent structure suggests that key residues in the kinase domain of mTOR may only shift into a catalytic position after the complex binds its essential activator, the small GTPase Rheb27. Similar co-crystallization approaches have also established the basis of mTORC1 inhibition by FKBP12–rapamycin and PRAS40, both of which bind the FKBP12–rapamycin binding domain of mTOR to partially occlude substrate entry into the kinase active site18,27. Further structural analysis of mTORC1 in the presence of its substrates and regulators may offer additional insights into the mTORC1 mechanism and function.
In contrast to mTORC1, mTORC2 retains the ability to phosphorylate its substrates upon acute rapamycin treatment. As with mTORC1, the core of mTORC2 is formed by mTOR and mLST8, the latter of which is required for mTORC2 stability and function19,28 (Fig. 1c). In lieu of RAPTOR, however, mTORC2 is defined by the unrelated scaffolding protein RICTOR29,30, which binds MAPK-interacting protein 1 (mSIN1)31,32,33, DEPTOR (as in mTORC1)24 and protein associated with rictor 1 or 2 (PROTOR1/2) to form the complex34,35. Of note, mSIN1 has a phospholipid-binding pleckstrin homology domain, which may help mTORC2 assemble on the plasma membrane36. Recent cryo-electron microscopy reconstructions of mTOR bound to mLST8, RICTOR and mSin1 show that mTORC2 also dimerizes to adopt a ‘lozenge’ shape37,38. These structures further suggest that RICTOR blocks the FKBP12–rapamycin complex binding site on mTOR, thereby rendering mTORC2 insensitive to acute inhibition by rapamycin. Nonetheless, prolonged rapamycin treatment can inhibit mTORC2 signalling by sequestering the cellular pool of mTOR into rapamycin-bound complexes that cannot nucleate new mTORC2 (refs39,40).
Functions of the mTOR signalling pathway
Activation of mTOR marks cellular entry into a ‘growth’ regime characterized by increases in both cell size and cell number. To keep pace with metabolic demand in these growing cells, mTORC1 and mTORC2 initiate biosynthetic cascades to support anabolism and cell proliferation.
Roles of mTORC1
mTORC1 phosphorylates substrates that increase the production of proteins, lipids, nucleotides and ATP while limiting autophagic breakdown of cellular components. Here, we review the major substrates and effectors downstream of mTORC1 (Fig. 2a). Many of these effectors were first identified through phosphoproteomic analyses in rapamycin-treated mammalian cell lines. However, this approach is far from comprehensive: mTORC1 function is exquisitely sensitive to the physiological and pharmacological context, and certain mTORC1 substrates are resistant to inhibition by rapamycin41,42,43. We posit that future studies using novel and specific mTORC1 inhibitors may uncover additional substrates and mTORC1-dependent processes.
Fig. 2: Targets of mTORC1 and mTORC2 signalling.
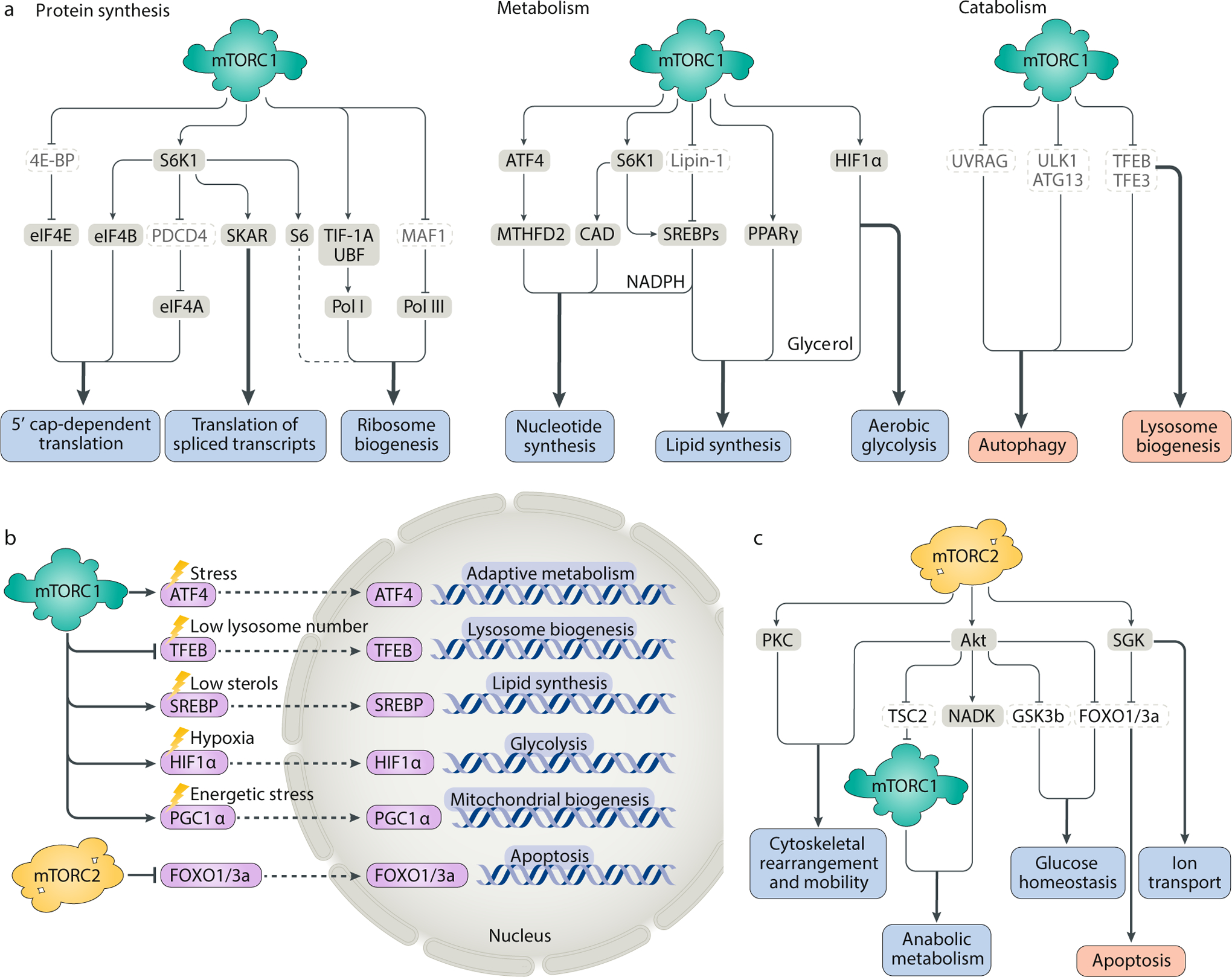
A. mTOR complex 1 (mTORC1) activation initiates a downstream anabolic programme that enhances the production of proteins, lipids, nucleotides and other macromolecules while inhibiting catabolic processes, such as autophagy and lysosome biogenesis. B. By regulating the expression or nuclear localization of transcription factors, mTORC1 and mTORC2 control the expression of genes that promote organelle biogenesis or alter metabolic flux through biosynthetic pathways. Although these transcription factors can be independently activated by specific, acute cellular stress signals (for example, hypoxia inducible factor 1α (HIF1α) can be directly activated by hypoxia and ATF4 can be directly activated by endoplasmic reticulum stress), mTORC1 and mTORC2 toggle the activation of these factors in a coordinated manner to support growth and proliferation. Thus, activation of mTORC1 can simultaneously activate ATF4, the sterol regulatory element binding proteins (SREBPs), HIF1α and yin–yang 1 (YY1)−peroxisome proliferator-activated receptor-γ (PPARγ) coactivator 1α (PGC1α) to drive diverse processes involved in cellular growth, all while blocking lysosomal biogenesis through transcription factor EB (TFEB). C. mTORC2 activates the AGC family kinases protein kinase C (PKC), Akt and serum- and glucocorticoid-induced protein kinase (SGK) to regulate the cytoskeleton, metabolism and ion transport and promote cell survival. CAD, carbamoyl-phosphate synthetase 2, apartate transcarbamoylase, dihydroorotase; 4E-BP, 4E-binding protein; eIF4, eukaryotic translation initiation factor 4; GSK3b, glycogen synthase kinase 3b; MTHFD2, methylenetetrahydrofolate dehydrogenase 2; PDCD4, programmed cell death 4; Pol I/Pol III, RNA polymerase I/RNA polymerase III; S6K1, p70 S6 kinase 1; TFE3, transcription factor E3; TIF-1A, transcription initiation factor 1A; TSC2, tuberous sclerosis complex 2; UBF, upstream binding factor; ULK1, unc-51-like autophagy-activating kinase 1.
Activation of protein synthesis
Protein synthesis is the most energy-intensive and resource-intensive process in growing cells44. It is therefore tightly regulated by mTORC1, which promotes protein synthesis by phosphorylating the eukaryotic initiation factor 4E-binding proteins (4E-BPs) and p70 S6 kinase 1 (S6K1) (Fig. 2a). In its unphosphorylated state, 4E-BP1 suppresses translation by binding and sequestering eukaryotic translation initiation factor 4E (eIF4E), an essential component of the eIF4F cap-binding complex. Upon phosphorylation by mTORC1, 4E-BP1 releases eIF4E and enhances 5′ cap-dependent translation of mRNAs45,46,47.
In concert with PDK, which phosphorylates the activation loop (T229), mTORC1 phosphorylates S6K1 on its hydrophobic motif (T389) to stimulate kinase activity48,49 (Fig. 2a). S6K1 subsequently phosphorylates its namesake target, ribosomal protein S6, a component of the 40S subunit. The function of S6 phosphorylation remains ambiguous: ablation of all five phosphorylation-target serine residues on S6 does not impair organismal viability or translation efficiency50, although some evidence suggests that S6 phosphorylation may promote transcription of genes involved in ribosomal biogenesis51. More directly, S6K1 and mTORC1 upregulate transcription of rRNA, the dominant component of newly-assembled ribosomes, by enhancing the activity of RNA polymerase I and RNA polymerase III through phosphorylation of the regulatory factors upstream binding factor (UBF)52, transcription initiation factor 1A (TIF-1A)53 and MAF1 (refs54,55). S6K1 also enhances protein synthesis by activating eIF4B (ref.56), a positive regulator of cap-dependent translation, and by degrading the eIF4A inhibitor programmed cell death 4 (PDCD4)57. In addition, S6K1 associates with SKAR at exon junction complexes to boost the rate of translation elongation in spliced transcripts58 (Fig. 2a).
Although 4E-BP1 and S6K1 both contribute to the regulation of global translation, recent evidence indicates that 4E-BP1 has a more prominent role. Deletion of S6K1 in mouse liver and muscle cells does not reduce global translation59,60; likewise, rapamycin treatment, which preferentially inhibits S6K1 over 4E-BP1, produces only a weak effect on global translation. By contrast, transcriptome-scale ribosome profiling reveals that mTOR inhibition dramatically suppresses translation of mRNAs carrying 5′ terminal oligopyrimidine motifs in a 4E-BP-dependent manner61,62. These terminal oligopyrimidine transcripts encode much of the translation machinery, including ribosomal proteins, suggesting yet another route by which mTORC1 may modulate protein synthesis.
Biomass accumulation: lipid and nucleotide synthesis and energetic homeostasis
As cells increase in size, they must generate lipids to sustain biogenesis of new membranes. Accordingly, mTORC1 drives lipid synthesis through two axes centred on the transcription factors sterol regulatory element binding protein 1/2 (SREBP1/2) and peroxisome proliferator-activated receptor-γ (PPARγ) (Fig. 2a). When sterol levels are low, the SREBPs translocate from the endoplasmic reticulum membrane to the nucleus, where they upregulate genes for de novo lipid and cholesterol synthesis63. Activated mTORC1 promotes this SREBP transcriptional programme by phosphorylating the SREBP inhibitor lipin 1 to exclude it from the nucleus64. Although the mechanism remains unclear, mTORC1 may also enhance the nuclear translocation and processing of the SREBPs in an S6K1-dependent manner65,66 (Fig. 2b). In addition, inhibition of mTORC1 has been shown to impair the expression of lipid homeostasis genes controlled by the nuclear receptor PPARγ67.
To maintain DNA replication and rRNA synthesis in proliferating cells, mTORC1 regulates the supply of one-carbon units for nucleotide biosynthesis. Recent work has shown that mTORC1 activates the transcription factor ATF4 and its downstream target, mitochondrial tetrahydrofolate cycle enzyme methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), to drive de novo purine synthesis68. Through its effector S6K1, mTORC1 also promotes phosphorylation and activation of carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, dihydroorotase (CAD), the rate-limiting enzyme in pyrimidine biosynthesis69,70. This mTORC1-dependent tuning of the nucleotide pool is crucial for anabolic balance and homeostasis. Indeed, in cells in which mTORC1 is hyperactive, uncoupling nucleotide biogenesis from nucleotide demand with a guanylate synthesis inhibitor leads to DNA damage, as limiting nucleotides are preferentially funnelled into rRNA to sustain high rates of ribosomal biogenesis and protein synthesis71. Because mTORC1 dysregulation is a signature of many cancers, inhibition of nucleotide synthesis may allow us to selectively target a metabolic vulnerability in transformed cells.
Besides its direct effects on biosynthetic enzymes, mTORC1 also potentiates growth by dictating large-scale changes in the metabolic fates of glucose. To generate energy and carbon units, mTORC1 upregulates the transcription factor hypoxia inducible factor 1α (HIF1α), which increases expression of glycolytic enzymes and favours glycolysis over oxidative phosphorylation66,72 (Fig. 2a,b). mTORC1-dependent activation of the SREBPs also increases flux through the pentose phosphate pathway, providing NADPH and carbon-rich precursors for lipid and nucleotide synthesis66. Finally, because biomass accumulation demands vast reserves of energetic currency, mTORC1 enhances translation of nuclear-encoded mitochondrial transcripts through 4E-BP1 to expand the ATP production capacity of the cell73. mTORC1 may additionally stimulate mitochondrial biogenesis by driving formation of the yin–yang 1 (YY1)−PPARγ coactivator 1α (PGC1α) transcriptional complex74.
Repression of catabolism and autophagy
In order to prevent a futile cycle in which newly synthesized cellular building blocks are prematurely broken down again, mTORC1 suppresses catabolic autophagy (Fig. 2a). To that end, mTORC1 applies inhibitory phosphorylation marks to unc-51-like autophagy-activating kinase 1 (ULK1) and ATG13, two key early effectors in the induction of autophagy75,76,77. In complex with 200-kDa FAK family kinase-interacting protein (FIP200) and ATG101, ULK1 and ATG13 drive formation of the autophagosome78. mTORC1 phosphorylation of ULK1 and ATG13 blocks this process, allowing proteins and organelles — including some that may be redundant or damaged — to accumulate in the cell rather than be degraded and recycled. Under nutrient-replete conditions, mTORC1 also phosphorylates UVRAG, which normally associates with the HOPS complex to assist in trafficking and fusion, as well as Rab7 activation. By disrupting this interaction, mTORC1 inhibits autophagosome maturation and the conversion of endosomes into lysosomes, thereby acting as a check on both the early and late stages of autophagy79.
Inhibition of mTORC1 by nutrient deprivation or rapamycin treatment flips the cell into a ‘starvation’ regime, shunting resources away from biosynthesis and towards autophagy. In interphase cells, turning off the mTORC1 molecular switch restores autophagosome initiation and permits nuclear translocation of both the transcription factor EB (TFEB) and the related transcription factor E3 (TFE3), which activate genes for lysosomal biogenesis in a coordinated fashion80,81,82 (Fig. 2a,b). Newly formed lysosomes then break down proteins and release constituent monomers back to the cytoplasm to regenerate the pool of cellular amino acids, enabling reactivation of the mTORC1 pathway after prolonged starvation83. Importantly, this coupling between nutrient status and autophagy is disrupted during mitosis, when CDK1 inhibits both mTORC1 and autophagosome formation to protect the genome from degradation after dissolution of the nuclear envelope84.
Recent studies demonstrate that the feedback loop between the lysosome and mTORC1 is crucial for cell survival in nutritionally sparse environments. For example, pancreatic cancer cell lines that rely on macropinocytosis for nutrients stop proliferating when ablation of the transporter SLC38A9 traps essential amino acids inside the lysosome, impairing autophagic reactivation of mTORC1 (ref.85). Strikingly, a similar fitness defect is observed in nutrient-deprived cells that lack the autophagy receptor nuclear fragile X mental retardation-interacting protein 1 (NUFIP1), which recruits ribosomes to the autophagosome upon mTORC1 inhibition86. Defects in ribosome degradation appear to block reactivation of the mTORC1 pathway, while supplementation of exogenous nucleotides can restore growth87. These data suggest that ribosomes may serve as a major storage depot for amino acids and ribonucleotides and thus imply that mTORC1 may trigger selective ‘ribophagy’ to maintain cell viability under nutritional stress88. How mTORC1 balances bulk versus selective autophagy89, how it exerts control over the kinetics of its own reactivation in starved cells and the functional importance of this reactivation are not fully understood. As lysosome–mTORC1 communication is essential in certain conditions in several tumour models, addressing these questions may shed light on the lysosome as a signalling organelle and guide new approaches for the treatment of cancer and metabolic disease.
Roles of mTORC2
The first direct substrate of mTORC2 was discovered serendipitously. While immunoblotting for T389 phosphorylation of the mTORC1 target S6K1 in RICTOR-depleted cells, researchers observed that mTORC2 knockdown did not affect S6K1 phosphorylation; instead, it suppressed a cross-reacting background band, which they identified as a homologous phosphorylation site on protein kinase Cα (PKCα)29 (Fig. 2c). A member of the AGC (PKA/PKG/PKC) family of protein kinases, PKCα is thought to act as a cytoskeletal regulator, although the mechanistic basis of this process remains unclear90. Accordingly, knockdown of RICTOR, mTOR or mLST8, but not RAPTOR, impairs the reorganization of the actin cytoskeleton network and inhibits chemotaxis and migration30,91; this phenotype, in turn, may account in part for the well-documented role of mTORC2 in the mobility and metastasis of cancer cells92,93.
Subsequent studies have revealed that mTORC2 also collaborates with PDK1 to activate other AGC family kinases, including several classes of PKCs94,95, the ion transport regulator serum- and glucocorticoid-induced protein kinase 1 (SGK1)96 and the oncogene Akt97. Akt is a central early effector in the PI3K pathway, where it mediates the cellular response to insulin and promotes proliferation. In that capacity, Akt rewires metabolism to resist stressors through the forkhead-box FOXO1/3a transcription factors98 and NAD kinase99 (Fig. 2b,c). As one of the most frequently mutated signalling nodes in cancer cells, Akt also governs the activity of glycogen synthase kinase 3b (GSK3b) to suppress apoptosis and modulate glucose homeostasis. In addition, Akt may mediate crosstalk between the mTORC1 and mTORC2 complexes by inactivating tuberous sclerosis complex 2 (TSC2), a strong inhibitor of mTORC1 activity100, and phosphorylating mSin1, an obligate component of mTORC2 (ref.101).
As yet, the relationship between mTORC2 and Akt is incompletely understood. Emerging evidence suggests that mTORC2 and Akt engage in mutually reinforcing layers of feedback phosphorylation that regulate localization and activity, although the effect of these marks — individually and cumulatively — is still unclear102. Moreover, unlike SGK1, Akt may not require mTORC2 for basal activation. Although mTORC2 kinase activity is necessary for phosphorylation of certain Akt substrates, such as FOXO1/3a, it is dispensable for others, including TSC2 and GSK3b (ref.19). Given that the FOXO proteins are regulated by both SGK1 and Akt, it is possible that SGK is, in this context, the more important mTORC2 effector, while Akt plays a subtler modulatory role.
Regulation of mTOR function
To mediate between cellular behaviour and the cellular environment, mTORC1 and mTORC2 integrate upstream signals, including nutrient levels, growth factor availability, energy and stress, to gate their own activation (Fig. 3). While the inputs and modes of regulation differ for each complex, we now recognize that mTORC1 and mTORC2 engage in substantial crosstalk — giving rise to signalling feedback loops with important consequences for health and disease.
Fig. 3: Upstream regulators of the mTOR signalling pathway.
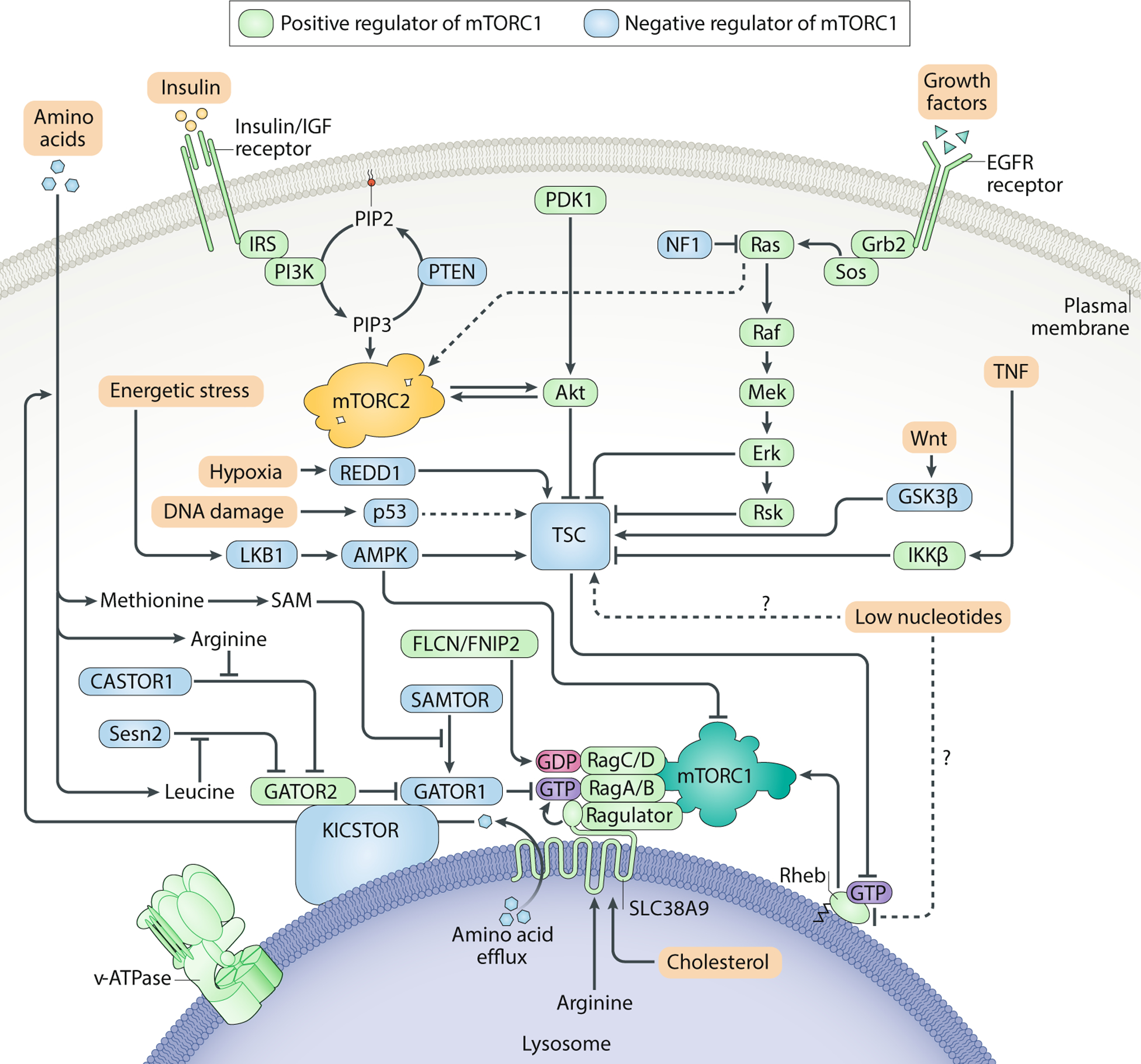
mTOR complex 1 (mTORC1) and mTORC2 integrate upstream environmental information to gate their own activation. Because mTORC1 controls cellular entry into an anabolic state that requires copious amounts of energy and macromolecules, activation of the complex should only occur when amino acids, insulin/growth factors, ATP and oxygen are all readily available. To ensure that all of these requirements are satisfied, mTORC1 must translocate to the lysosome by anchoring onto the Rag GTPases, which are only competent to recruit mTORC1 in the presence of amino acids. Once localized to the lysosomal surface, mTORC1 can be then be activated by the small GTPase Rheb in its GTP-bound state. Importantly, GTP loading of Rheb is promoted by growth factors and opposed by energetic stress or hypoxia. All of these inputs converge on tuberous sclerosis complex (TSC), which acts as a GAP for Rheb. mTORC2 is thought to be primarily regulated by growth factors. Although it is not clear where mTORC2 activation occurs, the pleckstrin homology domain on MAPK-interacting protein 1 (mSIN1) may recruit mTORC2 to the plasma membrane. Positive regulators of the mTORC1 pathway are shown in green, while negative regulators of mTORC1 are shown in blue. AMPK, AMP-activated protein kinase; CASTOR, cellular arginine sensor for mTORC1; EGFR, epidermal growth factor receptor; FLCN, folliculin; GATOR, GAP activity towards the Rags; Grb2, growth factor receptor-bound protein 2; GSK3, glycogen synthase kinase 3; IGF, insulin-like growth factor; IKKβ, inhibitor of nuclear factor κB kinase β; IRS, insulin receptor substrate; LKB1, liver kinase B1; Mek, MAPK/ERK kinase; NF1, neurofibromatosis type 1; PIP2, phosphatidylinositol (4,5)-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PTEN, phosphatase and tensin homologue; RSK, p90 ribosomal S6 kinase; SAM, S-adenosylmethionine; SAMTOR, S-adenosylmethionine sensor; Sos, son of sevenless; TNF, tumour necrosis factor.
Regulators of mTORC1
Cells must toggle mTORC1 activity in response to nutrient oscillations and other environmental changes stimulated by feeding or fasting. Because mTORC1 initiates a resource-intensive anabolic programme, it should only turn ‘on’ when energy, growth factors and macromolecular building blocks are all plentiful. To monitor and integrate these inputs, the mTORC1 pathway collects upstream signals at two sets of small G proteins, termed the Rheb and Rag GTPases (Fig. 3). Biochemical studies over the past decade have led to a model in which the nucleotide-loading state of the Rheb and Rag GTPases modulate, respectively, mTOR kinase activity103,104 and intracellular localization105,106 to promote cell growth. When the cellular environment is rich in cytokines, endocrine signals and ATP, Rheb maintains its active GTP-bound state on the surface of the lysosome and is competent to stimulate mTORC1 kinase activity107. However, mTORC1 can only co-localize with this population of GTP-Rheb when amino acids, glucose and other nutrients are readily available to activate the Rag heterodimer, which recruits mTORC1 from the cytoplasm to the lysosome. By funnelling all major environmental cues through this spatial ‘AND gate’, cells ensure that mTORC1 potentiates anabolism only when intracellular and extracellular conditions can support sustained growth.
Growth factors
mTORC1 acts as a downstream effector for growth factors and other mitogens, which often serve as proxies for broader paracrine and endocrine status. To regulate the mTORC1 pathway, these signals converge upon the tuberous sclerosis complex (TSC), a heterotrimeric signalling node upstream of Rheb that is composed of TSC1, TSC2 and TBC1D7 (ref.108). TSC acts as a GTPase-activating protein (GAP) for lysosomal Rheb, catalysing the conversion from the active Rheb-GTP state to the inactive GDP-bound state103,109. As a key ‘molecular brake’ for mTORC1 activation110, TSC is subject to many levels of regulation. Upon exposure to insulin, insulin/insulin-like growth factor 1 (IGF-1) activates Akt, which phosphorylates TSC2 at multiple sites to dissociate TSC from the lysosomal surface and relieve inhibition of Rheb and mTORC1 (ref.111,112,113,114). To tune the extent and duration of mTORC1 activation and restore TSC regulation after this stimulus, the mTORC1 substrate S6K1 then directly phosphorylates insulin receptor substrate 1 (IRS-1) as part of a negative feedback loop, blocking further insulin-mediated activation of the PI3K–Akt pathway115,116. Wnt and tumour necrosis factor (TNF) signalling also repress TSC activity, although the precise mechanism of this regulation is unclear117,118. In addition, TSC is subject to inhibitory phosphorylation from ERK119 and p90 ribosomal S6 kinase (RSK)120, two downstream substrates of the Ras receptor tyrosine kinase signalling pathway. Because mutations that activate the Ras and PI3K–Akt pathways occur in many cancers, TSC regulation of mTORC1 is often lost in oncogenic contexts, resulting in constitutive mTORC1 activity even in the absence of appropriate growth factor signals.
Independently of TSC and Rheb, growth factors can also modulate mTORC1 activity through PRAS40, an endogenous inhibitor of the mTORC1 complex. A substrate and component of mTORC1, PRAS40 associates with Raptor to abolish Rheb-driven mTORC1 activation in vitro22,23. However, in the presence of insulin, Akt phosphorylates PRAS40, leading to its sequestration by a cellular 14–3–3 scaffold protein and restoring mTORC1 kinase activity. How growth factor signals are coordinated through PRAS40 and Rheb, and the relative importance of each branch in different cellular contexts, remains an area of active study.
Energy and oxygen availability, and other cellular stresses
Under conditions of energy or oxygen scarcity, several factors work together to activate the TSC axis and suppress mTORC1 signalling. Periods of intense metabolic exertion or glucose withdrawal can deplete cellular stores of ATP, triggering the AMP-activated protein kinase (AMPK) complex, a master regulator of cellular energy charge. As an antagonist of most major ATP-consumptive processes, AMPK inhibits mTORC1 directly, by phosphorylating Raptor, and indirectly, by activating TSC2 (refs121,122,123). At the same time, by reprogramming metabolism away from anabolic pathways, AMPK relieves the pressure on mitochondrial respiration and reduces the chances of cellular damage from the generation of reactive oxygen species124.
Independently of AMPK, oxidative stress can also inhibit mTORC1 by upregulating REDD1, a small protein that activates TSC125,126. Other signs of cellular stress — ranging from organelle dysfunction to DNA replication stress — can further oppose mTORC1 activation127. For example, the endoplasmic reticulum unfolded protein response can inhibit mTORC1 by increasing transcription of the Sestrin proteins, key negative regulators that will be discussed in greater detail below128. Likewise, DNA damage induces various p53 target genes, including an AMPK subunit (AMPKβ), PTEN and TSC2, all of which can dampen mTORC1 activity to slow proliferation and protect genome integrity129.
Amino acids and other nutrients
Besides spurring growth factor release, feeding also replenishes the pool of intracellular nutrients. These nutrients, which constitute the basic molecular substrates for biology, include amino acids, nucleotides and vitamins, all of which may be partially or wholly derived from the diet. Among the major nutrients, amino acids play a dominant role in regulating the mTORC1 pathway130; indeed, Avruch and colleagues observed as early as 1998 that the amino acids leucine and arginine, in particular, are absolutely required for mTORC1 activation in mammalian cells131. How these amino acids communicate their availability to mTORC1, however, remained a complete mystery until 2008, when two groups independently reported the discovery of the Rag-GTPases as essential components of the nutrient sensing machinery105,106.
Unlike all other known small GTPases, the Rags are obligate heterodimers, configured such that RagA or RagB is bound to RagC or RagD. Anchored to the lysosome by the pentameric Ragulator complex (comprising p18, p14, MP1, C7orf59 and HBXIP, otherwise known as LAMTOR1–LAMTOR5)132,133,134, the Rags can be found in one of two stable conformations: an ‘on’ state, in which RagA/B is bound to GTP and RagC/D to GDP; and an ‘off’ state, in which the reverse is true. These stable nucleotide-loading states are maintained by intersubunit crosstalk between the Rags135, but they can be modulated by the amino acid and nutrient status through a series of upstream factors with GAP or GTP exchange factor activity towards the Rags. Emerging structural evidence shows that, under amino acid-replete conditions, Raptor grasps the ‘on-state’ Rags via a protruding ‘claw’136. This interaction recruits mTORC1 from the cytosol to the lysosome, allowing lysosomal Rheb to stimulate mTORC1 kinase activity. Thus, the Rags and Rheb define the two independent arms that converge to license the mTORC1 pathway (Fig. 3).
Drawing on work by several groups over the past decade, we now recognize that mTORC1 senses cytosolic and lysosomal amino acid concentrations through distinct mechanisms. Of the ‘nutrient sensing complexes’ that transmit cytosolic amino acid signals to the Rags, the most direct regulator of Rag status is the GAP activity towards the Rags 1 (GATOR1) complex137. GATOR1 is composed of three subunits — DEP domain-containing 5 (DEPDC5), nitrogen permease related-like 2 (NPRL2) and NPRL3 — with GAP activity residing in the NPRL2 subunit138. When cytosolic amino acid levels fall, GATOR1 experiences a poorly understood regulatory event that enables it to hydrolyse the GTP bound to RagA/B and inhibit the mTORC1 pathway139. In turn, GATOR1 is itself regulated by other upstream factors. The large KICSTOR complex, consisting of the proteins KPTN, ITFG2, C12orf66 and SZT2, tethers GATOR1 to the lysosome and is required for cellular sensitivity to amino acid deprivation140,141. Meanwhile, GATOR1 also physically interacts with GATOR2, a pentameric complex of WDR59, WDR24, MIOS, SEH1L and SEC13 (ref.137). Through unknown molecular mechanisms, the GATOR2 complex antagonizes GATOR1 function and acts as a potent positive regulator of mTORC1. Elucidating the link between GATOR2 and GATOR1 activity remains one of the most intriguing challenges in basic mTOR biology.
Recently, the question of GATOR2 function has attracted special attention because of the identification of two novel ‘amino acid sensors’, which relay the cytosolic availability of leucine and arginine to the mTORC1 pathway through interactions with GATOR2. Upon acute leucine starvation, the cytosolic leucine sensor Sestrin2 binds and inhibits GATOR2, preventing lysosomal recruitment of mTORC1 (ref.142). Refeeding restores leucine levels and allows the amino acid to bind a pocket on Sestrin2, dissociating the protein from GATOR2 to relieve mTORC1 inhibition142,143. Although the leucine-binding affinity of Sestrin2 dictates mTORC1 sensitivity to leucine deprivation in cell culture, Sestrin2 and its relatives Sestrin1 and Sestrin3 may also be effectors of leucine-independent stress pathways. In support of this hypothesis, the Sestrins are transcriptionally upregulated by ATF4 and the endoplasmic reticulum unfolded protein response128,144, and Sestrin overexpression alone is sufficient to suppress mTORC1 signalling in vitro145,146. By contrast, cellular arginine sensor for mTORC1 (CASTOR1) appears to be exquisitely sensitive to cytosolic arginine alone147,148. A protein that can exist either as a homodimer or as a heterodimer with CASTOR2, CASTOR1 also inhibits GATOR2 in the absence of arginine and dissociates from the complex when arginine is bound.
A second arginine sensor, SLC38A9, monitors amino acid levels inside the lysosomal lumen and defines the lysosomal branch of the nutrient sensing machinery149,150. SLC38A9 resides on the lysosomal membrane and transports neutral amino acids out of the organelle in an arginine-gated fashion85. This efflux activity may enable the products of autophagic protein degradation to reactivate the mTORC1 pathway after prolonged starvation. Synthesizing structural and biochemical evidence, we posit that the binding of lysosomal arginine to the first transmembrane helix of SLC38A9 frees the N terminus of the protein from the central pore151. This domain can then collaborate with Ragulator to push the Rags into the active state by promoting GTP loading of RagA/B152. Through a separate mechanism, the lysosomal v-ATPase, which maintains the pH gradient of the lysosome, has also been reported to interact with the Rag–Ragulator complex to influence the nucleotide-loading state of the Rags153. Finally, the folliculin (FLCN)–FNIP2 complex acts as a GAP for RagC/D to sustain mTORC1 activation in the presence of amino acids154,155. By modulating the status of RagC/D, FLCN–FNIP2 may also recruit and enhance phosphorylation of the transcription factors TFEB/TFE3, although it is unclear whether this process is mTORC1 independent156,157. If FLCN–RagC/D–TFEB/TFE3 does indeed constitute a distinct axis, loss of FLCN could amplify the TFEB/TFE3 transcriptional programme, an oncogenic signature in some cancers158,159, allowing us to reconcile FLCN’s status as a tumour suppressor in vivo with its activating role in the mTORC1 pathway.
The recent discovery of an S-adenosylmethionine (SAM) sensor, named SAMTOR, has shown that mTORC1 responds not only to amino acids (for example, leucine and arginine) but also to their metabolic by-products — in this case, a key methyl donor derived from methionine. Unlike Sestrin2 and CASTOR1, which oppose GATOR2 signalling when their cognate amino acids are absent, SAMTOR negatively regulates mTORC1 by binding GATOR1 and KICSTOR under methionine or SAM deprivation160. Restoration of SAM levels dissociates SAMTOR from these complexes and stimulates mTORC1 activity.
At present, we do not know how other amino acids impact mTORC1 activation, nor do we understand what role, if any, the general amino acid sensors GCN2 and ATF4 play in acute mTORC1 signalling cascades. While longer-term amino acid deprivation is thought to feed from GCN2 back to mTORC1 through transcriptional upregulation of ATF4 and the Sestrins, it is not clear whether GCN2 and ATF4 regulate mTORC1 in transiently starved cells. Moreover, we still lack mechanistic explanations for how several known metabolic inputs impinge on the pathway. For example, although acute withdrawal of glucose inhibits mTORC1 at least partially through activation of AMPK, a study in AMPK-null cells has demonstrated that glucose deprivation also signals through the Rag-GTPases161, reinforcing earlier evidence that glucose can signal independently of both AMPK and TSC162. Similarly, depletion of purine nucleotides inhibits mTORC1, perhaps as an indicator of replication stress, but it is not clear whether this inhibition is driven by TSC or by degradation of Rheb163,164. One recent study suggests that phosphatidic acid may activate mTOR signalling as a proxy for fatty acid availability165, while another implicates glutamine in Rag-independent reactivation of mTORC1 (ref.166). Cholesterol has also been shown to activate mTORC1 through a complex composed of SLC38A9 and the Niemann–Pick C1 protein281. Whether mTORC1 senses other metabolites essential for cell growth, such as vitamins or inorganic ions, remains an open question; equally unclear is how these nutritional requirements might diverge in cell types or organisms with different dietary and metabolic needs (Box 1).
Box 1: Cell-specific and organism-specific regulatory mechanisms across evolution in mTORC1 signalling.
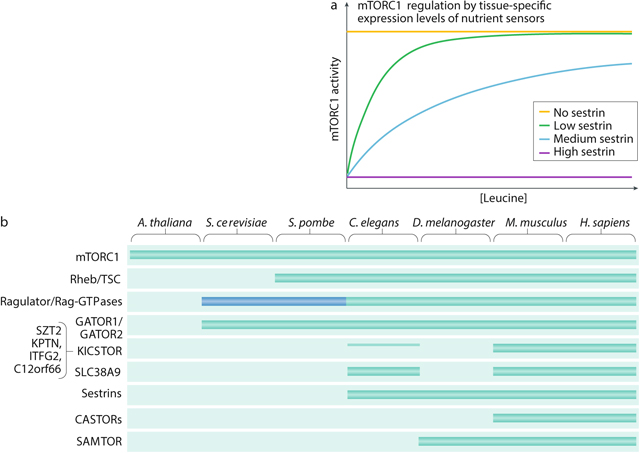
In order to align mTOR complex 1 (mTORC1) activity with tissue function, some specialized human cells may respond to unique inputs, adjust the weighting of upstream signals262 or regulate mTORC1 through non-canonical mechanisms. In muscle cells, mechanical stimuli have been shown to activate mTORC1 (refs263,264,265), whereas in primary osteoclasts, amino acid deprivation can abrogate mTORC1 signalling without dissociating the complex from the lysosome266. We postulate that specialized cells can also adapt to their niches by tuning expression of nutrient sensors. For example, in tissues where physiologically relevant leucine concentrations are relatively high, cells might selectively increase expression of Sestrin2 to raise the leucine threshold for mTORC1 activation. Conversely, cells that are protected from leucine fluctuations might abolish Sestrin2 expression altogether to render mTORC1 insensitive to leucine deprivation. Thus, differential expression of Sestrin could modulate mTORC1 sensitivity to leucine levels in a tissue-specific manner (see the figure, part a).
Although the core components of the nutrient sensing machinery — the Rag GTPases, Ragulator and the GAP activity towards the Rags (GATORs) — are conserved in metazoans (see note below), some of the direct amino acid sensors are absent in non-vertebrate lineages (conservation of mTORC1 pathway components in common model organisms is shown in the figure, part b). Based on sequence homology, Drosophila melanogaster retains Sestrin and the S-adenosylmethionine sensor SAMTOR but lacks both the lysosomal and cytosolic arginine sensors; meanwhile, Caenorhabditis elegans possesses SLC38A9 and Sestrin homologues but does not have a clear SAMTOR equivalent. The irregular pattern of conservation of the sensors may be linked to the distinct nutritional needs of each organism. In support of this idea, computational searches indicate that Saccharomyces cerevisiae, a model organism capable of synthesizing all 20 amino acids de novo, does not have any amino acid sensors and, consequently, does not require any individual amino acid for TORC1 activation. Instead, the S. cerevisiae TORC1 pathway may respond to the general availability of nitrogen and carbon sources267. Puzzlingly, S. cerevisiae also does not seem to require a Rheb homologue to activate TORC1 (ref.268), suggesting that its molecular circuitry may diverge sharply from that of other model organisms, including Schizosaccharomyces pombe269. The blue box (see the figure, part b) indicates that the EGO complex in yeast shares little sequence homology with Ragulator, although it appears to serve an analogous function.
Even nutrient sensors with recognizable homology may display functional differences in divergent species. Binding assays with radioactive leucine reveal that the D. melanogaster homologue of Sestrin (dSesn) has 5-fold lower affinity for leucine than the human protein142,270. We speculate that this molecular difference may allow dSesn to sense physiological leucine fluctuations in the D. melanogaster haemolymph, which has about a 5-fold to 10-fold higher amino acid concentration than human plasma270,271. Taken together with conservation patterns, these data also suggest an attractive hypothesis: perhaps organisms evolved or retained specific nutrient sensors to enable the TORC1 pathway to respond to limiting nutrients in their metabolic niches. However, because no unique sensors have yet been identified in non-human systems and the evolutionary lineage of the sensors is not well understood, it is difficult to draw correlations between evolutionary pressures and the functional architecture of the TORC1 nutrient sensing pathway. The discovery of novel nutrient sensors outside higher eukaryotes would clarify the evolutionary logic of the nutrient sensing axis and define new inputs into the TORC1 pathway. Moreover, sensors initially characterized in other species could be conserved in human cell types with specialized metabolic environments.
Notes. For reasons that remain unclear, the KICSTOR complex is the sole exception to this generalization. KPTN, ITFG2 and C12orf66 seem to drop out of the evolutionary tree in organisms more distal than mammals; SZT2, the largest component of the complex, may have a putative homologue in C. elegans but is not retained in flies or yeast. If KICSTOR serves as a molecular glue that holds human GATOR1 and GATOR2 together in a supercomplex, as one study has argued140, it is possible that it is dispensable in lower organisms where GATOR1 and GATOR2 are more constitutively bound to each other. Consistent with this hypothesis, the S. cerevisiae homologues for the GATORs, the SEACIT and SEACAT complexes, are indeed more tightly associated than their human counterparts and have been reported to form a supercomplex without any mediating proteins272.
A. thaliana, Arabidopsis thaliana; CASTOR, cellular arginine sensor for mTORC1; H. sapiens, Homo sapiens; M. Musculus, Mus musculus; TSC, tuberous sclerosis complex.
Regulators of mTORC2
In part because it has been difficult to tease apart the regulation of mTORC1 and mTORC2 with pharmacological agents, the activators of mTORC2 are still poorly defined. Even so, it is clear that mTORC2 is primarily regulated by growth factors through the PI3K pathway, with the unique mTORC2 component mSin1 acting as a key signal integrator (Fig. 3). Like other PI3K effectors, mSin1 possesses a pleckstrin homology domain, which autoinhibits mTORC2 kinase activity in the absence of insulin36. This inhibition is relieved by the binding of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), a product of insulin-induced or serum-induced PI3K activation36,167. PIP3 may recruit mTORC2 and Akt to the plasma membrane, where reciprocal phosphorylations between the two kinases modulate their localization and activation102. In several model systems, including Dictyostelium discoideum, this localization and activation is also regulated by the small GTPases Rac1, Rap1 and Ras, which bind to mTORC2 to direct chemotaxis and growth168,169,170. A recent study extends this paradigm to human cells by showing that mSin1 can recruit oncogenic Ras to directly catalyse mTORC2 kinase activity at the plasma membrane171. This finding connects mTORC2 to a major cancer pathway and reinforces its role in driving survival and proliferation.
Because mTORC1 downregulates insulin–PI3K–Akt signalling through feedback inhibition, it also engages in negative crosstalk with mTORC2 (ref.172). As previously described, mTORC1 can disrupt PI3K–Akt signalling through S6K1-dependent degradation of IRS1 (refs115,116); alternatively, mTORC1 can activate Grb10, a negative regulator of the insulin/IGF-1 receptor173,174. Both of these mechanisms have downstream implications for mTORC2 activity and may account for some of the paradoxical metabolic phenotypes associated with chronic rapamycin treatment (Fig. 4). Unexpectedly, mTORC2 is also activated by AMPK under energetic stress, suggesting that it may mediate cellular adaptation to oxygen-poor or nutrient-poor tumour environments in vivo175.
Fig. 4: mTOR signalling in metabolism.
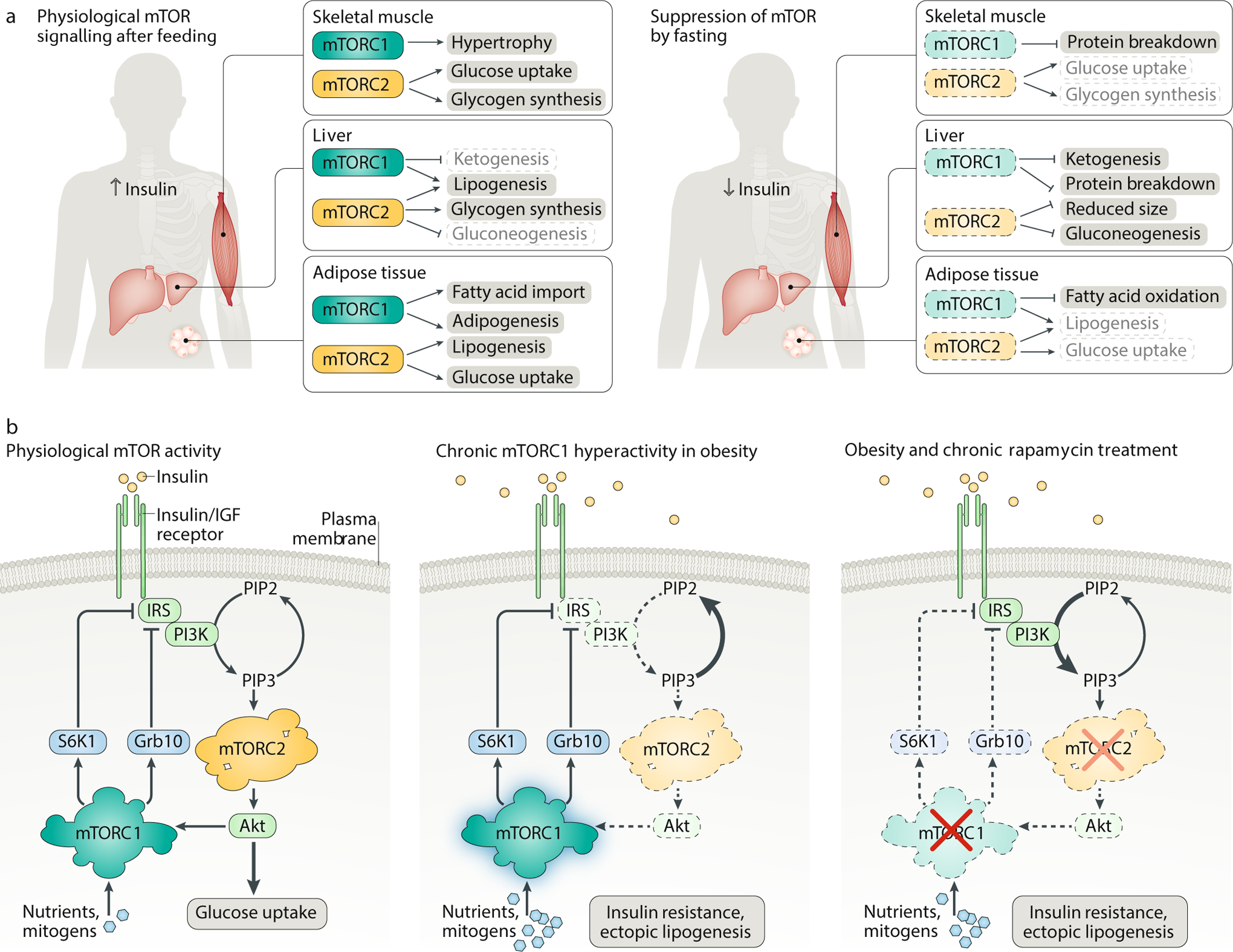
A. mTOR coordinates feeding and fasting with nutrient storage and mobilization. In the liver, skeletal muscle and adipose tissue, rising insulin levels after feeding activate both mTOR complex 1 (mTORC1) and mTORC2, promoting lipogenesis, glycogen synthesis and protein synthesis (left). During fasting, the nutrient, growth factor and insulin levels drop precipitously, tilting the metabolic balance in favour of gluconeogenesis, ketogenesis and lipolysis (right). B. Dysregulation of mTOR signalling in metabolic syndrome. Although the negative feedback loop between mTORC1 and mTORC2 is carefully balanced under physiological conditions (left), chronic hyperactivation of mTORC1 by excessive nutrients and mitogens can shut off PI3K–mTORC2 signalling, leading to insulin resistance, ectopic accumulation of lipids in the muscle and liver, and type 2 diabetes (middle). Rapamycin-based therapies have not been effective in diabetes patients with hyperactive mTORC1 signalling because prolonged rapamycin treatment also inhibits mTORC2 (right). Grb10, growth factor receptor-bound protein 10; IGF, insulin-like growth factor; IRS, insulin receptor substrate; PIP2, phosphatidylinositol (4,5)-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; S6K1, p70 S6 kinase 1.
mTOR in physiology and pathophysiology
Characterization of mTOR signalling nodes is a work in progress at the cellular level, but the functional regulation of the pathway becomes exponentially more complex at the organismal level, as mTOR must coordinate the storage and mobilization of nutrients and energy across different tissues. Unlike cells in culture, which are bathed in growth factors and nutrients and consequently maintain high mTOR activity, cells in vivo tend to display lower baseline activity and experience sharper fluctuations in mTOR activity upon fasting or feeding. Coordinating physiological responses with nutrient status requires the mTOR pathway to sense conditions within specialized niches and to enact tissue-specific anabolic or catabolic cascades. Appropriate regulation of mTOR is crucial for homeostasis and organismal health; conversely, imbalances in mTOR activity in various tissues can lead to metabolic dysregulation and disease.
mTOR in metabolic syndrome
As a critical regulator of glucose metabolism and lipogenesis across various tissues, the mTOR pathway is readily hyperactivated by overfeeding and underwrites many diseases of constitutive growth, including obesity and type 2 diabetes.
Insulin sensitivity and glucose homeostasis
To prevent the accumulation of nutrients in the blood, animals have evolved mechanisms to sequester macromolecules and energy after feeding. These processes are coordinated across different tissues by the release of insulin from the pancreas, which co-activates mTORC1 and mTORC2 to promote hypertrophy and growth (Fig. 4a). In skeletal muscle, insulin induces the uptake of glucose and enhances its storage as glycogen by stimulating the mTORC2–Akt axis176; at the same time, circulating amino acids are incorporated into new muscle biomass in an mTORC1-dependent manner.
By contrast, low levels of insulin following fasting induce autophagy in ‘dispensable tissues’ (that is, muscle and liver, as opposed to the brain), which break down protein stores to fuel gluconeogenesis in the liver. This catabolic programme has profound effects on metabolic organs: one study found that livers from mice fasted for 24 h decreased in weight by nearly 25%, with the difference arising not from changes in cell number but from reductions in cell size177. Strikingly, this fasting-induced shrinkage was abolished in mice with liver-specific knockouts of TSC1, Raptor or the autophagy gene Atg7, suggesting that the switch from anabolism to catabolism is primarily regulated by mTORC1 (refs177,178).
Substantial evidence now points to mTORC1 as a central mediator of organismal survival during nutrient restriction. For mice that cannot tune mTORC1 signalling, prolonged fasting — like the postnatal fast caused by disruption of the placental nutrient stream — can pose an insurmountable challenge. Unlike wild-type neonates, which rapidly inhibit mTORC1 after an initial drop in circulating glucose, mice expressing a constitutively active allele of RagA (RagA-GTP) are unable to suppress mTORC1 signalling during the perinatal fasting period161. Because these mutant mice fail to restrict their energy expenditure or trigger autophagy to supply free amino acids for gluconeogenesis, their plasma glucose levels plummet, leading to fatal hypoglycaemia within 1 day of birth. A similar perinatal lethality occurs in mice lacking the Sestrin proteins (upstream negative regulators of mTORC1)179 and in mice with defects in the autophagy machinery (downstream targets of mTORC1)180, demonstrating that mTORC1 activity must be tightly coupled to diet to maintain glucose homeostasis in vivo.
Adipocyte formation and lipid synthesis
Postprandial mTOR activation also promotes longer-term energy storage by increasing the synthesis and deposition of triglycerides in white adipose tissue (WAT). As the largest repository of energy in the body, WAT serves as a metabolic hub, tailoring its biosynthetic activity to fluctuations in mTOR signalling. In these cells, the mTORC1–S6K1–SREBP axis drives de novo lipogenesis64,65,66, while mTORC1 activation of PPARγ helps pre-adipocytes differentiate into mature tissue67,181 (Fig. 4a). S6K1 may also increase fatty acid import into adipocytes through a complex mechanism involving the glutamyl-prolyl-tRNA synthetase (EPRS)182. Consistent with the importance of mTORC1 in WAT, adipocyte-specific deletion of Raptor reduces WAT tissue mass and enhances lipolysis in mouse models183. Tantalizingly, Adi-Raptor KO mice are also resistant to diet-induced obesity184. Unfortunately, these defects in adipocyte expansion can drive fat deposits to accumulate in the liver instead, leading ultimately to hepatic steatosis and insulin resistance183.
While regulation of adipose tissue exerts second-order effects on other organs, mTOR also directly modulates lipid metabolism in the liver. Several groups have found that hepatic lipogenesis is impaired in both Raptor and Rictor-depleted mice, with mTORC2-dependent effects at least partially rescuable by constitutive activation of Akt64,185,186. In addition, mice with liver-specific hyperactivation of mTORC1 fail to fully stimulate the production of ketone bodies, which are synthesized from fatty acids to supply peripheral tissues with alternative energy packets during fasting177. Although the relationship between mTORC1 and ketogenesis is not entirely clear, insulin withdrawal likely inhibits mTORC1 phosphorylation of S6 kinase 2 (S6K2), which then enhances expression of ketogenic factors by freeing the transcription factor PPARα from its corepressor, nuclear receptor corepressor 1 (NCoR1). Similar ketogenic defects are also observed in aged mice, suggesting that long-term decline in liver function may stem from mTOR-driven dysregulation of lipid metabolism177.
Pharmacological interventions for metabolic disease
Many diseases of overfeeding, among them obesity and type 2 diabetes, produce a major and detrimental energy imbalance in the body. By generating a constant surplus of hormones, cytokines and nutrients, these diseases collapse the metabolic cycles that underwrite tissue homeostasis, forcing mTORC1 to remain in a persistent ‘on’ state. Constitutive mTORC1 signalling activates S6K1 and Grb10 to decouple the insulin/IGF-1 receptor from downstream PI3K pathway effectors, dampening the physiological response to insulin115,116,173,174 (Fig. 4b). Moreover, PI3K inhibition suppresses mTORC2–Akt to block glucose uptake and promote gluconeogenesis185,187, thereby further elevating the glycaemic load and exacerbating the ectopic fat deposition and glucose intolerance that constitute the hallmarks of metabolic syndrome.
Given that mTORC1 sits at the centre of a web of dysregulated metabolic signalling, it is tempting to imagine that inhibition of this node might reverse both the symptoms and underlying causes of obesity and diabetes. Lending support to these hopes, metformin, a first-line treatment for type 2 diabetes, has been shown to potently suppress mTORC1 by activating AMPK and TSC188; likewise, ablation of the mTORC1 effector S6K1 can protect against diet-induced obesity and enhance insulin sensitivity172. Unfortunately, direct pharmacological inhibition of mTORC1 yields more complex outcomes. Patients administered rapamycin experience more severe insulin resistance, perhaps because chronic rapamycin treatment disrupts not only mTORC1 but also the integrity of the mTORC2 complex, blunting the Akt-dependent insulin response40 (Fig. 4b). To bypass these adverse effects, it will be necessary to develop new, truly specific mTORC1 inhibitors, as well as tissue-specific modulators of mTORC1 function.
mTOR regulation of brain physiology and function
Within the brain, the mTOR pathway orchestrates a wide array of neuronal functions, temporally spanning every stage of development189,190. From framing basic cortical architecture to remodelling neuronal circuitry in response to experience, mTOR and its molecular accomplices shape both the signalling and the physical terrain of the brain (Fig. 5a,b). Not surprisingly, loss of mTOR regulation — through either genetic or chemical perturbations — has severe repercussions for neuronal function (Fig. 5a). Brain-specific knockouts of Raptor and Rictor display remarkably similar phenotypes, typified by microcephaly — via reductions in neuron size and number — and improper differentiation191,192. In addition, Raptor deletion in the brain also triggers early postnatal death191, while Rictor deletion leads to aberrant brain foliation and impaired dendrite extension192.
Fig. 5: mTOR signalling in the brain.
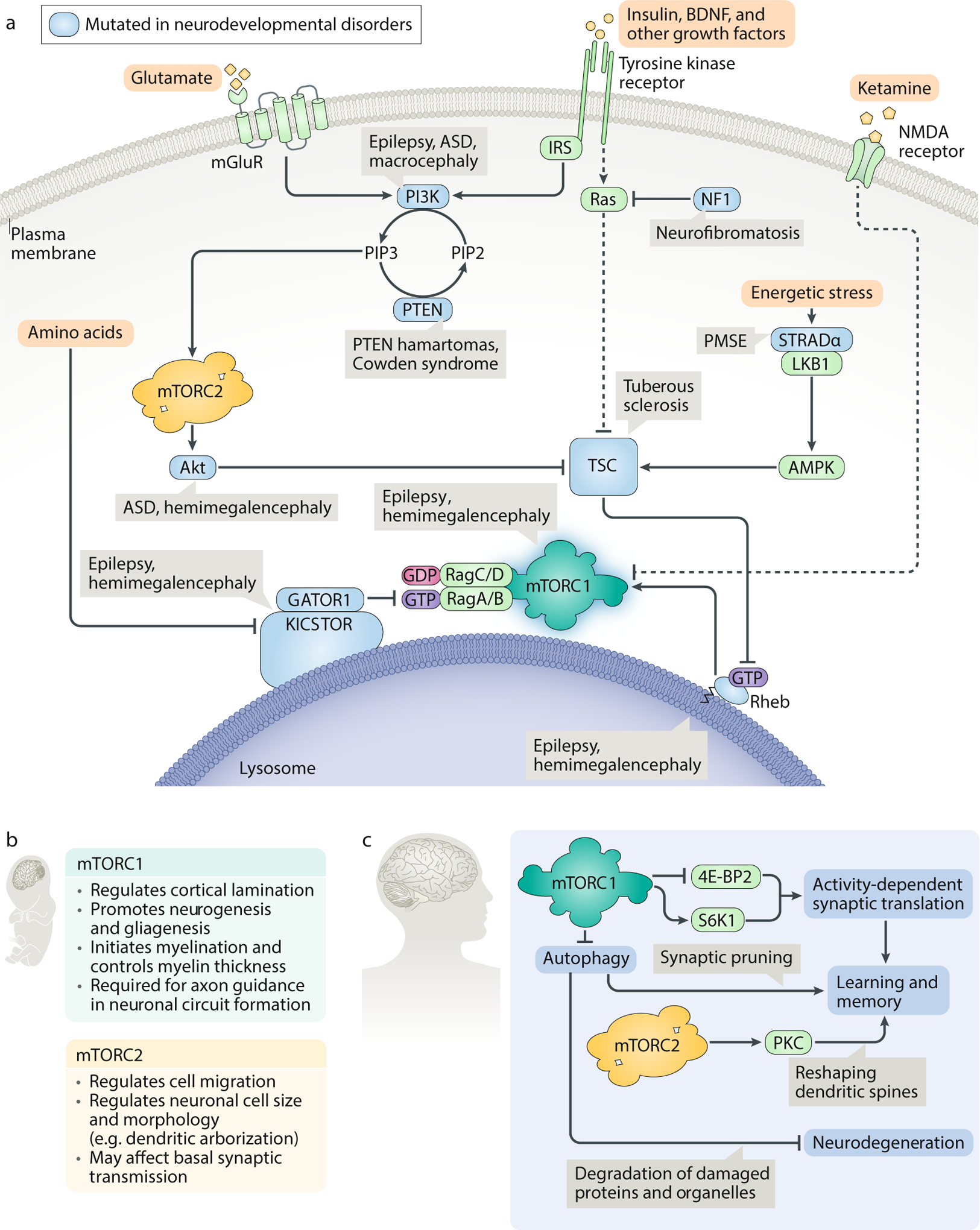
A. In the brain, mTOR complex 1 (mTORC1) signalling is activated not just by nutrients and insulin but also by several tissue-specific inputs, including the neurotransmitter glutamate and the neurotrophic growth factor brain-derived neurotrophic factor (BDNF). Dysregulation of the mTORC1 pathway is associated with a set of characteristic neurodevelopmental diseases, collectively termed ’mTORopathies’. Patients with mTORopathies suffer from severe epilepsy and may also display focal cortical dysplasia, macrocephaly or megalencephaly, cognitive and social defects, and benign tumours. Proteins from genes bearing mutations in neurodevelopmental diseases are shown in blue. B. Roles of mTORC1 and mTORC2 during neuronal development. Ablation of mTORC1 or mTORC2 in the nervous system perturbs cell and organ size and disrupts the cortical architecture of the brain. mTORC1 deletion also causes early postnatal lethality. C. Roles of mTORC1 and mTORC2 in postnatal maintenance of synaptic plasticity and homeostasis. mTORC1 regulates activity-dependent synaptic translation through its substrates eukaryotic initiation factor 4E-binding protein 2 (4E-BP2) and p70 S6 kinase 1 (S6K1) to strengthen or weaken a given neuronal circuit; moreover, it also promotes synaptic plasticity by pruning obsolete synapses through autophagy. Autophagy may additionally play a neuroprotective role by degrading misfolded proteins and damaged organelles. mTORC2 remodels the actin cytoskeleton in response to neuronal signal transmission and helps convert transient excitatory events into long-term memory. AMPK, AMP-activated protein kinase; ASD, autism spectrum disorder; GATOR, GAP activity towards the Rags; IRS, insulin receptor substrate; LKB, liver kinase B1; NF1, neurofibromatosis type 1; PIP2, phosphatidylinositol (4,5)-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PKC, protein kinase C; PMSE, polyhydramnios, megalencephaly and symptomatic epilepsy; PTEN, phosphatase and tensin homologue; STRADα, STE20-related kinase adapter protein-α; TSC, tuberous sclerosis complex.
mTOR in neurodevelopmental disorders
Hyperactive mTOR signalling, as observed in neurodevelopmental mTORopathies, is associated with characteristic defects (Fig. 5a). As a class, mTORopathies are caused by loss-of-function mutations in negative regulators of mTORC1, usually manifesting with some subset of the following symptoms: focal malformations in the brain, epileptic seizures, macrocephaly, autism spectrum disorder and benign tumours or cystic growths193. Perhaps the best-studied such disease is TSC, which arises when loss of either TSC1 or TSC2 induces constitutive mTORC1 activity. Patients with TSC often grow lesions that disrupt the laminar organization of the cortex, nucleating epileptogenic foci; these patients may also have enlarged neurons and ‘giant’ astrocytes193. Similar phenotypes are found in patients with inactivating mutations in the negative PI3K regulator PTEN194, the AMPK activator STRADα195 and the negative regulatory complexes GATOR1 and KICSTOR196,197,198,199,200, as well as those with activating mutations in Rheb or mTOR201,202,203. Given that mTORC1 has many roles in defining the morphology of the developing brain, the epilepsy that clinically distinguishes these disorders is likely seeded by prenatal neuronal mis-wiring204. However, acute rapamycin treatment can nonetheless suppress seizures caused by TSC1 loss in adult mice205, suggesting that mTOR hyperactivity can further stimulate ‘seizing’ in established neural circuits. Consistent with a model in which mTORC1 participates in multiple stages of epileptogenesis, recent speculation contends that the ketogenic diet, a validated therapy for treatment-refractory epilepsy, may work by depriving the mTORC1 pathway of activating nutrients206. Other, more direct mTOR inhibitors are currently in clinical trials as anti-epileptic agents207,208.
mTOR control of brain function via protein translation and autophagy regulation
Surprisingly little is known about the regulation of mTOR signalling in normal brain function and homeostasis. Unlike cell culture systems, the postnatal brain is mostly postmitotic, such that environmental inputs are consolidated not to stimulate growth or proliferation but rather to enact changes in neuronal morphology and connectivity. Although it is not clear which inputs are actually relevant in vivo, given that the brain is ‘nutritionally protected’ from acute fasting (that is, brain biomass and function are generally left intact for as long as possible under starvation, with the brain having first use of available glucose and ketone bodies), brain-derived neurotrophic factor (BDNF) has emerged as a major tissue-specific agonist of the neuronal mTOR pathway. As a PI3K activator, BDNF increases mTORC1 signalling near injured axons to encourage wound healing and repair209,210; in turn, BDNF release may itself be regulated by a feed-forward loop downstream of S6K1 (ref.211).
In collaboration with BDNF, mTOR regulates learning and memory by promoting translation at synapses through S6K1 and 4E-BP2 (ref.212) in a manner that is dependent on neuronal activity (Fig. 5c). This localized translation is rapamycin-sensitive and is crucial for the remodelling of dendritic spines that accompanies long-term potentiation213. Strikingly, animal models lacking TSC or 4E-BP2 recapitulate some of the social and cognitive abnormalities associated with autism spectrum disorder, suggesting that dysregulation of synaptic translation may affect higher-order brain functions214,215. In accordance with this paradigm, synaptic translation has also been linked to depression and psychiatric mood disorders. The NMDA receptor antagonist ketamine, a fast-acting antidepressant, has been shown to boost mTORC1 activity at the synapse, with psychiatric relief coinciding with an increase in synaptic protein, dendritic spine density and synaptic function211,216. In animal models, the Sestrin inhibitor NV-5138 appears to mediate similar improvements by directly activating mTORC1, independent of other upstream signals217. However, while these lines of evidence implicate mRNA translation in diverse aspects of synaptic plasticity and brain health, we still do not know which neuronal mRNAs are regulated by the mTORC1 pathway in response to specific stimuli, nor do we understand how mTORC1 and its substrates localize protein synthesis within individual neurons.
In recent years, it has become increasingly clear that translation is not the only mTORC1 output required for plasticity. In order to adjust the strength of a neuronal circuit, mTORC1 must simultaneously promote the building of new proteins at some synapses and the degradation of excess synaptic machinery at others. The latter process calls for local inhibition of the mTOR pathway, which triggers macroautophagy218 (Fig. 5c). Consistent with the apparent importance of autophagy in cognitive function (see Box 2), constitutive mTOR hyperactivity has been shown to compromise synaptic pruning and contribute to autism spectrum disorder-like social deficits in TSC-deficient mice219. Rapamycin treatment was sufficient to rescue these defects, but only when the autophagy pathway remained intact. While these findings are quite preliminary, taken in sum with the apparent efficacy of mTORC1 activators, like ketamine, they suggest that modulation of the mTORC1 pathway in the brain may hold promise as a therapeutic strategy to improve cognitive performance and memory in certain disease states.
Box 2: mTOR, autophagy and neurodegeneration.
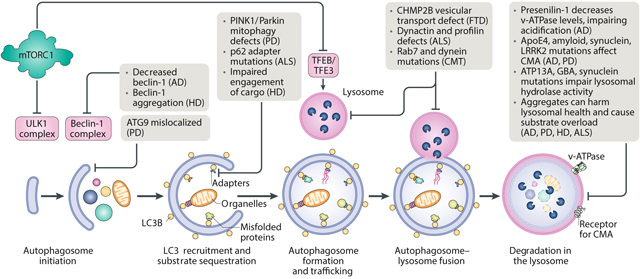
Genetic evidence implicates autophagy — and its major regulator, mTOR complex 1 (mTORC1) — in several devastating neurodegenerative disorders273 (see the figure). These disorders, which include Alzheimer disease (AD), Parkinson disease (PD), Huntington disease (HD), amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), lead to the progressive, permanent destruction of neurons, wreaking havoc on cognition and motor control. Although most cases of neurodegeneration arise sporadically, increasing in frequency with age, certain heritable mutations can boost disease incidence and severity within families, with many such mutations mapping to genes associated with proteostasis and lysosomal function. Indeed, failures in autophagic clearance have emerged as a key hallmark of neurotoxic cell death. In AD, as in several of its pathological cousins, misfolded, ubiquitylated proteins appear to clog autophagic vacuoles, which subsequently accumulate in dystrophic neurites274,275. Because neuronal cells cannot divide to dilute unwanted macromolecules or organelles and must rely on autophagy, any jam in autolysosome clearance propagates through the entire endocytic machinery and may compound metabolic and immunological traumas that lie far afield from the initial amyloid stressor. Multiple groups have confirmed that deletion of essential autophagy genes in the brain is sufficient to induce neurodegeneration even in the absence of disease proteins276,277, supporting a model that puts autophagy — and not amyloids — at the core of neurodegenerative disease.
The recent failure of drugs for AD targeting amyloid-β and tau in clinical trials has demonstrated that reduction of protein aggregates alone has little effect on cognitive function. Given the massive financial and societal costs of neurodegeneration, there is an urgent need for new therapies that delay or reverse disease progression through alternative mechanisms. Based on preclinical evidence, rapamycin may be a promising lead. Induction of autophagy through rapamycin treatment has been shown to eliminate aggregates and improve memory and behaviour in six different mouse models of AD, as well as several models of PD278,279,280. Moreover, as we will discuss below, rapamycin-mediated inhibition of mTOR may also reverse some of the cellular effects of ageing, the most important risk factor for neurodegeneration237. It should be noted, however, that rapamycin does not penetrate the blood–brain barrier with ease and only partially blocks mTORC1 phosphorylation of autophagy regulator unc-51-like autophagy-activating kinase 1 (ULK1)42; in addition, chronic application of rapamycin for neuroprotection would likely disrupt major pathways inside and outside the brain. These caveats suggest that future therapeutic strategies may need to establish a precise balance of neuronal mTOR activity to maintain homeostasis — a goal that will require us to develop a more nuanced understanding of when, why, where and how mTOR acts in the brain.
CMA, chaperone-mediated autophagy; CMT, Charcot–Marie–Tooth disease; TFE3, transcription factor E3; TFEB, transcription factor EB.
mTOR in cancer
Although the mTOR kinase itself is rarely mutated in cancer, it is readily hijacked by upstream oncogenic nodes, including those in the PI3K–Akt pathway and the Ras-driven MAPK pathway. As a result, mTOR signalling is hyperactive in up to 80% of human cancers220, in which context it plays a pivotal role in sustaining cancer cell growth and survival (Fig. 6a). Because tumour microenvironments are poorly vascularized and subject to severe nutritional restrictions, loss of the mTORC1 nutrient sensing machinery may help cancer cells evade metabolic checks on anabolism and proliferation. Thus, mutations in all three components of the GATOR1 complex have been implicated in glioblastomas137, while RagC and FLCN mutations have been found in follicular lymphoma and Birt–Hogg–Dubé syndrome, respectively221,222. Meanwhile, hyperactivation of mTORC2 can aggravate negative cancer prognoses by activating Akt and by supporting the cytoskeletal transformations that underlie metastasis92,93.
Fig. 6: mTOR in cancer and ageing.
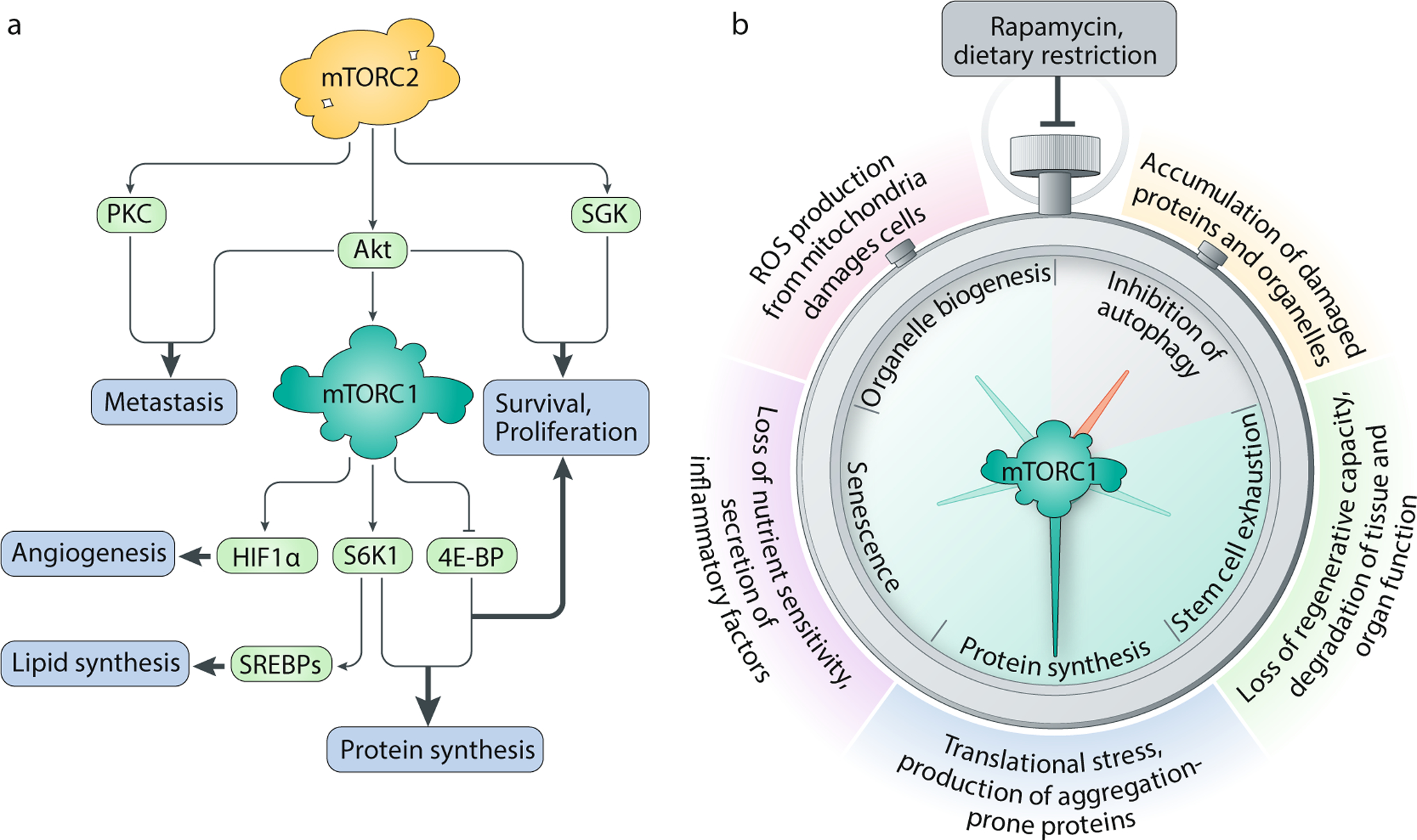
A. mTOR complex 1 (mTORC1) and mTORC2 participate in cancer pathogenesis by underwriting biosynthetic programmes and promoting proliferation and survival. Emerging evidence also implicates mTORC2 activity in metastatic transformations. B. Modulation of mTORC1 signalling in ageing cells may enable us to slow the molecular clock. mTORC1 activates processes that may accelerate cellular and tissue ageing, including protein synthesis, mitochondrial energy production and entry into senescence. Chronic mTORC1 activation also blocks autophagic clearance of damaged cellular components. Inhibition of this pathway — by either rapamycin treatment, genetic inactivation of mTORC1 or dietary restriction — has been shown to extend lifespan and improve physiological performance across a range of model organisms. 4E-BP, 4E-binding protein; HIF1α, hypoxia inducible factor 1α; PKC, protein kinase C; ROS, reactive oxygen species; SGK, serum- and glucocorticoid-induced protein kinase; S6K1, p70 S6 kinase 1; SREBP, sterol regulatory element binding protein.
To date, mTOR inhibitors have met with limited success as chemotherapeutic agents. The first generation of clinical rapamycin derivatives, known as ‘rapalogs’, were approved for advanced renal cell carcinomas in the late 2000s. Outside certain exceptional contexts223, these rapalogs have proved more cytostatic than cytotoxic, perhaps because they only partially block 4E-BP-dependent translation and fail to inhibit the pro-survival pathways regulated by mTORC2–Akt61,224. Inhibition of mTORC1 also drives autophagy, which has been shown to nourish cells in nutrient-poor tumour microenvironments225. A second generation of catalytic mTOR inhibitors (for example, Torin1, PP242, Ku-0063794) competes with ATP to occupy the kinase active site and sidesteps many of these issues by inhibiting all known substrates of mTORC1 and mTORC2 (refs41,226,227). Despite some concerns about tissue toxicity because of their broad effects, early clinical data suggest that catalytic mTOR inhibitors can be tolerated at effective doses228. However, prolonged treatment with these inhibitors can lead to a metabolic retrenchment that allows cancer cells to reactivate Akt without positive input from mTORC2, highlighting resistance as a key problem that must be tackled by next-generation therapies229,230,231.
mTOR in ageing
In line with a growing body of genetic and pharmacological evidence, mTOR activity is now recognized as a major driver of ageing — a process defined here as a progressive decline in physiological function that increases vulnerability to disease and death (Fig. 6b). Genetic inhibition of the mTORC1 pathway through depletion of mTOR or Raptor has been shown to extend lifespan in organisms as diverse as yeast232, nematodes233,234, flies235 and mammals236; in a similar vein, rapamycin treatment also promotes longevity across a wide swathe of the evolutionary tree237,238,239,240. Tantalizingly, rapamycin appears to prolong not just lifespan but also healthspan — the length of time that an organism enjoys efficient biological performance, free of disease or disability — suggesting that mTORC1 inhibition may slow ageing by reversing molecular changes associated with cellular deterioration241.
What are these mTORC1-sensitive molecular changes that affect ageing? One clue may come from dietary restriction, the only other intervention that produces a comparable and conserved increase in lifespan. Dietary restriction reduces nutrient intake without incurring malnutrition, pushing mTORC1 towards a catabolic regime. Indeed, dietary restriction is thought to counter ageing by acting through the mTORC1 pathway, as dietary restriction on top of chemical or genetic inhibition of mTORC1 fails to confer any additional longevity benefit in flies, worms and yeast232,235,242. Intriguingly, dietary restriction of a single amino acid, methionine, is sufficient to increase lifespan in flies243, implying that restrictions on protein synthesis may have a particularly important anti-ageing effect. Consistent with this observation, loss of the translation effector S6K1 extends lifespan in worms and mice, perhaps by halting the production of misfolded or aggregated proteins242,244. By reducing the energetic burden of translation, mTOR inhibition also relieves oxidative stress and prevents the accumulation of harmful metabolic by-products, leading to broad improvements in cellular function73.
In parallel with its downregulation of translation, mTOR inhibition restores autophagic capacity, which undergoes an age-related decline in many tissues245. Autophagy degrades obsolete or damaged cellular components and salvages them for ‘spare macromolecular parts’. Through this process, aged cells refresh their molecular equipment and clear damaged proteins and organelles, which have been implicated in age-related diseases from cardiomyopathy to neurodegeneration. Underscoring the importance of autophagy in healthy ageing, direct activation of autophagic flux can significantly increase lifespan and healthspan in mice246. Conversely, mTOR inhibition fails to extend lifespan in ATG-deficient worms, indicating that mTOR modulates longevity at least partly through autophagy-dependent mechanisms247,248.
mTORC1 has also been implicated in ageing at the tissue level. Several groups have shown that persistent mTORC1 signalling contributes to the exhaustion of stem cell pools, hindering tissue self-renewal in aged organisms249,250. mTORC1 hyperactivity is also a distinctive feature of senescent cells, which permanently arrest in the G0 phase of the cell cycle and undergo morphological alterations that eliminate sensitivity to amino acid and growth factor deprivation251. Exploiting translational programmes downstream of mTORC1, senescent cells synthesize and secrete pro-inflammatory cytokines to exacerbate ageing-related declines in fitness and tissue function252,253. Rapamycin treatment attenuates this inflammatory phenotype, although it is unclear whether mTORC1 inhibition can rescue cell cycle arrest or aid in the clearance of senescent cells.
Even though mTOR inhibitors are well validated as geroprotective agents in animal models, the potential side effects of chronic dosing (particularly insulin resistance and immunosuppression) have thus far precluded their use in healthy elderly humans. Recent studies, however, suggest that these concerns are not insurmountable. Because side-effect profiles for mTOR inhibitors have largely been inferred from patients undergoing cancer therapy or organ transplantation, they tend to reflect intense, high-dose regimens. Far lower doses are needed for anti-ageing benefits. Taking advantage of this distinction, one group found that intermittent dosing of rapamycin in mice could extend their lifespan without inciting glucose intolerance254,255. Another reported that low doses of mTOR inhibitors could actually improve immune function in elderly patients256. Efforts to harness mTOR inhibition as an anti-ageing strategy will have to build on these studies to define safe and effective doses in human cohorts.
Conclusions and perspectives
Perched at the interface between organisms and their environments, the mTOR pathway toggles the balance of anabolism and catabolism in response to contextual signals and guides nearly every aspect of metabolic function. Recent work has clarified the logical structure of the pathway and drawn the lysosome into renewed focus; structural advances have also allowed us to see, mechanistically, how key mTOR signalling nodes transduce nutritional information into molecular action. Building on careful in vivo studies, we have made remarkable progress in cataloguing the inputs and effectors of the mTOR pathway across various tissues and metabolic states, enhancing our understanding of mTOR signalling in health and disease.
Nonetheless, certain open questions remain stubbornly unresolved. Given that mTORC1 activation occurs at the lysosomal surface, how does the complex capture and phosphorylate its downstream substrates, which, with the exception of TFEB/TFE3, do not maintain lysosomal subpopulations? It is possible that lysosomal interactions with the endoplasmic reticulum, the Golgi and the plasma membrane may help bring mTORC1 into contact with some of its substrates257,258,259. Alternatively, while efforts to visualize the dynamics of the substrate search have not been conclusive260,261, we speculate that activated mTORC1 could exit the lysosome to phosphorylate targets at other loci in the cell, perhaps carrying Rheb in tow. Moving forward, we also seek an integrated understanding of mTOR signalling in specific tissues. Although many components of the mTOR pathway have been identified, it is not clear which regulatory inputs are dominant in any particular physiological milieu. In order to develop new therapeutics that evade some of the metabolic side effects of existing mTOR inhibitors, we hope to identify complex-specific or tissue-specific modulators of mTOR activity and establish them as targets for rational drug development.
One emerging theme from the study of mTOR dysregulation in human disease is that these pathologies are not just linked by a common aetiological basis — they also intersect with each other in mutually reinforcing ways. Just as excessive mTOR activity can lead to metabolic syndrome, obesity accelerates molecular ageing, which in turn amplifies the risk of neurodegenerative disease and cancer. Thus, even though the complexity and breadth of the mTOR signalling network increases the risk of toxicity, the unique spectrum of mTOR-dependent processes is also one of its most powerful advantages as a therapeutic target. More so than other strategies to delay ageing or counter disease, mTOR inhibition disrupts a wide variety of degenerative processes with a single intervention. Further insights into this fundamental pathway may ultimately lead to new treatments for currently intractable diseases and transform our ability to regulate health and homeostasis.
References
- 1.Vezina C, Kudelski A & Sehgal SN Rapamycin (AY-22,989), a new antifungal antibiotic I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot 28, 721–726 (1975). [DOI] [PubMed] [Google Scholar]
- 2.Martel RR, Klicius J & Galet S Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can J. Physiol. Pharmacol 55, 48–51 (1977). [DOI] [PubMed] [Google Scholar]
- 3.Eng CP, Sehgal SN & Vezina C Activity of rapamycin (AY-22,989) against transplanted tumors. J. Antibiot 37, 1231–1237 (1984). [DOI] [PubMed] [Google Scholar]
- 4.Houchens DP, Ovejera AA, Riblet SM & Slagel DE Human brain tumor xenografts in nude mice as a chemotherapy model. Eur. J. Cancer Clin. Oncol 19, 799–805 (1983). [DOI] [PubMed] [Google Scholar]
- 5.Bierer BE et al. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl Acad. Sci. USA 87, 9231–9235 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung J, Kuo CJ, Crabtree GR & Blenis J Rapamycin–FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69, 1227–1236 (1992). [DOI] [PubMed] [Google Scholar]
- 7.Brown EJ et al. A mammalian protein targeted by G1-arresting rapamycin–receptor complex. Nature 369, 756–758 (1994). [DOI] [PubMed] [Google Scholar]
- 8.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P & Snyder SH RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78, 35–43 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Sabers CJ et al. Isolation of a protein target of the FKBP12–rapamycin complex in mammalian cells. J. Biol. Chem 270, 815–822 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Heitman J, Movva NR & Hall MN Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253, 905–909 (1991). [DOI] [PubMed] [Google Scholar]
- 11.Cafferkey R et al. Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol. Cell Biol 13, 6012–6023 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunz J et al. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 73, 585–596 (1993). [DOI] [PubMed] [Google Scholar]
- 13.Helliwell SB et al. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol. Biol. Cell 5, 105–118 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keith CT & Schreiber SL PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science 270, 50 (1995). [DOI] [PubMed] [Google Scholar]
- 15.Kim DH et al. GβL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 11, 895–904 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Kim DH et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Hara K et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Yang H et al. mTOR kinase structure, mechanism and regulation. Nature 497, 217–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guertin DA et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev. Cell 11, 859–871 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Schalm SS, Fingar DC, Sabatini DM & Blenis J TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol 13, 797–806 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Nojima H et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem 278, 15461–15464 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Sancak Y et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Vander Haar E, Lee S-I, Bandhakavi S, Griffin TJ & Kim D-H Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol 9, 316–323 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Peterson TR et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137, 873–886 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yip CK, Murata K, Walz T, Sabatini DM & Kang SA Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 38, 768–774 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aylett CH et al. Architecture of human mTOR complex 1. Science 351, 48–52 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Yang H et al. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552, 368–373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang Y et al. Disruption of the scaffolding function of mLST8 selectively inhibits mTORC2 assembly and function and suppresses mTORC2-dependent tumor growth in vivo. Cancer Res. 79, 3178 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarbassov DD et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol 14, 1296–1302 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Jacinto E et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol 6, 1122–1128 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Frias MA et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol 16, 1865–1870 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Jacinto E et al. SIN1/MIP1 maintains rictor–mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Yang Q, Inoki K, Ikenoue T & Guan KL Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 20, 2820–2832 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pearce LR et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J 405, 513–522 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo SY et al. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor β expression and signaling. J. Biol. Chem 282, 25604–25612 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Yuan H-X & Guan K-L The SIN1-PH domain connects mTORC2 to PI3K. Cancer Discov. 5, 1127–1129 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that PIP3 activates mTORC2 by binding the pleckstrin homology domain of mSin1, which otherwise autoinhibits the kinase.
- 37.Chen X et al. Cryo-EM structure of human mTOR complex 2. Cell Res. 28, 518–528 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stuttfeld E et al. Architecture of the human mTORC2 core complex. eLife 7, e33101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarbassov DD et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Lamming DW et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thoreen CC et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem 284, 8023–8032 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang SA et al. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341, 1236566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choo AY, Yoon S-O, Kim SG, Roux PP & Blenis J Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl Acad. Sci. USA 105, 17414–17419 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buttgereit F & Brand MD A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J 312, 163–167 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brunn GJ et al. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 277, 99–101 (1997). [DOI] [PubMed] [Google Scholar]
- 46.Gingras AC et al. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 13, 1422–1437 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hara K et al. Regulation of eIF-4E BP1 phosphorylation by mTOR. J. Biol. Chem 272, 26457–26463 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Pullen N et al. Phosphorylation and activation of p70s6k by PDK1. Science 279, 707 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Burnett PE, Barrow RK, Cohen NA, Snyder SH & Sabatini DM RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl Acad. Sci. USA 95, 1432–1437 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruvinsky I et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 19, 2199–2211 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that a mouse bearing serine to alanine substitutions at all five phosphorylatable serines in ribosomal protein S6 does not experience any global defects in translation.
- 51.Chauvin C et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 33, 474 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Hannan KM et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol. Cell Biol 23, 8862–8877 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mayer C, Zhao J, Yuan X & Grummt I mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 18, 423–434 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michels AA et al. mTORC1 directly phosphorylates and regulates human MAF1. Mol. Cell Biol 30, 3749 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shor B et al. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J. Biol. Chem 285, 15380–15392 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holz MK, Ballif BA, Gygi SP & Blenis J mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123, 569–580 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Dorrello NV et al. S6K1- and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314, 467–471 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Ma XM, Yoon SO, Richardson CJ, Julich K & Blenis J SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 133, 303–313 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Mieulet V et al. S6 kinase inactivation impairs growth and translational target phosphorylation in muscle cells maintaining proper regulation of protein turnover. Am. J. Physiol. Cell Physiol 293, C712–C722 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Pende M et al. S6K1–/–/S6K2–/– mice exhibit perinatal lethality and rapamycin-sensitive 5ʹ-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell Biol 24, 3112–3124 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hsieh AC et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thoreen CC et al. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Horton JD, Goldstein JL & Brown MS SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest 109, 1125–1131 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peterson TR et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Porstmann T et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8, 224–236 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Duvel K et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; Using transcriptional and metabolic profiling, this study demonstrates that mTORC1 activates HIF1α and the SREBPs to induce glycolysis, lipid synthesis and the pentose phosphate pathway.
- 67.Kim JE & Chen J Regulation of peroxisome proliferator-activated receptor-γ activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 53, 2748–2756 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM & Manning BD mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ben-Sahra I, Howell JJ, Asara JM & Manning BD Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robitaille AM et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339, 1320–1323 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Valvezan AJ et al. mTORC1 couples nucleotide synthesis to nucleotide demand resulting in a targetable metabolic vulnerability. Cancer Cell 32, 624–638 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.He L et al. mTORC1 promotes metabolic reprogramming by the suppression of GSK3-dependent Foxk1 phosphorylation. Mol. Cell 70, 949–960 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zid BM et al. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 139, 149–160 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cunningham JT et al. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α transcriptional complex. Nature 450, 736–740 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Kim J, Kundu M, Viollet B & Guan KL AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol 13, 132–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hosokawa N et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with Kim et al. (2011), this study shows that mTORC1 directly regulates initiation of autophagy by applying inhibitory phosphorylations to components of the ULK1 complex.
- 77.Ganley IG et al. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem 284, 12297–12305 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dikic I & Elazar Z Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol 19, 349–364 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Kim YM et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 57, 207–218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Settembre C et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martina JA, Chen Y, Gucek M & Puertollano R MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roczniak-Ferguson A et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal 5, ra42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu L et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Odle RI et al. An mTORC1-to-CDK1 switch maintains autophagy suppression during mitosis. Mol Cell, 10.1016/j.molcel.2019.10.016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wyant GA et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171, 642–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wyant GA et al. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360, 751–758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guo JY et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 30, 1704–1717 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kraft C, Deplazes A, Sohrmann M & Peter M Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol 10, 602–610 (2008). [DOI] [PubMed] [Google Scholar]
- 89.An H & Harper JW Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol 20, 135–143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Larsson C Protein kinase C and the regulation of the actin cytoskeleton. Cell Signal. 18, 276–284 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Liu L, Das S, Losert W & Parent CA mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev. Cell 19, 845–857 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schmidt KM et al. Inhibition of mTORC2/RICTOR impairs melanoma hepatic metastasis. Neoplasia 20, 1198–1208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morrison Joly M et al. Two distinct mTORC2-dependent pathways converge on Rac1 to drive breast cancer metastasis. Breast Cancer Res. 19, 74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ikenoue T, Inoki K, Yang Q, Zhou X & Guan KL Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 27, 1919–1931 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li X & Gao T mTORC2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Rep. 15, 191–198 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.García-Martínez JM & Alessi DR mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J 416, 375 (2008). [DOI] [PubMed] [Google Scholar]
- 97.Sarbassov DD, Guertin DA, Ali SM & Sabatini DM Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science 307, 1098–1101 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Webb AE & Brunet A FOXO transcription factors: key regulators of cellular quality control. Trends Biochem. Sci 39, 159–169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hoxhaj G et al. Direct stimulation of NADP+ synthesis through Akt-mediated phosphorylation of NAD kinase. Science 363, 1088–1092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Inoki K, Li Y, Zhu T, Wu J & Guan KL TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol 4, 648–657 (2002). [DOI] [PubMed] [Google Scholar]
- 101.Humphrey SJ et al. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 17, 1009–1020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ebner M, Sinkovics B, Szczygieł M, Ribeiro DW & Yudushkin I Localization of mTORC2 activity inside cells. J. Cell Biol 216, 343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Inoki K, Li Y, Xu T & Guan KL Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Long X, Lin Y, Ortiz-Vega S, Yonezawa K & Avruch J Rheb binds and regulates the mTOR kinase. Curr. Biol 15, 702–713 (2005). [DOI] [PubMed] [Google Scholar]
- 105.Sancak Y et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim E, Goraksha-Hicks P, Li L, Neufeld TP & Guan KL Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol 10, 935–945 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with Sancak et al. (2008), this study defines the Rag-GTPases as a second upstream signalling branch that transduces amino acid availability to mTORC1. Sancak et al.’s study also shows that mTORC1 localizes to the lysosome under amino-acid-replete conditions.
- 107.Dibble CC & Manning BD Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol 15, 555–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dibble CC et al. TBC1D7 is a third subunit of the TSC1–TSC2 complex upstream of mTORC1. Mol. Cell 47, 535–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tee AR, Manning BD, Roux PP, Cantley LC & Blenis J Tuberous sclerosis complex gene products, tuberin and hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol 13, 1259–1268 (2003). [DOI] [PubMed] [Google Scholar]
- 110.Valvezan AJ & Manning BD Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat. Metab 1, 321–333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Garami A et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466 (2003). [DOI] [PubMed] [Google Scholar]
- 112.Manning BD, Tee AR, Logsdon MN, Blenis J & Cantley LC Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol. Cell 10, 151–162 (2002). [DOI] [PubMed] [Google Scholar]
- 113.Menon S et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Demetriades C, Doumpas N & Teleman AA Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156, 786–799 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with Menon et al. (2014), this study shows that insulin-mediated activation of Akt releases TSC from the lysosome, thereby freeing lysosomal Rheb to activate mTORC1.
- 115.Harrington LS et al. The TSC1–2 tumor suppressor controls insulin–PI3K signaling via regulation of IRS proteins. J. Cell Biol 166, 213–223 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shah OJ, Wang Z & Hunter T Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol 14, 1650–1656 (2004). [DOI] [PubMed] [Google Scholar]
- 117.Inoki K et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126, 955–968 (2006). [DOI] [PubMed] [Google Scholar]
- 118.Lee DF et al. IKKβ suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130, 440–455 (2007). [DOI] [PubMed] [Google Scholar]
- 119.Ma L, Chen Z, Erdjument-Bromage H, Tempst P & Pandolfi PP Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121, 179–193 (2005). [DOI] [PubMed] [Google Scholar]
- 120.Roux PP, Ballif BA, Anjum R, Gygi SP & Blenis J Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl Acad. Sci. USA 101, 13489–13494 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gwinn DM et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shaw RJ et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6, 91–99 (2004). [DOI] [PubMed] [Google Scholar]
- 123.Inoki K, Zhu T & Guan KL TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 (2003). [DOI] [PubMed] [Google Scholar]
- 124.Herzig S & Shaw RJ AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol 19, 121–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brugarolas J et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 18, 2893–2904 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.DeYoung MP, Horak P, Sofer A, Sgroi D & Ellisen LW Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14–3–3 shuttling. Genes Dev. 22, 239–251 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Demetriades C, Plescher M & Teleman AA Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat. Commun 7, 10662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Saveljeva S et al. Endoplasmic reticulum stress-mediated induction of SESTRIN 2 potentiates cell survival. Oncotarget 7, 12254–12266 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Feng Z et al. The regulation of AMPKβ1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1–AKT–mTOR pathways. Cancer Res. 67, 3043–3053 (2007). [DOI] [PubMed] [Google Scholar]
- 130.Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM & Meijer AJ Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem 270, 2320–2326 (1995). [DOI] [PubMed] [Google Scholar]
- 131.Hara K et al. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem 273, 14484–14494 (1998). [DOI] [PubMed] [Google Scholar]
- 132.Sancak Y et al. Ragulator–Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bar-Peled L, Schweitzer LD, Zoncu R & Sabatini DM Ragulator is a GEF for the Rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Su MY et al. Hybrid structure of the RagA/C–Ragulator mTORC1 activation complex. Mol. Cell 68, 835–846 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shen K, Choe A & Sabatini DM Intersubunit crosstalk in the Rag GTPase heterodimer enables mTORC1 to respond rapidly to amino acid availability. Mol. Cell 68, 821 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rogala KB et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science 366, 468–475 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bar-Peled L et al. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shen K, Valenstein ML, Gu X & Sabatini DM Arg-78 of Nprl2 catalyzes GATOR1-stimulated GTP hydrolysis by the Rag GTPases. J. Biol. Chem 294, 2970–2975 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shen K et al. Architecture of the human GATOR1 and GATOR1–Rag GTPases complexes. Nature 556, 64–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Peng M, Yin N & Li MO SZT2 dictates GATOR control of mTORC1 signalling. Nature 543, 433–437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wolfson RL et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 543, 438–442 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wolfson RL et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351, 43–48 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Saxton RA et al. Structural basis for leucine sensing by the Sestrin2–mTORC1 pathway. Science 351, 53–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ye J et al. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev. 29, 2331–2336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chantranupong L et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 9, 1–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Parmigiani A et al. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 9, 1281–1291 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chantranupong L et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 165, 153–164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Saxton RA, Chantranupong L, Knockenhauer KE, Schwartz TU & Sabatini DM Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 536, 229–233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rebsamen M et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519, 477–481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang S et al. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347, 188–194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lei HT, Ma J, Sanchez Martinez S & Gonen T Crystal structure of arginine-bound lysosomal transporter SLC38A9 in the cytosol-open state. Nat. Struct. Mol. Biol 25, 522–527 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shen K & Sabatini DM Ragulator and SLC38A9 activate the Rag GTPases through noncanonical GEF mechanisms. Proc. Natl Acad. Sci. USA 115, 9545–9550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zoncu R et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 334, 678–683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Petit CS, Roczniak-Ferguson A & Ferguson SM Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J. Cell Biol 202, 1107–1122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tsun ZY et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 52, 495–505 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wada S et al. The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 30, 2551–2564 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Villegas F et al. Lysosomal signaling licenses embryonic stem cell differentiation via inactivation of Tfe3. Cell Stem Cell 24, 257–270.e8 (2019). [DOI] [PubMed] [Google Scholar]
- 158.Perera RM et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Di Malta C et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 356, 1188–1192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gu X et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358, 813–818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Collectively with Wolfson et al. (2016), Saxton et al. (Science, 2016), Chantranupong et al. (2016) and Saxton et al. (Nature, 2016), this study describes the molecular action of the cytoplasmic ‘nutrient sensors’, which bind leucine, arginine and SAM and communicate their availability to the GATOR complexes.
- 161.Efeyan A et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493, 679–683 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kalender A et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 11, 390–401 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hoxhaj G et al. The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep. 21, 1331–1346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Emmanuel N et al. Purine nucleotide availability regulates mTORC1 activity through the Rheb GTPase. Cell Rep. 19, 2665–2680 (2017). [DOI] [PubMed] [Google Scholar]; Together with Hoxhaj et al. (2017), this study argues that purine nucleotide availability regulates mTORC1 activity, either by activation of TSC upon acute deprivation and degradation of Rheb under prolonged deprivation (Hoxhaj et al.) or through disruption of Rheb farnesylation (Emmanuel et al.).
- 165.Menon D et al. Lipid sensing by mTOR via de novo synthesis of phosphatidic acid. J. Biol. Chem 292, 6303–6311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jewell JL et al. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gan X, Wang J, Su B & Wu D Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem 286, 10998–11002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Khanna A et al. The small GTPases Ras and Rap1 bind to and control TORC2 activity. Sci. Rep 6, 25823 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Saci A, Cantley Lewis, C. & Carpenter Christopher L. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol. Cell 42, 50–61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Senoo H et al. Phosphorylated Rho–GDP directly activates mTORC2 kinase towards AKT through dimerization with Ras–GTP to regulate cell migration. Nat. Cell Biol 21, 867–878 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kovalski JR et al. The functional proximal proteome of oncogenic Ras includes mTORC2. Mol. Cell 73, 830–844 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with Khanna et al. (2016), Saci et al. (2011), and Senoo et al. (2019), this study implicates small GTPases in upstream regulation of mTORC2 activity.
- 172.Um SH et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205 (2004). [DOI] [PubMed] [Google Scholar]
- 173.Hsu PP et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yu Y et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332, 1322–1326 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kazyken D et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal 12, eaav3249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Cross DA, Alessi DR, Cohen P, Andjelkovich M & Hemmings BA Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 (1995). [DOI] [PubMed] [Google Scholar]
- 177.Sengupta S, Peterson TR, Laplante M, Oh S & Sabatini DM mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468, 1100–1104 (2010). [DOI] [PubMed] [Google Scholar]
- 178.Komatsu M et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol 169, 425 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Peng M, Yin N & Li MO Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 159, 122–133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kuma A et al. The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036 (2004). [DOI] [PubMed] [Google Scholar]
- 181.Tontonoz P, Hu E & Spiegelman BM Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 79, 1147–1156 (1994). [DOI] [PubMed] [Google Scholar]
- 182.Arif A et al. EPRS is a critical mTORC1–S6K1 effector that influences adiposity in mice. Nature 542, 357–361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lee PL, Tang Y, Li H & Guertin DA Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol. Metab 5, 422–432 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Polak P et al. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 8, 399–410 (2008). [DOI] [PubMed] [Google Scholar]
- 185.Hagiwara A et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 15, 725–738 (2012). [DOI] [PubMed] [Google Scholar]
- 186.Yuan M, Pino E, Wu L, Kacergis M & Soukas AA Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J. Biol. Chem 287, 29579–29588 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Puigserver P et al. Insulin-regulated hepatic gluconeogenesis through FOXO1–PGC-1α interaction. Nature 423, 550–555 (2003). [DOI] [PubMed] [Google Scholar]
- 188.Howell JJ et al. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 25, 463–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lipton JO & Sahin M The neurology of mTOR. Neuron 84, 275–291 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Graber TE, McCamphill PK & Sossin WS A recollection of mTOR signaling in learning and memory. Learn Mem. 20, 518–530 (2013). [DOI] [PubMed] [Google Scholar]
- 191.Cloetta D et al. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J. Neurosci 33, 7799–7810 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Thomanetz V et al. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol 201, 293–308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Crino PB mTOR signaling in epilepsy: insights from malformations of cortical development. Cold Spring Harb. Perspect. Med 5, a022442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pilarski R et al. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J. Natl Cancer Inst 105, 1607–1616 (2013). [DOI] [PubMed] [Google Scholar]
- 195.Puffenberger EG et al. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain 130, 1929–1941 (2007). [DOI] [PubMed] [Google Scholar]
- 196.Dibbens LM et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat. Genet 45, 546 (2013). [DOI] [PubMed] [Google Scholar]
- 197.Weckhuysen S et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 57, 994–1003 (2016). [DOI] [PubMed] [Google Scholar]
- 198.Yuskaitis CJ et al. A mouse model of DEPDC5-related epilepsy: neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol. Dis 111, 91–101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Basel-Vanagaite L et al. Biallelic SZT2 mutations cause infantile encephalopathy with epilepsy and dysmorphic corpus callosum. Am. J. Hum. Genet 93, 524–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nakamura Y et al. Constitutive activation of mTORC1 signaling induced by biallelic loss-of-function mutations in SZT2 underlies a discernible neurodevelopmental disease. PLOS ONE 14, e0221482 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Reijnders MRF et al. Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat. Commun 8, 1052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Allen AS et al. De novo mutations in epileptic encephalopathies. Nature 501, 217–221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.D’Gama AM et al. Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 21, 3754–3766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gallent EA & Steward O Neuronal PTEN deletion in adult cortical neurons triggers progressive growth of cell bodies, dendrites, and axons. Exp. Neurol 303, 12–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Abs E et al. TORC1-dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann. Neurol 74, 569–579 (2013). [DOI] [PubMed] [Google Scholar]
- 206.McDaniel SS, Rensing NR, Thio LL, Yamada KA & Wong M The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 52, e7–e11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/show/NCT01713946 (2017).
- 208.Brandt C et al. The novel, catalytic mTORC1/2 inhibitor PQR620 and the PI3K/mTORC1/2 inhibitor PQR530 effectively cross the blood–brain barrier and increase seizure threshold in a mouse model of chronic epilepsy. Neuropharmacology 140, 107–120 (2018). [DOI] [PubMed] [Google Scholar]
- 209.Hsu W-L et al. Glutamate stimulates local protein synthesis in the axons of rat cortical neurons by activating α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and metabotropic glutamate receptors. J. Biol. Chem 290, 20748–20760 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Park KK et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322, 963 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Autry AE et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Takei N et al. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J. Neurosci 24, 9760–9769 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Henry FE, Hockeimer W, Chen A, Mysore SP & Sutton MA Mechanistic target of rapamycin is necessary for changes in dendritic spine morphology associated with long-term potentiation. Mol. Brain 10, 50–50 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Tsai PT et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Gkogkas CG et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Li N et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that the behavioural effects of ketamine on murine depression models are mediated by rapid activation of mTOR and an ensuing increase in synaptic translation, synaptic protein levels and synapse number.
- 217.Kato T et al. Sestrin modulator NV-5138 produces rapid antidepressant effects via direct mTORC1 activation. J. Clin. Invest 129, 2542–2554 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Nikoletopoulou V, Sidiropoulou K, Kallergi E, Dalezios Y & Tavernarakis N Modulation of autophagy by BDNF underlies synaptic plasticity. Cell Metab. 26, 230–242 (2017). [DOI] [PubMed] [Google Scholar]
- 219.Tang G et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Menon S & Manning BD Common corruption of the mTOR signaling network in human tumors. Oncogene 27, S43–S51 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Okosun J et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat. Genet 48, 183–188 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nickerson ML et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt–Hogg–Dube syndrome. Cancer Cell 2, 157–164 (2002). [DOI] [PubMed] [Google Scholar]
- 223.Wagle N et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 4, 546–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Tabernero J et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J. Clin. Oncol 26, 1603–1610 (2008). [DOI] [PubMed] [Google Scholar]
- 225.Palm W et al. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162, 259–270 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Feldman ME et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLOS Biol. 7, e1000038 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Garcia-Martinez JM et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J 421, 29–42 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ghobrial IM et al. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: a phase I dose escalation study in patients with relapsed or refractory multiple myeloma, non-Hodgkin lymphoma, or Waldenström’s macroglobulinemia. Am. J. Hematol 91, 400–405 (2016). [DOI] [PubMed] [Google Scholar]
- 229.Rodrik-Outmezguine VS et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 1, 248–259 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Rodrik-Outmezguine VS et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 534, 272–276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Fan Q et al. A kinase inhibitor targeted to mTORC1 drives regression in glioblastoma. Cancer Cell 31, 424–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study finds that a ‘third-generation’ mTOR inhibitor that links a rapalog with an ATP-competitive catalytic inhibitor (described in Rodrik-Outmezguine et al. (2016)) is more effective than either first-generation or second-generation mTOR inhibitors in driving regression of treatment-resistant glioblastomas. The ‘third-generation’ inhibitor combats resistance caused by activating mutations in either the kinase domain or the FKBP12–rapamycin binding domain of mTOR.
- 232.Kaeberlein M et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310, 1193–1196 (2005). [DOI] [PubMed] [Google Scholar]
- 233.Vellai T et al. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426, 620 (2003). [DOI] [PubMed] [Google Scholar]
- 234.Jia K, Chen D & Riddle DL The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131, 3897–3906 (2004). [DOI] [PubMed] [Google Scholar]
- 235.Kapahi P et al. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol 14, 885–890 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wu JJ et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 4, 913–920 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Harrison DE et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents the first report of a pharmacological intervention — rapamycin treatment — capable of extending the maximal lifespan in mammals of both sexes.
- 238.Powers RW 3rd, Kaeberlein M, Caldwell SD, Kennedy BK & Fields S Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 20, 174–184 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Robida-Stubbs S et al. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 15, 713–724 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bjedov I et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 11, 35–46 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Bitto A et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. eLife 5, e16351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Hansen M et al. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6, 95–110 (2007). [DOI] [PubMed] [Google Scholar]
- 243.Grandison RC, Piper MDW & Partridge L Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 462, 1061–1064 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Selman C et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326, 140–144 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Hansen M, Rubinsztein DC & Walker DW Autophagy as a promoter of longevity: insights from model organisms. Nat. Rev. Mol. Cell Biol 19, 579–593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Fernandez AF et al. Disruption of the beclin 1–BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Toth ML et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 4, 330–338 (2008). [DOI] [PubMed] [Google Scholar]
- 248.Hansen M et al. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLOS Genet. 4, e24 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Chen C, Liu Y, Liu Y & Zheng P mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal 2, ra75 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Yilmaz OH et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 486, 490–495 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Carroll B et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J. Cell Biol 216, 1949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that mTORC1 activation is constitutive and insensitive to deprivation of growth factors or nutrients in senescent human fibroblasts, perhaps due to defects in membrane potential. Strikingly, this study also found that starvation coupled to mTOR inhibition selectively promotes senescent cell death.
- 252.Laberge RM et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol 17, 1049–1061 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Herranz N et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol 17, 1205–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Arriola Apelo SI et al. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell 15, 28–38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Arriola Apelo SI, Pumper CP, Baar EL, Cummings NE & Lamming DW Intermittent administration of rapamycin extends the life span of female C57BL/6J mice. J. Gerontol. A Biol. Sci. Med. Sci 71, 876–881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Mannick JB et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med 10, eaaq1564 (2018). [DOI] [PubMed] [Google Scholar]
- 257.Lim C-Y et al. ER–lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann–Pick type C. Nat. Cell Biol 21, 1206–1218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Hao F et al. Rheb localized on the Golgi membrane activates lysosome-localized mTORC1 at the Golgi–lysosome contact site. J. Cell Sci 131, jcs208017 (2018). [DOI] [PubMed] [Google Scholar]
- 259.Korolchuk VI et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol 13, 453–460 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Manifava M et al. Dynamics of mTORC1 activation in response to amino acids. eLife 5, e19960 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ahmed AR et al. Direct imaging of the recruitment and phosphorylation of S6K1 in the mTORC1 pathway in living cells. Sci. Rep 9, 3408 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Naito T, Kuma A & Mizushima N Differential contribution of insulin and amino acids to the mTORC1–autophagy pathway in the liver and muscle. J. Biol. Chem 288, 21074–21081 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Frey JW, Jacobs BL, Goodman CA & Hornberger TA A role for Raptor phosphorylation in the mechanical activation of mTOR signaling. Cell Signal. 26, 313–322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.You JS et al. The role of raptor in the mechanical load-induced regulation of mTOR signaling, protein synthesis, and skeletal muscle hypertrophy. FASEB J. 33, 4021–4034 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Jacobs BL et al. Eccentric contractions increase the phosphorylation of tuberous sclerosis complex-2 (TSC2) and alter the targeting of TSC2 and the mechanistic target of rapamycin to the lysosome. J. Physiol 591, 4611–4620 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Wang A et al. Activity-independent targeting of mTOR to lysosomes in primary osteoclasts. Sci. Rep 7, 3005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Stracka D, Jozefczuk S, Rudroff F, Sauer U & Hall MN Nitrogen source activates TOR (target of rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J. Biol. Chem 289, 25010–25020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study finds that nitrogen sources can activate TORC1 in prototrophic yeast strains in a transient, Rag-dependent manner, although sustained activation by glutamine may feed through an alternative mechanism.
- 268.Urano J, Tabancay AP, Yang W & Tamanoi F The Saccharomyces cerevisiae Rheb G-protein is involved in regulating canavanine resistance and arginine uptake. J. Biol. Chem 275, 11198–11206 (2000). [DOI] [PubMed] [Google Scholar]
- 269.Otsubo Y & Yamamato M TOR signaling in fission yeast. Crit. Rev. Biochem. Mol. Biol 43, 277–283 (2008). [DOI] [PubMed] [Google Scholar]
- 270.Wolfson RL & Sabatini DM The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab. 26, 301–309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Piyankarage SC, Augustin H, Grosjean Y, Featherstone DE & Shippy SA Hemolymph amino acid analysis of individual Drosophila larvae. Anal. Chem 80, 1201–1207 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Algret R et al. Molecular architecture and function of the SEA complex, a modulator of the TORC1 pathway. Mol. Cell Proteom 13, 2855 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Nixon RA The role of autophagy in neurodegenerative disease. Nat. Med 19, 983 (2013). [DOI] [PubMed] [Google Scholar]
- 274.Nixon RA et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol 64, 113–122 (2005). [DOI] [PubMed] [Google Scholar]
- 275.Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR & Lee VMY Lewy body-like α-Synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem 288, 15194–15210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Komatsu M et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 (2006). [DOI] [PubMed] [Google Scholar]
- 277.Hara T et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 (2006). [DOI] [PubMed] [Google Scholar]
- 278.Kaeberlein M & Galvan V Rapamycin and Alzheimer’s disease: time for a clinical trial? Sci. Transl. Med 11, eaar4289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Dehay B et al. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci 30, 12535 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Santini E, Heiman M, Greengard P, Valjent E & Fisone G Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal 2, ra36 (2009). [DOI] [PubMed] [Google Scholar]
- 281.Castellano BM et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann–Pick C1 signaling complex. Science 355, 1306–1311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]