


Abstract
Protein arginine methyltransferase 5 (PRMT5) catalyzes the symmetric di-methylation of arginine residues in histones H3 and H4, marks that are generally associated with transcriptional repression. However, we found that PRMT5 inhibition or depletion led to more genes being downregulated than upregulated, indicating that PRMT5 can also act as a transcriptional activator. Indeed, the global level of histone H3K27me3 increases in PRMT5 deficient cells. Although PRMT5 does not directly affect PRC2 enzymatic activity, methylation of histone H3 by PRMT5 abrogates its subsequent methylation by PRC2. Treating AML cells with an EZH2 inhibitor partially restored the expression of approximately 50% of the genes that are initially downregulated by PRMT5 inhibition, suggesting that the increased H3K27me3 could directly or indirectly contribute to the transcription repression of these genes. Indeed, ChIP-sequencing analysis confirmed an increase in the H3K27me3 level at the promoter region of a quarter of these genes in PRMT5-inhibited cells. Interestingly, the anti-proliferative effect of PRMT5 inhibition was also partially rescued by treatment with an EZH2 inhibitor in several leukemia cell lines. Thus, PRMT5-mediated crosstalk between histone marks contributes to its functional effects.
INTRODUCTION
Post-translational modifications of the N-terminal tails of histone proteins are involved in various chromatin-dependent processes, including transcriptional regulation, DNA damage repair and DNA replication. To regulate these cellular processes, histone modifications often act in combination, in a context-dependent manner, in what has been called a histone code (1). Indeed, histone modifications can promote, or antagonize, the deposition of other histone modifications. This crosstalk can occur on the same histone tail, often between adjacent or nearby histone residues, or on different histone tails (2). Well-characterized examples of these two types of crosstalk are the stimulation of GCN5-mediated histone H3K14 acetylation by H3S10 phosphorylation (3) and the influence of histone H2B monoubiquitination on H3K4 methylation (4,5).
Protein arginine methylation, catalyzed by a family of enzymes called Protein Arginine Methyltransferases (PRMTs), is attracting more and more attention, due to its involvement in many biological processes, including transcriptional regulation, RNA processing and signal transduction (6). The three types of PRMTs (Type I, Type II and Type III) catalyze asymmetric di-methylation, symmetric di-methylation and mono-methylation only, respectively, on arginine residues in histone and non-histone proteins. PRMT5 is the major type II enzyme in mammalian cells, catalyzing mono- and symmetric di-methylation on arginine residues in histones H2A and H4 at R3 and histone H3 at R2 and R8, as well as numerous non-histone proteins, including p53, BCL6 and Sm proteins (6–8). Together with its essential co-factor MEP50, PRMT5 critically regulates transcription, RNA splicing, cytokine signaling and DNA repair (9).
Methylation on histone arginine residues can promote the activation or repression of gene transcription. For example, PRMT5-mediated symmetric di-methylation on histone H4R3 and H3R8 is considered as repressive marks for gene expression (10); while the asymmetric di-methylation on H4R3 and H3R17, deposited by the type I enzymes PRMT1 and CARM1 (PRMT4), respectively, is often found on regulatory regions of active genes (10). A key issue is whether these marks are simply associated with the state of gene expression or exert an influence on the level of gene expression.
One way to address this issue for individual histone marks would be to identify crosstalk between a specific site of histone arginine methylation and other histone modifications. This has been demonstrated in several instances, with perhaps the best characterized being the antagonizing effect of H3R2me2a, catalyzed by the type 1 enzyme PRMT6, on tri-methylation of the nearby H3K4 residue, by MLL methyltransferases (11). Interestingly, the mono-methylation and symmetric di-methylation of H3R2 by PRMT5 seems to facilitate the deposition of H3K4me3 by MLL1 (12,13). Similarly, H3R8 can also be di-methylated symmetrically and asymmetrically; PRMT5-mediated H3R8me2s antagonizes the acetylation of H3K9 (14), while H3R8me2a blocks the binding of heterochromatin protein 1 (HP1) to methylated H3K9 (15). Trans-histone crosstalk, between H4 arginine methylation and H3 lysine methylation, has been demonstrated in neuronal cells, in which PRMT5-mediated H4R3me2s impairs the recruitment of MLL4, and thus decreases H3K4 tri-methylation (16).
In characterizing various effects of PRMT5 on gene expression, we found that the global level of H3K27 tri-methylation was markedly increased when PRMT5 was depleted or inhibited, in both normal and leukemic hematopoietic cells. We do not observe a direct impact of PRMT5 on the enzymatic activity of the PRC2 complex, but rather find that methylation of histone H3, at R2 and/or R8 by PRMT5, abrogates its subsequent methylation by PRC2 at K27. Given the contribution of H3K27me3 to gene silencing, we found that treating leukemia cells with an EZH2 inhibitor partially restored the expression of roughly half of the genes that were initially downregulated by PRMT5 inhibition, and one-quarter of these genes have increased H3K27me3 at promoter regions induced by PRMT5 inhibition, indicating that PRMT5 maintains the expression of a subset of genes by antagonizing PRC2-mediated transcriptional repression.
Growing evidence has suggested that PRMT5 is an oncogene, and a potential target in many types of human cancers, including leukemia and lymphoma (9,10). Interestingly, we found that the growth inhibitory effect of a PRMT5 inhibitor could be partially rescued by simultaneous exposure to an EZH2 inhibitor in several leukemia cell lines, suggesting that this crosstalk mechanism contributes to the cellular functions of PRMT5.
MATERIALS AND METHODS
Isolation of bone marrow and fetal liver cells from Prmt5 conditional KO mice
Prmt5 control and Prmt5 KO mice were described previously (17,18). Briefly, mice with 2 FloxP sites flanking exon 7 of the Prmt5 gene were crossed with either Mx1-cre Tg mice or Vav1-cre Tg mice (both strains were purchased from Jackson Laboratory). For deletion of Prmt5 in adult Bone Marrow (BM) cells, 2- to 3-month-old Prmt5fl/fland Mx1-Cre+;Prmt5fl/fl mice were given two consecutive intraperitoneal injections of poly (I:C) at 10 mg/Kg. BM cells were isolated from both tibias and femurs by flushing the cells out of the bone using syringes 7 days post first poly(I:C) injection. Hematopoietic Stem and Progenitor Cells (HSPCs) were purified by staining the cell surface lineage markers and c-kit, followed by FACS sorting of Lineage− and c-kit+ cells. PRMT5 control and deficient Lineage- Fetal Liver (FL) cells were isolated from E14.5 Prmt5fl/fland Vav1-Cre+;Prmt5fl/fl embryos, followed by staining of cell surface lineage markers and FACS sorting.
Leukemia cell lines, primary patient samples and drug treatment
Leukemia cell lines, HEL, Molm13, Nomo-1 and Monomac6 were maintained in RPMI1640 supplemented with 10% Fetal Bovine Serum; MV4–11 cells were maintained in IMDM with 10% Fetal Bovine Serum; and OCI-AML3 cells were cultured in MEM-alpha with 20% Fetal Bovine Serum.
The primary patient samples were obtained from Biospecimen Shared Resources at Sylvester Comprehensive Cancer Center, University of Miami Health System. Cells were cultured in IMDM containing 20% BIT, and supplemented with B-Mercaptoethanol (55 nM), Low Density Lipoproteins (40 µg/ml), human cytokines: IL6 (20 ng/ml), IL3 (20 ng/ml), G-CSF (20 ng/ml), GM-CSF (20 ng/ml), FLT3 (50 ng/ml) and SCF (50 ng/ml).
The PRMT5 specific inhibitor (GSK3186000A) was provided by GlaxoSmithKline (GSK) through a Research Collaboration and License Agreement between GSK and University of Miami. The EZH2 inhibitor (GSK343) was purchased from Sigma-Aldrich. Stock solutions were prepared in DMSO and stored at −20°C. Final DMSO concentrations were kept below 0.1%.
Western blot and histone extraction
Protein samples were separated by electrophoresis on denaturing 4–12% premade polyacrylamide gels (Invitrogen) and blotted to PVDF membranes (Millipore). Membranes were blocked in TBST buffer plus 5% milk. Major antibodies: H3K27me3 (Cell Signaling, 9733), H3K27me2 (Millipore 07–452) and H3K27me1 (Millipore, 07–448), H3R8me2s (Epigentek A-3706–050), PRMT5 (Millipore 07–405), EZH2 (Cell Signaling 5246), Symmetric Di-methyl Arginine (Cell Signaling 13222), UTX (Cell Signaling 33510), EED (Millipore, 09–774), RBBP4 (Bethyl Laboratories A301–206A-T) and SUZ12 (Cell Signaling 1335947).
Histones were purified from mouse BM cells using Histone Extraction Kit (Active Motif, 40028) as per manufacturer's instructions. Successful purification of histones was confirmed by Coomassie staining before they were used for western blotting.
In vitro methylation assay
The methylation assay was performed in methylation buffer containing 10 mM Hepes pH 7.5, 100 mM NaCl, 0.25 mM Ethylenediaminetetraacetic acid (EDTA) (8.0), 10 mM DTT, 2.5 mM MgCl2 and 5% glycerol supplemented with 1uCi 3H-SAM (Amersham) or 12.5 µM cold SAM, with a four hour incubation at 30°C. The reaction was stopped, by adding Sodium dodecyl sulphate (SDS) loading buffer, and the proteins were resolved on SDS-polyacrylamide gel electrophoresis gels. HA-PRMT5/MEP50 complex was purified from transfected 293T cells by anti- HA immunoprecipitation. The recombinant PRMT5/MEP50 complex was purchased from Reaction Biology Corporation (HMT-22–148), and the EZH2 complex (containing EZH2, EED and SUZ12) was purchased from Active Motif (31337). Recombinant H2A, H3 and H3.1/H4 tetramers were purchased from New England Biolabs. The recombinant and HeLa nucleosomes were obtained from Active motif (31466) and Reaction Biology Corporation (HMT-35–130), respectively.
The histone H3 peptides (residues 1–31), wt or with symmetric di-methylation on R2, R8 or both, were custom synthesized by Genemed Synthesis Inc. The recombinant nucleosomes with or without symmetric di-methylation on R2, R8 or both were custom synthesized by EpiCypher Inc. The concentration of these peptides was determined by Pierce Quantitative Colorimetric Peptide Assay (Thermo Fisher Scientific, 23275); and the concentration of nucleosomes was determined be Pierce BCA Protein Assay (Thermo Fisher Scientific, 23225).
The methylation assay was also performed with MTase-glo™ Methyltransferase Assay kit, purchased from Promega (V7601), according to the Manufacturer's instruction. Briefly, H3 peptides or nucleosomes were diluted to different concentrations, and incubated with 250 ng EZH2 complex, together with 20 µM SAM for 4 h at 30°C. The methyltransferase activity was determined by luminescence using a microplate reader (BioTek, Synergy 2).
RNA sequencing and data analysis
Molm13 cells were treated with DMSO, EZH2i (1 µM), PRMT5i (1 µM) and EZH2i (1 µM) plus PRMT5i (1 µM) for 4 days. Cells from four independent experiments were collected and total RNA was purified using an RNeasy Plus Mini Kit (Qiagen). Library preparation and RNA-sequencing were completed at Oncogenomics Core Facility at the Sylvester Comprehensive Cancer Center. Samples were sequenced using 75 bp paired ends with an Illumina NextSeq 500 and subsequent sequencing reads (∼40 to 50 million per sample) were trimmed and filtered using Cutadapt. Fastq files were aligned to Ensembl 87: GRCh38.p7 human transcriptome using STAR aligner [v2.5.3a] and RSEM [v1.3.0] to obtain expected gene counts. GC nomination between sequencing runs was performed using EDAseq v.2.12.0 within lane GC normalization. Differential expression was determined using DESeq2 [v1.30.0] and R [v3.4.1] with a Benjamini-hochberg FDR cutoff of 0.05. Heatmaps were generated using Euclidean distances between sample blind, variance stabilized transformed counts from DESeq2.
For splicing analysis, rMATS v3.1.0 (-t paired —libType fr-firstrand) and STAR v2.6.0c were used to align and compare differential splicing events. Fastq files were aligned to hg38 and Gencode V28 was used for splicing annotations. Differential splicing events were filtered for significance q < 0.05 and inclusion level difference of at least 20%.
ChIP-sequencing and data analysis
Molm13 cells were treated with DMSO, EZH2i (1 µM) and PRMT5i (1 µM) for 4 days. Cells from two independent experiments were collected and subjected to ChIP-sequencing using antibody specific for H3K27me3 (Cell Signaling, 9733). Briefly, cells were fixed in 1% formaldehyde for 5 min at RT, and then lyzed with sonication buffer (16.7 mM Tris pH 8, 167 mM NaCl, 0.1% SDS, 1% Triton X-100, 1 mM EDTA) supplemented with proteasome inhibitor cocktail (Millipore, 535140). Chromatin was sonicated with Bioruptor Pico (Diagenode) for 14 cycles with 30 s on and off. Immunoprecipitation was performed with 4 µg of cell lysate and 2 µg of antibody against H3K27me3 overnight at 4°C, and the antibody-chromatin complex was then pulled down with 20 µl of magnetic Protein A beads (NEB S1425s) 20 ng of drosophila chromatin (Active Motif, 53083) was included for normalization. The beads were washed twice with each of the following buffers: high salt buffer (20 mM Tris pH 8, 500 mM NaCl, 0.1% SDS, 1% Triton X-100 and 2 mM EDTA); Li buffer (10 mM Tris pH 8, 0.25 M LiCl, 1% NP40, 1% sodium deoxycholate and 1 mM EDTA,); TE buffer (pH 8). The beads were then suspended in 30ul buffer (100 mM Tris pH 8, 250 mM NaCl, 1 mM EDTA and 0.1% SDS,) with 1 µl of Proteinase K (Invitrogen, AM2546) and incubated at 65°C for 4 h. The DNA in the supernatant was purified with Agencourt AMPure beads (Beckman Coulter A63880). The purified DNA was used for DNA library construction (NEB, E7645) and sequenced on Illumina Nextseq 500 platform.
To generate signal files normalized by drosophila spike-in, a bowtie2 index was generated from a combined GRCh38 (ENCODE) and BDGP6 (Ensembl) genome fasta files (bowtie2 = 2.3.3.1). Sequencing reads were then trimmed (cutadapt = 1.15) and aligned to the merged genome. Average counts per million reads (CPM) were calculated genome-wide in 100bp bins across all samples. An interpolated non-parametric local regression (LOWESS, python = 3.7, statsmodels = 0.10.1) was used to model differences between samples based on the binned signal values of spike-in chromatin. This model was then used to adjust the hg38 signal. Scripts can be found at https://github.com/diderote/lowSpike. Heatmaps were generated using deeptools (3.3.1).
Cell viability, cell cycle and synergy analysis
For drug synergy analysis, cells were seeded in 24-well plates at 80,000 cells/ml and treated for 4 or 6 days with different doses of drugs, alone or in combination. Cell viability was assessed by a CellTiter-Glo Luminescent Cell Viability Assay (Promega G7570), according to the manufacturer's protocol. Bliss synergy was calculated using the bioconductor package synergyfinder v1.0.0 using default parameters for calculating bliss independence (19).
For cell-cycle analysis, cells were treated with the indicated inhibitors for 4 days, cells were then fixed in 70% ethanol overnight at −20°C and stained in PI staining buffer containing 50 µg/ml PI, 100 µg/ml RNase A, 0.1% Triton X-100 and phosphate-buffered saline for 30 min at RT. Stained cells were then subject to FACS analysis by BD FACSCanto II.
RESULTS
PRMT5 depletion or inhibition upregulates global H3K27 tri-methylation in hematopoietic cells
To determine whether PRMT5-mediated arginine methylation exhibits cross-talk with histone lysine methylation, we extracted core histones from bone marrow (BM) cells of control and PRMT5 conditional knockout mice, and assessed the level of histone H3 K4, K9, K27 and K36 methylation and H4 K20 methylation by Western blotting (Figure 1A). We found that H3K27 di- and tri-methylation are significantly upregulated in PRMT5-null BM cells, while H3K27 mono-methylation and methylation of other lysine residues (H3K4, K9 and K36, and H4K20), are largely unaffected. We confirmed the upregulation of H3K27me3 in adult hematopoietic stem and progenitor cells (HSPCs) (Figure 1B) and lineage negative fetal liver (FL) cells (Figure 1C) isolated from PRMT5 control and depleted mice. Consistent with the increase in H3K27me3 in the PRMT5-null BM and FL cells, we also found marked upregulation of H3K27me3 in PRMT5 knockdown MV4–11 cells (Figure 1D), and in PRMT5 inhibitor-treated human leukemia cell lines, HEL (Figure 1E), MV4–11 and Molm13 (Figure 1F). Treating these cells with an EZH2 specific inhibitor completely abrogated H3K27 tri-methylation (Figure 1E and F), indicating that the EZH2-containing PRC2 complex is the major enzymatic complex catalyzing this modification in these leukemia cells.
Figure 1.
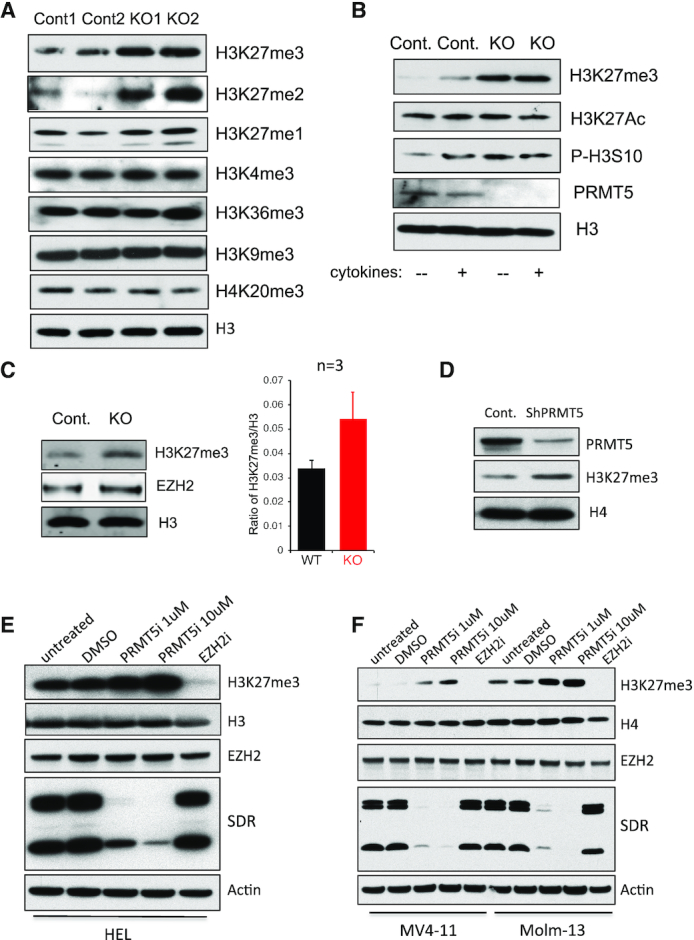
PRMT5 deletion or inhibition upregulates the global level of H3K27 tri-methylation in normal and malignant hematopoietic cells. (A) Histones were purified using Histone Extraction Kit from BM cells isolated from two control and two Mx1-Cre+ Prmt5 conditional KO mice 7 days post poly (I:C) injection. Mono-, di and tri-methylation on H3K27, as well as tri-methylation on H3K4, H3K9, H3K36 and H4K20 were determined by western blot. (B) Lineage- and c-kit+ HSPC cells were isolated from BM of day 7 control and Prmt5 KO mice. Cells were stimulated with or without cytokine mixture of murine SCF (100 ng/ml), IL-3 (20ng/ml), IL-6 (20ng/ml) and FLT3 ligand (100 ng/ml) for 10 min at 37°C. The level of PRMT5, H3K27me3, H3K27ac, H3S10ph and total H3 were determined by western blot with whole cell lysate. (C) Lineage- FL cells isolated from E14.5 Prmt5 control and Vav1-Cre+ KO embryos were subject to Western blot and an Odyssey Image System to determine the level of histone H3, H3K27me3 and EZH2 protein. A representative Western blot is shown at left, and a bar graph showing the increase in H3K27me3/H3 ratio at right (n = 3). (D) MV411 cells were transduced with lentiviruses expressing a scramble shRNA or shRNAs against PRMT5. Level of PRMT5 and H3K27me3 was shown with western blot, while H4 was used as a loading control. Leukemia cell lines HEL (E), MV411 and Molm13 (F) were treated with DMSO, PRMT5 Inhibitor 1 or 10 µM or EZH2 Inhibitor 1 µM for 4 days. Level of H3K27me3, EZH2 and PRMT5 was determined by western blot; while H3, H4 or β-actin was used as loading controls. The efficiency of PRMT5 Inhibitor was confirmed by the reduced level of cellular symmetric di-methylated arginine (SDR).
PRMT5 does not directly affect PRC2 enzymatic activity, but PRMT5-mediated H3 arginine methylation antagonizes H3K27 methylation by PRC2
Three core components of the PRC2 complex, EZH1/2, EED and SUZ12, are required for its enzymatic activity toward histone H3 (20). To explore whether these proteins can be directly methylated by PRMT5/MEP50, we utilized an invitro methylation assay and either recombinant PRMT5/MEP50, or PRMT5/MEP50 purified from transfected 293T cells. PRMT5 was unable to methylate EZH2, EED or SUZ12, even though it readily methylated recombinant histone H2A (Supplementary Figure S1A and B). To examine other mechanisms by which PRMT5 could control H3K27 di- and tri-methylation, we examined PRMT5 inhibitor-treated leukemia cells, and found no change in the protein expression level of any of the three PRC2 core components (Supplementary Figure S1C), nor did we find that PRMT5 activity level affects PRC2 complex formation, using immunoprecipitation and PRMT5 inhibitor-treated MV4–11 cells (Supplementary Figure S1D). We also determined that the level of PRC2 co-factors RBBP4 and JARID2, or the H3K27me3 demethylase UTX, did not change in PRMT5 inhibited cells (Supplementary Figure S1E).
To further explore whether PRMT5 activity affects PRC2 enzymatic activity, we purified the PRC2 complex from DMSO or PRMT5 inhibitor-treated MV4–11 cells using an antibody specific for EZH2, and determined its methyltransferase activity using an in vitro methylation assay. We found no change in the ability of PRC2 to methylate H3 in vitro, despite changes in the level of PRMT5 activity in the MV4–11 cells from which PRC2 was purified (Supplementary Figure S1F).
The PRMT5/MEP50 complex has been shown to methylate recombinant free histones H2A and H4 at R3, and H3 at R2 and R8 in vitro; however, how this methyltransferase complex modifies nucleosomal histones is still controversial. Surprisingly, we found that PRMT5/MEP50 primarily methylated H3 in mono- and oligo-nucleosomes isolated from HeLa cells in an in vitro methylation assay (Figure 2A). In contrast, the PRMT5/MEP50 failed to methylate H3 within recombinant nucleosomes, although H2A and H4 were weekly methylated, implicating that other histone modifications may be required before nucleosomal H3 can be methylated by PRMT5/MEP50. Supporting its effect on histone H3, we did observe a significant decrease in H3R8me2s levels after PRMT5 was knocked down by shRNAs (Figure 2B) or inhibited by chemical inhibitor (Figure 2C), in leukemia cells.
Figure 2.
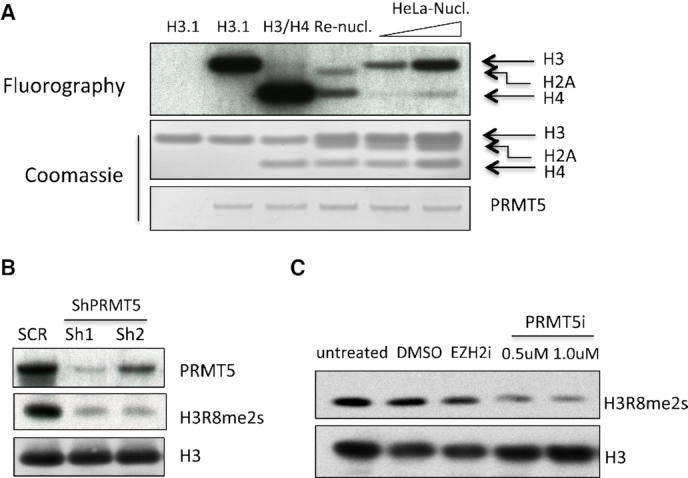
PRMT5 primarily methylates nucleosomal histone H3. (A). In vitro methylation assay was performed using recombinant PRMT5/MEP50 complex as enzyme, and recombinant H3.1, H3.1/H4 tetramer, mononucleosome or increased amount of mononucleosome purified from HeLa cells as substrates. The loading of histones and PRMT5 protein are shown by Coomassie staining. (B). MV4–11 cells were transduced by lentiviruses expressing control shRNAs or two different shRNAs against PRMT5. Levels of PRMT5 and H3R8 symmetric di-methylation was determined by western, H3 served as a loading control. (C). Molm13 cells were either untreated or treated with DMSO, PRMT5 inhibitor at 0.5 and 1 µM for 4 days. The level of H3R8me2s was determined by western blot analysis.
We next determined how PRMT5-mediated H3 arginine methylation could impact histone lysine methylation by EZH2 complex using in vitro methylation assays. As shown in Figure 3A and Figure S2A, pre-incubation of all three H3 variants (H3.1, H3.2 and H3.3) with PRMT5/MEP50 dramatically decreased their subsequent methylation by the EZH2 complex. Interestingly, auto-methylation of EZH2 was not affected by the presence of PRMT5/MEP50, suggesting that EZH2 enzymatic activity was intact (Supplementary Figure S2B). To exclude the possibility that the presence of PRMT5/MEP50 in the reaction could interfere with EZH2 enzymatic activity, we utilized biotinylated recombinant H3 as substrate, purified H3 by streptavidin beads after its methylation by PRMT5/MEP50 and then performed an EZH2 in vitro methylation assay using both unmodified H3 and pre-methylated H3 as substrates (Figure 3B). We observed a similar decrease in H3K27 methylation as shown in Figure 3A.
Figure 3.
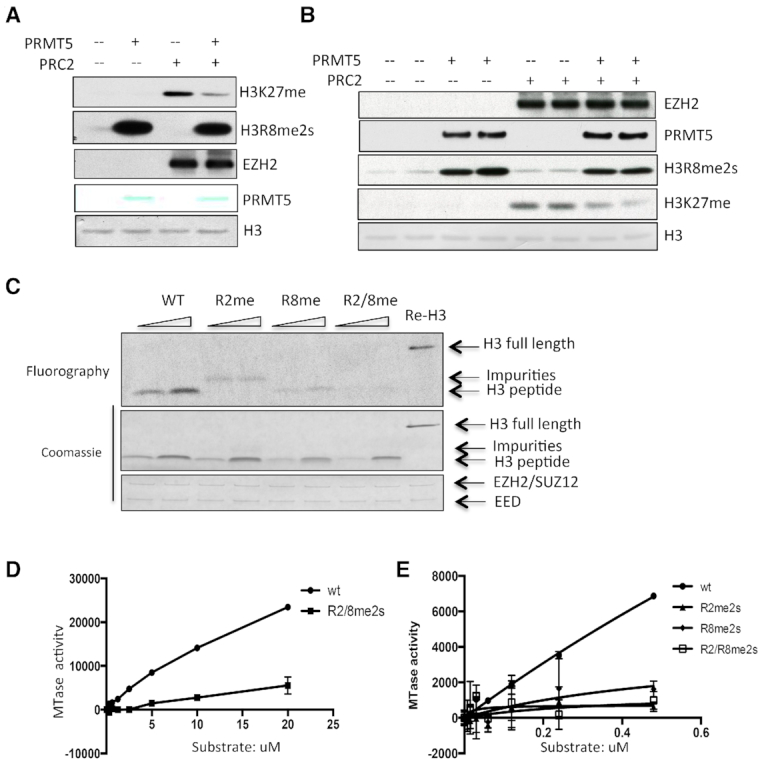
PRMT5-mediated H3R2/R8me2s impairs the deposition of methylation at H3K27 by PRC2. (A) Recombinant H3.1 was first methylated by PRMT5/MEP50 complex for 2 h at 30°C, and then, recombinant EZH2 complex was added to the reaction for another 2 h of incubation. EZH2 and methylation of H3K27 and H3R8 was determined by western with antibodies specific for EZH2, H3K27me1 and H3R8me2s, while loading of H3 and PRMT5 was detected by staining the membrane directly with membrane staining dye. (B) Sequential in vitro methylation assay was performed using biotinylated H3.1 as substrate. H3.1 was first methylated by recombinant PRMT5/MEP50 complex, and then the methylated H3 was purified by streptavidin beads, and incubated with recombinant EZH2 complex in the presence of 12.5 µM SAM. Methylation of H3K27 and H3R8, as well as loading of EZH2, PRMT5 and H3 was determined by western blot. (C) H3 peptides (amino acid 1–31) with or without symmetric di-methylation on R2, R8 or both were subjected to in vitro methylation assay with recombinant EZH2 complex, and the methylation of these peptides was determined by fluorography. (D) Different concentrations of WT H3 peptide and the peptide with symmetrically di-methylation on R2 and R8 were subject to in vitro methylation assay with constant concentration of recombinant EZH2 complex, and the methyltransferase activity was determined using MTase-Glo Methyltransferase Assay kit. (E) Different concentrations of wt mononucleosome and mononucleosomes with symmetric di-methylation on R2, R8 or both were subject to in vitro methylation assay with 250 ng EZH2 complex, and the enzyme activity was determined with the MTase-Glo Methyltransferase Assay kit.
To further determine the antagonizing effect between H3 R2 and R8 methylation and H3K27 methylation, we synthesized unmodified H3.1 peptide (from aa 1 to 31) and the same peptides that were symmetrically di-methylated on H3 R2, H3 R8 or on both. The correct post-translational modifications of these peptides were first confirmed by a dot blot assay, using antibodies specific for H3R2me2s and H3R8me2s (Supplementary Figure S2C); these peptides were then subjected to in vitro methylation assays. As shown in Figure 3C, methylation on R2 or R8, but especially on both residues, markedly impaired the methylation of these peptides by EZH2. We confirmed the impaired ability of EZH2 to methylate the modified peptides in a more quantitative manner, using the MTase-glo Methyltransferase Assay (Figure 3D). We next determined how methylation on these two arginine residues could affect the ability of the EZH2 complex to methylate histone H3 when packed in nucleosomes; we observed the same inhibitory effect on lysine methylation when we performed this assay using the modified nucleosomes (Figure 3E). At last, we determined whether arginine methylation affects the interaction between H3 and PRC2 using an in vitro binding assay, and recombinant un-modified H3 protein or H3 protein pre-methylated by PRMT5/MEP50 complex; we found no impact of arginine methylation of histone H3 on the affinity of the PRC2 complex for histone H3 (Supplementary Figure S2D). Taken together, the data show that arginine methylation of histone H3 at R2 and R8 by PRMT5 blocks its lysine methylation by the EZH2 complex at K27.
PRMT5-mediated histone arginine methylation maintains gene expression via blocking H3K27 tri-methylation by PRC2
PRMT5-mediated H4R3 and H3R8 methylation has been reported to generate repressive marks for transcription, likely through the recruitment of transcriptional co-repressors, including SIN3A, HDACs and MBDs (21). However, we have found that PRMT5 deletion, or inhibition, in hematopoietic cells leads to more than 50% of the altered genes being downregulated (17). To determine whether PRMT5 maintains gene expression via antagonizing PRC2-mediated H3K27 tri-methylation, we treated Molm13 cells with an EZH2 inhibitor, a PRMT5 inhibitor or a combination of both inhibitors and performed RNA-sequencing analysis. While EZH2 inhibition had a very mild effect on gene expression, affecting fewer than 100 genes that were either up- regulated (65%) or downregulated (35%) at least 2-fold, PRMT5 inhibition resulted in a 2-fold-change in the expression of ∼1300 genes, with roughly half being downregulated and half upregulated (Figure 4A). Pathway analysis of PRMT5 target genes reveals their involvement in cell cycle, DNA repair and signaling pathways (Figure 4B). In contrast to what we reported for normal HSPCs, we did not observe upregulation of a p53 gene signature, even though p53 is wild-type in these cells (17). The top 20 differentially expressed genes in each treatment group, compared to DMSO control group, are listed in Supplemental Tables S1–3.
Figure 4.

PRMT5 maintains gene expression via blocking EZH2-mediated H3K27 methylation. (A) Molm13 cells were treated with DMSO, EZH2 inhibitor (1 µM) or PRMT5 inhibitor (1 µM) or combination of both inhibitors at 1 µM for 4 days. Heat maps show the differentially expressed coding genes at 2-fold cut-off. (B) Pathway analysis of PRMT5 inhibitor-induced up- and downregulated genes. (C) Venn diagrams show the overlaps of 2-fold downregulated genes between: left: PRMT5i-treated and PRMT5i/EZH2i combined treatment groups; Middle: EZH2i-treated and PRMT5i-treated groups. Right: overlap of 2-fold upregulated genes between PRMT5i-treated and PRMT5i/EZH2i combined treatment groups. (D) Two representative RNA-sequencing tracks showing that the PRMT5i upregulated gene is not affected by combined treatment of EZH2i; while the PRMT5i downregulated gene is partially restored by EZH2i. (E) Heat map shows the differentially expressed genes between PRMT5i-treated and PRMT5i and EZH2i combined treatment groups.
Consistent with crosstalk between PRMT5 and EZH2/PRC2 function, we found that treating cells with an EZH2 inhibitor restored or partially restored the expression of 53% (335 genes) of the PRMT5i-induced 2-fold downregulated genes, while 42% of PRMT5i-induced upregulated genes were no longer 2-fold upregulated in the presence of the EZH2 inhibitor (Figure 4C). Figure 4D shows an example of restored downregulated genes (HIST1H1B), and an unchanged upregulated gene (STC3) in the combined treatment group, compared to the PRMT5 inhibitor-treated group. Not surprisingly, when the PRMT5 inhibitor-induced gene expression profile was directly compared to the combined treatment cells, we found that the majority of the differentially expressed genes (about 70%) between these two groups were upregulated genes (Figure 4E). We confirmed our RNA-seq results for several genes, using Real-time PCR; three representative PCR results are shown in Figure S3A.
To examine the upregulation of H3K27me3 in PRMT5 inhibited cells in a more site-specific manner, we performed ChIP-sequencing analysis in DMSO-, EZH2i- and PRMT5i-treated Molm13 cells. Consistent with our western blot results, PRMT5 inhibition led to a global increase in H3K27me3, and also a significant increase in H3K27me3 at the TSS region of genes (Supplementary Figure S3B and C). The H3K27me3 mark occupied 1/3 of all TSS regions in this AML cell line, and majority of these tri-methylation peaks were abrogated by the EZH2 inhibitor (Supplementary Figure S3C); however, treating cells with the EZH2 inhibitor resulted in only moderate changes in the gene expression profile, suggesting that loss of H3K27me3 at TSS does not result in changes in gene expression in most cases. Most importantly, we found increased H3K27me3 level at the promoter regions of a quarter of the genes (88 out of the 335 genes) that were initially downregulated by PRMT5i and restored or partially restored by EZH2i (Figure 5A). Two representative ChIP-seq tracks in Figure 5B show that PRMT5 inhibition leads to increase in the height of H3K27me3 peak at the TSS region of IGFBP2 (as well as IGFBP5, which is not expressed in this cell line), and an increase in the width of the H3K27me3 peak at the TSS of PTPN13. The changes in this histone mark are reflected by changes in the expression of these two genes assessed by qPCR (Figure 5B).
Figure 5.

PRMT5 inhibition upregulates H3K27me3 level at the promoter regions of a subset of PRMT5 target genes. (A) Heat map shows the H3K27me3 peaks at the TSS region of the 335 genes that were initially downregulated by PRMT5i and rescued or partially rescued by EZH2i in treated Molm13 cells. (B) Left: representative ChIP-sequencing tracks shows H3K27me3 peaks at the TSS region of IGFBP2/5 and PTPN13. Right: bar graph shows the expression level of IGFBP2 and PTPN13 determined by qPCR.
PRMT5 plays a key role as a regulator of RNA splicing, so we examined alternative splicing in the PRMT5 inhibited cells, using DMSO treated cells as control. We found ∼6000 aberrant splicing events, affecting ∼1500 genes. The majority (70%) of these events is exon skipping (Supplementary Figure S4A). There is modest overlap between the differentially expressed genes and the genes whose splicing is altered (Supplementary Figure S4B).
PRMT5-mediated repression of H3K27 tri-methylation contributes to its cell-cycle effects
PRMT5 is highly expressed in many human cancers, and it appears to be indispensable for cell proliferation and survival (9,10). Treating a variety of human leukemia cells with a PRMT5 inhibitor impairs their proliferation (18), so we hypothesized that the PRMT5-mediated repression in H3K27m3 contributes to its growth inhibitory effects. We treated several AML cell lines (MV411, Molm13, Nomo-1, HEL, MonoMac6 and OCI-AML3) and two primary AML patient cells (the information of these two patients is listed in Supplementary Table S4) with the PRMT5 inhibitor alone, the EZH2 inhibitor alone, or the combination of the two inhibitors, and determined cell viability using the CellTiter-Glo Luminescent Cell Viability Assay, after 4 or 6 days of treatment. The synergistic or antagonistic effect of these two inhibitors was analyzed using the Bliss Model (22). As shown in Figure 6A and Supplementary Figure S5, in four of the six leukemia cell lines tested, and one of the two primary AML cells, that proliferated significantly in vitro, the EZH2 inhibitor provided partial rescue of the PRMT5 inhibitor-induced impaired proliferation. The strongest antagonizing effect was seen in the Molm13 and Nomo-1 cells, while an additive effect was seen in the MV4–11 and MonoMac6 cells (Figure 6A and Supplementary Figure S5). The antagonistic effect of these two inhibitors on cell proliferation was also observed by counting the number of alive cells at the end of 4- or 6-day treatment (Figure 6B). Clearly, PRMT5-mediated crosstalk between histone arginine and lysine methylation contributes only partially to the changes observed in cell proliferation rates. We next analyzed cell cycle in Molm13 cells, using PI staining and FACS cell cycle analysis (Figure 6C and D), and found that the EZH2 inhibitor partially rescued the decreased percentage of cycling cells (cells in the S/G2/M phases of the cell cycle) that occurs secondary to PRMT5 inhibition, even though the EZH2 inhibitor alone had no effect on cell-cycle progression.
Figure 6.

PRMT5-mediated repression of H3K27 tri-methylation contributes to its cell-cycle effects. (A) Several leukemia cell lines and 2 AML patient samples were treated with different concentration of PRMT5i or EZH2i alone, or different combination of these two inhibitors for 6 days. Cell viability was determined by Celltiter-Glo assay. The synergy of these two inhibitors was determined by Bliss model using the Synergyfinder software. Negative synergy scores represent overall antagonizing effect of these two inhibitors; while positive scores mean synergistic effect. (B) Molm13 cells were treated with DMSO, EZH2i, PRMT5i and the combination of these two inhibitors, cells were counted at day 4 and day 6. n = 4. (C) Molm13 cells were treated with DMSO, PRMT5/EZH2 inhibitors for 4 days. Cells were fixed in ethanol and subjected to PI staining for cell-cycle analysis. Percentage of cells in S and G2/M phase was plotted here. n = 4. (D). A representative FACS plot of cell cycle is shown here. Ei: EZH2 inhibitor; P5i: PRMT5 inhibitor; Numbers on the X-axis indicate the concentration of PRMT5 inhibitor in uM; and EZH2 inhibitor was used at 1 µM in Figure 6B–D.
DISCUSSION
We have identified an important crosstalk between PRMT5-mediated arginine methylation and PRC2-mediated lysine methylation at the N-terminal tail of histone H3 that helps explain the ability of PRMT5 to positively regulate gene expression. PRMT5 inhibition or depletion leads to a global increase, as well as an increase at the promoter region of a subset of PRMT5-target genes, in H3K27 tri-methylation. The extensive biochemical and genetic analysis we performed demonstrates that the symmetric di-methylation of histone H3 on R2 and R8 antagonizes the ability of the PRC2 complex to accomplish H3K27 tri-methylation. This crosstalk mechanism exerts a substantial effect on PRMT5-mediated regulation on gene expression and cell proliferation (Figure 7).
Figure 7.

PRMT5-mediated histone arginine methylation antagonizes the transcriptional repression by PRC2. A cartoon showing how PRMT5-mediated histone arginine methylation could antagonize the transcriptional repression by PRC2.
To elucidate the mechanism of how PRMT5 deficiency upregulates H3K27me3, we found no evidence that PRMT5 disrupted the PRC2 complex or directly methylated any of its components. Furthermore, PRC2 methyltransferase activity is normal in PRMT5-deficient cells, and EZH2 auto-methylation occurs normally in the presence of active PRMT5. Thus, it does not seem that PRMT5 alters PRC2 activity directly to regulate H3K27me3 levels in the cell. Rather, it appears that histone H3 R2 and R8 methylation can impair the ability of PRC2 to mediate H3K27 tri-methylation. Recombinant histone H3 that has been pre-methylated by PRMT5/MEP50, cannot be fully methylated by PRC2, and the symmetric di-methylation of R2 and/or R8 in an H3 peptide or protein that is packaged in nucleosomes, impairs K27 methylation by PRC2, suggesting that H3 arginine methylation directly impairs H3 methylation on K27. We did not observe disruption of the interaction between histone H3 and the PRC2 complex in vitro, which suggests that the effect is likely mediated by a direct interference with the deposition of the methyl mark on K27. Histone modifications, such as H3K27me3 and H3K9me3, have been shown to have allosteric effect on EZH2 methyltransferase activity (23), which could explain how H3R2/R8me2s affects PRC2 activity. Further investigation on how PRMT5-mediated histone arginine methylation regulates the activity of PRC2 on chromatin will shed additional light on mechanisms of gene repression.
PRMT5 has been shown to methylate histones H2A and H4 at R3 and H3 at R2 and R8. Although, active PRMT5/MEP50 complex can readily methylate free H2A and H4 (24), it primarily methylates H3 when histones are packed in nucleosomes purified from HeLa cells in vitro (Figure 2A). The inability of PRMT5/MEP50 to methylate H3 in the recombinant nucleosome, suggests that other histone modification(s) may be required for this methyltransferase to methylate nucleosomal H3. The PRMT5/MEP50 core complex has been reported to interact with other co-factors, including pICLn, COPR5 and RioK1 (25–27), which function to determine the substrate specificity of PRMT5; thus, it is possible that the methylation of nucleosomal H2A or H4 requires other important co-factors, in addition to MEP50. Nonetheless, histone H2A has been shown to be methylated in the cytoplasm of mouse ES cells (28), and we found that PRMT5 prefers H4, rather than H3, in the H3/H4 tetramer (Figure 2A). Therefore, it is also possible that H2A and H4 are methylated on R3 before their deposition onto the chromatin. In addition, PRMT5 shuttles between cytoplasmic and nuclear compartments, and the subcellular localization of PRMT5 is cell type specific (29–31); thus, the function of PRMT5-mediated histone arginine methylation could be cell context-dependent. It is likely that H3 arginine methylation increases and H3K27me3 decreases upon PRMT5 translocation into the nucleus. However, it is still unclear how upstream signaling regulates PRMT5 shuttling between these two cell compartments.
PRMT5 and its effects on histone methylation are generally considered to be components of the gene repression machinery. Indeed, PRMT5 is found in several repressive complexes, including SIN3A, HDAC and NuRD complexes (21). However, several recent studies have highlighted the role of PRMT5 in both up- and downregulation of gene expression (12–13,32–33). We have identified another mechanism by which PRMT5 can maintain gene activation, via blocking the deposition of H3K27me3. We demonstrated that a subset of PRMT5-target genes is regulated through this crosstalk mechanism, using gene expression profiling. Our genome mapping of H3K27me3 reveals that PRMT5 inhibition leads to a global increase in H3K27me3 level, as well as increased H3K27me3 at the promoter regions of a fraction of these PRMT5 target genes.
It remains elusive why the PRMT5i and EZH2i have antagonizing effects on the proliferation of some AML cell lines, and an additive effect on others (Figure 6A). We explored whether PRMT5 protein levels or subcellular localization could determine the combinational effect of these inhibitors, but failed to see any correlation. Certainly, many other factors may influence the response, such as the cell-specific components of the PRC2 complex, the genomic distribution of H3K27me3, or the presence or absence of non-histone substrates that are methylated on arginine or lysine by PRMT5 or EZH2. Further investigation is clearly needed to delineate the basis for the variable effect seen.
As PRMT5 is often overexpressed in tumor cells, and PRMT5 inhibitors are currently in phase I clinical trials for treating cancer patients, this crosstalk mechanism may have important therapeutic implications. Furthermore, EZH2 deletion or loss-of-function mutations are seen in certain cancer types, including myeloid leukemia, T-ALL and malignant peripheral nerve sheath tumors, etc., and it will be important to determine how EZH2 genetic loss or mutation affects the tumor cell response to PRMT5 inhibitors. We attempted to explore this issue by depleting EZH2 using shRNAs in Molm13 and Nomo-1 cells, however, neither cell could proliferate or survive with a reduced level of EZH2 protein, even though a biologically effective concentration of EZH2 inhibitor had little effect on the proliferation of either cell line. These data suggest that EZH2 may have methyltransferase-independent functions, which are important for the survival and proliferation of AML cells. Indeed, similar observations have been reported by several other groups (34,35). While MTAP deficiency may predict for a better response to PRMT5 and PRMT1 joint inhibition (36), we do not yet know which mutations will predict for a clinically relevant response to combination treatment that targets PRMT5 and H3K27 methylation. Further investigation of combination epigenetic-targeted therapies are warranted in AML and other cancers as well.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Oncogenomics Shared Resource at Sylvester Comprehensive Cancer Center for RNA-sequencing services and the Biostatistics and Bioinformatics Shared Resource for data analysis. We also thank members of the Nimer lab and Lossos lab for reading this manuscript and providing thoughtful suggestions and comments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Cancer Institute (NCI) [5R01CA166835to SDN, 1P30CA240139to the Sylvester Comprehensive Cancer Center]. Funding for open access charge: NCI [RO1 CA166835-01 to S.N.].
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Strahl B.D., Allis C.D.. The language of covalent histone modifications. Nature. 2000; 403:41–45. [DOI] [PubMed] [Google Scholar]
- 2. Suganuma T., Workman J.L.. Crosstalk among histone modifications. Cell. 2008; 135:604–607. [DOI] [PubMed] [Google Scholar]
- 3. Walter W., Clynes D., Tang Y., Marmorstein R., Mellor J., Berger S.L.. 14-3-3 interaction with histone H3 involves a dual modification pattern of phosphoacetylation. Mol. Cell Biol. 2008; 28:2840–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dover J., Schneider J., Tawiah-Boateng M.A., Wood A., Dean K., Johnston M., Shilatifard A.. Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J. Biol. Chem. 2002; 277:28368–28371. [DOI] [PubMed] [Google Scholar]
- 5. Nakanishi S., Lee J.S., Gardner K.E., Gardner J.M., Takahashi Y.H., Chandrasekharan M.B., Sun Z.W., Osley M.A., Strahl B.D., Jaspersen S.L. et al.. Histone H2BK123 monoubiquitination is the critical determinant for H3K4 and H3K79 trimethylation by COMPASS and Dot1. J. Cell Biol. 2009; 186:371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blanc R.S., Richard S.. Arginine Methylation: The Coming of Age. Mol. Cell. 2017; 65:8–24. [DOI] [PubMed] [Google Scholar]
- 7. Lu X., Fernando T.M., Lossos C., Yusufova N., Liu F., Fontan L., Durant M., Geng H., Melnick J., Luo Y. et al.. PRMT5 interacts with the BCL6 oncoprotein and is required for germinal center formation and lymphoma cell survival. Blood. 2018; 132:2026–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Antonysamy S. The structure and function of the PRMT5:MEP50 complex. Subcell. Biochem. 2017; 83:185–194. [DOI] [PubMed] [Google Scholar]
- 9. Stopa N., Krebs J.E., Shechter D.. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol. Life Sci. 2015; 72:2041–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Greenblatt S.M., Liu F., Nimer S.D.. Arginine methyltransferases in normal and malignant hematopoiesis. Exp. Hematol. 2016; 44:435–441. [DOI] [PubMed] [Google Scholar]
- 11. Guccione E., Bassi C., Casadio F., Martinato F., Cesaroni M., Schuchlautz H., Luscher B., Amati B.. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007; 449:933–937. [DOI] [PubMed] [Google Scholar]
- 12. Migliori V., Muller J., Phalke S., Low D., Bezzi M., Mok W.C., Sahu S.K., Gunaratne J., Capasso P., Bassi C. et al.. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol. 2012; 19:136–144. [DOI] [PubMed] [Google Scholar]
- 13. Chen H., Lorton B., Gupta V., Shechter D.. A TGFbeta-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene. 2017; 36:373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pal S., Vishwanath S.N., Erdjument-Bromage H., Tempst P., Sif S.. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell Biol. 2004; 24:9630–9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rothbart S.B., Krajewski K., Nady N., Tempel W., Xue S., Badeaux A.I., Barsyte-Lovejoy D., Martinez J.Y., Bedford M.T., Fuchs S.M. et al.. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 2012; 19:1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dhar S.S., Lee S.H., Kan P.Y., Voigt P., Ma L., Shi X., Reinberg D., Lee M.G.. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012; 26:2749–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu F., Cheng G., Hamard P.J., Greenblatt S., Wang L., Man N., Perna F., Xu H., Tadi M., Luciani L. et al.. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J. Clin. Invest. 2015; 125:3532–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hamard P.J., Santiago G.E., Liu F., Karl D.L., Martinez C., Man N., Mookhtiar A.K., Duffort S., Greenblatt S., Verdun R.E. et al.. PRMT5 regulates DNA repair by controlling the alternative splicing of Histone-Modifying enzymes. Cell Rep. 2018; 24:2643–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He L., Kulesskiy E., Saarela J., Turunen L., Wennerberg K., Aittokallio T., Tang J.. Methods for High-throughput drug combination screening and synergy scoring. Methods Mol. Biol. 2018; 1711:351–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Margueron R., Reinberg D.. The polycomb complex PRC2 and its mark in life. Nature. 2011; 469:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karkhanis V., Hu Y.J., Baiocchi R.A., Imbalzano A.N., Sif S.. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 2011; 36:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ianevski A., He L., Aittokallio T., Tang J.. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics. 2017; 33:2413–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moritz L.E., Trievel R.C.. Structure, mechanism, and regulation of polycomb-repressive complex 2. J. Biol. Chem. 2018; 293:13805–13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burgos E.S., Wilczek C., Onikubo T., Bonanno J.B., Jansong J., Reimer U., Shechter D.. Histone H2A and H4 N-terminal tails are positioned by the MEP50 WD repeat protein for efficient methylation by the PRMT5 arginine methyltransferase. J. Biol. Chem. 2015; 290:9674–9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pesiridis G.S., Diamond E., Van Duyne G.D.. Role of pICLn in methylation of Sm proteins by PRMT5. J. Biol. Chem. 2009; 284:21347–21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lacroix M., El Messaoudi S., Rodier G., Le Cam A., Sardet C., Fabbrizio E.. The histone-binding protein COPR5 is required for nuclear functions of the protein arginine methyltransferase PRMT5. EMBO Rep. 2008; 9:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guderian G., Peter C., Wiesner J., Sickmann A., Schulze-Osthoff K., Fischer U., Grimmler M.. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J. Biol. Chem. 2011; 286:1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tee W.W., Pardo M., Theunissen T.W., Yu L., Choudhary J.S., Hajkova P., Surani M.A.. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010; 24:2772–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ancelin K., Lange U.C., Hajkova P., Schneider R., Bannister A.J., Kouzarides T., Surani M.A.. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat. Cell Biol. 2006; 8:623–630. [DOI] [PubMed] [Google Scholar]
- 30. Shilo K., Wu X., Sharma S., Welliver M., Duan W., Villalona-Calero M., Fukuoka J., Sif S., Baiocchi R., Hitchcock C.L. et al.. Cellular localization of protein arginine methyltransferase-5 correlates with grade of lung tumors. Diagn. Pathol. 2013; 8:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gu Z., Li Y., Lee P., Liu T., Wan C., Wang Z.. Protein arginine methyltransferase 5 functions in opposite ways in the cytoplasm and nucleus of prostate cancer cells. PLoS One. 2012; 7:e44033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tarighat S.S., Santhanam R., Frankhouser D., Radomska H.S., Lai H., Anghelina M., Wang H., Huang X., Alinari L., Walker A. et al.. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia. 2016; 30:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chung J., Karkhanis V., Baiocchi R.A., Sif S.. Protein arginine methyltransferase 5 (PRMT5) promotes survival of lymphoma cells via activation of WNT/beta-catenin and AKT/GSK3beta proliferative signaling. J. Biol. Chem. 2019; 294:7692–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee S.T., Li Z., Wu Z., Aau M., Guan P., Karuturi R.K., Liou Y.C., Yu Q.. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol. Cell. 2011; 43:798–810. [DOI] [PubMed] [Google Scholar]
- 35. Kim J., Lee Y., Lu X., Song B., Fong K.W., Cao Q., Licht J.D., Zhao J.C., Yu J.. Polycomb- and Methylation-Independent roles of EZH2 as a transcription activator. Cell Rep. 2018; 25:2808–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fedoriw A., Rajapurkar S.R., O’Brien S., Gerhart S.V., Mitchell L.H., Adams N.D., Rioux N., Lingaraj T., Ribich S.A., Pappalardi M.B. et al.. Anti-tumor activity of the type I PRMT Inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP Loss. Cancer Cell. 2019; 36:100–114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.