


Abstract
Genome editing has recently made a revolutionary development with the introduction of the CRISPR–Cas technology. The programmable CRISPR-associated Cas9 and Cas12a nucleases generate specific dsDNA breaks in the genome, after which host DNA-repair mechanisms can be manipulated to implement the desired editing. Despite this spectacular progress, the efficiency of Cas9/Cas12a-based engineering can still be improved. Here, we address the variation in guide-dependent efficiency of Cas12a, and set out to reveal the molecular basis of this phenomenon. We established a sensitive and robust in vivo targeting assay based on loss of a target plasmid encoding the red fluorescent protein (mRFP). Our results suggest that folding of both the precursor guide (pre-crRNA) and the mature guide (crRNA) have a major influence on Cas12a activity. Especially, base pairing of the direct repeat, other than with itself, was found to be detrimental to the activity of Cas12a. Furthermore, we describe different approaches to minimize base-pairing interactions between the direct repeat and the variable part of the guide. We show that design of the 3′ end of the guide, which is not involved in target strand base pairing, may result in substantial improvement of the guide's targeting potential and hence of its genome editing efficiency.
INTRODUCTION
The CRISPR-associated nucleases Cas9 and Cas12a (formerly known as Cpf1 (1)) are distinct types of crRNA-guided DNA endonucleases that have been developed into powerful genome editing tools (2–6). Cas9 and Cas12a have rapidly become popular tools for a broad spectrum of genetic engineering applications (7–11), based on the successful heterologous expression of these Cas nucleases and on the relatively easy adjustment of their specificity through exchanging their crRNA guides. The formation of functional crRNAs relies on the conversion of precursor RNA (pre-crRNA) to mature crRNA. In the case of Cas9, the repeat parts of the pre-crRNA are recognized by partly complementary trans-acting crRNAs (tracrRNA). In the presence of Cas9, base pairing between the pre-crRNA repeats and the tracrRNA anti-repeats results in local dsRNA fragments that are specifically cleaved by RNaseIII. After cleavage, the crRNA-tracrRNA pair remains stably bound by Cas9 (12) (Supplementary Figure S1A). To allow for easy crRNA adjustment for Cas9, a synthetic loop has been introduced to connect the crRNA repeat part with the tracrRNA anti-repeat fragment, resulting in a single-guide RNA (sgRNA) (13). In case of Cas12a, however, tracrRNA and RNaseIII are not involved in crRNA maturation. Cas12a directly associates with the pre-crRNA, most likely through recognizing the typical pseudoknot-type hairpin structure of the repeat fragments, after which maturation of the crRNA is catalysed by a dedicated catalytic ribonuclease domain of Cas12a (1,14–16) (Figure 1A).
Figure 1.

Analysis of Cas12a cRNA performance. Cas12a guide bound to target, workflow schematic and measurement of Cas12a activity using crRNAs with 14 different spacers. (A) Cas12a binds the repeat of the pre-crRNA, which forms a typical pseudoknot structure (blue). It recognizes a TTTV PAM (Yellow) and forms an R-loop with the spacer part of the crRNA (red) and the target strand of the DNA (green). Cleavage occurs mostly after positions 18 on the non-target strand and 23 on the target strand. (B) Target plasmid construction starts with a PCR, to incorporate a certain spacer (green box labelled ‘Spacer’) downstream from the leader and the first repeat (orange diamond) and a matching target sequence (green box labelled ‘Target’) on opposite sides of the origin of replication (ori) and mrfp gene. A second SacI-SalI fragment consists of the chloramphenicol resistance marker (cat) and the second repeat (orange diamond). The target plasmid is obtained by digestion of both fragments (SacI and SalI) and ligation. The plasmid is then transformed to an E. coli strain containing a plasmid pCas12a that allows for expression of FnCas12a with an ssrA tag upon induction by l-rhamnose. After transformation, cells are grown overnight in liquid medium, followed by inoculation in fresh medium containing L-rhamnose to induce Cas12a. Fluorescence of mRFP, which is expressed constitutively, is then measured to assess cleavage efficiency. (C) The different spacer sequences (Sp1–Sp14). The seed sequence is indicated by the green shade. (D) Cleavage efficiency of Cas12a shown in terms of fluorescence loss for 14 different spacers in pTarget3. Average values from three biological replicates are shown, with error bars representing SD. (E) Alignment highlighting differences between the sequence of Sp8 and Sp12 (red), and between Sp12 and Sp14 (blue).
A general issue for the application of both Cas9 and Cas12a nucleases appears to be the unpredictable success of crRNA design and target selection, often resulting in designing 3–4 crRNAs for target a single gene. On the one hand, this problem may be caused by differences in local chromatin structure that may severely affect the accessibility of chromosomal targets (17,18). On the other hand, it may be caused by the nucleotide composition of the variable parts of the crRNAs. Based on genome-wide guide library screens, different algorithms and scoring systems have been developed to predict crRNA performance of Cas9 (18–26). The secondary structure of the crRNA has been proposed to be a major player in crRNA performance (27), potentially resulting in poor cleavage activity (28). Also, in case of Cas12a, the editing efficiency varies substantially depending on the design of the crRNA (29). In an attempt to predict the guide functionality, a recent analysis of crRNA-dependent targeting activity of Cas12a from Acidaminococcus sp. (AsCas12a) and Lachnospiraceae bacterium (LbCas12a) has been used to compose an algorithm for crRNA design (29).
Although it is known that the spacer sequence of the crRNA may affect target cleavage efficiency, the molecular basis of this phenomenon remains unclear. An important feature of the Cas9 and Cas12 crRNAs is the formation of well-conserved secondary structures of their invariable sequences, that most likely allows for specific protein-RNA recognition and eventually for stable association of the crRNA and its partner nuclease. In case of Cas12a, perturbations in the hairpin/pseudoknot at the 5′ part of the crRNA (Figure 1A) most likely interfere with the complex formation of Cas12a and its crRNA guide. Hence, predicting cleavage efficiency solely based on spacer-target complementarity is insufficient, as also potential disruption of the pseudoknot should be taken into account. Unfortunately, the reliability of currently available tools for predicting the secondary structure of individual small RNA molecules is relatively low.
In this study, we aimed to reveal the molecular basis of the aforementioned variability of crRNA guide performance of Cas12a. We initially established a sensitive and robust mRFP-based fluorescence-loss assay in Escherichia coli to monitor the in vivo targeting efficiency of Cas12a from Francisella tularensis subsp. novicida (FnCas12a). This system was used to analyse how different spacer sequences and (pre-)crRNA variations affect target cleavage efficiency. We found that the effect on target cleavage by a single nucleotide change in a spacer often depends on its surrounding nucleotides. This observation suggests that these effects are not caused by direct nucleotide-protein interactions as previously proposed (29), but rather by the formation of distinct secondary structures of the closely related crRNA variants. Interestingly, we found that efficient targeting requires only 19 nucleotides of base pairing between the crRNA and the target strand (Supplementary Figure S2), even though position 20 can base pair as well (16). We also found that the last nucleotides of the spacer (position 20 and onwards) can be rationally modified to shift the folding equilibrium from an inappropriate fold, which decreases its efficiency, towards the optimal pseudoknot structure, resulting in the conversion of poorly-performing crRNAs to crRNAs with improved target cleavage efficiency. Our findings contribute to a better understanding of spacer-sequence dependent cleavage efficiencies, and provide design strategies to improve crRNA performance in general, and in Cas12a in particular.
MATERIALS AND METHODS
Strains and media
Escherichia coli DH10B T1R (Invitrogen) was used as host for cloning, plasmid propagation and fluorescence assays. Bacteria were generally cultured on LB medium (10 g/l peptone (Oxoid), 5 g/l yeast extract (BD), 10 g/l NaCl (Acros)) at 37°C. When required, media were supplemented with kanamycin (kan; 50 mg/l) and/or chloramphenicol (cam; 35 mg/l). Fluorescence assays were performed on M9TG medium (1x M9 salts (Sigma), 10 g/l tryptone (Oxoid), 5 g/l glycerol (Acros)). Induction of the FnCas12a was done with l-rhamnose (2 g/l).
Plasmids
The commercial pRham N-His SUMO Kan from Lucigen was made compatible for Ligation Independent Cloning (LIC) by polymerase chain reaction (PCR) (BG7802 and BG7803). The fncas12a gene was PCR amplified (BG7709 and BG7710) and cloned into pRham_LIC using a standard LIC protocol. The pRham-FnCas12a-DAS was made using pRham-FnCas12a as a base. The pRham-Cas12a was digested with BamHI-HF (NEB) and SpeI-HF (NEB). A fragment was created with a variant of the ssrA tag behind the Cas12a coding sequence (AANDENYADAS; see below) by PCR with Q5 polymerase (NEB), using BG8998 and BG10140 as primers and pRham-FnCas12a as template. The PCR fragment was digested with BamHI-HF and SpeI-HF and ligated into the pRham-FnCas12a digest using T4 ligase (NEB) to generate pCas12a.
Target plasmids pTarget1 to pTarget8 are generated from two fragments. The fragments are generated by PCR with Q5 polymerase (NEB). The first fragment contains the cat gene (chloramphenicol resistance) from pACYC184 flanked by a SalI site—terminator (Target1 – F1)/mature repeat (Target2 – F1)/full repeat (Target3 – F1) on one side and SacI on the other. The second fragment contains the P15A ori from pACYC184 and an mrfp gene. A SacI site—PAM—target is attached by PCR on the one end while the other end the crRNA is attached followed by a SalI site. Depending on whether a full repeat is required (Target1 – F2) or a mature repeat (Target4 – F2), a different template is used. Ligating the Target1 – F2 to Target1/2/3 – F1 yields pTarget1/2/3 respectively and ligating Target4 – F2 to Target1/2/3 – F1 yields pTarget4/5/6. Fragments were digested with SalI-HF (NEB) and SacI-HF (NEB), and ligated with T4 ligase (NEB). pTarget7 is generated by ligating Target7 – F1 to Target7 – F2, while pTarget8 is a ligation of Target8 – F1 and Target8 – F2. pTarget9 is a ligation of Target8 – F1 and Target9 – F2. These plasmids are assembled by Golden Gate cloning with SapI (NEB) and T4 ligase (NEB). An overview of the cloning details, including primer sequences, can be found in the supplementary data (Supplementary sequence 1, Supplementary Tables S1 and S2).
Fluorescence loss assay
The targeting activity of programmable nucleases in bacteria is generally measured either by transformation efficiency assays (based on the recovery of viable transformants), or by plasmid loss assays (based on loss of plasmid over time). In both cases, the fraction of bacteria harbouring a plasmid is assessed by plating in parallel on both selective and non-selective medium. However, apart from being labour intensive, we consider these methods not accurate enough to distinguish small differences in cleavage efficiency. Major drawbacks of transformation efficiency assays include inconsistency of bacterial competence and differences in plasmid purity and concentration, resulting in low accuracy. Plasmid loss assays do not suffer from these artefacts, but still require plating and colony counting. The duration of the expression of the Cas12a nuclease is quite essential, as resolution is lost either at high plasmid clearance rates or during extended Cas12a exposure. While the extended Cas12a exposure increases sensitivity, the information on plasmid cleavage efficiency is lost after full plasmid clearance.
For these reasons, we developed a robust screening approach that allows for accurate detection of variations in the copy number of a target plasmid. Apart from a chloramphenicol resistance marker (cat), the target plasmid (pTarget) contains a constitutively expressed reporter gene (mrfp), a short CRISPR array with a single spacer sequence, and a matching target sequence downstream of a 5′-TTTV PAM motif (hereafter referred to as ‘target’) (Figure 1B; Table 1; Supplementary Table S2). To ensure the differences in mRFP fluorescence are a direct result of Cas12a cleavage activity, and not, for example, caused by blocking of read-through transcription, the mrfp gene is isolated by two terminators. In addition, targeting occurs outside of the mrfp transcription region. The target plasmids were individually transformed to E. coli cells harbouring a second plasmid (pCas12a) that encodes FnCas12a (hereafter referred to as Cas12a). The rate of Cas12a-mediated clearance of the target plasmid is detected as loss of mRFP fluorescence, directly reflecting the spacer-based targeting efficiency.
Table 1.
pTarget construct design
| Name | 5′-spacer flank | 3′-spacer flank |
|---|---|---|
| pTarget1 | Leader-full repeat (L-FR) | SalI-Terminator (|T|) |
| pTarget2 | Leader-full repeat (L-FR) | SalI-Mature repeat-spacing-terminator (MR) |
| pTarget3 | Leader-full repeat (L-FR) | SalI-Full repeat-spacing-terminator (FR) |
| pTarget4 | Mature repeat (MR) | SalI-Terminator (|T|) |
| pTarget5 | Mature repeat (MR) | SalI-Mature repeat-spacing-terminator (MR) |
| pTarget6 | Mature repeat (MR) | SalI-Full repeat-spacing-terminator (FR) |
| pTarget7 | Mature repeat (MR) | Terminator (|T|) |
| pTarget8 | Mature repeat (MR) | Mature repeat-spacing-terminator (MR) |
| pTarget9 | Full repeat (FR) | Mature repeat-spacing-terminator (MR) |
Compared to plasmid loss or transformation assays, the here-established fluorescence-loss assay allows for distinguishing between efficient (good) and less-efficient (bad) crRNAs with high accuracy and ease. The fluorescence builds up as long as the plasmid is retained and the bacteria are still producing protein. The higher the activity of the Cas12a, the higher the loss of fluorescence. The effect of exposure to Cas12a is terminated by bacteria reaching the stationary phase where protein production eventually stops, so timing is less essential compared to a plasmid loss assay. Even when all the plasmid is lost, differences in targeting activity can still be extrapolated from fluorescence values. Important to note, however, is that the dilution of the preculture to the final culture dictates the exposure time. It is therefore essential that the dilution is carried out very precisely. If more sensitivity is required, the cells can be further diluted to allow for a longer period of plasmid loss.
Since targeting of the plasmid may cause escape mutants, we needed to limit the exposure of the target plasmid to Cas12a. Chemically competent E. coli DH10B harbouring the pCas12a plasmid were transformed with target plasmid and recovered in LB. After recovery, the bacteria were diluted 1:100 to 200 μl M9TG medium supplemented with kan/cam from the transformation mix, and grown in a 2 ml 96-well masterblock (Greiner) covered with a gas-permeable membrane at 37°C overnight. Presence of both kan and cam will ensure the bacteria retain both plasmids. After overnight growth, the bacteria were diluted 10−4 (two steps of 10−2) into 200 μl fresh M9TG medium supplemented with kan/cam, kan, or kan and with 2 g/l l-rhamnose and grown at 37°C overnight in a master block covered with a gas-permeable membrane. The cultures were cooled down to room temperature and diluted 5× in 1× PBS pH 7.4. As controls, non-targeted plasmid with mRFP (pTS001) and non-targeted plasmid without mRFP (pACYC184) were used alongside non-inoculated M9TG medium. The latter served both as a negative control for growth and a blank for fluorescence and light scattering. 100 μL of the diluted cultures was measured on a Synergy MX microplate reader. Fluorescence measurements were performed with an excitation at 584 nm with a bandwidth of 9 nm, emission at 607 nm with a bandwidth of 9 nm and a gain of 120. Fluorescence loss of (x) was calculated as follows:
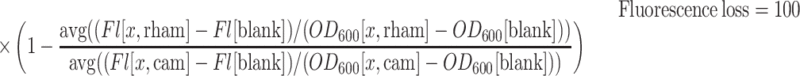 |
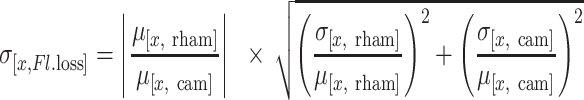 |
Fine-tuning of the assay
To allow for accurate comparative analyses of crRNA performance, fine-tuning of the assay has been performed at three levels. (I) Synchronizing cells: The time available for the plasmid clearance is crucial for the final fluorescence. For the best possible comparison of targeting performance of different crRNAs, the bacteria harbouring both plasmids (pCas12a and pTarget) were synchronized by growing them to the stationary phase in a pre-culture in the presence of antibiotics to select for maintenance of both plasmids. (II) Minimize targeting when undesired: Simultaneous selection of pTarget maintenance (through presence of chloramphenicol) and targeting of pTarget by background Cas12a activity, would allow for growth of escape mutants and hence selection of false negatives in the fluorescence loss assay. Indeed, under these conditions we observed sabotage of Cas12a activity in different ways: by deletion of the entire spacer through recombination of the two flanking repeats, by recombination of the ribosome binding site of the cas12a gene, and by introduction of a transposon into the cas12a coding region (not shown). To control the timing of Cas12a targeting, the cas12a gene expression is controlled by the rhamnose-inducible PrhaBAD promoter. (III) Limit the lifespan of Cas12a: To further reduce leaky expression of Cas12a, an ssrA degradation tag is fused to the C-terminus of the protein. While the native ssrA tag (‘AANDENYALAA’) almost completely abolishes the Cas12a activity, a less efficient tag variant (‘AANDENYADAS’) (30,31) was found to limit both the Cas12a residence time and its leaky expression (Supplementary Figure S3). It also reduces the protein levels of Cas12a in the cell while being induced. A large excess of Cas12a would cause crRNAs to bind because of the high effector protein concentration rather than a high affinity; in that case moderate affinity might cause the fluorescence to drop to background levels, resulting in loss of resolution for the high efficiency spacers. When the Cas12a concentration is limited, we can distinguish moderate from good spacers.
Without Cas12a induction and without antibiotics pressure on the target plasmid, a very low basic level of fluorescence loss is observed for most spacers (Supplementary Figure S4). A low, non-induced fluorescence loss indicates that the plasmid is not targeted severely during the synchronisation, and therefore, escape mutants have no significant growth advantage. Some of the highly efficient spacers do show substantial fluorescence loss without induction of Cas12a and without addition of cam. The presence of cam enforces target plasmid maintenance, and although we see a reduction in fluorescence compared to less efficient spacers, the reduction is only marginally under these conditions, and no escape mutants were observed.
In vitro cleavage assay
Pre-crRNA was made by in vitro transcription with the HiScribe™ T7 High Yield RNA Synthesis Kit (NEB). FnCas12a was diluted to 0.4 nM in 2× NEBuffer 4 with 2 mM Mg2+, instead of 20 mM Mg2+. Reactions were started by adding 0.2 nM pre-crRNA in a 1:1 ratio. Reactions were incubated at 37°C and sampled at t = 0, t = 5 and t = 10 min. Sampled reactions were quenched by adding 1 μl of quenching solution (9% SDS, 50 mM EDTA) to 10 μl of sample. Samples were heated to 95°C for 5 min and cooled to 12°C. Potassium dodecyl sulphate was pelleted by centrifugation and the 10 μl of supernatant was mixed with 2× RNA Loading Dye (NEB) and analysed on a 1.5 mm 5% acrylamide gel containing 7 M urea. The gel was run on a Bio-Rad Mini-PROTEAN Tetra Cell system at 15 mA until the bromophenol blue was at the bottom. RNA cleavage products were stained by SYBR gold and visualised on a Bio-Rad Gel Doc XR+.
RESULTS
Sequence of the spacer affects cleavage efficiency
A set of 10 crRNAs with different spacers of similar composition (Sp1–10; Figure 1C) was initially used for their in vivo functionality to allow Cas12a to target complementary plasmid-borne sequences in E. coli. The spacer transcripts had a 5′ leader sequence, followed by the spacer that was flanked by a full repeat on both ends (L-FR-Sp-FR; pTarget3; Table 1), which most closely resembles the original, native array (1). The observed targeting functionality (fluorescence loss) of nine spacers was in the range of 60–85%, with the exception of the poorly performing Sp8 (0%) (Figure 1D). To reveal the molecular basis of this phenomenon, four related spacers were designed (Sp11-14). This resulted in an interesting set of three closely related spacers with major differences in performance (Figure 1D, E). The most dramatic difference was observed between Sp8 and Sp12 that, although they differ only by four nucleotides, perform either very badly (Sp8, no fluorescence loss) or very well (Sp12, among the fastest). It should be noted that, with this set of crRNAs, similar trends in targeting efficiencies were observed for FnCas12a and AsCas12a (Supplementary Figure S5).
Fluctuations in cleavage efficiency with single nucleotide changes
Since Sp8 and Sp12 have the same seed (nucleotides 1–5), the composition of the seed sequence appears not to play a role in the observed differences in cleavage efficiency. Sp8 and Sp12 differ in only four nucleotides (at positions 10, 11, 19, 20; Figure 1E). To reveal which of these nucleotides are responsible for the major difference in targeting efficiency, we made a library to systematically test all 16 combinations (four nucleotide positions and two possible nucleotides per position) between Sp8 and Sp12. In addition, to shed light on the influence of the 3′ end of the pre-crRNA, libraries were generated in two different CRISPR designs. In one library the L-FR-Sp-FR (pTarget3; Table 1) crRNA design (Figure 2A) was used, whereas another library was made using the L-FR-Sp-|T| (pTarget1; Table 1) crRNA design (Supplementary Figure S6A), which minimizes the influence of the 3′ end. Overall, similar trends were observed for Sp8 variants in the two libraries. For the Sp8 variants, position 19 appeared of least influence on crRNA performance, and in Figure 2B the base at this position is referred to as ‘K’ (G or U) for clarity purposes. Screening of the library indicated that positions 10 and 11 are almost solely responsible for the low activity observed for Sp8 (Figure 2B). Surprisingly, only spacers containing the combination of A10 and U11 showed very low efficiency, independent of variation at position 20. Even changing either one of the positions, A10C or U11A, yielded moderate to high cleavage efficiency. The presence of the A10-U11 pair in the efficient Sp2 (Figure 1C, D), strongly suggests that its negative impact in Sp8 is not position dependent (interaction between the crRNA and the Cas12a protein), but rather context dependent (intramolecular interactions in the crRNA).
Figure 2.

Comparison of Sp8, Sp12 and Sp14. (A) Cleavage efficiency shown in terms of fluorescence loss for different Sp8 variants in pTarget3. Average values from three biological replicates are shown, with error bars representing SD. The wild type and mutated Sp8 nucleotide sequences are shown in upper case black and lowercase red letters, respectively. In brackets [NNNN] are the nucleotides in position 10, 11, 19 and 20. (B) A 3D representation of the data in panel A. Cleavage efficiency is represented by the intensity of the colour red for different spacers shown in a 4 letter code, which are the nucleotides in positions 10, 11, 19 and 20. K (G or U) at position 19 is constant and the average was taken to generate the red intensity for each spacer variant. The wild type and mutated Sp8 nucleotide sequences are shown in black and red, respectively. Each corner of the cube represents a certain spacer sequence, and moving along either of the 3 axes changes the sequences at one nucleotide position only. (C) Cleavage efficiency shown in terms of fluorescence loss for different Sp12 variant in pTarget3. In brackets [NNN] are the nucleotides at positions 5, 7 and 9. (D) A 3D representation of the data in panel C similar to panel B. (E) Predicted crRNA structures for Sp8, Sp12 and Sp14. Folding of an active crRNA is given on the left and the folding based on prediction are given on the right. Each spacer is in equilibrium with its active state and inactive state as indicated by the arrow. Thicker arrows represent that the equilibrium is shifted more towards a certain state.
Another major difference in cleavage efficiency was observed for the related spacers Sp12 and Sp14 (Figure 1D). These sequences differ at only three positions (5, 7 and 9; Figure 1E), so we made a variant library to pinpoint the determining nucleotides for both the L-FR-Sp-FR (pTarget3) design (Figure 2C) and the L-FR-Sp-|T| (pTarget1; Supplementary Figure S6B). Both in Sp12 and Sp14, position 5 is most important, while the impact of position 7 depends on its surrounding nucleotides, and position 9 appears to be the least influential on activity (Figure 2B). The highly efficient Sp12 was severely affected by changing position 5 (U5C) (Figure 2D). The analysis of both libraries revealed similar trends for most variants, indicating that cleavage efficiencies can be substantially influenced by a single nucleotide change within a given position in a spacer. As noted earlier, however, the presence of C5 in the efficient Sp6 (Figure 1C, D), suggests that the negative impact of C5 in Sp14 is not position dependent. Rather, this effect may depend on the surrounding nucleotides. These examples of context dependence indicate that differences in crRNA secondary structure correlate with fluctuating Cas12a cleavage efficiencies.
To check the potential involvement of crRNA secondary structure, attempts were made to predict the folding of the pre-crRNAs of Sp8, Sp12 and Sp14 (Figure 2E) with RNA secondary structure prediction tools RNAfold (32) and mFold (33). While the folding energies obtained by both tools are not exactly the same, the generated structures are similar. Instead of the pseudoknot that is required for an active Cas12a–crRNA complex, the analysis suggested a strong alternative structure of the pre-crRNA (Sp8) that is formed by base pairing between the leader and the spacer. The alternative structure of the Sp8 pre-crRNA is stabilised by a long stem, composed of mainly A•U pairs. Interrupting this stem, immediately causes the equilibrium to shift to the active fold (A10C or U11A) (Figure 2C, D). In Sp12, the alternative structure is neither stabilized by a very long nor a very strong stem, but the U5C mutation changes that drastically. While the Sp12 [cUA] variant had almost no activity left, the activity could be partially restored by U7A (Figure 2C, D). Without the U5C mutation, the U7A should sufficiently destabilize a stem that requires no further destabilisation, and indeed we saw limited effect of U7A in that case (Figure 2C–E). The A9C mutation has a counter-intuitive effect on Sp12. While the A9C will shorten the stem and destabilize it in the Sp12 [cUA] variant, it has a slight negative effect on the other three variants ([UUA], [UaA] and [caA]) (Figure 2C). Whereas our current understanding is inadequate to explain this phenomenon, it might be related either to alternative base pairing or to other inactive folds.
Sequences directly flanking the spacer affect pre-crRNA processing
When the mature crRNA is bound to the Cas12a protein, the repeat has a pseudoknot structure (16). Disrupting this structure is anticipated to affect the binding of the pre-crRNA to the Cas12a protein. To demonstrate this, an in vitro processing assay was performed (Figure 3A, B), revealing that the 164 nt pre-crRNA of pTarget3 is not cleaved very well at the first repeat for Sp8. Other spacers (Sp1, Sp4, Sp12) showed good processing of both repeats. This agrees well with the model that the lack of pseudoknot formation does affect binding and processing of the pre-crRNA, resulting in reduced levels of mature crRNA and, hence, impaired cleavage efficiency by Cas12a.
Figure 3.

Cleavage efficiency for spacers with different upstream and downstream sequences. (A) According to our hypothesis, the Sp1, Sp4 and Sp12 will have efficient processing for both repeats. The Sp8 causes misfolding of the pseudoknot and thereby impedes the processing of the first repeat. (B) In vitro pre-crRNA processing of Sp1, Sp4, Sp8 and Sp12 at different time intervals. The marker lane on the left shows three bands from the low range ssRNA ladder (NEB). The bands of 127 nt, 66 nt and 37 nt indicate processing of the pre-crRNA that would lead to a targeting RNP. The processing of the second repeat (103 nt, 61 nt) gives rise to a non-targeting RNP. (C) Two relatively inefficient spacers were tested in different pre-crRNA architectures (pTarget1 - pTarget6): Sp8 (red bars) and Sp12-variant [cUA] (blue bars). Average values from three biological replicates are shown, with error bars representing SD. A 20 nt leader-end is shown in orange, a full or mature repeat in blue, a 24 nt spacer sequence in red, a SalI restriction site in yellow, a spacing sequence in green and the terminator is shown in purple. Spacers were flanked on the 5′ end with a leader-end sequence and full repeat, or they were flanked by a mature repeat only. Within each category were spacers containing various downstream sequences. Sequences such as, only a terminator (|T|), a mature repeat-spacing sequence-terminator (MR) and a full repeat-spacing sequence-terminator (FR).
We then set out to systematically test the influence of different upstream and downstream sequences on the spacer cleavage efficiency of two bad spacers, the original Sp8 (Figure 1C) and a poorly performing Sp12-variant ([cUA]; Figure 2C). For both spacers, six different constructs were made and tested (pTarget1 to pTarget6; Figure 3C). The presence of a leader sequence upstream of the repeat-spacer resulted in a major reduction of the Cas12a cleavage activity, with similar trends for both spacers. In contrast, different sequences downstream of the spacer led to relatively minor fluctuations in cleavage efficiency for both spacers (Figure 3C). This indicated that the sequence context of the precursor and/or mature crRNA may seriously impact its performance. In this case, omitting the upstream leader sequence that probably disturbs the formation of the desired pseudoknot structure (Figure 2E) resulted in substantial restoration of crRNA performance.
crRNA folding can cause unstable pseudoknot formation
Assuming that at the pre-crRNA stage, correct folding of the pseudoknot is key for appropriate docking in Cas12a and hence for eventual cleavage efficiency, functionality of a guide correlates with its potential to form a pseudoknot. In the case of Sp8 where the leader is impeding pseudoknot formation, the nucleotides that caused the impediment, could be substituted or even omitted. When the crRNA nucleotides causing pseudoknot disruption are actually involved in base pairing with the target strand (Figure 1A), they cannot be changed or omitted. An alternative strategy to enhance pseudoknot formation nonetheless, would be to force those nucleotides to base pair with nucleotides at the crRNA 3′ end instead. Since adding secondary structure may cause issues of its own, it was assessed whether crRNA performance could be influenced by masking certain parts of the crRNA through designed intra-molecular base pairing with its own 3′-sequence. To increase the chance of back-folding, the distance between the 3′ tail and the masked part should be as small as possible. Therefore, instead of adding 5 nucleotides to the spacer, we replaced the last 5 nucleotides. Although it is known that that Cas12a does not need the full 23 nt spacer (1), it was important to ensure that shortening the base pairing of spacer and target to 19 bp did not influence the activity. Hence, we conducted a pilot experiment with pTarget3-Sp4, where the target (not the crRNA) was complementary to the crRNA up to position 19. Under these conditions, 19 bp appeared to be sufficient for maximal cleavage efficiency (Supplementary Figure S2). This was confirmed by a more elaborate experiment with different crRNA lengths (Supplementary Figure S8).
Next, we generated a new spacer that had as few (predicted) base pairs in the first 19 nucleotides as possible, approximately 50% GC-content and no single base stretches longer than 3 (Back-fold lib A). We then created a library of crRNAs in pTarget1 (Figure 4A) with variable spacer tail sequences to mask specific positions, ranging from the direct repeat (DR; position –3) to position 11 of the spacer (selected examples are depicted in Figure 4C–E).
Figure 4.

Cleavage efficiency for folding library. (A) A library of designs has been made in pTarget1. Panels C–E will only show the boxed part. (B) Cleavage efficiency shown in terms of fluorescence loss for different crRNA variants from Back-folding library A (Back-fold lib A). Average values from three biological replicates are shown, with error bars representing SD. (C–E) The intended folds of crRNAs of Back-fold lib [DR], Back-fold lib [2] and Back-fold lib [7]. The number in brackets indicates the starting position of the base pairing.
Back-fold lib A [DR] (Figure 4C) results in base pairs between positions 1–6 with positions 20–25. The SalI site starting at position 26 then base pairs with the last three nucleotides of the direct repeat (DR). The rest of the library started masking at position [n] and had the last base pair 4 nucleotides downstream. The nucleotide at position 25 was designed to force a mismatch with the nucleotide at [n–1], in an attempt not to extend the base pairing beyond 20–24; for example, the design of crRNA Back-fold lib [2] was such that positions 2–6 base paired with positions 20–24, and that the nucleotide at position 1 [2-1] mismatched with that at position 25. Likewise, in Back-fold lib A [4] the masking started at position 4, ended with position 8, and the nucleotide at position 3 [4-1] had a mismatch with the nucleotide at position 25. To minimize the chance of the spacer 3′ end base pairing with the 3′ end of the transcript, the crRNA design was L-FR-Sp-|T| (pTarget1). In the generated library, spacers were designed from random sequences and selected for showing minimal predicted secondary structure, ∼50% GC and unique library members.
For Back-fold lib A [DR] (Figure 4C), we observed that base pairing with the direct repeat (almost) abolished Cas12a activity (Figure 4B). Mismatching of position 25 and position 1, as is the case in Back-fold lib A [2], negated this effect completely (Figure 4B/4D). Unexpectedly, there appears to be no trend in cleavage efficiency between the different designs of Back-fold lib A [–1] to [11] (Figure 4B). Cleavage efficiencies fluctuated between 40 and 60% for all constructs within one library. In particular, Back-fold lib A [–1] showed no diminished activity even though the -1 position is considered to be part of the pseudoknot with a U•U pair (16). Possibly, the likelihood of base pairing between the 5′ and 3′ ends of the crRNA is reduced because of the distance between the positions. We also tested two other folding libraries made from the selection of spacer bases (Supplementary Figure S7), and again did not observe a trend between masking of a specific position and cleavage efficiency, apart from the [DR] constructs.
Hence, base pairing with the direct repeat does completely abolish Cas12a activity, supporting the model that inadequate pseudoknot formation is detrimental for crRNA functionality. On the other hand, these results show that crRNA structures that mask spacer positions [–1] to [11] do not abolish Cas12a activity; as discussed below, this may be useful for rescue of bad crRNAs.
Rescuing ‘impaired’ spacers
Certain spacer sequences may cause crRNAs to adopt a fold that disrupts the pseudoknot structure, resulting in hampered Cas12a binding and poor targeting activity. In an effort to rescue such spacers, we adjusted the 3′ end of the crRNA sequences such that any base pairing with the direct repeat is mitigated. This should favour pseudoknot formation. To test this, two additional crRNAs were designed. One crRNA has a spacer length of 19 nucleotides and base pairing between spacer positions 6–12 and the complementary nucleotides in the direct repeat, which cause an alternate fold that is unlikely to associate properly with the Cas12a protein, resulting in the ‘impaired’ spacer (Figure 5B; impaired). The other crRNA has the same spacer of 19 nucleotides with an additional five nucleotides at the 3′ end, that are designed such that they may allow for intramolecular base pairing with the spacer sequence from 9 to 14, avoiding the pseudoknot disruption, and thus converting the ‘impaired’ spacer into a ‘rescued’ one (Figure 5B; rescued). Since every nucleotide in the transcript may influence the folding, the spacers were flanked with a mature repeat at the 5′ end. Downstream of the spacer, a flanking sequence was included either with a terminator (pTarget7-IS and pTarget7-IS-rescued), or with a second repeat and a terminator (pTarget8-IS and pTarget8-IS-rescued). While the former will retain its terminator, the latter will be recognised and further processed by Cas12a, so it does not contain a mature repeat-terminator sequence during DNA targeting.
Figure 5.

Rescuing ‘impaired’ spacers. (A) Cleavage efficiency is shown in terms of fluorescence loss for various crRNA variants. The mature repeat is shown in blue, the 1–19 nt of spacer sequence in red, the 20–24 nt positions are shown in yellow. The crRNAs are categorised as ‘impaired’ and ‘rescued’ either with (pTarget7) or without (pTarget8) a 3′ terminator sequence. Average values from three biological replicates are shown, with error bars representing SD. (B) Detail of the structures in (A).
The ‘impaired’ spacer, either with or without terminator, has a lower cleavage efficiency than its ‘rescued’ counterpart (Figure 5A). Unlike aforementioned designs (Figure 3B), the spacers with terminator overall had a much lower efficiency than the ones with a mature repeat. The difference is that the terminator was positioned slightly closer to the R-loop in pTarget7 (Figure 5A) than it was in the pTarget1 or pTarget4 constructs (Figure 3B), due to the SalI site in the latter two designs. The terminator was even closer for the ‘impaired’ spacer than it is for the ‘rescued’ spacer, since the former was 5 nucleotides shorter.
To further prove that the observed increase in cleavage efficiency (Figure 5A) was not just caused by an increase of spacer length but rather by improved folding, we constructed two libraries with randomized tails using the ‘impaired’ spacer as a base. Each library contained the ‘impaired’ spacer of 19 fixed nucleotides and 5 variable nucleotides (NNNNN) at positions 20–24, followed by either the terminator (pTarget7-IS-N5) or a mature repeat (pTarget8-IS-N5). Within each library, 47 colonies were randomly selected and their cleavage efficiency was assessed (Figure 6A). Out of the 47, we selected 12 spacers that covered the whole range of cleavage efficiencies. Those were sequenced and their RNA structures were predicted by RNAfold (32). Again, some general trends are observed that support the correlation between pseudoknot formation and crRNA performance (fluorescence loss). In case of low efficiency spacers, base pairing with the direct repeat appears to be enhanced, hampering formation of the pseudoknot. In case of relatively efficient spacers, the proper pseudoknot structure is not challenged due to intra-spacer base pairing involving the variable positions (20–24) and (part of the) positions 6–12 (Figure 6B, C). The sequences that show intermediate efficiency seemed to correspond to spacers of which the pseudoknot/alternate states are not favoured either way.
Figure 6.

Cas12a activity of two ‘impaired’ spacer (IS) libraries varying position 20–24. (A) Cleavage efficiency shown in terms of fluorescence loss of 47 randomly selected colonies for pTarget7-IS-N5 and pTarget8-IS-N5. Non-fluorescent clones were omitted. The target and first 19 nucleotides of the spacer are the same as Figure 5. (B) Cleavage efficiency shown in terms of fluorescence loss from sequenced variants of pTarget7-IS-N5 with the given possible crRNA structure of variants 1, 16 and 46. The crRNAs contain a mature direct repeat (blue), a spacer (red), a variable sequence at position 20–24 (yellow) and a terminator (purple). (C) Cleavage efficiency shown in terms of fluorescence loss from sequenced variants of pTarget8-IS-N5 with the given possible crRNA structure of variants 1, 9 and 34. The crRNAs contain a mature direct repeat (blue), a spacer (red), a variable sequence at position 20–24 (yellow), a spacing sequence (green) and a terminator (purple). The cleavage position of the crRNA processing is indicated with an orange arrow.
To estimate the contribution of pre-crRNA misfolding to the complete Cas12a cleavage efficiency, we selected 16 spacers from a previous study (34) that performed poorly as judged from relatively low indel formation, despite the fact that the accessibility of corresponding human target genes was good. Apart from the original guides, tailored pre-crRNA designs were made (as described above) with nucleotides 20–24 base pairing with nucleotides 11–15 on every spacer and with a mismatch between nucleotides 10 and 25. Under control of the U6 promoter, transcription starts at the beginning of the full repeat and it ends at a polyU tail. A direct mimic in bacteria was impossible as the E. coli RNA polymerase does not stop at polyU when it is not preceded by a strong stem-loop. The closest approximation is adding the polyU tail to the spacer and mimic the termination by processing of a second mature repeat (FR-Sp-MR; pTarget9). This way, the crRNAs were most comparable to the mammalian situation. The tailored versions were made in pTarget8 (MR-Sp-MR; Figure 5). Most of the spacers performed well in the assay (Figure 7E). Spacers I, N, and to a certain extend also Spacer H, underperformed, and were not rescued by our tailoring approach. On the other hand, spacers K and O (Figure 7A, C) did show substantial improvement after tailoring (Figure 7B, D). While poor crRNA performance is clearly not explained solely by secondary structure in the pre-crRNA, implementing our design of folding the spacer tail back onto the spacer itself does not impede Cas12a activity (0–5%-point). Sometimes it results in a moderate improvement (crRNAs B, C, E, L: 5–20%-point), and sometimes in a major one (crRNAs K,O: >20%-point). While the targeting by spacer E was not impressive even after tailoring, the relative increase in efficiency was substantial (72%).
Figure 7.

Tailoring pre-crRNA design for reported ‘bad’ spacers. (A) Predicted structure of spacer K pre-crRNA as used by Kim et al. 2018. (B) Tailored pre-crRNA design of spacer K. (C) Predicted structure of spacer O pre-crRNA as used by Kim et al. 2018. (D) Tailored pre-crRNA design of spacer O. (E) Performance of the reported ‘bad’ spacers in the fluorescence loss assay. Original versions are in red and tailored versions are blue.
Limits of imposing structure onto the crRNA
Varying the flanking sequences has significantly improved the efficiency (plasmid targeting/fluorescence loss) of the poor performing Sp8 from 0–8% (pTarget1–3) to 40–50% (pTarget4–6) (Figure 3B). Still, it did not have the efficiency of well performing spacers Sp4 or Sp9 (75–85% fluorescence loss; Figure 1D). In the aforementioned designs, strong indications for undesired secondary structures were found in precursors of poorly performing crRNA variants. However, incompatible secondary structures that disrupt association with Cas12a and/or RNA–DNA binding, might also occur within mature crRNAs. As an example, we again focused on Sp8, the mature crRNA of which can potentially form two alternative structures that would disrupt pseudoknot formation (Figure 8A). By changing the tail of the spacer, we could mask the spacer nucleotides involved in the unfavourable alternate structures. Whereas minimal base pairing of the spacer nucleotides occurred in the aforementioned Back-fold lib (Figure 4), the nucleotides in the tail of these new designs have to compete with nucleotides in the direct repeat for base pairing with the spacer. In contrast to the aforementioned ‘impaired’ spacer (Figure 6), the tail's target nucleotides are positioned closer to the direct repeat. Short distance interactions are more likely to occur than long distance interactions, so five nucleotides in the spacer tail are not expected to compete effectively with the nucleotides in the direct repeat. Instead, these tail nucleotides are more likely to interact with matching nucleotides in the adjacent spacer part. Therefore, additional designs were made (MR-Sp-MR construct in pTarget8) in an attempt to improve the masking, and hence the performance of Sp8 (Figure 8B, C). Compared to aforementioned designs with long a loop (8 nt) and a short stem (5 nt) (Figure 4E), we here used pTarget8 to systematically test designs for intramolecular spacer/spacer-tail interactions, by varying both the loop size (4, 5 or 6 nt) and the stem size (4 × 3 bp, 3 × 4 bp or 2 × 5 bp) (Figure 8C). Because the spacer folding should be reversible to allow for base pairing with a complementary DNA target, all stems are interrupted by single nucleotide bulges and end with a mismatch. In case of the 4 and 5 nt loops, alternative designs (Figure 8C; alternative (alt)) were made to analyse to effect of base pairing to the seed region.
Figure 8.

Imposing structure onto the Sp8 crRNA. (A) The Sp8 has two alternate, inactive forms involving the first 11 nucleotides of the spacer and part of the 5′ repeat. The mature repeat (blue) is followed by 5 nucleotides of seed region (green) and 14 residual nucleotides of Sp8 (red). The tail (yellow) were replaced by other nucleotides depending on the construct. (B) Scatterplot of folding energy of the crRNA against the fluorescence loss. Numbers indicate the construct depicted in Figure 8C. The colour gradient corresponds to the fluorescence loss and correspond to the gradient colours of Figure 8C. Value ‘A’ corresponds with the Sp8 active form of Figure 8A. (C) Fluorescence loss in pTarget8 and structure of the designs for the rescue of Sp8. The fluorescence loss, depicted by a circle with gradient colour, and the numbers are linked to Figure 8B. The tail (yellow) added to the spacer folds back onto the spacer with different loop and stem sizes, always intermitted by a single nucleotide bulge. The tails that cover the seed extensively are also shortened by one stem to yield the alternative (alt) form. The grey circle indicates the activity of Cas12a in observed fluorescence loss, while the blue bar graph depicts the theoretical folding free energy of the ensemble omitting the repeat. To judge the stem strength at a glance, at the G•C pairs are marked in red and the A•U pairs in blue.
Compared to the control (Sp8 without tail, in pTarget5) that results in 50% fluorescence loss (Figure 8B (‘A’)), 10 out of 14 tested designs indeed showed improvement in cleavage efficiency of up to 14% point (Figure 8B). Two designs with relatively weak folds (e.g. [stem 3/loop 6]) did not result in improved efficiency, probably due to pseudoknot disruption. Likewise, the two designs that result in the strongest secondary structures [stem-5/loop-5] and [stem-4/loop-5] did not result in enhanced activity levels (43% and 53%), most likely because the stable fold of the crRNA’s spacer hampers efficient DNA targeting. Although no solid correlation was observed between Cas12a activity and the overall folding energy of the spacer/spacer-tail part of the crRNA, the distribution of base pairing strength (G•C-pairs versus A•U-pairs) did seem to be important. Hairpin formation is more likely to occur when the loop is small, and when there is stronger base pairing near the loop (32). This is in agreement with the difference in performance of designs [stem-3/loop-5; alternative] and [stem-3/loop-6] (Figure 8C). Although the overall folds are very similar, the former has improved activity (61% versus 52%) probably due to a smaller loop (5 nt versus 6 nt) and a stronger loop-adjacent stem (3 G•C-pairs versus 2 G•C pairs). Hence, the loop-size and stem composition determine the equilibrium of different crRNA folds, reflecting the overall cleavage activity.
Comparison of designs that only differ in their tail length (Figure 8C; alt), might reflect an effect of reduced masking of the seed region. In case of stem-3 variants no differences are observed, but in case of the stem-4 variants there seems to be a difference, possibly due to the longer tails covering the seeds more extensively (4 out of 5 seed bases). Relatively weak base pairing between the tail and the seed in [stem-4/loop-4] is rather well tolerated (59% in seed-masked versus 62% in seed-free), but stronger base pairing (3 G•C pairs instead of 2) in [stem-4/loop-5] appears to be penalised (53% in seed-masked versus 64% in seed-free) (Figure 8C).
DISCUSSION
High efficiency is an important criterium for genome editing applications by CRISPR-associated nucleases. The efficiency of genome editing depends on (I) delivery of Cas effector proteins and their crRNAs (35), (II) crRNA performance (this study), (III) accessibility (chromatin structure) of the target site (17), (IV) accuracy of the Cas nuclease (36), and (V) the host's mechanism to repair the generated double stranded break (DSB) (37). In this study, we set out to reveal the molecular basis of variable crRNA functionality in targeting a plasmid in E. coli by Cas12a.
As has been reported previously (29,34), we observed major fluctuation in crRNA functionality when analyzing a small set of Cas12a crRNAs (Figure 1D) and derived variants (Figure 2A, C). Our working hypothesis is that the pseudoknot structure of the pre-crRNA is required for appropriate binding by Cas12a, and hence for processing to mature crRNA and formation of a cleavage-compatible Cas12a/crRNA complex. The hairpin within this pseudoknot structure is formed through intramolecular base pairing of the palindromic part of the repeat. Structural variation can occur when flanking sequences (the upstream leader or the downstream spacer) compete for base pairing with the nucleotides in the repeat, resulting in alternative secondary structures. The relative strength of these competing structures will determine the equilibrium of the mixture of functional and non-functional crRNAs. In agreement with this assumption, it was found that the leader sequence combined with the sequence of the bad crRNA (Sp8) potentially adopts an alternative fold in which base pairing occurs between the Sp8 spacer and part of the direct repeat (Figure 2E). In the broader sense, any sequence adjacent to the repeat may cause the pre-crRNA to fold into a structure that is not recognised by Cas12a. In case of a CRISPR array, this includes interactions with other upstream and downstream sequences (Figure 2E).
Based on these insights, we then assessed whether it would be possible to avoid disruption of the pseudoknot structure by a competing spacer sequence, through designing base pairing of the spacer with a spacer-tail (i.e. positions 20–24, which are not involved in DNA targeting). We designed a library of constructs with a variable spacer tail that potentially could fold back onto the spacer at different positions (Figure 4). As expected, cleavage activity is abolished if the repeat is targeted with a strong competing complementary sequence, disrupting the pseudoknot structure. Interestingly, base pairing of the spacer-tail with the spacer-seed appeared not to affect Cas12a activity, indicating that after association with Cas12a, the designed stem-loop of the spacer part is ‘dissolved’ again. This spacer-tail design strategy can be very useful to rescue bad spacers, especially in editing efforts in which there is little flexibility with respect to the target site (e.g. base editing). To test this, a poorly-performing ‘impaired’ spacer was designed such that base pairing may occur between seven nucleotides of the spacer (positions 6–12) and the repeat, potentially resulting in formation of an undesired alternative structure (Figure 5). In two different crRNA constructs, the addition of a short spacer-tail (positions 20–24) that is complementary to spacer positions 9–14 indeed resulted in increased Cas12a cleavage efficiency, most likely through specific folding the tail back onto the spacer, thereby destabilizing the alternate structure and enhancing pseudoknot formation.
Further improvement of the Sp8 was achieved in a similar way as the ‘impaired’ spacer. By folding the tail of the spacer back onto the spacer itself, the efficiency was increased from below 50% (Figure 3B) to 64% (Figure 8). However, the targeting efficiency did not reach the top levels of e.g. Sp9 or Sp12 in pTarget3, of which detected fluorescence loss is 75–80% (Figure 1D). How far the improvement can be extended should be addressed in further studies.
By systematically analyzing the functionality of (pre-)crRNA variants, we have shown that in the context of both precursor and mature crRNA, the repeat can base pair with upstream and downstream sequences. Overall, our experimental findings and computational analyses (Supplementary data) support that the repeat's hairpin structure is a key requirement for high cleavage efficiencies. We introduced three crRNA features that are significantly correlated with experimentally determined plasmid loss efficiencies: pseudoknot stem-forming (high base pairing potential), pseudoknot loop-accessibility (low base pairing potential), and reversible spacer folding (base pairing potential of the first 19 nucleotides). Unfortunately, the currently available RNA folding algorithms can neither make an accurate prediction of all potential base pair possibilities, nor can they predict the (equilibrium constants between) different possible folding states. Nevertheless, we have tried to predict whether spacers get trapped in an undesired inactive state (no canonical pseudoknot), or rather in a desired active state (with canonical pseudoknot) (see Supplementary Figure S9). By doing so, we hoped to make a first step towards developing an algorithm for designing functional spacers for Cas12a. Although analysis of the used procedure mainly resulted in an indication of potentially incorrect folding of the crRNAs, it did not provide the desired robust estimation of their functionality (targeting efficiency).
In conclusion, although it may be difficult to pinpoint exactly why one spacer is more efficient than another, we can use our findings to propose some general guidelines for design of Cas12a-associated crRNAs. (I) Keep the pseudoknot structure intact. Folding of the pre-crRNA has a major impact on the DNA cleavage activity of Cas12a. Most importantly, the pseudoknot that corresponds to the palindromic part of the repeat should be formed correctly. Any stretch of RNA that is complementary to (part of) this repeat sequence may interfere with the pseudoknot formation. This includes any region upstream and downstream of the crRNA, as well as the spacer itself. Upstream and downstream regions are to be omitted as much as possible: the shorter the crRNA design the better (15). (II) Avoid base pairing between pseudoknot and spacer by imposing a structure onto the pre-crRNA. In case RNA structure prediction programs suggest base pairing between the repeat and the spacer, potential problems of such undesired base pairing can be reduced by introducing a 5–16 nt tail at the 3′ end of the 19 nt spacer. Too strong base pairing by the tail may result in an irreversible fold that will impede proper docking of Cas12a and/or formation of the R-loop configuration; on the other hand, too weak base pairing does not allow for the intended re-structuring of the pre-crRNA. As a rule of thumb, the overall strength of the spacer structure (not including pseudoknot) should range from –5 to –11 kcal/mol. Locally, high-GC stems towards the loop are less problematic than high-GC stems at the seed. Base pairing with the seed is not advised, unless the strength of the structure requires it. Tuning can be accomplished by introducing mismatches. Based on our observations, well-performing crRNAs may be obtained by designing stems of 3–4 bp, including 1–2 GC pairs, interspaced with a single mismatch. The hairpin loop should preferably be 4–5 nucleotides long, while the seed is kept free from base pairing. The shortest construct that can meet these guidelines is 19 nucleotides of target sequence of which the last 4–5 constitute the loop. Positions 20–29 should then form two interrupted stems of 4 bp, ending with a mismatch. Although spacers of which nucleotides 1–19 have the potential to base pair with the repeat or the seed may never reach the efficiency of other spacers, it should be possible to design a spacer-tail to gain sufficient functionality. (III) Introduce a well-structured RNA at the 3′ end. Ending with a terminator allows for the most accurate prediction of secondary structures, as the nucleotides of the terminator are unlikely to base pair anywhere else. Terminators should be separated from the direct repeat by at least 24 nucleotides of spacer to avoid steric hindrance. It should be kept in mind that the 3′ tail of the terminator may still elongate the terminator stem, which should be avoided. Ending with the highly structured mature repeat (last 19 nt of the direct repeat), a separation sequence that forms a hairpin, as well as the aforementioned highly structured terminator, maximizes the predictability of the pre-crRNA without the need for extra nucleotides attached to the spacer. Since the separation sequence used in this study does not form a strong hairpin, unlike the terminator, it could end up back-folding to the spacer. A terminator directly following the second mature repeat might solve such an issue, although we have not assessed the impact on the processing of the second repeat. (IV) Avoid intra-array complementarity. In case of a multiplex approach to target different sequences simultaneously, multiple spacers can be combined in a single array. However, such a design adds to the unpredictability of its fold and, therefore, of the efficiency of individual spacers. Should the application of the Cas12a demand an array, one needs to take care that the consecutive spacers do not interact with each other. Imposing a fold onto the pre-crRNA may be utilised to keep the spacers from interacting, but this does not work for two consecutive spacers with high homology.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Jorik Bot for experimental support at an early stage of this project.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Netherlands Organization for Scientific Research (NWO) by a TOP grant [714.015.001]; TTW grant [15804 to J.v.d.O.]; Veni grant [016.171.047 to R.H.J.S.]; Innovation Fund Denmark (Programme Commission on Strategic Growth Technologies) (to J.G.). Funding for open access charge: Nederlandse Organisatie voor Wetenschappelijk Onderzoek Stichting voor de Technische Wetenschappen [714.015.001].
Conflict of interest statement. None declared.
REFERENCES
- 1. Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., Van Der Oost J., Regev A. et al.. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR–Cas system. Cell. 2015; 163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doudna J.A., Charpentier E.. The new frontier of genome engineering with CRISPR–Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 3. Kim H., Kim J.S.. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014; 15:321–334. [DOI] [PubMed] [Google Scholar]
- 4. Sander J.D., Joung J.K.. CRISPR–Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014; 32:347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cox D.B.T., Platt R.J., Zhang F.. Therapeutic genome editing: prospects and challenges. Nat. Med. 2015; 21:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mohanraju P., Makarova K.S., Zetsche B., Zhang F., Koonin E. V., Van Der Oost J.. Diverse evolutionary roots and mechanistic variations of the CRISPR–Cas systems. Science. 2016; 353:556–568. [DOI] [PubMed] [Google Scholar]
- 7. Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S. et al.. High-Resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015; 163:1515–1526. [DOI] [PubMed] [Google Scholar]
- 8. Shalem O., Sanjana N.E., Zhang F.. High-throughput functional genomics using CRISPR–Cas9. Nat. Rev. Genet. 2015; 16:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang W., Ye C., Liu J., Zhang D., Kimata J.T., Zhou P.. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS One. 2014; 9:e115987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou H., Liu B., Weeks D.P., Spalding M.H., Yang B.. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 2014; 42:10903–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu W.Y., Lebbink J.H.G., Kanaar R., Geijsen N., Van Der Oost J.. Genome editing by natural and engineered CRISPR-associated nucleases. Nat. Chem. Biol. 2018; 14:642–651. [DOI] [PubMed] [Google Scholar]
- 12. Deltcheva E., Chylinski K., Sharma C.M., Gonzales K., Chao Y., Pirzada Z.A., Eckert M.R., Vogel J., Charpentier E.. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011; 471:602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fonfara I., Richter H., BratoviÄ M., Le Rhun A., Charpentier E.. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016; 532:517–521. [DOI] [PubMed] [Google Scholar]
- 15. Zetsche B., Heidenreich M., Mohanraju P., Fedorova I., Kneppers J., Degennaro E.M., Winblad N., Choudhury S.R., Abudayyeh O.O., Gootenberg J.S. et al.. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017; 35:31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Swarts D.C., van der Oost J., Jinek M.. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR–Cas12a. Mol. Cell. 2017; 66:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uusi-Mäkelä M.I.E., Barker H.R., Bäuerlein C.A., Häkkinen T., Nykter M., Rämet M.. Chromatin accessibility is associated with CRISPR–Cas9 efficiency in the zebrafish (Danio rerio). PLoS One. 2018; 13:e0196238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chari R., Mali P., Moosburner M., Church G.M.. Unraveling CRISPR–Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods. 2015; 12:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang T., Wei J.J., Sabatini D.M., Lander E.S.. Genetic screens in human cells using the CRISPR–Cas9 system. Science (80-.). 2014; 343:80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B.L., Xavier R.J., Root D.E.. Rational design of highly active sgRNAs for CRISPR–Cas9-mediated gene inactivation. Nat. Biotechnol. 2014; 32:1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ren X., Yang Z., Xu J., Sun J., Mao D., Hu Y., Yang S.J., Qiao H.H., Wang X., Hu Q. et al.. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Rep. 2014; 9:1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Malina A., Katigbak A., Cencic R., Maïga R.I., Robert F., Miura H., Pelletier J.. Adapting CRISPR/Cas9 for functional genomics screens. Methods Enzymol. 2014; 546:193–213. [DOI] [PubMed] [Google Scholar]
- 23. Moreno-Mateos M.A., Vejnar C.E., Beaudoin J.D., Fernandez J.P., Mis E.K., Khokha M.K., Giraldez A.J.. CRISPRscan: designing highly efficient sgRNAs for CRISPR–Cas9 targeting in vivo. Nat. Methods. 2015; 12:982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu H., Xiao T., Chen C.H., Li W., Meyer C.A., Wu Q., Wu D., Cong L., Zhang F., Liu J.S. et al.. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015; 25:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wong N., Liu W., Wang X.. WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015; 16:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. et al.. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR–Cas9. Nat. Biotechnol. 2016; 34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chu V.T., Graf R., Wirtz T., Weber T., Favret J., Li X., Petsch K., Tran N.T., Sieweke M.H., Berek C. et al.. Efficient CRISPR-mediated mutagenesis in primary immune cells using CrispRGold and a C57BL/6 Cas9 transgenic mouse line. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:12514–12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thyme S.B., Akhmetova L., Montague T.G., Valen E., Schier A.F.. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nat. Commun. 2016; 7:11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim H.K., Song M., Lee J., Menon A.V., Jung S., Kang Y.M., Choi J.W., Woo E., Koh H.C., Nam J.W. et al.. In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat. Methods. 2017; 14:153–159. [DOI] [PubMed] [Google Scholar]
- 30. Flynn J.M., Levchenko I., Seidel M., Wickner S.H., Sauer R.T., Baker T.A.. Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:10584–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McGinness K.E., Baker T.A., Sauer R.T.. Engineering controllable protein degradation. Mol. Cell. 2006; 22:701–707. [DOI] [PubMed] [Google Scholar]
- 32. Lorenz R., Bernhart S.H., Höner zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim H.K., Min S., Song M., Jung S., Choi J.W., Kim Y., Lee S., Yoon S., Kim H.. Deep learning improves prediction of CRISPR-Cpf1 guide RNA activity. Nat. Biotechnol. 2018; 36:239–241. [DOI] [PubMed] [Google Scholar]
- 35. Hu P., Zhao X., Zhang Q., Li W., Zu Y.. Comparison of various nuclear localization signal-fused Cas9 proteins and Cas9 mRNA for genome editing in Zebrafish. G3 Genes, Genomes, Genet. 2018; 8:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Strohkendl I., Saifuddin F.A., Rybarski J.R., Finkelstein I.J., Russell R.. Kinetic basis for DNA target specificity of CRISPR–Cas12a. Mol. Cell. 2018; 71:816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aird E.J., Lovendahl K.N., St. Martin A., Harris R.S., Gordon W.R.. Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun. Biol. 2018; 1:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.