


Abstract
Nucleosomal histones are barriers to the DNA repair process particularly at DNA double-strand breaks (DSBs). However, the molecular mechanism by which these histone barriers are removed from the sites of DNA damage remains elusive. Here, we have generated a single specific inducible DSB in the cells and systematically examined the histone removal process at the DNA lesion. We found that histone removal occurred immediately following DNA damage and could extend up to a range of few kilobases from the lesion. To examine the molecular mechanism underlying DNA damage-induced histone removal, we screened histone modifications and found that histone ADP-ribosylation was associated with histone removal at DNA lesions. PARP inhibitor treatment suppressed the immediate histone eviction at DNA lesions. Moreover, we examined histone chaperones and found that the FACT complex recognized ADP-ribosylated histones and mediated the removal of histones in response to DNA damage. Taken together, our results reveal a pathway that regulates early histone barrier removal at DNA lesions. It may also explain the mechanism by which PARP inhibitor regulates early DNA damage repair.
INTRODUCTION
Cells continuously encounter genotoxic stress that causes numerous DNA lesions on a daily basis (1). Among these lesions, DNA double-strand break (DSB) is one of the most deleterious types of lesions that need to be precisely repaired. Even if one DSB is not repaired, it will cause genomic instability and may induce tumorigenesis (2).
During evolution, cells have developed a sophisticated system to detect and repair DSB efficiently. Although DSB repair pathways have been well studied over the past few decades, the majority of such studies mainly focused on DNA metabolism at the sites of DSB. Notably, in eukaryotes, in addition to genomic DNA, a large number of proteins, such as nucleosomal histones, play important roles in DNA damage repair (3). Interestingly, by blocking the direct access to genomic DNA, histones act as barriers for transcription or replication machineries and therefore need to be efficiently removed from transcription and replication sites (4). Similarly, DNA damage repair machinery also needs direct access to the damaged DNA and the existence of nucleosomal histones at DNA lesions could be a barrier for successful repair of DSB. Thus, histones need to be evicted from DNA lesions for DSB damage repair (5,6). However, the underlying molecular mechanism of histone removal at DNA lesions remains elusive.
During the transcription and replication, signatory posttranslational modifications occur on histones (7), which are recognized by other functional partners as well as by chaperones for subsequent removal or deposition of histones (8–10). To date, numerous histone modifications have been identified to regulate transcription and replication (7,11,12). However, only a few of them have been implicated in DNA damage repair (13,14). One prominent histone modification that is linked to DNA damage repair is phosphorylation (15). In response to DSBs, histone H2AX, a variant of canonical H2A, is phosphorylated by a group of PI3-like kinases including ATM, ATR, and DNA-PK (16–18). Phosphorylation of H2AX occurs on Ser139, which serves as a platform to assemble and stabilize a group of DNA damage repair factors at the vicinity of DSBs before releasing them to broken DNA ends for repair (19). In addition to phospho-H2AX (aka γH2AX), H2A is also ubiquitinated at Lys13 and Lys15 following DSBs (20,21). It has been shown that a number of ubiquitin E3 ligases, such as RNF8 and RNF168, mediate DSB-induced H2A ubiquitination (ubH2A) (22). These ubiquitination events are downstream of H2AX phosphorylation as these E3 ligases including RNF8 and RNF168 are recruited to DSBs via γH2AX (23). Moreover, similar to γH2AX, ubH2A mediates the recruitment of DNA damage response factors to the vicinity of DSBs (22). Current evidence also supports histone H1 as the likely substrate of ubiquitination (24).
In addition to γH2AX and ubH2A, histones are also poly(ADP-ribosyl)ated at multiple sites by poly(ADP-ribose) polymerases (PARPs) in response to both single-stranded breaks (SSBs) and DSBs mediated DNA damage (25–30). Poly(ADP-ribosyl)ation (PARylation) is a unique posttranslational modification, occurring within seconds following DNA damage (31,32). It mediates early and fast recruitments of a number of DNA damage response factors to DNA lesions. As PARP1, the founding member of PARP family enzymes, is very abundant in nucleus, it is likely to serve as a key sensor to detect DNA lesions (33). This early and fast modification is also quickly digested by dePARylating enzymes such as PARG (34), so that DNA repair machinery will be able to access the broken DNA ends. Similar to other known histone modifications, PARylation regulates chromatin status (35). In its unique chemical structure, each ADP-ribose molecule contains two phosphate moieties that bring huge amount of negative charges to PAR chain. These negative charges may relax chromatin at DNA lesions by repelling adjacent negatively charged DNA molecules (36,37). Moreover, chromatin remodelers such as ALC1 are able to recognize PARylation at DNA lesions for chromatin remodeling in response to DNA damage (38,39). However, it still remains elusive if PARylation directly mediates histone removal at DNA lesions. Here, we have set up an inducible DSB system to characterize histone eviction at a single DSB site and revealed a PARylation-dependent pathway that mediates the recruitment of chaperones for histone removal at DNA lesions.
MATERIALS AND METHODS
Generation of an inducible DSB system
To introduce an I-SceI recognitionand cutting site in the X chromosome,long (∼5 kb) and short (∼3 kb) arms of from the X chromosome were amplified by PCR and inserted into the pKO-neo plasmid. The construct was then transfected in to HCT116 cells. Positive clones were selected by G418 selection. The cells from the single clone were further amplified and subjected to Southern blot for validation. The cells were then infected with a lent viral vector expressing I-SceI with a GRtag and the expression of I-SceI-GR was controlled by a tet-on promoter. The cells were then treated with doxycycline (1 μg/μl) to induce the expression of I-SceI-GR. At last, cells were treated with triamcinoloneacetonide (0.1 μM) to induce the translocation of I-SceI-GR from cytoplasm to nucleus to generate a single DSB.
Cell culture, transfection and laser micro-irradiation
HCT-116 and U2OS cell lines were used in this study. These cells were cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum and 1% penicillin (100 U/ml)/streptomycin (100 μg/ml) and incubated with 5% CO2 at 37°C in moist condition. Cells were transfected with the plasmids and siRNA using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's instructions. The sequences used for knock down were as follows: PARP1: GAGGAAGGUAUCAACAAAU; CAF1: GCACAGUCAUCAUUGAUUU; SSRP1: GCCAUGUCUACAAGUAUGA; ASF1A: CCGCAGGAAGGCAUAUGUUUGUA; RNF8: UGCGGAGUAUGAAUAUGAA; SPT16: GGCGGAAAGGAGAAGAUGA; Lig 4: GAACCACAAAGAUGUCACA; BRCA2: GAGGUGGAUCCUGAUAUGU; DAXX: GCCAGAGGAAGCAGUAGUU; and ANP32E: GGCUAAUGUGGAACUAAGU. For relocation kinetics analysis, we adopted laser microirradiation to generate DNA damage in cells. U2OS cells were transfected with GFP-tagged histone chaperones (ANP32B, ANP32E, ASF1A, ASF1B, CAF1, DAXX,SSRP1, SPT16, HIRA, RBBP7 and NASP) and then cultured on 35 mm glass-bottom dishes (MatTek Corporation). Laser micro-irradiation was performed using an inverted fluorescence microscope with the Micropoint Laser Illumination and Ablation System (Photonic Instruments).High energy UV laser (170 mJ at 10 Hz) was generated and focused on the nucleus through the light path of the microscope to yield DNA damage.
Reagents
The following inhibitors used in this study were purchased from Selleckchem: ATM inhibitor (KU60019), ATR inhibitor (VE822), DNA-PK inhibitor (NU7441) and PARP inhibitor (Olaparib). Antibodies against H2B (17–10054), H3.3 (MABE872), H3 (06-755), H4 (05-858), H2A.X (ABE1960) and H2A.Z (ABE1348) were purchased from Millipore.
Plasmid construction and lentivirus package
For the construction of theI-SceI-GR expressing vector, I-SceI-GR fragment amplified from the I-SceI-GR expressing vector (17654, Addgene) was inserted into pCW tet-on plasmid for lentivirus package. For lentivirus production, 293T cells were cultured in 10-cm dishes and 60% confluent cells then transfected with pCW-I-SceI-GR, pMD2g and psPAX2 plasmids. After 40 hours of incubation, the packaged virus-containing medium was harvested for subsequent use.
Extraction of cytoplasmic and nuclear proteins
Cells were resuspended in 1 ml of ice-cold PBS and then centrifuged at2000 rpm for 5 min at 4°C. After removing all supernatant, 6 volumes of cytoplasm extraction buffer (10 mM HEPES pH 7.5; 40 mM KCl; 2 mM MgCl2; 10% glycerol;1 mM NaPPi; 1 mM NaVO4; 1 mM NaF; 1 mM PMSF) was added to the pellet. Cells were crushed 25–30 times on ice and centrifuged at 2000 rpm for 5 min to collect the nuclei. The supernatant was then transferred to a new tube and centrifuged at 14 000 rpm for 15 min to collect the supernatant (cytoplasmic extract) for further analysis. To harvest nuclear fraction, 3 ml of 0.35 M sucrose buffer was added into 14 ml round bottom centrifuge tube. The nuclei pellet was resuspended in 5 volumes of 0.25 M sucrose buffer and then centrifuge tube was tilted to carefully remove the resuspended pellet in a layer over the 0.35 M sucrose buffer to clean the pellet. The tube was again centrifuged at 1500 g for 5 min at 4°C and supernatant was discarded. The pellet was finally re-suspended in buffer (10 mM HEPES pH 7.5; 500 mM NaCl; 1% Triton-X100;10% glycerol; 1 mM NaPPi; 1 mM NaVO4; 1 mM NaF; 1 mM PMSF) and vortexed for 15 min at 4°C followed by centrifugation at 14 000 rpm for 10 min and final, the supernatant was obtained a nuclear extract for further analysis.
Immunofluorescence and DNA FISH
Cells were treated with TA at the indicated incubation time, washed with PBS, fixed by 4% paraformaldehyde for 20 min and then permeabilized with 0.5% Triton X-100 for 10 min at room temperature. Following blocking with 5% goat serum and PBS washing, cells were incubated overnight in the primary antibody at 4°C. After completion of primary antibody incubation period cells were washed and incubated with FITC-conjugated secondary antibody for 1 h at room temperature in the dark. And then nuclei were counterstained with DAPI for 5 min followed by the addition of anti-fade mounting medium and finally cells, results were analyzed by using a fluorescence microscope. For the generation of DNA FISH probe, about 1-kilo base pairs downstream of I-the SceIsite were amplified by PCR and purified. PCR products were then subjected to biotin labeling according to the protocolof a nick translation kit (18247-015, Invitrogen). Following the IF steps, cells were post fixed with fresh 3% PFA for 10 min at room temperature after incubation with the corresponding secondary antibodies. Cells were washed twice in 2× SSC for 5 min and permeabilized in freshly made 0.1 M HCl/0.7% Triton for 10 min on ice. Cells were washed twice in 2× SSC for 5 min again and denatured in 50% formamide/2× SSC (pH 7.2) for 30 min at 80°C.Samples were washed by ice-cold 2× SSC for three times and hybridized with denatured probe overnight at 42°C. After four times of the wash and block,cells were incubated with fluorescently labeled streptavidin in blocking buffer for 40 min at room temperature. Finally, samples were stained with DAPI and observed using an inverted fluorescence microscope.
Chromatin immunoprecipitation (ChIP) and qPCR
For each group, 1 × 106 cells were cultured in one dish for each group. Added 37% formaldehyde to the medium and the final concentration is 1%. Cells were incubated at room temperature for 10 min and then the unreacted formaldehyde was quenched by adding 10× glycine to the medium at a final concentration of 125 mM for 5 min. The Medium was then discarded and washed with cold PBS twice. 1 ml ice-cold PBS containing protease inhibitor cocktail was added to the dishes and cells were scraped from each dish into a tube. Cells were pelleted by centrifuge and supernatant was removed. Cells were resuspended in 0.5 ml of SDS lysis buffer containing protease inhibitor cocktail. Cells were then sonicated on ice by using QSONICA sonicator for 10 min. The output of power is 30%. The sheared DNA was from 200 to 1000 bp based on this condition. The solution was then subjected to centrifuge at 12 000 g at 4°C for 10 min to remove insoluble material. 100 μl sheared DNA was moved to a new tube and mixed with 900 μl dilution buffer. 60 μl of protein G agarose beads were added to the solution and incubated for 1 h at 4°C with rotation. The beads were then collected at 3000–5000 × g for 1 min. Remove 10 μl of the supernatant was removed from the solution as input and stored at −20°C. The supernatant was moved to a new tube and incubated with immunoprecipitating antibody or irrelevant IgG at 4°C for overnight. 60 μl of protein G agarose beads were added to the solution for 1 h at 4°C with rotation. Beads were collected and sequentially washed twice with low salt buffer, high salt buffer, LiCl wash buffer and TE buffer. Beads were washed with each buffer twice. Then beads were incubated with 200 μl elution buffer at room temperature for 30 min. Supernatant and the stored input were added with 8 μl of 5 M NaCl and incubated at 65°C overnight to reverse the DNA–protein crosslink. 1 μl of RNase A was added to the supernatant and incubated for 30 min at 37°C. The supernatant was added with 4 μl of 0.5 M EDTA, 8 μl of 1 M Tris–HCl and 1 μl proteinase K and incubated at 45°C for 2 h to remove the proteins. The DNA was then purified following phenol: chloroform extraction and precipitation with 0.1 volume of 3 M sodium acetate, 1 μl glycogen and 2 volumes 100% EtOH. The purified DNA was eluted in 50 μl TE buffer and stored at −20°C. The immunoprecipitated DNA samples were analyzed by qPCR with primers listed in Supplemental Table 1. Primers for qPCR were designed based on the official nucleotide sequences from the NCBI database. Primer 3 online software was used to design the primers. CFX Connect™ Real-Time System (Bio-Rad) was used for qPCR amplification. The qPCR reactions were performed by 15 μl of 1:1 diluted iTaq™ Universal SYBR® Green Supermix (Bio-Rad) with three technical replicates. The cycling conditions were as follows: initial heat activation 95°C for 3 min, [95°C for 10 s, 55°C for 20 s] (40 cycles), and post-run melt-curve analysis was used to check amplicon purity. For the analysis of ChIP data on relative histone enrichment at the inducible DSB, the Ct value of each antibody group was subtracted from the Ct value of the correspondent IgG group. Then the ΔCt was calculated to fold enrichment. The results were normalized by untreated cells at each locus. For other ChIP analyses, we used the percent input method to estimate the quantity of enriched DNA (40,41) and included the IgG control as a dash line in each histogram.
Quantification of relative DSBs in I-SceI-GR system
We used qPCR assays to quantify the DSBs induced by I-SceI-GR. Paired primers used to amplify a DNA fragment are across the I-SceI site. If the site is digested by the I-SceI-GR, the primers cannot amplify the DNA fragment. The amplification of control groups (without TA treatment) was set as one, and the abundance of DSBs induced by I-SceI is calculated as a percentage compared to the control group. In addition, P7 primers amplified a fragment 16 kb downstream of the I-SceI site, which was used as an internal reference control for qPCR.
Southern blot
Genomic DNA of the model cells was isolated using a kit (Invitrogen, USA) for genomic DNA extraction. The genomic DNA was digested with SpeI overnight at 37°C and was subjected to agarose electrophoresis in a 0.7% agarose gel. Then, the DNA was transferred from the gel to the membrane. For the hybridization of the probes, the probes were labeled with 32P. One probe is targeting 500 bp upstream of the stop codon of the neo resistance gene, and the other probe is targeting the area of 500 bp which is 600 bp upstream of the second SpeI site (Chr X 102744527). After five times of stringent wash, the membrane was subjected to autoradiography.
Western blot
Cells were exposed to the indicated experimental conditions and time points before lysis using the SDS lysis buffer. The protein concentration was determined by the Bio-Rad protein assay kit (Bio-Rad, USA). The samples were then subjected to electrophoresis and transferred to the PVDF membrane followed by blocking in 5% milk in TBST buffer and then probed with indicated antibodies. The blots were visualized using an enhanced chemiluminescence detection system (Amersham, USA) and β-actin was used as a loading control.
Immunoprecipitation and dot blotting
U2OS cells were lysed with NETN-100 buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 2 mM EDTA, 0.5% Nonidet P-40) on ice for 30 min. Soluble fractions were subjected to immunoprecipitation and dot blotting and probed with anti-PAR antibody (Trevigen). For detecting histone PARylation, cells were lysed with 0.5% SDS and the lysates were diluted to 0.1% SDS for immunoprecipitation with anti-histone antibodies. The samples were subjected to dot blotting assay using anti-PAR antibody. To validate the recognition of SSRP1 to PARylated histone, cells were lysed with NETN-100 buffer and the lysates were immunoprecipitated with anti-SSRP1 or anti-FLAG antibodies. The immunoprecipitated proteins were eluted from the beads by 0.5% SDS and diluted to 0.1% SDS for the second round of immunoprecipitation with the anti-PAR antibody and blotting with anti-H2B antibody.
Statistical analysis
Data are represented as mean ± standard deviation (SD) obtained from three independent experiments. The significance of differences was evaluated by Student's t-test. *Statistically significant (P < 0.05). **Statistically significant (P < 0.01).
RESULTS
An inducible DSB in human genome
To examine the removal of histone barrier at DNA lesions, we established an inducible DSB system using HCT116, a diploid colon cancer cell line with wild type p53. HCT116 was derived from a male patient, thus has one X chromosome and one Y chromosome. Using conventional gene-targeting approach, we inserted one I-SceI site into the intergenic region in the long arm of the X chromosome with flanking genes transcribed actively (Figure 1A). With Southern blot, we confirmed that only one I-SceI site was inserted into the targeted region of genomic DNA (Supplemental Figure S1A). Next, we generated a plasmid construct encoding I-SecI fused with an N-terminal HA tag and a C-terminal glucocorticoid receptor (GR). The expression of the fusion protein is controlled by Doxycycline (Dox) induction system where cells express the inserted protein only after Dox treatment (Figure 1B and C, Supplemental Figure S1B). Because of the cytoplasmic nature of GR, the expressed fused protein was sequestered exclusively in the cytoplasm. However, when we treated cells with synthetic GR ligand triamcinolone acetonide (TA), it was recognized by GR and induced the relocation of HA-I-SceI-GR from the cytoplasm to the nucleus within 20 min. I-SceI is an endonuclease in the mitochondria of S. cerevisiae recognizing specific 18-nt sequence, which does not exist in the human genome. Of note, we inserted single copy of I-SceI site into the only X chromosome present in the male diploid cells to ensure the generation of a single DSB site. Thus, following the TA treatment, I-SceI generated only one DSB in the X chromosome in each cell, and we validated the I-SceI-induced DSB with qPCR. Using genomic DNA as the template, we designed a primer pair complementary to each side of the DSB. With mock treatment, this pair of primers amplified a DNA fragment from the genomic DNA template in a qPCR assay. However, following the TA treatment, with a control set of primers as the internal reference that is 16 kb downstream of the I-SceI site, we found that the DSB specifically abolished PCR amplification of DNA regions surrounding the I-SceI site (Figure 1D). Based on qPCR the results, we quantitatively measured the kinetics of I-SceI-induced DSB, and found that I-SceI site in most of the cells are digested within ∼30 min of the TA treatment (Figure 1D, Supplemental Figure S1C). Moreover, we stained the cells with a DNA FISH probe marking the I-SceI site. Since 53BP1 is a surrogate marker of DSB, we examined and found that the I-SceI site was positively stained by anti-53BP1 and anti-H2AX antibodies (Figure 1E), suggesting that the I-SceI-induced DSB has occurred.
Figure 1.

I-SceI induces a solo DSB in HCT116 cells. (A) A schematic diagram shows that an I-SceI site is inserted into the X chromosome of HCT116 cells using gene targeting approach. I-SceI is fused with a glucocorticoid receptor (GR) and an HA tag, and the expression of the fusion protein is controlled by Doxycycline (Dox). The relocation of the fusion protein from the cytoplasm to the nucleus is dictated by triamcinolone acetonide (TA) treatment. (B) Translocation of I-SceI-GR from cytoplasm to nucleus in response to the TA treatment. The relocation kinetics of I-SceI-GR was measured by immunofluorescence staining staining with anti-HA antibody following TA treatment. The percentage of positive nuclear staining is shown in the histograms. (C) Localization of I-SceI-GR was examined by Western blotting with indicated antibodies. GAPDH and Histone 4 (H4) were used as controls of protein loading for cytosol and nuclear fractions, respectively. (D) I-SceI-GR induced DSB was examined by qPCR. The primers at the ‘0’ position flank both sides of DSB. The primer set #7 at 16 kb downstream of the DSB was used as the reference control. (E) Upon TA induction, I-SceI-GR induces a DSB in the nucleus. DNA probe was used to map the I-SceI site (red dot), which was also stained with anti-γH2AX antibody or anti-MDC1 antibody to indicate the DSB (green dot).
Histone removal at vicinity of the I-SceI-induced DSB
To examine the histone status during DNA damage repair, we designed 10 sets of primers flanking both sides of the I-SceI-induced DSB from 0 to 16 kb away (Figure 2A). Using ChIP assays with anti-H2B and anti-H4 antibodies, we examined the nucleosomal histone status and found that histone removal occurred very quickly at the DSB. Histone removal was observed immediately after the TA treatment and, we found that histone removal has occurred and the kinetics of histone removal was highly similar to that associated with the kinetics of the I-SceI-induced DSB (Figure 2B) repair. We also examined the recruitment of Ku70 as a DNA damage marker by ChIP assay, and Ku70 could be rapidly recruited to the I-SceI-induced DSB (Supplemental Figure S2A). Similarly, the relative level of γH2AX was clearly increased at the vicinity of the I-SceI-induced DSB as well (Supplemental Figure S2B). Moreover, we found that nucleosomal histone was evicted up to the 4 kb away from the DSB (Figure 2C) in both directions. In addition to canonical histones, histone variants are involved in DSB repair. The typical example is H2AX, a variant of H2A. In response to DSB, H2AX is phosphorylated by PI-3 like kinases, including ATM, ATR, and DNAPK, at Ser139. The phosphorylated H2AX (aka H2AX) provides the platform to anchor DNA damage repair factors to repair DNA lesions (19). Thus, we examined the status of H2AX at the I-SceI-induced DSB. Consistent with earlier studies (42,43), we found that H2AX was removed from the center of DSB with a kinetics and range similar to other canonical histones, suggesting that H2AX does not exist at the center of DSB and may only mediate the recruitment of DSB repair factors to the vicinity of DSBs (Figure 2D) (44). In addition, we also examined the removal of H2A.Z and H3.3. And again, in agreement with the earlier studies (45,46), we found the eviction of these histone variants from the I-SceI-induced DSB similar to canonical histones, suggesting that all histone species are evicted quickly and non-specifically at DSBs (Figure 2D). To evaluate the impact of cell cycle on histone removal, we synchronized the cells at G1/S boundary using double thymidine block-release and examined the removal of histone H2B and H4 with ChIP assay. The results show that histone removal occurred in both G1 and S phase cells, suggesting that cell cycle during the interphase has a little impact on histone removal at the DSB (Supplemental Figure S3). In addition, we treated cells with Lig4 siRNA or BRCA2 siRNA to suppress either NHEJ or HR, or used MRE11 inhibitor to suppress DSB end resection (47–49). However, we did not find obvious suppression of histone removal (Supplemental Figure S4). These indicate that histone removal is not directly regulated by NHEJ, HR or DNA end resection.
Figure 2.
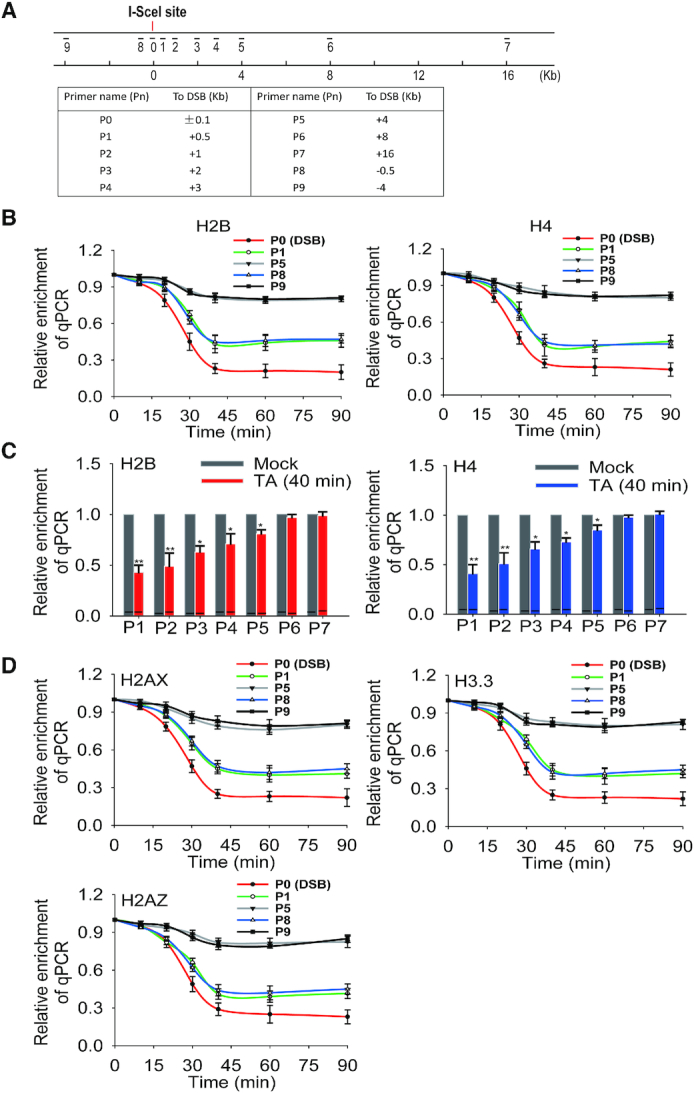
Histone eviction at vicinity of I-SceI-induced DSB. (A) A schematic diagram shows the positions of primer sets for ChIP assays with respect to DSB site. The sequences of the primers used in this study were listed in Supplemental Table 1. (B) Removal of nucleosomal histones at the vicinity of I-SceI-induced DSB. The histones H2B and H4 eviction was examined by ChIP assays with anti-H2B or anti-H4 antibodies and qPCR with different sets of primers. The results were analyzed and shown as mean ± SD. (C) Relative enrichment of H2B and H4 are summarized in the histograms. Data were shown as mean ± SD. * P < 0.05, ** P < 0.01. (D) Removal of histones H2AX, H3.3 and H2AZ at the DSB. ChIP assays were carried out with anti-H2AX, anti-H3.3 and anti-H2AZ antibodies for the histone removal analyses. The results were analyzed and shown as mean ± SD.
Poly(ADP-ribosyl)ation facilitates the early phase histone removal at the DSB
Next, we explored the regulation mechanism of histone removal at DNA lesions. Since histones are modified at N-terminal or C-terminal tails by posttranslational modifications including poly(ADP-ribosyl)ation, phosphorylation and ubiquitination in response to DNA damage, we examined if these posttranslational modifications regulates histone removal at the I-SceI-induced DSB. We treated the cells with PARP inhibitor olaparib to suppress poly(ADP-ribosyl)ation. Interestingly, we found that olaparib treatment transiently delayed histone removal at the I-SceI-induced DSB (Figure 3A). Moreover, we knocked down PARP1 using siRNA or overexpressing PARG to suppress endogenous PARylation, and obtained similar results (Figure 3A, Supplemental Figure S5, and Supplemental Figure S6), suggesting that poly(ADP-ribosyl)ation plays an important role in the early phase histone removal at DNA lesions.
Figure 3.
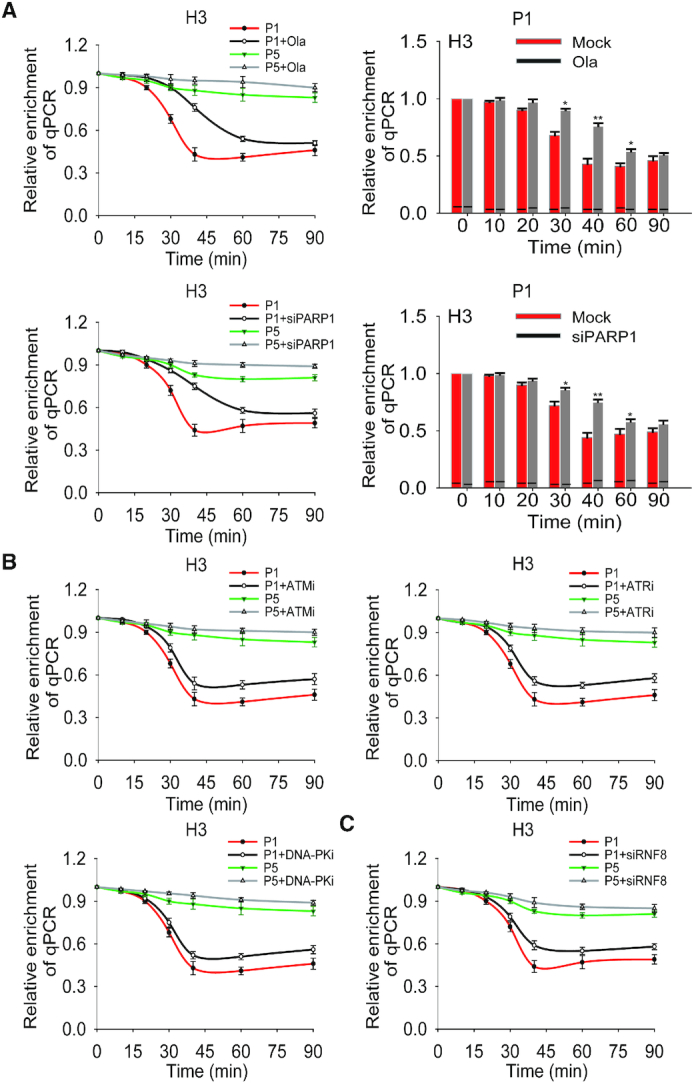
PARP inhibition delays histone removal at the I-SceI-induced DSB. (A) The impact of PARylation in histone removal at the I-SceI-induced DSB. The cells were treated with or without 1 μM olaparib or PARP1 siRNA. The histone removal was examined by ChIP assays with anti-histone H3 antibody. The qPCR assays were performed with primer set #1 and #5 (shown as P1 and P5 in Supplemental Table 1) at indicated time points. The results of qPCR from P1 were summarized in the histogram and shown as mean ± SD (right panel) (*P < 0.05; ** P < 0.01). (B) The effect of PI-3 like kinase-mediated phosphorylation on histone removal at the I-SceI-induced DSB. The histone removal was examined by ChIP assays following cells were treated with or without ATM inhibitor (ATMi, 5 μM KU60019), ATR inhibitor (ATRi, 1 μM VE822) or DNA-PK inhibitor (DNA-PKi, 1 μM NU7441). Results were analyzed and shown as mean ± SD. (C) The impact of ubiquitination on histone removal at the I-SceI-induced DSB. The cells treated with or without RNF8 siRNA were used for assessing histone removal by ChIP assays. Relative enrichment was shown as mean ± SD.
Since ATM, ATR and DNA-PK catalyze histone phosphorylation in response to DNA damage, we treated cells with specific inhibitors to individually suppress the enzymatic activities of ATM, ATR and DNA-PK. We found that these inhibitors treatment did not delay the histone removal kinetics, but slightly impaired overall histone removal (Figure 3B and Supplemental Figure S5). Earlier studies have shown that histone phosphorylations, especially γH2AX governs histone ubiquitination in response to DSBs. Since DNA damage-induced histone ubiquitination is mainly regulated by ubiquitin E3 ligase RNF8 (50,51), we knocked down RNF8 by siRNA (Supplemental Figure S5) and found that lacking RNF8, similar to the inhibition of PI-3 like kinases, knocking down RNF8 slightly suppressed the overall histone removal but did not affect the removal kinetics (Figure 3C).
Moreover, it has been reported that other histone modifications including acetylation and methylation may also be enriched at the vicinity of DNA lesions. Here, we examined these histone modifications at the I-SceI-induced DSB but did not observe their enrichment (Supplemental Figure S7). Collectively, these results show that poly(ADP-ribosyl)ation regulates the early phase histone removal at DNA lesions.
Histone chaperones mediate histone removal at DNA lesions
Next, we further explored the molecular mechanism underlying histone removal at DNA lesions. Since histone removal and deposition are mediated by histone chaperones, we examined which histone chaperone(s) may participate in DNA damage repair. We separately expressed GFP tagged histone chaperones, including ANP32B, ANP32E, ASF1A, ASF1B, CAF1, DAXX, the FACT complex (including SSRP1 and SPT16), HIRA, Rbap46, and sNASP, in U2OS cells and exposed them to laser microirradiation. Among these histone chaperones, only ASF1A, CAF1 and the FACT complex reached to DNA lesions (Figure 4A), suggesting that these histone chaperones may be involved in DNA damage repair. Among these three chaperones, only the FACT complex is able to quickly relocate to DNA lesions (Figure 4A), suggesting that the FACT complex may participate in the early phase DNA damage response. Next, we knocked down each chaperone by siRNA (Supplemental Figure S8) and examined histone removal at the I-SceI-induced DSB site. Consistent with the relocation kinetics to DNA lesions, only loss of the FACT complex impaired early phase histone removal (Figure 4B and Supplemental Figure S9A), suggesting that the FACT complex may be the histone chaperone for early phase histone removal. Moreover, we observed epistasis on histone removal following co-depletion of SSRP1 and PARP1 (Supplemental Figure S9B).
Figure 4.

Histone chaperones are recruited to DNA lesions. (A) The relocation kinetics of SSRP1, SPT16, ASF1A, and CAF1 to DNA lesions. GFP tagged histone chaperones were expressed in U2OS cells. The relocation was monitored in a time course following laser microirradiation. The relative EGFP intensity is shown as mean ± SD. (B) Loss of the FACT complex impairs histone removal from the DSB. The histone chaperone SSRP1 knockdown cells were harvested following I-SceI-induced DSB and subjected to ChIP assay using anti-H2A, anti-H2B, anti-H3 or anti-H4 antibodies. The samples were examined by qPCR with primer set #1 and #5 to examine histone removal. Relative enrichment of histones at the DSB was shown in the graphs. **P < 0.01.
Since some histone variants are not as abundant as the canonical histones, their chaperones may not be easily detected at DNA lesions using laser microirradiation. Thus, we further used the inducible DSB system to examine the potential recruitment of those chaperones. Interestingly, we found that ANP32E and DAXX could be mildly enriched at DNA lesions (Supplemental Figure S10A). However, like ASF1A and CAF1, loss of either ANP32E or DAXX did not affect early phase histone removal (Supplemental Figure S10B).
The FACT complex recognizes histone PARylation at DNA lesions
Since both PARylation and the FACT complex regulate early phase histone removal at DNA lesions, we dissected the functional correlation between PARylation and the FACT complex during DNA damage response (52). The human FACT complex contains two subunits SPT16 and SSRP1 (53). In the laser microirradiation assays, we found that both SPT16 and SSRP1 were quickly recruited to DNA lesions (Figure 4A). Interestingly, when cells were treated with olaparib to suppress PARylation, the recruitment of SPT16 and SSRP1 were suppressed (Figure 5A), suggesting that the FACT complex recognizes PARylation signals at DNA lesions. We also used the inducible DSB system to examine the recruitment of SSRP1 and found that the recruitment of SSRP1 was clearly suppressed following PARP inhibitor treatment (Supplemental Figure S11).
Figure 5.

The FACT complex recognizes PARylation at DNA lesions. (A) The FACT complex recognizes PARylation at DNA lesions. U2OS cells expressing GFP-SSRP1 or GFP-SPT16 were pretreated with or without 1 μM olaparib for 2 hours, followed by laser microirradiation. The relocation kinetics of GFP-tagged SSRP1 and SPT16 were monitored in a time course. Results were shown as mean ± SD. (B) SSRP1 mediates the recruitment of the FACT complex to DNA lesions. The DD mutants of SSRP1 and SPT16 with an N-terminal GFP tag were expressed in U2OS cells. The recruitment kinetics was examined. (C) Mapping the domain of SSRP1 that recognizes PARylation. GFP-SSRP1 with deletion of the MD domain (MD) or the C-terminal region (CTR) was expressed in U2OS cells. The recruitment kinetics was examined. (D) Histones are PARylated following DNA damage. 293T cells were treated with or without 1 mM MMS. Damage-induced histone PARylation was examined with indicated antibodies. (E) SSRP1 recognizes PARylated histones. 293T cells were treated with 1 mM MMS in the presence or absence of olaparib (1 μM). SSRP1-associated PARylated histones were analyzed with sequential IP with anti-SSRP1 and anti-PAR antibodies and Western blot with anti-histone antibodies (left panel). Immunoprecipitation assays were carried out with anti-SSRP1 antibody and analyzed by Western blot with anti-histone antibodies (right panel). (F) The C-terminal region (CTR) of SSRP1recognizes PARylated histones. Full-length SSRP1 and the CTR mutant were expressed in 293T cells. Cell lysates were examined by IP and Western blot with indicated antibodies. The input of H2B was used as a protein loading control. (G) The C-terminal region (CTR) of SSRP1 is a PAR-binding motif. Recombinant GST tagged SSRP1 and its mutants were generated from E. coli and incubated with PAR. PAR was detected by dot blotting with the anti-PAR antibody. (H) The CTR of SSRP1 mediates histone removal. The cells were treated with SSRP1 siRNA and simultaneously expressing siRNA-resistant full-length SSPR1 or CTR. Histone removal was examined by ChIP assays with anti-H2B or anti-H3 antibody.
SPT16 contains an N-terminal domain (NTD), a dimerization domain (DD), a middle domain (MD) and a C-terminal domain (CTD), whereas SSRP1 has an N-terminal dimerization domain (DD), a middle domain (MD) and a C-terminal region (CTR) (53). SPT16 forms a heterodimer with SSRP1 via the interactions between the DD of each protein. Next, we generated the internal DD domain deletion mutants (ΔDD) in either SPT16 or SSRP1 to abolish the complex formation (Supplemental Figure S12A), and expressed the mutants in U2OS cells (Supplemental Figure S13 and Supplemental Figure S12B). With laser microirradiation, we found that lacking the interaction region in SSRP1 did not affect the recruitment, whereas the ΔDD mutant of SPT16 abolished its relocation to DNA lesions (Figure 5B), suggesting that it is SSRP1 that mediates the whole complex to DNA lesions. We also used siRNA to knockdown SSRP1 and expressed siRNA-resistant SSRP1 or the ΔDD mutant of SSRP1 in the cells, and found that the full-length SSRP1 but not the ΔDD mutant rescued the recruitment of SPT16 to the laser strip (Supplemental Figure S12B). To identify the PARylation recognition region(s) in SSRP1, we deleted each domain of SSRP1 and found that lacking the CTR of SSRP1 abolished for the recruitment (Figure 5C)
Moreover, we treated 293T cells with MMS to induce global DNA damage response. In agreement with earlier studies (26,54), histones were PARylated (Figure 5D), and recognized by wild type SSRP1 (Figure 5E), but not by the CTR mutant (Figure 5F). Moreover, PARylated histones such as H2B were still part of nucleosome or at least in the histone dimer form (Supplemental Figure S14). We also generated recombinant full length SSRP1, the CTR, and CTR mutant proteins. With an in vitro binding assay, we found that the CTR could directly bind to PAR (Figure 5G). These results suggest that ADP-ribosylation facilitates the recognition of histones by CTR of SSRP1 following DNA damage.
Finally, we expressed full-length SSRP1 or the CTR mutant in SSRP1 knocked down cells, and found that only the full length SSRP1 but not the CTR mutant restored the early phase histone eviction at the I-SceI-induced DSB site (Figure 5H). Moreover, we replaced the CTR with the WWE motif of RNF146, another PAR-binding motif. This chimera was able to relocate to DNA lesions and mediates early phase histone removal (Supplemental Figure S15). In addition, we also performed laser microirradation assay to validate the role of SSRP1 and PARylation on histone removal at DNA lesions. Following SSRP1 siRNA, PARP1 deficient cell or PARP inhibitor treatment, the removal of histone H2B was remarkably suppressed, indicating that both SSRP1 and PARylation facilitate the removal of histones at DNA lesions during early DNA damage response (Supplemental Figure S16) (55). We also examined the role of SSRP1-mediated histone removal in DNA damage repair. Using GFP reporter assays, we found that the CTR of SSRP1 plays an important role in both NHEJ and HR (Supplemental Figure S17)
Taken together, these results suggest that SSRP1 recognizes histone PARylation and mediates early phase histone eviction at DNA lesions (Figure 6).
Figure 6.
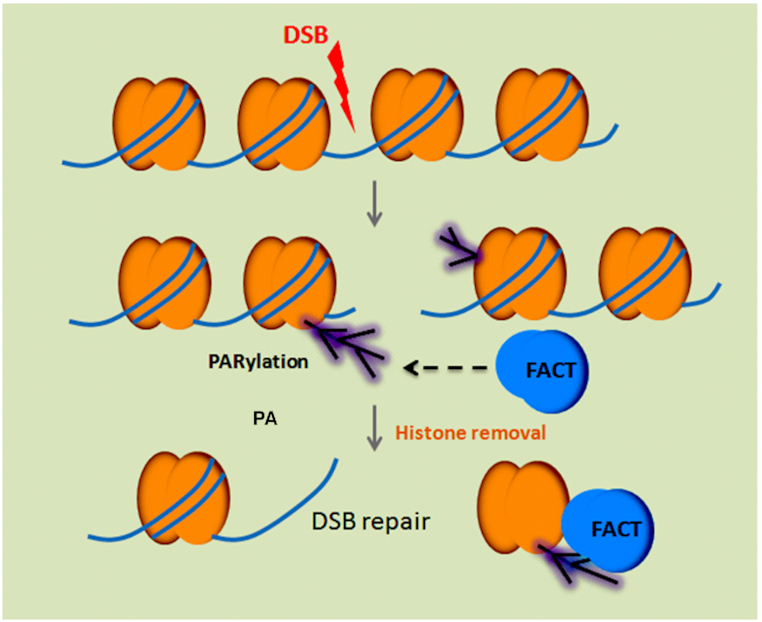
A schematic diagram to depict that the FACT complex recognizes PARylation and mediates histone removal. Following DNA damage, histones are PARylated and recognized by histone chaperone the FACT complex for their removal, which facilitates DNA repair processing.
DISCUSSION
In this study, we symmetrically examined histone removal at DNA lesions, which is an important step prior to their successful repair. Histones are a barrier for the repair DNA lesions as they may occupy the sites of DNA damage and block the access of damage site for repair factors. Thus, eviction of histones at DNA lesions occur extremely fast so that lesions can be timely repaired. In our study, we found that the kinetics of histone removal is almost identical to that of DSB induction. Moreover, we have shown that the removal of histones extends up to a few kb away from the sites of DNA damage. It is consistent with earlier studies on DSB repair (6). Since the histone removal is not regulated by cell cycle or different DSB repair mechanisms, it is possible that it regulates the high order of the chromatin architecture and indirectly facilitates DSB repair. In addition, we found that H2AX was also removed from the center of DSB, although H2AX is a surrogate marker of DSB. In fact, our results are in agreement with early studies using high-resolution fluorescence staining showing that H2AX only localizes on the peripheral of the DSB response region instead of the repair center (56). Thus, like other canonical histones, H2AX is also removed from the sites of DNA lesions for DSB repair to occur.
Moreover, we have shown that PARylation regulates early phase histone eviction. PARylation represents the first wave of the signal generated at DNA lesions, and histones are major PARylation substrates in response to DNA damage (37). The early phase histone removal mediated by PARylation may ensure proper DNA damage repair. It has been shown that PARP inhibitor treatment impairs timely DNA damage repair (57). One of the underlying mechanisms could be due to the delay in histone removal by PARP inhibitor treatment. PARylation sites on histones have been mapped and all the canonical histones are PARylated in response to DNA damage (27,29,30). Since there are quite a few sites of PARylation on histones, it is very likely that these modifications are interchangeable and play a redundant role in histone removal. Since PARylation itself brings a huge amount of negative charges to the chromatin, it may repel negatively charged DNA and facilitate histone removal (36,58). However, the release of histones from the chromatin needs histone chaperones. Here, we identified the FACT complex as the histone chaperone that recognizes their PARylation as an early signal for histone removal at DNA lesions. In the FACT complex, the CTR of SSRP1 interacts with PARylated histones. It has been reported that the CTR of SSRP1 facilitates the FACT complex to bind to nucleosome (53). Here, we provide evidence that the CTR specifically recognizes PARylated histones. With the known structure of ortholog of SSRP1, we performed structure modeling and reveal a possible PAR-binding site in the CTR (Supplemental Figure S18). However, the binding details should be further examined by structural analysis. Of note, the FACT complex also recognizes unmodified H2A/H2B dimer or H3/H4 dimer. Thus, ADP-ribosylation on histone is not essential for the interaction between the FACT complex and nucleosomal histones (Figure 5E). It only facilitates the histone eviction at the early phase of DNA damage response. Nevertheless, our results clearly demonstrate a pathway of histone removal at the sites of DNA damage (Figure 6). In addition to the FACT complex, other histone chaperones are also relocated to DNA lesions, but not mediated by PARylation. Thus, it is possible that PARylation only mediates early phase histone eviction, and other mechanisms mediate PARylation-independent histone eviction. Some of the chaperones (e.g. ANP32E) have been shown to mediate histone removal at DNA lesions (46,59). Moreover, other non-canonical histone chaperones (e.g. APLF, Swi/Snf, Ino80, and NURD) have been revealed over the past 20 years (60–63), and some of them (e.g. APLF) can even recognize PARylation at DNA lesions (64). Thus, it is possible that these histone chaperones function together with the FACT complex to remove histones at DNA lesions.
In addition to PARylation, DNA damage-induced phosphorylation and ubiquitination also regulate histone removal (6). However, different from PARylation, phosphorylation and ubiquitination do not significantly affect the kinetics of histone removal rather slightly impair overall histone removal. Since ATM, ATR, and DNA-PK may have a redundant role in DNA damage response, it is possible that suppression of each enzyme may not allow us to observe obvious defects on histone removal. Similarly, in addition to RNF8, other ubiquitin E3 ligases may compensate for the loss of RNF8 and facilitate histone removal at DNA lesions (51). Compared to PARylation, both phosphorylation and ubiquitination occur at the later stage of DNA damage response. Thus, it is possible that phosphorylation and ubiquitination may collaborate with PARylation for mediating histone eviction. In particular, suppression of PARylation only transiently delay the histone removal, suggesting that other signals may compensate for the loss of PARylation and activate histone removal. Since histones can be phosphorylated and ubiquitinated in response to DNA damage, specific histone chaperones may recognize these modified histones for their release from the sites of DNA damage (65,66). Future analysis of phosphorylation and ubiquitination at DNA lesions may reveal other histone removal pathways.
In addition to histone removal, some histones variants are also enriched at DNA lesions (67,68). However, it is not contradicted with our observation. In our system, we examined the early phase histone eviction during the initial damage response. We found that all the histones were removed at the sites of DNA damage, suggesting that most histones at DNA damage sites could be barriers for DSB repair. Meanwhile, specific histone variants such as H3.3 and H2A.Z may promote DSB repair (67,68). However, these histone variants are not the major histone species. Thus, it is possible that all the histones have to be removed non-specifically in the first step. Then, the specific histone species can be selectively deposited at DNA lesions to facilitate DSB repair in the next step. If canonical histones are not removed in the beginning, it is difficult for the enrichment of those specific histone variants at DNA lesions in the later stage.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Zhenkun Lou, Andre Nussenzweig and Jeremy Stark for sharing invaluable reagents.
Author contributions: X.Y. conceived and designed the project. G.Y., Y.C., X.L. S.H.C. and J.W. performed the experiments. G.Y. and S.H.C analyzed data. G.Y. organized figures. G.Y., A.K.S. and X.Y. wrote the manuscript. All authors read and reviewed the manuscript.
Notes
Present address: Yibin Chen, Bellicum Pharmaceuticals Inc., 2130 West Holcombe Boulevard, Suite 800, Houston, TX 77030, USA.
Present address: Jiaxue Wu, Life Science Institute, Fudan University, Shanghai, P.R. China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [CA130899, CA240392 and CA233664 to X.Y., in part] X.Y. is a recipient of Research Scholar Award from Tower Cancer Research Foundation and Translational Research Award from Pancreatic Cancer Action Network. Founding for open access charge: NIH [CA130899].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jackson S.P., Bartek J.. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Richmond T.J., Davey C.A.. The structure of DNA in the nucleosome core. Nature. 2003; 423:145–150. [DOI] [PubMed] [Google Scholar]
- 4. Grunstein M. Histone function in transcription. Annu. Rev. Cell Biol. 1990; 6:643–678. [DOI] [PubMed] [Google Scholar]
- 5. Ransom M., Dennehey B.K., Tyler J.K.. Chaperoning histones during DNA replication and repair. Cell. 2010; 140:183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hauer M.H., Gasser S.M.. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017; 31:2204–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bannister A.J., Kouzarides T.. Regulation of chromatin by histone modifications. Cell Res. 2011; 21:381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Avvakumov N., Nourani A., Cote J.. Histone chaperones: modulators of chromatin marks. Mol. Cell. 2011; 41:502–514. [DOI] [PubMed] [Google Scholar]
- 9. Burgess R.J., Zhang Z.. Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Biol. 2013; 20:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Q., Zhou H., Wurtele H., Davies B., Horazdovsky B., Verreault A., Zhang Z.. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell. 2008; 134:244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bar-Ziv R., Voichek Y., Barkai N.. Chromatin dynamics during DNA replication. Genome Res. 2016; 26:1245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ehrenhofer-Murray A.E. Chromatin dynamics at DNA replication, transcription and repair. Eur. J. Biochem. 2004; 271:2335–2349. [DOI] [PubMed] [Google Scholar]
- 13. Vidanes G.M., Bonilla C.Y., Toczyski D.P.. Complicated tails: histone modifications and the DNA damage response. Cell. 2005; 121:973–976. [DOI] [PubMed] [Google Scholar]
- 14. Tjeertes J.V., Miller K.M., Jackson S.P.. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009; 28:1878–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M.. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998; 273:5858–5868. [DOI] [PubMed] [Google Scholar]
- 16. Ward I.M., Chen J.J.. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001; 276:47759–47762. [DOI] [PubMed] [Google Scholar]
- 17. Burma S., Chen B.P., Murphy M., Kurimasa A., Chen D.J.. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001; 276:42462–42467. [DOI] [PubMed] [Google Scholar]
- 18. Stiff T., O’Driscoll M., Rief N., Iwabuchi K., Lobrich M., Jeggo P.A.. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004; 64:2390–2396. [DOI] [PubMed] [Google Scholar]
- 19. Celeste A., Fernandez-Capetillo O., Kruhlak M.J., Pilch D.R., Staudt D.W., Lee A., Bonner R.F., Bonner W.M., Nussenzweig A.. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 2003; 5:675–651. [DOI] [PubMed] [Google Scholar]
- 20. Mattiroli F., Vissers J.H., van Dijk W.J., Ikpa P., Citterio E., Vermeulen W., Marteijn J.A., Sixma T.K.. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012; 150:1182–1195. [DOI] [PubMed] [Google Scholar]
- 21. Gatti M., Pinato S., Maspero E., Soffientini P., Polo S., Penengo L.. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle. 2012; 11:2538–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Uckelmann M., Sixma T.K.. Histone ubiquitination in the DNA damage response. DNA Repair (Amst.). 2017; 56:92–101. [DOI] [PubMed] [Google Scholar]
- 23. Polo S.E., Jackson S.P.. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Gene Dev. 2011; 25:409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thorslund T., Ripplinger A., Hoffmann S., Wild T., Uckelmann M., Villumsen B., Narita T., Sixma T.K., Choudhary C., Bekker-Jensen S. et al.. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015; 527:389–393. [DOI] [PubMed] [Google Scholar]
- 25. Bonfiglio J.J., Fontana P., Zhang Q., Colby T., Gibbs-Seymour I., Atanassov I., Bartlett E., Zaja R., Ahel I., Matic I.. Serine ADP-ribosylation depends on HPF1. Mol. Cell. 2017; 65:932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leidecker O., Bonfiglio J.J., Colby T., Zhang Q., Atanassov I., Zaja R., Palazzo L., Stockum A., Ahel I., Matic I.. Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat. Chem. Biol. 2016; 12:998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vivelo C.A., Leung A.K.. Proteomics approaches to identify mono-(ADP-ribosyl)ated and poly(ADP-ribosyl)ated proteins. Proteomics. 2015; 15:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rouleau M., Aubin R.A., Poirier G.G.. Poly(ADP-ribosyl)ated chromatin domains: access granted. J. Cell Sci. 2004; 117:815–825. [DOI] [PubMed] [Google Scholar]
- 29. Zhang Y., Wang J., Ding M., Yu Y.. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat. Methods. 2013; 10:981–984. [DOI] [PubMed] [Google Scholar]
- 30. Martello R., Leutert M., Jungmichel S., Bilan V., Larsen S.C., Young C., Hottiger M.O., Nielsen M.L.. Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat. Commun. 2016; 7:12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haince J.F., McDonald D., Rodrigue A., Dery U., Masson J.Y., Hendzel M.J., Poirier G.G.. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008; 283:1197–1208. [DOI] [PubMed] [Google Scholar]
- 32. Yang G., Liu C., Chen S.H., Kassab M.A., Hoff J.D., Walter N.G., Yu X.C.. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018; 46:3446–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lautier D., Lagueux J., Thibodeau J., Menard L., Poirier G.G.. Molecular and biochemical features of poly (ADP-ribose) metabolism. Mol. Cell. Biochem. 1993; 122:171–193. [DOI] [PubMed] [Google Scholar]
- 34. Barkauskaite E., Jankevicius G., Ahel I.. Structures and mechanisms of enzymes employed in the synthesis and degradation of PARP-Dependent protein ADP-Ribosylation. Mol. Cell. 2015; 58:935–946. [DOI] [PubMed] [Google Scholar]
- 35. Beneke S. Regulation of chromatin structure by poly(ADP-ribosyl)ation. Front. Genet. 2012; 3:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Poirier G.G., de Murcia G., Jongstra-Bilen J., Niedergang C., Mandel P.. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. PNAS. 1982; 79:3423–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu C., Vyas A., Kassab M.A., Singh A.K., Yu X.. The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 2017; 45:8129–8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gottschalk A.J., Timinszky G., Kong S.E., Jin J., Cai Y., Swanson S.K., Washburn M.P., Florens L., Ladurner A.G., Conaway J.W. et al.. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. PNAS. 2009; 106:13770–13774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ahel D., Horejsi Z., Wiechens N., Polo S.E., Garcia-Wilson E., Ahel I., Flynn H., Skehel M., West S.C., Jackson S.P. et al.. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science. 2009; 325:1240–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haring M., Offermann S., Danker T., Horst I., Peterhansel C., Stam M.. Chromatin immunoprecipitation: optimization, quantitative analysis and data normalization. Plant Methods. 2007; 3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Milne T.A., Zhao K., Hess J.L.. Chromatin immunoprecipitation (ChIP) for analysis of histone modifications and chromatin-associated proteins. Methods Mol. Biol. 2009; 538:409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Iacovoni J.S., Caron P., Lassadi I., Nicolas E., Massip L., Trouche D., Legube G.. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010; 29:1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Clouaire T., Rocher V., Lashgari A., Arnould C., Aguirrebengoa M., Biernacka A., Skrzypczak M., Aymard F., Fongang B., Dojer N. et al.. Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Mol. Cell. 2018; 72:250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kinner A., Wu W., Staudt C., Iliakis G.. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008; 36:5678–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alatwi H.E., Downs J.A.. Removal of H2A.Z by INO80 promotes homologous recombination. EMBO Rep. 2015; 16:986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gursoy-Yuzugullu O., Ayrapetov M.K., Price B.D.. Histone chaperone Anp32e removes H2A.Z from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair. PNAS. 2015; 112:7507–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Critchlow S.E., Bowater R.P., Jackson S.P.. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol. 1997; 7:588–598. [DOI] [PubMed] [Google Scholar]
- 48. Xia F., Taghian D.G., DeFrank J.S., Zeng Z.C., Willers H., Iliakis G., Powell S.N.. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. PNAS. 2001; 98:8644–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Garcia V., Phelps S.E., Gray S., Neale M.J.. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature. 2011; 479:241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jackson S.P., Durocher D.. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell. 2013; 49:795–807. [DOI] [PubMed] [Google Scholar]
- 51. Schwertman P., Bekker-Jensen S., Mailand N.. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016; 17:379–394. [DOI] [PubMed] [Google Scholar]
- 52. Gao Y., Li C., Wei L., Teng Y., Nakajima S., Chen X., Xu J., Leger B., Ma H., Spagnol S.T. et al.. SSRP1 cooperates with PARP and XRCC1 to facilitate single-strand DNA break repair by chromatin priming. Cancer Res. 2017; 77:2674–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Winkler D.D., Luger K.. The histone chaperone FACT: structural insights and mechanisms for nucleosome reorganization. J. Biol. Chem. 2011; 286:18369–18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Messner S., Altmeyer M., Zhao H., Pozivil A., Roschitzki B., Gehrig P., Rutishauser D., Huang D., Caflisch A., Hottiger M.O.. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010; 38:6350–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Strickfaden H., McDonald D., Kruhlak M.J., Haince J.F., Th’ng J.P., Rouleau M., Ishibashi T., Corry G.N., Ausio J., Underhill D.A. et al.. Poly(ADP-ribosyl)ation-dependent transient chromatin decondensation and histone displacement following laser microirradiation. J. Biol. Chem. 2016; 291:1789–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chapman J.R., Sossick A.J., Boulton S.J., Jackson S.P.. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012; 125:3529–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. O’Connor M.J. Targeting the DNA damage response in cancer. Mol. Cell. 2015; 60:547–560. [DOI] [PubMed] [Google Scholar]
- 58. Wei H., Yu X.. Functions of PARylation in DNA damage repair pathways. Genomics Proteomics Bioinformatics. 2016; 14:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Formosa T. The role of FACT in making and breaking nucleosomes. Biochim. Biophys. Acta. 2012; 1819:247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mehrotra P.V., Ahel D., Ryan D.P., Weston R., Wiechens N., Kraehenbuehl R., Owen-Hughes T., Ahel I.. DNA repair factor APLF is a histone chaperone. Mol. Cell. 2011; 41:46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schwabish M.A., Struhl K.. The Swi/Snf complex is important for histone eviction during transcriptional activation and RNA polymerase II elongation in vivo. Mol. Cell. Biol. 2007; 27:6987–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Klopf E., Paskova L., Sole C., Mas G., Petryshyn A., Posas F., Wintersberger U., Ammerer G., Schuller C.. Cooperation between the INO80 complex and histone chaperones determines adaptation of stress gene transcription in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 2009; 29:4994–5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang Y., Ng H.H., Erdjument-Bromage H., Tempst P., Bird A., Reinberg D.. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999; 13:1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li G.Y., McCulloch R.D., Fenton A.L., Cheung M., Meng L., Ikura M., Koch C.A.. Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. PNAS. 2010; 107:9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pavri R., Zhu B., Li G., Trojer P., Mandal S., Shilatifard A., Reinberg D.. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell. 2006; 125:703–717. [DOI] [PubMed] [Google Scholar]
- 66. Wu M.Y., Lin C.Y., Tseng H.Y., Hsu F.M., Chen P.Y., Kao C.F.. H2B ubiquitylation and the histone chaperone Asf1 cooperatively mediate the formation and maintenance of heterochromatin silencing. Nucleic Acids Res. 2017; 45:8225–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xu Y., Ayrapetov M.K., Xu C., Gursoy-Yuzugullu O., Hu Y., Price B.D.. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell. 2012; 48:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Frey A., Listovsky T., Guilbaud G., Sarkies P., Sale J.E.. Histone H3.3 is required to maintain replication fork progression after UV damage. Curr. Biol. 2014; 24:2195–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.