


Abstract
Base editing (BE) is a powerful tool for engineering single nucleotide variants (SNVs) and has been used to create targeted mutations in cell lines, organoids and animal models. Recent development of new BE enzymes has provided an extensive toolkit for genome modification; however, identifying and isolating edited cells for analysis has proven challenging. Here we report a ‘Gene On’ (GO) reporter system that indicates precise cytosine or adenine base editing in situ with high sensitivity and specificity. We test GO using an activatable GFP and use it to measure the kinetics, efficiency and PAM specificity of a range of new BE variants. Further, GO is flexible and can be easily adapted to induce expression of numerous genetically encoded markers, antibiotic resistance genes or enzymes, such as Cre recombinase. With these tools, GO can be exploited to functionally link BE events at endogenous genomic loci to cellular enzymatic activities in human and mouse cell lines and organoids. Thus, GO provides a powerful approach to increase the practicality and feasibility of implementing CRISPR BE in biomedical research.
INTRODUCTION
Base editing (BE) is a powerful genome engineering tool that harnesses Cas9-mediated gene targeting to induce specific point mutations in DNA or RNA (1). Base editors consist of (i) a partially enzymatically disabled Cas9 protein (Cas9n, or ‘nickase’) to enable genomic targeting, (ii) a fused nucleobase deaminase to catalyze transition mutations and in some cases, (iii) one or more uracil glycosylase inhibitor (UGI) domains, which enhance base conversion by mitigating endogenous DNA repair activity (2). Cytosine base editors (CBEs) use APOBEC or AID deaminase domains to induce C>T (or G>A) mutations (3,4) while adenine base editors (ABEs), use an engineered bacterial protein, TadA, to introduce A>G (or T>C) changes (5). In theory, more than half of all pathogenic point mutations can be introduced or reversed by BE (2,5–7), making it a powerful approach to interrogate disease-associated single nucleotide variants (SNVs). Indeed, numerous studies have highlighted the power of BE to engineer defined alterations in cell lines, organoids, and in vivo in a diverse array of model systems (2–3,6,8–11).
Unlike Cas9, which shows remarkable efficacy in creating homozygous disruptive mutations following DNA double-strand breaks (DSBs), BE is relatively inefficient. BE activity depends on the level of enzyme expression, sequence context of the target site, cellular DNA repair, and likely other unexplained dependencies. Identifying and enriching BE events in cells is critical to streamline the use of these tools for biomedical research. To date, a number of BE reporters have been described, each using distinct mechanisms, but all ultimately based on the induction or suppression of GFP fluorescence. Hence, identification of base edited, live cells by BE reporters has thus far relied on fluorescence-based imaging or cell sorting (FACS) of a single fluorophore, somewhat limiting their broad application across different cell systems.
Here, we describe a flexible ‘Gene On’ (GO) functional reporter system that enables detection and enrichment of BE activity in living cells. We show that GO can be used to directly and quantitatively compare the efficiency, off-target activity, and PAM selectivity of existing and novel BE enzymes. Most importantly, because GO is based on translation initiation, it is not limited to the regulation of GFP, but enables the induction of different fluorescent and bioluminescent markers, antibiotic selection or functional enzymes, such as Cre-recombinase. Thus, GO is a specific, flexible and functional BE reporter that can streamline the application of base editing activity in primary and immortalized cell lines and organoids.
MATERIALS AND METHODS
Cloning
GFPGO-PGK-Neo lentiviral construct was generated by InFusion assembly (Clontech # 638909) of a custom GFP(ACG) gBlock cassette (Supplementary Table S1) into AcsI/AgeI-digested SGEN (Addgene #111171) backbone. AdGO-PGK-Neo and AdGO2-PGK-Neo viral constructs were generated by InFusion assembly of custom gBlock cassettes into EcoRI/NsiI-digested GFPGO backbone. GFPGO2 PAM variant constructs were generated by amplification of 99mer oligonucleotides with For and Rev primers (Supplementary Table S1) and InFusion assembly into EcoRI-digest GFPGO vector. mUGISGO and mUGISGO2 were generated by InFusion assembly of IRES and codon-optimized mScarlet-I polymerase chain reaction (PCR) amplicons into GFPGO backbone. A second version of mUGISGO was generated incorporating silent mutations in the first 100 bp of GFP to eliminate potential CTG alternate start sites. A PGK-Neo cassette was inserted downstream of the mScarlet-I cDNA using by InFusion assembly into SalI-digested mUGISGO vector. The mouse U6-sgRNA cassette (12) was generated using a custom gBlock from IDT and cloned into ‘all-in-one’ following PCR amplification and InFusion assembly. Luc2GO, BlasGO and CreGO constructs were generated by InFusion assembly of Luc2/Blas/CreGO PCR amplicons into digested LRT2B backbone containing mU6-sgRNA cassette and an sgRNA recipient site downstream of the human U6 promoter. Cre2GO was generated by replacing the first ∼450 bp of Cre with a custom gBlock (IDT) in an AscI/BamHI-digested CreGO backbone. FNLS-NG was generated using a custom gBlock of codon-optimized sequence incorporating Cas9-NG (13) mutations and assembled using InFusion into digested FNLS backbone (6). FNLS-HiFi was generated using InFusion assembly of 5′ and 3′ FNLS PCR amplicons, incorporating the R691A mutation (14) in the junction of these fragments. FNLS-HF1 and FNLS-HiFi-NG were generated by InFusion assembly of PCR amplicons from FNLS, HF1RA (6), FNLS-HiFi and FNLS-NG. All sgRNAs were cloned into the BsmBI site of either LRT2B or dual sgRNA recipient vectors (BlasGO and Cre2GO) using annealed oligonucleotides (Supplementary Table S2). Prime editing gRNA (pegRNAs) and nicking gRNA were designed based on guidelines described in Anzalone et al. (15) and cloned as a gBlock into the BsmBI/EcoRI site of LRT2B (6). All other viral vectors used were previously described (6). All plasmids described have made available at the non-profit repository Addgene.
Cell lines
HEK293T (ATCC CRL-3216), MDA-MB-231 (ATCC HTB-26), DLD1 (ATCC CCL-221) and NIH3T3 (ATCC CRL-1658) cell lines were purchased from the ATCC. Stocks were tested for mycoplasma routinely every 6 months and maintained in Dulbecco's Modified Eagle's Medium (DMEM, Corning # 10-013-CV) containing 1% Pen/Strep (Corning #30-002-Cl) and 10% FBS (231 and DLD1) or 10% FCS (3T3s) at 37C with 5% CO2. Mouse embryonic fibroblasts (MEFs) from the indicated genotypes were isolated at E13.5 and expanded for two passages before transfection. P53KO MEFs were generated by transient transfection of an LCG (Lenti-Cas9-GFP) plasmid containing a p53 gRNA using PEI and selected for p53 disruption by treatment with Nutlin3a at 10 µg/ml for 5–10 days. Absence of persistent Cas9 expression was confirmed by Western Blot. MEFs were maintained in DMEM containing 1% Pen/Strep and 10% FBS. Antibiotic selection was performed in complete media as follows: Puromycin (2 days at 1–2 µg/ml), Blasticidin (5 days at 5–10 µg/ml), Neomycin (3–4 days at 400 µg/ml).
HEK293T transfection
HEK293T cells were plated at 60% confluence in a 12-well and 16 h later transfected with 1 µg of editor expression vector and 0.5 µg LRT2B plasmid containing gRNA of interest in 100 µl DMEM using a 1:3 DNA:PEI ratio. Cells were washed 12 h later in complete media. Cells were assayed and collected 2–7 days post-transfection. For PE3, 0.5 µg of nicking gRNA in LRT2B was transfected 24 h after the primary transfection.
DLD1 transfection
DLD1 cells were plated at 60% confluence in a 12 well and 16 h later transfected with 3uL Lipofectamine 2000 (Thermo Fisher Scientific #11668030) and 1.5 ug of editor expression vector according to manufacturer's protocols.
Lentiviral transduction
HEK293T cells were plated at 85–90% confluence in a 6-well and 24 h later transfected with 2.5 µg of the vector of interest, 1.25 µg of PAX2 and 0.625 µg VSVg in 75 µl DMEM using a 1:3 DNA:PEI ratio. Cells were washed 24 h later in complete media and lentiviral supernatants were collected at 24, 48 and 72 h post-media replacement. HEK293T cells were cleared out by centrifugation and lentiviral supernatants were diluted 1:4–1:10 prior to target cell infection. Target cells were seeded at 40–60% confluence and incubated with the lentiviral supernatant dilution containing 8 µg/ml Polybrene overnight (16 h). Twenty-four hours later we replaced the viral supernatant dilution with complete medium and cells were assayed or subject to selection 48 h post-transduction.
Flow cytometry
We used a Thermo Fisher 2018 Attune NxT flow cytometer. Cells were trypsinized at the indicated time points and resuspended in 300 µl of complete medium. Flow cytometry assays were carried out in round-bottom 96-well plates and data were acquired at a flow rate of 500 µl/min. At least 25 000 events from the single cell population gating were recorded, and all experiments were performed in triplicates coming from independent transductions. Weill Cornell Flow cytometry core performed sorting using a BD FACS Aria II Cell Sorter and Sony MA900 cell sorter.
Luciferase activity
Cells were cultured in 96-well flat-bottom plates in 200 µl of complete media. Cells were stimulated by Dual-Glo Luciferase Assay System (Promega) and luminescence measured on 96-well plate reader.
Organoid culture, transfection and transduction
Murine small intestine organoids from the indicated genotypes were isolated and maintained as previously described (16). Isolation of murine pancreatic ductal organoids was done modifying previously described protocol (17). Briefly, pancreas was minced and washed in Hanks’s Balanced Salt Solution (Corning), and then incubated for 30 min at 37°C with Collagenase V to release the ducts. After washing twice with DMEM/10% FBS media, ducts were resuspended in ‘basal media’ [Advanced DMEM/F12 (Corning) containing 1% penicillin/streptomycin, 1% glutamine, 1.25 mM N-acetylcysteine (Sigma Aldrich # A9165-SG) and B27 Supplement (Gibco)] and mixed 1:10 with factor reduced (GFR) Matrigel (BD Biosciences). Forty microliters of the resuspension was plated per well in a 48-well plate and placed in a 37°C incubator to polymerize for 10 min. To culture ductal pancreatic organoids the ‘basal media’ described above was supplemented with 10 nM Gastrin (Sigma), 50 ng/ml EGF (Peprotech), 10% RSPO1-conditioned media, 100 ng/ml Noggin (Peprotech), 100 ng/ml FGF10 (Peprotech) and 10 mM Nicotinamide (Sigma). Note: Culture freshly isolated organoids in pancreatic organoid media (POM) containing 10 µM Rock inhibitor (Y2732) during 72–48 h. For subculture and maintenance, media was changed on organoids every 2 days and organoids were passaged 1:3 every 5 days. To passage, the growth media was removed and the Matrigel was resuspended in cold basal media and transferred to a 15-ml Falcon tube. Organoids were mechanically disassociated using a P1000 and pipetting 40 times. Five milliliters of cold phosphate-buffered saline were added to the tube and cells were then centrifuged at 1200 rpm for 5 min and the supernatant was aspirated. Cells were then resuspended in GFR Matrigel and plated as above. Organoids were transfected or transduced as previously described (18). Antibiotic selection was performed in complete media as follows: Blasticidin S (7 days at 5 µg/ml) and Neomycin (7 days at 100 µg/ml). All animal experiments were approved by the Weill Cornell Medicine Institutional Animal Care and Use Committee (IACUC) under protocol 2014-0038.
PCR amplification for sequencing
Target genomic regions of interest were amplified by PCR using primers in Supplementary Table S3. Endogenous off-target loci chosen were those with the highest GUIDEseq read counts (19) that also contain ‘edit-able’ cytosines in the gRNA. PCR was performed with Herculase II Fusion DNA polymerase (Agilent Technologies, Palo Alto, CA, USA, #600675) or EconoTaq PLUS (Lucigen #30033-2) according to the manufacturer's instructions using 200 ng of genomic DNA as a template and under the following PCR conditions: 95°C × 2 min, 95°C - 0:20 → 58°C - 0:20 → 72°C - 0:30 × 34 cycles, 72°C × 3 min (6). PCR products were purified using QIAquick PCR Purification Kit (Qiagen Cat.# 28106).
Sequencing
Sanger sequencing reactions were conducted at Eton Bioscience (Union, NJ, USA). Sanger sequencing chromatograms were visualized using Geneious version 10.2.6 created by Biomatters and analyzed using EditR (19). Targeted amplicon library preparation and NGS sequencing (MiSeq; 2 × 250bp) were performed at GENEWIZ, Inc. (South Plainfield, NJ, USA) and analyzed using CRISPResso2 (20). Raw MiSeq fastq files have been deposited in the sequence read archive (SRA) under accession PRJNA588416.
Statistical analysis
All statistical tests used throughout the manuscript are indicated in the appropriate figure legends. In general, to compare two conditions, a standard two-tailed unpaired t test was used, assuming variance between samples. In most cases, analyses were performed with one-way or two-way ANOVA, with Tukey's correction for multiple comparisons. Unless otherwise stated, each replicate represents an independent cell transfection or independently transduced cell line. All statistics are reported in Supplementary Table S4.
RESULTS
Development of a specific cytosine base editing reporter: CyGO
To detect cytosine BE events in real time in living cells, we sought to create a reporter that would become activated upon APOBEC-mediated C>T conversion, but not following Cas9-mediated indels. Protein translation requires an initiating AUG codon immediately downstream of a Kozak sequence (e.g. gccRccAUGG) in messenger RNA (mRNA) transcripts. Therefore, we hypothesized that the de novo creation of an ATG codon (from ACG) at the start of a cDNA would enable translational initiation and production of a specific protein detectable in individual cells with active cytosine BE (Cytosine Gene On, or ‘CyGO’; Figure 1A).
Figure 1.

GO detects and reports cytosine and adenine base editing with high sensitivity and specificity. (A) Schematic showing cytosine BE (CyGO) system containing (i) a cDNA with a silent ‘ACG’ start site (ii) FNLS base editor consisting of Cas9n tethered to APOBEC and (iii) sgRNA targeting the 5′ ‘ACG’ for cytosine deamination (sgGO). (B) Fluorescence imaging of DLD1s transduced with GFPGO, sgGO-tdTomato and Cas9 or FNLS. (C) Flow cytometry analysis of DLD1, 231 and 3T3 cells transduced with GFPGO and Cas9, FNLS or no editor (WT) 3 days after sgGO-tdTomato infection. GFP+ cells were gated on tdTomato+ cells. (D) Time course flow cytometry of GFP induction after sgGO-tdTomato transduction. (E) Sanger sequencing chromatograms of GFPGO locus in 3T3s containing GFPGO and Cas9, FNLS or no editor (WT) 3 days post-sgGO-tdTomato infection. (F) Correlation of C>T editing with percentage of GFP+ cells (gated on total cells). (G) (top) Sanger sequencing chromatograms depicting GFPACG target sequence in HEK293T with integrated GFOGO reporter under transfected BE and prime editing (PE) conditions. For PE, guide-linked RNA template and reverse transcriptase-fused Cas9 enzyme (PE2) (15) were used to generate the target ATG and secondary non-coding (G>C) change two base pairs upstream from the target cytosine to confirm PE-specific activity. Nicking sgRNA was transfected 24 h after first transfection for PE3. (bottom) Flow cytometry analysis of BE and PE transfected HEK293Ts with integrated GFPGO reporter 7 days after transfection. (H) (top) Schematic displaying an all-in-one CyGO containing (i) sgGO, (ii) GFP ‘ACG’ gene and (iii) constitutive marker mScarlet-I (mUGISGO). Including a GFP tagging 5′ nuclear localization signal and modified 5′ targeting sgRNA (sgGO2)-generated mUGISGO2. (bottom, left) Fluorescence microscopy of DLD1s expressing FNLS 3 days post infection with mUGISGO or mUGISGO2. (bottom right) Fluorescence and brightfield (BF) imaging of small intestinal murine organoids infected with mUGISGO 3 days after FNLS or no editor (WT) transfection. Dashed lines indicate auto-fluorescent lumen. (I) Schematic of adenine BE (AdGO) system containing (i) a cDNA with a premature ‘TAG’ or ‘TGA’ stop codon, (ii) ABE base editor and (iii) sgRNA targeting the 5′ cDNA at the early stop codon (sgAdGO). AdGO2 and sgAdGO2 were designed with a modified 5′ region. (J) Flow cytometry analysis of 231s expressing ABE and AdGO or AdGO2 quantifying GFP activation 3- and 8-days post sgAdGO-tdTomato or sgAdGO2-tdTomato infection. GFP+ cells were gated on tdTomato+ cells. (K) Sanger sequencing traces of AdGO and AdGO2 loci from 231s expressing ABE and AdGO or AdGO2 6 days after sgAdGO-tdTomato or sgAdGO2-tdTomato infection.
To test this idea, we generated a ‘silent’ GFPACG construct and integrated it in human and mouse cells via lentiviral transduction; as expected, cells transduced with a SFFV-GFPACG-PGK-NeoR (GFPACG) lentivirus showed no detectable GFP fluorescence (Supplementary Figure S1a). We then introduced an sgRNA targeting the 5′ region of GFPACG (sgGO) in a tdTomato-P2A-BlasR lentivirus (6) into GFPACG cells expressing either Cas9 or an optimized cytidine base editor (FNLS) (together, referred to as (GFPGO) (6). While Cas9/sgGO efficiently targeted the GFPACG cDNA, inducing indels in more than 60% of cases, we did not detect GFP fluorescence above background levels (Figure 1B and C; Supplementary Figure S1b). In contrast, FNLS-expressing cells showed a robust, cell line dependent, induction of GFP fluorescence in immortalized mouse fibroblasts (NIH3T3s) and human cancer cell lines, (DLD1s and MDA-MB-231s) (231s) (Figure 1B and C; Supplementary Figure S1c and 2a). In each cell line, editing occurred rapidly, approaching saturation within the first 48 h following sgGO transduction (Figure 1D). Targeted sequencing revealed a strong linear correlation of C>T (ACG>ATG) base substitutions with the percentage of GFP positive cells (Figure 1E and F), confirming the direct relationship between BE and GFP signal. As expected, there was no positive association of Cas9 or CBE-induced indels with GFP fluorescence (Figure 1F and Supplementary Figure S1b). Editing detection was not limited to a single GFPGO reporter or guide design. Changing the GO targeting sequence (sgGO2) and reporter construct (GFPGO2) enabled induction of a nuclear localized GFP (Supplementary Figure S1d and e), allowing more simple quantification of edited cells by image-based detection.
In addition to detection of BE events, GO can be adapted to quantify prime editing (PE), whereby targeted DNA alterations changes are incorporated using a guide-linked RNA template and reverse transcriptase-fused Cas9 enzyme (15). To ensure any GFP induction was due to template-guided repair, we introduced a second, non-coding (G>C) change two base pairs upstream from the target cytosine, in the Kozak sequence. In these experiments, editors (FNLS or PE2) and guide RNAs were transfected into HEK293T cells carrying a lentiviral integrated GO reporter. Flow cytometry analysis of cells one week following transfection showed clear induction of GFP-positive cells, relative to enzyme or guide only controls (Supplementary Figure S3a). As expected, PE showed ∼70% reduced efficiency compared to BE (Figure 1G), and was not increased by the use of an optimized sgRNA scaffold or addition of the second ‘nicking’ guide RNA (PE3) (Supplementary Figure S3b). Sanger sequencing confirmed the introduction of both G>C and C>T changes in GFP-sorted PE cells (Figure 1G).
To simplify delivery of the reporter components, we generated an all-in-one vector containing sgGO or sgGO2 under a murine U6 promoter (12), latent GFPGO or GFPGO2 cDNA and constitutively expressed marker (i.e. mScarlet-I) to identify transduced cells (mU6-GFPACG-IRES-mScarlet-I with GO or GO2, ‘mUGISGO/GO2’; Figure 1H). As described in the GFPGO/GO2 ‘two-vector’ systems, introduction of the all-in-one reporter mUGISGO or mUGISGO2 in FNLS-expressing cells showed rapid induction of GFP fluorescence (40–60%) not seen in cells with Cas9 or no editor (Figure 1H; Supplementary Figure S3c and d). As a final validation of the reporter system, we introduced the mUGISGO reporter into mouse intestinal organoids and observed GFP activation in mScarlet-I-positive cells only after transient transfection with FNLS (Figure 1H). Together, these data show that CyGO reliably and quantitively detects CBE-mediated C>T transitions but not Cas9-based indels in human and mouse cell lines and similarly detects base editing activity in organoids.
Adenine base editing activates modified GFPGO: AdGO
Adenine base editors catalyze the deamination of adenine to inosine, which is recognized as guanine during DNA replication, thus generating A>G transitions (5). We tested the flexibility of the GO reporter in detecting adenine base editing. We engineered a stop codon, TGA, at the 5′ end of a GFP cDNA (AdGO) to prevent translation extension, and a corresponding sgRNA targeting the 5′ region of GFPTGA (sgAdGO) (Figure 1I). Thus, A>G base substitution by an adenine base editor (ABE) at this site, creates a TGG (Trp) codon, preventing early translation termination and allowing GFP expression. In 231 cells stably expressing (i) ABE, (ii) sgAdGO, (iii) and the AdGO reporter, we detected robust GFP activation (Figure 1J; Supplementary Figure S3e and wardf); Cells lacking the base editor showed no GFP fluorescence (Supplementary Figure S3e-f). Again, GFP activation by adenine base editing was not limited to a single reporter/sgRNA combination as GFP activation was detected in a parallel reporter with a modified target (AdGO2) and sgRNA (sgAdGO2) (Figure 1J). AdGO and AdGO2 reporters were gRNA specific in that only their respective sgRNA resulted in GFP expression (Figure 1J) which corresponded to A>G base substitutions at the respective AdGO/GO2 locus (Figure 1K). Together, these data show that the GO reporter system can precisely detect both cytosine, adenine and prime editing in living cells.
GO enables a quantitative comparison of PAM flexibility and fidelity by BE enzymes
To date, efforts to measure PAM preferences of variant BE enzymes have relied on changing gRNA target sequences to accommodate the location of a given endogenous PAM site. This may introduce gRNA dependent effects on editing efficiencies when comparing PAM-flexible editors. To provide a system to directly compare editing efficiency with different PAM sites using the same sgRNA, we engineered three versions of the GFPGO2 reporter containing distinct PAM sites (NGG, NGC and NGT) (Figure 2A). We introduced these PAM reporters into 231, DLD1 and 3T3 cell lines expressing a series of codon-optimized BE enzymes containing Cas9 variants that have been reported to recognize NGN PAMs (Figure 2B) (13,21). As expected, FNLS showed efficient editing of targets with NGG PAMs in all cell lines, but minimal editing of other targets (Figure 2C and Supplementary Figure S4a). Similarly, xFNLS (containing xCas9 (21)) showed equivalent editing at NGG targets in 2/3 cell lines, and increased modification of NGT targets in the same cells, though not in 3T3s. FNLS-NG (containing Cas9-NG (13)) had the broadest editing scope, showing equivalent editing at NGG, NGT and NGC sites (Figure 2C). These data are consistent with recent reports quantifying DNA cleavage and BE of Cas9, xCas9 and Cas9-NG variants across different genomic loci (22,23). The relative efficiency of FNLS-NG was confirmed at an endogenous genomic locus (Apc.1405) for two overlapping sgRNAs that share the same target cytosine with NGG and NGT PAMs (Figure 2D).
Figure 2.

GO quantifies base editing activities of BE enzymes with various PAM specificities and fidelities. (A) Panel of GFPGO2 reporters with different PAM recognition sequences: NGG, NGT, NGC designed with constant 20 bp complementary target sequence at 5′ end of GFP cDNA for targeting by sgGO2-tdTomato. (B) Schematic of BE PAM variant enzymes included in experimental panel. (C) Flow cytometry analysis of GFP activation in (left) 231 and (middle) 3T3 cell lines expressing the panel of editors (B) and infected with each of the PAM GFPGO2 reporters 6 days after transduction with sgGO2 (notated by GFPGO2 PAM site). GFP+ cells were gated on tdTomato+ population. (right) Schematic showing the endogenous Apc locus (codon 1405) in 3T3 cells that contains a cytosine targetable by two nearly identical sgRNA with adjacent and distinct PAM recognition sequences, NGG and NGT. 3T3 cell lines expressing the panel of editors (B) were transduced with either of two nearly identical sgRNAs (NGG or NGT). Deep sequencing analysis of C>T editing events as a fraction of total reads at the Apc.1405 locus in the targeted panel of cells (bottom). (D) A panel of sgRNAs targeting the 5′ sequence of the GFPGO cDNA modified from sgGO were designed to include one to three mismatches across its complementary sequence. (E) Schematic of high fidelity BE enzymes included in experimental panel, including novel HiFi-NG enzyme created by combining mutations indicated to improve fidelity, and/or PAM flexibility. (F) 231 and 3T3 cells stably expressing GFPGO and panel of high-fidelity BE enzymes 3 and 6 days post-transduction with the panel of mismatch sgGOs in the LRT2B vector. GFP+ cells were gated on tdTomato+ cells.
In addition to assessing PAM selectivity, GO can be used to directly compare the fidelity of BE enzyme variants. Using a series of sgRNAs with one, two or three mismatched bases (Figure 2D), GO revealed significantly improved target specificity of three new high-fidelity variants: FNLS-HF1 and FNLS-HiFi, and FNLS-HiFi-NG (Figure 2E and F; Supplementary Figure SD4b and c) (14,24–25). In particular, HiFi-FNLS-NG, combines high specificity with PAM flexibility at both reporter and endogenous targets, while retaining relatively high on-target activity (Figure 2F; Supplementary Figure S4b and c). It is interesting to note that individual BE enzymes do not always perform similarly across different cell lines, with regards to PAM specificity and off-target activity. The reason/s for this are unknown, but we expect that the ease of direct BE measurement with flexible reporters such as this, without the requirement for targeted deep sequencing, will provide a feasible setting in which to explore the factors that govern BE activity and fidelity across different cell systems.
GO activates a variety of genetically encoded reporters
As translation initiation at ATG (or less commonly, CTG) is a universal feature of protein coding genes, induction by GO should be generally applicable to almost all genetically encoded reporters (26). To test the flexibility of GO in reporting BE by a fluorescent protein other than GFP, we generated a ‘silent’ mScarlet-IACG reporter (Figure 3A) engineered to have the same sequence as the 5′ end of GFPGO which is targeted by sgGO. We generated an all-in-one vector containing sgGO, the mScarlet-I cDNA with 5′ 20 bp complementary sequence to sgGO and a constitutively expressed neomycin resistance gene (ScarGO). As described for GFP, mScarlet-I expression was not detectable in WT 231 cells, but was efficiently induced in G418-resistant cells expressing FNLS (Figure 3A). ScarGO induction efficiency was similar to the GFPGO reporters in each of the three cell lines evaluated (3T3s, DLD1s and 231s) (Figure 3A; Supplementary Figure S2b and S5a-b). We next tested GO induction of a non-fluorescent marker by replacing the 5′ coding sequence of Luciferase 2 (Luc2) with the sgGO target sequence and silent ‘ACG’ start site (Luc2GO, Figure 3B). Transduction of both 231 cells and immortalized murine embryonic fibroblasts (MEFs) revealed a dramatic induction of Luciferase activity only in cells expressing FNLS (Figure 3B). Thus, GO can be adapted to a variety of genetically encoded reporter constructs, providing a flexible option for identification of BE activity in different cell systems and induction of a bioluminescent reporter that will enable imaging of in vivo BE activity.
Figure 3.

GO is highly modular and can be functionally exploited to enrich base editing activity. (A) (top) Schematic of an all-in-one mScarlet-I GO reporter with (i) modified 5′ end in the mScarlet-I cDNA to include the silent start site ‘ACG’ within the 20bp sgGO complementary sequence, (ii) sgGO and (iii) neomycin selection gene (together, ScarGO). (bottom, left) Fluorescence microscopy of 231s expressing no editor (WT) or FNLS 3 days after infection with ScarGO. (bottom, right) Flow cytometry of 231s expressing no editor (WT) or FNLS 3- and 8-days after infection with ScarGO. (B) (top) Schematic of an all-in-one-Luciferase GO reporter with (i) a modified 5′ end in the Luciferase2 cDNA to include the silent start site ‘ACG’ within the 20 bp sgGO complementary sequence and (ii) sgGO (together, Luc2GO). (bottom) Luminescence in 231s and immortalized MEFs expressing no editor (WT) or FNLS 3 days after infection with and without Luc2GO. (C) (top) Schematic of an all-in-one Blasticidin GO reporter with (i) modified 5′ end in the Blasticidin (Blas) resistance gene to include silent start site ‘ACG’ within the 20-bp sgGO complementary sequence (ii) sgGO and (iii) an endogenous targeting sgRNA under an independent U6 promoter (together, BlasGO). (bottom) GEMSA staining/colony forming assay of Blas-treated DLD1 cells with FNLS or no editor (WT) infected with BlasGO containing sgRNAs targeting EMX1 or no sgRNA. (D) Deep sequencing analysis of corresponding loci quantifying editing events as a fraction of total reads in DLD1 cells with BlasGO (containing gRNAs EMX1.259, FANCF.S1, CTNNB1.S33) (left) transduced or (right) transfected with and without FNLS or Blas treatment. (E) Quantification of editing events for two off-target sites corresponding to two endogenous gRNAs EMX1.259, FANCF.S within BlasGO in DLD1 cells (left) transduced or (right) transfected with FNLS (F) Ratio of on target to off target editing events for the OT sites with editing >1% in DLD1 cells transduced or transfected with FNLS. (G) Brightfield images of murine pancreatic organoids with inducible base editor enzyme infected with BlasGO and treated with and without Blas. (H) Sanger sequencing chromatograms of CR8 locus in pancreatic organoids expressing BE enzyme and infected with BlasGO (containing no gRNA or sgRNA targeting CR8) with and without Blas treatment. (I) Sanger sequencing chromatograms of Apc.1529 locus in pancreatic organoids expressing BE enzyme and infected with BlasGO (containing no gRNA or sgRNA targeting Apc.1529) with and without Blas treatment.
GO enriches endogenous base editing by induction of antibiotic resistance
Isolation of base edited cells with fluorescent reporters require cell sorting, which may be challenging for some primary cell types or organoid models or due to limited access to specialized equipment. We reasoned that induction of an antibiotic resistance gene would provide a simple and affordable way to select cells with active BE. To test this idea, we designed a silent ‘ACG’ Blasticidin (Blas)-resistance cDNA in an ‘all-in-one’ system containing sgGO under the control of a murine U6 promoter, and an empty sgRNA scaffold downstream of the human U6 promoter where user-specific sgRNAs can be integrated (12) (BlasGO, Figure 3C). We next cloned three well-characterized sgRNAs (EMX1.S259, FANCF.S1 and CTNNB1.S33) into the BlasGO reporter and transduced DLD1 cells. We chose DLD1 cells for these experiments as they show relatively low BE activity compared to other cell lines we have examined (Figure 1C), and thus represent a ‘difficult-to-edit’ cell system. DLD1 cells stably expressing FNLS showed robust outgrowth in the presence of Blas, and Blas-resistant cells had an average 2-fold increase in the frequency of C>T editing at each cytosine of the endogenous targets (Figure 3D). As we have previously observed in DLD1 cells (6), we also identified a large number of indels and non-C>T edits, though the relative frequency of each was highly target/sgRNA dependent (Supplementary Figure S5c).
Stable viral expression of BE enzymes drives efficient gene modification, even in the absence of selection; however, it may induce ongoing off-target effects (27), or cause immune-rejection of cells transplanted into recipient animals (28). Transient transfection of editors can bypass these effects but may also significantly reduce editing efficiency in difficult to transfect cell systems. Indeed, owing to low (5–10%) transfection efficiency and the integration of the BlasGO reporter in only a sub-population of cells, FNLS transfected BlasGO-DLD1 cells showed minimal evidence of editing in the bulk (unselected) population (Figure 3D and Supplementary Figure S5d); In most cases, around 1%. However, Blas-selected cells showed a dramatic 10- to 120-fold enrichment of C>T editing at each of the three endogenous target sites (EMX1.S259, FANCF.S1 and CTNNB1.S33) (Figure 3D and Supplementary Figure S5d). To determine whether this selection approach also increases the frequency of off-target mutagenesis, we measured editing at validated sgRNA-dependent off target (OT) sites for EMX1.S259 and FANCF.S1 sgRNAs (24). In cells transduced with FNLS, we found a considerable amount of cytosine editing at EMX1-OT2 (45%) and FANCF-OT1 (16%). Remarkably, despite similar enrichment for on-target editing in transfected cells, off-target editing was dramatically reduced (Figure 3E). For off-target sites with editing activity >1%, the ratio of on-target to off-target editing within the gRNA targeting window increased up to 50-fold (Figure 3F). Together, this suggests the transduction of BlasGO sgRNAs and transient expression of BE enzymes is an effective strategy to isolate edited cells without continual enzyme expression and in otherwise inefficient settings of BE activity.
We have previously used BE in organoids to engineer missense or nonsense mutations in Pik3ca or Apc, respectively (6,29). In these cases, modified organoids have a proliferative advantage under restrictive culture conditions and edited populations can be enriched by functional selection. However, isolation of rare BE events that provide no selective advantage in organoids is challenging, as it is difficult to either sort or clone primary cells. To test the functionality of BlasGO in selecting organoids with BE activity, we transduced pancreatic organoids carrying a doxycycline (dox) inducible BE enzyme (6) with BlasGO and treated organoids with Blas in the presence and absence of dox. As expected, only dox-treated (BE expressing) organoids survived Blas treatment (Figure 3G). We next transduced organoids with a BlasGO vector carrying a second sgRNA targeting an intergenic region on mouse chromosome 8 (CR8). Similar to what we observed in DLD1 cells, bulk (unselected) populations showed no evidence of targeted editing, while Blas-selected organoids had approximately 40% C>T editing, as measured by EditR software (19) (Figure 3H and Supplementary Figure S5e). Similarly, an sgRNA targeting Apc (Apc.1529;(6)) showed no detectable editing prior to selection, while Blas-selected organoids had ∼44% altered alleles (Figure 3I and Supplementary Figure S5e). Thus, in both cell lines and organoids, GO offers a practical and simple approach to engineer and study ‘selection-neutral’ mutations.
Surrogate Cre-mediated recombination with Cre2GO
For modeling disease, BE offers a range of possibilities when combined with existing genetic systems, such as Cre/LoxP. Linking these two distinct gene targeting events would provide a means to ensure both BE and Cre are active in the same cells. In theory, initiation of Cre expression by GO could trigger LoxP recombination only in cells with active BE, and those cells would have enriched editing at other endogenous genomic sites. To directly test this, we generated MEFs carrying a R26-LSL-tdTomato allele (30) that induces red fluorescence following Cre-mediated recombiation. Transduction of the all-in-one CreGO vector induced tdTomato in more than 70% of MEFs expressing FNLS, but also in nearly 40% of parental cells, suggesting leakiness of Cre expression in the absence of BE (Figure 4A and B; Supplementary Figure S6). We noted that in the first 100 amino acids of Cre there were a number of in-frame ATG (Met) and CTG (Leu) codons that could serve as alternate translation start sites (26). We therefore mutated each CTG>CTC (Leu>Leu) and each ATG>AGT (Met>Ser), creating Cre2GO (Figure 4A). Masking each of the potential start codons had a dramatic impact, effectively eliminating Cre activation in the absence of BE (<0.5% tdTomato-positive), but maintaining high levels of Cre induction (>80% tdTomato-positive) in FNLS-expressing cells (Figure 4B).
Figure 4.
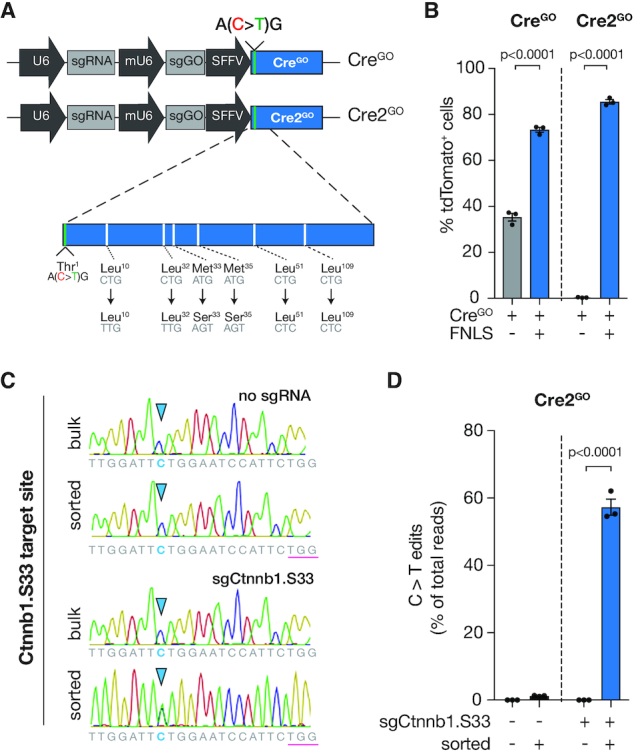
Surrogate Cre-mediated recombination using Cre2GO BE reporter. (A) (top) Schematic of all-in-one Cre recombinase GO reporter with (i) modified 5′ end in the Cre-recombinase cDNA to include silent start site ‘ACG’, (ii) sgGO and (iii) an endogenous targeting sgRNA (CreGO). (bottom) Schematic of modified CreGO reporter with masked, synonymous alternative start sites (Cre2GO). (B) Flow cytometry of immortalized MEFs containing an R26-LSL-tdTomato allele expressing FNLS-P2A-GFP or no editor and CreGO or Cre2GO reporters. FNLS expressing cells were gated on tdTomato+ cells within the GFP+ population. WT cells are gated on tdTomato+ cells. (C) R26-LSL-tdTomato MEFs expressing FNLS and Cre2GO with sgRNA targeting endogenous Ctnnb1.S33 or no sgRNAwere sorted for tdTomato expression. Sanger sequencing chromatograms are displayed for the bulk and sorted populations at the Ctnnb1.S33 locus. (D) Deep sequencing analysis of C>T edits at the Ctnnb1.S33 locus in MEFs described in C.
To test the enrichment of endogenous BE by surrogate Cre activity, we generated a Cre2GO reporter with a tandem gRNA targeting Ser33 of mouse Ctnnb1. We simulated restricted BE activity by low MOI transduction with FNLS-P2A-GFP (∼10% GFP-positive), and sorted tdTomato-positive cells after 6 days. As observed in GFPGO and BlasGO, bulk (unsorted) cells showed low levels of Ctnnb1.S33 editing (∼1%), while tdTomato-positive cells had enriched target cytosine editing to greater than 55% (Figure 4C and D).
Together, these data show that GO can tether base editing to antibiotic resistance or cellular enzymatic activity, which can be used to functionally enrich endogenous BE events.
DISCUSSION
Here, we describe a flexible BE reporter system that enables the identification and enrichment of cells and organoids with active cytosine or adenine BE using fluorescence, bioluminescence, antibiotic resistance or enzymatic markers. In conjunction with the expanding range of optimized BE enzymes, GO vectors can streamline the application of base editing for the generation of targeted genome manipulation in vitro and potentially in vivo.
We first demonstrated the flexibility and quantitative nature of GFPGO to profile a range of new BE variants, identifying the activity and specificity of PAM-flexible and high-fidelity enzymes. Most efforts to characterize PAM specificity and off-target activity use endogenous target sites that may influence BE activity due to local chromatin features and/or other unknown variables. These tests are critical for defining the real-world effect of specific sgRNAs, but are limiting for direct, quantitative comparisons of BE enzymes. Our experiments revealed cell-type and enzyme-dependent effects on editing efficiency at NGG and NGN PAM sites. While we tested a relatively small panel of BE enzymes and cell lines, GO could be deployed to quickly compare features newly generated enzymes in mammalian cells, optimize editing conditions across different cellular contexts, or broadly classify BE capability of large numbers of cell systems.
A number of recent studies have described BE reporters that induce or inhibit GFP expression by distinct mechanisms. For instance, DOMINO detects base editing by loss of GFP fluorescence (31), while BE-FLARE (32) and TREE (33) generate a fluorescent shift from BFP to GFP after cytidine deamination of codon 66. Finally, Martin et al. established a panel of GFP reporters that restore fluorescence upon C> T base editing at three target codons within GFP (34). Using an adaptable translation initiation approach, we demonstrate the activation of a range of non-GFP reporters. Activating fluorescence markers other than GFP (i.e. mScarlet-I) enables use of GO in systems already utilizing GFP or similarly emitting fluorophores. Alternatively, activating antibiotic resistance with GO facilitates enrichment of endogenous base editing event without the need for flow-based sorting. This is particularly useful in cell systems that are difficult or tedious to sort, such as primary cell types or organoids. It also enables enrichment of endogenous mutations that do not have a natural positive selection, thus increasing the practicality of using BE to model large numbers of disease variants. While we validated this approach using BlasGO, in theory the same strategy could be used for other selection markers such as NeoR, PuroR or HygroR.
While GO enables the robust enrichment of endogenous BE events, in most cases, we were unable to achieve more than 50–70% target editing. In some cases, this was due to non-C>T editing (i.e. C>A or C>G) or the generation of insertions and deletions (indels) at the target site (Supplementary Figure S5). It is also possible that our current version of GO is a particularly good sgRNA/target combination, increasing the likelihood of reporter activation with even low levels of BE activity. It is also important to consider that BE at endogenous target sites is highly dependent on the quality of each individual sgRNA and target site accessibility (35,36). In all, we evaluated GO as a surrogate marker for BE at six endogenous loci (three human, three mouse), and in each case saw marked enrichment of target editing. However, as each sgRNA is unique, there will be some targets that remain difficult to edit and enrich. Given these inherent variables, it will not be possible for any individual reporter to accurately reflect the potency of all sgRNAs. However, the use of sgRNA mismatches or different sgRNA sequences (i.e. GO2) to reduce reporter efficiency may improve enrichment strategies for difficult targets.
BE has enormous potential for in vivo modeling of pathogenic mutations but given the variability in editing efficiency across different cell types, it is still not clear in which contexts BE will work effectively. Directly tethering BE to activation of Luc2 or Cre will provide a clear readout for in vivo editing efficiency and will enable close integration with established Cre/LoxP systems. This is particularly timely as the first transgenic BE animal models are now available (37).
GO is a rapid and robust series of genetic tools (Supplementary Figure S7) that can be used to detect and quantify base editing activity, efficiency and kinetics. With simple modifications, GO can be adapted to initiate expression of a range of functional reporters, and thus, expands the usability and application of current base editing technologies to include enriching endogenous base editing and triggering a secondary enzymatic activity. We expect GO-based systems will enhance the feasibility of base editing as a go-to genome engineering approach, particularly in difficult-to-manipulate model systems.
DATA AVAILABILITY
Raw MiSeq fastq files have been deposited in the sequence read archive (SRA) under accession PRJNA588416.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Dow lab for advice and comments on preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authors' contributions: A.K. and M.F. designed and performed experiments, analyzed data and wrote the paper. J.Z., B.D., M.P.Z. and S.G. performed experiments and/or analyzed data. LED designed and supervised experiments, analyzed data and wrote the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health/National Cancer Institute [R01CA229773-01A1; T32 CA203702 to M.P.Z., in part]. Funding for open access charge: National Institutes of Health/National Cancer Institute [R01CA229773-01A1].
Conflict of interest statement. L.E.D. is a scientific advisor and hold equity in Mirimus Inc.
REFERENCES
- 1. Rees H.A., Liu DR.. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018; 19:770–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu DR.. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016; 533:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Komor A.C., Zhao K.T., Packer M.S., Gaudelli N.M., Waterbury A.L., Koblan L.W., Bill Kim Y., Badran A.H., Liu D.R.. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017; 3:eaao4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hess G.T., Tycko J., Yao D., Bassik M.C.. Methods and applications of CRISPR-Mediated base editing in Eukaryotic genomes. Mol. Cell. 2017; 68:26–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R.. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature. 2017; 551:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zafra M.P., Schatoff E.M., Katti A., Foronda M., Breinig M., Schweitzer A.Y., Simon A., Han T., Goswami S., Montgomery E. et al.. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018; 36:888–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y., Gao R., Wu J., Xiong Y.C., Wei J., Zhang S., Yang B., Chen J., Yang L.. Comparison of cytosine base editors and development of the BEable-GPS database for targeting pathogenic SNVs. Genome Biol. 2019; 20:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R.. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018; 36:843–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.-E., Lee H., Chung E. et al.. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018; 36:536–539. [DOI] [PubMed] [Google Scholar]
- 10. Song C.-.Q., Jiang T., Richter M., Rhym L.H., Koblan L.W., Zafra M.P., Schatoff E.M., Doman J.L., Cao Y., Dow L.E. et al.. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng. 2019; 4:125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chadwick A.C., Wang X., Musunuru K.. In vivo base editing of PCSK9 (proprotein convertase subtilisin/kexin type 9) as a therapeutic alternative to genome editing. Arterioscler Thromb. Vasc. Biol. 2017; 37:1741–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vidigal J.A., Ventura A.. Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nat. Commun. 2015; 6:8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H. et al.. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018; 361:1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J. et al.. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018; 24:1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A. et al.. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019; 576:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O’Rourke K.P., Ackerman S., Dow L.E., Lowe S.W.. Isolation, culture, and maintenance of mouse intestinal stem cells. Bio Protoc. 2016; 6:e1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huch M., Bonfanti P., Boj S.F., Sato T., Loomans C.J., van de Wetering M., Sojoodi M., Li V.S.W., Schuijers J., Gracanin A. et al.. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013; 32:2708–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schatoff E.M., Goswami S., Zafra M.P., Foronda M., Shusterman M., Leach B.I., Katti A., Diaz B.J., Dow L.E.. Distinct CRC-associated APC mutations dictate response to Tankyrase inhibition. Cancer Discov. 2019; 9:1358–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kluesner M.G., Nedveck D.A., Lahr W.S., Garbe J.R., Abrahante J.E., Webber B.R., Moriarity B.S.. EditR: a method to quantify base editing from sanger sequencing. CRISPR J. 2018; 1:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E. et al.. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019; 37:224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z. et al.. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018; 556:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller S.M., Wang T., Randolph P.B., Arbab M., Shen M.W., Huang T.P., Matuszek Z., Newby G.A., Rees H.A., Liu D.R. et al.. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat. Biotechnol. 2020; doi:10.1038/s41587-020-0412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim H.K., Lee S., Kim Y., Park J., Min S., Choi J.W., Huang T.P., Yoon S., Liu D.R., Kim H.H. et al.. High-throughput analysis of the activities of xCas9, SpCas9-NG and SpCas9 at matched and mismatched target sequences in human cells. Nat. Biomed. Eng. 2020; 4:111–124. [DOI] [PubMed] [Google Scholar]
- 24. Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K.. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016; 529:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rees H.A., Komor A.C., Yeh W.H., Caetano-Lopes J., Warman M., Edge A.S.B., Liu D.R.. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017; 8:15790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakahigashi K., Takai Y., Kimura M., Abe N., Nakayashiki T., Shiwa Y., Yoshikawa H., Wanner B.L., Ishihama Y., Mori H.. Comprehensive identification of translation start sites by tetracycline-inhibited ribosome profiling. DNA Res. 2016; 23:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim S., Kim D., Cho S.W., Kim J., Kim J.S.. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014; 24:1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thomas C.E., Ehrhardt A., Kay M.A.. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003; 4:346–358. [DOI] [PubMed] [Google Scholar]
- 29. Schatoff E.M., Goswami S., Zafra M.P., Foronda M., Shusterman M., Leach B.I., Katti A., Diaz B.J., Dow L.E.. Distinct colorectal Cancer-Associated APC mutations dictate response to tankyrase inhibition. Cancer Discov. 2019; 9:1358–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R. et al.. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010; 13:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Farzadfard F., Gharaei N., Higashikuni Y., Jung G., Cao J., Lu T.K.. Single-Nucleotide-Resolution computing and memory in living cells. Mol. Cell. 2019; 75:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coelho M.A., Li S., Pane L.S., Firth M., Ciotta G., Wrigley J.D., Cuomo M.E., Maresca M., Taylor B.J.M.. BE-FLARE: a fluorescent reporter of base editing activity reveals editing characteristics of APOBEC3A and APOBEC3B. BMC Biol. 2018; 16:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Standage-Beier K., Tekel S.J., Brookhouser N., Schwarz G., Nguyen T., Wang X., Brafman D.A.. A transient reporter for editing enrichment (TREE) in human cells. Nucleic Acids Res. 2019; 47:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin A.S., Salamango D.J., Serebrenik A.A., Shaban N.M., Brown W.L., Harris R.S.. A panel of eGFP reporters for single base editing by APOBEC-Cas9 editosome complexes. Sci. Rep. 2019; 9:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Daer R.M., Cutts J.P., Brafman D.A., Haynes K.A.. The impact of chromatin dynamics on Cas9-Mediated genome editing in human cells. ACS Synth. Biol. 2017; 6:428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jensen K.T., Floe L., Petersen T.S., Huang J., Xu F., Bolund L., Luo Y., Lin L.. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017; 591:1892–1901. [DOI] [PubMed] [Google Scholar]
- 37. Annunziato S., Lutz C., Henneman L., Bhin J., Wong K., Siteur B., van Gerwen B., de Korte-Grimmerink R., Zafra M.P., Schatoff E.M. et al.. In situ CRISPR-Cas9 base editing for the development of genetically engineered mouse models of breast cancer. EMBO J. 2020; e102169.doi:10.15252/embj.2019102169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw MiSeq fastq files have been deposited in the sequence read archive (SRA) under accession PRJNA588416.