

Abstract
Alu retrotransposons account for more than 10% of the human genome, and insertions of these elements create structural variants segregating in human populations. Such polymorphic Alus are powerful markers to understand population structure, and they represent variants that can greatly impact genome function, including gene expression. Accurate genotyping of Alus and other mobile elements has been challenging. Indeed, we found that Alu genotypes previously called for the 1000 Genomes Project are sometimes erroneous, which poses significant problems for phasing these insertions with other variants that comprise the haplotype. To ameliorate this issue, we introduce a new pipeline – TypeTE – which genotypes Alu insertions from whole-genome sequencing data. Starting from a list of polymorphic Alus, TypeTE identifies the hallmarks (poly-A tail and target site duplication) and orientation of Alu insertions using local re-assembly to reconstruct presence and absence alleles. Genotype likelihoods are then computed after re-mapping sequencing reads to the reconstructed alleles. Using a high-quality set of PCR-based genotyping of >200 loci, we show that TypeTE improves genotype accuracy from 83% to 92% in the 1000 Genomes dataset. TypeTE can be readily adapted to other retrotransposon families and brings a valuable toolbox addition for population genomics.
INTRODUCTION
Mobile element insertions (MEIs) are ubiquitous and important contributors to genomic variation between and within species (1–3). Active ME families continuously generate new insertions which segregate among individuals. Individual MEI results in structural variants between genomes that can lead to more complex chromosomal rearrangements through non-homologous recombination between parts or copies of the same ME family (4–7). Both MEI and ME-mediated rearrangements represent a substantial source of genomic instability, which has been implicated in more than 100 human cases (8). Conversely, ME activity also contribute to the emergence of adaptive genetic novelties (9–13).
In humans, recently mobilized MEs mostly include members of the LINE-1, Alu, and SVA families. Together these elements make up over a quarter of the human genome, but few remain polymorphic, i.e. being either present or absent between two genomes (14,15). Such polymorphic MEIs (pMEIs) account for hundreds to thousands of loci per individual (2,15–16). The extent of pMEIs segregating in the human population is yet to be determined, but Alu is known to be the most common source of human pMEIs. For instance, close to 20 000 Alu copies have been identified as segregating among 2504 humans sampled as part of the 1000 Genomes Project (1000 GP) (2,17).
Alu elements are powerful markers for genetic and evolutionary studies of human populations. As non-autonomous retrotransposons, Alus amplify through a copy-and-paste mechanism utilizing LINE-1 machinery (18) and are inherently incapable of precise excision, providing identical-by-descent loci virtually free of homoplasy (19). Accordingly, Alus have been shown to track human population history (16,20–22). Like most MEIs, Alu insertions in humans are usually thought of as neutral variants that achieve fixation in the population mostly through genetic drift (23,24). Alus are known to contribute to both Mendelian and complex diseases (8,25–26), in fact, >70 de novo Alu insertions are identified as causal variants. Furthermore, polymorphic Alu insertions have been identified as candidate causative variants in common polygenic diseases (27), and a handful have been shown to alter mRNA splicing (28). Finally, worldwide reference pMEI datasets such as those produced by 1000 GP (2) can be used in conjunction with gene expression data (e.g. RNA-seq) to identify loci associated with changes in gene expression (29,30). Together these studies suggest that pMEIs, and Alus in particular, play an important, yet still underappreciated role in human phenotypic variation.
Recognizing the abundance and biological significance of MEIs, a growing number of software packages have been developed in the past few years to detect and map pMEIs in whole-genome resequencing (WGS) data relative to a reference genome (31). For studies of human pMEIs, Tea (32), Retroseq (33), Mobster (34), Tlex2 (35), RelocaTE2 (36), STEAK (37), MELT (17), TranSurVeyor (38), polyDetect (39), ERVcaller (40), TEBreak (41) and AluMine (42) are among the most recent software tools available. The algorithmic refinement dedicated to accurately detecting pMEIs, and Alus in particular, in WGS data has led to an increase of the quality of the calls. Notably, the accurate detection of the presence or absence of a specific Alu at a precise breakpoint has improved substantially in recent years (17,40,43).
Although the discovery of Alu and other pMEI alleles is generally benchmarked extensively when these methods are evaluated, far less attention has been paid to individual biallelic genotyping, i.e. determining whether the insertion is a homozygote or heterozygote for each pMEI locus. Genotyping accuracy is critical for phasing insertion polymorphisms with single nucleotide polymorphisms (SNPs), relating insertions with expression quantitative trait loci (eQTL) and identifying disease-risk loci using genome-wide association studies (GWAS). Similarly, accurate genotypes are necessary to infer the effects of drift and selection on allele frequencies. However, genotyping accuracy of pMEI released with the 1000 GP dataset (2) was solely estimated comparing calls to 250-bp reads (concordance estimated to 98%), which are too short to capture a typical Alu insertion (∼300 bp). To our knowledge, only a handful pipelines, including MELT (17), polyDetect (39), ERVcaller (40), TEBreak (41) and AluMine (42) are the only maintained tools that directly allow genotyping for non-reference pMEIs. Moreover, MELT and AluMine appear to be the only software currently offering the option to directly genotype reference pMEIs (i.e. elements present in the reference genome but segregating in the population, often at higher frequencies than non-reference pMEIs). However, neither MELT nor other tools have been subject to a systematic assessment of their genotyping performance. Given the ever-growing number of resequencing efforts, there is a pressing need to develop highly accurate genotyping tools to enhance the diverse methods available to detect pMEIs.
To fill this gap, we have developed a new bioinformatics pipeline, TypeTE, which improves the genotyping of pMEIs discovered by tools like MELT in whole genome resequencing data. Our method is based on the accurate recreation of both the presence and absence of pMEI alleles before the remapping of reads for genotyping. We apply TypeTE to both low- and high-coverage sequence data from the phase 3 of the 1000 GP (2) and the Simons Genome Diversity Project (SGDP) (44), respectively. We benchmarked the results against a unique collection of more than 200 PCR-based genotyping assays, which shows that applying TypeTE significantly improves genotype accuracy. By applying TypeTE to all polymorphic Alu insertions discovered in 445 human samples used for the 1000 GP phase 3 (low-coverage WGS) and the Genetic European Variation in Disease Consortium (GEUVADIS) (45), we provide a new genotype dataset in the VCF format (variant call format; see Data Availability section) that will facilitate the functional and evolutionary analysis of polymorphic Alu insertions.
MATERIALS AND METHODS
Pipeline implementation
Non-reference MEI
TypeTE-non-reference is designed to genotype insertions absent in the reference genome (Figure 1A, Supplementary Figure S1). Based on the information provided in a VCF file (and based on the format produced by MELT), the location and orientation of each Alu insertion are first collected. For each breakpoint, reads that are mapped in a window of 500 bp (250 bp upstream and downstream of the breakpoint) are extracted for each sample from its alignment file (BAM). The mates of discordant reads (mapping somewhere else in the genome) are also extracted from the BAM file of each individual. The reads from all individuals at each studied locus are then combined, and a local de-novo assembly with all the reads is attempted using SPAdes v3.11.1 (46). Minia (v2.0.7) (47) is used as an alternate assembler when SPAdes fails to generate an assembly of the sequences (‘scaffolds.fasta’). If the locus do not provide enough reads for a complete assembly of the Alu insertion, the genomic locations where the discordant read mates are mapped are identified and intersected with the respective RepeatMasker track (we used the coordinates version hg19 for 1000 GP data and hg38 for the SGDP data; RepeatMasker track generated using Repbase version 20140131 for the UCSC genome browser). Using a majority rule, the most likely Alu subfamily consensus for the copy inserted at that locus is identified. To verify orientation and identify target site duplications (TSDs), a hallmark of Alu insertions, homology-based searches are performed. First, the identified Alu consensus of a given locus is searched with blastn (v. 2.6.0+) against the assembled contigs. Then, a second blastn is performed using the sequence of the reference genome (500 bp window centered on the pMEI breakpoint) against the assembled contigs. The contig with the highest score when searched against the Alu and the reference sequence is selected and searched for target site duplications flanking the MEI. To identify the strand of the MEI, the sequence flanking the insertion in the contig is further compared with the reference sequence. For each MEI, the two alleles are reconstructed as follows: a new, larger window of ±500 bp is extracted upstream and downstream of the breakpoint predicted by MELT. This represents the ‘absence’ allele. To recreate the ‘presence’ allele, TypeTE first removes the predicted TSDs from the extracted reference sequence and inserts the fully assembled MEI with its two TSDs in the correct orientation. If the assembly fails to generate a complete sequence of the MEI with flanking TSDs, the TSD predicted by the transposable element (TE) detection program (in our case MELT) is duplicated and placed at the 5′ and 3′ end of the consensus MEI in the composite allele.
Figure 1.

Overview of the TypeTE pipeline. TypeTE is divided in two main scripts. The first (A) genotypes non-reference insertion (TypeTE-nonref) and the second (B) genotypes reference pMEI (TypeTE-ref). (A) TypeTE-ref creates the reference allele (REF) by extracting ±500 bp from the Alu predicted breakpoint. The alternate allele (ALT), corresponding to the pMEI presence is made by (1–2) removing the predicted TSD from the ±500 bps extracted sequence. Then, for each locus, read pairs (including discordant mates) are extracted from the individual bam files and are pooled for local assembly (3). If TSDs are identified in the assembly, the sequence is then inserted onto the flanking (4). In case the assembly is incomplete, the Repbase consensus for the predicted TE family is inserted instead (4). (B) The REF allele is created after extraction of ±500 bp from the 5′ and 3′ ends of the adjusted Alu position (including TSDs). The ALT allele is then created removing the Alu sequence and 1 TSD from the same extracted sequence. (C) Genotyping. For each locus, read-pairs of each sample are extracted in a 500 bps window centered on the predicted breakpoint. For each sample, these reads are then mapped to the two alleles and genotype likelihood are computed.
Reference TE
TypeTE-reference determines genotypes of Alus present in the reference genome that are absent in other individuals (Figure 1B, Supplementary Figure S2). In this case, no read extraction is necessary to reconstruct the insertion allele. However, the exact coordinates and TSDs of each pMEI present in the reference genome are reassessed as follows: the breakpoints identified from MELT for the location of the reference TE are further refined using the corresponding RepeatMasker annotation track to identify the exact location and orientation of each TE inserted in the reference genome. At first, the Alus sequences found within ±50 bp of the predicted MELT breakpoints are extracted from the reference genome. If none is found within that boundary, Alus within ±110 bp of the predicted breakpoints are collected. However, we did not find any difference in the number of elements identified after increasing the boundary up to 200 bp. The flanking sequence of the Alu insertion is also extracted and the TSDs’ coordinates are identified whenever possible. Then, based on these new coordinates, a region of ±500 bp upstream and downstream of the 5′ and 3′ end of the Alu locus is extracted from the reference genome to represent the ‘presence’ allele. The ‘absence’ allele is defined by removing the Alu sequence as well as one TSD from the extracted locus.
Genotyping
TypeTE automatically generates input files and parallelizes the method developed by Wildschutte et al. (48), called insertion-genotype, to genotype each Alu insertion in every individual. Briefly, read-pairs with at least one read mapping to the target locus are extracted and mapped against the reconstructed ‘presence’ and ‘absence’ alleles using bwa (v. 0.7.16a) (49). The number of reads that align to each allele, and their associated mapping quality values are tabulated and likelihoods for the three possible genotype states are calculated (50). Reads that map equally well to the empty and insertion alleles are assigned a mapping quality of 0 by bwa (49) and do not contribute to this calculation. Additionally, read pairs are required to partially align to the repeat sequence and pairs that align entirely within the target repeat sequence are ignored, since these reads may not be specific to the targeted locus. By default, the genotype with the highest likelihood is chosen, but the resulting likelihoods may optionally be used as inputs to downstream programs which estimate genotypes based on patterns across multiple samples and sites. After genotyping, individual per-sample VCFs are concatenated.
Evaluation of the 1000 GP genotypes quality and TypeTE performance
Genotype calling
In order to evaluate the quality of the Alu genotype calls available in the 1000 GP phase 3 structural variants (SV) dataset ([(2)], average depth of coverage 7.4X), we gathered the genotypes available for both non-reference (indicated by ‘<INS:ME:ALU>’ in the available VCF file) and reference (tagged with ‘SV_TYPE = DEL_ALU’). We ran TypeTE-reference and TypeTE-non-reference on the same loci as well as MELT-discovery (non-reference) and MELT-deletion (reference) using its version 2.1.4 (referred to as MELT2 for the remainder of the manuscript) in order to take into account, the most recent changes added to its genotyping module. Additionally, we tested the performances of TypeTE with samples from the SGDP (44), which has higher coverage (average 42X).
With the 1000 GP data, we ran TypeTE and MELT2 on 445 CEU, TSI, GBR, FIN and YRI individuals also present in the GEUVADIS dataset (RNA-seq) (45). In the 1000 GP VCF file released by Sudmant et al. (2), Alu genotypes were produced by MELT (version 1) for non-reference insertions. However, polymorphic reference Alu insertions were first discovered along with other genomic deletions with a set of SV detection tools (BreakDancer, Delly, CNVnator, GenomeSTRiP, Variation-Hunter, SSF and Pindel), then genotyped with the same algorithm as any other SV (2). Because the sample size used in this study is smaller than the 1000 GP original (n = 445 versus n = 2504), MELT2 – which first need to identify pMEI hallmarks before genotyping them – did not re-identify all the loci genotyped by the 1000 GP and TypeTE. Also, probably because of changes in the newer version, some Alu breakpoints were slightly different between the Sudmant et al. (2) dataset and the MELT2 output. Thus, in order to reconcile and compare the three datasets (1000GP, MELT2, TypeTE), bedtools intersect (v.1.5) (51) was used with a window of ±30 bp around each original 1000 GP Alu breakpoint. Finally, the predicted genotypes were compared to PCR assays of 108 non-reference and 43 reference loci in 42 individuals from the CEU population (see next section).
For the SGDP data, reference and non-reference polymorphic Alu insertions were called using MELT2 in 14 publicly available individuals from the South Asian population for which we had access to DNA. The genotypes of the loci discovered were then determined using TypeTE and a subset was compared to PCR-based genotypes previously obtained for the same 14 individuals (9 non-reference and 67 reference loci) (20).
PCR typing in a subset of 1KGP and SGDP dataset
Non-reference (108) and reference (43) Alu loci identified by the 1000 GP were tested in 42 CEU individuals represented in a 30-trio reference panel of the CEPH (Centre d’Étude du Polymorphisme Humain; HAPMAPPT01, Coriell Institute for Medical Research). Primers flanking the Alu insertion sites were selected using Primer3 (52). PCR amplifications were performed using OneTaq Hot Start Quick-Load 2x Master Mix (New England BioLabs) using 3-step PCR (initial denaturation: 94°C, 15′, (94°C, 15″; 57°C, 15″; 68°C, 30″) for 30 cycles; final extension 68°C, 5′). Sequences for 20 new primer pairs are available in Supplementary Table S1; the remainder are available in (27). Accuracy was evaluated by replication in duplicate samples and by evaluating the number of Mendelian errors in related individuals of the trios. In the SGDP dataset, non-reference (9) and reference (67) Alu loci were previously genotyped by PCR in 14 South Asian samples (20). Primers around each Alu insertion were selected using Primer3 (52). PCR amplification was performed using three-step PCR (initial denaturation: 94°C, 3′; (94°C, 15″; 60°C, 15″; 72°C, 30″) for 30 cycles; final extension 72°C, 5′) in 1X PCR buffer (10 mM Tris, pH 8.3, 50 mM KCl, 1.5 mM MgCl2) with 200 uM dNTPs, 10 pmol each primer, and 1 U Taq polymerase. Annealing temperature was adjusted for each primer set. DMSO (5–10%) was used to improve amplification for some loci. Three detailed examples of the PCR genotyping are given in Supplementary Figure S6 and the details about all loci genotyped by PCR are available in the Supplementary Table S2.
Effect of genotype corrections on per sample Alu insertion discovery
In some cases, new genotyping changed the presence/absence status of an Alu insertion for a given genome. Only considering pMEI presence/absence, we define a false positive call (FP) as a case in which an Alu copy is called present, either homozygote or heterozygote in one sample, while the PCR reports it absent. A false negative (FN) is recorded when an Alu is called absent (homozygote absent) while it is called as either homozygote present or heterozygote by PCR. True positive (TP) and true negative (TN) are the same calls (presence/absence), respectively, being validated by PCR. For each dataset and method, we calculated the sensitivity (ability of the method to discover a pMEI: TP/(TP + FN)), the precision (or positive predictive value: TP/(TP + FP)) as well as the F1 score as described by Rishishwar et al. (43), which corresponds to the harmonic mean of sensitivity and precision and summarizes the overall performance of each method.
Estimation of mappability scores
The mappability scores are downloaded for the GRCh37/hg19 version of reference assembly for 100mers (ftp://hgdownload.soe.ucsc.edu/gbdb/hg19/bbi/wgEncodeCrgMapabilityAlign100mer.bw). The downloaded file is processed (53) and is converted to bed format (54). These data are stored in an indexed mysql table. The mappability scores for genomic regions in the flanking region (±250 bp) of the predicted Alu breakpoint for non-reference insertions and flanking region (±250 bp) of the reference Alu insertions are extracted from the table, and the mean of the mappability scores is recorded in a dedicated table and is provided with the output files.
Calculation of local read depth
The average read depth at genomic regions in the flanking region (±250 bp) of the predicted Alu breakpoint for non-reference insertions and flanking region (±250 bp) of the reference Alu insertions is calculated using samtools (Version: 1.4.1). Only reads with a mapping quality of 20 or more (mapped with > 99% probability) and bases with a quality of 20 or more (base call accuracy of > 99%) are counted.
Inbreeding coefficient (Fis) estimates
In order to assess how genotype quality affects common population genetics summary statistics, we computed the per locus inbreeding coefficient (Fis) for the loci assayed by PCR. Fis is a common metric used in population genetics to assess the excess (Fis < 0) or the depletion (Fis > 0) in heterozygotes relative to the expected genotypes proportion at Hardy-Weinberg equilibrium. Allele frequencies were calculated using the genotypes produced by each method (1000 GP, MELT2, TypeTE and PCR) as follows:
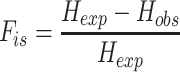 |
with Hexp = 2pq, p = presence allele insertion frequency, q = (1− p)Hobs is the observed number of heterozygotes.
All statistical analyses were carried out with R version 3.5.1 (R Core Team 2018).
RESULTS
Concordance of the 1000 GP phase 3 genotypes with PCR assays
Alu genotype predictions of the 1000 GP phase 3 release, called using MELT (version 1) for non-reference loci and a combination of SV tools for reference insertions were collected (2). To assess the accuracy of the genotypes, we compared the 1000 GP genotype predictions to a dataset of 108 non-reference and 43 reference Alu loci genotyped by PCR in 42 individuals (Figure 2A, Table 1, see also Materials and Methods). Presence of both ‘presence’ and ‘absence’ alleles (with and without Alu) were confirmed by the presence of bands of expected size in the agarose gel electrophoresis and in most cases further validated by Sanger sequencing of PCR amplicons (see Materials and Methods). To further ensure genotype accuracy, PCR assays were performed for all trios of the CEPH CEU panel (n = 30). In every case, we observed genotypes consistent with the Mendelian transmission of alleles from parents to offspring (see Materials and Methods). Thus, the PCR-based assays provide highly reliable genotypes. Upon comparing the PCR-based results to the genotype predictions provided by the 1000 GP phase 3 release, we found an overall concordance rate (total number of individual genotype predictions identical to the PCR-based genotypes/total number of predictions) of 83.31% (3649/4380) for non-reference Alu insertions and of 80.72% (1248/1590) for reference Alu insertions. Considering the PCR-based assays as the most reliable genotypes, these observations suggest that the genotypes predicted for the phase 3 of the 1000 GP suffer from a substantial level of errors.
Figure 2.

Comparison of the predicted genotypes in the 1000 GP dataset with PCR-assays in 42 CEU individuals. Each vertical bar represents one locus, and match or error regarding the genotype for each individual are piled up on the Y axis and color coded according to the legend. NA values (no genotype predicted or failed PCR) are removed from the plot).
Table 1.
Genotype prediction accuracy (%) for each category of insertions when compared with PCR generated genotypes
| 1000 GP | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Non-reference insertions (n = 108 × 42) | Reference insertions (n = 43 × 42) | |||||||||
| hom ref (0) | het (1) | hom alt (2) | NAs | overall | hom ref (2) | het (1) | hom alt (0) | NAs | Overall | |
| 1000 GP | 98.92% | 98.28% | 23.49% | -* | 83.31% | 97.71% | 90.00% | 41.84% | -* | 80.72% |
| MELT 2.1.4 | 99.02% | 92.27% | 68.01% | 1.90% | 87.95% | 98.63% | 37.97% | 26.62% | 5.37% | 71.00% |
| TypeTE | 98.44% | 89.28% | 93.61% | 1.54% | 92.14% | 91.46% | 84.54% | 87.82% | 1.04% | 91.56% |
| SGDP | ||||||||||
| non-reference insertions (n = 9 × 14) | reference insertions (n = 67 × 14) | |||||||||
| MELT 2.1.4 | 92.31% | 94.87% | 9.09% | 0.00% | 79.57% | 97.93% | 88.26% | 2.95% | 0.00% | 70.13% |
| TypeTE | 92.31% | 94.87% | 100.00% | 0.00% | 94.44% | 91.19% | 87.58% | 91.14% | 0.00% | 91.01% |
NA: Not applicable as no genotypes reported.
*No NA genotype are recorded in the Sudmant et al. (2015) dataset.
TypeTE pipeline overview
In order to improve the quality of Alu genotyping by short read sequencing analysis, we developed TypeTE, which allows the re-genotyping of both reference and non-reference Alu insertions. The pipeline is divided into two main modules. The non-reference module predicts genotypes of Alu insertions absent from the reference genome, while the reference module predicts genotypes of Alu insertions present in the reference genome (Figure 1). Details about the implementation of each module are given in the Materials and Methods section as well as in Supplemental Figure S1 and Supplemental Figure S2. The basic principle of TypeTE is to recreate the most accurate sequences for both the ‘presence’ and ‘absence’ alleles and remap reads to these reconstructed alleles to infer genotypes. TypeTE currently uses as input a VCF file such as that typically produced by TE discovery tools like MELT to locate each individual TE insertion. Inorder to reconstruct the pMEI alleles, the pipeline performs an independent analysis of each predicted TE insertion breakpoint, retrieve a consensus sequence for the TE inserted, identifies its target site duplication and the strand of insertion. After allele reconstruction (see Materials and Methods), reads mapping to each insertion locus are extracted from individual alignment file (BAM) and mapped against the reconstructed alleles for genotyping, using an automated and parallelized version of the method developed by Wildschutte et al. (48). Upon completion of the pipeline, a new VCF file with the corrected pMEI position, genotypes and genotypes likelihoods is then produced as an output.
Benchmarking TypeTE
In order to assess the accuracy of the predictions made by TypeTE, we ran the pipeline on a subset of 445 individuals of European and African ancestry included in the 1000 GP dataset (see Materials and Methods). These samples were selected because they are both represented in the 1000 GP (WGS) and GEUVADIS (RNA-seq) datasets, which we reasoned would be particularly useful for functional and evolutionary analyses of pMEIs. We also compared the performance of TypeTE with a recent version of MELT (version 2.1.4, hereafter abbreviated as MELT2) using the packages MELT-discovery (non-reference pMEI) and MELT-deletion (reference pMEI) on the same dataset. TypeTE and MELT2 genotypes were then compared to 108 non-reference and 43 reference pMEI for which we collected or generated PCR genotypes.
For non-reference insertions, we found that the genotypes predicted by MELT2 were more concordant with the PCR-based genotypes than those originally produced by the 1000 GP (obtained with MELT1), reaching a concordance rate of 87.95% due to an additional 131 individual genotypes matching PCR results (versus 83.31% for 1000GP/MELT1; +131/4298 accurate genotypes). Note that the total number of genotypes considered correspond to the total number of predictions available and does not take into account missing genotypes (unascertained genotypes based on the genotype likelihoods). TypeTE further increased the concordance of the genotype predictions, achieving a rate of 92.14% (+325/4313 accurate genotypes compared to original 1000 GP release). For reference insertions, MELT2 showed the lowest concordance rate of all methods tested, with only 71% of the genotypes matching the PCR results (versus 80.72% 1000GP/SV-dedicated tools; −374/1504 individual genotypes). TypeTE performed better with reference insertions than it did with non-reference pMEI, achieving 91.56% concordance (+141/1575 genotypes). Based on these results, we consider that TypeTE delivers consistently the most accurate pMEI genotypes regarding the different methods tested.
We further tested the genotyping performance of MELT2 and TypeTE using the SGDP data (44), which benefits from deeper sequencing coverage than the 1000 GP data (average read depth: 42X versus 7.4X). We tested the concordance of the predicted genotypes with 67 reference and 9 non-reference Alu loci across 14 SDGP individuals previously genotyped by PCR (20). MELT2 showed a concordance rate of 70.13% for reference loci, while TypeTE matched the PCR results for 91.01% of the predicted genotypes (+181 correct genotypes; Figure 3 and Table 1). For the nine non-reference loci that were experimentally genotyped, the concordance rate was 78.57% for MELT2 and 94.44% for TypeTE (+20 correct genotypes). It is worth mentioning that the sample size for non-reference SGDP pMEI is rather small (N = 10) relative to the other comparisons (N > 60 in all other cases). Nevertheless, and as observed with the 1000 GP, TypeTE performed consistently well with both non-reference and reference loci for the SGDP data.
Figure 3.

Comparison of the predicted genotypes in the SGDP dataset with PCR-assays in 14 South Asian individuals. Each vertical bar represents one locus, and match or error regarding the genotype for each individual are piled up on the Y axis and color coded according to the legend. NA values (no genotype predicted or failed PCR) are removed from the plot).
In order to analyze in more details the genotyping performance of each method, we calculated the concordance rate by genotype category (0 or (0/0): homozygote absent, 1 or (0/1): heterozygote, 2 or (1/1): homozygote present) corresponding to the percent of correct genotypes in one category to the total number of calls for this category (Table 1). Additionally, we report the percentage of unascertained loci (NA genotypes) for each method.
We then investigated how the concordance between predicted and PCR genotypes is distributed across loci and individuals by calculating the average concordance rate per locus (total number of correct genotypes at a locus/total number of individuals with a predicted genotype). Regardless of the genotype category (reference/non-reference), TypeTE showed higher average concordance rate per locus, as well as lower variance for this value, than the other methods (Figure 4). The greatest improvement was when the genotypes of reference insertions were compared to MELT2, where the concordance rate of TypeTE is always significantly higher (Tukey's HSD, P < 0.05).
Figure 4.

Average error rate per locus across methods and datasets. *: significant difference, Tukey's HSD, P < 0.05; NS: not significant.
For each locus assayed by PCR in the 1000 GP dataset, we also examined whether read mappability and local read coverage affected genotyping predictions for TypeTE. We did not find a significant correlation between genotype concordance and the mappability score (ranging: 0.1–1) computed in a 500-bp window around the pMEI breakpoints (Supplementary Figure S3. Pearson's product–moment correlation, r = 0.20, P = 0.281 for non-reference loci and r = 0.13, P = 0.414 for reference loci). We also found that the depth of coverage for a given locus, which ranged from 4.7× to 10× across these loci, was not correlated to genotype concordance for neither reference (r = 0.12, P = 0.4538) or non-reference insertions (r = –0.01, P = 0.957) (Supplementary Figure S4). We conclude that at least for the loci tested by PCR, the level of repetitiveness of the flanking sequence of individual Alu insertions and the local read depth do not appear to influence the genotyping performance of TypeTE.
Effect of genotype corrections on variant discovery
Different methods can assign different genotypes for some loci due to the inherent differences in their approach or due to locus-specific features. For example, a heterozygous locus for the presence of Alu can be genotyped either as homozygous presence or absence by different methods. We first converted the biallelic genotypes into presence/absence calls in order to assess sensitivity, precision (positive predictive value), and the overall detection accuracy, summarized by the F1 score (harmonic mean of sensitivity and precision, see Materials and Methods) for each method considering PCR results as delivering true genotypes. TypeTE received the highest F1 score in each dataset (1000 GP or SGDP) and for both types of insertion (reference or non-reference) (Figure 5). The small number of loci tested for the SGDP-non-reference dataset (n = 9) did not allow us to find significant differences between the methods; however, we show that the increased F1 score of TypeTE with the 1000 GP non-reference loci is due to a significant increase of the sensitivity compared to the other methods. The higher F1 score of TypeTE with reference insertions from both 1000 GP and SGDP datasets is driven by higher precision (TP/(TP + FP)). In comparison with the existing methods, overall genotyping accuracy for TypeTE (F1 score) was higher for both reference and non-reference Alu insertions.
Figure 5.

Effect of method and dataset on variant discovery performance. Sensitivity, precision and F1 score are compared for each dataset (1000 GP and SGDP) according to the type of insertion (non-reference vs reference) and the genotyping method used (1000 GP, MELT2.1.4 and TypeTE). Error bars: 95% confidence interval. Non-overlapping intervals denotes a significant difference between scores.
Influence of re-genotyping on population genetics statistics
To illustrate the importance of accurately genotyping Alus, we calculated the population-wise inbreeding coefficient (Fis) for each locus in 42 individuals of the CEU cohort (1000 GP) and 14 individuals of the South Asian cohort (SGDP). Compared to the original 1000 GP and MELT2 genotypes, the Fis values calculated with TypeTE genotypes are concordant with the ones based on PCR genotypes. These results are even more striking when only reference loci are considered: while TypeTE and PCR estimates of Fis are centered at 0, MELT2 and 1000 GP genotypes suggest a clear deviation of most loci from Hardy-Weinberg equilibrium (Figure 6). We note that estimates of the Fis are more variable using the SGDP data, which can be explained by its smaller sample size and a higher population substructure (e.g. Wahlund effect due to individuals from distinct castes or tribes grouped together) than the 1000 GP dataset. These examples further highlights the importance of accurate genotyping for the correct inference of population genetics parameters.
Figure 6.

Per locus inbreeding coefficient (Fis). The Fis is estimated for each locus using the alleles frequencies given by each method (1000 GP: original 1000 GP genotypes, MELT2, TypeTE and PCR assays) and for each of the 1000 GP (n = 42 individuals) and SGDP (n = 9 individuals) datasets. Red dashed-line: expected Fis at Hardy–Weinberg equilibrium (Fis = 0).
Influence of the dataset quality on genotype prediction
To discover factors specific to each dataset that influence genotype prediction, we compared the results obtained in the 1000 GP dataset (average depth of 7.4X) with the results from analysis of the SGDP (average depth of 42X) to the respective PCR genotypes. The provenance of the dataset did not influence the variant discovery abilities (pMEI present or absent in a given individual) of MELT2 and TypeTE (Supplementary Figure S5). However, we found that the percentage of unascertained loci differed between the 1000 GP and SGDP datasets. Between ∼1% and 5.4% of genotypes were not ascertained by either MELT2 and TypeTE in the 1000 GP dataset, probably due to low coverage. Conversely, all SGDP loci are called in every individual for the SGDP dataset (Table 1). Even though the number of loci and the sequencing coverage varied between datasets, influencing MELT2 performances, the genotyping accuracy of TypeTE was sustained across datasets.
DISCUSSION
The purpose of TypeTE is to provide automatic and reliable genotyping of pMEIs using short paired-end reads from either whole genome or targeted sequencing. To our knowledge, MELT (17) is currently the only tool with continued support and documentation that allows direct genotyping of both reference and non-reference pMEI. While its performance for variant discovery has made it a popular tool for pMEI mapping, to our knowledge its performance at genotyping has never been comprehensively tested. Moreover, there was no formal testing of the genotype quality of the pMEIs reported in the phase 3 release of the 1000 GP (2). Here, we collected the results of more than 150 locus-specific PCR genotyping assays to test ∼ 1% of all Alus (151/13 963) originally released by the 1000 GP, as well as those produced by a recent version of MELT (v. 2.1.4) and those inferred by TypeTE.
Combining reference and non-reference Alus, our results indicate that ∼18% of the genotypes reported by the 1000 GP are different from PCR-derived genotypes we collected and produced, and which we consider the most reliable, based on duplicate samples and expected Mendelian segregation in related individuals. These results are at odds with a previous estimate of 98% genotype concordance observed for the non-reference insertions using a PCR-free approach based on 250 bp reads (2). However, we believe that this method is less accurate than PCR genotyping to capture a full-length Alu insertion, which is usually larger (∼300 bp) than the read size. Genotypes reported by the 1000 GP were predicted using the first version of MELT for non-reference loci, but genotyping methods developed for other structural variation (indels, inversions, etc.) were used for reference insertions. While MELT2 appears to offer a noticeable improvement over its first version for genotyping non-reference Alus, its overall genotyping performance is diminished when applied to reference loci, with genotyping errors reaching more than 20% when compared to the PCR. For both categories of loci, we observe that most errors are caused by the underestimation of homozygous genotypes carrying the alternative allele, relative to the reference genome (Table 1). We also note that for non-reference insertions, MELT’s genotyping algorithm benefited from improvements deployed in the version tested (v2.1.4) compared to its original release, in particular to detect homozygous insertion (1/1). However, this increased sensitivity to detect pMEI alleles from read alignments seems to be accompanied by a reduced power to detect ‘absence’ alleles for reference insertions (MELT-deletion module). Such errors are consequential for population genetics analysis because they lead to inaccurate estimation of population genetics parameters. For example, calculation of the inbreeding coefficient (Fis) shows that the original release of the 1000 GP genotypes was overestimating heterozygotes, leading to negative and likely inaccurate values of Fis (Figure 6). Genotypes obtained with MELT2 improve these estimates for non-reference insertions, but the results appear less accurate when computed from a small sample and they are more inaccurate for reference insertions. This difference is critical given that reference insertions are more likely to segregate at higher frequencies than non-reference pMEI. The aforementioned issues underscore the need for a tool specifically dedicated to the genotyping of pMEI.
Toward this goal, we have developed TypeTE and applied it to genotype both reference and non-reference Alu insertions in a sample of the 1000 GP and SDGP datasets. Our benchmarking results show that TypeTE has an average concordance rate of 91% or greater with PCR-based genotyping. Importantly, TypeTE maintains a genotyping accuracy >84% under all genotyping scenarios. While TypeTE performs better than MELT1 and MELT2 for non-reference insertions, the most significant improvement is for reference insertions. In particular, the genotypes predicted by 1000 GP and MELT2 never reached >41.8% concordance with the experimental results when the PCR called a homozygote absence (0/0); by contrast, TypeTE predicted these genotypes with >87% concordance in the two datasets tested (1000 GP and SGDP). Consequently, calculation of Fis based on TypeTE genotypes shows better concordance with that based on PCR-derived genotypes, and fits the neutral expectation as we observe no deviation from Hardy-Weinberg equilibrium for a single human population (55).
The principal difference between TypeTE and MELT derives from characteristics of the actual data on which the genotyping is performed. While both methods implement the core genotyping algorithm described by Li et al. (50), TypeTE relies on a strategy based on re-alignment of the reads against both presence and absence alleles before computation of the genotype likelihoods, an approach initially introduced by Wildschutte et al. (48). Furthermore, TypeTE facilitates the genotyping with no user intervention by using as input the VCF file produced by MELT (or virtually the output of any other pMEI detection software transformed in VCF with the necessary loci information, see online TypeTE manual) to generate a new VCF delivering the predicted genotypes and their likelihood. TypeTE also uses recently developed assemblers (SPAdes (46) and Minia (47)) and use reads from all individuals for a locus for local Alu assembly which, in our hands, showed a higher rate of assembly than the CAP3 assembler (48,56). In addition, TypeTE can still genotype pMEIs if a de-novo assembly is impossible: if an incomplete Alu is assembled, TypeTE substitutes it with the exact consensus sequence based on the information provided by discordant and split reads assigned to that location (see Materials and Methods). This step enables the reconstruction of alleles and possibly compensate the genotyping errors associated with lack of coverage. The reconstruction of alternative alleles (either by local assembly or consensus-based)—a major difference with MELT—appear to significantly improves the accuracy of Alu genotyping. Finally, TypeTE predicts the TSD accompanying each insertion and the pMEI orientation, which ensures optimal reconstruction of the two alleles. Collectively these implementations enable TypeTE to generate highly accurate Alu insertion genotypes.
We further tested whether the quality of the starting dataset, in particular its sequencing depth, influenced TypeTE’s performance. By comparing results between the 1000 GP and SGDP datasets, which use different sequencing depth (on average 7.4X and 42X, respectively), we found that TypeTE performs equally regardless of coverage depth, at least for reference insertions, for which we had enough loci to compare between datasets. Using both non-reference and reference Alu insertions genotyped with TypeTE in the 1000 GP dataset, we also showed that the average sequence coverage of the region flanking these loci does not seem to influence genotyping accuracy. Thus, TypeTE can support the analysis of large population dataset without stringent or highly uniform coverage requirements.
While TypeTE offers significant improvements over MELT, it failed to genotype accurately as small proportion of the loci we experimentally assayed (16/227). Neither low sequencing coverage nor mappability issues could be readily implicated as hindering genotyping at these loci. We believe that other locus-specific idiosyncrasies prevent the ability of TypeTE to produce an accurate allele call for these particular elements. For instance, earlier tests on the pipeline showed that a 1-bp insertion at the end of the element in one allele or a slight error in the TSD prediction could dramatically affect the re-mapping and genotype predictions. A specific assessment of the bioinformatic methods aimed to identify TSDs should be able to improve this type of issues. Identifying boundaries of Alu insertion in low complexity (especially A-rich) regions is challenging due to individuals and populations variations (57) in the length of the poly-A tail of the element, and according to our tests, RepeatMasker often fails to identify the exact boundaries of such reference elements. Even though our pipeline in principle considers such subtle sequence variation, at least for one locus, we found that the TSD was overlapping the annotated poly-A region. Implementing changes to identify similar instances could mitigate genotyping miscalls for those loci. Additionally, our ability to evaluate the concordance of genotype predictions in low-complexity and highly repetitive regions was restrained to PCR-accessible loci. Because of this technical limitation, our analysis filtered out these regions, that are also extremely difficult to map or genotype with short reads. We have also noticed that altering the parameters or method for local de novo assembly can improve the assembly of certain pMEI. An automated approach to customize the assembly parameters for each locus that failed with the standard approach would enhance the reconstruction of non-reference TE sequences. Identifying proper orientation of insertions is also crucial in accurately genotyping the insertions and we are also contemplating a read-based approach to identify the orientation of insertions in addition to the current assembly-based approach. Collecting more benchmarking data might allow us to characterize more finely these issues and to adapt the pipeline accordingly. Notwithstanding these peculiar instances, TypeTE has the lowest error rate of all methods tested and as such it represents a valuable advance in the field.
The task and challenges of pMEI genotyping have been largely overlooked in the literature. Yet we show here that inaccurate genotyping of pMEIs can significantly bias population genetics inferences. It is presumably because of these issues that reference pMEIs have been ignored altogether in previous population genomics studies using pMEIs (58–60). By improving genotyping accuracy for both reference and non-reference insertions, TypeTE will enhance future population genomics studies using pMEI as markers or variants. Notably, our results now offer a dataset of genotyped Alu insertions for 445 samples of the 1000 GP that is complemented by a wealth of functional data including RNA-seq (45), DNA methylation (61), DNase I accessibility (62) and ATAC-seq (63,64). We anticipate that these resources will open new avenues to explore the cis-regulatory influence of pMEIs in humans (30). The modularity of TypeTE allows one to easily combine new assemblers to improve the reconstruction of each pMEI, but it is also possible to skip this step and only use consensus sequence of MEI to speed up the computation time. The design of TypeTE makes it compatible with any data produced by pMEI detection tools and future updates are scheduled to genotype insertions from any other retroelement families in virtually any species.
DATA AVAILABILITY
TypeTE and its documentation is freely available in the Github repository https://github.com/clemgoub/TypeTE. Updated vcf files with TypeTE Alu genotypes for the 445 individuals included in the 1000GP/GEUVADIS project are publicly available at Dryad. https://datadryad.org/stash/dataset/doi:10.5061/dryad.dbrv15dx1.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35 GM122550, R01 GM059290 to C.F., GM118335, GM059290 to L.B.J.]. Funding for open access charge: National Institutes of Health [R35 GM122550, R01 GM059290 to C.F., GM118335, GM059290 to L.B.J.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kidwell M.G., Lisch D.. Transposable elements as sources of variation in animals and plants. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:7704–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M.H.-Y. et al.. An integrated map of structural variation in 2, 504 human genomes. Nature. 2015; 526:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Underwood C.J., Henderson I.R., Martienssen R.A.. Genetic and epigenetic variation of transposable elements in Arabidopsis. Curr. Opin. Plant Biol. 2017; 36:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jurka J., Kohany O., Pavlicek A., Kapitonov V.V., Jurka M.V.. Duplication, coclustering, and selection of human Alu retrotransposons. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Song M., Boissinot S.. Selection against LINE-1 retrotransposons results principally from their ability to mediate ectopic recombination. Gene. 2007; 390:206–213. [DOI] [PubMed] [Google Scholar]
- 6. Xing J., Zhang Y., Han K., Salem A.H., Sen S.K., Huff C.D., Zhou Q., Kirkness E.F., Levy S., Batzer M.A. et al.. Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 2009; 19:1516–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thomas J., Perron H., Feschotte C.. Variation in proviral content among human genomes mediated by LTR recombination. Mob. DNA. 2018; 9:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hancks D.C., Kazazian H.H. Jr. Roles for retrotransposon insertions in human disease. Mob. DNA. 2016; 7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oliver K.R., McComb J.A., Greene W.K.. Transposable elements: powerful contributors to angiosperm evolution and diversity. Genome Biol. Evol. 2013; 5:1886–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chuong E.B., Elde N.C., Feschotte C.. Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 2017; 18:71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wallace A.D., Wendt G.A., Barcellos L.F., de Smith A.J., Walsh K.M., Metayer C., Costello J.F., Wiemels J.L., Francis S.S.. To ERV is human: a phenotype-wide scan linking polymorphic human endogenous retrovirus-K Insertions to complex phenotypes. Front. Genet. 2018; 9:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horváth V., Merenciano M., González J.. Revisiting the relationship between transposable elements and the eukaryotic stress response. Trends Genet. 2017; 33:832–841. [DOI] [PubMed] [Google Scholar]
- 13. Jangam D., Feschotte C., Betrán E.. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 2017; 33:817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mills R.E., Andrew Bennett E., Iskow R.C., Devine S.E.. Which transposable elements are active in the human genome. Trends Genet. 2007; 23:183–191. [DOI] [PubMed] [Google Scholar]
- 15. Hancks D.C., Kazazian H.H. Jr. Active human retrotransposons: variation and disease. Curr. Opin. Genet. Dev. 2012; 22:191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stewart C., Kural D., Strömberg M.P., Walker J.A., Konkel M.K., Stütz A.M., Urban A.E., Grubert F., Lam H.Y.K., Lee W.-P. et al.. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 2011; 7:e1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. 1000 Genomes Project Consortium Gardner E.J., Lam V.K., Harris D.N., Chuang N.T., Scott E.C., Pittard W.S., Mills R.E., Devine S.E.. The mobile element locator tool (MELT): population-scale mobile element discovery and biology. Genome Res. 2017; 27:1916–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dewannieux M., Esnault C., Heidmann T.. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003; 35:41–48. [DOI] [PubMed] [Google Scholar]
- 19. Doronina L., Reising O., Clawson H., Ray D.A., Schmitz J.. True homoplasy of retrotransposon insertions in primates. Syst. Biol. 2019; 68:482–493. [DOI] [PubMed] [Google Scholar]
- 20. Watkins W.S., Rogers A.R., Ostler C.T., Wooding S., Bamshad M.J., Brassington A.-M.E., Carroll M.L., Nguyen S.V., Walker J.A., Prasad B.V.R. et al.. Genetic variation among world populations: inferences from 100 Alu insertion polymorphisms. Genome Res. 2003; 13:1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jurka J., Bao W., Kojima K.K.. Families of transposable elements, population structure and the origin of species. Biol. Direct. 2011; 6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rishishwar L., Tellez Villa C.E., Jordan I.K.. Transposable element polymorphisms recapitulate human evolution. Mob. DNA. 2015; 6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boissinot S., Davis J., Entezam A., Petrov D., Furano A.V.. Fitness cost of LINE-1 (L1) activity in humans. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:9590–9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cordaux R., Lee J., Dinoso L., Batzer M.A.. Recently integrated Alu retrotransposons are essentially neutral residents of the human genome. Gene. 2006; 373:138–144. [DOI] [PubMed] [Google Scholar]
- 25. Larsen P.A., Hunnicutt K.E., Larsen R.J., Yoder A.D., Saunders A.M.. Warning SINEs: Alu elements, evolution of the human brain, and the spectrum of neurological disease. Chromosome Res. 2018; 26:93–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hueso M., Cruzado J.M., Torras J., Navarro E.. ALUminating the path of atherosclerosis progression: Chaos theory suggests a role for alu repeats in the development of atherosclerotic vascular disease. Int. J. Mol. Sci. 2018; 19:E1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Payer L.M., Steranka J.P., Yang W.R., Kryatova M., Medabalimi S., Ardeljan D., Liu C., Boeke J.D., Avramopoulos D., Burns K.H.. Structural variants caused by insertions are associated with risks for many human diseases. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E3984–E3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Payer L.M., Steranka J.P., Ardeljan D., Walker J., Fitzgerald K.C., Calabresi P.A., Cooper T.A., Burns K.H.. Alu insertion variants alter mRNA splicing. Nucleic Acids Res. 2019; 47:421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang S., Zang C., Xiao T., Fan J., Mei S., Qin Q., Wu Q., Li X., Xu K., He H.H. et al.. Modeling cis-regulation with a compendium of genome-wide histone H3K27ac profiles. Genome Res. 2016; 26:1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goubert C., Zevallos N.A., Feschotte C.. Contribution of unfixed transposable element insertions to human regulatory variation. 2019; 3 October 2019, preprint: not peer reviewed 10.1101/792937. [DOI] [PMC free article] [PubMed]
- 31. Goerner-Potvin P., Bourque G.. Computational tools to unmask transposable elements. Nat. Rev. Genet. 2018; 19:688–704. [DOI] [PubMed] [Google Scholar]
- 32. Lee E., Iskow R., Yang L., Gokcumen O., Haseley P., Luquette L.J. 3rd, Lohr J.G., Harris C.C., Ding L., Wilson R.K. et al.. Landscape of somatic retrotransposition in human cancers. Science. 2012; 337:967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keane T.M., Wong K., Adams D.J.. RetroSeq: transposable element discovery from next-generation sequencing data. Bioinformatics. 2013; 29:389–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thung D.T., de Ligt J., Vissers L.E.M., Steehouwer M., Kroon M., de Vries P., Slagboom E.P., Ye K., Veltman J.A., Hehir-Kwa J.Y.. Mobster: accurate detection of mobile element insertions in next generation sequencing data. Genome Biol. 2014; 15:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fiston-Lavier A.-S., Barrón M.G., Petrov D.A., González J.. T-lex2: genotyping, frequency estimation and re-annotation of transposable elements using single or pooled next-generation sequencing data. Nucleic Acids Res. 2015; 43:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen J., Wrightsman T.R., Wessler S.R., Stajich J.E.. RelocaTE2: a high resolution transposable element insertion site mapping tool for population resequencing. PeerJ. 2017; 5:e2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Santander C.G., Gambron P., Marchi E., Karamitros T., Katzourakis A., Magiorkinis G.. STEAK: A specific tool for transposable elements and retrovirus detection in high-throughput sequencing data. Virus Evol. 2017; 3:vex023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rajaby R., Sung W.-K.. TranSurVeyor: an improved database-free algorithm for finding non-reference transpositions in high-throughput sequencing data. Nucleic Acids Res. 2018; 46:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baboon Genome Analysis Consortium Jordan V.E., Walker J.A., Beckstrom T.O., Steely C.J., McDaniel C.L., St Romain C.P., Worley K.C., Phillips-Conroy J., Jolly C.J. et al.. A computational reconstruction of phylogeny using insertion polymorphisms. Mob. DNA. 2018; 9:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen X., Li D.. ERVcaller: identifying polymorphic endogenous retrovirus and other transposable element insertions using whole-genome sequencing data. Bioinformatics. 2019; 35:3913–3922. [DOI] [PubMed] [Google Scholar]
- 41. Sanchez-Luque F.J., Kempen M.-J.H.C., Gerdes P., Vargas-Landin D.B., Richardson S.R., Troskie R.-L., Jesuadian J.S., Cheetham S.W., Carreira P.E., Salvador-Palomeque C. et al.. LINE-1 evasion of epigenetic repression in humans. Mol. Cell. 2019; 75:590–604. [DOI] [PubMed] [Google Scholar]
- 42. Puurand T., Kukuškina V., Pajuste F.-D., Remm M.. AluMine: alignment-free method for the discovery of polymorphic Alu element insertions. Mob. DNA. 2019; 10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rishishwar L., Mariño-Ramírez L., King Jordan I.. Benchmarking computational tools for polymorphic transposable element detection. Brief. Bioinform. 2016; 18:908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mallick S., Li H., Lipson M., Mathieson I., Gymrek M., Racimo F., Zhao M., Chennagiri N., Nordenfelt S., Tandon A. et al.. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 2016; 538:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lappalainen T., Sammeth M., Friedländer M.R., ’t Hoen P.A.C., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G. et al.. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013; 501:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D. et al.. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012; 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chikhi R., Rizk G.. Space-efficient and exact de Bruijn graph representation based on a Bloom filter. Algorithms Mol. Biol. 2013; 8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wildschutte J.H., Baron A., Diroff N.M., Kidd J.M.. Discovery and characterization of Alu repeat sequences via precise local read assembly. Nucleic Acids Res. 2015; 43:10292–10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011; 27:2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Quinlan A.R., Hall I.M. et al.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G.. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012; 40:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kent W.J., Zweig A.S., Barber G., Hinrichs A.S., Karolchik D.. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 2010; 26:2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Neph S., Kuehn M.S., Reynolds A.P., Haugen E., Thurman R.E., Johnson A.K., Rynes E., Maurano M.T., Vierstra J., Thomas S. et al.. BEDOPS: high-performance genomic feature operations. Bioinformatics. 2012; 28:1919–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hosking L., Lumsden S., Lewis K., Yeo A., McCarthy L., Bansal A., Riley J., Purvis I., Xu C.-F.. Detection of genotyping errors by Hardy–Weinberg equilibrium testing. Eur. J. Hum. Genet. 2004; 12:395–399. [DOI] [PubMed] [Google Scholar]
- 56. Huang X., Madan A.. CAP3: a DNA sequence assembly program. Genome Res. 1999; 9:868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen J.-M., Stenson P.D., Cooper D.N., Férec C.. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum. Genet. 2005; 117:411–427. [DOI] [PubMed] [Google Scholar]
- 58. Wang L., Rishishwar L., Mariño-Ramírez L., King Jordan I.. Human population-specific gene expression and transcriptional network modification with polymorphic transposable elements. Nucleic Acids Res. 2016; 45:2318–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang L., Norris E.T., Jordan I.K.. Human retrotransposon insertion polymorphisms are associated with health and disease via gene regulatory phenotypes. Front. Microbiol. 2017; 8:1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rishishwar L., Wang L., Wang J., Yi S.V., Lachance J., King Jordan I.. Evidence for positive selection on recent human transposable element insertions. Gene. 2018; 675:69–79. [DOI] [PubMed] [Google Scholar]
- 61. Pai A.A., Bell J.T., Marioni J.C., Pritchard J.K., Gilad Y.. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet. 2011; 7:e1001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Degner J.F., Pai A.A., Pique-Regi R., Veyrieras J.-B., Gaffney D.J., Pickrell J.K., De Leon S., Michelini K., Lewellen N., Crawford G.E. et al.. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012; 482:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kumasaka N., Knights A.J., Gaffney D.J.. Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet. 2016; 48:206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kumasaka N., Knights A.J., Gaffney D.J.. High-resolution genetic mapping of putative causal interactions between regions of open chromatin. Nat. Genet. 2019; 51:128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TypeTE and its documentation is freely available in the Github repository https://github.com/clemgoub/TypeTE. Updated vcf files with TypeTE Alu genotypes for the 445 individuals included in the 1000GP/GEUVADIS project are publicly available at Dryad. https://datadryad.org/stash/dataset/doi:10.5061/dryad.dbrv15dx1.