

Abstract
Rheumatoid arthritis is a chronic autoimmune syndrome associated with several genetic, epigenetic, and environmental factors affecting the articular joints contributing to cartilage and bone damage. Although etiology of this disease is not clear, several immune pathways, involving immune (T cells, B cells, dendritic cells, macrophages, and neutrophils) and nonimmune (fibroblasts and chondrocytes) cells, participate in the secretion of many proinflammatory cytokines, chemokines, proteases (MMPs, ADAMTS), and other matrix lysing enzymes that could disturb the immune balance leading to cartilage and bone damage. The presence of autoantibodies preceding the clinical onset of arthritis and the induction of bone erosion early in the disease course clearly suggest that initiation events damaging the cartilage and bone start very early during the autoimmune phase of the arthritis development. During this process, several signaling molecules (RANKL-RANK, NF-κB, MAPK, NFATc1, and Src kinase) are activated in the osteoclasts, cells responsible for bone resorption. Hence, comprehensive knowledge on pathogenesis is a prerequisite for prevention and development of targeted clinical treatment for RA patients that can restore the immune balance improving clinical therapy.
1. Introduction
Rheumatoid arthritis (RA) is a chronic, inflammatory syndrome comprised of various disease phenotypes. RA is characterized by aggressive synovial hyperplasia causing destruction of articular joints. A combination of genetic, epigenetic, and environmental factors is responsible for the onset and development of RA. An array of susceptible genes (human leukocyte antigen (HLA) class II and more than 100 susceptibility loci including PTPN22, PADI4, TRAF1, and CTLA4), nongenetic factors (sex hormones, smoking, periodontal infection, and microbiota), immune (macrophages, dendritic cells, mast cells, neutrophils, T cells, and B cells) and nonimmune (fibroblasts and chondrocytes) cells, and inflammatory mediators (autoantibodies, cytokines, chemokines, and proteases) are collectively involved in the inflammatory processes targeting the cartilage and bone effectuating functional loss of joints (Figure 1) [1].
Figure 1.
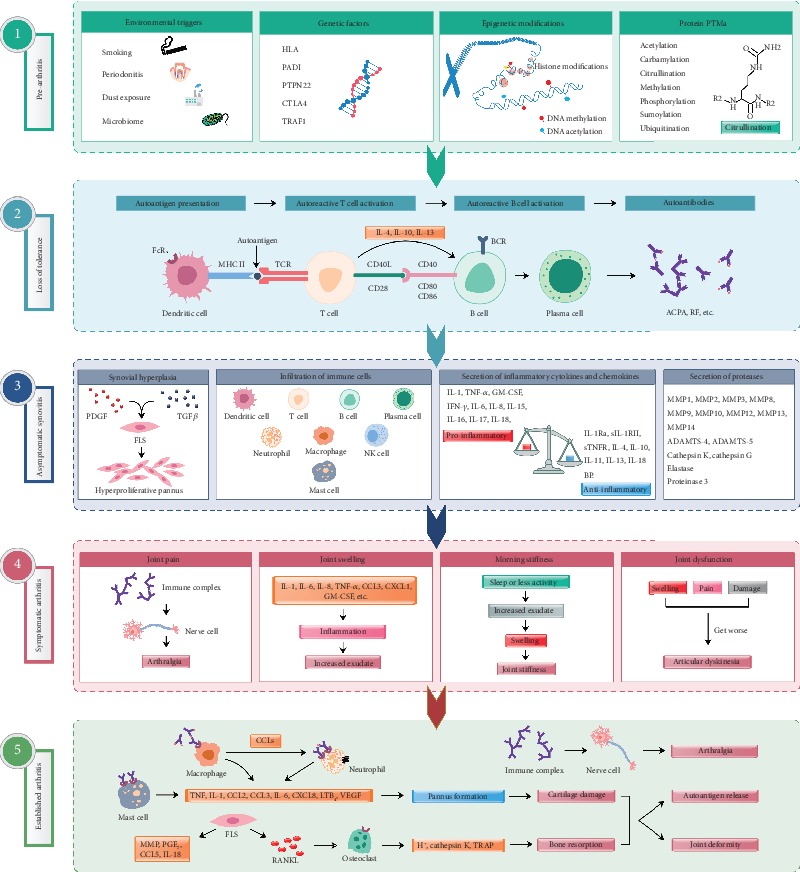
Different phases in RA pathogenesis. (1) Genetic, epigenetic, and environmental factors contribute to arthritis progression. Multiple environmental risk factors (for example, smoking, pollutants, or microbes), when come in contact with the mucosal sites, are most likely responsible for causing local inflammatory events and immune system activation inducing epigenetic modifications and protein posttranslational modifications (PTMs) [59], before crossing the threshold to trigger disease in genetically vulnerable people. (2) Dendritic cells presenting altered self or related peptides to T cells (breakdown of tolerance mechanisms) leads to the activation of T and B cells effectuating synthesis of cytokines and autoantibodies. Progressively, these autoantibodies are produced more and more, which recognize several neoepitopes by the process of epitope spreading, and gets overt during the onset of the clinical disease [1]. (3) Disease development involves autoimmune responses against both posttranslationally modified and unmodified self-antigens, which starts many years before the subclinical synovitis and appearance of clinical symptoms [60]. (4) Autoantibodies induced during this preclinical phase can also be responsible for bone erosion and pain. Before the onset of inflammation, these alterations could reduce overall functions of the joints. After the autoantibodies start binding to different epitopes and form immune complexes, inflammation in the synovium and development of arthritis ensue. (5) Antibody-induced cartilage and bone changes, if minor, resolve without any considerable damages. However, if left untreated or in the presence of continuous external stimuli, these changes can give rise to chronic inflammation, joint destruction, and disability [59]. Arthritis is associated with both local as well as systemic pathological manifestations. ACPA: anticitrullinated protein antibody; ADAMTS: a disintegrin and metalloproteinase with thrombospondin motifs; BCR: B cell receptor; CCL: c-c motif chemokine ligand; CXCL: C-X-C motif chemokine ligand; CTLA4: cytotoxic T-lymphocyte-associated protein 4; FcR: Fc receptor; FLS: fibroblast-like synoviocytes; GM-CSF: granulocyte-macrophage colony-stimulating factor; HLA: human leukocyte antigen; IFN-γ: interferon gamma; IL: interleukin; IL-1Ra: interleukin-1 receptor antagonist; IL-18BP: interleukin-18-binding protein; LTB4: leukotriene B4; MMP: matrix metalloproteinase; MHC II: major histocompatibility complex class II; NK cell: natural killer cell; PADI: peptidyl arginine deiminase; PDGF: platelet-derived growth factor; PGE2: prostaglandin E2; PTPN22: protein tyrosine phosphatase nonreceptor type 22; RANKL: receptor activator of nuclear factor kappa B ligand; RF: rheumatoid factor; sIL-1RII: soluble interleukin 1 receptor II; sTNFR: soluble tumor necrosis factor receptors; TCR: T cell receptor; TGFβ: transforming growth factor β; TNF-α: tumor necrosis factor α; TRAF1: TNF receptor-associated factor 1; VEGF: vascular endothelial growth factor.
The synovium is one of the major target tissues in RA [2]. During joint inflammation, macrophage-like synoviocytes (MLS) and fibroblast-like synoviocytes (FLS) proliferate to form the pannus, which invades and destroys the cartilage. These cells are the major sources of factors that can promote inflammation and joint destruction. Autoantibodies contribute to the inflammatory process by acting as the mediator of joint inflammation and bone erosion [3]. 50–80% of RA patients have autoantibodies depending on the duration of the disease. Autoantibodies can initiate inflammatory effector pathways, which affect chondrocytes and the cartilage causing release of extracellular matrix (ECM) components. In this context, glycosylation of autoantibodies is crucial. Decreased IgG-Fc sialylation is associated with RA and osteoclastogenesis, while an increase in sialylation decreased inflammatory bone loss [4]. Bone erosion and loss of physical function in arthritis begin early and progress along with disease severity. The main triggers of bone erosion are an inflamed synovium, proinflammatory cytokines, autoantibodies, and receptor activator of nuclear factor κB ligand (RANKL). Breakdown of self-tolerance causes activation of immune and nonimmune cells resulting in the production of inflammatory mediators. Fibroblasts expressing RANKL together with macrophage colony-stimulating factor (M-CSF) promote differentiation of preosteoclasts into bone-resorbing osteoclasts; this process is initiated at the junction of the cartilage and bone. Targeting T and B cells, proinflammatory mediators, signaling molecules, and synovium-specific targets are pursued as new treatment options [5]. In this review, we discuss about disease pathways contributing to cartilage and bone damage to facilitate research on developing targeted drugs.
2. T Cells
Increasing evidences demonstrate that RA development results due to an imbalance between CD4+ T cell subsets [6, 7]. Under physiological conditions, T cells are tolerant toward self-antigens [8, 9]. The presence of T cells in the inflamed synovium, association of arthritis with HLA loci, and transfer of disease using T cells in rodent models suggest the importance of T cells in arthritis pathogenesis. However, difficulty in identifying consistent T cell effector mechanisms and not so convincing results from T cell-targeted therapies question this view. Currently, CD4+ T cells reactive with citrulline were identified and the detection rate was high during the first 5 years after RA diagnosis. Although this finding provides a theoretical basis for the correlation between citrulline-specific CD4+ T cells and RA, the mechanisms of T cell activation and its role in promoting joint inflammation need further investigation. Recent studies identified a defective glycolytic process present in T cells from RA patients, which causes glucose to be diverted into the pentose phosphate pathway, driving the accumulation of NADPH and ROS consumption. With an excess of reducing equivalents, T cells are unable to activate the relevant redox kinase, which enabled bypassing the regulatory checkpoint of the G2/M cell cycle that is conducive for their excessive proliferation [10].
Citrulline-specific CD4+ T cells of Th1 memory phenotype are higher in RA patients [11]. Upon stimulation with many cytokines, CD4+ T cells differentiate into Th1, Th2, and Th17 cells secreting different cytokines, while an excessive production of Th17 cells associates with disease severity in many autoimmune diseases. Th17 cells can also mediate osteoclast activation and synovial neovascularization causing bone erosion. Overexpression of Th17-specific transcription factor, retinoic acid-related orphan receptor (RORγt), not only induced a high expression of chemokine receptor 6 (CCR6) but also promoted CD4+ T cell migration into the affected joints through the CCR6-specific chemokine ligand 20 (CCL20) pathway [7].
On the other hand, Treg cells by secreting IL-10 and TGF-β maintain lymphocyte homeostasis and tolerance. When a RA mouse model was administered with sialic acid-binding Ig-like lectin-9, Th17 cell differentiation was reduced and Treg cells proliferated, which attenuated joint inflammation and bone damage. In this context, Foxp3 plays a crucial role by affecting the glycolysis and metabolism of Treg cells through the phosphatidylinositol 3-kinase/protein kinase B/rapamycin target protein (PI3K/Akt/mTOR) signaling pathway [12].
It has been suggested that PTPN22 encoding a tyrosine phosphatase contributes to the immune tolerance by limiting the signaling events after recognition of autoantigens and weak agonistic antigens by T cell antigen receptors (TCRs) of naive and effector T cells, while not hindering the response to foreign antigens [13]. This observation suggests that inactivation of the PTPN22 allele can amplify the effector or memory T cells, which possibly could enhance the development of an autoimmune disease. More importantly, the PTPN22-related alleles have a stronger interaction with arthritis-susceptible HLA-DR alleles. Therefore, an in-depth functional study of PTPN22 gene polymorphisms in arthritis development may further improve our understanding of RA pathogenesis.
3. Dendritic Cells
During homeostasis, dendritic cells (DCs) are involved in the maintenance of immune regulation and tolerance. However, in RA by presenting self-peptides, they trigger differentiation and activation of the auto-reactive T cells as well as innate immune effector functions [14]. In RA patients, increased numbers of DCs are present in the synovial fluid and tissues. Interestingly, tolerogenic DCs (TolDCs) can be generated by genetic and pharmacological modifications or by using cytokines in an antigen-specific manner. Induction of such immune tolerance mechanisms is a promising approach to treat or prevent autoimmune disorders. Many application methods to achieve antigen-specific therapy were reviewed recently [15]. Autologous self-antigen-loaded TolDCs are capable of deleting or reprogramming auto-reactive T cells and were used for the treatment of experimental arthritis with more promising results [16].
4. B Cells and Autoantibodies
Importance of B cells in the pathogenesis of RA has been studied and discussed extensively. Initially, the role of B cells in arthritis was appreciated mainly in terms of autoantibodies because of their importance in clinical diagnosis and prognosis and also as inflammatory mediators. However, B cells can also contribute to disease pathogenesis through antibody-independent mechanisms [17] including antigen presentation, modulation of T and dendritic cell functions, and production of proinflammatory and regulatory cytokines, facilitating the tertiary lymphoid tissue formation in target organs and possibly tissue repair as well. In recent years, the clinical efficiency of B cell-targeted therapies has revealed the pathogenic properties of B cells clearly in several inflammation-dependent diseases [18]. Recent studies have provided insights into the enrichment of memory B cell subsets distinguished by the expression of Fc-like receptor 4 (FcRL4) in the joints and mucosa-associated lymphoid tissues of RA patients. Interestingly, antibodies produced from FcRL4+ B cells have high binding capacity to citrullinated autoantigens [19].
Autoantibodies are highly prevalent and detectable in RA patients' sera several years before the clinical symptoms appear [20]. During the time of disease onset, epitope spreading [21], avidity maturation [22] and proinflammatory IgG-Fc glycosylation phenotype [23] of ACPAs were found to occur. Interestingly, the presence of desialylated ACPAs is more during active joint inflammation and transfer of sialic acid-enriched antibodies attenuated experimental arthritis [24]. Alterations in N-glycome induce Fc conformational changes that have direct influence on antibody effector and immunoregulatory functions. Furthermore, germ line-encoded antibodies were identified to be important in experimental arthritis and self-antigen-specific B cells were neither deleted nor anergized [25]. Autoantibodies after binding to their target antigens trigger downstream inflammatory cascades either directly or in the form of immune complexes [26]. At this effector phase of arthritis, activation of different pathways of complement [27, 28] and FcγR-bearing immune cells contributes to cartilage destruction either directly or by promoting the secretion of inflammatory cytokines and matrix lysing enzymes (Figure 2) [29]. At the same time, antibodies binding to collagen type II (CII) can also induce target damage independent of inflammatory mediators or cells [30]. These antibodies disrupt the integrity of the cartilage matrix by promoting impaired cartilage formation, inhibiting cartilage fibril generation, and disassembling CII fibrils in the ECM. In addition, anti-CII antibodies induced pain prior to and after the appearance of arthritis symptoms and involved in immune complex-mediated activation of neurons [31]. Upon passive transfer, purified anti-CII antibodies from RA patients induced arthritis in naive mice [32], which demonstrated their pathogenicity. Unlike anti-CII antibodies, ACPAs might be nonpathogenic [33]. Conversely, ACPAs were shown to mediate osteoclastogenesis [34] and be responsible for bone loss prior to the onset of clinical arthritis [35]; ACPAs induced pain [36] and FLS migration through activation of phosphoinositide 3-kinase [37]. Immune complexes or ACPAs from RA patients induced TNF-α production in peripheral blood mononuclear cells and macrophages [38]. Furthermore, ACPAs were suggested to be agonists for a receptor-mediated response, but this notion is still controversial [39]. Hence, more studies are needed to understand all the possible roles of ACPAs in RA.
Figure 2.
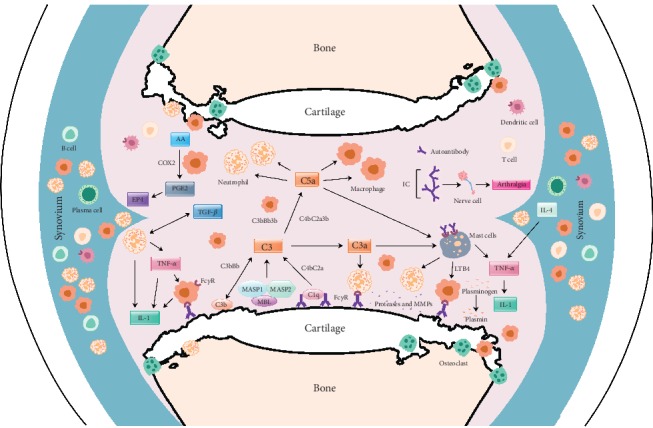
Likely interactions of molecules and factors in the antibody mediated joint inflammation. Upon binding to joint antigens or deposited as immune complexes on the cartilage surface, autoantibodies initiate inflammation-dependent and inflammation-independent activities, which culminate in the direct damage to the cartilage and bone. Activation of complement cascades by autoantibodies leads to the release of anaphylatoxins (C3a, C5a), attracting FcR-bearing immune cells to the inflammation foci, which in turn get more activated and secrete cytokines that can further activate resident nonimmune cells in the joint. All these cells in the inflamed joint secrete more inflammatory mediators and extracellular matrix lysing enzymes that could destroy the cartilage and bone. AA: arachidonic acid; C1q, C2a, C3, C3a, C4b and C5a and B (factor B): complement components; CCL3: chemokine (C-C motif) ligand 3; COX2: cyclooxygenase-2; EP4: prostaglandin receptor; MASP: mannose-associated serine protease; MBL: mannose-binding lectin; IC: immune complex; IL: interleukin; LTB4: leukotriene B4; FcγR: Fc gamma receptors; PGE2: prostaglandin E2; TGF: transforming growth factor; TNF: tumor necrosis factor.
5. Macrophages
Macrophages are central in perpetuating arthritis development by stimulating neovascularization, clearing apoptotic immune cells, and promoting the proliferation of fibroblasts and secretion of proteases. Based on the expression of surface molecules, cytokine secretion, and arginine metabolism, macrophages are classified into pro- (M1) and anti-inflammatory (M2) phenotypes [40]. The synovial macrophages of RA patients are of M1 phenotype, which highly express proinflammatory proteins PHD3, CCR2, MMP12, and TNF-α with a concomitant low expression M2-type polarization markers [41]. In addition, the level of synuclein A (activin A) encoded by the proinflammatory INHBA gene is significantly elevated in RA patients produced by activated macrophages that can mediate M1-type polarization [42]. Such polarized M1 macrophages secrete a large number of proinflammatory cytokines (IFN-γ, TNF-α, IL-1, and IL-6), chemokines (CCL5, CXCL-1, and CXCL-10), and various matrix lysing enzymes, which in turn activate fibroblasts and osteoclasts; aid in the recruitment of neutrophils, monocytes, and lymphocytes; and trigger a series of inflammatory reactions that accelerate inflammation and cause destruction to the articular cartilage. In RA, such a high activation of macrophages increases the expression of toll-like receptors (TLR2, TLR3, TLR4, and TLR7) and promotes the synovial inflammation and cartilage destruction by producing enzymes, cytokines, and other inflammatory factors [43]. In addition, autophagy of macrophages plays an essential role in the pathogenesis of RA [44], which can increase the number of osteoclasts contributing to enhanced bone resorption activity.
6. Neutrophils
In RA, neutrophils may alter immune regulation by increasing their cell survival and mobility, having anomalous inflammatory activity, increasing oxidative stress, releasing of neutrophil extracellular traps (NETs), and also by interacting with resident FLS in the synovium to promote inflammatory and antigen-presenting phenotype [45]. High levels of NETs are present in the serum, synovial tissue, rheumatoid nodules, and skin of ACPA+ RA patients. The formation of NETs requires two major biochemical activities. First, the inactivation of the PTPN22 enzyme is considered to be necessary for the production of NETs, which may be involved in the removal of the nuclear envelope and NET components [46]. Second, NETs are composed of DNA and histones, which can be acted upon by peptidyl arginine deiminase type IV (PADI4) causing citrullination. This process prevents histone methylation and transcription leading to chromatin depolymerization, a central event in the NET formation [46]. In addition, NETs contain PAD deposits that promote the formation of citrulline products. Interestingly, the level of NETs in the plasma are highly specific (92%) and sensitive (91%) during diagnosis of early RA patients.
7. Fibroblasts
Normal FLS in the synovial intimal lining layer has important functions in the maintenance of joint homeostasis by secreting hyaluronan, lubricin, and plasminogen activator; controlling synovial fluid volume and normal inflammatory responses; and regulating leukocyte trafficking and in the maintenance of the joint capsule. In arthritis, FLS are hyperproliferative and an impaired apoptosis could also promote accumulation of FLS in the joints. In RA, FLS produces cytokines and proteases, apart from acquiring an aggressive, tumor-like phenotype because of transcriptional mechanisms of imprinting and epigenetic changes, which could mediate cartilage destruction and drive joint inflammation [47]. IL-17 is one of the crucial factors in transforming FLS into an invasive RA-FLS type and may directly assist in FLS-mediated progression of RA by significantly increasing its activation, migration, and invasive potential. RA-FLS also secretes many proangiogenic factors like fibroblast growth factor, vascular endothelial growth factor (VEGF), hypoxia-inducible factors (HIFs), and IL-18, which promote new blood vessel formation, pannus growth, and inflammation.
8. Chondrocytes
Chondrocytes are unique to the articular cartilage, which maintain an equilibrium between synthesis and breakdown of extracellular matrix under physiological conditions. A chondrocyte secretome contains extracellular matrix proteins, cytokines, growth factors, enzymes, and their inhibitors as well as many other protein components having different target specificities [48]. Cytokines trigger chondrocytes to release more cytokines and matrix metalloproteinases (MMPs) that can degrade the cartilage and also inhibit generation of tissue inhibitors of metalloproteinases (TIMPs). IL-1β released during inflammation can increase the catabolic activities of chondrocytes by inhibiting the spontaneous calcium signaling as well as in altering signaling in the cell cycle and Rho GTPases present within the chondrocytes. Dependent on the release of proinflammatory cytokines from the synovium, chondrocytes are activated to participate in cartilage damage. At the same time, they could also act as the source of proinflammatory cytokines, which in turn increases catabolic events in the cartilage while suppressing anabolic tissue repair and remodeling processes.
9. Role of Cytokines
Cytokines are involved in many inflammatory events related with the regulation of inflammation, autoimmune responses, synovitis, and articular joint destruction. Many crucial cytokines (IL-1, IL-6, IL-10, IL-12, IL-15, IL-17, IL-18, TNF-α, TGFβ, IL-23, etc.) [49] (Table 1) and all the four family of chemokines (CXC, CC, C, and CX3C) [50] (Table 2) are contributing to the joint inflammation. Successful amelioration of signs and symptoms of arthritis in patients with TNF-α-neutralizing agents has transformed RA treatment strategies quite significantly, which facilitated further research to target other inflammatory cytokines like IL-1, IL-6, and IL-17. However, targeting cytokines should be done with caution because they are pleiotropic, redundant, and multifunctional apart from having antagonistic and synergistic functions between them. Many RA patients are still reported to be refractory to anti-TNF-α therapy and TNF inhibitors were found to be more effective in clinical trials than in daily clinical practice. Moreover, anti-inflammatory cytokines also can promote joint inflammation [51]. Pro- and anti-inflammatory cytokines and chemokines involved in joint inflammation/resolution, their cellular sources, target cells, and major functions are summarized in Tables 1 and 2, respectively.
Table 1.
Pro- and anti-inflammatory cytokines in RA.
| Cytokines | Sources | Affected cells | Types | Major functions |
|---|---|---|---|---|
| Proinflammatory | ||||
| TNF-α | Fibroblast, endothelial cell, and macrophage | Synoviocyte, chondrocyte, osteoclast, and endothelial cell | None | Promotes pannus tissue formation; synovial inflammation; synthesis of MMPs, PGE2, and collagenase; vasospasm, joint destruction; osteoclastogenesis; and bone resorption [61]. |
| IFN-γ | NK cell, T cell, macrophage, B cell, and dendritic cell | T cell, B cell, macrophage, endothelial cell, and APC | None | Activates antigen-presenting cells, upregulates the expression of MIC molecules on monocytes, and aggravates the local inflammatory responses in RA. |
| IL-1 | Fibroblast, macrophage, endothelial cell, synovial cell, and chondrocyte | Synovial cell, macrophage, neutrophil, B cell, T cell, osteoclast, synaptic cell, and chondrocyte | IL-1α, IL-1β, IL-18, IL-1, F5-10, and IL-33 | Promotes TNF-α and IL-6 production, vasospasm, synaptic cell and chondrocyte synthesis, release of PGE2, secretion of MMPs, proliferation of synovial cells, and osteoclast differentiation and activity. Acts synergistically with TNF-α to damage the cartilage. |
| IL-2 | T cell, monocyte, dendritic cell, and synovial cell | T cell, B cell, macrophage, and NK cell | None | Activates and maintains T cell differentiation and proliferation, mediates phosphorylation of STAT5, and triggers active transcription in Treg cells [12]. |
| IL-6 | Osteoblast, stromal cell, T cell, B cell, fibroblast, endothelial cell, monocyte, and keratinocyte | Th17 cell, B cell, osteoclast, macrophage, neutrophil, and synoviocyte | None | Regulates osteoclast formation and bone resorption, promotes proliferation and differentiation of activated B cells, enhances the effects of IL-1 and TNF-α, maintains differentiation and homeostasis of Th17 cells, triggers synthesis of acute-phase proteins, and induces synovial neovascularization. |
| IL-8 | Monocyte, FLS, macrophage, synoviocyte, synovial lining cell, and endothelial cell | Neutrophil, NK cell, T cell, synovial cell, chondrocyte, and macrophage | None | Chemotactic and activation factor for neutrophils, stimulates NK and T cells to express γ-interferon and FasL, respectively, promotes apoptosis of T and B cells, stimulates synovial cells, and chondrocytes to produce NO, PGE2, MMPs, TNF-α, and IL-6. |
| IL-15 | Macrophage, fibroblast, endothelial cell, dendritic cell, myocyte, epithelial cell, and astrocyte | CD4+ and CD8+ T cell, monocyte, macrophage, and NK cell | None | Promotes the accumulation and activation of T cells into the joints; secretion of other cytokines (TNF-α), adhesion molecules, proteases, and monocyte chemotactic factors (MCP-1 and IL-8); and expression of IL-17 and MMPs (MMP-1 and MMP3). It is associated with bone destruction and secretion of RF in RA [62]. |
| IL-17 | CD4+ Th17 cell, NK cell, neutrophil, eosinophil, monocyte, and mast cell | T cell, fibroblast, synovial cell, macrophage, neutrophil, osteoclast, synaptic cell, chondrocyte, endothelial cell, and epithelial cell | IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F | By binding to IL-17RA/RC receptors, stimulates production of TNF-α, IL-1β, IL-6, IL-8, IL-23, chemokines, nitric oxide, prostaglandins, GM-CSF, and MMPs; promotes RANKL expression, NF-κB activation, and vasospasm formation; activates osteoblasts and osteoclasts by regulating the RANKL/RANK/OPG signaling pathway; and inhibits the synthesis of proteoglycans and collagen [63]. |
| IL-18 | Macrophage, synoviocyte | Th1 cell, chondrocyte, synovial cell | None | Promotes expression of mononuclear factors and TNF-α. |
| IL-23 | Macrophage, dendritic cell, and B cell, | T cell, dendritic cell, and macrophage | None | Induces formation and differentiation of Th17 cells, differentiation of osteoclasts, e production of IL-6, TNF-α, IL-1β, and chemokines (CXCL-1, GCP-2, and CXCL-8). |
|
| ||||
| Anti-inflammatory | ||||
| IFN-α | Leukocyte, dendritic cell | T cell, monocyte, macrophage, NK cell, synoviocyte, and B cell | None | Inhibits synthesis of collagen, expression of MHC antigens, proliferation of B cells, release of prostaglandins from monocytes, and bone resorption and disrupts the balance between collagen and fibronectin. |
| IFN-β | FLS, macrophage, dendritic cell, and osteoclast | T cell, dendritic cell, macrophage, NK cell, fibroblast, and osteoclast | None | Reduces the production of TNF-α, IL-1β, and IL-6 and increases the secretion of IL-1Rα and IL-10, modulates signaling molecules involved in the NF-κB pathway, and inhibits osteoclastogenesis and the negative feedback pathway of c-fos [64]. |
| IL-1Ra | Monocyte, macrophage, neutrophil, epithelial cell, and fibroblast | Monocyte, macrophage, lymphocyte, epithelial cell, and fibroblast | None | Abolishes IL-1 activation. |
| IL-4 | Th2 cell, mast cell, basophil, and dendritic cell | B cell, T cell, macrophage, dendritic cell, chondrocyte, FLS, osteoclast, and APC | None | Inhibits the production of MMPs, IL-1β, IL-6, IL-8, IL-12, IL-17, and TNF-α and promotes the production of IL-1Rα and soluble TNF receptors, directs B cells to produce antibodies like lgG1 and lgE, and promotes the expansion of Th2 cells and adhesion of macrophages to vascular endothelial cells [65]. |
| IL-10 | Th2 cell, B cell, macrophage, mast cell, monocyte, synovial cell, and chondrocyte | B cell, Th1 cell, macrophage, APC, and monocyte | None | Inhibits the expression of MHC class II antigens, proliferation of Th1 and B cells, and production of IFN-γ, TNF-α, IL-1, lL-3, GM-CSF, COX-2, and MMPs and promotes the production of IL-1Rα, soluble tumor necrosis factor receptor, and TIMPs. |
| IL-13 | B cell, T cell, and macrophage | B cell, monocyte, and endothelial cell | None | Inhibits ADCC and production of inflammatory cytokines (IL-1, IL-6, IL-8, and TNF-α), induces expression of IL-1Ra, and promotes B cell differentiation and antibody synthesis. |
| IL-25 (also called as IL-17E) | Th17 cell, Th2 cell, mast cell, epithelial cell, and macrophage | Th17 cell, NK cell, type 2 myeloid cell, Th9 cell, basophil, eosinophil, mast cell, endothelial cell, macrophage, dendritic cell, and CD4+ T cell | None | Inhibits IL-17A and IFN-γ production and induces the expression of various chemokines and cytokines (IL-4, IL-5, and IL-13). Expression of IL-25 in articular cartilage inhibits the synthesis of cartilage and bone stimulates the release of NO and IL-6 production. |
|
| ||||
| Cytokine with dual effects | ||||
| TGF-β | FLS, macrophage, monocyte, NK cell, and T cell | Fibroblast, epithelial cell, Th17 cell, Treg cell, and macrophage | None | Proinflammatory: chemotactic to fibroblasts, promotes fibroblast proliferation, epithelial cell differentiation, and the stability of Th17 cells. |
| Anti-inflammatory: in the absence of IL-6, promotes the differentiation and proliferation of Treg cells and inhibits activation of IFN-γ, TNF-α, macrophages, and antibody production. | ||||
Abbreviations: ADCC: antibody-dependent cellular cytotoxicity; APC: antigen-presenting cells; COX-2: cyclooxygenase-2; CXCL: chemokine (C-X-C motif) ligand; FLS: fibroblast-like synoviocytes; GCP-2: granulocyte chemotactic protein-2; GM-CSF: granulocyte-macrophage colony-stimulating factor; IFN: interferon; MHC: major histocompatibility complex; MCP-1: monocyte chemoattractant protein-1; MIC: macrophage inhibitory cytokine; MMP: matrix metalloproteinase; NO: nitric oxide; OPG: osteoprotegerin; PGE2: prostaglandin E2; RANK: receptor activator of nuclear factor-κB; RANKL: receptor activator of nuclear factor kappa Β ligand; TIMP: tissue inhibitors of metalloproteinase; TNF: tumor necrosis factor.
Table 2.
Various chemokines in RA development.
| Members | Sources | Ligand(s) | Target(s) | Major functions |
|---|---|---|---|---|
| CXC | ||||
| CXCL1/groα | FLS, chondrocyte, macrophage, endothelial cell, synovial lining cell, and osteoclast | CXCR2, DARC | Neutrophil, macrophage, and fibroblasts | Enhances collagen deposition on RA fibroblasts, thereby fibrillating the synovium. |
| CXCL4/PF4 | Macrophage, monocyte, and T cell | CXCR3 | Neutrophil, fibroblast, monocyte, and Th17 cell | Prevents monocyte apoptosis, induces differentiation of macrophages and enhances monocyte phagocytosis and oxygen radical production, exacerbates synovial inflammation, and promotes its chronicity by attracting and activating monocytes to the inflamed tissue. |
| CXCL5/ENA-78 | Monocyte, fibroblast, synovial lining cell, macrophage, and endothelial cell | CXCR2, DARC | Neutrophil, Th17 cell, and FLS | Potent chemotactic factor for neutrophils and also an angiogenic factor. Increased levels of CXCL5 contribute to enhanced levels of RANKL expression. |
| CXCL6/GCP-2 | Fibroblast, endothelial cell | CXCR2 | Neutrophil, vascular endothelial cell, and chondrocyte | Plays an important role in neutrophil migration and angiogenesis, promotes angiogenesis, and aggravates joint inflammation. |
| CXCL7/CTAP-III | Macrophage, monocyte | CXCR2 | Neutrophil, connective tissue cell | It has an angiogenic effect that affects many aspects of connective tissue metabolism, including the proliferation of synovial fibroblasts and stimulation of synovial fibrosis. |
| CXCL8/IL-8 | Synoviocyte, macrophage, synovial lining cell, endothelial cell, and monocyte | CXCR1, CXCR2, and DARC | Neutrophil, osteoblast, NK cell, T cell, synovial cell, and chondrocyte, macrophage | Regulates leukocyte adhesion molecule expression and acts as a mediator of angiogenesis. |
| CXCL9/Mig | Synoviocyte, macrophage, neutrophil, monocyte, endothelial cell, fibroblast, and keratinocyte | CXCR3 | T cell, eosinophil, monocyte, and NK cell | Promotes recruitment of activated T lymphocytes and mast cell precursors and their migration to the site of inflammation. |
| CXCL10/IP-10 | Synoviocyte, leukocyte, FLS, chondrocyte, neutrophil, monocyte, endothelial cell, fibroblast, and keratinocyte | CXCR3 | Macrophage, FLS, T cell, eosinophil, monocyte, and NK cell | Induces osteoclast differentiation and mediates RANKL expression in synovial CD4+ T cells. |
| CXCL12/SDF-1 | Synoviocyte, endothelial cell, osteoblast, and stromal cell | CXCR4 | T cell, monocyte, endothelial cell, osteoclast, and stromal cell | Involved in the recruitment of CD4+ T cells to the RA synovium, synovial inflammation, and synovial lymphoid neogenesis. It binds to endoglin, which mediates the development of integrin-dependent leukocyte transendothelial migration, osteoclastogenesis, and bone erosion, leading to radiological progression of RA. |
| CXCL13/BLC | Fibroblast, endothelial cell, follicular dendritic cell, CD4+ T cell, monocyte, and macrophage | CXCR5 | B cell, Tfh cell, Th17 cell, and Treg cell | Coordinates the migration and preferential sequestration of B and T cells in the inflammatory areas. |
| CXCL16 | Macrophage, fibroblast, dendritic cell, monocyte, B cell, T cell, smooth muscle cell, and endothelial cell | CXCR6 | T cells, NK cell, B cell, FLS, plasma cell, and APC | Mediates leukocyte infiltration into synovial tissue involving the MAP kinase (MAPK) pathway and also in lymphocyte recruitment and lymph node organization. |
|
| ||||
| CC | ||||
| CCL2/MCP-1 | Synoviocyte, fibroblast, macrophage, and osteoblast | CCR2, CCR10, | Monocyte, T cell, NK cell, basophil, macrophage, and osteoclast | Induces angiogenesis, recruits macrophages to the joints, and involves in TNF-mediated osteoclast differentiation. |
| CCL3/MIP-1α | Synoviocyte, monocyte, fibroblast, FLS, neutrophil, macrophage, lymphocyte, basophil, mast cell, and dendritic cell | CCR1, CCR5, and CCR3 | Monocyte, T cell, B cell, NK cell, basophil, eosinophil, and dendritic cell | Induces leukocyte chemotaxis, promotes the circulation of T cells into the inflammatory tissues, activates proinflammatory cytokines and RANKL by modulating activation of the PI3K/AKT signaling pathway, and upregulates CD4+ T cells. |
| CCL5/RANTES | Fibroblast, T cell, endothelial cell, chondrocyte, monocyte, and macrophage | CCR1, CCR3, and CCR5 | Monocyte, T cell, NK cell, eosinophil, and basophil | Induces MMP-1 and MMP-13 expression as well as collagenase activity by activating RA synovial fibroblasts and may promote IL-1β-induced bone destruction. |
| CCL13/MCP-4 | Chondrocyte, fibroblast, epithelial cell, and endothelial cell | CCR2, CCR3 | Eosinophil, monocyte, T cell, FLS, and basophil | Stimulates proliferation of synovial fibroblasts. |
| CCL18/PARC | Neutrophil, dendritic cell | Unknown | T cell, APC, B cell, and macrophage | Promotes the recruitment of T cells by APC. |
| CCL20/MIP-3α | Synovial lining cell, monocyte | CCR6 | Th17 cell, B cell, monocyte, dendritic cell, osteoblast, and osteoclast | Recruits IL-17-producing CCR6+ Th17 cells into the synovium and promotes osteoblast proliferation and osteoclast differentiation and may cooperate with the RANKL to affect bone formation and resorption. |
| CCL25/TECK | Dendritic cell, epithelial cell, endothelial cell, and macrophage | CCR9 | Macrophage, monocyte, and T cell | CCL25 may be involved in the differentiation of monocytes into macrophages, especially in RA. |
|
| ||||
| C | ||||
| XCL1 | CD8+ T cell, CD4+ Th1 cell, and NK cell | XCR1 | Monocyte, FLS, T cell, thymocyte, B cell, NK cell, neutrophil, and CD8+ dendritic cell | Promotes T cell chemotaxis, stimulates the accumulation of T cells in arthritis joints, and downregulates production of MMP-2 by synovial fibroblasts. |
|
| ||||
| CX3C | ||||
| CX3CL1/Fractalkine | Monocyte, macrophage, FLS, endothelial cell, and dendritic cell | CX3CR1 | T cell, CD16+ monocyte, macrophage, FLS, and osteoclast | Mediates angiogenesis, enhances the adhesion of senescent T cells to synovial fibroblasts, costimulates the production of proinflammatory cytokines, and regulates cytoskeletal structure, proliferation, and migration of synovial fibroblasts. |
Abbreviations: APC: antigen-presenting cell; BLC: B lymphocyte chemoattractant; CTAP-III: connective tissue activating protein-III; CX3CL1: Fractalkine; CXCR: CXC chemokine receptor; DARC: duffy antigen receptor for chemokines; ENA-78: epithelial-neutrophil activating protein-78; FLS: fibroblast-like synoviocytes; GCP-2: granulocyte chemotactic protein-2; groα: growth-related gene product α; IL-8: interleukin-8; IP-10: interferon-γ-inducible protein; Mig: monokine induced by IFN-γ; MAPK: mitogen-activated protein kinase; MCP: monocyte chemoattractant protein; MIP: macrophage inflammatory protein; PARC: pulmonary- and activation-regulated chemokine; PF4: platelet factor 4; RANKL: receptor activator of nuclear factor kappa Β ligand; RANTES: regulated upon activation normally T cell expressed and secreted; SDF-1: stromal cell-derived factor; TECK: monocyte differentiation and chemotaxis to chemokine ligand 25; Tfh cell: follicular helper T cell; XCL1: lymphotactin.
10. MMPs, ADAMTS, and TIMPs
Several proteinases like MMPs, ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs), neutrophil elastase, and cathepsins (G and B) can damage the cartilage directly. Depletion of proteoglycans from articular cartilage is an initial event in RA development leading to the degradation of the collagen fibrils. It is of interest to note that CII-reactive monoclonal antibodies upon passive transfer induced a significant amount of proteoglycan depletion within 72 hours [30]. Many MMPs (MMP 1-3, MMP 7-9, MMP-13, and MT1-MMP) preferentially split the bond between Asn341-Phe342 of aggrecan. Conversely, ADAMTS1, ADAMTS4, and ADAMTS5 cleave the Glu373-Ala374 bond in addition to other sites in the G2-G3 domains of proteoglycans. Thus, both the MMP and ADAMTS enzymes contribute to aggrecan degradation during arthritis development. TIMPs are endogenous blockers of MMPs and regulators of matrix turnover, tissue reorganization, and cellular activity. Sources, targets, and receptors/ligands and major functions of MMPs, ADAMTS, and TIMPs involved in arthritis pathogenesis are summarized in Table 3.
Table 3.
MMPs, ADAMTS, and TIMPs in arthritis pathogenesis.
| Enzymes/inhibitors | Category | Enzymes | Sources | Targets | Major functions |
|---|---|---|---|---|---|
| Metalloproteinases | |||||
| MMP-1 | Collagenases | Collagenase-1, interstitial collagenase | Monocyte, fibroblast, smooth muscle cell, chondrocyte, macrophage, endothelial cell, and keratinocyte | Collagens (I–III, VII, VIII, and X), gelatin, aggrecan, L-selectin, IL-1β, entactin, ovostatin, MMP-2, and MMP-9 | Releases MMP-9, promotes Akt dephosphorylation, degrades collagens I, II, II, III, VI, and IX and proteoglycans [66, 67]. |
| MMP-2 | Gelatinases | Gelatinase-A, 72 KDa gelatinase | Synoviocyte, CD34+ vascular endothelial cells, CD68+ macrophage, CD14+ monocyte, and stromal cell | Gelatin, collagens I, III, IV–VII, IX and X, Laminin, elastin, Fibronectin, proteoglycan, pro-MMP-13 | Increases VEGF expression and angiogenesis, promotes angiogenesis and directs degradation of cartilage matrix. |
| MMP-3 | Stromelysins | Stromelysin-1 | FLS, chondrocyte, synovial lining cell, endothelial cell | Collagens (III–V and IX), gelatin, aggrecan, perlecan, decorin, laminin, elastin, casein, osteonectin, ovostatin, entactin, plasminogen, MBP, IL-1β, MMP-2, TIMP-2, MMP-7, MMP-8, MMP-9, MMP-13, proteoglycans, and fibronectin | Degrades cartilage connexin, fibronectin, and major components of cartilage such as collagen type IV, VII, IX, and XI, activates collagenase secreted by synoviocytes, directly destroys cartilage tissue, activates interstitial collagenase, and promotes the formation of vasospasm [68]. |
| MMP-8 | Collagenases | Collagenase-2, neutrophil collagenase | Neutrophil, plasma cell, fibroblast, dentin cell, endothelial cell, bronchial epithelial cell, keratinocyte, and macrophage | Collagens (I–III, V, VII, VIII, and X), gelatin, aggrecan, and fibronectin | Degrades type I, II, and III collagens, activates MMP-2 and MMP-3, regulates the activity of TNF-α, IL-1β, and T cell membrane proteins CD2, CD4, and CD8. |
| MMP-9 | Gelatinases | Gelatinase-B, 92 kDa gelatinase | Monocyte, synoviocyte, CD34+ vascular endothelial cell, CD68+ macrophage, and neutrophil | Collagens (IV, V, VII, X, and XIV), gelatin, entactin, aggrecan, elastin, fibronectin, osteonectin, plasminogen, MBP, and IL-1β | Participates in the degradation of ECM including types IV and V collagen, proteoglycans, elastin, and gelatin and in ECM remodeling. |
| MMP-10 | Stromelysins | Stromelysin-2 | FLS, chondrocyte, and osteoblast, | Collagens (III–V), gelatin, casein, aggrecan, elastin, MMP-1, and MMP-8 | Activates MMP-1, MMP-7, MMP-8, and MMP-9 and 13 prototypes, enhances the dissolution of cartilage collagen by activating procollagenases, and enhances cartilage collagen lysis caused by IL-1 and oncostatin M. |
| MMP-12 | Other enzymes | Macrophage metalloelastase | Macrophage, monocyte, and chondrocyte | Collagen IV, gelatin, elastin, casein, fibronectin, vitronectin, laminin, entactin, fibrinogen, fibrin, and plasminogen | Degrades type IV collagen, fibronectin, laminin, vitronectin, proteoglycan, chondroitin sulfate, and myelin basic protein and activates MMP-2 and MMP-3. |
| MMP-13 (interstitial collagenase) | Collagenases | Collagenase-3 | Chondrocyte, FLS, and macrophage | Collagens (I–IV, IX, X, and XIV), gelatin, plasminogen, aggrecan, perlecan, fibronectin, osteonectin, and MMP-9 | Degrades collagen fibers of types I, II, III, V, and XI as well as basement membrane proteoglycans. |
| MMP-14 | MT-MMP | MT1-MMP, MT-MMP-1 | Macrophage, myeloid cell, FLS, and CD68+ osteoclast | Collagens (I–III), gelatin, casein, fibronectin, laminin, vitronectin, entactin, proteoglycans, and MMP-2 and 13 | Promotes FLS to invade cartilage. MT1-MMP degrades collagen types I, II, and III, laminin-1 and laminin-5, fibronectin, vitronectin, fibrin, and aggrecan, and activates pro-MMP-2 and pro-MMP-13 on the cell surface [69, 70]. |
|
| |||||
| ADAMTS [71, 72] | |||||
| ADAMTS-1 | Aggrecanases | METH-1, Aggrecanase-3 | Chondrocyte, macrophage | Aggrecan, versican | Cleaves the proteoglycan versican. |
| ADAMTS-4 | Aggrecanases | Aggrecanase-1 | FLS, chondrocyte | Aggrecan, crevican, COMP, decorin, fibromodulin, and versican | Cleaves aggrecan. |
| ADAMTS-5 | Aggrecanases | Aggrecanase-2, ADAMTS-11 | FLS, stromal cell | Aggrecan | Cleaves aggrecan. |
| ADAMTS-7 | None | ADAMTS-7B | FLS, chondrocyte | COMP, α2M | Degrades cartilage oligomeric matrix protein (COMP). |
| ADAMTS-9 | None | KIAA1312 | FLS | Aggrecan, versican | Degrades aggrecan and has the potential to cleave other cartilage molecules. |
| ADAMTS-12 | None | None | FLS, chondrocyte | Aggrecan, COMP, and α2M | Degrades COMP. |
|
| |||||
| Inhibitors | |||||
| TIMP-1 | Glycoprotein | None | Macrophage, connective tissue cell, chondrocyte, FLS, T cell, and monocyte | MMP-3, MMP-9, MMP-14, MMP-16, MMP-19, MMP-24, ADAM10, and pro-MMP-9 | Weak inhibition of MMP-14, MMP-16, MMP-19, and MMP-24 and ADAM10. Inhibits pro-MMP interactions with pro-MMP-9, formation of synovial blood vessels, activation of MMP-3 and 9, and synovial vascular invasion in RA. |
| TIMP-2 | Glycoprotein | None | Chondrocyte, FLS, T cell, and monocyte | ADAM12, pro-MMP-2, and MMP-9 | Inhibits all the MMPs (prevents overactivation of MMP-9), ADAM12, and pro-MMP interactions with pro-MMP-2. |
| TIMP-3 | Glycoprotein | None | FLS, chondrocyte, macrophage, and monocyte | ADAM10, ADAM12, ADAM17, ADAM28, and ADAM33; ADAMTS-1-2 and 4-5; pro-MMP-2,9; and MMP-2, MMP-9, and MMP-13 | Inhibits all the MMPs and ADAM10, ADAM12, ADAM17, ADAM28, and ADAM33; ADAMTS-1, ADAMTS-4, and ADAMTS-5, ADAMTS-2 (weak); and pro-MMP interactions with pro-MMP-9 and pro-MMP-2. |
| TIMP-4 | Glycoprotein | None | FLS, endothelial cell, T cell, and monocyte (less) | ADAM17d, ADAM28, and ADAM33 and pro-MMP-2 | Inhibits most of the MMPs and ADAM17d and ADAM28, ADAM33 (weak), pro-MMP interactions with pro-MMP-2) and development of arthritis. |
Abbreviations: ADAMTS: a disintegrin and metalloproteinase with thrombospondin motifs; COMP: cartilage oligomeric matrix protein; GEP: granulin-epithelin precursor; TIMP: tissue inhibitor of metalloproteinases; vWFCP: von Willebrand factor cleaving protease; α2M: α2-macroglobulin.
11. Signaling Pathways Affecting Bone Destruction
Bone erosion starts during the early phase of arthritis development causing deformity of the articular joints, which affects quality of patients' life. Molecular mechanisms underlying differentiation and activation of bone-eroding cells, osteoclasts, are well documented, and several signaling pathways are contributing to osteoclast maturation and activation causing joint destruction (Figure 3).
Figure 3.
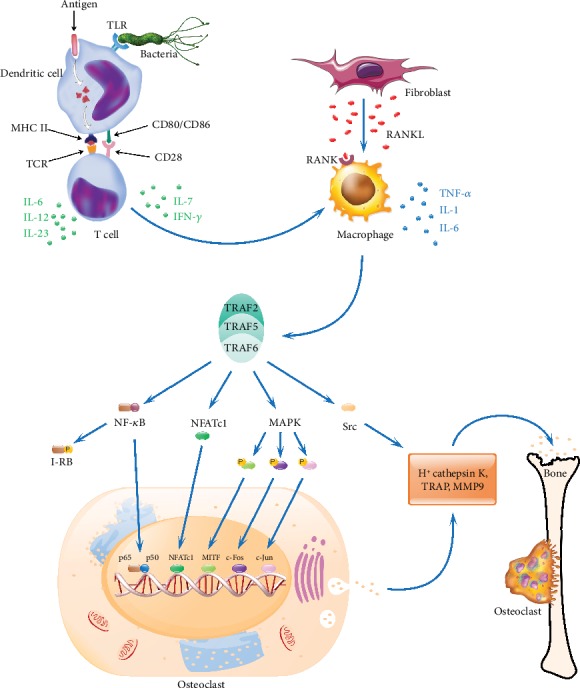
Signaling pathways in osteoclast activation. During RA pathogenesis, antigen-presenting cells after uptake of an autoantigen or pathogenic molecules process and present antigenic determinants on their cell surface in conjunction with arthritis-permissible HLA/MHC class II molecules, which activate differentiation of T cells into different subphenotypes. The activated T cells secrete various cytokines like IL-6, IL-7, IL-10, IL-12, IL-17, IL-23, and IFN-γ. These cytokines modulate macrophages to secrete various pro- and/or anti-inflammatory cytokines and other inflammatory mediators. Upon exposure to the inflammatory cytokines, fibroblast-like synoviocytes express RANKL, which binds with its receptor (RANK) present on the cell surface of activated macrophages initiating the RANK/RANKL pathway through TRAF 2, 5, and 6 proteins, which leads to the activation of downstream NF-κB, MAPK, NFATc1, and Src signaling cascades. These factors after translocation initiate the expression of genes like TRAP, CtsK, and MMP-9 in the nucleus, which promote osteoclastogenesis and bone resorption. TLR: toll-like receptor; TCR: T cell receptor; CtsK: cathepsin K; I-κB: inhibitor of the NF-κB transcription factor; IL: interleukin; IFN-γ: interferon gamma; MAPK: mitogen-activated protein kinase; MHC II: major histocompatibility complex II; MITF: microphthalmia-associated transcription factor; MMP 9: matrix metalloproteinase 9; NFATc1: nuclear factor of activated T cells, calcineurin-dependent 1; NF-κB: nuclear factor kappa B; p50 and p65: REL-associated proteins (also called NF-κB1 and RelA) involved in NF-κB heterodimer formation and nuclear translocation and activation; RANK: receptor activator of nuclear factor-κB; RANKL: receptor activator of nuclear factor κB ligand; Src: intracellular non-receptor tyrosine kinase; TRAF: TNF receptor-associated factor; TRAP: tartrate-resistant acid phosphatase. c-Jun and c-Fos form the early response transcription factor, AP-1.
12. RANKL/RANK Pathway
RANKL (also called TNFSF11, OPGL, TRANCE, and ODF) and its receptor RANK are indispensable regulators of bone repair and remodeling processes. Several hormones and cytokines induce RANKL production in osteoblasts and synovial fibroblasts. After binding with RANK, RANKL triggers the recruitment of an adaptor molecule TRAF-6 resulting in the activation of signaling molecules like NF-κB, c-Jun N-terminal kinase (JNK), AKT/PKB, ERK, Src, and p38 mitogen-activated protein (MAP) kinases and the transcription factor, and nuclear factor of activated T cells, calcineurin-dependent 1 (NFATc1) [52]. Hence, the RANKL/RANK signaling pathway is a potential therapeutic target in osteolytic diseases. Denosumab (RANKL-specific human monoclonal antibody) is currently used for treating osteoporosis, osteosarcoma, multiple myeloma, and bone metastasis [53]. Although denosumab is highly specific to RANKL and has a good effect on bones, safety concerns still exist. On the other hand, the RANKL/RANK pathway is having important functions in osteoblasts as well. Vesicular RANK, secretion product of matured osteoclasts, by binding to osteoblast-derived RANKL facilitates bone formation by initiating RANKL reverse signaling leading to the activation of Runt-related transcription factor 2 (Runx2) [54].
13. NF-κB Signaling Pathway
Initiation of the RANKL/RANK pathway causes NF-κB activation, which contributes to osteoclast differentiation. After NF-κB stimulation, several TNF-receptor- (TNFR-) related factors associate with the cytoplasmic domain of RANK. Among them, TRAF-6 is indispensable for osteoclast formation and activation [55], while NF-κB p50 and p52 subunits modulate RANKL and TNF-α-induced differentiation of osteoclast precursors. Mice deficient in p50 and p52 proteins are osteopetrotic. NF-κB-activating upstream catalytic (IKK-α and IKK-β) and noncatalytic (IKK-γ also known as NEMO) subunits of IκB kinase are also crucial in the generation of osteoclasts. RelB is the NF-κB-inducing kinase (NIK) downstream subunit, which is also responsible for osteoclast differentiation.
14. MAPKs
Mitogen-activated protein kinase (MAPK) lineage consists of p38-MAP kinases (p38-MAPK α, β, γ, and δ isoforms), c-Jun N-terminal kinases (JNK1-3), and extracellular signal-regulated kinases (ERK1-2). RANKL stimulation activates many of these kinases, which regulate different cellular responses. When a specific inhibitor (SB203580) or a natural product from teasel for p38-MAPKα and β was used, significant inhibition of osteoclast formation but not its functions was observed. RANKL-activated p38-MAPKs can directly phosphorylate STAT1 and regulate the expression of target genes. In addition, JNKs and their upstream kinase MKK7 are also involved in osteoclastogenesis. However, neither JNK1 nor JNK2 deficiency led to significant bone defects. Mice deficient in both JNK1 and JNK2 have embryonic lethality during midgestation [56]; hence, a conditional knock-out in the bone marrow might address the importance of JNKs in osteoclastogenesis. AP-1 and related genes (c-Jun, JunB, c-Fos, and Fra but not JunD), controlled by JNKs, are also crucial for osteoclast differentiation and maturation. ERK is another MAPK subunit getting activated upon RANKL stimulation, which regulates the survival and differentiation of both osteoclasts and osteoblasts. However, several receptor systems in many organs operate via NF-κB and MAPK pathways; hence, targeting these pathways might not be optimal for treating bone damage.
15. NFATc1
Stimulating with RANKL leads to the induction of several genes mediating osteoclast differentiation and function, including cellular fusion, polarization, and secretion of acid hydrolases. Some of these genes are transcribed by another key factor, NFATc1 [57], which is a downstream target of RANKL. NFATc1 expression causes osteoclast differentiation and induction of osteoclast-related genes including TRAP, cathepsin K, and calcitonin receptors in association with c-Fos. NFATc1-deficient cells are defective in osteoclastogenesis. In the absence of RANKL, overexpression of NFATc1 induced osteoclast precursor cell differentiation into TRAP+ osteoclast-like cells. Moreover, c-Jun signaling is also critical in the regulation of NFATc1 activation. Transgenic mice specifically expressing dominant-negative form of c-Jun in the osteoclasts exhibit severe osteopetrosis. Thus, NFATc1 acts as an important mediator in coupling RANK signaling events to osteoclast differentiation.
16. Src Kinase
Src is a protein-tyrosine kinase involved in the cell development, division, relocation, and survival. The resorption process of osteoclasts depends on their attachment and movement on the surface of the bones to form a sealing zone [58]. Targeted interference in c-Src gene expression led to the development of osteopetrosis, and it is a critical factor in RANKL-induced activation of protein tyrosine kinase 2 (Pyk2) and αvβ3 integrin assembly, which is essential for the adhesion and skeleton organization. Binding of RANK and RANKL meditates the recruitment of TRAF-6 and c-Src. Subsequently, TRAF-6 enhances c-Src activation causing phosphorylation of signaling molecules like E3 ubiquitin-protein ligase, and Cbl. Src complexes with Pyk2, Cbl, and ADAP (adhesion and degranulation promoting adaptor protein, also called SLAP-130 or Fyb). Phosphorylation of these signaling molecules is a prerequisite for integrin-mediated osteoclast functions. Hence, targeting c-Src might be a viable future treatment strategy for osteoporosis and higher bone resorption observed in RA patients.
17. Conclusions
Interplay between multiple factors engender aberrations in immune recognition and activation causing initiation of molecular pathways targeting cartilage and bone. Various immune and nonimmune cells are crucial during this process. Resident and infiltrating cells in the joints proliferate and secrete proinflammatory cytokines, chemokines, and matrix lysing enzymes that could destroy the joints leading to functional loss. Moreover, different signaling cascades are activated during osteoclast activation and differentiation that are involved in the bone resorption activity. Hence, targeting a single effector molecule is insufficient to block cartilage and bone damage in arthritis. Since RA is an immune-mediated disorder, therapeutics restoring immune balance certainly can improve clinical therapy.
Acknowledgments
KSN would like to thank the Southern Medical University, Guangzhou, China, for the start-up grants (C1034211, C1051004) and international exploration grant (C1051427).
Additional Points
Key Messages. Rheumatoid arthritis is a multi-factorial syndrome involving interactions between genetic, epigenetic and environmental factors. Initiation events damaging joints start very early during the autoimmune phase of arthritis development. Comprehensive knowledge on pathogenesis is a prerequisite for developing optimal treatment and potential drugs. Targeting a single effector molecule is insufficient to block cartilage and bone damage in arthritis. Since rheumatoid arthritis is an immune-mediated disorder, therapeutics restoring immune balance can improve clinical therapy.
Conflicts of Interest
The authors declare no conflict of interest.
Authors' Contributions
All the three authors contributed in preparing this manuscript.
References
- 1.McInnes I. B., Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet. 2017;389(10086):2328–2337. doi: 10.1016/S0140-6736(17)31472-1. [DOI] [PubMed] [Google Scholar]
- 2.Orr C., Vieira-Sousa E., Boyle D. L., et al. Erratum: Synovial tissue research: a state-of-the-art review. Nature Reviews Rheumatology. 2017;13(10):p. 630. doi: 10.1038/nrrheum.2017.161. [DOI] [PubMed] [Google Scholar]
- 3.Fang Q., Ou J., Nandakumar K. S. Autoantibodies as diagnostic markers and mediator of joint inflammation in arthritis. Mediators of Inflammation. 2019;2019:22. doi: 10.1155/2019/6363086.6363086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harre U., Lang S. C., Pfeifle R., et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nature Communications. 2015;6(1):p. 6651. doi: 10.1038/ncomms7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burmester G. R., Feist E., Dorner T. Emerging cell and cytokine targets in rheumatoid arthritis. Nature Reviews Rheumatology. 2014;10(2):77–88. doi: 10.1038/nrrheum.2013.168. [DOI] [PubMed] [Google Scholar]
- 6.Yudoh K., Matsuno H., Nakazawa F., Yonezawa T., Kimura T. Reduced expression of the regulatory CD4+ T cell subset is related to Th1/Th2 balance and disease severity in rheumatoid arthritis. Arthritis and Rheumatism. 2000;43(3):617–627. doi: 10.1002/1529-0131(200003)43:3<617::AID-ANR19>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 7.Komatsu N., Okamoto K., Sawa S., et al. Pathogenic conversion of Foxp 3+ T cells into TH17 cells in autoimmune arthritis. Nature Medicine. 2014;20(1):62–68. doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- 8.Malmstrom V., Backlund J., Jansson L., Kihlberg J., Holmdahl R. T cells that are naturally tolerant to cartilage-derived type II collagen are involved in the development of collagen-induced arthritis. Arthritis Research. 2000;2(4):315–326. doi: 10.1186/ar106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Herwijnen M. J., Wieten L., van der Zee R., et al. Regulatory T cells that recognize a ubiquitous stress-inducible self-antigen are long-lived suppressors of autoimmune arthritis. Proceedings of the National Academy of Sciences. 2012;109(35):14134–14139. doi: 10.1073/pnas.1206803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z., Shen Y., Oishi H., et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Science Translational Medicine. 2016;8(331):331ra38–331ra38. doi: 10.1126/scitranslmed.aad7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.James E. A., Rieck M., Pieper J., et al. Citrulline-specific Th1 cells are increased in rheumatoid arthritis and their frequency is influenced by disease duration and therapy. Arthritis & Rhematology. 2014;66(7):1712–1722. doi: 10.1002/art.38637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu L., Barbi J., Pan F. The regulation of immune tolerance by FOXP3. Nature Reviews Immunology. 2017;17(11):703–717. doi: 10.1038/nri.2017.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salmond R. J., Brownlie R. J., Morrison V. L., Zamoyska R. The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nature Immunology. 2014;15(9):875–883. doi: 10.1038/ni.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wehr P., Purvis H., Law S. C., Thomas R. Dendritic cells, T cells and their interaction in rheumatoid arthritis. Clinical and Experimental Immunology. 2019;196(1):12–27. doi: 10.1111/cei.13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shakya A. K., Nandakumar K. S. Antigen-specific tolerization and targeted delivery as therapeutic strategies for autoimmune diseases. Trends in Biotechnology. 2018;36(7):686–699. doi: 10.1016/j.tibtech.2018.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Schinnerling K., Soto L., Garcia-Gonzalez P., Catalan D., Aguillon J. C. Skewing dendritic cell differentiation towards a tolerogenic state for recovery of tolerance in rheumatoid arthritis. Autoimmunity Reviews. 2015;14(6):517–527. doi: 10.1016/j.autrev.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 17.Shen P., Fillatreau S. Antibody-independent functions of B cells: a focus on cytokines. Nature Reviews Immunology. 2015;15(7):441–451. doi: 10.1038/nri3857. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann K., Clauder A. K., Manz R. A. Targeting B cells and plasma cells in autoimmune diseases. Frontiers in Immunology. 2018;9:p. 835. doi: 10.3389/fimmu.2018.00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka Y., Ohira T. Mechanisms and therapeutic targets for bone damage in rheumatoid arthritis, in particular the RANK-RANKL system. Current Opinion in Pharmacology. 2018;40:110–119. doi: 10.1016/j.coph.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Rantapää-Dahlqvist S., de Jong B. A. W., Berglin E., et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis & Rheumatism. 2003;48(10):2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 21.van der Woude D., Rantapaa-Dahlqvist S., Ioan-Facsinay A., et al. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Annals of the Rheumatic Diseases. 2010;69(8):1554–1561. doi: 10.1136/ard.2009.124537. [DOI] [PubMed] [Google Scholar]
- 22.Suwannalai P., van de Stadt L. A., Radner H., et al. Avidity maturation of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis & Rheumatism. 2012;64(5):1323–1328. doi: 10.1002/art.33489. [DOI] [PubMed] [Google Scholar]
- 23.Rombouts Y., Ewing E., van de Stadt L. A., et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Annals of the Rheumatic Diseases. 2015;74(1):234–241. doi: 10.1136/annrheumdis-2013-203565. [DOI] [PubMed] [Google Scholar]
- 24.Ohmi Y., Ise W., Harazono A., et al. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nature Communications. 2016;7(1, article 11205) doi: 10.1038/ncomms11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao D., Khmaladze I., Jia H., et al. Pathogenic autoreactive B cells are not negatively selected toward matrix protein collagen II. Journal of Immunology. 2012;187(9):4451–4458. doi: 10.4049/jimmunol.1101378. [DOI] [PubMed] [Google Scholar]
- 26.Ludwig R. J., Vanhoorelbeke K., Leypoldt F., et al. Mechanisms of autoantibody-induced pathology. Frontiers in Immunology. 2017;8:p. 603. doi: 10.3389/fimmu.2017.00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bemis E. A., Norris J. M., Seifert J., et al. Complement and its environmental determinants in the progression of human rheumatoid arthritis. Molecular Immunology. 2019;112:256–265. doi: 10.1016/j.molimm.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holers V. M., Banda N. K. Complement in the initiation and evolution of rheumatoid arthritis. Frontiers in Immunology. 2018;9:p. 1057. doi: 10.3389/fimmu.2018.01057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nandakumar K. S. Targeting IgG in arthritis: disease pathways and therapeutic avenues. International Journal of Molecular Sciences. 2018;19(3):p. 677. doi: 10.3390/ijms19030677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nandakumar K. S., Bajtner E., Hill L., et al. Arthritogenic antibodies specific for a major type II collagen triple-helical epitope bind and destabilize cartilage independent of inflammation. Arthritis and Rheumatism. 2008;58(1):184–196. doi: 10.1002/art.23049. [DOI] [PubMed] [Google Scholar]
- 31.Farinotti A. B., Wigerblad G., Nascimento D., et al. Cartilage-binding antibodies induce pain through immune complex-mediated activation of neurons. The Journal of Experimental Medicine. 2019;216(8):1904–1924. doi: 10.1084/jem.20181657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petkova S. B., Konstantinov K. N., Sproule T. J., Lyons B. L., Awwami M. A., Roopenian D. C. Human antibodies induce arthritis in mice deficient in the low-affinity inhibitory IgG receptor Fc gamma RIIB. The Journal of Experimental Medicine. 2006;203(2):275–280. doi: 10.1084/jem.20051951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada H., Ozawa T., Kishi H., et al. Cutting edge: B cells expressing cyclic citrullinated peptide-specific antigen receptor are tolerized in normal conditions. Journal of Immunology. 2018;201(12):3492–3496. doi: 10.4049/jimmunol.1800826. [DOI] [PubMed] [Google Scholar]
- 34.Harre U., Georgess D., Bang H., et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. The Journal of Clinical Investigation. 2012;122(5):1791–1802. doi: 10.1172/JCI60975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleyer A., Finzel S., Rech J., et al. Bone loss before the clinical onset of rheumatoid arthritis in subjects with anticitrullinated protein antibodies. Annals of the Rheumatic Diseases. 2014;73(5):854–860. doi: 10.1136/annrheumdis-2012-202958. [DOI] [PubMed] [Google Scholar]
- 36.Wigerblad G., Bas D. B., Fernades-Cerqueira C., et al. Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Annals of the Rheumatic Diseases. 2016;75(4):730–738. doi: 10.1136/annrheumdis-2015-208094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun M., Rethi B., Krishnamurthy A., et al. Anticitrullinated protein antibodies facilitate migration of synovial tissue-derived fibroblasts. Annals of the Rheumatic Diseases. 2019;78(12):1621–1631. doi: 10.1136/annrheumdis-2018-214967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laurent L., Clavel C., Lemaire O., et al. Fcγ receptor profile of monocytes and macrophages from rheumatoid arthritis patients and their response to immune complexes formed with autoantibodies to citrullinated proteins. Annals of the Rheumatic Diseases. 2011;70(6):1052–1059. doi: 10.1136/ard.2010.142091. [DOI] [PubMed] [Google Scholar]
- 39.Toes R., Pisetsky D. S. Pathogenic effector functions of ACPA: where do we stand? Annals of the Rheumatic Diseases. 2019;78(6):716–721. doi: 10.1136/annrheumdis-2019-215337. [DOI] [PubMed] [Google Scholar]
- 40.Self-Fordham J. B., Naqvi A. R., Uttamani J. R., Kulkarni V., Nares S. MicroRNA: Dynamic regulators of macrophage polarization and plasticity. Frontiers in Immunology. 2017;8:p. 1062. doi: 10.3389/fimmu.2017.01062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palacios B. S., Estrada-Capetillo L., Izquierdo E., et al. Macrophages from the synovium of active rheumatoid arthritis exhibit an activin A-dependent pro-inflammatory profile. The Journal of Pathology. 2015;235(3):515–526. doi: 10.1002/path.4466. [DOI] [PubMed] [Google Scholar]
- 42.Massague J. TGFβ signalling in context. Nature Reviews Molecular Cell Biology. 2012;13(10):616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cuda C. M., Pope R. M., Perlman H. The inflammatory role of phagocyte apoptotic pathways in rheumatic diseases. Nature Reviews Rheumatology. 2016;12(9):543–558. doi: 10.1038/nrrheum.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vomero M., Barbati C., Colasanti T., et al. Autophagy and rheumatoid arthritis: current knowledges and future perspectives. Frontiers in Immunology. 2018;9:p. 1577. doi: 10.3389/fimmu.2018.01577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Neil L. J., Kaplan M. J. Neutrophils in rheumatoid arthritis: breaking immune tolerance and fueling disease. Trends in Molecular Medicine. 2019;25(3):215–227. doi: 10.1016/j.molmed.2018.12.008. [DOI] [PubMed] [Google Scholar]
- 46.Vermeren S., Miles K., Chu J. Y., Salter D., Zamoyska R., Gray M. PTPN22 is a critical regulator of Fcγ receptor-mediated neutrophil activation. Journal of Immunology. 2016;197(12):4771–4779. doi: 10.4049/jimmunol.1600604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartok B., Firestein G. S. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunological Reviews. 2010;233(1):233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanchez C., Bay-Jensen A. C., Pap T., et al. Chondrocyte secretome: a source of novel insights and exploratory biomarkers of osteoarthritis. Osteoarthritis and Cartilage. 2017;25(8):1199–1209. doi: 10.1016/j.joca.2017.02.797. [DOI] [PubMed] [Google Scholar]
- 49.McInnes I. B., Buckley C. D., Isaacs J. D. Cytokines in rheumatoid arthritis - shaping the immunological landscape. Nature Reviews Rheumatology. 2016;12(1):63–68. doi: 10.1038/nrrheum.2015.171. [DOI] [PubMed] [Google Scholar]
- 50.Koch A. E. Chemokines and their receptors in rheumatoid arthritis: future targets? Arthritis and Rheumatism. 2005;52(3):710–721. doi: 10.1002/art.20932. [DOI] [PubMed] [Google Scholar]
- 51.Nandakumar K. S., Holmdahl R. Arthritis induced with cartilage-specific antibodiesis IL-4-dependent. European Journal of Immunology. 2006;36(6):1608–1618. doi: 10.1002/eji.200535633. [DOI] [PubMed] [Google Scholar]
- 52.Han Z., Boyle D. L., Chang L., et al. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. The Journal of Clinical Investigation. 2001;108(1):73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raje N., Terpos E., Willenbacher W., et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: an international, double-blind, double-dummy, randomised, controlled, phase 3 study. The Lancet Oncology. 2018;19(3):370–381. doi: 10.1016/S1470-2045(18)30072-X. [DOI] [PubMed] [Google Scholar]
- 54.Ikebuchi Y., Aoki S., Honma M., et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature. 2018;561(7722):195–200. doi: 10.1038/s41586-018-0482-7. [DOI] [PubMed] [Google Scholar]
- 55.Jang H. D., Hwang H. Z., Kim H. S., Lee S. Y. C-Cbl negatively regulates TRAF6-mediated NF-κB activation by promoting K48-linked polyubiquitination of TRAF6. Cellular & Molecular Biology Letters. 2019;24(1):p. 29. doi: 10.1186/s11658-019-0156-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang L., Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 57.Zhou C., You Y., Shen W., et al. Deficiency of sorting nexin 10 prevents bone erosion in collagen-induced mouse arthritis through promoting NFATc1 degradation. Annals of the Rheumatic Diseases. 2016;75(6):1211–1218. doi: 10.1136/annrheumdis-2014-207134. [DOI] [PubMed] [Google Scholar]
- 58.Kikuta J., Ishii M. Osteoclast migration, differentiation and function: novel therapeutic targets for rheumatic diseases. Rheumatology. 2013;52(2):226–234. doi: 10.1093/rheumatology/kes259. [DOI] [PubMed] [Google Scholar]
- 59.Catrina A. I., Svensson C. I., Malmstrom V., Schett G., Klareskog L. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nature Reviews Rheumatology. 2017;13(2):79–86. doi: 10.1038/nrrheum.2016.200. [DOI] [PubMed] [Google Scholar]
- 60.Smolen J. S., Aletaha D., Barton A., et al. Rheumatoid arthritis. Nature Reviews Disease Primers. 2018;4(1):p. 18001. doi: 10.1038/nrdp.2018.1. [DOI] [PubMed] [Google Scholar]
- 61.Gradaigh D. O.’., Ireland D., Bord S., Compston J. E. Joint erosion in rheumatoid arthritis: interactions between tumour necrosis factor, interleukin 1, and receptor activator of nuclear factor B ligand (RANKL) regulate osteoclasts. Annals of the Rheumatic Diseases. 2004;63(4):354–359. doi: 10.1136/ard.2003.008458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McInnes I. B., Leung B. P., Sturrock R. D., Field M., Liew F. Y. Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nature Medicine. 1997;3(2):189–195. doi: 10.1038/nm0297-189. [DOI] [PubMed] [Google Scholar]
- 63.Hwang S. Y., Kim J. Y., Kim K. W., et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Research & Therapy. 2004;6(2):R120–R128. doi: 10.1186/ar1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takayanagi H., Kim S., Matsuo K., et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416(6882):744–749. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- 65.Krabben A., Wilson A. G., de Rooy D. P., et al. Association of genetic variants in the IL4 and IL4R genes with the severity of joint damage in rheumatoid arthritis: a study in seven cohorts. Arthritis and Rheumatism. 2013;65(12):3051–3057. doi: 10.1002/art.38141. [DOI] [PubMed] [Google Scholar]
- 66.Eck S. M., Blackburn J. S., Schmucker A. C., Burrage P. S., Brinckerhoff C. E. Matrix metalloproteinase and G protein coupled receptors: co-conspirators in the pathogenesis of autoimmune disease and cancer. Journal of Autoimmunity. 2009;33(3-4):214–221. doi: 10.1016/j.jaut.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burrage P. S., Mix K. S., Brinckerhoff C. E. Matrix metalloproteinases: role in arthritis. Frontiers in Bioscience. 2006;11:529–543. doi: 10.2741/1817. [DOI] [PubMed] [Google Scholar]
- 68.Mehana E. E., Khafaga A. F., El-Blehi S. S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: an updated review. Life Sciences. 2019;234, article 116786 doi: 10.1016/j.lfs.2019.116786. [DOI] [PubMed] [Google Scholar]
- 69.Chen Z., Wang H., Xia Y., Yan F., Lu Y. Therapeutic potential of mesenchymal cell-derived miRNA-150-5p–Expressing exosomes in rheumatoid arthritis mediated by the modulation of MMP14 and VEGF. Journal of Immunology. 2018;201(8):2472–2482. doi: 10.4049/jimmunol.1800304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zucker S., Pei D., Cao J., Lopez-Otin C. Membrane type-matrix metalloproteinases (MT-MMP) Current Topics in Developmental Biology. 2003;54:1–74. doi: 10.1016/s0070-2153(03)54004-2. [DOI] [PubMed] [Google Scholar]
- 71.Lin E. A., Liu C. J. The role of ADAMTSs in arthritis. Protein & Cell. 2010;1(1):33–47. doi: 10.1007/s13238-010-0002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu C. J. The role of ADAMTS-7 and ADAMTS-12 in the pathogenesis of arthritis. Nature Clinical Practice Rheumatology. 2009;5(1):38–45. doi: 10.1038/ncprheum0961. [DOI] [PMC free article] [PubMed] [Google Scholar]