

Abstract
This chapter reviews the use of standard transmission electron microscopy (TEM) of plastic-embedded material, as well as protocols for the immunolabeling of cryosections, in the analysis of viral interactions with cells. It focuses particularly on the assembly of two types of enveloped viruses: (1) the beta herpesvirus—human cytomegalovirus (HCMV), and (2) the primate lentiviruses—the simian and human immunodeficiency viruses (SIV and HIV). The chapter discusses the ways EM is used to identify morphological features of the various stages in the assembly of virus particles, to distinguish immature and mature particles, or to analyze steps involved in the acquisition of lipid membranes by enveloped viruses. In addition, it demonstrates the way immunolabeling allows the quantification of viral components, even in individual virus particles, and comparisons between particles at different locations in the cell or at different stages in viral assembly. Together with the newly developed methods for electron tomography and correlative immunofluorescence studies and EM, huge potential exists to unravel more details about virus assembly in the near future.
Due to their small size, viruses can only be clearly visualized by electron microscopy (EM). Consequently, our understanding of the replication of many viruses has been greatly enhanced by high‐resolution EM studies. Here we describe how transmission EM of plastic‐embedded material and immunolabeling studies can be used to analyze the interactions of viruses with their host cells. We will focus particularly on the assembly of two types of enveloped viruses: the beta‐herpesvirus, human cytomegalovirus (HCMV), and the primate lentiviruses, the simian and human immunodeficiency viruses (SIV and HIV, respectively).
I. Introduction
Viruses are responsible for numerous diseases ranging from the common cold to smallpox, from childhood infections such as measles and chicken pox to major epidemics like AIDS, which now affects more than 40 million people worldwide and kills more than 3 million a year.1 Outbreaks of highly pathogenic viruses such as Ebola, which causes a lethal hemorrhagic fever, the SARS coronavirus, or, most recently, avian influenza have raised widespread public concern. Other viruses are implicated as causative agents for some cancers.
The virus particles themselves are tiny, with the sizes of most virions at the limit of the resolution of even the best light microscopes. Most viruses are on the order of 50–200 nm in diameter, though the smallest parvoviruses can measure less than 30 nm, while poxviruses or rhabdovirus particles like the vesicular stomatitis virus can be 0.3‐ to 0.4‐μm long. Filoviruses such as Ebola or Marburg virus or filamentous forms of the influenza virus have diameters of about 80–100 nm, but can reach lengths of several μm. Viruses can be localized by fluorescence methods using specific antibodies directed against viral proteins, or viral components coupled to the green fluorescent protein (GFP) or its derivatives. This allows virus‐infected cells to be analyzed by FACS methods or by immunofluorescence staining to reveal the distribution of major viral components, but these techniques cannot reveal much about structure. Viruses can be visualized by fluorescence microscopy as small spots, though the resolution is rarely sufficient to determine whether fluorescent spots represent virions, aggregates of viral proteins, or subviral particles. When carefully controlled, fluorescence methods can sometimes allow virus particles to be counted (Pizzato et al., 1999), while more recent multicolor fluorescence coincidence detection methods allow quantitative measurements of the antigenic composition of individual virions (Li et al., 2004). However, a detailed analysis virus structures requires higher resolution methods such as electron microscopy (EM). At the simplest level, negative staining of virus preparations can reveal some features of virus structure, size, and heterogeneity, and this can be of diagnostic value (Curry et al., 2006). More detail is revealed when virus preparations are rapidly frozen and the vitrified specimens examined by cryo‐EM. When combined with data from X‐ray diffraction studies, or with electron tomography or single‐particle analysis of isolated virions, highly detailed virus structures can be derived at near atomic resolution (Briggs 2006, Grunewald 2003).
As obligate intracellular parasites, viruses are dependent on living host cells for their replication. The viruses must first identify and adhere to a suitable host cell, which occurs through interactions of viral surface components or envelope glycoproteins with specific cellular receptors. The viral DNA or RNA genome must then access the relevant replication machineries of the host cells (Marsh and Helenius, 2006). Enveloped viruses have to fuse with a cellular membrane, allowing a naked virus core to be released into the cytoplasm. Further uncoating reactions and/or transport of the core to the cell's nucleus may also be required. Even the complex poxviruses, which carry their own machineries enabling them to carry out transcription and replication in the cytoplasm, need to access the appropriate location inside the cell, for example, in the vicinity of the endoplasmic reticulum (Schramm and Krijnse Locker, 2005). Once access to the replicative machineries has been established, the viral genetic material induces the host cell to synthesize the components required for the production of new virus particles, as well as virus‐specific enzymes or regulatory proteins that may reprogram the cells' activities. New virus particles are assembled, and, for enveloped viruses, acquire a lipid membrane by budding through a cellular membrane. Many cellular organelles have been shown to be involved in viral replication and assembly, including the plasma membrane, components of the biosynthetic pathway [endoplasmic reticulum, the Golgi apparatus or the trans‐Golgi network (TGN)], and endosomes. These compartments are exploited in different ways and to varying extents by different viruses. Some viruses subvert the host cells' normal functions, perhaps generating new virus‐induced structures or modifying normal cellular organelles to produce extensive “virus factories.” These processes are most effectively studied with the electron microscope. The characteristic morphologies allow the easy identification of mature and assembling virus particles in cells, and the effects on the cell's own structure can be detected alongside. The EM techniques are even more powerful when combined with immunolabeling protocols, in particular immunolabeling of cryosections.
Here we review the use of standard transmission electron microscopy (TEM) of plastic‐embedded material, as well as protocols for immunolabeling of cryosections, in the analysis of viral interactions with cells. As examples, we will focus particularly on the assembly of the human and simian immunodeficiency viruses (HIV and SIV, respectively) and on the more complex herpesvirus, the human cytomegalovirus (HCMV).
II. Analysis of Virus Assembly on Plastic Sections
Since many viruses have characteristic structures that are easily identified in the electron microscope, studies of ultrathin sections of plastic‐embedded preparations of virus infected cells by TEM can be remarkably informative. Indeed, most of the early investigations into viral life cycles used plastic‐embedded specimens (Dales, 1973). Various stages of the viral life cycles have been documented by EM on plastic sections, including virus entry pathways using clathrin‐coated pits (Helenius et al., 1980) or caveolae (Kartenbeck 1989, Stang 1997) and transport of viruses to the nucleus (Sodeik et al., 1997). Such studies require very high input numbers of virus particles, and since often only a fraction of the visible virus particles are actually infectious, nonproductive pathways might also be visualized. The EM data therefore have to be interpreted alongside biochemical and pharmacological investigations.
TEM has also been used to study the generation of new virions, and the most striking images are obtained during analysis of virus assembly in infected cells. Many viruses induce spectacular changes in the host cell's morphology, for example modifying cellular structures to establish specialized replication structures or “viral factories” (Novoa et al., 2005). EM can be used to identify morphological features of the various stages in the assembly of virus particles, distinguish immature and mature particles, or analyze steps in the acquisition of lipid membranes by enveloped viruses. For example, a range of EM techniques, including analysis of plastic‐embedded material, are being used to determine the origin of the membranes wrapping the different intracellular forms of vaccinia virus (Heuser 2005, Schramm 2005, Sodeik 2002). In addition, EM can be used to study the organelles involved in virus production and to determine whether and how viral infection modifies cellular compartments or generates new structures.
A. Epon Embedding of Infected Cell Preparations: Technical Considerations
As for all EM studies, cell preparation techniques must aim to preserve the structure of the virus‐infected cells as close to the living state as possible. Standard fixation protocols employ aldehyde fixatives to immobilize proteins and osmication to stabilize membrane structures. We now use a mixture of 2% formaldehyde (from paraformaldehyde), which is small and penetrates easily into cells, and 1.5% glutaraldehyde, a more potent fixative that cross‐links cell components, in 0.1‐M phosphate buffer, pH 7.4. To minimize disturbance to the cells, it is best to prepare a double‐strength fixative (i.e., 4% formaldehyde/3% glutaraldehyde) at the temperature of the cells and add this directly into the culture medium. High‐pressure freezing, followed by freeze‐substitution with aldehyde fixatives may give even better preservation (Murk 2003, Studer 2001). However, many viruses are pathogenic and must be inactivated by fixation before the infected cells can be handled safely outside safety hoods or containment laboratories.
For enveloped viruses, the preservation of membrane structures in the preparations is also important. We obtain good results using a post‐fixation step in reduced osmium (1% osmium tetroxide, OsO4, in 1.5% potassium ferricyanide, K3Fe(CN)6, for 1 h on ice). Membrane structures are especially well preserved if the cells are also treated with tannic acid, which acts as a mordant between osmium‐treated structures and lead stains, improving the contrast and fine delineation of various cellular structures, but especially membranes (Simionescu and Simionescu, 1976). Cells are washed and incubated in a low molecular weight tannic acid (gallotannin, 1%) in 0.05‐M sodium cacodylate buffer for 45 min at room temperature, followed by a brief treatment (5–10 min) with 1% sodium sulfate, dehydrated and embedded in an epoxy resin such as Epon 812. Ultrathin sections are contrasted with a lead citrate stain before viewing in the EM.
B. Assembly of the Human Cytomegalovirus
The beta‐herpesvirus HCMV is a complex double‐stranded DNA virus whose 230 kilo base‐pair (kB) genome encodes around 200–250 open reading frames, depending on the virus strain (Britt and Boppana, 2004). Virus particles consist of an inner icosahedral capsid enclosing the DNA genome. This is coated by a layer of at least 15 different tegument proteins and wrapped in an outer lipid envelope containing the viral membrane proteins. The HCMV genome encodes around 40 proteins with features of transmembrane glycoproteins, ranging from the abundantly expressed glycoprotein complexes (gM/gN, gB, and gH/gO/gL) to regulatory proteins, viral Fc receptors, and chemokine receptor‐like proteins (Britt and Boppana, 2004). The gB and gH/gO/gL complexes are essential for viral infectivity, functioning in virus attachment to the host cell and membrane fusion (Britt and Mach, 1996).
The assembly of HCMV takes place at specific replication sites both in the nucleus and in the cytoplasm of infected cells, often leading to large increases in cell size (cytomegaly). Distinct stages in the replication of HCMV have been documented by EM, as illustrated in Fig. 1 . The viral capsid proteins move into the nucleus, where they assemble into 100‐nm‐diameter icosahedral cores that are then filled with the viral DNA to produce nucleocapsids (Fig. 1A). The nucleocapsids are thought to leave the nucleus by budding through the inner membrane of the nuclear envelope, before fusing and de‐enveloping at the outer nuclear membrane, so that naked capsids are released into the cytoplasm. This idea is supported by EM analysis, where enveloped capsids can sometimes be observed in dilations of the nuclear envelope (Fig. 1B). In the cytoplasm, the particles acquire a layer of tegument proteins, a process that often occurs at characteristic cytoplasmic virus factories (Fig. 1C), identified as accumulations of electron‐dense tegument proteins in the vicinity of the centrioles/microtubule organizing center. The virus factories also tend to contain various membrane vesicles and are often surrounded by enlarged mitochondria. The virus particles acquire their final envelope by budding into characteristic crescent‐shaped membrane vesicles (Fig. 1D), and simultaneously become enclosed in small membrane‐limited vacuoles. Fusion of these vacuoles with the plasma membrane leads to the release of HCMV virions into the extracellular space. The morphology of the fully assembled extracellular virus particle (150‐ to 200‐nm diameter) shows the viral envelope, coated with a layer of the viral glycoprotein complexes (Fig. 1E). Three types of particles can be distinguished on HCMV‐infected cells, including enveloped nucleocapsids, empty capsid particles that lack the DNA genome [giving rise to noninfectious enveloped particles (NIEPs)], and cytoplasmic aggregates of tegument proteins, which produce enveloped “dense bodies” (see Fig. 1F). On infected human fibroblasts, we have also occasionally observed HCMV particles within or budding into multivesicular late endosomes (Fig. 1G). Various HCMV glycoproteins, including several viral chemokine receptor‐like proteins are also located on endosomes and multivesicular bodies and may be incorporated into the viral membrane during the budding step (Fraile‐Ramos 2001, Fraile‐Ramos 2002).
Fig. 1.
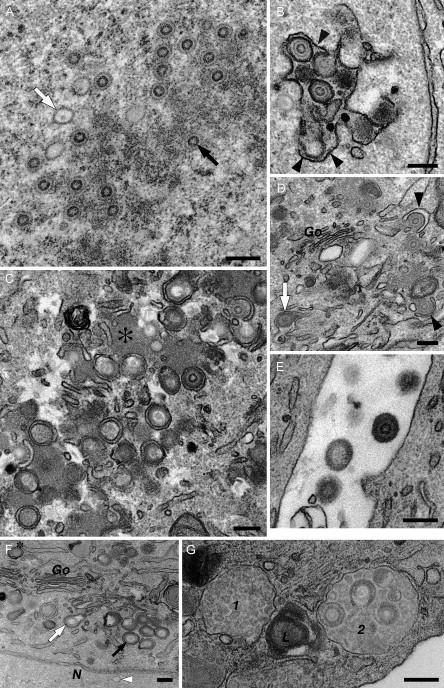
Stages of HCMV assembly. HCMV assembly was documented in human embryonic lung or foreskin fibroblast cells fixed in 2% formaldehyde/1.5% glutaraldehyde and postfixed with reduced OsO4 and tannic acid. (A–E) Human embryonic lung cells. (A) The nucleus contains empty icosahedral capsids (e.g., white arrow) or nucleocapsids enclosing a darker ring composed of the viral DNA (black arrow). (B) The capsids bud into the lumen of the nuclear envelope, acquiring a first membrane from the inner nuclear envelope (black arrowheads). This membrane is lost during fusion with the outer membrane of the nuclear envelope, so that naked capsids are released into the cytoplasm. (C) A cytoplasmic “virus factory” composed of electron‐dense accumulations of viral tegument proteins (asterisk) and various membrane vesicles. HCMV capsid particles acquire a layer of tegument and then proceed to wrap into the membrane vesicles. (D) Detail of the envelopment of tegumented capsids at cytoplasmic membranes near the Golgi complex (Go). The membrane vesicles are deformed into characteristic crescents (black arrowheads). This leads to the production of mature enveloped HCMV particles within cytoplasmic vacuoles (white arrow). (E) Fusion of these vesicles with the plasma membrane releases HCMV particles into the extracellular space. (F and G) Human foreskin fibroblasts. (F) Three types of enveloping particles in the cytoplasm. The white and black arrows indicate envelopment of an empty capsid and a DNA‐filled nucleocapsid, respectively, while the asterisk marks an enveloped tegument aggregate or “dense body.” Note also the empty capsid (white arrowhead) in the nucleus (N), which lacks the tegument layer. (G) Two multivesicular late endosomes (1, 2) and a lysosome containing multilamellar membranes (L). Endosome 2 contains several assembled HCMV virions. Images were taken from cells infected for 72 h (A and B), 96 h (E, F, and G), or 120 h (C and D). Scale bars = 200 nm.
C. Assembly of SIV Virions and Virus‐like Particles
Retroviruses, including the primate lentiviruses HIV and SIV, are single‐stranded RNA viruses with a much smaller genome and simpler structure than the herpesviruses. The two copies of the unspliced 9.8‐kB RNA genome contain three major genes, gag, pol, and env; these code for the structural proteins, viral enzymes (reverse transcriptase, integrase, and protease), and the envelope glycoprotein (Env), respectively. There are also sequences for a small number of accessory proteins (Freed, 2004). Like other enveloped viruses, HIV and SIV acquire their membrane by budding through a cellular membrane (Fig. 2A ). Hence all of the components that make up an infectious virus particle need to be brought together at this membrane budding site in a coordinated manner (Pelchen‐Matthews et al., 2004). Virus assembly is driven primarily by the Gag polyprotein that contains all of the structural proteins of the virus, namely the matrix protein (MA) that forms a protein shell immediately beneath the viral membrane, the capsid protein (CA) that condenses into the truncated cone‐shaped core that is characteristic of HIV and SIV particles (see Fig. 2A), and the nucleocapsid (NC) that stabilizes the viral RNA genome. Gag alone is able to bind membranes, to deform them into a bud and later a stalked “lollipop”‐type structure, and the presence of Gag can be visualized as a thick, membrane‐associated, electron‐dense layer at the budding site (Fig. 2A). Eventually, a scission reaction releases the budding virion from the host cell, and, concomitant with or shortly after release, the viral protease cleaves the Gag polyprotein, allowing CA and NC to condense into the cone‐shaped viral core of mature virions. Indeed, cells transfected with the gag gene alone will produce virus‐like particles (VLPs) with morphological features identical to immature SIV buds (Fig. 2B), but in the absence of the protease these particles cannot mature.
Fig. 2.
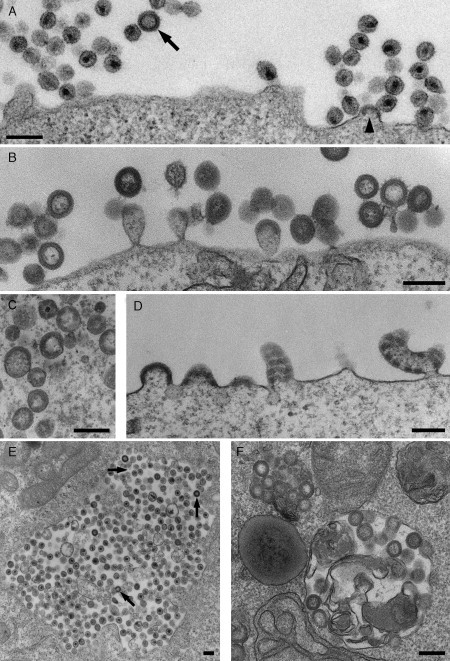
Assembly of SIV viruses and VLPs. (A) CEMx174 T2 cells acutely infected with a SIVmac251‐derived virus produce many virions at the cell surface. A budding virus can be recognized by the electron‐dense Gag layer accumulating under the membrane (arrowhead). The Gag layer is also visible in the immature virus particle (arrow). (B) VLPs with the morphology of immature viruses and budding figures accumulate at the surface of COS cells that have been transfected with the SIV Gag protein. (C and D) COS cells expressing a chimeric SIV Gag‐GFP show more irregular VLPs (C) or cell surface‐budding figures (D). The particles lack the thin electron‐dense line next to the VLP lumen, and the electron‐dense Gag protein layer is frequently interrupted due to steric interference by the GFP moiety. (E and F) Intracellular vacuoles containing viruses or VLPs can occasionally be found in CEMx174 cells infected with SIV NC‐MAC (LaBranche et al., 1995) (E) or COS cells expressing SIV Gag (F), respectively. Arrows in (E) indicate virus particles with immature morphology. Scale bars = 200 nm.
We have analyzed VLP assembly driven by the Gag protein in COS cells transfected with the SIVmac239 gag gene, or with a chimeric Gag‐GFP construct. In both cases, the cells released VLPs, which could be isolated and purified on sucrose gradients and which contained intact membrane envelopes as judged by their resistance to digestion with proteinase K. The Gag‐GFP particles could be visualized by fluorescence microscopy as brightly fluorescent spots at the cell surface and as larger aggregates at intracellular sites.
On Gag‐transfected cells, VLPs were easily detected by EM, both budding from the plasma membrane and free in the medium surrounding the cells. Detailed analysis revealed a number of differences between particles budding from Gag or Gag‐GFP‐transfected cells. VLPs produced by Gag‐transfected cells had a fairly homogeneous size distribution of 110–125 nm and consisted of a membrane envelope with an internal electron‐dense protein layer and an electron‐lucent center (Fig. 2B). Characteristically, the protein layer ended with a sharp line of electron density next to the lumen, similar to the appearance of immature virions budding from SIV‐infected cells (cf. Fig. 2A, arrow). Particles from Gag‐GFP‐transfected cells showed a wider size distribution (110–160 nm, with occasional even larger particles) and significant shape heterogeneity, including oval or elongated particles (Fig. 2C). The electron‐dense layer at the budding sites appeared thicker (31 ± 7 nm as compared to 23 ± 3 nm for Gag buds) and lacked the electron‐dense line next to the particle lumen. In addition, the protein coat often showed a banded pattern with a regular periodicity of about 50–60 nm, both under the plasma membrane and in the budding or budded particles (Fig. 2C and D). Such interruptions in the protein layer were never seen for Gag VLPs and are likely to result from steric interference to Gag packing by the GFP moiety, which represents approximately one‐third of the chimeric Gag‐GFP protein. Aberrant Gag‐GFP‐budding structures have also been observed for HIV (Larson 2005, Muller 2004), though budding particles of normal appearance were seen in cells coexpressing Gag‐GFP together with unmodified Gag. Such cells still release green fluorescent VLPs, allowing correlative light microscopy/EM analysis (Larson et al., 2005).
As for other retroviruses, the budding of SIV and HIV Gag is dependent on an intact “late” or L‐domain (Freed, 2002) that recruits cellular factors required for the scission and release of the budding particles (Morita and Sundquist, 2004). Again EM analysis has been useful in defining the stages of budding arrest, and the effects of mutations in the viral late domains or of dominant negative versions of the cellular ESCRT (endosomal sorting complexes required for transport) proteins (Pornillos 2003, von Schwedler 2003).
The distribution of virus buds can also be useful in defining the sites of virus assembly. For example, SIV and HIV assemble primarily at the plasma membrane of T cells, T‐cell lines, or a variety of model tissue culture cell lines. However, intracellular vacuoles filled with virus particles can also occasionally be observed, both on SIV‐infected T cells (Fig. 2E) and on COS cells expressing high levels of the SIV Gag protein (Fig. 2F). This is similar to the assembly of HIV in primary human monocyte‐derived macrophages, which accumulate large vacuoles packed with virus particles (Gendelman et al., 1988). But on these static EM images, it is unclear where these intracellular viruses have assembled: virions near the cell surface might have been formed at intracellular locations and secreted, while intracellular virions could have been taken up by the cell, for example, by endocytosis. The presence of budding profiles or immature virions (Fig. 2E) can give an indication that viruses are actually assembling at a particular site, but such images are comparatively rare. Although the oligomerizing Gag protein can easily be identified on plastic sections, the presence of other viral proteins, such as Env, in the viruses is more difficult to establish. A further limitation of TEM of Epon sections is that it can be hard to identify the nature of the intracellular virus‐containing structures, for example, whether they are part of the endosome/lysosome system, or whether they represent structures modified by the virus, such as swollen Golgi cisternae or enlarged TGN vesicles. These questions can in part be addressed by immunolabeling of infected cells with antibodies against various intracellular markers. At least in the case of HIV assembly in macrophages, immunolabeling methods have demonstrated that the intracellular virus‐containing vacuoles contain some markers for late endosomes and lysosomes (Pelchen‐Matthews 2003, Raposo 2002).
III. Immunolabeling of Ultrathin Cryosections: Applications of the Tokuyasu Technique to Study Virus Assembly
Many viral structural proteins are expressed at high levels and tend to be incorporated into virus particles at high copy number. For example, HIV may contain up to 5000 Gag molecules per particle (Briggs et al., 2004). In addition, as these proteins tend to be highly immunogenic, a range of antibodies or sera against viral components are available commercially or through research facilities such as the NIH AIDS Research and Reference Reagent Program. Many antibodies recognize the viral proteins in their native state and give strong signals in immunolabeling reactions. To identify cellular compartments, it is necessary to perform double labeling experiments with antibodies directed against specific marker proteins, and this requires postembedding immunolabeling protocols.
Some highly expressed viral proteins have been immunolocalized on Araldite‐embedded sections (Landini 1987, Landini 1991) or on Epon etched by treatment with, for example, hydrogen peroxide (Hensel et al., 1995), but the harsh conditions required during cell preparation (fixation in high concentrations of aldehyde fixatives, osmication, dehydration, and embedding in plastic) and etching destroy many of the antibody‐binding sites. Hence this method is not generally recommended. Embedding in more hydrophilic resins such as Lowicryl or LR White retains more antigenic epitopes, but the preservation of cellular membranes in these resins is poor. This is a particular disadvantage if enveloped viruses are studied, as the interaction with the cell's membrane systems is critical to virus production. By far the best option for antigen preservation is to maintain the biological material in its hydrated state, which is achieved by using vitreous ice as the embedding medium for cryosectioning. Cellular membranes are well preserved in this technique—they tend to be negatively stained and appear as white lines.
A. Immunolabeling of Cryosections
The use of cryosections for immunolocalization studies was originally proposed by Tokuyasu (Tokuyasu and Singer, 1976) and is described in more detail in Tokuyasu (1997). Subsequently, the method has been further improved and modified and is now routinely used in many laboratories. Good descriptions of the technique can be found in Liou 1996, Raposo 1997. The following section focuses and expands on some of the steps involved.
1. Preparation of Cryosections
For the standard immunolabeling of cryosections, it is still necessary to fix the infected cells and tissues with aldehyde fixatives to immobilize proteins, though milder fixations largely avoiding glutaraldehyde can also give good results. Methods for high‐pressure freezing followed by freeze‐substitution with aldehyde fixatives and rehydration before processing for cryosectioning are also being developed and may give still better cell preservation (Koster and Klumperman, 2003). However, when working with infected cell preparations, the fixation step also provides a useful way to inactivate the viruses. We routinely use a fixative containing 4% formaldehyde (prepared from paraformaldehyde and added to the cells as a double strength, i.e., 8%, fixative as detailed above). As glutaraldehyde is a cross‐linking fixative, it can improve cell preservation and the sectioning properties of the embedded, frozen cells, even at low concentrations (≤0.2%), but even these low amounts can destroy some antigenic epitopes. Fixed cells are rinsed in a glycine‐containing buffer before embedding in gelatin, and embedded cell pellets are dissected into blocks suitable for sectioning, infiltrated with 2.3‐M sucrose as a cryoprotectant and frozen onto specimen pins by plunging into liquid N2. Specimen blocks can be stored in a nitrogen bank for extended times, allowing further analysis at later dates or when new antibodies become available.
With the availability of stable nitrogen‐cooled chambers for the new generation of ultramicrotomes (Leica Microsystems, Vienna, Austria) and the introduction of diamond knives specifically for cryosectioning (e.g., the Diatome cryoimmuno 35° knife with its large diamond pick‐up platform), ultrathin sectioning for EM has become feasible in any EM laboratory. Sections with a nominal thickness of 50–70 nm with silver to pale gold interference colors tend to be the most useful. Further improvements have been introduced in the methods for section retrieval with mixtures of sucrose and methylcellulose (Liou et al., 1996), while the ability to store sections for up to a year on Formvar and carbon‐coated grids (Griffith and Posthuma, 2002) makes immunolabeling experiments more productive.
2. Immunolabeling
For immunolabeling, grids are removed from the slides on which they were stored and placed—sections down—on buffered 2% gelatin, which is then melted. From the gelatin, grids are successively floated on droplets of the various incubation solutions in phosphate‐buffered saline (PBS), comprising:
– Quench buffer (50‐mM glycine buffer, perhaps containing 50‐nM NH4Cl) to inactivate any residual fixative. Rinse in PBS.
– A protein solution to block nonspecific sites. We normally use 1–2% bovine serum albumin (BSA), but fish‐skin gelatin or fetal calf serum have also been used.
–The primary antibody: Droplets of 5–25 μl of antibody in a 1–2% BSA‐containing carrier buffer. For some antibodies, nonspecific binding can be reduced by the inclusion of acetylated BSA (Aurion, Wageningen, The Netherlands) in this step only.
– Extensive washing in buffers containing BSA.
– The colloidal gold probe to react with the primary antibody, followed by more washes.
– A brief fixation (5–10 min) with 1–2% glutaraldehyde to stabilize the immune complexes.
– Finally, samples are washed extensively with distilled water, stained briefly in neutral uranyl acetate (Tokuyasu, 1978), rinsed in water, and embedded in uranyl acetate–methylcellulose (Raposo et al., 1997).
3. Gold Probes for Immunolabeling
While it is possible to stabilize colloidal gold directly with the desired primary antibody, gold‐labeled second antibody reagents are more convenient to use, and many are now commercially available. Gold probes conjugated with second antibodies against a variety of species are available, as are colloidal gold particles stabilized with protein A, protein G, or streptavidin (to detect biotinylated compounds) or with lectins such as concanavalin A or wheat germ agglutinin.
We prefer to use protein A‐gold (PAG) for localization studies as it is very versatile and allows double‐staining immunolabeling even with primary antibodies from the same species (see below). Although protein A has up to four immunoglobulin binding sites, in practice, only one site tends to be available in a suitable orientation to bind the IgG Fc region. Protein A reacts strongly with antibodies from a number of species, including rabbit, human (but not human IgG3), and some mouse IgGs. However, for rat or goat primary antibodies, and for most mouse antibodies, a bridging antibody is required. This can give the appearance of clustered or clumpy labeling. Protein A conjugates can also be used to stain cells expressing Fc receptors: as the protein A‐binding site on antibodies is located in the Fc region, PAG will only bind to those antibody molecules bound to their specific antigens (with their protein A‐binding site exposed) and not those that have been captured by Fc receptors, where this site is hidden.
Gold particles suitable for EM are in the range of 5–20 nm, while ultrasmall particles can be visualized after silver enhancement (see Chapter 22 by Cheutin et al., this volume). Labeling efficiencies tend to be higher for the smaller gold particles (Robinson et al., 2000), while large probes (15–20 nm) should only be used with high‐affinity antibodies directed against abundant epitopes. On the other hand, the larger 15‐nm‐gold particles are easier to see in the EM and this can be used to focus the investigator's attention to features of interest such as virus‐infected cells or virus assembly compartments within these cells; modern digital cameras can be calibrated to enhance the contrast so that accumulations of gold particles can be detected even at low magnification and used to “home in” on the labeled structures. By contrast, 5‐nm‐gold particles can only be seen clearly at primary magnifications of >25,000×, they are easier to see on weakly contrasted sections, and can be missed if the contrast is high or if they are located near more electron‐dense structures.
B. An Example of Immunolabeling for EM: Assembly of SIV VLPs and Virions
Some of our own studies on the assembly of VLPs from the SIV Gag protein illustrate applications of immunolabeling of cryosections (Fig. 3 ). The VLP‐budding profiles in COS cells transfected with the Gag‐GFP chimera were labeled with antibodies directed against the SIV MA or CA proteins (Fig. 3A and B, respectively), or with an antiserum against GFP (Fig. 3C). These antibodies show finely distinct labeling patterns, demonstrating the resolution of the EM‐immunolabeling method: antibodies against MA label the buds immediately beneath the membrane, consistent with the location of MA at the periphery of the VLPs, while anti‐CA antibodies stain more centrally over the electron‐dense Gag layer that is polymerizing under the membrane. Both the anti‐MA and anti‐CA antibodies are mouse IgG1s requiring a rabbit anti‐mouse IgG‐bridging antibody (an extra incubation step between the primary antibody and PAG in the protocol above). This gives the possibility to amplify the signal, as several bridging antibody molecules may bind to the primary antibody, and results in a more clustered distribution of the gold particles. The rabbit anti‐GFP antibody interacts directly with PAG, and no bridging antibody was used (Fig. 3C). Labeling here is by single gold particles. This is particularly striking over an area of flat polymerizing Gag‐GFP, where the striations in the Gag‐GFP layer are decorated with evenly spaced gold particles, located just below the electron‐dense Gag layer. Since the length of an antibody molecule is ∼9 nm, gold particles would be expected to be located within 10 nm of the antigen in labeling reactions with a single primary antibody. If a bridging antibody is used, this will reduce the space resolution. While the orientation of the primary and bridging antibodies relative to the section may vary, for quantitative analysis it is generally assumed that the antigen of interest is located within ∼20 nm of the surface of the gold particle.
Fig. 3.
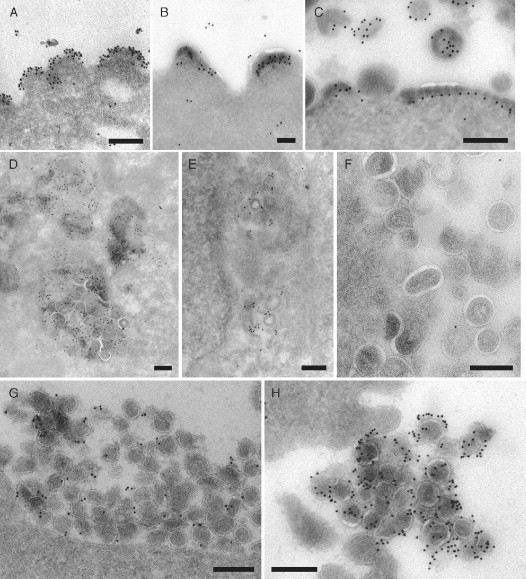
Analysis of SIV assembly by immunolabeling of cryosections. (A, B, and C) SIV Gag‐GFP VLPs budding from the surface of transfected COS cells. The buds were immunolabeled with mouse monoclonal antibodies against SIV MA (mAb KK59 in A) or CA (mAb KK64 in B, antibodies provided by Dr. K. Kent through the NIBSC Centralised Facility for AIDS Reagents, Potters Bar, UK) and a rabbit anti‐mouse IgG‐bridging antibody (Dako UK Limited, Ely, UK), or with a rabbit polyclonal antiserum recognizing GFP (C; Living Colors™ peptide antibody, Clontech Laboratories, Inc., Mountain View, CA, USA). All antibodies were detected with 10‐nm PAG. (D, E, and F) COS cells expressing SIV Gag and assembling VLPs in intracellular vacuoles resembling multivesicular bodies were immunolabeled with mouse monoclonal antibodies against CD63 (Fraile‐Ramos et al., 2001) or against LBPA (Kobayashi et al., 1998, provided by Dr. J. Gruenberg, University of Geneva, Switzerland) and 10‐nm PAG. The LBPA antibody labels intracellular (E), but not cell surface (F) VLPs. (G and H) Virus particles at the surface of CEMx174 cells chronically infected with SIVmac239 expressing an Env glycoprotein with a long cytoplasmic domain (G) or SIVmac239/251T with a cytoplasmically truncated Env protein (H) were stained with an antibody against SIV Env SU (Edinger et al., 2000, provided by Dr. R. W. Doms, University of Pennsylvania, Philadelphia) and 10‐nm PAG. The immunolabeling suggests that these two virus strains incorporate different amounts of the Env protein. PAG reagents were purchased from The Cell Microscopy Center, University Medical Center, Utrecht, The Netherlands. Scale bars = 200 nm.
Because of their characteristic structures, VLPs can be recognized in transfected cells even without immunolabeling, especially for the Gag VLPs, where more regular particles are formed. Since VLPs and budding figures are observed both at the plasma membrane and in some intracellular vacuoles, immunolabeling with antibodies against cellular markers can be used to identify these intracellular structures. Here, the VLP‐containing vacuoles were labeled by antibodies against the late endosomal protein CD63 (Fig. 3D) or the lipid lysobisphosphatidic acid (LBPA) (Kobayashi et al., 1998), which accumulates on the internal membranes of late endosomes and lysosomes (Fig. 3E). The presence of these markers identifies the intracellular vacuoles as late endosomes. Some VLPs appear to be directly labeled with gold particles, suggesting that CD63 or LBPA can themselves be incorporated into the budding virion membrane. By contrast, VLPs budded at the cell surface are not stained by the anti‐LBPA antibody (Fig. 3F), indicating that there may be biochemical differences between VLPs budded at the cell surface or at the membrane of endosomes.
We have also examined the assembly of various SIV strains that differ only in their cell surface Env. As for other retroviruses, the SIV Env glycoprotein is composed of two subunits, the surface glycoprotein (SU, gp120), which is noncovalently attached to the transmembrane subunit (TM, gp41). TM contains a long cytoplasmic domain of around 150–180 amino acids, depending on the virus strain. This long cytoplasmic domain is required for virus transmission in vivo, but when some SIV isolates, such as SIVmac239, are passaged in human tissue culture cell lines, they spontaneously truncate the cytoplasmic domain and express an Env protein with a shorter cytoplasmic tail of only around 20 amino acids. Truncation of the Env TM cytoplasmic domain can increase Env incorporation into virions (Johnston 1993, Zingler 1993). We have examined CEMx174 cells chronically infected with SIVmac239 or with a variant expressing an Env protein with a truncated TM protein (SIVmac239/251Tail). Ultrathin cryosections were stained with an antibody against the V3 loop on the SIV SU protein (Edinger et al., 2000). As shown in Fig. 3G and H, there was only sparse gold labeling of the budded SIVmac239 particles (Fig. 3G), while virions with the truncated Env TM protein were much more strongly labeled (Fig. 3H). Consistent with the exposure of Env at the surface of the particles, the virions were decorated with PAG on the outside of the virions. Indeed, the abundant Env glycoproteins on these particles was visualized as a fuzzy electron‐dense layer (Fig. 3H). Since gold particles appeared as discrete spots on the images, they could be counted to give an estimate of the Env densities on virions. This suggested that, while SIVmac239 viruses were labeled with only 1.2 gold particles per virion (Fig. 3G), the short tail variant (SIVmac239/251Tail) contained 8.7 gold particles per virion (Fig. 3H), suggesting that this SIV variant incorporates more than seven times as much Env as the wild‐type SIVmac239, in agreement with the analysis of purified SIV particles by HPLC or electron tomography studies, which reveal ∼8‐ to 10‐fold higher incorporation of truncated Env proteins into SIV (Chertova 2002, Zhu 2003). However, immunolabeling of cryosections offers the possibility of analyzing the composition of individual virus particles, rather than bulk populations, and may be used to compare, for example, viruses budded at intracellular sites or at the cell surface.
The gold particle counts also give a rough indication of the efficiency of antigen detection on immunolabeled cryosections: if SIVmac239 virions with a truncated cytoplasmic domain, such as the SIVmac239/251Tail, contain about 79 Env trimers per virion (Zhu et al., 2003) and virus particles are, approximately, cut in half on 50‐ to 70‐nm cryosections, then the gold particle counts above represent a labeling efficiency of around 7%. This is in broad agreement with the values previously determined for the labeling of the Semliki Forest virus spike protein complex in infected baby hamster kidney cells (Griffiths and Hoppeler, 1986), or for labeling of 5′‐nucleotidase at the cell surface (Howell et al., 1987). The high labeling of virions here probably results from the use of a rabbit anti‐mouse bridging antibody in the staining reaction, which gives some amplification, and from the fact that the stained virus particles are located in the extracellular space rather than embedded in the cytoplasmic matrix. The penetration of gold probes into cryosections depends on the density of the organelles, allowing reagents to penetrate well into some structures, while access to dense structures such as secretory granules may be severely restricted (Slot et al., 1989; Chapter 21 by Ladinsky and Howell, this volume). Labeling efficiencies vary for different antibody/antigen combinations, and depend on the size of the gold probes (higher labeling efficiencies are obtained with smaller gold probes), while steric effects may reduce the access of antibodies or gold probes at sites of high antigen density. While labeling efficiencies of around 10% or less can be acceptable for the study of viruses or virus‐infected cells, where viral proteins are expressed in abundance, inefficient detection can be a problem in the study of cellular antigens that may be present at much lower copy number.
C. Protocols for Double‐Staining Immunolabeling
More information can be obtained in double‐labeling studies using different sized gold particles to detect several distinct antigens. In studies of virus assembly, it can be useful to localize a viral structural protein and a cellular marker to define the virus assembly compartments or to detect multiple viral antigens. For example, in HIV the levels of CA and Env can be compared and the composition of individual virus particles at different locations in the cell analyzed. To ensure specificity of the labeling, these studies have to be more carefully designed and controlled than single antibody immunolabeling reactions.
1. Immunofluorescence Staining of Semithin Cryosections
It is often useful to screen the samples by immunofluorescence staining before embarking on more complex investigations. This is conveniently done with semithin (0.5 μm) cryosections that are cut with glass knives at −80°C, picked up on a droplet of 2.3‐M sucrose, and placed on glass slides. The slides should be marked by scoring small (5‐mm diameter) circles to indicate the area where the sections will be placed. A number of sections from different specimens can be placed side‐by‐side and screened together with the same antibody. Immunolabeling is performed on the slides by placing drops of solutions over the sections. Fixative is quenched by incubation in a solution of glycine and NH4Cl as above. For sections fixed in glutaraldehyde fixatives, a brief incubation in a freshly prepared solution of sodium borohydride is also required to reduce the autofluorescence of glutaraldehyde‐fixed tissue. Normally, there is no need to permeabilize the cells, as many organelles are cut open during the sectioning; however, if desired, buffers containing detergents such as Triton X‐100 or Saponin may be used. When these samples are stained, good images without “out‐of‐focus” fluorescence from adjacent layers in the cell can be obtained, even with a standard fluorescence microscope. Immunofluorescence thus allows a rapid assessment of the specimens: it can give an indication of the proportion of virus‐infected cells and, in situations where virus assembly occurs in a restricted location, of the number of cell profiles sectioned through the area of interest. The immunofluorescence patterns will give a first indication of the location of structures of interest, antibody reactivities can be conveniently assessed and antibodies titered, while double‐staining studies can give a first indication of the compartments involved.
An example of screening by immunofluorescence is shown in Fig. 4 . Here, sections of human primary monocyte‐derived macrophages infected for 13 days with HIV‐1BaL were stained with a rabbit serum to the HIV MA protein and a fluorescent anti‐rabbit antibody. The staining reveals that some of the cells contain intracellular accumulations of the virus (Fig. 4A). In addition, the sections were incubated with a monoclonal antibody recognizing the tetraspanin CD9 and anti‐mouse Alexa Fluor‐488 (Fig. 4B). CD9 is found primarily at the plasma membrane and is here seen to outline a number of cells. In addition, some cells show granular deposits of intracellular CD9 staining. In the HIV‐infected cell, the intracellular virus accumulation (MA‐stained) colocalizes with the CD9 marker.
Fig. 4.
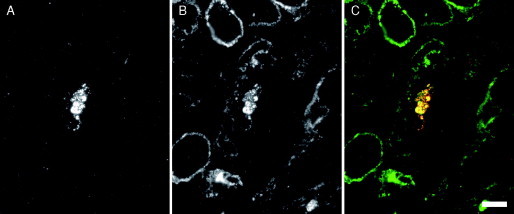
Screening semithin cryosections by immunofluorescence. Cryosections (0.5‐μm thick) from macrophages infected for 13 days with HIV‐1BaL were immunolabeled with a rabbit antiserum against HIV‐1 MA (UP595, provided by Dr. M. Malim, King's College London) and a monoclonal antibody against CD9 (mouse monoclonal anti‐CD9, Serotec, Kidlington, Oxford, UK), and the bound antibodies were detected with goat anti‐rabbit Alexa Fluor‐594 and goat anti‐mouse Alexa Fluor‐488 secondary antibodies (Invitrogen Molecular Probes, Paisley, UK). (A) Red fluorescence. MA staining reveals an accumulation of HIV viruses in one of the cells. (B) Green fluorescence. CD9 staining. (C) Merged image shows colocalization in yellow. Scale bar = 10 μm.
2. Double‐Staining Protocols with PAG Reagents
The simplest double immunolabeling protocols are achieved when staining with two primary antibodies from different species is followed by the relevant gold‐labeled secondary antibodies; this can sometimes be performed in just two incubations with mixed primary and secondary reagents, respectively. Double staining with PAG reagents requires sequential incubations, but it does sometimes permit double staining even with primary antibodies from the same species. These double‐staining protocols rely on the fact that the protein A‐binding site on antibodies is easily inactivated by a brief incubation in glutaraldehyde (5–10 min in 1–2% glutaraldehyde; we have found that a similar incubation in formaldehyde fixatives is not sufficient to block later cross‐reactions). With this in mind, a number of double‐ or even triple‐staining protocols can be devised (Table I ). Antibodies that are able to bind protein A directly allow the simplest staining sequences, but protocols for labeling two mouse antibodies, even if one requires a bridging antibody, can also be developed. For example, labeling with a mouse IgG1, a rabbit anti‐mouse bridging antibody and a small PAG particle can be followed by glutaraldehyde fixation, quenching and then a blocking incubation with an isotype‐matched (again IgG1) irrelevant antibody to saturate any remaining sites on the bridging antibody that have affinity for mouse antibodies. Following another fixation step to inactivate PAG‐binding sites on the blocking antibody, the sections can be labeled with a mouse IgG2a or IgG2b followed by direct staining with a larger PAG particle. It is still not possible to double stain with two antibodies from the same species if they both require a bridging antibody (e.g., two weakly binding mouse IgG1s).
Table I.
Double‐Staining Protocols with PAG
| Four‐step staining |
| (two primary antibodies, both able to bind PAG directlya) |
| First primary antibody/PAG‐1 ‐GA‐b Second primary antibody/PAG‐2 |
| Five‐step staining |
| (one primary antibody requiring a bridging antibody, the second, from a different species able to bind PAG directly) |
| First primary antibody/Bridging antibody/PAG‐1 ‐GA‐ Second primary antibody/PAG‐2 |
| OR First primary antibody/PAG‐1 ‐GA‐ Second primary antibody/Bridging antibody/PAG‐2 |
| Six‐step staining |
| (two primary antibodies from different species, both requiring a bridging antibody) |
| First primary antibody/Bridging antibody/PAG‐1 ‐GA‐ Second primary antibody/Bridging antibody/PAG‐2 |
| (two mouse primary antibodies, the first requiring a rabbit anti‐mouse IgG‐bridging antibody) |
| First primary antibody/Bridging antibody/PAG‐1 ‐GA‐ Isotype‐matched blocking antibodyc ‐GA‐ Second primary antibody/PAG‐2 |
Strong protein A‐binding antibodies include (among others) rabbit antibodies, human IgG1 and 2 (not IgG3) and some mouse IgG 2a, 2b. Antibodies from rat, goat, or sheep do not generally bind to protein A.
GA denotes the fixation to inactivate protein A‐binding sites. This consists of 5–10 min in 1–2% glutaraldehyde, followed by washing in PBS, quenching in 50‐mM glycine buffer, as well as a preincubation in BSA‐containing blocking buffer.
Same isotype as the first primary antibody.
The choice of the size of gold particles used is central to the success of a double‐staining immunolabeling experiment. Since all colloidal gold preparations contain some spread of particle sizes, combinations of 5‐ and 15‐nm gold particles give the most unambiguous distinction between the labels. Commercial preparations of 10‐ and 15‐nm gold both contain a small proportion of 12‐nm particles, so it may be difficult to identify the antibody associated with some individual particles. Given the ease with which large gold particles can be located at the EM, the biggest gold particles should be used to label structures of particular interest, for example, virus particles or a marker for a rare compartment. Due to steric interference and masking of sites, the efficiency of immunolabeling is reduced for the second or later antibody incubations, and hence the most critical staining reactions should be done first. For steric reasons, there is also less cross‐reaction if the smallest gold particles are used before larger conjugates in the staining sequence. Further restrictions arise if one antibody is sensitive to the glutaraldehyde fixation step: such an antibody then has to be used as the first primary antibody. To allow interpretations of the labeling patterns, antigen distributions should be carefully documented in single‐staining experiments, and all staining experiments need to be carefully controlled, for example, by using the “wrong” first antibodies or omitting reagents. The most informative controls are provided by cell lines lacking an antigen of interest, for example, after depletion by small interfering RNAs (siRNAs) or, when studying virus assembly, by uninfected cells, by pre‐absorbing antibodies with their antigen, and by omitting the second primary antibody.
D. Double‐ and Triple‐Labeling Studies of HIV Assembly in Macrophages
As already mentioned, HIV assembly in primary human monocyte‐derived macrophages occurs in large, apparently intracellular, vacuoles with various complex morphologies. Some vacuoles are filled with viruses, while others contain scattered virus particles in irregular or interconnected structures. These structures can be stained with antibodies against markers for late endosomes (Pelchen‐Matthews et al., 2003) or for the major histocompatibility class II complex (Raposo et al., 2002), suggesting that they may be related to late endosomes or equivalent to the MIIC, the compartment where major histocompatibility class II molecules are loaded with peptides. Often only a fraction of the macrophages in a culture are infected with virus, which may be found in a restricted location in the cell, so screening samples by immunofluorescence (see Fig. 4 and above) is essential. Here, the preliminary immunofluorescence studies suggested that the virus vacuoles contain the tetraspanin CD9. This was further investigated in EM double‐labeling experiments. CD9 is located primarily at the plasma membrane, with rare staining also of intracellular vacuoles (cf. Fig. 4B), hence we wanted to mark the location of CD9 with large gold particles (PAG 15 nm). However, the CD9 antibody was extremely sensitive to glutaraldehyde fixation, with even a 5‐min incubation in 1% glutaraldehyde eliminating most of the signal. Thus, we used a protocol where staining with anti‐CD9/rabbit anti‐mouse bridging antibody/15‐nm PAG was followed by a rabbit anti‐HIV MA antiserum and 5‐nm PAG. Virus‐containing vacuoles were strongly labeled for CD9, which was frequently located over the virus particles in the lumen of the vacuole or over small vesicles resembling the intralumenal vesicles of multivesicular bodies or “exosomes” (Fig. 5A ). The identity of the virus particles was confirmed by their staining with 5‐nm PAG.
Fig. 5.
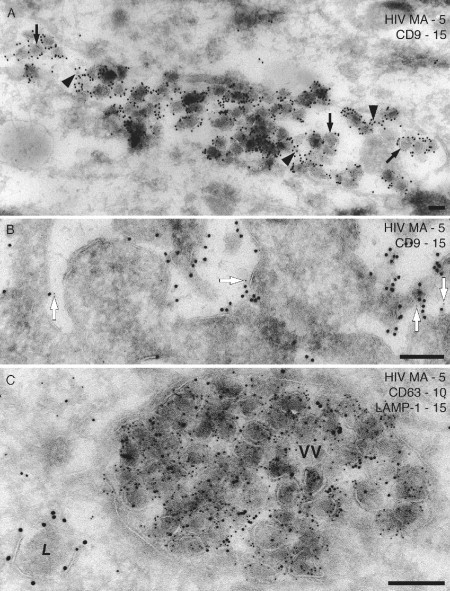
Double‐ and triple‐staining immunolabeling with PAG. (A and B) Ultrathin cryosections of 7‐day HIV‐1BaL infected (A) or uninfected macrophages (B) were stained for CD9 (mouse monoclonal anti‐CD9, rabbit anti‐mouse bridging antibody and 15‐nm PAG) followed by the rabbit antiserum against HIV‐1 MA and 5‐nm PAG. (A) An intracellular vacuole containing many virus particles is stained strongly for CD9, both on the virus particles (black arrows) and on smaller intralumenal vesicles (black arrowheads). (B) CD9 labeling at the cell surface of uninfected macrophages. A small number of 5‐nm PAG particles are seen close to 15‐nm PAG (white arrows), indicating a minor amount of cross‐reaction in this double‐labeling protocol. (C) Triple staining with a mouse monoclonal anti‐HIV MA antibody (4C9, Ferns et al., 1987, provided by Drs. R. B. Ferns and R. S. Tedder through the NIBSC Centralised Facility for AIDS Reagents) and 5‐nm PAG, the mouse monoclonal anti‐CD63 and 10‐nm PAG and a rabbit anti‐LAMP‐1 (provided by Dr. S. Carlsson, Umea University, Sweden) and 15‐nm PAG. The large vacuole is packed with virus particles that are also labeled for CD63, but there is little labeling for LAMP‐1. By contrast, a lysosome (L) nearby is strongly labeled for LAMP‐1 on its limiting membrane. Scale bars = 200 nm.
In this double‐staining protocol, we observed some cross‐reaction of the second gold reagent (5‐nm PAG) with the first antibody (anti‐CD9). This was particularly noticeable when we examined CD9 labeling at the cell surface of uninfected macrophages (which lacked the MA antigen). A number of 5‐nm gold particles were still observed, and most of these were located close to (within 20 nm of) a 15‐nm PAG particle (Fig. 5A, arrows). Such cross‐reaction would not occur if the smaller gold were used first, as a larger gold identifying the second antigen would be sterically blocked from accessing any surviving PAG‐binding sites in the first antibody layer. Given the distinct pattern with which the anti‐MA antibody labels virus particles, the cross‐reacting gold particles could easily be identified in this study. However, this example serves to illustrate the importance of careful controls in the immunolabeling reactions.
Figure 5C shows an example of a triple labeling with three sizes of gold particles. Here sections from HIV‐1BaL‐infected macrophages were stained sequentially with three antibodies that are all able to bind PAG directly. Virus particles are identified with a mouse monoclonal antibody against HIV‐1 MA and 5‐nm PAG, the 10‐nm PAG marks the late endosome protein CD63, while a rabbit antiserum and 15‐nm PAG were used to detect the lysosomal marker, LAMP‐1. Virus particles are clearly decorated with the 5‐nm gold particles, and often also labeled with the 10‐nm PAG identifying CD63. Indeed, biochemical studies have shown that CD63 is incorporated into the envelope of infectious HIV virions produced by macrophages (Nguyen 2003, Pelchen‐Matthews 2003). This is important because for many viruses, including HIV, only a fraction of the visible particles are infectious, and EM techniques alone cannot distinguish infectious from morphologically normal noninfectious virus particles. Cryosection immunolabeling allows the analysis of virus components, for example, the content of Envs that are essential for infectivity. Nonetheless, it is important to study statistically relevant numbers of virus particles, and the data are best analyzed in conjunction with complementary virological and biochemical studies. Finally in Fig. 5C, the 15‐nm gold particles identifying LAMP‐1 are only rarely seen in the virus vacuole, but nearby an electron‐dense vacuole, probably a lysosome, is clearly labeled on its limiting membrane. These studies support the idea that the compartment where HIV assembles in macrophages is related to late endosomes but is distinct from lysosomes (Pelchen‐Matthews 2003, Raposo 2002).
E. Localization of Other Cellular and Viral Components
The immuno‐EM method described so far focused mainly on detecting protein antigens with suitable antibodies. Some antibodies are also available against lipid antigens, as shown above for LBPA (Kobayashi et al., 1998) (see Fig. 3). The standard aldehyde fixatives described adequately immobilize proteins, but some lipids may relocate during the sectioning and labeling procedures. Hence lipid localization studies can benefit from high‐pressure freezing to prepare samples (Mobius et al., 2002). Cryosections are cut from the frozen specimens and collected with pick‐up solutions supplemented with aldehyde fixatives and uranyl acetate to stabilize the membranes. For lipid localizations, the cryosections should be labeled immediately, and it is also advisable to perform the staining incubations on ice to prevent or at least slow the relocation of the lipid components (Downes et al., 2005). In addition to antibodies, various lipid‐binding probes have been described that are useful for EM localization studies, including toxins such as perfringolysin O, which binds cholesterol (Mobius et al., 2002). Other probes have been developed using protein domains that bind specific phosphoinositides (Downes et al., 2005).
In addition, in situ hybridization techniques are being adapted to detect viral DNA or RNA genomes at the EM level (Risco and Carrascosa, 1999). Although EM in situ hybridization reactions on ultrathin frozen sections appear to be the most sensitive (Le Guellec, 1998), the ultrastructural preservation can be poor, and many investigators favor postembedding methods with Lowicryl or LR White resin‐embedded material. In situ hybridization studies localizing adenovirus or Herpes Simplex virus DNA have been described (Goto 2001, Puvion‐Dutilleul 1989, Puvion‐Dutilleul 1998). Similarly, Lowicryl‐embedded preparations of infected cells were used to detect HIV or SIV genomes in virus particles or budding figures and at cytoplasmic sites near the plasma membrane of infected cells (Canto‐Nogues et al., 2001). Viral RNA was also detected in the nucleus of peripheral blood mononuclear cells even before they exhibited morphological evidence of infection.
F. Quantification of Gold Particle Distributions
Immunolabeling is extremely useful as an aid to the identification of structures and to establish patterns of colocalization with marker proteins. In addition, gold particles appear as discrete spots on electron micrographs and so can be counted and their distributions analyzed for a more quantitative description of the experimental data. This is particularly useful for poorly expressed antigens or in situations where the gold labeling is sparse and dispersed. When gold particle distributions on a set of photographs are compared with the distribution of random points (e.g., intersections on an overlaid lattice), relative labeling indexes can be calculated and used to identify significantly labeled compartments (Mayhew 2002, Mayhew 2004), and different experimental samples can be compared under various conditions.
Many of the new generation of CCD cameras that are being developed for EM are equipped with particle identification programs that can facilitate the recognition and counting of gold particles of various sizes and the analysis of particle distributions and densities. This can again be useful to compare labeling over different compartments or on specimens prepared under different conditions. More complex methods of analyzing particle distributions using, for example, nearest neighbor algorithms and clustering analysis are also being developed (Hancock and Prior, 2005).
Given the ease with which gold particles can be detected and counted, it is important to be aware of the pitfalls associated with gold particle analyses. Nonspecific labeling needs to be clearly defined, for example on preparations lacking the relevant antigen, and quantified. Variations in the labeling efficiencies for different antibody antigen combinations or gold particle sizes must be accounted for. In particular, variations in the ability of different sized gold particles to penetrate cryosections (Slot et al., 1989), which may depend on the density of the structure to be analyzed, can give rise to different labeling efficiencies over different structures. For example, an antigen at the cell surface may be more accessible, and hence more strongly labeled, than the same molecule within an intracellular organelle, leading to large variations in labeling efficiencies (Griffiths and Hoppeler, 1986); hence, in some situations, labeling densities can only be used to compare variations in antigen concentration in similar organelles, and EM studies should be supported by, for example, quantitative biochemical analysis from cell fractionation.
IV. Future Developments
As illustrated above, the traditional methods of analyzing plastic‐embedded preparations of virus‐infected cells, together with the more powerful application of immunolabeling of ultrathin cryosections can provide detailed information on the sites and mechanisms of assembly of viruses. Often, limitations arise due to small numbers of infected cells, which make finding areas of interest laborious and time consuming. This task may be facilitated when a CCD camera is available to allow systematic scanning across the specimens.
The development of stable specimen holders and tilt stages and the associated computer software is making EM tomography more widely available (see Part V, this volume), and these methods should also be applied to the analysis of virus‐infected cells. Tomography can reveal membrane connections and might be particularly revealing in studies of surface connections of budding viruses or of budding mutants. Tomography methods have also been used on thick cryosections (Koster and Klumperman, 2003; Chapter 21 by Ladinsky and Howell, this volume), revealing more structural details. However, colloidal gold probes do not penetrate into thick cryosections. Ultrasmall gold reagents are available, which can penetrate much further into sections and can be visualized by silver enhancement. A range of ultrasmall gold reagents against various antibodies is already available. Some of these are linked to Fab fragments, while reagents based on even smaller antibody fragments such as camelid heavy‐chain variable domains (Spinelli et al., 2000) are being developed. A related technology is the development of gold nanoparticles with defined chemical structures and therefore more uniformity than gold colloids. These particles can be covalently linked to various binding reagents, including antibodies, Fab fragments, ligands, or haptens, and show good stability and a long shelf life. Like ultrasmall gold reagents, gold nanoparticles can penetrate more deeply into sections, and can be visualized by silver enhancement or using gold‐based autometallography reactions that may be compatible with biological materials.
A particularly exciting development is correlative light microscopy and EM. Already applications have been described for GFP‐labeled HIV (Larson et al., 2005) or HCMV (Sampaio et al., 2005). Cells growing on gridded coverslips can be transfected, infected, or microinjected with GFP chimeras, or engineered virus strains. After imaging in a fluorescent microscope, possibly with live‐cell recording of the fluorescent constructs, cells are fixed and embedded, and the same cells can be relocated in the EM and may be imaged, even by tomography. Similar techniques are being developed for preparing cryosections from monolayers of cells (Oorschot et al., 2002), and for relocating the fluorescent cells on immunolabeled cryosections (Koster and Klumperman, 2003). Correlative methods may also make use of reagents such as FluoroNanogold® (nanogold particles as above, that have been modified with a fluorescent probe, either fluorescein or one of the Alexa Fluor® dyes). Such reagents can be used to label cells, or even cryosections, for analysis both at the fluorescent light microscope level and subsequently, after silver or gold enhancement, at the EM level. Indeed, ultrathin cryosections may be stained with FluoroNanogold® and then silver‐enhanced and viewed at the EM, with significantly higher labeling efficiencies than can be obtained with colloidal gold probes (Takizawa and Robinson, 2003). Fluorescent quantum dots are also reported to be easily visible in the EM and may have similar applications (Giepmans et al., 2005).
V. Summary and Conclusions
Here we have demonstrated how EM, and particularly immunolabeling of cryosections, constitute powerful methods to analyze virus assembly. Particles with characteristic structures are easily identified, while immunolabeling allows quantification of viral components, even in individual virus particles, and comparisons between particles at different locations in the cell or at different stages in viral assembly. For many viruses, a range of specific antibodies are available, while reagents against cellular markers are widely commercially available. Together with the newly developed methods for electron tomography and correlative immunofluorescence studies and EM, huge potential exists to unravel more details of virus assembly in the near future.
Acknowledgments
We thank our collaborators Dr. Thomas Kledal, Dr. Jim Hoxie, and Dr. Wolfgang Ballensiefen for providing infected cell preparations, Dr. Lindsay Hewlett for help with the HCMV EM experiments, and Dr. Lucy Collinson for critical comments on the manuscript. We are grateful to the NIBSC Centralised Facility for AIDS Reagents, Potters Bar, UK and to Drs. Jean Gruenberg, Bob Doms, Michael Malim, and Sven Carlsson for providing antibody reagents. This work was supported by the UK Medical Research Council.
Footnotes
UNAIDS Epidemic Update, November 2005.
References
- Briggs J.A., Grunewald K., Glass B., Forster F., Krausslich H.G., Fuller S.D. The mechanism of HIV‐1 core assembly: Insights from three‐dimensional reconstructions of authentic virions. Structure. 2006;14:15–20. doi: 10.1016/j.str.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Briggs J.A., Simon M.N., Gross I., Krausslich H.G., Fuller S.D., Vogt V.M., Johnson M.C. The stoichiometry of Gag protein in HIV‐1. Nat. Struct. Mol. Biol. 2004;11:672–675. doi: 10.1038/nsmb785. [DOI] [PubMed] [Google Scholar]
- Britt W.J., Boppana S. Human cytomegalovirus virion proteins. Hum. Immunol. 2004;65:395–402. doi: 10.1016/j.humimm.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Britt W.J., Mach M. Human cytomegalovirus glycoproteins. Intervirology. 1996;39:401–412. doi: 10.1159/000150510. [DOI] [PubMed] [Google Scholar]
- Canto‐Nogues C., Hockley D., Grief C., Ranjbar S., Bootman J., Almond N., Herrera I. Ultrastructural localization of the RNA of immunodeficiency viruses using electron microscopy in situ hybridization and in vitro infected lymphocytes. Micron. 2001;32:579–589. doi: 10.1016/s0968-4328(00)00053-6. [DOI] [PubMed] [Google Scholar]
- Chertova E., Bess J.W., Jr, Crise B.J., Sowder I.R., Schaden T.M., Hilburn J.M., Hoxie J.A., Benveniste R.E., Lifson J.D., Henderson L.E., Arthur L.O. Envelope glycoprotein incorporation, not shedding of surface envelope glycoprotein (gp120/SU), is the primary determinant of SU content of purified human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 2002;76:5315–5325. doi: 10.1128/JVI.76.11.5315-5325.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry A., Appleton H., Dowsett B. Application of transmission electron microscopy to the clinical study of viral and bacterial infections: Present and future. Micron. 2006;37:91–106. doi: 10.1016/j.micron.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales S. Early events in cell‐animal virus interactions. Bacteriol Rev. 1973;37:103–135. doi: 10.1128/br.37.2.103-135.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes C.P., Gray A., Lucocq J.M. Probing phosphoinositide functions in signaling and membrane trafficking. Trends Cell Biol. 2005;15:259–268. doi: 10.1016/j.tcb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Edinger A.L., Ahuja M., Sung T., Baxter K.C., Haggarty B., Doms R.W., Hoxie J.A. Characterization and epitope mapping of neutralizing monoclonal antibodies produced by immunization with oligomeric simian immunodeficiency virus envelope protein. J. Virol. 2000;74:7922–7935. doi: 10.1128/jvi.74.17.7922-7935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferns R.B., Tedder R.S., Weiss R.A. Characterization of monoclonal antibodies against the human immunodeficiency virus (HIV) gag products and their use in monitoring HIV isolate variation. J. Gen. Virol. 1987;68:1543–1551. doi: 10.1099/0022-1317-68-6-1543. [DOI] [PubMed] [Google Scholar]
- Fraile‐Ramos A., Kledal T.N., Pelchen‐Matthews A., Bowers K., Schwartz T.W., Marsh M. The human cytomegalovirus US28 protein is located in endocytic vesicles and undergoes constitutive endocytosis and recycling. Mol. Biol. Cell. 2001;12:1737–1749. doi: 10.1091/mbc.12.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraile‐Ramos A., Pelchen‐Matthews A., Kledal T.N., Browne H., Schwartz T.W., Marsh M. Localization of HCMV UL33 and US27 in endocytic compartments and viral membranes. Traffic. 2002;3:218–232. doi: 10.1034/j.1600-0854.2002.030307.x. [DOI] [PubMed] [Google Scholar]
- Freed E.O. Viral late domains. J. Virol. 2002;76:4679–4687. doi: 10.1128/JVI.76.10.4679-4687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed E.O. HIV‐1 and the host cell: An intimate association. Trends Microbiol. 2004;12:170–177. doi: 10.1016/j.tim.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Gendelman H.E., Orenstein J.M., Martin M.A., Ferrua C., Mitra R., Phipps T., Wahl L.A., Lane H.C., Fauci A.S., Burke D.S., Skillman D., Meltzer M.S. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony‐stimulating factor 1‐treated monocytes. J. Exp. Med. 1988;167:1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans B.N., Deerinck T.J., Smarr B.L., Jones Y.Z., Ellisman M.H. Correlated light and electron microscopic imaging of multiple endogenous proteins using quantum dots. Nat. Methods. 2005;2:743–749. doi: 10.1038/nmeth791. [DOI] [PubMed] [Google Scholar]
- Goto T., Kohno T., Nakano T., Fujioka Y., Morita C., Sano K. An improved procedure of electron microscopic in situ hybridization for detecting adenovirus DNA. J. Electron Microsc. (Tokyo) 2001;50:73–76. doi: 10.1093/jmicro/50.1.73. [DOI] [PubMed] [Google Scholar]
- Griffiths G., Hoppeler H. Quantitation in immunocytochemistry: Correlation of immunogold labeling to absolute number of membrane antigens. J. Histochem. Cytochem. 1986;34:1389–1398. doi: 10.1177/34.11.3534077. [DOI] [PubMed] [Google Scholar]
- Griffith J.M., Posthuma G. A reliable and convenient method to store ultrathin thawed cryosections prior to immunolabeling. J. Histochem. Cytochem. 2002;50:57–62. doi: 10.1177/002215540205000106. [DOI] [PubMed] [Google Scholar]
- Grunewald K., Desai P., Winkler D.C., Heymann J.B., Belnap D.M., Baumeister W., Steven A.C. Three‐dimensional structure of herpes simplex virus from cryo‐electron tomography. Science. 2003;302:1396–1398. doi: 10.1126/science.1090284. [DOI] [PubMed] [Google Scholar]
- Hancock J.F., Prior I.A. Electron microscopic imaging of Ras signaling domains. Methods. 2005;37:165–172. doi: 10.1016/j.ymeth.2005.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A., Kartenbeck J., Simons K., Fries E. On the entry of Semliki forest virus into BHK‐21 cells. J. Cell Biol. 1980;84:404–420. doi: 10.1083/jcb.84.2.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensel G., Meyer H., Gartner S., Brand G., Kern H.F. Nuclear localization of the human cytomegalovirus tegument protein pp150 (ppUL32) J. Gen. Virol. 1995;76:1591–1601. doi: 10.1099/0022-1317-76-7-1591. [DOI] [PubMed] [Google Scholar]
- Heuser J. Deep‐etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J. Cell Biol. 2005;169:269–283. doi: 10.1083/jcb.200412169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell K.E., Reuter‐Carlson U., Devaney E., Luzio J.P., Fuller S.D. One antigen, one gold? A quantitative analysis of immunogold labeling of plasma membrane 5′‐nucleotidase in frozen thin sections. Eur. J. Cell Biol. 1987;44:318–327. [PubMed] [Google Scholar]
- Johnston P.B., Dubay J.W., Hunter E. Truncations of the simian immunodeficiency virus transmembrane protein confer expanded virus host range by removing a block to virus entry into cells. J. Virol. 1993;67:3077–3086. doi: 10.1128/jvi.67.6.3077-3086.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartenbeck J., Stukenbrok H., Helenius A. Endocytosis of simian virus 40 into the endoplasmic reticulum. J. Cell Biol. 1989;109:2721–2729. doi: 10.1083/jcb.109.6.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Stang E., Fang K.S., de Moerloose P., Parton R.G., Gruenberg J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature. 1998;392:193–197. doi: 10.1038/32440. [DOI] [PubMed] [Google Scholar]
- Koster A.J., Klumperman J. Electron microscopy in cell biology: Integrating structure and function. Nat. Rev. Mol. Cell Biol. 2003;(Suppl.):SS6–SS10. [PubMed] [Google Scholar]
- LaBranche C.C., Sauter M.M., Haggarty B.S., Vance P.J., Romano J., Hart T.K., Bugelski P.J., Marsh M., Hoxie J.A. A single amino acid change in the cytoplasmic domain of the simian immunodeficiency virus transmembrane molecule increases envelope glycoprotein expression on infected cells. J. Virol. 1995;69:5217–5227. doi: 10.1128/jvi.69.9.5217-5227.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landini M.P., Severi B., Cenacchi G., Lazzarotto T., Lindenmeier W., Necker A. Human cytomegalovirus structural components: Intracellular and intraviral localization of p38. Virus Res. 1991;19:189–198. doi: 10.1016/0168-1702(91)90045-w. [DOI] [PubMed] [Google Scholar]
- Landini M.P., Severi B., Furlini G., Badiali De Giorgi L. Human cytomegalovirus structural components: Intracellular and intraviral localization of p28 and p65–69 by immunoelectron microscopy. Virus Res. 1987;8:15–23. doi: 10.1016/0168-1702(87)90036-0. [DOI] [PubMed] [Google Scholar]
- Larson D.R., Johnson M.C., Webb W.W., Vogt V.M. Visualization of retrovirus budding with correlated light and electron microscopy. Proc. Natl. Acad. Sci. USA. 2005;102:15453–15458. doi: 10.1073/pnas.0504812102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guellec D. Ultrastructural in situ hybridization: A review of technical aspects. Biol. Cell. 1998;90:297–306. [PubMed] [Google Scholar]
- Li H., Zhou D., Browne H., Balasubramanian S., Klenerman D. Molecule by molecule direct and quantitative counting of antibody‐protein complexes in solution. Anal. Chem. 2004;76:4446–4451. doi: 10.1021/ac049512c. [DOI] [PubMed] [Google Scholar]
- Liou W., Geuze H.J., Slot J.W. Improving structural integrity of cryosections for immunogold labeling. Histochem. Cell Biol. 1996;106:41–58. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- Marsh M., Helenius A. Virus entry: Open Sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhew T.M., Griffiths G., Lucocq J.M. Applications of an efficient method for comparing immunogold labelling patterns in the same sets of compartments in different groups of cells. Histochem. Cell Biol. 2004;122:171–177. doi: 10.1007/s00418-004-0685-x. [DOI] [PubMed] [Google Scholar]
- Mayhew T.M., Lucocq J.M., Griffiths G. Relative labelling index: A novel stereological approach to test for non‐random immunogold labelling of organelles and membranes on transmission electron microscopy thin sections. J. Microsc. 2002;205:153–164. doi: 10.1046/j.0022-2720.2001.00977.x. [DOI] [PubMed] [Google Scholar]
- Mobius W., Ohno‐Iwashita Y., van Donselaar E.G., Oorschot V.M., Shimada Y., Fujimoto T., Heijnen H.F., Geuze H.J., Slot J.W. Immunoelectron microscopic localization of cholesterol using biotinylated and non‐cytolytic perfringolysin O. J. Histochem. Cytochem. 2002;50:43–55. doi: 10.1177/002215540205000105. [DOI] [PubMed] [Google Scholar]
- Morita E., Sundquist W.I. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- Muller B., Daecke J., Fackler O.T., Dittmar M.T., Zentgraf H., Krausslich H.G. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J. Virol. 2004;78:10803–10813. doi: 10.1128/JVI.78.19.10803-10813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murk J.L., Posthuma G., Koster A.J., Geuze H.J., Verkleij A.J., Kleijmeer M.J., Humbel B.M. Influence of aldehyde fixation on the morphology of endosomes and lysosomes: Quantitative analysis and electron tomography. J. Microsc. 2003;212:81–90. doi: 10.1046/j.1365-2818.2003.01238.x. [DOI] [PubMed] [Google Scholar]
- Nguyen D.G., Booth A., Gould S.J., Hildreth J.E. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J. Biol. Chem. 2003;278:52347–52354. doi: 10.1074/jbc.M309009200. [DOI] [PubMed] [Google Scholar]
- Novoa R.R., Calderita G., Arranz R., Fontana J., Granzow H., Risco C. Virus factories: Associations of cell organelles for viral replication and morphogenesis. Biol. Cell. 2005;97:147–172. doi: 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oorschot V., de Wit H., Annaert W.G., Klumperman J. A novel flat‐embedding method to prepare ultrathin cryosections from cultured cells in their in situ orientation. J. Histochem. Cytochem. 2002;50:1067–1080. doi: 10.1177/002215540205000809. [DOI] [PubMed] [Google Scholar]
- Pelchen‐Matthews A., Kramer B., Marsh M. Infectious HIV‐1 assembles in late endosomes in primary macrophages. J. Cell Biol. 2003;162:443–455. doi: 10.1083/jcb.200304008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelchen‐Matthews A., Raposo G., Marsh M. Endosomes, exosomes and Trojan viruses. Trends Microbiol. 2004;12:310–316. doi: 10.1016/j.tim.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Pizzato M., Marlow S.A., Blair E.D., Takeuchi Y. Initial binding of murine leukemia virus particles to cells does not require specific Env‐receptor interaction. J. Virol. 1999;73:8599–8611. doi: 10.1128/jvi.73.10.8599-8611.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O., Higginson D.S., Stray K.M., Fisher R.D., Garrus J.E., Payne M., He G.P., Wang H.E., Morham S.G., Sundquist W.I. HIV Gag mimics the Tsg101‐recruiting activity of the human Hrs protein. J. Cell Biol. 2003;162:425–434. doi: 10.1083/jcb.200302138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puvion‐Dutilleul F., Besse S., Pichard E., Cajean‐Feroldi C. Release of viruses and viral DNA from nucleus to cytoplasm of HeLa cells at late stages of productive adenovirus infection as revealed by electron microscope in situ hybridization. Biol. Cell. 1998;90:5–38. doi: 10.1016/s0248-4900(98)80230-x. [DOI] [PubMed] [Google Scholar]
- Puvion‐Dutilleul F., Puvion E. Ultrastructural localization of viral DNA in thin sections of herpes simplex virus type 1 infected cells by in situ hybridization. Eur. J. Cell Biol. 1989;49:99–109. [PubMed] [Google Scholar]
- Raposo G., Kleijmeer M.J., Posthuma G., Slot J.W., Geuze H.J. Immunogold labeling of ultrathin cryosections: Application in immunology. In: Herzenberg L.A., Weir D.M., Herzenberg L.A., Blackwell C., editors. “Handbook of Experimental Immunology”. Blackwell Science, Inc.; Oxford, UK: 1997. pp. 1–11. [Google Scholar]
- Raposo G., Moore M., Innes D., Leijendekker R., Leigh‐Brown A., Benaroch P., Geuze H. Human macrophages accumulate HIV‐1 particles in MHC II compartments. Traffic. 2002;3:718–729. doi: 10.1034/j.1600-0854.2002.31004.x. [DOI] [PubMed] [Google Scholar]
- Risco C., Carrascosa J.L. Visualization of viral assembly in the infected cell. Histol. Histopathol. 1999;14:905–926. doi: 10.14670/HH-14.905. [DOI] [PubMed] [Google Scholar]
- Robinson J.M., Takizawa T., Vandre D.D. Enhanced labeling efficiency using ultrasmall immunogold probes: Immunocytochemistry. J. Histochem. Cytochem. 2000;48:487–492. doi: 10.1177/002215540004800406. [DOI] [PubMed] [Google Scholar]
- Sampaio K.L., Cavignac Y., Stierhof Y.D., Sinzger C. Human cytomegalovirus labeled with green fluorescent protein for live analysis of intracellular particle movements. J. Virol. 2005;79:2754–2767. doi: 10.1128/JVI.79.5.2754-2767.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm B., Krijnse Locker J. Cytoplasmic organization of POXvirus DNA replication. Traffic. 2005;6:839–846. doi: 10.1111/j.1600-0854.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- Simionescu N., Simionescu M. Galloylglucoses of low molecular weight as mordant in electron microscopy. I. Procedure, and evidence for mordanting effect. J. Cell Biol. 1976;70:608–621. doi: 10.1083/jcb.70.3.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot J.W., Posthuma G., Chang L.Y., Crapo J.D., Geuze H.J. Quantitative aspects of immunogold labeling in embedded and in nonembedded sections. Am. J. Anat. 1989;185:271–281. doi: 10.1002/aja.1001850220. [DOI] [PubMed] [Google Scholar]
- Sodeik B., Ebersold M.W., Helenius A. Microtubule‐mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1997;136:1007–1021. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeik B., Krijnse‐Locker J. Assembly of vaccinia virus revisited: De novo membrane synthesis or acquisition from the host? Trends Microbiol. 2002;10:15–24. doi: 10.1016/s0966-842x(01)02256-9. [DOI] [PubMed] [Google Scholar]
- Spinelli S., Frenken L.G., Hermans P., Verrips T., Brown K., Tegoni M., Cambillau C. Camelid heavy‐chain variable domains provide efficient combining sites to haptens. Biochemistry. 2000;39:1217–1222. doi: 10.1021/bi991830w. [DOI] [PubMed] [Google Scholar]
- Stang E., Kartenbeck J., Parton R.G. Major histocompatibility complex class I molecules mediate association of SV40 with caveolae. Mol. Biol. Cell. 1997;8:47–57. doi: 10.1091/mbc.8.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer D., Graber W., Al‐Amoudi A., Eggli P. A new approach for cryofixation by high‐pressure freezing. J. Microsc. 2001;203:285–294. doi: 10.1046/j.1365-2818.2001.00919.x. [DOI] [PubMed] [Google Scholar]
- Takizawa T., Robinson J.M. Ultrathin cryosections: An important tool for immunofluorescence and correlative microscopy. J. Histochem. Cytochem. 2003;51:707–714. doi: 10.1177/002215540305100602. [DOI] [PubMed] [Google Scholar]
- Tokuyasu K.T. A study of positive staining of ultrathin frozen sections. J. Ultrastruct. Res. 1978;63:287–307. doi: 10.1016/s0022-5320(78)80053-7. [DOI] [PubMed] [Google Scholar]
- Tokuyasu K.T. Immunocytochemistry on ultrathin cryosections. In: Spector D.L., Goodman R.D., Leinwand L.A., editors. “Cells, a Laboratory Manual: Volume 3: Subcellular Localization of Genes and Their Products”. Cold Spring Harbor Laboratory Press; Woodbury, New York: 1997. p. 131.131. 131.127. [Google Scholar]
- Tokuyasu K.T., Singer S.J. Improved procedures for immunoferritin labeling of ultrathin frozen sections. J. Cell Biol. 1976;71:894–906. doi: 10.1083/jcb.71.3.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler U.K., Stuchell M., Muller B., Ward D.M., Chung H.Y., Morita E., Wang H.E., Davis T., He G.P., Cimbora D.M., Scott A., Krausslich H.G. The protein network of HIV budding. Cell. 2003;114:701–713. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- Zhu P., Chertova E., Bess J., Jr., Lifson J.D., Arthur L.O., Liu J., Taylor K.A., Roux K.H. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA. 2003;100:15812–15817. doi: 10.1073/pnas.2634931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingler K., Littman D.R. Truncation of the cytoplasmic domain of the simian immunodeficiency virus envelope glycoprotein increases env incorporation into particles and fusogenicity and infectivity. J. Virol. 1993;67:2824–2831. doi: 10.1128/jvi.67.5.2824-2831.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]