

TABLE OF CONTENTS
ESID 2014 ORAL PRESENTATIONS.........................................................................................................S139 – S170
ESID 2014 POSTER PRESENTATIONS......................................................................................................S170 – S504
TOPIC: EDUCATIONAL DAY..…..............................................................................................................S170 – S189
TOPIC: INFLAMMATION...…..................................................................................................................S189 – S201
TOPIC: AUTOIMMUNITY AND DYSREGULATION........................................................................................S201 – S235
TOPIC: INNATE IMMUNITYIMMUNITY.....................................................................................................S235 – S275
TOPIC: THERAPY.................................................................................................................................S275 – S313
TOPIC: OTHERS...................................................................................................................................S313 – S385
TOPIC: B CELL.....................................................................................................................................S385 – S432
TOPIC: T CELL.....................................................................................................................................S432 – S475
TOPIC: LATE BREAKING ABSTRACTS.......................................................................................................S475 – S504
INGID 2014 ORAL PRESENTATIONS........................................................................................................S504 – S508
INGID 2014 POSTER PRESENTATIONS.....................................................................................................S508 – S512
IPOPI 2014 POSTER PRESENTATIONS.....................................................................................................S512 – S515
ESID 2014 Oral Presentations
ESID-0092 Staphylococcal Disease in Humans with Inherited TIRAP/MAL Deficiency and Impaired Antibody Response to the TLR2 Agonist LTA
L. Israel1, P. Arkwright2, J.L. Casanova1, A. Puel 1
1Human Genetics of Infectious Diseases, Imagine, PARIS, France
2Royal Manchester Children’s Hospital, University of Manchester, Manchester, United Kingdom
The human genetic basis of staphylococcal disease is largely unknown, but patients with inherited MyD88 or IRAK-4 deficiency are prone to staphylococcal disease. We describe here eight individuals with inherited deficiency of TIRAP (also called MAL), an adaptor acting downstream from TLR2 and TLR4. These individuals are homozygous for a loss-of-function TIRAP allele. The four-year-old proband suffered from life-threatening staphylococcal pneumonia. The other seven individuals identified were adult relatives of the proband, aged between 16 and 50 years, who had never presented any serious infection. Responses to the TLR2 agonists PAM2CSK4, PAM3CSK4, and FSL-1, and the TLR4 agonist lipopolysaccharide (LPS) were impaired in the fibroblasts, granulocytes and monocytes of all TIRAP-deficient individuals tested. However, the whole-blood response to staphylococcal lipoteichoic acid (LTA), another TLR2 agonist, was impaired only in the index case. This defective response was due to a lack of anti-LTA antibodies in the patient's plasma specific to the index case, and it was reversible by the addition of exogenous monoclonal anti-LTA Abs. The combined effect of inherited TIRAP deficiency and a lack of anti-LTA Abs therefore accounts for staphylococcal disease in this patient. We thus provide here the first description of human inherited TIRAP deficiency. Our results further suggest that human TIRAP-dependent TLR2 immunity is important for the control of staphylococcal infection in children lacking anti-LTA antibodies, but that TIRAP is otherwise redundant in host defense.
ESID-0280 A New Genetic Disorder Predispose Mendelian Susceptibility to Mycobacterial Diseases
X.F. Kong 1, T. Lasseau1, C. Trouillet2, A. Bousfiha3, C. Aytekin4, C. Deswarte5, S. Boisson-Dupuis1, G. Vogt5, P. Gros6, S. Tangye7, F. Geissmann2, J. Bustamante8, J.L. Casanova1
1St. Giles Laboratory of infectious disease, The Rockefeller University, New York, USA
2Division of Immunology Infection and Inflammatory Diseases, King's College London Medical School, London, United Kingdom
3Clinical Immmunology Unit, Medical School. King Hassan II University, Casablanca, Morocco
4Department of Pediatric Immunology, Dr. Sami Ulus Maternity and Children's Health and Diseases Training and Research Hospital, Ankara, Turkey
5Laboratoire de Génétique Humaine des Maladies Infectieuses INSERM U1163, Institut Imagine, Paris, France
6Department of Biochemistry, McGill University, Montreal, Canada
7Immunology Program, Garvan Institute of Medical Research, Sydney, Australia
8Centre d'Etudes des Déficits Immunitaires (CEDI), Hôpital Necker Enfants-Malades, Paris, France
Mendelian susceptibility to mycobacterial diseases (MSMD) is a rare syndrome, and the known genetic etiologies impair the IFN-?/IL-12 axis. We report three patients with MSMD from two consanguineous families, whose whole blood display normal responses to IFN-? and IL-12. Whole-exome sequencing (WES) and Sanger sequencing revealed two different essential splicing variations of an immune-related gene. Family segregation suggested an autosomal recessive inheritance. These mutations were not found in any public database. RT-PCR revealed abnormal splicing of the candidate gene, with certain exons skipped in all cell types tested, including EBV-B cells, SV-40 fibroblast and PBMCs. In vitro transfection of plasmids showed that the mutant splicing forms abolished the protein production. This gene encodes for a transmembrane protease and expresses the highest levels in monocytes and dendritic cells (DCs). Functional experiments revealed its specific substrate was accumulated at elevated levels in both primary and immortalized cells. Two chemicals were able to inhibit the enzymatic activity of the candidate gene and led to an accumulation of the substrate in EBV-B cells from healthy controls. The accumulation of the substrate is detrimental to certain immune cells, resulting in an absence of CD1c+ DC in these patients. These findings suggest a new complete protease defect in MSMD.
ESID-0131 Abolished Type I IFN Immunity Underlies Severe Influenza Disease in Humans
M. Ciancanelli 1, S.X.L. Huang2, H. Garner3, P. Luthra4, L. Israelsson5, Y. Itan1, C. Basler4, D. Chaussabel5, L. Abel6, I. Pellier7, F. Geissmann3, H.W. Snoeck2, S.Y. Zhang1, J.L. Casanova1
1Laboratory of Human Genetics of Infectious Disease, The Rockefeller University, New York, USA
2Columbia Center for Translational Immunology, Columbia University Medical Center, New York, USA
3Centre for Molecular & Cellular Biology of Inflammation, King's College London, London, United Kingdom
4Microbiology, Icahn School of Medicine at Mount Sinai, New York, USA
5Systems Immunology, Benaroya Research Institute, Seattle, USA
6Laboratory of Human Genetics of Infectious Disease Necker Branch, Institut National de la Santé et de la Recherche Médicale INSERM-U1163, Paris, France
7Oncology Research Center Nantes-Angers, INSERM U892, Angers, France
Life-threatening influenza disease in otherwise healthy individuals and its absence in patients with inborn errors of adaptive immunity both remain unexplained. We performed whole exome sequencing on one child born to healthy non-consanguineous parents, who was hospitalized for three weeks in intensive care with acute respiratory distress due to the 2009 pandemic H1N1 strain of influenza. This child carries two loss-of-function alleles of FLU1. Although they are present in normal numbers, neither the patient's peripheral blood mononuclear cells nor her plasmacytoid dendritic cells produce any detectable IFN-a in response to ?ex?vivo stimulation with a variety of agonists and viruses, including influenza virus. Gene array and quantitative PCR demonstrate a profound lack of type I IFN expression in both cell types infected with influenza virus. The uncontrolled replication of influenza virus in the patient's fibroblasts in?vitro is rescued by wild-type FLU1 expression or by exogenous IFN-a/ß. We further differentiated pulmonary epithelial cells from induced pluripotent stem cells from the patient. Preliminary results suggest that a lack of IFN-a/ß production allows for rapid influenza virus replication in type II pneumocytes in particular. These findings indicate that human IFN-a/ß immunity is required for protective immunity against influenza virus, via plasmacytoid dendritic cells or pulmonary epithelial cells, or both, but is otherwise largely redundant in host defense. They also provide evidence that severe influenza in otherwise healthy children may result from single-gene inborn errors of intrinsic and/or innate immunity.
ESID-0407 Identification of IRF3 Mutations in Adult Patients with Herpes Encephalitis
T. Mogensen 1, N. Mork1, L.L. Andersen2, E. Kofod-Olsen1, T. Orntoft3, L. Ostergaard1, R. Hartmann2, C.S. Larsen1, M. Christiansen4
1Infectious Diseases, Aarhus University Hospital Skejby, Aarhus N, Denmark
2Molecular Biology, Aarhus University, Aarhus C, Denmark
3Molecular medicine, Aarhus University Hospital Skejby, Aarhus N, Denmark
4Clinical Immunology, Aarhus University Hospital Skejby, Aarhus N, Denmark
Background: An increasing number of mutations in the Toll-like receptor (TLR) 3 signaling pathway resulting in impaired antiviral interferon (IFN) responses has been demonstrated to contribute to the development of herpes encephalitis (HSE). In this study whole exome sequencing (WES) was performed to identify mutations associated with susceptibility to HSE.
Methods: 13 adults with a previous history of HSE were included, and patient DNA and PBMCs were isolated. WES was performed and analyzed by bioinformatics. Antiviral responses to relevant stimuli (HSV-1, the TLR3-agonists PolyIC, and Sendai virus) were measured by RT-qPCR to evaluate the functional immunological impact of identified mutations.
Results: In two HSE patients we identified two separate rare heterozygous mutations in the transcription factor IRF3 in the TLR3 pathway, which has not previously linked to HSE. Both mutations were located in the trans-activating domain of the molecule and predicted to be deleterious. When PBMCs from these two patients were stimulated with the relevant stimuli, the levels of IFNß as well as the IFN stimulated gene CXCL10 were reduced compared to an age- and gender matched controls, whereas TNFa production was intact.
Conclusion: The identification of these novel IRF3 mutations and their functional consequence contributes to the knowledge of the molecular and genetic mechanisms underlying HSE and is important for understanding disease pathogenesis. A better insight into these aspects may impact on clinical practice, including genetic testing, prophylactic antiviral treatment, and possibly, initiation of early treatment with both acyclovir and IFN in patients with HSE.
ESID-0069 Two Independent Killing Mechanisms of Candida Albicans by Human Neutrophils: Evidence from Innate Immunity Defects
R. Gazendam 1, J.L. Hamme1, A. Tool1, M. Houdt1, M. Herbst2, J.G. Liese2, F.L. Veerdonk3, D. Roos1, T.K. van den Berg1, T.W. Kuijpers4
1Blood Cell Research, Sanquin, Amsterdam, Netherlands
2Peadiatric infectious Diseases and Immunology, University Children Hospital, Wurzburg, Germany
3Nijmegen Center for Infection Immunity and Inflammation, Radboud University, Nijmegen, Netherlands
4Emma Childrens Hospital, University of Amsterdam, Amsterdam, Netherlands
Invasive fungal infections, accompanied by high rates of mortality, represent an increasing problem in medicine. Neutrophils are the major effector immune cells in fungal killing. Based on studies with neutrophils from patients with defined genetic defects, we provide evidence that human neutrophils use two distinct and independent phagolysosomal mechanisms to kill Candida albicans. The first mechanism for the killing of unopsonized Candida albicans was found to be dependent on Complement Receptor 3 (CR3), the signaling proteins phosphatidylinositol-3-kinase (PI3K) and CARD9, but was independent of NADPH oxidase activity. The second mechanism for the killing of opsonized Candida albicans was strictly dependent on Fc-gamma receptors, protein kinase C (PKC) and Reactive Oxygen Species (ROS) production by the NADPH oxidase system. Each of the two pathways of Candida killing required Syk tyrosine kinase activity, but dectin-1 was dispensible for both of them. These data provide an explanation for the variable clinical presentation of fungal infection in patients suffering from different immune defects, including dectin-1 deficiency, CARD9 deficiency or Chronic Granulomatous Disease (CGD).
ESID-0090 Inherited IL-17RC Deficiency in Patients with Chronic Mucocutaneous Candidiasis
Y. Ling 1, S. Cypowyj2, A. Belkadi1, C. Aytekin3, M. Galicchio4, Y. Camcioglu5, A. Ikinciogullari6, F. Dogu6, R. Levy1, M. Migaud1, B. Boisson2, A. Bolze2, Y. Itan2, C. Picard1, L. Abel1, J. Bustamante1, J.L. Casanova1, A. Puel1
1Laboratory of Human Genetics of Infectious Diseases Necker Branch INSERM 1163, University Paris Descartes Imagine Institute, PARIS, France
2St. Giles Laboratory of Human Genetics of Infectious Diseases Rockefeller Branch, Rockefeller University, New York, USA
3Department of Pediatric Immunology, Dr. Sami Ulus Maternity and Children's Health and Diseases Training and Research Hospital, Ankara, Turkey
4Allergy and Immunology Service, Victor J. Vilela Children's Hospital, Rosario Santa Fe, Argentina
5Division of Infectious Diseases Clinical Immunology and Allergy Department of Pediatrics, Cerrahpasa Medical Faculty Istanbul University, Istanbul, Turkey
6Department of Pediatric Immunology and Allergy, Ankara University Medical School, Ankara, Turkey
Chronic mucocutaneous candidiasis (CMC) is characterized by recurrent or persistent infections of the skin, nails, oral and genital mucosae by Candida albicans. Autosomal recessive IL-17RA and ACT1 deficiencies and autosomal dominant IL-17F deficiency, each reported in a single kindred, underlie chronic mucocutaneous candidiasis (CMC) in otherwise healthy patients. We report three unrelated patients, aged 6, 11, and 33 years, with autosomal recessive IL-17RC deficiency and isolated CMC. The patients are homozygous for different nonsense alleles, which prevent the expression of IL-17RC. The defect is complete, abolishing cellular responses to IL-17A and IL-17F homo- and heterodimers. However, in contrast to the IL-17RA- or ACT1- deficient patients tested, the response to IL-17E (IL-25) was maintained in the IL-17RC-deficient patients. These experiments of nature indicate that human IL-17RC is essential for mucocutaneous immunity against C.?albicans, but otherwise largely redundant.
ESID-0104 Mucosal Immune Dysregulation in Omenn Syndrome
B. Cassani 1, R. Rigoni2, V. Maina3, V. Marrella1, G. Pesole4, P. Vezzoni1, F. Grassi2, J.R. Mora5, A. Villa1
1IRGB, CNR, Milan, Italy
2Dipartimento di Biologia e Genetica per le Scienze Mediche, Universita' degli Studi, Milan, Italy
3Dep. Human Genome, Humanitas Clinical and Research Center, Milan, Italy
4Dipartimento di Bioscienze Biotecnologie e Biofarmaceutica, Universita' di Bari, Bari, Italy
5Gastrointestinal Unit, Massachusetts General Hospital Harvard Medical School, Boston, USA
Omenn syndrome (OS) is a rare monogenic disorder associating immunodeficiency and autoimmune-like manifestations that can present in the form of inflammatory bowel disease (IBD)-illness. We studied the intestinal homeostasis in the Rag2R229Q/R229Q murine model, closely recapitulating the human disease. Constant signs of inflammation, in both small and large intestines, were detected in mutant mice. Analysis of immune cell infiltrates revealed high numbers of CD4+T cells and, intriguingly, of Foxp3+ Treg cells, as a unique feature of the lamina propria compartment. An increased expression of the gut homing receptors, CCR9 and a4ß7, on peripheral memory CD4+ T cells confirmed the abnormal lymphocyte trafficking to the gut in OS. A pro-inflammatory profile, characterized by a Th1/Th17 skewing, distinguished immune responses occurring in OS intestine, and underlined the increased susceptibility to TNBS-induced colitis in mutant mice. Remarkably, similar pattern was also evident in the periphery. On the contrary, B cells were poorly present in the gut of OS mice and the fecal level of IgA was significantly reduced, correlating with augmented intestinal permeability. Metagenomic analysis revealed substantial changes in the composition of the gut microbial communities in the mutant mice, with an overall decrease in the bacterial biodiversity. Importantly, gut flora depletion following antibiotic treatment significantly reduced both local and systemic inflammatory immune responses, suggesting that microbial factors play a critical role in the pathogenesis of autoimmune disease associated with hypomorphic RAG defects.
ESID-0719 IL21 Deficiency Results in Early-Onset Inflammatory Bowel Disease and Common Variable Immunodeficiency–Like Disease
E. Salzer 1, A. Kansu2, H. Sic3, P. Majek4, A. Ikinciogullari5, F.E. Dogu5, N.K. Serwas1, E. Santos-Valente1, W.F. Pickl6, I. Bilic1, S.A. Ban1, Z. Kuloglu2, A.?M. Demir2, A. Ensari7, J. Colinge4, M. Rizzi3, H. Eibel3, K. Boztug1
1Kaan Boztug Laboratory, CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria
2Gastroenterology, Ankara University, Ankara, Turkey
3Hermann Eibel Laboratory, Center for Chronic Immunodeficiency, Freiburg, Germany
4Jacques Colinge Laboratory, CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria
5Immunology, Ankara University, Ankara, Turkey
6Christian Doppler Laboratory for Immunomodulation and Institute of Immunology, Center for Pathophysiology Infectiology and Immunology, Vienna, Austria
7Pathology, Ankara University, Ankara, Turkey
Alterations of the immune homeostasis in the gut can result in development of inflammatory bowel disease (IBD), as recently elucidated by Mendelian forms of IBD affecting IL10 or the IL10-receptor complex. However, also different types of primary immunodeficiency disorders may be associated with intestinal inflammation, often as one of their leading clinical presentations.
In this study, we investigated a large consanguineous family with three children suffering from early-onset IBD manifesting in the first year of life, leading to death in infancy in 2 of them. We performed combined Homozygosity mapping and exome sequencing to identify the molecular cause of the disorder.
A homozygous mutation in IL21 (c.T147C, p.Leu49Pro) was discovered, showing perfect segregation with the disease. In vitro assays demonstrated that mutant IL21Leu49Pro does not induce STAT3 phosphorylation or immunoglobulin class-switch recombination (CSR). The detected mutation in IL21 resulted in reduced numbers of circulating CD19 B-cells (including IgM+ naïve and class-switched IgG memory B cells), accompanied by an increase in transitional B cells. Ex vivo stimulation of patient B cells with wildtype IL21 and CD40 ligand resulted in normal CSR comparable to healthy individuals.
We here identify human IL21 deficiency as a novel genetic etiology of early-onset IBD associated with CVID-like primary immunodeficiency. In this disease, alternatively to allogeneic hematopoietic stem cell transplantation, recombinant IL21 may represent a 'targeted' experimental treatment strategy.
ESID-0360 CTLA-4 Deficiency – A Novel Autosomal-Dominant Immune Dysregulation Syndrome
D. Schubert 1, C. Bode1, R. Kenefeck2, T.Z. Hou2, J. Wing3, S. Unger1, A. Bulashevska1, B.?A. Grüning4, B.?S. Petersen5, A.A. Schäffer6, N. Frede1, G. Dueckers7, S. Seneviratne2, M. Kanariou8, A. Rensing-Ehl1, U. Salzer1, K. Warnatz1, F. Emmerich9, T. Cathomen9, P. Fisch10, M. Rakhmanov1, A. Franke5, S. Sakaguchi3, L.S.K. Walker2, D.?M. Sansom2, B. Grimbacher1
1Center for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany
2UCL Institute of Immunity and Transplantation, Royal Free Campus, London, United Kingdom
3Department of Experimental Immunology, Osaka University, Osaka, Japan
4Department of Computer Science, University of Freiburg, Freiburg, Germany
5Institute of Clinical Molecular Biology, Christian-Albrechts-University of Kiel, Kiel, Germany
6National Library of Medicine, National Institutes of Health, Bethesda, USA
7Pediatric Immunology and Rheumatology, HELIOS Kliniken Krefeld, Krefeld, Germany
8Department of Immunology and Histocompatibility, “Aghia Sophia” Children's Hospital, Athens, Greece
9Institute for Cell and Gene Therapy, University Medical Center Freiburg, Freiburg, Germany
10Department of Pathology, University Medical Center Freiburg, Freiburg, Germany
Common Variable Immunodeficiency (CVID) is not only a primary immunodeficiency with low immunoglobulin levels, absent vaccination responses and recurrent infections; it is often an immune dysregulation syndrome with complications like autoimmunity, lymphoproliferation and (autoimmune) enteropathy. We have investigated a large autosomal-dominant CVID family with five patients who presented with low antibody levels and recurrent infections in combination with autoimmune thrombocytopenia, Hashimoto thyroiditis, splenomegaly, granuloma formation and severe autoimmune enteropathy. In order to identify the genetic cause, we performed whole exome sequencing in fourteen family members. We identified a heterozygous stop codon mutation in Exon 1 of the co-inhibitory molecule CTLA-4 which is an essential mediator of regulatory T cell (Treg) function. Screening an additional 80 patients with a comparable clinical phenotype, we identified three more families (five patients) with novel splice site and missense mutations in CTLA-4. Reduced numbers of naïve T cells, a progressive loss of B cells and a lack of ?d-T cells were observed in peripheral blood of the patients. Furthermore, CTLA-4 expression levels were strongly reduced under basal conditions as well as upon activation of Tregs and conventional T cells. Interestingly, Tregs (FoxP3 positive) were present in normal to elevated numbers. However, those Tregs were not functional in Treg suppression assays and had strongly reduced activity in ligand binding and transendocytosis assays. Taken together, the genetic and immunological data indicate that heterozygous mutations in CTLA-4 are causing an immune dysregulation syndrome with hypogammaglobulinemia, enteropathy and autoimmunity.
ESID-0767 Human CTLA4 Haploinsufficiency Causes Severe Immune Dysregulation
G. Uzel 1, H.S. Kuehn2, Q. Weiming 3, B. Lo1, E.K. Deenick4, J.E. Niemela2, D.T. Avery4, J.N. Schickel5, D.Q. Tran6, J. Stoddard2, Y. Zhang1, D.?M. Frucht3, K.V. Rao1, H.C. Su1, L.D. Notarangelo7, K.N. Olivier1, J. McElwee8, S. Pittaluga9, J.B. Oliveira10, E. Meffre5, T.A. Fleisher2, S.M. Holland1, S.G. Tangye4, M.J. Lenardo1
1NIAID, National Institutes of Health, Bethesda, USA
2CC, National Institutes of Health, Bethesda, USA
3CDER, Food and Drug Administration, Bethesda, USA
4Immunology Department, Garvan Institute of Medical Research, Sydney, Australia
5Department of Immunobiology, Yale University School of Medicine, New Haven, USA
6Department of Pediatrics, University of Texas Medical School, Houston, USA
7Children’s Hospital, Harvard Medical School, Boston, USA
8Merck Research Laboratories, Merck and Co., Boston, USA
9NCI, National Institutes of Health, Boston, USA
10IMIP, Instituto de Medicina Integral Prof. Fernando Figueira, Recife-PE, Brazil
Adaptive immune responses are balanced by signals delivered by co-stimulatory and regulatory receptors. Cytotoxic T lymphocyte antigen-4 (CTLA-4) is key to this process due to its essential inhibitory role. We sought to determine the immune consequences of novel germline genetic defects in CTLA4 in humans.
We used whole exome and Sanger sequencing to identify underlying genetic defects in patients with lymphocytic inflammatory lesions of the brain, lung and gut with or without hypogammaglobulinemia.
Monoallelic deleterious mutations in CTLA4 were found in 7 patients from 4 unrelated families. Expression of CTLA-4 protein by activated T cells, as well as of CTLA4 and FOXP3 mRNA by regulatory T (Treg) cells from affected patients were significantly reduced compared to healthy controls. A 77 year-old family member, who carried the family mutation but no clinical phenotype had normal CTLA4 and FOXP3 mRNA in her Treg cells, suggesting variable penetrance. siRNA knockdown and complementation studies identified the pathogenetic mechanism as haploinsufficiency. While Ctla4 +/- mice have no obvious phenotype, CTLA4 +/- humans have impaired Treg cell function. Consistent with this, hyperactivation of effector T cells and infiltration of non-lymphoid organs by activated lymphocytes characterize this condition. Progressive loss of circulating B cells with an increase of predominantly autoreactive CD21loCD38lo B cells may relate to their hypogammaglobulinemia.
Heterozygous human CTLA4 mutations cause severe immune dysregulation and defective B cell homeostasis. Inherited human CTLA4 deficiency highlights the differences between human and murine disease and demonstrates the stringent regulation of CTLA-4 required for normal T and B lymphocyte homeostasis.
ESID-0096 IL-17RA and ADA2 Deficiency in Siblings with Recurrent Infections and Chronic Inflammation
F. Angelini 1, F. Fellmann2, J. Wassenberg3, M. Perreau4, N. Arenas Ramirez5, G. Simon6, O. Boyman7, O. Demaria8, S. Christen-Zaech8, M.T. Luder3, D. Hohl8, A. von Scheven-Gete3, M. Belfiore2, M. Gilliet8, P.Y. Bochud9, Y. Perrin3, M. Beck Popovich10, P.A. Bart4, D. Martinet2, M. Hofer1
1Immunology Allergy and Rheumatology Unit Department of Pediatrics, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland
2Service of Medical Genetics, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland
3Department of Pediatrics Immunology Allergy and Rheumatology Unit, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland
4Immunology and Allergy Unit Department of Medicine, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland
5Department of Immunology, University Hospital Zurich, Zurich, Switzerland
6Rhumatology Unit, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland
7Rhumatology Unit, University Hospital Zurich, Zurich, Switzerland
8Department of Dermatology, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland
9Infectious Diseases Unit Department of Medicine, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland
10Hematology-Oncology Unit Department of Pediatrics, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland
Background. Data on patients affected by chronic mucocutaneous candidiasis underscore the importance of Interleukin-17 Receptor A (IL-17RA) in mucocutaneous immunity. Little is known about the role of ADA2 in regulation of immune response, although recent reports linked ADA2 deficiency with inflammation and vasculitis.
Methods. We report two siblings (Child 1 and Child 2) who have suffered since childhood from recurrent mucocutaneous infections by C.?albicans and S.?aureus, chronic systemic inflammation and vasculitis only partially responsive to systemic steroid treatment. Array-CGH analysis showed a homozygous carrier state for a 770?kb deletion on chr22q11.1 encompassing IL-17RA, CECR1 and XKR3. Child 1 died at the age of 16 years. Child 2 displayed variable skin and scalp lesions and at the age of 18 years developped retinal vasculitis, venous occlusions and bleeding. None of them had brain involvement or stroke. Child 2 is currently treated with infliximab, a TNF-a blocker. Immunological studies were carried out by flow cytometry, ELISA and RIA.
Results. We found a lack of IL-17RA expression wich implies a dysfunction of the IL-17 signaling pathway, confering susceptibility to recurrent mucocutaneous infections. Interestingly, we detected an in?vitro and ?in?vivo upregulation of pro-inflammatory cytokines, (IL-1ß and TNF-a), consistent with the persistent systemic inflammation.
Conclusions. The present report reveals a likely causal link between ADA2 deficiency, chronic inflammation and vasculitis corroborating a potential role of ADA2 in modulating immunity and inflammation. In addition it underscores the usefulness of whole genetic analyses combined with immunological investigation in patient with immunodeficiency.
ESID-0732 A Novel Primary Immunodeficiency Disorder with Multiple Defects of the Lymphoid System Caused by Perturbed NF-KAPPA B Signaling
K. Willmann 1, S. Klaver1, F. Dogu2, E. Santos-Valente1, W. Garncarz1, I. Bilic1, E. Mace3, E. Salzer1, C. Domínguez Conde1, H. Sic4, P. Májek1, P.P. Banerjee3, G.I. Vladimer1, S. Haskologlu2, M.G. Bolkent2, A. Küpesiz5, A. Condino-Neto6, J. Colinge1, G. Superti-Furga1, W.F. Pickl7, M.C. van Zelm8, H. Eibel4, J.S. Orange3, A. Ikinciogullari2, K. Boztug1
1CeMM, Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria
2Department of Pediatric Immunology and Allergy, Ankara University Medical School, Ankara, Turkey
3Center for Human Immunobiology, Baylor College of Medicine and Texas Children's Hospital, Houston Texas, USA
4Centre of Chronic Immunodeficiency, University Medical Centre Freiburg, Freiburg, Germany
5Department of Pediatric Hematology, Akdeniz University Medical School, Antalya, Turkey
6Department of Immunology, Institute of Biomedical Sciences University of São Paulo, São Paulo, Brazil
7Christian Doppler Laboratory for Immunomodulation and Institute of Immunology, Center for Pathophysiology Infectiology and Immunology Medical University of Vienna, Vienna, Austria
8Department of Immunology, Erasmus MC University Medical Center, Rotterdam, Netherlands
We studied two index patients from a consanguineous pedigree suffering from recurrent bacterial, viral and Cryposporidium infections. Combining SNP array based homozygosity mapping with exome sequencing, we identified an underlying biallelic mutation in the MAP3K14 gene encoding the NF-?B inducing kinase NIK. In silico analysis predicted and functional assays confirmed loss of kinase activity leading to defective activation of both canonical and non-canonical NF-?B signaling in patient cells.
Patients carrying loss-of function NIK exhibited B-cell lymphopenia, decreased frequencies of class-switched memory B-cells, reduced somatic hypermutation and hypogammaglobulinemia. The observed B-cell defects were due to impaired survival, as B-cells showed lower expression of the prosurvival factor BCL-2. While overall T-cell numbers were normal, follicular helper cells were reduced, likely due to defective ICOSL upregulation on B-cells. Memory T-cells were perturbed, showing insufficient expression of IL-7 receptor, and antigen-specific T cell proliferation was blocked. Natural killer (NK)-cell numbers were decreased and although they largely acquired appropriate developmental markers, NK-cells exhibited defective activation and impaired formation of NK-cell immunological synapses. Collectively, our data illustrate the non-redundant role of NIK for maintenance and function of human immunity, and demonstrate unexpectedly complex aberrations of lymphoid immunity in NIK deficiency.
ESID-0288 A Novel Immunodeficiency Caused by a Mutation in Transferrin Receptor 1 that Disrupts Iron Transport
H.H. Jabara 1, S.E. Boyden2, J. Chou1, N. Ramesh1, M.J. Massaad1, L. Notarangelo1, M.?D. Fleming3, W. Al-Herz4, L.M. Kunkel2, R.S. Geha1
1Immunology, Children's Hospital, Boston, USA
2Genetics and Genomics, Children's Hospital, Boston, USA
3Pathology, Children's Hospital, Boston, USA
4Pediatrics, Kuwait University, Kuwait, Kuwait
Multiple patients in a consanguineous family suffered from combined immunodeficiency, intermittent neutropenia, thrombocytopenia, and mild anemia. The patients had normal T and B cell numbers, but impaired T and B cell proliferation and immunoglobulin production. Genetic analyses revealed in the patients, but not in unaffected family members or controls, a homozygous p.Y20H missense mutation in TFRC, which encodes transferrin receptor 1 (TFR1). The mutation disrupted the TFR1 internalization motif critical for transferrin endocytosis. TFR1 surface expression was markedly increased in the patients' lymphocytes and fibroblasts, but only modestly in their normoblasts, and TFR1 internalization was decreased in the patients' lymphocytes. Transduction of wild-type TFRC restored transferrin uptake by the patients' fibroblasts and addition of iron citrate in?vitro rescued the patients' T and B cells defects. The metalloreductase six-transmembrane epithilial antigen of the prostate (STEAP3), which possesses an internalization sequence similar to that of TFR1, was selectively expressed in normoblasts and associated with TFR1. Overexpression of Steap3, but not of an internalization defective Steap3 mutant, rescued transferrin uptake in the patients' fibroblasts, suggesting that STEAP3 provides an accessory TFR1 endocytosis signal that spares the patients from severe anemia.
ESID-0451 Autosomal-Recessive Agammaglobulinemia Due to Homozygous Mutations in Artemis: Do We Need a Modifier?
T. Volk 1, I. Reisli2, A. Björkman3, U. Pannicke4, P. Fisch5, A.A. Schäffer6, A. Bulashevska1, B.?A. Grüning7, D. Pfeifer8, S. Güner2, E.H. Sayar2, L. Hammarström9, A. Durandy10, U. Salzer1, Q. Pan-Hammarström3, M. Rizzi1, K. Schwarz4, B. Grimbacher1
1University Medical Center Freiburg and University of Freiburg, Centre for Chronic Immunodeficiency (CCI), Freiburg, Germany
2Department of Pediatric Immunology and Allergy - Necmettin Erbakan University, Meram Medical Faculty, Konya, Turkey
3Department of Laboratory Medicine, Karolinska Institutet, 171 77 Stockholm, Sweden
4University Ulm, Institute for Transfusion Medicine, Ulm, Germany
5Medical Center - University of Freiburg, Center for Pathology, Freiburg, Germany
6Department of Health and Human Services, National Center for Biotechnology Information - National Institutes of Health, Bethesda Maryland 20894, USA
7Department of Computer Science - University of Freiburg, Bioinformatics Group, Freiburg, Germany
8Department of Hematology and Oncology, University Medical Center, Freiburg, Germany
9Department of Clinical Immunology and Transfusion Medicine, Karolinska University Hospital, Stockholm, Sweden
10Université Paris V René-Descartes, Unité 768 Institut National de la Santé et de la Recherche Médicale (INSERM), Paris, France
Primary immunodeficiencies are rare diseases with very heterogeneous clinical phenotypes.
We studied three Turkish children born to first degree consanguineous parents who presented with hypogammaglobulinemia, low peripheral B cell counts (1% of leucocytes) and recurrent respiratory infections during infancy. By whole exome sequencing and homozygosity mapping we sought to identify a novel genetic defect. Mutations in genes known to cause autosomal recessive agammaglobulinemia or CVID were not detected. Homozygosity mapping revealed one large linkage interval on chromosome 2.
We identified two homozygous missense mutations which perfectly segregate with the phenotype: One mutation in RIF1 resides within the linkage region while the second mutation in DCLRE1C (Artemis) is outside of the linkage interval.
Functional assays indicate increased radiosensitivity, an oligoclonal TCR repertoire, increased alternative end-joining during class switch recombination and almost absent V(D)J recombination in patients’ cells. Complementation assays will show which of the two mutations causes the observed phenotype or whether the combination of both mutations gives rise to the immunodeficiency observed in these patients.
Our laboratory findings are consistent with previously published patients suffering from combined immunodeficiency due to hypomorphic Artemis mutations. However, the clinical phenotype of the patients only fulfils the diagnostic criteria of CVID with low B cells, as T cell numbers are normal.
We therefore conclude that clinical, functional, and genetic analysis must be combined to obtain a precise diagnosis and provide appropriate care for patients suffering from primary immunodeficiencies.
ESID-0806 CLEC16A Associates with Human Common Variable Immunodeficiency and Influences Murine B Cell Survival and Function
J. Li1, S. Jørgensen 2, S.M. Maggadottir1, M. Bakay1, K. Warnatz3, J. Glessner1, U. Salzer3, R.E. Schmidt4, E. Resnick5, S. Goldacker3, M. Buchta3, T. Witte4, L. Padyukov6, F. Atschekzei4, J.T. Elder7, R.P. Nair7, K.E. Sullivan8, J.S. Orange9, S. Schreiber10, W. Lieb11, B. Fevang12, P. Aukrust12, H. Chapel13, C. Cunningham-Rundles5, A. Franke10, T.H. Karslen2, B. Grimbacher3, H. Hakonarson1, L. Hammarström14, E. Ellinghaus10
1Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, USA
2K.G. Jebsen Inflammation Research Centre Research Institute of Internal Medicine Division of Cancer MedicineSurgery and Transplantation, Oslo University Hospital Rikshospitalet, Oslo, Norway
3Center for Chronic Immunodeficiency, University Hospital of Freiburg, Freiburg, Germany
4Clinic for Immunology and Rheumatology, Hannover Medical School, Hannover, Germany
5Institute of Immunology and Department of Medicine, Mount Sinai School of Medicine, New York, USA
6Rheumatology Unit, Karolinska Institutet and Karolinska University Hospital Solna, Stockholm, Sweden
7Department of Dermatology, University of Michigan Ann Arbor, Michigan, USA
8Division of Allergy and Immunology, The Children’s Hospital of Philadelphia, Philadelphia, USA
9?Section?of Immunology Allergy and Rheumatology, Department of Pediatric Medicine Texas Children’s Hospital, Houston, USA
10Institute of Clinical Molecular Biology, Christian-Albrechts-University Kiel, Kiel, Germany
11Institute of Epidemiology and Biobank popgen, Christian-Albrechts-University of Kiel, Kiel, Germany
12?Section?of Clinical Immunology and Infectious diseases, Oslo University Hospital Rikshospitalet, Oslo, Norway
13Department of Clinical Immunology, Nuffield Department of Medicine University of Oxford, Oxford, United Kingdom
14Department of Laboratory Medicine, Division of Clinical Immunology and Transfusion Medicine Karolinska University Hospital Huddinge, Stockholm, Sweden
Common variable immunodeficiency disorder (CVID) has a prevalence of 1:25,000 in European populations, and presents a clinically important form of immunodeficiency. In this primary immune deficiency, inadequate quantity and quality of immunoglobulins results in susceptibility to bacterial infections. CVID is of heterogeneous clinical presentation and the underlying genetic mechanism is poorly understood. We conducted an association analysis of 123,127 common single nucleotide polymorphisms (SNPs) across 778 CVID cases and 10,999 healthy controls of European ancestry through dense genotyping of immune-related loci on the immunochip. In this largest CVID genetic study performed to date, we identified the first non-HLA CVID risk locus at CLEC16A (rs17806056, P=2.0×10-9). CLEC16A has been reported to be associated with multiple autoimmune disorders. Although the function of CLEC16A is largely unknown, it encodes a C-type lectin-like domain protein that is expressed in B-cells, dendritic cells and natural killer cells. Because of the critical role of B cell function in CVID development, we further examined B cell phenotype in Clec16a knock down (KD) mice generated by crossing Clec16aloxP mice with UBC-Cre-ER-LBD-tg mice. We detected significantly reduced fractions of CD19+ B cells after induction of Clec16a KD compared to control group in two independent experiments (P=0.041 and P=2.3×10-4, respectively) and altered immunoglobulin production with increased IgM secretion (P=0.003). These findings suggest an impact of CLEC16A on B cell functional properties. The reported association of CLEC16A with multiple autoimmune disorders presents a compelling link between these disorders and autoimmunity commonly seen in CVID.
ESID-0640 BCR-Mediated Canonical NF-KB Signaling is Disturbed in a Subgroup of CVID Patients
B. Keller 1, I. Stumpf1, K. Warnatz1
1Centre for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany
Introduction:
Common variable immunodeficiency (CVID) is the most prevalent antibody deficiency syndrome in humans. In over 90% of the patients the aetiology of the disease is still unknown. Disturbed activation of signals downstream of the T- or B-cell receptor (BCR) might play a role in a subgroup of patients.
Objective:
To analyse the canonical NF-?B signaling pathway in CVID patients.
Methods:
Degradation of I?Ba and phosphorylation of p65 were determined in B-cell subpopulations of CVID patients with a reduction of switched memory B cells (CVID smB-) after different stimuli. The results were correlated with the capacity of B-cell activation and proliferation.
Results:
BCR-mediated degradation of I?Ba and phosphorylation of p65 was impaired in a subgroup of CVID patients. Interestingly, most of them presented with an accumulation of CD21low B cells referring to group CVID smB- 21low. CD40 and TLR9-mediated degradation NF-KB signaling was comparable to healthy controls, excluding a general defect in the canonical NF-?B pathway. Since PMA-induced degradation of I?Ba was not affected the dysregulation localizes up-stream of PKCß.Furthermore, the up-regulation of NF-?B-related activation markers, like CD69 and CD25 and proliferation was impaired, while NF-?B-independent markers were not affected, suggesting a direct impact of disturbed NF-?B signaling on the functional impairments of B cells in these patients.
Conclusion:
Dysregulated canonical NF-?B signaling downstream of the BCR in a subgroup of CVID patients might result from and contribute to the prevailing B-cell dysfunction in these patients. So far, the underlying mechanism for this impairment remains elusive.
ESID-0192 AP3D Deficiency Defines a New Type of Hermansky-Pudlak Syndrome
S. Ammann1, A. Schulz2, I. Krägeloh-Mann3, M. Schöning3, H. von Bernuth4, U. zur Stadt5, K. Lehmberg5, S. Ehl 1, H. Hennies6
1University Medical Center and University of Freiburg, Center for Chronic Immunodeficiency, Freiburg, Germany
2University Medical Center Ulm, Department of Pediatrics, Ulm, Germany
3University Medical Center Tübingen, Department of Pediatrics, Tübingen, Germany
4University Medical Center Charité, Department of Pediatrics, Berlin, Germany
5University Medical Center Hamburg Eppendorf, Center for Diagnostic, Hamburg, Germany
6University of Cologne, Cologne Center for Genomics, Cologne, Germany
Hermansky-Pudlak syndrome (HPS) is a heterogenous disease with albinism, bleeding disorder and cellular storage disorder. HPS2 is associated with neutropenia and immunodeficiency. We present a patient with oculocutaneous albinism, severe psychomotor retardation with seizures, mild facial dysmorphism, microcephaly, hip dysplasia and reduced hearing. He had increased susceptibility to airway infections and persistent hepatosplenomegaly. Immunological screening investigations revealed chronic neutropenia, an inverted CD4/CD8 ratio and elevated IgE. Consistent with the suspected diagnosis of Hermansky-Pudlak-Syndrome Type 2 (HPS2), NK and T cell degranulation and cytotoxicity were absent. However, genetic analysis revealed no mutations in AP3B1 (HPS2) or other genes associated with immunodeficiency and albinism such as LYST (Chediak-Higashi-Syndrome), RAB27A (Griscelli-Syndrome 2) or LAMTOR3 (P14 deficiency). Whole exome sequencing identified a homozygous late frame-shift mutation in AP3D1 leading to a premature stop codon. A natural mouse mutant with an Ap3d1 null mutation (Mocha Mice) shares the neurological, hematological and hypopigmentation phenotype of our patient and shows the storage pool deficiency characteristic of HPS. Our patient did not show abnormal bleeding and had asymptomatic seroconversion to EBV and CMV without developing signs of HLH. AP3d is a subunit of the heterotetramer adaptor-protein 3 (AP3) complex existing in a ubiquitously expressed form and a primarily neuronally expressed form. The AP3d subunit is essential for both forms, whereas AP3ß3A affected in HPS2 can is substituted by AP3ß3B in the neuron-specific heterotetramer, explaining the severe neurological phenotype of our patient. AP3d deficiency causes a new variant of HPS in humans, which we propose to classify as HPS2b.
ESID-0597 The Extended Clinical Phenotype of 58 Patients with Dock8 Deficiency
K.R. Engelhardt1, E.M. Gertz A.A. Schäffer2, S. Keles3, E.C. Sigmund 4, R. Ceja5, A. Sassi6, L. Graham7, M.J. Masaad8, F. Mellouli9, I. Ben-Mustapha10, M. Khemiri9, S.S. Kilic11, A. Etzioni12, A.F. Freeman13, J. Thiel I. Schulze4, W. Al-Herz14, A. Metin15, O. Sanal I. Tezcan16, M. Yeganeh17, T. Niehues G. Dueckers18, S. Weinspach19, T. Patiroglu E. Unal20, M. Dasouki21, M. Yilamaz F. Genel C.Aytekin N. Kutukculer A. Somer M. Kilic I. Reisli Y. Camcioglu22, A.?R. Gennery A.J. Cant23, A. Jones H.B. Gaspar24, P.D. Arkwright25, M.C. Pietrogrande26, Z. Baz27, S. Al-Tamemi28, V. Lougaris29, G. Lefranc30, A. Megarbane31, J. Boutros N. Galal32, M. Bejaoui9, M.R. Barbouche33, R.S. Geha T. A. Chatila5, B. Grimbacher4
1Dept. of Immunology and Molecular Pathology, Royal Free Hospital and University College London, London, United Kingdom
2Department of Health and Human Services, National Center for Biotechnology Information National Institutes of Health, Bethesda, USA
3Department of Pediatrics, David Geffen School of Medicine at the University of California, Los Angeles, USA
4Center for Chronic Immunodeficiency (CCI), University Medical Center Freiburg, Freiburg, Germany
5Division of Immunology Allergy and Rheumatology Department of Pediatrics, David Geffen School of Medicine at the University of California, Los Angeles, USA
6Laboratory of Vaccinology and Molecular Genetics, Pasteur Institute of Tunis and University of Tunis el Manar, Tunis el Manar, Tunisia
7Royal Free Hospital and University College London, Dept. of Immunology and Molecular Pathology, London, United Kingdom
8Division of Immunology, The Children's Hospital, Boston, USA
9Department of Pediatrics, Bone Marrow Transplantation Center, Tunis, Tunisia
10Laboratory of Immunology Vaccinology and Molecular Genetics, Pasteur Institute of Tunis and University, Tunis el Manar, Tunisia
11Department of Pediatric Immunology, Faculty of Medicine Uludag University, Bursa, Turkey
12Meyer's Children Hospital Rambam Health Care Campus and Rappaport Faculty of Medicine, Technion-Israel Institute of Technology, Haifa, Israel
13Laboratory of Clinical Infectious Diseases National Department of Health and Human Services, Institute of Allergy and Infectious Diseases National Institutes of Health, Bethesda, USA
14Department of Pediatrics Faculty of Medicine, Kuwait University and Allergy and Clinical Immunology Unit, Kuwait City, Kuwait
15Pediatric Immunology Unit, SB Ankara Diskapi Children's Hospital, Ankara, Turkey
16Pediatric Immunology Unit, Hacettepe University Children's Hospital, Ankara, Turkey
17Children's Medical Center Tehran University of Medical Sciences, Immunology Asthma and Allergy Research Institute, Tehran, Iran
18Zentrum für Kinder- und Jugendmedizin, HELIOS Klinikum Krefeld, Krefeld, Germany
19Department of Pediatric Oncology, Hematology and Clinical Immunology Center of Child and Adolescent Medicine Heinrich-Heine-University Düsseldorf, Düsseldorf, Germany
20Department of Pediatrics, Division of Pediatric Hematology and Immunology/ Oncology Faculty of Medicine Erciyes University, Kaysery, Turkey
21Department of Pediatrics, University of Kansas Medical Center, Kansas City, USA
22_, Cukurova University Adana/ Division of Pediatric Immunology Behcet Uz State Hospital Izmir /Department of Pediatric Immunology Dr. Sami Ulus Maternity and Children's Health and Diseases Training and Research Hospital Ankara/ Ege University Faculty of Medicine Department of Pediatrics Izmir/ Division of Infectious Diseases and Immunology Istanbul Medical Faculty/ Firat University Elazig/ Konya Necmettin Erbakan University Division of Pediatric Allergy and Immunology/ Division of Pediatric Allergy-Immunology and Infectious Diseases Cerrahpasa Medical Faculty Istanbul University,, Turkey
23Institute of Cellular Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, United Kingdom
24Department of Immunology, Great Ormond Street Hospital, London, United Kingdom
25Royal Manchester Children's Hospital, University of Manchester, Manchester, United Kingdom
26Department of Pediatrics, University of Milan Fondazione Policlinico IRCCS, Milan, Italy
27Department of Pediatrics, St George Hospital University Medical Center, Beirut, Lebanon
28Department of Pediatrics, Sultan Qaboos University, Muscat, Oman
29Department of Clinical and Experimental Sciences University of Brescia Spedali Civili di Brescia, Pediatrics Clinic and Institute for Molecular Medicine A.Nocivelli, Brescia, Italy
30University Montpellier 2 and CNRS, Institute of Human Genetics, Montpellier, France
31Medical Genetics Unit, Saint Joseph University, Beirut, Lebanon
32Specialized Pediatric Hospital, Primary Immunodeficiency Clinic Cairo University, Cairo, Egypt
33Laboratory of Immunology Vaccinology and Molecular Genetics, Pasteur Institute of Tunis and University of Tunis el Manar, Tunis el Manar, Tunisia
Background: Mutations in DOCK8 cause a combined immunodeficiency (CID) also classified as an autosomal-recessive form of the hyper-IgE syndromes (HIES). Early diagnosis of DOCK8 deficiency is of clinical importance due to a difference in prognosis and management.
Objectives: To study the mutational spectrum of DOCK8 deficiency and report on the frequency of specific clinical findings. Identification of clinical features distinguishing DOCK8 deficiency from other forms of HIES.
Methods: Fifty-eight patients from 45 families with a combined immunodeficiency and the phenotype of autosomal-recessive HIES with and without DOCK8 mutations were studied. Regression was used to compare clinical data from 35 patients with DOCK deficiency with 10 AR-HIES patients without a DOCK8 mutation and 64 patients with STAT3 mutations.
Results: DOCK8-deficient patients had a median IgE of approximately 5,800?IU and eosinophil levels were usually (83%) at least 1000/?µl. Bacterial (87%), viral (72%), and fungal (67%) infections were frequently observed. Abscesses (63%) and allergies (72%) were common clinical problems. In contrast to STAT3 deficiency, there were few pneumatoceles, bone fractures, and teething problems. Mortality was high (31%). A combination of five clinical features was helpful in distinguishing patients with DOCK8 mutations from those with STAT3 mutations.
Conclusions: We propose the following diagnostic guidelines for DOCK8 deficiency:
Possible: HIES diagnosis plus hypereosinophilia and upper respiratory tract infections Absence of parenchymal lung abnormalities, retained primary teeth, and minimal trauma fractures.
Probable: Above features plus consanguinity, severe viral infections, allergies, and/or low IgM levels.
Definitive: Biallelic mutation in DOCK8 and/or lack of full-length protein expression.
ESID-0810 Clonal Tracking After Gene Therapy Reveals Composition, Fate and Activity of Hematopoietic Stem Cells and T-Cells
L. Biasco1, S.(. Scala1, N. Cieri3, L.B. Ricci1, F. Dionisio1, C. Baricordi1, S. Scaramuzza1, A. Calabria1, S. Giannelli1, C. Cancrini4, A. Scarselli4, L.(. Naldini1, E. Montini1, C. Bonini3, A.(. Aiuti 1
1San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET), Milan, Italy
2Università Vita-Salute San Raffaele, Milan, Italy
3San Raffaele Scientific Institute, Milan, Italy
4Ospedale Pediatrico Bambin Gesù, Rome , Italy
5University of Rome “Tor Vergata”, Rome, Italy
Study of hematopoietic stem cell (HSC) activity and lympho-hematopoietic dynamics after gene therapy (GT) with HSC or T cells are of paramount importance for the design of innovative therapies for primary immunodeficiencies. We are conducting a clinical trial for Wiskott-Aldrich Syndrome based on the infusion of genetically engineered HSC which represent a unique setting where each vector-marked progenitors and its blood cell progeny is traceable ?in?vivo by a vector integration site (IS). We unraveled the timing and nature of short, intermediate and long term HSC output showing that progenitor output occurs in distinct waves during the first 6-9 months after transplantation reaching a “homeostatic equilibrium” by 12 months after GT. We estimated that 1900-7000 transduced HSC clones were stably contributing to the progenitor repertoire for up to 3 years after infusion of gene corrected CD34+ cells. We also performed a comprehensive study of T-cells dynamics and plasticity in ADA-SCID patients who had received gene corrected peripheral blood T cells in the absence of progenitor cell infusion. We found that vector-positive putative naïve cells were detected for more than 11 years but the vast majority were actually the recently discovered T memory stem cell (Tscm). We characterized Tscm through phenotypic and functional assays and measured plasticity and hierarchical relationships by clustering of identical vector IS among T-cell subpopulations. Collectively, our data constitute the first molecular tracking of individual hematopoietic clones in humans providing an unprecedented detailed analysis of HSC and Tscm activity and dynamics ?in?vivo.
ESID-0234 Treatment of Post-HSCT Immune Deficiency by Infusion of Ex Vivo Generated T-Cell Precursors
I. André-Schmutz 1, C. de Chappedelaine2, S. Susini2, E. Elkaïm2, S. Mouraud2, B. Ternaud3, A. Gabrion3, J. Blondeau3, C. Reimann4, F. Touzot3, M. Cavazzana3
1Human Lympho-Hematopoiesis Lab, Imagine - INSERM U1163, PARIS, France
2Human Lympho-Hematopoiesis Lab, Imagine - INSERM U1163, PARIS, France
3Biotherapy Department, Necker-Enfants Malades Hospital, PARIS, France
4Center for Child and Adolescent Medicine, Universitätsklinikum Freiburg, Freiburg, Germany
Slow T-cell reconstitution is a major clinical concern after HLA-partially incompatible hematopoietic stem cell transplantation (HSCT). The aim of this project was to demonstrate the ability of a soluble chimeric protein (composed of the notch ligand DL4 and the constant part of a human IgG) to induce the T-cell commitment of a CD34+ cell population derived from different sources. The ultimate concept is that injection into the patient of this ?ex?vivo T-cell-committed cell population can significantly accelerate the reconstitution of the adaptive immune system compartment after a partially HLA-incompatible HSCT.
We exposed cord blood and adult CD34+ cells to DL-4 in the presence of a mixture of cytokines. Three to seven-day exposure was sufficient to induce the generation of CD7+ T cell precursors. At these time points, the potential of T-cell differentiation was maximal. Furthermore, these CD7+ precursors had the phenotypic and molecular features of early thymic T-cell precursors as shown by the qPCR analysis of the transcription of several T-cell specific genes. These results prompted us to further investigate their T-cell potential in two different xenotransplantation models (NSG mice, either irradiated adult or non irradiated neonates). The results obtained with CB-derived T-cell precursors evidenced faster and more robust T-cell reconstitution than that obtained with non-manipulated cells. Human T cells generated in NSG mice were polyclonal and functional. All our efforts are now focused on translating this experimental protocol into a phase I/?II clinical trial involving several transplantation units in France.
ESID-0747 Human JAGN1 Deficiency Causes Disturbed Myeloid Cell Homeostasis and Severe Congenital Neutropenia
K. Boztug 1, P.?M. Järvinen2, E. Salzer1, T. Racek2, S. Mönch2, W. Garncarz1, E.M. Gertz3, A. Antonopoulos4, S. Haslam4, L. Schieck5, J. Puchalka2, J. Diestelhorst2, G. Appaswamy5, B. Lescoeur6, R. Giambruno1, J.W. Bigenzahn1, U. Elling7, D. Pfeifer8, K. Welte5, G. Brandes9, R. Sherkat10, J. van der Werff ten Bosch11, N. Rezaei12, A. Etzioni13, C. Bellanné-Chantelot14, G. Superti-Furga1, J.M. Penninger7, K.L. Bennett1, J. von Blume15, A. Dell4, J. Donadieu16, C. Klein2
1CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria
2Department of Pediatrics, Dr. von Hauner Children’s Hospital Ludwig-Maximilians University, Munich, Germany
3Computational Biology Branch National Center for Biotechnology Information, National Institutes of Health, Bethesda, USA
4Department of Life Sciences, Imperial College London, London, United Kingdom
5Department of Pediatric Hematology/Oncology, Hannover Medical School, Hannover, Germany
6Department of Hematology, Hospital R Debré, Paris, France
7IMBA Institute of Molecular Biotechnology of the Austrian Academy of Sciences, Vienna, Austria
8Department of Hematology/Oncology Genomics Core Laboratory, University Medical Center Freiburg, Freiburg, Germany
9Department of Cell Biology, Hannover Medical School, Hannover, Germany
10Infectious Diseases Research Center Department of Infectious Diseases, Isfahan University of Medical Sciences, Isfahan, Iran
11Department of Pediatrics, Hospital of the Free University of Brussels, Brussels, Belgium
12Research Center for Immunodeficiencies Pediatrics Center of Excellence Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran
13Division of Pediatrics and Immunology, Rappaport School of Medicine Technion, Haifa, Israel
14Genetics Department, AP-HP Pitié-Salpêtrière Hospital Pierre and Marie Curie University, Paris, France
15Max-Planck Institute of Biochemistry, Martinsried, Germany
16Histocytology Reference Center, Armand Trousseau Children’s Hospital Pediatric Hematology Unit, Paris, France
We here identify a novel genetic cause of severe congenital neutropenia (SCN) due to biallelic mutations in the gene encoding Jagunal homolog 1 (JAGN1). Genome-wide linkage analysis in an index family from Northern Africa with 5 children suffering from SCN identified a single candidate interval on chromosome 3. This interval contained a total of 30 genes, including JAGN1. Sanger sequencing revealed a homozygous mutation c.3G>A in JAGN1 leading to disruption of the defined start of translation.
Analysis of 90 SCN patients identified 9 distinct homozygous mutations in JAGN1 in 14 SCN patients, thus accounting for approximately 10% of SCN patients. The clinical phenotype was variable and included failure to thrive, developmental delay and bone skeletal abnormalities. Notably, all JAGN1-deficient patients showed partial or complete refractoriness rh-GCSF therapy.
JAGN1 is the human ortholog of a gene originally identified in Drosophila melanogaster critical for oogenesis and ER reorganization.
JAGN1-mutant human granulocytes showed enlarged ER structures and paucity of secretory vesicles. In line with the presumed defects in ER function, we found that JAGN1-mutant neutrophils exhibited anomalous N-glycomic profiles including markedly reduced fucosylation. JAGN1-deficient neutrophils showed increased apoptosis in response to TNFa and staurosporine. Interaction proteomics of JAGN1 identified a limited number of interaction partners including Coat Protein I (COPI) complex members (COPA, COPB2, and COPG2), suggesting a role for JAGN1 in vesicular trafficking from Golgi to ER. Taken together, JAGN1 emerges as a hitherto unrecognized factor necessary in differentiation and survival of neutrophil granulocytes and a novel gene implicated in SCN.
ESID-0571 The Extended Spectrum of Leukocyte Adhesion Deficiency-1: Single Institution Long Term Follow-Up of 12 Cases
K. Walkovich 1, D.B. Kuhns2, D.A. Long Priel2, S.M. Holland3, G. Uzel3
1Pediatrics Hematology/Oncology, University of Michigan C.S. Mott Children's Hospital, Ann Arbor, USA
2Fredrick National Laboratory for Cancer Research, Clinical Services Program Leidos Biomedical Research Inc., Fredrick, USA
3National Institute of Allergy & Infectious Disease NIH, Laboratory of Clinical Infectious Diseases, Bethesda, USA
Leukocyte adhesion deficiency type-1 (LAD-1) is a primary immune disorder characterized by defective leukocyte chemotaxis, migration and adhesion. LAD-1 is defined by bi-allelic mutations in ITGB2, the gene encoding the beta2 (ß2) integrin CD18, that lead to impairment in ß2 integrin expression, aß2-integrin heterodimer formation and/or function. Without functional integrin dimers, neutrophils fail to adhere to the endothelium/ transmigrate to tissues, leading to a state of tissue neutropenia with the absence of pus, leukocytosis, and delayed wound healing. The same process that facilitates tissue neutrophil emigration also mediates homotypic adhesion between lymphocytes. To understand the spectrum of clinical features, we examined the clinical profiles of 12 patients followed at the National Institutes of Health over the last two decades. To better understand the relationship between mutation and disease, we evaluated their genetic and immune features. In our cohort, we identified several novel ß2 integrin mutations. Four patients had uni-allelic reversion mutations in a small subset of CD8 T cells, making revertant mosaicism a relatively common event in LAD-1. Patients had predominantly bacterial infections involving lymph nodes, skin, lungs and perianal tissue. Candida esophagitis was observed frequently, but other fungal infections were rare. Periodontitis, pyoderma gangrenosum-like skin ulcerations, HPV infection and autoimmunity were prevalent. Inverted CD4/CD8 ratio, increased circulating MAIT cells, poor specific antibody responses and diminished NK cell function were among the observed and previously underappreciated laboratory findings. Notably, the disease spectrum evolves over time and can involve colitis, warts, autoimmunity, which represent previously underappreciated features of LAD-1.
ESID-0057 Personalized Therapy for X-Linked Agammaglobulinemia (XLA) by Oligonucleotide-Induced Splice-Correction
C.I.E. Smith 1, B. Bestas1, P.?M.?D. Moreno1, B. Berglof1, K.E.M. Blomberg1, D.K. Mohammad1, A.F. Saleh2, T. Sutlu3, K.E. Lundin1, J.Z. Nordin1, S. Kharazi3, R. Månson3, S. EL Andaloussi1, M.J. Gait2, J. Wengel4
1Laboratory Medicine, Karolinska Institutet, Stockholm, Sweden
2Laboratory of Molecular Biology, Medical Research Council, Cambridge, United Kingdom
3Medicine, Karolinska Institutet, Stockholm, Sweden
4Department of Physics Chemistry and Pharmacy, University of Southern Denmark, Odense, Denmark
X-linked agammaglobulinemia (XLA) is an inherited immunodeficiency resulting from mutations in the BTK gene severely blocking B cell development. Some XLA-causing mutations affect splicing of BTK pre-mRNA. Here, we assess the treatment potential of antisense, splice-correcting oligonucleotides (SCOs) targeting a mutated BTK transcript. Both the SCO design and chemical properties were optimized for this purpose. In order to study the potential of the SCOs, we engineered a novel, Bacterial Artificial Chromosome (BAC)-transgenic mouse carrying a splice-defective human BTK gene with a mutation originally described in an XLA family. To avoid interference of the orthologous mouse protein, mice were bred onto a Btk knockout background. Thus, our transgenic mouse provides a unique, humanized animal model to study treatment strategies for diseases resulting from splice-site mutations. For the first time, we conclusively demonstrate the capacity of SCOs to correct aberrantly spliced BTK in B lymphocytes, including pro-B cells. The corrected BTK mRNA restored expression of functional BTK protein. This was shown both by enhanced lymphocyte survival and reestablished BTK activation upon B cell receptor stimulation. As a final proof-of-concept we were able to correct the mutation in primary patient cells. Thus, our approach may represent a versatile, future, personalized medicine for XLA.
ESID-0282 Long-Term Continuous Intra-Erythrocyte Infusion of Dexamethasone Reduces Neurological Symptoms in Ataxia Telangiectasia Patients
A. Soresina1, R. Micheli 2, V. Leuzzi3, A. Molinaro4, M. Maffeis1, D. D'Agnano3, P. Ferremi5, M. Marini5, M. Magnani6, L. Chessa7, A. Plebani1
1Dpt of Pediatrics, Pediatrics Clinic & Ins.for Molecular Medicine A.NocivelliUniversity of Brescia, Brescia, Italy
2Unit of Child Neurology and Psychiatry, Spedali Civili and Università di Brescia, Brescia, Italy
3Dpt of Pediatrics and Child Neurology and Psychiatry, Sapienza Università di Roma, Roma, Italy
4School in Reproductive and Developmental Science, Università di Trieste and Università di Brescia, Brescia, Italy
5Service of Immunoematology and Trasfusional Medicine, Spedali Civili and Università di Brescia, Brescia, Italy
6Dpt of Biomolecular Sciences, Università di Urbino, Urbino, Italy
7Dpt of Clinical and Molecular Medicine, Sapienza Università di Roma, Roma, Italy
Background. Ataxia-teleangiectasia (AT) is a rare devastating neurodegenerative disease presenting with early onset ataxia, oculocutaneous teleangiectasias, immunodeficiency, radiosensitivity, and proneness to cancer. In a previous phase II study we showed that 6 monthly infusions of autologous erythrocytes loaded with dexamethasone (EryDex) were effective in improving the neurological impairment in young AT patients. Present paper report on the result of the extension of this study for an additional 24 month-period.
Methods. After the end of first trial, 4 subjects continued to be treated with monthly EryDex infusions for further 24 months and their clinical outcome was compared with that of 7 age-matched subjects who had stopped the treatment after the first 6 infusions. The protocol included serial assessment of ataxia (by International Cooperative Ataxia Rating Scale) and adaptive behavior (by Vineland Adaptive Behavior Scales), and clinical and laboratory tests revealing treatment- and steroid-dependent adverse effects if present.
Results. Patients in the extended study experienced a continuous neurological improvement with respect to their pre-treatment status while control subjects showed a progressive neurological deterioration (according to the natural history of the disease) after the discontinuation of the treatment. The delivery system we adopted proved to be safe and well-tolerated and none of the side effects usually associated with the chronic administration of corticosteroids was observed during the entire trial.
Conclusions. These preliminary promising results call for a wide-scale controlled study on protracted treatment of AT patients with dexamethasone loaded erythrocytes.
ESID-0647 A Whole Transcriptome Sequencing Pipeline for the Identification and Characterisation of Disease-Causing Genes in Primary Immunodeficiency Diseases
V. Menzel 1, M. Faßhauer2, U. Sack1, M. Borte2, S. Borte3
1Translational Center for Regenerative Medicine, University Leipzig, Leipzig, Germany
2Department of Pediatrics, ImmunoDeficiencyCenter Leipzig (IDCL) at Hospital St. Georg Leipzig, Leipzig, Germany
3Department of Laboratory Medicine, Karolinska Institutet, Stockholm, Sweden
Enabling a correct genetic diagnosis in newly-presenting patients with primary immunodeficiency diseases (PID) remains an obstacle in routine clinical practice. With the development of next-generation sequencing (NGS) methods, mainly exome (DNA) sequencing and the analysis of single nucleotide polymorphism and single-nucleotide variations (SNP/SNV) has been applied to PID patients in recent years. Here we show that whole transcriptome (RNAseq) sequencing, which covers SNP/SNV information as well as differential gene expression analysis, is sufficient to establish a functional molecular diagnosis in PID patients. We have developed and optimized a RNA-sequencing pipeline from peripheral blood cells and have included a cohort analysis of CVID and CID patients.
Our results indicate that the expression level of cell-lineage-specific (signature) marker genes can indicate whether a particular cell type (e.?g. B cells) is altered in the frequency in peripheral blood. Furthermore, SNPs and SNVs which are detected via Exome-sequencing analysis, can also be validated via RNAseq analysis. Due to allele-specific expression of genes, SNPs/?SNVs contained in the DNA can be totally absent, reduced or exclusively present in the transcriptome. However, SNPs present in the genome – but not in the transcriptome - are unlikely to be disease-causing. Similarly to genome-wide association studies (GWAS), RNAseq allows identifying unknown PID genes either being related directly to the pathology of primary immunodeficiencies, or affecting more complex networks implied in the global immunologic homeostasis. This genetic typing of SNP/SNV, GWAS and expression analysis allows a precise specification of complex genetic diagnoses and is an important step towards an individualized therapy.
ESID-0051 Cohort-Wide Identification of Morbid Alleles with the Human Gene Connectome
Y. Itan 1, S.Y. Zhang1, L. Abel2, J.L. Casanova1
1Human Genetics of Infectious Diseases, The Rockefeller University, New York, USA
2Human Genetics of Infectious Diseases, INSERM, Paris, France
Introduction: to determine the disease-causing allele(s) underlying primary human inborn errors, high-throughput genomic methods are applied and provide thousands of gene variants per patient. We recently reported a novel approach, the “human gene connectome” (HGC) – the set of all in silico-predicted biologically plausible routes and distances between all pairs of human genes. The HGC is a powerful approach for prioritizing gene variants by biological distance from known disease-causing genes (http://lab.rockefeller.edu/casanova/HGC)
Objectives and Aims: there is currently no available method for automating the selection of candidate disease-causing mutant alleles in the absence of a known morbid gene in at least one patient with the disease of interest, posing a major bottleneck in the field in high-throughput clinical genomics which we aim to resolve.
Methods and Results: We hypothesized that within a cohort of patients with the same Mendelian disease, the cluster that contains the key disease-causing gene for each patient is the HGC-predicted biologically smallest cluster. We then developed and applied a Mendelian clustering algorithm, which estimates the biologically smallest HGC-predicted cluster that contains one allele per patient. By that we (i) approximated a solution for an NP-complete algorithmic problem (i.?e. not possible to solve on a large scale by a computer), and (ii) estimated and statistically validated a set of disease-causing alleles in a whole exome sequencing cohort herpes simplex encephalitis patients
Conclusions: the unbiased approach described above should facilitate the discovery of morbid alleles in patients with primary inborn errors that lack a genetic etiology.
ESID-0334 Whole Exome Sequencing for Primary Immunodeficiencies – Experience From the Asian Primary Immunodeficiency (APID) Network
P. Lee 1, J. Yang1, W.K. Liew2, A.H. Latiff3, M.K. Thong4, A.?C.W. Lee5, O. Jirapongsananuruk6, K.K. Al-Saad7, N. Kechout8, K.W. Chan1, C.Y. Chong1, T.L. Lee1, H.K. Ho1, W.W. Tu1, W.L. Yang1, Y.L. Lau1
1Department of Paediatrics and Adolescent Medicine, Li Ka Shing Faculty of Medicine The University of Hong Kong, Hong Kong, Hong Kong China
2Department of Paediatrics, KK Women’s and Children Hospital, Singapore, Singapore
3Department of Paediatrics, Pantai Hospital Kuala Lumpur, Kuala Lumpur, Malaysia
4Department of Paediatrics, Faculty of Medicine University of Malaya, Kuala Lumpur, Malaysia
5Parkway Cancer Centre, Parkway Mount Elizabeth Hospital, Singapore, Singapore
6Division of Allergy and Immunology Department of Pediatrics, Faculty of Medicine Siriraj Hospital Mahidol University, Bangkok, Thailand
7Department of Pediatrics, Salmanyia Medical Complex, Manama, Bahrain
8Department of Immunology, Institut Pasteur d’Algérie, Algiers, Algeria
Background: Next generation sequencing (NGS) revolutionizes the discovery of genetic causes for PID. For most PID, genetic etiologies can be established by candidate gene strategy. However, this becomes increasingly laborious given the growing phenotypic and genotypic complexity of PID.
Objective: We sought to develop a discovery and diagnostics NGS platform for investigating patients with undefined PID.
Method: Our study is based on 1,051 patients referred to APID from 68 hospitals since 2001. Disease-causing mutations were identified in 509 patients from a panel of 75 PID genes. Patients who lacked a genetic diagnosis despite extensive investigations were selected for whole exome sequencing (WES) using Illumina HiSeq and in-house bioinformatics pipeline.
Results: Mutations were identified in 29/48 patients (60.4%) from 2009-2014. These include newly characterized PID genes such as PIK3CD, TTC37 and TTC7A. STAT1 mutations were found in patients with penicilliosis, or type I DM and CID. Homozygous G6PC3 and CD79A mutations were readily identified in consanguineous kindreds affected by congenital neutropenia and agammaglobuinemia, respectively. PNP deficiency and MHC class II (RFXANK) deficiency rarely described in Asians were diagnosed. IL2RG mutations were identified in atypical T-B-NK+ and TlowB+NK+ SCID, and SH2D1A mutation in agammaglobulinemia without lymphoproliferative disease. WES revealed DCLRE1C and CIAS1 mutations which had been missed by Sanger sequencing. Furthermore, an unexpected NLRP12 mutation co-existing with a CIAS1 mutation was identified in a patient with CINCA.
Conclusion: NGS is a powerful tool which facilitates cost-effective investigations of PID, particularly in multi-centered network involving patients with diverse ethnic backgrounds and disease heterogeneity.
ESID-0620 High Diagnostic Yield by Exome Sequencing of 275 Primary Immunodeficiency Patients
A. Stray-Pedersen 1, H.S. Sorte1, P.?S. Samarakoon1, L. Forbes2, T. Gambin3, O.K. Rodningen1, I.C. Hanson4, L.M. Noroski2, C. Davis4, F. Seeborg4, S.K. Nicholas4, J.W. Caldwell5, N.Y. Chokshi4, D. Bayer4, H.C. Erichsen6, T.G. Abrahamsen6, G.E. Tjonnfjord7, P. Aukrust8, B. Fevang8, M.S. Caldirola9, L.A. Pedroza10, S.N. Jhangiani3, S. Gu3, A. Patel11, D. Muzny3, R.A. Gibbs12, W.T. Shearer4, R. Lyle1, J.S. Orange4, J.R. Lupski3
1Department of Medical Genetics, Oslo University Hospital, Oslo, Norway
2Department of Pediatrics, Center for Human Immunobiology of Texas Children's Hospital Baylor College of Medicine, Houston Texas, USA
3Department of Molecular and Human Genetics, Baylor-Hopkins Center for Mendelian Genomics Baylor College of Medicine, Houston Texas, USA
4Department of Pediatrics ?Section?of Immunology Allergy and Rheumatology, Baylor College of Medicine and Texas Children's Hospital, Houston Texas, USA
5?Section?Pulmonary Critical Care Allergy and Immunologic Diseases, Wake Forest Baptist Medical Center, Winston-Salem NC, USA
6Department of Pediatrics, Oslo University Hospital, Oslo, Norway
7Institute of Clinical Medicine, University of Oslo, Oslo, Norway
8?Section?of Clinical Immunology and Infectious Diseases, Oslo University Hospital, Oslo, Norway
9Department of Immunology, Ricardo Gutierrez Children's Hospital, Buenos Aires, Argentina
10Department of Genetics and Immunology, Universidad San Francisco de Quito, Quito, Ecuador
11Medical Genetics Laboratories Molecular and Human Genetics, Baylor College of Medicine, Houston Texas, USA
12Human Genome Sequencing Center, Baylor College of Medicine, Houston Texas, USA
Primary immunodeficiencies (PIDD) constitute a heterogeneous group of genetic diseases affecting the immune system. Symptoms can range from mild to severe and life threatening. Severity and treatment options depend on the genetic aetiology. Genetic diagnosis and subtype classification are complicated by overlapping phenotypes, and >250 primary immunodeficiency genes have been reported, and few have been offered for diagnostic genetic testing. New sequencing technologies have the potential to improve the diagnostic yield. We examined the utility of exome sequencing to detect SNPs and CNVs in PIDDs. As of May 2014, 275 patients with extensive immunological and genetic testing from 241 families have been recruited from Texas Children Hospital (Houston, USA) and Oslo University Hospital (Norway). Strategies for genetic analysis were tailored based on clinical data, immunophenotyping and family history, for 5/?6 of the families only the proband was subjected to exome sequencing. Initially, exome data were systematically screened for variants in reported and potential PIDD genes. In addition, a computational copy number variant prediction pipeline was applied to enable identification of potential disease-causing genetic imbalances from the exome data. For CNVs customized chromosomal microarray with exon wise coverage of all PIDD genes were used. Analysis of the first 126 families identified a definitive molecular PIDD diagnosis in 1/?3 of the patients. An additional 1/?3 had interesting variants in relevant genes, requiring further investigations determine their clinical importance. This project shows that exome sequencing of primary immmunodeficiency patients improves genetic diagnostic yield, which is important to direct targeted and curative therapy.
ESID-0721 Europe Immunoglobulin Map
A. Sediva 1, E. Förster-Waldl2, I. Meyets3, V. Mulaosmanovic4, E. Naumova5, J. Keletic6, J. Litzman7, S. Velbri8, M. Seppanen9, E. Oksenhendler10, K. Warnatz11, M. Kanariou12, L. Marodi13, M. Keogan14, I. Quinti15, T. Prokofjeva16, R. Duobiene17, F. Hentges18, E. deVries19, M. Pac20, S. Lopes da Silva21, M. Bataneant22, P. Ciznar23, T. Avcin24, T. Espanol25, A. Gardulf26, H. Chapel27, M. Belevtsev28, K. Mironska29, A. Shcherbina30, D. Koruga31, M. Hoernes32, I. Reisli33
1Dept. of Immunology, 2nd School of Medicine Charles University and University Hospital Motol, Prague, Czech Republic
2Univ. Klinik für Kinder- und Jugendheilkunde Center for Congenital Immunodeficiencies, Medizinische Universität Wien, Wien, Austria
3Department of Pediatrics, University Hospital Gasthuisberg, Leuven, Belgium
4University Clinical Center, Sarajevo Children's Hospital, Sarajevo, Bosnia and Herzegovina
5Clinical Immunology, Alexandrovska University, Sofia, Bulgaria
6Department of Pediatrics Division of Clinical Immunology, University Hospital Center, Zagreb, Croatia
7Dept. Clinical Immunology Allergology, St. Ann's University Hospital Brno, Brno, Czech Republic
8Clinical Immunology, Tallinn Childrens' Hospital, Tallin, Estonia
9Department of Medicine Division of Infectious Diseases Immunodeficiency Unit, Helsinki University Central Hospital, Helsinki, Finland
10Department of Clinical Immunology, Hôpital Saint-LouisUniv Paris Diderot Sorbonne, Paris, France
11Centre of Chronic Immunodeficiency, University Medical Center Freiburg University of Freiburg, Freiburg, Germany
12Center for Primary Immunodeficiencies - Paediatric Immunology, 'Aghia Sophia' Children's Hospital, Athens, Greece
13Department of Infectious and Pediatric Immunology, University of Debrecen Medical and Health Science Center, Debrecen, Hungary
14Department of Immunology, Beaumont Hospital, Dublin, Ireland
15Department of Molecular Medicine, Sapienza University of Rome, Rome, Italy
16Pediatric Immunology, Children's Clinical University Hospital, Riga, Latvia
17Center of Pediatrics, Children‘s Hospital Affiliate of Vilnius University Hospital, Vilnius, Lithuania
18Unit of Immunology and Allergology, Centre Hospitalier de Luxembourg, Luxembourg, Luxembourg
19Department of Paediatrics, Jeroen Bosch Hospital, 's-Hertogenbosch, Netherlands
20Department of Immunology, Children’s Memorial Health Institute, Warsaw, Poland
21Department of Immunology and Allergology, Hospital de Santa Maria, Lisbon, Portugal
22Department of Pediatrics, Victor Babes University of Medicine and Pharmacy, Timisoara, Romania
231st Pediatric Department, Comenius University, Bratislava, Slovakia
24Department of Allergology Rheumatology and Clinical Immunology, University Children's Hospital University Medical Centre, Ljubljana, Slovenia
25Immunology Unit, Vall d'Hebron University Hospital, Barcelona, Spain
26Division of Clinical Immunology Department of Laboratory Medicine, Karolinska InstitutetKarolinska University Hospital, Stockholm, Sweden
27Primary Immunodeficiency Unit, Nuffield Department of Medicine University of Oxford, Oxford, United Kingdom
28Immunology Laboratory Research Department, Belarusian Research Center for Pediatric Oncology Hematology and Immunology, Minsk, Belarus
29Department of Immunology, University Children's Hospital, Skopje, Macedonia
30Immunology, Federal Research and Clinical Center for Pediatric Hematology Oncology and Immunology, Moscow, Russia
31SPwID, Supporting Persons with Primary Immunodeficiency, Belgrade, Serbia
32Division of Immunology Haematology and BMT, University Children's Hospital Zurich, Zurich, Switzerland
33Department of Pediatric Hematology, Necmettin Erbakan University Meram Faculty of Medicine, Konya, Turkey
Background and Methods: ESID Primary Immunodeficiencies Care in Development Working Party, in order to improve diagnosis and care of patients with primary immunodeficiencies (PIDs), initiated the pan-European survey on a use and availability of immunoglobulins in European countries which resulted in Euro Immunoglobulin Map. In each European country one contact person has been appointed to answer the survey questionnaire. Some effort to map the situation started already in 2006, and since 2011 a regular survey reflects the development in immunoglobulin usage all over the Europe.
Results: The survey brings an unique and interesting view on a situation with immunoglobulin treatment of PIDs in individual European countries and further shows the trend and changes in this area. Since 2012 the situation improved mainly for Eastern European countries. IVIG products are available in practically all European countries even if the spectrum of available preparations and the access of PID patients to IVIG therapy might differ. SCIG are being more and more used, spreading from North and West to East and South. In 2014 there are only very few European countries without an access to SCIG. The introduction of SCIG therapy is more pronounced in pediatric population. Europe Immunoglobulin Map also shows subtly different strategies in use of immunoglobulins and substantial differences in price and spectrum of immunoglobulin preparations in individual countries. Conclusion: This pan-European survey has shown to be a very useful and interesting tool to map, harmonize and facilitate an availability of immunoglobulins as a basic treatment for PIDs.
ESID-0611 The PCID Study: Update and Baseline Features of the First Fifty Patients
C. Speckmann 1, A. Uhlmann1, S. Doerken1, M. Wolkewitz2, S. Fuchs1, A. Aiuti3, M. Albert4, W. Al-Herz5, L. Allende6, T. Avcin7, C. Cancrini8, A. Cant9, S. Hambleton9, B. Gaspar10, S. Ghosh11, A. Gennery9, L. Gonzalez-Granado6, F. Hauck4, M. Hoenig12, D. Moshous13, T. Niehues14, C. Picard13, J. Reichenbach15, N. Rieber16, M. Seidel17, P. Soler-Palacin18, P. Stepensky19, A. Worth10, S. Ehl1
1Center for Chronic Immunodeficiency, University Freiburg Medical Center, Freiburg, Germany
2Institute of Medical Biometry and Medical Informatics, University Freiburg Medical Center, Freiburg, Germany
3PCID Study consortium, PCID Study consortium, Milano, Italy
4PCID Study consortium, PCID Study consortium, Munich, Germany
5PCID Study consortium, PCID Study consortium, Kuwait, Kuwait
6PCID Study consortium, PCID Study consortium, Madrid, Spain
7PCID Study consortium, PCID Study consortium, Lubiljana, Slovenia
8PCID Study consortium, PCID Study consortium, Rome, Italy
9PCID Study consortium, PCID Study consortium, Newcastle, United Kingdom
10PCID Study consortium, PCID Study consortium, London, United Kingdom
11PCID Study consortium, PCID Study consortium, Düsseldorf, Germany
12PCID Study consortium, PCID Study consortium, Ulm, Germany
13PCID Study consortium, PCID Study consortium, Paris, France
14PCID Study consortium, PCID Study consortium, Krefeld, Germany
15PCID Study consortium, PCID Study consortium, Zurich, Switzerland
16PCID Study consortium, PCID Study consortium, Tübingen, Germany
17PCID Study consortium, PCID Study consortium, Graz, Austria
18PCID Study consortium, PCID Study consortium, Barcelona, Spain
19PCID Study consortium, PCID Study consortium, Jerusalem, Israel
Combined immunodeficiencies (CID) are a heterogeneous group of T-cell disorders manifesting with infections, immune dysregulation or malignancies. They can be delineated from SCID by absence of severe viral or opportunistic infections in the first year of life.
The genetic and clinical heterogeneity of CID make individual decisions on the appropriate indication and time point of HSCT difficult. We have therefore initiated a prospective multi-center cohort study on patients with evidence of T-cell deficiency and at least one associated clinical manifestation (severe infection, immune dysregulation or malignancy). We intend to enroll 200 patients with such a profound combined immunodeficiency (PCID) phenotype.
The primary objective of the study is to provide natural history data on patients with PCID, irrespective of whether they undergo HSCT or not. Further goals are to determine survival, frequency of severe events and quality of life 5 years after study inclusion.
We report clinical, immunological and genetic observations of the first 50 enrolled patients. A genetic diagnosis was established in 50% of the patients. Half of these have variants of leaky SCID (e.?g. ADA, IL2RGC, PNP or RAG), the other half has a variety of genetic disorders including mutations in Caspase-10, Coronin-1A, DOCK8, NBS1, PI3Kd, RMRP and ZAP-70. We will review their immunological phenotype as well as their infectious and immunological complications documented at study entry. About a third of these patients have been transplanted within a year after enrolment. We will review these therapeutic decisions and provide an initial analysis of current practice in treating these patients.
ESID-0655 Incidence and Prevalence of Congenital Neutropenia in France
J. donadieu 1, B. Beaupain1, M. Pasquet2, P. Labrune3, S. Cohen Beaussant4, C. rigaud5, L. Faivre6, N. Mahlaoui7, F. Bachelerie8, C. Bellanné Chantelot9
1Hémato oncology, hopital Trousseau, PARIS cedex 12, France
2Hémato oncology, CHU Toulouse, Toulouse, France
3Pediatric dep, Hop A Béclére, Clamart, France
4Pediatry, CHU Besançon, Besancon, France
5Pediatrics, CHU Limoges, Limoges, France
6Genetic dpt, CHU Dijon, DIJON, France
7Immunology Hematology, Ceredih Hop Necker, Paris, France
8Cermit, CNRS, Clamart, France
9Genetic Laboratory, Hopital Pitié Salpétriére, Clamart, France
Introduction:. Estimation of the incidence and prevalence of congenital neutropenia (CN) is poorly documented in nationwide study.
Objective: To calculate the incidence at birth and to estimate the prevalence of congenital neutropenia
Methods: The french chronic neutropenia registry (FCNR) prospectively enrolled patients with chronic neutropenia, with multiple sources of information - all pediatric hemato Immune units in france, - reference Centers - diagnostic lab. To calculate incidence at birth, only cases birth between 1995 and 2005 are taken in consideration, as the completeness has been validated for this period. Genetic abnormalities are currently tested and the result is used to define each disease.
Results: 196 patients with CN has been identified. The incidence at birth in france of CN is 2.4/10-5 (95% CI : 2.04 -2.75) which represent a mean number of 19 new cases /year in a country with about 800 000 birth/year. Expected prevalence, under the assumption of 50 years life expectancy, is 1.5 10-5 inhabitants and represent 971 cases in a country of 65 106 inhabitants, while 524 cases are currently included in our registry. The respective proportion of genetic sub type are SBDS in 24%, ELANE in 17% (cyclic 8% permanent 9%), SLC37A4 in 7%, TAZ in 5%. CXCR4, GATA2, G6PC3, TAZ, VPS13B, 16ORF57,Jagn1 HAX1 are present in less 2% while 35% of the case remains undetermined.
Conclusion: This study offer an estimation of the major descriptive epidemiological parameter in congenital neutropenia and the relative frequency of several congenital neutropenia.
ESID-0419 Analysis of Immunoglobulin Treatment in the ESID Patient Registry. Do European Clinicians Dose SCIG Differently from IVIG?
B. Gathmann 1, G. Kindle for the ESID Registry Working Party2, S. Doerken1, C. Hermann3, N. Nikolov4
1Center for Chronic Immunodeficiency (CCI) Clinical Research Unit, University Medical Centre Freiburg and University of Freiburg, Freiburg, Germany
2The ESID Registry is based on contributions by the following national registries: CEREDIH (France) REDIP (Spain) PID-NET (Germany) UKPIN (UK) IPINET (Italy) AGPI (Austria) the Netherlands Czech Republic. Additional contributions are received from the following countries: Turkey Poland Ireland Portugal Belgium Switzerland Slovakia Sweden Slovenia Croatia Serbia Greece Belarus Russia Hungary Romania Ukraine Estonia Lithuania Egypt Israel. Center for Chronic Immunodeficiency (CCI) Clinical Research Unit, University Medical Centre Freiburg and University of Freiburg, Freiburg, Germany
3Baxter Innovations, Baxter, Vienna, Austria
4Baxter Healthcare SA, Baxter, Zuerich, Switzerland
Background: The US Food and Drug Administration (FDA) recommends dose adjustments of 37%-53% when switching from intravenous (IV) to subcutaneous (SC) immunoglobulin (IG) replacement, while the European Medicines Agency (EMA) recommends 1:1 dosing. It is a matter of debate whether the average dose in adult patients differs in respect to the route of administration and whether patients receive higher doses after switching from IVIG to SCIG.
Methods: Data reported in the ESID Registry between 2004 and 2014 were provided through the PPTA (www.pptaglobal.org) for analysis. 4,026 treatment intervals from 2,886 patients were available for analysis. 1,409 of these had a diagnosis of CVID, followed by agammaglobulinaemia (n=414) and IgG subclass deficiency (n=170). Every patient contributed one single value to each evaluation. If several intervals were available for a patient, the mean dose value was used.
Results: Median monthly doses in mg/kg were similar in SCIG patients: 427 SCIG vs. 437 IVIG (Figure?1). We could not observe a significant dosing difference in the 118 patients, who had switched from IVIG to SCIG (437 vs. 431 mg/kg, p=0.676).
When comparing countries, doses varied significantly. They were highest in the United Kingdom (500 mg/kg), France (449 mg/kg) and the Netherlands (443 mg/kg). In contrast, median doses in Italy and Germany were relatively low (397 and 398 mg/kg) (Figure?2).
Conclusions: The ESID Registry data showed that clinically, SCIG and IVIG doses were equivalent. Switching from IVIG to SCIG in general was not associated with an increase of the dose.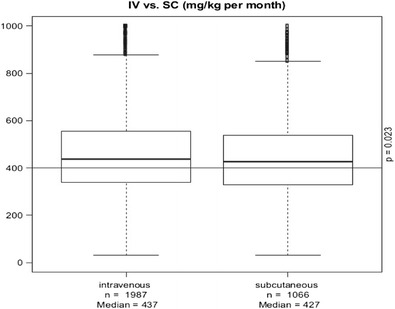
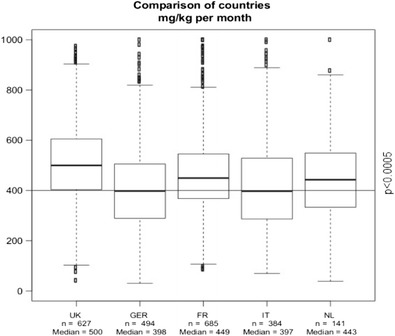
ESID-0418 The ESID Online Registry: A Major Leap Forward
B. Gathmann 1, M. Kanariou2, J.M. van den Berg2, J. Drabwell2, D. Edgar2, A. Farrugia2, G. Kindle2, N. Mahlaoui2, N. Matamoros Flori2, A. Soresina2, V. Thon2, B. Wolska2, W. Vach3, S. Ehl2
1The ESID Registry Steering Committee, The ESID Registry is based on contributions from the following national registries: CEREDIH (France) REDIP (Spain) PID-NET (Germany) UKPIN (UK) IPINET (Italy) AGPI (Austria) the Netherlands Czech Republic. Additional contributions are received from the following countries: Turkey Poland Ireland Portugal Belgium Switzerland Slovakia Sweden Slovenia Croatia Serbia Greece Belarus Russia Hungary Romania Ukraine Estonia Lithuania Egypt Israel., 's-Hertogenbosch, Netherlands
2The ESID Registry Steering Committee, ESID, 's-Hertogenbosch, Netherlands
3Center for Medical Biometry and Medical Informatics, University of Freiburg, Freiburg, Germany
Since 2004, ESID runs a European-wide online registry for PID being the largest Registry worldwide with 19,366 patients (May 2014).
In 2012, the ESID Registry Steering Committee decided that the Registry should be redesigned, in order to improve both data quality and user friendliness, and promote epidemiological, clinical and translational research.
The technical backbone and user interface have been overhauled and allow three levels of registration: Level 1 is mandatory and aims at basic epidemiological features with yearly updates on survival and treatment. A genetics module will be implemented to be filled in by the genetics laboratories for consistent terminology. Level 2 foresees more detailed clinical and laboratory information, while Level 3 provides a platform for disease-specific projects.
Criteria for the clinical diagnosis of PID with unknown genetic cause have been elaborated and have to be confirmed for patients at registration. Built-in checks now validate all fields for consistency and completeness, and users are guided through the data entry process.
Additional datasets are being programmed, data links to the Spanish and Italian registries have to be created, and the software will be 'rolled out' to the UKPID and LASID registries. Importantly, data output for the centres will be extended, including an automatized annual report with benchmarking figures of the centre relative to national and European figures.
We would like to use the ESID 2014 Meeting as an opportunity to present the new Registry to the ESID community and encourage all PID centres to report their patients.
ESID-0342 Pulmonary Manifestations in Adult Patients with Chronic Granulomatous Disease
H. Salvator 1, N. Mahlaoui2, E. Catherinot1, B. Pilmis3, B. Dunogue3, M.?A. Gougerot-Pocidalo4, O. Lortholary3, A. Fischer2, L.J. Couderc1
1Respiratory Diseases Unit, Foch Hospital, Suresnes, France
2CEREDIH (French National Reference Center for PID), Necker-Enfants Malades Hospital, Paris, France
3Infectious Diseases Unit, Necker-Enfants Malades Hospital, Paris, France
4Immunitary dysfunction unit, Bichat Hospital, Paris, France
Rationale
Chronic granulomatous disease (CGD) is characterized by recurrent infections and inflammatory events, frequently affecting the lungs. Thanks to better childhood management, improvement of life expectancy allows most patients to reach adulthood.
Objective
To describe the pattern of pulmonary manifestations occurring during adulthood in CGD patients.
Methods
Retrospective study of the French CEREDIH registry (national reference center for primary immunodeficiencies), focusing on pulmonary complications in adult patients (=16 years old) with CGD.
Results
Medical data were obtained for 67 CGD adult patients: 70% X-linked and 22% autosomale recessive. Median age at last follow-up was 25 years (17-59).
Pulmonary complications affected two-third of adult patients. Their incidence was significantly higher than in childhood (mean annual rate 0.22 vs 0.07, p=0.01).
Infectious risk persisted despite anti-infectious prophylaxis. Invasive fungal infections were frequent (0.11/year/patient) and asymptomatic in 37% of the cases. Their diagnosis often required lung biopsy (10/30).
Non-infectious respiratory events concerned 28% of adult patients and were more frequent in patients with the X-linked form. They were associated with a concomitant fungal infection in 40% of the cases. Radiological pattern was classified as circumscribed nodules/parenchymal consolidation (11/20) or interstitial lung disease (9/?20). Lung biopsies mainly showed granuloma and neutrophilic/eosinophilic microabcesses. Immune-modulator therapies were required in most cases (70%), mainly corticosteroids (10/14), thalidomide (4/?14) and hydroxychloroquine (4/?14).
Conclusion
Respiratory manifestations remain major complications of CGD in adulthood. Specific pulmonary monitoring should be proposed in order to identify new events and to manage their impact in the long term.
ESID-0303 Euroflow-PID Strategy for Identification of Circulating Peripheral Blood B-Lymphocyte Subsets in Primary Immunodeficiencies
M. Perez-Andres 1, M.C. Zelm2, E. Blanco1, E. Lopez-Granados3, A.K. Kienzler4, T. Kalina5, M. Vlkova6, M. Van der Burg2, J.J.M. Van Dongen2, A. Orfao1
1Serv Cytometry-Dep Medicine, CICancer-Univ Salamanca, Salamanca, Spain
2Immunology, Erasmus MC, Rotterdam, Netherlands
3La Paz Univ Hosp-IdiPAZ, Clinical Immunology Unit, Madrid, Spain
4Nuffield Dep Medicine Experimental Medicine Div, Univ. Oxford, Oxford, United Kingdom
5Pediatric Hematology and Oncology, Charles University in Prague, Prague, Czech Republic
6Masaryk University and St Anne`s University Hospital Brno, Institute of Clinical Immunology and Allergology, Brno, Czech Republic
A detailed classification of peripheral blood (PB) B-cells may improve clinical management of primary antibody deficiencies (PAD). Accordingly, combinations were built, tested and optimized in several rounds, leading to a final panel of either two 8-colour tubes or one 11-colour tube. Standardized and sensitive methods for sample preparation and flow cytometry data acquisition allowed systematic analysis of up to 10×106 leukocytes (5×105 B-cells) per tube.
PB naive B-cells showed distinct degrees of maturation with a heterogeneous phenotypic profile from the CD21-/CD24hi/CD38hi/smIgM++/smIgD+ immature/transitional subset to CD21+/CD24low/CD38low/smIgM+/smIgD++ fully mature naive B-cells, with different levels of CD5 along maturation. Anti-Ig isotypic markers and CD27 were included to discriminate IgM++D+, IgG+, IgA+, and IgM-D+ memory B cells (normal adult range: 8-55, 8-35, 8-35 and <0.1-2 cells/uL, respectively), and IgA+, IgM+D-, IgG+, IgM-D+, and smIg- PB plasma cells (0.5-4.6, 0.2-0.8, 0.3-2.0, <0.1-1.6, 0.2-1.0 cells/uL). Also, the distribution of IgG1, IgG2, IgG3, IgA1 and IgA2 within the memory B-cells (21-33, 4-20, 3-10, 8-25, 2-11 cells/uL) and plasma cells (0.3-0.8, 0.2-1.2, 0.1-0.2, 0.4-3.8, 0.4-0.9 cells/uL) was evaluated. Additional subsets of memory B-cells were found based on the CD27/?CD21 pattern of expression, with percentages of CD21+CD27+ cells between 40%-65% of IgG3+ to 80%-85% of IgG2+ B-cells.
Simultaneous usage of these combinations with the new EuroFlow Infinicyt software tools allowed assessment of B-cell maturation and dissection of >30 B-cell subsets, and the identification of distinct B-cell defects and maturation blockades. Further studies in large patient series are ongoing to evaluate the potential contribution to diagnostic classification and monitoring of PAD.
ESID-0133 Auto-Inflammation, Immunodeficiency and Lymphangiectasia in Patient Missing Lubac Component
B. BOISSON 1, E. Laplantine2, K. Dobbs3, N. Tarantino2, M. Hazen3, G. Hopkins3, D. Likun3, M. Chrabieh4, C. Picard4, E. Tsitsikov3, S.Y. Pai3, W. Al-Herz5, J.L. Casanova1, A. Israel2, L. Notarangelo3
1St. Giles Laboratory of Human Genetics of Infectious Diseases, The Rockefeller University, New York, USA
2Laboratory of Molecular Signaling and Cell Activation, Pasteur Institute, Paris, France
3Division of Immunology and The Manton Center for Orphan Disease Research, Harvard Medical School, Boston, USA
4Laboratory of Human Genetics of Infectious Diseases, Imagine Institute, Paris, France
5Department of Pediatrics, Al-Sabah Hospital, Kuwait City, Kuwait
We investigated one patient from consanguineous kindred with early-onset multiorgan inflammation, increased susceptibility to infections, severe lymphopenia, and antibody deficiency. This patient further developed systemic lymphangiectasia. This patient presents some common clinical features related to recently reported HOIL-1 deficient patient without any mutation in this gene
Due to absence of known etiology explaining her clinical phenotype, we set out to identify this genetic disorder with a hypothesis of autosomal recessive transmission model. We used the Genome-Wide Human SNP Array and whole exome sequencing to investigate this family.
Patient was found to carry loss-of-expression and loss-of-function mutations in another ubiquitin ligase belonging to a complex involved in the formation of ubiquitin chains (LUBAC). The disruption of this complex in this patient results in the impairment of the NF-kappaB activation in response to IL-1beta and to a lesser extent TNF-alpha in fibroblast cells. By contrast, the patients` leukocytes are constitutively hyperactivated ex-vivo and display enhanced responses to IL-1beta but not TNF-alpha.
Autosomal recessive deficiency in LUBAC involved in the formation of ubiquitin chains confirms a new class of disorder with unbalanced cellular responses to pro-inflammatory cytokines, characterized by the paradoxical association of auto-inflammation and recurrent infection, and the surprising development of lymphangiectasia.
ESID-0667 PGM3 Mutations Cause a Congenital Disorder of Glycosylation with Severe Combined Immunodeficiency, Congenital Neutropenia and Skeletal Dysplasia
A. Stray-Pedersen 1, H.S. Sorte2, P.H. Backe3, L. Mørkrid3, N.Y. Chokshi4, H.C. Erichsen5, T.G. Abrahamsen5, A. Rønnestad5, L.R. Forbes4, T. Gambin1, S. Jhangiani1, D.?M. Muzny6, K.B.P. Elgstøen7, M. Bjørås8, C. Speckmann9, S. Ehl9, A. Patel10, K. Raymond11, P. Hall12, C. Martinez13, E. Merckoll14, J. Westvik14, G. Nishimura15, O.K. Rødningen2, C.F. Rustad2, R. Gibbs6, J.R. Lupski1, J.S. Orange4, E. Lausch9, I.C. Hanson4
1Baylor-Hopkins Center for Mendelian Genomics of the Department of Molecular and Human Genetics, Baylor College of Medicine, Houston Texas, USA
2Department of Medical Genetics, Oslo University Hospital, Oslo, Norway
3Department of Medical Biochemistry, Oslo University Hospital, Oslo, Norway
4Department of Pediatrics ?Section?of Immunology Allergy and Rheumatology, Baylor College of Medicine and Texas Children's Hospital, Houston Texas, USA
5Department of Pediatrics, Oslo University Hospital, Oslo, Norway
6Human Genome Sequencing Center, Baylor College of Medicine, Houston Texas, USA
7Department of Medical Biochemistry, Oslo University Hospital, Oslo, USA
8Department of Microbiology, Institute of Clinical Medicine University of Oslo, Oslo, USA
9Department of Pediatrics, Freiburg University Hospital, Freiburg, Germany
10Medical Genetics Laboratories Molecular and Human Genetics, Baylor College of Medicine, Houston Texas, USA
11Department of Laboratory Medicine and Pathology, Mayo College of Medicine, Rochester Minnesota, USA
12Emory Genetics Laboratory Department of Human Genetics, Emory University, Decatur Georgia, USA
13Texas Children’s Cancer and Hematology Center Department of Pediatrics Center for Cell and Gene Therapy, Baylor College of Medicine, Houston Texas, USA
14Department of Radiology, Oslo University Hospital, Oslo, Norway
15Department of Pediatric Imaging, Tokyo Metropolitan Children's Medical Center, Tokyo, Japan
Human phosphoglucomutase 3 (PGM3) catalyzes the conversion of GlcNAc-6-P into GlcNAc-1-P during the synthesis of UDP-GlcNAc, a sugar nucleotide critical to multiple glycosylation pathways. We identified three unrelated children with recurrent infections, congenital leukopenia including neutropenia, B and T cell lymphopenia, and progression to bone marrow failure. Whole exome sequencing demonstrated deleterious mutations in PGM3 in all three subjects, delineating their disease to be due to an unsuspected congenital disorder of glycosylation (CDG). Functional studies of the disease associated PGM3 variants in E.?coli cells demonstrated reduced PGM3 enzyme activity for all mutants tested. Two of the 3 children had skeletal anomalies resembling Desbuquois dysplasia with short stature, brachydactyly, dysmorphic facial features, and intellectual disability, however these additional features were absent in the third child, showing the clinical variability of the disease. Two children received hematopoietic stem cell transplantation with cord blood and matched-related donor bone marrow; both had successful engraftment and correction of neutropenia and lymphopenia. We define PGM3-CDG as a treatable immunodeficiency, document the power of whole exome sequencing in gene discoveries for rare disorders, and illustrate the utility of genomic analyses in studying combined and variable phenotypes.
ESID-0539 Hypomorphic Homozygous Mutations in Phosphoglucomutase3 Impair Immunity and Increase Serum IGE Levels
A. Sassi1, M. Fliegauf2, L. Yang 2, G. Wu3, S.M. Haslam3, F. Mellouli4, T. Patiroglu5, E. Unal5, M.?A. Ozdemir5, Z. Jouhadi6, K. Khadir6, L. Ben-Khemis1, M. Ben-Ali1, I. Ben-Mustapha1, L. Borchani7, M. Khemiri8, A. Maul-Pavicic2, M. Rakhmanov2, P. Henneke2, H. Kraus2, H. Eibel2, U. Kölsch9, S. Nadifi10, M. Nilsson11, M. Bejaoui4, A.A. Schäffer12, C.I.E. Smith13, A. Dell3, M.R. Barbouche1, B. Grimbacher2
1Vaccinology and Molecular Genetics, Pasteur Institute of Tunis and University Tunis EI Manar, Tunis, Tunisia
2Center for Chronic Immunodeficiency (CCI), University Medical Center Freiburg, Freiburg, Germany
3Department of Life Sciences, Imperial College London, London, United Kingdom
4Pediatrics Department, Bone Marrow Transplantation Center, Tunis, Tunisia
5Division of Pediatric Hematology and Oncology, Faculty of Medicine of Erciyes University, Kayseri, Turkey
6Department of Pediatric Infectious Diseases, CHU IBN ROCHD of Hassan II University, Casablanca, Morocco
7Laboratory of Venoms and therapeutic Molecules, Institut Pasteur de Tunis, Tunis, Tunisia
8Pediatrics Department A, Children’s Hospital of Tunis, Tunis, Tunisia
9Labor Berlin and Institute of Medical Immunology, Charité and Campus Virchow Klinikum, Berlin, Germany
10Department of Genetics, Hassan II University, Casablanca, Morocco
11Department of Immunology, Uppsala University Rudbeck Laboratory, Uppsala, Sweden
12National Center for Biotechnology Information, National Institutes of Health, Bethesda, USA
13Clinical Research Center and Department 34 of Laboratory Medicine, Karolinska University Hospital Huddinge, Huddinge, Sweden
Recurrent bacterial and fungal infections, eczema and elevated serum IgE levels characterize patients with the hyper-IgE syndrome (HIES). Known genetic causes are mutations in Tyk2, STAT3 and DOCK8, all involved in lymphocyte signal transduction.
In HIES patients who do not carry mutations in Tyk2, STAT3 or DOCK8, we have recently identified three disease-causing homozygous mutations in PGM3 which encodes phosphoglucomutase-3. PGM3 is a crucial enzyme in the glycosylation pathway and catalyzes a key step in the synthesis of uridine diphosphate N-acetylglucosamine, which is the substrate required for the biosynthesis of N-glycans. The mutations cause amino acid changes in PGM3 (p.Leu83Ser; p.Glu340del; p.Asp502Tyr) which ultimately lead to reduced levels of tri-antennary and tetra-antennary N-glycans in leukocytes from affected individuals. In addition, T-cell proliferation and differentiation were impaired,and most patients had developmental delay and/or psychomotor retardation.Thus, hypomorphic PGM3 mutations characterize a novel type of HIES, associated with defective glycosylation albeit the exact pathogenic mechanism remained obscure.
To investigate the disease mechanism, we generated a genetic rescue model by stably expressing a non-mutant form of PGM3 in EBV-transformed B-lymphocytes from patients and controls using retroviral IRES-EGFP vectors. In this approach, employing lectin binding assays, our results indicate that the glycosylation defect could be at least partially reversed. With detailed glycomic analyses we will identify glycosylation-defective proteins including CD23, CD21, gp130, IgE and IgG which might be involved in the disease progression.
ESID-0189 Systematic Neonatal Screening for Severe Combined Immunodeficiency and Severe T-Cell Lymphopenia: Analysis of Cost-Effectiveness Based on French Real Field Data
N. MAHLAOUI1, M. CLEMENT2, C. MIGNOT1, C. LE BIHAN3, H. RABETRANO2, L.Y. HOANG2, A. FISCHER1, M. AUDRAIN4, I. DURAND-ZALESKI 2
1CEREDIH (French National Reference Center for PID), Necker-Enfants Malades Hospital, Paris, France
2URC Eco (Clinical Research Unit in Health Economics), Assistance Publique-Hôpitaux de Paris Hôtel Dieu Hospital, Paris, France
3Medical information unit, Necker-Enfants Malades Hospital, Paris, France
4Immunology Laboratory, Nantes University Hospital, Nantes, France
Background and Objective: In preparation for making the case for inclusion of severe combined immunodeficiency (SCID) in a European-wide newborn screening (NBS) program, we explored the costs incurred and potentially saved by early management of SCID.
Methods:
Test costs: A micro-costing study prospectively documented the resources used in a laboratory piloting a NBS test on Guthrie cards using the T-cell Receptor Excision Circles (TRECs) quantification method.
Treatment costs: SCID patients admitted at the national reference center for primary immunodeficiency in France between 2006 and 2010 were included. Costs of admissions were estimated from actual national production costs. We estimated the costs for patients who underwent an early versus delayed hematopoietic stem cell transplantation (HSCT) (=age 3 vs. > age 3 months, respectively).
Results: Unit cost of the test varied between €4.69 and €6.79 for 33,800 samples per year depending on use and saturation of the equipment. Of the 30 included patients, 27 underwent HSCT after age three months. At one year post-HSCT, 10 of these 27 patients had died and all three early-transplanted patients survived. The medical costs for HSCT after age 3 months were €195,776 (IQR=€165,884-€257,160) versus €86,179 (range: €59,014-€272,577) when performed before age 3 months.
Conclusion: Early detection of SCID would reduce the cost of treatment by €100,000 per case. Assuming a €5 unit cost per test, the incidence required to break even is 1:20,000; however if the survival advantage of HSCT before 3 months is confirmed, universal screening is likely to be cost-effective.
ESID-0809 Human SHP-1 Mutation Causes an Autoinflammatory and Immunodeficiency Phenotype
V. Bigley 1, R. Dickinson1, L. Jardine1, P.P.,S.(. Milne1, H. Griffin2, M. Santibanez-Koref2, M. Haniffa1, A. Cant3, S. Hambleton3, A. Gennery3, M. Collin1
1Human Dendritic Cell Lab, Institute of Cellular Medicine Newcastle University
2Institute of Genetic Medicine Newcastle University
3Department of Paediatric Immunology and Infectious Disease, Newcastle upon Tyne Hospitals NHS Foundation Trust
Primary immunodeficiency offers a unique and powerful means of dissecting the development and functionality of human immune cells ?in?vivo. The biological consequences and genetic basis of immunodeficiencies due to loss or dysfunction of lymphocytes and granulocytes are well described, but the antigen presenting arm of the immune system has been under studied. Our recent work identifying IRF8 and GATA2 mutations, resulting in loss of monocytes and dendritic cells, have demonstrated the importance of antigen presentation in maintaining a regulated immune system
We have recently identified a more specific disorder of monocyte homeostasis; CD16+ (non-classical) monocyte deficiency, in a consanguineous family with two affected offspring. Clinical features presenting from early childhood included an immunodeficient picture with recurrent pneumocystis Jirovecii Pneumonia and candidiasis, autoinflammatory features including pneumonitis, and a consumptive anaemia requiring splenectomy. Pathological features included macrophage accumulation in alveolar spaces, iron-laden macrophages in liver, extramedullary haematopoiesis and a lack of splenic B cell follicles, low circulating B cell numbers. Whole exome sequencing identified a protein destabilizing, autosomal recessive mutation in Src Homology region 2 domain containing Phosphatase 1 (SHP-1/PTPN6). In vitro studies reveal that SHP-1 R492W results in an unstable protein demonstrated by the presence of mRNA but absence of protein in leukocytes. Expressed in the haematopoietic system, this phosphatase is involved in negative regulation of tyrosine kinase pathways. Clinical and pathological features resemble those of the homozygous Ptpn6me ‘motheaten’ mouse’ with autoimmune and inflammatory dysregulation leading to a complex disorder of both monocyte and lymphoid homeostasis and function
ESID-0538 The Clinical Manifestations, Management and Outcomes of Activated PI3K-Delta Syndrome (APDS) in UK and Ireland Cohort
T.I. Coulter 1, A. Chandra2, T.R. Leahy3, J. Curtis4, C.M. Bacon5, J. Babar6, G. Farmer7, S. Savic8, S.O. Burns9, A.?M. Jones10, P.J. Gavin3, C.F. Feighery1, P. Forsyth11, R. Herriot12, J.?D. Edgar13, M. Abinun14, G. Markelj15, M. Ibrahim16, H. Baxendale2, R. Doffinger2, D. Kumararatne2, A.?M. Condliffe4, A.J. Cant14, S. Nejentsev4
1Department of Immunology, School of Medicine Trinity College Dublin, Dublin, Ireland
2Department of Clinical Biochemistry and Immunology, Addenbrooke’s Hospital, Cambridge, United Kingdom
3Department of Paediatric Infectious Diseases and Immunology, Our Lady's Children's Hospital Crumlin, Dublin, Ireland
4Department of Medicine, University of Cambridge, Cambridge, United Kingdom
5Northern Institute for Cancer Research, University of Newcastle and Department of Cellular Pathology Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom
6Department of Radiology, Cambridge University Hospitals NHS Foundation Trust, Cambridge, United Kingdom
7Department of Paediatrics, Raigmore Hospital, Inverness, United Kingdom
8Department of Clinical Immunology and Allergy, St James’s University Hospital, Leeds, United Kingdom
9Institute of Immunity and Transplantation University College London, Department of Immunology Royal Free London NHS Foundation Trust and Great Ormond Street Hospital for Children NHS Foundation Trust., London, United Kingdom
10Department of Paediatric Immunology, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
11Department of Haematology, Raigmore Hospital, Inverness, United Kingdom
12Department of Immunology, NHS Grampian, Aberdeen, United Kingdom
13Regional Immunology Service, The Royal Hospitals Queen's University, Belfast, United Kingdom
14Primary Immunodeficiency Group, Institute of Cellular Medicine Newcastle University, Newcastle upon Tyne, United Kingdom
15Department of Allergology Rheumatology and Clinical Immunology, University Children's Hospital University Medical Center, Ljubljana, Slovenia
16Department of Immunological Medicine, School of Medicine King’s College Hospital NHS Foundation Trust, London, United Kingdom
Activated PI3K-d syndrome (APDS) is a recently described autosomal dominant Primary Immune Deficiency due to gain-of-function mutation in catalytic subunit of phosphoinositide 3-kinase d encoded by PIK3CD gene.
We describe 22 individuals (9 families) attending immunology services in UK and Ireland with genetically defined APDS due to E1021K mutation in PIK3CD.
All patients suffered from recurrent respiratory tract infections since early childhood, with 73% developing early-onset bronchiectasis. Significant herpes, cryptosporidium and BCG-vaccine related infections were also encountered.
Lymphoproliferative manifestations, persistent lymphadenopathy, hepatosplenomegaly and nodular lymphoid hyperplasia, often occurred concurrently and complicated 73% of cases. 27% of patients had a paediatric-onset autoimmune or inflammatory condition and two patients developed lymphoma. Lymphoma, autoimmunity and inflammatory disease were only found in individuals with a history of lymphoproliferative manifestations.
95% of cohort had an antibody deficiency including 8 cases of hypogammaglobulinaemia. 73% had elevated IgM levels. Of note often the clinical complications were more severe than expected for the degree of antibody deficiency demonstrated.
Most patients required immunoglobulin replacement therapy (90%), with 2 cases ultimately managed by Haematopoietic Stem Cell Transplant.
Two deaths occurred within the cohort (from lymphoma at 19yo, respiratory failure at 39yo). Family histories revealed numerous early deaths from chronic respiratory disease in relatives .
Our APDS cohort had a significant burden of bronchiectasis, lymphoproliferation and immune-mediated disease (91%). While most patients demonstrated primary antibody defects worrying many developed infections and complications often associated with combined immune deficiency. The clinical complications and incidence of early death in APDS supports aggressive management.
ESID-0211 A Human Immunodeficiency Caused by Mutations in the Phosphoinositide 3-Kinase (PI3K) Regulatory Subunit P85 Alpha Gene PIK3R1 and the Subsequent Hyperactivation of PI3K Signaling
M. Deau1, L. Heurtier1, P. Frange2, F. Suarez3, M. Cavazzana1, C. Picard4, A. Durandy1, A. Fischer2, S. Kracker 1
1Human Lymphohematopoiesis Laboratory, Imagine, PARIS, France
2Unité d’Immunologie et Hématologie Pédiatrique, Hôpital Necker Enfants-Malades, PARIS, France
3Service d'Hématologie Adultes, Hôpital Necker Enfants-Malades, PARIS, France
4Center for Primary Immunodeficiencies, Hôpital Necker Enfants-Malades, PARIS, France
Primary immunodeficiencies can be caused by hyperactivation of phosphatidylinositol-3-OH kinase (PI3K) signaling pathway as gain-of-function mutations in PIK3CD encoding for the p110d subunit results in activated PI3K-d syndrome (APDS). Class IA PI3K molecules are composed of a p110 catalytic subunit (p110a, p110ß or p110d) and a regulatory subunit (p85a, p55a, p50a, p85ß or p55?) that regulates p110's stability, cellular localization and function. Using whole-exome sequencing we identified two heterozygous splice-site mutations in the PIK3R1 gene coding for the p85a subunit of phosphatidylinositol-3-OH kinase (PI3K) in four patients (from three unrelated families) diagnosed with hypogammaglobulinemia. Both mutations resulted in deletion of exon 10 and thus a shortened p85a protein lacking part of the PI3K p110-binding domain. The hypothetical loss of p85a-mediated inhibition of p110 activity was supported by the observation of increased phosphorylation of the kinase Akt (a known downstream signaling molecule for PI3K) in T cell blasts. T cell blasts displayed enhanced activation-induced cell death, which could be inhibited by addition of the PI3Kd inhibitor IC87114 and low naïve T and memory B cell counts were observed in patients’ peripheral blood. Therefore, patients carrying the PIK3R1 splice-site mutations presented a phenotype reminding that of APDS patients carrying gain-of-function mutations in PIK3CD. Our results suggest that various defects in the PI3K-triggered pathway can cause primary immunodeficiencies.
ESID-0685 A Short-Term Treatment Summary Using M-TOR Inhibitors in Patients with Germline PIK3CD Mutations: Prospects for Future
G. Uzel1, J. Stoddard1, B. Martinez1, A. Agharahimi1, A.A. Hussey1, L.R. Folio2, S. Pittaluga3, T.A. Fleisher1, S.G. Tangye 4, S.M. Holland1
1NIAID, National Institutes of Health, Bethesda, USA
2CC, National Institutes of Health, Bethesda, USA
3NCI, National Institutes of Health, Bethesda, USA
4Immunology Department, Garvan Institute of Medical Research, Sydney, Australia
We have recently reported germ line dominant gain of function mutations in PIK3CD, encoding for the p110d catalytic subunit of phosphoinositide 3-kinase (PI3K1 p110d) and leading to immunodeficiency and immune dysregulation. Spectrum of findings including lymphoproliferation, and obstructive lymphadenopathy, increased incidence of lymphomas (often EBV-associated), EBV and CMV viremia, autoimmune cytopenias, recurrent sinopulmonary infections, bronchiectasis and chronic lung disease prompted us to search for treatment intervention.
Given the constituent activation of PI3K-AKT-MTOR pathway, we aimed at pharmacological inhibition of MTOR when clinically indicated. Sirolimus or everolimus, specific inhibitors of mTORC-1, were used to treat 7 patients at a dose range of 2.9 - 3.4 mg/m2 for sirolimus or 1 mg/m2 everolimus and aiming for a serum trough level of 9-15 ng/ml, for a duration of 2-40 months. Laboratory, radiologic and clinical data were analyzed to assess treatment response.
We were able to control lymphadenopathy and lymphoproliferation as documented by CT scan or PET imaging, controlled cytopenias, while resolving the incidence of obstructive symptoms such as endobronchial infections and obstructive sleep apnea. Immunologic parameters that were followed and favorably improved included increased percentage of naïve CD4+ T cells, decreased CD10+ and increased CD27+ B cell percentages, decreased percentage of PD1 and Ki67 positive lymphocytes. We were also able to detect a readily diminished baseline S6 phosphorylation in circulating B cells.
Results of this short term and open labeled treatment approach that was initiated due to clinical indications and emerging needs gives us future prospects into pharmacologically manipulating this enzyme.
ESID-0410 IL12 Signaling Deficiency is a Novel Cause of the Autoimmune Lymphoproliferative Syndrome
S. Nabhani1, S. Ginzel1, M. Hagit2, S. Revel-Vilk3, D. Harlev2, B. Fleckenstein4, R. Thiele5, H.J. Laws1, A. Borkhardt1, P. Stepensky3, U. Fischer 1
1Department of Pediatric Oncology Hematology and Clinical Immunology, Heinrich Heine University Medical Faculty, Düsseldorf, Germany
2Pediatric Hematology Unit, Shaare Zedek Medical Center, Jerusalem, Israel
3Hadassah Hebrew University Medical Center, Shaare Zedek Medical Center, Jerusalem, Israel
4Department of Clinical and Molecular Virology, Friedrich-Alexander-University Erlangen-Nürnberg, Erlangen, Germany
5Department of Computer Science, Bonn-Rhein-Sieg University of Applied Sciences, Sankt Augustin, Germany
The autoimmune lymphoproliferative syndrome (ALPS) is typically caused by mutations in the Fas death receptor pathway (FAS, FASLG or CASP10 genes), but for 20-30% of cases the genetic defect is unknown. Whole-exome sequencing identified a homozygous R212* mutation in the IL12/?IL23 receptor-component IL12RB1 in a patient who presented with classical ALPS symptoms (chronic non-malignant, noninfectious lymphadenopathy, splenomegaly, hepatomegaly, elevation of DNT cells, autoimmune cytopenias with polyclonal hypergammaglobulinemia, persistently increased vitamin B12 and IL10 levels) for more than 16 years without known ALPS mutations. Fas receptor mediated apoptosis was functional in?vitro upon stimulation with recombinant Fas ligand (FasL). The mutation led to IL12RB1 protein truncation and loss of cell surface expression. IL12 signaling was abrogated as demonstrated by deficient downstream STAT4 phosphorylation and IFN? production. Low FasL expression on T-cells and low soluble FasL plasma levels were probably due to lack of IFN? mediated transcriptional activation of FasL. Upregulation of FasL mRNA and protein by T-cells in response to prolonged IL12 stimulation was deficient. In contrast to healthy controls, prolonged IL12 stimulation did not trigger apoptosis in the IL12RB1 deficient as well as two FasL deficient patients. Heterozygous carriers of the IL12RB1 or the FasL mutation showed an intermediate response. Whereas heterozygous FasL mutation carriers were asymptomatic, heterozygous IL12RB1 mutation carriers were sub-clinically affected (showing e.?g. moderate elevation of DNT cells). Our data demonstrate that IL12 employs Fas signaling to achieve T-cell apoptosis and reveal IL12 signaling deficiency as a new cause of ALPS via its impact on FasL expression.
ESID-0411 Abnormally Differentiated CD4+ OR CD8+ T-Cells with Phenotypic and Genetic Features of Double Negative T-Cells in Human FAS Deficiency
A. Rensing-Ehl 1, S. Völkl2, C. Speckmann3, M.R. Lorenz4, J. Ritter5, A. Janda6, M. Abinun7, H. Pircher8, B. Bengsch9, R. Thimme9, I. Fuchs1, S. Ammann1, A. Allgäuer2, K. Kentouche10, A. Cant7, S. Hambleton7, C. Bettoni da Cunha11, S. Huetker12, I. Kühnle12, A. Pekrun13, M.G. Seidel14, M. Hummel5, A. Mackensen2, K. Schwarz4, S. Ehl1
1Center for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany
2Department of Internal Medicine 5, University Hospital Erlangen, Erlangen, Germany
3Center for Paediatrics and Adolescent Medicine, University Medical Center Freiburg, Freiburg, Germany
4Institute for Transfusion Medicine, University of Ulm, Ulm, Germany
5Institute of Pathology, Charité – University Medicine Berlin, Berlin, Germany
6Center for Paediatrics and Adolescent Medicine, University Medical Center and University of Freiburg, Freiburg, Germany
7Department of Paediatric Immunology, Great North Children`s Hospital, Newcastle upon Tyne, United Kingdom
8Institute of Immunology, University Medical Center Freiburg, Freiburg, Germany
9Department of Internal Medicine II, University Medical Center Freiburg, Freiburg, Germany
10Department of Paediatrics, Jena University Hospital, Jena, Germany
11Department of Pediatric Hematology and Oncology, Hannover Medical School, Hannover, Germany
12Department of Pediatric and Adolescent Medicine, University Medical Center Göttingen, Göttingen, Germany
13Department of Paediatric Haematology/Oncology, University Medical Center Bremen, Bremen, Germany
14Paediatric Haematology-Oncology, Medical University of Graz, Graz, Austria
Accumulation of CD3+ TCRab+CD4-CD8- double negative T (DNT) cells is a hallmark of autoimmune lymphoproliferative syndrome (ALPS). DNT origin and differentiation pathways remain controversial. Here we show that human ALPS DNT have features of terminally differentiated effector memory CD45RA+ (TEMRA) cells, but are CD27+CD28+KLRG1- and do not express the transcription factor T-bet. This unique phenotype was also detected among CD4+ or CD8+ ALPS TEMRA cells. TCRb deep sequencing revealed a significant fraction of shared CDR3 sequences between ALPS DNT and both CD4+ and CD8+TEMRA cells. Moreover, in ALPS patients with a germline FAS mutation and somatic loss of heterozygosity, in whom biallelic mutant cells can be tracked by absent Fas expression, Fas negative T-cells accumulated not only among DNT, but also among CD4+ and CD8+TEMRA cells. These data indicate that in human Fas deficiency DNT can not only derive from CD8+, but also from CD4+ T-cells. Furthermore, defective Fas signalling leads to aberrant transcriptional programs and differentiation of subsets of CD4+ and CD8+ T-cells. Accumulation of these cells before their double negative state appears to be an important early event in the pathogenesis of lymphoproliferation in ALPS patients.
ESID-0688 Next Generation Sequencing Discovers Novel Genes Causative for CVID and Lymphoma in CVID
P. Lugar 1, S. Dave1
1Medicine, Duke University Medical Center, Durham, USA
Objective: CVID is the most common primary immune deficiency characterized by clinically diverse phenotypes of immune dysregulation and susceptibility to infections. The genetic causes of the disease remain largely unknown. We sought to define the gene-coding mutations underlying CVID and the genetic susceptibility to lymphoma.
Methods: We performed exome-sequencing on 218 well-characterized CVID patients using a solution-based method (Agilent) to generate over 5 billion sequencing reads on the Illumina platform. We compared these to 6258 additional exomes from control patients without CVID.
Results: We identified missense, nonsense and frameshift mutations in 16 candidate genes in CVID, but not in control subjects. In particular, we identified novel mutations in NOTCH1, that was recurrently mutated in CVID. Nearly 50% of the patients with NOTCH1 mutations developed lymphoma. Separately, we sequenced over 212 cases of B cell lymphomas including diffuse large B cell lymphoma, Burkitt lymphoma and Mantle cell lymphoma. We found overlapping patterns of mutations among CVID patients with lymphoma and these B cell lymphomas, suggesting a shared inherited susceptibility to both immune deficiency and lymphoma in these patients. Many of the other genes we identified have not previously been implicated in immune deficiency.
Conclusions: Our study identifies a number of novel candidate gene mutations in CVID and the genetic susceptibility to lymphoma.
ESID-0619 Exploring T-Cell Disturbances in Common Variable Immunodeficiency Disorders
A. Serra-Caetano 1, R.R. Barbosa1, S.P. Silva2, M.P. Barbosa3, A.E. Sousa1, S.L. Silva2
1Centro de Imunodeficiências Primárias, Instituto de Medicina Molecular. Faculdade de Medicina / Universidade de Lisboa, Lisboa, Portugal
2Centro de Imunodeficiências Primárias. Serviço de Imunoalergologia. Hospital de Santa Maria / Centro Hospitalar Lisboa Norte, Instituto de Medicina Molecular. Faculdade de Medicina / Universidade de Lisboa, Lisboa, Portugal
3Serviço de Imunoalergologia, Hospital de Santa Maria / Centro Hospitalar Lisboa Norte, Lisboa, Portugal
T-cell imbalances are frequently observed in Common Variable Immunodeficiency Disorders (CVID), despite the archetypal definition being defective antibody production. Moreover, T-cell disturbances usually persist under immunoglobulin replacement therapy. Autoimmune manifestations are frequent, which may be related to regulatory T-cell (Treg) alterations.
We investigated the Treg compartment in 31 CVID patients and 30 healthy controls (HC). CVID patients featured significantly lower CD25 levels despite preserved frequency of Foxp3+ cells within CD4, pointing to dissociated expression of these Treg markers. Accordingly, we observed an expansion of the Foxp3+ CD25neg population in direct correlation with T-cell activation in CVID.
Since ?c-cytokines up-regulate CD25 expression, we hypothesized that an impaired ability to respond to these major homeostatic cytokines may underlie reduced CD25 expression. We selected a group of CVID patients with low CD25 expression and evaluated STAT5 phosphorylation (pSTAT5) levels, a downstream signaling event, both ex-vivo and upon whole-blood short-term stimulation with optimal IL-7, IL-2 or IL-15 doses. We observed no major alterations in ex-vivo pSTAT5 levels. However, Treg from CVID patients showed significantly lower responses to IL-2 than to IL-15, in contrast to HC pattern. Regarding other memory CD4 subsets, the circulating CXCR5+ (Folicular T-helper, Tfh) population was significantly expanded in CVID, and showed marked reduction in CD25 expression. Notably, Tfh response to IL-7 was significantly impaired, and pSTAT5 comparable to HC levels were only reached upon IL-15 stimulation.
Overall, we document distinct impairments in response to IL-7, IL-2 and IL-15 that may be instrumental to understand T-cell disturbances in CVID.
ESID-0551 Long-Term Safety of Infliximab in CVID-Related Inflammatory Bowel Disease
N. Verma 1, A.?D. Webster1, S.L. Seneviratne1, S.O. Burns1, M. Dziadio1, A. Symes1, S. Workman1, B. Grimbacher1, R. Chee1
1Immunology, Royal Free Hospital, London, United Kingdom
Introduction
Common Variable Immunodeficiency (CVID) associated bowel disease can present with chronic diarrhoea with histological features mimicking Inflammatory Bowel Disease (IBD). Infliximab, an anti-TNFa monoclonal antibody has been widely used for short term treatment of IBD.
Method
We report 4 CVID patients with chronic diarrhoea whose symptoms were effectively controlled with the long-term use of Infliximab without significant side-effects.
Results
Three patients (2 females aged 61 and 67 yrs, and one 44?yr old male), all with >10 year history of CVID, were commenced on Infliximab over 10 years ago. Another female patient (aged 63 yrs) started Infliximab one year ago. Pre-treatment biopsy showed inflammatory changes in the large bowel, and in three cases also in the small bowel consistent with CVID-related IBD, with absent mucosal plasma cells. Three of the patients had evidence of cytomegalovirus reactivation in the bowel when using a sensitive immuno-staining technique to identify CMV antigens; two of these patients were given short-term treatment with valganciclovir with marked improvement in diarrhoea. Subsequent regular Infliximab therapy led to a marked long-term improvement in stool frequency. None of the patients have developed potential complications of Infliximab, in particular mycobacterial infection, malignancy or autoimmune disorders.
Conclusion
Infliximab, when used in a similar manner to that recommended in the UK national protocol for IBD, has been effective in controlling CVID-related IBD. It appears to be a safe long-term therapy in CVID without the side effects seen with more traditional steroid therapy.
ESID-0081 TCR Alpha/Beta Depletion for Stem Cell Transplantation in Primary Immunodeficiency Diseases: Results and Controversy
D. Balashov 1, M. Maschan1, P. Trakhtman1, A. Shcherbina2, Y. Skvortsova1, L. Shelikhova1, A. Livshits1, E. Boyakova1, V. Tsetlina1, J. Muzalevsky1, E. Gutovskaya1, E. Kurnikova1, G. Novichkova1, A. Maschan1
1Bone Marrow Transplantation, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
2Clinical Immunology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
Aim of our study was to examine the effect TCRaß-graft depletion for HSCT from URD or haploidentical donors in patients with PID.
25 patients (m:f = 20 : 5; median age 3,7 years [0,2-17,0]) with different PIDs [CGD (n=2), HIgM (n=1), WAS (n=6), HLH (n=4), SCID (n=4), WHIM (n=1), NBS ( n=2), ?-LP ( n=1), SCN (n=2), STAT1 (n=1) and unidentified PID (n=1)] were transplanted with TCRaß/CD19-depletion of the grafts. Stem cell source was peripheral blood from URD or haploidentical donors.
23 patients received Treosulfan-based RIC (TREO-FLU) with top-up of Alemtuzumab or ATG. Additional 2 patients with NBS received conditioning with low doses of BU, CY, FLU and ATG. GVHD prophylaxis included Tacrolimus /Tacrolimus + MTX.
The median number of TCRaß+ cells in the graft was 10,7 × 103/kg (0,8 × 103/kg – 12,5 × 104/kg).
Engraftment occurred in 23 patients, except STAT1 [n=1] and SCN [n=1]. There were 5 graft rejections. All non-engrafted/rejected patients were retransplanted.
Acute GVHD grade I-II occurred in 40 % of patients; we had no patients with GVHD grade III-IV or chronic GVHD.
Two patients died due to infectious complications after second HSCT and 1 patient (NBS) relapsed with lymphoma.
TCR aß/CD19 depletion –alternative technology for clinical application in haploidentical or unrelated transplantation. Application of this approach dramatically reduced TRM and risk of severe GVHD, but the rate of graft rejection was high with the use of RIC. To overcome this obstacle, we plan to enhance condition regimen with CY and Melphalan in high risk of graft rejection.
ESID-0018 Immunological and Metabolic Correction After Lentiviral Vector Mediated Haematopoietic Stem Cell Gene Therapy for ADA Deficiency
B. Gaspar 1, K. Buckland1, C. Rivat1, C. Booth1, K. Gilmour1, K. Cornetta2, D. Carbonaro3, M. Schmidt4, D.B. Kohn3, A.J. Thrasher1
1Institute of Child Health, UCL, London, United Kingdom
2IUVPF, Indiana University, Indianapolis, USA
3Department of Microbiology Immunology and Molecular Genetics, UCLA, Los Angeles, USA
4National Center for Tumor Diseases and German Cancer Research Center, Heidelberg University, Heidelberg, Germany
Adenosine deaminase deficiency leads to severe combined immunodeficiency. We developed a lentiviral vector in which a codon optimised version of the human ADA gene is driven by an internal EFS promoter and undertook a phase I/?II trial. Inclusion criteria allowed enrolment of patients even if a fully matched unrelated donor was available.
5 patients aged between 1.2-4.5 yrs were treated. Busulfan i.v. at a single dose of ~5 mg/kg was given as conditioning. A mean of 5 x 10e6 CD34+ cells/kg were returned with a mean vector copy number (VCN) in the transduced population of 4.2 (range: 2.4-6.1).
The procedure was well tolerated by all patients. At a mean follow up of 361 days, there has been significant immunological recovery. All patients have shown a rise in total T cell counts from a mean of 204 cells/ul pre gene therapy to a mean of 940 cells/ul. CD4+ cells have risen from a mean of 94 cells/ul pre gene therapy to a mean of 416 cells/ul (range: 200-690). Mitogen responses to PHA have normalised in all patients and there is naive T cell recovery. Gene marking is detectable in the periphery with a mean VCN of 0.41 in PBMCs. Integration site analysis shows some expansions but no persistence of expanded clones. All patients are clinically well and have been able to stop some prophylactic antibiotics.
Lentiviral vector mediated gene therapy for ADA deficiency is well tolerated and allows effective recovery of immunological and metabolic parameters with no significant adverse events thus far
ESID-0134 Single Centre Retrospective Analysis of 33 Patients with X-SCID Transplanted with Parental HLA- Haploidentical Grafts
M. Hoenig 1, D. Kleila1, C. Schuetz1, I. Furlan1, M. Wiesneth2, K.M. Debatin1, W. Friedrich1, A. Schulz1
1Pediatrics, University Medical Center, Ulm, Germany
2Institute for Clinical Transfusion Medicine and Immunogenetics Ulm, German Red Cross Blood Transfusion Service Baden Württemberg-Hessen and Institute of Transfusion Medicine, Ulm, Germany
X-linked Severe Combined Immunodeficiency (X-SCID) is caused by genetic defects in the common gamma chain (IL2RG) of the receptors for IL-2,-4,-7,-9,-15 and IL-21. We retrospectively analysed data from 33 patients with IL2RG-deficiency subsequently transplanted with T-cell depleted HLA-haploidentical parental grafts in our institution between 1982 and 2005. Overall survival (OS) was 82% (27/33) with a median follow up of 10.8 years (1.4 - 24.6). 19 pts were transplanted with and 14 pts without conditioning. OS was 84% and 79% respectively. Four pts died due to infectious complications (CMV, BCG, EBV), in two pts with respiratory failure no infectious agent was identified; no pt died due to GvHD. T-cell engraftment was found in all pts except one who failed to engraft paternal cells without conditioning but was successfully retransplanted with a maternal graft. Successful retransplantations with conditioning were performed in two pts 6 and 12 years after the first attempt without conditioning because of recurrent infections (in spite of Ig-substitution) and autoimmune hepatitis respectively. Stable B-cell function without the need for Ig-substitution was noted in 12/16 long term survivors (75%) after conditioning and in merely 3/?11 pts (27%) treated without conditioning. For pts with X-SCID without an HLA-compatible donor, haploidentical transplantation with and without conditioning is a therapeutic option with excellent survival. The probability of posttransplant B-cell function can be increased by conditioning.
ESID-0404 Reduced Diversity of the Immune Receptor Repertoire as New Pathophysiological Marker for CVID
H. IJspeert 1, G.J. Driessen2, E. Simons1, I. Pico-Knijnenburg1, D. van Zessen3, V.A.S.H. Dalm4, P.?M. van Hagen4, J.J.M. van Dongen1, A.P. Stubbs3, M. van der Burg1
1Immunology, Erasmus MC University Medical Center, Rotterdam, Netherlands
2Pediatrics, Erasmus MC University Medical Center, Rotterdam, Netherlands
3Bioinformatics, Erasmus MC University Medical Center, Rotterdam, Netherlands
4Internal Medicine, Erasmus MC University Medical Center, Rotterdam, Netherlands
Common variable immunodeficiency (CVID) is the most prevalent form of primary idiopathic hypogammaglobulinemia. We previously defined five B-cell maturation patterns based on immunophenotypic and molecular characteristics, representing immunologically homogenous groups with different pathophysiology (Driessen et?al. Blood 2011). Pattern 1 and 2 were proposed to be linked to a B-cell production or early maturation defect, whereas patterns 3 to 5 were linked to germinal or post-germinal center defects. We hypothesized that CVID patients with an early B-cell defect (pattern 1-2) would have a reduction in the naive B-cell receptor repertoire.
Naïve B cells were sorted from peripheral blood of 17 CVID patients (2-5 patients per B-cell maturation pattern) and healthy controls. B-cell receptor rearrangements (IGH) were amplified using 6 replicates and sequenced using 454 sequencing. Subsequently, the diversity of the naive repertoire was calculated by determining the coincidental sequences using our Galaxy pipeline for repertoire analysis (http://galaxyproject.org/).
CVID patients with B-cell maturation pattern 1 and 2 had significantly lower B-cell receptor diversity compared to CVID patients with pattern 3 to 5 (p
In conclusion, CVID patients with an early B-cell defect have reduced antigen receptor repertoire diversity. This indicates that antigen receptor repertoire diversity might represent a new pathophysiological marker.
ESID-0123 Assessment of Nutritional Status in Agammaglobilinemia: Results of a Multicenter Study
M. Raimondi 1, R.M. Dellepiane1, P. Pavesi1, A. Soresina2, M. Carrabba3, S. Martino4, G. Russo5, G. Patuzzo6, C. Pignata7, F. Specchia8, M. Duse9, R. Gallizzi10, V. Moschese11, G.L. Marseglia12, M.C. Pietrogrande13, C. Agostoni13
1Pediatrics University of Milan, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milano, Italy
2Pediatrics University of Brescia, Spedali Civili di Brescia, Brescia, Italy
3Internal Medicine University of Milan, Fondazione IRCCS Ca'Granda Ospedale Maggiore Policlinico, Milano, Italy
4Pediatrics University of Torino, Ospedale Infantile Regina Elena, Torino, Italy
5Pediatrics haematology and oncology University of Catania, Azienda Policlinico Vittorio Emanuele, Catania, Italy
6Clinical Immunology University of Verona, Policlinico G.B. Rossi, Verona, Italy
7Pediatrics University of Napoli, Azienda Ospedaliera Universitaria Federico II, Napoli, Italy
8Pediatrics University of Bologna, Policlinico S.Orsola-Malpighi, Bologna, Italy
9Pediatrics University of Roma, Istituto universitario "La Sapienza", Roma, Italy
10Pediatrics University of Messina, Azienda G.Martino, Messina, Italy
11Pediatrics University of Roma Tor Vergata, Policlinico Tor Vergata, Roma, Italy
12Pediatrics University of Pavia, Policlinico San Matteo, Pavia, Italy
13Pediatrics University of Milano, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milano, Italy
Primary antibody deficiencies are a group of disorders characterized by an unusual susceptibility to infections and malnutrition. Little is known on nutritional status in primary immunodeficiencies, since only one study was published on this topic (1).
The aim of the multicenter study is general assessment of nutritional status of patients with primary agammaglobulinemia (XLA or AAR).
Sixty-five patients (all males, aged 2-57 years, median age 18,5 years) were enrolled referring to eleven Italian Centers (IPINET). Short questionnaire to evaluate physiological data and nutritional habits was subministered. Anthropometric indicies (weight, height, BMI, skinfolds and circumferences) and blood tests (IGF-1, prealbumin, albumin, blood glucose, insulin, total cholesterol, HDL, LDL, apolipoprotein A and B, triglycerides) were measured. All data were collected in standardized Case Report Form.
Fourty-nine patients showed weight and BMI percentiles between 25 ° and 97 °. Weight and BMI percentiles at enrollment seemed similar or higher that the corresponding percentiles at the time of diagnosis. The laboratory blood tests values showed no significant difference compared to the reference values for each age class.
No signs of malnutrition were found in tested patients. We speculate that an adequate immunoglobulin replacement therapy in patients with Agammaglobulinemia may help in reducing the risk of malnutrition by supplying protein chains and trough the reduction of infections.
(1) KOUHKAN A ET AL. A study of malnutrition in Iranian patients with primary antibody deficiency. Iran J Allergy Asthma Immunol 2004; 3(4):189-96
ESID-0808 T Cell Depleted Reduced Intensity Haematopoietic Stem Cell Transplantation for Immune Dysfunction Due to GATA 2 Mutation
K. Sturgess 1, E. Thoulouli2, M. Slatter3, G. Jackson1, A. Cant3, S. Hambleton3, V. Bigley1, M. Collin1
1Northern Centre for Bone Marrow Transplant, Newcastle upon Tyne Hospitals NHS Foundation Trust
2Department of Haematology, Central Manchester University Hospitals
3Department of Paediatric Immunology and Infectious Disease, Newcastle upon Tyne Hospitals NHS Foundation Trust
Primary immunodeficiency offers a unique and powerful means of dissecting the development and functionality of human immune cells ?in?vivo. The biological consequences and genetic basis of immunodeficiencies due to loss or dysfunction of lymphocytes and granulocytes are well described, but the antigen presenting arm of the immune system has been under studied. Our recent work identifying IRF8 and GATA2 mutations, resulting in loss of monocytes and dendritic cells, have demonstrated the importance of antigen presentation in maintaining a regulated immune system
Here we report successful outcomes in four patients with GATA2 mutation treated with T cell depleted reduced intensity allogeneic stem cell transplantation. Patients received fludarabine-based conditioning combined with melphalan, busulfan or treosulfan and ?in?vivo T cell depletion with alemtuzumab 50-60 mg. Three were transplanted with unrelated donors with 10/10, 9/?10 (HLA-C) and 8/?10 (HLA-A and DQ) antigen matches and one was transplanted from a GATA2 wild-type HLA-matched sibling. Two patients had pulmonary alveolar proteinosis (PAP) at transplant and one was on continuous oxygen and overnight non-invasive ventilation.
All patients are alive and well between 2-4 years after transplant. No patients suffered graft versus host disease and all have 100% donor myeloid engraftment. Respiratory status has significantly improved in the two patients with PAP. Non mhycobacterial infection and papillomata due to HPV infection have resolved. Significantly, one patient with carcinoma in situ stage 3 had complete resolution of a lesion. One patient who gave birth prematurely to her first child has successfully carried a healthy infant to term. These results suggest that T cell depleted reduced intensity transplant is feasible and safe in patients with GATA2 mutation and is associated with minimal graft versus host disease combined with rapid restoration of immunodeficiency.
ESID 2014 Poster Presentations
Topic: Educational Day
ESID-0564 Acute Primary Infection with Mouse Cytomegalovirus Represents a Natural Animal Model for Virus-Associated Secondary Hemophagocytic Lymphohistiocytosis
E. Brisse 1, T. Mitera1, M. Imbrechts1, C.H. Wouters2, G. Andrei3, P. Matthys1
1Laboratory of Immunobiology, Katholieke Universiteit Leuven, Leuven, Belgium
2Laboratory of Pediatric Immunology, University Hospital Gasthuisberg KULeuven, Leuven, Belgium
3Laboratory of Virology and Chemotherapy, Katholieke Universiteit Leuven, Leuven, Belgium
Hemophagocytic lymphohistiocytosis (HLH) is a complex, life-threatening immune disorder, characterized by systemic inflammation, an out-of-control cytokine storm and wide-spread organ damage. In contrast to primary HLH, secondary HLH occurs without any known mutation in granule-mediated cytotoxicity, in a context of infections, malignancies or autoimmune/autoinflammatory disorders. Both subtypes are generally precipitated by an infectious agent, predominantly a member of the Herpesviridae. Exploiting this knowledge, we created an animal model for virus-associated secondary HLH by administering mouse cytomegalovirus (MCMV) to immunocompetent wild-type mice. Infected mice displayed the clinicopathologic features of HLH as set forth in the Histiocyte Society diagnostic guidelines: fever, cytopenia, hemophagocytosis, hyperferritinemia and elevated serum levels of soluble CD25. Additionally, mice developed lymphadenopathy, coagulopathy, liver dysfunction and hypercytokinemia. Strikingly, MCMV-infected interferon (IFN)-gamma-deficient mice were more prone to develop HLH-like symptoms, challenging the major pathogenic role attributed to IFN-gamma in most mouse models of primary HLH. As secondary HLH is known to occur in patients with primary infection or reactivation of human cytomegalovirus, our research demonstrates MCMV infection of wild-type mice as a promising natural model with utility for elucidating the poorly understood pathogenesis of secondary HLH and exploring novel treatment options.
ESID-0762 Soluble MHC CLASS II Antigens as Potent Regulators of the Immune Response
K. Bakela 1, N. Kountourakis2, M. Aivaliotis2, I. Athanassakis1
1Biology, University of Crete, HERAKLION, Greece
2Imbb, FORTH, HERAKLION, Greece
Soluble MHC-II (sMHCII) molecules are present in body fluids of healthy individuals and are considered to be involved in the maintenance of self tolerance, while they are also related to various diseases. Their concentration increases during ?in?vivo antigen-specific tolerogenic stimulation and it was recently shown that exosome-mediated tolerance is MHC-II dependent. At the cellular level, sMHCII proteins compete with membrane MHCII for TCR binding. Soluble MHC-II proteins were isolated from serum of HSA-tolerant mice and purity was verified through ELISA, SDS-PAGE, and Western blot analysis using specific a-class II antibodies. SDS-PAGE revealed one band at ~60?kDa which was found to be highly glycozylated. Mass spectrometry showed that the soluble molecules are loaded with antigenic peptides. Soluble MHCII suppressed an antigen-specific immune response by reducing the number of active cells as tested by 3??dR incorporation assays. In fact, they increased CTLA+/CD4+ numbers while reducing CD28+/CD4+ T-cells as tested by FACS analysis. ELISA experiments showed that sMHCII induced secretion of IL-10, while increasing the expression of CD25 surface marker in control CD4+ T-cells. Evaluation of TCR signaling in CD4+ cells indicated downregulation of phosphorylated forms of ZAP-70 and LAT kinases in the presence of sMHCII. Finally, the results showed that sMHCII could negatively regulate antigen-specific immune response ?in?vivo as well as in?vitro by inducing immunosuppressive CTLA4+ and CD25+ T-cell populations and immunosuppressive mediators, playing thus an important role in immune homeostasis.
ESID-0261 Unravelling Cellular and Molecular Mechanisms of Skin Degeneration in the Hypomorphic RAG2 Mouse Model of Omenn Syndrome
V. Maina 1, A. Keren2, S. Mantero1, E. Fontana3, P.L. Poliani3, A. Gilhar2, B. Cassani4, A. Villa4
1Telethon Institute for Gene Therapy, Scientific Institute and University Hospital San Raffaele, Milan, Italy
2Skin Research Laboratory Faculty of Medicine, Technion-Israel Institute of Technology, Haifa, Israel
3Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy
4-, Milan Unit Istituto di Ricerca Genetica e Biomedica Consiglio Nazionale delle Ricerche, Milan, Italy
Several skin disorders are characterized by a strong inflammatory and autoimmune component, considered the leading cause of the disease, together with genetic and environmental factors. Skin degeneration is a common clinical sign in several PIDs. Omenn syndrome is a paradigmatic example as severe erythroderma is the clinical hallmark, and alopecia and vitiligo are reported in patients. Rag2R229Q/R229Q mouse faithfully recapitulates all the clinical and laboratory findings observed in OS. Here, we propose the Rag2R229Q/R229Q mouse as a model to study the pathogenesis of different forms of skin degeneration. Oligoclonal activated T cell infiltration and Langerhans cells’ ectopically distribution and impaired migration characterize Rag2R229Q/R229Q skin. In particular, in Rag2R229Q/R229Q vitiligo skin, CD4+ and CD8+ cells are present around and within the damaged hair follicles, correlating with INFa increase. By FACS analysis, CD8+ T and NK cells predominate in vitiligo whereas CD4+ T cells infiltrate psoriatic Rag2R229Q/R229Q skin. Consistently, immunostaining and real time analysis reveal the lack of tyrosinase and tyrosinase related protein in Rag2R229Q/R229Q affected skin. In Rag2R229Q/R229Q psoriatic skin, PMN and pDCs infiltration correlates with INFa increase. Moreover, IL12p40 expression increases in Rag2R229Q/R229Q unaffected and vitiligo skin, while IL23p19 and IL6 increase in psoriatic skin. Reg3? is expressed only in Rag2R229Q/R229Q psoriatic skin, together with high levels of S100A8, S100A9 and IL1ß. Interestingly, Treg cells significantly accumulated in affected skin. Overall, these results are instrumental to understand the mechanisms underlying skin degeneration and to identify novel biological markers as useful targets in the clinical setting.
ESID-0170 The Psychological Health of X-Linked Carriers of Chronic Granulomatous Disease (CGD) in the United Kingdom
A. Battersby 1, F. McKendrick2, M.S. Pearce3, C. Cale4, H. Braggins5, D. Goldblatt6, A.?R. Gennery1
1Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
2Department of Health Psychology, Newcastle upon Tyne Hospitals Foundation Trust, Newcastle upon Tyne, United Kingdom
3Institute of Health and Society, Newcastle University, Newcastle upon Tyne, United Kingdom
4Clinical Immunology, Great Ormond Street Hospital, London, United Kingdom
5CGD Society, Great Ormond Street Hospital, Newcastle upon Tyne, United Kingdom
6Institute of Child Health, University College London, London, United Kingdom
Background
CGD is a rare primary immunodeficiency.70% of UK cases are X-linked (XL). XL-carriers are usually confirmed after a relative's diagnosis and have several risk factors for psychological health problems;a chronically ill relative, their own medical problems including an association with SLE. SLE is associated with anxiety and depression.
Methods
XL-CGD carriers were recruited via known cases.Participants completed validated psychological health questionnaires; Hospital Anxiety and Depression Score (HADS)
Pediatric Inventory for Parents (PIP).
Results were compared to population norms and published data from comparable groups. A HADS score>7 (anxiety/depression) is abnormal[1].Symptom frequency was compared with parents of CF children[2].HAD mean scores were compared with SLE patients[3].
PIP quantifies distress of parenting chronically unwell children with frequency (PIP-F) and severity (PIP-S) sub-scores.PIP's validation in oncology patients provides comparison data[4].
Results
HADS: 54 completed
No significant difference found between CGD carrier mothers and other carrier relatives.
66% XL-CGD had carriers greater than borderline anxiety,significantly more than CF parents.
| HADS | CGD Carriers | SLE(high-pain) | SLE(low-pain) |
| Anxiety | 9.6 | 9 | 4 |
| Depression | 5 | 8 | 3 |
PIP: 45 completed.
CGD scores were similar to oncology parents, but had higher PIP-F (p=0.006).
No correlation seen between PIP and anxiety/depression symptoms (r<0.3)
Discussion
CGD carriers had significant anxiety rates, greater than CF parents.Lack of correlation between HADS and PIP suggests anxiety was not solely related to their child's disease.
This study highlights that XL-CGD carriers suffer unrecognised but potentially significant psychological health problems which may impact upon their own lives and ability to cope with the needs of their children.
ESID-0213 A Case of Isolated Cerebral Nocardiosis Caused by a Defect in the IL12-IFNG Axis
D. Bogaert 1, V. Debacker2, S. Callens3, D. Ommeslagh3, M. Dullaers4, K.Y. Vermaelen5
1Pediatric Pulmonology and Immunology Clinical Immunology Research Lab, University Hospital Ghent, Ghent, Belgium
2Clinical Immunology Research Lab, University Hospital Ghent, Ghent, Belgium
3Pulmonary Medecine, University Hospital Ghent, Ghent, Belgium
4Clinical Immunology Research lab, University Hospital Ghent, Ghent, Belgium
5Pulmonary Medicine Clinical Immunology Research Lab, University Hospital Ghent, Ghent, Belgium
Introduction: Human nocardiosis is considered an opportunistic infection as it is usually associated with immunocompromising conditions, although it may occur in immunocompetent patients. We report a 34-year-old caucasian male who presented with seizures provoked by a cerebral Nocardia farcinica abscess. His past medical history was uneventful and secondary immunodeficiency was ruled out. Similar to Mycobacterium, the immune response against Nocardia is primarily cell-mediated. We therefore suspected a defect in the IL12/?IL23-IFN? axis.
Objective: To identify the primary immunological defect.
Methods: Routine immunological assessment was done. Additional tests were performed on stimulated patient and healthy control PBMCs. Cytokine concentrations were measured with ELISA. Flow cytometry was used to analyze IL12Rß1 surface expression and STAT4 phosphorylation.
Results: Routine immunological workup only revealed mild lymphopenia with mildly decreased to normal lymphocyte reactivity. In contrast, in?vitro tests uncovered a profoundly blunted IFNg release in response to stimulation with purified protein derivate and/or IL12. The intrinsic capacity to secrete IL12p70 or IFNg however was intact. IL12Rß1 surface expression on patient T cells was comparable with those of healthy control cells, although IL12-induced STAT4 phosphorylation was partially reduced.
Conclusion: Nocardiosis has been reported in a few pediatric patients with IL12p40 or IL12Rß1 deficiency. Here we present evidence for a functional defect in the IL12R signaling pathway with conserved IL12Rß1 expression levels in an adult patient with cerebral nocardiosis. Further immunogical investigations and subsequential genetic analysis of candidate genes are ongoing. The patient was treated with long-term antibiotics and neurosurgery, and is currently free of infection.
ESID-0188 Acquired Partial Lipodystrophy: A Rare Clinical Presentation of a Complement Deficiency
D. Bogaert 1, M. Dullaers2, J. Vandewalle3, P. Stordeur4, E. Govaere5, F. Haerynck6
1Pulmonary Diseases, University Hospital Ghent Medical Research Building (MRB), Ghent, Belgium
2Clinical Immunology Research lab, University Hospital Ghent, Ghent, Belgium
3Pediatric Nephrology and Rheumatology, University Hospital Ghent, Ghent, Belgium
4Immunochemistry Lab Immunobiology Clinics, Erasme Hospital Université Libre de Bruxelles, Brussels, Belgium
5Pediatric Immunology and Allergology, OLV Hospital, Aalst, Belgium
6Pediatric Immunology and Pulmonology, University Hospital Ghent, Ghent, Belgium
Lipodystrophies encompass a group of inherited and acquired syndromes characterized by loss of adipose tissue. Each type has an unique pathogenesis, clinical picture and pattern of adipose-tissue distribution.
We present an 8-year-old boy with Barraquer-Simons' syndrome, an acquired partial lipodystrophy (APL) syndrome accompanied by C3 nephritic factor (C3 NeF). At the age of 5 years he developed idiopatic thrombocytopenia (ITP). One year later, he had pneumococcal pneumonia with septicemia. Moreover, he suffers from recurrent warts on feet and hands. During previous years, his facial appearance progressively changed due to gradual loss of subcutaneous fat. An underlying complement deficiency was suspected. He revealed persistently low serum levels of complement factor C3 and profound decreased activity of the alternative pathway with slightly decreased hemolytic activity of the classical pathway. Additional tests revealed the presence of autoantibody C3 NeF. Serum levels of other complement factors were normal. There were no abnormalities in phagocytic, cellular or humoral immunity.
Barraquer-Simons' syndrome is an APL characterized by autoimmune-mediated loss of adipose tissue from the face, neck, arms and trunk. C3 NeF induces the lysis of adipocytes expressing complement factor D. Moreover, C3 NeF binding with C3 convertase prolongs its half life leading to excessive consumption of C3 causing susceptibility to encapsulated bacteria and glomerulonephritis. The patient is treated with prophylactic antibiotics and received antipneumococcal and antimeningococcal vaccines.
Conclusion: C3 Nephritic factor is not only associated with overconsumption of the alternative complement pathway but can also induce lysis of adipocytes leading to partial lipodystrophia.
ESID-0479 Role of Natural Killer Cells in Humans: Phenotype of Patients with NK Cell Deficiency in Adults with Primary Hypogammaglobulinemia in the French DEFI Study
M. Ebbo 1, L. Gerard2, F. Vély3, C. Farnarier3, E. Oksenhendler2, N. Schleinitz1, E. Vivier4
1Médecine Interne, hôpital de la Conception - Aix Marseille Université, Marseille, France
2Immunopathologie Clinique, hôpital Saint-Louis, Paris, France
3Immunologie, hôpital de la Conception, Marseille, France
4CNRS UMR 7280 - INSERM U1104, Centre d'Immunologie de Marseille-Luminy, Marseille, France
Natural Killer (NK) cells have been shown to exert antiviral and antitumoral activities. Nevertheless most of data derive from mouse models and functions in human host defense remain unclear. Well-defined and selective inherited defect in NK cells are lacking and data from large human cohorts are rare. To better characterize the role of NK cells in humans, we study a large cohort of 457 patients with common variable immunodeficiency (CVID) from the French DEFI study. We compared phenotypes of 99 patients with severe NK cell lymphopenia (<50/mm3), 118 patients with mild NK cell lymphopenia (50-100/mm3) and 240 patients with normal NK cell count (>100/mm3). A higher prevalence of granuloma (25.3% vs 13.6% and 8.8%), lymphoid hyperplasia (35.4% vs 24.6% and 19.2%) and auto-immune cytopenia (25.3% vs 12.7% and 14.6%) was observed in patients with severe NK cell lymphopenia as compared to others groups. Pulmonary infections (63.6% vs 59.3% and 44.2%), invasive infections (68.7% vs 60.2% and 48.8%) and digestive diseases (51.5% vs 38.1% and 32.5%) were also more frequent in this population. Interestingly, no difference was noted for viral infections or neoplasia. CD4+ T cell counts were lower only in 25% of patients with NK cell lymphopenia, suggesting that this factor does not account alone for the observed phenotype. In conclusion, patients with CVID and NK cell deficiency present a peculiar phenotype. The particular high frequency of pathological events in this subgroup argues for non-redundant immune functions of NK cells in humans when the adaptive immune response is not optimal.
ESID-0210 Complement Component C8 Deficiency
H. Martínez-Banaclocha 1, M.R. Moya-Quiles1, J. Eguía-Núñez1, P. Martínez-García1, G. Salgado-Cecilia1, E.M. Novoa-Bolívar1, M.V. Bernardo-Pisa1, M.C. García-Calatayud1, M.R. Álvarez-López1, A.?M. García-Alonso1
1Servicio de Inmunología, Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain
Similar to the other deficiencies of terminal complements components, deficiency of the eight component of the complement is frequently associated with recurrent neisserial infections, especially meningitis caused by Neisseria meningitidis. This disorder has rarely been diagnosed in the Spanish population.
We report a case of a 20 year-old-man, with non consanguineal parents, who has suffered from recurrent bronchitis and sinusitis since birth. In 1999 and in 2001 the patient suffered meningococcal septicaemia. In both cases, the microbiological cultures were positive for Neisseria meningitidis serotype B. No family history of meningitis or other severe infections could be elicited.
In 2001 radial immunodiffusion serological blood tests revealed undetectable functional activity of both the classical (CH100) and alternative (AP100) complement pathways and a very low level of C8 in patient's serum, whereas levels of C3, C4, C5, C6, C7 and C9 were within the normal range. Not others abnormalities in humoral and cellular immunity arm were detected. In 2013, sequence analysis of the C8B gene revealed a previously described homozygous single C>T exchange in exon 9 (c.1282C>T), leading to a stop codon, which provoked the truncation of the C8B protein (p.R248X). Sequencing of PCR-amplified exon 9 from DNA of the family revealed that the patient´s mother and sisters are heterozygous for the mutation.
Up to date, the patient presents frequent mild respiratory and gastrointestinal infections, folliculitis and urticaria.
This report highlights the importance of complement screening in cases of recurrent meningococcal infections in order to consider adequate clinical recommendations and genetic counselling.
ESID-0278 Chronic Mucocutaneous Candidiasis, Recurrent Herpetic Infections and Suppurative Eyelid Infections in a Patient Carrying a Novel Gain-of-Function Mutation in the STAT1 DNA-Binding Domain
G. Giardino 1, E. Cirillo1, V. Gallo1, R. D'Assante1, M. Paciolla2, G. Ruggiero1, M.V. Ursini2, R. Carsetti3, A. Puel4, C. Pignata1
1Department of Translational Medical Sciences, Università "Federico II", Naples, Italy
2International Institute of Genetics and Biophysics, CNR, Naples, Italy
3Research Center, Ospedale Pediatrico Bambino Gesù (IRCCS), Rome, Italy
4Laboratory of Human Genetics of Infectious Diseases Necker Branch, Inserm U980 and University Paris Descartes Necker Medical School, Paris, France
Chronic mucocutaneous candidiasis (CMCC) is characterized by noninvasive persistent Candida infections. Gain-of-function mutations in signal transducer and activator of transcription 1 (STAT1) cause impaired STAT1 dephosphorylation, diminished IL-17-producing T-cells, and CMCC. We report on a 17-year-old boy with CMCC. At 7 years of age he developed mucocutaneous candidiasis. At 8 years he suffered from severe varicella infection and since 11 he experienced recurrent herpetic infections involving genitals and limbs, recurrent abscesses and suppurative eyelid infections. Familial history was negative. The physical examination revealed oral thrush, onichomycosis, suppurative eyelid infection (figure), furunculosis and parodontitis. Azole-sensitive Candida Albicans grew from oral lesions, nails and esophageal mucosa cultures. Prophylactic treatment with fluconazole resulted in a decrease of frequency and severity of fungal infections. Laboratory evaluation revealed normal white blood cell, T- and B-lymphocyte counts, T-lymphocyte proliferation, Ig and IgG subclasses serum levels and responses to vaccines. HIV serology was negative and IgE levels were persistently elevated (684 KU/L). Transitional (8.2%), mature (79.8%) and memory (12%) B cell levels were normal. Memory B cells mostly included IgM and only few switched cells (88 and 12%, respectively). CD4 and CD8 naïve and memory T cells were normal. IL-17-producing T-cell numbers were 0%. Toll like receptor stimulation resulted in high levels of IL-10, IL1 beta, TNF alpha, IFN-gamma, IL-6 and IL-8. Full-length sequencing of STAT1 genomic DNA identified a T387A heterozygous mutations in the DNA-binding domain, not identified in the unaffected parents. This mutation has not been previously reported.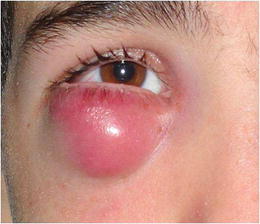
ESID-0128 IL-17RA Deficiency Underlying Chronic Mucocutaneous Disease (CMCD): Genetic and Clinical Features in 7 Patients from Turkey and Japan
R. Levy 1, S. Okada2, Y. Camcioglu3, J. Toubiana4, N. Hyakuna5, M. Migaud1, A. Takaki2, O. Ohara6, M. Kobayashi2, J.L. Casanova7, A. Puel1
1Necker Branch INSERM 1163 University Paris Descartes Imagine Institute, Laboratory of Human Genetics of Infectious Diseases, Paris, France
2Department of Pediatrics, Hiroshima University Graduate School of Biomedical & Health Sciences, Hiroshima, Japan
3Division of Infectious Diseases Clinical Immunology and Allergy Department of Pediatrics, Cerrahpasa Medical Faculty Istanbul University, Istanbul, Turkey
4Department of Pediatrics, Necker Hospital AP-HP and University Paris Descartes, Paris, France
5Center of Bone Marrow Transplantation, Faculty of Medicine University of the Ryukyus, Okinawa, Japan
6Department of Human Genome Research, Kazusa DNA Research Institute, Chiba, Japan
7Rockefeller Branch the Rockefeller University, St. Giles Laboratory of Human Genetics of Infectious Diseases, New York, USA
Chronic mucocutaneous candidiasis (CMC) is characterized by persistent symptomatic infection of the nails, skin and mucosae mostly caused by Candida albicans. Some patients with CMC have no other prominent clinical manifestations. This rare condition is often referred to as CMC disease (CMCD). Whereas, autosomal dominant STAT1 gain of function represents the major genetic etiology of this PID, complete autosomal recessive IL-17RA deficiency in one patient and partial autosomal dominant IL-17F deficiency in one multiplex kindred were the first two genetic etiologies to be published in 2011. Following a whole exome sequencing approach in a cohort of 284 patients with CMCD, we found seven additional patients, in four families, bearing homozygous variants in IL17RA (Fig.?1): five patients of Turkish origin in three kindreds, three of whom born to first cousin parents; P1 and P2 born to non consanguineous Japanese parents. The familial segregation confirmed an autosomal recessive pattern of inheritance in all cases. Three children were found to be homozygous for the c.196C>T (P1;P2) and c256C>A (P7) nonsense mutations whereas the other 4 patients were found to bear homozygous misense (P3; P4) and frameshift (P5; P6) variants in the key functional SEFIR domain of IL-17RA. None of these mutant alleles were found in the public databases. All patients presented with early onset of mucocutaneous candidiasis. Other clinical findings included Staphylococcus aureus dermatitis (P3;P4); possible pulmonary tuberculosis (P7); bacterial pneumonia (P3;P4;P5); seborrheic dermatitis (P3;P4). The IL-17RA protein expression and functional validation is currently in progress in patients’ PBMCsandfibroblasts.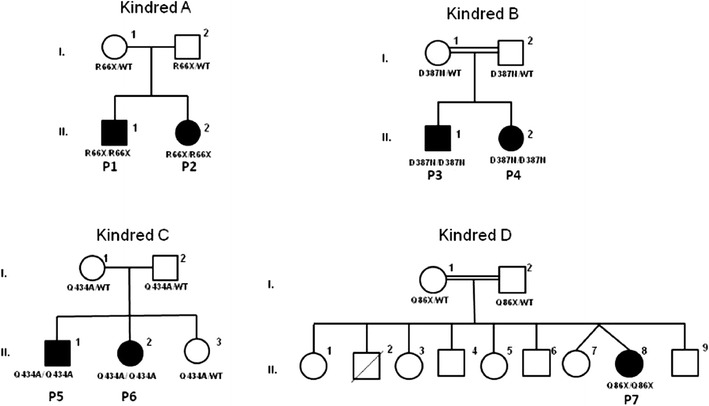
ESID-0178 Impaired Development of IL-17-Mediated Immunity in a Patient with a Novel STAT1 Mutation
B. Soltész 1, B. Tóth1, G. Csorba1, S. Taskó1, A.K. Sarkadi1, L. Maródi1
1Department of Infectious and Pediatric Immunology, University of Debrecen, Debrecen, Hungary
Chronic mucocutaneous candidiasis disease (CMCD) is characterized by persistent or recurrent infection of the skin, nails, and oral- or genital mucosae. The most common cause of CMCD is STAT1 gain-of-function mutation affecting primarily of the coiled-coil domain of signal transducer and activators of transcription 1 (STAT1) and mutation in the DNA-binding domain (DBD) is less frequent. We have studied a 26 year-old Hungarian female with severe CMCD from early childhood. Sanger sequencing was used to analyse sequences of coding exons by using genomic DNA. We detected a previously unknown heterozygous c.1219C>G (L407V) mutation in STAT1 affecting the DBD of the protein. Non-adherent white blood cells were activated with anti-CD3 and treated with IL-23, IL-1ß, IL-6, TGF-ß and IL-2 cytokines. After 5 days, low proportions of IL-17A- and IL-22-producing CD4+ T cell lymphocytes were detected by intracellular staining and flow cytometry. Candida-induced secretion of IL-22 by mononuclear cells and small concentrations of IL-22 cytokine by differentiated T cells were measured by ELISA. These data suggest that the new mutant allele may result in impaired differentiation of non-adherent CD4+ T cells to CD4+/IL-17+ and CD4+/IL-22+ cells causing susceptibility to Candida infection on body surfaces.
ESID-0099 Long-Term Outcome of 19 Patients with Aid-Hyper IGM Syndrome
J. Boursiquot 1, P.?M. Bédard1, L. Côté2, J.P. Drolet1, R. Gagnon1, J. Hébert1, J. Longtin2, H. Senay2, N. Verreault1, A. Lavoie1
1Clinical Immunology and Allergy, CHU de Québec - Pavillon CHUL, Quebec City, Canada
2Microbiology and Infectious Diseases, CHU de Québec - Pavillon CHUL, Quebec City, Canada
Introduction
HyperIgM syndrome is characterized by absence of IgG and IgA but normal or elevated serum concentration of IgM. Mutations in activation-induced cytidine deaminase (AID) result in Hyper-IgM syndrome (HIS) type 2. Little is known about the long-term prognosis of these patients.
Objective
To study natural history, complications and prognosis of patient with AID-HIS.
Methods
We retrospectively reviewed the medical records of diagnosed AID-HIS patients.
Results
We collected the data of 19 patients from 14 different families. They all shared the same mutation on the AID gene (c.334C>T; p.Arg112Cys). Median age was 52 years old. Median age at diagnosis was 11 years-old. Median follow-up time was 39 years for a total of 680 patient-years. One patient died of pulmonary hemorrhage. All patients but one were treated with parenteral immunoglobulins. Prophylactic antibiotics were actively used in 6 patients.
Common infections reported were: otitis (95%), sinusitis (84%), pneumonia (74%), pharyngitis (53%), meningoccal meningitis (37%) and mastoiditis (26%). Fourteen patients had a chest CT-scan performed. Bronchiectasis (36%), micronodules (36%) and pleuritis (29%) were the most frequent lung abnormalities. Twelve patients had pulmonary function tests. Two patients had restrictive pattern and 2 patients had obstruction. Other complications included adenopathies (42%), splenomegaly (26%) cancer (11%) and hepatic fibrosis (11%).
Conclusion
Although long-term survival is good, type 2 HIS patients are at risk of non-infectious complications including pulmonary sequelae which clinicians should be aware of. As different gene mutations exist for AID, further genotype-phenotype correlation studies would be of interest.
ESID-0711 Altered Central and Peripheral B-Cell Tolerance Checkpoints in Wiskott-Aldrich Syndrome Patients
M.C. Castiello 1, F. Pala1, M. Bosticardo1, N. Chamberlain2, H. Morbach2, S. Scaramuzza1, F. Ferrua3, M.H. Albert4, M.G. Roncarolo1, L. Naldini1, A. Aiuti1, E. Meffre2, A. Villa5
1San Raffaele Telethon Institute for Gene Therapy, San Raffaele Scientific Institute, Milan, Italy
2Department of Immunobiology, Yale University School of Medicine, New Haven CT, USA
3Pediatric Immunohematology and Bone Marrow Transplantation Unit, San Raffaele Scientific Institute, Milan, Italy
4Ludwig Maximilians University Munich, Dr. von Hauner Children's Hospital, Munich, Germany
5Milan Unit, IRGB CNR, Milan, Italy
Wiskott-Aldrich Syndrome (WAS) is a severe X-linked primary immunodeficiency characterized by micro-thrombocytopenia, eczema, infections and high susceptibility to autoimmune manifestations and tumors. Autoimmunity has been mainly attributed to the breakdown of self tolerance sustained by dysfunction of both natural T regulatory and effector T cells. We recently showed that B-cell compartment of WAS patients is perturbed with an overrepresentation of CD21low B cells, enriched in autoreactive clones, and increased B-cell activation factor (BAFF) levels. To evaluate the role of WAS protein (WASp) in B-cell tolerance checkpoints, we analyzed the frequency of autoreactive clones in different B-cell subpopulations by amplifying and cloning immunoglobulin chain genes from single B cells of four WAS patients. We found a decreased frequency of autoreactive new emigrant/transitional B cells in WAS patients, suggesting a hyperfunctional central B-cell checkpoint in the absence of WASp. This finding is associated with an increased B-cell activation after in?vitro stimulation of B-cell receptor. In contrast, high frequency of polyreactive and Hep2 reactive clones were found in mature naïve B cells of WAS patients, indicating a defective peripheral B-cell checkpoints. In order to evaluate whether hematopoietic stem cell gene therapy (HSC-GT) can correct B-cell tolerance checkpoints in WAS patients, we are analyzing central and peripheral checkpoints in patients after HSC-GT (ongoing trial, sponsored by TIGET, Italy).
In conclusion, for the first time we provide evidence that WASp plays an important role in the maintenance of B-cell tolerance in humans.
Acknowledgements: supported by grant Telethon and Minister of Health RF-2009
ESID-0446 Targeted Re-Sequencing of a CVID/?Agammaglobulinemia Cohort
N. Frede 1, M. Buchta1, B. Grimbacher1
1Centre of Chronic Immunodeficiency, University Medical Centre Freiburg, Freiburg, Germany
Antibody deficiencies constitute the most common primary immunodeficiencies. During recent years, a growing number of mutations underlying hypo- and agammaglobulinemias have been identified. To facilitate the establishment of a genetic diagnosis, we developed a next-generation sequencing panel for targeted re-sequencing, comprising 27 genes. We analysed 46 patient samples employing Agilent’s HaloPlex and Illumina’s MiSeq technologies for target enrichment and sequencing. The average reading depth constituted 600 reads. 97.78% of the target regions were covered at least 20 times, while 96.73% had a coverage of greater than 100x. In the 46 patients a total of 2308 variants were called. Out of these 541 were missense mutations, 3 frameshift mutations, as well as one nonsense and one splice site mutation.
We identified a compound heterozygous mutation in LRBA (c.T2836fs/c.G1420fsx) as well as three novel mutations in CTLA4 (T124P, R70W and a splice site mutation). Furthermore, we detected two frameshift mutations as well as one missense mutation in another yet undisclosed candidate gene. Finally, there are 16 additional novel, previously undescribed variants (missense mutations) in 9 different genes, which require further work-up for evaluation. We have since designed a second version of this CVID/?Agammaglobulinemia re-sequencing chip, for which we will present the data in October.
Hence, targeted re-sequencing led to a significant reduction in costs (€ 250 per sample; € 5 per gene) and required working time (approx. 3-4 weeks from gDNA to interpreted result for up to 50 genes) and, thus, proved to be an important alternative to Sanger sequencing.
ESID-0152 B-Precursor Acute Lymphoblastic Leukemia in a Patient with X-Linked Agammaglobulinemia
A. Hoshino 1, Y. Okuno2, M. Migita3, H. Ban3, X. Yang1, N. Kiyokawa4, S. Kojima2, O. Ohara5, H. Kanegane1
1Pediatrics, University of Toyama, Toyama, Japan
2Pediatrics, Nagoya University, Nagoya, Japan
3Pediatrics, Japanese Red Cross Kumamoto Hospital, Kumamoto, Japan
4Pediatric Hematology and Oncology Research, National Research Institute for Children Health and Development, Tokyo, Japan
5Human Genome Research, Kazusa DNA Research Institute, Kisarazu, Japan
Background: X-linked agammaglogulinemia (XLA) is caused by mutation in the BTK gene encoding Burton’s tyrosine kinase protein. XLA is characterized by the development arrest at the pro B-cell to pre B-cell stage. B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is the most common malignancy in children, and its pathogenesis is explained by the impairment of lymphoid development and additional impairment of events. Given that impairment of lymphoid development is related to leukemogenesis, there is a possibility that XLA patients may have increased susceptibility to BCP-ALL. We here describe a first report of BCP-ALL patient associated with XLA.
Patient and Methods: The patient with recurrent bacterial infections was diagnosed as having an XLA with a BTK mutation (c.1051A>G, p.R307G) at the age of 6 years, and regular intravenous immunoglobulin (IVIG) replacement therapy was initiated. A routine hematological study, which was performed before IVIG replacement therapy, disclosed lymphoblasts in the peripheral blood by chance at 10 years of age. The bone marrow examination was consistent with BCP-ALL, and he received chemotherapy. To identify the secondary mutation, whole-exome sequencing was performed using DNA from bone marrows at diagnosis and during remission.
Results: MLL2 (c.8740delC, p.H2914fs) mutation was yielded as tumor specific variants, which is recurrently mutated in diffuse large B cell lymphoma.
Conclusions: Our report provides the first evidence of germline BTK mutation and acquired MLL2 mutation affecting leukemogenesis, and provides a new insight into germline mutations in leukemia development.
ESID-0065 Retinoic Acid Imporoves Defective TLR-Signalling in Common Variable Immunodeficiency-Derived B Cells
R. Indrevær 1, K. Holm1, P. Aukrust2, L. Osnes3, E. Naderi1, B. Fevang2, H. Blomhoff1
1Department of Biochemistry Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway
2?Section?of Clinical Immunology and Infectious Diseases, Oslo University Hospital Rikshospitalet, Oslo, Norway
3Institute of Immunology, Oslo University Hospital Rikshospitalet, Oslo, Norway
Common variable immunodeficiency (CVID) is characterized by reduced levels of serum immunoglobulines, particularly of the IgG-subclasses, resulting in a high incidence of infections. Although the etiology of CVID is largely unknown, impaired toll-like receptor (TLR)9-signalling has been identified, and reduced levels of serum vitamin A have also been shown. In the present study we have explored the ability of the vitamin A derivate all-trans retinoic acid (RA) to restore the defective TLR-signalling in CVID-derived B cells. We demonstrate that RA nearly normalizes the diminished proliferation and IL-10 secretion in CVID-derived B cells stimulated via the TLRs TLR9 and RP105, and that RA also improves the defective IgG-secretion in these cells. To elucidate the mechanisms involved in the reduced IgG-secretion, we determined the impact of RA on the expression of Blimp-1 and AID, proteins crucial for plasma cell generation and isotype switching, respectively. We found that RA induced the expression of Blimp-1 to the same extent in both CVID-derived- and normal B cells, whereas the RA-induced expression of AID was impaired in the CVID-derived B cells. This suggests that the main defect in CVID-derived B cells is related to the isotype switching machinery rather than plasma cell development. Although RA was not able to fully restore the defective IgG-secretion in CVID-derived B cells, its ability to double the level of IgG induced upon TLR9/?RP105-stimulation may still be of clinical relevance. Our results also support initiation of clinical trials on supplementation of vitamin A in combination with TLR9/?RP105-stimulators for treatment of CVID.
ESID-0137 Profound Symptomatic Hypogammaglobulinemia: A Rare Late Complication After Rituximab Treatment for Immune Thrombocytopenia. Report of 3 Cases and Systematic Review of the Literature
R. Levy 1, L. Galicier2, M. Mahévas3, D. Boutboul2, J. Moroch4, V. Loustau3, C. Guillaud3, L. Languille3, O. Fain5, P. Bierling3, M. Khellaf3, E. Oksenhendler2, B. Godeau3
1INSERM U1163, Imagine, PARIS, France
2Clinical Immunology, Hôpital Saint-Louis Assistance Publique Hôpitaux de Paris Université Paris Diderot Paris VII, PARIS, France
3Internal Medicine department, French national reference center for autoimmune cytopenias Hôpital Henri Mondor Assistance Publique Hôpitaux de Paris Université Paris Est Créteil, PARIS, France
4Anatomopathology department, Hôpital Henri Mondor Assistance Publique Hôpitaux de Paris Université Paris Est Créteil, PARIS, France
5Internal Medicine department, Hôpital Jean-Verdier Assistance Publique-Hôpitaux de Paris Université Paris XIII, PARIS, France
B-cell depletion with rituximab (RTX) is widely used in autoimmune diseases and as a second-line therapy in patients with immune thrombocytopenia (ITP). The incidence of RTX-induced hypogammaglobulinemia is challenging to appreciate due to a heterogeneous follow-up but also because of confounding factors such as concomitant immunosuppressive treatments. We retrospectively analyzed 189 ITP patients treated with RTX in 3 French referral centers. We conducted a systematic review of the literature on 32 studies (2001-2014) reporting the use of RTX in the setting of ITP. We also searched for case reports of hypogammaglobulinemia following RTX in ITP.
We observed 3/?189 patients who developed symptomatic hypogammaglobulinemia more than 2 years after RTX (initial Ig level was normal). All 3 patients presented recurrent severe infections. In 2 patients, the outcome suggested CVID. In the third one, the peripheral blood was devoid of CD19+CD20+ B-cells and B-cell precursors were impaired in bone marrow (Fig 1). Among 1245 ITP patients who received RTX for ITP in the literature, gammaglobulins were monitored prior to and after RTX in 351 cases (28%). This enabled to identify 21 patients who presented secondary hypogammaglobulinemia, 14 of whom having received concomitant dexamethasone. Finally, we found 4 case reports of patients with ITP and symptomatic hypogammaglobulinemia that was possibly related to RTX. This large analysis led us to recommend the monitoring of serum Ig level both before and repeatedly after RTX initiation for the treatment of ITP. Physicians should be aware of this rare but severe complication of RTX.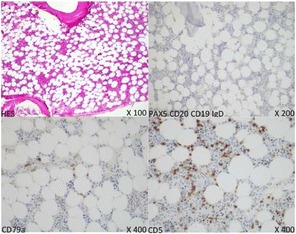
ESID-0359 29-Year Follow Up of Two Siblings with HLA Class II Deficiency
D. Pleguezuelo 1, C. Gianelli1, S. Sobre Casas1, P. Gonzalez-Urra1, A. De Andres1, J.L. Castañer1
1Clinical Immunology, Hospital Universitario Ramon y Cajal, Madrid, Spain
We describe a 29-year follow up of two siblings from a consanguineous Spanish family, living in Ceuta, a Spanish enclave in North-Africa, presenting a homozygous mutation in RFXANK gene which results in a 26 base-pair deletion and HLA class II deficiency, who are doing well without bone marrow transplantation (BMT).
Brother and sister were referred to our Clinic in order to evaluate a family history of premature deaths, repeated respiratory tract infections and diarrhea in the setting of a persistent reversal of the CD4+/CD8+ ratio and absence of HLA-DR expression on lymphocytes.
At diagnosis the brother (24y) showed normal immunoglobulin levels, whereas the sister (10y) had undetectable IgA. CD4+ T cells ranged from 7% to 15% of lymphocytes. In both patients HLA class II was unexpressed and HLA class I was diminished on fresh collected peripheral blood lymphocytes and monocytes. After 48?h incubation with IFN-gamma, a flow-cytometry analysis showed absence of expression of HLA-DR and 5-6 times lower HLA class I expression than age-matched healthy donors.
The brother presented with bronchiectasis, eccemas, two immune thrombocytopenic purpura episodes, otitis, sinusitis, and pneumonias, decreasing its incidence dramatically with age and supportive treatment (IVIG and cotrimoxazole prophylaxis). Although coeliac disease was not diagnosed persistent diarrhea was controlled with gluten-free diet. The sister presented with diarrhea and lower respiratory infections without pneumonias.
BMT was considered but not performed in neither the brother nor the sister because of their ages at diagnosis and their amazing follow-up without remarkable infections with supportive treatment.
ESID-0506 Mutation of Phosphoinositide-3-Kinase Regulatory Subunit Alpha 1, Leading to Hyperactivation of Phosphoinositide-3-Kinase, Results in an Immunodeficiency Resembling APDS
D.A. Parry1, S. Maisuria2, G. Argumugacani2, C.V. Logan1, P.?M. Wood2, A. Cant3, H.C. Gooi4, C.A. Johnson1, S. Nejentsev5, G. Doody6, S. Savic 2
1Institute of Biomedical and Clinical Sciences, University of Leeds, Leeds, United Kingdom
2Clinical Immunology and Allergy, St. James's University Hospital, Leeds, United Kingdom
3Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
4Allergy and Immunology, Kings College Hospital, London, United Kingdom
5Department of Medicine, University of Cambridge, Cambridge, United Kingdom
6Institute of Cancer and Pathology, University of Leeds, Leeds, United Kingdom
Introduction
Hyperactivating mutations in PIK3CD (MIM 602839), encoding the catalytic class IA phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) subunit p110d, cause novel immunodeficiency named activated-PI3K-delta immunodeficiency syndrome (APDS). We describe 3 patients from 2 unrelated families, with a heterozygous mutation in PIK3R1 resulting in a clinical phenotype remarkably similar to APDS.
Methods
Genetic mutation analysis by whole exom and Sanger sequencing. pAKT levels were assayed by flow cytometry and immunofluoresence
Results
All cases presented with recurrent sinopulmonary and ear infections from early childhood. Bronchiectasis was established in one patient by the age of 3 years. All had splenomegaly, two developed haematological malignancies. IgA and IgG2 subclass deficiency, normal/elevated levels of IgM and impaired anti-pneumococcal response found in all at presentation. Defective immunoglobulin class-switch recombination was demonstrated in one patient.
A splice donor variant in PIK3R1 (NM_181523.2:c.1425+1G>A) was found in the two related patients (Family A). The sporadic case had a heterozygous mutation in the neighbouring nucleotide in the same splice donor site (NM_181523.2:c.1425+2T>A). This resulted in removal of exon 11 from the mature transcript leading to in the in-frame removal of codons for amino acids 434-475 (NP_852664.1), which lie between two SH2 domains in a region required for regulation of p110 activity.
HEK and T-cell lines transfected with mutated forms of PIK3R1 demonstrate increased pAKT compared to wild type. pAKT appeared more highly expressed in the patients B cells compared to healthy controls following stimulation via BCR.WASp plays an important role
Conclusion WASp plays an important role
Mutations in PIK3R1 we described here lead to increased PI3K activity and APDS-like phenotype.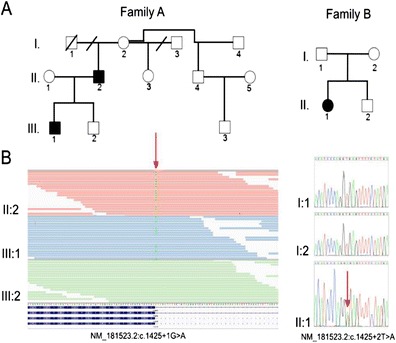
ESID-0167 Correlation Between FCGR3A-158 Polymorphism and Hypogammaglobulinaemia in Hematological Patients After Rituximab Treatment
E. Villegas 1, L. García-Maño2, A. Gutiérrez-García2, N. Martínez-Pomar1, J. Iglesias1, N. Matamoros1
1Immunology, Hospital Universitari Son Espases, Palma de Mallorca, Spain
2Hematology, Hospital Universitari Son Espases, Palma de Mallorca, Spain
INTRODUCTION
Rituximab is a monoclonal IgG1 antibody directed against the CD20 antigen. It is used to treat B-cell malignancies and induces almost a complete depletion of normal B lymphocytes in peripheral blood. In some cases, rituximab treatment causes prolonged hypogammaglobulinaemia. It has been reported that the presence of valine (V) at position 158 of FCGR3A (CD16a) has a higher affinity to human IgG than the phenylalanine (F) allele. The hypogammaglobulinaemia after rituximab treatment could be correlated with this polymorphism.
OBJECTIVES
The aim of this study is to demonstrate the possible correlation between immunoglobulin levels after rituximab treatment and the FCGR3A-158 polymorphism.
MATERIAL AND METHODS
This study included 60 hematological patients that received rituximab maintenance treatment. The subjects was tested for the FCGR3A-158 polymorphism by allele specific polymerase chain reaction (AS-PCR) and classified in three groups: F/?F, F/?V and V/?V.
RESULTS
Statistical analysis demonstrated that immunoglobulin levels were significantly lower in the homozygous V/?V group. On the other hand, when the overall genotype distribution of the control subjects was compared with that of the patients with hypogammaglobulinaemia, a significant skewing was observed. The FCGR3A-158V/V genotype was present at significantly higher frequency in these patients.
CONCLUSIONS
This study demonstrates than the levels of immunoglobulin after rituximab treatment are correlated with the FCGR3A-158 polymorphism. The confirmation of this result may imply the introduction of these studies as a diagnostic test and provide a more accurate rituximab treatment to avoid secondary hypogammaglobulinaemia.
ESID-0616 Positive Outcome of Stem Cell Transplantation for IL-21R Deficiency in the Absence of Liver Disease
B. Keller 1, P. Stepensky 2, O. Abuzaitoun3, A. Shaag4, B. Yaacov4, S. Unger1, M. Seidl5, M. Weintraub2, O. Elpeleg 4, K. Warnatz 1
1Centre for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany
2Pediatric Hematology-Oncology and Bone Marrow Transplantation, Hadassah Hebrew University Medical Center, Jerusalem, Israel
3Nablus Speciality Hospital, Nablus Speciality Hospital, Nablus, Palestine (via Israel)
4Monique and Jacques Roboh Department of Genetic Research, Hadassah Hebrew University Medical Center, Jerusalem, Israel
5Centre for Chronic Immunodeficiency and Department of Pathology, University Medical Center Freiburg, Freiburg, Germany
IL-21 is a potent cytokine regulating a multiplicity of immune functions. Here we report about a new patient with combined immunodeficiency (CID) due to a homozygous mutation in the IL-21 receptor (IL-21R) leading to absent IL-21R surface expression and signaling. Similar to previously reported patients, the patient presented with hypogammaglobulinemia and Pneumocystis jirovecii pneumonia. Importantly, this patient did not suffer from liver disease, implying that in the previously reported patients with IL-21R deficiency the sclerosing cholangitis was probably secondary to cryptosporidial infection.
Cellular immunodeficiency was not readily detected by extended screening (TRECs, immunophenotype, naïve CD4 T cells and T-cell proliferation). Even in-vitro T-cell function and numbers of circulating T-follicular-helper-cell were normal. However, a severe reduction of class-switched memory B cells and reduced serum IgG but increased IgM levels demonstrated the significance of IL-21 in the germinal center reaction and class switch in particular. Accordingly, B-cell proliferation and plasmablast differentiation were abrogated after IL-21/CD40 stimulation in-vitro and the up-regulation of CD25 and CD69 was disturbed. Elevated transitional B cells suggest an unexpected role of IL-21/IL21R in early B-cell development.
Given the presentation with Pneumocystis jiroveci pneumonia, the early diagnosis and the reported poor prognosis after secondary complications our patient underwent successful hematopoietic stem cell transplantation. The fact that IL-21R deficiency is missed as a form of profound combined immunodeficiency by standard testing, emphasis the role of targeted panel sequencing of CID genes for rapid diagnosis and timely definitive treatment.
ESID-0387 Prolonged Survival in Two Patients with a New Homozygous STIM1 Mutation
H. Schaballie1, R. Rodriguez2, E. Martin2, G. Frans1, L. Moens1, C. Lenoir2, X. Bossuyt1, S. Latour2, I. Meyts 3, C. Picard4
1Microbiology and Immunology, Catholic University of Leuven, Leuven, Belgium
2Laboratory of Lymphocyte Activation and Susceptibility to EBV infection INSERM UMR1163, Imagine Hôpital Necker - Enfants Malades, Paris, France
3Pediatric Immunodeficiencies, University Hospitals Leuven, Leuven, Belgium
4Laboratory of Human Genetics of Infectious Diseases INSERM UMR 1163, Imagine Hôpital Necker - Enfants Malades, Paris, France
Autosomal recessive mutations in the Ca2+ sensing protein stromal interaction molecule 1 (STIM1) result in a phenotype with combined immunodeficiency, immune dysregulation and non-progressive myopathy. Immune dysregulation was marked by auto-immune cytopenia in all 6 cases reported in the literature. Other manifestations include enamel defects, anhydrosis and iris hypoplasia. Only 2 patients are currently alive after being treated by autologous hematopoietic stem cell transplantation at the age of 1.2 and 6 years.
We present two siblings, born to consanguineous parents from Moroccan origin, with a new homozygous missense STIM1 mutation. The 21-yr old boy suffered from failure to thrive, recurrent bacterial and viral infections, bronchiectasis, hepatosplenomegaly, enamel hypoplasia, psoriasis, disabling myopathy and anhydrosis. He also had colitis with recurrent diarrhoea since the age of 10. His 8-yr old sister presented with recurrent and invasive bacterial infections which improved on intravenous immunoglobulin therapy at the age of 4. Furthermore, she suffered from dermatitis, enamel hypoplasia, dental abscesses, brittle nails and myopathy. The patients did not experience auto-immune cytopenia and had no iris hypoplasia. Sanger sequencing of STIM1 gene revealed a homozygous c.494C>A mutation in exon 4 (p.P165Q). An abolished TCR-mediated calcium flux and reduced STIM1 protein expression were demonstrated in the patients' T cells.
In conclusion, STIM1 deficiency must be considered in the evaluation of all patients with immunodeficiency and myopathy, even in patients who survive infancy and in patients without signs of immune dysregulation.
ESID-0461 Hematopoietic Cell Transplant for Autoimmune Lymphoproliferative Syndrome: A Single Center Experience
S. Chandrakasan 1, S. Kuerbitz2, J. Bleesing1
1Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children's Hospital Medical Center, Cincinnati, USA
2Stem cell transplantation Showers Family Center for Childhood Cancer and Blood Disorders, Akron Children's Hospital, Akron, USA
Autoimmune lymphoproliferative syndrome (ALPS) is a disorder of defective apoptosis, characterized by autoimmunity and lymphoproliferation. While most patients can be managed with immunosuppressive therapy, a proportion of patients develop severe refractory autoimmunity and/ or lymphoma, requiring rescue by Hematopoietic Cell Transplantation (HCT).
Methods: We analyzed the outcomes of 4 patients, ranging in age from 15 months to 17 years. Three patients had germ-line mutations in Fas, while the 4th had a somatic Fas mutation. Indications for HCT were refractory cytopenias, bone marrow aplasia, lymphoma, recurrent infections, and marked eosinophilia. Three received reduced intensity conditioning with campath, fludarabine and melphalan. One patient underwent myeloablative conditioning with busufan, cyclophosphamide and ATG. Two patients received matched unrelated donor (URD) HCT and two received single locus mismatched URD HCT.
Result: All patients achieved full donor chimerism, while grade II to IV acute GVHD developed in three patients. Significant autoimmune cytopenia was noted in two patients. Three patients survive at a median follow-up of 54 months, while the 4th patient died due to refractory GVHD and marked eosinophilia leading to coagulopathy and ARDS. Of the three surviving patients one has achieved complete T and B cell immune reconstitution, and two have achieved partial to complete T cell reconstitution without B cell reconstitution thus far.
Conclusion: HCT can correct immune defects associated with ALPS. However, managing post-HCT complications is challenging, inviting careful consideration of HCT guidelines for ALPS.
ESID-0306 Hematopoietic Stem Cell Transplantation for Primary Immune Deficiencies in France: First Overview by the Registry of Stem Cell Transplant for Immune Deficiencies in Europe (SCETIDE)
V. COURTEILLE 1, J. Provot1, B. Neven2, F. Suarez3, Y. Bertrand4, V. Barlogis5, F. Fouyssac6, S. Blanche2, N. Mahlaoui1, A. Fischer1
1CEREDIH, Necker-Enfants Malades Hospital, Paris, France
2Paediatric immunology and hematology Department, Necker-Enfants Malades Hospital, Paris, France
3Adult Hematology Department, Necker-Enfants Malades Hospital, Paris, France
4Paediatric immunology and hematology Department, Institut d'Hématologie et d'Oncologie Pédiatrique (IHOP), Lyon, France
5Paediatric hematology Department, La Timone Hospital, Marseille, France
6Paediatric immunology and hematology Department, University Hospital, Nancy, France
Introduction:
SCETIDE registry was created 30 years ago on behalf of the EBMT Inborn Error Working Party in order to improve knowledge on HSCT and care for patients with SCID and other PIDs (last report by Gennery AR et?al. JACI, 2010).
Objective and Methods:
The French National Reference Centre for PID (CEREDIH) at Necker hospital, Paris, has developed and supported a new online registry sytem (www.scetide.org/) including currently 3250 patients transplanted since 1968. French member centers were asked to register their new transplants and comparison with other registries enabled us to identify other hospitals which agreed to report their patients in SCETIDE.
As of May 2014, 860 patients (1015 transplants, 137 patients had a second HSCT) have been registered by 18 centres since 1972, mainly by Necker hospital with 655 patients (76%), Lyon (71 patients, 8%), Marseille (34 patients, 8%), Nancy (27 patients, 3%). Median age at transplant was 3.1 years. Half of patients had a predominantly T-cell deficiency (mainly SCID, 31%). Donor was identical sibling in 233 patients (27%), other related in 329 transplants (45%) and unrelated in 204 patients (23%). Source of HSCs was BM for 710 patients (83%), PBSC for 71 patients (8%) and CB in 60 transplants (7%). Overall survival is 60%.
Conclusion:
This is the first French nationwide overview of HSCT for PIDs. Key epidemiological indicators are very comparable to the whole European registry (3250 patients). Further detailed analyses (including survival) will be presented.
ESID-0680 Targeted Gene Therapy in the Treatment of X-Linked Hyper-IGM Syndrome
C.Y. Kuo 1, M.?D. Hoban2, A.V. Joglekar3, D.B. Kohn4
1Division of Allergy & Immunology Department of Pediatrics, University of California Los Angeles, Los Angeles, USA
2Department of M.I.M.G., University of California Los Angeles, Los Angeles, USA
3Department of Biology, California Institute of Technology, Pasadena, USA
4Departments of Pediatrics and M.I.M.G., University of California Los Angeles, Los Angeles, USA
X-linked hyper-IgM syndrome is characterized by absent IgG, IgA, and IgE with normal/elevated IgM due to defects in the CD40 ligand gene essential for immunoglobulin class switch recombination. Previous studies using gene therapy in murine models resulted in abnormal lymphoproliferation due to unregulated expression of the gene. Targeted genome modification may be a safe and effective alternative providing definitive treatment.
Custom TALENs targeting the 5’ untranslated region of the CD40L gene were created and their function validated using surveyor endonuclease assays. Allelic disruption levels of up to 31% in K562 cells were achieved. To evaluate the capacity for targeted integration of a cassette at the cut site, K562 and Jurkat cells were electroporated with the TALEN pair and a donor molecule containing a promoterless GFP reporter gene flanked by homology arms that parallel the cut site. In/Out PCR using a forward primer within the GFP region and a reverse primer in the genomic DNA demonstrated proper integration into K562 cells. Targeted insertion of the GFP reporter should also provide a measure of physiologic induction of CD40L expression. Thus, expression of the targeted GFP reporter was evaluated in Jurkat cells (which naturally express ˜60% CD40L), with levels of up to 10% detected by FACs and increasing to 22% upon phytohemagglutinin activation.
These results demonstrate that site-specific gene insertion using TALENs at CD40L is achievable and sets the stage for further work demonstrating that physiologic expression of the endogenous CD40L gene could provide a viable therapy for immune reconstitution in XHIM.
ESID-0545 Novel GMP-Compatible Protocol Employing an Allogeneic B-Cell Bank for Clonal Expansion of Allospecific Natural Regulatory T-Cells
S. Landwehr-Kenzel 1, F. Issa2, S.H. Luu3, M. Schmück3, A. Zobel3, H. Lei3, A. Thiel3, N. Babel3, K. Wood2, H.D. Volk3, P. Reinke3
1Berlin-Brandenburg Center for Reg. Therapies Dept. Ped. Pneumology and Immunology, Charité University Berlin, Berlin, Germany
2Transplantation Research Immunology Group Nuffield Department of Surgical Sciences, John Radcliffe Hospital, Oxford, United Kingdom
3Berlin Brandenburg Center for Regenerative Therapies, Charite Universitätsmedizin Berlin, Berlin, Germany
The adoptive transfer of natural regulatory T-cells (nTreg) has evolved as new therapeutic approach to reshape undesired immune reactivity in autoimmunity and transplantation towards “tolerance”. First clinical trials using polyclonal nTreg for adoptive T cell transfer demonstrated safety and hints of efficacy. However, low frequencies of (allo)antigen-specific nTreg among the polyclonal cell pool and a broad antigen non-specific suppression considerably limit the therapeutic potential. Recently, antigen-specific isolation and subsequent expansion of (allo)antigen-specific nTreg has successfully been achieved but the absolute nTreg yield is relatively low.
Here, we describe now a novel GMP-compatible expansion protocol for the generation of alloantigen-specific nTreg cell products based on the stimulation of nTreg by allogeneic activated B-cells of an EBV-free B-cell bank, designed to cover the majority of HLA-types. Phenotypically, the final alloantigen-specific nTreg product shows preserved characteristics of natural regulatory T cells and did not differ to polyclonally expanded nTreg pools. T-cell receptor repertoire analyses by next-generation-sequencing revealed impressive expansion by several log-steps of even very low-abundance alloantigen-specific nTreg clones. Functionally, we found superior target?allogen-specific suppression capacities in?vitro. Moreover, using a chimeric, clinically relevant humanized skin graft mouse model, we found HLA-specific suppression by alloantigen-specifically expanded nTreg to be significantly more effective as compared to polyclonally expanded cells.
In summary, we demonstrate functional superiority of allogen-specific nTreg products in preventing graft versus host disease. At the same time, we describe a novel approach for the generation of (allo)antigen-specific nTreg products which is characterized by high replicability and easy transferability to full GMP-standards.
ESID-0632 Correcting RAG1 Deficiency Using CRISPR/CAS9
L. Ott de Bruin 1, Y.N. Lee1, K. Musunuru2, L.D. Notarangelo1
1Immunology, Boston Children's Hospital, Boston, USA
2Department of Stem Cell and Regenerative Biology, Harvard university, Cambridge, USA
Background:
Mutations in the Recombination Activating Gene 1 (RAG1) are associated with heterogeneity of the clinical and immunological phenotype. Treatment for RAG1 deficiency is based on allogeneic hematopoietic stem cell transplantation, but suboptimal results have been observed after transplants from donors other than HLA-matched siblings. We recently reported a cellular platform to investigate genotype-phenotype correlation in human RAG1 (hRAG1) deficiency. In this system, Abelson-murine leukemia virus (A-MuLV) transformed Rag1 -/- pro-B cells that contain an inverted GFP cassette are transduced with a retroviral vector encoding for wild-type or mutant hRAG1, and recombination activity is assessed by GFP expression.
We are now using this cellular platform to correct hRAG1 mutations with (CRISPR)/Cas9. Cas9, a hRAG1-specific guide RNA (gRNA) and a suitable repair single stranded DNA oligonucleotide (ssODN) are being delivered to the cells.
Methods:
A-MuLV pro-B cells with hRAG1 mutations are nucleofected with gRNA, Cas9 and wild-type hRAG1 ssODN. GFP expression and induction of VDJ rearrangements at the Igh locus serve as evidence of gene correction.
Results:
Using surveyor assay and NGS, we have demonstrated that the CRISPR/Cas9 system is able to introduce DNA double strand breaks in the hRAG1 transgene. In a first experiment, attempting to correct the R394W (c.C1180T;c.C1180T) hRAG1 mutation, FACS analysis showed increase in the proportion of GFP+ cells (13.3% vs 0.06%).
Conclusion:
Our data suggest that CRISPR/Cas9 is able to introduce double strand breaks in the hRAG1 locus. We are currently using this strategy to correct a series of mutations in the nonamer binding region of hRAG1.
ESID-0535 Excellent Outcome but Delayed CD4+ T Cell Reconstitution Following Stem Cell Transplant with Serotherapy for Chronic Granulomatous Disease
E. Rivers 1, P. Hiwarker2, R. Hough3, C. Cale1, H.B. Gaspar2, R. Chiesa4, P. Amrolia4, W. Qasim2, P. Veys4, A. Worth1
1Immunology, Great Ormond Street Hospital for Children, London, United Kingdom
2Cellular & Molecular Immunology, Institute of Child Health University College London, London, United Kingdom
3Bone Marrow Transplant, University College London Hospitals, London, United Kingdom
4Bone Marrow Transplant, Great Ormond Street Hospital for Children, London, United Kingdom
33 stem cell transplants (SCTs) in 30 patients with Chronic Granulomatous Disease (CGD) were analysed from a single centre treating patients at 2 sites (Paediatric and Adolescent units) between 2002-2013. Outcomes were compared to 118 SCTs performed for Primary Immunodeficiency (PID) (2005-2010). The median age of transplant was higher in CGD patients (8.6 v 1.3 years). The majority of patients received reduced intensity/toxicity conditioning (CGD 71.4%, PID 57.7%). 45% of SCTs were conditioned using reduced busulphan/fludarabine/serotherapy, and consequently there was a higher incidence of serotherapy use in the CGD cohort (84.9% v 54.2%). Overall survival was 93.3% (82.2% in PID cohort). 3 patients had graft rejection and all were successfully transplanted by a second procedure. Grade 2-4 GVHD was seen in 21.2%.
CGD patients showed significantly slower CD3+CD4+ T cell reconstitution compared to other PID patients (median count at 1-year post: 0.42 v 0.75x109/L, p=0.014). No differences were seen between CD3+CD8+, or CD19+ populations. Significantly slower CD4+ reconstitution was retained when analysis was limited to serotherapy treated patients and cord transplants were excluded (0.25 v 0.75x109/L, p=0.002). This delay was also seen when CGD patients were compared to all SCT recipients (not exclusively PID) over 2compared to all SCT recipients (not exclusively PID) years of age, receiving serotherapy conditioned SCT (n=65).
Outcome of patients with CGD treated by SCT is now excellent, however CD3+CD4+ T cell reconstitution appears slower than for other conditions. As CGD is treated at a younger age we will learn whether this is a disease specific phenomena or an age of transplant confounder.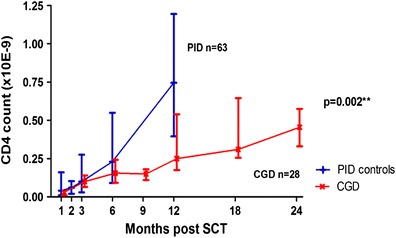
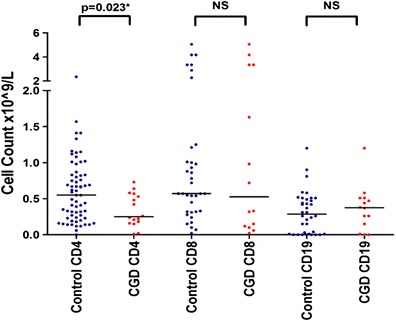
ESID-0385 Stem Cell and T-Cell Gene Therapy Using Sin-Lentiviral Vector in Type 3 Familial Hemophagocytic Lymphohistiocytosis
T. Soheili 1, J. Riviere1, E. Verhoeyen2, A. Durand1, F. Sepulveda1, G. de Saint Basile1, A. Galy3, A. Fischer1, I. André-Schmutz1, M. Cavazzana1
1U1163 Inserm, Imagine Institute, Paris, France
2ENS, Lyon University, Lyon, France
3U951 Inserm, Genethon, Evry, France
Familial hemophagocytic lymphohistiocytosis (FHL) is characterized by prolonged fever, hepatosplenomegaly and pancytopenia. Type 3 of FHL (FHL3), which accounts for 30-35% of all FHLs results from mutatios in UNC13D gene encoding Munc13-4 protein. Munc13-4 controls fusion of lytic granules with the plasma membrane in cytotoxic lymphocytes and its defect leads to impaired cytotoxic activity in T and NK lymphocytes. The only curative method for FHL3 is allogenic hematopoietic stem cell (HSC) transplantation. For those patients without compatible bone marrow donor, gene therapy could represent a therapeutic option. As Munc13-4’s function is to allow proper cytotoxic activity in mature CD8+ T cells, both HSCs and CD8+ T cells may constitute targets for gene correction.
Our study is based on a comparative analysis to investigate the efficacy and safety of stem cell and T-cell gene therapy for FHL3. To this end we have generated SIN-lentiviral constructs expressing Munc13-4 and used them to produce either VSVG or T-cell specific pseudotyped lentiviral vectors. We first demonstrated that transduction of FHL3 patients’ CD8 effector cells restored their cytotoxic function to near-normal levels. Moreover we noted that the overexpression of Munc 13-4 in normal human HSCs did not alter in?vitro differentiation of these cells towards T-cells.
These preliminary results will be followed by ?ex?vivo gene transfer experiments in Munc13-4 deficient “Jinx” mice to further investigate the functional restoration and toxic effects. This strategy, if approved, could offer a safe therapeutic method for FHL3 patients and other genetic or acquired dysfunctions of T-lymphocytes.
ESID-0120 Valutation of Antibody Response to PEG-ADA in Patients with Adenosine Deaminase Deficiency
R. Baffelli 1, L. Notarangelo2, M. Zucchi1, F. Bolda1, A. Beghin1, A. Caruso3, F. Porta2, A. Lanfranchi1
1Stem Cell Lab - Children's Hospital, Spedali Civili Brescia, Brescia, Italy
2Oncohematology and BMT Unit - Children's Hospital, Spedali Civili Brescia, Brescia, Italy
3Microbiology and Virology Unit - Children's Hospital, Spedali Civili Brescia, Brescia, Italy
Adenosine Deaminase (ADA) deficiency is an inherited autosomal recessive immunodeficiency which represents about 10-15% of SCID.
Immunodeficiency is due to accumulation of toxic metabolites that can’t be degraded, collected in plasma and transported into cells where they reach high levels.
These patients required decisive therapeutic protocols to reconstitute the immune function.
Bone marrow transplantation is the first choice of treatment but, in absence of a HLA identical donor, enzyme replace therapy with Peg-ADA is the faster and more effective method for elimination of toxic metabolites.
In our center 27 ADA patients were found and we following therapy of 17 of these, 10 undergoing BMT, 7 in treatment with Peg-ADA.
The metabolic correction takes place normally in 2-4 months. The recovery of immune competence is variable but results in a rapid recovery in number of lymphocytes. However, long-term evaluation showed a gradual decline of T lymphocyte.
Long term follow up shows that Peg-ADA patients are detoxified 0.0098 micromol/mlRBC dAdenosine (healthy 0.004±0.001micromol/mlRBC) but have fewer lymphocytes (300/mmc, range:360-1500/mmc).
4 of 7 patients after an initial increase of levels of antibodies have maintained very low levels.
3 of these patients, in treatment from 15, 16 and 17 years, we have found high amount of antibodies (75 UI 470 UI, 24 UI) and correlates with decrease in circulating lymphocytes and ADA plasma activity.
Based on our patients, presence of neutralizing antibodies reduces activity of circulating pegADA enough to allow the increase of toxic metabolites and impair immune recovery and effectiveness of the therapy.
ESID-0598 Combined Newborn Screening for Familial Hemophagocytic Lymphohistiocytosis and Severe T and B Cell Immunodeficiencies
S. Borte 1, M. Meeths2, M. Nordenskjöld3, L. Hammarström1, J.I. Henter2, U. von Döbeln1, Y.T. Bryceson4
1Department of Laboratory Medicine, Karolinska Institutet, Stockholm, Sweden
2Department of Women’s and Children’s Health, Karolinska Institutet, Stockholm, Sweden
3Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden
4Department of Medicine, Karolinska Institutet, Stockholm, Sweden
Newborn screening assays using T-cell receptor (TRECs) and kappa-deleting recombination excision (KRECs) circles have successfully been applied to the identification of patients with severe combined immunodeficiencies (SCID) and primary agammaglobulinemias. The decision-matrix based on the Wilson&Jungner criteria for future newborn screening assays proposed familial hemophagocytic lymphohistiocytosis (FHL) to be a potential candidate for expended screening assays of severe primary immunodeficiency diseases. We describe a common genetic inversion in the UNC13D gene – frequent in the northern European countries - that causes FHL type 3, which is amenable for newborn screening. In a triplex newborn screening assay that works with regular punches from routine Gurthrie cards, we present a novel combined assay for the screening of TREC, KREX and UNC13D wild-type copies. Using a retrospective cohort analyses we show, that this assay enables a rapid, cost-efficient and reliable method to detect SCID, XLA and FHL type 3 all in one newborn screening assay.
ESID-0143 Hereditary Folate Malabsorption Due to Novel Compound Heterozygous Mutations in SLC46A1 Exhibiting Severe Combined Immunodeficiency
N. Chida 1, M. Yamada2, K. Kishimoto3, R. Kobayashi3, K. Kobayashi3, O. Ohara4, T. Ariga2
1Department of Dentistry for Children and Disabled Person Graduate School of Dental Medicine, Hokkaido University, Sapporo, Japan
2Department of Pediatrics Graduate School of Medicine, Hokkaido University, Sapporo, Japan
3Department of Pediatrics, Sapporo Hokuyu Hospital, Sapporo, Japan
4Department of Human Genome Technology, Kazusa DNA Research Institute, Chiba, Japan
Hereditary folate malabsorption (HFM) is a rare autosomal recessive disorder characterized by intestinal folate malabsorption and impaired folate transport into the central nervous system. Typical HFM patients suffer from megaloblastic anemia, combined immunodeficiency, and neurologic abnormalities. A 4 month-old female infant presented with Pneumocystis pneumonia and developmental delay. The findings of megaloblastic anemia, thrombocytopenia, absent T cell proliferative response, hypogammaglobulinemia, and low serum folate level prompted us to study the responsible gene for HFM. Two novel compound heterozygous mutations of SLC46A1 encoding proton-coupled folate transporter (PCFT) were identified in this patient: c.566G>T (p.G189V) and c.1166-285G>T (p.A389GfsX20). G189 is a conserved residue located in the fifth transmembrane domain of PCFT directly involved in the binding and translocation of folate and proton. The latter mutation in intron 3 was shown to generate a cryptic splice donor site for a 168-bp insertion between exon 3 and 4, leading to premature termination in the middle of the insertion. Transient transfection study demonstrated both mutants were associated with markedly reduced PCFT expression, indicating these mutations are associated with loss of function of PCFT. This patient achieved recovery of her immunological and neurophysiological status with parenteral administration of folinic acid which was initiated from 4 months of age. HFM should be recognized as one of the primary immunodeficiency diseases which need early diagnosis and treatment. Optimal dose of folinic acid is still a matter of controversy especially with regard to achieving normal neurological development. A large scale long-term follow-up study is required to elucidate this issue.
ESID-0317 Atypical Combined Immune Deficiency Associated with Skin Granulomata and Chronic NK Cell Proliferative Disease Linked to Heterozygous Compound Mutations in RAG2 Gene
V. Daza 1, C. Bethune2, D. Tucker3, P. Roberts3, C. Dearden4, E. Morris1, K.C. Gilmour5, S. Bibi6, S. Loughlin6, S.O. Burns1
1University College London Institute of Immunity and Transplantation and Department of Immunology, Royal Free London NHS Foundation Trust, London, United Kingdom
2Department of Immunology, Derriford Hospital NHS Foundation Trust, Plymouth, United Kingdom
3Department of Haematology, Torbay Hospital South Devon Healthcare NHS Foundation Trust, Torquay, United Kingdom
4Department of Haemato-Oncology, The Royal Marsden NHS Foundation Trust, London, United Kingdom
5Department of Immunology, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
6North East Thames Regional Genetics Laboratory, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
Loss of function RAG mutations in humans cause immunodeficiency and dysregulation with variable clinical phenotype, ranging from severe combined immunodeficiency and Omenn syndrome to milder immunodeficiencies, in some cases associated with granuloma formation.
We report a 19 year-old male who presented at the age of 15 with cutaneous granulomas on his leg which were resistant to treatment. Although he did not receive systemic immunosuppression he was found to have panhypogammaglobulinaemia and T-cell lymphopoenia without a significant past history, except for one hospital admission for a respiratory tract infection at the age of 13 years. Molecular analysis revealed three heterozygous amino acid–changing variations in RAG2 gene, which were detected by next generation sequencing using a SureSelct custom capture library, confirmed by Sanger sequencing. We identified c.104C>T (p.Gly35Val) previously reported as heterozygous mutation in SCID; as well as two single nucleotide variants, one of which is predicted to be damaging in Silico.
He was noted to have an elevated NK cell count at initial testing that rose over time, associated with a fall in neutrophil numbers and hemoglobin. A subsequent bone marrow aspirate demonstrated a clonal NK cell infiltration with associated red cell aplasia. He is currently maintained on methotrexate with immunoglobulin replacement therapy and antimicrobial prophylaxis but continues to require regular blood transfusions. Bone marrow transplantation is planned.
RAG mutations can present outside early childhood with combined immunodeficiency and granulomas. To our knowledge chronic NK cell proliferative disease has not been described, extending the clinical spectrum of this genetic defect.
ESID-0046 Genotype-Phenotype Correlation of Recombination and DNA Repair Activity of Disease Causing Mutations in Human DCLRE1C
K. Felgentreff 1, Y.N. Lee1, F. Frugoni1, L. Du1, M. van der Burg2, B. Sleckman3, L.D. Notarangelo1
1Division of Immunology, Boston Children's Hospital, Boston, USA
2Department of Immunology, Erasmus MC, Rotterdam, Netherlands
3Department of Pathology and Immunology, Washington University School of Medicine, St Louis, USA
The endonuclease Artemis, encoded by DCLRE1C, is a key player in the non-homologous end-joining (NHEJ) pathway. Besides its specific function in hairpin opening during the V(D)J recombination process critical for lymphocyte development, it is also involved in the repair of DNA double strand breaks. Patients with Artemis deficiency usually present with Severe Combined Immunodeficiency (SCID) and radiosensitivity, but milder variants are also known.
To test for function of Artemis mutants, Artemis-/- pro-B cells were immortalized with the Abelson murine leukemia virus (A-MuLV) and engineered to contain an inverted GFP cassette flanked by recombination signal sequences, and then transduced with a retroviral vector enconding for either wild-type or each of >40 mutant human Artemis proteins. GFP expression was used to measure recombination activity; DNA repair proficiency was studied by analyzing H2AX de-phosphorylation after ionizing irradiation (IR).
Residual recombination and considerable DNA repair activity were observed even in the absence of Artemis. Frameshift mutations disrupting the metallo-ß-lactamase or ß-CASP region of the protein were deleterious, but mutations (including deletions) in the C-terminal domain retained considerable activity. Patients reported with an atypical/leaky presentation of SCID, were mostly found to be compound heterozygous for a null and a hypomorphic allele. These leaky mutations retained up to 80-100% of WT activity for both recombination and DNA repair, possibly reflecting also protein overxpression in the system.
In summary, we report on a cellular model that allows rapid analysis of functional activity associated with human DCLRE1C mutations.
ESID-0543 PCR Array Based Approach to Find IL-6/STAT3 Mediated Downstream Target Genes Involved in the Skeletal Pathogenesis of Hyper-IGE Syndrome
S. Goel 1, D. suri2, S. Singh2, R. Minz1, A. Rawat2, S. Sehgal1, B. Saikia1
1Immunopathology, Post Graduate Institute of Medical Education and Research, Chandigarh, India
2pediatrics, Post Graduate Institute of Medical Education and Research, Chandigarh, India
Introduction:
Autosomal dominant Hyper-IgE Syndrome (AD-HIES)is associated with skeletal and dental abnormalities in addition to immunological defects and caused by mutation in STAT3 gene. The role of STAT3 in the pathogenesis of the skeletal manifestations is however poorly elucidated.
Objective:
To study the expression of genes involved in bone morphogenesis inSTAT3 related HIES patients by PCR Array.
Materials and Methods:
PBMC’s from healthy controls (HCs, n=2), mutation proven HIES patients (n=2), STAT3-/-PC-3 and STAT3+/+LNCaP cells were stimulated with IL-6 (30 ng/ml) for 2 hrs. Total RNA was extracted and quantitative PCR Array was performed to study the relative mRNA expression of 43 selected genes.
Results:
Expression of RORyt, V.D3 receptor, SOCS3, Osteopontin, LRP5 and gp130 wasmore than 2-fold higher in HCs and LNCaP cells after stimulation with IL-6 whereas their expressions were unchanged in mutation positive patients and PC-3 cells.Similarly, RUNX2 expression was also up-regulated in HCs whereas along with DKK2 and Epigen, it was down-regulated in mutation positive patients.Osteopontin showed a 13-fold up regulation in HCs but no change was observed in HIES patients when compared to respective un-stimulated cells. Moreover, expression of Osteopontin in HIES patients was only 0.08 fold compared to HCs and suggests its defective regulation through IL-6 mediated STAT3 signaling.
Conclusion:
Osteopontin has diverse but significant role in the regulation of immune and developmental systems. Altered expression of Osteopontin through STAT3 might be involved in the manifestation of skeletal abnormalities in HIES.
ESID-0358 Hypomorphic Mutation in DOCK8 Causes Combined Immune Deficiency Without Hyper-IGE
A.K. Kienzler 1, P.A. van Schouwenburg1, I. Marwah1, J.M. Taylor2, K. Thomson2, J.C. Taylor3, E. Blair4, H. Chapel1, S.Y. Patel1
1NDM Experimental Medicine Division, University of Oxford, Oxford, United Kingdom
2Oxford NHS Regional Molecular Genetics Laboratory, Oxford University Hospitals NHS Trust, Oxford, United Kingdom
3Oxford Biomedical Research Centre, Wellcome Trust Centre for Human Genetics, Oxford, United Kingdom
4Department of Clinical Genetics, Oxford University Hospitals NHS Trust, Oxford, United Kingdom
We report an eight year old girl presenting with an undefined combined immunodeficiency characterised by severe bacterial respiratory tract infections leading to bronchiectasis, decreased serum IgG and IgM, absent vaccination responses, CD4 lymphopenia and absent TRECs. We undertook whole exome sequencing of the trio (patient and parents) and screened for variants present in 204 genes associated with primary immunodeficiencies. We identified a previously unreported single nucleotide insertion in DOCK8 leading to a premature stop codon (c.6019dupT, p.Tyr2007fs) on the maternal allele predicted to lead to the absence of the last 81 N-terminal amino acids. In addition we found a large deletion (Chr9:g.204193-g.343954) encompassing exons 1-14 on the paternal allele. Loss-of-function mutations in DOCK8, encoding a guanine-nucleotide exchange factor for CDC42 and RAC, cause the most common form of autosomal-recessive hyper-IgE syndrome. Typically, patients present with eosinophilia, recurrent sino-pulmonary infections, severe cutaneous viral infections, severe food allergies, and massively elevated serum IgE levels; the latter three absent in our patient. Although it is recognized that patients may present with a spectrum of disease related symptoms, molecular mechanisms explaining phenotypic variability in patients with DOCK8 mutations need to be defined. To date all reported DOCK8 mutations cause null alleles and consequently DOCK8 expression is completely absent. In contrast, in our patient we have confirmed expression of a truncated DOCK8 protein. Therefore, the less severe phenotype in our patient might be accounted for by residual function of a hypomorphic DOCK8 allele. Functionality of the truncated DOCK8 protein is currently investigated.
ESID-0526 Phenotypic and Genetic Heterogeneity in Congenital Neutropenia – Exome Sequencing Reveals Overlap with Other Primary Immunodeficiency Disorders
N. Kohistani 1, J. Puchalka1, T. Racek1, N. Rezaei2, E. Unal3, S. Unal4, R. Skinner5, A. Aglaguel6, F. Ailal6, C. Klein1
1Dr. von Hauner Children's Hospital, Ludwig Maximilians University, Munich, Germany
2Children's Medical Center, Tehran University of Medical Sciences Research Center for Immunodeficiencies, Tehran, Iran
3Faculty of Medicine, Erciyes University, Kayseri, Turkey
4Division of Pediatric Hematology, Hacettepe University, Ankara, Turkey
5Department of Paediatric and Adolescent Haematology and Oncology and Children's BMT Unit, Great North Children's Hospital, Newcastle upon Tyne, United Kingdom
6Clinical Immunology Unit Hospital-University Center Ibn Rochd, King Hassan II University, Casablanca, Morocco
Congenital neutropenia syndromes are a rare and heterogeneous disease entity. Despite recent progress, the genetic cause remains elusive in 30-50% of patients.
In an attempt to find novel disease causing mutations we performed whole exome sequencing in 55 trios (patient and both parents), 14 incomplete trios (one parent missing), and 10 singleton patients. All patients were referred for genetic workup of congenital neutropenia. We identified 15 patients with mutations in known neutropenia-related genes such as G6PC3 (n=5), ELANE (n=2), JAGN1 (n=2), CSF3R, CXCR4, HAX1, GFI1, SBDS, and VPS45 (n=1 in each case). This series also includes several novel mutations (see table). Of note, in three cases we found potentially disease-causing mutations in genes involved in distinct immunological diseases: IL7R (Pat #1; compound heterozygous: c.376A>G, c.1165G>C), PTPRC (Pat #2; homozygous: c. 2539A>G) and PSTPIP1 (Pat #3; heterozygous: c.748G>A). Mutations in IL7R and PTPRC are known to cause severe combined immunodeficiency (SCID). PSTPIP1 mutations cause the autoinflammatory disease PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum and acne). Patients #1 and #2 came to clinical attention because of recurrent bacterial infections and low neutrophil counts, they also had mild lymphopenia. Patient #1 had also thrombocytopenia and splenomegaly. Pat #3 presented with hepatosplenomegaly, lymphadenopathy and recurrent inflammatory episodes.
Our findings suggest that congenital neutropenia is a phenotypic trait overlapping with several other genetically defined diseases of the immune system. Multiple other non-recurring genetic variants were identified that may serve as a reference dataset for the potential future identification of other genes associated with congenital neutropenia.
| Gene | Transcript | Genotype | Variation | Novel | Patient count |
| G6PC3 | NM_138387 | Homozygous | c.2T>G, p.M1R | Yes | 1 |
| c.583-G,p.Glu195fs | Yes | 1 | |||
| c.758G>A, p.R253H | Yes | 1 | |||
| c.323C>T, p.P108L | Yes | 2 | |||
| ELANE | NM_001972 | Heterozygous | c.640-G, p.Gly214fs | No | 1 |
| c.688-G, p.Asp230fs | No | 1 | |||
| JAGN1 | NM_032492 | Homozygous | c.297C>G, p.Y99* | No | 1 |
| c.485A>G, p.Q162R | No | 1 | |||
| CSF3R | NM_000760 | Homozygous | c.922C>T, p.R308C | No | 1 |
| CXCR4 | NM_003467 | Heterozygous | c.954-C,p.R308C | Yes | 1 |
| GFI1 | NM_005263 | Heterozygous | c.1208A>G, p.K403R | No | 1 |
| HAX1 | NM_006118 | Homozygous | c. 180-A, p.Gu60fs | No | 1 |
| SBDS | NM_016038 | Homozygous | c.258+2T>C | No | 1 |
| VPS45 | NM_007259 | Homozygous | c.712G>A, p.E238K | No | 1 |
ESID-0361 Successful Interferon-Alpha Treatment of a Herpes Simplex-Triggered Eyelid Tumor in DOCK8 Deficiency
C. Papan 1, B. Hagl1, V. Heinz1, M.H. Albert1, O. Ehrt2, J. Sawalle-Belohradsky1, J. Neumann3, M. Ries4, P. Bufler1, A. Wollenberg5, E.D. Renner1
1University Children's Hospital at Dr. v. Haunersches Kinderspital, Ludwig Maximilian University, München, Germany
2Department of Ophthalmology, Ludwig Maximilian University, München, Germany
3Department of Pathology, Ludwig Maximilian University, München, Germany
4Children's Hospital, Klinikum Memmingen, Memmingen, Germany
5Department of Dermatology and Allergology, Ludwig Maximilian University, München, Germany
Background: DOCK8 (dedicator of cytokinesis 8) deficiency is the most common form of autosomal-recessive hyper-IgE syndrome caused by loss-of-function mutations in the gene DOCK8, presenting as combined immunodeficiency with eczema, food allergies, recurrent respiratory tract and skin infections, and an elevated risk of malignancies. Stem cell transplantation is the treatment of choice but acute management of recurrent viral skin infections can be challenging.
Case presentation: We report a girl of consanguineous origin who suffered from eczema, multiple food allergies, asthma and recurrent severe infections since early infancy. At 15 years of age DOCK8 deficiency was molecularly defined (homozygous c.3120+1g>t leading to exon 25 skipping). She was started on intravenous immunoglobulin substitution therapy and antibacterial and antiviral prophylaxis. At 17 years of age she presented with a slowly progressing tumorous swelling of the eyelids. The diagnosis of a herpes simplex virus (HSV)-triggered blepharoconjunctivitis was established by high levels of HSV type 1 DNA (150000 geq/ml) in the biopsy while malignancy was ruled out. Initial therapy consisting of vancomycin, acyclovir, methylprednisolone, and high-dose intravenous immunoglobulins failed. Literature search suggested that interferon-alpha (IFN-a) might overcome DOCK8 deficiency by interfering with viral replication and augmenting antigen cross-presentation and NK cell cytotoxicity. IFN-a application resulted in a rapid and long-term regression of the tumor without relevant adverse events.Conclusion: IFN-a has the potential to be a good therapeutic option in DOCK8 deficiency, especially for patients ineligible for stem cell transplantation or those in need of acute intervention.Reference: J Allergy Clin Immunol. 2014 May;133(5):1456-8.
ESID-0715 T-Cells and B-Cells Abnormalities in 7 Children with Ataxia Telangiectasia with ATM Mutations
S. Sharapova 1, I. Gyryanova1, S. Vahliarskaya2, O. Pashchenko2, I. Kondratenko2, M. Belevtsev3
1Research, Belarusian Research Center for Pediatric Oncology Hematology and Immunolog, Minsk region, Belarus
2Clinical immunology, Russian Children's Clinical Hospital, Moscow, Russia
3Research, Belarusian Research Center for Pediatric Oncology Hematology and Immunology, Minsk region, Belarus
Introduction: Ataxia telangiectasia (AT) is an autosomal recessive multisystem DNA repair disorder caused by mutations in the ataxia telangiectasia mutated (ATM) gene. In our study we aimed to analyze T- and B-cell subsets in a cohort of patients with ataxia-telangiectasia and reveal abnormalities in their immune system.
Materials and Methods: The study included seven patients (5 boys and 2 girls, aged from 2 to 9 years) with confirmed ataxia-telangiectasia diagnosis by ATM gene sequencing from Belarus and Russia. Flow cytometric analysis of the T- and B-cell subsets including naïve and memory (CD4+/CD8+CD45RA+/CD45RO+), recent thymic emigrants (CD4+CD31+CD45RA+), regulatory T-cells (CD4+CD127-CD25+), transitional (CD19+CD38++IgM++), (CD21lowCD38high), naïve (CD19+CD27-IgM+/IgD+), switched memory (CD19+CD27+IgM-/IgD-), marginal zone B cells (CD19+CD27+IgM+/IgD+) and putative regulatory B cells (CD19+CD24highCD38high). All results were processed using methods of non-parametric statistics and compared with healthy controls.
Results: Total lymphocyte generation was decreased compared to control (P=0.003). As a result, decreased absolute number of all T- and B-cell subsets was revealed in our study. Percentage of T-cells CD3+ (P=0.005), CD8+ (P=0.004), CD4+/CD8+ naive (P<0.0001, P=0.006), Tregs (P=0.013), RTE (P<0.0001), B cells (CD27+IgM-) (P=0.004), putative Bregs (P=0.004) was decreased.
Percentage of CD3+HLA-DR P<0.0001, CD4+/CD8+CD45RO+(P<0.0001, P<0.001), CD21lowCD38high (P=0.0036) was increased compared to control.
Conclusions: Immunodeficiency in ataxia telangiectasia patients affects both T- and B-cells. We observed severe T-cell deficiency in AT patients because of the almost complete absence of naïve T-cells and RTE and significant dysregulation of B-cell subsets that appear as a high expansion of functionally immature B-lymphocytes and lack of putative regulatory B-cells.
Topic: Inflammation
ESID-0351 Lack of Trophoblast-Specific Isoforms of CD74 May Protect Pregnancy
W. Alabdulmenaim 1, H. Al Ssadh1, N. Fernández1
1School of Biological Sciences, University of Essex, Colchester, United Kingdom
For a successful pregnancy, the maternal immune system must not only protect the allogenic fetus from rejection but the host must also maintain its defence against harmful pathogens. Recent reports have shown that CD74, a membrane-bound protein involved in antigen presentation is involved in infection and chronic inflammatory conditions. CD74 also acts as a Macrophage Migration Inhibitory Factor receptor. We have set out to study the role of IFN-? and LPS in the regulation of CD74 protein in trophoblast derived cell lines (JEG-3 and ACH3P) associated with successful pregnancy. Methods: The expression of CD74 was assessed by RT-PCR, flow cytometry, western blot and confocal fluorescence microscopy. Results: Our results from RT-PCR have indicated that IFN-? and LPS up regulate the expression of CD74 mRNA. Flow cytometry provides data showing an up-regulated intracellular expression of CD74 in contrast to the cell surface expression on both cell lines using IFN-? and LPS. Although, no protein band for CD74 on both cell lines using IFN-? whereas, was detected at molecular weight 35?kDa using LPS at western blot results. Conclusion: CD74 is up regulated using LPS but not IFN-?. This is at variance with expression of most immunological receptors, which are upregulated with IFN-?. Thus, the lack of specific isoforms of CD74 expression on cell surface of human trophoblast cells in response to infection and pro-inflammatory cytokine suggest a protective role in pregnancy.
ESID-0536 Disturbed Systemic Inflammatory Response in Patients with 22Q11.2 Deletion (DIGEORGE) Syndrome
D. Aresvik 1, K. Lima2, T. Overland3, T.E.(. Mollnes4, T.G.(. Abrahamsen5
1Department of Paediatric Research, Oslo University Hospital, Oslo, Norway
2Division of Medicine, Akershus University Hospital, Lørenskog, Norway
3Women and Children’s Division, Oslo University Hospital, Oslo, Norway
4Research Laboratory, Nordland Hospital and University of Tromsø, Bodø and Tromsø, Norway
5Women and Children’s Division, Oslo University Hospital and University of Oslo, Oslo, Norway
6Department of Immunology, Oslo University Hospital, Oslo, Norway
7University of Tromsø, Tromsø, Norway
8University of Oslo, Oslo, Norway
Background and aim: The 22q11.2 deletion syndrome (DS), also known as DiGeorge syndrome, is a genetic disorder with an estimated incidence of 1:4000 births. These patients may suffer from disorders of many organ systems, with cardiac malformations, thymic hypoplasia/aplasia, hypoparathyroidism, cleft palate and psychiatric disorders being the most frequent. The incidence of autoimmune diseases is increased in older patients. The aim of the present study was to examine whether the 22q11.2 DS patients have a disturbed systemic inflammatory reaction as compared to controls.
Methods: Patients with a proven deletion of chromosome 22q11.2 by in situ hybridization (FISH) test or multiplex ligation-dependent probe amplification assay (MLPA) (n=57) and a group of age and sex matched healthy controls (n=54) were included in the study. Serum levels of 27 cytokines, chemokines and growth factors were analysed using a multiplex cytokine assay. For comparison of two groups, the Mann-Whitney U test was used.
Results: The 22q11.2 DS patients had distinctly and significantly raised serum levels of granulocyte colony-stimulating factor (G-CSF) (p<0.001) and interferon-inducible protein 10 (IP-10) (p<0.001) compared to healthy controls. In contrast, the levels of interleukin-1 receptor antagonist (IL-1ra) (p=0.027), IL-12 (p=0.041), IL-17 (p=0.018) and basic fibroblast growth factor (FGF basic) (p=0.010) were slightly, but significantly lower in the 22q11.2 DS patients compared to controls.
Conclusion: The present data support a disturbed systemic inflammatory balance in patients with 22q11.2 DS as compared to healthy controls. This might have implications for the pathogenesis of the syndrome and development of disease manifestations such as autoimmunity.
ESID-0016 Early Switch from Inflammatory to Anti-Inflammatory Cytokine in Chlamydia Trachomatis-Infected Macrophages Initiates Chlamydia Chronic Course
A. Azenabor 1, J. Cintron-Cuevas1, H. Schmitt1, V. Bumah1
1Biomedical Sciences, University of Wisconsin-Milwaukee, Milwaukee, USA
The reason for the subtle inflammatory change during Chlamydia trachomatis infection is unclear. Our findings explain how macrophage pro-inflammatory response switches to anti-inflammatory response during C.?trachomatis infection of mixed culture of macrophages and Jurkat T-cells. We assessed the establishment of productive infection in individual or mixed cell culture models and determined the status of C.?trachomatis in the cells by monitoring the proportions of the estimated IFUs that shed HSP-60 or MOMP. Also, the time-course expression of IL-12, IL-10 and IFN-? or IL-12R, IL-10R, and IFN-?-R was assessed. Finally, we determined the early changes in cytokine elaboration during intracellular Ca2+ impairment. There was productive infection in all individual and mixed cell culture models. The proportions of IFU that shed HSP-60 was heightened in infected THP-1/Jurkat mixed culture model, while shedding of MOMP was higher in infected macrophage/Jurkat mixed culture and infected macrophages only. There was profound early elaboration of IL-10, varying significantly from IL-12 and IFN-? in all infected cells except in the case of Jurkat; where all cytokine elaboration was down-regulated. The IL-10-receptor was up-regulated in infected macrophage/Jurkat cells and THP-1/Jurkat cells compared with other models in which IL-12 and IFN-?-receptors were predominantly expressed. The levels of IL-12 and IL-10 were up-regulated in 1 hr or 3 hr respectively following the impairment of intracellular Ca2+ in macrophage/Jurkat model. The implication of these findings is that chlamydia mediates an early switch from inflammatory to anti-inflammatory function in macrophages by down-regulating IFN-? in Jurkat cells, thus evoking a chronic infection course.
ESID-0146 TXNIP Suppresses Endotoxic Shock via Inhibiting Nitric Oxide Synthesis
I. Choi 1
1Immunotherapy Research Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon, Korea
Thioredoxin-interacting protein (TXNIP) has multiple functions, including tumor suppression and involvement in cell proliferation and apoptosis. However, its role in the inflammatory process remains unclear. In this report, we demonstrate that TXNIP-/- mice are significantly more susceptible to lipopolysaccharide (LPS)-induced endotoxic shock. In response to LPS, TXNIP-/- macrophages produced significantly higher levels of nitric oxide (NO) and inducible nitric oxide synthase (iNOS), and an iNOS inhibitor rescued TXNIP-/- mice from endotoxic shock-induced death, demonstrating that NO is a major factor in TXNIP-mediated endotoxic shock. Subsequently, when TXNIP-/- mice were injected with LPS or E.?coli, IL-1ß and IL-18 production was reduced with increased S-nitrosylation of NLRP3 compared to wild-type controls. Taken together, these data demonstrate that TXNIP is a novel molecule that links NO synthesis and NLRP3 inflammasome activation during endotoxic shock.
ESID-0205 NLRP3 and MEFV Gene Variants in PFAPA Patients in Slovenia
D. Perko1, M. Debeljak 2, N. Toplak1, T. Avcin1
1Department of Allergology Rheumatology and Clinical immunology, University Children's Hospital University Medical Centre Ljubljana, Ljubljana, Slovenia
2Unit for Special Laboratory Diagnostics, University Children's Hospital University Medical Centre Ljubljana, Ljubljana, Slovenia
Objectives: The aim of the study was to assess the frequency of MEFV and NLRP3 gene variants in patients with PFAPA syndrome from Slovenia in order to determine whether genes involved in other autoinflammatory diseases might play a role in PFAPA pathogenesis.
Patients and methods: We collected clinical and laboratory data of PFAPA patients, who were followed at the University Children's Hospital Ljubljana. All 10 exons of MEFV and 9 exons of NLRP3 were directly sequenced.
Results: 30 PFAPA patients were tested. Mean age at the syndrome onset was 2.1±1.3 and at diagnosis 4.2±1.8 years. 19(63%) patients were male and 11(37%) were female. Mean duration of episode was 3.5 days, mean interval between the episodes was 3.5 weeks. Most common symptoms beside fever were pharyngitis and cervical adenitis in 90% and aphtosis in 63%.10 patients (33%) were found to have 11 heterozygous variants. 6 patients have Q703K variant in NLRP3, one E148Q in MEFV and one combination of I591T in MEFV and Q703K in NLRP3. Novel variant in NLRP3, P200T, was identified in one patient. One girl was found to have known variant in NLRP3 gene, S726G, which is associated with CINCA syndrome. This girl has had typical PFAPA symptoms, but also epilepsy and mild developmental delay.
Conclusion: Five different MEFV and NLRP3 gene variants were identified in 10(33%) PFAPA patients with MEFV variants found in 2 and NLRP3 variants in 9. Our results indicate genetic heterogeneity of PFAPA population and possible overlap with other periodic fever syndromes.
ESID-0320 The Clinical Utility of Serum IGG4 Measurements
M. Dziadzio 1, S.L. Seneviratne1, J. Harvey1, N. Verma1, F. Tahami1, R. Chee1
1Immunology, Royal Free Hospital, London, United Kingdom
IgG4-related disease (IgG4RD) is a fibro-inflammatory condition characterized by tumefactive lesions with histopathological evidence of infiltrating polyclonal IgG4+ plasma cells and, in approximately 60-70% of cases, elevated serum IgG4 which is however not specific for IgG4RD.
We investigated the clinical utility of IgG4 measurements in patients’ sera analysed by our immunology laboratory (RID assay). A retrospective analysis of IgG4 levels obtained over a period of 5 years was performed. The results were correlated with the histology results from individual patients, where available.
The outcomes from a total of 164 samples (99 males, 65 females) were analysed. Serum IgG4 was raised (IgG4>1.3 g/?l) in 27 (16.4%) samples, range: 1.4–10 g/?l. The clinical indications given were: pancreatitis 4/?33 (positive/ total samples), autoimmune pancreatitis 4/?28, suspected IgG4RD 6/?13, PSC 4/?15, retroperitoneal fibrosis 1/?9, cholestasis 3/?5, biliary stricture 1/?5, aortitis 0/?5, pancreatic carcinoma 1/?4, cholangiocarcinoma 1/?4, cholangitis 0/?2, other or not known 2/?41.
88/164 patients had biopsies. only 6/?13 patients with suspected IgG4RD underwent biopsy; 3/?6 had histologically confirmed IgG4RD, localised to the kidneys (serum IgG4: 1.64- 5.0 g/?l).
Of the remaining 82 patients biopsied, serum IgG4 was raised in 13 and normal in 69. IgG4 staining was done in 11/82: it was positive (autoimmune pancreatitis) in 1/?13; negative in 10/69.
In summary, only 2.4% of our patients had confirmed IgG4RD: all had raised serum IgG4. Although serum IgG4 remains the only screening test for IgG4RD, isolated elevated IgG4 levels are non-specific and not diagnostic. Characteristic histological findings are necessary to confirm the diagnosis of IgG4RD.
ESID-0708 Can the Granule Release Assay Help Distinguish Between Primary Versus Secondary Haemophagocytic Lymphohistiocytosis (HLH)?
B. Malani1, A. Rao1, K. Gilmour 2
1Haematology, Great Ormond Street Hospital, London, United Kingdom
2Immunology, Great Ormond Street Hospital, London, United Kingdom
HLH is a clinical diagnosis with systemic features of a hyper-inflammatory syndrome. The presence of a mutation confirms a diagnosis of Familial (primary) HLH. Secondary HLH, is a retrospective diagnosis, based on exclusion of known mutations and the presence of a trigger. Degranulation assays quantifying CD107a surface expression on NK cells and CTLs, have a sensitivity of 96% and a specificity of 88% for primary HLH. Its utility in distinguishing primary versus secondary HLH is unclear.
We conducted a retrospective audit of the Granule Release Assay (GRA) requested in 68 children at Great Ormond Street Hospital. Children with perforin/ SAP/XIAP mutations were excluded.
16% had an abnormal GRA with mutations in Munc 13-4, Munc 18-2 and STX11 genes.
15% had an abnormal GRA and no mutation was identified. 70% are alive, 2 post HSCT.
69% had a normal GRA and no mutation was identified. 85% are alive, 6 post HSCT.
Indications for HSCT in children without mutations (both abnormal and normal GRA), were a clinical diagnosis of primary HLH, systemic onset JIA, bone marrow failure and leishmaniasis (post-mortem).
Degranulation assays may not help clinicians distinguish between primary and secondary forms of HLH. They can be normal in some children with primary HLH as defined by meeting the HLH criteria and clinical need for HSCT. In the absence of a genetic mutation, irrespective of GRA result, a decision to initiate therapy or to proceed to HSCT should be based on a comprehensive exclusion of known triggers and consideration of alternative diagnoses.
ESID-0027 Association Between Primary Immunodeficiency Diseases and Vasculitis Syndrome
T. Hara 1, M. Ishimura1, H. Takada1, T. Kusuda1, Y. Nakashima1, K. Murata1, S. Kanno1, H. Nishio1
1Pediatrics, Kyushu University Hospital, Fukuoka, Japan
We previously conducted a nationwide survey of primary immunodeficiency disease (PID) patients in Japan (Ishimura et?al. J Clin Immunol 2011). At the survey, we found 4 patients with PID (chronic granulomatous disease: CGD, Wiskott-Aldrich syndrome: WAS, complement component 9 deficiency: C9 deficiency, and familial Mediterranean fever: FMF) complicated by Kawasaki disease among vasculitis syndrome. Kawasaki disease in CGD was characterized by the lack of high fever as well as by the occurrence of coronary aneurysm. In addition to our survey case, WAS was reported to be associated with necrotizing vasculitis and aneurysm. C1, C2 and C4 deficiencies are associated with the development of vasculitis, while FMF-related vasculitis syndrome includes Henoch–Schönlein purpura, polyarteritis nodosa, protracted febrile myalgia, Behçet’s disease, and cutaneous vasculitis. However, it remains to be determined whether Kawasaki disease observed in a patient with C9 deficiency or FMF was a causal or a chance association. Indeed, there was no association between E148Q MEFV variant and Kawasaki disease. Although hyper-IgE syndrome (STAT3 deficiency) with vasculitis was not found in our survey, STAT3 deficiency is frequently complicated by vasculitis and coronary or brain aneurysms. Patients with CGD, WAS and hyper-IgE syndrome are susceptible to extracellular bacteria such as S.?aureus through the deficient superoxide production, defective migration of innate immune cells, and Th17 cell defect, respectively. We will discuss possible explanations for the occurrence of Kawasaki disease in these PIDs.
ESID-0132 Severe Defect in Regulatory T Cell Homeostasis in a Murine Model for Familial Hemophagocytic Lymphohistiocytosis
S. Humblet-Baron 1, S. Bornschein1, L. Van Eyck1, C. Wouters2, F. Baron3, P. Matthys4, A. Liston1
1Department of Microbiology and Immunology VIB, University of Leuven, Leuven, Belgium
2Pediatric Rheumatology, University of Leuven, Leuven, Belgium
3GIGA-I3 Laboratory of Cell and Genetic Therapy, University of Liège, Liège, Belgium
4Laboratory of Immunobiology Rega Institute, University of Leuven, Leuven, Belgium
Familial hemophagocytic lymphohistiocytosis (FHL) is a severe clinical condition characterized by massive inflammation. This condition involves uncontrolled expansion of CD8 T cells, activation of macrophages (histiocytes) and florid cytokine storm. In primary FHL the disease origin is genetic, involving defects in granule-mediated cytotoxicity, such as Perforin. The main murine model of FHL exploits this genetic origin, utilizing Perforin KO mice. Unmanipulated perforin-deficient mice are asymptomatic, however infection with lymphocytic choriomeningitis virus (LCMV) results in a disease progression that closely parallels the manifestations observed in FHL patients. We specifically studied the role of regulatory T cells (Tregs) in this inflammatory disorder. While resting perforin KO mice harbored normal Treg numbers, we observed that following LCMV infection Treg numbers were dramatically decreased in perforin KO compared to WT mice. Our data showed a profound Treg homeostasis dysregulation due to decreased IL-2 availability in perforin KO mice. The lower IL-2 levels could be attributed to both lower IL-2 secretion by CD4+ T cells and increased IL-2 consumption by activated CD8+ expressing very high level of CD25 (IL-2 receptor) at their surface. The IL-2 deprivation created a toxic environment to Tregs, with transferred wildtype Tregs failing to survive or reduce disease. Together, these data demonstrate that reduced Treg number due to an IL-2 homeostasis defect may contribute to the massive inflammation found in FHL.
ESID-0490 Mevalonate Kinase Deficiency: About 2 Cases
F. Ailal 1, I. Benhsaien1, L. Jeddane1, J. Najib1, A.A. Bousfiha1
1Clinical Immunology Unit, Faculty of Medicine and Pharmacy, Casablanca, Morocco
Mevalonic aciduria (MVA) and hyper-Immunoglobulinemia D syndrome (HIDS) represent the two extremes of the clinical spectrum of disease caused by mevalonate kinase deficiency (MVK), the first enzyme involved in the biosynthesis of cholesterol. We report two cases. The first patient, aged 5, has a partial deficiency with clinically recurrent fever associated with tonsillitis, cervical lymphadenopathy and oral ulcers. IgA level was very high at 11 g/?L, IgD increased to 174 mg/L and mevalonic aciduria was positive at 19.1?mmol / mol. The genetic study confirmed the disease. Evolution is good on anti-inflammatory treatment.
The second patient, 9 year old, presents in addition to recurrent fevers, recurrent pneumonia and inflammatory lymphadenopathy, failure to thrive and mental retardation. This is mevalonic aciduria with very high IgD (1320) and increased mevalonaturia to 3557?mmol / mol. Despite treatment with anti-inflammatory, patient keep showing febrile seizures.
Mevalonate kinase deficiency should be sought in patients with recurrent fevers even with symptoms suggesting infections and after exclusion of conventional PID.
ESID-0103 Recurrent Aphthous Stomatitis in Childhood
S.S. Kilic 1, G. Uner1
1Pediatric Immunology, Uludag University Faculty Of Medicine, BURSA, Turkey
Introduction: Recurrent aphthous stomatitis (RAS) is characterized by periodic painful, single or multiple ulcers which heal spontaneously. Despite their high prevalence, etiopathogenesis remains unclear.
Objective: We aimed to disclose the etiological causes of the RAS and contribute to the development of treatment methods by examining common predisposing factors.
Method: Study and control groups consisted of respectively 65 RAS patients and 47 controls of whom CBC, ANC, serum iron, folic acid, vitamin A, E and B12 , Herpes simplex virus IgM, helicobacter pylori IgA and CMV IgM levels were measured. Nutrition, dental hygiene, food allergy, smoking habits were assessed by questionnaire method.
Results: Minor aphthous stomatitis was detected in 95.4% of the patients. In terms of the location, 16.9% (n = 11) of the patients had RAS in the same place. Oral hygiene was poor in the patient group. Monotypic diet was determined in 43.1% of the patients and 4.3% of the controls (p = 0.000). Smoking was observed in 1.5% of the patients and 4.3% of the controls. There were no differences in terms of the level of serum vitamin A, E, B12, folic acid, iron between the patients and controls. No infectious origin leading to RAS was detected in patients.
Conclusion: In our study, we found that the oral hygiene was worser and monotypic nutrition habits were more common among the study group. Therefore, to clarify the etiology of RAS which affects such a large community, we need to conduct more comprehensive prospective studies in large number of patients.
ESID-0058 Global Lung Hypermethylation Can Be Involved in Altered Inflammatory Response Pattern, in Intrauterine Undernourishment
M.M. Ballico1, M.H.C. Carvalho1, N.O.S. Camara2, R.G. Landgraf3, M.?A. Landgraf 1
1Departamento de Farmacologia, Instituto de Ciências Biomédicas/USP, São Paulo, Brazil
2Departamento de Imunologia, Instituto de Ciências Biomédicas/USP, São Paulo, Brazil
3Laboratory of Inflammation and Vascular Pharmacology, Universidade Federal São Paulo - Diadema, Diadema, Brazil
Introduction: It has been demonstrated that adverse environmental factors in the prenatal period cause changes in the normal pattern of growth and development of the fetus. Indeed, failure of the maternal-placental supply to match fetal nutritional demand results in a range of fetal adaptations developmental changes. In this work, we have investigated the impact of intrauterine undernutrition on the inflammatory markers, and the correlation of these markers with the lung dysfunction; the lung global methylation pattern was also investigated.
Methods: All procedures used in this study were approved and performed in accordance with guidelines established by the ethics committee of the ICB/USP (CEUA–67/2013). Female Wistar rats were randomly divided into 2 groups: nourished (NR; ad libitum diet) and undernourished (UR; 50% food restriction). At 8-week-of age, lung global methylation pattern was determinated, and corticosterone, leptin, IL-1ß, TNF-a, IL-5 and IL-13 were evaluated by Bioplex, in serum from NR and UR offspring. The response to LPS intratracheal instillation (1.5 mg/g/20 mL) was also evaluated in both groups.
Results: We did not observe difference in corticosterone, leptin, IL-1ß and TNF-a levels, when compare UR and NR offsprings. Both the global lung methylation (150%) as IL-5 (375%) and IL-13 (83%) levels were higher in UR than NR offsprings. In NR offspring, inflammatory cell infiltration into airways after LPS instillation was increased (x3.5 times), compared to UR offspring.
Conclusion: These preliminary results indicate that epigenetics alterations can be involved in altered inflammatory response pattern presented by UR offspring.
Financial support: FAPESP (2012/?51104-8, 2010/?01404-0) and CNPq
ESID-0080 Toll Like Receptor-4 Expression is Reduced in Acute Lung Inflammation in Intrauterine Undernourishement
A.?M. Balbino1, T.C. Torres1, L. Fernandes1, M.?A. Landgraf2, R.G. Landgraf 1
1Laboratory of Inflammation and Vascular Pharmacology, Federal University of São Paulo - Campus Diadema, Diadema, Brazil
2Department of Pharmacology, Institute of Biomedicas Sciences - USP, São Paulo, Brazil
Intrauterine undernourishment (UR) can induce a range of fetal adaptations, which can lead to permanent alterations in adult life, such as reduced inflammatory response. In this study, we standardized a model of UR in mice and investigated the impact of UR on the Toll Like Receptor-4 (TLR4) expression. The correlation of UR with the development of lung inflammatory response was also studied. All procedures used in this study were approved by the ethics committee of the UNIFESP(CEP–1666/09). Female C57Bl/6 mice were randomly divided into 2 groups: nourished (NR, ad libitum diet) and undernourished (UR, 25% food restriction). After birth, each litter was left with the mother for 28 days. 5-6 male C57BL/6 mice at 8-9 wk of age were used for each group. Control group was given saline intranasally (i.n.,30 µL). Experimental groups were given LPS (i.n.,1.5 µg/g/30 µL). The bronchoalveolar lavage fluid (BALF) was collected 6?h after instillation to evaluate cellular infiltration in lung. Lungs were harvested for measurement: the cellular infiltration; TLR-4 expression and long-form leptin receptor by western blot. UR mice stimulated by LPS were presented significantly reduced in total cell (45.9%) and neutrophils (76.2%) in BALF when compared to UR group. Besides, cellular infiltration in the peribronchial area was also significantly reduced (37.8%). Expression of TLR4 and leptin receptor was reduced (45% and 39.8%) when compared to NR group. Our preliminary results suggest that UR mice dowregulates leptin receptor TLR4 expression and and modulates acute lung inflammatory response stimulated by LPS.
Financial support: FAPESP (2010/?01404-0, 2012/?51104-8) and CNPq.
ESID-0749 Immunomodulatory, Anti-Inflammatory and Anti-Tumor Activities of Nigella Sativa
A.F. Majdalawieh 1, R.I. Hmaidan1, R.I. Carr2
1Biology Chemistry and Environmental Sciences, American University of Sharjah, Sharjah, United Arab Emirates
2Microbiology and Immunology, Dalhousie University, Halifax, Canada
AIMS: The potential immunomodulatory effects of Nigella sativa are investigated with regard to splenocyte proliferation, macrophage function, and NK anti-tumor activity using BALB/?c and C57/BL6 primary cells. Methods: Splenocyte proliferation was assessed by [3H]-thymidine incorporation. ELISA was performed to assess cytokine secretion by splenocytes and macrophages, and Griess assay was performed to evaluate NO production by macrophages. Using YAC-1 lymphoma cells, the potential of Nigella sativa extract to promote the cytotoxic activity of NK cells was also examined by JAM assay. Results: Our findings reveal that the aqueous extract of Nigella sativa significantly enhances splenocyte proliferation in a dose-responsive manner. The aqueous extract of Nigella sativa favours the secretion of Th2, versus Th1, cytokines by splenocytes. The secretion of IL-6, TNFa, and NO; key pro-inflammatory mediators, by primary macrophages is significantly suppressed by the aqueous extract of Nigella sativa, indicating that Nigella sativa exerts anti-inflammatory effects. Finally, experimental evidence indicates that the aqueous extract of Nigella sativa significantly enhances NK cytotoxic activity against YAC-1 tumor cells, suggesting that the documented anti-tumor effects of Nigella sativa may be, at least in part, attributed to its ability to serve as a stimulant of NK anti-tumor activity. Conclusions: Our data present Nigella sativa as a traditionally used herb with potent immunomodulatory, anti-inflammatory, and anti-tumor properties. We anticipate that Nigella sativa ingredients may be employed as effective therapeutic agents in the suppression and regulation of diverse immune reactions implicated in various conditions and diseases including cancer.
Keywords: Anti-Inflammatory, Anti-Tumor, Herbal Medicine, Immunomodulation, Nigella sativa
ESID-0558 Neonatal-Onset Multisystem Autoinflammatory Disease. Case Report
Z. Marin Montoya 1, M.?A. Yamazaki Nakashimada1, S.E. Espinoza Padilla2, A. Contreras Verduzco3, M.?D.?M. Saez de Ocariz4
1Immunology, Instituto Nacional de Pediatria, Mexico, Mexico
2Immunodeficiency Investigation, Instituto Nacional de Pediatria, Mexico, Mexico
3Allergy, Instituto Nacional de Pediatria, Mexico, Mexico
4Dermatology, Instituto Nacional de Pediatria, Mexico, Mexico
We report a 6-years-old female, born in Copala, Guerrero, Mexico, from a healthy 33-years-old mother and 48-years-old father with no consanguinity. One healthy 8-years-old brother. No history of early deaths, allergies, rheumatological or neurological diseases in the family.
A generalized urticarial rash was noticed since the first day of life. Unquantified fever since the first months of life which exacerbated the rash was treated with antibiotics. History of upper respiratory infections about 3 ??- 4 times per year. Presented diffuse abdominal pain, 3-4 times a month, sometimes associated with diarrheal stools. She has presented inconsistently knee and ankle pain and arthralgias. In November, she presented symptoms’ exacerbation with fever of 38 - 38.5 °C every day, mainly in the evening and night, generalized maculo-papular rash without predominance of onset time, headache, nausea and conjunctival hyperemia with blurred vision without being accompanied by vomiting.
She was examined by an ophthalmologist who diagnosed bilateral grade I papilledema.
Neurologist confirmed bilateral papilledema and chronic recurrent headache. Audiological test reported bilateral normal hearing.
The laboratories showed normocytic hypochromic anemia, leukocytosis at the expense of neutrophils, high platelet count and elevated acute phase reactants.
The skin biopsy revealed a superficial neutrophilic perivasculitis and some eosinophils without deep vessel involvement.
Steroid treatment was started. We are currently looking for the genetic diagnosis.
ESID-0150 Defect of Suppression of Inflammasome-Independent Interleukin-8 Secretion from SW982 Synovial Sarcoma Cells By Familial Mediterranean Fever-Derived Pyrin Mutations
J. Masumoto 1, R. Sugiyama2, K. Agematsu3, K. Migita4, J. Nakayama5, S. Mokuda6, F. Ogura6, K. Haraikawa6, C. Okumura6, S. Suehiro6, S. Morikawa6, Y. Ito6
1Department of Pathology, Ehime University School of Medicine, Toon, Japan
2Department of Molecular Pathology, Shinshu University School of Medicine, Matsumoto, Japan
3Department of Infectious Immunology, Shinshu University Graduate School of Medicine, Matsumoto, Japan
4Clinical Research Centre, Nagasaki Medical Centre, Nagasaki, Japan
5Department of Molecular Pathology, Shinshu University Graduate School of Medicine, Matsumoto, Japan
6Department of Pathology, Ehime University School of Medicine, Ehime, Japan
Familial Mediterranean fever (FMF) is a recessive inherited autoinflammatory syndrome. FMF patients have a mutation in the Mediterranean fever (MEFV) gene, encoding pyrin. Although pyrin is known to regulate the inflammasome, a platform for processing interleukin (IL)-1ß, the function in inflammasome-absent cells has not been fully elucidated. Then, we constructed expression plasmids of wild-type (WT) pyrin and mutated pyrin, such as E148Q, M694I, M694V, and E148Q+M694I, and transfected these expression plasmids into SW982 synovial sarcoma cells, which did not express two inflammasome components, ASC and caspase-1. IL-8 and IL-6 were spontaneously secreted from the culture supernatant of SW982 cells without any stimulation, whereas IL-1ß and TNF-a could not be detected even when stimulated with lipopolysaccharide. IL-8 but not IL-6 secretion from SW982 cells was largely suppressed by WT pyrin, but less suppressed by mutated pyrin, which appeared to become weaker in the order of E148Q, M694I, E148Q+M694I, and M694V mutations. As for IL-8 and IL-6, similar results were obtained using stable THP-1 cells expressing the WT pyrin or mutated pyrins, such as M694V or E148Q, when stimulated by LPS. In addition, IL-8 secretion from mononuclear cells of FMF patients was significantly higher than that of healthy volunteers when incubated on a culture plate. Thus, our results suggest that IL-8 secretion from SW982 synovial sarcoma cells suppressed by pyrin independently of inflammasome is affected by pyrin mutations, which may reflect the activity in FMF arthritis.
ESID-0563 Undefined and Fatal Meningoenchephalitis in a Patient with Combined Immune Deficiency: A Case Report
L. mendonca 1, M.T. Barros1, N. Siqueira de Castro1, C.M. Kokron1, F. Mascarenhas1, L.A. Marcondes Fonseca1
1clinical immunology and allergy, HCFMUSP, Sao Paulo, Brazil
Encephalitis is defined by the presence of inflammation of the brain in association with clinical evidence of neurological dysfunction. On the etiology it can be due to infection (most common cause is viruses) or non-infection (post vaccine or auto immune). The primary immunodeficiency is defined as a genetic basis that leads any alteration in the immune system (innate or adaptative) predisposing to infections, auto immunity and malignancies. Combined immunodefiency are potentially fatal disease with alteration in T and B cell function. The objective of this paper is to relate a case of a patient with encephalitis and our difficulty to establish the etiology. L.S.N., 25 years old female patient followed by the combined immunodeficiency presents with an acute arthritis and erythema nodosum and with acute seizures. The liquor analysis and the CT are attached (figure 1 and 2). The patient underwent an uncontrolled epileptic status that required intubation and sedation to control. She received broad-spectrum antibiotics, anti-fungal, anti-viral, tuberculostatic treatment and corticosteroids and despite everything she died. We tried to determine the correct etiology but wasn’t possible.
| Liquor analysis | Results |
| Cells | 132 |
| Linf | 44% |
| Mon | 26% |
| Neutrophyls | 27% |
| Glicosis | 40 |
| Lactate | 32,9 |
| ADA | 26,8 |
| PCR toxoplasmosis | Negative |
| Antigen criptococo | Negative |
| PCR Tuberculosis | Not detected |
| BAAR | Negative |
| Herpes simplex | Negative |
| Syphilis | Negative |
| PCR CMV | Negative |
ESID-0022 MEFV Mutations in Egyptian Children with Familial Mediterranean Fever: Relation to Disease Manifestations
R. ElHawary1, S. Elanwary2, S. Meshaal 1
1Clinical and Chemical Pathology Department, Cairo University, Cairo, Egypt
2Pediatrics Department, Cairo University, Cairo, Egypt
Familial Mediterranean Fever (FMF); an auto-inflammatory disease that is inherited as autosomal recessive trait, is characterized by episodic, self-limiting attacks of fever, along with abdominal pain, sterile serositis and arthritis.
FMF is caused by outbursts of inflammatory cytokines. Mutations in Mediterranean Fever Gene (MEFV) lead to a pro-inflammatory state. The inflammation observed in FMF is characterized by neutrophil influx to peripheral tissues and increased serum levels of acute-phase reactant proteins and cytokines.
Methods: 424 patients with clinical manifestations suggestive of FMF were enrolled in this study in the period Jan 2011- March 2014. Mutations in MEFV gene were studied by reverse hybridization using the FMF Strip Assay™, Viennalab. Possible correlation between clinical presentations and detected mutations was investigated.
Results: MEFV mutations were detected in 175 patients among whom 78 had compound heterozygous or homozygous mutations. The most common mutations were: M694I (49/78) 63% of cases followed by V726A (39%) and M680I(G/?A) (23%). The most frequent complaint was Fever (100%) abdominal pain (81%) followed by chest pain (54%) and joint pain (43%).
None of the mutations detected showed a significant correlation with the disease manifestations or age of onset of the disease. In this study, we have reported a cohort of 97 patients with classical symptoms of FMF; most of them responded to Colchicine treatment, although only one heterozygous mutation was detected in MEFV (most common was E148Q 31%). Although those patients may have other periodic fevers, the patients clinically appeared to have FMF and significant numbers responded to colchicine.
ESID-0577 HSCT Rescues the Immunological and Vascular Phenotype of ADA2-Deficiency
L. Van Eyck 1, M.S. Hershfield2, D. Pombal3, S.J. Kelly2, N.J. Ganson2, L. Moens4, G. Frans4, H. Schaballie5, G. De Hertogh6, J. Dooley3, X. Bossuyt4, C. Wouters7, A. Liston3, I. Meyts8
1Department of Immunology and Microbiology, VIB Autoimmune genetics laboratory, Leuven, Belgium
2Duke University Medical Center, Duke University, Durham, USA
3Department of Immunology and Microbiology KU Leuven, VIB Autoimmune genetics laboratory, Leuven, Belgium
4Department of Immunology and Microbiology Experimental Laboratory Immunology, KU Leuven, Leuven, Belgium
5Department of Immunology and Microbiology Childhood Immunology, KU Leuven University Hospitals Leuven, Leuven, Belgium
6Department of Pathology, KU Leuven, Leuven, Belgium
7Department of Immunology and Microbiology Childhood Immunology, KU Leuven, Leuven, Belgium
8Departmenf of Immunology and Microbiology Department of Pediatrics, KU Leuven University Hospitals Leuven, Leuven, Belgium
Recessive loss-of-function mutations in CECR1 (cat eye syndrome chromosome region, candidate 1) encoding adenosine deaminase 2 (ADA2) were recently identified as the cause of a syndrome including recurrent lacunar infarcts, vasculopathy and inflammation.
We identified two siblings from non-consanguineous parents with a homozygous p.Arg169Gln mutation in ADA2 resulting in plasma ADA2-deficiency. The clinical phenotype was dominated by immunodeficiency with autoimmunity and lymphoproliferation. In the index patient pancytopenia and lymphoproliferation were refractory to treatment with mycofenolate, sirolimus, meracptopurine, ciclosporin, tacrolimus. In the younger brother recurrent and complicated viral infections were present as well as inflammatory bowel disease only responsive to sirolimus. Both patients showed hypogammaglobulinemia and subtle defects in both B as well as T cell compartment.
The index patient received hematopoietic stem cell transplantation (HSCT) from a healthy sibling after conditioning with oral busulfan and cyclophosphamide. The immediate post-transplant period was complicated by a pineal gland hemorrhage. Moreover, there was delayed engraftment of platelets and neutrophils. Late veno-occlusive disease occurred but responded well to supportive measures. At 5y follow-up, he is well and off medication. His immunological reconstitution is good and he has not shown any further vascular event.
To conclude: immunodeficiency and auto-immunity can dominate the clinical picture of ADA2 deficiency. We show that HSCT corrects the metabolic defect and the immunological and vascular phenotype.
ESID-0639 Novel Mutations in MVK Associated with Hyperimmunoglobulinemia D with Periodic Fever Syndrome Phenotype
D. Moraes Vasconcelos 1, E. Fujihira1, J.B. Oliveira1, A.A. Jesus2, C. Silva2, A.P.?M. Castro2, M.B. Dorna2, L. Watanabe2, A. Pontillo1, N. Chuffi-Barros1, C.M.?A. Jacob2, M.M.S. Carneiro-Sampaio2, A.J. Duarte1
1Dermatology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
2Pediatrics, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
Mevalonate kinase deficiency (MKD) is a rare AR inborn error of cholesterol and nonsterol isoprenoid biosynthesis caused by mutations in the MVK gene. It can originate two distinct clinical phenotypes: hyperimmunoglobulinemia D with periodic fever syndrome (HIDS) and mevalonic aciduria.
Case 1: A 7- year-old (yo) male of a consanguineous family with a history of intermittent fever and hepatosplenomegaly since six months-old. The patient had recurrent respiratory and gastrointestinal infections and received treatment to tuberculosis with 4yo. Physical examination revealed cervical adenitis, hepatosplenomegaly and rash on extremities. Laboratory findings included mild anemia and elevated inflammatory markers, elevated IgA and IgD levels even in the absence of an inflammatory attack. Genomic sequencing of MVK gene revealed a homozygous c.709A>T mutation leading to T237S.
Case 2: A 6 yo male who presents recurrent fever since the age of one month. Abdominal pain, oropharyngeal hyperemia, massive cervical adenopathy and failure-to-thrive are associated to the febrile episodes. Immunological investigation showed a mild anemia with a hemoglobin electrophoresis suggestive of thalassemia, elevated polyclonal IgA levels, elevated expression of IL1B and normal double negative T cells. Multiple lymphnode biopsies showed reactive adenopathy pattern and the myelogram was normal. The genomic sequencing of the MVK gene revealed a mutation in c.62 C>T, leading to a protein that has A21V; a second mutation c.155 G>A, leading to S52N.
Clinical and laboratory characteristics of the patients’ inflammatory attacks are compatible with HIDS. The diagnosis was confirmed by demonstration of the mutations in the MVK gene.
ESID-0084 Magnetic Resonance with Diffusion Weighted Imaging: A New Method to Detect Active Lung Inflammation in Patients with Primary Antibody Deficiencies
C. Milito 1, G. Serra2, F. Pulvirenti1, C. Di Iorio1, F. Fraioli2, A. Marra3, I. Quinti1
1Molecular Medicine, Sapienza University of Rome, Rome, Italy
2Radiological Oncological and Pathological Sciences, Sapienza University of Rome, Rome, Italy
3Clinical Medicine, Sapienza University of Rome, Rome, Italy
Patients with primary antibody deficiency suffer from infectious and non infectious pulmonary complications. The need to recurrently detect lung alterations poses concerns regarding the radiation dose in patients with increased radio sensitivity.Following these considerations, we introduced Diffusion Weighted Imaging in our diagnostic approach for patients with primary antibody deficiency. We hypothesized that Magnetic Resonance Imaging with Diffusion Weighted Imaging might represent a useful radiation free technique in the distinction between inactive lung abnormalities versus areas of active inflammation.We performed a single center longitudinal study on 18 patients using chest magnetic resonance imaging and pulmonary function tests at baseline and after 18 months. Diffusion weighted imaging sequence was included. All studies were evaluated by Bhalla modified score.Bronchial abnormalities were the prevalent abnormalities recorded. Total parenchymal and bronchial scores negatively correlated with FEV1. Only the bronchial wall thickening and the mucus plugging scores showed an improvement during follow up. Consolidations and nodules were recorded in 40% out of patients. Nodule dimension was stable in the majority of patients, while dimensions generally improved. Half of consolidations and one third of nodules had diffusion weighted imaging restrictions associated to FEV1 reduction, granulomatous and lymphadenopathy manifestations, and an expansion of CD21low B cells.Magnetic Resonance Imaging with BLADE sequences was a reliable technique in the in the follow up of bronchial and parenchymal alterations in patients with antibody deficiencies. The addition of Diffusion Weighted Imaging sequences might provide valuable records to detect the presence of areas of active inflammation in these patients.
ESID-0122 Clinical, Genetic and Immunological Characterization of a Patient with the New PSTPIP1-Associated Autoinflammatory Disease Hyperzincemia with Hypercalprotectinemia
I. Sologuren1, E. Ruiz-Ortiz2, Y. Barrios3, E. Herrera-Ramos1, M. Martínez-Saavedra1, N. González-Quevedo1, R. López-Almaraz4, E. Colino5, J. Aróstegui2, C. Rodríguez Gallego 1
1Immunology, Hospital Universitario de Gran Canaria Dr. Negrín, Las Palmas de Gran Canaria, Spain
2Immunology, Hospital Clínic, Barcelona, Spain
3Immunology, Hospital Universitario de Canarias, Sta Cruz de Tenerife, Spain
4Pediatric, Hospital Universitario de Canarias, Sta Cruz de Tenerife, Spain
5Pediatric, Hospital Materno-Infantil, Las Palmas de Gran Canaria, Spain
The field of autoinflammatory diseases has greatly expanded since the term autoinflammation was coined in 1999. However, in many patients the molecular basis of the disease remains still elusive.
The patient suffered from recurrent perianal and gluteal abscesses by Gram-negative bacteria between the age of 17 days and 4 months, as well as from a hepatic abscess by E.?coli when he was 21 month-old. He had intermittent neutropenia and thrombocytopenia and persistent anemia.
Routine immunological evaluation at the age of 4 months, including ELANE sequencing and bone marrow studies, was normal. Whole blood and PBMC cultures showed low responses to several TLR agonists. However, high levels of pro-inflammatory cytokines could be detected in unstimulated cultures. Reevaluation of the clinical data pointed to a persistent, non-periodic, autoinflammatory disorder. The clinical course resembled that of five patients with hypercalprotectinemia and hyperzincemia (Hz/Hc) previously reported (Lancet 2002; 360:1742-5). He had persistent fever, with persistently elevated C reactive protein (75-206 mg/L) values, lymphadenopathy, hepatosplenomegaly and severe growth delay. He was shown to present with extremely high levels of Zn and the alarmins calprotectin (S100A8/A9) and calgranulin (S100A12), compared with controls and with patients with other autoinflammatory disorders. Genetic analysis showed that the patient had a new mutation in PSTPIP1 (E250K). Interestingly, other mutations in PSTPIP1 cause the syndrome of pyogenic arthritis, pyoderma gangrenosum and acne (PAPA).
At present, thirteen patients with this novel autoinflammatory disorder have been diagnosed (unpublished results). The clinical course is sharply different from that observed in PAPA syndrome.
ESID-0283 A Homozygous Silent Mutation of the IL-10RA Gene in an Infant with Perianal Fistula
G. Segundo 1, C.P. Barros1, K.P. Fernandes1, E.R. Resende1
1Pediatrics, Universidade Federal de Uberlândia, Uberlândia, Brazil
Case report: LSC, male, product of consanguinity parents. At age 2mo started a recurrent diahrrea and enterocolitis. Ate age 4mo, he presented an intestinal perforation and resection of 36?cm of jejune. At age 6 months was admitted in our service with mal-nutrition, perianal fistula, fissures and plicomas. He received elementary diet with improvement of nutrition and partial control of diarrhea. At age 1 year, he started recurrent infections, and worsened diarrhea and perianal fistulas. Colonoscopy and biopsy showed: chronic ulcerated inflammation polipoid granulation tissue. He was started on prednisone, azathioprine, and infliximabe without response. He needed to internalize several times to control his general state. At age 2y3mo he was submitted a new procedure to minimize perianal fistula. We hypothesized IL-10RA deficiency. From the material obtained IL10RA gene (NM_001558.3) was PCR amplified and the exons (±10 base pairs) were sequenced. In this investigation was found a homozygous variant c.537G>A (p.T179T) in IL10RA gene. This variant, although do not cause change in the amino acid (silent mutation), occurs in the last base of exon 4, which may affect exonic splicing and cause disruption of the IL10 receptor, with a loss of function of the pathway. The patient maintains the inflammatory intestinal disease without response to conventional treatment, with several hospitalizations. Bone marrow transplantation was indicated and now he waits for the same.
Conclusion: The patient presents a new silent mutation in IL10RA gene in association with a severe clinical of very earlier onset inflammatory bowel disease
ESID-0111 Screening for Primary Immunodeficiencies in Paediatric Patients with Inflammatory Bowel Disease. Is It Worth?
P. Soler-Palacin 1, E. Busquets2, D. Alvarez-de-la-Sierra3, A. Martín-Nalda1, O. Segarra2, M. Alvarez2, C. Figueras1, M. Martinez-Gallo3
1Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d'Hebron Institut de Recerca Vall d'Hebron Universitat Autonoma de Barcelona, Barcelona, Spain
2Pediatric Gastroenterology Hepatology and Nutrition Division, Hospital Universitari Vall d'Hebron Institut de Recerca Vall d'Hebron Universitat Autonoma de Barcelona, Barcelona, Spain
3Immunology Division, Hospital Universitari Vall d'Hebron Institut de Recerca Vall d'Hebron Universitat Autonoma de Barcelona, Barcelona, Spain
Background: Inflammatory bowel disease (IBD) in children makes up a diverse group of diseases. A proportion of patients with early-onset IBD may suffer from several primary immunodeficiency diseases (PID). Objective: to screen paediatric patients with IBD to discard potential PID that could be the underlying cause of the intestinal manifestations.
Patients and methods: Patient selection was done according to the diagnostic criteria established by the IBD working-group of the ESPGHAN. Serum immunoglobulin levels, HIV serology and total leucocyte count were studied using regular methods. T, B and NK lymphocyte subsets, oxidative burst assay and HLA-DR, TCRaß/?d, and CD45RA+/RO+ expression was analysed by flow cytometry. In vitro production by PBMCs of TNFa after LPS and IL-10 stimulation was done to explore interleukin (IL)-10 signalling defects.
Results: Thirty-nine patients were included (22 male, median age 10.5 years). Crohn disease was diagnosed in 64%, ulcerative colitis in 28% and very early-onset colitis in 8%. There were 2/?39 family cases. No individual cases of PID were found in our population. However, when screening IL-10 signaling defects, significantly decreased production of TNFa in response to LPS was found in 9 patients, but 6/?9 were receiving TNFa inhibitors. Two out of three “real” non producers were early-onset severe colitis. All patients were able to inhibit TNFa production by adding IL-10.
Conclusions: Using this first step immunological screening no PID causing IBD has been found in our series. However, as previously reported, some patients with severe clinical phenotype may have immunological disturbances that should be further evaluated.
ESID-0629 Case Report: Hyperthyroidism in a Patient with Granulomatous CVID
K. van Bilsen 1, V.A.S.H. Dalm1, S. Neggers2, M.W. van der Ent1, G.J.A. Driessen3, A.J. van der Lely2, M. Van der Burg4, P.?M. van Hagen4
1Internal Medicine/Clinical Immunology, Erasmus MC University Medical Center, Rotterdam, Netherlands
2Internal Medicine/Endocrinology, Erasmus MC University Medical Center, Rotterdam, Netherlands
3Pediatrics, Erasmus MC University Medical Center, Rotterdam, Netherlands
4Immunology, Erasmus MC University Medical Center, Rotterdam, Netherlands
Introduction: Thyroid disease is a frequently occuring comorbidity in patients with CVID. We describe a female who developed hyperthyroidism based on granulomatous disease. Furthermore we describe the frequency of thyroid diseases in our cohort of 78 CVID patients.
Methods: 1. A case report of a woman with granulomatous CVID who developed granulomatous thyroiditis.
2. Description of the patients with thyroid disease in our CVID cohort.
Patients: 1. A 39-year old woman who was diagnosed with granulomatous CVID at the age of 27, presented at our outpatient clinic with signs of hyperthyroidism. Physical examination revealed an enlarged thyroid with a painful nodule in the right thyroid lobe. Thyroid biochemistry showed a TSH level of 0.075 mU/L and an fT4 level of 28.4 mU/L. Pathological examination revealed granulomatous inflammation. Systemic analysis by CT-scan showed progression of systemic lymph nodes. Immunological examination showed a decreased number of total B cells, normal absolute counts of naive and natural-effector B cells and a decreased number of memory B cells. No serum thyroid antibodies were detected in order to rule out autologic anti-thyroid antibodies or anti-thyroid antibodies in therapeutic IgG. In course of time patient's thyroid biochemistry normalized spontaneously.
2. In our CVID cohort of 78 patients, 7 have thyroid disease.
Conclusions: 1. To our best knowledge this is the first case-report describing granulomatous CVID associated with hyperthyroidism mimicking granulomatous thyroid disease (De Quervain thyroiditis).
2. The prevalence of thyroid disease in our CVID cohort is 8.9 percent.
ESID-0695 Granulomatous Disease in Common Variable Immunodeficiency
L.S. Kamphuis1, D.J. Kwekkeboom1, G.J. Driessen1, M. van der Burg1, J.A. van Laar1, M.C. van Zelm1, J.J. van Dongen1, V.A. Dalm1, M. van Hagen 1
1IMMUNOLOGY, ERASMUS MC, ROTTERDAM, Netherlands
Introduction. Granulomatous disease occurs in 10-22% of the patients with CVID. The presence of granulomas in CVID is associated with a worse prognosis and increased prevalence of lymphoproliferative disorders.
Patients and Methods. In this study we present retrospective data of imaging of granulomatous disease in CVID using somatostatin receptor scintigraphy (SRS). We analyzed 37 CVID patients with SRS.
Results. SRS was negative in 11 patients (35%), 5 patients (14%) had one lesion and 21 patients (51%) had more lesions. Most inflammatory sites were in the lungs (46%), but localizations in inguinal and salivary glands were common as well (both 10%). In 4 SRS positive patients a biopsy confirmed granulomatous inflammation.
Angiotensin converting enzyme (ACE) levels were elevated (>68 U/?l) in 8 out of 24 CVID patients. 7 of these 8 patients were SRS positive (88%). Therefore elevated levels of serum ACE are suggestive for granulomatous CVID and could be a potential marker for granulomas in CVID. In 24 out of 28 patients, soluble interleukin-2 receptor (sIL-2R) serum levels were elevated. Contradictionary to ACE, no correlation was found between granulomatous CVID and sIL2-R. Analysis of the peripheral B cell compartment in CVID patients showed no differences between CVID patients with and without granulomas.
Conclusions. According to our data the percentage of granulomatous disease in CVID is underestimated, 65% of the CVID patients in this study had lesions suspected for granulomas. SRS provides a useful imaging technique to determine granulomatous organ involvement in patients with CVID.
ESID-0337 Granulomatous Lymphocytic Interstitial Lung Disease (GLILD) in Common Variable Immunodeficiency (CVID): A Role for Cytomegalovirus
D. Webster 1, S. Seneviratne2, N. Verma2, M. Rodriguez-Justo3, J. Paterson4, L. Ambrose5, J. Hurst6, L. Gamez-Diaz7, B. Grimbacher7, S. Workman2
1Clinical Immunology, University College London, London, United Kingdom
2Clinical Immunology, Royal Free Hospital, London, United Kingdom
3Pathology, University College London, London, United Kingdom
4UCL Advanced Diagnostics Laboratory, University College London, London, United Kingdom
5Institute of Immunity and Transplantation, University College London, London, United Kingdom
6Respiratory Medicine, University College London, London, United Kingdom
7Centre for chronic immunodeficiency, University of Freiburg, Freiburg, Germany
Background: Granulomatous lymphocytic interstitial lung disease (GLILD) is a serious complication of unknown aetiology in patients with common variable immunodeficiency (CVID).
Methods: Patients with GLILD (with Institutional Review Board permission) were screened for cytomegalovirus (CMV) in blood and tissues using PCR and sensitive immuno-histology. Specific CMV T cell function and LPS-responsive beige-like anchor (LRBA) expression was also tested.
Results: Four CVID patients with GLILD, confirmed on lung biopsy, had CMV in saliva and urine but were not viraemic. Immuno-histology showed that the cellular infiltrate was predominantly CD4+ T cells and B cells, while vesicle-like accumulations of CMV antigen were seen in the cytoplasm of bronchiolar epithelial cells. Circulating T cells from all 4 patients expressed intracellular IFN-? when stimulated with CMV derived peptides, but the two patients with the most severe GLILD failed to secrete IFN-? during peptide stimulation. PHA stimulated PBMCs of the three patients were LRBA deficient using Western blotting, but sequencing has so far failed to find a disease causing mutation in the coding gene. The clinical outcomes up to two years of treatment with valganciclovir and steroids will be presented.
Conclusion: The data suggests a novel mechanism where CMV inhaled from saliva is taken up by the bronchiolar epithelia and stimulates a chronic lymphocytic reaction. The chronic excretion of virus from salivary glands and kidneys suggests inadequate control of CMV, possibly related to the LRBA deficiency. This is the first report directly linking CMV with GLILD in CVID, adding complexity to the management of this complication.
ESID-0372 Atypical Presentation of Cryopirynopathy in Two Family Members – Have We Really Catched Disease-Causing Mutation?
B. Wolska-Kusnierz 1, D. Rowczenio2, E. Wiesik-Szewczyk3, E. Bernatowska4
1Immunology, The Children's Memorial Health Institute, Warsaw, Poland
2Centre for Amyloidosis and Acute Phase Proteins, National Amyloidosis Centre, London, United Kingdom
3Immunology and Clinical Allergoloy Department, Military Institute of Medicine, Warsaw, Poland
4Immunology Department, Children's Memorial Health Institute, Warsaw, Poland
Introduction. Cryopyrin-associated periodic syndrome is autoinflammatory condition caused by NLRP3/CIAS1 gene mutations encoding for cryopyrin, a major component of the inflammasome, leading to an excessive production of interleukin-1beta (IL-1ß). Clinical manifestations include recurrent episodes of systemic inflammation involving skin, joints, eyes and the central nervous system and shows variable penetrance regarding disease severity and symptoms.
Objective. To enhance the difficulties in correct interpretation of genetic results in context of atypical clinical phenotype of patients.
Material. We present the unique clinical and laboratory presentation of two patients ( mother and son) with mutation in NLRP3 gene.
Results. Recurrent episodes of fever, muscle aches, headaches and transient rashes are observed in both patients. 6-year-old boy presents with sleep disorders and epilepsy and mild hearing loss was found in hi 34-old mother. There is no increase in sedimentation rate or C-reactive protein on both patients; but increased levels of serum amyloid A (SAA) and calprotectin were noticed during flares. There is no abnormalities in blood count in a boy, but mother presents with chronic, severe microcytic anemia. Molecular genetic testing showed a homozygous V198M mutation in NLRP3 gene.
Conclusions. The significance of the low-penetrance V198M mutation in NLRP3 gene in two family members with atypical phenotype remains unclear. If the remarkable response to interleukin 1 (IL-1) blockade would be obtained in both patients it would confirms that their clinical features are indeed mediated by IL-1. It would enhance our knowledge in regards to the expending spectrum of clinical phenotype of cryopirynopathies.
ESID-0658 Implementation of Home Measurement of CRP Levels in Diagnosis and Monitoring of Childen with Autoinflammatory Diseases
B. Wolska-Kusnierz 1, J. Jezierska1, E. Bernatowska1
1Immunology, The Children's Memorial Health Institute, Warsaw, Poland
Introduction. Recognition of autoinflammatory diseases (AIDs) in the last decade is growing rapidly. About 250 children is under care with suspicion or diagnosis of AIDs in our centre and monitoring of inflammatory markers is fundamental in their diagnosis and monitoring of treatment,.
Objective. To check the practical usefulness of home CRP monitoring in patients during diagnosis or monitoring of autoinflammatory diseases.
Material. Eight patients with genetically confirmed or suspicion of AIDs ( 2 FMF, 2 TRAPS, 1 MWS, 2 PFAPA, 1 suspicion of AIDs) were monitored at home with both use of diary of symptoms/treatment and regular CRP measurement. CRP levels were checked by parents at home using ready-to-use system on a finger-prick blood sample of their child.
Results. Ready–to-use system provided patients with very fast, easy to perform and safe CRP measurement, having obtained reliable results. Children and their parents found avoidance of frequent outpatient clinic visits and painful collection of blood from veins extremely important. Regular, up to twice a day measurement of CRP greatly facilitated the diagnosis and monitoring of patients' treatment. It proved to be extremely useful in an appropriate modification of treatment with the use of steroids in PFAPA or IL1-blockers in TRAPS.
Conclusions. The results of introductory pilot study of home measurement of CRP levels in children with autoinflammatory diseases are encouraging. The opportunity of fast, regular monitoring of inflammatory marker in home conditions improved both diagnostic-therapeuthical process as well as quality of children's life.
Topic: Autoimmunity and Dysregulation
ESID-0264 Immunomodulatory Therapy for Severe Autoimmune Polyendocrinopathy Type-1 (APS-1)
M. Abinun 1, S. Hodges2, T. Cheetham3, M. Ognjanovic4, N. Hopper5, A. Burt6, K. Wood6, D. Lilic7
1Paediatric Immunology, Great North Children's Hospital, Newcastle upon Tyne, United Kingdom
2Paediatric Gastroenterology, Great North Children's Hospital, Newcastle upon Tyne, United Kingdom
3Paediatric Endocrinology, Great North Children's Hospital, Newcastle upon Tyne, United Kingdom
4Paediatric Nephrology, Great North Children's Hospital, Newcastle upon Tyne, United Kingdom
5Paediatrics, Sunderland Royal Hospital, Sunderland, United Kingdom
6Histopathology, Royal Victoria Infirmary, Newcastle upon Tyne, United Kingdom
7Institute for Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
We report on treatment with combined immunsuppressive and lymphocyte-depletion (monoclonal antibodies, MoAb) therapies in a child with unusually severe autoimmune polyendocrinopathy syndrome type 1 (APS-1).
He presented at the age of 2?yr with autoimmune hepatitis type 2 (LKM-2+ve; biopsy-proven), oesophageal and nail candidiasis, insulin-dependent diabetes mellitus; subsequently developing exocrine pancreatic insufficiency, autoimmune hypothyroidism, pernicious anaemia and poor growth. AIRE mutation confirmed (also in sister, with hypoparathyroidism only).
In spite of combined immunosuppressive treatment with steroids, azathioprine and tacrolimus, chronic autoimmune hepatitis progressed; at the age 7?yr he had anti-CD20 MoAb rituximab (375 mg/m^2 x4 doses) which had no benefit clinically nor on liver biopsy 6 months post-treatment. He was subsequently treated with anti-CD52 MoAb alemtuzumab (1 mg/kg total dose) resulting in 4 years of clinical stability on maintenance prednisolone (5 mg/day), azathioprine and tacrolimus. He was on prolonged antimicrobial prophylaxis (septrin, itraconazole, aciclovir) and immunoglobulin replacement, and had no infectious complications. At age 12?yr he developed a severe and prolonged episode of autoimmune enteropathy with electrolyte and fluid dysbalance, which eventually responded to sirolimus.
Recently, at the age of 13 yr, he presented with renal failure; renal biopsy showed an immune complex-mediated process affecting glomeruli and the tubulo-interstitial compartment. Sirolimus was stopped, and he was treated with plasma exchange, methylprednisolone pulses and rituximab (750 mg/m^2 x2) with only transient benefit. We are planning a further course of alemtuzumab, with the aim to preventing progression to end-stage renal failure, dialysis and/or renal transplantation.
ESID-0550 The Immunophenotype of Bone Marrow Lymphocytes in Children with Immune Trombocytopenic Purpura
S. Alavi 1, Z. Aryan2, M. Ebadi2
1Pediatric Congenital Hematologic Disorders Research Center, shahid Beheshti Medical University, Tehran, Iran
2Molecular Immunology Research Center Children’s Medical Center Department of Immunology, Tehran University of Medical Sciences, Tehran, Iran
Purpose: Primary immune thrombocytopenic purpura (ITP), caused by immune system dysfunction, is recognized as the leading cause of thrombocytopenia in pediatric patients. Nonetheless, inadequate studies have been performed on bone marrow immunophenotyping of these children. In this study, we aimed to investigate the immunophenotype of bone marrow lymphocytes in children with ITP.
Patients and methods: Between 2008 to 2012, 35 children with ITP and 26 age and sex matched healthy controls were recruited. All participants underwent bone marrow aspiration. Appropriate B cell, T cell and myeloid lineage monoclonal antibodies were employed to determine the immunophenotype of these patients.
Results: CD10, CD19 and CD20, all indicative of premature and mature B cell markers, were significantly greater in children with ITP. CD22, mainly expressed on mature B cells was slightly, but not significantly reduced in the patients’ group (p=0.42). On the other hand, T cell markers including CD2, CD3, CD5 and CD7 were underexpressed. CD33, a specific marker for myeloid lineage, was underexpressed in the patients’ group (5.6±4.7 vs. 12.9±7.3, p<0.001). Noteworthy, the immunophenotype did not significantly differ between acute and persistent cases.
Conclusion: Overall, a phenotype characterized by increased pre-B cell markers along with decreased T cell immunophenotypic markers was observed in bone marrow lymphocytes of children with ITP in the present study. Further larger-scale studies are recommended to confirm our findings, as precise mapping of the immunophenotype of lymphocytes in these patients would pave the road to improved diagnosis and treatment.
ESID-0469 Chronic Neutropenia with Multiple Autoimmunity and CD4 Lymphopenia in a Malay Child: A New Entity?
A. ALI 1, I.J. ABDUL HAMID2, I.H. ISMAIL3, A.H. ABDUL LATIFF4, L.M. NOH1
1Department of Pediatric, Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia
2Advanced Medical & Dental Institute, Universiti Sains Malaysia, Penang, Malaysia
3Department of Pediatric, Universiti Putra Malaysia, Serdang, Malaysia
4Pantai Hospital Kuala Lumpur, Bangsar, Kuala Lumpur, Malaysia
Introduction: Autoimmune presentation in primary immunodeficiency diseases (PID) is a known association. We are presenting a possible new entity of PID with perplexing autoimmune phenomena and CD4 lymphopenia
Case Presentation: NA, a 7 years old Malay girl from non-consanguineous marriage presented with recurrent abscesses, sinopulmonary infections, oral candidiasis and failure to thrive from the age of 3 months old. Immunological workout from earlier follow-up showed mild CD4 lymphopenia with hypergammaglobulinaemia and low C4 level. NBT and lymphocyte proliferation test were normal. At the age of 3 ½ years, she was noted to be mildly neutropenic where cyclical neutropenia was suspected, but it normalized. Despite Bactrim prophylaxis, she continued to have recurrent infection including chronic otitis media. Repeated immunological workout at 6 years old showed similar immunological profile with severe persistent neutropenia. Antinuclear antibodies(ANA), anti double-stranded DNA antibodies(anti-dsDNA), direct Coombs test and perinuclear anti-neutrophil cytoplasmic antibodies(p-ANCA) turned positive by the age of 6 ½ years old. Nevertheless, examinations were normal and she remained asymptomatic for autoimmune diseases or vasculitis with normal urine and renal investigations. She was treated for possible PID with autoimmune phenomena, oral Prednisolone was commenced followed by intravenous immunoglobulin therapy. Post therapy, her CD4 counts and autoantibodies had normalized with improving neutrophil count and remained infection free.
Conclusion: This case possibly describing a new entity of PID with autoimmune phenomena. Further functional immunological and molecular investigation need to be explored.
ESID-0214 Immunologic and Molecular Characterization of Three Patients with Autoimmune Lymphoproliferative Syndrome by Somatic FAS Mutations (ALPS-SFAS)
A. Martínez-Feito1, J. Melero2, S. Mora-Díaz1, C. Rodríguez-Vigil3, R. Elduayen4, I. De Diego5, J.T. Ramos5, R. del Orbe-Barreto6, M. Riñón7, L.M. Allende 1
1Immunology, Hospital Universitario 12 de Octubre, Madrid, Spain
2Immunology, Hospital Universitario Infanta Cristina, Badajoz, Spain
3Pediatric Hemato-Oncology, Hospital Universitario Miguel Servet, Zaragoza, Spain
4Hematology, Hospital Universitario Infanta Cristina, Badajoz, Spain
5Pediatrics, Hospital Universitario de Getafe, Madrid, Spain
6Hematology, Hospital Universitario de Cruces, Bilbao, Spain
7Immunology, Hospital Universitario de Cruces, Bilbao, Spain
Introduction: Autoimmune lymphoproliferative syndrome (ALPS) is a genetic disorder of lymphocyte apoptosis. It is characterized by autoimmune features and lymphoproliferation. Most ALPS patients are associated with germline or somatic dominant FAS mutations.
Aim: Immunologic and molecular characterization of three ALPS-sFas patients.
Patients and Methods: We present three patients who fulfilled clinical and immunological parameters for ALPS. We measured the following ALPS-biomarkers: DN T-cells, plasmatic IL-10, sFas ligand, sCD25, vitamin B12 and immunoglobulins. Fas-induced apoptosis were assessed using agonistic anti-Fas monoclonal antibody (Apo1.3) both in T-cell blasts and in DN T-cells. Analysis of germline FAS, CASP10 and somatic FAS mutations were performed in DNA extracted from peripheral blood or from isolated DN T-cells, respectively.
Results: Patient1 and patient2 showed normal Fas-induced apoptosis using T-cell blasts and abnormal Fas-induced apoptosis using DN T-cells with increased levels of ALPS-biomarkers. We confirm sFAS mutations in these patients. However, patient3 showed abnormal Fas-induced apoptosis using T-cell blasts and abnormal Fas-induced apoptosis using DN T-cells with increased ALPS-biomarkers. Due to defective apoptosis in T-cell blast, we sequenced CASP10 gene and found the germline Y446C substitution, previously found as a polymorphism (allelic frequency 1,8% in Caucasian population). Additionally, we sequenced FAS from DN T-cells and found a sFAS mutation.
Conclusions: ALPS-sFAS are the second most common genetic etiology of ALPS. It could be caused by the concurrent effect of mutations hitting different genes involved in Fas function. Specifically, we think that the CASP10 variation also influenced the clinical phenotype due to the sFAS mutation in patient 3.
ESID-0533 Analysis of Natural Regulatory T Cells (TREGS) in Patients with 22Q11.2 Deletion Syndrome
S. Di Cesare 1, P. Ariganello1, P. Puliafito1, M. Mandolesi1, M.L. Romiti1, A. Aiuti2, C. Cancrini1
1DPUO University Department of Pediatrics, Bambino Gesù Children's Hospital, Rome, Italy
2Pediatric Immunology San Raffaele Telethon Institute for Gene Therapy (TIGET), San Raffaele Hospital, Milan, Italy
Introduction Chromosome 22q11.2 deletion is the most common chromosomal deletion syndrome and is found in patients with DiGeorge Syndrome. Partial DiGeorge (pDGS) patients present with mild/?moderate lymphophenia, recurrent infections and immunological dysregulation. A decreased frequency of Tregs has been reported in limited cohorts of patients with pDGS. Tregs play a crucial role in self-tolerance.
Aim To evaluate Tregs in a cohort of pDGS patients.
Patients/Methods Tregs, identified as CD4+CD25highCD127lowFOXP3+, were analyzed by flow cytometry in twenty-five pDGS patients. The analysis of Treg-specific demethylated region (TSDR) was performed by qPCR after genomic DNA extraction.
Results/Discussion Tregs absolute counts were decreased in pDGS, probably secondary to lymphopenia, but normal frequency distribution was observed, in contrast with some previously reported data. FOXP3 is considered a specific marker for Tregs, but it can be expressed also in activated T cells. The analysis of TSDR in the FOXP3 locus constitutes up to now the best specific Tregs marker, because it defines a FOXP3 stable expression.
We found a good correlation between the epigenetic TSDR analysis and Tregs frequency in pDGS, except for one patient with persistent EBV infection, but a functional defect of Tregs can be excluded only by “in?vitro” suppression assays.
In conclusion, we observed no significative difference between Tregs frequency in pDGS children and healthy controls. A larger cohort of pDGS patients should be evaluated to confirm these preliminary results. Tregs suppression assay should be performed to determine their functionality and additional markers should be added to identify the T regs subsets.
ESID-0413 Genetic Causes of Early-Onset IBD: Scanning Via NGS
D. August 1, N. Frede1, M. Buchta1, L. Gámez-Días1, D. Kotlarz2, C. Klein2, B. Petersen3, A. Franke3, M. Laaß4, E.O. Glocker5, B. Grimbacher6
1AG Grimbacher, Centre for Chronic Immunodeficiency, Freiburg, Germany
2Department of Pediatrics Dr. von Hauner Children’s Hospital, Ludwig-Maximilians-Universität Munich, Munich, Germany
3Institute of Clinical Molecular Biology, University Hospital SH Campus Kiel, Kiel, Germany
4Children’s Hospital, Technical University Dresden, Dresden, Germany
5Institute of Medical Microbiology and Hygiene, University of Freiburg, Freiburg, Germany
6AG Grimbacher, Centre for Chronic Immunodeficiency, Freiburg, Germany
Inflammatory Bowel Disease (IBD) is a heterogeneous group of chronic inflammatory disorders of the intestinal tract with an incidence of 1-10/100.000 in children in western countries. Patients with an onset of symptoms prior to the age of 10 are classified as early-onset IBD, thus taking in account the distinct phenotype in these patients. In the last years, several monogenetic mutations have been identified as disease-causing, which led to the implementation of hematopoietic stem cell therapy as a potential cure for these patients. However, most of the early-onset IBD patients remain with unknown genetic cause. Identification of new disease-causing mutations will contribute to a better understanding of the pathophysiology and will support a fast and accurate diagnosis as a basis for an optimal treatment.
Currently many genome scanning techniques are used for the identification of new genes associated with disease. However, many of them are expensive and time consuming. Here, we scanned 31 candidate genes for IBD in 44 patients with the diagnosis of early-onset IBD by Next Generation Sequencing (Haloplex enrichment and Illumina Sequencing). We will provide preliminary findings including the listing of candidate genes and a work-flow how this approach can facilitate genetic diagnosis in patients with early-onset IBD.
ESID-0574 Rapamicyn Treatment Improves the Frequency of Circulating Tregs and Their Suppressive Function, in Immune Dysregulation Polyendocrinopathy Enteropathy X-Linked (IPEX) Syndrome with Unusual Presentation
F. Barzaghi 1, L. Passerini2, G. Barera3, M.P. Cicalese4, L. Albarello5, A. Mariani6, A. Aiuti1, R. Bacchetta1
1Department of Paediatric Immunohematology and Division of Regenerative Medicine Stem Cells and Gene Therapy, San Raffaele Telethon Institute for Gene Therapy, Milan, Italy
2Division of Regenerative Medicine Stem Cells and Gene Therapy, San Raffaele Telethon Institute for Gene Therapy, Milan, Italy
3Department of Paediatrics, San Raffaele Scientific Institute, Milan, Italy
4Department of Paediatric Immunohematology, San Raffaele Scientific Institute, Milan, Italy
5Department of Pathology, San Raffaele Scientific Institute, Milan, Italy
6Department of Gastroenterology, San Raffaele Scientific Institute, Milan, Italy
A 10-year-old boy came to our attention for recurrent vomiting since he was 9-year-old. Despite the atypical presentation, Immune-dysregulation-polyendocrinopathy-enteropathy-X-linked syndrome was suspected due to severe ulcerative gastritis with chronic inflammatory infiltrates in the gut mucosa and high serum levels of anti-harmonin auto-antibodies. Detection of FOXP3 gene mutation (c.210+1 G>C) confirmed the diagnosis.
Total enteral nutrition by aminoacidic formula was started, together with immunosuppressive therapy with Rapamycin, based on the rescue of suppressive function after in?vitro exposure of the patient's T cells with Rapamycin. Weight, height, and nutritional indexes improved, and food was progressively reintroduced. Six months later, endoscopic and histological evaluation of the gastric and duodenal mucosa showed a marked improvement as compared to pre-treatment. Expression of FOXP3 on CD4+ cells was still reduced as compared to that of normal donors, however within the CD4+CD25+CD127- regulatory T cells (Tregs) FOXP3 expression was increased as compared to the value before therapy. Importantly, the suppressive activity of the patient's FOXP3 mutated Tregs that was markedly impaired at diagnosis, was at least partially restored during Rapamycin treatment. In conclusion, we report of a case of unusual IPEX in which clinical and histological responses to Rapamycin are associated with increased circulating Tregs and improved suppressive function, which were never reported in the presence of FOXP3 mutation. He is now clinically stable, without signs of other autoimmune manifestations. Further studies are on going to understand whether the positive outcome is related to the specific mutation present in this patient and could be long-lasting.
ESID-0774 Hemophagocytic Lymphohistiocytosis Associated with Severe Combined Immunodeficiency
M. Bataneant 1, A. Pascalau2, C. Jinca1, E. Boeriu1, G. Miculschi3, L. Ritli3, S. Arghirescu1, M. Baica2, L. Marodi4, M. Serban1
1IIIrd Pediatric Clinic, University of Medicine and Pharmacy "Victor Babes", Timisoara, Romania
2IIIrd Pediatric Clinic, Children's Hospital "Louis Turcanu", Timisoara, Romania
3Pediatric Clinic, Children’s Hospital “Gavril Curteanu”, Oradea, Romania
4Pediatric infectology and immunology, University of Debrecen Medical and Health Science Center, Debrecen, Hungary
Introduction. Hemophagocytic lymphohistiocytosis (HLH) is an uncommon disease of childhood, usually secondary to infections, malignancy or autoimmunity and in more rare cases familial. It is characterized by fever, splenomegaly, cytopenia, hypertriglyceridemia, reduced NK cell activity, increased ferritin and sCD25.
Case presentation. Male, 3 months old, BCG vaccinated, with normal physical and motor development, was admitted with suspicion of hemochromatosis due to high level of transaminasis, ferritin and negative viral serology. Mother had two brothers who died in infancy due to infections. The disease started at 2 months of age with cough and fever which didn’t respond to antibiotics. Clinical examination revealed a good general status, rush on the face and trunk, cough, polypneea, without crackles, splenomegaly. Pulmonary X ray evidenced multiple right paratracheal opacities. Laboratory investigations showed Hb=7,4g%, Platelet = 39.000/mmc, lymphocyte = 1780/?mmc (5400-7200), TGO=3119u/l (0-84), TGP=339u/l (0-60), Ferritin = 5866 µg/l (30-400), trigyceridemia=5,5 mM/l (0,7-1,7). Hemochromatosis was excluded and was taken into discussion hemophagocytic lymphohistiocytosis. Immunological investigations performed showed severe T lymphopenia (0,1%) compatible with severe combined immunodeficiency (SCID) diagnosis, genetically confirmed as ?c SCID. Gastric lavage revealed BK. Under tuberculostatic treatment liver and pulmonary manifestations improved. The patient was unrelated matched bone marrow transplanted.
Conclusions. Sometimes the clinical and biological picture is misleading in SCID and very rare the clinical picture is dominated by hemophagocytic lymphohistiocytosis syndrome, in our case determined by BCG-itis. The link between HLH and SCID is probably reduced number and activity of NK cells, predisposing to infections and exaggerated histiocyte activation.
ESID-0621 PRKDC Mutations Associated with Immunodeficiency, Granuloma and Aire-Dependent Autoimmunity
A.L. Mathieu1, E. Veronese2, G.I. Rice3, F. Fouyssac4, Y. Bertrand5, C. Picard6, J.E. Walter7, L. Notarangelo8, C. Schuetz9, D. Moshous10, M. Van Der Burg11, H. Kemp12, C. Malcus13, I. Rouvet13, N. Fabien13, C. Caux2, J.P. De Villartay10, T. Walzer1, A. Belot 14
1U1111, INSERM, Lyon, France
2U1052, INSERM, Lyon, France
3Genetic Medicine, University of Manchester, Manchester, United Kingdom
4Pediatric Hematology, CHU Nancy, Nancy, France
5Pediatric Hematology, Hospices Civils de Lyon, Lyon, France
6Pediatric Immunology, IMAGINE, Paris, France
7Division of Allergy/Immunology Massachusetts General Hospital for Children, Harvard Medical School, Boston, USA
8Division of Immunology Boston Children's Hospital and Harvard Stem Cell Institute, Harvard Medical School, Boston, USA
9Department of Pediatrics and Adolescent Medicine, University Medical Center Ulm, Ulm, Germany
10Pediatric Immunology Necker, IMAGINE, Paris, France
11Deptartment of Immunology Erasmus MC, University Medical Center Rotterdam, Rotterdam, Netherlands
12Department of Human Metabolism, The Medical School University of Sheffield, Sheffield, United Kingdom
13Immunobiology, Hospices Civils de Lyon, Lyon, France
14Pediatric Rheumatology & Nephrology, Hospices Civils de Lyon, Lyon, France
Background PRKDC encodes for DNA-dependent protein kinase catalytic subunit (DNA-PKcs), a kinase that forms part of a complex (DNA-PK) crucial for DNA double-strand break (DSB) repair and V(D)J recombination. In mice, DNA-PK also interacts with the transcription factor AIRE (autoimmune regulator) to promote central T cell tolerance.
Objective We sought to understand the causes of an inflammatory disease with granuloma and autoimmunity, associated to decreasing T and B cell counts over time diagnosed in two unrelated patients.
Methods Genetic, molecular, and functional analyses were performed to characterize an inflammatory disease evocative of a combined immunodeficiency.
Results We identified PRKDC mutations in both patients. These patients exhibited a defect in DNA DSB repair and V(D)J recombination. Circulating T cells had a skewed cytokine response typical of Th1 and Th2 profiles. Moreover, mutated DNA-PKcs failed to promote AIRE-dependent transcription of peripheral tissue antigens in?vitro. The latter defect correlated ?in?vivo, with the production of anti-Calcium Sensing Receptor (anti-CaSR) autoantibodies, which are usually found in AIRE-deficient patients. In addition, 9 month after bone marrow transplantation, Patient 1 developed Hashimoto thyroiditis suggesting that organ-specific autoimmunity is linked to non-hematopoietic cells such as AIRE-expressing thymic epithelial cells.
Conclusion Deficiency of DNA-PKcs, a key AIRE partner, can present as an inflammatory disease with organ-specific autoimmunity and these findings highlight the essential role of DNA-PKcs in regulating autoimmune responses and maintaining AIRE-dependent tolerance in human.
ESID-0422 Inherited Multiple Autoimmunity: Molecular and Clinical Characterization of IPEX and IPEX-Like Syndromes
S. Ciullini Mannurita 1, M. Vignoli1, G. Colarusso1, R. Bacchetta2, M. Cecconi3, A. Tommasini4, A. Gennery5, S.M. Holland6, A.J. Cant5, E. Gambineri1
1Department of "NEUROFARBA" section of Child's Health, University of Florence A.Meyer Children's Hospital, Florence, Italy
2Division of Regenerative Medicine Stem Cells and Gene Therapy, San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET), Milan, Italy
3Human Genetic Laboratories, Ospedale Galliera, Genoa, Italy
4Paediatric Immunology Laboratory, IRCCS Burlo Garofalo, Trieste, Italy
5Department of Paediatric Immunology, Newcastle upon Tyne Hospitals, Newcastle upon Tyne, United Kingdom
6Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases National Institutes of Health, Bethesda, USA
IPEX is characterized by early-onset severe autoimmune enteropathy, eczema and endocrinopathy. Increasing number of patients affected by multiple autoimmunity resembling IPEX, without FOXP3 mutations have contributed to define an IPEX-like phenotype.
We set to identify clinical and laboratory indicators to better define this disease spectrum.
Overall, we analyzed 76 patients with suspect of IPEX syndrome. Considering patients clinical features, Tregs and FOXP3 expression by flow cytometry, FOXP3, CD25 and/or STAT5b genes were sequenced. Only in 16 patients we identified FOXP3 mutations. Absent expression of CD25 was detected in two patients with CD25 mutations. and one patient was described as a STAT5b deficiency. In the IPEX-like cohort immune mediated enteropathy, frequently associated with failure to thrive, was the key clinical feature. In patients without enteropathy, endocrinopathy and cytopenias are the main clinical manifestations. The age of onset is variable: a group of patients presents diarrhoea within the first year of life and behave similarly to the IPEX cohort, another group had a delayed onset characterized by multiple autoimmune disease (skin and endocrinopathy being less predominant than to autoimmune cytopenias and infections), a third group had only early onset diarrhoea with no other symptoms (IBD-like?). Elevated IgE and dysgammaglobulinemia are often reported.
Outputs from this study define disease subgroups facilitating specific molecular studies in order to better understand and delineate the mechanisms of tolerance failure in patients with complex autoimmune disease. The final objectives would be the definition of new disease entities and ultimately discovery of new genes involved in immune-dysregulation diseases.
ESID-0684 The FOXP3 Gene Mutation in Infancy with Immune Dysregulation, Polyenocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome in Hungary
I. Csürke 1, F. Rieux-Laucat2, A. Szabó3, F. Dicso1
1Pediatric Department, CountyTeaching Hospital, Nyiregyháza, Hungary
2Laboratory, Hopital Necker Enfant Malades, Páris, France
3Pediatric Department, Semmelweis University, Budapest, Hungary
Immune dysregulation,polyendocrinopathy,enteropathy,X-linked (IPEX) syndrome is a rare monogenic, primer immunodeficiency (PID),characterized by multi-organ autoimmunity caused by the mutation in FOXP3 gene, which is the master gene of regulatory T cells(Treg).
In this case report we will present a patient with IPEX syndrome causing by a missense mutation of FOXP3 gene.The male infant was born on the 36.gest.week,with 2600g from an uneventful pregnancy.On the 4th day of life a heart murmur has been detected,VSD revealed by echocardiography.The first presentation of this condition was intractable,watery diarrhoea, and also diffuse, maculo-papular skin lesions have been appeared on his body at 5?day of age.When he was 10?day old heart failure has been developed and the baby was transferred to ICU.The laboratory tests suggested a severe perinatal infection; therefore combined antibiotic therapy has been administered.On the third week of life regarding to the high blood sugar levels, intensified insulin treatment was introduced.One month of age he had severe anemia which required red blood cell transfusion.By nutrition with special formula was not changed profuse diarrhea,skin disorders,difficult diabetes control, anemia, thrombocytopaenia,elevated total IgE levels and male gender raised the suspicion of IPEX. Based on clinical picture commenced on immunosuppressive therapy.The flowcytometry and genetic examinations confirmed (missense mutation in the last exon of the Foxp3 gene: c.1208 g>t; p.G403W) the clinical diagnosis.At the age of 3 months we were able to confirm genetically the IPEX syndrome.
This case is the first demonstrated of IPEX syndrome in Hungary.
ESID-0689 Autoimmune Lymphoproliferative Syndrome (ALPS). A Mexican Case Report
P. de Baro-Alvarez 1, A. Partida-Gaytan2, O. Saucedo-Ramirez1, G. López-Herrera2, L. Berrón-Ruíz2, B.E. del Río-Navarro1
1Alergia e Inmunología Clínica Pediátrica, Hospital Infantil de México Federico Gómez, México, Mexico
2Unidad de Investigación en Inmunodeficiencias Primarias, Instituto Nacional de Pediatría, México, Mexico
Introduction Autoimmune lymphoproliferative syndrome (ALPS) is a disorder of apoptosis in which the inability of lymphocytes to die leads to non-malignant massive lymphadenopathy, hyper-splenism, and autoimmune cytopenias, of childhood onset. The immunological findings include polyclonal hypergammaglobulinemia and expansion of T cells lacking either CD4 or CD8 on their surface (double-negative T cells). The most frequent genetic causes are mutations in gene encoding for CD95 (FAS), CD95L (FASL), and caspase 10 (CASP10), though recently many ALPS-like syndromes with genetical heterogeneity have been described.
Case report We present a 13 year old male, with history of adenoamigdalectomy at age 6 years because of recurrent infections, appendectomy and axilar and inguinal node biopsy at age 10 years with unspecific follicular hyperplasia. At age 11 years he suffered from recurrent acute otitis media (AOM), requiring bilateral ventilation tubes and mastoidectomy. Beause of persistence of AOM, important lymph node enlargement, hepatosplenomegaly and citopenias (neutropenia & thrombocytopenia) multidisciplinary diagnostic work up was done; infectious and oncological causes were discarded. Immunological approach discarded humoral defect but revealed increased double negative T cells (TCRabCD3+CD4-CD8-), increased vitamin B12 levels and evidence of autoimmunity integrating clinical diagnosis of ALPS (according to revised 2010 criteria). Immunosuppressive therapy was started with excellent clinical response, though molecular diagnosis is still pending.
Conclusion Primary immunodeficiencies, although frequently present with recurrent or atypical infections, clinical presentation is not restricted to such phenomena. Lymphoproliferation and autoimmunity also reflect defects on the immune system and are recognized as PIDs. The knowledge of such clinical heterogeneity may aid in prompter diagnosis.
ESID-0675 Relationship Between Primary Antibody Deficiency and Autoimmunity: Effect of TACI and PTPN22 Mutations on the Expression of B Regulatory, T Regulatory and T Follicular-Helper Cells
E. Ballanti 1, S. Di Cesare2, M. Chiriaco3, F. Specchia4, R. Miniero5, S. Ferrari6, D. Lucente5, M. Romiti7, C. Cancrini3, R. Perricone9, P. Rossi3, G. Di Matteo8, V. Moschese7
1UOC Reumatologia, Policlinico Tor Vergata, Rome, Italy
2DPUO DPUO Department of University Hospital Pediatrics, Ospedale Pediatrico Bambino Gesu', Rome, Italy
3DPUO Department of University Hospital Pediatrics, Ospedale Pediatrico Bambino Gesu', Rome, Italy
4Dip. di Pediatria, S. Orsola-Malpighi University Hospital, Bologna, Italy
5Department of Pediatrics, University Magna Graecia of Catanzaro, Catanzaro, Italy
6Medical Genetics Unit, S. Orsola-Malpighi University Hospital, Bologna, Italy
7Medicina dei Sistemi, University of Tor Vergata, Rome, Italy
8Medicina dei Sistemi, University of Tor Vergata, Roma, Italy
9Rheumatology, Allergology and Clinical Immunology, University of Rome Tor Vergata, Rome, Italy
Autoimmune manifestations are often observed in patients suffering from primary antibody deficiencies (PAD), with immune thrombocytopenia purpura and autoimmune haemolytic anaemia representing the most common conditions. The pathogenesis of autoimmunity in these patients remains to be elucidated. An increased incidence of autoimmunity has been demonstrated in CVID patients with C104R TACI mutation. The 1858T allele of protein tyrosine phosphatase non-receptor type 22 (PTPN22) exhibits a consistent associations with sporadic autoimmune disease but has been recently demonstrated to be associated with PAD and coexistent autoimmune diseases. The cellular and immunological defects that link the PTPN22 or TACI mutations to autoimmunity and/or immunodeficiency remain to be elucidated. A genetic variation in PTPN22 and/or TACI could be associated to altered function of T and B lymphocytes. The effect of TACI and PTPN22 mutations on the expression of B regulatory (B reg), T regulatory (T reg) and T follicular-helper (TFH) cells has been evaluated in a cohort of pedriatic patients with hypogammaglobulinemia with or without TACI and/or PTPN22 mutation and in their adult relatives with or without autoimmune disorders. Preliminary data indicate reduction of B reg cells and increased amount of TFH cells. These results are consistent with data from other two recent works that demonstrated activation of autoreactive B cells in patients with a single TACI or PTPN22 mutation.
The emerging concept that PAD are not only attributed to B cell-intrinsic defects but also to functional impairments in other immune cell lineages, opens up new roads for the understanding of these conditions.
ESID-0373 MBL Deficiency Doen't Predispose to Systemic Lupus Erythematosus in Algerian Population
K. Djenouhat 1, H.I.B.?A. Ait Hamoudi2, M. Boucelma3, F. Haddoum4
1Immunology, Institut Pasteur of Algeria, Algiers, Algeria
2Immunology, CHU Mustapha, Algiers, Algeria
3Internal medecine, CHU Bab El Oued, Algiers, Algeria
4Nephrology, CHU Parnet, Algiers, Algeria
Introductionand aim: Circulating MBL is synthesized by liver, monocytes, and leukocytes, and functions as the initial factor in the lectin pathway of complement. Variants of MBL gene have been associated with autoimmune disorders. The aim of this study was to explore whether MBL deficiency is associated with susceptibility to systemic lupus erythematosus (SLE) in Algerian population.
Patients and methods: A total of 83 SLE patients and 52 healthy controls were enrolled in the study. Sex ratio is 1male for 10 females for both populations
Result and discussion: Antigenic MB was performed by human ELISA kit and functional MBL measurement by functional MBL/MASP-2 assay has been measured for the quantitative measurement of active human MBL according to the manufacture's recommendations(The Hycult Biotech human, Netherlands ). Dosage of C1q by radial immunodiffusion and dosages of C3 and C4 components were done by mini-neph (Binding site, U.K.).
Without considering the state of disease, 14.45% of SLE patients have very low amounts of MBL, half of them were in relapse. However, when assays are redone during remission, we noticed no statistical difference between SLE patients and healthy controls (6.02% vs 5.76%, p>0.05).
Results of the present study indicate, on the one hand, that MBL deficiency frequency in Algerian population is similar to Caucasian one; and in the second hand, there is no possible risk of SLE in patients with MBL deficiency even without performing MBL genotyping.
ESID-0309 Recurrent Myelitis in CVID
L. Marinangeli 1, R. Morariu2, S. Gambini2, V. Pedini2, M.G. Danieli2
1Allergologia ed immunologia, Immunologia Clinica, Ancona, Italy
2Medicina Interna, Clinica Medica, Ancona, Italy
The acute myelitis is an inflammation of the neurons and nerve bundles that pass through the spinal cord, and can be determined by multiple causes.
We describe two cases of women with a history of recurrent myelitis. The first, 71 years old, presented with sensory ataxia, paraparesis and dysesthesia of the lower limbs, weakness of the right hand, neurogenic bladder.
The second, 57 years old, presented with atasso-spastic gait, tetra-hyperreflexia, difficulty in emptying the bladder.
In both cases laboratory evaluation showed decreased serum levels of all immunoglobulin isotypes (IgG 384 mg/dl, IgA 10 mg/dl, IgM 11 mg/dl in the first patient; IgG 545 mg/dl, IgA 53 mg/dl, IgM 34 mg/dl in the second one) and low specific antibody responses.
These elements have allowed us to formulate the diagnosis of common variable immunodeficiency (CVID).
Both patients were successfully treated with high dose of intravenous immunoglobulin (IVIg) 2 g/?Kg on two consecutive days monthly: the first patient for three months, then as remission-maintaining drug, we decided to treat her with subcutaneous immunoglobulin (SCIg); the second patient maintains intravenous administration at a dose of 30 g/?month.
At three-years follow-up control, the response to treatment was good. No relapses occurred.
Myelitis is a rare manifestation of CVID, a disease that is associated in 20% of cases with systemic autoimmune disorders.
The intravenous and subcutaneous immunoglobulin treatment is also effective in protecting the recurrence of such event.
ESID-0272 Human CD33 Deficiency
C.B. Geier 1, D. Bra1, A. Linder1, M.M. Eibl1, H.M. Wolf1
1Immunologische Tagesklinik, Immunologische Tagesklinik, Vienna, Austria
CD33 is an immunoglobulin superfamily protein expressed on cells of the myeloid lineage. It contains two Ig-like extracellular domains and two immunoreceptor tyrosine-based inhibitory motifs (ITIM). Although the whole spectrum of CD33 remains unclear, present studies suggest a role as an inhibitory receptor.
In the present study we describe a patient with complete CD33 deficiency. The patient presented with a history of recurrent angioedema.C1 inhibitor deficiency type I and II were excluded, no mutation in F12 gene was found. The Patients’ myeloid lineage completely lacked CD33 surface expression. Molecular analysis revealed a homozygous 4 nt deletion which resulted in a frameshift and premature stop codon. The patents’ unrelated parents and sister are heterozygous and healthy.
In the CD33 deficient patient monocytes and granulocytes were normal in number, degranulation of activated basophiles and eosinophils stimulated with FMLP and measurement of oxidative burst by flow cytomerty was within the normal range. Measurement of IL8 and IL-1RA production by CD33 deficient mononuclear revealed an enhanced spontaneous IL8 production. LPS and Fc?R mediated IL8 production was close to the upper limit of healthy controls. IL1-RA release was comparable to normal controls. Down-regulation of CD193, chemokine receptor 3, on basophils activated via CD33 was reduced in the patient.
In the absent of functional CD33, dysregulation of proinflamatory mediators might play a role in the pathogeneses of angioedema in our patient.
ESID-0219 Recurrent Salmonella Infections Associated with Cutaneous Vasculitis and Abdominal Tuberculosis in Interleukin-12 Receptor Beta 1 Deficiency: Two Case Reports
F. GENEL 1, S. BAHCECI ERDEM1, N. GULEZ1, E. OZBEK1, I. DEVRIM1, J. BUSTAMANTE2, J.L. CASANOVA2
1Department of Pediatrics, Dr. Behcet Uz Children's Hospital, IZMIR, Turkey
2Laboratory of Human Genetics of Infectious Diseases, Institut National de la Sante et de la Recherche Medicale, Paris, France
Patients with interleukin-12 receptor beta 1 (IL-12Rbeta1) deficiency have increased susceptibility to mycobacterial and salmonella infections.
Case 1: An 18 month old boy born to third-cousin parents was admitted to our hospital with fever and a 2 month history of left axillary lymphadenomegaly. Salmonella spp was isolated in blood culture. Flow cytometry revealed absence of IL-12Rbeta1 expression on activated T cells. IL-12Rbeta1 sequence analysis revealed a homozygous mutation C198R. In follow up recurrent salmonella infection observed and subcutaneous interferon gamma treatment were initiated. At age 5 years, he experienced palpable purpuric skin lesions on the lower extremities. Stool culture yielded Salmonella spp. Clinical signs and symptoms of vasculitis resolved after ceftazidime and interferon gamma treatment. At age 10 years, a syndrome of abdominal pain with fever led to a laparotomy and abdominal tuberculosis was diagnosed. Antituberculous therapy was initiated.
Case 2: A 3.5 year old boy from non-consanguineous family admitted to our hospital with fever arthralgia, purpuric eruptions on the lower extremities and recurrent salmonella infection. Genetic analysis revealed a homozygous mutation in IL12Rb1 gene, g.10568_10574delins8 (c.557_563delins8) conferring a complete defect. Anti-bacterial therapy and interferon gamma treatment provided good response.
As a result recurrent salmonella infections and mycobacterial infections should alert clinicians for an underlying genetic defect of the IL-12/IFN gamma axis.
ESID-0223 Haematopoietic Stem Cell Transplantation May Resolve the Immune Deficit Associated with STAT-3 Deficient Hyper-IGE Syndrome
S. Harrison 1, M. Slatter1, Z. Nademi1, S. Jolles2, W. Al-Herz3, T. Flood4, A. Cant4, R. Doffinger5, C. Glocker6, B. Grimbacher7, M. Abinun1, A. Gennery1
1Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
2Immunodeficiency Centre for Wales, University Hospital of Wales, Cardiff, United Kingdom
3Allergy and Clinical Immunology Unit, Al-Sabah Hospital, Kuwait City, Kuwait
4Paediatric Immunology Allergy and Infectious Diseases, Great North Children's Hospital, Newcastle upon Tyne, United Kingdom
5Department of Clinical Biochemistry and Immunology, Cambridge University, Cambridge, United Kingdom
6Center for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany
7Institute of Immunity and Transplantation, University College London, London, United Kingdom
Background: STAT3-deficient hyper-IgE syndrome (HIES) is a rare autosomal dominant primary immunodeficiency, typified by recurrent staphylococcal infections, skin, bone and lung abnormalities, eosinophilia, and IgE serum levels >2000 IU/ml1. Negative results following haematopoietic stem cell transplantation (HSCT) reported in 20002 perpetuated largely supportive treatment thereafter1.
Methods: A retrospective review of STAT3-deficient patients who underwent HSCT.
Results: A 24-year-old woman with recurrent infections from birth and frequent hospitalisation was transplanted 18 years ago. Despite 100% donor chimerism, the procedure was deemed unsuccessful following IgE increase, but subsequent improvement is manifest by infrequent hospital admissions, fewer infections and improved skin and lung function. IgE levels remained elevated (3314 kU/L, Figure?1). She no longer requires IVIG, but shows an IL17 production, which indicates incomplete Th17 reconstitution, IFN-? and IL12 response and production are normal.
A 22-year-old man suffered frequent severe bronchopneumonia, bronchiectasis resulting in lobectomy, osteopenia causing pathological fractures, and repeated hospital admissions, negatively impacting development. At 9 years post-HSCT, he had 100% donor chimerism with reduced IgE from 8055 to118 kU/L, with significant improvement in lung function testing, decreased infections, skin inflammation, and hospital admissions, despite impaired IL17 production. He has relinquished disability living allowance and is now in employment.
A 13-year-old patient showed improved lung function 1 year post-HSCT, with normal IL17 production.
Discussion: These 3 patients demonstrated improvement in immunological disease and social parameters. HSCT may cure the underlying immune deficit in HIES, and should be considered for a select group of patients3. Appropriate selection criteria need to be defined.
ESID-0222 Identification of Autoantibodies in Carriers of X-Linked Chronic Granulomatous Disease by Autoantigen Microarray Analysis
K. Abernethy 1, A. Battersby1, M.S. Pearce2, D. Barge3, J. Walter4, L. Notarangelo2, C. Cale5, D. Goldblatt6, A. Gennery1
1Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
2Division of Immunology, Boston Children's Hospital, Boston, USA
3Flowcytometry Laboratory, Royal Victoria Infirmary, Newcastle upon Tyne, United Kingdom
4Division of Allergy/Immunology, Massachusetts General Hospital for Children, Massachusetts, USA
5Clinical Immunology, Great Ormond Street Hospital, London, United Kingdom
6Institute of Child Health, University College London, London, United Kingdom
Introduction: X-linked chronic granulomatous disease (XL-CGD), a rare primary immunodeficiency due to mutations in CYBB causes reduction in, or absence of, phagocyte respiratory oxidative burst. Clinical manifestations include infection susceptibility, granuloma formation and inflammatory/autoimmune disease. XL-CGD carriers have dual leukocyte populations, expressing wild type or mutant CYBB. Discoid lupus has been observed carriers, with increasing evidence that autoimmune phenomena are more common. Routine autoantibody screening by immunofluorescence is usually negative. We investigated the frequency and nature of autoantibodies in carriers using a high-throughput method.
Methods: Serum from 43 XL-CGD carriers, identified via the UK CGD registry, was tested for IgG antibodies against a panel of 95 auto-antigens. Microarray technology exhibits greater sensitivity for autoantibody detection than traditional techniques, permitting rapid screening of a large set of autoantibody specificities. Positivity was defined as normalised fluorescence intensity greater than the mean plus 2 standard deviations of normal controls.
Results: 42/43 (97.7%) carriers were positive for at least one IgG autoantibody. 11/43 (25.6%) were positive for >25% of auto-antigens, most frequently glycated-albumin, heparan HSPG, nuculoporin-62 and C1q. Auto-antibodies against Nup62, GP2, SP100, Collagen IV, Vimentin and myelin-associated glycoprotein were significantly increased in carriers compared to normal controls. We observed a significant negative correlation (r=-0.48, p=0.002) between the frequency of positive autoantibodies and neutrophil oxidative burst measured by Dihydrorhodamine Flow Cytometric Assay.
Discussion: Microarray data may identify potential biomarkers to assist in identifying carriers at risk of autoimmune disease. Further work is required to confirm the significance of these findings using ELISA techniques.
ESID-0579 Aicardi Goutieres Syndrome: New Insights on a Novel Set of Inborn Errors of Immunity
D. Vairo 1, R.M. Ferraro1, D. Moratto1, S. Pizzi2, M. Cattalini3, S. Orcesi4, A. Tincani5, F. Facchetti6, J. Galli7, A. Plebani3, Y. Crow8, L.D. Notarangelo, 9, P. Plevani2, E. Fazzi7, S. Giliani1
1c/o Spedali Civili, A.Nocivelli Institute for Molecular Medicine, Brescia, Italy
2Biochemistry, University of Milan, Milan, Italy
3University of Brescia, Pediatric Clinic, Brescia, Italy
4Unit of Child Neurology and Psychiatry, IRCCS "C. Mondino National Institute of Neurology" Foundation, Pavia, Italy
55Allergology and Immunology Clinic, University of Brescia, Brescia, Italy
6Pathology, University of Brescia, Brescia, Italy
7Unit of Child Neurology and Psychiatry, University of Brescia, Brescia, Italy
8Manchester Centre for Genomic Medicine, University of Manchester, Manchester, UK
Aicardi–Goutières Syndrome (AGS) is an inherited neurological disorder characterized by early onset in infancy, basal ganglia and white matter calcifications, leukoencephalopathy, and elevated CSF interferon-a (IFN-a) levels. The inappropriate induction of Type I interferon-mediated immune response let to include AGS in Type I interferonopathy. Causative mutations have been described in several genes encoding intracellular enzymes involved in nucleic acid metabolism. The mutations cause the intracellular accumulations of nucleic acids which trigger a chronic cell intrinsic antiviral Type I IFN response
In this study, we explored the immune setting in a patient with mutation in the gene TREX1, six patients mutated in RNASEH2B gene and a patient mutated in IFIH1 gene. Flowcytometry and immunoblot analysis show abnormal activation of the IFNa signaling. All subjects show higher expression of STAT1 protein and an increase of STAT1 phosphorylation after IFNa stimulation. Patients show an endogenous up-regulation of inflammatory cytokines and chemokines, high levels of CXCL10, both as mRNA and protein, responsible for the recall of leukocyte in the inflammation sites and an increased levels of antiviral MxA. Finally, AGS patients show a pro-inflammatory activity of IL10, resulting in an abnormal activation of STAT1 that lead to an up-regulation of genes IFNa dependent.
Taken together these data show an active dysregulation role mediated by IFNa on the immune system which results in a constitutive anti viral phenotype
ESID-0768 Late Diagnosis of IPEX in a Carrier of a Disease-Associated Mutation in FOXP3
L.D. Notarangelo 1, D. Moratto2, M. Chiarini3, S. Masneri2, C. Mazza2, J.L. Casanova4, B. Boisson4, L.D. Notarangelo5, S. Giliani2
1Pediatric Oncoematology, Spedali Civili, Brescia, Italy
2Cytogenetic and Clinical Genetic, A.Nocivelli Institute for Molecular Medicine, Brescia, Italy
3Laboratory of Biotechnology Diagnostics Department, Spedali Civili, Brescia, Italy
4St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller University, NY, USA
5Childrens' Hospital, Harvard medical School Division of Immunology, Boston, USA
The patient is the second child of healthy and probably consanguineous parents, whose firstborn died at 5 months of age for sepsis after having presented with eczema, lymphoadenomegaly and chronic diarrhea. At the age of 27 he was analyzed by whole exome sequencing (WES) in search for the underlying cause of the clinical phenotype. Since no candidate genes were found filtering for autosomal recessive traits, the analysis was extended to the X-chromosome showing a C481S mutation in the FOXP3 gene, that has already been associated to typical IPEX phenotype.
Clinical history of this patient started at 3 months of age with oral candidiasis; at 6 months he was hospitalized because of a protracted diarrhea, eczema and failure to thrive. Since the first year of age he was put on a cow milk-free diet because of a diagnosis of milk protein intolerance, and on antimicrobial prophilaxis and IVIG therapy because of several episodes of wheezing, pneumonias and persistent diarrhea. The subsequent clinical course has been characterized by several episodes of steroid responsive thrombocytopenia, alopecia (7 y.o.), autoimmune thiroiditys (13 y.o.) and occasional pulmonary infections. At present he is in good general condition, with no major infection; he still presents alopecia and he is on antimicrobial prophilaxis, IVIG and immunosuppressive therapy.
Laboratory findings showed a prevalence of effector cells in the T-lymphocyte compartment and a partially restricted T-cell-receptor repertoire. Regulatory T cells were reduced and showed an impaired Foxp3 expression, confirming the detrimental effect of the reported mutation.
ESID-0638 FLH Type 5 Caused by a Novel Mutation in STXBP2 Gene: An Unusual Cause of Failure to Thrive and Diarrhea in Infancy
F. Haerynck 1, R. De Bruyne2, M. Dullaers3, R. Uwera2, B. Callewaert4, E. De Baere4, M. Van Winckel2, S. Van Biervliet2, S. Vandevelde2, V. Bordon5
1Pediatric Pulmonology and Immunology, University Hospital Ghent, Ghent, Belgium
2Pediatric Gastroenterology and Hepatology, University Hospital Ghent, Ghent, Belgium
3Department of Respiratory Medicine- Clinical Immunology Research Lab, University Hospital Ghent, Ghent, Belgium
4Center of Medical Genetics, University Hospital Ghent, Ghent, Belgium
5Pediatric Hematooncology and Bone marrow transplant Center, University Hospital Ghent, Ghent, Belgium
Familial hemophagocytic lymphohistiocytosis (FHL) is caused by genetic defects in cytotoxic granule components or their fusion machinery, leading to impaired natural killer cell and/or T lymphocyte degranulation and/or cytotoxicity. STXBP2, also known as MUNC18-2, has recently been identified as the disease-causing gene in FHL type 5 (FHL-5).
We represent a 9-month old boy with a previous history of recurrent infections, failure to thrive and chronic diarrhea and an acute presentation of irritability, fever, hepatosplenomegaly, ascites, pancytopenia, low fibrinogen, elevated ferritin and triglycerides and increased soluble CD25 (24200 pg/ml) compatible with hemophagocytic lymphohistiocytosis (HLH). No haemophagocytosis was seen on bone marrow and brain MRI showed demyelinisation of the white matter. Further diagnostic work-up revealed slightly decreased NK cell activity and normal expression of perforin. According to the history of failure to thrive, intractable diarrhea, hypogammaglobulinemia and HLH, a mutation analysis of STXBP2 and UNC13D genes was performed: two mutations were identified in STXBP2 gene: 902+5G>A mutation in intron10 affecting RNA splicing and a novel mutation c.421 del G in exon 6 (p.Glu141ArgfsX68) leading to a frame shift. Treatment was started following HLH-2004 protocol and finally, the patient underwent hematopoietic stem cell transplantation (HSCT) with good immunological response. However, diarrhea continues requiring parenteral nutrition.
FLH-5 should be considered in the differential diagnosis of a child presenting with HLH in a context of chronic diarrhea and failure to thrive. In FLH-5, partial conservation of NK cell function can be observed, as is seen in our proband.
ESID-0347 Identification of a Novel Homozygous FAS Death Ligand (FASLG) Mutation Affecting the Intracellular Protein Domain that Causes Autoimmune Lymphoproliferative Syndrome (ALPS)
A. Hönscheid1, S. Nabhani1, P.T. Oommen1, J. Schaper2, M. Kuhlen1, H.J. Laws1, A. Borkhardt1, U. Fischer1
1Department of Pediatric Oncology Hematology and Clinical Immunology Medical Faculty, Heinrich Heine University, Düsseldorf, Germany
2Department of Diagnostic and Interventional Radiology Medical Faculty, Heinrich Heine University, Düsseldorf, Germany
The Autoimmune Lymphoproliferative Syndrome (ALPS) is a rare disorder, which is characterized by chronic, non-malignant lymphoproliferation, autoimmunity and an elevated risk of malignancies due to defective lymphocyte apoptosis. The FasL/?Fas pathway initiates apoptosis in lymphocytes thereby controlling immune responses. Worldwide only 400 ALPS cases are described. In two thirds the genetic defect is identified. Most cases are associated with germline or somatic mutations in the FAS gene. Only four disease-causing variants in the FASLG gene are described to date.
We report on two siblings of a Libyan family who were born to consanguineous parents. The seven years old boy (ALPS#1) and his four years old sister (ALPS#2) presented both with massive lymphadenopathy, hepatosplenomegaly, hypergammaglobulinemia, increased levels of double negative T cells (TCRaß+, CD4-CD8-) and vitamin B12 (for detailed ALPS characteristics see table).
We performed conventional sequencing of FAS and FASLG genes and identified a novel homozygous 1?bp insertion in the intracellular domain of the FASLG gene in both patients, which results in absent FasL protein expression as confirmed by western blot and flow cytometry. In vitro stimulation of activated patients` lymphocytes with recombinant FasL showed normal apoptosis verifying an intact Fas pathway and pointing to the lacking FasL expression as the cause for the clinical diagnosis of ALPS in both patients.
Our present findings report a new mutation in the FASLG gene, which is inherited in an autosomal recessive mode and has to be taken into account for genetic screening in ALPS patients lacking FAS mutations.
Table?Clinical and immunological characteristics
| ALPS#1 | ALPS#2 | reference | |
| Clinical phenotype | |||
| age (years) | 7 | 4 | |
| lymphadenopathy | + | + | |
| hepastosplenomegaly | + | + | |
| Autoimmunity | |||
| Clinical features | thrombocytopenia | / | |
| Antibodies against | CD16, CD177,GPIIb/IIIa,GPIa/IIa | CD16, GPIIIb/IIIIa,GPIa/IIa | |
| Immunological phenotype | |||
| TCRab+, CD4-/CD8- DNT cells [% of CD3+ cells] | 17,39 | 29,17 | 5 |
| B220+ [% of DNT] | 70,52 | 79,54 | 8-40 |
| CD3+, CD25+ [%] | 1,43 | 1,05 | 1-5 |
| CD3+, HLA-DR+ [%] | 2,51 | 1,91 | 6,0-7,0 |
| CD3+CD25/HLA-DR ratio | 0,57 | 0,55 | 3 |
| CD19+, CD27+ [%] | 4,58 | 5,27 | 16 |
| CD4/CD8 ratio | 0,8 | 0,7 | 1,2-1,6 |
| IgG [mg/dL] | 4198 | 3657 | 572-1474 |
| IgA [mg/dL] | 491 | 984 | 34-305 |
| IgM [mg/dL] | 72 | 86 | 31-208 |
| Vitamin B12 [pg/mL] | >2000 | >2000 | 197-866 |
ESID-0300 High Dimensional Single-Cell Characterization of Human Toll-Like-Receptor Activation: Potential Clinical Applications in Systemic Lupus Erythematosus (SLE)
E. Hsieh 1, W. O'Gorman2, E. Savig2, P.F. Gherardini2, I. Balboni3, M. Davis4, G.P. Nolan4
1Allergy and Immunology, Stanford University, Palo Alto, USA
2Immunology, Stanford University, Palo Alto, USA
3Rheumatology, Stanford University, Palo Alto, USA
4Microbiology and Immunology, Stanford University, Palo Alto, USA
Aberrant Toll-Like-Receptor (TLR) activation can result in the development of autoimmunity and/or immunodeficiency. TLRs are differentially expressed between immune cell subsets, but a comprehensive analysis of how regulators across the different cell types respond in a system-wide manner has previously not been possible. We used mass cytometry (CyTOF) to characterize TLR activation in healthy individuals, establishing a reference map against which to compare pathological processes. We applied this TLR reference framework to further understand deregulated immunomodulatory cytokine networks in te pathogenesis of systemic lupus erythematosus (SLE).
In healthy donor blood samples, TLR activation induced distinct signaling patterns and downstream cytokine signatures in different immune cell subsets. SLE patient blood samples, however, demonstrated an altered cytokine signature with increased MCP-1 (CCL2) production by CD14hi monocytes in the absence of any stimulation. In addition, stimulation of healthy donor blood samples with SLE patient plasma increased MCP-1 production, suggesting that plasma factors induce monocyte activation and production of MCP-1. We also observed decreased MCP-1 production after patients initiated immunosuppression, suggesting that MCP-1 may be useful as a disease biomarker and a potential therapeutic target.
ESID-0056 DOCK8 Deficient Patients Have a Breakdown in Peripheral B Cell Tolerance and Defective Regulatory T Cells
E. Janssen 1, H. Morbach2, S. Ullas1, J. Bannock2, C. Massad2, L. Menard2, I. Barlan3, G. Lefranc4, H. Su5, M. Dasouki6, W. Al-Herz7, S. Keles8, T. Chatila1, R. Geha1, E. Meffre2
1Immunology, Boston Children's Hospital, Boston, USA
2Immunobiology, Yale University School of Medicine, New Haven, USA
3Pediatric Allergy and Immunology, Marmara University, Istanbul, Turkey
4Laboratory of Molecular Immunogenetics, IMGT University of Montpellier and CNRS Institute of Human Genetics, Montpellier, France
5Human Immunological Diseases Unit, National Institutes of Health, Bethesda, USA
6Genetics Endocrinology and Metabolism, University of Kansas Medical Center, Kansas City, USA
7Pediatrics, Kuwait University, Kuwait City, Kuwait
8Pediatric Immunology and Allergy, Meram Medical Faculty, Konya, Turkey
Background: Dedicator of Cytokinesis 8 (DOCK8) deficiency is characterized by recurrent sinopulmonary and cutaneous infections, eosinophilia, elevated serum IgE levels, and a high incidence of allergic and autoimmune manifestations.
Objective: Our objective was to determine the role of DOCK8 in the establishment and maintenance of human B cell tolerance.
Methods: Autoantibodies were measured in the plasma of DOCK8 deficient patients by autoantigen microarray, ELISA, and HEp-2 cell staining. The antibody coding genes from new emigrant/transitional and mature naive B cells from DOCK8 deficient patients were cloned and assessed for their ability to bind self-antigens. Regulatory T (Treg) cells in the peripheral blood were analyzed by flow cytometry, and their function was tested by examining their capacity to inhibit the proliferation of CD4+CD25- T effector (Teff) cells.
Results: DOCK8 deficient patients had increased levels of diverse autoantibodies in their plasma. We determined that central B cell tolerance did not require DOCK8 as evidenced by the normal low frequency of polyreactive new emigrant/transitional B cells in DOCK8 deficient patients. In contrast, autoreactive B cells were increased in the mature naïve B cell compartment, exposing a defective peripheral B cell tolerance checkpoint. We also determined that Treg cells were decreased and exhibited impaired suppressive activity in DOCK8 deficient patients.
Conclusions: Our data support a critical role for DOCK8 in Treg cell homeostasis and function and the enforcement of peripheral B cell tolerance.
ESID-0020 Clinical Presentation and Long-Term Outcome of Anti-Interferon-Gamma-Autoantibodies Associated Infections: Case Series of 10 Patients
T. Kampitak 1, K. Jutivorakool1, C. Suankratay1
1Medicine, Chulalongkorn University, Bangkok, Thailand
Background: Anti-interferon-?-autoantibodies have been recently demonstrated a causal role of opportunistic infections in Asian adults without human immunodeficiency virus (HIV) infection. Associated infections typically involve disseminated infections caused by nontuberculous mycobacteria (NTM) and Salmonella. However, data regarding clinical presentation and long-term outcome of affected patients remain lacking.
Methods:
Opportunistic infections associated with anti-interferon-?-autoantibodies in otherwise immunocompetent patients occurred during 2008-2013 in Chulalongkorn University Hospital, Bangkok, Thailand were reviewed.
Results:
Ten patients carrying anti-interferon-?-autoantibodies with opportunistic infections were identified.. High-titer anti-interferon-?-autoantibodies were detected in all patients but none in patients with disseminated tuberculosis or healthy controls. The infections usually first manifested in middle age, (39-69, mean 52 years). Most patients (80%) were men. All patients presented with NTM (80% disseminated), particularly rapid growing mycobacteria (70%), with or without other opportunistic infections. Infections with multiple mycobacterium species were present in half of patients. Co-infections included Mycobacterium tuberculosis, Salmonella, cryptococcosis, Penicillium marneffei and varicella-zoster virus. The 3 most commonly involved organs were lymph node, lung and bone, respectively. NTM bacteremia was found in 3 patients. Reactive dermatoses, mainly Sweet’s syndrome, were present in majority of patients (80%) while co-existing autoimmune diseases were notably absent. Although all but one patient survived, recurrent infections were common once active treatments were discontinued.
Conclusion:
Anti-interferon-?-autoantibodies associated infections are characterized by disseminated NTM and other opportunistic infections in HIV-uninfected adult patients. Reactive skin disease, in particular Sweet’s syndrome, is the common characteristic presenting feature. Although most patients have non-fatal prognosis, long-term active therapy is generally required due to frequent recurrence after treatment withdrawal.
ESID-0055 B-Cell Immunophenotyping of Large Cohort of CVID Patients Revealed Subcluster of Patients with Distinct Clinical Features and Similar Cellular, Humoral and Genomic Abnormalities
V. Kanderova 1, J. Stuchly1, M. Vlkova2, I. Hermanova1, M. Trkova3, O. Hrusak1, A. Sediva4, J. Litzman2, E. Fronkova1, T. Kalina1
1CLIP (Childhood Leukaemia Investigation Prague), Charles University 2nd Faculty od Medicine and Motol University Hospital, Prague, Czech Republic
2Dpt Clinical Immunology and Allergology, St Anne’s University Hospital and Faculty of Medicine Masaryk University, Brno, Czech Republic
3Cytogenetics, GENNET - Genetics and Reproduction Medicine center, Prague, Czech Republic
4Dpt Immunology, Charles University 2nd Faculty od Medicine and Motol University Hospital, Prague, Czech Republic
Common Variable Immunodeficiency (CVID) is a heterogeneous disorder characterized by low levels of IgG, IgA, and/or IgM, impaired antibody response after antigen challenge and the resulting bacterial infections. The aim of the study was to examined the detailed phenotype of peripheral blood cellular compartments and define the subgroups of CVID patients which may share the same pathological mechanisms.
Previously reported unsupervised method of probability binning (Kalina 2009) was used to create hierarchical tree according to B-cell similarities. The cohort of 98 patients and 47 healthy donors split into 11 clusters and additional one with missing B-cells. Only one B-cell cluster (cluster 5, n=14), consisting only of patient samples, presented with characteristically aberrant phenotype of B-cells (more than 30% CD21low cells from naive B-cells) as well as T-cells (reduced population of naive and enlarged population of exhaused CD4+ T-cells). The cytokines in plasma showed higher levels of IFNgamma, IL-2 and IL-10 (p<0.05) in cluster 5 patients. Moreover cluster 5 patients suffer from splenomegaly and autoimmunity (thrombocytopenia). All these features together with higher expression of CD70 and CD69 suggested their overall immune system activation. In addition, whole-exome sequencing (verified by PCR) appeared single nucleotide polymorphisms in CD21 and IFI44 genes whose functional consequences are currently being tested. Preliminary, in?vitro apoptosis was pronounced in cluster 5 B-cells when stimulated with anti-TLR9 (ODN2006) and IFNalpha.
The standardised flow cytometry profiling of large group of CVID patients defined a new subgroup that only partially overlaps with Freiburg Ia class.
NT/11414-5, NT/13271, P302/?12/G101, UNCE204012, GAUK64391
ESID-0615 Cytokines as Autoantibody Targets in APECED
K. Kisand 1, J. Kärner1, A. Ranki2, K. Krohn2, P. Peterson1
1Molecular Pathology, Institute of Biomedicine and Translational Medicine University of Tartu, Tartu, Estonia
2Department of Dermatology Allergology and Venereology, Institute of Clinical Medicine University of Helsinki, Helsinki, Finland
Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) is a rare genetic autoimmune disease, caused by Autoimmune Regulator (AIRE) gene mutations. The first disorders usually appear in infancy, typically with chronic mucocutaneous candidiasis (CMC), followed by hypoparathyroidism and adrenocortical failure . The exact timing and features vary greatly, but other endocrinopathies and ectodermal manifestations usually develop later at lower frequencies. The syndrome is characterized by the presence of high-titer organ-specific autoantibodies that are often targeted against intracellular enzymes. These autoantibodies are most likely epiphenomena to tissue destruction. However, there are multiple autoantibody reactivities in APECED that have not shown correlation with any of the manifestations in these patients.
The aim of this study was to detect autoantibodies to a wide variety of cytokines in APECED patients using luciferase based immunoprecipitation assay and cytokine neutralization, and to correlate the autoantibody pattern to the phenotype.
We found that the prevalence of autoantibodies to type I interferons (IFN) and T helper (Th)17-related cytokines was the highest. Other cytokines were recognized only occasionally. In addition to the well-known correlation of anti-IL-22 and anti-IL-17F with CMC, we found that the neutralizing capacity of antibodies against type I IFNs correlated with type I diabetes in APECED patients. However, patients with autoantibodies to IL-6 showed no increased susceptibility for staphylococcal infections as shown for non-APECED patients with similar autoantibodies.
Although it is not understood why the cytokine autoantibodies develop in APECED patients, our data suggest that they can modulate the disease in many ways and can be even protective in certain circumstances.
ESID-0674 Autoimmune Neutropenia of Infancy- The 25-Years Experience of Children’s Memorial Health Institute in Warsaw
M. Klaudel-Dreszler 1, B. Pietrucha2, E. Heropolitanska-Pliszka2, B. Wolska-Kusnierz2
1Gastroenterology Hepatology and Feeding Disorders, Children's Memorial Health Institute, Warsaw, Poland
2Immunology, Children's Memorial Health Institute, Warsaw, Poland
Autoimmune neutropenia of infancy (AIN) is characterized by severe neutropenia, usually mild clinical course and spontaneous remission.
Aim: Clinical course, immunological aberrations and efficacy of treatment has been analysed.
Patients: Since 1987 till 2012, 100 patients (54 girls, 46 boys) have been diagnosed and treated due to AIN. The diagnostics was based on abnormal ANC within 6 months, typical smears of bone marrow, antigranulocytic antibodies(AGABs) assays.
Results:
We established the diagnosis of AIN in 35 pts due to detection of AGABs and in 65 pts due to typical clinical course and bone marrow smears. The most sensitive was GIFT- positive in 60% of pts, MAIGA was positive in 40%, GAT in 31%. Mean age at disease onset was 7 months in first group and 11 months in the second one. Mean ANC at diagnosis was 120/µl(0 – 500). All children developed upper respiratory tract infections, 50% had sepsis, pneumonia or perianal abscess, 48% chronic gingivitis, 20% staphylococcal lymphadenitis. Monocytosis was found in 85% of pts, leukopenia in 20%, policlonal hypergammaglobulinemia in 45%. AIN has resolved in 66 pts so far: in 40% of pts before 3 yo and in 11 pts over 6 yo. Antibiotic prophylaxis was implemented in infants and over 12 mo in case of severe infections. Only one child required G-CSF in low dose.
Conclusions:
AIN is the most frequent cause of chronic neutropenia under 5 years of age.
AIN may last longer, up to 9 years, and have severe clinical course, requiring G-CSF treatment.
ESID-0394 Caspase-10 Deficiency in a Child with Pulmonary Hypertension
A.Y. Kreins 1, D.T. Groot1, S.O. Burns2, A. Thrasher3, N. Martinez-Alier1
1Paediatric Infectious Diseases and Immunology, Evelina London Children's Hospital, London, United Kingdom
2Department of Immunology, University College London Institute of Immunity and Transplantation, London, United Kingdom
3Department of Infection & Immunity, University College London Institute of Child Health, London, United Kingdom
We report a patient who suffered from recurrent viral respiratory tract infections and worsening, chronic respiratory symptoms. Imaging and lung biopsy showed fibrosis and inflammation supporting a diagnosis of bronchiolitis obliterans with organizing pneumonia (BOOP). Despite high-dose corticotherapy, the patient progressively developed severe pulmonary hypertension. She also displayed persistent cytopaenias, lymphadenopathy and splenomegaly, which are not typically seen in BOOP. A diagnosis of autoimmune lymphoproliferative syndrome (ALPS) was eventually considered. ALPS is a rare genetic disorder of lymphocyte apoptosis. It is characterized by chronic lymphadenopathy, splenomegaly and multilineage cytopaenias. These clinical features are found in many other conditions, making the diagnosis of ALPS challenging. An assay testing for defective lymphocyte apoptosis in this patient was ambiguous, but further investigations revealed increased double negative T cells (DNT). Genetic testing allowed the identification of a germline mutation in caspase-10, confirming the diagnosis of ALPS-CASP10. Rituximab and mycophenolate mofetil were initiated and the patient's symptomatology quickly improved. Systemic autoimmunity, in particular pulmonary lesions, is rare in ALPS. Pulmonary hypertension secondary to autoimmune lung disease has never been reported in these patients. This case report highlights the need to test for ALPS in every child with unexplained lymphadenopathy, splenomegaly and cytopaenias, as more targeted treatments should be started as early as possible to control the patient's disease.
ESID-0305 Recurrent Fever in a Turkish Girl Caused by NLRP12 Gene Mutation
M.?A.A. Kusters 1, S.S. Henriet2, M.E. van Gijn3, A. Simon4, M. van der Flier2
1Pediatrics, Radboud University Medical Center, Nijmegen, Netherlands
2Pediatrics, Radboud University Medical Center and Radboud Institute of Molecular Life Sciences, Nijmegen, Netherlands
3Medical Genetics, University Medical Center Utrecht, Utrecht, Netherlands
4Internal Medicine, Radboud University Medical Center and Radboud Institute of Molecular Life Sciences, Nijmegen, Netherlands
Familial Mediterranean Fever (FMF) is the most frequent and best known of all hereditary auto-inflammatory disorders. Diagnosis of hereditary auto-inflammatory disorders is based on a combination of clinical criteria, age of onset and clinical response to medication such as colchicines, and genetic analysis. Clinical diagnosis of FMF can usually be confirmed by a mutation in the MEFV gene; in = 30% of PFS patients no MEFV mutation is found. A growing number of new hereditary autoinflammatory syndromes has been described in recent years.
We present a 10-years old Turkish girl who had previously been diagnosed as FMF. From the age of four, recurrent episodes of fever lasting for a few days combined with abdominal pain, fatigue and headaches occurred. Appendectomy was performed at four years of age because of suspected appendicitis, but microscopy did not confirm this. Genetic analysis of the MEFV gene showed no mutations. Medical treatment with colchicine did not result in clinical improvement. Next generation sequencing was performed to analyse 19 known autoinflammatory disease causing genes, revealing a NLRP12 gene mutation: heterozygotic c.1054C>T p. (Arg352Cys). This pathogenic missense mutation is causative of familial cold autoinflammatory syndrome type 2.
Our case demonstrates how the clinical phenotype of familial cold autoinflammatory syndrome type 2 can present with episodes of fever resembling FMF. This suggests that more patients diagnosed with FMF but without MEFV gene mutations may in fact suffer from familial cold autoinflammatory syndrome type 2 due to NLRP12 mutations.
ESID-0432 Role of Aire in the Induction of Human Tolerogenic Dendritic Cells
K.L. Crossland 1, S. Pearce2, M. Abinun3, T. Cheetham4, C.M.U. Hilkens5, D. Lilic1
1Primary Immunodeficiency Group Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
2Institute of Genetic Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
3Dept of Paediatric Immunology, Great North Childrens Hospital, Newcastle upon Tyne, United Kingdom
4Dept of Paediatric Endocrinology, Great North Childrens Hospital, Newcastle upon Tyne, United Kingdom
5Musculoskeletal Research Group Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
Loss of tolerance leads to uncontrolled immunity that can result in autoimmune mediated damage and disease. Mechanisms underlying tolerance induction and loss of tolerance are still poorly understood. An important model for studying human autoimmunity is the Autoimmune Polyendocrinopathy Syndrome type 1 (APS1) aka Autoimmune Polyendocrinopathy Candidiasis Ectodermal Dystrophy (APECED), where loss-of-function mutations of the Autoimmune Regulator (AIRE) gene result in debilitating, potentially lethal organ specific autoimmunity. The AIRE protein plays a crucial role in the induction of central tolerance by promoting ectopic expression of tissue-specific antigens in medullary thymic epithelial cells (mTECs), thus enabling removal of self-reactive T lymphocytes during thymocyte development. AIRE expression has recently been detected in peripheral cells, including myeloid dendritic cells (DC), suggesting that AIRE may also have a role in inducing peripheral tolerance. To assess this, in a model of human interleukin-4 / GM-CSF induced monocyte-derived DCs, we investigated the expression of AIRE in inflammatory DC (infDC) and tolerogenic DC (tolDC) in healthy individuals and APS1 patients. We demonstrate that AIRE is expressed in moDCs and is up-regulated during the differentiation from monocytes to moDC. We show for the first time that: 1) tolDCs from healthy individuals express AIRE; 2) that AIRE expression is greater in tolDC than inflDC; 3) and that tolDC can also be generated from APECED monocytes. Our findings contribute to the better understanding of the role AIRE may have in the induction and function of tolerogenic dendritic cells and its relevance to the maintenance of peripheral tolerance.
ESID-0610 Role of Autoimmunity in Hepatitis C Virus Infection: A Case Report and a Brief Review of Literature
F.M.S. Lima 1, L.O. Mendonça1, C.M. Kokron1, J. Kalil1, P.T. Cordova1, M.T. Barros1
1Immunology and Allergy, Hospital das Clinicas da FMUSP, Sao Paulo, Brazil
Hepatitis C virus (HCV) is an important public health problem worldwide. Immunological complications are found in 40-74% of patients with HCV, as well as the prevalence of HCV infection is higher among individuals with these conditions, suggesting a pathogenetic virus influence. Understanding the association between HCV and autoimmune manifestations, may light to virus testing, beyond to indicate antiviral e immunomodulatory treatment. This case report is about a 56year old male patient with skin scaly erythematous lesions and stellate hypo pigmented spots, muscle atrophy of the extremities and peripheral neuropathy march with diffuse muscle weakness associated with a positive history of hepatitis C. Biopsy of skin lesions showed inflammatory features, some with fibrinoid necrosis. Electromyography demonstrated myopathy and neuropathy pattern. The patient was submitted to HCV infection treatment, but had several collateral effects that required the avoidance of the drugs. Immunosuppressive medications were also administrated, reaching partial control of symptoms with cyclosporine. As a systemic pathology, autoimmunity may occur even without hepatic manifestations. Studies indicate that peripheral nervous system disorders have been observed as complications of HCV infection. Nerve biopsy revealed vasculitic neuropathy. HCV RNA has been detected in peripheral nerve, muscle and brain tissue of patients. Analyzing skin vasculitic lesions from test patients, but not control subjects, the HCV virion was found in association with IgM and IgG. HCV alone was detected in some vessel walls, and in skin and ductal epithelium, and vascular endothelium in inflamed, but not normal skin.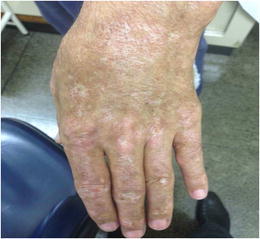
ESID-0216 Autoimmune Haemolytic Anemia (AIHA) in Common Variable Immunodeficiency (CVID): Immunological and Clinical Features
V. Lougaris 1, M. Baronio1, D. Moratto1, S. Masneri1, T. Lorenzini1, M. Andolini1, M. Vitali1, G. Tampella1, A. Plebani1
1Department of Clinical and Experimental Sciences, Pediatrics Clinic & Ins.for Molecular Medicine A.NocivelliUniversity of Brescia, Brescia, Italy
Autoimmune haemolytic anemia (AIHA) represents one of the known autoimmune complications in Common Variable Immunodeficiency (CVID). Nonetheless, treatment is not yet standardized and outcome may not be favorable. We decided to investigate the immunological and clinical features of 5 CVID patients with AIHA and compared the findings with 45 CVID patients without autoimmune cytopenias in order to better characterize this subgroup of CVID patients. All CVID patients included in this study are alive under regular replacement treatment. CVID patients with AIHA present lower peripheral B cell numbers (p=0,028), with altered B cell maturational pattern: expansion of CD21lo B cells and reduction of memory B cells, both switched and unswitched, when compared to CVID patients without autoimmune cytopenias. Immunoglobulin serum leveles at diagnosis were not significantly different among the two groups of CVID patients. Splenomegaly was present in all CVID patients with AIHA (p<0,001); two of these patients underwent splenectomy due to lack of response to medical treatment. Interestingly, we observed a high prevalence of granulomas in CVID patients with AIHA (40%) (p<0,001), while they were not found in any of the CVID patients without autoimmune cytopenias. Associated conditions within the CVID group with AIHA included celiac disease (20%), B cell lymphoma (20%) and invasive infections (60%).
Our findings suggest that CVID patients with AIHA represent a distinct subgroup within CVID with particular clinical and immunological features.
ESID-0061 Spectrum of Perforin Gene Mutations in Familial Hemophagocytic Lymphohistiocytosis 2 (FHL2) Patients from India
M. Madkaikar 1, S. Mhatre1, M. Desai2, K. Ghosh3
1Paediatric Immunology and Leukocyte Biology, National Institute of Immunohaematology ICMR, Mumbai, India
2Immunology and Hematology, Bai Jerbai Wadia Hospital for Children, Mumbai, India
3Director, National Institute of Immunohaematology ICMR, Mumbai, India
Inherited perforin deficiency is a rare autosomal recessive disorder that causes severe form of hemophagocytic lymphohistiocytosis (HLH) also called as FHL2. Perforin deficiency accounts for approximately 30-35% of FHL cases. The aim of this study was to analyze the nature of mutations in Indian patients with FHL2 and to study the genotype-phenotype correlation and utilize this knowledge for prenatal diagnosis.
13 patients fulfilling HLH criteria of Histiocyte Society with absent or low perforin expression on NK cells by flow cytometry were included in the study. The entire coding region and intronic splice sites of the PRF1 gene were sequenced from the genomic DNA of these patients.
10 patients had early presentation with severe classical clinical manifestations and 3 had delayed onset with unusual presenting features viz Hodgkin's lymphoma, tuberculosis and acute lymphoblastic leukemia. Sequence analysis revealed 11 (8 novel and 3 known) different mutations (8 missense mutations, 1 nonsense mutation 1 insertion mutation and 1 indel mutation). Missense mutation Trp129Ser in heterozygous state was present in all the 3 patients with delayed onset of disease. 12 patients with expired due to progressive disease except one patient who is currently in remission with HLH directed therapy. This data was useful in providing prenatal diagnosis by chorionic villous sampling (CVS) in 3 affected families.
A wide heterogeneity was observed in the nature of mutations in Indian FHL2 patients. Molecular characterization of PRF1 gene not only helped in confirmation of diagnosis but also in genetic counseling and pre-natal diagnosis in affected families.
ESID-0706 Ectopic Aire Expression in Thymic Cortex Reveals Inherent Properties of Aire as a Medullary Stromal Factor
M. Matsumoto 1, H. Nishijima1
1Division of Molecular Immunology, Institute for Enzyme Research, Tokushima, Japan
Thymocyte development requires a serial input from surrounding stromal element including thymic epithelial cells (TECs). Thymic cortex is a site to ensure MHC restriction for developing thymocytes, whereas thymic medulla plays an important role for eliminating autoreactive T cells and for producing Tregs in order to establish self-tolerance. Aire within medullary TECs (mTECs) functions as an essential regulator in the latter platoform. We asked whether Aire is absolutely required to exist within the medulla, or Aire could also function within the cortex for establishing self-tolerance. In order to test this question, we established a semi-knockin strain (ß5t/Aire-KI) on NOD background in which Aire is expressed under control of the promoter of ß5t, a thymoproteasome expressed in the cortex, through BAC technology. Aire protein was successfully expressed within cortical TECs (cTECs) as a typical nuclear dot protein in ß5t/Aire-KI, indicating that cTECs are permissive for Aire protein expression. Although Aire has been implicated for the transcriptional activator of may tissue-restricted antigen (TRA) genes, cTECs ectopically expressing Aire did not confer the transcriptional expression of either Aire-dependent or Aire-indipendent TRA genes. We crossed ß5t/Aire-KI onto Aire-deficient NOD mice, thereby generating a strain in which Aire expression is confined to cTECs. NOD mice expressing Aire only within cTECs succumbed to autoimmunity as seen in Aire-deficient NOD mice. Our results suggest that Aire must be present within mTECs in order to function as a regulator of autoimmunity.
ESID-0336 Complement Activation in a Group of Patients with IGG 4 Related Disease at Hospital Das Clíncas – University of São Paulo
L. mendonca 1, F. Mascarenhas1, C. Kokron1, J. Kalil1, M. Barros1
1clinical immunology and allergy, HCFMUSP, Sao Paulo, Brazil
The IgG4 related disease, only recently known, is characterized by dense infiltration tissue, rich in lymphocytes and IgG4 positive plasma cells , storiform fibrosis, and often IgG4 increase in serum measures. The first description was associated with autoimmune pancreatitis and is currently considered a systemic disease. The IgG4 it’s not considered to initiate the complement activation either can’t activate the FC region. Our objective is to review clinical and laboratory records of patients followed with the diagnosis of IgG4-related disease. As in the cases described above , the vast majority of patients are male, and two cases with renal disease ( glomerulonephritis interstitial ) are men. Between all the patients, only two cases described above ,1 and 3 had systemic involvement as the first manifestation of the disease. The others presented with involvement of only one body of which only one, case 2 had progression to systemic disease . The tumor Kuttner and glomerulonephritis are the first manifestations observed in case 4 and 2, respectively. About the laboratorial measures, only the first case , the parotid with localized disease , had normal serum complement , the other bands were undetectable C4 fraction. Patients with more severe systemic disease , case 1 and case 4, had consumption of c3 and c4 . Regarding inflammatory markers , as seen in the graph, the cases 1 and 2 are responsible for the biggest titles of ESR, probably related to severity and extent of disease.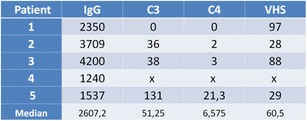
ESID-0698 Type 1 Diabetes Mellitus with Negative Specific Autoantibodies in Common Variable Immunodeficiency
T. Milota 1, J. Kayserova1, P. Kralickova2, B. Obermannova3, S. Pruhova3, A. Klocperk4, M. Podrazil4, A. Sediva4
1Department of Immunology, Faculty Hospital Motol, Prague 5, Czech Republic
2Department of Clinical Immunology and Allergology, University Hospital Hradec Kralove, Hradec Kralove, Czech Republic
3Department of Pediatrics, University Hospital Motol, Praha, Czech Republic
4Department of Immunology, University Hospital Motol, Praha, Czech Republic
Common variable immunodeficiency (CVID) is a heterogenous group of disorders characterised by an impaired immunoglobulin production and dysregulation of the immune system. Granulomatous diseases, malignity and especially autoimmune disorders with 20-25% prevalence belong to the most frequent complications beside recurrent infections. On the contrary association CVID and Type 1 diabetes mellitus (T1DM) is very rare, described only in few case reports.
T1DM is an organ specific autoimmune disease related to destruction of beta cells in pancreatic islets, followed by insulin deficit leading to hyperglycaemia, hallmark of T1DM. T1DM is usually coupled with the production of specific autoantibodies. In spite of this the destruction is mainly caused by T- lymphocyte populations. The presence of autoantibodies is not essential.
In this review we present 2 case reports with severe humoral immunodeficiency as part of CVID, both of them developed T1DM during the course of the disease. The autoantibodies associated with T1DM have never been detected, the diagnosis of T1DM always preceded CVID by several years.
On the example of our patients we show unusual association CVID and T1DM as complicated autoimmune disease. T1DM related autoantibodies are not necessary for pathogenesis of this disease, which can develop even in the terrain of heavily impaired antibody production. The absence of autoantibodies can complicate early assessment of the corect diagnosis. Early and correct diagnosis followed by the consistent and efficient therapy can markedly improve prognosis and course of CVID as well as the compensation of T1DM.
ESID-0233 Autoimmune Lymphoproliferative Syndrome Due to CASP10 Mutation Presenting with Severe Lymphopenia: A Case Report
A.P. Grammatikos1, C. Cale2, S.O. Burns3, M.?A.A. Ibrahim 1
1Department of Immunological Medicine Division of Asthma Allergy & Lung Biology School of Medicine, King’s College Hospital NHS Foundation Trust King’s Health Partners King’s College London, London, United Kingdom
2Department of Clinical Immunology, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
3Institute of Immunity and Transplantation and Department of Immunology, Royal Free London NHS Foundation Trust University College London, London, United Kingdom
Autoimmune lymphoproliferative syndrome (ALPS) is a rare inherited disorder associated with non-malignant lymphoproliferation and autoimmunity due to defects in the apoptosis pathway. Mutations in FAS are most commonly seen in these patients, while FASL and CASP10 mutations account for less than 1% of all cases.
We discuss a case in which the initial presentation was an episode of cutaneous leishmaniasis. This patient was found to be pan-T cell lymphopaenic on routine blood tests and did not have a history of recurrent or persistent infections. Her only medical history was of autoimmune hypothyroidism.
She was found to have a T cell count of 0.3 x109 (0.8-2.5 x109). B and NK cell counts were normal. Immunological testing revealed a very low (5%) naïve CD4+ T cell population. T cells exhibited normal proliferative responses and B cells produced sufficient amounts of antibodies. Antibody responses to vaccinations were all normal. No anti-lymphocyte antibodies were detected in her. Immunophenotyping revealed an increased percent of circulating alpha-beta-TCR+ double negative cells (13.9%), which is consistent with ALPS. Subsequent sequencing revealed a mutation in gene CASP10 in exon 9 (c.1216A>T or p.ILE406LEU), confirming ALPS.
CASP10 mutations have only been reported in a few patients of ALPS and this case provides some insight into this rare disorder. Although being classically described as a lymphoproliferative disorder, lymphopenia can be a presenting feature of ALPS and it should be included in the differential diagnosis in these patients.
ESID-0802 Histopathological Characterization of HLA Class I Deficiency Granulomatosis
D. Moraes Vasconcelos 1, N. Mortari2, O.H.M. Leite2, I. Nisida2, A.?C. Nicodemo2, C. Pagliari3, M. Sleiman4, J. Zimmer4, M.I.S. Duarte3
1Dermatology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
2Infectious and Parasitic Diseases, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
3Pathology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
4Laboratoire d'Immunogénétique-Allergologie, CRP-Santé Luxembourg, Luxembourg, Luxembourg
HLA class I deficiency is a rare disease (less than 30 reported cases in the world) with remarkable clinical and biological heterogeneity. This syndrome is caused by defects in TAP-1, TAP-2 and Tapasin (MIM 604571), preventing normal cell surface expression of HLA class I molecules, and thus antigen presentation to cytotoxic CD8 T cells and NK cells are defective, leading to dysregulation of the immune response and granulomatous vasculitis. Female, 38YO, born to non-consanguineous parents, presenting lower limb lesions for 10 years. Clinical features began with relapsing acute otitis media, requiring surgical debridement of mastoiditis in 1998. After the procedure painful erythemato-papular lesions appeared, progressing to ulcerated pustular lesions affecting the face, oral mucosa (hard palate fistulizing to sinuses) and right leg. The ulcers progress in the legs, painful and progressive, relapsing after antimicrobial treatment.
Histopathology: 2002: Chronic inflammation with loose granulomas, saves subcutaneous tissue. Grocott, AFB, PAS negative; 2004: Chronic granulomatous dermatitis, without necrosis; 2008: Granulomatous dermatitis, epithelioid palisade surrounding necrotic center; non-leukocytoclastic vasculitis with intimal proliferation; 2011: Granuloma with caseous necrosis; 2012: Immunohistochemistry shows lots of CD1a+ APCs in the epidermis, macrophages in the dermis and sparse NK cells throughout the entire skin. T cells (CD4 and CD8) well represented, with several B cells interspersed. Among T cells, lots of IL-17+ T cells, some FoxP3+, Caspase 3 and granzyme+ and IFN-gamma. Several APC cytokines, such as IL-1, IL-6 and IL-12, and some TNF+ cells. Regulatory cytokines well expressed with IL-4, IL-10 and TGF-beta throughout the lesions.
ESID-0714 Odontological Treatment in Patients with Hereditary Angioedema
R.C. Bonatto Vilarim1, S. Oliveira Martins1, M.P. Siqueira Melo Peres1, L. Oliveira Mendonça2, T.A. Bezerra 3, F. Loffredo D'Ottaviano2, M. Domingues-Ferreira3, A.A. Motta2, D. Moraes Vasconcelos3
1Odontology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
2Immunology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
3Dermatology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
Introduction: Odontological treatment in patients with hereditary angioedema can cause acute symptoms such as swelling in the face or other parts of the body and even death by asphyxia.
Materials and Methods: Retrospective study that evaluated the odontological procedures and symptoms caused by dental treatment in 15 patients with hereditary angioedema who underwent treatment in the Hospital das Clínicas Odontological Division of the University of São Paulo.
Results: Fifteen patients (9 women and 6 men) were submitted to dental treatment. The procedures were: 11 supragingival scaling and 4 subgingival scaling, 19 dental extractions, 3 endodontic treatments and 14 dental restorations. All supragingival scaling, one subgingival scaling, the endodontic treatments and restorations were performed without prophylaxis. All extractions and three of the subgingival scaling were performed under previous prophylaxis: danazol, tranexamic acid, fresh frozen plasma and C1 - INH inhibitor. No patient developed edema of the upper airways. Two patients underwent to complex dental extractions, mucoperiosteal flap and osteotomy for removal of three teeth. The patients developed facial edema, one of them presented postoperatory nausea and swelling in the arm. Patients who underwent simple extractions showed no swelling or other symptoms. The difference of edema in patients undergoing simple and complex surgery was significant P: 0.0130.
Conclusion: The development of edema was related to complex surgical dental procedures. Prophylaxis was effective in preventing the symptoms of angioedema in simple surgical procedures. Non-surgical dental procedures did not require prophylaxis. A study to assess which dental procedures require prophylaxis is necessary.
ESID-0555 Chronic Mucocutaneous Candidiasis and Autoimmunity Without Molecular Diagnosis
I. Moreira 1, A. Ginaca1, P. Carabajal1, S. Caldirola1, M.I. Gaillard1, D. Di Giovanni1, A. GómezRaccio1, L. Bezrodnik1
1Immunology Group, Hospital de Niños R Gutierrez, Buenos Aires, Argentina
Clinical Case: 16 yo girl, background of chronic thrush and effusive media otitis since 3 months of age. At 2 yo she started with recurrent nail, scalp and skin candida infection, partial improvement with oral antifungal treatments and extended sequelae scars. At 6 yo she started with antifungal prophylaxis, continuous free of fungal infections until 13 yo when she was admitted to hospital because of acute hepatitis, esophageal candidiasis and staphylococcus bacteremia. After improvement with intravenous antifungal and antibacterial treatment she was derived to our center. Neither other infections, ectodermal dystrophy nor endocrinopathy. No family history of autoimmunity nor immunodeficiency. Negative HIV tests. She presented hyper IgG (2500 mg/dl) with normal IgA, M and E. Adequate antibody response to protein antigens but impaired specific polysaccharide response. Bcell compartment with low memory and elevated transitional cells. No severe CD4 Tcells lymphopenia with CD4/CD8 inversion. Mild elevation of double negative Tcells (3.3%) and diminished Treg cells (0.8%). Normal lymphocyte proliferation, DHR test and soluble FAS-ligand. Negative intradermal reaction to candidin. Normal expression of IL12 and IFNg receptors. Since 13 yo she developed high ANA titers with nuclear dots and centromere pattern and progressively positivity of anti CENP B, Sp100, smooth muscle, LC1 and parietal cells Ab. No mutation in AIRE, STAT1 and CARD9 genes. Actually she is under antibiotic and antifungal prophylaxis, meprednisone and azathioprine. She is without new infections but persists with liver involvement.
Conclusion: Progressive autoimmune laboratory features together with chronic mucocutaneous candidiasis let us suppose a disease of immune dysregulation.
ESID-0376 Modular Transcriptional Repertoire Analysis in Down Syndrome Thymus
C. Moreira-Filho 1, S.Y. Bando1, F.B. Bertonha1, F.N. Silva1, L.D.A.F. Costa1, M. Carneiro-Sampaio1
1Pediatrics, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
In Down syndrome (DS) genomic dysregulation caused by trisomy 21 causes thymic structural and functional abnormalities. DS patients present a small and abnormal thymus, characterized by lymphocyte depletion, cortical atrophy, and loss of corticomedullary demarcation.
Here we characterized trisomy 21- driven transcriptional alterations in human thymus through gene coexpression network (GCN) analisys for differentially expressed genes (DE networks) and for the complete set of valid transcripts (CO networks). We used corticomedullary thymic sections, obtained during heart surgery from 10 DS and 10 karyotipically normal individuals (CT), age- (2-18 months-old) and gender-matched. A network-based approach for GCN analysis allowed the study of interactions between all the system's constituents based on community detection. Changes in connectivity observed for hierarchically important hubs in DS and CT gene networks corresponded to sub-network changes, i.?e. module (communities) changes. Distinct communities of highly interconnected gene sets were topologically identified for DS and CT networks. Trisomy 21 gene dysregulation in thymus may therefore be viewed as the breakdown and altered reorganization of functional modules. The high modularity of DS-DE network contrasts with its reduced connectivity, thus indicating a certain degree of disorganization of modular interactions caused by gene dosage imbalance. As a whole, the results indicate that GCNs' functional and topological modules correspond, and that trisomy 21 may be interpreted as the breakdown and altered reorganization of functional modules.
In conclusion, alterations observed in DS networks (connectivity, modularity, and communities' structure) reflect chromosome 21 dysregulation in thymus which leads to a deficiency in immune response.
ESID-0400 Report of 3 Cases with Mendelian Susceptibility to Mycobacterial Infections and Hemophagocytic Lymphohistiocytosis
R. Muriel Vizcaino 1, N. Ramirez2, L. Santos2, M.?A. Yamazaki3
1Research Unit in primary immunodeficiencies, NATIONAL INSTITUTE OF PEDIATRICS, Mexico City, Mexico
2Cinvestav, Instituto Politécnico Nacional, Mexico City, Mexico
3Immunology, NATIONAL INSTITUTE OF PEDIATRICS, Mexico City, Mexico
Background: Secondary hemophagocytic lymphohistiocytosis (HLH) may be triggered by infections with or without the context of primary immunodeficiency. Viruses are the most common triggers of HLH, however many other microorganisms have been reported. There are reports (55 cases through 2012) of HLH as a rare but deadly manifestation of mycobacterial infection. Whereas Mendelian Susceptibility to Mycobacterial Infections (MSMD) have frequently disseminated or severe infection by mycobacteria, coupled with the defect IL-12/INF-? axis (involved in cytotoxicity), may lead to an increased incidence of HLH in these patients, compared with general population. However, to our knowledge there are no clinical reports of HLH associated with MSMD.
Clinical cases: We present three patients with extrapulmonary mycobacterial infection and a defect in the IL-12/INF-? axis, two with an IL12RB1 deficiency, and one with a probable STAT-1 defect, who developed HLH during their evolution.
Discussion: Secondary HLH is associated mainly with cancer, infections, immune disorders or uncontrolled autoimmune diseases. There seems to be increased susceptibility to develop HLH in patients with defects in IL-12/INF-? axis. To date three of the ten patients who have been diagnosed in our hospital with this defect, have developed HLH. Although there are reports of HLH secondary to mycobacterial infection, is it a rare trigger. Whence the association of HLH and MSMD, could be secondary to convergent mechanisms as the immune defect itself (defects in cytotoxicity and immune dysregulation) and disseminated or severe mycobacterial infection.
ESID-0329 A Case of Late Onset FHL with Missense Mutation in UNC13D Gene
N. NAKANO 1, F. OCHI1, K.O.Z.O. NAGAI1, T. YASUMI1, K. MORITANI1, M. ISHIMAE1, M. EGUCHI1, H. TAUCHI1, E. ISHII1
1Pediatrics, Ehime University Graduate School of Medicine, Matsuyama, Japan
Familial hemophagocytic lymphohistiocytosis(FHL) has typically been the suspected diagnosis when hemophagocytic lymphohistiocytosis (HLH) develops in early infancy,therefore late-onset case is very rare.Herein we present the first reported instance of late-onset FHL3 in Japan.
The patient is a 7-year-old boy.He had developed HLH in response to bronchial pneumonia . EB virus, cytomegalovirus , and mycoplasma were negative. Symptoms improved promptly whereasby high-dose steroid pulse therapy , but he was repeated relapse during steroidtapering. He had no mutations of the XLP1, XLP2, Fas gene . Wheras he was normal in NK cell activity at the first medical examination, we was subjected to discrimination of FHL. Analysis of the molecules involved in granule transport of CTL / NK cells show that expression of Munc13-4 protein of patient were reduced , cytotoxic activity and degranulation of CTL of patient had both decreased. Further analysis in UNC13D gene revealed compound heterozygous nonsense/missense mutations,he was diagnosis as FHL type3. He received unrelated bone marrow transplantation but he died of severe chonic extensive GVHD one year after transplantation.
In FHL type2,there is a tendency that the onset may be delayed at the patient of missense mutation more than of nonsense mutation that was associated with reduced cytotoxic activity.
Similar study has not been made because in Japan missense mutation cases in FHL3 have not been reported. Functional analysis of protein is needed in the future , but we thought the degree of residual activity of protein function is closely related to age of onset.
ESID-0656 A Rare Presentation of CVID with an Orbital Inflammatory Mass
C. James1, O. Ndum 1, K. Ruda- Wessell2, E. Toller-Artis2, R. Hostoffer3, H. Tcheurekdjian3
1Internal Medicine, University Hospitals-Case Medical Center, Cleveland, USA
2Allergy/Immunology, University Hospitals Richmond Regional Medical Center, Cleveland, USA
3Allergy/Immunology, Case Western Reserve University, Cleveland, USA
Introduction
Ophthalmic conditions, such as granulomatous uveitis, retinal vasculitis, and multifocal choroiditis, in common variable immunodeficiency (CVID) are rare; however, ocular manifestations as the presenting symptom of CVID have not been documented. We present a patient with no history of immunodeficiency, who was found to have an ophthalmic inflammatory mass, leading to a diagnosis of CVID, successfully treated via intravenous immunoglobulin (IVIg) therapy.
Methods
A 41 year-old female presented with right maxillary sinus pressure and sharp pain behind her right eye. MRI showed a retrobulbar mass in the right orbit, extending to the apex. Surgical biopsy revealed lymphocytic inflammation and fibrosis.
Results
Initial evaluation showed anti-nuclear antibody titer of 1:40 but was otherwise unremarkable. Treatment with azathioprine, prednisone, and mycophenolate mofetil failed, and pain persisted with unchanged retrobulbar mass. Further work-up demonstrated decreased serum IgG of 562 mg/dL (700-1600 mg/dL), normal IgA (139 mg/dL), normal IgM (162 mg/dL), and poor pneumococcal immunization response with minimal protection against 4 of 14 serotypes. She was diagnosed with CVID and treated with IVIg 1000 mg/kg every 4 weeks. She reported 66% reduction in eye pain after two courses. IVIg dosing was then titrated to 1000 mg/kg every 3 weeks. Pain resolved with nearly 40% decrease in retrobulbar mass size.
Conclusion
Ophthalmic symptoms with CVID are often identified after confirming the diagnosis. This report highlights the first known case of an ocular inflammatory mass in a woman with no other signs of clinical immunodeficiency as the initial manifestation of CVID, and successful treatment with IVIg.
ESID-0462 Inflammatory Bowel Disease in Japanese Patients with XIAP Deficiency
N. Nishida 1, X. Yang1, A. Hoshino1, H. Kanegane1
1pediatrics, University of Toyama, Toyama, Japan
Objective: X-linked lymphoproliferative syndrome (XLP) is caused by SH2D1A and XIAP mutation. Patients with XLP mainly present with hemophagocytic lymphohistiocytosis (HLH); however, up to 20% of patients with XIAP deficiency are described to be associated with inflammatory bowel disease (IBD). Recently, it was reported that XIAP mutations were observed in male patients with pediatric-onset Crohn's disease. To elucidate relationship between XIAP deficiency and IBD, we demonstrated the clinical and immunological features of IBD in Japanese patients with XIAP deficiency.
Method: The clinical information was retrospectively collected from the XIAP-deficient patients associated with IBD. Immunological study was performed in some patients.
Result: Ten patients (approximately 40%) of Japanese patients with XIAP deficiency were complicated with IBD. The onsets of IBD were 2 to 16 years of age. Intriguingly, 3 patients present only IBD. Nearly all patients were treated with biological drugs and two of them received colectomy. One patient underwent cord blood transplantation and he achieved remission of IBD. In families with the same mutation, one sibling showed IBD, but the other did not show.
Conclusion: Regarding IBD, there was no genotype-phenotype correlation. There may have additional factor related with the occurrence of IBD. In Japan, the frequency of IBD in XIAP-deficient patients was higher than those in U.S. and Europe. This data suggested the racial differences of IBD occurrence in XIAP deficiency.
ESID-0088 Don’t Stop Search Endocarditis in Patients Under Immunosuppressive Treatment
F. Pulvirenti 1, C. Milito1, A. Pesce1, A. Ciervo2, I. Quinti2
1Molecular Medicine, Sapienza University of Rome, Rome, Italy
2Infectious Parasitic and Immune mediated diases, Italian National Health Istitute, Rome, Italy
A 70-year-old female with Systemic Lupus Erythematosus had fever and chills associated with arthritis, fatigue, lymphadenopathy, skin purple lesions and erythema nodosum. The patient was under therapy with methotrexate, adalimumab and methyl prednisone because of her rheumatic disease. Fever continued despite the increase of steroid therapy and antibiotics therapy.
Laboratory tests showed pancytopenia and high erythrocyte sedimentation rate while other inflammatory markers were normal. A skin biopsy showed dermal perivascular inflammatory infiltrate with lymphohistiocytic cells and eosinophilic and neutrophilic granulocytes.
An extensive infectious diseases laboratory workup, including sets of blood cultures and sierological tests, was performed with a negative outcome. A trans-thoracic echocardiogram was performed and no vegetation was remarkable; a trans-esophageal echocardiogram showed a small posterior leaflet mitral valve vegetation. The patient was tested for the main etiologic agents of culture-negative endocarditis by PCR. Because of positivity of both Coxiella burnetii and Borrelia burgdorferi DNA, Chronic Q fever and Lyme disease were diagnosed. An association of doxycycline and hydroxychloroquine was started. After 10 days the fever disappeared and the patient had a progressive improvement of arthritis and skin lesions.
Co-infection between C.burneti e B.burgorferi was rarely reported but pathogenically possible because these pathogens can be transmitted by the same species of arthropods.
This case report emphasizes the importance of excluding an infection as a cause of unusual arthritis. The use of PCR is mandatory in patients under immunosuppressive therapy since serological response could be missing.
ESID-0600 Autoimmune Cytopenias in Siblings with Familiar Occurrence of CVID (Case Report)
S. Raffac 1, J. Gabzdilova2, M. Mitnikova2, E. Barlova1, N. Stecova2
1immunology, University Hospital of L.Pasteur, Kosice, Slovakia
2hematology, University Hospital of L.Pasteur, Kosice, Slovakia
Background: Familiar occurrence of CVID is rare, representing 10-15% of cases. Common symptoms of CVID are recurrent respiratory infections, autoimmune disorders occur in 20% of cases. In addition, hematologic autoimmunity (ITP- 7%, AIHA 4%, autoimmune neutropenia 1%) often precedes the diagnosis CVID. Case report: We present a familiar occurrence of CVID siblings with autoimmune cytopenias. Woman, 26 years old, diagnosed with CVID hospitalized for attack severe AIHA. Given the negativity Coombs testing was considered by non-immune cause hemolysis, the present hepatosplenomegaly due to performed a liver biopsy. When HD CS prevent a sufficient response to treatment , therefore, applied HD IVIG with rapidly improving blood counts and subsequent regular application of IVIG 0.4 g/?Kg b.w. Her brother, 25 years old, with CVID and ITP hospitalized for relapsing ITP with signs of bleeding diathesis in severe thrombocytopenia (PLT 3x109 / L). Outpatient administration of IVIG (140 g) and CS (prednisone 100 mg) was without sufficient effect. Therefore, the patient was hospitalized and applied treatment with HD CS (methylprednisolone i.v. 1 g) with continual decreasing dosage. During treatment was applied the substitution of IVIG, that lead to improving blood counts parameters.Summary: AIHA in CVID may be associated with negativity Coombs test. These include possible hemolysis by NK cells independent of antibody, the presence of small numbers of red blood cell bound IgG, which are below the threshold of the Coombs' test, low-affinity autoantibodies, IgA and IgM autoantibodies. If clinical suspicion is high, early immunosuppresion treatment and specialized testing may be needed.
ESID-0507 RAS-Associated Autoimmune Leukoproliferative Disease (RALD): First Argentinian Patient
D. Cabanillas1, A. Fynn2, P. del Palacio3, L. Perez4, J.B. Oliveira5, L. Regairaz 1
1Immunology, Hospital de Niños Sor Maria Ludovica, La Plata, Argentina
2Hematology, Hospital de Niños Sor Maria Ludovica, La Plata, Argentina
3Immunology, Hospital Sor Maria ludovica, La Plata, Argentina
4Immunology, Hospital de Niños Garrahan, CABA, Argentina
5Immunology, Instituto de Medicina Integral Professor Fernando Figueira, Recife, Brazil
RALD are ALPS-like disorders caused by mutations in NRAS and KRAS genes.
We report the first Argentinian patient with RALD by NRAS mutation
The patient is a 2-years-old girl, without consanguinity in the family. When she was 3 months old she developed CMV infection (possitive IgM and RCP) that resolved with valanciclovir, BCGitis and muguet. She was referred to our unit with high suspiction of primary immunodeficiency. During the follow up she developed chronic, non-malignant lymphoproliferation (persistent lymphomonocytocis, hepatosplenomegaly), and autoimmunity (hemolytic anemia (Coombs: ++++) and thrombocytopenia, with good response to steroids. She was evaluated by hematologist with high suspiction of juvenile myelomonocytic leukemia (JMML). Immunological assesment showed hipergammaglobulinemia, normal lymphocyte subsets and normal “double-negative” T cells (<2%). Evaluation of the intrinsic pathway of apoptisis revealed defective IL-2 withdrawal-induced apoptosis. An heterozygous mutation (G12D) in NRAS confirmed the diagnosed of RALD.
RALD must be suspected in patients with autoimmunity, immune cytopenia and lymphadenopathy or hepatosplenomegaly who do not meet with the well-defined diagnostic criteria of ALPS or JMML.
ESID-0179 A New Case of Autoimmune Lymphoproliferative Syndrome (ALPS) Caused by a Homozygous FAS Ligand Gene (FASLG) Mutation
R. Ruiz-García 1, S. Mora-Díaz1, L. Martínez-Lostao2, G. Lozano-Sánchez3, E. Paz-Artal1, J. Ruiz-Contreras4, L.I. Gonzalez-Granado4, A. Anel2, D. Moreno3, L.M. Allende1
1Inmunología, Hospital Universitario 12 de Octubre, Madrid, Spain
2Bioquímica Biología Molecular y Celular, Universidad de Zaragoza, Zaragoza, Spain
3Pediatría, Hospital Materno-Infantil Carlos Haya, Málaga, Spain
4Inmunodeficiencias Pediatría, Hospital Universitario 12 de Octubre, Madrid, Spain
Introduction:
Clinically, Autoimmune Lymphoproliferative Syndrome (ALPS) is characterized by chronic nonmalignant lymphoproliferation, autoimmune cytopenia, susceptibility to malignancy, increased number of double-negative (DN) T-cells and defective apoptosis. Most cases of ALPS are associated with germline or somatic FAS mutations (ALPS-FAS or ALPS-sFAS).
Objectives:
We report a new homozygous FAS ligand gene (FASLG) mutation in an ALPS patient (ALPS-FASLG).
Patients and Results:
We describe two patients born to consanguineous parents who presented a severe form of ALPS. The patient 1 was admitted into hospital because generalized lymphadenopathy and hepatosplenomegaly, his sibling (patient 2) died at the age of 6 years with massive hepatosplenomegaly. DN T-cells, levels of plasma IL-10, sFAS ligand, sCD25, vitamin B12 and immunoglobulin G were determined as ALPS biomarkers. All ALPS biomarkers tested were markedly increased. However, sFAS ligand could not be detected in the patient 1’s plasma. The FASLG gene was sequenced, and its expression was analyzed by Western blotting. Activation-induced cell death (AICD) was also impaired in the patient 1. To confirm the defect in FasL functionality in the patient 1, FasL-negative 293T cells were transfected with either the WT or the mutated FASLG and were used as effectors in cytotoxicity assays. The results were compared with other ALPS-FASLG patient (patient 3, previously described by our group).
Conclusion:
Here we described the third case of ALPS caused by homozygous mutations in FASLG and autosomal recessive inheritance. AICD assays and plasmatic sFAS ligand levels could help to easily diagnose this rare form of ALPS-FASLG.
ESID-0585 The Balance Between Regulatory T Cells and TH17 Cells in Autoimmunity in Children with Primary Humoral Immunodeficiencies
M. Rutkowska 1, A. Szaflarska2, M. Lenart1, A. Pituch-Noworolska1, M. Siedlar1
1Departmen of Clinical Immunology, Polish-American Institute of Pediatrics Jagiellonian University Medical College, Krakow, Poland
2Departmen of Clinical Immunology, University Childrens' Hospital Wielicka 265, Krakow, Poland
Common variable immunodeficiency (CVID) and selective IgA deficiency (SIgAD) are the most common primary antibody deficiencies. Approximately 20% to 30% of patients with CVID or SIgAD develop autoimmune disorders. However, clinical features of the autoimmune phenotype in these patients might be atypical and cause the delay of final diagnosis. Recent studies highlight the role of Th17 cells in the development of autoimmune diseases. Moreover, it was shown that the differentiation factors of Th17 cells reveal a link with induction of Foxp3+ regulatory T cells (Treg).
The studied group included 26 patients with CVID and 28 with SIgAD. Children in whom immunodeficiency diseases were excluded formed control group (n=30). The Treg and Th17 cell levels were analyzed by flow cytometry.
The significant difference in the absolute number and percentage between Treg and Th17 cells was observed only in control group. In CVID and SIgAD patients the levels of both populations were comparable. The level of Treg was decreased in patient with CVID and SIgAD in comparison to control group, however only in SIgAD patients observed differences were statistically significant. The percentage of Treg was significantly decreased in CVID children with autoimmune disease in comparison to control group and to remaining CVID patients.
Obtained results suggest the possible role of Treg cells in development of autoimmune diseases in children with primary immunodeficiencies. Moreover, our study may have clinical implications, since the diagnosis of autoimmune disease in CVID and SIgAD patients remains difficult due to the atypical clinical and immunological features.
ESID-0789 Familial Mediteranian Fever and Primary Immune Deficiency
M. sadeghi shabestari 1, P. habibi1
1pediatrics, children hospital, Tabriz, Iran
Case presentation:
A 27-month boy admitted to children’s hospital with chief compliant of fever and cervical lymphadenopathy. In the past medical history, he had documental FMF by genetic analysis (E167D Homozygot) under treatment of colchicines and History of recurrent outpatient treatment for periods of fever and abdominal pain.
On admission, the patient had a temperature of 38°C axillary, bilateral cervical adenopathy, Erythematous pharynx, purulent PND, and gingivitis.
Lab values include a CBC which showed neutropenia (ANC = 900) and reverse ratio of (CD4 25%, CD8 43%) in flowcytometery, but Immune glubolins and iso – hemaglutinis test result were within normal limits. Bone marrow aspiration showed moderate arrest in Myelopoiesis. The throat culture was positive for Enterobacter and Multiple CBC values confirmed persistent Neutropenia during admission.
Conclusion:
The newly added category to primary immune deficiency is inflammatory disorders such as FMF.
The presented patient diagnosed as congenital neutropenia (variant of WASP), the correlation between immune deficiency and FMF confirmed, and the patient treated with GCSF and IVIG and discharged with well condition.
Key words:
FMF, Immunodeficiency
ESID-0803 Autoantibodies to IL-17A May Be Correlated with the Severity of Mucocutaneous Candidiasis in APECED Patients
A.K. Sarkadi 1, S. Taskó1, G. Csorba1, B. Tóth1, M. Erdos1, L. Maródi1
1Department of Infectious and Pediatric Immunology, University of Debrecen, Debrecen, Hungary
The relative roles of autoantibodies against IL-17-type cytokines in susceptibility to chronic mucocutaneous candidiasis (CMC) in patients with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) remain poorly defined. The purpose of this longitudinal study was to analyze the relationship between the occurrence of mucocutaneous candidiasis and levels of anti-IL-17A, anti-IL-17F and anti-IL-22 autoantibodies. We studied six APECED patients with various disease manifestations. Clinical data were collected during regular follow-up. Anti-endocrine organ antibody levels, clinical chemistry and immunology parameters were determined in routine laboratory assays on freshly isolated serum. Levels of autoantibodies against IL-17A, IL-17F, IL-22, IFN-a and IFN-?, and cytokine release by Candida-exposed blood cells were determined by ELISA. Mutations were analyzed by sequencing genomic DNA. Four patients carried the germline c.769C>T homozygous nonsense mutation, which results in R257X truncation of the AIRE protein, and two patients were compound heterozygous for the c.769C>T/c.1344delC mutation. We found high levels of antibodies against IL-17A in the samples of one patient presenting CMC since infancy and low or undetectable anti-IL-17A levels in the sera of five patients without candidiasis. By contrast, levels of autoantibodies against IL-17F and IL-22 were higher in all patients than in healthy controls. Levels of cytokine release by Candida-exposed PBMC were low or negligible in all patients. We suggest that anti-IL-17A antibodies may play an important role in the predisposition to candidiasis of APECED patients. Data also suggest that the impaired release of IL-17-type cytokines by blood cells may be an element of the immunopathology of CMC in APECED patients.
ESID-0436 Unexpected Hypomorphic JAK3 Deficiency in a Child Presenting with Skin Granuloma and Chronic Diarrhoea
A. Scarselli 1, S. Di Cesare1, G. Di Matteo1, A. De Matteis1, P. Ariganello1, M.L. Romiti1, H.B. Gaspar2, A. Aiuti3, K. Gilmour4, C. Cancrini1
1Department of Pediatrics (DPUO), Pediatric Hospital Bambino Gesu', Roma, Italy
2Molecular Immunology Unit, UCL Institute of Child Health, London, United Kingdom
3Pediatric Immunohematology and Bone Marrow Transplant Unit and San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET), Scientific Institute HS Raffaele, Milan, Italy
4Molecular Immunology Unit, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
A 2-year-old female, born from consanguineous parents, was referred for persistent lymphopenia during hospitalization due to chronic diarrhoea, pleuro-pneumonia and EBV infection. She presented persistent skin lesions from the age of 16 months. The skin biopsy revealed a non-infectious, non-necrotizing granulomatous dermatitis. The immunological investigations showed a marked T-cell reduction, increased B-cells and normal NK-cells. Reduced thymopoiesis and impaired lymphocytes response to antigens and mitogens were found. Functional humoral immunity was impaired although the immunoglobulins were normal for age and she presented high level of antibody titres to chickenpox, that she had at the age of 2 months. JAK3 expression evaluated by western blot and STAT5-phosphorylation on peripheral blood lymphocytes, on CD4+CD45RO+ subset as well as on EBV-transformed B-cell line were reduced. Gene sequencing revealed the homozygous missense mutation p.R403H in JAK3 gene. The same mutation was described in human malignancies and in a single SCID-patient where it was present in a compound heterozygous. During the follow-up, a bilateral focal tenosynovitis appeared on both ankles. Residual JAK3-activity could explain the milder clinical presentation (normal growth, no severe infections, normal immunoglobulin and circulating NKs) although environmental and epigenetic factors could not be excluded. This case confirms the high heterogeneity of JAK3-deficiency and suggests clinicians to consider JAK3-hypomorphic mutation in all case of T-lymphopenia. Moreover, since cutaneous alterations may be one of the earliest sign of many Primary Immunodeficiencies, the persistence of undefined skin lesions should raise the suspicion of an underlying immune defect.
ESID-0516 Long-Term Continuous Remission After Allogeneic Hematopoietic Stem Cell Transplantation in ALPS/?CVID Overlap Syndrome Due to Lipopolysaccharide Responsive Beige-Like Anchor Protein (LRBA) Deficiency
M.G. Seidel 1, T. Hirschmugl2, W. Schwinger1, L. Gamez-Diaz3, N. Serwas2, A. Deutschmann4, G. Gorkiewicz5, W. Zenz4, C. Windpassinger6, M. Speicher6, B. Grimbacher3, C. Urban1, K. Boztug2
1Pediatric Hematology Oncology, Medical University of Graz, Graz, Austria
2Research Center for Molecular Medicine, CEMM, Vienna, Austria
3Centre for Chronic Immunodeficiency CCI, University Medical Center, Freiburg, Germany
4General Pediatrics, Medical University of Graz, Graz, Austria
5Inst. of Pathology, Medical University of Graz, Graz, Austria
6Inst. of Human Genetics, Medical University of Graz, Graz, Austria
In addition to previously described 11 patients with LRBA deficiency, we report an extended phenotype and the clinical course of a severe immune dysregulation syndrome in two girls from a consanguineous family of Kurdish origin. Symptoms included cytopenias (chronic immune thrombocytopenia [ITP] in one and autoimmune hemolytic anemia [AIHA] in the other girl), exocrine pancreas insufficiency, intractable enteropathy, and lymphoproliferative disorder (LPD), starting at pre-school age in both patients. Immunoglobulin levels were borderline normal in both girls before initiation of immunosuppressive treatment. TCRabCD4-CD8- (double-negative) T-cells were slightly increased in peripheral blood and >30% in a lymph node biopsy of the older patient. Nine years ago at 10 years, she underwent allogeneic hematopoietic stem cell transplantation from her HLA-identical mother on the grounds of progressive pancytopenia, LPD, and invasive infections, at that time without genetic diagnosis, leading to clinical remission. Years later, ITP recurred, albeit being responsive to low-dose romiplostim (5 µg/kg every 4-6 weeks to maintain >70.000ptl/µL) without substantial reduction of quality-of-life. The now 11 years-old younger sister was successfully treated with rituximab for AIHA five years ago but suffers from ongoing enteropathy requiring total parenteral nutrition. To elucidate the genetic cause, homozygosity mapping was combined with whole-exome sequencing, enabling detection of a novel mutation in LRBA (c.7162delA; p.T2388fs). As expected from this deleterious frame-shift mutation, protein expression was abolished. Heterozygous family members are asymptomatic. These results indicate that allogeneic hematopoietic stem cell transplantation may persistently ameliorate LRBA deficiency.
ESID-0796 Clinical Insight of New FAS Gene (TNFSRF6) Mutation
V. Selmanovi? 1, T. Avcin2, A. Dizdarevic-Omercahic1, M. Debeljak3, D. Milicic-Pokrajac4, A. Cengic5, M. Mackic-Djurovic6, M. Sadikovic1
1Allergology Rheumatology and Clinical Immunology, Children's Hospital University Clinical Center Sarajevo, Sarajevo, Bosnia and Herzegovina
2Allergology Rheumatology and Clinical Immunology, Department for Allergology RheumatoChildren's Hospital University Clinical Center Ljubljana Slovenia, Ljubljana, Slovenia
3Center for Medical Genetics, University Children's Hospital University Medical Center Ljubljana Slovenia, Ljubljana, Slovenia
4Nephrology department, 1Children's Hospital University Clinical Center Sarajevo Bosnia and Herzegovina, Sarajevo, Bosnia and Herzegovina
5Allergology Rheumatology and Clinical Immunology, 1Children's Hospital University Clinical Center Sarajevo Bosnia and Herzegovina, Sarajevo, Bosnia and Herzegovina
6Center for Cytogenetics, Medical School Sarajevo University of Sarajevo Bosnia and Herzegovina, Sarajevo, Bosnia and Herzegovina
Background: Autoimmune Lymphoproliferative Syndrome (ALPS) is a disorder of abnormal lymphocyte survival caused by dysregulation of the FAS apoptotic pathway. Defective apoptosis can lead to lymphoproliferation, autoimmunity and malignancy. Most patients (60-70%) have germline mutation in FAS gene (TNFRSF6), usually inherited in an autosomal dominant fashion. We describe 3 sister's offspring, one diagnosed as ALPS (Figure?1).
Patient 1. Boy, age 9,5y diagnosed as ALPS, based on chronic nonmalignant lymphoproliferation, elevated peripheral DNTcells, defective apoptosis by in?vitro assay; positive autoimmune cytopenia, elevated polyclonal hypergammaglobulinemia, plasma IL-10 and vitamin B12, positive family history of lymphoproliferation. FAS gene mutation, exon 6 (TNFSRF6) 536T>G (p.Leu179Arg). Now 16y, healthy.
Patient 2. Girl, age 13y diagnosed as multicystic pulmonary disease and tuberculosis. Age 14y, wound after lipoma removal was not healing for 8 months. Age 15y, died due to undefined systemic disease complicated with macrophage activation syndrom. Differential diagnosis: pulmonary-renal syndrom (Wegener?), SLE? complement deficiencies? lymphoproliferative diseases presented with pulmonary problems accompanied with systemic symptoms, glomerulonephritis? Could it be ALPS related disease?
Patient 3. Girl, age 2,5y diagnosed hemolytic-uremic sy. Due to positive family anamnesis, genetically tested: FAS gene mutation exon 6 (TNFSRF6) 536T>G (p.Leu179Arg). Four years follow up now: mild splenomegaly and cervical lyphadenopathy, raised vitamin B12, intermitently positive autoantibodies. Healthy.
Conclusion: First, we described a FAS gene mutation not found in ClinVar database as patogenic so far. Second, ALPS patients have family members with the same genetic alterations and an absent or very mild clinical phenotype, or could it be opposite?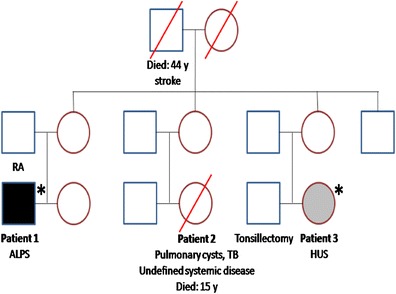
ESID-0110 Current Genetic Techniques to Diagnose FOXP3 Mutation in Three Boys Who Died 25 Years Ago and its Usefulness for Preconception Counselling
P. Soler-Palacín 1, E. Cobos-Carrascosa1, O. Colobran2, B. Valle-del-Barrio1, M. Martinez-Gallo2, A. Martin-Nalda1, S. Lois3, C. Figueras1
1Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d'Hebron Institut de Recerca Vall d'Hebron Universitat Autonoma de Barcelona, Barcelona, Spain
2Immunology Division, Hospital Universitari Vall d'Hebron Institut de Recerca Vall d'Hebron Universitat Autonoma de Barcelona, Barcelona, Spain
3Research Unit in Translational Bioinformatics in Neurosciences, Universitat Autonoma de Barcelona, Barcelona, Spain
Introduction: Preconception council (PC) is an essential tool for relatives of children with primary immunodeficiencies (PID). In the last 20 years, the discovery of new mutations has permitted the diagnosis of several diseases with fatal prognosis as IPEX (Immune disregulation, polyendocrinopathy, enteropathy, X-linked) syndrome. We present the case of three 3 brothers who died 25 years ago of an undiagnosed severe disease whose sisters sought for PC.
Patients and methods: medical charts from all deceased patients were reviewed. Genetic study of FOXP3 gene was performed by direct sequencing.
Clinical case: 5 siblings (3 boys, 2 girls) from no consanguineous parents; maternal uncle died at the age of 3 months because of hepatitis and diarrhea. Case 1 (1963): died at the age of 2 months because of infectious meningoencephalitis. Case 2 (1986): died at 13 months of age due to Klebsiella spp. sepsis, persistent diarrhea and severe skin lesions; blood count revealed no abnormalities. Case 3 (1988): several bacterial and fungal sepsis leading to death, severe eczema and diarrhea, IgE > 2000 IU/L and eosinophilia.
Twenty-five years later, their two sisters sought for PC, DNA from case 2 necropsy was studied and the molecular analysis revealed a novel mutation in hemizygosity in the FOPX3 gene (c.1099T>G p.Phe367Val). Both the mother and the older sister were carriers of the same mutation. We are working in this mutation’s impact by in silico studies.
Conclusion: The use of current molecular diagnostic techniques in necropsy specimens from undiagnosed cases may allow PC in patients’ relatives.
ESID-0187 Autoimmune Complications in CVID in Adults
J. Strakova 1
1Clinical Immunology and Allergology, University Hospital Martin, Martin, Slovakia
Introduction : Autoimmunity is common in CVID. Patients : 20 adults with CVID. In 12 (60%) of them autoimmunity was found: LIP:1, Splenomegaly with hypersplenism: 2, Lymphadenopathy :1, Autoimmune cytopenia : 2 GIT :7, Autoimmune thyreoiditis : 4, Allopetia : 2, RA:1. Cummulation of more autoimmune complications in CVID in a single patient : Female, 50 y. CVID was diagnosed in 2006 (in her 42y.) when hospitalized for LIP. IG therapy with good response. Lab. findings ( before IG replacement ) : IgG 3.4?g /l, igA 0.09?g /l, IgM O.26?g /l, on IVIG therapy : IgG 7.0?l g /l. At the onset of the disease present were : generalized LAP, splenomegaly, sprue – like disease, chronic autoimmune thyreoditis. The recent 2 y. decreasing platelets and enlarging of the spleen were observed. Treatment with high dose IVIG, corticosteroids, biological therapy with no response. Due to splenomegaly (20.5x14.5x6-10 cm, weight 1234 g) with hypersplenism, splenectomy was indicated. Histologically hypersplenism, granulomatous proliferation associated with CVID, no malignancy. Discussion : The single treatment of CVID is immunoglobulin replacement therapy. Regular supplementation of IG can prevent /reduce infections, but cannot prevent/or cure the autoimmune complications of CVID. In adults there is a risk of cummulation od more autoimmune complications (together with other comorbidities). Patients with autoimmunity in CVID have worse prognosis then those without autoimmunity. Conclusion : We hope that the future will bring a better understanding of immune dysregulation in CVID and therefore new therapeutic strategies in CVID patients.
ESID-0031 Novel Mutation of FOXP3 Gene to the First Reported Patient with Immunodysregulation, Polyendocrinopathy and Enteropathy X-Linked Syndrome in Greece
S. Tantou 1, I. Orfanou2, M. Tzanoudaki3, C. Picard4, F. Rieux-Laucat5, M. Kanariou6
1Specialized Center & Referral Center for Primary Immunodeficiencies – Paediatric Immunology, ''Aghia Sophia'' Children's Hospital, Athens, Greece
2First Dept of Paediatrics Medical School University of Athens, "Aghia Sophia" Children's Hospital, Athens, Greece
3Specialized Center & Referral Center for Primary Immunodeficiencies – Paediatric Immunology, "Aghia Sophia" Children's Hospital, Athens, Greece
4Study center of Primary Immunodeficiencies, Necker-Enfant Malades Hospital Assistance Publique - Hôpitaux de Paris (AP-HP)Sorbonne Paris Cité Paris Descartes University Imagine Institut INSERM UMR 1163, Paris, France
5Imagine Institut INSERM UMR 1163, Sorbonne Paris Cité Paris Descartes University, Paris, France
6Specialized Center & Referral Center for Primary Immunodeficiencies – Paediatric Immunology, "Aghia Sophia" Children's Hospital Athens Greece, Athens, Greece
Introduction:
Immunodysregulation, Polyendocrinopathy and Enteropathy X-linked (IPEX) Syndrome is a rare Primary Immunodeficiency, caused by mutations in FOXP3, which encodes FOXP3 protein essential for T regulatory cells. Until 2012, 63 hemizygous mutations had been published, while the exact correlation of genotype-phenotype has not been clarified.
Case Report:
A case of infant, who, to our knowledge, is the first IPEX-patient ever diagnosed in Greece is reported. He is the first child of a Greek father and a Chinese mother. His symptoms started during infancy when he was presented with a rash, the cause of which was thought to be milk allergy. After some months, diarrhea was noted and Type 1 diabetes mellitus was diagnosed at 3 years of age and he was admitted to our Hospital. Immune investigations revealed reduced expression of FOXP3, elevated levels of IgE and positive anti-enterocyte antibodies. DNA analysis confirmed the diagnosis of IPEX which was caused by a novel hemizygous missense FOXP3 mutation (.C1161G, p.H387Q). At the time, he commenced on rapamycin, while HSCT has been recommended.
Conclusion:
Early diagnosis of IPEX syndrome is critical for the outcome and patients’ survival. Essential diagnostic tool remains the detection of the causal mutation. Novel mutations contribute to better understanding of such a rare disease.
ESID-0180 Molecular Characterization of a Novel Intronic Mutation in the SH2D1A Gene
B. Tóth 1, B. Soltész1, E. Gyimesi2, G. Csorba1, A. Veres3, A. Lányi3, G. Kovács4, M. Erdos1, L. Maródi1
1Department of Infectious and Pediatric Immunology, University of Debrecen, Debrecen, Hungary
2Immunology, University of Debrecen, Debrecen, Hungary
3Institute of Immunology, University of Debrecen, Debrecen, Hungary
42nd Department of Pediatrics, Semmelweis University, Budapest, Hungary
X-linked lymphoproliferative disease (XLP) is a well-defined innate error of immunregulation characterized by Epstein-Barr virus-induced fatal infectious mononucleosis (FIM), hypogammaglobulinemia, vasculitis, and predisposition to lymphoid malignancy. XLP is caused by mutation either in the gene of the signaling lymphocyte activation molecule (SLAM)-associated protein (SAP) (SH2D1A) or the X-linked inhibitor of apoptosis protein (XIAP). We describe here a novel c.137+5G>A intronic mutation in SH2D1A in association with FIM in an 8-year-old male patient. Genetic sequencing of blood cell-derived cDNA in his maternal step brother carrying the same genomic mutation, revealed a 22?bp deletion in the SAP cDNA. Immunoblot analysis and flow cytometry performed in this patient showed the lack of SAP protein expression in peripheral blood lymphocytes. These data suggest that the novel c.137+5G >A intronic mutation result is loss of function of SH2D1A protein and leads to typical XLP phenotype.
ESID-0066 A Late Onset Interleukin-1-Receptor Antagonist Deficiency with a Novel Mutation
E. Ulusoy 1, N.E. Karaca1, H. El-Shanti2, E. Kilicoglu1, G. Aksu1, N. Kutukculer1
1Department of Pediatrics, Ege University Faculty of Medicine, Izmir, Turkey
2Shafallah Medical Genetics Center, Doha, Qatar
Background: Interleukin-1-receptor antagonist (IL-1Ra) deficiency (DIRA) is a rare autoinflammatory disorder, involving neonatal onset of pustulosis, periostitis, and sterile osteomyelitis. The underlying genotypic abnormality involves a recessive mutation in IL1RN, which encodes for IL-1Ra.
Observations: We report a 12-year-old girl who presented with later onset of 1 year age, older than the other children with DIRA reported before. She developed pustular lesions healing with scar formation at 1 year of age. She had been hospitalized four times because of these lesions in the last 11 years. At the age of 12, she was hospitalized for arthralgia of the knees, elbows and ankles and arthritis of the left knee. Physical examination revealed hyperpigmented scars on the right cheek, bilateral inguinal and paraumblical regions, and paronychia of the thumbs. She had contracture of left knee, episcleritis and failure to thrive. Chest radiography showed widening of the anterior ribs and scoliosis. The skeletal survey showed periostitis with osteolysis, irregular widening of proximal radius and heterotypic ossification of hip joint. Skin biopsy demonstrated neutrophil infiltration of epidermis, subcorneal and subepidermal pustular dermatosis. Genetic analysis revealed a novel mutation (p.R26X) in IL1RN confirming the clinical diagnosis of DIRA. We obtained both clinical and laboratory remission with canacinumab ( interleukin-1-beta inhibitor) treatment.
Conclusion: It is important to consider that pustular dermatosis of DIRA may present not only in neonatal period, but also in older ages. Identification of these patients early in life impacts on treatment, prognosis, and quality of life.
ESID-0412 Lymph Nodes of CVID Patients with Lymphadenopathy are Characterized by Dysmorphic Germinal Centers, Impaired Class-Switch and Reduced Plasma Cells
S. Unger 1, M. Seidl2, K. Schrenk2, C. Wehr1, S. Goldacker1, A. Schmitt-Gräff2, M. Werner2, K. Warnatz1
1University Medical Center Freiburg, Center for Chronic Immunodeficiency, Freiburg, Germany
2University Medical Center Freiburg, Institute of Pathology, Freiburg, Germany
Introduction:
Hypogammaglobulinemia and recurrent bacterial infections are hallmarks of common variable immunodeficiency (CVID). Since numbers of circulating class-switched memory B cells and plasmablasts are severely reduced in most patients, the germinal center (GC) reaction as the source of both populations is thought to be disturbed in many CVID patients.
Objective:
Immunohistomorphologic and flowcytometric investigation of CVID derived lymph nodes.
Methods:
Immunohistochemical studies were performed on lymph node biopsies from ten CVID patients with lymphadenopathy and immune-competent controls. Seven CVID patients were additionally investigated by flowcytometry.
Results:
All CVID patients were capable of forming normal GCs, however, numerous irregular-shaped GCs were also present. The percentage of ill-defined GCs correlated positively with circulating CD21low B cells and negatively with circulating CXCR5+ memory CD4+ T cells. Disturbed dark/?light zone polarization and fragmented FDC networks were characteristic features of dysmorphic GCs. Flowcytometry revealed a relative expansion of GC and GC founder B cells in 3/?7 patients. While IgM+ plasma cells (PCs) were still present in most cases, class-switch to IgA was abrogated in 8/?10 patients. In 4/?6 patients IgG+ PCs located only inside the GC, compatible with an insufficient emigration of post GC cells in some CVID patients.
Conclusions:
Formation of GC is intact in CVID patients with lymphadenopathy, unlike the early defect in GC formation seen in ICOS- or CD40L deficiency. Histological hallmarks are the severely reduced PC count and the presence of ill-defined GCs.
ESID-0718 Intramedullar Lymphoproliferation in a Young Adult with Autoimmune Lymphoproliferative Syndrome
J. Van Der Werff Ten Bosch 1, V.K. Rao2, I. Maric3, M. De Waele4, H. De Raeve5
1Pediatrics, UZ Brussel, Brussels, Belgium
2Molecular Development ?Section?Laboratory of Immunology Division of Intramural Research, National Institute of Allergy and Infectious Diseases National Institutes of Health, Bethesda, USA
3Department of Laboratory Medicine Clinical Center, National Institutes of Health, Bethesda, USA
4Department of Laboratory Haematologie, UZ Brussel, Brussels, Belgium
5Department of Anatomopathology, UZ Brussel, Brussels, Belgium
A 20 years old female presented with chronic bone pain. She had been diagnosed with Autoimmune Lymphoproliferative Syndrome at the age of 4, caused by a mutation in the splice site of the third exon of the TNFSFR6 (FAS) gene. Salient clinical features at that time were cytopenias associated with significant lymphoproliferation leading to splenectomy before the diagnosis of ALPS. Her symptoms improved markedly on Fansidar, but the lymphadenopathies reappeared as soon as the administration of the drug was stopped. At the moment of presentation, there were no other clinical problems.
The main clinical features were pain in the hands, the lower back, the ankles and especially both knees. Furthermore, the patient suffered from fatigue and abdominal pain. There were no other signs of lymphoproliferation. The blood sample showed, besides the known features of ALPS, an elevated sedimentation rate. Alfa/?beta Double negative T (DNT) cells were 35%. Imaging studies were performed and an MRI showed signs of intra osseous lymphoproliferation. A bone marrow biopsy revealed extensive population of DNT cells. Molecular analysis excluded a monoclonal malignant population. Interestingly, the patient had no other signs of lymphoprliferation at the time of diagnosis. The patient was treated with corticosteroids and mycofenolate-mofetil with an intermediate response. At this time, she is lost for follow up.
In a recent report at 2014 USCAP meeting 5/?30 bone marrows revealed diffuse interstitial lymphocytosis from the cohort of ALPS patients at NIH. Hence, clinicians should beware of this unusual finding of nonmalignant intra medullary lymphoproliferation in ALPS.
ESID-0444 Hematological Stem Cell Transplantation in ADA2 Deficiency
J. van Montfrans 1, A. van Royen- Kerkhof1, M. Bierings2, I. Aksentijevich3, A. Zavialov4, Q. Zhou3
1Pediatric Immunology and Infectious Diseases, University Medical Center Utrecht, Utrecht, Netherlands
2Pediatric Hematology and Oncology, University Medical Center Utrecht, Utrecht, Netherlands
3Inflammatory Disease ?Section?NHGRI, NIH, Bethesda, USA
4Turku Centre for Biotechnology, University of Turku, Turku, Finland
ADA2 deficiency is a recently reported cause of cytopenias and inflammatory vascular- and skin disease. We report two siblings with recently diagnosed ADA2 deficiency (homozygous R169Q mutation in CECR1), one of whom underwent HSCT in 2003.
Patient A presented at age 6 months with livedo reticularis, hepato-splenomegaly, hypercoagulability, granulopenia and complete red cell aplasia. He underwent HSCT for a presumed diagnosis of atypical Blackfan-Diamond anemia using a matched unrelated donor after myeloablative conditioning. He showed rapid immune reconstitution with resolution of cytopenias, skin lesions, hepatosplenomegaly and hypercoagulability. He has had 100% donor chimerism in peripheral blood and has been off medications for 9 years. Serum ADA2 levels are now within the normal range for age. A recent MRI of the brain was negative for vasculopathic changes. HSCT corrected the ADA2 mutation in blood cells, but not in (somatic) buccal mucosa cells.
His sibling (B) presented in 2009?at age 6 years with hepatosplenomegaly, hypogammaglobulenia and profound lymphopenia. He developed unexplained fever, livedo and cerebral stroke at the age of 10, which prompted us to sequence CECR1. Patient B is now using etanercept and is in a stable clinical condition, although with persisting lymphopenia and low grade inflammation. HSCT is being considered.
We conclude that cytopenia, livedo and hepatosplenomegaly can be corrected by HSCT in ADA2 deficiency. The absence of vasculopathy and resolution of hypercoagulability after HSCT suggests that correction of ADA2 levels in blood reduces macrophage activation and endothelial disruption, that likely cause vasculopathy and stroke in ADA2 deficiency.
ESID-0510 IPEX-Like Syndrome: New IL2RALFA Gene Mutation
M. Vignoli 1, S. Ciullini-Mannurita1, G. Guariso2, E. Gambineri1
1Dept of NEUROFARBA section of Child's Health, University of Florence- Meyer Children's Hospital, Florence, Italy
2Dept of Women’s and Children’s Health Unit of Paediatric Gastroenterology, University Hospital of Padova, Padova, Italy
IL2Ra encodes the a-subunit (CD25) of the IL2 receptor complex. CD25 is constitutively expressed at high levels by CD4+CD25+FOXP3+ regulatory T cells. Its binding with IL2 is the first step in the formation of the high affinity receptor complex and promotes the transcription of FOXP3.
For its role in FOXP3 espression the analysis of IL2Ra gene has been performed in patients with clinical features resembling IPEX that were negative for FOPX3 mutations, and few IPEX-like cases have been reported harboring IL2Ra mutations.
We have characterized a new IL2Ra mutation in a 2 months-old male patient which presented early-onset severe diarrhoea, eczema and elavated IgE levels.
The sequence analysis revealed a missense variation in exon 2 that is predicted to lead to the replacement of a cysteine. The region encoded by exon 2 constitutes one of the two sushi domains that are essential for IL2-IL2Ra interaction and the cysteine is involved in a disulfide-bond formation that is important for the correct 3D-structure folding.
The molecular data suggest that the nucleotide variation can lead to the degradation of the a subunit after its synthesis, because of the misfolded protein structure, and to the absence of the receptor expression.
This hypothesis is supported by cytofluorimetric analysis that revealed total absence of cell surface CD25 expression both in freshly-isolated and activated CD4+ T cells and presence of intracytoplasmic CD25 expression immediately after T cells activation.
The molecular diagnosis allowed the little patient to undergo an early and successful hematopoietic stem cell transplantation.
ESID-0757 Evaluation of FAS, FASLG and IL10 AS FAS Mutation Biomarkers in a Family with Autoimmune Lymphoprolipherative Syndrome (ALPS)
B.?M. Abramczuk1, M.?A. Teocchi 1, V.D. Ramalho1, T.N. Mazzola1, M.M. vilela2
1Center for Investigation in Pediatrics-CIPED, Faculty of Medical Sciences University of Campinas, Campinas SP, Brazil
2Pediatrics Immunology Division and Center for Investigation in Pediatrics -CIPED, Faculty of Medical Sciences University of Campinas, Campinas SP, Brazil
Introduction: ALPS is characterized by a chronic accumulation of non-malignant lymphoid cells, increased circulating TCRa/?ß+CD4-CD8- cells and autoimmune manifestations.
Methods: We studied an ALPS-FAS patient, his mother, a maternal uncle and a maternal aunt, all of them with a heterozygous substitution in the splice donor site of intron 4, but with different clinical manifestations. The patient´s clinical history includes organomegaly, hemolytic anemia, hypergammaglobulinemia and glomerulonephritis. His mother had congenital herpes infection and leukopenia. His aunt and uncle had glomerulonephritis that declined without specific treatments. The uncle also had hemolytic anemia. The quantification of soluble FAS ligand (sFASL) and interleukin (IL)-10 was performed on frozen serum samples by ELISA. FAS and FASL mRNA relative expression in peripheral blood mononuclear cells were evaluated with real-time quantitative PCR.
Results: We observed high sFASL and IL-10 concentrations in the serum of the ALPS-FAS patient and his mother, but normal levels in the serum of the uncle and the aunt. The family showed a depressed FASLG mRNA relative expression in comparison with the controls (p=0.0381, reference gene: HPRT1 or GAPDH+HPRT1). The FAS mRNA relative expression was similar in both groups.
Conclusions: In spite of the well-known apoptotic defect in ALPS patients, mutational consequences are poorly understood. The fact that the same mutation in distinct, but related, subjects causes severe, moderate, mild or absent clinical manifestations is still unexplained. Our study reinforces the role of biomarkers as an important tool to predict ALPS clinical manifestations and to clarify the molecular signalling pathways affected by the FAS mutation.
ESID-0255 Deep Immunophenotyping Approaches in Common Variable Immunodeficiency
J. Wanders 1, S. Burns1, H. Stauss1, L. Walker1, D. Sansom1
1Department of Immunology, UCL Institute of Immunity and Transplantation, London, United Kingdom
Common variable immunodeficiency (CVID) is the most frequently diagnosed primary immunodeficiency. However single gene defects have only been reported in a relatively small fraction of CVID patients. Both B and T cell defects have been identified in patients providing markers for improved diagnosis.
B cell sub-populations have been extensively investigated in CVID and characteristically patients show reduced class switched memory B cell populations. Importantly, there is also a strong requirement for T cell interactions with B cells in order to generate effective class switching. Moreover, autoimmunity is frequently seen in CVID suggesting in some patients there may be much more generalised immune dysregulation beyond antibody deficiency. We are therefore developing a more integrated immunophenotyping approach to characterise different T cell, Treg, B cell, and APC populations whilst concentrating on pathways which are relevant to cellular function.
We have developed multicolor flow cytometric phenotyping panels capable of identifying Treg and Tfh populations, which, together with standard B cell phenotyping, give an improved picture of immune status. To date, we have seen changes in expression patterns in several markers in the Tfh and Treg panels in CVID patients. In combination with genome sequencing of our patients this provides a useful approach to identifying cell type specific changes to narrow down functionally significant genetic mutations.
ESID-0349 Deep Sequencing of the TCR? CDR3 Region Reveals a Unique Set of T-Cells that Are Closely Associated with Enteropathy in Common Variable Immunodeficiency (CVID)
G. Wong 1, M. Klinger2, D. Millar1, P. Mistry1, H. Clifford3, A. Huissoon3, M. Faham2, M. Cobbold1
1MRC Centre for Immune Regulation, University of Birmingham, Birmingham, United Kingdom
2Sequenta Inc, Sequenta Inc, San Franciso, USA
3West Midlands primary immunodeficiency centre, Birmingham Heartlands Hospital, Birmingham, United Kingdom
Introduction: Abnormal T-cell oligoclonality was previously demonstrated in CVID by spectratyping but its biological role is not clear. The T-cell receptor (TCR) ß-chain complementarity determining region 3 (CDR3) sequence represents a unique molecular signature of a T-cell and by parallel sequencing of this region we aim to better characterise the nature of the hyperexpanded T-cells in CVID.
Methods: Fifteen healthy donors, six patients with X-linked agammaglobulinaemia (XLA) and forty-two CVID patients were recruited into the study. Primary sequences of the CDR3 regions were generated from peripheral blood mononuclear cells using next-generation sequencing technique. Results were verified using the High V-Quest bioinformatics tool on the IMGT webpage and homology of CVID clonotypes analysed using Clustal-? (EMBL).
Results: Compared to healthy controls, the number of DNA clonotypes were lower in CVID patients with inflammatory complications (p=0.0041). However, the TCR repertoire diversity between healthy controls and CVID were similar by Shannon Entropy (p=0.3894). We identified a number of large/hyperexpanded TCRß clonotypes (>0.01%) that were shared amongst CVID patients but were not found in XLA or healthy controls (n=189). Homology analysis of these clonotypes revealed distinct clusters, which were closely associated with the clinical complication of enteropathy (p=0.0026).
Conclusion: Our data suggest that enteropathy in CVID could be driven by a specific set of T-cells. Further work will focus on isolating and determining the putative antigenic targets of these T-cells in order to test the hypothesis that they are responsible for the enteropathy in CVID.
ESID-0190 X-Linked Dysgammaglobulinemia Associated with Somatically Reverted Memory T Cells in a Family with X-Linked Lymphoproliferative Syndrome Type 1
X. Yang 1, N. Nishida1, A. Hoshino1, K. Goi2, T. Kanzaki3, K. Yoshida4, H. Muramatsu5, S. Ogawa4, S. Kojima5, H. Kanegane1
1Department of Pediatric, University of Toyama, Toyama, Japan
2Department of Pediatric, University of Yamanashi, Chuo, Japan
3Department of Rheumatology and Collagen Diseases, Yamanashi Prefectural Central Hospital, Kofu, Japan
4Department of Pathology and Tumor Biology, Kyoto University, Kyoto, Japan
5Department of Pediatric, Nagoya University Graduate School of Medicine, Nagoya, Japan
Background: X-linked lymphoproliferative syndrome type 1 (XLP1) caused by SH2D1A gene mutation is a rare but severe immunodeficiency characterized by extreme vulnerability to Epstein-Barr virus (EBV) infection, dysgammaglobulinemia, and a high incidence of lymphoma. SH2D1A encodes the adaptor molecule SLAM-associated protein (SAP), which is expressed in T and natural killer cells and is required for cytotoxicity against EBV-infected B cells. The clinical presentation of XLP1 is variable, and many efforts have been done to find the relationship between the phenotype and genotpye. Nonetheless, it is still unkonwn why some XLP1 patients present less severe clinical manifestations even after EBV infection.
Study design: Three male patients in a family present hypogammaglobulinemia with normal B cell number and one case with a past history of EBV-positive lymphoma. We perfromed the whole exome sequncing in these patients.
Result: Gene analysis disclosed that these patients exhibited a missense mutation (2T>C) in the SH2D1A gene. SAP expression by flow cytometry showed nearly deficient; however, a small fraction of SAP-positive cells were identified in T cells, especially in CD45RO+ (memory) fractions of CD4+ and CD8+ T cell. This apparent reversion to SH2D1A positivity occurrend in CD8+ memory T cells in all three cases and also in CD4+ memory T cell in one case.
Conclusion: Somatic reversion of SH2D1A gene is selectively observed in memory T cells, and this may be associated with a mild phenotype of XLP1 patient.
ESID-0523 Successful Management of APECED with Rituximab
M. York 1, R. Sargur1, A. Shrimpton1, W. Egner1
1Clinical Immunology and Allergy, Sheffield Teaching Hospitals, Sheffield, United Kingdom
Background
Autoimmune Polyendocrinopathy, Candidiasis and Ectodermal Dystrophy (APECED) is a rare, autosomal recessive disease caused by mutations in the Autoimmune Regulator (AIRE) gene.
Clinical Case
We present the case of a 27-year-old female diagnosed with APECED Syndrome. She has a complex history of recurrent candidiasis, antibody deficiency, lymphocytic interstitial pneumonitis, bronchiectasis and multiple endocrine abnormalities including autoimmune hepatitis. Despite immunosuppression with Azathioprine, Hydroxychloroquine and oral steroids, she developed deteriorating lung function, recurrent exacerbations of bronchiectasis and candidiasis requiring treatment with high dose Fluconazole and IV Caspofungin on a periodic basis. A multi-disciplinary approach to her management was taken involving Immunology, Respiratory and Rheumatology teams and treatment with the anti-CD20 monoclonal antibody Rituximab was commenced. It is now a year since she started treatment with Rituximab and her symptoms have improved.
Conclusion
Treatment of APECED with Rituximab has rarely been described in the literature. This case demonstrates the importance of a multi-disciplinary approach in managing complex patients with APECED. Treatment with Rituximab can be considered in patients with APECED who remain uncontrolled despite conventional immunosuppression.
ESID-0006 Disturbed Treg Homeostasis in Familial Hemophagocytic Lymphohistiocytosis
S. Humblet-Baron1, S. Bornschein1, A. Liston 1
1VIB, University of Leuven, Leuven, Belgium
Foxp3-positive regulatory T (Treg) cells are a crucial immunosuppressive population of CD4 T cells, yet the homeostatic processes and survival programs that maintain the Treg cell pool are poorly understood. We report that peripheral Treg cells markedly alter their proliferative and apoptotic rates to rapidly restore numerical deficit through an interleukin 2-dependent and costimulation-dependent process. By contrast, excess Treg cells are removed by attrition, dependent on the Bim-initiated Bak- and Bax-dependent intrinsic apoptotic pathway. The antiapoptotic proteins Bcl-xL and Bcl-2 were dispensable for survival of Treg cells, whereas Mcl-1 was critical for survival of Treg cells, and the loss of this antiapoptotic protein caused fatal autoimmunity. These normal homeostatic processes are disturbed in the mouse model of Familial hemophagocytic lymphohistiocytosis (FHL), where excessive IL-2 consumption by activated CD8 T cells results in IL-2-deprivation of Treg and a resulting collapse in Treg numbers. Together, these data define the active processes by which Treg cells maintain homeostasis via critical survival pathways, and how these processes can be distorted in primary immunodeficiencies.
ESID-0256 Aberrant TCR and MTORC1 Signalling Pathways Characterize Lymphopenia-Induced Autoimmunity in Omenn Syndrome
V. Maina 1, A. Anselmo2, A. Villa3, B. Cassani3
1Telethon Institute for Gene Therapy, Scientific Institute and University Hospital San Raffaele, Milan, Italy
2Humanitas Clinical and Research Center Rozzano, Milan, Italy
3Milan Unit Istituto di Ricerca Genetica e Biomedica Consiglio Nazionale delle Ricerche, Milan, Italy
In lymphopenic conditions, compensatory homeostatic T-cell proliferation takes place to re-establish normal immune homeostasis. However, chronic recurrence of this process might generate autoimmunity. To understand the molecular pathways at the interface between immune and metabolic regulation leading to loss of self-immune tolerance, we analyzed the TCR signalling and mTOR energy-sensing pathway in the RAG2R229Q/R229Q mouse model of Omenn Syndrome, associating profound immunodeficiency and autoimmunity, closely recapitulating the human disease. RAG2R229Q/R229Q T cells have reduced early ERK phosphorylation and defective mTORC-1 activation upon anti-CD3/CD28 stimulation compared to control. Interestingly, the rapid STAT3 activation in mutant T cells correlates with the Th17-skewed phenotype. Conversely, mutant Treg cells display higher induction of ERK and STAT3 phosphorylation compared to naturally anergic control Treg. Consistently, mutant Foxp3+ cells display CD44hiCD45RBloCD62L- activated memory phenotype, similar to non-Treg population. Furthermore, stimulated RAG2R229Q/R229Q Treg upregulate expression of CD25 and Foxp3 markers. Remarkably, besides the major input provided by the sustained TCR signalling, a defective mTORC-1 activation is evident in mutant Tregs, correlating with their impaired suppressive function and failure to prevent gut inflammation in a Rag1-/- colitis model. Consistently, marked lymphocyte infiltration and pro-inflammatory cytokine production are found in the intestinal lamina propria and skin of RAG2R229Q/R229Q mice, in spite of the predominant presence of Tregs in these sites.
Overall, results from this study may provide mechanistic insights into the molecular and metabolic events underlying autoimmunity and lymphoproliferative disease and be therefore instrumental for the development of novel targeted immunotherapy strategies.
Acknowledgements: Grant FIRB-MIUR n. RBFR12I3UB
ESID-0384 The Human Nei Endonuclease VIII-Like 3 (NEIL3) is a Novel Gene Associated with the Development of Auto-Antibodies
M. Massaad 1, D. Tsuchimoto2, J. Chou1, T. Ohsumi3, J. Zhou4, H. Jabara1, J. Kane1, K. Schmitz5, K. Torisu2, Y. Nakabeppu2, P. Kang6, E. Choueiry7, A. Megarbane7, M. Mizui8, M. Kyriacos5, G. Tsokos8, W. El-Herz9, L. Notarangelo1, S. Wallace4, R. Geha1
1Medicine/Immunology, Boston Children's Hospital, Boston, USA
2Medicine, Kyushu University, Fukuoka, Japan
3Molecular Biology, Massachusetts General Hospital, Boston, USA
4Microbiology and Molecular Genetics, University of Vermont, Burlington, USA
5Genetics, Boston Children's Hospital, Boston, USA
6Pediatric Neurology, Boston Children's Hospital, Boston, USA
7Medicine, Universite Saint Joseph, Beirut, Lebanon
8Medicine, Harvard Medical School, Boston, USA
9Pediatric Medicine, Kuwait university, Kuwait, Kuwait
We identified the genetic cause of a combined immunodeficiency in a consanguineous family with history of severe recurrent infections, autoimmunity, chronic diarrhea, and death.
The patients had normal numbers of T/?B cells and normal T cell function. However, B cell proliferation and immunoglobulin production were decreased. Furthermore, the patients had high levels of serum auto-antibodies. Whole genome sequencing (WGS) identified a mutation in a highly conserved residue in the base excision repair enzyme NEIL3. The mutation does not affect protein expression but abolishes enzymatic activity. To determine the effect of loss of NEIL3 function, we studied Neil3-/- mice. Neil3-/- mice had no overt immune defect, however, they had high levels of serum auto-antibodies, and developed autoimmune kidney damage following treatment with the TLR3 ligand polyI:C. We later identified an unrelated asymptomatic individual with the same mutation in NEIL3, and found that she had high levels of serum auto-antibodies. LRBA lies in the same region of homozygosity as NEIL3. Further analysis of the WGS data identified a homozygous duplication of exons49-53 of LRBA. We confirmed this duplication at the level of mRNA/?cDNA, and showed that it resulted in loss of LRBA expression.
We identified NEIL3 as a novel gene associated with the development of auto-antibodies. While the mutations in both NEIL3 and LRBA contribute to the patient’s autoimmunity, the infections and colitis are likely due to the mutation in LRBA. This mutation would have been missed by whole exome sequencing, underlining the importance of WGS for the detection of disease-causing structural variations.
ESID-0393 Role of Aire and Dectin Receptors at Fungal Synapse; A Proposed Mechanism of Cytokine Response by Macrophages Upon Hyphae Recognition
J. Tavares de Albuquerque 1, P. BANERJEE2, M. BARBOSA-CARVALHO3, C. ARSLANIAN3, F. WEILER4, M. DA SILVA4, M. LAZARETTI4, L. PEDROZA5, J. ORANGE6, A. CONDINO-NETO7
1Department of Immunology, Instituto de Ciências Biomédicas University of São Paulo, São Paulo, Brazil
2Department of Pediatrics, Center for Human Immunobiology Texas Children's Hospital, Houston, USA
3Department of Immunology, Instituto de Ciências Biomédicas University of São Paulo, Sao Paulo, Brazil
4Department of Immunology, Federal University of Sao Paulo Brazil, Sao Paulo, Brazil
5Department of Immunology, Universidad San Francisco de Quito, Quito, Ecuador
6Department of Pediatric Medicine Immunology Allergy & Rheumatology, Center for Human Immunobiology Texas Children's Hospital, Houston, USA
7Department of Immunology, nstituto de Ciências Biomédicas University of São Paulo, Sao Paulo, Brazil
Introduction: Autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is an autoimmune disease caused by mutation in autoimmune regulator gene (AIRE). Patients with AIRE mutation are susceptible to yeast infection. To investigate this phenomenon, we previously established that AIRE could regulate Syk dependent Dectin-1 pathway for yeast recognition. Dectin-2 receptor, however, directly recognizes hyphae and shares the same Syk dependent pathway to elicit an immune response.
Objective: To evaluate interactions among several proteins during fungal synapse formation, and immune response against hyphae in AIRE deficient macrophages.
Methods: Following 24?h of hyphae stimulation, cytokine secretion was measured in AIRE shRNA treated THP-1 cell line and partially differentiated macrophages from APECED patient. Further, THP-1 cells were stimulated with hyphae for 30?min and immunoprecipitated for Dectin-2 and probed for AIRE and Syk. Using Volocity software, kinetics of synaptic accumulation of AIRE, Syk, Dectin-1 and Dectin-2 was also determined from confocal micrographs of THP-1 cells.
Results: Upon hyphae stimulation, AIRE knockdown THP-1 cells or AIRE deficient patient macrophages secreted IL-6 and TNF-a, but, at lower levels in comparison to respective control groups. In hyphae activated THP-1 cells, AIRE physically interacted with Dectin-2, Syk, and CARD9. AIRE also colocalized with Dectin-2 and Syk at the fungal synapse. Colocalization between both Dectin receptors and AIRE at the fungal synapse reached its peak at 20 min. Dectin-1 accumulation at the synapse, however, reached saturation at 30 min.
Conclusions: We suggest that AIRE could modulate the physiology of fungal synapse formation, which later orchestrates effective immune response against hyphae.
Financial support: CNPq AND FAPESP
Topic: Innate Immunity
ESID-0185 The Health of Carriers of X-Linked Chronic Granulomatous Disease (CGD) in the United Kingdom
A.?C. Battersby 1, D. Goldblatt2, M.S. Pearce3, H. Braggins4, C. Cale5, D. Barge6, A.?R. Gennery1
1Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
2Institute of Child Health, University College London, London, United Kingdom
3Institute of Health and Society, Newcastle University, Newcastle upon Tyne, United Kingdom
4CGD Society, Great Ormond Street Hospital, London, United Kingdom
5Clinical Immunology, Great Ormond Street Hospital, London, United Kingdom
6Blood Sciences Flowcytometry Laboratory, Newcastle upon Tyne Hospitals Foundation Trust, Newcastle upon Tyne, United Kingdom
Background
: X-linked (XL) CGD, a rare primary immunodeficiency due to CYBB mutations leading to defective gp91PHOX, results in decreased/absent respiratory oxidative burst. High rates of discoid lupus have been reported in XL-carriers. Current literature about XL-CGD carrier health is limited to small series and anecdotal reports. We undertook a large in-depth study of XL-carriers.
Methods
: XL-CGD families were identified and XL-carriers recruited. Participants completed detailed questionnaires about their medical health with particular attention to SLE and gastrointestinal (GI) symptoms. Affected and unaffected groups were compared by t-tests to see if age or neutrophil oxidative burst (NOB) were significant.
Results:
71 recruited (study remains open)
Mean age 43 years (3-77).
Mean NOB 50% (9-94%) with a lower value associated with recurrent skin abscesses (p=0.002)
Photosensitivity, mouth ulcers and joint symptoms occurred in >60%.
GI symptoms present in >50%.10 had undergone invasive investigations. 4 diagnosed with Inflammatory Bowel Disease (IBD).
| Affected | Unaffected | |
| Recurrent Ulcers | 54 | 17 |
| Raynaud's | 25 | 46 |
| Joint Pain | 45 | 26 |
| Photosensitivity | 52 | 19 |
| GI Symptoms | 37 | 34 |
| Recurrent Skin Abscess | 6 | 65 |
Age was not significantly different in affected/unaffected groups.
Discussion
: The study's strength is the detail collected. It is the largest, most comprehensive study of XL-CGD carriers to date.
The main finding is significant medical problems are more common and diverse than previously considered. The overlap with IBD has not been previously described, highlighting the association between CGD colitis and IBD.
XL-CGD carriers are at greater risk of medical problems than previously thought. Symptoms do not correlate well with NOB or age. Greater awareness of potential medical problems is required.
ESID-0182 Quality of Life (QOL) is Reduced in X-Linked Carriers of Chronic Granulomatous Disease (CGD)
A. Battersby 1, M.S. Pearce2, D. Goldblatt3, H. Braggins4, C. Cale5, A.?R. Gennery1
1Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
2Institute of Health and Society, Newcastle University, Newcastle upon Tyne, United Kingdom
3Institute of Child Health, University College London, Newcastle upon Tyne, United Kingdom
4CGD Society, Great Ormond Street Hospital, Newcastle upon Tyne, United Kingdom
5Clinical Immunology, Great Ormond Street Hospital, Newcastle upon Tyne, United Kingdom
Background
: X-linked (XL) CGD is a rare primary immunodeficiency due to mutations in CYBB leading to defective gp91PHOX (an NADPH oxidase complex subunit) resulting in decreased or absent respiratory oxidative burst. XL-carriers have a high incidence of discoid lupus. Other symptoms are anecdotally reported. Carrier status is usually confirmed after a relative has been diagnosed with CGD.
Methods
: XL-CGD families were identified and carriers recruited. Participants completed a validated QoL questionnaire; the SF36v2.Means for each domain were compared with published UK norms using a one-sample t-test.
Results
49/71 XL-carriers (34 mothers) completed the SF36v2.
QoL was reduced in all domains.
| Domain | CGD Carriers | UK Norms (female age 35-54)[1] | P-value |
| Physical Function | 79.28 (29.4) | 89.4(18.3) | 0.0099 |
| Role Physical | 74.35(32.2) | 84.0(32.0) | 0.00235 |
| Bodily Pain | 66.24(30.5) | 79.4(22.0) | 0.002 |
| General Health | 56.31(28.5) | 74.1(20.3) | 0.00 |
| Vitality | 44.98(25.5) | 58.2(19.9) | 0.002 |
| Social Function | 69.68(29.5) | 86.7 (20.5) | 0.001 |
| Role Emotional | 71.88(31.3) | 80.3 (33.6) | 0.0341 |
| Mental Health | 63.16(16.8) | 71.6(17.8) | 0.005 |
Table?1: SF36 Mean Scores for CGD XL-Carriers and UK Norms
Discussion
: This study demonstrates that QoL is reduced in XL-CGD carriers compared to population norms.
There may be several reasons for this. We have demonstrated elsewhere that XL-CGD carriers have more medical problems than previously described. The majority of participants in this study (70%) were mothers of CGD children: stresses associated with caring for a child with CGD may adversely affect QoL.
The importance of this study is that XL-CGD carriers have not previously been shown to have significant problems yet these findings demonstrate that QoL is significantly reduced when compared with similarly aged women.
ESID-0783 Immunodeficiency with Hyper-Immunoglobulin M Syndrome
M. Ben Khaled 1, S. Thraya1, M. Ouederni1, L. Bouzidi1, N. Dhouib1, R. Hassouna1, A. Haoua1, F. Mellouli1, M. Bejaoui1
1Immuno-Hematology Pediatric Service, Bone Marrow Transplant Center, Tunis, Tunisia
Introduction: Hyper-Immunoglobulin M (HIGM) syndrome is a rare disorder characterized by recurrent infections associated with decreased serum immunoglobulin (Ig) G, IgA and IgE, and normal to increased IgM levels. Several genetic defects account for this syndrome. Other primary immunodeficiency disorders (PID) can also present with Ig levels suggestive of HIGM.
Objective: To define PID associated with HIGM syndrome and to describe clinical and biological features of such disease.
Method: Retrospective study over 16 years (March 1998 - March 2013), including patients with PID associated with HIGM syndrome followed within the National Center of Bone-Marrow Transplant of Tunisia.
Results: Forty-one patients were included in this study (25 boys and 16 girls) of which 21 were born to consanguineous parents. Their average age was 77 months (3–360). The mean age at onset of disease was 29 months (2-168). Similar history in the family was found in 17 cases. The disorders were divided into: Common variable immunodeficiencies (n=2), primary HIM syndromes (n=25), ataxia-telangiectasia (n=13) and ataxia-telangiectasia-like (n=1). The most common clinical signs included: respiratory tract infections (n=29), otitis (n=18), chronic diarrhoea (n=12), lymphoid hyperplasia (n=10) ocular telangiectasia (n=12), ataxia (n=13) and autoimmune manifestations (n=3). The level of serum IgG was high in 31 patients and normal in ten others. The mean rate of IgM was 6,6 g/?l (0,46-29,9). Eight patients died during follow-up.
Conclusion: Immunodeficiency with HIM is relatively common in Tunisia. Front of an immunological phenotype suggestive of hyper IgM syndrome, clinical manifestations may suggest the diagnosis.
ESID-0782 Hyper Immunoglobulin E in Primary Immunodeficiency Disorders
M. Ben Khaled 1, S. Thraya1, M. Ouederni1, L. Bouzidi1, N. Dhouib1, A. Haoua1, R. Hassouna1, F. Mellouli1, M. Bejaoui1
1Immuno-Hematology Pediatric Service, Bone Marrow Transplant Center, Tunis, Tunisia
Introduction: High levels of immunoglobulin E are ordinarily found in atopy and parasitosis. The association of hyper IgE syndrome (HIGE) with recurrent infections suggests diagnosis of immune deficiency.
Objective: To determine primary immunodeficiency disorders (PID) associated with HIGE and to describe clinical and biological features of these diseases.
Methods: Retrospective study over 16 years (1998 - 2013), including patients with PIDs associated with HIGE followed within the National Center of Bone-Marrow Transplant of Tunisia.
Results: Thirty-six patients were included (30 boys and six girls) of which 17 were born to consanguineous parents. Their average age at diagnosis was 57 months (1–180). The mean age at onset of disease was 19 months (1 -132). Similar history in the family was found in 16 cases. The disorders were divided into: Immunoglobulin A deficiency (n=3), Omenn syndromes (n=3), cyclic neutropenia (n=2), Chronic granulomatous Disease (n=2), Buckley syndrome (n=23). The most common clinical signs included: allergic manifestations (n=12), buccal candidiasis (n=6), lymphadenopathy (n=7), otitis (n=17), respiratory tract infections (n=20), cutaneous infections (n=25) and diarrhea (n=6). The mean rate of IgE was 7974 g/?l(229-55940).Treatment depends on the type of the primary disorder. Six patients died during follow-up.
Conclusion: HIGE was found in several types of PIDs. The causes were varied in our series: primitive (Job syndrome), infectious or atopic. Further researches focusing on the mechanisms of these genetic defects in IgE regulation is needed for both the understanding of these diseases as well as the application of these understandings to therapeutics.
ESID-0314 Insight Into BCG Infection in Pid Patients, Vaccinated at Birth with BCG Vaccine – 30 Years of Experience
E. Bernatowska 1, B. Wolska – Kusnierz1, M. Pac1, E. Heropolitanska-Pliszka1, B. Piatosa2, J. van Dongen3, M. van der Burg3
1Department of Immunology, The Children's Memorial Helath Institute, Warsaw, Poland
2Histocompatibility Laboratory, The Children's Memorial Helath Institute, Warsaw, Poland
3Department of Immunology, Erasmus MC, Rotterdam, Netherlands
Introduction: High reactogenic BCG Danish vaccine was replaced in Poland by BCG Moreau strain in 1955. Frequency of disseminated BCG infection, in children with primary immunodeficiencies vaccinated with BCG Moreau manufactured by Biomed, Poland were estimated.Patients and methods: One thousand five hundred sixty three cases of primary immunodeficiencies were diagnosed in the Department of Immunology, Children's Memorial Health Institute in Warsaw between 1980-2013.Results: Different forms of SCID were recognized in 62 children. Disseminated BCG infection was ascertained only in SCID patients with phenotype T-B+NK-; in 7 out of 16 patients with ?c deficiency, in 1 out of 2 with JAK3 deficiency and in 2 out of 31 patients with unknown mutation. Two SCID patients died because of BCG infection. There was not disseminated BCG infection in group of T-B+NK+SCID patients, including 19 RAG1/?2, 2 Artemis, and 1 ZAP and Cernunnos deficiency respectively. Mendelian susceptibility to mycobacterial diseases (MSMD) was detected in four patients: IFNGR1 deficiency and IL-12 deficiency each in two patients. All of them developed BCG infection, one IL-12 deficient patient died. In the group of primary immunodeficiencies regarded to be less prone to Mycobacterium infections; CGD n=56, HIES n=20 and XL-HIGM n=9 patients no case of tuberculosis or disseminated BCG infection have been diagnosed.
Conclusion: Disseminated BCG infection appeared only in SCID patients with lack of NK cells. Not only presence of NK cells play a key role in protection againts BCG infection, but also usege of a high profile of safety vaccine.
ESID-0242 Distribution of the 1637DELC Allele Among MBL2 Genotypes
H. Bjarnadottir 1, M. Arnardottir1, B.R. Ludviksson1
1Immunology, Landspitali - The National University Hospital of Iceland, Reykjavík, Iceland
Complement activation via the lectin pathway (LP) is mediated by five pattern recognition proteins (PRPs). These are mannan-binding lectin (MBL), collectin-11 (CL-11), ficolin-1-3. MBL insufficiency is associated with susceptibility to infectious diseases. The frequency of MBL insufficiency (AX/O and O/?O genotypes) has been determined to be 7.9% among Icelandic blood donors (IBD). This is relatively high among healthy individuals and suggests that MBL might be a redundant molecule. Ficolin-3 is the most abundant of the PRPs in serum. The 1637delC mutation in the FCN3 gene causes ficolin-3 deficiency. If MBL insufficiency is compensated for by ficolin-3, then it is conceivable that ficolin-3 levels in MBL insufficiency are higher than in sufficient cases. Therefore, we hypothesize that the1637delC allele should be rare or not found among AX/O and O/?O genotypes.The aim was to investigate the disribution of the 1637delC allele among MBL2 genotypes.The cohort consisted of blood donors and individuals that had been referred to our lab for MBL evaluations (N=637). MBL deficiency variants in exon 1 were determined in addition to the downregulating allele X in the MBL2 promoter using melting curve analysis. The 1637delC allele was determined by RFLP-PCR.The resulting MBL2 genotypes were grouped into insufficient producers of MBL (AX/O and O/?O) (N=106) and sufficient (A/?A and AY/O) (N=531). Twenty 1637delC heterozygotes were detected in the sufficient group (3.8%), whereas the allele was not found in the insufficient group (p=0.0426). The results support our hypothesis that MBL insufficient individuals are not carriers of the 1637delC allele.
ESID-0398 Detection of Chronic Granulomatous Disease X Linked Carriers Through Funtional Studies in Peripheral Blood
L. BLANCAS 1, N. Yamazaki2, S. Espinosa1, F.J. Espinosa1
1Research Unit for immunodeficiencies, Instituto Nacional de Pediatría, Mexico City, Mexico
2Immunology departement, Instituto Nacional de Pediatría, Mexico City, Mexico
Detection of X linked chronic granulomatous (XL-CGD) carriers is necessary amongst maternal women relatives of X-LCGD patients. The objective is offering a genetic counseling. In LatinAmerica there is not accessible to perform sequencing gene in all hospitals for their detection. In most of the cases XL-CGD carriers show two populations of neutrophils and monocytes, one functional and other non functional. This principle is usefull to detect them.
The objective of the study was to detect the CGD-XL carriers through123 dihydrorodamine assay (DHR). Secondary we measured the gp91phox expression in them with a bimodal pattern. Also we correlated the results of both assays in carriers.
Amongst the probable carriers we performed DHR in peripheral blood. Each assay was done at the same time in a healthy control. A carrier through DHR was defined as a positive histogram with a bimodal pattern. We measured gp91phox expression in monocytes through flow cytometry in carriers (and healthy control) detected by DHR.
We found 27 women as a carriers through a bimodal pattern in DHR. In all we observed two patterns of gp91phox expression in monocytes, one with expression and other without expression (dot-plot).
We conclude that both assays are useful tools for the detection of carriers of XL-CGD. Also both studies have a correlation in the carriers. We suggest to performed in LatinAmerica hospitals in which molecular studies are not accessible. We take in account that in women with a normal DHR, the carrier status can not be discarded, in that cases sequencing is necessary.
ESID-0047 Frequent Non-Infectious Gastrointestinal Abnormalities Among Patients with Chronic Granulomatous Disease
A. Broides 1, R. Mohammed2, B. Reid2, C.M. Roifman2, E. Grunebaum2
1ambulatory care unit, Soroka University Medical Center and Ben-Gurion University of the Negev, Beer-Sheva, Israel
2Pediatrics, Division of Immunology and Allergy. Hospital for Sick Children. University of Toronto, Toronto, Canada
Background: In addition to infections, chronic granulomatous disease (CGD) is characterized by increased inflammation that may contribute to various gastrointestinal (GI) abnormalities.
Objective: To better characterize the non-infectious GI abnormalities among CGD patients and the effects of different treatments.
Methods: Analysis of medical records of all patients with CGD treated by the immunology service at the Hospital for Sick Children, Toronto, Ontario, between 2000 and 2012. The diagnosis of CGD was established by nitroblue tetrazolium reduction and/or neutrophil oxidative burst index (NOBI) measured by dihydrorhodamine oxidation.
Results: Eleven male patients suffering from CGD were included in the study. All patients had one or more non-infectious GI abnormality including colitis (72.7%) and oral aphthous ulcers (36.3%). Failure to thrive occurred in 5 patients (45.4%), all with associated colitis. Bone marrow transplantations (BMT) using HLA-identical sibling donors were performed in 4 patients, with 75% long-term (>3 years) survival. In these 3 patients, the inflammation-mediated GI manifestations that was present before BMT resolved. In contrast, 6 of 7 patients who did not receive BMT (p=0.033) continued to suffer from GI disease resulting in failure to thrive, GI bleeding and life threatening small bowel perforation and often required immune suppressive medications.
Conclusions: Inflammatory GI manifestations, particularly colitis, are very frequent in CGD and are often associated with significant morbidity. Allogeneic BMT, particularly if an HLA-matched sibling donor is available should be considered in these patients.
ESID-0643 Epidemiology of Severe Infections in Neutropenic Patients with ELANE Mutation
J. donadieu 1, G. Plat2, B. Beaupain3, C. Bellanné Chantelot4
1Hémato oncology, hopital Trousseau, PARIS cedex 12, France
2Hémato oncology, CHU Toulouse, Toulouse, France
3Hémato oncology, Hopital Trousseau, Paris, France
4Genetic Laboratory, Hopital Pitié Salpétriére, Paris, France
Background: This study evaluates, in patients with ELANE neutropenia, the rate of severe infections and their determinants.
Methods: Severe infections are any life threatening bacterial or fungal infections. The data of all the 119 patients carrying an ELANE mutation were analyzed. Median follow-up was 13.9 years. These patients are diagnosed with cyclic (n =46) or permanent neutropenia (n=73). Only periods without treatment with GCSF, outside of a leukemic transformation, were taken into account.
Results: A total of 214 severe infectious episodes were observed: septicemia n=16; Cellulitis n=95; Pneumopathy n=80; Mastoiditis n=12, Pyeloneph n=11 Omphalitis n=11; others sites n=17. A microbiologic documentation was found in less than 50% of episodes: Staph Aureus n=37; Staph Epid n=3, Strepto A n=8, Strepto pneumoniae n=8; Pseudomonas aeruginosa n=18; E Coli n=18; Others n=10; Aspergillosis n=4; Candida n=5. Globally, fungal infections represent 3.8% of the episodes. The 10-year and 20-year risk of severe infection was respectively 43% (95% CI: 34-52%) and 64% (95% CI 54-74%). The median ratio of severe infections per year was 0.13/year, but is highly depend on the age, the first year of life having the highest rate. The cyclic or permanent pattern of neutropenia has an impact on age of first infection, but did not affect the cumulative risk of severe infection by age of 20 years. Monocytosis and elevated immunoglobulin levels are correlated with the rate of severe infection.
Conclusion: This study provided an objective measure of the risk of severe infection in patients with ELANE neutropenia.
ESID-0186 A Case of Dendritic Cell, Monocyte, B and NK Lymphoid Deficiency (DCML)
M. Dullaers 1, T. Kerre2, F. Coppieters3, D. Sichien4, N. De Cabooter5, V. Debacker5, R. Speeckaert6, P. Coucke7, E. Debaere7, F. Haerynck8, B.N. Lambrecht4, K.Y. Vermaelen9
1Clinical Immunology Research Lab, University Hospital Ghent, Ghent, Belgium
2Hematology, University Hospital Ghent, Ghent, Belgium
3Center for Medical Genetics, University Hospital Ghent, Ghent, Belgium
4Lab for Immunoregulation and Mucosal Immunology VIB, Ghent University, Ghent, Belgium
5Clinical Immunology Research lab, University Hospital Ghent, Ghent, Belgium
6Dermatology, University Hospital Ghent, Ghent, Belgium
7Center for Medical Genetics, Ghent University, Ghent, Belgium
8Pediatric Immunology and Pulmonology, University Hospital Ghent, Ghent, Belgium
9Pulmonary Medicine Clinical Immunology Research Lab, University Hospital Ghent, Ghent, Belgium
Primary immunodeficiencies with a defect in monocytes, dendritic cells (DC) and lymphocytes have recently been described as a result of IRF8 or GATA-2 mutations. IRF8 mutations cause a syndrome characterized by atypical mycobacterial infections and myeloproliferation. Mutations in GATA-2 were reported to cause a similar syndrome, referred to as DC, monocyte, B and NK lymphoid (DCML) deficiency or MonoMac syndrome.
Here we describe a patient with typical manifestations of DCML, which were diagnosed at the age of 22 with Mycobacterium kansasii mediastinal lymphadenitis and massive palmoplantar HPV papillomas. He had a history of thrombocytopenia and monocytopenia and his bone marrow showed signs of myelodysplasia.
Peripheral blood flow cytometry revealed a virtual absence of monocytes and conventional and plasmacytoid DCs and decreased numbers of NK and B cells. Immunostainings on skin biopsies confirmed preservation of epidermal langerhans cells and tissue macrophages. S100 positive dendritic-shaped cells were abundant in the mediastinal lymph nodes. In contrast to reported DCML patients, Flt3L and CD34+ progenitors were not increased in peripheral blood. Sequencing of IRF8 and GATA-2 genes did not identify a disease-causing mutation, nor did whole exome sequencing. Known intronic mutations in GATA-2 were excluded and cDNA analysis of GATA-2 is ongoing.
The patient received an allogeneic stem cell transplantation with 10/10 HLA-matched unrelated donor. By 6 months post-transplant his warts had completely resolved and circulating monocytes and lymphocytes normalized. He developed severe chronic GVHD of the skin and esophagus and lungs, which is now slowly resolving under immunosuppressive treatment.
ESID-0787 Defective Respiratory Burst: A Primary Neutrophil Disorder and Secondary to Bacterial Infections
Z. El-Sayed1, D. El-Ghoneimy 1, M. Sallam2, N. Shawky1
1Pediatrics, Ain Shmas University, Cairo, Egypt
2Clinical pathology, Ain Shmas University, Cairo, Egypt
Background: Defective respiratory burst is the underlying disorder in CGD, but whether it is associated with other primary immunodeficiency diseases (PID) or secondary to bacterial infections, remains to be evaluated.
Patients and Methods: This is a case-control study which included 19 patients with diagnosed PID and 81 patients with recurrent infections. In addition to clinical evaluation, complete blood counts, phagocytic and candidicidal lytic indices and Dihydro rhodamine (DHR) test were done for patients as well as 50 healthy age and sex-matched healthy subjects as a control group. Serum IgA level was measured for those with recurrent infections and normal DHR test.
Results: DHR test was defective in 31% of PID patients, where it was 24% in one patient with a picture consistent with X-linked CGD. The remaining 5 patients (26%) had DHR test that ranged between 2.1% and 82.5%; they were 4 patients with ataxia telangiectasia (AT), one patient with T–B–NK+ SCID, and one patient with Hermansky Pudlak Syndrome. Among patients with recurrent infections, 11 patients (14%) were found to have defective DHR test. Of these patients, 6 (7.4%) had clinical features and DHR test consistent with the diagnosis of CGD. In the remaining 5 (6.6%) patients, the DHR test ranged between 66.7% and 85% and became normal after recovery of infections.
Conclusion: Defective neutrophil respiratory burst can be identified in PID patients. However, reversible defective DHR test is not uncommon during acute bacterial infections in otherwise normal children. Hence, DHR test results should be interpreted with caution.
ESID-0036 Clinical and Laboratory Workup of a Patient with Whim Syndrome
A. ElMarsafy1, R. ElHawary 2, S. Meshaal2, M. ElSharkawy3, R. ELKady1, D. AbdelAziz1, N. Galal1, J. Boutrous1, A. Mostafa2
1Pediatrics, kasr alainy school of medicine, Cairo, Egypt
2Clinical pathology, kasr alainy school of medicine, Cairo, Egypt
3Chemical pathology, kasr alainy school of medicine, Cairo, Egypt
Background:WHIM syndrome is a rare congenital immunodeficiency disorder, it is an acronym for some of the characteristic symptoms of the disorder (W)arts, (H)ypogammaglobulinemia, (I)nfections, and (M)yelokathexis. (Liu et?al 2012)
(Myelokathexis refers to neutropenia resulting from retention of mature neutrophils and increased neutrophil apoptosis in the BM)
Aim of the study: To describe the clinical and laboratory workup of a patient with suspected WHIM syndrome.
A male patient from a non-consanguineous marriage presented with diarrhea following Rota virus vaccination, followed by perianal lesions and recurrent otitis.
He had no organomegaly and no lymphadenopathy. Blood picture showed persistent neutropenia and lymphopenia. His serum immunoglobulins levels were all low. Flow cytometric analysis of his blood cells showed CD3 lymphopenia and marked decrease in the memory CD19 (CD19+CD27+ cells were 0.8%).
Work up for differential diagnosis for other causes of neutropenia was started.
Bone marrow aspiration showed marked hyperplasia, with many cells having signs of apoptosis (hypercondensation of chromatin, hyper segmentation of nucleus and cytoplasmic vaculation)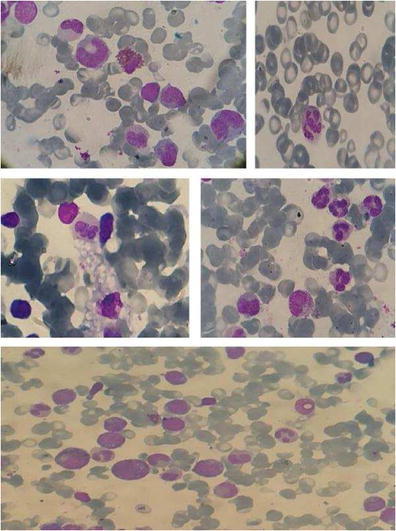
Fluorescence insitu hybridization (FISH) for XY chromosomes showed no maternal engraftment causing graft versus host disease.
A diagnosis of WHIM syndrome became very likely; however genetic confirmation by sequencing the chemokine CXC4 receptor gene is mandatory.
ESID-0198 Identification of Two New Gain-of-Function STAT1 Mutations in the DNA-Binding Domain
G. Frans 1, L. Moens1, H. Schaballie1, G. Wuyts1, D. Dillaerts1, X. Bossuyt1, J. Ceuppens2, I. Meyts3
1Microbiology and Immunology, Catholic University of Leuven, Leuven, Belgium
2Clinical Immunology, University Hospitals Leuven, Leuven, Belgium
3Pediatrics, University Hospitals Leuven, Leuven, Belgium
Introduction: Chronic mucocutaneous candidiasis (CMC) is caused by inborn errors of interleukin-17 immunity. Recently, gain-of-function mutations in the coiled-coil and DNA-binding domain (DBD) of STAT1 were shown to cause autosomal dominant CMC. Here, we describe two CMC patients in whom a mutation in the DBD was identified.
Results: Patient 1 is an 8-year old girl who was diagnosed with CMC at the age of 2 based on failure to thrive, aphtous stomatitis and Candida esophagitis. The finding of thin hair, coarse facial features, and delayed teeth eruption led to the diagnosis of hypothyroidism. She has a heterozygous K388E missense mutation in the DBD.
Patient 2 is a 47-year old man with CMC and Candida-induced esophageal stenosis. He suffers from recurrent bronchial infections and bronchiectasis. Symptoms were present from birth. A novel heterozygous R321S missense mutation in the DBD was identified.
Both patients demonstrated prolonged phosphorylation of STAT1 after IFN-? stimulation and failed to produce IL-17 in response to PHA, Candida albicans, and Staphylococcus aureus.
Conclusion: A mutation in the DBD of STAT1 was identified in both patients. Nevertheless, their phenotype was different with hypothyroidism and bronchiectasis as the major clinical problems in addition to CMC. This clinical heterogeneity remains a major pitfall in the diagnosis of STAT1 gain-of-function mutations. Laboratory evaluation of STAT1 phosphorylation and IL-17 production could provide a useful tool in identifying affected patients.
ESID-0014 XLP-Associated Cerebral Vasculitis Successfully Treated with Bone Marrow Transplant
K. Frith 1, T. O'Brien2, S. Tangye3, R. Sutton4, J. Ziegler1, P. Gray1
1Department of Immunology and Infectious Diseases, Sydney Children's Hospital, Randwick, Australia
2Kids Cancer Institute, Sydney Children's Hospital, Randwick, Australia
3Immunology and Immunodeficiency Group, Garvan Institute of Medical Research, Sydney, Australia
4Children's Cancer Institute Australia for Medical Research, University of NSW, Randwick, Australia
Vasculitis is rare in X-linked lymphoproliferative disease (XLP) caused by mutations in SH2D1A. To date, there have been 3 reports of cerebral vasculitis complicating XLP with no survivors.
We present a 10 year old boy with XLP1 due to a mutation in SH2D1A. His complicated history includes restrictive arthropathy at presentation, two episodes of acute respiratory distress requiring mechanical ventilation, aplastic anaemia, gastritis, dysgammaglobulinemia and NK dysfunction.
He developed infrequent headaches and intermittent blurred vision, and MRI brain scan showed multiple areas of old infarction. MRA was normal, however cerebral angiography confirmed diffuse cerebral vasculitis effecting small and medium-sized vessels in all vascular territories with a fusiform pseudoaneurysm affecting the right posterior inferior cerebellar artery. CSF had a mild lymphocytic pleiocytosis with an extremely high protein level (6.75 g/?L). CSF viral studies including EBV and CMV PCR were negative, however HHV-7 was repeatedly identified by PCR.
After initial treatment with systemic corticosteroids and aspirin thromboprophylaxis he underwent an urgent 6/?6 matched cord blood transplant with reduced toxicity conditioning. After failed engraftment a second transplant using a 5/?6 cord and conditioning with busulphan and fludarabine engrafted successfully with 100% donor chimerism. 250+ days post HSCT there have been no further neurological symptoms and CSF protein is normalising, while the CSF HHV-7 has disappeared. Unfortunately, recovery has been complicated by the development of anti-glomerular basement membrane disease and the therapy for this has prohibited performance of repeat angiogram.
This is the first report of successful haematopoietic stem cell transplantation in XLP-associated cerebral vasculitis.
ESID-0206 A Report of Chronic Granulomatous Disease
J. Eguía-Núñez 1, G. Salgado-Cecilia1, H. Martínez-Banaclocha1, M.R. Moya-Quiles1, S. Alfayate-Miguélez2, A.I. Menasalvas-Ruiz2, A. Bernal-Ramos1, A. Minguela-Puras1, M.R. Álvarez-López1, A.?M. García-Alonso1
1Servicio de Inmunología, Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain
2Servicio de Aislados, Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain
Chronic granulomatous disease (CGD) is a primary immunodeficiency disorder of phagocytes resulting by severe recurrent bacterial and fungal infections. CGD is caused by mutations in genes that encode the NADPH oxidase, the enzyme that generates microbicidal oxygen radicals. This mechanism involves p22phox, p40phox, p47phox, p67phox, gp91phox and rac components.
We report a case of a 4 year-old boy from moroccan family with consanguineal parents who was hospitalized in 2010 by severe necrotizing pneumonia with radiologic abnormalities, including bilateral infiltrates, mediastinal lymph nodes and hepatosplenomegaly. He had a sister and two brothers; one of them suffered recurrent infections and died at the age of 21 months by meningitis and hepatic affectation.
In the flow cytometric immunological test we observed an interruption of oxidative capacity in neutrophils (PMN) measured by respiratory burst through oxidation of dihydrorhodamine (DHR) and release of superoxide anion through inhibition of reduction of ferrocytochrome c and not membrane expression of peptide gp91phox of the flavocytochrome b558 in PMN. At that time, we observed lymphocytosis and serum IgG and IgM were increased. Not others abnormalities in humoral and cellular immunity were detected. The DHR test showed that mother’s PMN were normal in 70% and gp91phox was expressed in 76% of mother’s PMN. We are doing the genetic studies for definite diagnosis and genetic counselling.
During the first year after diagnosis, the patient was treated with Interferon Gamma-1b, Cotrimoxazole and Itraconazole. Up to date, the patient only receives Cotrimoxazole and Itraconazole prophylaxis with positive development and without severe infections.
ESID-0220 Clinical and Immunological Features of Patients with Complement Deficiencies: A Single Centre Experience
F. GENEL 1, N. GULEZ1, L. SKATTUM2, L. TRUEDSSON2
1Department of Pediatrics, Dr. Behcet Uz Children's Hospital, Izmir, Turkey
2Clinical Immunology and Transfusion medicine, University and Regional Laboratories Region Skåne, Lund, Sweden
The complement system is a group of plasma and cell surface proteins all of which are needed for normal function of complement in innate and adaptive immunity. Complement deficiencies are associated with increased infection susceptibility and increased risk for autoimmune diseases. The aim of the study was to evaluate the clinical, immunological features and outcomes of patients with complement deficiencies. Sixteen patients from 9 families with complement deficiency diagnosed between 2003-2014?at Dr. Behcet Uz Children's Hospital were reviewed.
Age at diagnosis was ranged between 5-21 years. Six patients with C1 inhibitor deficiency, 5 patients with type 1 and 1 patient with type 2, presented with cutaneous swelling and/or abdominal pain. Three patients from a single family were diagnosed having properdin deficiency there meningococcal infection was seen in two siblings. Four patients from two families with factor I deficiency presented with recurrent respiratory tract infections and/or vasculitic eruptions. One of these patients also developed immune complex glomerulonephritis and meningitis. C8 deficiency was diagnosed in two siblings as a result of the analysis following meningococcal infection in one of them. One patient with C1q deficiency presented with recurrent pneumococcal meningitis and pneumonia.
Early recognition and diagnosis of complement deficiencies can facilitate prevention of life-threatening complications, such as upper airway obstruction in C1 inhibitor deficiency and also prevent severe bacterial infections such as meningitis by considering adequate clinical recommendations such as prophylactic antibiotics and vaccines.
ESID-0221 A Rare Cause of Bacterial Meningitis and Recurrent Pneumonia: Congenital Asplenia
S. BAHCECI ERDEM1, F. GENEL 1, E. OZBEK1, N. GULEZ1, B. ERDUR1
1Department of Pediatrics, Dr. Behcet Uz Children's Hospital, Izmir, Turkey
The absence of a spleen is a well-known risk factor for severe bacterial infections, especially due to encapsulated bacteria. Congenital asplenia can be part of multiple congenital abnormalities as in heterotaxy including Ivemark syndrome with congenital anomalies of the heart or great vessels or it can be isolated, which is extremely rare.
16 month old girl from a consanguineous family admitted with pulmonary infection and a history of a ventricular septal defect. Her medical history included pneumonia at the age of 5 months, urinary tract infection at the age of 8 months and bacterial meningitis at the age of 9 months. Past history revealed that her mother's three siblings were died of septicemia and meningitis.
Her total neutrophil and lymphocyte counts, lymphocyte phenotyping by flow cytometry, serum C3, C4 and IgA levels were normal. IgG and IgM levels were slightly decreased. Peripheral blood smears showed numerous Howell-Jolly-bodies. On abdominal ultrasound no spleen could be detected and a scintigraphic study supported the diagnosis of congenital asplenia.
The patient had been vaccinated against meningococci, pneumococci, and Haemophilus influenzae type B, and penicillin prophylaxis was administered. During a 18 month follow-up, she was asymptomatic for severe infections.
As a result early recognition and diagnosis of congenital asplenia can facilitate prevention of life-threatening infections by considering adequate clinical recommendations such as antibiotic prophylaxis and vaccination.
ESID-0292 Angioedema Due to C1 Inhibitor Deficiency: An Argentine Experience
A. GINACA 1, P. MARTINEZ1, A. GOMEZ RACCIO1, M. DOMINGUEZ1, L. BEZRODNIK1
1Immunology unit, Hospital de Niños Ricardo Gutierrez, Buenos Aires, Argentina
C1inhibidor deficiency (C1INHD) is characterized by recurrent episodes of skin-mucosal, non-painful, non-pruritic edema, affecting face, limbs, gastrointestinal tract and genitalia; laryngeal edema is the most severe complication. Hereditary form is an autosomal dominant disorder, although there are de novo mutations. The acquired one may be due to autoantibodies against C1INH or hyperactivation of classical complement pathway.
Aim: to describe clinical and laboratory findings in C1INHD patients
Patients(p): 13p with quantitative C1INHD. METHODS: classical complement pathway activity (CH50) by Kent-Fife; serum levels of C3 and C4 by nephelometry, C1INH and C1q by radial immunodiffusion. After diagnosis of index case , we studied the family.
Results: 12p from 8 families showed hereditary C1INHD: 8p were index cases and 4p were detected by family screening, 7 women, median age of symptoms onset : 30 months (range 12-120 ), median age at diagnosis: 120 months (range 67-468). Clinical manifestations : 11p symptomatic: 10p angioedema, 8p abdominal pain, 3p vomiting, 2p dehydration, 2p diarrhea, 1p figurative urticaria. 1p asymptomatic. All showed normal C3 and C1q levels. Mean C4=7 mg/dl (VN: 15-35), mean C1INH=51 mg/l (VN 196-382), 91% showed low CH50. One of them has no family history. 1p presented acquired C1INHD with urticaria, angioedema, C4 and C1q decreased.
Conclusions: Very low ??C4 with normal C3 are highly suggestive of C1INHD in patients with angioedema. Low C1q let us to differentiate hereditary from acquired deficiency. Onset of symptoms was observed at early age, arriving later at diagnosis. Family screening could prevent severe complications
ESID-0174 C1 Inhibitor Upregulates its Own MRNA Expression Level: Clue to Possible Autoregulatory Loop
L. Grodecká 1, T. Kovácová2, P. Lockerová3, B. Ravcuková3, J. Litzman1, T. Freiberger3
1Institute of Clinical Immunology and Allergology, Masaryk University - Faculty of Medicine, Brno, Czech Republic
2Molecular Immunology and Microbiology, Central European Institute of Technology - Masaryk University, Brno, Czech Republic
3Molecular Genetics Laboratory, Centre for Cardiovascular Surgery and Transplantation, Brno, Czech Republic
C1 inhibitor (C1inh) deficiency is a primary cause of type I and type II hereditary angioedema (HAE). Though HAE is transmitted as an autosomal dominant trait (generally only one allele of the SERPING1 gene is mutated), the C1inh level in type I patients' plasma is decreased to 5-30% of normal. Type II patients tend to have normal or elevated level of the protein. Surprisingly, the mRNA expression is decreased in both HAE types and the decrease was shown for both wild type and mutant alleles in most of the tested patients. Searching for an explanation, we found that the alpha-1-antitrypsin (an archetype of Serpin protein family) regulates its own mRNA expression by a positive autoregulatory loop. In order to test the possibility of similar regulation of C1inh expression, we measured the change of SERPING1 mRNA level in HepG2 cell cultures upon addition of C1inh protein to culture media. As C1inh we used HAE therapeutic Berinert P (CSL Behring). We performed thorough RT-qPCR experiments using three housekeeping genes at the same time. These results showed an upregulation of SERPING1 mRNA by 21% (p = 0.008, Mann-Whitney test). The only possible protein contamination known to the Berinert P manufacturer was beta-haptoglobin which we used as one of the controls. However, its addition to the media did not show any significant effect on the SERPING1 expression. Our results suggest that C1inh expression could be regulated by a positive autoregulatory loop, what might explain its specificities seen in HAE patients.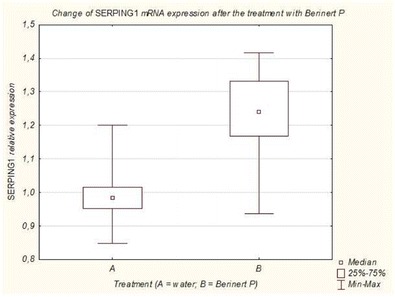
ESID-0750 Absent TLR-Mediated CD62L Shedding in a 62-Year Old Man
M. Guckian 1, I. MacLennan2, M. Drayson2, T.K. Krishna3, L. Diwakar3, M. Cobbold2, A. Huissoon3
1Immunology, Heartlands Hospital, Birmingham, United Kingdom
2Clinical Immunology, Medical School, Birmingham, United Kingdom
3Clinical Immunology, Heartlands Hospital, Birmingham, United Kingdom
Interleukin-1 receptor-associated kinase 4 (IRAK4) deficiency and myeloid differentiating factor 88 (MyD88) deficiency syndromes are monogenic, autosomal recessive primary immune-deficiency disorders resulting in innate immune defects. They are typically diagnosed in children with recurrent pneumococcal and other deep-seated infections, and susceptibility to infection appears to decrease during teenage years.
We describe a 65-year-old man, investigated for an uncharacterized immunodeficiency, documented over a 53-year period. His family history was unremarkable, with no reported parental consanguinity. He initially presented aged 12 with staphylococcal septic arthritis. Subsequently he suffered lobar pneumonia (14), spinal abscess (15), cerebellar abscess (36) and streptococcal meningitis (aged 54 years) and further staphylococcal arthritis episodes. No mycobacterial infections were reported. Patient had remained well on prophylactic amoxicillin for 11 years.
Full blood and white cell differential counts, lymphocyte subsets, IgG, IgA and complement function were normal. Serum IgM was consistently raised, but CD154 expression was normal. Protein and conjugate vaccine responses were normal, with reduced Pneumococcal vaccine responses. No Howell-Jolly bodies were detected, and type 1 cytokine analysis showed only a partial reduction in IL-12 production (7% of normal control). However CD62L shedding in response to lipopolysaccharide stimulation was absent.
Antibiotics and pneumococcal vaccination have prevented further serious invasive infections. If confirmed, this will be the oldest index patient newly-diagnosed with a TLR-related defect. IRAK-4/MyD88 deficiency should be considered in adults with a strong history of unusually severe or recurrent infection. CD62L shedding is a simple and robust screening test.
ESID-0520 Identification and Molecular Characterization of Two New Mutations Causing C5 Deficiency in Three Un-Related North-African Families
R. Colobran 1, C. Franco-Jarava1, A. Martin-Nalda2, L. Minguell2, N. Baena3, J.F. Delgado4, R. Pujol-Borrell1, P. Soler-Palacin2, M. Hernandez-Gonzalez1
1Immunology, Vall d'Hebron University Hospital, Barcelona, Spain
2Pediatric Infectious Diseases and Immunodeficiencies Unit, Vall d'Hebron University Hospital, Barcelona, Spain
3Laboratory of Genetics, Corporació Sanitària i Universitària Parc Taulí, Sabadell, Spain
4Immunology, Corporació Sanitària i Universitària Parc Taulí, Sabadell, Spain
Complement[S1] C5 deficiency (C5D) is a rare autosomal recessive defect associated to recurrent Neisseria spp. infections. We report the cases of three unrelated patients of North African origin, (P1), (P2) and (P3) who had suffered of septicemia by uncommon serotypes (E29 and Y[S2] ) of Neisseria meningitidis. C3 and C4 were normal but activation through the classical (CH50) and the alternative pathways (AP50) was reduced. In addition, neither C5 nor its fragment sC5b9 could be detected. Haemolytic activity was restored by the addition of purified C5 to patients’ sera. Sequencing of C5 showed that P1 carried a homozygous deletion of 2 nucleotides in exon 21 (c.2607_2608delAA) leading to a frameshift and a truncated protein of 872 aa (compared to normal 1676 aa reference sequence) and P2 and P3 carried a homozygous deletion of 3 nucleotides in exon 9 (c.960_962delCAA) leading to deletion of one aa (Asn320). All mutations were novel, and in all families the parents were identified as heterozygous carriers. These results illustrate the molecular heterogeneity of this disorder. Systematic studies will be required to establish the real prevalence of this deficiency in the different populations including this little studied African group. As with other immunodeficiencies, the analysis of new cases and genotype phenotype correlations can contribute to understand better the role of complement in protection from infection.
ESID-0381 Dyskeratosis Congenita: A Rare Syndromic Cause of Haemophagocytic Lymphohistiocytosis (HLH)
L. Hinds 1, F. Shackley1, H. Clements2, B. Tesi3, S. Chiang3, Y. Bryceson3, T. Flood4, C. Waruiru1
1Department of Immunology & Infectious Diseases, Sheffield Children's Hospital NHS Foundation Trust, Sheffield, United Kingdom
2Department of Paediatrics, Sherwood Forest Hospitals NHS Foundation Trust, Mansfield, United Kingdom
3Department of Women's & Children's Health, Karolinska Institutet, Stockholm, Sweden
4Department of Immunology, Great North Children's Hospital, Newcastle, United Kingdom
Haemophagocytic lymphohistiocytosis may be a presenting feature of rare inherited syndromes associated with primary immunodeficiency. We present a case of Dyskeratosis Congenita, a diagnosis established following detailed investigation of an episode of EBV-driven HLH.
A 4-year-old boy presented with prolonged fever, lymphadenopathy, vasculitic rash, hepatosplenomegaly, deranged liver function and clotting. He fulfilled the criteria of HLH, with pancytopaenia, elevated ferritin, hypertriglyceridaemia and low fibrinogen. Haemophagocytosis was not detected in a bone marrow aspirate; lymph node histology was normal. He had evidence of primary EBV infection (peak VL 869,030 copies/?ml). The HLH process aborted spontaneously.
Predating this was a history of life-threatening neonatal sepsis with pancytopaenia, recurrent chest infections and unexplained enteropathy. At 7 years of age, despite appropriate antibiotics, he developed extensive bronchiectasis, and commenced immunoglobulin supplementation. He was small in stature (height 0.4th centile), with fine hair, peg-shaped teeth, nail dystrophy, splenomegaly, but no significant learning difficulties.
He had persistently low natural killer (NK) cells in peripheral blood, normal IgG, low IgA, low IgM and unsustained specific antibody responses. Assays excluded XLP, XIAP, familial HLH mutations, ALPS and Schwachmann-Diamond syndrome. His cytotoxic lymphocyte activity showed pathologically low NK cell-mediated cytotoxicity. Further genetic analysis revealed a hemizygous mutation (c.838A>C;p.S280R) in DKC1, associated with an X-linked form of Dyskeratosis Congenita.
While the association between Dyskeratosis Congenita and bone marrow failure is well-known, the link with HLH has not been described. Treatment depends on the degree of bone marrow failure and this patient is under consideration for haematopoietic stem cell transplant.
ESID-0572 Nasal Epithelial Microrna Expression in Infants with Respiratory Syncytial Virus Disease - MIR-125A and MIR-429 Differentially Expressed According to Disease Severity
C. Inchley 1, T. Sonerud1, H.O. Fjærli1, B. Nakstad1
1Department of Pediatric and Adolescent Medicine, Akershus University Hospital, Lørenskog, Norway
Introduction
Most children are exposed to respiratory syncytial virus (RSV) during the first two years of life, with mild upper airways disease the most common presentation. 2 – 3% are admitted to hospital with lower airways infection, some requiring oxygen, extra fluids or mechanical ventilation. We have investigated the expression microRNA, emergent regulators of the immune system, in the nasal epithelium of children with RSV infection to identify differences between those with mild, and those with severe disease.
Methods
Nasal cytology specimens were collected from RSV positive children and healthy controls under 12 months of age. RSV positive children were grouped into mild, moderate and severe disease severity groups. A microRNA microarray identified candidate differentially expressed microRNA. 32 microRNA underwent qPCR verification. Expression profiles were compared between groups using one-way ANOVA with post-hoc testing.
Results
In the final qPCR analysis there were 19 control, 16 mild, 9 moderate and 19 severe disease samples. Compared to control, miR-125a and miR-429 were downregulated in mild disease (p = 0.03 and 0.02, respectively), and in moderate disease (p = 0.02 and 0.09, respectively), but not in severe disease (p = 0.3 and 0.3, respectively).
Discussion
We have profiled microRNA expression in nasopharyngeal cytology brushings of RSV-positive infants. miR-125a has important functions within innate immunity and macrophage function. The lack of downregulation of miR-125a and miR-429 in severe disease may help explain differences in disease phenotypes on infection with RSV, and the role of these microRNA in respiratory virus infection should be further investigated.
ESID-0492 Chronic Granulomatous Disease in Morocco
L. Ait Baba1, F. Ailal 2, N. El Hafidi3, M. Hubeau4, F. Jabot-Hanin4, N. Benajiba5, Z. Aadam1, F. Conti4, C. Deswarte4, L. Jeddane2, A. Aglaguel6, O. El Maataoui7, J. El Bakkouri7, J. Najib2, R. Martinez-Barricarte8, L. Abel8, N. Habti9, R. Saile1, J.L. Casanova8, J. Bustamente4, A.A. Bousfiha2, H. Salih Alj1
1Laboratory of Biology and Health URAC34—Metabolic and Immunologic pathology Research Team, Faculty of Science of Ben M'sik King Hassan II University, Casablanca, Morocco
2Clinical Immunology Unit, Faculty of Medicine and Pharmacy, Casablanca, Morocco
3Children Hopsital, Avicennes University Hospital, Rabat, Morocco
4Laboratory of Human Genetics of Infectious Diseases Necker Branch, Institut National de la Santé et de la Recherche Médicale U980 Imagine institute, Paris, France
5Department of Pediatrics, Al Farabi Hospital Mohamed VI University Hospital, Oujda, Morocco
6Immunology Laboratory, Faculty of Science and Technology Hassan II University - Mohammedia, Mohammedia, Morocco
7Laboratory of Immunology, Averroes University Hospital, Casablanca, Morocco
8St. Giles Laboratory of Human Genetics of Infectious Diseases Rockefeller Branch, The Rockefeller University, New-York, USA
9Laboratory of Genetics and Cellular Enegineering, Faculty of Medicine and Pharmacy, Casablanca, Morocco
Purpose Chronic granulomatous disease (CGD) is characterized by an inability of phagocytes to produce reactive oxygen species (ROS. CGD patients are known to suffer from recurrent bacterial and/or fungal infections from the first year of life onwards. From 2009 to 2013, 12 cases of CGD were diagnosed in Morocco.We describe here these Moroccan cases of CGD.
Methods We investigated the genetic, immunological and clinical features of 12 Moroccan patients with CGD from 10 unrelated kindreds.
Results All patients were children suffering from recurrent bacterial and/or fungal infections. All cases displayed impaired NADPH oxidase activity in nitroblue tetrazolium (NBT), dihydrorhodamine (DHR) or 2',7' dichlorofluorescein diacetate (DCFH-DA) assays. Mutation analysis revealed the presence of four different CYBB mutations in four kindreds, a recurrent NCF1 mutation in three kindreds, and a new NCF2 mutation in one kindred. A large deletion of CYBB gene has been detected in a patient. The causal mutation in the remaining one kindred was not identified.
Conclusion The clinical features and infectious agents found in these patients were similar to those in CGD patients from elsewhere. The results of mutation analysis revealed a high level of genetic and allelic heterogeneity among Moroccan CGD patients. The small number of patients in our cohort probably reflects a lack of awareness of physicians. Further studies on a large cohort are required to determine the incidence and prevalence of the disease, and to improve the description of the genetic and clinical features of CGD patients in Morocco.
ESID-0488 Poikiloderma with Neutropenia, Clericuzio Type: About 4 Cases
A. Aglaguel1, J. El Bakkouri2, S. Sekhraoui3, L. Jeddane4, N. Habti5, H. Abdelghaffar1, A.A. Bousfiha4, F. Ailal 4
1Immunology Laboratory, Faculty of Science and Technology Hassan II University - Mohammedia, Mohammedia, Morocco
2Laboratory of Immunology, Averroes University Hospital, Casablanca, Morocco
3Department of Pediatrics, Mohamed V Hospital, El Jadida, Morocco
4Clinical Immunology Unit, Faculty of Medicine and Pharmacy, Casablanca, Morocco
5Laboratory of Genetics and Cellular Enegineering, Faculty of Medicine and Pharmacy, Casablanca, Morocco
Introduction: Poikiloderma with neutropenia (PN) is a rare autosomal recessive disease. It is expressed by early poikiloderma, hyperkeratosis of the extremities, short stature, nail dystrophy and recurrent viral or bacterial infections.
Observations: We report 4 cases, born from consanguineous parents. Skin dermatitis began in the first year of life showing generalized skin dyschromia, sclero-atrophy, cheeks hyperemia and telangiectasia with photosensitivity, palmoplantar hyperkeratosis and nail involvement. They also presented recurrent respiratory infections. Physical examination showed a failure to thrive and facial dysmorphism. Laboratory analyses revealed isolated neutropenia in 3 patients, and pancytopenia in a patient with normal myelogram.
Discussion: Approximately 36 cases of PN were identified in the literature, including 14 Indians described by Clericuzio . They presented early poikiloderma , non-cyclic neutropenia and / or neutrophil dysfunction , abnormal nails, palmoplantar hyperkeratosis , recurrent infections , failure to thrive and facial dysmorphism . Parental consanguinity and familial cases have been reported. A mutation in the C16orf57 gene has recently been reported.
Conclusion: The Clericuzio syndrome is a rare autosomal recessive disease that combines genodermatosis and neutropenia . The risk for squamous cell carcinoma was reported.
ESID-0700 Chronic Granulomatous Disease Revealed by BCGITIS
H. Ouair1, M. Kourime2, L. Jeddane 2, J. Najib2, A.A. Bousfiha2, F. Ailal2, H. Salih Alj1
1Laboratory of Biology and Health URAC34—Metabolic and Immunologic pathology Research Team, Faculty of Science of Ben M'sik King Hassan II University, Casablanca, Morocco
2Clinical Immunology Unit, Faculty of Medicine and Pharmacy, Casablanca, Morocco
Chronic granulomatous disease (CGD) is a primary immunodeficiency due to defect in neutrophil oxidative metabolism ; which is characterized by severe and recurrent bacterial and fungal infections and by the formation of granulomas. The mycobacterial infections are rarely reported in this immune deficiency and are most often associated with the BCG vaccination.
We report an infant of 10 months-old showing isolated BCGitis with reactivation of BCG and huge axillary adenopathy. The diagnosis of CGD was done later following the onset of staphylococcal cervical lymphadenopathy. This is a gp91 phox deficiency due to CYBB mutation (c.897 + 1G > T). Evolution is good on antibacillary, antibacterial and antifungal prophylaxis.
The association of mycobacterial infections with CGD has been neglected in countries where exposure to mycobacteria is low. CGD should be considered in children with BCGitis and BCGosis , and in some patients with severe tuberculosis, even in the absence of typical infections of the CGD , which may occur thereafter. Similarly, BCG vaccination should be formally contraindicated in patients with diagnosed or suspected CGD and their siblings before exclusion of CGD diagnosis.
ESID-0712 Family with Hereditary Angioedema Type I Caused by Novel Mutation
M. Jesenak 1, P. Banovcin1, T. Freiberger2
1Centre for Hereditary Angioedema Department of Pediatrics, Comenius University in Bratislava Jessenius Faculty of Medicine in Martin, Martin, Slovakia
2Genetic laboratory, Centre of Cardiovascular and Transplantation Surgery, Brno, Czech Republic
Hereditary angioedema (HAE) is a rare, life-threatening disorder caused by C1-inhibitor (C1-INH). Three subtypes of HAE have been described. HAE type I is characterized by decreased concentration and function of C1-INH, whereas in HAE type II the concentration can be within normal range but the protein is non-functional.
We report a family with 11 members affected by HAE. Four males died before determination of HAE diagnosis due to laryngeal oedema. In the rest of the living family members (3 males and 4 females), the dominant location of angioedema was gastrointestinal tract. Laboratory examination revealed significant decline of C4, C1-INH concentration and function. Molecular genetic analysis was also performed. Mutation c.1371_1373delTGC, p.(Ala459del) in exon 8 of the SERPING1 gene was detected using PCR and sequencing. No other mutation was found in coding regions and adjacent intronic sequences of the gene, including large gene rearrangements analysed by fluorescent multiplex PCR. This frameshift deletion has not been described so far, however, there are several factors in favour of presumption that it is rather a pathogenic variant. Missense mutations in neighbouring codons leading to HAE (II) phenotype have been published, all missense variants in codon 459 are predicted as damaging using SIFT and Polyphen2 prediction tools and mutation detected in our family segregates clearly with a clinical and laboratory phenotype. Based on the laboratory findings, this mutation can be connected with the typical laboratory pattern of HAE type I although it is close to the typical region of HAE type II.
ESID-0243 Aorto-Oesophageal Fistula in a Patient with Chronic Mucocutaneous Candidiasis (CMC) and a Novel STAT1 Mutation
M. Ponsford 1, A. Roberts1, T. El-Shanawany1, R. Doffinger2, J. Thaventhiran3, E. Carne1, S. Jolles1
1University Hospital of Wales, Immunodeficiency Centre for Wales, Cardiff, United Kingdom
2Addenbrooke's Hospital, Department of Clinical Biochemistry & Immunology, Cambridge, United Kingdom
3Addenbrooke's Hospital, CIMR Medicine, Cambridge, United Kingdom
Background
Chronic mucocutaneous candidiasis (CMC) is characterised by candida infections of the skin, nails and mucous membranes. Mutations in STAT1 have been identified in autosomal dominant CMC and are associated with defective Th1 and Th17 responses. We describe a 48?yr old male with a novel mutation in the coiled coil domain of STAT1 c.846A>T who had childhood bronchiectasis, chronic mucosal candidiasis and presented with haematemesis and malaena. Endoscopy identified an aorto-oesophageal fistula (AOF) with associated thoracic aorta pseudo-aneurysm on computed-tomography (CT) scanning. This was on a background of a requirement for repeated oesophageal balloon dilatation for refractory benign oesophageal stricture that had shown only a limited response despite use of a metal stent in 2012 following a localised oesophageal perforation.
Methods
STAT1 sequencing was established and identified the c.846A>T mutation in both the patient and his son. Endoscopy, CT scan and white cell scan were undertaken. Functional studies of the effect of the novel mutation are ongoing.
Results
The imaging studies reveal probable fungal aortitis involving the aortic arch complicating the urgent endovascular thoracic aortic stent repair (Figure?1). The patient was switched from oral itraconazole to intravenous caspofungin with reduction in inflammatory markers, however fungal clearance of the infected stent and graft even with prolonged intravenous antifungal therapy remains challenging.
Conclusion
The development of an aorto-oesophageal fistula in combination with imaging evidence for probable fungal oesophagitis and aortitis demonstrate the risks of developing life threatening complications despite anti-fungal prophylaxis in CMC.
ESID-0044 BCG-OSIS Causing Excessive Hyperferritinemia in a Patient with NEMO Mutation
N.E. Karaca 1, E. Ulusoy1, G. Aksu1, J. Bustamante2, J. Feinberg2, J.L. Casanova2, N. Kutukculer1
1Department of Pediatrics, Ege University Faculty of Medicine, Izmir, Turkey
2Laboratoire de Génétique Humaine des Maladies Infectieuses INSERM U980, Faculté de Médecine Necker, Paris, France
Background: X-linked hypomorphic mutations of nuclear factor-?B essential modulator (NEMO) result in a wide range of clinical phenotypes, including anhidrotic/hypohidrotic ectodermal dysplasia with immunodeficiency and mendelian susceptibility to mycobacterial disease (MSMD).
Observations: 2-year-old male, born to non-consanguineous parents, presented with fever, generalized lymphadenopathies, hepatosplenomegaly, elevated acute phase reactants (APR) and hypergammaglobulinemia at 8-months of age. Mycobacterium bovis was isolated from lymph node and liver biopsies. Lymphocyte subsets and phagoburst tests were normal, ruling-out severe combined immunodeficiency and chronic granulomatous disease. He was suspected to have a defect in IL12/?IFN-? pathway. Treatment with recombinant IFN-?, isoniaside, chlaritromycine and rifabutine were started. During follow-up, functional and genetic analysis of IL-12Rß1 and IFN-?R were found to be normal; lymphoproliferation and hepatosplenomegaly regressed, whereas CRP and ESR remained high, with increasing ferritin levels. At 2-years-of age, he admitted with darkening color of the skin, coarse hair and hepatosplenomegaly. Laboratory examinations revealed anemia, low IgG/high IgM, elevated APR and extremely high ferritin (14300 µg/?L) levels. Bone marrow biopsy excluded hemophagocytic-lymphohistiocytosis. Hepatic biopsy excluded iron deposition in the liver, but it revealed M.?bovis-associated-granulomatous hepatitis. Genetic analysis showed a defect (c.74_77delACGT) in exon 2 of NEMO gene. Skin biopsy confirmed the presence of sweat glands. Despite roboust anti-mycobacterial treatment, he deceased due to disseminated mycobacterial infection leading to multi-organ failure.
Conclusion: It must be kept in mind that patients with NEMO defect may present without ectodermal dysplasia, and high ferritin, which is a marker of systhemic inflammation, must suggest hepatic mycobacterial disease, especially in patients with MSMD.
ESID-0548 A Mouse Model of X-Linked Neutropenia
M. Keszei 1, M. Baptista1, L. Westerberg1
1Department of Microbiology Tumor and Cell biology, Karolinska Institutet, Stockholm, Sweden
Wiskott-Aldrich Syndrome Protein (WASP) is a key actin cytoskeleton regulator in hematopoietic cells. WASP mutations manifest in a spectrum of X-linked immunodeficiencies. While loss-of-function mutations in WASP cause the more common Wiskott-Aldrich syndrome(WAS) and X-linked thrombocytopenia(XLT), gain-of-function mutations in the autoregulatory GTPase binding domain of WASP cause a rare form of severe congenital neutropenia(SCN). Although a plethora of neutrophil deficiencies originate from impaired actin dynamics, the deep understanding of the etiology and outcome of these diseases are often limited by the few patients identified. High homology between the human and murine WASP provided us with a unique opportunity to examine the WASP mediated X-linked neutropenia(XLN) in a knock-in mouse model. Murine XLN WASP(I296T) exhibits the molecular features of human XLN WASP mutant proteins: such as increased susceptibility to proteases and increased ability to induce actin polymerization. More importantly, murine XLN neutrophils exhibit altered villi structure, surface expression of Integrin alpha M, and defective reactive oxygen production, which indicates impaired neutrophil function. Similarly to other murine models of neutropenias (such as ELA2,HAX1, or AP3B1) XLN mice do not exhibit severe congenital neutropenia without induction, which highlights differences between mice a human myelopoiesis. The XLN murine model suggests that an intricate interplay between WASP and its interacting small Rho-GTPases, Cdc42 and Rac, governs key elements of neutrophil function. Our investigation will provide extensive insight into the pathomechanism of XLN and it will contribute to identify key therapeutic targets to treat XLN and other neutrophil defects.
ESID-0244 LPS-Modified Outer Membrane Vesicles (fmOMVs) Provide Protective Innate Immunity Against Influenza Infection Prior to the Induction of Vaccine-Induced Adaptive Immune Responses
D. Kim 1, E. Bae1, T. Lee1, C. Kim1, S. Lee1, M. Yeom1, W. Na1, D. Song1, S. Kim1
1Viral Infectious Disease Research Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon, Korea
Influenza has been a serious health problem due to the high mutation and transmission rates as well as the pathogenicity of the influenza viruses. Since the conventional vaccines are type-specific and require several weeks for the induction of protective immunity, novel antiviral agents are needed to overcome these limits of the vaccines. In this study, we evaluated the antiviral potential of LPS-modified outer membrane vesicles (fmOMVs), which are natural bacterial vesicles containing diverse innate immune stimulators, using a murine influenza model. While the conventional vaccine exhibited subtype or strain-specific efficacy, pretreatment of fmOMV conferred rapid protection against various subtypes of influenza viruses. Notably, coadministration of fmOMV with the influenza antigens significantly increased the antigen-specific immune response, and provided protective immunity before the induction of the vaccine-induced antibody response. These results demonstrate that fmOMV is an active and immediate immunomodulator capable of controlling influenza virus infection, as well as an adjuvant for the enhancement of adaptive immunity.
ESID-0289 G6PC3 Deficiency: A Primary Immune Deficiency Beyond Just Neutropenia
A. KIYKIM 1, S. BARIS1, A.O. ÖZEN1, E. KARAKOÇ-AYDINER1, D. ÇIÇEKKÖKÜ1, J. SAWALLE-BELOHRADSKY2, K. BOZTUG3, C. KLEIN2, I.B. BARLAN1
1Pediatric Allergy and Immunology, Marmara University Pendik Training and Research Hospital, Istanbul, Turkey
2Gene Center, Dr. von Hauner Children's Hospital Ludwig-Maximilians-University, Munich, Germany
3Department of Paediatrics and Adolescent Medicine, Center for Congenital Immunodeficiencies (CCID) Medical University of Vienna, Vienna, Austria
Glucose 6 phosphatase catalytic subunit 3 (G6PC3) deficiency was recently defined as a new SCN (severe congenital neutropenia) subgroup remarkable with congenital heart defects, urogenital malformations, endocrine abnormalities and prominent superficial veins. Here, we report 3 patients with G6PC3 deficiency presenting with recurrent diarrhea, failure to thrive and sinopulmonary infections leading to bronchiectasis. In PI and II, a combined immune deficiency was suspected due to early onset disease with lymphopenia, neutropenia and thrombocytopenia, variable reductions in lymphocyte subpopulations and favorable response to IVIG therapy. In these patients neutropenia was evident from infancy, whereas it was not recognized until 13 years of age in PIII. Apart from the neutropenia, all 3 patients had intermittent thrombocytopenia, anemia and lymphopenia (persistently low in PI and II, in the lower range of normal in PIII). All three patients had failure to thrive and some of the classical syndromic features of G6PC3 deficiency; cardiac abnormalities and visibility of superficial veins in all, endocrinologic problems in PI and PIII and urogenital abnormalities in PII. Flow cytometric examination revealed diminished numbers of CD4+CD31+CD45RA+ thymic naive cells in the peripheral blood suggesting low thymic output. Our experience suggests that a diagnosis of congenital neutropenia may not be as straightforward in such patients with combined lymphopenia, neutropenia and thrombocytopenia. A high index of suspicion and the other syndromic features of G6PC3 were clues to diagnosis of those cases. Screening of all CID with neutropenia may help to uncover the whole spectra of deficiency of G6PC3.
ESID-0741 High IL-6 Yet Never Elevated CRP in Two Newborns - Incidentally Found Inhibitory and Precepetating Neonatal IL-6 Autoantibodies
U. Koelsch 1, S. Heller2, D. Hüsemann3, N. Unterwalder1, M. Bauer2, C. Meisel1, M. Nauck4, H.U. Thomalla4, H. Bernuth von2, C. Bührer5
1Immunology, Labor Berlin – Charité-Vivantes, Berlin, Germany
2Paediatric Pneumology and Immunology, Charité Children's Hospital, Berlin, Germany
3Children Clinic, Werner-Forßmann Clinics, Eberswalde, Germany
4Children Clinic, J.-Kentman Clinics, Torgau, Germany
5Neonatology, Charité Children's Hospital, Berlin, Germany
CRP and IL-6 were assessed in a preterm (35+4) (N1) and a term newborn (N2) due to preterm rupture of membranes (>18?h pre delivery). In both, immediately post delivery IL-6-levels were elevated (2921 pg/ml (N1) and 360 pg/ml (N2)) but CrP levels were normal. Follow up CrP levels in both newborns were always normal but IL-6-levels persisted high at discharge after 12 and 10 days respectively (900 pg/ml (N1) and 592 pg/ml (N2)). Antibiotic treatment was stopped after CrP-levels were found to have never increased after 7 (N1) and 4 days (N2). Interestingly in both neonates during a course of a Rotavirus-infection at day 44 and 8 of life again IL-6 levels increased (2522 pg/ml (N1), 607 pg/ml (N2)) despite missing concurrent clinical symptoms or CrP-increase.
Serum of both neonates blocked IL-6-dependent STAT3-phosphorylation, indicating inhibitory antibodies. Elevated IL-6-levels without infection and inhibitory IL-6-antibodies were also found persisting in both mothers (70 pg/ml (N1) and 59,7 pg/ml (N2)). IL-6-Antibodies were not detectable anymore after 6 months in both infants, whereas they persisted in the mothers. We therefore conclude the here characterized antibodies preventing IL-6-degradation and signalling being maternal auto-antibodies.
IL-6 is a key cytokine for acute-phase-responses that is broadly considered as very early sign of infection in neonates. We cannot distinguish whether the here observed phenomenon of highly elevated IL-6 yet no elevated CrP was triggered by an infection or by perinatal stress. Since IL-6auto-antibodies may occur as frequent as 1:1000 in healthy adults increased IL-6-levels without subsequent adequate rise of CrP in newborns may also occur incidentally.
ESID-0357 WASp Regulates NK Cell Killing of Tumor Cells
J. Kritikos 1, C. Dahlberg1, M. Baptista1, A.K. Wagner1, J. Orange2, H. Brauner1, L. Westerberg1
1Microbiology Tumor and Cell Biology, Karolinska institutet, Stockholm, Sweden
2Texas Children's Hospital, Baylor College of Medicine, Houston TX, USA
NK cells play a key role in tumor surveillance by forming an immunological synapse with the target cells. The Wiskott-Aldrich syndrome protein (WASp) coordinates receptor signaling to changes in the actin cytoskeleton and regulates the dynamic assembly of the immunological synapse. Loss-of-function mutations in WASp result in the severe primary immunodeficiency Wiskott-Aldrich syndrome with a high incidence of tumors. We have investigated how WASp deficiency affects NK cell maturation and function in tumor surveillance. We found that WASp-deficient NK cells have a reduced capacity to kill MHC class I negative cells ?in?vivo and this is associated with more rapid growth of a B16 melanoma tumor cell line in WASp deficient mice. To determine the cause of decreased killing ?in?vivo, we examined maturation and functionality of WASp-deficient NK cells in?vitro. We found a decreased proportion of mature CD11b+CD27- NK cells in WASp-deficient mice as well as increased proportion of CD69+ and KLRG1+ NK cells, suggesting altered threshold for activation. We also observed a decreased formation of synapses between WASP-deficient NK cells and YAC-1 target cells, compared to WT NK cells, along with less actin polarization towards the synapse. Wildtype NK cells activated by YAC-1 cells or through stimulation via the activating receptor NKp46 showed degranulation of CD107+ granules and IFNgamma production. In contrast, WASp-deficient NK cells showed markedly reduced degranulation and IFNgamma production. This degranulation defect could be completely rescued with IL-2 stimulation. Together, our results suggest that WASp is required for NK cell-mediated killing of tumor cells.
ESID-0106 Innate Immunity in CVID: Neutrophil Migration and Phagoburst Activity, Adhesion Molecules (CD11A, CD18), Natural Killer Cell Functions and NKT Cells
N. Kutukculer 1, E. Azarsiz1, N.E. Karaca1, G. Koturoglu1, G. Aksu1
1Department of Pediatrics, Ege University Faculty of Medicine, Izmir, Turkey
Defective adaptive immune responses, with impairment of T/?B cell maturation and activation, are common features in common variable immune deficiency patients (CVID). However, recently attention has been focused on innate immune system defects as a possible explanation for CVID heterogeneity.
Functional steps required for the activity of neutrophils and monocytes include migration (Migratest), and phagocytosis and killing of the target (Phagoburst test). Integrins CD11a/CD18 (LFA-1) which are found on leucocytes help leucocyte transmigration to inflammation area. In addition to above parameters, CD3-CD16+CD56+ (NK cells), Natural killer cell cytotoxicity, CD3-CD16+CD56+28+ cells, CD3-CD16+CD56+28- cells, natural killer T cells CD3+CD16+CD56+ (NKT) were evaluated in 20 CVID patients and 26 healthy controls.
Migratest (% of activated granulocytes) was found to be significantly low in CVID patients compared to controls although no significant difference was observed in percentages of phagocytic cells which produce reactive oxidants. Neutrophils and lymphocytes expressing CD11a were both significantly decreased in CVID patients. No difference was found for the evaluation of CD18+ cells in both groups.
In CVID group, CD28+ NK cells were significantly high while CD28- NK cells were significantly low compared to healthy controls. No significant difference was found in terms of NKT cells and CTLA-4+ cells between patient and control groups.
In conclusion; we tried to elucidate the contribution of innate immunity to the physiopathology of CVID and defects in migration and adhesion capacity of phagocytic cells were determined. In contrast, CD28+ NK cells were highly increased in CVID patients probably for compensating the above innate immunity defect.
ESID-0595 A New Homozygous Mutation in Complement Factor D Associated with Meningococcal Septicaemia and Absent Haemolytic Function of the Alternative Complement Pathway
S. Lear 1, J. Harvey2, D. Kumararatne3, J. Thaventhiran3
1Immunology, Cambridge University Hospital NHS Trust, cambridge, United Kingdom
2Immunology, Royal Free Hospital NHS Trust, London, United Kingdom
3Immunology, Cambridge University Hospital NHS Trust, Cambridge, United Kingdom
In 2012 we reported the case of a patient presenting with meningococcal septicaemia who was found to have a defect in the alternate complement pathway. Although levels of Factor D in the serum were normal as measured by ELISA, the lytic capacity as measured by the alternate complement pathway function (AP50) was absent. This defect was corrected with the addition of exogenous factor D, but not Factor B or properdin.
Further investigations have identified a novel mutation in Factor D. The patient and her brother are homozygous for this mutation and both parents are heterozygote carriers. The brother is clinically asymptomatic but analysis of the alternate complement pathway (AP50 test) shows a similar result to the index case.
We have generated mutant and wild-type-His tagged Factor D. The results of their assessment in enzymatic assays of catalytic function will be presented.
ESID-0497 Expression Pattern of NK Cell-Activating Receptors on NK Cells, NKT Cells and T Lymphocytes in Children with Severe and/or Recurrent Infection with Herpes Viruses
M. Lenart 1, A. Szaflarska2, M. Rutkowska1, A. Pituch-Noworolska1, M. Siedlar1
1Department of Clinical Immunology, Jagiellonian University Medical College Polish-American Institute of Pediatrics, Krakow, Poland
2Department of Clinical Immunology, University Children's Hospital, Krakow, Poland
The pathomechanism of severe and recurrent infections with Herpes viruses is still largely unknown. Antiviral response of NK cells, but also T lymphocytes and natural killer T (NKT) lymphocytes, is highly dependent on appropriate function of activating receptors, such as natural cytotoxicity receptors (NKp30, NKp44 and NKp46), C-type lectin-like receptors (NKp80 and NKG2D), Fc gamma receptors (i.?e. CD16), and signalling lymphocyte activation molecule (SLAM) receptors (i.?e. CD150, CD48, 2B4). The aim of this study was to analyse expression pattern of these receptors on NK cells, NKT cells, T lymphocytes, and their subsets, in children suffering from severe and/or recurrent infections with Herpes viruses: Herpes simplex virus (HSV), Epstein-Barr virus, and Varicella zoster virus; and age-matched healthy controls. In comparison to healthy controls, children with Herpes viral infections had significantly decreased expression of NKG2D receptor (2-fold difference), on CD16brightCD56dim and CD16dimCD56bright NK cells, in case of both percentage of cells expressing each receptor and the expression level measured as median fluorescent intensity (MFI). MFI of NKG2D receptor was also decreased in these patients on CD8+ NKT and CD8+ T lymphocytes. The patients had also significantly downregulated expression (MFI) of CD16, NKp46 and 2B4 receptors (2-fold difference for each receptor) on both subsets of NK cells. The differences of expression of these receptors were observed particularly in patients with HSV infections. Our results suggest that NKG2D, CD16, NKp46 and 2B4 receptors on cell populations engaged in antiviral response may play role in pathophysiology of severe and recurrent Herpes viral infections.
ESID-0225 Inflammatory Manifestations in a Single-Center Cohort of Patients with Chronic Granulomatous Disease
A. Magnani 1, P. Brosselin2, J. Beauté3, N. de Vergnes2, R. Mouy4, M. Debré4, F. Suarez5, O. Hermine5, O. Lortholary6, S. Blanche7, A. Fischer7, N. Mahlaoui7
1Département de Biothérapie Centre de Référence des Déficits Immunitaires Héréditaires CEREDIH Institut Imagine, Hopital Necker Enfants Malades, Paris, France
2Centre de Référence des Déficits Immunitaires Héréditaires CEREDIH Institut Imagine, Hopital Necker Enfants Malades, Paris, France
3Centre de Référence des Déficits Immunitaires Héréditaires CEREDIH, Hopital Necker Enfants Malades, Paris, France
4Service d'Immunohématologie et Rhumatologie Pédiatrique, Hopital Necker Enfants Malades, Paris, France
5Service d'Hématologie Adultes Centre de Référence des Déficits Immunitaires Héréditaires CEREDIH Institut Imagine, Hopital Necker-Enfants Malades, Paris, France
6Service de Maladies Infectieuses et Tropicales Centre de Référence des Déficits Immunitaires Héréditaires CEREDIH Institut Imagine, Hopital Necker-Enfants Malades, Paris, France
7Service d'Immunohématologie et Rhumatologie Pédiatrique Centre de Référence des Déficits Immunitaires Héréditaires CEREDIH Institut Imagine, Hopital Necker-Enfants Malades, Paris, France
Background: Chronic granulomatous disease (CGD) is a rare phagocytic disorder that results not only in infections but also in potentially severe inflammatory manifestations that can be difficult to diagnose and treat.
Objective: To describe inflammatory manifestations in a single-center cohort of patients with CGD.
Methods: Medical records of patients treated at Necker-Enfants malades Hospital (Paris, France) between 1968 and 2009 and registered at the French National Reference Center for Primary Immunodeficiencies (CEREDIH) were retrospectively reviewed.
Results: In a population of 98 patients, 221 inflammatory episodes were recorded in 68 individuals (69.4%). The episodes incidence rate was 0.15 per person-year (0.18 in patients with X-linked (XL) and 0.08 in autosomal recessive (AR) CGD). The most commonly affected organs were the gastrointestinal tract (88.2% of the patients), lungs (26.4%), urogenital tract (17.6%) and eyes (8.8%). Inflammation at other sites (the skin, central nervous system, tympanum) and autoimmune manifestations were recorded in 19.1% and 10.3% of the patients, respectively. Granuloma was found in 50% of the 44 histological analyses reviewed. The risk of inflammatory episodes was 2-fold higher in XL-CGD than in AR-CGD (relative risk=2.22, 95%CI=[1.43-3.46]).
Conclusions: Inflammatory manifestations in CGD are more frequent than generally reported and can be responsible of important morbidity. Results show for the first time that patients with XL-CGD have a two-fold higher risk of developing inflammatory episodes than patients with AR-CGD. Whereas the most commonly affected organ is the gastrointestinal tract, other sites can be involved - making the management of patients with CGD a complex, multidisciplinary task.
ESID-0199 Chronic Granulomatous Disease in Patients Reaching Adulthood: A Nationwide Retrospective Study of 80 Cases in France from CEREDIH
B. DUNOGUE1, B. PILMIS2, F. SUAREZ2, C. ELIE3, H. SALVATOR4, A. FISCHER2, S. BLANCHE2, O. HERMINE2, N. MAHLAOUI 2, O. LORTHOLARY2
1Infectious Diseases Unit, Necker-Enfants Malades Hospital, Paris, France
2CEREDIH (French National Reference Center for PID), Necker-Enfants Malades Hospital, Paris, France
3Clinical Research Unit, Necker-Enfants Malades Hospital, Paris, France
4Pneumology Unit, Foch Hospital, Paris, France
Introduction: Prognosis of CGD has now much improved, allowing a majority of patients to reach adulthood. Very few studies have focused on the long-term outcome of adult CGD patients.
Objective: To study the clinical course and sequelae at various time points (age 16, 20, 30 and beyond) of CGD patients, diagnosed before 16 years of age.
Method: A one-year French national retrospective study of patients screened through the CEREDIH national registry.
Results: Eighty CGD patients were included. Median ages at diagnosis and last follow-up were 2.52 and 23.9 years, respectively. Seventeen patients were older than 30 years at time of last follow-up. Seven patients had undergone HSCT.
A total of 553 infections requiring hospital care occurred. These characteristics as well as their annual frequency did not vary, before and after age 16.
A total of 224 inflammatory flares occurred in 71 patients. Their annual frequency did not significantly increase after age 16. Main sequelae were a small median height and weight and a mild chronic restrictive respiratory failure. At the age of 16 years, only 53% of patients were in high school. After 30 years, 9/?13 patients were working, and 2 had had children. Ten patients died during adulthood.
Conclusion: Adult CGD patients display similar characteristics and rates of severe infections and inflammatory flares as during their childhood. The high rate of handicap now becomes a matter of medical and social considerations. A careful follow-up is thus recommended in specialized centers.
ESID-0635 A New Mutation of IL12RB1GENE in a Patient with Mendelian Susceptibility to Mycobacterial Disease
M. mahloujirad 1, S.A. sarrafzade1, J.C. Bustamante2, J.L. Casanova3, A. Domingues2, C. Deswarte2, M. Mansouri4, M. moin1
1clinical immunology, ImmunologyAsthma and Allergy Research Institiute tehran university of medical science, Tehran, Iran
2Laboratoire de Génétique Humaine des Maladies Infectieuses IINSERM, Institut Imagine, paris, France
3Giles Laboratory of Human Genetics of Infectious Diseases, Howard Hughs Medical Institute, paris, France
4Department of Immunology and Allergy Mofid Children Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran
Mendelian Susceptibility to Mycobacterial Disease (MSMD) is a rare congenital syndrome that was first described in 1951. Patients with MSMD are susceptible to weakly pathogenic mycobacteria, such as BCG vaccine and environmental nontuberculous mycobacteria. The prevalence of MSMD is estimated to be as a total of 0.59 cases per million births. The causative genetic defects in the genes including: IFNGR1, IFNGR2, IL-12RB1, IL-12B, STAT1, CYBB, IKBUG, IRF8 and GATA2 have been reported in these patients. Here, we introduce a new mutation in a MSMD patient with disseminated BCG infection.
Case: The patient is a 2.5 years old girl and a child of consanguineous parents (first cousin). The disease was started four months after birth and she was referred to IAARI with cervical and axillary lymphadenitis and para-aortic lymphadenopathy. She had received BCG vaccine on the first day of her life. Anti tuberculosis medications were administered immediately.Primary and advanced screening tests for immunodeficiency were normal. Exome-sequencing analysis was done in INSERM Institute to identify the genetic defects of the patient. The results showed a new mutation in IL12RB1 gene (c.1172 del C.).
Conclusion: The genetic findings would be important in definite diagnosis, better decision for treatment strategy and prenatal diagnosis in PID pateints. This study on MSMD patients is continuing with collaboration of INSERM.
ESID-0578 Molecular Characterization of a Patient Affected by Leukocyte Adhesion Deficiency Type 1 (LAD-1) Successfully Treated with Adjunctive Immunoglobulin Therapy
J.L. Maravillas-Montero 1, M. Yamazaki-Nakashimada2, F. Rivas-Larrauri2, L. Berrón-Ruiz3, N. Ramírez-Alejo1, L. Blancas-Galicia3, S. Scheffler-Mendoza2, F.J. Espinosa-Rosales3, A. Olaya-Vargas4, L. Santos-Argumedo1
1Biomedicina Molecular, Centro de Investigacion y de Estudios Avanzados del IPN, Mexico City, Mexico
2Inmunología Clínica, Instituto Nacional de Pediatría, Mexico City, Mexico
3Unidad de Investigación en Inmunodeficiencias, Instituto Nacional de Pediatría, Mexico City, Mexico
4Oncología, Instituto Nacional de Pediatría, Mexico City, Mexico
Leukocyte adhesion deficiency (LAD) is an uncommon primary immunodeficiency. This disease is characterized by pronounced leukocytosis and several bacterial and fungal infections with a lack of leukocyte recruitment. A patient with a severe LAD-1 phenotype was analyzed by flow cytometry and functional assays to demonstrate the improper adhesive and phagocytic responses of his leukocytes. A single homozygous defect that involves a missense mutation (c.817G > A) that encodes for a G273R substitution in CD18 was identified. The adhesion and phagocytosis assays demonstrated the inability of patient's leukocytes to perform these functions. Expression of the LFA-1 (CD11a/CD18) on the co-transfected HEK 293 cells with the mutated form of CD18 was not detected. Finally, the patient has been treated with immunoglobulin as an adjunctive therapy with positive results. We propose that intravenous immunoglobulin treatment is safe and efficacious in LAD-1 patients before hematopoietic stem cell transplantation, and helpful in controlling severe infections. Subcutaneous immunoglobulin appeared to help wound healing in a refractory ulcer in this patient.
ESID-0792 Clinical and Genetic Features of CGD in Eastern and Central European Countries
G. Markelj 1, M. Debeljak2, S. Pa?ic3, A. Janda4, P. Ci?nár5, S. Sharapova6, E. Heropolitanska-Pliszka7, O. Paschenko8, A. Székely9, T. Freiberger10, M. Bataneant11, M. Erdos9, E. Bernatowska7, L. Marodi9, A. ?edivá4, M. Belevtsev6, I. Kondratenko8, T. Avcin1
1Department of Allergology Rheumatology and Clinical Immunology, University Children’s Hospital University Medical Centre, Ljubljana, Slovenia
2Department of Laboratory Diagnostics, University Children’s Hospital University Medical Centre, Ljubljana, Slovenia
3Department of Paediatric Immunology and Infectious Diseases, Mother and Child Health Institute, Belgrade, Serbia
4Department of Immunology, 2nd Medical School of Charles University & University Hospital Motol, Prague, Czech Republic
51st Paediatric Department, Comenius University Bratislava, Bratislava, Slovakia
6Immunology Laboratory, Belarusian research Centre for Paediatric Oncology and Haematology, Minsk, Belarus
7Department of Immunology, Children's Memorial Health Institute, Warsaw, Poland
8Clinical Immunology Department, Russian Children’s Clinical Hospital, Moscow, Russia
9Department of Infectious and Paediatric Immunology, University of Debrecen Medical and Health Science Centre, Debrecen, Hungary
10Centre of Molecular Biology and Gene Therapy, University Hospital Brno, Brno, Czech Republic
11Victor Babes University of Medicine and Pharmacy of Timisoara, Victor Babes University of Medicine and Pharmacy of Timisoara, Timisoara, Romania
Chronic granulomatous disease(CGD) is a rare primary immunodeficiency with mutations in NADPH oxidase enzyme complex which causes failure of phagocytic cells to kill microorganisms. We retrospectively analysed medical records of patients diagnosed with CGD in the last 35 years from immunological diagnostic centres from 9 central and eastern European countries (Estonia, Latvia, Croatia, Romania, Poland, Belarus, Ukraine, Czech Republic, Slovakia, Hungary, Serbia and Slovenia) and Russia. Genetic sequencing from patients‘DNA was performed in genetic centres in Ljubljana, Belarus and Netherlands for mutations in known genes involved in CGD pathogenesis: CYBB, CYBA, NCF1, NCF2. We included 126 patients with CGD in our cohort, 7 were female. The mean age at presentation of the disease was 12 months and at diagnosis 3,9 years. Lymphadenitis (43%), dermatitis(16%), enteritis(16%), pulmonary infections(13%), liver abscesses(4%) and septicaemia(6%) were the most common clinical presentation. Complications of BCG vaccination (28%) were the most common presenting infection. In total 917,4 years of follow-up in our cohort, the patients suffered 834 different severe infectious episodes (0.9 per year). Respiratory (25%), lymph node (25%) and gastrointestinal tract (18%) infections represented the most prevalent severe infections. We identified 99 different mutations out of 126 genes tested. In 90 patients we identified different mutations in CYBB gene, 2 unrelated patients had the same mutation in CYBA gene and in 5 patients had typical deletion in NCF1 gene. In our cohort we observed high incidence of BCG infections as a presenting symptom. Apart from high BCG infections patients included in our study had similar frequencies of infections and infecting microorganisms as patients described in previous series.
ESID-0293 Inherited IL-12R?2 Complete Deficiency Predisposes to BCG Disease and Tuberculosis
R. Martínez Barricarte 1, C. Aytekin2, J. Markle3, X.F. Kong4, B. Fleckenstein5, S. Tangye6, E. van de Vosse7, S. Boisson-Dupuis1, J. Bustamante8, J.L. Casanova1
1Casanova Lab, The Rockefeller University, New York, USA
2Department of Pediatric Immunology, Dr. Sami Ulus Maternity and Children's Health and Diseases Training and Research Hospital, Ankara, Turkey
3Casanova Lav, The Rockefeller niversity, New York, USA
4Casanova Lab, The Rockefeller niversity, New York, USA
5Institute for Clinical and Molecular Virology, University Erlangen-Nuremberg, Erlangen, Germany
6Immunology Program, Garvan Institute of Medical Research, Sydney, Australia
7Medical Center, Leiden University, Leiden, Netherlands
8Imagine Institute, University Paris Descartes, Paris, France
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare syndrome predisposing individuals to severe infection by non-pathogenic mycobacteria like BCG-vaccines and environmental mycobacteria in otherwise healthy individuals. These individuals are also susceptible to the more virulent mycobacteria M.?tuberculosis. To date, nine morbid genes (IFNGR1, IFNGR2, STAT1, IRF8, NEMO, CYBB, ISG15, IL12B and IL12RB1), all involved in the IFN-?-IL-12/IL-23 loop, have been identified. The most prevalent genetic cause underlying MSMD is complete autosomal recessive IL-12Rß1 deficiency resulting in abolished cellular responses to IL-12 and IL-23. We report here the first AR complete IL-12Rß2 deficiency found in a consanguineous Turkish family by whole exome sequencing. Three patients were found to carry a homozygous nonsense mutation at position Q138. This mutation leads to a complete lack of expression of IL-12Rß2?at the cell surface. In addition, we compared the capacity of cells from IL-12Rß2 mutant and IL-12Rß1 deficient patients to respond to IL-12 and IL-23. Like IL-12Rß1 deficient patient, IL-12Rß2 patient-derived cells do not respond to IL-12. Contrary to IL-12Rß1 deficient patients, their response to IL-23 was intact. One homozygous mutant individual suffered from localized BCG disease, another from bona fide tuberculosis, and the third one remains asymptomatic even though he was BCG vaccinated. All heterozygous family members were healthy. Overall, AR IL-12Rß2 deficiency is a novel genetic etiology of MSMD and tuberculosis, due to impaired IL-12-dependent induction of IFN-?.
ESID-0565 Familial Chronic Granulomatous Disease and Natural-Killer Cell Deficiency
M. Martínez de Saavedra 1, E. González Roca2, A. Muntasell3, J. Gómez-Sirvent4, M. López-Rodríguez1, M.C. Acevedo Gil1, Y. Florido-Ortega1, M. López-Botet3, J. Aróstegui2, C. Rodríguez-Gallego1
1Immunogy, Hospital Universitario de Gran Canaria Dr. Negrín, Las Palmas de Gran Canaria, Spain
2Immunogy, Hospital Clinic Universitari de Barcelona, Barcelona, Spain
3Immunogy, Hospital del Mar Medical Research Institute, Barcelona, Spain
4Pediatrics, Hospital Universitario Nuestra Señora de La Candelaria, Santa Cruz de Tenerife, Spain
Patients with Chronic Granulomatous Disease (CGD) show susceptibility to infections by catalase positive fungi and bacteria. X-CGD may, rarely, occur due to deletions involving CYBB (gp91phox).
A 18 month-old male diagnosed with inflammatory bowel disease, suffered from perihepatic abscess by S.?aureus (8 months-old) as well as cellulitis and osteomyelitis by Serratia marcescens (14 months-old). His 4 year-old brother had been diagnosed with Crohn's disease. Noteworthy, both patients suffered from severe infections by CMV.
Oxidative activity was negative in the two brothers, normal in their father and 90-95% normal in their mother. An inherited chrX 36971138-37956289 deletion, involving the genes FAM47C, PRRG1, LANCL3, XK, CYBB, DYNLT3, Cxorf27 and SYTL5, was found by means of Affymetrix SNP 6.0 array and Comparative Genomic Hybridization Array. Mother's granulocytes, monocytes and B lymphocytes expressed gp91phox in 95% of the cells, suggesting almost complete skewed X-chromosome inactivation. Values of NK cells were severely reduced in both patients and in the mother. Phenotypic studies of NK cells (CD56, CD16, perforin, granzyme A, granzyme B, NKG2D, NKP46, NKP30, NKp44, NKG2C, NKG2A, LILRB1, CD94, DNAM) showed that the observed NK cells were mostly immature; functional studies of NK cells (degranulation and NK citotoxicity) also showed a functional defect.
We report a family presenting with CGD; but NK deficiency, with impaired NK cell maturation, was also observed. CGD due to contiguous deletion syndrome is not accompanied by NK disorders, and patients are not particularly susceptible to viral infections. Overall, data would suggest an independent inheritance of both immunodeficiencies.
ESID-0589 Linking the Flt3 Signaling Pathway to Dendritic Cell Deficiencies in a Subset of Common Variable Immunodeficiency Patients
K. Medina 1, K. Gwin1, K. Allen1, S. Samant2, R. Pyle2, J. Dolence1, R. Abraham2
1Immunology, Mayo Clinic, Rochester, USA
2Dept of Medicine and Laboratory Pathology, Mayo Clinic, Rochester, USA
Common Variable Immunodeficiency (CVID) is a primary immunodeficiency disease characterized by low serum immunoglobulin, impaired antibody responses, and recurrent bacterial infections. Furthermore, many CVID patients develop autoimmune disease and malignancies. Together, these immune sequelae suggest that a subset of patients harbor genetic perturbations that impact innate and humoral immune function. Dendritic cells (DCs) play an essential role in bridging the innate and adaptive immune responses. Importantly, these cells play an important role in the maturation, differentiation, survival, and immune function of peripheral B, T, and NK cells. A subset of CVID patients exhibit DC deficiencies and impaired DC function has been linked to the pathophysiology of the disease. The Flt3 signaling pathway is critical for the generation, migration, and homeostasis of DCs. Alterations in serum levels of Flt3-ligand and surface expression of the Flt3 receptor on residual DCs were evaluated to determine if defects in the Flt3 signaling pathway represent a novel genetic variable and possible prognostic indicator for impaired immunity in some CVID patients. Variations in serum FL were documented but did not segregate patients with DC deficiencies. However, patients with increased serum FL had increased incidence of autoimmune cytopenias, autoimmune disease, and malignancies. DCs play a critical role in the detection of neoplastic lesions and maintenance of peripheral tolerance and elevation in serum FL has been documented in mice with DC deficiencies. Thus, further evaluation of defects in the Flt3 signaling pathway in CVID patients is warranted.
ESID-0425 A Novel STAT1 Gain-of-Function Mutation in Chronic Muco-Cutaneous Candidiasis Patient Associated to IL-17T Cells Defective Differentiation In Vitro Despite Normal Range Ex Vivo
N. Mekki 1, I. Ben-Mustapha1, L. Liu2, L. Boussofara3, S. Okada4, S. Cypowyj4, N. Ghariani3, W. Saidi3, J.L. Casanova2, J.L. Casanova4, A. Puel2, M.R. Barbouche1
1Laboratory of Transmission Control and Immunobiology of Infections (LR11IPT02), Institut Pasteur de Tunis, Tunis, Tunisia
2Laboratory of Human Genetics of Infectious Diseases Necker Branch, Necker Medical School INSERM U980 and University Paris Descartes, Paris, France
3Department of Dermatology, CHU Farhat Hached, Sousse, Tunisia
4St. Giles Laboratory of Human Genetics of Infectious Diseases Rockefeller Branch, The Rockefeller University, New York, USA
Introduction:
Chronic muco-cutaneous candidiasis (CMC) is characterized by recurrent or persistent infections of the skin and mucosae with Candida albicans. Recently discovered, heterozygous STAT1 gain-of-function (GOF) mutations are associated with sporadic or familial, autosomal dominant CMC in patients with few but diverse other clinical manifestations, including cerebral aneurysms and auto-immune thyroiditis. In these patients, STAT1 transcriptional activity in response to cytokines is increased due to an impaired nuclear dephosphorylation.
Case report:
We describe the first Tunisian patient bearing a novel GOF mutation located in the coiled-coil domain of STAT1. An 8-years old girl, displays multiple candidiasis lesions of the skin and mucosa. Because of the disseminated CMC and the absence of HIV or immunosuppression, CMCD was suspected and further investigation was performed. Patient’s genomic DNA sequencing of the STAT1 CCD found a heterozygous mutation leading to increased phosphorylation and activity of STAT1 in response to cytokines, including IFN-g in particular. Interestingly, unlike previously reported patients bearing GOF STAT1 alleles, this patient displays normal levels of IL-17-producing T cells as assessed ?ex?vivo from total leukocytes. However, like other patients his naïve CD4+ T cells show a decreased capacity to differentiate in?vitro into IL-17-producing T cells.
Discussion:
Our findings suggest that the impact of STAT1 GOF mutations on the development of IL-17T cells, as assessed ?ex?vivo, varies from mutation to mutation, from patient to patient, or both. Thus, the mucocutaneous immune response defects, perhaps better mimicked by in vitro differentiation are relevant to study.
ESID-0454 Mycobacterial Infections in Tunisian Primary Immunodeficiency Patients
N. Mekki 1, I. Ben-Mustapha1, M. Ben-Ali1, L. Boughammoura2, N. Guediche3, M.T. Sfar4, A. Sammoud5, M. Hchicha6, A. Mahfoudh7, J. Chemli8, M. Bejaoui9, M.R. Barbouche1
1Laboratory of Transmission Control and Immunobiology of Infections (LR11IPT02), Institut Pasteur de Tunis, Tunis, Tunisia
2Department of Pediatrics, CHU Farhat Hached, Sousse, Tunisia
3Department of Pediatrics, CHU Fattouma Bouguiba, Monastir, Tunisia
4Department of Pediatrics, CHU Tahar Sfar, Mahdia, Tunisia
5Department of Pediatrics, Children's Hospital Bab Saadoun Square, Tunis, Tunisia
6Department of Pediatrics, CHU Hedi Chaker, Sfax, Tunisia
7Department of Pediatrics pediatric emergency and intensive care, CHU Hedi Chaker, Sfax, Tunisia
8Department of Pediatrics, CHU Sahloul, Sousse, Tunisia
9Department of Peadiatric Immuno-Haematology, National Bone Marrow Transplantation, Tunis, Tunisia
Introduction:
Mycobacterial infections are observed in several primary immunodeficiency diseases (PIDs). The manifestations of these infections include local and disseminated BCG infection, pulmonary and extra-pulmonary tuberculosis.
Objective:
To study prevalence, clinical expression and microbiological findings in Tunisian PIDs patients with mycobacterial infections.
Methods:
We conducted a retrospective study in which we included PIDs patients diagnosed during the last two decades.
Results:
Among 565 PIDs patients diagnosed, 69 developed mycobacterial infection. Patients with Chronic Granulomatous Disease (CGD), Severe Combined ImmunoDeficiency (SCID) or Mendelien Susceptibility to Mycobacterial Disease (MSMD) are the most susceptible for mycobacterial infection. HLA Class II deficiency patients are less affected (3/?68). Among 105 CGD patients, 21 (20%) developed BCG associated infection including 13 BCG-osis and only 7 (6%) developed infection with M. tuberculosis. Among 71 SCID and Omenn Syndrome patients, 8% developed BCG-osis but none had M. tuberculosis infection. Among MSMD patients, all BCG vaccinated IL12B deficiency (6 patients) and IFNGR1 deficiency (4 patients) developed BCG-osis and all 8 IL12RB1 deficiency patients had either BCG-osis or tuberculosis. No environmental mycobacteria have been identified in any of these patients.
Discussion:
In Tunisia, tuberculosis is endemic and BCG vaccination at birth is universal. Mycobacterial infections, particularly BCG-osis, are frequent among PIDs patients. The clinical presentation and the disease course depend on strain and underlying PID. Immunological investigation for SCID, MSMD, CGD or other PIDs is recommended in children with mycobacterial infections. SCID newborn screening not yet available in Tunisia and delayed BCG vaccination may help decrease prevalence.
ESID-0390 A Case of DOCK8 Deficient Hyper-IGE Syndrome Presenting as Eczema and Lung Disease
S. Meléndez 1, B. Del Río1, J. Del Río1, A. Partida2
1Pediatric Allergy and Immunology, Hospital Infantil de México Federico Gómez, Ciudad de México, Mexico
2Pediatric Allergy and Immunology, Instituto Nacional de Pediatria, Ciudad de México, Mexico
Introduction
The hyper- IgE syndrome is a rare immunodeficiency characterized by high IgE levels, abscesses, eczema, and sinopulmonary infections. The autosomal dominant disease is characterized by mutations in STAT 3 and the autosomal recessive form by mutations identified in DOCK8.
Case presentation
A 8 years old female patient, with a history of chickenpox and lesions of atopic dermatitis since 4 years; at 6th year with Herpes Zoster. She was referred to our hospital due to pneumonia. On physical examination she was found with disseminated dermatosis with signs of scratching and hyperchromic lesions in genitals and limbs. Besides onychomadesis and finger clubbing. In a simple and contrasted CT was found with interstitial infiltrate and cylindrical bronchiectasis. On admission with eosinophilia (2310), plaquetosis, elevated GGT and transaminasemia. IgG positive serum for EBV. IgE (1480 mg/dl). Flow cytometry was performed, which reports CD3: 50%, 1254 (1200-4000), CD4: 23%, 587 (560-2600), CD8: 24%, 1254 (1200-4000), CD19: 33% (6-32%), CD16/?56: 7.18%. With these findings, diagnosis of AR hyper-IgE syndrome caused by DOCK8 mutation was suspected; sample was sent to establish mutation by sequence analysis. Currently treated with IVIG 500 mg/kg with clinical improvement. She is in the hematopoietic stem cells protocol waiting for donor.
Discussion
It has been estimated over 100 patients diagnosed with DOCK8 deficiency worldwide. .Once diagnosed, these patients are treated with antimicrobial agents, IVIG, in addition to monitoring cancer. Transplantation of hematopoietic stem cells is only curative treatment in this syndrome and should be considered as early as possible diagnosis.
ESID-0059 Chronic Granulomatous Disease; A Cohort of Egyptian Children
S. Meshaal 2, N. Galal1, R. Al kady1, D. Abdelaziz1, J. Boutros1, R. Elhawary2, A. Marsafy1
1Pediatrics, Cairo University, Cairo, Egypt
2Clinical Pathology, Cairo University, Cairo, Egypt
Chronic Granulomatous Disease(CGD) is an inherited disease that results from a defect in the phagocyticcells of the immune system. It is a heterogeneous disorder caused by defects inone of the five major subunits of NADPH complex. This is the first report from Egypt of a cohort of CGDpatients from a single tertiary referral center. There were twenty males andeleven females. The consanguinity rate was 74.2%. The mean age at diagnosis wasforty six months whereas the mean age of presentation was sixteen months.
The most common manifestations were softtissue abscesses in 82%, pneumonia in 79.3%, deep organ abscesses in 35.5%, gastrointestinal symptoms 34 .6%, lymphadenitis in 30.8 and osteomyelitis in 26.9%.
Although XLR-CGD universally constitutesthe most common expression of the disease, only 7 of our 31 patients belongedto this group. Those children’s mothers showed flowcytometric carrier patterns.Nineteen patients had autosomal recessive patterns based on having abnormal SI,positive family history and normal maternal granulocyte functions. Somepatients had abnormal SI but mothers were not available for testing and hencewere unclassified. The underlying mutation testing was done for three casesonly. There was no significant difference between both groups regarding age ofpresentation and age at diagnosis. The SI was significantly lower in the Xlinked group with a p value of 0.015.
Rare but fatal complications were alsoreported among patients as development of Hemophogocytic Lymphohistiocytosis(HLH) syndrome. Only one patient underwent bone marrow transplantation.
ESID-0581 Pediatric Reference Intervals for C3 Complement in Healthy Sudanese Children Determined by In-House Sandwich ELISA
W.A.A. mohammed 1, M. Kirschfink2, S.H. El-Safi3
1Clinical chemistry, Faculty of Medical Laboratory SciencesUniversity of Khartoum, khartoum, Sudan
2Immunochemistry, Institute of Immunology Heidelberg, Heidelberg, Germany
3Medical Microbiology & Parasitology, Faculty of Medicine University Khartoum, khartoum, Sudan
Background: C3 complement is the most commonly measured complement components serving as an important diagnostic tool in the investigation and monitoring patients with inflammatory diseases as well as autoimmune disorders such as systemic lupus erythematosus (SLE).
Objectives: As in most African countries with their individual ethnic populations reference values for most parameters are lacking, the aim of this study was to establish a reference interval for C3 in Sudanese healthy children.
Methods: A cost –effective and sensitive in house a sandwich enzyme-linked immunosorbent assay (ELISA) for the assessment of C3 levels was established and thoroughly validated. Performance characteristics, including analytical sensitivity, linearity, imprecision, and recovery, were assessed. Reference interval was established from 593 healthy Sudanese children volunteers aged from 6 months to 18 years. Reference intervals were generated according to CLSI C28-A3 statistical guidelines.
Results: The assay is highly sensitive, specific and shows excellent quantitative characteristics such as reproducibility, dilution linearity and recovery (98.3–101.9%). The working range be 2.062- 132 ng/mL (r2=0.999). The intra-assay CVs were <49% and the inter-assay CVs were <6%. The mean serum C3 concentration was 1.69 g/?l with a 95% confidence interval of 1.12-2.53 g/?l. There was no significant difference in the C3 concentration identified by gender (p=0.078).
Conclusions: For the first time C3 reference intervals were established for healthy Sudanese children, allowing correct interpretation of deficiency status of this important complement component in diseased children. The applied in-house ELISA is advantageous over commercially available assays because of its sensitivity and low costs.
ESID-0679 Histopathological Aspects of Acute-Form Paracoccidioidomycosis in IL-12BR1 Deficient Patients
D. Moraes Vasconcelos 1, A.E. Campeas2, M.S. Ferrão2, C.L. Yu2, C. Pagliari3, R.A. Brasil4, M.N. Sotto3, M.I.S. Duarte3
1Dermatology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
2Pediatrics, Instituto de Infectologia Emilio Ribas, São Paulo, Brazil
3Pathology, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
4Pathology, Instituto de Infectologia Emilio Ribas, São Paulo, Brazil
Paracoccidioidomycosis (PCM) is caused by the dimorphic fungus Paracoccidioides brasiliensis. PCM presents two clinical forms: acute form (AF) and chronic form (CF). CF presents pulmonary, skin and mucosal involvement. AF presents lymphadenopathy, hepatosplenomegaly and bone marrow dysfunction. We have previously described that AF PCM could be related to CD212 deficiency.
Two patients, one 18 yo male (1) and other 11 yo female (2) presented long-lasting fever and abdominal (and/or leg) pain with disseminated lymphadenopathy and hepatosplenomegaly. The biopsy of lymphnodes showed AF-PCM. Patient 1 had a previous history of BCGitis and disseminated salmonellosis, suggesting a defect of the IL-12/23 – IFN-g axis (published elsewhere). Patient 2 presented later pulmonary and pleural mycobacteriosis.
The histological evaluation of lymphnodes showed typical granulomas with epithelioid response, giant cells and central necrosis with several fungal forms. Immunohistochemistry showed a palisade of CD4+ T cells and just a few in the central area. Conversely, CD8+ T cells were much less representative. Immunostaining for macrophages showed large numbers of cells. The staining for cytokines showed almost no staining for IL-4. Moreover, the staining for TNF-a and for IFN-g was scarcely distributed. Conversely, IL-17 was clearly evident as well as TGF-b around granulomas.
There is an interesting clinical-pathological relationship between the clinical features, the evolution of the PCM after therapy and the histopathological aspects. These points are important to understand the benign features of the PCM in these patients and support the idea of therapy with IFN-g in case of a possible relapse of the infectious disease.
ESID-0395 Is the Functionality of Monocyte Derived Dendritic Cell from Chronic Mucocutaneous Candidiasis Patients Altered?
P. Ordonhez Rigato1, A. Santos Dias1, D. Moraes Vasconcelos 1, A.J. Duarte1
1Dermatology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
Chronic mucocutaneous candidiasis (CMC) is a syndrome with persistent or recurrent infection of the skin, nails, oral or genital mucosa by Candida albicans. The inefficient defense against Candida sp is a result of dendritic cell (DC) or T cell defect impairing Th17 and Th1 immune response. Monocyte derived dendritic cell (moDC) were cultured with C.?albicans to verify the functionality of these cells and the possibility to drive Th response in?vitro as a future immunotherapy in CMC.
MoDC from healthy individuals (CTRL) and CMC patients were generated in?vitro and phenotypically analyzed. The moDC were generated by adherence and cultured with IL-4 and GM-CSF in?vitro. At day 5 moDC received or not IL-1b, TNF-a, IL-6 or inactivated fungus; 48 hours after stimulation, cells were analyzed by flow cytometry. Proliferation of CD4 and CD8 T cell cultured with moDC was analyzed by CFSE assay.
Immature moDCs from CMC group expressed higher levels of CD1a than mature or inactivated yeast moDCs. Activation assessed by HLA-DR, CD40, CD80, CD83 and CD86 expression were higher in moDCs cultured with C.?albicans from CTRL than CMC. There was no evident modulation of CCR6, CCR7, CCR9, TLR2, TLR4 and TLR6. C-lectin receptors (CD206, CD209, DEC-1) were modulated by C.?albicans on moDCs from CMC and CTRL. The moDCs of CMC cultured with C.?albicans poorly induced proliferation of CD19+, CD4+ and CD8+ cells. Our preliminary data showed impaired function of moDC in CMC patients considering HLA-DR upregulation and B and T cell proliferation induction.
ESID-0664 Hereditary Angioedema: Report of 68 Cases
A.S. Grumach1, M. Domingues-Ferreira2, N. Chuffi-Barros2, T.A. Bezerra2, D.L. Bertolini2, R. Muniz Junior2, L.E. Prestes-Carneiro2, A.J. Duarte2, D. Moraes Vasconcelos 2
1Dermatology, Faculdade de Medicina do ABC, São Paulo, Brazil
2Dermatology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
Hereditary angioedema (HAE) is a disease characterized by attacks of angioedema affecting the subcutaneous tissue, as well as the respiratory and gastrointestinal tracts. It can be secondary to deficiency of the C1INH or Factor XII, therefore affecting complement, coagulation, fibrinolysis, and bradykinin pathway. The frequency of HAE is estimated in 1 in 10,000 or 50,000 people and respiratory involvement can lead to death by asphyxiation in 25-40% of untreated cases.
We collected clinical and laboratory data of 68 patients followed at the ADEE-3003: gender, age, age at diagnosis, clinical manifestations, time to diagnosis, triggering factors, treatment, family history and dosages of C esterase inhibitor (C1INH), of C4 and CH50 were assessed.
There was female predominance (44/68), with wide variation in age (8-70 years) with symptoms onset in childhood and adolescence in most cases. 17% of patients had at least one episode of edema, the triggers were trauma (17/68), stress (5/?68), menstrual cycle (5/?68) and the rest unidentified. Family history was positive in 44/68, leading to the discovery of several patients after the index case. Prophylactic therapy with androgens and/or fibrinolytics was used, and icatibant was successfully used to treat the most severe crises of the patients.
Although there are several reports of cases of HAE in other countries, this diagnosis is rarely recognized in our country. The family history was crucial in the investigation of the disease.
ESID-0606 Differential Methylation of a CPG Dinucleotide Upstream of RNASE7 is Associated with Staphylococcal Disease
A. Nieters 1, S. Rieg2, S. Hofmann3, W. Kern2, M. Elgizouli1
1University Medical Center Freiburg, Center for Chronic Immunodeficiency, Freiburg, Germany
2University Medical Center Freiburg, Department of Infectious Diseases, Freiburg, Germany
3University Medical Center Freiburg, Department of Dermatology, Freiburg, GermanyKnowledge of host factors that determine predilection to common infections in individuals with no identified immunodeficiency is limited. RNASE7 is an innate molecule with potent anti-bacterial properties expressed across several tissue types but best characterized as an integral defence component of healthy skin and other mucosal barriers. DNA methylation contributes to the regulation of gene expression and is subject to environmental influences. We investigated DNA methylation patterns of a cytosine-phosphate-guanine dinucleotide (CpG) 1500 base pairs upstream of RNASE7 in relation to local and systemic staphylococcal disease in adult patients recruited at the University Hospital Freiburg, Germany. RNASE7 methylation was assayed using pyrosequencing technology in DNA samples derived from lesional skin biopsies of 33 patients suffering from hidradenitis suppurativa, a chronic inflammatory skin predominantly caused by Staphylococcus aureus, compared to the healthy skin of 5 patients undergoing surgery for melanoma. Cases had a median methylation level of 77 percent compared to 90 percent in controls (Mann Whitney test, p value = 0.008). Based on this observation, we investigated the methylation pattern of the same RNASE7 locus in the whole blood DNA of 46 patients hospitalised for Staphylococcus aureus bacteraemia (SAB) compared to 17 healthy controls. Bacteraemia patients had a median methylation level of 87 percent and healthy controls 83 percent (Mann Whitney test, p value = 0.001). Our findings suggest a role for differential methylation of RNASE7 in staphylococcal disease and point to the utility of DNA methylation as a marker of disease.
ESID-0315 Primary Complement Deficiencies– A Retrospective Study
A. Moreira1, G. Franchini2, J. Vasconcelos2, L. Marques2, I. Lima2, J. Guimarães2, M. Guedes2, I. Lopes1, E. Neves 2
1Immunoallergology, Centro Hospitalar Vila Nova Gaia e Espinho EPE, Vila Nova Gaia, Portugal
2Immunology, Centro Hospitalar do Porto, Porto, Portugal
Background: Primary complement deficiencies (PCD) are rare with a prevalence of 0,03%. We aimed to characterize patients with PCD diagnosed in our department from 1990 to 2014.
Methods: Data was collected retrospectively: gender, age, symptoms, diagnosis delay (DD), immunological study, treatment and family history.
Results: Eighteen patients with a median age of 12 years (5-72) were studied. Hereditary angioedema (HAE) was diagnosed in 10 (9 females). Clinical onset median age (OMA) was 8,6 (±6,3) years and the mean DD was 1,8 years (±1,3). Six patients had a family history of angioedema and low levels of C4 were detected in all patients. Three also had decreased C1q levels. Nine had HAE type I and 1 HAE type II. Long-term prophylaxis was needed in three patients. C4 deficiency was diagnosed in 3 cases, C2 in 2 and C7, C8 and C9 in one patient each. Six of them were females. C4 deficiencies occurred in the same family (mother and 2 daughters). Two were asymptomatic and one had systemic lupus erythematosus and recurrent respiratory infections. Patients with C2 deficiency presented with severe infections (primary pneumococcal peritonitis and pseudomonas respiratory infection), in the second case associated with lupus. C7, C8 and C9 deficiencies were diagnosed in 3 children with severe meningococcal infections.
Conclusion: Gender, OMA and clinical features of HAE patients were similar to those referred in literature. Infections and autoimmune diseases were the main clinical presentations in the other PCD, as expected. Awareness is crucial for early diagnosis of these rare disorders
ESID-0773 Invasive Pulmonary Aspergillosis Diffusing to Contiguous Tissues in Chronic Granulomatous Disease: Clinical Features and Therapeutic Difficulties
M. OUEDERNI 1, S. THRAYA1, M. BEN KHALED1, N. DHOUIB1, R. HASSOUNA1, A. HAOUA1, F. MELLOULI1, M. BEJAOUI1
1Pediatric Immuno-hematology Unit, Bone Marrow Transplant Center, Tunis, Tunisia
Introduction: Chronic granulomatous disease (CGD) is characterized by recurrent life-threatening bacterial and fungal infections. Invasive pulmonary aspergillosis (IPA) is a major cause of morbidity and mortality in this disease.
Subjects: Herein, we describe clinical features and therapeutic difficulties of IPA in CGD patients followed within the National Center of Bone-Marrow Transplant of Tunis.
Results: Four girls with CGD were included, aged respectively 2, 3, 4 and 13 years. They received oral antifungal prophylaxis with Itraconazole (n=2) and Voriconazole (n=2). Clinical signs included dyspnea and fever (n=4), swelling and purulent tumefaction of the back (n=3), trunk pustullar lesions (n=1). Chest computed tomography showed multiple scattering nodules, alveolar condensations (n=4) diffusing to pleura (n=3), ribbings (n=1) and soft tissues with fistulization to the skin (n=3). Serum galactomannan antigen was positive in 2 cases. Culture on bronchoalveolar lavage fluid grew positive for aspergillus in 2 cases. Fluid and tissue specimens from skin abscess were positive to Aspergillus in 2 cases. Aspergillus species were Ornatus (n=1), Fumigatus (n=2), and Nidulans (n=1). Intravenous Voriconazole alone was inefficient in one patient. Prolonged association of voriconazole to Amphoterecine B in all patients led to regression of skin, pleural and bone lesions in 1 to 2 months. Regression of pulmonary lesions needed 4 months of intravenous treatment. No surgery was needed.
Conclusion: Without early and efficient antifungal therapy, pulmonary aspergillosis is further complicated by invasion of bone, soft tissues and skin. Primary prolonged combination therapy was essential in our patients to control IPA diffusing to contiguous tissues.
ESID-0562 Innate IGG Molecules and Innate B Cells Have Impact on Primary Immunodeficiencies and Immunotherapy
V. Oxelius 1
1Department of Pediatrics and Clinical Immunology, Institute of Laboratory Medicine, Lund, Sweden
Background: IgG was registered by markers of the immunoglobulin constant heavy G chains (IGHG)(Fc?)(GM) genes, inherited in Mendelian fashion and allelic exclusion. The distinguished alternative allotypes of ?3-, ?1- and ?2- chain genes have different structures and functions. The GM allotypes of ?3-?1- ?2 are found in fixed combination, *bfn, *bf-n,*gan and *ga-n, signaling 4 B cells: B*bfn, B*bf-n , B*gan and B*ga-n .The haplotypes constitute 10 individual diplotypes. Bacterias, virus and allergens, active and passive immunotherapy affect individual diplotypes differently.
GWAS and HapMap do not include GM genes and have missed IGHG genes as candidate genes.
Objective: To investigate IGHG(Fc?)(GM) genes, markers of innate IgG molecules and innate B cells, in primary immunodeficiencies and immunotherapy.
Methods: A new sensitive competitive ELISA method was used for IGHG gene mapping and quantities of innate IgG subclasses. A IGHG gene map and normal range of innate IgG subclass levels are given.
Results: Selective IGHG(Fc?)(GM) genes dominate in PIDs: Homozygous diplotypes: IGHG*bf-n/*bf-n in IgG2D and CVID , IGHG*ga-n/*ga-n in IgG3D, IgAD and WAS; Homozygous IGHG2*-n/*-n : in CVID, WAS, IgG2D, IgG3D and IgAD .The IGHG2*-n allele is dominating , in all PIDs.
In IVIG treatment of PID patients with homozygous IGHG2*-n/*-n, the mean trough levels of foreign IgG, during 8 months, were registered. The IgG2*n molecules were preserved about 4 times the amount of IgG1 variants.
Conclusion: IGHG2*-n, innate IgG2*-n molecules, of the IGHG*bf-n and IGHG*ga-n haplotypes, from B*bf-n and B*ga-n cells are linked to PIDs. During treatment with IVIG, IgG2*n is preserved.
ESID-0746 Level of GP 91 Phox Expression in CGD Carriers is Related with Differential B –Cell Impairment and Autoimmune Manifestations
P. Palma 1, N. Cotugno1, I. Salfa1, J. Serafinelli1, M. Chiriaco2, S. Di Cesare2, G. Di Matteo2, P. Rossi1, A. Finocchi1
1University-Hospital Pediatric Department, Bambino Gesù Children Hospital and University of Tor Vergata, Roma, Italy
2Department of Public Health and Cellular Biology, Bambino Gesù Children Hospital and University of Tor Vergata, Roma, Italy
Background: Chronic granulomatous disease (CGD) is a genetically heterogeneous primary immunodeficiency due to defective nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity . Although the defect mainly affect macrophage/phagocytic compartment, B-cell function is also impaired in CGD patients as we recently described (article in press). Such impairment could affect female X-linked CGD carriers, as well, who may display similar clinical manifestations to CGD patients with significant impact on their life.
Objective: We investigated 4 CGD mothers in order to analyze gp 91 phox expression, phenotype and proliferation capability due to the B cell compartment , compared to age matched healthy control (HC). Results: CGD mothers show an altered B-cell phenotype similar to those described in their siblings: higher total naïve (CD19+ CD27-IgD+) and lower total (CD19 + CD27+) and resting memory (CD19 + CD27 + CD21+) B cells levels. Furthermore, similar impairment is shown in proliferation experiments, where CGD carriers demonstrate a lower proliferation rate upon single stimuli compared to HC. These alterations relate to different gp91phox expression in B cells. Interestingly, CGD mothers with higher gp 91 phox expression in B cells seem to compensate these defects.
Conclusion: Our data show a partial impairment in terms of phenotype and proliferation of the B cell compartment in CGD carriers. These functional defects seem to be inversely related to the gp 91phox expression and autoimmune manifestations. These results agree with the recent evidence that female carriers, mostly considered unaffected, display conditions often underlying an autoimmune pathogenesis. Larger studies have to be performed to confirm the validity of our results.
ESID-0310 The B-Cell Side of Chronic Granulomatous Disease
N. Cotugno1, A. Finocchi1, A. Cagigi1, G. Di Matteo2, M. Chiriaco2, S. Di Cesare2, P. Rossi1, A. Aiuti1, P. Palma 1, I. Douagi3
1University Department of Pediatrics, Pediatric Hospital Bambino Gesu' and University of “Tor Vergata”, Roma, Italy
2Department of Public Health and Cellular Biology Tor Vergata” University Rome Italy, Pediatric Hospital Bambino Gesu' and University of “Tor Vergata”, Roma, Italy
3Department of Medicine Huddinge Karolinska Institutet Stockholm Sweden, Center for Hematology and Regenerative Medicine (HERM), Stockholm, Switzerland
Chronic granulomatous disease (CGD) is a primary immune deficiency characterized by a defect in reactive oxygen species (ROS) production. Although the impact of CGD mainly reflects on the macrophage/phagocytic compartment, B-cell responses are also impaired in CGD patients.
In order to investigate how defective gp91phox expression ROS production specifically impacts on B-cell, antibody responses and function, B-cell phenotype, proliferation capability and maintenance of long-term memory in CGD patients were examined.
Here we show that the memory B-cell compartment is impaired among CGD patients as indicated by reduced total (CD19+CD27+) and resting (CD19+CD27+CD21+) memory B-cells in parallel to increased naïve (CD19+CD27-IgD+) B-cell frequencies. Data on CGD carriers reveal that such alterations are related to gp91phox expression. Moreover, proliferative capabilities of B-cells upon selective in?vitro stimulation of B-cell receptor (BCR) or toll-like receptor 9 (TLR9) pathways were reduced in CGD patients compared to age matched healthy controls. Significantly lower measlesspecific antibodies and antibody secreting cells (ASC) were also observed, indicating a poor ability to maintain long term memory in these patients.
Altogether, our data suggest that CGD patients present a defective B-cell compartment in terms of frequencies of memory B-cells, response to in?vitro stimulation and maintenance of long-term antigen-specific memory.
ESID-0042 Clinical Spectrum and Mutation Detection in a Cohort of Greek Patients with Familial Mediterranean Fever
E. Papadopoulou-Alataki 1, S. Katafigiotis2, C. Trakatelli3, A. Alataki2, A. Lambropoulos2, M. Emboriadou1
1Fourth Department of Pediatrics Papageorgiou Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
2Molecular Biology Lab Papageorgiou Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
3Third Department of Internal Medicine Papageorgiou Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
Introduction. Familial Mediterranean Fever (FMF) is an autosomal recessive disorder characterized by recurrent attacks of fever and serositis. FMF gene (MEFV) is located on chromosome 16p encoding the pyrin protein.
Objective. To study the clinical spectrum of FMF patients along with MEFV mutations and investigate a potential association between genotype-phenotype.
Patients-Methods. Forty patients (17males) who fulfilled the diagnostic Tel Hashomer criteria, were enrolled in the study: 25 children aged from 2 to 14 years (median 8.5) and 15 adults from 16 to 45 years of age (median 30). All patients and 33 unaffected relatives from 21 unrelated families were tested by Reverse Hybridization assay for the molecular detection of the most common 12 MEFV mutations.
Results. Family history was positive in 47.5% of the patients. The delay of diagnosis ranged from 1 to 19 years. The number of febrile episodes along with abdominal pain ranged from 2 to 18 per year, duration: 1-6 days. In adult patients, chest pain during episodes and myoskeletal pain was present in 81% and 56% respectively. Only 50% of FMF children complained for chest or myoskeletal pain. The mutations detected were: M694V 62.5%, E148Q 25%, M680I(G/?C) 23%, R761H 7.5%, A744S 7.5%, P369S 5%, V726A 5%, K695R 2.5%. Among 40 patients, 4 were found homozygous, 24 heterozygous and 11 compound heterozygous. 21/33 unaffected relatives were found heterozygous.
Conclusions. The commonest mutation was M694V, associated with renal amyloidosis in two cases. No difference in clinical picture was found between patients with single or double MEFV mutation.
ESID-0165 Concentration OF IGG, IGA and IGM Antibodies Specific to Pneumococcal Capsular Polysaccharide in Adult Blood Donors
O. Hemmings1, S. Allen1, S. Harding2, A. Parker 3
1Clinical Chemistry, The Binding Site Group Ltd, Birmingham, United Kingdom
2Clinical Research and Development, The Binding Site Group Ltd, Birmingham, United Kingdom
3Head of Scientific Affairs, The Binding Site Group Ltd, Birmingham, United Kingdom
Background
We report the normal serum concentrations of pneumococcal antibodies in adult blood donors.
Methods Serum was obtained from blood donors (n=231; median age 43.5 years; range 18-90 years); 18-20 years (n=12; median=19.5 years), 21-30 years (n=78; median=25 years), 31-40 years (n=18; median =36 years), 41-50 years (n=37; median=46 years), 51-60 years (n=25; median=56 years), 61-70 years (n=29; median =67 years), 71-80 years (n=21; median=74 years), and 81-90 years (n=11; median =85 years). Exclusion criteria included no consent, known infection and/or elevated serum CRP concentrations (>10 mg/L). Serum was analysed utilising PCP IgG, IgA & IgM EIA kits (The Binding Site, Birmingham, UK).
Results
Median PCP IgG titres were significantly higher in ages 61-70 years (98.3 mg/L; p=0.0004), 71-80 years (110.4 mg/L; p<0.0001), and 81-90 years (66.5 mg/L; p=0.018) compared to those aged 18-60 years (median 34.7 mg/L).
PCP IgA median titres were significantly higher in ages 51-60 years (27.6 U/?mL; p=0.036), 61-70 years (35.1 U/?mL; p=0.0078), 71-80 years (43.2 U/?mL; p=0.0065), and 81-90 years (55.3 U/?mL; p=0.0034) compared to those aged 18-50 years (median 19.8 U/?mL).
The median PCP IgM titres decreased with age and there was a significantly lower median serum PCP IgM titre in patients aged 81-90 years (27.1 U/?mL) compared to those aged 18-80 years (median 54.5 U/?mL; p=0.0017).
Conclusion
Pneumococcal immunity for PCP IgG is reached between 60-80 years, is stable from 18-50 years and then rises with age for IgA and decreases progressively with age from 18-90 years for PCP IgM.
ESID-0618 Neutrophil Function in a Leucocyte Adhesion Deficiency Type I (LAD I) Patient
L. Perez 1, L. Regairaz2, J. Bustamante3, M. García4, D. Cabanillas2
1Service of Immunology and Rheumatology, Hospital de Pediatría "Prof. Dr. Juan P. Garrahan", Ciudad de Buenos Aires, Argentina
2Service of Immunology, Hospital de Niños "Sor María Ludovica", Ciudad de La Plata, Argentina
3Laboratory of Human Genetics of Infectious Diseases Necker Branch, Institut National de la Santé et de la Recherche Médicale, Paris, France
4Service of Infectology, Hospital de Niños "Sor María Ludovica", Ciudad de La Plata, Argentina
LAD I is an autosomal recessive disorder caused by mutations in the common chain CD18 of the beta2 integrin family. We present the characterization of a novel ITGB2 gene mutation in terms of residual level of ß2 integrin expression, adhesiveness and ligand binding ability.
A 2-year-old girl born to consanguineous parents presented separation of the umbilical cord on day 10 with ulceration and omphalitis by Escherichia coli, neck cellulitis, submaxilar adenitis, and leukocytosis.
CD11b and CD18 expression at baseline and after fmlp stimulation was determined by flow cytometry. Colorimetric assay for adherence to plastic and chemiluminescence measurements for respiratory burst (RB) studies induced by several stimuli were performed in purified neutrophils. Whole exome sequencing was applied to identify the causal variant. Final candidates were confirmed by Sanger sequencing and tested for familial segregation.
Results: CD11b and CD18 expression on patient neutrophils related to the control cells represented 6% and 19% respectively. CD18 was upregulated after exposure to fmlp. Neutrophil adhesiveness to plastic by PMA was completely abolished. OZy stimulated RB was substantially impaired while PMA and fmlp response showed a normal ability of neutrophil NADPH oxidase to oxygen radical generation. Genetic analysis conducted to identify c400G>T mutation in ITGB2 gene.
Conclusions: Neutrophil adhesiveness to plastic and ligand binding ability of CD18/?CD11b to OZy were impaired in spite of the residual level of ß2 integrin expression. The upregulation of CD18 expression by fmlp indicated that the patient cells have a conserved response to external stimuli.
ESID-0119 Familial C8 Beta Deficiency: A Long History
E. Clerici 1, R.M. Dellepiane1, C. Caccia Dominioni1, L.V. Beilis1, P. Pavesi1, M.C. Pietrogrande1
1Department of Paedriatics University of Milan, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milano, Italy
Complement component C8 is one of the five terminal complement components required for the formation of the membrane attack complex. Complete absence of C8 results in increate susceptibility to gram-negative bacteria such as Neisseria species, while heterozygous condition does not predispose to the infection.
We report 3 cases of functional and immunochemical deficiency of the complement component C8, diagnosed in three Albanian brothers. The eldest brother at 16 years presented with an episode of meningococcal sepsis. At the medical history parents reported that even the other two daughters have previously suffered from meningingococcal sepsis, the elder (14 year-old) 10 years before at 4 year, while the younger (6 year-old) 5 years before, at 18 month (a meningococcal sepsis complicated by fingers and toes necrosis, which required amputation). All family members (included parents) were subjected to functional analisys of complement, in consideration of an hereditary condition. The results of analysis evidenced undosable levels of CH50 in all three children, while both parents resulted normal. A more specific tests by SDS-page and Western blot techniques revealed complete absence of C8 ß (a variant of C8) in all three children. Parents level of C8 ß was reduced, but not absent, suggesting an heterozygous condition. Genetic exams to individuate the specific mutation are still in course of procedure. All the patients have been placed under continuous antibiotic prophylaxis and specific vaccine immunization.
In conclusion, this long history underlines the importance of investigating properly patient's medical and family history.
ESID-0365 A Novel Insertion Mutation in the IL2RG Gene
Z. Pistár 1, B. Soltész1, L. Maródi1, B. Tóth1
1Department of Infectious and Pediatric Immunology, University of Debrecen, Debrecen, Hungary
Nearly 50% of patients with severe combined immunodeficiency (SCID) have IL2RG mutation localised to the X chromosome. Blood samples of a 7 month-old male patient with T-B+NK- SCID and his mother were sent to our laboratory for gene sequencing. The patient died of a severe infection and we could not analyse the clinical phenotype. gDNA was extracted from blood white cells and mutational analysis was performed by amplifying exons 1–8 and the flanking intronic regions of IL2RG by PCR, and sequencing by using the Big Dye Terminator cycle sequencing kit and an ABI 3130 capillary sequencer. We found a novel, hemizygous c.295insA mutation in the patient’s sample. This insertion mutation caused frame shift and termination of transcription 11 codon downstream resulting in a valine-isoleucine change in amino acid position 99 of the protein (V99fsX11). In the mother’s gDNA sample a heterozygous c.295insA mutation was identified. This is the second mutation we have identified in the IL2RG in Hungary over the past 10 years suggesting under diagnosis of SCID and pointing to the need of neonatal SCID screening.
ESID-0678 Erdheim-Chester Non-Langerhans Cell Histiocytosis in a Patient with Mutation of the IL-12RB1 Gene - A Case Report
L.E. Prestes-Carneiro 1, T. Brito2, R.C.B. Gryschek3, N.M. Orii4, J.B.O. Filho4, A.J.S. Duarte4, D.?M. Vasconcelos4
1Department of Immunology and Infectious Diseases, Oeste Paulista University, Presidente Prudente São Paulo, Brazil
2Department of Pathology, Faculdade de Medicina da Universidade de São Paulo São Paulo Brazil, São Paulo, Brazil
3Department of Infectious and Parasitic Diseases, Faculdade de Medicina da Universidade de São Paulo São Paulo Brazil, São Paulo, Brazil
4Laboratory of Medical Investigation Unit 56, Faculdade de Medicina da Universidade de São Paulo São Paulo Brazil, São Paulo, Brazil
We report the case of a 28-year-old man, admitted in 2008 to the Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP), Sao Paulo, Brazil. His symptoms had started 3 years earlier with a history of pulmonary disease showing fair-pleural expansive lesions with bilateral fistula, intermittent fever, consumptive syndrome, hepatosplenomegaly, and generalized lymphadenopathy. At admission, he was emaciated and prostrated and physical examination showed mandibular stiffness to the lateral region of the neck and bilateral cervical lymphadenopathy. Laboratory findings showed signs of inflammation, with an ESR of 77 mm/h, C-reactive protein 257 mg/L, alfa1-acid glycoprotein 402 mg/dL and ferritin 1,623 ng/mL. Radiologic exams showed extensive pneumonia with pleural lesions and a significant decrease of the right upper thorax, hepatosplenomegaly and supra-aortic lymphadenopathy, as well. Immunophenotyping revealed a reduced number of CD8 T-cell subsets, CD19+ B-cells, and low activity of NK cells. Dihydrorhodamine assay showed a 90.5% reduction activity. He received empirically one year of claritromicin and cotrimoxazole for a chronic non laboratory identified actinomyces infection. A diagnosis of histiocytosis was previously suspected without laboratory confirmation. New pulmonary biopsies were obtained and a revision of the old ones was performed in HC-FMUSP. Histologic and immunohistochemistry results were compatible with the pulmonary form of Erdheim-Chester disease (ERD): CD1A (-); CD68 (histiocytes)(+); PSA (-); S-?100 Protein (-) and AE-1/AE-3 (epithelium) (+). An impaired expression of CD212 was found, with mutation of the gene confirmed by capillary sequencing. Treatment for ERD was established and his clinical condition improved remarkably.
ESID-0089 Effects of IVIG Replacement on Monocytes Subpopulations and Function in CVID Patients
V. Conti1, A. Prezzo1, F. Cavaliere1, M. Iacobini2, I. Quinti 1
1Molecular Medicine, Sapienza University of Rome, Rome, Italy
2Pediatrics, Sapienza University of Rome, Rome, Italy
Intravenous IgG (IVIG) therapy is widely used as replacement and anti-inflammation therapy in a variety of acute and chronic autoimmune diseases, but the mechanisms of action, especially of the antinflammatory activity, is still unclear. IVIGs induce significant modifications in the innate and adaptive compartment of the immune system including the monocyte/macrophage system.
A decrease of CD14+CD16++ monocytes was observed in CVID patients after IVIG infusion.
We investigated in a choort of 19 CVID patients effects of IVIG ?in?vivo administration on monocytes subpopulations, CD11b and Siglec9 expression and oxidative burst.
Phenotypic analysis was performed on whole blood samples by staining with monoclonal antibodies Siglec9 FITC, CD11b APC, CD14 PE and CD16 PerCP. Results were expressed as percentage of cells that stained positive for a given marker, or as its mean fluorescence intensity (MFI) within the defined population.
The quantitative evaluation of leukocyte oxidative burst was determined with PHAGOBURST kit (Glycotope Biotechnology).
After IVIG infusion we observed in CVID patients a significantly decrease of intermediate (CD14++CD16++) and non classical (CD14+CD16++) monocytes (p: 0,002 and 0,02 respectively). CD11b as well as Siglec9 MFI value is higher in classical monocytes of CVID patients than in healty donors (HD) monocytes (p: 0,003). The expression of CD11b and Siglec9 on classical monocytes decreases after IVIG infusion but not on intermediate monocytes (p: 0,003 and 0,0012 respectively). No differences were observed on monocytes oxidative burst after E. Coli stimulation between HD and CVID patients, while it decreased in CVID patients after IVIG infusion (p: 0,004).
ESID-0456 A Report of Complement Deficiency from Iran
E. Rashidi 1, M. Fazllolahi1, A. Mahdaviani2, A. Shafiei3, B. Mirsaeid Ghazi4, Z. Pourpak 1, M. Moin1
1Immunology Asthma and Allergy Research Institiute (IAARI), Tehran University of Medical Sciences, Tehran, Iran
2Pediatric Respiratory Diseases Research Center National Research Institute of Tuberculosis and Lung Diseases (NRITLD), Shahid Beheshti University of Medical Sciences, Tehran, Iran
3Department of Immunology and Allergy, Yazd University of Medical Sciences, Yazd, Iran
4Department of Immunology Bahrami Children's Hospital, Tehran University of Medical Sciences, Tehran, Iran
Genetic defects of complement proteins are rare, with an estimated prevalence of 0.03% for any inherited complement deficiency in the general population. The most common clinical presentations of these patients are recurrent infections with encapsulated bacteria, recurrent neisserial infections, and systemic autoimmune disease.We report a case series of four patients who referred to Immunology, Asthma and Allergy Research Institute with recurrent or unusual bacterial infections. The first case was a 3 year old boy with a history of 18 times hospitalization because of septic arthritis (at 8 months), recurrent sinusitis, recurrent pneumonitis, recurrent otitis media and meningitis (in 3 year old). The second one was a 19 month old boy presented with severe pneumonia (at 8 months) and periorbital cellulitis (at 19 months). The third case was a 2 year old girl with history of 2 episodes of meningitis (at 2 months and 2 year). The last one was a 24 year old woman with history of severe pneumonia (at age 24). These cases were screened for primary immunodeficiency. CH50 was undetectable or significant decreased and the other screening tests were normal. In the next step the level of complement components were measured. The first 3 cases had undetectable or markedly decrease C3 levels and the last one had undetectable C2. Although the complement deficiency is rare but evaluation of complement function is very important in patients with recurrent infections so that appropriate immunization and antibiotic prophylaxis can be prevent recurrent or life-threatening infections.
Complement deficiency, Primary Immunodeficiency, Iran
ESID-0476 Ectodermal Dysplasia with Immunodeficiency: AD-EDA-ID
L. Regairaz 1, D. Cabanillas1, P. del Palacio1, A. Bernasconi2, J. Rossi2, J. Yancoski2, S. Danielian2
1Immunology, Hospital Sor Maria Ludovica, La Plata, Argentina
2Immunology, Hospital Garrahan, CABA, Argentina
Ectodermal dysplasia with immunodeficiency (EDA-ID) presents with impaired development of skin appendices and host defense, resulting in severe bacterial disease. Molecular defects include hypomorphic mutations in NEMO (XL-EDA-ID) and hypermorphic mutations in IKBA (AD-EDA-ID). We report a patient with AD-EDA-ID.
Pre term girl without family history of immunodeficiency. She was hospitalized since birth with diagnosis of P.?aeruginosa sepsis. At 2 months old she was referred to our hospital with septic arthritis by Pseudomonas spp. and muguet. During hospitalization she developed multiple infections (arthritis, myositis, thigh abscess, chronic osteomyelitis in tibia by S.?aureus) mild respiratory distress, diarrhea and failure to thrive. Ectodermal Dysplasia was diagnosed by clinical features and skin biopsy. Laboratory studies showed persistent lymphocitocis, anemia and elevated reactive C protein during infection. Humoral assessment showed agammaglobulinemia. Lymphocyte subsets revealed high number of CD4CD45RA cells and low number of CD4CD45RO cells. Lymphocyte proliferation was low to PHA and OKT3. Expression of NEMO protein by Flow Citometry (FC) was normal. Monocyte's TNF- production was absent in response to LPS measured by FC. A heterozygous mutation (D31G) in IkBA gene confirmed the diagnosis of AD-EDA-ID. She was treated with gammaglobulin, fluconazol and antibiotics. At 7 months old she died of meningoencephalitis and sepsis by Acinetobacter while HLA studies were in process.
ESID-0763 Altered NK Cell Phenotype and Function in a Case of Complete Deficiency of the T-Cell Antigen Receptor Zeta Chain (CD247)
H. Reyburn 1, C. Aydogmus2, A. Blazquez Moreno3, S. Haskologlu4, S. López Cobo3, F. Cipe2, A. Babayigit2, M. Agúndez Llaca3, F. Dogu4, D. Dukovska3, E.M. García Cuesta3, J.R. Regueiro5, A. Ikinciogullari4, M. Vales-Gomez3
1Immunology and Oncology, Centro Nacional de Biotecnología, Madrid, Spain
2Pediatric Immunology, Istanbul Kanuni Sultan Süleyman Hospital, Istanbul, Turkey
3Immunology and Oncology, Centro Nacional de Biotecnologia, Madrid, Spain
4Pediatric Immunology-Allergy, Ankara University School of Medicine, Ankara, Turkey
5Immunology, Complutense University School of Medicine, Madrid, Spain
Introduction
The CD247 dimer is critical for assembly, vesicular transport and signalling through the TCR and several NK receptors. We have had the opportunity to study the NK cells of a new case of inherited CD247 deficiency as well as various family members.
Case report
A baby girl born to consanguineous parents developed fever and cytopaenia after routine vaccination at 2 months of age. Immunodeficiency was suspected because cytomegalovirus infection did not remit after standard therapy. Initial haematological examination suggested TCR deficiency and CD247 deficiency was identified by intracellular flow cytometry and confirmed by RT-PCR and sequencing as due to a c.2T>C (p.M1T) mutation.
Results
The patient had normal numbers of circulating NK cells, but the late stages of differentiation of these cells were impaired. In vitro, the ability of these NK cells to proliferate in mixed cultures in?vitro was severely limited, a phenotype likely related to T cell hypofunction, since addition of IL-2 to the cultures restored NK cell proliferation. Lastly, the levels of expression of NK receptors that associate with CD247 were markedly decreased and activation of the NK cell via these receptors was inefficient.
Conclusions
These data provide novel insights into the biology of human NK cells and also suggest that deficient NK cell function could contribute to the spectrum of pathology observed in these patients. Promotion of NK cell activation and expansion could be a viable strategy to improve the health of these patients until bone-marrow transplantation is carried out.
ESID-0026 Role of NOX2 in Pneumonia with Pseudomonas Aeruginosa
J.C. Ryu 1, J.H. Ryu2, J.H. Yoon2
1Brain Korea 21 PLUS Project for Medical Science, Yonsei University
2Research center for human natural defense system, Yonsei University
Pseudomonas aeruginosa is an opportunistic pathogen involved in nosocomial infections. While a number of studies have demonstrated the roles of reactive oxygen species (ROS) in host defense againt Pseudomonas aeruginosa infection, the implication of NADPH oxidase NOX2 of neutrohpil in this process has been overlooked. Here, we show that the contribution of NADPH oxidase NOX2 of neutrophil to the pathogenesis of Pseudomonas aeruginosa infection in murine lungs, addressing both the contribution to microbial killing and regulation of the inflammatory response.
After Pseudomonas aeruginosa injection into lung, gp91(phox)(-/-) mice demonstrated significant decrease in bacterial clearance from the lungs as compared with wild-type C57BL/6 mice. And the greater numbers of neutrophils in gp91(phox)(-/-) mice were associated with increased lung injury. Levels of BUN, AST in serum were also increased in gp91(phox)(-/-) mice.
Taken together our results indicate that the NOX2 provide a potential target to limit the pathological consequences of Pseudomonas aeruginosa infection.
ESID-0605 Novel Factor I Deficiency in a Patient with Leukocytoclastic Vasculitis
J.T. Bay 1, T.L. Katzenstein2, K. Kofoed3, M.?O. Skjødt1, P. Garred1, L. Schejbel1
1Laboratory of Molecular Medicine Dept. of Clinical Immunology, Rigshospitalet Copenhagen University Hospital, Copenhagen, Denmark
2Department of Infectious Diseases, Rigshospitalet Copenhagen University Hospital, Copenhagen, Denmark
3Department of Dermato-allergology, Gentofte Hospital University of Copenhagen, Copenhagen, Denmark
Introduction: Factor I is an important complement inhibitor, and deficiency of factor I is a rare disorder. The clinical presentation of complete factor I deficiency varies and include severe recurrent bacterial infections, glomerulonephritis and/or systemic lupus erythematosus, while subtotal factor I deficiency is associated with atypical haemolytic uremic syndrome.
Aim: We report a patient with homozygous factor I deficiency caused by a novel mutation in the CFI gene (MIM:217030).
Methods: The patient, a 28-years old woman with consanguineous parents, presented with recurrent leukocytoclastic vasculitis in the lower extremities with no associated systemic involvement, and without increased infection tendency. Initial testing showed low C3 concentration and a detailed complement evaluation was performed.
Results: We found decreased activity in all three complement pathways, normal levels of C2 and C4, decreased levels of C3 (0,35 g/?L, ref 0,77-1,38), C5 (33%, ref 72-171%) and factor H (47%, ref 69-154%), and severely decreased levels of factor B (<3%, ref 59-154%) and factor I (<5%, ref 60-152%). Sequencing revealed a homozygous missense mutation in exon 2 of the CFI gene leading to a C<T substitution (40266C>G, NG_007569, C54W), which is likely to impair the folding and/or secretion of the protein.
Conclusions: With the identification of a homozygous novel CFI mutation we confirmed the suspicion of factor I deficiency in a patient presenting with leukocytoclastic vasculitis as the only clinical feature. Even though the clinical symptoms of complete factor I deficiency varies among patients, this is an unusual presentation not previously described.
ESID-0534 Certain Elane Mutations Predispose to More Aggressive Course of Severe Congenital Neutropenia (SCN)
E. Deordieva1, T. Varlamova2, V. Bobrynina2, A. Kozlova1, G. Novichkova3, A. Shcherbina 1
1immunology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
2molecular biology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
3hematology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
ELANE gene mutations are the most common cause of severe congenital neutropenia, mutations occur in all exons, in most introns and 5’UTR. So far no significant correlation of the type and location of the mutation and severity of the disease have been found. Yet, Makaryan V. et?al. reported, that several mutation (e.?g. at the positions C151 and G214) lead to more severe phenotype as judged by poor response to G-CSF treatment and high frequency of MDS\AML development (Abstract 3275, ASH Meeting 2012).
Results: Here we report two patients with SCN harboring previously described ELANE mutations. The patient with ELANE mutation c.452G toC (Cys151Ser) had a very poor response to G-CSF and required a dose of 50 ug/kg/day to reach 1000 neutrophils\ul level. Moreover, this patient developed AML very early in life - at the age of 13 months, which required hematopoietic stem cell transplantation (HSCT). Second patient, with the ELANE mutation c.640 G>A, Gly 214Arg is also a very poor responder to G-CSF, upon treatment with up to 50 ug/kg/day of G-CSF maximal neutrophil counts achieved were only 500 cells\ul, accompanied with a lot of serious infectious complications. He underwent HSCT at the age of 8 months, and had no signs of AML/MDS at the time of transplant.
Conclusion: Our data confirms the observation that at least two mutations in ELANE should be considered unfavorable in terms of prognosis and patients should be considered for early HSCT. More studies of phenotype-genotype correlation in SCN are needed.
ESID-0114 Invasive Meningococcal Disease and Complement Pathway Deficiencies. Is Systematic Screening Warranted?
P. Soler Palacín 1, L. Minguell1, A. Martin-Nalda1, M. Hernandez2, C. Franco2, O. Colobran2, R. Pujol2, C. Figueras1
1Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d'Hebron Institut de Recerca Vall d'Hebron Universitat Autonoma de Barcelona, Barcelona, Spain
2Immunology Division, Hospital Universitari Vall d'Hebron Institut de Recerca Vall d'Hebron Universitat Autonoma de Barcelona, Barcelona, Spain
Background: Although the relationship between recurrent invasive meningococcal disease (IMD) and complement pathway deficiencies (CPD) is clear, there is scarce data about CPD prevalence in patients with a single episode of IMD in our media. Prompted by the detection of two cases of C5 deficiency we undertook a systematic search.
Objectives: To determine the prevalence of CPD in patients with a unique episode of IMD and their demographic, clinical and microbiological feature.
Patients and methods: Descriptive cross-sectional study including all patients under 18 years of age diagnosed with a single episode of IMD (January 1997-July 2013). IMD was defined as a positive meningococcal culture and/or PCR detection in peripheral blood and/or cerebrospinal fluid. Complement pathway assessment was performed through haemolytic assays (CH50, AP50) and Enzyme immunoassays (MBL and Ficolin-3).
Results: We included 80 children (M/?F ratio: 1.2; mean age: 27 months; caused by serogroup B (64), serogroup C (9), serogroup Y and serogroup E19 (1 each), 5 isolates could not be serogrouped. Only the initial index cases of C5 deficiency (serogroup Y and E19, respectively) were detected. Both were Mahgreb and consanguinity was present in one family. C5 deficiency was also diagnosed in the brother of the first case and parents from both cases were carriers of the mutation.
Conclusion: At this point of the study only the index cases caused by uncommon serogroups were associated with CDP. Therefore, it seems reasonable to limit this screening to selected cases. However, a national study has started to provide more strong data.
ESID-0330 VPS 45-Associated Primary Infantile Myelofibrosis – Update on Clinical and Laboratory Findings
P. Stepensky 1, A. Saada2, M. Cowan3, A. Tabib4, U. Fischer5, Y. Berkun6, H.A.N.I. Saleh7, N. Simanovsky8, A. Kogot-Levin2, H. Ganaiem6, A. Shaag2, S. Zenvirt2, A. Borkhardt5, N. Bryant3, D. Mevorach4, O. Elpeleg2, M. Weintraub1
1Pediatric Hemato-Oncology and Bone Marrow Transplantation, Hadassah Hebrew University Medical Center, Jerusalem, Israel
2Monique and Jacques Roboh Department of Genetic Research, Hadassah Hebrew University Medical Center, Jerusalem, Israel
3Institute of Molecular Cell & Systems Biology, College of Medical Veterinary and Life Sciences University of Glasgow, Glasgow, United Kingdom
4Rheumatology Research Center and Department of Medicine, Hadassah Hebrew University Medical Center, Jerusalem, Israel
5Department of Pediatric Oncology Hematology and Clinical Immunology, Medical Faculty Center of Child and Adolescent Health Heinrich Heine University, Dusseldorf, Germany
6Department of Pediatrics, Hadassah Hebrew University Medical Center, Jerusalem, Israel
7Pediatric Hemato-Oncology Unit, Augusta Victoria Hospital, Jerusalem, Israel
8Department of Radiology, Hadassah Hebrew University Medical Center, Jerusalem, Israel
Introduction: Severe congenital neutropenia as well as primary myelofibrosis are rare in infancy. Elucidation of the underlying mechanism is important as it can extend our understanding of human hematopoiesis.
Methods and results: Using homozygosity mapping followed by exome sequencing we identified a Thr224Asn mutation in the VPS45 gene in four infants from three unrelated consanguineous families who presented with life-threatening neutropenia with progressive bone marrow failure, defective platelet aggregation and myelofibrosis. The mutation segregated in the families, was not present in controls and affected a highly conserved codon. Introduction of the corresponding mutation into yeast resulted in reduced cellular levels of Vps45 and also of the cognate syntaxin Tlg2 which is required for membrane trafficking through the endosomal system. A defect in the endosomal-lysosomal pathway, the homologous system in human, was suggested by the absence of lysosomes in the patients' fibroblasts. Importantly, accelerated apoptosis was observed in the patients' myeloid cells in peripheral blood and bone marrow. Three children underwent stem cell transplantation which was successful in two cases.
Conclusions: The diagnosis of VPS 45- associated PMF should be considered in all children presenting with severe congenital neutropenia with subsequent development of pancytopenia. VPS 45- associated myelofibrosis is a novel primary immune deficiency which can be successfully corrected by HSCT if applied early in the course of disease using appropriate conditioning. Further investigation is necessary to better elucidate the molecular pathogenesis, clinical spectrum and appropriate therapeutic intervention in this unique form of primary immunodeficiency.
ESID-0473 Recurrent Malignancies in Autosomal Recessive Hyper-IGE Syndrome with DOCK8 Deficiency
N. Suratannon 1, M. van der Burg2, V. Dalm2, M. van Gijn3, J. J.M. van Dongen2, P. van Biezen4, E. Lugtenburg5, M.C. van Zelm2, G.J. Driessen6, P.?M. van Hagen4
1Pediatrics, Chulalongkorn University, Bangkok, Thailand
2Immunogloy, Erasmus MC, Rotterdam, Netherlands
3Medical Genetics, University Medical Center Utrecht, Utrecht, Netherlands
4Internal Medicine, Erasmus MC, Rotterdam, Netherlands
5Hematology, Erasmus MC, Rotterdam, Netherlands
6Pediatric Infectious Disease and Immunology, Erasmus MC, Rotterdam, Netherlands
Background: Autosomal recessive(AR) hyper-IgE syndrome(HIES) is caused by loss- of-function mutations in the DOCK8 gene. The patients are prone to develop recurrent viral infections, autoimmunity and malignancies. The pathophysiology is related to impair B cell, T cell or NKT cell development.
Methods: We identified a 24-year-old woman with AR-HIES caused by a DOCK8 mutation. Flow cytometric analysis of T,B,NK cell subset were performed. Genetic analysis was performed by exome sequencing.
Results: The patient had eczema and recurrent pulmonary infections. At age 19, she developed a large B cell non-Hodgkin’s lymphoma(stage IV). At age 22 she developed Hodgkin’s lymphoma(stage I) which recurrent one year after. Systemic infections of herpes simplex virus, molluscum contagiosum, condyloma acuminata, listeria meningitis and autoimmune hemolytic anemia were found. Flow cytometric analysis revealed increased numbers of CD3+ T cells and CD8+ T cells. Percentages of naive, memory and effector CD8+ T cells were as follows;7.2, 20.7 and 72.1%. CD4+ T cell subsets were normal. Low numbers of Va24+/Vß11+ NKT cell (<0.01% of CD3+ cells) and class switched memory B cells (CD27+IgD-IgM-) 11 cells/µl (13-122) were found. Genetic analysis revealed heterogenous DOCK8 gene mutation: deletion in exon 2-11 and c.5386C>T p(Arg1796) while both parents have the recessive inheritance. She was successfully treated with allogeneic bone marrow transplantation.
Conclusion: We present a DOCK8 deficient patient who developed two different consecutive hematological malignancies. Our findings suggest that dysregulation of CD8+ T cells and NKT cells play important roles on viral suspectibility and tumor survillence in DOCK8 deficient patient.
ESID-0183 Elane Gene Mutations in Hungarian Patients with Severe Congenital Neutropenia
A. Székely 1, B. Tóth1, B. Soltész1, G. Csorba1, L. Maródi1, M. Erdos1
1Department of Infectious and Pediatric Immunology, University of Debrecen, Debrecen, Hungary
Several genetic defects mostly mutations of the ELANE gene causing severe congenital neutropenia (SCN) have been described. We investigated possible mutations of the ELANE, HAX1, GFI1, CSF3R, G6PC3, WAS and CXCR4 genes in 12 patients with SCN. Genomic DNA was extracted from peripheral blood and the candidate gene sequence was amplified by PCR. Nucleotide sequences were determined with an ABI 3130 automated sequencer. All coding exons and flanking intronic sequences of ELANE, HAX1 and GFI1, and hotspot regions of CSF3R (exon 15), G6PC3 (exon 2,3,4), WAS (exon 8,9) and CXCR4 (exon 2) were sequencing. Six patients had recurrent heterozygous mutations in ELANE. In three patients we found a cytosine-adenine substitution (--199C>A) in the promoter of the ELANE gene. Missense mutation (c.182C>T; c.377C>T) was detected in two patients. We found a splice mutation (c.597+1G>A) in one patient. Our data indicate that ELANE mutations may be the most common genetic cause of SCN in Hungary.
ESID-0052 Clinical and Laboratory Profile of Chronic Granulomatous Disease at Bai Jerbai Wadia Hospital, Mumbai
P. TAUR 1, P. Keni1, M. Desai1, B. Agarwal2, N. Shah2, S. Udgire2, M. Madkaikar3, A. Dalvi3, M. Gupta3, S. Prabhu4
1Immunology, B.J.Wadia Hospital for Children, Mumbai, India
2Pediatric Hematology and Oncology, B.J.Wadia Hospital for Children, Mumbai, India
3Pediatric Immunology and Leukocyte Biology, NIIH (ICMR) KEM Hospital Mumbai, Mumbai, India
4General Pediatric, B.J.Wadia Hospital for Children, Mumbai, India
Introduction: Chronic Granulomatous Disease (CGD) is an inherited disorder that affects NADPH oxidase enzyme resulting in defective generation of super oxide by phagocytic cells.
Aims and Objectives: To analyze clinical profile, microbiological spectrum and treatment outcome in CGD.
Methods: Clinical records of 24 children were diagnosed as CGD between 2007 and 2013 were retrospectively analyzed.
Results: Mean age of first presentation was 1 year 5 months (Birth to 10 years) and of diagnosis was 2 years 11 months (2 months to 11 years).M: F ratio was 3.8:1. Pneumonia was present in all followed by abscess in 14/24(58.33%) patients.Osteomylites, diarrhea and UTI was positive in 16.6%, 12.5% and 8.2% respectively.Liver and lung abscess was noted in 1 patient.CNS affection was seen in 3 patients (12.5%).Organisms isolated were S.aureus (25%), Klebsiella (12.5%),salmonella (8.2%) and CONS (12.5%).Tuberculosis was positive in 66.67%;one case was positive for atypical mycobacteria infection. Commonest fungus isolated was Aspergillus in 6 patients(25%) whereas Candida was isolated on 4 occasions(16.67%).One patient had basidiobolus grown in pus at IV site infection.Penicilliosis was twice isolated from temporal and sternal abscess in one case. Consanguinity was seen in 29.17%.All children were put on prophylaxis of Septran and Itraconazole.The mean duration of follow up was 5.2 years.One has undergone BMT and is well.6 patients are lost to follow up and 1 died within one year of diagnosis.
Conclusion: CGD is not an uncommon problem in India.X – linked recessive inheritancies more common in India.Majority presented with pneumonia,abscess and osteomyelitis.All patients should be treated with prophylactic doses of Cotrimoxazole and Itraconazole.
ESID-0043 Clinical and Laboratory Profile of Patients with Severe Combined Immunodeficiency at a Tertiary Care Hospital, Mumbai
P. TAUR 1, P. Keni1, M. Desai1, A. Swami2, S. Mudaliar2, M. Madkaikar3, K. Ghosh3, A. Dalvi3, A. Mishra3, S. Mhatre3
1Immunology, B.J.Wadia Hospital for Children, Mumbai, India
2Pediatric Hematology and Oncology, B.J.Wadia Hospital for Children, Mumbai, India
3Pediatric Immunology and Leukocyte Biology, NIIH (ICMR) KEM Hospital Mumbai, Mumbai, India
Introduction: Severe Combined Immunodeficiency(SCID) represents a group of rare,sometimes fatal,congenital disorders characterized by little or no immune response. SCID is a pediatric emergency and can be cured by stem cell transplant.
Aims and Objectives: To study the clinical,laboratory profile and treatment outcome of patients presenting with SCID at Bai Jerbai Wadia Hospital for Children,Mumbai.
Methods: A retrospective study of 31 patients diagnosed as SCID was done.The clinical record and investigation sheet were analyzed.
Results: The mean age of presentation was 9.9 months(1 month- 4 years)with most of them presenting at 3-4 months of age.There was male predominance with M: F ratio 3.4:1.Consanguinity was noted in 45% patients.
Failure to thrive was most common presenting complaint accounting for 90% of the patients.The other major presenting complaints included skin rashes(51%),persistent or chronic diarrhea(45%),recurrent pneumonia(38%),recurrent pyogenic abscess(32%),otitis media(16%) and oral thrush(16%).3 patients develop BCGosis.
Common microbiological isolates were pseudomonas aeruginosa-4,E.coli-5,cryptosporidium-5,mycobacterium-3,acinetobacter-1 and CSF showed candida in one patient.
Low Absolute lymphocyte count(ALC) was noted in 25 children.The mean ALC was 1581.The lymphocyte subset analysis revealed 54.8%(17/31) B-T-NK+,22.6%(7/?31) B-T-NK- and 22.6%(7/?31)B+T- variety of SCID. Immunoglobulin levels were low in all patients.
Treatment: All patients received IV immunoglobulin,septran and fluconazole prophylaxis.None opted for stem cell transplantation.Only 4 cases are following regularly for last 2 years.Remaining were lost to follow up.
Conclusion: SCID is a medical emergency and should be diagnosed at earliest.Low ALC is an important clue to suspect SCID.LSSA allows us to diagnose SCID and predict possible molecular defect.There is an urgent need to set up BMT facilities for SCID patients.
ESID-0364 A Novel Missense Sequence Variant in the FOXP3 Gene
B. Tóth 1, S. Baráth2, B. Soltész1, Z.S. Pistár1, L. Maródi1
1Department of Infectious and Pediatric Immunology, University of Debrecen, Debrecen, Hungary
2Department of Clinical Immunology, University of Debrecen, Debrecen, Hungary
IPEX syndrome was proposed to result from mutations in the FOXP3. However, additional genetic causes including STAT1 GOF mutation have been proposed to cause IPEX-like syndrome. We have received blood samples of a young infant suspected to have IPEX, and his mother for analysis of Treg cells and gene sequences from a community hospital. The patient died and we could not study clinical phenotype. Investigation of CD4+CD25+bright FOXP3+ Treg cells was performed by using three color flow cytometry. The percentage of Tregs was decreased in the patient compared to that in a healthy control (Pt, 0.2%; control, 4.5%). However, the mean fluorescence intensity of FOXP3 was equal in patient and control Treg cells. gDNA sequences were analysed by amplifying exons 1–11 and the flanking intronic regions of FOXP3 by. We found a hemizygous c.1207G>T nucleotide change in the patient’s sample. This mutation caused glycine-tryptophan amino acid change of protein at position 403 (G403W). The same sequence variant was detected in heterozygous form in the mother’s gDNA sample. This single nucleotide change was not found in 100 alleles of healthy individuals. Considering the conservation of G403 amino acid, harmful efficacy of this variant was predicted by PolyPhen2 software analysis. The c.1207G>T variant localized to the DNA-binding domain of FOXP3, could affect the function of the protein. Taken together, our data suggest albeit do not directly prove that the novel c.1207G>T sequence variant of FOXP3 may be responsible for the manifestation of IPEX syndrome.
ESID-0525 Immune Status of Children with Chronic Granulomatous Disease
T. Uglova 1, S. Aleshkevich2
1clinical recearch, National Research Center for Pediatric Oncology Hematology and Immunology, Minsk, Belarus
2treatment, National Research Center for Pediatric Oncology Hematology and Immunology, Minsk, Belarus
Chronic granulomatous disease (CGD) is a primary immunodeficiency that affects phagocytes of the innate immune system and leads to recurrent or persistent intracellular bacterial and fungal infections and to granuloma formation. Chronic granulomatous disease is characterized by WBCs that cannot produce activated O2 compounds and by defects in phagocytic cell microbicidal function. This impairment in killing is caused by any of several defects in the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex, which generates the microbicidal respiratory burst.Manifestations include recurrent infections; multiple granulomatous lesions of the lungs, liver, lymph nodes, and GI and GU tract; abscesse; lymphadenitis. Diagnosis is by assessing O2 radical production in WBCs via a flow cytometric oxidative burst assay.
We examined the immune status of 18 children with CGD in the dynamics of the disease. The comparison group consisted of 37 children with poor health (PH), in which CGD has been excluded , and 18 children with severe chronic neutropenia (SCN). In children with CGD in the course of clinically significant infections immune status has not changed . The increase of CD3+ HLA- DR lymphocytes at children with CGD compared to PH and SCN (5.7; 2.2 and 2.3 respectively, p<0.01) and CD3+ CD16+ CD56+ lymphocytes (2.7, 0.9 and 0.35, respectively, p<0.01). The absolute number of B- lymphocytes was significantly lower among the children with CGD compared with SCN (0.5 and 1.3 respectively). IgA levels were higher in children with CGD (2.0 g/?l) than in children with PH (0.9 g/?l) and SCN (0.39), p<0.01. IgG levels were not significantly different.
ESID-0291 Nonclinical Studies to Assess Possible Effects of Antibodies to rHuPH20 on the Endogenous Counterpart
R. Veneziale 1, M.?A. Printz1, M. Jorge1, F. Araiza1, B.J. Sugarman1, H.P. Schwarz2, A.?M. Hoberman3, J. Cavagnaro4, G.I. Frost5, F. Drake6, D.C. Maneval1, H.M. Shepard7, P.J. Lapinskas1
1Pharmacology and Safety Assessment, Halozyme Therapeutics Inc, San Diego, USA
2Clinical Strategy BioTherapeutics, Baxter Innovations GmbH, Vienna, Austria
3Global Developmental and Reproductive and Juvenile Toxicology, Charles River Laboratories, Horsham, USA
4Principal, Access BIO, Boyce, USA
5Health Sector, Currently at Intrexon Corporation, San Diego, USA
6Alliance and Program Management, Halozyme Therapeutics Inc, San Diego, USA
7Research, Halozyme Therapeutics Inc, San Diego, USA
The recombinant human PH20 hyaluronidase, rHuPH20, is approved to facilitate absorption of subcutaneously co-administered agents by transiently depolymerizing hyaluronan in the extracellular matrix of the skin. Endogenous PH20 protein is known to be localized to sperm though there have been sporadic reports of PH20 expression in other tissues. Some primary immunodeficiency disease patients treated with subcutaneous IgG facilitated by rHuPH20 can produce antibodies to rHuPH20, so possible risks for anti-rHuPH20 antibodies associated with binding to endogenous PH20 were assessed. Regulatory guidances suggest that nonclinical studies can be informative in the evaluation of anti-therapeutic antibodies on an endogenous counterpart. Nonclinical safety assessment of rHuPH20 included a chronic toxicity study in monkeys and developmental and reproductive toxicity studies in mice. To further understand possible risks of anti-rHuPH20 antibodies, dedicated studies were conducted in the rabbit to assess male and female fertility, embryo-fetal development, and postnatal offspring development through adulthood, including developmental milestones, neurobehavioral endpoints and mating outcome. Anti-rHuPH20 antibodies generated in the nonclinical species were cross-reactive to the species-specific PH20. Anti-rHuPH20 antibodies also neutralized the PH20 enzyme activity in monkey and rabbit. Maternal transfer of anti-rHuPH20 antibodies to the offspring was observed. Prolonged exposure to anti-rHuPH20 antibodies had no effect on fertility or offspring development. Studies in multiple species did not detect safety signals associated with anti-rHuPH20 antibodies. Our approach demonstrates a comprehensive assessment of the potential for effects of antibodies on the endogenous protein
ESID-0681 Novel WASp Mutation in the ARP 2/?3 Binding Domain Results in Abolished In Vitro Actin Polymerisation, but a Mild X-Linked Thrombocytopenia Clinical Phenotype
A. Worth 1, J. Metelo2, D. Moulding2, S. Burns3, A. Thrasher2
1Immunology, Great Ormond Street Hospital, London, United Kingdom
2Molecular Immunology Unit, UCL Institute of Child Health, London, United Kingdom
3Immunology, Royal Free Hospital, London, United Kingdom
Mutations in the WASp gene lead to a spectrum of clinical phenotypes from full Wiskott Aldrich Syndrome (WAS) to clinically insignificant X-linked thrombocytopenia (XLT). WASp is a regulator of the actin cytoskeleton, initiating actin filament nucleation via the Arp2/?3 complex. Here we describe the clinical, immunological and biochemical consequences of a novel WASp mutation (W500R) in the Arp2/?3 binding domain. Patient A presented at 8 months with persistent thrombocytopenia (15-40x109/L), eczema and recurrent upper respiratory tract infections. He had normal lymphocyte subsets, normal vaccines responses (including polysaccharide), normal PHA T-proliferative response, but an impaired response to anti-CD3 stimulation. WASp protein expression was normal, but WASp gene sequencing revealed a missense mutation in a highly conserved residue in the VCA domain. Patient A has 5 male relatives with low platelet numbers but no evidence of immunodeficiency/immunedysregulation (WASp-W500R confirmed in grandfather). In vitro actin polymerisation activity of W500R-WASp was completely inhibited, but retained in WASp-A134T, a pathological missense mutation in the WASp EVH1 domain. WASp immunoprecipitation from patient derived LCL lysates revealed normal WIP, but impaired CDC42 binding (reverse pattern to WASp-A134T). Monocyte derived dendritic cells demonstrated impaired but not abolished podosome formation (% cells with podosomes; WT 66.5%, W500R 13.6%), and reduced podosomes per cells (W500R 12.3, WT 35.2). Despite abolition of WASp induced actin polymerisation, patients with WASp-W500R mutation have a mild XLT clinical phenotype and retain some ability to form podosomes. This demonstrates the functional importance of domains other than the VCA domain (e.?g. EVH1 domain) in WASp function.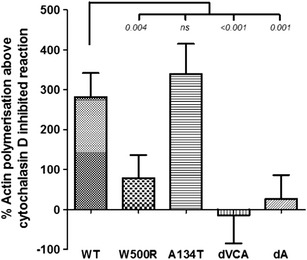
Figure?1: Quantification of actin polymerisation using a bead based in?vitro ATP induced actin polymerisation assay. WT - wild type, W500R, A134T - missense mutations, dVCA - WASp with entire VCA domain (p.430-502) deleted, dA - WASp with acidic tail sequence (p. 487-502) deleted.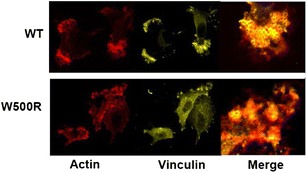
Figure?2: Monocyte derived immature dendritic cells wer plated on fibronectin coated coverslips and stained for f-actin and vinculin. Podosome formation was assessed using confocal microscopy. Images represent typical podosome positive cells.
ESID-0102 A Case of Neutrophil-Specific Granule Deficiency Presenting with Recurrent Pyogenic Infection of the Skin; Pathological and Molecular Analysis of a Novel C/?EBP? Mutation
A. Yachie 1, M. Muraoka1, T. Wada1, Y. Sakakibara1, T. Toma1, N. Akagi2, T. Yokota2
1Pediatrics, Kanazawa University Graduate School of Medical Sciences, Kanazawa, Japan
2Stem Cell Biology, Kanazawa University Graduate School of Medical Sciences, Kanazawa, Japan
Neutrophil-specific granule deficiency (SGD) is a rare autosomal recessive primary immunodeficiency with neutrophil dysfuntion. Defects in a myeloid-specific transcription factor, CCAAT/enhancer binding protein epsilon (C/?EBPe), have been identified in previously reported cases. The gene mutation results in impaired production of neutrophil granular proteins, such as lactoferrin and defensins. Patients with SGD usually present with severe, chronic cutaneous infections. Only limited number of cases has been reported to date and there is little information regarding the relationship between the clinical phenotype and the gene mutation. We recently experienced a 55 years-old female case of SGD who presented with life-long history of recalcitrant skin infections with ulcer and scar formation. The patient’s elder brother who died at the age of 11 had similar skin symptoms. Peripheral blood smear showed characteristic changes, including reduction of cytoplasmic granule and increased bilobed nuclei of the neutrophils, monocytosis, absence of eosinophils and increase of basophils. Flowcytometric examination revealed significant reduction of CD16 and abnormal expression of CD14 on the neutrophil surface. C/?EBPe gene examination revealed homozygous deletion of 6 base-pairs within leucine zipper domain of the molecule. The functional analysis of the mutant molecule showed marked reduction in transcriptional activity, but with relatively normal DNA binding capacity. Further analysis is underway to disclose the mechanism of reduced function of the molecule and its relevance to the characteristic clinical pictures of this rare disease.
ESID-0126 Clinical and Genetic Investigation of Two Sisters with Severe Recurrent Seborrheic Dermatitis
N.?B. Zurro 1, E.B. de Oliveira1, V.M. Dantas2, A. Condino-Neto1
1Immunology, Institute of Biomedical Sciences University of São Paulo, São Paulo, Brazil
2Immunology, Federal University of Rio Grande do Norte, Natal, Brazil
Case Report: We report on an unusual clinical case of two sisters (15 and 17 year old) born from a family of consanguineous parents. The 17 year old patient developed the first symptoms at 9 months of age with pioderma and skin abscess as a consequence of severe seborrheic dermatitis and exhibiting for the remaining 13 years, subsequent infectious episodes as otitis, pneumonia, skin infections and a relevant liver abscess due to S.?aureus. The younger sister, presented an abscess in the cervical lymph node region at 4 months of age. She developed recurrent otitis and pneumonia accompanied by several events of cystitis. Interestingly, both sisters presented recurrent severe seborrheic dermatitis (Figure?1).
Results: Evaluation of superoxide relase by the cytochrome c test in the oldest patient revealed that the monocytes (UNSTIMULATED: 1.170 PMA: 0.275) fail to produce superoxide after activation with PMA (30 nM). Additionally the evaluation of oxidative burst by DHR test in the younger sistert revealed that the neutrophils (UN: 38.21 PMA: 207.84) and monocytes (UN: 637.80 PMA: 719.85) are defective to reduce DHR afteractivation with 90 nM PMA. Sequencing of NCF1 gene revealed a frameshift deletion (?GT) at the beginning of exon 2 creating a premature stop codon (p.Tyr26fsX26), the same mutation was found in both sisters.
Conclusion: Severe and recurrent seborrheic dermatitis may be an unusual clinical manifestation in CGD.
Figure?1: Patient (13 years old) with severe seborrheic dermatitis.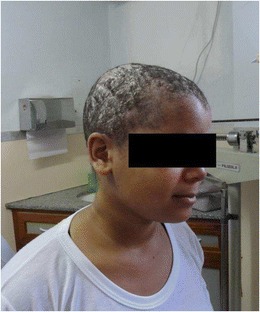
ESID-0144 Severe Enterovirus 71 Infection in Children with Toll-Like Receptor 3 Deficiency
C.Y. Kuo1, C.C. Lo2, H.K. Lim3, J.L. Casanova3, T.Y. Lin1, S.Y. Zhang3, C.L. Ku 2
1Department of Pediatrics, Chang Gung Children's Hospital, Taoyuan, Taiwan
2Graduate Institute of Clincal Medical Sciences, Chang Gung University, Taoyuan, Taiwan
3St. Giles Laboratory of Human Genetics of Infectious Diseases, The Rockefeller University, New York, USA
Background:
The spectrum of diseases of childhood caused by enterovirus 71 (EV71) is broad, ranging from asymptomatic infection or self-limited hand-foot-and-mouth diseases (HFMD) to life-threatening encephalitis. However, the molecular mechanisms underlying these different clinical presentations remain unknown. We hypothesized that severe EV71 infection in children might reflect an intrinsic host single gene defect of anti-viral immunity. We searched for mutations in Toll-like receptor 3 (TLR3), which have been previously found to be mutated in children with herpes simplex encephalitis (HSE), in young patients with severe EV71 infection.
Method:
We sequenced TLR3 and tested the impact of the mutations found. We tested dermal fibroblasts from our patient and other patients with known genetic defect in TLR3 or related genes, for their response to both poly(I:C) stimulation and EV71 infection.
Result:
We found that three children were heterozygous for the TLR3 W769X, E211K and R867Q mutations, which were shown to have severe impact on the TLR3 function. The patient’s fibroblasts with heterozygous W769X mutation had impaired but not abolished response to TLR3 agonist poly(I:C). We also showed that TLR3 was crucial in cellular defense against EV71 infection, as complete TLR3-deficient or TLR3 heterozygous W769X fibroblasts were highly vulnerable to EV71 infection.
Discussion:
TLR3 deficiency may underlie severe EV71 infection. Children with severe EV71 infection should be tested for inborn errors of TLR3 immunity. Children with such disorder may be at risk of viral diseases other than HSE.
ESID-0333 ACT1 Deficiency: A Defect in IL-17 Mediated Immunity Underlying Chronic Mucocutaneous Disease (CMCD) in a Multiplex Consanguineous Kindred
R. Levy 1, N. Mahlaoui2, M. Migaud1, J.L. Casanova3, A. Puel1
1Necker Branch INSERM 1163 University Paris Descartes Imagine Institute, Laboratory of Human Genetics of Infectious Diseases, PARIS, France
2Service d’Immuno-Hématologie Pédiatrique, CEREDIH Centre de Référence des Déficits Immunitaires Héréditaires Hôpital Universitaire Necker-Enfants Malades, PARIS, France
3Rockefeller Branch the Rockefeller University, St. Giles Laboratory of Human Genetics of Infectious Diseases, NEW YORK, USA
Chronic mucocutaneous candidiasis disease (CMCD) is characterized by persistent or recurrent symptomatic infection of the mucosae, skin, and nails mostly caused by Candida albicans, occurring in patients with usually no other prominent clinical manifestations. Whereas, heterozygous gain-of-function mutations of STAT1 were recently discovered and found as the main genetic etiology, autosomal recessive (AR) IL-17RA deficiency and partial autosomal dominant IL-17F deficiency were the first two genetic etiologies to be published in 2011. More recently, a fourth genetic defect has been identified in two siblings with AR CMCD and bearing a biallelic missense mutation in the adaptor molecule ACT1. This missense mutation, located in the SEFIR, impaired the homotypic interaction of ACT1 with the IL-17 receptors abolishing the response to IL-17A and IL-17F in fibroblasts and to IL-17E in leukocytes. In a large cohort of CMCD patients, we identified two additional patients, born to Algerian first cousins, suffering from early onset of CMC (oral thrush) and seborrheic dermatitis on scalp. In addition, one patient suffered from intertrigo on hands and feet. Whole-exome sequencing revealed a homozygous missense mutation of ACT1 (R542W). The familial segregation by Sanger sequencing confirmed an AR pattern of inheritance. This variant affects the SEFIR domain of the protein and was not found in any public databases or in our in-house exome database (>1800 exomes). This variant might impair the protein expression and/or be responsible for a defect in the homotypic interaction with IL-17 receptors. Functional validation is currently in progress in the patients’ PBMCs and fibroblasts.
ESID-0231 Gain-of-Function STAT1 Mutation Impairs STAT3 Function Predisposing To Chronic Mucocutaneous Candidiasis
J. Zheng1, F.L. van de Veerdonk2, K.L. Crossland3, S.P. Smeekens2, M. Abinun4, A.?R. Gennery4, J. Mann5, D. Lendrem3, M.N. Netea2, A.?D. Rowan3, D. Lilic 1
1Primary Immunodeficiency Group Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
2Internal Medicine, Radboud University, Nijmegen, Netherlands
3Musculoskeletal Research Group Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
4Paediatric Immunology, Great North Childrens Hospital, Newcastle upon Tyne, United Kingdom
5Fibrosis Research Group Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
STAT3 activation triggers transcription of interleukin (IL)-17 which is crucial for mounting protective immune responses against fungi. Several mutations affecting the STAT3/IL-17 pathway have been reported, resulting in selective susceptibility to fungal (Candida) infection, a hallmark of Chronic Mucocutaneous Candidiasis (CMC). In patients with autosomal-dominant (AD)-CMC we previously reported defective Th17 responses and identified an underlying gain-of-function (GOF) STAT1 mutation leading to hyper-phosphorylation of STAT1. How this affects STAT3 or leads to decreased IL-17 remains to be determined. In patients with AD-CMC, we assessed how GOF-STAT1 mutations affect STAT3 activation, DNA-binding, gene expression, cytokine production and the effect of epigenetic modification. We show that stimulation of STAT3 in the presence of GOF-STAT1 mutations leads to significantly reduced transcription of STAT3-inducible genes (RORC/?IL-17/IL-22/IL-10/c-Fos/SOCS3/c-Myc). This was not due to impaired STAT3 phosphorylation, altered nuclear translocation nor sequestration of STAT3 into STAT1/STAT3 heterodimers. DNA binding to a STAT-consensus binding site construct (hSIE) was intact but binding to an endogenous STAT3 DNA target was impaired. The reduced STAT3-dependent gene transcription could be normalized by inhibiting STAT1 activation with fludarabine or enhancing acetylation with histone deacetylase (HDAC) inhibitors trichostatin A or ITF2357. Silencing HDAC1, HDAC2 and HDAC3 indicated an important role for HDAC1. Impaired STAT3-dependent gene transcription likely underlies decreased Th-17 cytokine production, susceptibility to fungal infections and other pathology seen in AD-CMC patients and could define novel therapeutic approaches for this potentially lethal disease.
ESID-0705 Detection of Low Frequency Variants of NLRP3 in German "Mutation-Negative" CAPS Patients by Massive Parallel Sequencing
M. Lesche1, A. Kränkel1, A. Dahl1, J. Roesler2, A. Rösen-Wolff 2
1Deep Sequencing Group SFB655, BIOTEC TU Dresden, Dresden, Germany
2Department of Pediatrics, Universitätsklinikum "Carl Gustav Carus", Dresden, Germany
Somatic mosaicism of NLRP3 has been identified in a high percentage of 'mutation-negative' patients suffering from chronic infantile neurologic, cutaneous, articular (CINCA) syndrome. The aim of the present study was to detect and quantify low frequency variants of NLRP3 in German patients suffering from cryopyrin associated periodic fever syndromes (CAPS) including CINCA, Muckle-Wells syndrome (MWS), and familial cold autoinflammatory syndrome (FCAS). All exons of NLRP3 were amplified by PCR from genomic DNA isolated from PBMCs of healthy controls or CAPS patients who signed written informed consent. Thereafter, PCR products were analyzed by Illumina short read sequencing. For SNV calling a customized pipeline on basis of the GATK pipeline (1000 Genomes project) was utilized using a 40.000x coverage to assure sufficient sensitivity. In order to determine the accuracy of quantification, PCR products containing a known heterozygous mutation (T348M) were mixed with NLRP3 wildtype PCR products to obtain dilutions of the mutated sequences. We were able to exactly quantify (5%) the diluted low frequency mutation (T348M). In one CINCA patient a new variant (L359S) was detected in 30% of the DNA sequences that had not been identified by classical Sanger sequencing of an older sample. Up to now we could not detect low frequency NLRP3 variants in MWS or in FCAS patients. Massive parallel sequencing is a reliable method to quantify low frequency variants of NLRP3.
This study was supported by Novartis Pharma GmbH, Nürnberg, Germany (ARW); BMBF project NGSgoesHPC (ML), and SFB655 (DFG) (AD, AK).
ESID-0575 Hemophagocytic Lymphohistiocytosis in Two Patients with Interferon-Gamma Receptor Deficiency
B. Tesi 1, E. Sieni2, C. Neves3, F. Romano4, A.I. Cordeiro3, C. Canessa4, S. Chiang5, V. Cetica2, H. Schlums5, J.I. Henter1, M. Nordenskjöld6, A.P. Hsu7, S.M. Holland7, Y.T. Bryceson5, J.F. Neves3, C. Azzari4
1Childhood Cancer Research Unit Department of Women's and Children's Health, Karolinska Institutet Karolinska University Hospital, Stockholm, Sweden
2Department Pediatric Hematology Oncology, Meyer Children's University Hospital, Florence, Italy
3Primary Immunodeficiencies Unit Hospital Dona Estefânia, Centro Hospitalar de Lisboa Central, Lisbon, Portugal
4Division of Pediatric Immunology Department of Health Sciences University of Florence, Anna Meyer Children's Hospital Jeffrey Modell Center for Primary Florence Italy Immunodeficiencies, Florence, Italy
5Centre for Infectious Medicine Department of Medicine, Karolinska Institutet Karolinska University Hospital Huddinge, Stockholm, Sweden
6Clinical Genetics Unit Department of Molecular Medicine and Surgery and Center for Molecular Medicine, Karolinska Institutet Karolinska University Hospital, Stockholm, Sweden
7National Institute of Allergy and Infectious Diseases, NIH Bethesda, MD, USA
Hemophagocytic Lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome associated with mutations in genes required for lymphocyte cytotoxicity. Experiments in animal models have suggested a key role for interferon (IFN)-? in the pathogenesis of HLH. Therefore, IFN-? represents an appealing pharmacological target. We report a 2-month-old infant and a 4 year-old child first diagnosed with fulminant HLH and subsequently with disseminated Mycobacterium bovis following BCG vaccination or Mycobacterium tuberculosis infection, respectively. Remarkably, genetic analyses identified homozygous IFNGR2 deletions and homozygous missense IFNGR1 mutations in the respective patients, whereas no mutations were identified in genes required for lymphocyte cytotoxicity. In one patient, cellular analyses confirmed abrogated IFN-?-mediated STAT1 phosphorylation, whereas cytotoxic lymphocyte degranulation and perforin expression were normal. Interestingly, a paucity of peripheral blood NK cells was noted in both patients and NK cell activity was defective in the one tested. Our findings of fatal HLH in patients with IFN-? receptor-deficiency argue for the use of refined functional assays in the clinical diagnosis of HLH and indicate that mechanisms other than IFN-? can contribute to the pathophysiology of the disease. The latter observation may be of significance in the context of potentially treating HLH with anti-IFN-? therapy.
ESID-0117 Novel Compound Heterozygous Mutation in STAT2 in Two Patients Suffering from Severe Viral Infections
L. Van Eyck 1, G. Frans1, L. Moens1, D. Pombal1, I. Meyts1, A. Liston1
1Microbiology and immunology, KU Leuven, Leuven, Belgium
Introduction
STAT2-deficiency has been proposed as the cause of unusually severe viral illness in infancy in two siblings who developed disseminated vaccine-strain measles following routine immunization and of whom the infant sibling died from an unknown viral infection. STAT2-deficiency was associated with profound failure of type I interferon signaling, which is thought to be responsible for the observed defect in innate antiviral immunity.
Methods
Whole-exome sequencing was performed on a 9-year-old girl who presented with frequent and severe viral infections from birth including complicated measles following immunization with measles-mumps-rubella vaccine at the age of 12 months.
Results
We identified a compound heterozygous p.G522R and p.R506X mutation in STAT2 leading to STAT2-deficiency in the index patient. Beside vaccine-strain measles, she experienced frequent viral infections of varying severity including enteroviral meningitis, recurrent varicella zoster infection, persistent warts and mollusca contagiosa. She also showed persistently high Epstein-Barr virus copy numbers in blood and cerebrospinal fluid. The mutations were confirmed by Sanger sequencing in the index patient and in a male sibling who presented in a similar way. He had a relatively infection-free window from the age of 4 years on but then succumbed to an infection with an unidentified agent at the age of 7 years.
Conclusion
This finding confirms STAT2-deficiency as a cause for a defect in innate antiviral immunity. It demonstrates that, although the frequency of viral infections decreases with increasing age, patients may still be at risk for overwhelming viral illness at a later age.
ESID-0793 Necrotising Pneumococcal Pneumonia Due to Inhibitory Anti-IL6 Autoantibodies?
U. Koelsch1, N. Unterwalder1, N. Durmus2, M. Bauer3, T. Renner2, S. Seeliger2, S. Rose-John4, C. Meisel1, H. von Bernuth 3
1Immunology, Labor Berlin, Berlin, Germany
2Children's Hospital, Kliniken St. Elisabeth, Neuburg an der Donau, Germany
3Pediatric Pneumology & Immunology, Charité University Medicine, Berlin, Germany
4Institute of Biochemistry, Christian Albrecht University, Kiel, Germany
A previously healthy 6 years old boy presented with necrotising pneumococcal pneumonia, recurring pleural effusions, lobar pleural atelectasis, abscess formation and attenuated acute phase responses. Despite immediately started antibiotic therapy a lobar resection of the lung became necessary. The combination of severe pneumococcal pneumonia and attenuated acute phase response was suspicious for a defect in Toll-like receptor and IL-1 receptor signalling. However the patient’s granulocytes showed normal CD62L-shedding upon activation with agonists for TLR2/?6 (PAM2CSK4) and TLR4 (LPS). In addition IL-8, IL-10 and TNFa-production were normal upon stimulation with TLR-agonists and IL-1ß in whole blood. In contrast IL-6-production upon activation with TLR-agonists could neither be detected in the supernatant of whole blood nor in the supernatant of mononuclear cells of a healthy control incubated with patient´s serum. IL-6-specific sandwich ELISA detected a human anti-IL-6-IgG1-autoantibody. This autoantibody inhibited IL-6 mediated STAT3 phosphorylation. In summary we here describe the first child with severe pneumococcal infection in whom autoantibodies against IL-6 were detected. The role of anti-IL-6-autoantibodies remains to be investigated in its biological significance and implications up to now. However since IL-6-signalling plays an important role in triggering acute phase responses it may be hypothesized that autoantibodies against IL-6 contribute to low CrP-values and eventually lead to the development of severe infections.
Topic: Therapy
ESID-0274 Treatment Options for Severe Immunodeficiency Due to Gain-of-Function STAT-1 Mutation
M. Abinun 1, A. Cant1, L. Strain2, D. Lilic3
1Paediatric Immunology, Great North Children's Hospital, Newcastle upon Tyne, United Kingdom
2Northern Molecular Genetics Service, Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom
3Institute for Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
At ESID 1994, we reported a trial with interferon-gamma (IFN?) treatment (50 ug/m^2 3xweek for 4 months, followed by 100 ug/m^2/day) in a child with severe isolated chronic mucocutaneous candidiasis (CMC). There was no clinical benefit and treatment was stopped after 6 months because of fever, diarrhoea, vomiting, fatigue, thrombocytopaenia, leukopaenia, rise in transaminases and derranged clotting. However, cytokine production by peripheral blood mononuclear cells stimulated with Candida-antigens assessed before and after 2-4 months of IFN? therapy showed marked reduction of very high pre-treatment levels of IL-6 and moderate increase of IFN? levels, both returning to pre-treatment levels after cessation of IFN? treatment (1).
By the age 6 yr, she had debilitating nail, oral and oesophageal CMC, chronic lung disease, required surgical correction of oesophageal strictures, naso-gastric tube feeding, parenteral nutrition and progressive ‘combined immunodeficiency’ (decrease of serum IgA and M (on IVIg), CD3/CD4 T lymphocytes, and Candida-antigen induced proliferative responses in?vitro). During assessment for allogeneic haematopoietic stem cell transplantation (HSCT), she died following a liver biopsy.
In 2012, from a stored DNA sample we found a heterozygous mutation in the DNA-binding domain of STAT1 known to be gain-of-function (GOF) (exon 11; c.961A>G (p.Arg321Gly)).
The clinical spectrum of GOF-STAT1 mutations is heterogeneous. For the severe end of the spectrum, treatment options include G(M)-CSF (2) and allogeneic HSCT (3).
1. Abinun M et?al. Progress in immune deficiency V, Springer-Verlag Iberica, Barcelona, 255, 1995
2. Wildbaum G et?al. JACI 2013;132:761
3. Adave JC et?al. J Clin Imm 2013;33:1273
ESID-0739 Hematopoetic Stem Cell Transplantation (HSCT) for Primary Immunodeficiency Diseases (PIDS): Clinical Features and Outcome (A Single Center Experience)
A.Z. Akelma1, F. Dogu 2, S. Haskologlu2, F. Cipe3, C. Aytekin4, I. Reisli5, M. Yuksek6, A. Yildiran7, M. Aydogan8, D. Guloglu2, T. Kendirli9, A. Ikinciogullari2
1Pediatric Allergy and Immunology Unit, Kecioren Teaching and Research Hospital, Ankara, Turkey
2Department of Pediatric Allergy and Immunology, Ankara University Medical School, Ankara, Turkey
3Pediatric Immunology Unit, Kanuni Sultan Suleyman Research and Training Hospital, Istanbul, Turkey
4Pediatric Immunology Unit, Dr. Sami Ulus Maternity and Children's Health and Diseases Training and Research Hospital, Ankara, Turkey
5Department of Pediatric Allergy and Immunology, Necmettin Erbakan University Meram Medical School, Konya, Turkey
6Department of Pediatric Allergy and Immunology, Bulent Ecevit University Medical School, Zonguldak, Turkey
7Department of Pediatric Allergy and Immunology, Ondokuz Mayis University Medical School, Samsun, Turkey
8Department of Pediatric Allergy and Immunology, Kocaeli University Medical School, Kocaeli, Turkey
9Pediatric Intensive Care Unit, Ankara University Medical School, Ankara, Turkey
Background: Hematopoietic stem cell transplantation (HSCT) is the single best curative treatment defined for Primary immunodeficiency (PID) so far. In the current study, we aimed to assess the role of PID type, donor type and clinical status on HSCT success rates.
Materials and Method: We retrospectively reviewed the records of a total of 95 HSCTs procedures performed in 72 patients diagnosed (41 SCID and 31 Non-SCID) with PID between October 1997 and June 2014, in Ankara University Medical School Department of Pediatric Allergy and Immunology.
Results: When classified according to the source of donor, patients who had a HLA-matched sibling donor (MSD) in the SCID group had 91.6% those who had a matched related donor (MRD) had 85.7% and those who received a haploidentical donor transplantation had 63.6% survival rates following the transplantations. We assessed several potential risk factors associated with survival in SCID patients. Patients diagnosed over 6 months of age with a pre-existing pulmonary infection, requiring intensive care and/or mechanical ventilation had significantly lower survival rates.
Conclusions: Our results demonstrated once more that HSCT performed from matched family donor is a lifesaving treatment in various types of PIDs, either SCID or Non-SCID while haploidentic setting seems to be a reliable choice of treatment in SCID.
ESID-0053 Comparing the Use of Subcutaneous Versus Intravenous IG Replacement Therapy in Three Adult CVID Patients from Kuwait
M. Al-Ahmad 1, A. Maher1
1Allergy Department, Al-Rashed Allergy Center, Sulibikhat, Kuwait
Introduction: CVID is the most common symptomatic PID among adults. Both SCIG and IVIG are an available effective replacement therapy. No data available on SCIG use in the Arabian Gulf area
Methods: To compare efficacy, safety, and tolerability between IVIG and SCIG replacement therapy in three adult CVID patients
Results: We had two females, and one male, with a mean age of 26 years. All subjects had previously received IVIG treatment for average duration of 5 years, with a mean serum IgG trough level of 500 mg/dl. Patients were shifted to SCIG for many reasons. SCIG infusions were given under medical supervision at the hospital, on a weekly basis. The annual infection rate, like pneumonias, sinusitis, otitis media and others, have declined significantly after switching to SCIG in all three patients. There was also an increase in IgG plasma concentration following the switch, with more steady-state level (average 668.9 mg/dl). There was a statistically significant increase in the mean IgG trough level at weeks 16, 24, and 36 compared with the mean IgG trough level during IVIG treatment (p
Conclusion: Remarkably, all the 3 subjects were successfully switched to SCIG with significant improvement in annual rate of infections and more steady state of serum trough level of IG.
ESID-0072 Outcome of Hematopoietic Stem Cell Transplantation (HSCT) in Adolescents and Young Adults with Primary Immunodeficiencies
M. Albert 1, G. Notheis1, F. Hauck1, M. Führer1, C. Klein1, I. Schmid1
1Pediatric Hematology/Oncology/Immunology, Dr von Hauner Childrens Hospital, Munich, Germany
Adolescents and young adults (AYAs) with leukemia have a significantly worse outcome after allogeneic HSCT if compared to children due to an increased risk for transplant related mortality. This may be one of the reasons why AYAs with primary immunodeficiencies (PID) are less frequently referred for HSCT even though they share the same dismal natural disease outcome.
To assess the outcome of AYAs with PID we retrospectively analyzed a cohort of 17 consecutive AYAs (15-25 years) transplanted from MSD/MFD (n=3) or MUD (n=14) between 2004 and 2013 and compared them to 49 consecutive children with PID (17 MSD/MFD, 24 MUD, 8 MMFD) transplanted at the same center during that period.
Four of the AYAs and one pediatric patient had developed a malignancy before HSCT. Median age at transplant was 18 in AYAs and 3 years in the pediatric cohort. After a median follow up of 2,8 years 14/17 patients (82%) in the AYA and 42/49 (86%) in the pediatric cohort are alive and disease-free (p=0,74). Causes of death were IPS, aspergillosis, and adenovirus (1 each) in the AYA cohort and VOD (2), adenovirus (2), sepsis, and BCG (1 each) in the pediatric cohort. Acute GVHD °III-IV or extensive chronic GVHD was not observed.
Disease free survival after allogeneic HSCT for AYAs with PID is comparable to that of pediatric patients with the same diseases using current transplant protocols and supportive measures. Age above 15 years should therefore not be considered an a priori contraindication for HSCT in patients with PID.
ESID-0142 Practice of Intravenous Immunoglobulin Therapy Among Pediatric Patients in a Malaysia University Hospital
A. ALI 1, Y.K. LAI2, L.M. NOH1
1Department of Pediatric, Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia
2Department of Pharmacy, Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia
Introduction: Intravenous immunoglobulin (IVIG) is used for a growing range of clinical indications. The treatment is costly with limited supply; thus a need for clear evidence of whether the practice is indicated and effective.
Objective:To evaluate the utilization of IVIg among pediatric patients at Universiti Kebangsaan Malaysia Medical Center (UKMMC).
Methods: Five year (2009-2013) chart review of pediatric patients (from birth to 12 years) who received IVIg treatment was conducted.
Results: One hundred and seventeen patients were reviewed, with 52% were female. The clinical diagnoses included hematology (27%), neonatal sepsis (25%), autoimmune (21%), infection (other than neonate) 18%, neurology (8%) and immunodeficiency (2%). The indications for IVIg had level evidence of I in 42% of cases, while 30% of level IIa/IIb/IIc and 25% of level III. In regards to adverse reaction, one patient with Steven Johnson Syndrome had generalized tonic clonic seizures, while 9 patients with Kawasaki disease, 6 of them had new onset fever and 3 had worsening of the pre-existing rash. 73% of cases had complete resolution, 8% had chronic morbidity (5 patients with Acute Immune thrombocytopenia Purpura(ITP) progress to Chronic ITP and 4 patients with chronic demyelinating disease had residual neurological sequela) while 12% passed away (all due to severe sepsis mostly from premature cohort).
Conclusion: Practice of IVIg therapy among pediatric patients in UKMMC were mostly indicated with good level of evidence. There was minimal adverse effect documented with a satisfactory clinical outcome except for neonatal sepsis that had proven in late 2011 to have no clinical benefit.
ESID-0100 Rituximab Treatment in Two Patients with Anti-Interferon Gamma Autoantibody-Associated Disseminated Mycobacterial Infections
J.J. Bosco 1, K. Nicholls2, J. McComish2, K. Bleasel2, G. Unglik2, S. Choo3, K. Flanagan4, D.A. Fulcher5, G. Naselli6, E. Martinuzzi7, S.M. Holland8, S.K. Browne8, L.C. Harrison6
1Molecular Medicine Division, The Walter and Eliza Hall Institute of medical Research, Melbourne, Australia
2Department of Clinical Immunology and Allergy, Royal Melbourne Hospital, Melbourne, Australia
3Immunology Laboratory, Royal Children's Hospital, Parkville, Australia
4Infectious Disease Service, Launceston Hospital, Tasmania, Australia
5Immunopathology and Flow Cytometry Unit, Institute of Clinical Pathology and Medical Research, Westmead, Australia
6Molecular Medicine Division, The Walter and Eliza Hall Institute of Medical research, Melbourne, Australia
7Endocrinology Metabolism and Diabetes, INSERM, Paris, Australia
8Laboratory of Clinical Infectious Diseases, National Institutes of Health, Bethesda, Australia
Disseminated non-tuberculous mycobacterial (NTM) infections typically occur in the context of iatrogenic immunosuppression or inherited genetic mutations. Rarely, cytokine-specific autoantibodies have been described in patients with disseminated infections. Here, we present two unrelated healthy adult females with disseminated NTM infection associated with autoantibodies to interferon-gamma (IFN-?) that responded to B-cell depletion therapy with rituximab after an initial failure of conventional antimycobacterial treatment.
The two HIV-negative patients had Mycobacterium avium and Mycobacterium abscessus infections, respectively. They presented with subacute multifocal bone, lung and lymph node involvement. Routine studies revealed normal immunoglobulin concentrations, T-cell proliferation to mitogens and granulocyte oxidative burst.
Blood mononuclear cell stimulation analyses with cytokine bead array revealed normal IL-12p40 production and IFN-? production in response to exogenous IL-12. Both patients' mononuclear cells had normal tumour necrosis factor-a (TNF-a) production in response to IFN-? that was abrogated in the presence of autologous serum. By ELISA, both patients had high titre (>1:5000) anti-IFN-? IgG in the serum. The patients' IgG inhibited IFN-?-induced MHC class II protein upregulation on CD14+ monocytes.
Both patients suffered disease progression in spite of with appropriate antimycobacterial therapy. High dose IFN-? was empirically added for one patient. Following rituximab treatment, both patients had significant clinical improvement and successful withdrawal of other therapy. Disease resolution corresponded with decreased anti-IFN-? antibody titers.
Autoantibodies against cytokines may focally disrupt normal immunity and predispose to more severe and resistant NTM disease. In the latter circumstances, screening for anti-IFN-? autoantibodies may clarify the pathophysiology and allow targeted therapy.
ESID-0375 Persistent Hypogammaglobulinemia Following Rituximab (ANTI-CD20) Treatment After Pediatric Allogeneic Stem Cell Transplantation
R.G.M. Bredius 1, A. van der Zwan1, M.C. van Zelm2, M.M. van Ostaijen-ten Dam1, C.M. Jol-van der Zijde1, M.J.?D. van Tol1, A.?C. Lankester1
1Pediatrics, Leiden University Medical Center, Leiden, Netherlands
2Immunology, Erasmus MC, Rotterdam, Netherlands
Post-transplant lymphoproliferative disease (PTLD) following Epstein-Barr virus (EBV) reactivation is a potentially fatal complication after allogeneic hematopoietic stem cell transplantation (HSCT). Patients suffering from EBV reactivation are treated with anti-CD20 (Rituximab). This results in complete depletion of B-cells, which recover after several months upon disappearance of Rituximab from the circulation.
In total 34 patients were treated with Rituximab for EBV post-HSCT. Recipient age, HSCT related factors and B-cell counts at the time of Rituximab infusion did not seem to have an effect on Rituximab clearance. In children receiving a single Rituximab infusion (375 mg/m2), time to Rituximab clearance was highly variable ranging from 7 to 37 weeks, which correlated with the repopulation dynamics of B-cells. Rituximab had no impact on the recovery of CD4+ nor CD8+ T-cells. Three patients developed severe hypogammaglobulinemia, one with lymphadenopathy and mild arthritis, despite recovery of B-cells and T-cells of donor origin. All three showed a lack of switched memory B-cells without accumulation in other B-cell differentiation stages. Compared to controls no difference was detected in the capacity of T-cells to upregulate activation markers upon in?vitro stimulation. In cultures of PBMC, B-cells did produce low levels of IgG and IgA.
In conclusion, Rituximab treatment for EBV leads to intensive but transient B-cell depletion in the majority of patients, whereas a subgroup of patients develop severe irreversible hypogammaglobulinemia. It remains as yet unknown whether this is caused by an intrinsic B-cell defect or an imbalanced T-B cell collaboration.
ESID-0151 Adverse Reactions of Intravenous Immunoglobulin Replacement Therapy in Primary Immune Deficient Patients; A 12 Year Experience of a Single Center
Y. CAMCIOGLU 1, E. Ozek1, H. Çokugras1, N. Akcakaya1
1Pediatric allergy and Clinical Immunology, Istanbul UNiversity Cerrahpasa Medical Faculty, Istanbul, Turkey
IVIG is a biological product derived from donors blood, therefore there are some adverse reactions associated with IVIG administration. Adverse reaction rates were reported between %1 to %40, occuring within 72 hours after initiation of the infusion which can be categorized as mild, moderate or severe.
The aim of the presentation is to evaluate adverse reactions to IVIG in 138 patients with PID who had received IVIG in the department during a 12 year period (2000-2012). Twenty five patients had hypoglobulinemia, 12 XLA, 23 CVID, 5 HIGM, 4 HIES, 1?I.E. syndrome, 9 CID, 2 Griscelli syndrome, 2 IFN?R deficiency , 1Myd 88 deficiency and 5 AT.
During the 12 years, a total of 2845 infusions, 5115 bottles of IVIG were applied to the patients. A total of 52 (1,8%) infusions were associated with adverse reactions in 18 (13%) patients. The highest rate of adverse reactions occured in CVID patients who had low serum IgA levels.
The most common symptoms of mild adverse reactions were urticaria and fever observed in 28 patients (0,98%). Moderate adverse reactions such as vomiting, wheezing were observed in 19 (0,66%) patients. Severe reactions (anaphlaxis, hypotension ,bronchospasm) were observed only in 5 (0,17%) CVID patients.
Adverse reactions to IVIG infusion are not rare in patients with PID(13%) , severe reactions may occur especially in CVID patients with low IgA . Physicians and nurses should be aware of these reactions and vital signs must be observed during infusion in order to manage and prevent them.
ESID-0311 Thrombomimetic Treatment in a CVID-Associated Immune Thrombocytopenia
M. Carrabba 1, G. Fabio2
1Internal Medicine, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
2Internal Medicine, Università degli Studi & Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
Immune thrombocytopenia (ITP) is present in up to 7% of common variable immunodeficiency (CVID) patients. Splenectomy or immunosuppressive therapy with Rituximab are the recommended therapeutic option for patients with refractory ITP that fail corticosteroid and IVIg therapy.
Thrombopoietin receptor agonists (Eltrombopag and Romiplostim) have been approved for the second line treatment of ITP according to the American Society of Hematology 2011 evidence-based practice guideline. However, there are no data available so far on treatment of refractory ITP on CVID patients.
We present a 43-years old patient with CVID diagnosed in May 2012. At diagnosis, splenomegaly, lymphadenopathy and lung sarcoid like-disease were present. Lung disease recovered after high dose of corticosteroids, stopped in June 2013. Reduced percentages of switched memory B cells (1%) and low proportion of CD21low cells (11.9%) were found.
In February 2014, severe ITP (22x109/L) refractory to first line therapy developed. Eltrombopag was started at the dosage of 50?mg per day orally, after bone marrow trephine biopsy. Platelets count started to increase after 15 days of therapy.
All of treatments for ITP non-responders have either proven long-term adverse events such as septicemia after splenectomy or other complications of potent immunosuppressors mainly in immunocompromised patients.
Thrombomimetic medications are demonstrated to be effective and adverse effects have generally been mild, though they have been available for too short a time to comprehend fully long-term toxicities. However, the lack of data about their use in CVID patients is the main limit that needs to be passed through future confirmatory trials.
ESID-0518 Change of Route or Place for Administration Improves Satisfaction of Patients Receiving Immunoglobulins for Primary Immunodeficiency (Visages Study)
R. Jaussaud 1, B. Bienvenu2, E. Hachulla3, C. Hoarau4, M. Pasquet5, G. Cozon6, P. Clerson7, J.C. Crave8, F. Lefebvre8, P. Cherin9
1Internal Medicine Clinical Immunology and Infectious Disease, Robert Debré University Hospital, Reims, France
2Internal medicine, Côte de Nacre University Hospital, Caen, France
3Internal Medicine, Lille Claude Huriez University Hospital, Lille, France
4Clinical Immunology and Nephrology, Tours Bretonneau University Hospital, Tours, France
5Pediatric-Hematology and Oncology, Toulouse University Children Hospital, Tolouse, France
6Clinical Immunology and rheumatology, Lyon-Est University Hospital, Lyon, France
7Biostatistics, Orgametrie, Roubaix, France
8Medical, Octapharma France SAS, Boulogne-Billancourt, France
9Internal Medicine, Pitié-Salpêtrière University Hospital, Paris, FranceIntroduction
Quality of life (QoL) and patient satisfaction have never been explored in primary immunodeficiency (PID) patients treated by IgG replacement therapy (IgRT) in France.
Objective
To investigate if changes in route or place of IgRT conducted in real-life conditions improve patient’s satisfaction and QoL.
Methods
PID patients aged > 5 years receiving IgRT for =3 months entered the study. Infections rate, QoL, and satisfaction were prospectively evaluated using SF-36, CHQPF50 and Life Quality Index over a 12-month follow-up.
Results
154 PID patients (age 32.6±20.8 years, 52% females) receiving IgRT for 7.8±7.8 years entered the study. Route was intravenous Ig (IVIG) for 72 patients (47%) (HOSP: 76%) and subcutaneous Ig (SCIG) for 82 patients (53%) (HOME: 95%). 46% of patients had a previous history of switch from IVIG to SCIG and 12% from SCIG to IVIG. At entry, QoL were similar whatever the route or place for administration. Satisfaction was higher for SCIG and for home administration. Over the follow-up period, 23 patients (19%) changed the route or place for IgRT: 9 switched from IVIG to SCIG, 2 from SCIG to IVIG, 19 from hospital to home and 2 from home to hospital with significant increases of all dimensions of satisfaction (treatment interference, therapy related problems and therapy settings). Over the follow-up period, the incidence rate of severe infections was 0.13/patient-year
Conclusion
Visages is the largest study conducted in real-life in PID to date. Change from IVIG to SCIG and from hospital to home increases patient satisfaction regarding IgRT.
ESID-0602 ARDS Following Hematopoietic Stem Cell Transplantation in a CSF2RB-Deficient Pediatric Patient with Pulmonary Alveolar Proteinosis (PAP)
J. De Bruycker 1, H. Decaluwe1, S. Selleri1, D. Bérubé1, F. Le Deist2, I. Fernandez2, P. Teira1, E. Haddad1
1Pediatrics, University of Montreal, Montreal, Canada
2Microbiology and Immunology, University of Montreal, Montreal, Canada
We present a fatal evolution of ARDS following bone marrow transplantation (BMT) in a typical 11y old PAP patient with CSF2RB defect, and discuss the challenging procedure of BMT in this rare intrinsic defect of macrophages. This PAP was complicated by recurrent hemoptysis and severe pulmonary infections with fungi and atypical mycobacteria. The patient was treated by repeated BAL. Due to a poor quality of life, we decided to perform a BMT. She underwent a BMT from a match unrelated (10/10) donor with a RIC regimen (fludarabine, busulfan, alemtuzumab). On day +10 after transplantation, myeloid engraftment was reached with full donor chimerism. The procedure was initially uneventful. At day +21, she developed ARDS. Specific workup didn’t confirm any infectious pathogen. A pulmonary lavage didn’t improve the respiratory failure. Patient was treated with aggressive mechanical ventilation. Despite cyclophophamide, anakinra and repeated pulse of steroids, the patient eventually died from respiratory failure. There was no sign of GVHD and no significant proteinosis. Because of the very high level of GMCSF in serum and BAL before BMT, we suspected a neutrophil trapping occurring with the engraftment. Accordingly, neutrophil count in blood was found extremely high up to 29 x 10^9/L on day +48. This case, that seems to be the first reported BMT in a patient with CSF2RB-mutation PAP, illustrates how challenging this procedure can be. We suspect that very high levels of GMCSF could be responsible for the terrible evolution observed in our patient.
ESID-0254 Use of Human Immunoglobulin in Secondary Immunodeficiencies Associated with Hematological Malignancy: EPICURE Study
O. Benbrahim1, B. Grosbois2, J. Viallard 3, S. Choquet4, B. Royer5, J. Delain6, J. Crave6, P. Clerson7, V. Levy8
1Oncologie-Médicale, CHR Orléans, Orléans, France
2Service de Médecine interne, Hôpital Sud, Rennes, France
3Service de médecine interne et maladies infectieuses, Hôpital Haut Lêveque, Pessac, France
4Hématologie, Groupe Hospitalier Pitié Salpétrière, Paris, France
5Hématologie, CHU d'Amiens, Amiens, France
6Immunotherapy Intensive Care & Emergency Medicine, Octapharma France SAS, Boulogne-Billancourt, France
7Biostatistique, Orgamétrie, Roubaix, France
8Hématologie, Hôpital Avicenne, Paris, France
Introduction
Treatments for hematological malignancy are increasingly immunosuppressive and few data are available. The objective of EPICURE study is to describe indications and modalities of replacement therapy by immunoglobulin in secondary immune deficiency (SID) adult patients associated with hematological malignancies (including lymphoma).
Patients and Methods
EPICURE is an observational, prospective, longitudinal study involving 40 centers. To date 238 patients have been included. We report data of the first 97 patients monitored.
Results
The analysis focused on 97 patients (61 men, 36 women) with myeloma (N=21), chronic lymphocytic leukemia (N=29), non-Hodgkin B lymphoma either aggressive (N=15) or indolent (N=20) or other hematologic malignancies (N=12). Ninety patients had a total of 173 infectious episodes in last 12 months. In 46 cases (47%), treatment with immunoglobulin was administered subcutaneously at 97 ± 37 mg/kg weekly. Immunoglobulin subcutaneous injections started at hospital in 45/46 cases and relay at home was planned for all patients. In contrast, 51 patients started treatment with intravenous immunoglobulin at hospital (50/51 cases, 414 ± 263 mg/kg every 3.7 ± 0.7 weeks) and home treatment was rarely planned (1/?51 case). Physicians’ expectations are focused on infections prevention (96 cases), survival improvement (82 cases), lower hospitalizations rate (96 cases) and reduction of antibiotics consumption (92 cases).
Conclusion
Replacement immunotherapy was started subcutaneously in almost 50% of patients. Longitudinal monitoring will describe physician’s satisfaction, compare the occurrence of infections with and without treatment and define therapeutic profiles in order to improve care of SID patients.
ESID-0275 Hematopoetic Stem Cell Transplantation for LRBA Deficiency
F. Dogu 1, S. Haskologlu1, B. Dalgic2, O. Dur3, Z. Kuloglu4, A. Kansu4, A. Ikinciogullari1
1Department of Pediatric Immunology and Allergy, Ankara Univesity Medical School, Ankara, Turkey
2Department of Pediatric Gastroenterology, Gazi Univesity Medical School, Ankara, Turkey
3Department of Pediatrics, Ankara Univesity Medical School, Ankara, Turkey
4Department of Pediatric Gastroenterology, Ankara Univesity Medical School, Ankara, Turkey
Three papers published in 2012 demonstrate that deleterious mutations of LRBA cause CVID and autoimmunity, and are associated with inflammation. The manifestations of LRBA deficiency include hypogammaglobulinemia due to defective B-cell differentiation, recurrent infections and various autoimmune disorders. Here we present a new case of LRBA deficiency who was treated by HSCT after nonmyeloablative conditioning regimen. The patient was 12.5 years old girl of consanguineous parents. The first symptoms were chronic diarrhea and vomiting at 6 years of age and increased frequency of respiratory infections. Two years ago celiac disease was diagnosed and she was put one gluten-free diet without significant improvement. Stat5b deficiency was suspected with clinical findings of severe growth retardation, bronchiectasis and dry skin. Mutation analyses revealed homozygous C5505del T at LRBA gene and she was referred to our clinic. On physical examination severe growth retardation, clubbing, hepatosplenomegaly and fine crackles at both lung were detected. Immunological evaluation revealed decreased IgM levels with normal isohemagglutinin titre, normal,T , B ,NK cell counts and normal activation response to PHA and anti-CD3. As she has HLA fully matched sibling donor and multiple organ dysfunction (bronchiectasis, tubulopathy and severe colitis) non-myeloablative conditioning regimen consisted of BU/Flu/ATG applied and she received 8x106/kg CD34+ PBSC from her brother. She is now on posttransplant +9d. In conclusion LRBA deficiency should be considered in patients with early-onset colitis and autoimmunity. Long-term follow-up and additional reports of patients with LRBA mutations will delineate the full phenotypic spectrum of disease as well as the treatment options.
ESID-0049 Successful Hematopoietic Stem Cell Transplantation in an Infant with Griscelli Syndrome Type 2
R. Dorbeker 1, M.?A. Yamazaki-Nakashimada2, S.E. Espinosa-Padilla3, F.J. Espinosa-Rosales3, M. Perez-Garcia4, A. Olaya-Vargas4, S.O. Lugo-Reyes3, S.O. Lugo-Reyes3
1Pediatria, Hospital Central Sur de Alta Especialidad Petroleos Mexicanos, Mexico City, Mexico
2Inmunologia, Instituto Nacional de Pediatria, Mexico City, Mexico
3Unidad de Investigación en Inmunodeficiencias, Instituto Nacional de Pediatria, Mexico City, Mexico
4Unidad de Trasplante de Celulas Hematopoyeticas, Instituto Nacional de Pediatria, Mexico City, Mexico
Introduction:
Griscelli syndrome is a rare autosomal recessive disease characterized by pigmentary abnormalities, mainly silver-gray hair. 3 types of Griscelli syndrome have been described (type 1, 2 and 3)
Type 2 is caused by mutation in the RAB27 gene and is characterized by cutaneous hypopigmentation, immunodeficiency, and hemophagocytic lymphohistiocytosis. Its only curative treatment is the hematopoietic stem cell transplantation.
Case presentation:
We present an infant with Griscelli syndrome type 2, with background of one deceased sister diagnosed with Griscelli syndrome type 2 year and 8 months old. On admission, she presented silver-gray hair, without evidence of hemophagocytic lymphohistiocytosis or infectous disease. At age 4 months allogeneic hematopoietic stem cell transplantation (unrelated umbilical cord donor) was performed. The only complication she presented was cutaneous graft versus host disease treated with cyclosporine, prednisone, and mycophenolate mofetil. Now she has 3 years old, neurologically asymptomatic, without evidence of hemophagocytic lymphohistiocytosis and 100% graf chimerism.
Discussion:
The treatment of choice for patients with Griscelli syndrome type 2 is the stem cell transplantation, with a better prognosis before hemophagocytic lymphohistiocytosis is developed. In our institution, we have 3 cases of Griscelli syndrome type 2 with hematopoietic stem cell transplantation, this is the only succesfull. Our success lies on early diagnosis (1 month age) and early transplantation (less than 6 months of age).
ESID-0062 Isoagglutinin Depletion in Human Immunoglobulin Products: Donor Screening or a Specific Immunoaffinity Chromatography Step
I. El Menyawi 1, B. Siani1, M. Imboden1, A. Gaida1, I. Glauser1, R. Bolli1, K. Willimann1, S. Wymann1, A. Marques1, E. Widmer1
1Research & Development, CSL Behring, Bern, Switzerland
Introduction
Hemolytic events associated with intravenous IgG (IVIG) therapy are rare and occur predominantly in patients with a non-O blood group. Isoagglutinins (anti-A/B antibodies originating from donor plasma) are a potential causative factor. Two approaches to deplete isoagglutinins in IgG products were investigated: exclusion of plasma from donors with high isoagglutinin titres and introduction of a specific anti-A/B immunoaffinity chromatography (IAC) step in the IgG purification process.
Methods
Donor plasma pools and final IgG products were prepared according to the manufacturing process of Privigen®/Hizentra®. Donor screening was performed by a high-throughput anti-A assay; 30 industry-scale IgG lots were prepared from plasma pools with exclusion of donors with the highest anti-A isoagglutinin titres. In separate experiments, Anti-A/B isoagglutinins were depleted using an additional A/?B-trisaccharide-coupled IAC step at the end of the manufacturing process; 10 industry-scale IgG product lots were prepared using IAC. Final anti-A/B isoagglutinin titres were measured by the indirect agglutination test.
Results
Exclusion of donors with high anti-A titres resulted in a reduction of both anti-A and anti-B isoagglutinins by 1 titre step in the final product. Introduction of a specific IAC step reduced anti-A and anti-B titres by at least 2 titre steps in the final product, while other product characteristics remained unchanged.
Conclusion
Anti-A/B isoagglutinin reduction in IgG products is feasible on an industrial production scale, either by using anti-A donor screening or by introducing a specific IAC depletion step. Implementing one of these approaches may reduce the risk of hemolysis in IgG therapy.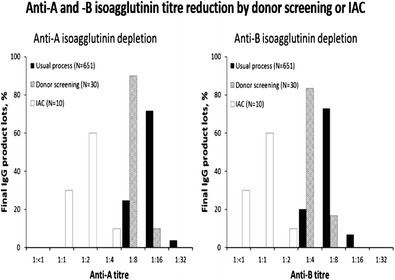
ESID-0493 Enteroviral Recurrent Meningo-Encephalitis in a Patient with X-Linked Agammaglobulinemia
M. Esquivel 1, A. Bravo Kleiman1, I. Moreira1, G. Seminario1, A. Gomez Raccio1, D. Di Giovani1, L. Bezrodnik1
1Immunology, Hospital de Niños R Gutierrez, Buenos Aires, Argentina
We report the case of a 6 years-old boy who was diagnosed as XLA (mutation in exon 6 of BTK) at 10 months-old after suffering from severe infections, and who was under gammaglobulin replacement therapy.
Although presenting good basal levels of serum IgG (more than 900 mg/dL), in September 2013, he started with recurrent episodes of meningitis. On November 2nd, he had the third episode and CSF enterovirus PCR was positive. So, weekly high dose of IVIG was prescribed.
On Nov 29th he relapsed with photophobia, fever and headache. Neurological examination revealed a mild paresis on right arm and leg and abnormal left auditive evoked potentials. CSF worsened again and he was diagnosed as Enteroviral recurrent meningo-encephalitis. As intensive IVIG therapy failed, on Dec 18th he started with daily intrathecal IG (ITIG) with a good response. On Dec 28th he was in clinical remission and CSF almost normalized . So, frequency of ITIG was decreased to every 48hs, but as he relapsed, IVIG/?ITIG was again indicated daily.
Attempting to find a curative strategy, he received pocapavir which is an investigational antienteroviral agent (orally, at 600 mg/d, for ten days). No adverse events. Frequency of IVIG/?ITIG was decreased to weekly with good tolerance. CSF normalized and on april 16th he was discharged home. Nowadays (may 24th), he remains asymptomatic and continues weekly IVIG and periodic controls.
ESID-0037 Hematopoietic Stem Cell Transplantation for Primary Immunodeficiency – A Twenty Years Experience in the Czech Republic
R. Formankova 1, P. Sedlacek1, P. Keslova1, P. Riha1, A. Sediva2, E. Mejstrikova1, O. Hrusak1, J. Litzman3, T. Freiberger4, J. Stary1
1Department of Pediatric Hematology and Oncology, Faculty Hospital Motol, Prague 5, Czech Republic
2Department of Immunology, Faculty Hospital Motol, Prague 5, Czech Republic
3Department of Clinical Immunology and Alergology, St. Anne´s University Hospital, Brno, Czech Republic
4Genetics Laboratory, Centre of Cardiovascular and Transplantation Surgery, Brno, Czech Republic
Background: Allogeneic hematopoietic stem cell transplantation (HSCT) is effective therapy of lethal forms of primary immunodeficiency (PID).
Methods: We present results of 62 HSCT in 56 children with PID performed between 1994-2013?at Pediatric HSCT Unit in Prague. 19 HSCT were performed in 16 patients with severe combined immunodeficiency (SCID) and 43 in 40 non-SCID patients. Non-SCID diagnosis were: Wiskott-Aldrich syndrome (WAS; 8), chronic granulomatous disease (CGD; 9/?8), CD40 ligand deficiency (6/?5), familial lymphohistiocytosis (FHL;11/10), other (9). 74% of patients were transplanted from unrelated donor. 55 of 62 grafts were unmanipulated, 8 patients were transplanted with umbilical cord blood. Conditioning regimens based on Busulfan (Bu) and Cyclophosphamide were replaced by less toxic regimens consisted of Bu or Treosulfan with Fludarabine in majority of patients since 2008.
Results: Primary engraftment was achieved in 98% of evaluable patients (49/50). Acute GVHD was documented in 41% of patients. Transplant related mortality was 23% (4 SCID, 9 non-SCID) due to infections (8), toxicity (4) and GVHD (1). OS of the whole group is 75%, no difference between SCID and non-SCID patients. 42/56 patients are alive and well with improved immunity at median follow-up 68 months (4-233) after HSCT. BCG infection was serious complication in most of SCID patients at the time of nationwide vaccination.
Conclusions: HSCT for PID lead to cure of disease and normal quality of life in most patients and prognosis of transplanted patients changes markedly according to time of transplant.
Supported MH CZ-DRO, University Hospital Motol, Prague, Czech Republic 00064203
ESID-0202 Intrathecal Amphotericin B Therapy in a Patient with X-Linked Chronic Granulomatous Disease and Refractory Cerebral Invasive Aspergillosis
V. Gallo1, E. Cirillo 1, G. Giardino1, R. D'Assante1, P. Spennato2, G. Cinalli2, C. Pignata1
1Translational Medical Sciences, Università "Federico II", Naples, Italy
2Pediatric Neurosurgery, A.O.R.N.Santobono-Pausilipon Hospital, Naples, Italy
Introduction: Invasive aspergillosis (IA) is one of the life-threatening complications in CGD patients, and represents a major cause of morbidity and mortality. The main therapy is the IV- injection of antifungal agents, as Voriconazole and Amphotericin-B. However, the mortality rate is very high even with the combination of two or more antifungal IV-drugs.
Objective: To provide a possible rescue therapy for cerebral IA refractory to conventional antifungal therapy in CGD patients.
Methods: We report on the case of a 17-year-old boy affected with X-CGD, who developed chronic pulmonary aspergillosis subsequently complicated with cerebral aspergillosis refractory to multiple pharmacological strategies. The patient underwent successful extensive neurosurgical treatment consisting of endoscopic third ventriculo-cysternostomy and lesion exeresis. Histopathology studies showed within the lesion abscessual areas with micelial hyfes and positive periodic acid-shiff and Grocott staining, confirming the fungal infection. A rescue therapy with intrathecal injection of Amphotericin B at 1 mg/kg/day every 3 days for two months by external ventricular drainage was started.
Results: After 5 weeks, an improvement of the neurological signs was noted; CSF mycological cultures and detection of fungal DNA by PCR were negative. No complications related to ventricular drainage or intrathecal injection of Amphotericin B, such as nerve irritations or bacterial infections, occurred during the treatment. However, in spite of the negative mycological findings, the clincal conditions remain severely compromised.
Conclusion: Intrathecal Amphotericin B in combination with systemic therapy could be a possible rescue and relatively safe treatment for selected, seriously ill patients with refractory cerebral IA and poor prognosis.
ESID-0308 The Use of Immunoglobulin in a Patient with Selective Deficiency of IGG3
S. Gambini 1, I. Savore1, R. Morariu1, L. Paolini1, M.G. Danieli1
1Medicina Interna, Clinica Medica, Ancona, Italy
Immunoglobulin G (IgG) subclass deficiency is a rare primary immunodeficiency syndrome with recurrent infections and associated autoimmune manifestations.
We here report the case of a 43 years old woman affected of post-traumatic panniculitis since 2010. She presented also complications like lymphedema and elephantiasis of forearm with frequent infections of subcutaneous tissue. She needed several hospitalizations with repeated antibiotic courses. A subsequent hospitalization was due to an acute respiratory failure supported by H. influenzae type B infection, which was followed by a post-infectious acute myelitis with residual paraparesis and neurogenic bladder. The patients was treated with a long-term steroid therapy that caused the onset of iatrogenic type II diabetes mellitus and hypertension. The patient then came to our attention.
The presence of severe recurrent infections in a subject with no others obvious causes led us to undertake an immunological research which detected decreased serum levels of IgG3 (170 mg/ l; normal values 410-1300 mg/ l).
We therefore initiated an immunoglobulin replacement therapy, initially intravenously at a dose of 30?g monthly then subcutaneously, view of the difficulty of finding venous access in the upper limbs. During SCIg treatment, serum IgG3 levels increased with no further infectious episodes or recurrence of myelitis.
There have been few reports of complex autoimmune disorders (myelitis, panniculitis) associated with IgG3 deficiency, in a patient with recurrent infections of previously unknown origin. The selective IgG3 deficiency could explain the complex clinical picture of our patients, in which the immunoglobulin replacement therapy indeed was a valid therapeutic option.
ESID-0218 Granulomatous and Lymphocytic Interstitial Lung Disease in a Patient with Common Variable Immunodeficiency
F. GENEL 1, D. CAN1, N. GULEZ1, H.T. NACAROGLU1, S. BAHCECI ERDEM1, C.S. KARKINER1
1Department of Pediatrics, Dr. Behcet Uz Children's Hospital, Izmir, Turkey
Common variable immunodeficiency is a primary immunodeficiency characterized by hypogammaglobulinemia and T-lymphocytes dysfunction. Approximately 10-15% of patients with CVID develop granulomatous/lymphocytic interstitial lung disease, which is frequently accompanied by splenomegaly, adenopathy, autoimmune cytopenias, gastrointestinal and hepatic disease.
A three year old girl was admitted to our hospital with a clinical history of recurrent upper respiratory infections, and splenomegaly. She had a marked decrease of all serum immunoglobulin isotypes and low specific antibody responses. The diagnosis of CVID was based on clinical and laboratory findings and IVIG therapy was started.
At the age of 6 years, she presented with cough. Thorax CT revealed mediastinal adenopathy, paranchymal multiple nodular opacities, ground glass opacities and bronchial wall thickening. Lung biopsy revealed non-necrotizing granuloma and lymphocytic interstitial pneumonia. A diagnosis of granulomatous/lymphocytic interstitial lung disease was made and 2 mg/kg/day prednisolone were given. Although the patient's symptoms improved and there was reduction in the extent of nodularity and ground glass opacification on the HRCT scan, relapses observed with the dose reduction of oral prednisolone during 6 years follow up. At the age of 12 years, in addition to pulmonary findings increase at hepatosplenomegaly and trombositopenia observed. Rituximab therapy was started and azathioprine was added. Platelet count rose to normal levels and pulmonary radiographic abnormalities decreased.
As a result physicians must be aware of non-infectious complications such as granulomatous/lymphocytic interstitial lung disease in patients with CVID and there is need to determine the best modality of therapy to treat CVID associated granulomatous/lymphocytic interstitial lung disease.
ESID-0369 Haematopoietic Stem Cell Transplantation for CD40 Ligand Deficiency: Results from an EBMT/?ESID Inborn Error Working Party Study
F. Ferrua 1, A. Janda2, V. Courteille3, M. Slatter4, M.H. Albert5, H. Al-Mousa6, B. Al-Saud6, D. Balashov7, Y. Bertrand8, V. Bordon9, W. Czogala10, F. Dogu11, A. Fasth12, R. Formankova13, J. Gozdzik10, C. Heilmann14, M. Hönig15, A. Ikinciogullari16, K. Kalwak17, S.J. Keogh18, G. Kriván19, A.?C. Lankester20, I. Meyts21, B. Neven22, F. Suarez23, N. Mahlaoui3, A. Fischer24, A.?R. Gennery4
1Department of Paediatric Immunology and HSCT, Great North Children's Hospital, Newcastle upon Tyne, United Kingdom
2Center for Chronic Immunodeficiency (CCI) University Medical Center, University of Freiburg, Freiburg, Germany
3CEREDIH (French National Reference Center for PID), Necker-Enfants Malades University Hospital, Paris, France
4Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
5Department of Pediatric Hematology/Oncology, Dr. von Haunersches Children's Hospital, Munich, Germany
6Department of Pediatrics, King Faisal Specialist Hospital & Research Center, Riyadh, Saudi Arabia
7Department of Bone Marrow Transplantation, Federal Research and Clinical Centre of Paediatric Haematology Oncology and Immunology named after Dmitry Rogachev, Moscow, Russia
8Institut d'Hematologie et d'Oncologie Pediatrique, Hospices Civils de Lyon, Lyon, France
9Pediatric Hematology and Oncology, Ghent University Hospital, Ghent, Belgium
10Department of Transplantation, Children's University Hospital, Krakow, Poland
11Department of Pediatric Immunology and Allergy, Ankara University School of Medicine, Ankara, Turkey
12Dept of Pediatrics, University of Gothenburg, Gothenburg, Sweden
13Department of Pediatric Hematology and Oncology, University Hospital Motol, Prague, Czech Republic
14Paediatric Clinic, Rigshospitalet, Copenhagen, Denmark
15Department of Pediatrics and Adolescent Medicine, University Medical Center, Ulm, Germany
16Department of Pediatric Immunology - Allergy and BMT Unit, Ankara University Medical School, Ankara, Turkey
17Department of Pediatric Hematology/Oncology and BMT, Wroclaw Medical University, Wroclaw, Poland
18Oncology Unit, The Children's Hospital Westmead, Westmead New South Wales, Australia
19Dep. of Pediatric Hematology and Haemopoietic Stem Cell Transplantation, United St. Istvan and St Laszlo Hospital, Budapest, Hungary
20Dep. of Pediatrics/Div. of Stem Cell Transplantation, Leiden University Medical Center, Leiden, Netherlands
21Department of Pediatric Immunodeficiencies, University Hospitals Leuven, Leuven, Belgium
22Unité d'immuno-hématologie pédiatrique, Hôpital Necker-Enfant Malades, Paris, France
23Hématologie Adulte, Hôpital Necker, Paris, France
24Scientific Director, Institut Imagine, Paris, France
Introduction: CD40 ligand (CD40L) deficiency, a rare X-linked primary immunodeficiency, causes recurrent sinopulmonary infections, Pneumocystis jirovecii and Cryptosporidium parvum infections and autoimmunity. Long-term survival with supportive therapy only is poor, hepatic disease being the major cause of death. Currently, the only curative treatment is haematopoietic stem cell transplantation (HSCT).
Methods: We retrospectively collected data on outcome of HSCT to improve future management. We present the preliminary analysis of 64 patients undergoing HSCT for CD40L deficiency in Europe between 1993-2013 in 18 different centres.
Results: Overall survival was 78%, and 88% in transplants performed after 2000 (figure 1) and in children <10 years [follow up (FU): 0.1–17.1 years]. Donors included 18 HLA-identical siblings, 52 unrelated volunteers and 3 haploidentical parents, for a total of 73 procedures (5 second HSCT, 4 boosts), of which 21 were T-cell depleted. In 51% of the transplants reduced-intensity conditioning was used. Most deaths occurred within 4 months of HSCT, predominantly from infections (44%). Pre-existing organ damage and severe GVHD were associated with poor outcome. Seven transplants were rejected. Donor chimerism >50% was observed in 72% of survivors at last FU, in some decreasing chimerism was detected over time. Among those with FU >1 year, immunoglobulin replacement and prophylactic antibiotics were stopped in 78% and 71% of patients respectively.
Conclusions: Our preliminary results show that HSCT can be curative in CD40L deficiency, with improved survival if performed early. Further studies however should determine the optimal conditioning regimen to favour long-term engraftment and full immunoreconstitution without increasing toxicity.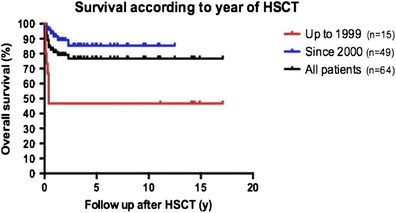
ESID-0465 Clinical and Immunological Correction of DOCK8 Deficient Patient by Allogeneic Hematopoietic Stem Cell Transplantation
N. Gulez 1, S. Aksoylar2, S.E. Bahceciler3, D. Can3, F. Genel3, S. Kansoy2
1Pediatric Immunology and Allergy, Dr. Behçet Uz Childen's Hospital, Izmir, Turkey
2Pediatric Heamatology and Oncology Department Pediatric Bone Marrow Center, Ege University Medical Faculty, Izmir, Turkey
3Pediatric Immunology and Allergy, Dr. Behcet Uz Children's Hospital, Izmir, Turkey
Hyper IgE syndrome (HIES) is a rare primary immunologic disorder affecting multisystem. Autosomal-recessive hyper-IgE syndrome (AR-HIES) is a combined immunodeficiency recently found to be associated with mutations of DOCK8. Eczema, sinopulmonary infections, chronic viral infections of the skin, blood eosinophilia and markedly elevated serum IgE are characteristic features in all patients with DOCK-8 deficiency. The only definitive treatment option is allogeneic hematopoietic stem cell transplantation (HSCT). We report a patient with early severe manifestation of DOCK8 deficiency, who underwent allogeneic HSCT at the age of 6 years following a myeloablative conditioning regimen from an unrelated HLA 9/?10 matched donor. The transplant course was complicated venooclusive disease and severe Grade III acute GVHD after transplantation. Eleven months after transplantation the patient is in excellent clinical condition with immunosupressive therapy of chronic GVHD. Eczematous rash and chronic viral skin infections have subsided, associated with normalization of IgE levels and absolute numbers of eosinophils. Chimerism analysis shows stable full donor chimerism. DOCK8 deficiency can be successfully cured by allogeneic HSCT. This treatment option should be considered early after diagnosis, as opportunistic infections and malignancies that occur more frequently during the natural course of the disease are associated with higher morbidity and mortality.
ESID-0070 No Evidence of Prion Infection in Recipients of UK Sourced Immunoglobulin
M. Helbert 1, M. Helbert1, C. Bangs1, T. Garcez1, J. Ironside2
1Immunology, Manchester Royal Infirmary, Manchester, United Kingdom
2National CJD Research & Surveillance Unit, University of Edinburgh, Edinburgh, United Kingdom
During the bovine spongiform encephalopathy (BSE - mad cow disease) outbreak in the 1990s, abnormal prions were transmitted through beef to humans. In some humans, this caused variant Creutzfeldt-Jakob disease (vCJD). Subsequently, abnormal prion transmission has been shown to occur following transfusion of red cells and, probably, through purified factor VIII.
A collective risk assessment of haemophiliacs in 2004 indicated that these patients required special measures to minimise the risk of onward infection. These have included quarantining of endoscopes and restricted access to some procedures. On the other hand, individualised risk assessments for PID patients did not indicate that any measurable risk and no PID patients required special precautions.
In the 2000s, the number of new cases of vCJD fell dramatically. However, the best evidence available suggests that the background abnormal prion infection rate in the UK is approximately 500/million.
A surveillance study for abnormal prion transmission in PID patients who received UK sourced immunoglobulin was established in 2006. The study has followed more than 60 patients who were exposed to implicated batches of immunoglobulin. None of these patients have developed clinical features of vCJD. Analysis of more than 100 tissue samples from these individuals has not detected abnormal prion infection.
We will describe practical steps that we have learnt can be used now to reduce the future impact of outbreaks of infectious agents in the plasma supply.
ESID-0386 Tolerability of Subcutaneous Gamma Globulin in Two Patients with Ataxia Telangiectasia
V. Hernandez-Trujillo 1, W. Blouin1, J. Calderon1
1Division of Allergy and Immunology, Miami Children's Hospital, Miami, USA
Rationale: Some patients with Ataxia Telangiectasia (AT) require gamma globulin therapy for hypogammaglobulinemia. Tolerability of subcutaneous gamma globulin has not been previously reported in patients with AT.
Case Discussion: Two patients with AT, hypogammaglobulinemia and poor antibody response to specific antigens presented for routine evaluation. One patient was a 17 year old male had active sarcoid skin disease, advanced neurological degeneration, chronic Epstein Barr Virus infection and respiratory infections. The second patient was a 17 year old female with AT and scoliosis. Both patients had poor response to protein and polysaccharide vaccines.
Results: The first patient had pre-treatment IgG level of 674 mg/dl. He was started on IVIG 500 mg/kg/month. He was then transitioned to weekly subcutaneous gamma globulin therapy of 125 mg/kg/week with a post-treatment level of 850 mg/dl. The second patient had pre-treatment IgG of 130 mg/dl. She received two doses of IVIG at 500 mg/kg, then transitioned to weekly subcutaneous dose of 125 mg/kg/week with a post-treatment level of 938 mg/dl. Both patients tolerated the infusions well without pre-medication. Neither patient reported site reactions despite thin body habitus.
Discussion: Patients with Ataxia Telangiectasia and hypogammaglobulinemia with poor specific antibody response tolerated use of 20% subcutaneous gamma globulin. Both were able to achieve adequate post-treatment levels
ESID-0805 Should Immunoglobulin Be Dosed by Actual Body Weight in Obese Patients?
J. Hodkinson 1, M. Lucas2, M. Lee3, M. Harrison4, M. Lunn4, H. Chapel2
1Medical Affairs, Biotest (UK) Ltd, Birmingham, United Kingdom
2Department of Clinical Immunology, University of Oxford, Oxford, United Kingdom
3School of Public Health, University of California Los Angeles, Los Angeles, USA
4Neuroimmunology, National Hospital for Neurology and Neurosurgery, London, United Kingdom
Background
It has been suggested that the dose of normal immunoglobulin (Ig) should be restricted in obese patients for pharmacokinetic (PK), safety and economic reasons. However, there is little data to support the assertion that doses of Ig should be capped for patients with a BMI greater than 30.
Objective
To provide data to shed light on this question, a retrospective audit was undertaken.
Methods
31 obese patients were matched with a clinically equivalent but lean patient across a range of indications. The data was collected at two centres which determine dose based on clinical outcome, whether that is infection prevention or effective immunomodulation. The trough level and increment achieved for a given weight–adjusted dose was compared between the obese and lean cohorts.
Results
It was shown that on average obese patients achieved a higher trough and increment compared with the lean patients. This data indicates that there is a genuine difference in the PK of Ig in obese and lean patients at a population level. However when individual matched pairs were analysed it was shown that obesity could not reliably predict that a reduced dose would be sufficient to achieve the desired clinical outcome.
Conclusion
Despite significant PK differences between the obese and lean cohorts, individual patient variability necessitates that a general recommendation for dosing in obese patients is not appropriate, and obese patients should be dosed on an individual basis to clinical outcome.
ESID-0426 JS Where Are We Now? A Patient Perspective a Further Two Years On
D. Hughes 1, F. Ashworth1
1Clinical Immunology and Allergy Unit, Sheffield Teaching Hospital, Sheffield, United Kingdom
The Mid Staffordshire NHS Foundation Trust Public Inquiry (DOH 2013) recommended the recognition of the central importance of nurses in the delivery of safe, compassionate care.
The values embedded within the report underpin the key principles when J.S was previously presented at INGID (2012); however this has now been formalised as compassionate care in nursing.
The presentation explores the complex issues surrounding the patient’s quality of life with chronic long term conditions. J.S has previously been on home therapy which has had a dramatic improvement on his quality of life however his circumstances have changed and he is now struggling with compliance for a variety of reasons in his home life. This has caused falling immunoglobulins levels and repeated chest infections that ended in a ten day hospital admission.
We will present the nursing care interventions that have been put into place for J.S to improve his compliance with treatment and remove the burden of care that he has felt on home therapy. Our outcomes will demonstrate that we have learnt that the patient’s choice between SCIG/?IVIG therapies changes over time. Supporting people to manage their own health- an introduction to patient activation`
(DOH, 2014) has identified similar issue to PID patients. Using this publication we’ll explore the issues concerning J.S, based upon his knowledge, skill and confidence for managing his own health and health care. We must make the quality of care as important as the quality of treatment, (Jeremy Hunt, Health Secretary, 2013).
ESID-0798 Hematopoietic Stem Cell Transplantation for the Patients with Activated PI3K-Delta Syndrome
K. Imai 1, Y. Tsujita2, K. Mitsui-Sekinaka3, N. Mitsuiki1, T. Takashima1, T. Okano1, Y. Aoki4, F. Kimoto5, M. Inoue5, F. Iwasaki6, T. Kaneko7, T. Waragai8, H. Sano8, A. Kikuta8, T. Morio4, S. Nonoyama3
1Department of pediatrics, Tokyo Medical and Dental University, Tokyo, Japan
2Department of pediatrics, National Defense Medical College, Saitama, Japan
3Department of Pediatrics, National Defense Medical College, Saitama, Japan
4Department of Pediatrics, Tokyo Medical and Dental University, Tokyo, Japan
5Department of Hematology/Oncology, Osaka Medical Center and Research Institute for Maternal and Child Health, Osaka, Japan
6Department of Haematology, Kanagawa Children's Medical Centre, Kanagawa, Japan
7Department of Haematology/Oncology, Tokyo Metropolitan Children's Medical Centre, Tokyo, Japan
8Department of Pediatrics, Fukushima Medical University, Fukushima, Japan
Activated phosphatidylinositol-3-OH kinase (PI3K)-delta syndrome (APDS) is a newly described primary immunodeficiency syndrome characterized by predominantly antibody deficiency (PAD) and pulmonary disease due to germ line activating mutations of PIK3CD gene. Because of progressive lymphopenia, they suffered from opportunistic infections, and massive lymphoid organ hyperplasia and B cell lymphoma are frequent in later life. Thus hematopoietic stem cell transplantation (HSCT) is a treatment of choice as reported in previous reports.
We identified 11 APDS patients with E1021K mutation in Japan from 175 PAD patients. Six patients of APDS were treated by hematopoietic stem cell transplantation (HSCT) for the massive lymphoid organ hyperplasia and recurrent infections. In five patients, HSCT were successful after reduced intensity conditioning regimens including fludarabine and alkyrating agents. One patient was rejected and retransplanted 1 year later. One splenectomized patient was died due to pneumococcal sepsis after 2 years of HSCT. The other 4 patients were transplanted successfully and alive. Thus HSCT could be a treatment of choice for this disease.
ESID-0641 Complementary Alternative Medicine in PID: Approved By a Large Majority by the Patients but Often Ignored by the Doctors
A. Servettaz1, D. Lambert1, V. Gauche1, P. Schmitt1, V. Laurant-Noel1, R. Jaussaud 1
1Médecine interne maladies infectieuses immunologie clinique, CHU de Reims, Reims, France
The place dedicated to Complementary alternative medicines (CAM) in PID by the patients is still unknown.
To estimate the frequency and reasons for use of CAM in adult patients suffering from PID, in addition to IgG replacement therapy (IgRT).
A prospective study was conducted between January 1 and August 31, 2013, in the Champagne-Ardenne area, France. An anonymous questionnaire was sent to all the adult patients treated by IgRT. Patients were asked about their PID, on socio-demographic elements, on the CAM.
Among 33 eligible patients (28 CVID, 2 LOCID, 2 Bruton, 1 Good), 29 have fully completed the questionnaire. 48.3% have recourse to CAM. No differences on age, sex, place of residence and age of the PID were found between users and non-users of CAM. Among the patients of low level of instruction, 35.3% use CAM, versus 66.7% who have a level equivalent to the high school diploma (p = 0.09). Traditional Chinese medicine, homeopathy (64.3%) and natural products (64.3%) are the most frequently used. Half the users resorts to it at least once a week. The main reason advanced is to exercise a better control over the disease (72%). For 71.4%, the action is complementary to the IgRT. 28.6% consider CAM essential. The symptoms making resort in CAM are infections (57.1%), asthenia (50%), stress (42.9%), pain (35.7%). 78.6% have informed their GP, but only 50% spoke to the specialist about it. Almost all patients (93.1%) wish the establishment of consultation dedicated in CAM in our centre.
ESID-0268 Screening Protocols to Detect Respiratory Infections in PID: Findings from a European Survey and Subclinical Infection Working Group
S. Jolles 1, S. Sánchez-Ramón2, I. Quinti3, P. Soler-Palacin4, C. Agostini5, B. Florkin6, L.J. Couderc7, N. Brodszki8, A. Jones9, H. Longhurst10, K. Warnatz11, F. Haerynck12, A. Matucci13, E. de Vries14
1Cardiff Institute of Infection and Immunity, University Hospital of Wales, Cardiff, United Kingdom
2Department of Immunology, Hospital General Universitario Gregorio Marañón, Madrid, Spain
33rd Department of Internal Medicine and Clinical Immunology, Sapienza University of Rome, Rome, Italy
4Pediatric Infectious Diseases and Immunodeficiencies Unit, Vall d’Hebron University, Barcelona, Spain
5Department of Clinical and Experimental Medicine, University of Padua, Padua, Italy
6CHR de la CITADELLE, Site CITADELLE, Liege, Belgium
7Université de Versailles St Quentin en Yvelines, Hopital FOCH, Versailles, France
8The Children’s Hospital, Skåne University Hospital, Lund, Sweden
9Institute of Child Health, Great Ormond Street Hospital, London, United Kingdom
10Department of Immunology, Barts and The London National Health Service Trust, London, United Kingdom
11Centre of Chronic Immunodeficiency, University Medical Centre Freiburg, Freiburg, Germany
12Pediatric Pulmonary Medicine and Immune Disorders, Ghent University Hospital, Ghent, Belgium
13Department of Internal Medicine, University of Florence, Florence, Italy
14Department of Paediatrics, Jeroen Bosch Hospital, Hertogenbosch, Netherlands
Patients with primary antibody deficiency suffer progressive lung disease due to underlying subclinical infection despite adequate levels of replacement immunoglobulin. Findings from a meeting of UK and EU experts (Paris, June 2013) suggested that subclinical infection was not adequately monitored and screening protocols vary by centre, disease and patient. Various screening measures, including lung function testing (FEV1, FVC), CT, MRI, and induced sputum with exacerbations were used across different centres. Whilst CT scanning was widely reported to be useful in the assessment of lung disease, only 4/?14 centres regularly performed CT scans in patients without confirmed lung disease.
To better understand which screening protocols are used across Europe a survey was conducted to identify how screening type and timing differ between centres and patient groups. The survey covered centres located in the UK, Italy, Spain, France, Germany, Belgium, Sweden and the Netherlands. Data was collected in adult and paediatric patients with PID to assess antibody levels, lung/?upper airway infections, screening methods, including lung function tests; induced sputum with exacerbations; CT; MRI; and treatments used to combat underlying infection (e.?g., antibiotics, IgG therapy).
It is hoped that the survey results will improve our understanding of the differences in screening protocols across Europe, identify key screening tests that should be used to monitor subclinical infection and provide guidance on how and when they should be used. This may represent a starting point for development of a European standard of best practice for detecting and monitoring subclinical infection in PID patients.
ESID-0162 Is There a Role for the Measurement of Specific Antibody Levels in Patients with Primary Antibody Deficiency Receiving Immunoglobulin Replacement?
R. Jones 1, M. Moody1, C. Selwood1, P. Williams1, A. Heaps1, T. El-Shanawany1, S. Jolles1
1Immunodeficiency Centre for Wales, University Hospital of Wales, Cardiff, United Kingdom
Background
The diagnosis of primary antibody deficiency (PAD) relies on laboratory assessment of antibodies. While IgG level determination is standardised, assessing antibody deficiency by interpretation of vaccination responses remains challenging. Conjugated (tetanus and Haemophilus influenzae type B [HIB]) and unconjugated (pneumococcal polysaccharide) vaccines are used to test T-dependent and T-independent antibody responses, respectively. For individual pneumococcal serotypes, protective antibody levels are not well established (range 0.35–1.3 mg/L); neither is the proportion of serotype-specific antibodies above cut-off that confirm normal immune response. We aimed to determine the relationship between trough IgG levels and levels of antibodies specific to tetanus, HIB, pneumococcus and pneumococcal serotypes.
Methods
In this retrospective single-centre study, a cohort of patients with PAD (N=123) receiving IgG replacement was stratified into trough IgG ranges of <7, 7–9, and >9 g/?L. The proportion of patients with protective tetanus, HIB, and pneumococcal antibody levels was calculated.
Results
Overall, the proportion of patients with protective tetanus- and HIB-specific antibody levels was 99% and 93%, respectively, while protective levels of pneumococcal antibodies were achieved in only 70% of patients. The proportion of patients protected against pneumococcal disease depended on the chosen cut-off levels. Using a cut-off of 50 mg/L, 22% of patients with IgG levels <7 g/?L, 69% of patients with 7–9 g/?L, and 76% of patients >9 g/?L were protected.
Conclusion Higher trough IgG levels achieved with replacement therapy correlated with higher specific-antibody titres. These findings may have implications for individual dose adjustment in selected patients.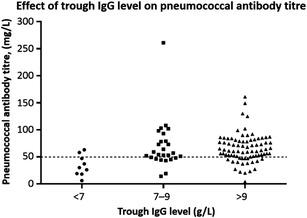
ESID-0230 A New Method of Screening for Antibody Deficiency Using Calculated Globulin (CG)
S. Jolles 1, R. Borrell1, S. Zouwail2, A. Heaps1, H. Sharp1, M. Moody1, C. Selwood1, P. Williams1, S. Holding3, T. El-Shanawany1
1University Hospital of Wales, Immunodeficiency Centre for Wales, Cardiff, United Kingdom
2University Hospital of Wales, Department of Biochemistry, Cardiff, United Kingdom
3Hull Royal Infirmary, Immunology Department, Hull, United Kingdom
Diagnostic delay is a major problem for patients with primary antibody deficiency (PAD) and although strategies using education, computer sorting codes and the ’10 warning signs’ aimed at reducing this time are employed, delays of many years remain frequent before a diagnosis is made and treatment is commenced. Unfortunately the very successful neonatal screening for severe combined immunodeficiency (SCID) using TRECs for T cells and KRECs for B cells will not detect the majority of PAD.
There continues to be a pressing clinical need for a means to screen for PAD to avoid diagnostic delay and the consequences of delayed treatment in terms of end organ damage including bronchiectasis and mortality.
We describe a new method for screening for antibody deficiency using calculated globulin (CG). CG is usually tested as part of a liver function test profile in both primary and secondary care and determines the serum globulin concentration, of which immunoglobulins are a major component. The main use of CG hitherto has been to detect paraproteins when the level is high. This study investigated the potential to use low levels of calculated globulin to detect antibody deficiency.
The study has defined cut-off values for the lower limits of the reference range to detect both primary and secondary antibody deficiency and new paraproteins associated with immune-paresis. CG is cheap (0.1 Euro), widely available and under-utilised. As a proof of principle we have used CG to detect antibody deficient patients, shortening diagnostic delay and time to treatment with immunoglobulin replacement therapy.
ESID-0209 Experience from Liver Transplantation in Six Patients with Primary Immunodeficiency
S.F. Jørgensen 1, M.E. Macpherson2, K. Bjøro3, T.H. Karslen3, A. Foss4, B. Fevang2, P. Aukrust2, I. Nordøy2
1Institute of internal medicine, Oslo University Hospital, Oslo, Norway
2?Section?of Clinical Immunology and Infectious Diseases, Oslo University Hospital, Oslo, Norway
3?Section?of Transplantation Medicine, Oslo University Hospital, Oslo, Norway
4?Section?of Transplantation Surgery, Oslo University Hospital, Oslo, Norway
Introduction: An unknown proportion of patients with primary immunodeficiency have liver disease of various etiologies, and some progress to end-stage liver disease. Reports on the outcome after liver transplantation in these patients are scarce.
Methods: Descriptive demographic and clinical data were retrieved from the patient records and the Nordic Liver Transplant Registry.
Results: Five patients with Common variable immunodeficiency (CVID) (Patients 1-5) and 1 patient with Bruton's ammaglobulinemia (Patient 6) aged 19-62 years, underwent liver transplantation at Oslo University Hospital between 1993 and 2013. Patients 1-4, transplanted from 2009 – 2013, are still alive. Patient 1 and 2 presented with hepatopulmonary syndrome, Patient 3 developed acute-on-chronic liver failure due to portal vein thrombosis, and Patient 4 had cryptogenic, decompensated, cirrhosis. None of these patients have had rejection or relapse of their liver disease to date (median follow-up 3,2 [1,4-4,4]). Reported complications after transplantation for patient 1-4 include Clostridium difficile colitis (1) Pneumocystis jirovecii pneumonia (1), and CMV reactivations and penile cancer (1). Patients 5 and Patient 6 who received liver allografts in 1998 and 1993, respectively both had chronic hepatitis C infection. They died after 0.1 and 1.8 years due to cerebral Aspergillus infection and Gram negative sepsis respectively.
Conclusions: Patients with primary immunodeficiency can be successfully liver transplanted. Enhanced survival from 2009-2013 may be due to improved immunosuppression and better timing of transplantation. Patients with primary immunodeficiency should not be excluded from liver transplantation based on their immunodeficiency alone.
ESID-0158 Safety and Tolerability of Hizentra® in Patients with Primary Immunodeficiency in Japan, Europe, and the US
H. Kanegane 1, K. Imai2, M. Yamada3, H. Takada4, T. Ariga3, T. Hara4, M. Rojavin5, W. Hu6, A. Hubsch7, S. Nonoyama8
1Department of Pediatrics Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, Toyama, Japan
2Department of Community Pediatrics Perinatal and Maternal Medicine, Tokyo Medical and Dental University, Tokyo, Japan
3Department of Pediatrics, Hokkaido University Graduate School of Medicine, Sapporo, Japan
4Department of Pediatrics, Graduate School of Medical Sciences Kyushu University, Fukuoka, Japan
5Research & Development, CSL Behring LLC, King of Prussia, USA
6Research & Development, CSL Limited, Melbourne, Australia
7Research & Development, CSL Behring AG, Berne, Switzerland
8Department of Pediatrics, National Defense Medical College, Tokorozawa, Japan
Background
Patients with primary immunodeficiency (PID) require life-long IgG replacement therapy. Hizentra®, a 20% subcutaneous IgG solution, is increasingly prescribed. This report aims at characterizing the safety and tolerability profiles of Hizentra® in clinical research PID patients in Japan, Europe, and the US.
Methods
Data were extracted from 4 studies: 3 open-label, prospective, multicenter, pivotal Phase III studies carried out in Japan, Europe, and the US, and a follow-up study in Japan.
Results
In Japan, Europe, and the US, patients were enrolled for up to 48, 64 and 40 consecutive weeks, respectively. Overall, 125 patients received 5,208 weekly infusions. While all patients in Japan and the US and most patients in Europe (98%) experienced at least one adverse event (AE), almost all AEs were mild or moderate in intensity; only one AE caused infusion interruption. In total, 1,853 treatment-related AEs were reported with rates of 0.219, 0.098, and 0.632 AE/infusion in Japan, Europe, and the US, respectively. As expected for subcutaneous administration, the most common treatment-related AEs were reactions at the infusion site (0.205, 0.058, 0.591 AE/infusion in Japan, Europe, and the US, respectively); systemic AEs (headache [0, 0.007, 0.016] and pruritus [0, 0.008, 0]) were very uncommon. The differences in recorded local AEs originate mainly from different assessment methods, time points and scales. Of 45 serious AEs reported, only 1 (encephalitis) was considered possibly study-medication related and its occurrence can be alternatively explained.
Conclusion
These results confirmed the positive safety profile of Hizentra® in patients with PID worldwide.
ESID-0754 Treatment with Subcutaneous Immunoglobulins in Infants with Severe Atopic Eczema Associated with Hypogammaglobulinemia
J. Kayserova 1, H. Kubesova1, A. Polouckova1, R. Zachova1, A. Sediva1
1Dpt. of Immunology, 2nd School of Medicine Charles University and University Hospital Motol, Prague, Czech Republic
Infant form of atopic dermatitis (AD) appears between 2nd and 6th month of age and has spread spectrum of clinical manifestations that vary from mild local to severe generalized form.
In our department, we investigated children with severe generalized form of AD with limited effect of local treatment consisted of topical corticosteroids and emollients and with recurrent impetiginization. In majority of these patients, distinct IgG transient hypogammaglobulinemia in infancy was detected (IgG <3.65 g/?L, mean 1.82 g/?L, range 0.57 – 3.43 g/?L ). Serum levels of IgA and IgM isotypes were also decreased or in normal range. The serum IgE level varies between 3.5 – 11 668 IU/mL (mean 1159.3). B-cell immunodeficiency was excluded.
We initialized the treatment with subcutaneous immunoglobulins in dose 0.1-0.2 g/?kg/month in 19 infants. In 15/19 patients, we observed prompt improvement of the eczema, decreased requirement of topical corticosteroids and antibiotics and the increase of serum IgG levels (mean 3.67 g/?L, range 2.57 – 5.15 g/?L). We also observed significant decrease of total serum IgE level in 9/?19 patients.
Immunological examination is fully indicated in severe AD form in infants. Established IgG hypogammaglobulinemia requires immunoglobulin substitution therapy leading to the modulation of the immune system and to decreased infections. Finally, it results in the improvement of eczema.
ESID-0195 Subcutaneous Immunoglobin Replacement Therapy – Flexible Dosing
S. Duraisingham1, J. Dempster1, L. Lorenzo1, S. Grigoriadou2, M.S. Buckland2, H.J. Longhurst 2
1Immunology department, Bart's Health NHS Trust, London, United Kingdom
2Immunopathology department, Bart's Health NHS Trust, London, United Kingdom
Background
Standard treatment for patients with antibody deficiency involves 3–4 weekly intravenous or weekly subcutaneous IgG (SCIg) replacement therapy. Recent data suggest that treatment efficacy is sustained under less frequent subcutaneous dosing regimens. The clinical outcomes following weekly SCIg treatment were compared with those provided by SCIg infusions received every 10–14 days in a real-world setting.
Methods
This retrospective analysis included all adult patients receiving SCIg who were attending the Barts Health NHS Trust Immunodeficiency Clinic in London, UK as of May 2013 (N=92). SCIg doses and trough levels, infusion frequency, and infections/complications were reviewed.
Results
The majority of patients received weekly SCIg (83/92 patients); only 10% of patients received less frequent infusions (every 10–14 days). Most patients self-administered IgG at home, especially those receiving weekly infusions. There was no relationship between infusion frequency and patient indication (primary and secondary antibody deficiencies). The median 4-weekly dose received by patients infused every 10–14 days was lower than that of patients on a weekly regimen (0.387 g/?kg/4 weeks [95% confidence interval 0.320–0.571] vs 0.617 g/?kg/4 weeks [0.386–1.375], p=0.0002); however, mean trough IgG levels were comparable between the two groups. No difference in prevalence of bronchiectasis was recorded between the two groups and preliminary data suggest that both groups experienced serious and non-serious infections with similar frequency.
Conclusion
SCIg appears equally efficacious when administered weekly and at 10–14?day intervals. In future, increased flexibility in SCIg dosing may allow individualisation of SCIg dosing for each patient.
ESID-0091 Primary vs. Secondary Antibody Deficiency: Clinical Features and Infection Outcomes of Immunoglobulin Replacement
S.S. Duraisingham1, M.S. Buckland1, J. Dempster1, L. Lorenzo1, S. Grigoriadou1, H.J. Longhurst 1
1Immunology, Barts Health NHS Trust, London, United Kingdom
Background
Secondary antibody deficiency can occur as a consequence of haematological malignancies, chemotherapy or immunosuppressive medications. The prevalence of secondary antibody deficiencies is increasing but published data to inform the clinical management of these patients is lacking. Here we review our cohort of patients receiving immunoglobulin (Ig-) replacement and compare primary and secondary antibody deficiency patients.
Methods
A retrospective review of 167 adult patients receiving Ig-replacement attending the Immunodeficiency clinic was conducted after obtaining written consent. The causes of immunodeficiency, diagnosis delay, clinical and laboratory features, and infection frequency before and after Ig-replacement were compared for those with primary (n=126) or secondary antibody deficiency (n=39).
Results
Both primary and secondary antibody deficiency groups experienced comparable delays in diagnosis and had a similar prevalence of bronchiectasis (37% vs. 28%). The most common causes of secondary antibody deficiency were chemotherapy for B cell lymphoma, the use of Rituximab, corticosteroids or immunosuppressive medications. Secondary antibody deficiency patients had higher baseline levels of serum IgM and IgA and a greater percentage of switched memory B cells. The secondary antibody deficiency group received a lower dose of Ig-replacement (g/?kg/4-weeks) (mean ±95% CI: 0.59±0.10 vs. 0.80±0.12, *p=0.011), but maintained similar IgG trough levels (9.92±0.63 vs. 10.62±0.50, p=0.171). Secondary antibody deficiency patients experienced more serious infections before treatment, but both groups had a significantly reduced frequency of serious and non-serious infections following Ig-replacement.
Conclusion
Patients with secondary antibody deficiency experience similar problems with diagnosis delay and bronchiectasis as primary antibody deficiency patients, and similarly benefit from Ig-replacement.
ESID-0702 Temozolomide-Induced Immunosuppression and Anticancer Activity is Associated with Development of Allergy: Management by Desensitization
D. Maddox 1
1Allergy and Immunology, Mayo Clinic, Rochester, USA
Temozolomide is a highly reactive cancer chemotherapeutic agent capable of forming covalent adducts with a wide variety of endogenous proteins. Effective in brain cancers, clinical use of this agent is often attended by development of allergic symptoms, most commonly skin rashes, especially urticaria. We present here a series of cases in which allergy to temozolomide has been managed by drug desensitization. These desensitizations have been accomplished in the span of a single day, in the outpatient Allergy Laboratory of the Mayo Clinic in Rochester, Minnesota. Details will be presented regarding the dose escalation strategy, and the clinical responses of the cases during the course of desensitization. We have been successful in achieving tolerance to the drug in all but one case. There have been no severe episodes of anaphylaxis induced by the protocol used, and administration of the drug has been exclusively oral.
ESID-0573 Reduced Intensity Conditioning Allogenteic Hematopoietic Cell Transplantation Outcomes for 160 Patients with Primary Immune Deficiencies
R. Marsh 1, D. Bellman1, M. Jordan1, M. Grimley1, A. Kumar1, S. Jodele1, K.C. Myers1, S. Chandra1, T. Leemhuis1, P.A. Mehta1, J.J. Bleesing1, M.B. Jordan1, S.M. Davies1, A.H. Filipovich1
1Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children's Hospital Medical Center, Cincinnati, USA
Introduction: Reduced intensity conditioning (RIC) regimens are increasingly used for the hematopoietic cell transplantation (HCT) of patients with primary immune deficiencies (PID). Data regarding large patient groups are limited.
Methods: We analyzed outcomes of 160 PID patients who underwent allogeneic RIC HCT using alemtuzumab, fludarabine, and melphalan at Cincinnati Children's Hospital from 2004-May, 2013. Kaplan Meier curves were generated to estimate one year and long-term event-free (EFS) survival (event=death or retransplant). Cox proportional hazard regression models were constructed using backward selection to examine the effects of the following covariates on overall survival: age, diagnosis (HLH/XLP, SCID, CID/CVID, Other), donor/recipient HLA match, occurrence of grade 3-4 acute graft versus host disease (GVHD), occurrence of mixed chimerism (MC), and alemtuzumab schedules. Covariates with p<0.10 were included in the final model. Significance was considered for p<=0.05.
Results: One-year and long-term EFS were estimated at 74% (CI 67-81%) and 62% (CI 54-70%), respectively. Overall survival was adversely influenced by a diagnosis category of Other (HR 3.21, CI 1.54-6.71, p=0.002), donor/recipient HLA mismatch (HR 3.11, CI 1.12-8.67m p=0.030), and grade 3-4 acute GVHD (HR 1.95, CI 1.01-3.77, p=0.045). The occurrence of MC appeared to positively influence survival (HR 0.50, CI 0.25-0.99, p=0.048).
Conclusion: Our experience suggests a 1 year EFS of 74% for PID patients treated with alemtuzumab, fludarabine, and melphalan RIC HCT. Donor/recipient HLA mismatch and grade 3-4 GVHD significantly negatively affect overall survival. There appears to be an advantage for patients who develop mixed chimerism, likely due to the associated lack of GVHD.
ESID-0025 The Role of C1 Esterase Inhibitor Levels in CVID
I. Melamed 1, A.L.E.X. Testori1, A. Mcdonalnd1, M. Heffron1
1Research, IMMUNOE Health Centers, Centennial, USA
Objective:
CVID is the most common PID . IVIG is relatively safe, with headaches and fatigue being the most common side effects. C1 esterase inhibitor (C1-INH) is the major inhibitor of the classical pathway (3-4). Recent studies suggest some anti-inflammatory function for this molecule, possibly explaining the effects of C1-INH in diseases other than hereditary angioedema (5).
Design:
A prospective study evaluating C1 INH in 73 patients diagnosed with CVID . C1-inhibitor functional assay results are reported as % of mean normal activity. Quantitative assays report C1-INH concentration in mg/dL.
Method:
The primary endpoint was the C1 INH levels with a correlation to the severity of AEs occurring after IVIG therapy, which included: headache, fatigue, nausea, and pain.
Results:
14 of these patient had low levels of C-?1 INH protein, varying from 6 to 20 mg/dl (mean of 16 +/_ 2, with normal range 21-39 mg/dl) and/or reduced enzyme function ranging from 20% to 65% ( mean of 44% +/- 12% , with 67% of mean normal enzymatic function being the lower limit of what is considered normal enzyme activity).
We found a correlation between AE and C1-INH levels.
Conclusion:
We propose that a subset of patients with CVID is characterized by lower serum C-?1 INH protein concentration and/or enzymatic C1-INH function. This implies that these patients could be more vulnerable to side effects of IVIG therapy. As a mechanism, we hypothesize that the infusion of IGGs may trigger consumption of complement proteins and of C1-INH, precipitating inflammatory reactions, including aseptic meningitis.
ESID-0019 The Benefit of Gammaplex 5% IVIG for Patients Not Able to Tolerate 10% IVIG
I. Melamed 1, M. Heffron1, A. McDonald1
1Research, IMMUNOE Health Centers, Centennial, USA
Objective:
Tolerability of IVIG is very good. However, a subpopulation of patients have difficulty tolerating a high concentration product, such as a 10% IVIG formulation, due to systemic adverse events (AEs), such as headaches, fatigue, fever and muscle aches. While many patients experience fewer systemic AEs with SCIG, it requires weekly infusions and most patients experience infusion site reactions.
Design:
A retrospective chart review of 12 patients switched to a 5% solution of IVIG from a higher concentration due to the occurrence of non-serious AEs.
Method:
Via descriptive statistics, primary endpoint: number and severity of adverse events occurring between 5% IVIG and 10% IVIG, which included: headache, fatigue, nausea, arthralgia, muscle spasms, pain. Secondary endpoint: trough levels of IVIG.
Results:
Severity of adverse events with the 5% IVIG infusions was generally of 1+ or 2+ in nature vs. the 2+, 3+ and 4+ severity reported with the 10% infusions. Severity of 6 common AEs was reduced with a switch to a 5% concentration without compromising infection-preventing trough levels of IgG.
Conclusion:
We observed 5% IVIG was better tolerated in patients previously having side effects with 10% IVIG and may be a valid alternative for patients who poorly tolerate infusion with a 10% solution. Investigation of the mechanism responsible for better tolerability with 5% IVIG vs 10% is needed.
ESID-0584 Membranoproliferative Glomerulonephritis in a Patient with X-Linked Agammaglobulinemia: Case Report
R. Mijanovic 1, S. Andrejevic1, A. Kezic2, S. Pasic3, J. Markovic-Lipkovski4, B. Bonaci-Nikolic1
1Clinic for allergology and clinical immunology, Clinical centre of Serbia, Belgrade, Serbia
2Clinic for nephrology, Clinical centre of Serbia, Belgrade, Serbia
3Department for clinical immunology and allergology, Institute for Health Protection of Mother and Child of Serbia, Belgrade, Serbia
4Institute of pathology, Medical faculty of Belgrade, Belgrade, Serbia
X-linked agammaglobulinemia (XLA) is a primary immunodeficiency (PID) characterized by agammaglobulinemia requiring replacement treatment with immunoglobulin. The association of XLA and membranoproliferative glomerulonephritis (MPGN) is unexpected and, to our knowledge, only three cases were previously published.
Case Report: A 28 year-old man was diagnosed with XLA when he was 3-years old and he has been treated with parenteral immunoglobulin since then. Twenty-two years later he was diagnosed as sprue-like enteropathy and gluten free diet was introduced. During last year he gradually developed edema of lower limbs. Laboratory studies revealed nephrotic range proteinuria, hypoalbuminemia with normal renal function and serum complement levels. Viral infections were excluded by PCR studies (HIV, HCV), and HBsAg was also negative. A renal biopsy was performed and histology presented glomeruli with membranoproliferative patterns, with segmental thickening of glomerular basal membrane, without significant interstitial inflammation or tubular atrophy. The patient was diagnosed with MPGN. The preparation of immunoglobulin was replaced by different brand, and ramipril was started at a dose of 2.5 mg/day. After one month the patient maintained a normal blood pressure and normal renal function, and the range of proteinuria was decreased from 3?g to 1.5 g/?day. The dose of ramipril was increased to 5 mg/day. After two months proteinuria was below 1?g per day.
Conclusion: Immunosuppressive therapy is expected to increase incidence of infections especially in patients with PID. We herein present the first case of MPGN in a patient with XLA with favorable clinical response not treated with corticosteroids.
ESID-0512 Haploidentical TCR Alfa/?Beta and CD19 Depleted Stem Cell Transplant for Primary Immunodeficiency – 4 Cases
B. Morillo-Gutierrez 1, Z. Nademi2, J. Dunn3, D. Barge4, S. Hambleton2, M. Abinum2, T. Flood2, A.J. Cant2, A.?R. Gennery2, M. Slatter2
1Department of Paediatric Immunology, Newcastle upon Tyne Hospital NHS Foundation Trust, Newcastle Upon Tyne, United Kingdom
2Department of Paediatric Immunology, Institute of Cellular Medicine Newcastle University. Newcastle upon Tyne Hospital NHS Foundation Trust, Newcastle Upon Tyne, United Kingdom
3Hematopoietic Stem cell Laboratory, Newcastle upon Tyne Hospital NHS Foundation Trust, Newcastle Upon Tyne, United Kingdom
4Regional Immunology Laboratory, Newcastle upon Tyne Hospital NHS Foundation Trust, Newcastle Upon Tyne, United Kingdom
BACKGROUND
For many patients with Primary immunodeficiency (PID), Hematopoietic stem cell transplant (HSCT) is the only curative treatment available. If there is no HLA identical donor the use of HLA-haploidentical family donors is problematic due to the increased occurrence of graft versus host disease (GVHD) and graft rejection particularly for patients with forms of PID other than severe combined immune deficiency. Various T-cell depletion strategies have been used, but some can lead to slow immune reconstitution with the consequent risk of morbidity and mortality from infection. Depletion of T cell receptor alpha and beta (TCRaß) and CD 19+ cells, with selection of TCR?d T-cells and preservation of NK, myeloid and plasmacytoid dendritic cells which aid engraftment and also decrease the risk of infection, has been shown to be a promising new technique.
CASE SERIES We report four children with different PIDs treated successfully using this technique (see figure 1 for transplant details).
CONCLUSION
To our knowledge these are the first 4 patients reported in the UK who have had a successful haploidentical TCRaß and CD19 depleted HSCT. All 4 had viral reactivation but are alive and well 2 -18 months post transplant, with chimerism >95% and good immune reconstitution. Pending the results of larger series, this could be a promising technique in haploidentical HSCT with early and sustained engraftment, early immune recovery and low risk of GVHD for PID patients.
ESID-0772 Rotavirus Vaccine Serotype Infection in SCID Patients Following Introduction of Universal Rotavirus Vaccination in UK – 4 Cases
B. Morillo-Gutierrez 1, A. Worth2, M. Slatter3, Z. Nademi3, T. Flood3, M. Valappil4, S. Waugh5, J. Breuer6, B. Gaspar7, A. Gennery3
1Department of Paediatric Immunology, Newcastle upon Tyne Hospital NHS Foundation Trust, Newcastle Upon Tyne, United Kingdom
2Department of Immunology, Great Ormond Street Hospital, London, United Kingdom
3Department of Paediatric Immunology, Institute of Cellular Medicine Newcastle University. Newcastle upon Tyne Hospital NHS Foundation Trust, Newcastle Upon Tyne, United Kingdom
4Department of Laboratory Medicine, Newcastle upon Tyne Hospital NHS Foundation Trust, Newcastle Upon Tyne, United Kingdom
5Department of Laboratory Medicine, Newcastle upon Tyne Hospital NHS Foundation Trust, Newcastle Upon Tyne, United Kingdom
6Department of Virology, Camelia Botnar Laboratories Great Ormond Street Hospital, London, United Kingdom
7Molecular and Cellular Immunology section, UCL Institute of Child Health, London, United Kingdom
BACKGROUND
Chronic infection with rotavirus following vaccination with live-attenuated virus strain in SCID was described in US after introduction of the vaccine in routine childhood vaccination schedules in 2009; some patients developed life-threatening infections; therefore, SCID was listed as a contraindication to receiving this vaccination. The recommendation to vaccinate infants between 6-8 weeks of life, before most infants were diagnosed with SCID; further established the need of a newborn screening programme for SCID, subsequently successfully introduced in many US states from May 2010 . In UK, rotavirus live vaccine was included routinely in the infant vaccination schedule from 1 July 2013; however, no newborn screening program for SCID has been implemented in the UK to date.
CASE SERIES
We report four infants who received rotavirus live vaccine before being diagnosed with SCID and had subsequently rotavirus vaccine-serotype isolated in stools. Two patients presented with severe failure to thrive requiring parenteral nutrition. All presented with chronic diarrhoea. To date, two patients have been successfully transplanted and have cleared the infection.
CONCLUSION
These are the first 4 patients reported in the UK with SCID and rotavirus vaccine-serotype enteric infection following the introduction of the rotavirus live vaccination. As this infection can lead to significant nutritional deficit and life-threatening complications, we reiterate that rotavirus live vaccine should not be given to those infants with SCID. This will be easier to avoid once SCID newborn screening is implemented.
ESID-0127 Registry Study to Collect Long-Term Safety Data from Female Subjects Who Become Pregnant During Treatment with HYQVIA® (IGHY)
B. McCoy1, N. Nikolov2, A. Nagy1, C. Hermann1, R.I. Schiff 3
1Bioscience, Baxter Healthcare, Vienna, Austria
2BioScience, Baxter Healthcare SA, Zurich, Switzerland
3Bioscience, Baxter Healthcare, Westlake Village, USA
Introduction: IgHy (recombinant human hyaluronidase [rHuPH20]-facilitated subcutaneous [SC] infusion of immunoglobulin [IG]) is used as replacement therapy in adults (=18 years) with primary immunodeficiency (PID) syndromes, myeloma, or chronic lymphocytic leukemia. This is a non-interventional, prospective, uncontrolled, open-label, multicenter, post-authorization study in the EEA to evaluate the safety of IgHy in women who become pregnant during treatment. The outcome of the pregnancy as well as the physical and neurological development of the infant will be monitored. This abstract describes the study design to facilitate the enrollment of patients treated in clinical settings.
Study: All females who become pregnant during IgHy treatment (treated while pregnant or in the 30 days prior to conception) will be encouraged to participate. As soon as the patient becomes aware of the pregnancy, treatment with IgHy will be stopped. Subjects will receive an alternative gammaglobulin treatment and all other medical care will be performed as is standard. The overall duration of the study is approximately 6 years and there is no minimum sample size pre-specified. Data from medical records on the routine assessments during the pregnancy will be collected. In those subjects consenting to blood drawing, samples will be taken to assess antibodies against rHuPH20 every 3 months. Data will be assessed on the development of the fetus in utero, and on the growth and development of the infant for 2 years after delivery. Descriptive methods will be used to analyze and report data from this IgHy pregnancy registry.
ESID-0098 Non-Interventional Post-Authorization Safety Study (PASS) on the Long-Term Safety of HYQVIA® (IGHY)
K. Fielhauer1, N. Nikolov2, C. Hermann1, A. Nagy1, R.I. Schiff 3
1Bioscience, Baxter Healthcare, Vienna, Austria
2Bioscience, Baxter Healthcare SA, Zurich, Switzerland
3Bioscience, Baxter Healthcare, Westlake Village, USA
Introduction: IgHy (recombinant human hyaluronidase [rHuPH20]-facilitated subcutaneous [SC] infusion of immunoglobulin [IG]) is a novel treatment that has been approved as a replacement therapy in adults (=18 years) with primary immunodeficiency (PID) syndromes, myeloma, and chronic lymphocytic leukemia. This is a non-interventional, prospective, uncontrolled, multi-center, open label, post-authorization safety study to be conducted in the EEA to acquire additional data (including the assessment of anti-rHuPH20 antibodies) on the long-term safety of IgHy. This abstract describes the study design to facilitate the evaluation of patients treated in a clinical setting.
Study: This study is expected to enroll 80 to120 adult patients who have been prescribed IgHy. Treatment regimens will be prescribed at the discretion of the attending physician. The recruitment period will be 3 years and the overall duration of the study is expected to be approximately 6 years. Data from laboratory assessments (including the optional measurement of anti-rHuPH20 antibody titers) performed on a regular basis during routine clinical visits will be collected as available. Data to be documented includes safety (AEs and SAEs), dose, frequency and rates of administration, number of needle sites, total IgG levels, health-related quality of life, and health resource use. Data will be obtained from medical records, questionnaires, and any subject diaries or home treatment records maintained by the patient. If a subject becomes pregnant, IgHy treatment will be stopped and the subject will be invited to participate in the Pregnancy Registry. Descriptive methods will be used to analyze and display data from this study.
ESID-0139 Post-Authorization Safety Surveillance of KIOVIG (Human Normal Immunoglobulin 10% Liquid, Baxter) in an Open, Uncontrolled, Non-Interventional, Observational Cohort Study in Immunodeficiency and Autoimmune Diseases
S. Misbah1, P. Soler-Palacín2, B. McCoy3, W. Engl3, V. Empson3, N. Nikolov 4
1Academic Centre L4, John Radcliffe Hospital, Oxford, United Kingdom
2Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d'Hebron, Barcelona, Spain
3BioScience, Baxter Healthcare, Vienna, Austria
4BioScience, Baxter Healthcare SA, Zurich, Switzerland
Background: Immunoglobulin preparations are used in patients with antibody deficiencies and autoimmune disorders. KIOVIG is a liquid 10% human IgG preparation for intravenous infusion and is licensed for subcutaneous use in some countries.
Methods: Patients receiving KIOVIG were treated according to local protocols. Infusion rates were 0.5 mL/kg/h-6 mL/kg/h, increasing as tolerated. Dose was dependent on indication. Subjects were followed for 6±1 weeks to 12±2 months depending on indication. Efficacy, adverse events (AEs) and infusion rates were recorded.
Results: Eighty-eight subjects were enrolled at 20 sites: 39 had immunodeficiency disease (ID) and 49 had autoimmune disease (immune thrombocytopenia [ITP]:26, Kawasaki disease:9, Guillain-Barré syndrome:14). In ID, the mean serious bacterial infection rate was 0.18 infection/patient/year; monthly infection rates were 0.17 for IgG-pretreated subjects and 0.50 for IgG-naïve subjects. In ITP, the median platelet count was 7x109/L at diagnosis and 356x109/L during observation. In Guillain-Barré syndrome, all subjects had a disability score =Grade 2?at screening; at the end of observation, 2 were Grade 0 (healthy), 5 were grade 1 (showed only minor symptoms), and 7 still had symptoms rated =Grade 2. In Kawasaki disease, all 9 subjects had elevated temperatures at screening and 8/?9 were afebrile after treatment. One serious AE related to Kiovig (urticaria) occurred in one subject with ID; none occurred in subjects with autoimmune disease. No severe suspected adverse drug reactions were reported for any indication.
Conclusions: Efficacy and safety of KIOVIG in this observational study under routine clinical conditions corroborated results reported in previous clinical studies.
ESID-0271 Kiovig (Human Normal Immunoglobulin 10% Liquid) in Pediatric Immune Thrombocytopenia (ITP): Safety and Efficacy Data from a Prospective, International, Non-Controlled, Observational Study
B. Blazek1, C. Hermann2, H. Leibl2, B. McCoy2, D. Gelmont3, N. Nikolov 4
1Faculty Hospital Prague-Motol, University Hospital Ostrava, Ostrava, Czech Republic
2BioScience, Baxter Healthcare, Vienna, Austria
3BioScience, Baxter Healthcare, Westlake Village, USA
4BioScience, Baxter Healthcare SA, Zurich, Switzerland
Background: Immunoglobulin (IG) products have been shown to be effective in certain immune-mediated disorders such as ITP. The efficacy and safety of KIOVIG in adults (aged =18 years) with chronic ITP has been shown previously (Varga, 2006).
Methods: Pediatric patients with acute ITP (platelet counts <30x109/L) and/or needing treatment to prevent bleeding complications were enrolled. KIOVIG was administered intravenously at an initial rate of 0.5 mL/kg/h, but increased gradually to 6 mL/kg/h if well tolerated. Subjects were followed for 6±1 weeks. Platelet counts, adverse drug reactions (ADRs) and serious adverse events (SAEs) for subjects (aged =18 years) are reported here.
Results: Twenty-five pediatric patients with acute ITP (<30x109/L) were enrolled. The median platelet count at the time of diagnosis was 6x109/L (range 1-28). Of the 25 subjects, 80% (20/25) had at least one bleeding event at the time of their first KIOVIG infusion and 84% (21/25) had no new bleeds after the first treatment. At study termination, 96% (24/25) of subjects reached platelet counts of >30x109/L; this level was reached within a median time of 3 days after the first KIOVIG infusion. The median maximum level of platelet count during observation was 356x109/L (range 41-887). Suspected ADRs, generally mild/?moderate, occurred in 48% (12/25) of subjects; no SAEs were related to KIOVIG.
Conclusions: The use of Kiovig in pediatric patients with ITP in the real world setting is comparable to what has been observed in clinical studies, and corroborates the therapeutic profile of KIOVIG under routine clinical conditions.
ESID-0776 Outcome and Complications After Allogenic Haematopoietic Stem Cell Transplantation in Major Histocompatiblity Complex Class II Deficiency
M. OUEDERNI 1, R. HASSOUNA1, M. BEN KHALED1, S. THRAYA1, N. DHOUIB1, A. HAOUA1, F. MELLOULI1, M. BEJAOUI1
1Pediatric Immuno-hematology Unit, Bone Marrow Transplant Center, Tunis, Tunisia
Background: haematopoietic stem cell transplantation (HSCT) is the only curative option for patients with major histocompatibility complex class II (MHCII) deficiency; however, the overall cure rate is lower than in other immunodeficiencies. This study details complications and outcome of MCHII deficiency after allogeneic HSCT.
Subjects and Methods: This is a retrospective study of 13 patients who underwent allogenic HSCT for MHCII deficiency between 2007-2013.
Results: All patients received bone marrow graft from identical related donors. The median age at HSCT was 17 months (7 months to 6,4 years). No primary graft failure was noted. Viral infections were detected in 8 patients (61,5%) between 21-85 days post HSCT (median 28d): Cytomegalovirus (n=6), Adenovirus (n=2). 11 out of 13 patients (84,6%) developed acute graft-versus-host disease (aGvHD) between 6-18 days (madian13d). No chronic GvHD occurred. Seven patients developed sinusoidal obstruction syndrome between 2–25 days (median 9d). Seven of 13 patients (53, 4%) died after HSCT ranging from 8-114 days (median 59d). Primary causes of death included: infections (viral =3, bacterial =2), aGvHD (n=1), bronchiolitis obliterans (n=1). Patient age exceeding 2 years at HSCT was found significantly associated to higher mortality (p=0,004, Odds ratio=7).
Conclusion:
Despite the availability of HLA-identical related donor, MHC class II deficiency is linked with poor prognosis. Age at transplantation is a limiting factor, multiplying the risk of death by 7 if higher than two years and suggesting the role of pre-HSCT viral infectious status. Thus, improving HSCT results would be through early diagnosis and prompt management.
ESID-0478 Efficacy and Tolerability of Privigen in Secondary Immunodeficiencies – Results from a Multicenter Observational Study
H.R. Slawik1, M. Plath1, M. Reiser2, B. Otremba3, P. Lotichius4, D. Pfruender 4
1, Onkologische Schwerpunktpraxis, Augsburg, Germany
2, Praxis internistischer Onkologie und Hämatologie, Köln, Germany
3, Onkologische Praxis, Oldenburg, Germany
4, CSL Behring, Hattersheim, Germany
Introduction: Privigen is a 10% polyvalent human IgG preparation for intravenous administration (IVIG) using L-proline as stabiliser.
Objective: To investigate the efficacy and tolerability of Privigen in a large sample of patients with secondary immunodeficiencies.
Methods: Interim analysis of an observational study in 128 German centers (cut-off date July 9, 2013).
Results: 1,166 patients (632 males, 534 females; mean age 67 years) received a total of 10,783 Privigen infusions. The mean observation period was 9.4 months. The average monthly dose was 14.0?g (0.2 g/?kg body weight). The majority of the patients had hematologic diseases; in 24 patients, other reasons for the secondary immunodeficiency were reported (mainly solid tumors). Efficacy was judged as very good or good in 93.8%, moderate in 5.4% and insufficient in 0.8% of evaluable cases (n=1,082). Patients who had not received any IVIG treatment prior to study entry and who received =6 infusions of Privigen for =120 days (infusion intervals =20 d and =60 d) experienced significantly fewer infections during the study than before: The mean annualized infection rate dropped from 5.6 to 1.5 (n=91; p< 0.0001). Tolerability was judged as very good or good in 92.5%, moderate in 4.7% and insufficient in 1.8% of all cases (1.0% missing data). Adverse events possibly or probably related to Privigen were reported for 158 of the 10,783 infusions (1.5%); 12 events were considered serious (0.1%).
Conclusions: Privigen significantly reduced infection rates in secondary immunodeficiencies. Tolerability was very good or good in the majority of patients.
ESID-0136 Analysis of Immunoglobulin (IG) Treatment in Germany: Do Doctors Differentiate Dosages Between Different Forms of Administration in Primary Immune Deficient (PID) Patients?
M. Borte 1, D. Huscher2, N. Zessack3, D. Pittrow4, M. Hensel5, R. Gold6, W. Kirch7, M. Stangel8, X. Ye9, P. Faivre10, C. Sommer11, M. Reiser12, M. Fasshauer1, U. Baumann13
1Paediatric Rheumatology Immunology and Infectiology, Hospital St. Georg, Leipzig, Germany
2Epidemiology, German Rheumatism Research Centre Berlin and Rheumatology Charité University Hospital, Berlin, Germany
3Medical Affairs, Baxter GmbH, Unterschleissheim, Germany
4Institute for Clinical Pharmacology, Medical Faculty Technical University Carl Gustav Carus, Dresden, Germany
5Q5 14-22, Mannheimer Onkologie Praxis, Mannheim, Germany
6Department for Neurology, St. Josef-Hospital Ruhr University, Bochum, Germany
7Institute for Clinial Pharmacology, Medizinische Fakultät Carl Gustav Carus an der TU Dresden, Dresden, Germany
8Department for Neurology, Medizinische Hochschule Hannover, Hannover, Germany
9Medical Affairs, Baxter HealthCare, Deereld IL, USA
10Medical Affairs, Baxter HealthCare SA, Zurich, Switzerland
11Department for Neurology, University Hospital, Wuerzburg, Germany
12Praxis Internistische Onkologie, (PIOH), Cologne, Germany
13Paediatric Pulmonology Allergy and Neonatology, Hanover Medical School, Hanover, Germany
Background: This study examines whether there are dosage differences for PID patients receiving intravenous (IV) compared to subcutaneous (SC) Ig treatment. European Medicines Agency recommends monitoring IgG trough levels which lead to dose equivalence amongst SC and IV, while Food and Drug Administration recommends an increase in dose of 37-53% when moving from IV to SC due to lower bioavailability of serum IgG observed with the SC route of administration.
Methods: Patients were identified from the German SIGNS registry (NCT01287689), a prospective observational study which assesses long-term Ig use in patients with immune deficiencies. Mann-Whitney test was used for comparing continuous variables.
Results: Of the 629 patients registered as of 07 May 2014, a total of 169 PID patients were identified: 65 children (<18 yrs) and 104 adults. 72% of the children received SC vs 14% of adults at baseline (p<0.001). Mean 4-week dosage for children was 404 (IV) vs 576 mg/kg (SC) (p=0.432) and 270 (IV) vs 760 mg/kg (SC) for adults (p=0.001). IgG trough levels for children at last available visit were 7.2 (IV) and 8.3 g/?l (SC) (p=0.059), for adults 5.3 (IV) and 6.7 g/?l (SC) (p=0.083).
Conclusions: The data show that in adult PID patients, dosages of SC are significantly higher compared to IV – according to treatment recommendations indicating an underdosement of the adult population receiving IV IG. Further analysis and research is needed to determine whether this pattern is also observed in other patient populations.
ESID-0259 Radiosensitivity and Blimp-1 Expression in Patients with Common Variable Immune Deficiencies
S. Ferrari 1, V. Conti2, A. Prezzo2, F. Cavaliere2, R. Zuntini1, I. Quinti2
1Genetics, S. Orsola Malpighi Hospital, Bologna, Italy
2Molecular Medicine, Sapienza University of Rome, Rome, Italy
In a cohort of 18 CVID patients radiosensitivity was evaluated at different time points (T0, T1, T24) after a 4?Gy irradiation by measuring the rate of H2AX histone phosphorylation (?H2AX) on T lymphocytes unstimulated or cultured with Staphylococcus aureus enterotoxin B with or without caffeine and Ku-55933. A constitutive H2AX phosphorylation was evident in resting T cells of HD and CVID patients. Irradiation induced a prompt increase of ?H2AX expression that reached its maximum expression at T1 in all HD and in 15/18 CVID patients. At time point T24 the level of ?H2AX was below the constitutive level. The addition of caffeine and Ku-55933 inhibited ?H2AX expression to the level found in AT patients. We then investigated if irradiation could induce Blimp-1 activation in T cells of HD and CVID patients at the same time points. In HD, immediately after irradiation Blimp-1 was over-expressed, before the ?H2AX appearance, then it decreased at T1 and T24. In CVID, the Blimp-1 expression was delayed in that it peaked at T1 post-irradiation at the time of ?H2AX expression, and it was still evident at T24 when the ?H2AX expression was lost. In HD, AID expression was evident starting from T1, and then it continued to increase thereafter to T24. In CVID, a low AID expression was detectable only at T24.
Our observations showed that Blimp-1 is the first signal of DNA DBS after irradiation preceding the H2AX phosphorylation. This phenomenon was weak and delayed in CVID patients.
ESID-0085 Adequate Patient’s Outcome Achieved with Short Immunoglobulin Replacement Intervals in Severe Antibody Deficiencies
C. Milito 1, F. Pulvirenti1, G. Barilaro1, A. Pesce1, M. Digiulio1, C. Di Iorio1, L. Bonanni1, I. Quinti1
1Molecular Medicine, Sapienza University of Rome, Rome, Italy
The optimal immune globulin replacement dosages required to minimize infections in patients with Primary Antibody Deficiencies are not definitely established with a wide variation in treatment practices. Subgroups of patients may benefit from treatment with higher or lower dosages of immunoglobulins. The aim of our study was to verify the efficacy of a new rationale for individualized immunoglobulin utilization and to elucidate the effects of care on patient outcomes to identify what works, in which setting and under what conditions.
A single centre interventional study on 119 patients with Primary Antibody Deficiencies was performed to determine for each patient the best interval between intravenous immune globulins injections and the Ig cumulative monthly dosage to maintain IgG trough levels >500 mg/dL, to reduce major infections and adverse events.
Ninthly eight per cent of patients achieved the study objective. Patients with low switched memory B cells and IgA serum levels and/or affected by bronchiectasis and/or enteropathy and/or adverse events despite pre-medications achieved our objective by shortening the administration intervals to 2-weeks or to 1-week without increasing the monthly cumulative dosage. The adverse events were reduced by administrating low Ig dosages in a single setting. Two patients with severe enteropathy needed to receive both intravenous and subcutaneous immunoglobulins to reach a satisfactory outcome.
Patients with fewer complications achieved the study objective with immune globulin replacement administered with the widely used interval of 3 or 4 weeks.
The exact timing and optimal immunoglobulin prophylaxis regimen might be tailored according to clinical and immunological markers.
ESID-0086 Acute and Compensated Chronic Haemolysis in Patients with Antibody Deficiencies on Immunoglobulin Replacement Treatment
F. Pulvirenti 1, C. Milito1, C. Di Iorio1, G. Girelli1, I. Quinti1
1Molecular Medicine, Sapienza University of Rome, Rome, Italy
IgG replacement with intravenous or subcutaneous immune globulins is a lifelong replacement therapy in patients with primary antibody deficiencies. Haemolysis after immune globulins has been described in patients receiving high dosages while haemolysis after immune globulin administration at replacement doses has been considered of little clinical significance. Differently from acute haemolysis, mild haemolytic reactions can be easily missed and the true incidence of such reactions is difficult to document without careful clinical and laboratory follow-up.
We analysed the clinical consequences of possible passive transfer of blood group antibodies after immune globulin administration in 162 patients with primary antibody deficiencies on Ig substitutive treatment in a single centre observational study.
Immune globulins administered at replacement dosages caused severe acute haemolysis in 3/?156 patients due to irregular alloantibodies. Anti-A antibodies caused mild haemolysis in 5/?156 patients. In addition, immune globulins contributed to establish a condition of poorly symptomatic compensated chronic haemolysis possibly leading to splenomegaly and causing adverse events. Polyvalent immune globulin preparations examined contained multiple clinically significant antibodies that could have unexpected haemolytic consequences, as anti-C and anti-c, whose research and titration are not required by the European Pharmacopoeia. Thus, in terms of safety the issue of acute and chronic haemolysis in long term recipients of immunoglobulin treatment administered at replacement dosages should be more widely recognized.
ESID-0285 Immune Reconstitution in SCID Patients with Diseminated BCG Infection After HSCT: A-Single Center Experience
N. Ramirez Uribe 1, S. Espinosa-Padilla2, F. Espinosa-Rosales2, F. Mujica-Guzman2, L. Romero-Guzman3, M. Pérez-García1, A. Olaya-Vargas1, E. Medina-Torres2
1Hematopoietic stem cell transplant Unit, Instituto Nacional de Pediatría, Mexico City, Mexico
2Research Primary Immune deficiency Unit, Instituto Nacional de Pediatría, Mexico City, Mexico
3Laboratory of Hematology, Instituto Nacional de Pediatría, Mexico City, Mexico
Introduction: Hematopoietic stem cell transplantation (HSCT) constitutes the standard therapy for Severe Combined Immune deficiency (SCID) patientes given their significant impaired immunity leads them to early deaths. The overaching goal is to recover a normal immunologic function that will leads protection against infections and other diseases, including autoimmunity and malignancies. In addition tuberculosis still an endemic disease in Mexico and BCG vaccine profilaxis is administered in all newborn. Under this circumstances must SCID will present simultaneously with BCG complications.
Methods: We included patients diagnosed with SCID and diseminated BCG, in wich were performed allogenic HSCT and had immune reconstitution approach. We recollect immunophenotyping lymphocytes results performed by flow cytometry during a period of one year, with measures at 1, 3, 6, 9 and 12 months.
Results: Five patients were included in the study. The age of range at the HSCT were between 12 and 28 months. Four patients received umbilical cord blood and one was a matched related donor. One patient had complete chimerism, 3 had partial and one had primary graft failure. One out of 3 patients with partial chimerism lost engraftment secondary to an infection. All of them still alive. We onbtained normal numbers at CD3+, CD8+ CD16+56?at month, CD19+ at 3 months and CD4+ at 6 month. In 3 of the patients the antifimic was withdraw succesfully.
Discussion: We observed immune reconstitution in all the patientents with engrafment even if it was with partial chimerism. The diseminated BGC remisión can be interpreted as normal immune function.
ESID-0701 Four Cases of Rapid Push Administration of Immunoglobulin
A. Richter 1
1Immunology, Queen Elizabeth Hospital, Birmingham, United Kingdom
BACKGROUND:
Rapid push is a subcutaneous technique where immunoglobulin (Ig) is given by manual injection, using a butterfly needle and syringe. Four cases are described where rapid push was the patient preferred technique for the treatment of hypogammaglobulinaemia (?Ig), recurrent chest infections and failure to respond to vaccination. In each case, 2?g (10mls) Hizentra was drawn up into a 10?ml syringe and administered through a 21G butterfly needle over 4-7 minutes.
CASES:
| Case | Diagnosis | Rationale | Dose | Ig dynamics | Outcome | Side effects | Training time |
| 1 | CLL – secondary ?Ig | Minimise fluctuations in Ig, no pump | 2g 3x week | IgG ?4.28 g/?l -8.33 g/?l after 4 weeks | Reduction in infection rate at 1 year | None | 3 hours |
| 2 | CVID | Flexibility and quick injection time | 2g 4x week | IgG ?3.2 g/?l to of 6.78 g/?l at 3 weeks and 7.18 g/?l at 4 weeks | Infection free since, returned to work | 2 mild local redness, settled within 8 hours | 3 hours |
| 3 | Rheumatoid arthritis, secondary ?Ig | Simplicity, no pump | 2?g 4x week | IgG ?2.7 g/?l to 5.9 g/?l at 3 weeks and 6.9 g/?l at 3 months | Infection, stopped prophylactic antibiotics | None | 2 hours |
| 4 | Granulomatous polyangiitis, secondary ?Ig | Simplicity, no pump | 2?g 4x week | Awaiting results | Awaiting results | None. Loading dose 4?g (20mls) also well tolerated | 2 hours |
CONCLUSION: These cases found the rapid push technique to be a safe, efficient alternative for administrating Ig replacement therapy, that achieved rapid normalisation of immunoglobulin trough levels. Patients have commented on the simplicity of the process, ease of training and speed and flexibility of administration. The applicability to home treatment, low ancillary costs and short training time make rapid push a cost effective option.
ESID-0627 Vanishing Bile Duct Syndome Following Haematopoietic Stem Cell Transplantation for Severe Combined Immunodeficiency
G. Ristic 1, D. Vujic2, S. Pasic1, S. Djuricic3, Z. Zecevic2, M. Zecevic2
1Immunology, Mother and Child Health Care Institute, Belgrade, Serbia
2Bone Marrow Transplant, Mother and Child Health Care Institute, Belgrade, Serbia
3Pathology, Mother and Child Health Care Institute, Belgrade, Serbia
Introduction: Vanishing bile duct syndrome (VBDS) is a group of acquired disorders associated with progressive destruction and disappearance of the intrahepatic bile ducts and cholestasis. Hepatic graft-versus-host disease may be complicated with VBDS, refractory to all available therapies and eventually leading to end-stage liver disease.
Objective: To investigate clinical severity, cause and treatment options in patient transplanted for SCID.
Methods: Retrospective case note search.
Results: A male child with X-linked severe combined immunodeficiency disease (X-SCID) confirmed prenatally by molecular analysis. He underwent haplo-identical haematopoietic stem cell transplantation (HSCT) at the age of 6 months. His older brother with X-SCID died at the age of 15 months due to disseminated BCG infection. He had non pretransplant conditioning. Cyclosporine was administered for GVHD prophylaxis after HSCT. Acute GVHD with skin, gut, and liver involvement was observed from day +17. Immunosuppressive therapy included: corticosteroids and cyclosporine. The patient received antibiotics, antiviral and antifungal agents, as well as IVIg substitution therapy. After 5 months of intensive treatment, the skin changes resolved and the bowel recovered, but chronic hepatic damage with severe cholestasis due to vanishing bile duct syndrome remained.
Conclusions: Treatment of VBDS may require plasmapheresis in case of hyper-viscosity or a high elevation in low-density lipoprotein and sometimes even liver transplant because of the end-stage liver disease. Ultimate treatment is control of the graft-vs-host disease.
ESID-0191 Patient-Reported Overall Well-Being as a Measure of Wear off Effect in IVIG-Treated Patients with Primary Immune Deficiency
M. Rojavin 1, J.P. Lawo2, A. Hubsch3
1Research & Development, CSL Behring LLC, King of Prussia, USA
2Research & Development, CSL Behring GmbH, Marburg, Germany
3Research & Development, CSL Behring AG, Berne, Switzerland
RATIONALE
Wear-off effect in primary immune deficiency (PID) presents clinically as an increased susceptibility to infections and a decreased quality of life towards the end of intravenous immunoglobulin (IVIG) dosing cycles. Although it has been reported anecdotally, frequency and intensity were not yet quantified.
METHODS
Patients with PID in two prospective open-label Phase III studies recorded daily their perception of overall well-being (OWB) using a 5-point scale from 1 (very poor) to 5 (very well). Wear-off was defined as a drop =1 in OWB on >3 days of a dosing cycle’s last week compared to the mean score recorded on Week 2.
RESULTS
Data from 86 patients on 4-weekly IVIG schedule (615 dosing cycles) and from 33 patients on 3-weekly schedule (315 cycles) were analyzed. On average, OWB increased over 3–5 days following infusion, remained constant for 2 weeks and decreased during the rest of the cycle; variations were small (Figure). Thirty-seven patients (43%) on 4-week schedule and 20 patients (60%) on 3-week schedule experienced wear-off at least once. Overall, wear-off was recorded in approximately 10% of all cycles with OWB mean decreases from Week 2 to the last week of the cycle of 1.189 (from 4.287±0.742 to 3.098±0.748) and 1.151 (from 3.87±0.789 to 2.719±0.954) for the 4-week and 3-week schedules, respectively.
CONCLUSION
Wear-off effect is characterized by a decreased OWB during the last week of 3- or 4-weekly IVIG dosing cycles. More frequent administration and/or a switch to subcutaneous replacement therapy would probably be beneficial for patients experiencing wear-off.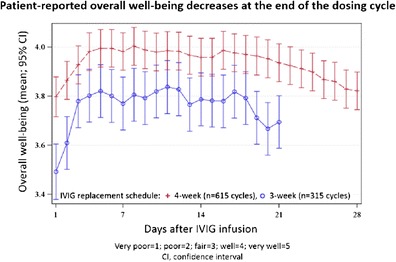
ESID-0260 Antibodies to Recombinant Human Hyaluronidase (RHUPH20) in PID Subjects Treated with HyQvia are Qualitatively Similar to Those in a Subset of the Normal Healthy Population
S. Rosengren 1, L. Huang2, L. Jadin3, G. Wei2, M.?A. Printz4, J.K. McVey5, J. Souratha1, D. Conway6, D.B. Muchmore7, H.P. Schwarz8, R.I. Schiff9, B.J. Sugarman1
1Translational Technology, Halozyme Therapeutics, San Diego, USA
2New Molecular Entities, Halozyme Therapeutics, San Diego, USA
3Pathobiology, Halozyme Therapeutics, San Diego, USA
4Bioanalytical, Halozyme Therapeutics, San Diego, USA
5Quality Assurance & Regulatory Affairs, Baxter, Deerfield, USA
6Clinical Operations, Halozyme Therapeutics, San Diego, USA
7CDMA, Halozyme Therapeutics, San Diego, USA
8Clinical Strategy Biotherapeutics, Baxter, Vienna, Austria
9Clinical Strategy Biotherapeutics, Baxter, Westlake Village, USA
Subcutaneous immunoglobulin 10% with rHuPH20 (HyQvia) constitutes a novel approach to immunoglobulin delivery in subjects with primary immunodeficiency (PID). In a Phase III trial of HyQvia, rHuPH20-reactive antibodies were present in 15/83 evaluable subjects at least once during treatment, and persisted at least 16 weeks in 7/?83 subjects. Neutralizing antibodies were not observed in these subjects. Titers decreased in most study subjects despite continued HyQvia treatment. There was no temporal association between rHuPH20-reactive antibodies and adverse events. In a normal healthy population, 5-6% of individuals also tested positive for rHuPH20-reactive antibodies in two separate studies of 692 adult volunteers and 961 plasma donors, respectively, and these antibodies persisted for at least 5 months in 5 of 5 subjects monitored longitudinally. The prevalence of rHuPH20-reactive antibodies was higher in older individuals; however, no signal of infertility or autoimmune/chronic inflammatory conditions associated with the presence of antibodies could be identified in this normal population. PH20-reactive antibodies affinity-purified from 4 PID trial subjects with the highest titers as well as from 4 healthy subjects with pre-existing titers were of isotypes IgM, IgG, and IgA in similar ratios. Characteristics of antibody binding to endogenous and recombinant PH20 and sperm were comparable for the antibodies from patients and healthy individuals. These results indicate that 1) rHuPH20-reactive antibodies that developed in a subset of PID subjects treated with HyQvia were qualitatively similar to those existing in the normal untreated population, and 2) the presence of such antibodies was not associated with any discernable clinical sequelae.
ESID-0527 Primary Immunodeficiency with Defects in Glycosylation, Is There Any Role of Interferon Gamma as Part of the Treatment?
S. Scheffler-Mendoza 1, L. Blancas-Galicia1, M. Yamazaki-Nakashimada2
1Immunodeficiency Research Unit, Instituto Nacional de Pediatría, Mexico City, Mexico
2Clinical Immunology Department, Instituto Nacional de Pediatría, Mexico City, Mexico
There are primary immunodeficiencies with defect of neutrophil glycosylation. There were described these defects in the glycogen storage disease type 1b (GSD-1b) and in G6PC3 deficiency. GSD-1b is a heterogeneous inborn error of metabolism characterized by disturbed glucose homeostasis, neutropenia and neutrophil dysfunction. It is caused by deficiency in a glucose-6-phosphate transporter. It has been described that neutrophils in GSD-1b have diminished respiratory burst and phagocytic activities. GSD-1b could course with clinical phenotype as Chronic Granulomatous Disease (CGD). We report a patient with GSD-1b; successfully treated with Granulocyte Colony Stimulating Factor (G-CSF), interferon gamma (IFN) and prophylactic antibiotics.
CASE REPORT
A 3 year-old female patient was born at term from consanguineous parents with 2 healthy brothers. She coursed with hepatomegaly and hypoglycemia. Liver biopsy revealed diagnosis of GSD-1b. She had multiple infections including gastroenteritis, respiratory tract infections, severe pneumonia, septic shock, skin abscess, and oral and genital ulcers. She was diagnosed with neutropenia treated with G-CSF, but she persisted with severe infections. We performed 1,2,3-Dihidrorodamine assay showing lower production of reactive oxygen species. We added to the treatment IFN gamma at 50 mg/m2 doses subcutaneously three times per week. After 1 year of treatment the patient presented clinical improvement and a decrease in the frequency and severity of the infections.
CONCLUSION
GSD-1b patients have neutrophil dysfunction secondary to abnormalities in chemotaxis, diminished bactericidal and respiratory burst activities. We propose the use of IFN gamma to enhance the neutrophil function and also the use of prophylactic antibiotics as in CGD.
ESID-0482 Cost-Effectiveness of Intravenous Immunoglobulin (IVIG) Treatment in Patients with B-Cell Immunodeficiencies
I.N. Smirnova1, T.G. Kosacheva2, N.?B. Kuzmenko3, A. Shcherbina 4, G.A. Novichkova5
1Immunology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
2Immunology, Clinical Children’s Hospital ?9, Moscow, Russia
3Immunology, Federal Research and Clinical Center for Pediatric Hematology Oncology and Immunology, Moscow, Russia
4imunology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
5Hematology, Federal Research and Clinical Center for Pediatric Hematology Oncology and Immunology, Moscow, Russia
IVIG is the main therapeutic modality in humoral PID. Multiple studies showed its effectiveness in reducing mortality and morbidity.
In the light of high cost of the IVIG we compared medical and other costs, related to the disease in patients with humoral PIDs with or without IVIG treatment.
23 patients (ages 2-16 years) with B-cell deficiencies (73% X-linked agammaglobulinemia, all genetically confirmed, 27% common variable immunodeficiency) were included in the study.
We analyzed medical and other disease-related costs during 2 years preceding the diagnosis (without IVIG therapy) retrospectively and prospectively during 2 years on regular IVIG therapy.
The patients analyzed fell into 3 categories:
The 1st group – patients who have experinsed several severe infections before therapy, an average time to diagnosis 3 years. In this group costs on IVIG treatment were 2,5 times higher than before the diagnosis.
The 2nd group - patients with very late diagnosis (average time to diagnosis 7 years), who have had multiple severe infections before the IVIG treatment and acquired chronic lung complications. In this group the costs before IVIG the treatment were higher, than on IVIG treatment.
3rd group - diagnosis within the first year of life (due to preceding family history). The comparative analysis was not possible, but it was noted that these patients had no history of serious infections before or while on IVIG therapy.
In conclusion, regular IVIG therapy not only leads to improved quality of life. In some cases it brings down the disease-related costs and is economically advantageous.
ESID-0487 Effect of Rituximab Treatment of Granulomatous-Lymphocytic Interstitial Lung Disease (GLLID) in Patients with Primary Immunodeficiency (PID)
Y.A. Rodina1, I.N. Smirnova1, D.S. Abramov1, Y.V. Olshanskaya2, M.M. Doubrovin3, A. Shcherbina 1
1Immunology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
2Cytogenetics, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
3Radiology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
Introduction: GLLID is characterized by typical morphological and radiological features, and is a frequent complication of common variable immunodeficiency (CVID) and other PID, which is associated with poor disease prognosis. Standard immunosuppressive therapy (glucocorticoids, cytostatics ) is effective only partially and is accompanied with multiple side effects long-term.
Objective: To evaluate Rituximab efficacy in treatment of GLLID in patients with PID.
Patients and Methods: We followed five patients with GLLID: three patients with CVID (ages 5, 11 and 12 years), 2 patients with genetically confirmed DiGeorge syndrome (ages 3 and 11 years), mean post-treatment follow-up 7.5 months. GLLID diagnosis was confirmed via biopsy, in all cases lesion contained high numbers of CD20+ B-cells. Four patient had pulmonary insufficiency. All patients received long-term IVIG, prophylactic antimicrobial therapy, without effect with regard of pulmonary process. 4 patients previously received corticosteroids, azathioprine, cyclosporine and sirolimus with partial, short-term effect.
Results: Rituximab was administered at a dose of 375 mg/m2 weekly for 4 weeks, then 1 infusion at 3 and 6 months after initiation of therapy. No side effects were noted in the group. After 1 month of treatment there was a dramatic improvement of radiological features and symptoms of lung insufficiency, which persisted upon follow-up.
Conclusions: Rituximab is effective in the treatment of GLLID in patients with PID and is well tolerated. Long-term efficacy and safety is yet to be assessed.
ESID-0501 Sirolimus is Effective in Treatment of Patients with Autoimmune Lymphoproliferative Syndrome (ALPS)
E.V. Deripapa1, I.N. Smirnova1, T.A. Varlamova2, V.O. Bobrynina2, O.A. Shvets1, D.?D. Baidildina3, I.V. Fisiun3, I.I. Kalinina3, M.?A. Maschan4, A. Shcherbina 1, A.A. Maschan3
1Immunology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
2Molecular genetics, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
3Hematology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
4HSCT, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
ALPS is a primary immunodeficiency resulting in T-lymphocyte dysregulation and defective FAS - mediated apoptosis. Among the typical features are increased number of double-negative (DN) T-lymphocytes, lymphadenopathy, splenomegaly and autoimmune complications, primarily cytopenias.
Objective: To evaluate sirolimus efficacy in treatment of ALPS.
Patients and Methods: We analyzed effectiveness of Sirolimus therapy in 12 patients ages 4 months -17 years with ALPS (time of observation 3 month- 2.5 years, mean 18 mnths). The diagnosis was made based on typical symptoms of lymphoproliferation, autoimmune cytopenias, increased numbers of DN lymphocytes, and identification of germ-line FAS mutation (6 cases).We used sirolimus at a starting dose of 3 mg/m2, subsequently the dose was adjusted to achieve therapeutic plasma concentrations of 8-15 ng/ml.
Results: 4 patients received sirolimus as a monotherapy at the onset of their symptoms, 8 patients started sirolimus treatment in addition to various immunosuppressive therapy of severe cytopenias, which has been previously started.
In all patients studied we observed full and persistent therapeutic effect: normalization of the spleen and lymph nodes sizes, normalization of hematological parameters, immunoglobulin levels, drastic reduction of DN T lymphocytes. During the observation period all patients were taken of other immunosuppressive therapy without relapse of symptoms. Therapy was well tolerated: side effects were observed in one case (moderate feet swelling). 16 y.o. patient, while on sirolimus therapy, developed acute varicella, complicated by V.zoster meningitis, which might or might not be the result of sirolimus treatment.
Conclusions: Sirolimus is an effective and safe in treatment of patients with ALPS.
ESID-0013 Concomitant Use of Corticosteroid with Antimicrobials for Liver Abscesses in Patients with Chronic Granulomatous Disease
M.S. Oh1, K. Shin 1
1Pediatrics, Jeju National University School of Medicine, Jeju, Korea
Chronic granulomatous disease (CGD) is a rare inherited disorder caused by a defective NADPH oxidase enzyme and characterized by recurrent bacterial and fungal infections. Although liver abscess is a common manifestation of CGD, the management of liver abscesses in CGD patients is not well-defined. In addition, the generalized guideline for treating liver abscesses does not necessarily apply to CGD patients. Corticosteroids are commonly used to control granulomatous complications in patients with CGD, such as inflammatory gastrointestinal and genitourinary lesions. Corticosteroids have also been used in combination with antimicrobials to treat refractory infections in patients with CGD. However, corticosteroids are capable of suppressing symptomatic inflammation and all potential infections must be adequately covered prior to corticosteroid initiation. We report three typical CGD cases with liver abscesses refractory to conventional treatments that were successfully treated with the concomitant use of corticosteroids and antimicrobials. However, it remains unclear whether corticosteroid therapy is required for liver abscesses refractory to conventional treatments. Based on our observations, concomitant use of corticosteroid in combination with optimal antimicrobials should be considered for refractory liver abscesses in CGD.
ESID-0569 Acute HLH in Griscelli-Syndrome with Mixed Donor and Recipient Chimerism After Matched Unrelated Bone-Marrow Transplantation
M. Sirin 1, F. Kropp1, M. Hoenig1, G. Dueckers2, C. Schuetz1, T. Niehues2, A. Schulz1
1Pediatrics and Adolescent Medicine, University Medical Center Ulm, Ulm, Germany
2Pediatric Immunology and Rheumatology, HELIOS Children's Hospital Krefeld, Krefeld, Germany
We report on a girl of northern African origin who was diagnosed with Griscelli-Syndrome at the age of 14 months. She had typical clinical findings, no signs of neurological impairment and degranulation defect of NK/CTL (natural killer-cells and cytotoxic T-lymphocytes). She underwent stem cell transplantation (SCT) in October 2012 of matched unrelated donor (MUD) before any signs of hemophagocytic lymphohistiocytosis (HLH) occurred. For conditioning she received treosulfan, fludarabine, thiotepa and alemtuzumab. The course of this SCT was uneventful with initially complete donor chimerism. During the first year post-transplantation she developed an increasing mixed chimerism which could not be prevented by repeated DLIs. 14 months after transplantation she developed a mastoiditis with facial palsy. Donor fraction at this time: 40% (30% in T-cells, 10% in MNCs). Instantly she developed an acute flare up of HLH fulfilling 7/?8 criteria according to the diagnostic guidelines. Immunosuppressive therapy (dexamethasone, CSA and alemtuzumab) was initiated and the patient clinically responded well to this therapy with normalization of her body temperature and inflammatory markers. Unexpectedly though she developed persistent bone-marrow aplasia with aleukocytosis and need for frequent transfusions of erythrocytes and thrombocytes. Four weeks after occurence of the episode of acute HLH we performed a second PBSCT from the same MUD. Conditioning this time was with targeted intravenous busulfan, fludarabine and alemtuzumab. GvHD-prophylaxis was CSA and MMF. Hematological reconstitution was in time, without severe toxicity or infections. 4 months after second SCT she shows complete donor chimerism and self-limiting grade 2 cutaneous aGvHD.
ESID-0135 The Assessment of Adherence to Guidelines in the Use of Intravenous Immunoglobulin as an Indicator of Health Care Quality in a Tertiary Pediatric Hospital
A. Fernandez-Polo1, P. Soler Palacín 2, I. Gaso-Gago1, C. González-Guerrero1, A. Martin-Nalda2, C. Figueras2, J. Cutillas1
1Pharmacy Department, Hospital Universitari Vall d'Hebron. Institut de Recerca Vall d'Hebron. Universitat Autònoma de Barcelona, Barcelona, Spain
2Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d'Hebron. Institut de Recerca Vall d'Hebron. Universitat Autònoma de Barcelona, Barcelona, Spain
Background and aims: To assess the indications of intravenous immunoglobulin (IVIg) treatment in a tertiary pediatric hospital and whether the clinical indications matched with the evidence-based clinical guidelines.
Methods: observational, cross-sectional study during 2012. All patients in treatment with IVIg were included. The British Health Department Clinical Guideline for Immunoglobulin Use (2nd Edition 2008, Update 2011) was used as a reference guide for priority indications (Red, Blue, Grey and Black).
Results: 193 children were included (109 male), median age was 4.01 years (range 1 day-19.78 years). Data was collected from 849 administrations and 10989.03?g IVIg were used. Infusions were delivered in hospitalized patients in 51.70% (439) and 48.29% (410) in Day Care Hospital. Use of IVIg was: 17.34% (34) stem cell transplantation, 12.24% (24) primary and 9.69% (19) secondary antibody deficiency, 7.14% (14) immune thrombocytopenia, 7.14% (14) solid organ transplantation and 18.36% (36) ‘Others’. Immunoglobulin use by colour-coded prioritisation (patients): Red 46.43% (91), Blue 24.49% (48), Grey 6.12% (12), Black 0.51% (1) and ‘Others’ 22.45% (44). Total volume of IVIg used for Grey and Black indications was 13.16%. There was only one black (sepsis) and 2 grey indications in 12 patients (post-exposure prophylaxis of varicella and antibody mediated rejection of SOT).
Conclusions: The highest IVIg use (patients and volume) were immunological disorders, followed by haematological indications and, to a lesser extent, transplantation. Mainly colour-coded priorization indication was Red or Blue (70% of patients). There is an on-going multicentric study to better assess current immunoglobulin prescribing practices in pediatric patients
ESID-0063 Clinical Studies Evaluating the Pharmacokinetics, Efficacy, Safety and Tolerability of a New Human Plasma-Derived Intravenous Immunoglobulin Product ("NEWGAM 10%") in Patients with Primary Immunodeficiency Disease
M. Borte 1, S. Wietek2, B. Pyringer3, T.E. Svae4, J. Moy5, I. Melamed6
1Klinik für Kinder-und Jugendmedizin, Klinikum St. Georg, Leipzig, Germany
2Corporate Medical & Scientific Affairs, Octapharma Pharmazeutika Produktionsges.m.b.H., Vienna, Austria
3Clinical R&D, Octapharma Pharmazeutika Produktionsges.m.b.H., Vienna, Austria
4Scientific & Medical Affairs, Octapharma USA, Hoboken, USA
5Department of Immunology & Microbiology, Rush University, Chicago, USA
6Immunology, IMMUNOe Health Centers, Centennial, USA
Background: “NewGam” is a novel high-purity glycine-stabilised human normal immunoglobulin (10% IVIG) product from Octapharma. It is manufactured using two dedicated steps for pathogen safeguarding (solvent/detergent treatment and nanofiltration).
Methods: Prospective, open-label, non-controlled phase III study NGAM-01 enrolled 51 children and adults with primary immunodeficiencies receiving 0.2-0.8 g/?kg “NewGam” at 3- or 4-week intervals for one year. Primary objective was assessing “NewGam’s” efficacy in preventing serious bacterial infections (SBIs). Determination of the pharmacokinetic (PK) profile and evaluation of “NewGam’s” safety profile were secondary objectives. Extension study NGAM-05 assessed the safety and tolerability of high infusion rates (up to 0.14 mL/kg/min) in 21 patients from NGAM-01 for three months.
Results: Number of SBIs per patient exposure year, PK profile and additional efficacy parameters (other infections, days with use of antibiotics, absent from school or work, and hospitalised due to infection per patient exposure year) were in line with figures reported for other new IVIGs launched during the last decade. In NGAM-01 only few infusions were accompanied by pre-medication, and 90% were administered at maximum speed allowed after 7th infusion (0.08 mL/kg/min); in NGAM-05, 85% of infusions were administered at maximum speed allowed after 3rd infusion (0.14 mL/kg/min).
Conclusion: “NewGam” demonstrated a PK profile and efficacy performance equivalent to other IVIGs commercialised during the last decade. With only a few adverse events revealed, it may display a very positive safety feature over some of the currently licensed IVIGs. Administration of “NewGam” even at infusion rates of 0.14 mL/kg/min was generally well tolerated.
ESID-0035 Efficacy and Safety of Self-Administered Subcutaneous Immunoglobulin in Polish Children with Primary Immunodeficiency Diseases
I. Stelmach 1, K. Bal1, A. Sobocinska1, M. Zaczeniuk1, M. Podsiadlowicz-Borzecka1
1Department of Pediatrics and Allergy, N. Copernicus Hospital, Lodz, Poland
BACKGROUND
Intravenous immunoglobulin (IVIG) infusions are currently standard therapy for children with primary immune deficiency (PID). This was an observational study to investigate the efficacy and safety of a subcutaneously administrated immunoglobulin (SCIG) in children with PID, switched from IVIG to SCIG.
METHODS
We studied 18 patients, aged 2- 18 years. Children use IVIG therapy at a dose 400 mg/kg every 4 weeks; after their final IVIG infusion, children were switched to SCIG at weekly dose of 100 mg/kg. At each visit (every 4 weeks, during the year of IVIG and during the year of SCIG therapy), the number of upper and lower respiratory tract infections (RTI), the IgG serum levels (g/?L) and the frequency of treatment- related adverse events of the IgG therapy were evaluated.
RESULTS
We noticed: i) higher serum level of IgG in SCIG group (median, upper- lower quartiles: 9.3, 7.7- 11.8) compared with IVIG group (median, upper- lower quartiles: 6.5, 5.8- 8.5), ii) less frequent lower RTI in SCIG group (median, upper- lower quartiles: 1.0, 0.75- 1.0) compared with IVIG group (median upper- lower quartiles 3.0, 1.0- 3.25) and iii) less frequent treatment- related adverse events: 0% in SCIG group vs 50 % of patients in IVIG group (p = 0.0005).
CONCLUSION
Analysis of home care use of Ig in PID in children revealed that the SC route should be introduced as fast as possible to reduce the number of RTI and to maintain the stable level of IgG. That allows the safe and effective treatment for children with PID.
ESID-0450 Intravenous Immunoglobulin Therapy Affects the Number and the Function of Immune Cells In Common Variable Immunodeficiency
I. Stelzer 1, H.P. Brezinschek2, R. Fuchs3, H. Mangge4, W. Graninger2, U. Demel2
1Clinical Institute of Medical and Chemical Laboratory Diagnostics, Medical University of Graz, Graz, Austria
2Division of Rheumatology and Immunology, Medical University of Graz, Graz, Austria
3Institute of Pathophysiology and Immunology, Medical University of Graz, Graz, Austria
4Clinical Institute of Medical and Chemical Laboratory Diagnostics Research Unit on Lifestyle and Inflammation-associated Risk Biomarkers, Medical University of Graz, Graz, Austria
Background: Although, intravenous immunoglobulin therapy (IVIg) is used for treatment of e.?g. autoimmune diseases, the immunomodulatory mechanisms ?in?vivo remain to be clarified. Since IVIg therapy is frequently performed in common variable immunodeficiency, we aim to investigate the ?in?vivo immunomodulatory effects of IVIg per se to elucidate underlying mechanisms.
Methods: All participants gave their informed consent (EK decision number 21-292 ex 09/10). Hematological parameters and leukocyte subpopulations were counted on a hematocytometer. Lymphocyte subpopulations, B-lymphocyte subpopulations, apoptotic/necrotic cells, and circulating CD34+/CD45dim hematopoietic stem and progenitor cells (CPCs) were analyzed by flow cytometry. Additionally, proliferative response to mitogens (phytohaemagglutinin and concanavalin A) was determined. Data were analyzed by paired student´s t-test or by Wilcoxon test for paired samples depending on the distribution of data.
Results: The absolute counts of leukocytes, neutrophil granulocytes, monocytes, lymphocytes, eosinophil granulocytes and CPCs were significantly reduced after IVIg therapy. In addition, absolute counts of lymphocyte subpopulations (CD3+, CD3+/CD8+, CD3+/CD4+, CD3+/CD16+/CD56+, CD19+ cells) were significantly decreased and also a significant impact on the response of lymphocytes to concanavalin A and phytohaemagglutinin was seen. Furthermore, significantly decreased absolute counts of viable, apoptotic and necrotic cells were determined. Interestingly, the percentage of necrotic cells decreased, whereas the percentage of viable cells increased significantly after IVIg therapy.
Conclusion: In summary we conclude that the ?in?vivo immunomodulatory mechanisms of IVIg influence both the number and the function of immune cells. Based on our findings, further investigations of ?in?vivo immunomodulatory mechanisms of IVIg therapy including autoimmune diseases are planned.
ESID-0338 Use of Whole Exome Sequencing in the Decision Making Process Regarding Stem Cell Transplantation
P. Stepensky 1, U. Fischer2, B. Keller3, H. Saleh4, O. Abuzaitoun5, S. Nabhani2, S. Revel Vilk1, A. Borkhardt2, M. Weintraub1, K. Warnatz3, O. Elpeleg6
1Pediatric Hematology-Oncology and Bone Marrow Transplantation, Hadassah Hebrew University Medical Center, Jerusalem, Israel
2Department of Pediatric Oncology Hematology and Clinical Immunology, University Children’s Hospital Medical Faculty Heinrich-Heine-University, Dusseldorf, Germany
3Center for Chronic Immunodeficiency, University Hospital, Freiburg, Germany
4Pediatric Hemato-Oncology Unit, Augusta Victoria Hospital, Jerusalem, Israel
5Pediatrics, Nablus Speciality Hospital, Nablus, Palestine (via Israel)
6Monique and Jacques Roboh Department of Genetic Research, Hadassah Hebrew University Medical Center, Jerusalem, Israel
Introduction: A genetic diagnosis is crucial for clinical decision about different aspects of HSCT in patients with primary immune deficiency (PID). The recent introduction of exome sequencing (ES) enhances our ability to cope with the great genetic heterogeneity.
Materials and methods: Twenty three patients from 18 families were included. The patients presented with various forms of PID. Twenty two patients were the product of consanguineous marriages. Clinical, genetic and immunological evaluations were part of a trilateral collaboration.
Results: Four groups of patients were evaluated. The first group included six children with SCID. In the second group were four children with four new forms of profound combined immune deficiency. Three of them underwent HSCT based on the results of ES and one is waiting for HSCT. A third group of patients (four children) were diagnosed with severe congenital neutropenia (SCN). A mutation in the VPS45 gene was identified, defining a novel form of SCN. Three children underwent HSCT. The fourth group comprised five children from four families with a clinical picture of autoimmune lymphoproliferative syndrome. 2/?5 kids were listed for HSCT based on the results of ES. Disease causing mutations were identified in all the patients, usually novel mutations in known disease associated genes; in four families the mutations affected first-in-human genes.
Conclusions: The results of our approach underscore the importance of ES in the identification of new disease-causing genes, as well as in the delineation of known monogenetic syndromes, providing the transplant physician with pertinent information in the decision-making process regarding HSCT.
ESID-0148 Evaluate the Appropriateness of Short Term Immunoglobulin Prophylaxis for IGG Subclass Deficiency Pediatric Patients
Y. Tanaka 1, K. Gotoh1, Y. Ohtsu1, N. Tsumura1, T. Matsuishi1
1Pediatrics, Kurume University, Fukuoka, Japan
Patients with immunoglobulin G subclass deficiency or decline sometimes suffer from recurrent bacterial and respiratory infections, including otitis media, pneumonia. In adulthood low levels of IgG3 is the most common IgG subclass abnormality. On the other hand, in childhood IgG2 deficiency is the more major than IgG3 abnormality. IgG2 deficiency patients often experience severe and recurrent infections, because IgG2 works on encapsulated bacteria like S.?pneumonia and H.?influenzae type B. But there is no established management that be shown the methods and indication of antibiotic prophylaxis or appropriate intravenous immunoglobulin (IVIG) to prevent infection for these patients. We performed IVIG replacement (with monthly 200?300 mg/kg/dose ) for 6 pediatric patients (the age of 18?30 months) of IgG2 deficiency disease whom poorly reaction to antimicrobial agents. All of patients, recurrent infections were reduced during IVIG prophylaxis, and the IgG2 levels were normalized in 7? 12 months. Further 4 patients recurrent infections were disappeared in 10?12 months. We have finished IVIG prophylaxis to all IgG2 deficiency patients after 12 months past from starting IVIG prophylaxis. But all patients did not have recurrent infections. At the past investigation, the normalization of the IgG2 levels of IgG2 deficiency patient takes time for 1 to 5 years. Our study suggests that recurrent infection of IgG2 deficiency children may disappear earlier than other reports, and may be able to over the IVIG therapy in more short-term.
ESID-0752 Special Features of Evolution of Allogeneic Bone Marrow Transplantation Without Pre-Transplant Conditioning in a Severe Combined Immunodeficiency
S. THRAYA 1, F. Mellouli1, M. Ben Khaled1, M. Ouederni1, A. Haoua1, R. Hassouna1, M. Bejaoui1
1Pediatric Immuno-Hematology department, Bone Marrow Transplant Center, Tunis, Tunisia
Introduction: Bone marrow transplantation is usually preceded by intensive chemotherapy and radiation therapy designed to completely eliminate recipient immune-competent cells that might reject the donor bone marrow.
Objective: To describe infectious and immunological features in allogeneic bone marrow transplantation without pre-transplant conditioning in a patient with Omenn syndrome.
Observation: M.T is a five-month old boy with an Omenn syndrome who received a genoidentical allogeneic bone marrow transplantion (BMT) from his brother without preconditioning regimen or Graft-versus-host disease (GVHD) prophylaxis, according to the recommendations of the EBMT. On day 6, the patient developed an acute GVHD. This GVHD affected the skin, the liver, the gastrointestinal tract, and was mastered by a heavy immunosuppressive therapy (Anti-Lymphocyte Serum, corticosteroides and cyclosporin). He developed on day 9, a bronchoalveolitis to parainfluenza III, and on day 13, a methicillin-resistant Staphylococcus aureus skin infection, complicated with a sepsis. Engraftment was complete, with donor chimerism detected by molecular biological analysis. Cellular immunity was reconstituted, and because of a deficit in the humoral immune response (residual IgG level=5 g/?l), several infections of various origins marred the clinical evolution of the patient. On day 147, the patient presented a chronic GVHD, with cutaneous lesions and On day 210, he reactivates the hepatic and digestive GVHD. Despite adequate immunosuppressive therapy, the patient died as a result of its GVHD on day 246.
Conclusion:Without pre-transplant conditioning, engraftment is possible. However, the serious risk of GVHD is very important. Preventive treatment with cyclosporine and methotrexate should be established to achieve such a graft.
ESID-0434 Subcutaneous Immunoglobulin Overcome Intravenous Immunoglobulin Limitation in Protein Losing Enteropathy
A. Trizzino 1, A. Trizzino1, C. Mosa1, A. Macaluso2, L. Valore1, P. D'Angelo1
1Pediatric Oncology Hematology, ARNAS CIVICO, Palermo, Italy
2Pediatric Specialization School, Università degli Studi di Palermo, Palermo, Italy
Primary intestinal lymphangectasia is a rare disorder characterized by impaired lymphatic drainage that causes lymphatic vessels engorgement and rupture leading to lymph fluid leakage in to the bowel lumen. Not only Albumin but also other proteins with a slow turnover rate (IgG) are affected. The disorder is sometimes associated with abnormalities of both umoral and cellular immune system (Lymphocytes leakage), recurrent and opportunistic infections that may be associated with increased morbidity and mortality.
A 13 years old boy was admitted for chronic sinusitis. A complete blood count revealed a decreased number of lymphocytes (Ly 600/mm3). The total serum protein was 4.3 g/?L and albumin was 1.7 g/?L. His serum IgG were below normal levels (195 mg/dl). Physical examination disclosed edema of the right lower limb, genitals and left forearm. A stool examination showed increased levels of a-1antitripsin. Capsule endoscopy revealed lymphangectasia involving the entire small bowel.
Conservative treatment with a low fat diet supplemented with medium chain triglycerides, steroids and octreotide therapy failed. Albumin and intravenous immunoglobulin (909 mg/Kg/month) were effective only in the short term. Every two weeks IVIg administration was not able to maintain serum IgG concentration at 500 mg/dl or greater. Subcutaneous 20% Ig twice a week administration (872 mg/Kg/month) was started and IgG levels finally approached physiologic concentrations. Subcutaneous route of administration is as safe and effective as IVIg and is also associated to a different pharmacokinetic of exogenous immunoglobulin permitting that more stable and physiologic levels of IgG were achieved in our patient.
ESID-0431 Endobronchial Balloon-Assisted Drainage to Cure Pulmonary Abscess in a Chronic Granulomatous Disease Patient
A. Trizzino 1, A. Trizzino1, C. Mosa1, S. Messina2, P. Dones3, P. Vitulo4, P. D'Angelo1
1Pediatric Oncology Hematology, ARNAS CIVICO, Palermo, Italy
2Pediatrics, Policlinico Universitario, Palermo, Italy
3Pediatric Infectious diseases, ARNAS CIVICO, Palermo, Italy
4Pneumology, ISMETT, Palermo, Italy
Chronic granulomatous disease (CGD) is a rare immunodeficiency characterized by impaired intracellular microorganisms killing due to defects of NAPH-oxidase complex. Pneumonia and pulmonary abscess are the commonest infections up to diagnosis and during follow-up in the italian cohort.
G.F. is a 5 years old boy. His past medical history was relevant for scalp foruncolosis at birth and frequent infections in his early childhood (pneumonitis and lymph node abscesses). Eight days before admission he complained of acute onset of fever and productive cough. A diagnosis of right lobar pneumonia was made and Ceftriaxone was started. A chest CT scan showed apical right lung consolidation. Bronchoscopy showed a severe purulent bronchitis; in the BAL, a strain of Criptococcus neoformans was isolated. Neutrophil respiratory burst was depressed (DHR123 5%). Molecular analysis confirmed X-Linked CGD. A multi-drug antibiotic and antifungal regimen was promptly started but only a partial clinical response was obtained. Septic fever and raised CRP persisted. Endobronchial balloon-assisted drainage (EBAD) was performed under fluoroscopic guidance in 4 steps with patient sedated and ventilated with laryngeal mask: anatomical location of the lesions using a flexible bronchoscope; balloon tipped guide wire inserted into the afferent bronchus and pushed into the cavity; balloon inflation and bronchial lumen dilation; bronchial drainage of the lesions' content.
Few days after EBAD CRP normalized and fever recessed. Aggressive and prolonged management of infections is necessary in CGD patients. EBAD technique has been successfully employed for the first time in our GCD patient with a difficult lung cavitated lesion.
ESID-0435 Primary Immunodeficiency (PID). From Comunication of Diagnosis to Illness Experience in a Pediatric and Adult Small Group of Patients
A. Trizzino 1, S. Battiato1, C. Taormina1, P. Guadagna1, A. Trizzino1, C. Mosa1, P. D'Angelo1
1Pediatric Oncology Hematology, ARNAS CIVICO, Palermo, Italy
PID is a heterogeneous group of diseases. Generally, patients and families experience confusion and ambiguity in diagnostic and therapeutic process.
Looking at patient and family functioning we have useful information about relationship with physicians, information processing of the disease and treatment, approaching difficulties of care.
We started a psychological support to patients and families from diagnosis trough the various steps of treatment. The main instrument was the semi-structured clinical interview. The aim of the study was investigate experience of illness, identify any psychological distress, and anticipate development of psychosomatic symptoms associated to chronic disease.
Patients and families have difficulty to openly share information about their own disease. Interviews showed a significant ambiguity in diagnosis communication, lack of clarity about the chronicity, poor understanding both general clinical condition and actual dynamics of the disease:
Parents: difficulties in disease understanding and acceptance, problems to lead their children, attempt to hide chronicity.
(Younger) adults: attempt to hide disease with other people, firm closure in relationships, personal experience of shame.
Timeliness and clarity are essential in diagnosis communication. Most patients had received diagnosis in the childhood without further update of information through their psychosomatic development. The presence of a treatment reference center and an ongoing psychological support in the chronicity, during different steps growth, significantly reduce the presence of specific disorders related to disease awareness, also facilitate social adaptation. This preliminary survey will lead us to transform the communication of the diagnosis in a dynamic process that involve physicians, psychologists, patients and families.
ESID-0356 Subcutaneous Immunoglobulin as Replacement Therapy of Children with Primary Immunodeficiencies
T. Uglova 1, S. Aleshkevich2
1clinical recearch, National Research Center for Pediatric Oncology Hematology and Immunology, Minsk, Belarus
2treatment, National Research Center for Pediatric Oncology Hematology and Immunology, Minsk, Belarus
Objective. Analysis of the efficacy and tolerance of subcutaneous immunoglobulin "Gammanorm» (Octapharma AG, Sweden) in pediatric patients with primary antibody deficiencies
Materials and methods. A total of 8 patients aged 7-18 years with congenital disorders of antibody formation received Gammanorm for 52 weeks. Before therapy, all patients received 52 weeks of intravenous immunoglobulin Octagam. Gammanorm administered weekly at a dose of 0.1 g/?kg (0.4 g/?kg/month). IgG levels in serum were determined on day 0 and 4, 12, 24, 36, 52 weeks after initiation of therapy. Efficacy criteria - the level of IgG in serum and frequency of infectious episodes. Criteria for tolerance - the frequency of serious adverse events, the frequency and severity of systemic and local adverse reactions.
Results. IgG level in the serum after intravenous injection OKTAGAM was 4,19±0,66 g/?l , a month after the start of therapy Gammanorm - 7,12±0,8 g/?l and remained stable throughout the observation period. No significant differences in IgG level in serum in patients during therapy Gammanorm according to age (p=0.786) and diagnosis (p=0.823).
All patients had a higher level of serum IgG after administered Gammanorm (Me=6.9 g/?l) compared to the previous levels of IgG during IVIG therapy (Me=4,2 g/?l).
The frequency of infectious episode/patient/year decreased from 7.0 when using Octagam to 2.1 when using Gammanorm.
During the year, no serious adverse events and no systemic adverse events of mild to moderate severity were registered.
Conclusion. Subcutaneous administration of the drug GAMMANORM in patients with PID at home is safe and effective.
ESID-0307 Delayed B Cell Immune Reconstitution Three Years After Rituximab in Pediatric Patients
J. van der Spek 1, A. van Royen- Kerkhof1, J.J. Boelens2, J. van Montfrans1
1Pediatric Immunology and Infectious Diseases, University Medical Center Utrecht, Utrecht, Netherlands
2Blood and Marrow Transplantation Program, University Medical Center Utrecht, Utrecht, Netherlands
Background Rituximab, a chimeric monoclonal antibody depleting CD20-expressing cells, is used to treat Hematopoietic Cell Transplantation (HCT)-associated complications, autoimmune diseases, and malignancies. Persistent hypogammaglobulinemia is an increasingly recognized long-term side effect of rituximab. We investigated the incidence of prolonged impaired B cell function after rituximab treatment in children.
Methods We retrospectively studied all children in the UMC Utrecht who received rituximab for complications post-HCT or autoimmune disease between 2004-2012. We evaluated the cumulative incidence of reconstitution of B cell numbers, distribution and function, and determined the incidence of impaired B cell function three years after rituximab. We defined normal B cell function as IgG >4 g/?l =6 weeks after cessation of immunoglobulin supplementation.
Results We analyzed 18 post-HCT and 14 autoimmune disease patients, who received a median of 3.5[1-12] and 4.0[2-4] rituximab infusions (375 mg/m2) and had 1162.5[507-1848] and 1304[442-2306] days of B cell follow-up, respectively. Baseline characteristics were not significantly different between the indication groups. Figure?1 shows the cumulative incidence of normal B cell function. Three patients in each indication group (18.8% of 32 patients), all of whom received 4x375mg/m2, had impaired B cell function three years after rituximab. All required immunoglobulin supplementation. Age, B cell numbers before rituximab, number of rituximab infusions, and medication use after rituximab were no significant predictors of delayed immune reconstitution.
Conclusion Approximately 20% of children receiving rituximab showed impaired B cell function after three years. Future studies with larger patient groups should establish a more detailed risk profile of these patients.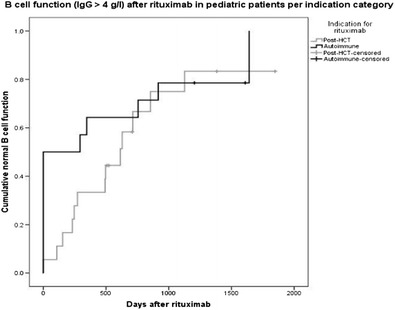
ESID-0101 Prenatal Genetic Analysis in Fetus from Carrier Mothers Associated with Primary Immunodeficiency Diseases
W.I. Lee 1, J. Huang1, K. Yeh1, P. Cheng2, T. Jaing3, S. Lin4, L. Chen1, L. Ou1, T. Yao1
1Pediatric allergy immunology and rheumatology, Chang Gung Memorial and Children's hospital, Taoyuan, Taiwan
2Obstetrics/Gynecology, Chang Gung Memorial and Children's hospital, Taoyuan, Taiwan
3Pediatric Hematology and Oncology, Chang Gung Memorial and Children's hospital, Taoyuan, Taiwan
4Department of Pediatrics, Chang Gung Memorial and Children's hospital, Taoyuan, Taiwan
Background: Primary immunodeficiency diseases (PIDs) are a rare disorder. Early optimal intervention increase survival rate. To those with identified genetic defects, prenatal genetic analysis form maternal amniotic fluid cells (AFCs) or/and chorionic villus samplings (CVS) provides more precise diagnosis for her offspring to take adequate action.
Methods: After mothers’ consents, obstetrics doctors took their AFCs or/and CVS at safe gestations for pre-natal genetic diagnosis. We performed PCR to amplify their genetic DNA or/and complement DNA to analyze candidate genes.
Results: During 2007-2013, nine carrier mothers [3 with Wiskott-Aldrich syndrome (WAS), 4 with incontinentia pigmenti (IP), 1 with X-linked chronic granulomatous disease (CGD), 1 with X-linked agammaglobulinemia (XLA), and 1 with severe combined immunodeficiency (SCID)] received 12 times pre-natal genetic analysis. In 3 offspring born to WAS carrier mothers, 1 was affected male infancy and transplanted by unrelated cord blood stem cells (UCBSCT) and 2 were female carriers. In 5 offspring born to 4 IP mothers, one male and one female were normal, but three females were IP. In 2 offspring born to SCID mothers, one female was RAG2 mutation and the other was normal. The X-linked CGD mother had an affected infancy successfully transplanted by UCBSCT. Unfortunately, we identified an XLA carrier mother after her boy had hypogammaglobuliemia and died of pseudomonas sepsis.
Conclusions: For early treatment for PIDs infancy, prenatal genetic diagnosis is mandatory to those carrier mothers with identified genetic defects, although newborn Guthrie cards screening for T cell receptor excision circles (TRECs) is available after birth.
ESID-0631 Safety and Tolerability of Human Immune Globulin Subcutaneous, 20% (IGSC 20%): Analysis of a Phase 2/?3 Study in Patients with Primary Immunodeficiencies (PID) in Europe
G. Kriván 1, B. Dérfalvi2, F. Dicso3, W. Engl4, B. McCoy4, H. Leibl4, R.I. Schiff5, L. Yel5, and the 20% IGSC Study Group5
1N/A, Szent László Hospital, Budapest, Hungary
2N/A, Semmelweis University, Budapest, Hungary
3N/A, András Jósa Teaching Hospital, Nyíregyháza, Hungary
4N/A, Baxter BioScience, Vienna, Austria
5N/A, Baxter BioScience, Westlake Village, USA
Introduction: IGSC 20% is a ready-for-use, highly-purified human immunoglobulin G (IgG) liquid preparation for subcutaneous administration. We report analysis of a phase 2/?3 study of IGSC 20% in patients aged =2 years with PIDs in Europe.
Methods: Epoch 1: IGSC 16% or intravenous IG 10% (IGIV) was administered at pre-study doses every 3 months. Epoch 2: IGSC 20% was administered weekly for 12 months (epoch 1 doses). Primary endpoint: rate of validated acute serious bacterial infections (SBIs).
Results: As of May 2014, all patients completed the study: 49 patients (age 2–67 years) started the study; 1 patient discontinued due to pregnancy in epoch 1; 45 patients completed the study through epoch 2 (discontinuations due to: local pain [n=1]; personal reasons [n=2]). At interim analysis (October 2013), during IGSC 20% treatment (n=48), 1 acute SBI (pneumonia, moderate severity) was reported. The infection rate/?patient-year was 4.29 with IGSC 20%. There were no serious adverse events considered related to any treatment. The local adverse drug reaction (ADR) rate was 0.055/infusion (ADRs in 16/48 [33.3%] patients); all mild in severity. One patient incurred the majority of local ADRs under IGSC 20% (local ADR rate excluding this patient: 0.032/infusion; ADRs in 15/47 [31.9%] patients). No severe systemic ADRs during IGSC 20% treatment were reported. Of 1812 IGSC 20% infusions, 99.6% were completed without interruption or slowing the rate.
Conclusion: IGSC 20% was well-tolerated, with no dose adjustments needed from pre-study immunoglobulin dose. Data from the final analysis (12-month treatment period) will be presented.
ESID-0226 Efficacy/Safety/Tolerability of Human Immune Globulin Subcutaneous, 20% (IGSC 20%): Interim Analysis of a Phase 2/?3 Study in Patients with Primary Immunodeficiencies (PID) in North America
D. Suez 1, I. Melamed2, I. Hussain3, M. Stein4, S. Gupta5, K. Paris6, W. Engl7, B. McCoy7, R.I. Schiff8, L. Yel8
1N/A, Allergy Asthma & Immunology Clinic PA, Irving, USA
2N/A, IMMUNOe Health Centers, Centennial, USA
3N/A, Vital Prospects Clinical Research Institute PC, Tulsa, USA
4N/A, Allergy Associates of the Palm Beaches, North Palm Beach, USA
5N/A, University of California Irvine, Irvine, USA
6N/A, LSU Health Sciences Center Children’s Hospital of New Orleans of New Orleans, New Orleans, USA
7N/A, Baxter BioScience, Vienna, Austria
8N/A, Baxter BioScience, Westlake Village, USA
Introduction: IGSC 20% is a subcutaneously administered, ready-for-use, liquid preparation of highly purified human immunoglobulin G (IgG). We report the first interim analysis of a phase 2/?3 study of IGSC 20% in patients aged =2 years with PID in North America.
Methods: In epoch 1 (13 weeks), IgG 10% was administered intravenously at prestudy doses every 3-4 weeks. In epochs 2-4, IGSC 20% was administered weekly as follows: epoch 2 (~12-16 weeks)—145% of the weekly equivalent epoch 1 dose; epoch 3 (12 weeks)—dose adjusted per AUC assessments in epochs 1-2; epoch 4 (40 weeks)—dose adapted individually per epoch 3 serum IgG trough levels. The primary endpoint is the rate of validated acute serious bacterial infections (SBIs).
Results: As of January 2014, 75 patients (age 3-83 years) started the study: 48 completed epoch 1 (1 discontinued). During IGSC 20% treatment (n=47) in epochs 2-3, no acute SBI episodes were reported; the total infection rate was 3.20/patient-year. There were no serious adverse events with IGSC 20% treatment. The local adverse drug reaction (ADR) rate was 0.045/infusion; all were mild/?moderate in severity. No severe systemic ADRs were reported with IGSC 20 Of 705 IGSC 20% infusions, 99.9% were completed without slowing the infusion rate or interrupting/stopping administration. Mean serum IgG trough level under IGSC 20% (1-week interval; n=30) was 15.2 g/?L.
Conclusion: In the interim analysis of this study, IGSC 20% provided an effective and well-tolerated therapy. This study is ongoing to confirm efficacy/safety/tolerability/pharmacokinetics of IGSC 20% (12-16-month treatment period).
ESID-0215 Long Term Safety, Efficacy, and Tolerability of Recombinant Human Hyaluronidase [RHUPH20]-Facilitated Subcutaneous Infusion of Immunoglobulin G (IGG) (HYQVIA; IGHY) in Adults with Primary Immunodeficiencies (PI)
R.L. Wasserman1, M. Stein2, I. Melamed3, L. Kobrynski4, J.A. Grant5, S. Gupta6, W. Engl7, H. Leibl7, L. Yel 8, R.I. Schiff8
1N/A, DallasAllergyImmunology, Dallas, USA
2N/A, Allergy Associates of the Palm Beaches, North Palm Beach, USA
3N/A, IMMUNOe Health Centers, Centennial, USA
4N/A, Emory University, Atlanta, USA
5N/A, University of Texas Medical Branch, Galveston, USA
6N/A, University of California Irvine, Irvine, USA
7N/A, Baxter BioScience, Vienna, Austria
8N/A, Baxter BioScience, Westlake Village, USA
Introduction: IGHy allows subcutaneous IgG administration at the same frequency and bioavailability as intravenous (IV) IgG (IGIV). We report efficacy, safety and tolerability of IGHy in patients aged =18 years treated for up to 3 years.
Methods: Fifty-nine patients received IGIV for 3 months followed by IGHy at 3-, or 4-week intervals for approximately 18 months; 52 were treated for up to an additional 21 months at the same dose and interval. rHuPH20 was infused at 75U/g IgG followed by IgG at 108% of the IV dose. rHuPH20 was discontinued after up to 3 years of exposure; patients were followed for an additional 6–12 months. Assessments included rates of adverse events (AEs), serious AEs (SAEs), and infections; tolerability; and rHuPH20 antibody levels.
Results: Maximum IGHy exposure for patients was 3 years (139 patient-years). Over the full study course, rates of temporally-associated AEs/patient-year were 3.01 (local) and 2.83 (systemic; excluding infections). No SAEs were related to IGHy. The annual infection rate was 2.98/patient-year. Of 2450 IGHy infusions administered (including ramp-up), 98% required no administration changes due to tolerability concerns or AEs. Twelve patients had non-neutralizing anti-rHuPH20 antibody titers =1:160 on 1 or more occasion with no associated AEs; titers declined despite continuing rHuPH20 in 11 of 12 patients. No patients developed neutralizing anti-rHuPH20 antibodies.
Conclusion: In patients treated with IGHy for up to 3 years, infection rates were low and AE rates were comparable to previously reported rates for subcutaneously-administered IgG, but with infusion volumes and rates equivalent to IGIV.
ESID-0788 Subcutaneous Immunoglobulins in Prevention of Hereditary Angioedema Attacks
R. Zachová 1, M. Sobotková1, T. Milota1, A. ?edivá1, J. Bartunková1
1Institute of Immunology, 2nd Medical School Charles University Prague, Prague 5, Czech Republic
Introduction
Hereditary angioedema (HAE) is a rare genetic disorder resulting from deficiency ( type I) or dysfunction ( type II) of C1 inhibitor (C1-INH) and belongs to bradykinin-mediated angioedemas. C1-INH regulates the activation of the complement, kinin-kallikrein, coagulation and fibrinolytic systems. It is characterised by reccurent episodes of angioedema which most often affects the skin or mucosal tissues of upper respiratory and gastrointestinal tracts. Angioedema attacks can be serious and disfiguring as well as causing abdominal pain resembling emergency. Laryngeal edema may cause fatal asphyxiation. It may greatly impair the physical and mental health and the quality of life of the patient. Triggers of the attacks are stress, mechanical stimulation , hormonal changes ( estrogenes) or infection.
Methods
In our group of 45 patiens from the Center for HAE we follow 6 HAE patients with frequent HAE attacks. We observed high linkage of attacks to infections in this group. We found mild hypogamaglobulinemia or immunoglobulin-subclass deficiency in HAE patiens with frequent HAE attacks due to infection. After introduction of subcutaneous immunoglobulins into prophylactic treatment of this patiens the number of attacks dropped . The patients were educated in self - administrations of subcutaneous immunoglobulins and in home treatment .
Conclusions.
Subcutaneous immunoglobulins self administered in home treatment by patiens with frequent HAE attacks due to recurrent infections led to decrease of frequency of HAE attacks in our group of patients.
ESID-0779 Long-Term Safety and Efficacy of Retroviral-Mediated Gene Therapy for ADA-SCID
M.P. Cicalese 1, F. Ferrua1, R. Pajno1, F. Barzaghi1, M. Frittoli1, L. Castagnaro1, S. Giannelli2, F. Dionisio2, I. Brigida2, A. Tabucchi3, F. Carlucci3, A. Hogg4, K. Singh4, A. Campanile4, R. Philipson4, J. Appleby4, M.G. Roncarolo5, A. Aiuti6
1TIGET and Pediatric Immunohematology, San Raffaele Scientific Institute, Milano, Italy
2TIGET, San Raffaele Scientific Institute, Milano, Italy
3UOC Clinical Pathology, University of Siena and Azienda Ospedaliera Universitaria Senese, Siena, Italy
4Rare Diseases, GlaxoSmithKline, Stevenage, United Kingdom
5TIGET and Pediatric Immunohematology, San Raffaele Scientific Institute and VIta Salute University, Milano, Italy
6TIGET and Pediatric Immunohematology, San Raffaele Scientific Institute and Univ. of Rome Tor Vergata, Milano, Italy
SCID due to adenosine deaminase deficiency (ADA-SCID) is characterized by impaired lymphoid development and function, and systemic manifestation of metabolic toxicity. Eighteen children have been treated with infusion of autologous bone marrow (BM) CD34+ cells transduced with a gammaretroviral vector encoding ADA combined with reduced intensity conditioning. Treatment was administered in the context of two pilot studies (n=3), a phase I/?II clinical trial (n=12), or compassionate use program (n=3). Median age at treatment was 1.7 years (range 0.5 to 6.1). As of last follow up (December 2013), all patients are alive and well. The median follow up in the patients included in the long-term study (AD1115611) is 7 years (range 2.6 - 13.3). Three subjects required reintroduction of ERT and 2 of them received allogeneic BM transplantation. In the 15 patients who remain off ERT, vector transduced cells continue to be stably detected in multiple lineages in the BM and peripheral blood, with higher levels in lymphoid cells, due to selective advantage. Purine metabolites levels in RBC remain low, indicating effective systemic detoxification. T cell counts, including CD4+ naïve T cells, and T-cell proliferation to anti-CD3 have progressively increased after gene therapy. IVIg infusions have been discontinued in 12 patients, with positive response to vaccinations. Moreover, a reduction in the rate of severe infections was observed. No event of insertional oncogenesis has been detected during the follow up. In summary, these data confirm the long-term safety and efficacy of gene therapy for ADA-SCID. Since 04/2012 GSK has become the study sponsor.
ESID-0437 Hematopoietic Stem Cell Transplantation for Patients with Primary Immune Deficiencies in Europe: An Update by the European Registry (SCETIDE)
V. COURTEILLE 1, J. Provot1, B. Neven2, F. Suarez3, A. Gennery4, P. Veys5, R. Bredius6, A. Lankester6, N. Mahlaoui1, A. Fischer1
1CEREDIH, Necker-Enfants Malades Hospital, Paris, France
2Paediatric immunology and hematology Department, Necker-Enfants Malades Hospital, Paris, France
3Adult Hematology Department, Necker-Enfants Malades Hospital, Paris, France
4Institute of Cellular Medicine Paediatric Immunology Dept, Great North Children's Hospital, Newcastle, United Kingdom
5Bone Marrow Transplant unit, Great Ormond Street Hospital, London, United Kingdom
6BMT Centre, Leiden University Hospital, Leiden, Netherlands
Introduction:
SCETIDE registry was created 30 years ago on behalf of the EBMT Inborn Error Working Party in order to improve knowledge on HSCT and care for patients with severe combined immunodeficiencies (SCIDs) or other primary immunodeficiencies (PIDs). This project has given original and recognized results (last report by Gennery AR et?al. JACI, 2010).
Objective and Methods:
Current technologies now allow a better data management and a greater mutualisation of skills and know-how. To this aim the French National Reference Centre for PID (CEREDIH) at Necker hospital, Paris, developed and supported a new online registry sytem (www.scetide.org). Member centers were asked to register their new transplants and comparison with other registries enabled to identify other hospitals which agreed to report their patients in SCETIDE.
Results:
As of May 2014, 3251 patients have been reported by 77 centres in 25 countries. Main participating centres are Paris Necker hospital (20%), Newcastle (10%) and London GOSH (10%).
A total of 3747 transplants were reported between 1968 and 2013, mostly for SCID (33%) and other T cell deficiencies (20%), HLH (17%), WAS (9%), phagocytic disorders (12%) and other PIDS (9%).
Donor was identical sibling in 21%, other related in 32% and unrelated in 31%. Source of HSCs was BM in 63% of HSCT, PBSC in 17% and CB in 9%. Overall survival is 60%.
Conclusion:
SCETIDE is the largest worldwide PID registry. Further detailed analyses, including survival according to groups of PID and types of transplant, will be presented.
ESID-0028 Gene Therapy of X-CGD: Past, Present and Future
J. Schwäble1, B. Goebel1, G. Santilli2, M. Rothe3, U. Modlich4, J. Reichenbach5, H. Serve1, M. Grez 6, G. Galy7, A. Thrasher2
1Department of Medicine Hematology/Oncology, Goethe University Frankfurt, Frankfurt, Germany
2Department of Immunology, Institute of Child Health, London, United Kingdom
3Experimental Hematology, Hannover Medical School, Hannover, Germany
4Department of Virology, Paul-Ehrlich-Institute, Frankfurt, Germany
5Division of Immunology, Children Hospital Zurich, Zurich, Switzerland
6Gene Therapy Unit, Georg-Speyer-Haus, Frankfurt am Main, Germany
7Molecular Immunology and Biotherapies, GENETHON, Evry, France
Chronic granulomatous disease (CGD) is a rare inherited immunodeficiency characterized by life-threatening infections due to a functional defect in phagocytic cells. Although the disease can be cured by allogenic bone marrow transplantation, this treatment requires an HLA-identical donor. The genetic modification of autologous hematopoietic stem cells is an option for patients lacking a donor. We have treated a total of 4 X-linked CGD patients with gene modified cells. Gene therapy provided clear clinical benefit, with subjects recovering from refractory infections. The patients remained stable and free of severe infections for several months. Long term outcome was compromised by clonal dominance leading to a myelodysplastic syndrome in 3 out of the 4 patients. To improve safety and efficacy, we developed a SIN lentiviral vector for gene therapy of X-CGD. In this vector the codon-optimized gp91phox transgene is driven by a myeloid-restricted promoter. Extensive pre-clinical testing showed: (i) the G1XCGD vector restores superoxide production in murine and human cells to therapeutically relevant levels in?vitro and ?in?vivo, (ii) no clonal dominance was observed in?vitro or ?in?vivo as determined by the retrieval frequency of retroviral integration sites. Based on this work the G1XGD vector has been approved for clinical use by the regulatory authorities for a Phase I/?II clinical trial in London and Frankfurt. The trial (EudraCT Nr. 2012-000242-35) is sponsored by Genethon and supported by the Net4CGD collaborative project (EC-FP7-GA n°305011). The trial is a multicentre study with additional sites scheduled in Zürich, Paris, Los Angeles Bethesda and Boston.
ESID-0343 Allogeneic Hematopoetic Stem Cell Transplantation to Treat Refractory Inflammatory Bowel Disease in an X-CGD Carrier with Non-Random X-Inactivation
F. Hauck 1, A. Klenk1, P. Bufler2, I. Schmid1, S. Koletzko2, C. Klein1, M.H. Albert1
1Hematology and Oncology, Dr. von Hauner Children's Hospital, Munich, Germany
2Gastroenterology and Hepatology, Dr. von Hauner Children's Hospital, Munich, Germany
Five genetic traits are known to cause chronic granulomatous disease (CGD), but the most common one is X-linked CGD (X-CGD) due to hemizigous mutations in CYBB. CGD is characterized by invasive bacterial and fungal infections accompanied by granulomatous inflammation. Inflammatory bowel disease (IBD) can be an additional or isolated manifestation. Allogenetic hematopoietic stem cell transplantation (alloHSCT) is the standard curative treatment.
X-CGD carriers normally show random X-chromosome inactivation and retain 50% of NADPH-oxidase activity. Nevertheless, they can develop aphthous stomatitis and discoid lupus erythematosus. In rare cases, X-CGD carriers present with non-random X-chromosome inactivation and, depending on the remaining NADPH-oxidase acitivity, can develop invasive bacterial and fungal infections (activity <10%) or autoinflammatory manifestations (activity 10-15%).
We report on an X-CGD carrier with non-random X-chromosome inactivation and NADPH-oxidase activity of 12%. At the age of twelve years, she developed severe Crohn-like IBD that was refractory to treatment with exclusive enteral nutrition and immunosuppression including anti-TNFa medication. As here clinical course aggravated over time, she received alloHSCT from a matched unrelated donor (10/10) after conditioning with submyeloablative targeted busulfan, fludarabine and alemtuzumab at the age of 18 years. She engrafted at day +15 without any major complications and no signs of graft versus host disease (GVHD). Currently at day +60, her colitis has significantly improved with remaining bowel wall thickening.
To our knowledge, this is the first case of an X-CGD carrier with non-random X-chromosome inactivation and IBD who has been successfully treated with alloHSCT.
ESID-0687 U.S. Clinical Gene Therapy Trials for Adenosine Deaminase-Deficient Severe Combined Immune Deficiency (ADA-SCID)
D. Carbonaro Sarracino 1, K. Shaw1, R. Sokolic2, A. Davila1, E. Garabedian2, C. Silvin2, P. Barman1, S. de Oliveira3, X. Jin1, S. Geiger1, M. Smogorzewska4, J. Jagadeesh2, G. Crooks5, T. Moore3, A. Wayne6, M. Hershfield7, B. Gaspar8, A. Thrasher8, F. Candotti2, D. Kohn9
1Microbiology Immunology & Molecular Genetics, University of California Los Angeles, Los Angeles, USA
2Genetics and Molecular Biology Branch, National Human Genome Research Institute, Bethesda, USA
3Pediatrics, University of California Los Angeles, Los Angeles, USA
4Research Immunology/Bone Marrow Transplant, Childrens Hospital Los Angeles, Los Angeles, USA
5Pathology and Pediatrics, University of California Los Angeles, Los Angeles, USA
6Hematologic Diseases ?Section?Pediatric Oncology Branch, National Human Genome Research Institute, Bethesda, USA
7Medicine and Biochemistry, Duke University School of Medicine, Durham, USA
8Institute of Child Health, University College London, London, United Kingdom
9Microbiology Immunology & Molecular Genetics and Pediatrics, University of California Los Angeles, Los Angeles, USA
Autologous hematopoietic stem cell transplantation (HSCT) of gene-modified cells (gene therapy) has shown clinical benefit for ADA-SCID when combined with non-myeloablative conditioning and enzyme replacement therapy (ERT) cessation. In a Phase II study (2009-2012) recently closed to enrollment (NCT00794508), patients received autologous CD34+ cells modified with the MND-ADA g-retroviral vector after conditioning with busulfan (4 mg/kg) and ERT cessation (n=10). Patients were treated between 3 months and 15 years of age (median=11.5 months) and follow-up ranges from 18 and 60 months. With the exception of the oldest patient (at 15y), all others remain off ERT with normalized PBMC ADA activity. All nine remaining off ERT show normal proliferative responses to mitogens and three of nine (3 of 5 infants) were able to discontinue IVIg. MND-ADA is detected in PBMC (0.1-2.6 VCN) and in granulocytes (0.01-0.3 VCN) at most recent visit. A new Phase I/?II trial was opened in May 2013 (NCT01852071) in which patients have received autologous CD34+ cells modified with the self-inactivating lentiviral vector EFS-ADA after conditioning with busulfan (4 mg/kg) and ERT cessation at 30 days post-transplant (n=6). Patients were treated at 4 and 42 months old and the three who are past 30 days remain off ERT. The patients with the longest follow-up have normal or higher PBMC ADA activity (n=2). Monitoring for insertional oncogenesis is on-going in both studies and has not detected any monoclonal proliferative events. These results demonstrate the efficacy and safety of gene therapy for ADA SCID.
Topic: Others
ESID-0325 Assessment of Primary Immunodeficiency Disorders Among Suspected Children in East Delta of Egypt
M. Abdelaziz Almalky 1, S. hussein1, S. Abdelsalam1, M. Abdelsalam1, M. Atfy1, A.?M.?A.L. Zidan2
1pediatrics, Faculty of medicine Zagazig University, Zagazig, Egypt
2Clinical pathology, Faculty of medicine Zagazig University, Zagazig, Egypt
Background: PIDs are rare genetic disorders that affect the development and/or function of the immune system. The prevalence of these disorders in our population is still unknown. The aim of this study is to identify various types of PIDs in suspected children at Zagazig University hospitals in east delta of Egypt, their characteristic features, clinical manifestations and laboratory profiles.
Patients and methods; all patients under 15 years of both sex admitted or consulted at Zagazig University hospital who were suspected to be PID were included in this study. After taking history and thorough clinical examination, CBC with differential, Measurement of serum immunoglobulins (IgA, IgG, IgM, IgE), measurement of basic panel of lymphocytes (CD3,CD4,CD8,CD19, CD16,56) by flowcytometry.Other investigations were ordered as needed.
Results: Fifty patients were diagnosed from July 2011 to December 2013. The spectrum of PIDs in our center was as follow: predominantly antibody deficiency was the most common category by 19 cases (38%) followed by combined immunodeficiency 10 cases (20%) then well defined syndromes by 9 cases (18%), auto inflammatory disorders by 4 cases (8%), and complement disorders by 3 cases (6%), Phagocytic disorders by 3 cases (6%) and lastly immune dysregulation by 2 cases (4%). None of our cases were diagnosed as disorder in innate immunity. Median age of onset of symptoms was 7 months; the median time of diagnosis lag was 24 months. Pneumonia was the most common presentation. Consanguinity rate was 60%. Mortality rate was 20%.
Conclusion: PIDs are not rare but underestimated in our center.
ESID-0474 Clinical Features and Mutation Analysis of 51 Iranian Patients with Hyper Immunoglobulin E Syndrome
M. Tavassoli1, M. Ghadami2, H. Abolhassani 3, M. Kokabee1, N. Parvaneh1, M. Yeganeh1, B. Mirminachi1, M. Movahedi4, M. Gharagozlou4, N. Rezaei1, A. Aghamohammadi1
1Research Center for Immunodeficiencies, Pediatrics Center of Excellence Children's Medical Center Tehran University of Medical Sciences, Tehran, Iran
2Department of Immunology, School of Medicine Tehran University of Medical Sciences, Tehran, Iran
3Division of Clinical Immunology Department of Laboratory Medicine, Karolinska Institutet at the Karolinska University Hospital Huddinge, Tehran, Iran
4Department of Allergy and Clinical Immunology, Pediatrics Center of Excellence Children's Medical Center Tehran University of Medical Sciences, Tehran, Iran
Background: Hyper IgE syndrome (HIES) consist a group of primary immunodeficiency disorders characterized by recurrent staphylococcal abscesses, sinopulmonary infections and chronic eczema as well as markedly elevated IgE levels. Most of HIES have been found to be inherited as autosomal dominant due to mutations in signal transducer and activator of transcription 3 gene (STAT3) whereas some of HIES cases have evidence of mutation in dedicator of cytokinesis 8 (DOCK8) and Tyrosine Kinase2 (TYK2) genes those have been identified with autosomal recessive pattern of heritage.
Methods: A prospective clinical evaluation was conducted on 51 HIES patients with severe atopic dermatitis (SCORing Atopic Dermatitis >50) and IgE level >2000 IU/ml to show their clinical and immunologic features as well as the result of mutation analysis. The phenotypic severity by National institute of health (NIH) HIES score were calculated to predict STAT3 mutation.
Results: The measurement of NIH HIES score showed the mean of 47.1±17.3 (range 10-78) and the mean of SCORAD was 68.4±15.4. From 51 patients, STAT3 gene mutation was identified in 17 cases from 16 unrelated families including 8 new mutations. DOCK8 deficiency was documented in only one female case. Patients were classified based on the damaged domain of final protein. Age at the time of diagnosis and delay diagnosis in Src homology 2 (SH2) group were significantly higher compared to DNA binding domain (DNA-B) group.
Conclusion: The results of this study highlight the importance of mutation analysis in evaluation of HIES patients.
Keywords: Hyper IgE syndrome, STAT3, DOCK8, Mutation
ESID-0500 Immunologic Evaluation of Patients with Recurrent Infections
S. Yousefzadegan1, A. Nadjafi1, H. Abolhassani2, S. Mansouri1, B. Mirminachi 1, M. Tavakol3, N. Rezaei4, A. Aghamohammadi1
1Research Center for Immunodeficiencies, Pediatrics Center of Excellence Children's Medical Center Tehran University of Medical Science, Tehran, Iran
2Division of Clinical Immunology Department of Laboratory Medicine, Karolinska Institutet at the Karolinska University Hospital Huddinge, Tehran, Iran
3Chronic Respiratory Diseases Research Center, National Research Institute of Tuberculosis and Lung Diseases (NRITLD) Shahid Beheshti University of Medical Sciences, Tehran, Iran
4Molecular Immunology Research Center, Tehran University of Medical Science, Tehran, Iran
Abstract
Background: Recurrent infections seem to be a common complaint in children whom are referring to general practitioners and pediatricians' offices. Diagnostic evaluation and appropriate treatment of these individuals during their follow-up period of illnesses, in hospital and/or at outpatient setting, are very important. The aim of this study was to evaluate the frequency of primary immunodeficiencies (PID) among patients with recurrent infections.
Methods: One-hundred patients with a history of recurrent infections were enrolled in the study. Comprehensive laboratory tests including immunological investigations were performed in all individuals.
Results: Among all studied patients, 21 cases (21%) were finally diagnosed as PID, 18 (18%) as allergy, 16 (16%) as secondary immunodeficiency and 15 (15%) as anatomical or functional disorders. No underlying defects have been found in the other patients. The most common presentations were lower respiratory tract infections (31%) and upper respiratory tract infections (30%), followed by severe systemic infections (13%), gastrointestinal infections (11%), soft tissue infections or abscess (11%) and skin infections (4%). The rate of parental consanguinity (p=0.001) and soft tissue infections (p=0.004) were significantly higher in PID patients compared to other causes of recurrent infections.
Conclusions: The real rate of PID as a cause of recurrent infection is much more than routinely noted in a selected group of pediatric patients; so, appropriate immunological tests should be done as early as possible in children with recurrent infections in certain conditions.
Keywords: Recurrent infection, Primary immunodeficiency, Children
ESID-0716 Characterization of Crohn Disease in X-Linked Inhibitor of Apoptosis Protein-Deficient Male Patients and Female Symptomatic Carriers
C. Aguilar 1, C. Lenoir2, N. Lambert3, B. Bègue4, N. Brousse5, D. Canioni5, D. Berrebi6, M. Roy7, S. Gerart2, H. Chapel8, T. Schwerd9, L. Siproudhis10, M. Schappi11, A. Al-Ahmari12, A. Yamaide13, M. Mori14, L. Galicier15, B. Neven16, J. Routes17, H. Ulhig18, S. Koletzko9, S. Patel8, H. Kanegane19, C. Picard20, A. Fischer21, N. Cerf Bensussan22, F. Ruemmele23, J.P. Hugot24, S. Latour1
1Lymphocyte Activation and susceptibility to EBV, Imagine, Paris, France
2Lymphocyte Activation and susceptibility to EBV, Imagine, PARIS, France
3CEDI, Necker Hospital APHP, PARIS, France
4UMR1163, Imagine, PARIS, France
5Pathology Department, Necker Hospital APHP, PARIS, France
6Pathology Department, Robert Debré Hospital, PARIS, France
7INSERM UMR 843, Paris Diderot University, PARIS, France
8Primary Immunodeficiency Unit, Oxford University, OXFORD, United Kingdom
9University of Munich Medical Center, University of Munich, MUNICH, Germany
10Gastroenterology Unit, Rennes Hospital, RENNES, France
11Pediatrics center, Medical University Center Geneva, GENEVA, Switzerland
12Department of Pediatric Hematology/Oncology, King Faisal Specialist Hospital, RIYADH, Saudi Arabia
13Department of Pediatrics, City UNiversitary Mecial Center, Yokohama, Japan
14Division of Allergy and Rheumatology, Chiba CHildren's hospital, Chiba, Japan
15Immunology Unit, Saint Louis Hospital, Paris, France
16Department of Immunology and Haematology, Necker Hospital APHP, Paris, France
17Departement of pediatrics, Medical College of Wisconsin, Milwaukee, USA
18Translational Gastroenterology Unit, Oxford University, OXFORD, United Kingdom
19Department of Pediatrics, University of Toyama, Toyoma, Japan
20CEDI, Necker Hospital APHP, Paris, France
21Department of Immunology and Haematology, Necker Hospital APHP and Imagine, Paris, France
22INSERM UMR 1163, Imagine, Paris, France
23Gastroenterology pediatric unit, Necker Hospital APHP, Paris, France
24INSERM UMR 843, Paris Diderot University, Paris, France
Crohn disease is an inflammatory bowel disease (IBD) with a complex mode of inheritance. Although nucleotide binding and oligomerization domain containing 2 (NOD2) is the strongest risk factor, the cause of Crohn disease remains unknown in the majority of the cases. IBD has been reported in some X-linked inhibitor of apoptosis (XIAP)-deficient patients.
Methods: We performed a phenotypical and histologic analysis of the IBD affecting 17 patients with hemizygous mutations in XIAP, including 3 patients identified by screening 83 patients with pediatric-onset IBD. The X chromosome inactivation was analyzed in female carriers of heterozygous XIAP mutations, including 2 adults with IBD.
Results: Clinical presentation and histology of IBD in patients with XIAP deficiency overlapped with those of patients with Crohn disease. The age at onset was variable (from 3 months to 41 years), and IBD was severe and difficult to treat. In 2 patients hematopoietic stem cell transplantation fully restored intestinal homeostasis. Monocytes of patients had impaired NOD2- mediated IL-8 and monocyte chemoattractant protein 1 production, as well as IL-10, in response to NOD2 and Toll-like receptor 2/?4 costimulation. The 2 heterozygous female carriers of XIAP mutations with IBD displayed abnormal expression of the XIAP mutated allele, resulting in impaired activation of the NOD2 pathway.
Conclusion: IBD in patients with XIAP deficiency is similar to Crohn disease and is associated with defective NOD2 function in monocytes. Importantly, we report that it is not restricted to male patients because we identified 2 symptomatic female heterozygous carriers of XIAP mutations.
ESID-0753 Diagnosis and Management of PID.....Our Experience from Mumbai
J. AHMED 1, R. merchant1
1pediatric, nanavati hospital, Mumbai, India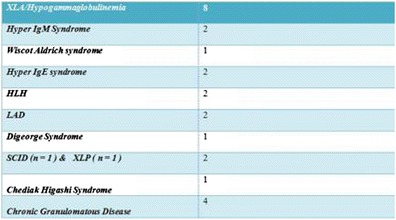
we present our experience in managing PID patients at tertiary centre in Mumbai
XLA:out of eight ,5 received regular IVIG replacement therapy and showed good growth and less morbidity. 2 children received IVIG from the beginning and are free from any morbidities. One developed Chron’s disease at the age of 6 years despite on IVIG replacement therapy. One not on regular transfusion succumbed to fulminant sepsis and other is left with major respiratory morbidities. cost of therapy is prohibitive to majority and with no social support system , these patients depend heavily on charitable and social organization of medical aid.
Hyper IGM Syndrome :- We transplanted 1 child with a novel CD40 Ligand mutation,is now off transfusion and symptom free with 80% chiemrism .2nd patient developed auto immune hemolytic anemia. Getting a matched donor is still problem in India due to lack of centralized HLA registry
Hyper IgE:Our both Hyper IgE were diagnosed late after their first decade, one had severe eczema and Second patient was diagnosed at the age of 13 due to persistent autoimmune hemolytic anemia. Late diagnosis and low awareness is key issue . Treatment of co morbidities is major issue with no permanent treatment available.
HLH:Our both patient had secondary HLH , triggered by infection. Perforin levels were normal. Awareness and Lab support is necessary for diagnosis.
CGD :- All our CGD patients were diagnosed due to non resolving pneumonia. These patients were put on long term oral prophylaxis of antibiotics and antifungal to prevent recurrences. Compliance with therapy is important
ESID-0417 Immune Evaluation of a Cohort of Pediatric Patients Presenting with Lymphoma. Is There an Underlying Primary Immunodeficiency?
C. Bianchi1, A. Esteve2, J. Parareda3, M. Piquer4, A.?M. Plaza4, M. Juan5, L. Alsina 4
1Pediatrics, Hospital Sant Joan de Déu, BARCELONA, Spain
2Allergy and Clinical Immunology, Fundació Sant Joan de Déu, BARCELONA, Spain
3Oncology, Hospital Sant Joan de Déu, BARCELONA, Spain
4Allergy and Clinical Immunology, Hospital Sant Joan de Déu, BARCELONA, Spain
5Immunology Department – CDB, Hospital Clínic, BARCELONA, Spain
INTRODUCTION: Lymphoma can be a manifestation of a primary immunodeficiency (PID), such as Common Variable Immunodeficiency (CVID), X-Linked Proliferative Syndromes (XLP) and Autoimmune Lymphoproliferative Syndrome (ALPS), besides particular syndromic forms of PID. Our aim was to rule out a PID in a cohort of pediatric patients diagnosed with lymphoma.
METHODS: All patients<18yo seen in the Oncology outpatient clinic of a tertiary hospital between July 2011 and July 2013 with the diagnosis of lymphoma, were referred to the clinical immunologist for evaluation of possible PID, which included: chart review, physical exam and laboratory with: T and B cell subphenotyping and function, immunoglobulin levels and vaccine responses to diphtheria, tetanus, pneumococcus, and SAP and XIAP expression for males, which were performed 2 years after lymphoma diagnosis.
RESULTS: A total of 27 lymphoma patients were included in the study, 19 males. Median age at diagnosis was 7.4 years (3-16yo). None had a past-medical history of recurrent infections or autoimmunity, nor had syndromic features. 18/27 (66%) lymphomas were of B cell origin (14 Burkitt). The immunological study revealed no primary immunodeficiency, yet 12 patients (44%) presented an immunoglobulin (Ig) secretion alteration (mostly of IgG and/or IgM) at diagnosis or during treatment. In 4 patients, hipoIgG has not resolved during follow-up (>2 years post-lymphoma), but they suffer no infections, vaccine responses and B cell phenotyping are normal, discarding CVID. 2 patients have persistent hipoIgM.
CONCLUSION: In pediatric patients with lymphoma, hypogammaglobulinemia (IgG, IgM) can be observed at diagnosis, during treatment and follow-up, with no underlying PID.
ESID-0690 Clinical Features of 22Q11 Deletion Syndrome Patients from Mexico. Mexican Group for Immunodeficiencies Collaboration
A. Alvarez Cardona 1, S. Espinosa Padilla2, F. Espinosa Rosales3
1Centro de Ciencias de la Salud, Universidad Autonoma de Aguascalientes, Aguascalientes, Mexico
2Jeffrey Modell Diagnostic Center, Instituto Nacional de Pediatria, Mexico DF, Mexico
3Presidency, Fundación Mexicana para Niñ@s con Inmunodeficiencias Primarias, Mexico DF, Mexico
Introduction and Rationale.
22q11.2 deletion syndrome (22q11DS) is a common disorder that presents in 1:4000 births, with several series described everywhere little is known about the 22q11DS patients from Mexico.
Clinical features include heart disease, hypocalcemia, typical facies and immunodeficiency.
The more frequently used diagnostic test is fluorescence in situ hybridization (FISH).
From the last 30 years the Mexican Group For Primary Immunodeficiency (MEXGID) has been thriving with several centers treating 22q11DS patients.
By describing the clinical features of 22q11DS from Mexico the knowledge about the disorder will widen as an initial effort for further research and collaboration.
Methods
Through an open invitation to all the members of the MEXGID and interested attending physicians we aim to describe the clinical features of 22q11DS patients from Mexico.
Study type: retrospective and descriptive analysis.
The criteria for 22q11DS will be: positive FISH for 22q11.2 deletion and any clinical features.
Information will be registered about clinical features, laboratory tests and demographic data from patients.
Appropriate informed consent will be obtained from parents or caregivers.
Data will be presented as frequencies or categories according to its distribution and processed with Prism Graphpad for OS for further analysis.
ESID-0645 The First Decade of Customised Genetic Testing for PIDS in New Zealand
R. Ameratunga 1, S.T. Woon1
1VIM, Auckland hospital, Auckland, New Zealand
Primary immune deficiency disorders (PIDs) are a group of diseases associated with a genetic susceptibility to recurrent infections, malignancy, autoimmunity and allergy. The molecular basis of many of these disorders has been identified in the last two decades. Most are inherited as single gene defects. Identifying the underlying genetic defect plays a critical role in patient management including diagnosis, identifying atypical presentations, family studies, providing prognostic information, prenatal diagnosis and is useful in defining new diseases. New Zealand is a geographically isolated developed country in the south pacific. In 2004 we developed a dedicated customized genetic testing service for PID patients in New Zealand. This accredited diagnostic program offers rapid turnaround times for genetic tests and minimizes the risk of laboratory errors. Here we review the first decade of customized genetic testing for PIDs in New Zealand.
ESID-0240 Primary Immunodeficiency Disease in China
Y. An 1, X. Zhao1
1Rheumatology and Immunology, Children's Hospital of Chongqing Medical University, Chongqing, China
Primary immunodeficiency disease was recognized as an important branch of clinical immunology in China almost in the early 1980s when Pediatric Immunology section was formed, Several PID centers have joined in the network including Children’s Hospital of Chongqing Medical University, Fudan University, University of Hong Kong an so on. With fund from National Natural Science Foundation of China and National Health and Family Planning Commission, more than 1300 cases of PID patients were reported, the estimate PID incidence rate were much lower that former reports and the cases focus on common XLA, WAS,CGD, XHIM and SCID. In our center of Children’s Hospital of Chongqing Medical University, with a total of 26 pathogenic genes were identified and more than 285 mutations were detected from 337 candidate patients, in which WASP (103/337), BTK (66/370), CYBB (37/337), CD40L (22/337) defects and SCID (16/337) were with higher incidence. In addition, some newly identified PID were also detected such as DOCK8, BIRC4, FOXP3 and somatic NRAS mutation, in which thirty patients (WAS, XHIM and X-CGD) have got HSC transplantation. A registry net (http://piddata.gotoip2.com/login.php) as well as education and information for the public website for PID (www.pidchina.org) were established recently. Although the diagnosed PID cases in China increased, a great number of patients with definite gene mutations have not get HSC transplantation. Achieving boundless multi-center, cross-discipline collaboration, newborn screening, HSC transplantation and more fund supports were needed for better outcome of PID patients in China.
ESID-0692 Beta-2-Microglobulin Deficiency: More Than a MHC-Class-I Deficiency Syndrome
Ö. ARDEN?Z 1, S. Unger2, H. Onay3, C. Keck4, C. Cianga5, B. Gerceker6, B. Martin7, I.L.K.A. Fuchs2, U. Salzer2, S. Ammann2, A. Ikinciogullari8, D. Güloglu8, T. Dereli6, R. Thimme7, S. Ehl2, K. Schwarz9, A. Schmitt-Graeff4, P. Cianga5, P.A.U.L. Fisch4, K. Warnatz2
1Internal Medicine Allergy and Clinical Immunology, Ege University Medical Faculty, Izmir, Turkey
2Center for Chronic Immunodeficiency, University Medical Center Freiburg and University of Freiburg, Freiburg, Germany
3Medical Genetics, Ege University Medical Faculty, Izmir, Turkey
4Pathology, Institute of Pathology University Medical Center Freiburg, Freiburg, Germany
5Immunology, Grigore T. Popa University of Medicine and Pharmacy, Iasi, Romania
6Dermatology, Ege University Medical Faculty, Izmir, Turkey
7Internal Medicine II, University Medical Center Freiburg, Freiburg, Germany
8Pediatric Immunology and Allergy, Ankara University School of Medicine, Ankara, Turkey
9Institute for Transfusion Medicine University of Ulm, Institute for Clinical Transfusion Medicine and Immunogenetics Ulm, Baden-Württemberg-Hessen, Germany
Background: MHC-class-I deficiency syndromes are mostly caused by TAP1 or TAP2 deficiencies. Recurrent respiratory tract infections (RTI) and in some ulcerating granulomatous skin disease are the main components of clinical presentation.
Objective: Describing molecular and immunological features of beta-2-microglobulin (b2m) deficiency diagnosed in two siblings with Turkish origin.
Methods: Genetic defect was detected by analyzing the candidate gene. Flow-cytometry, functional tests and immunohistochemistry were applied.
Results: The female was referred with recurrent RTIs and ulcerating skin lesions and nasal perforation. Both siblings developed bronchiectasis, while the brother was mainly asymptomatic. A novel homozygous splice site mutation c.67+?G>T in intron 1 of the B2M gene was detected. Not only polymorphic MHC-class-I molecules, but also other b2m-containing molecules including CD1a-c and FcRn were absent from the surface of b2m-deficient cells. Both siblings showed undetectable levels of b2m, low albumin and IgG serum levels, compatible with the absent FcRn expression. In both siblings, CD8+ gd T cells were expanded and NK failed to kill MHC-class-I-negative target cells, which resembles findings in TAP-deficiency syndrome. The heterozygous parents were normal for all tested parameters except for B2M mRNA expression.
Conclusion: Herein we describe for the first time the immunological effect of b2m-deficiency in humans in more detail. Although it shares clinical and laboratory abnormalities with TAP-deficiency, b2m-deficiency has a broader effect on the immune system caused by the absent surface expression of additional b2m-associated molecules. Absent b2m serum levels, hypogammaglobulinemia and hypoalbuminemia are suggestive hallmarks in the differential diagnosis.
ESID-0207 Evaluation of the T-Cell Receptor Excision Circles Assay Performances for Severe Combined Immunodeficiency Neonatal Screening on Guthrie Cards in a French Single Centre Study
M. AUDRAIN 1, S. MIRALLIE2, N. BOURGEOIS1, V. SEBILLE3, H. RABETRANO4, I. DURAND-ZALESKI4, C. PIERRES5, N. MAHLAOUI6, A. FISCHER6, C. THOMAS7
1Immunology Laboratory, Nantes University Hospital, Nantes, France
2Neonatal Screening Laboratory, Nantes University Hospital, Nantes, France
3Biometry Platform, Nantes University Hospital, Nantes, France
4URC-Eco, APHP Hotel Dieu, Paris, France
5Clinical research Unit, Nantes University Hospital, Nantes, France
6CEREDIH (French National Reference Center for PID), Necker-Enfants Malades Hospital, Paris, France
7Pediatric Immunology, Nantes University Hospital, Nantes, France
Objective:
To evaluate the performances of the TREC quantification assay for neonatal screening of SCID in a French University Medical Centre and to estimate the cost of the test.
Methods:
Repeatability and reproducibility were tested. Then 5028 unselected and de-identified DBS from newborns, 8 DBS from SCID patients were analyzed. Quantitation of TRECs was performed by real time quantitative PCR as described by Gerstel-Thompson et?al. PCR reaction was performed either on 96-plates with manual distribution or on 384-plates with automated distribution. Cut-off values were established using the 99th percentile method for manual and automated method separately. Cost was estimated using activity-based micro-costing.
Results and discussion:
Intra-assay and inter-assay CV was fewer than 2.5% for extraction and PCR. Our calculated cut-off led to re-test 0.22%-0.66% of samples that remained equivocal or inconclusive depending on the method used. All SCID patients but one (ZAP70 deficient) had undetectable TRECs. In a CDC exercise including diluted samples and using our cut-off values, one expected normal sample was classified as equivocal. By using a lower cut-off, this sample was well-classified, while the all re-test rate fell to 0.04%. By doing so, we would not miss the SCID patients except for the ZAP70 deficiency.
The test cost varied between €3.9 and €5.7 if testing more than 50,700 samples per year with the manual method or between €3.1 and €4.3 with the automated method.
ESID-0039 Do Elevated Serum IGM Levels Have to Be Included in Probable Diagnosis Criteria of Patients With Ataxia-Telangiectasia?
E. Azarsiz 1, N.E. Karaca1, N.C. Gunaydin1, N. Gulez2, C. Ozturk3, G. Aksu1, F. Genel2, N. Kutukculer1
1Pediatric Immunology, Ege University Faculty of Medicine, Izmir, Turkey
2Department of Pediatric Immunology, Dr Behcet Uz Children’s Hospital, Izmir, Turkey
3Department of Pediatrics, Tepecik Training Hospital, Izmir, Turkey
Ataxia-telangiectasia (AT) is a rare, neurodegenerative genetic disorder with progressive neurological abnormalities, telangiectasias and immunodeficiency. Delay in diagnosis/misdiagnosis is probable due to its wide clinical heterogeneity in infancy. Patients have sinopulmonary infections and decreased immunoglobulins (Ig). 10% of patients with normal/elevated IgM are often misdiagnosed as hyperIgM syndrome. Definitive diagnosis is made by showing disease causing mutation. on ATM (ataxia telangiectasia mutated) gene that is known to play a role in class switch recombination. We evaluated twenty patients (13,8+4,1 years) retrospectively to search out whether there was any other findings for the probable diagnosis criteria which were defined by European Society for Immunodeficiencies. Twelve patients were born to consanguineous parents. Two mothers had malign breast neoplasm. Patients often (80%) had infections. and had low IgG (45%), IgA (65%), elevated IgM (60%), low CD3+CD4+ T lymphocyte frequency (45%). The mean initial AFP concentration was 191,9+140,1 ng/mL. High AFP levels showed no statistically relationship with the raised IgM values.
High IgM concentrations may be the only presenting feature in AT and may be misdiagnosed as hyper-IgM syndrome. In case of missense mutation or complete absence of ATM gene, defective repair of double-stranded DNA breaks caused by V(D)J recombination of immunoglobulin may yield B lymphocyte maturation deficiency and high IgM concentrations. We need more investigation about CSR in these patients, however, if we take the high frequency of cases with high IgM (60%) into consideration, we can speculate that high IgM concentration may accepted as a probable diagnosis criteria in near future.
ESID-0124 Purine Nucleoside Phosphorylase (PNP) Deficiency: Children's Hospital of Brescia Experience
R. Baffelli 1, M. Zucchi1, L. Notarangelo2, F. Bolda1, A. Beghin1, A. Caruso3, F. Porta2, A. Lanfranchi1
1Stem Cell Lab - Children's Hospital, Spedali Civili Brescia, Brescia, Italy
2Oncohematology and BMT Unit - Children's Hospital, Spedali Civili Brescia, Brescia, Italy
3Microbiology and Virology Unit - Children's Hospital, Spedali Civili Brescia, Brescia, Italy
Purine nucleoside phosphorylase (PNP) deficiency is a rare autosomal recessive metabolic disorder which represents about 4% of all SCID.
Affected individuals have a history of recurrent infections; autoimmune disorders are also frequent and include autoimmune hemolytic anemia, idiopathic thrombocytopenia, autoimmune neutropenia, lupus and central nervous vasculitis.
Variation in clinical presentation may cause difficulties in initial classification.
8 patients arrived at our center with suspected symptoms of PNP deficiency, 7 from another center.
Enzymatic and molecular diagnosis were performed in our lab.
For 2 of these, enzymatic diagnosis wasn't possible because they were transfused.
5 patients showed borderlines values that suggest a possible transfusion not reported, only 1 patient (M.L.) showed zero enzymatic activity.
To confirm or exclude PNP deficiency, molecular diagnosis has been performed for all patients.
M.L. was confirmed as PNP deficit, with a homozygous mutation called E89K due to a single base exchange, 3 patients showed polymorphism but no disease causing mutation and 4 not showed mutation.
4 patients continued to be followed at our center, M.L. underwent allogeneic bone marrow transplantation from unrelated donor 4 months after diagnosis. After 7 years the patient is well, engraftment is complete and the enzymatic values of PNP is in normal range.
Three were diagnosed after: B.G. has been identified as SCID T-B+NK- gamma chain, M.E. bone marrow aplasia and T.H. primary immunodeficiency. All three have undergone bone marrow transplantation (table). PNP-deficiency is extremely rare disease, less than 100 patients have been diagnosed and the symptoms are similar to other immunodeficiencies.
| Patient | Enzymatic Values (healthy >300U/gHb) | Mutation | Age at BMT (months) | Type of BMT | Follow up (months) | Last Engratment |
| B.G. | 213.5 | NO | 8 | HLA-identical siblings | 36 |
CD3+: 100% CD15+: 0% |
| C.G. | Not performed | NO | patient returned to his center after exclusion of PNP deficiency | |||
| F.A. | 284 |
Polymorphism c.60C>T |
patient returned to his center after exclusion of PNP deficiency | |||
| M.E | Not performed |
Polymorphism c.151C>A |
103 | Matched unrelated donor | 39 |
PBL: 100% PMN: 100% |
| S.D. | 255 | NO | ||||
| T.H. | 269 | NO | 32 | Haploidentical | Deceased after 1 month |
CD3+: 0% CD15+: 0% |
| T.G. | 310.5 |
Polymorphism c.60C>T |
patient returned to his center after exclusion of PNP deficiency | |||
| M.L. | 0 |
Polymorphisms c.151C>A c.171C>T Mutation: c.265G>A p.E89K |
26 | Matched unrelated donor | 84 |
PBL: 100% PMN: 100% |
ESID-0113 Analysis of Mutation in Adenosine Deaminase Gene: Description of 5 New Mutation Cases and Review of a Single Centre
R. Baffelli 1, L. Notarangelo2, M. Zucchi1, F. Bolda1, A. Beghin1, A. Caruso3, F. Porta2, A. Lanfranchi1
1Stem Cell Lab - Children's Hospital, Spedali Civili Brescia, Brescia, Italy
2Oncohematology and BMT Unit, Spedali Civili Brescia, Brescia, Italy
3Microbiology and Virology Unit - Children's Hospital, Spedali Civili Brescia, Brescia, Italy
ADA deficiency is a rare condition and majority of patients presenting SCID phenotype; 15-20% of patient has phenotype 'delayed' or 'late onset'. Mutations are classified in 5 classes (0, I, II, III e IV) based on residual enzyme activity. More than 60 mutations were described and affecting entire ADA gene.
After screening enzymatic test, molecular diagnosis was performed by sequencing of 12 exons of ADA gene.
Ethnic, genotypic and phenotypic characteristics are described in table. New mutations are relative to patients 1,14,17and19. Patient 1 was heteroallelic for mutation [c.275delC] in exon3 with effect F61fsX87 and for mutation [c.1104G>T] in exon11 with effect E337X due of replacement in transcript of a glutamic acid Glu (GAA) at codon 337 with a stop codon (TAA). Patient 14 was homozygous for a one base deletion [c.91delT] with effect G31fsX41. This mutation is compatible with total absence of activity in?vitro. Patient 17 is homozygous for mutation [c.976C>A] in exon10 with effect T294K. Residual enzyme activity in E.coli is 0.001% of wild type (group I). Patient 19 was heteroallelic for mutation previously described (H15D) and for mutation [c.1065delA] in exon10; this mutation leads a stop codon (R324fsX325). In patients with known mutations, 5 are homozygous for R211H, 4 of these are gypsy (only 2 related). D123fsX132 and R156C mutations were detected in 3 unrelated patients from single region (Apulia).
Our study confirms heterogeneity of ADA mutations. R211H mutation is the most frequent but its presence in unrelated gipsy family might suggest a founder effect.
| Pt | Mutation | Geographical origin of patients | Phenotype | Age at diagnosis (months) | ||
| 1 | F |
p.F61fsX87 p.E337X |
c. 275deIC c.1104G>T |
Tuscany Italy |
SCID | 4.4 |
| 2 | F |
IVS3 G+1>A p.R16C |
c.561C>T |
Apuglia Itay |
SCID | 6.8 |
| 3 | M |
p.H15D p.R235Q |
c. 138C>G c. 704G>A |
Lombardy Italy |
SCID | 3.8 |
| 4 | F |
p.R101Q p.A329V |
c. 397G>?A | Ukraine | SCID | 10.6 |
| 5 | F |
p.H15D p.H15D |
c. 138C>G c. 138C>G |
Apulia Italy |
SCID | 1.2 |
| 6 | M |
p.R211H p.R211H |
c. 727G>A c. 727G>A |
Gipsy | SCID | 5 |
| 7 | F |
p.G31fsX41 p.G31fsX41 |
c. 91deIT c. 91deIT |
Tunisia | SCID | 2 |
| 8 | M |
p.R211H p.R211H |
c. 727G>A c. 727G>A |
Gipsy | SCID | 2 |
| 9 | F |
p.H15D p.V129M |
c. 138C>G c. 480G>A |
Republic of Macedonia | SCID | 5 |
| 10 | M |
p.R211H p.R211H |
c. 727G>A c. 727G>A |
Gipsy | SCID | 3.7 |
| 11 | F |
p.S291L p.E319fsX321 |
c. 967C>T c. 1051_1055deIAAGAG |
Calabria Italy |
SCID | 0 |
| 12 | F |
p.G216R p.G216R |
c. 741G>A c. 741G>A |
Bulgaria | SCID | 6 |
| 13 | F |
p.H15D p.S333fsX338 |
c.138C>G c. 1091_1092deITA |
Bulgaria/Campania Itaty | SCID | 9 |
| 14 | M |
p.H15D p.S333fsX338 |
c. 138C>G c. 1091_1092deITA |
Lombardy/Calabria Italy | SCID | 1.7 |
| 15 | M |
p.R211H p.R211H |
c. 727G>A c. 727G>A |
Gipsy | SCID | 3.2 |
| 16 | F |
p.R211H p.R211H |
c. 727G>A c. 727G>A |
Gipsy | SCID | 3.7 |
| 17 | F |
p.R156C p.D123fsX132 |
c. 561C>T c. 459deIG |
Apuglia Italy |
SCID | 3.3 |
| 18 | M |
p.R156C p.E319fsX321 |
c. 561C>T c. 1051_1055deIAAGAG |
Tuscany Italy |
SCID | 2.1 |
| 19 | M |
p.V129M ? |
c.480G>A |
Campania Italy |
delayed onset | 13 |
| 20 | F |
p.D123fsX132 p.D123fsX132 |
c. 462deIG c. 462deIG |
Apulia Italy |
SCID | 3 |
| 21 | M |
p.T294K p.T294K |
c. 976C>A c. 976C>A |
Senegal Africa |
SCID | 1 |
| 22 | F |
p.H15D p.R324fsX325 |
c. 138C>G c. 1065deIA |
Lombardy Italy |
SCID | 4 |
| 23 | M |
p.V129M p.S291L |
c. 480G>A c. 967C>T |
Lazio Italy |
SCID | 24 |
| 24 | F |
p.V177M p.E319fsX321 |
c. 624G>A c. 1051_1055deIAAGAG |
Lazio Italy |
SCID | 3 |
| 25 | M |
p.V177M p.E319fsX321 |
c. 624G>A c. 1051_1055deIAAGAG |
Lazio Italy |
SCID | 36 |
| 26 | M |
p.R235W p.R235W |
c. 798c>t c. 798c>t |
Serbia | SCID | 3 |
| 27 | M |
p.R211H p.R211H |
c. 727G>A c. 727G>A |
Gipsy | SCID | |
ESID-0547 Primary Immunodeficiency and Infection in Intensive Care Unit
A. Baldolli 1, A. Seguin2, G. Maigne1, N. Martin Silva1, B. Bienvenu1
1Internal Medicine, University Hospital, Caen, France
2Intensive Care, University Hospital, Caen, France
Introduction :
Diagnosis of primary immunodeficiency (PID) in adults is rare and may be supported by the 6 ESID signs for adult. No established strategy for PID detection in medical intensive care unit.
Objective :
To evaluate the occurrence of PID in ICU patients.
Methods :
We conducted a retrospective study from 01/2011 to 12/2012 in the ICU of our University Hospital, to estimate the prevalence of PID. Patients aged 18 to 65, hospitalized for a severe or opportunistic infection or idiopathic hemophagocytic syndrome, without any known warning sign or predisposing factor, were included. HIV serology, complement dosage, immunoglobulin quantification, complete blood count and body CT-scan were performed. A consultation in the Internal Medicine department was scheduled 4 to 8 weeks after ICU discharge.
Results :
1596 patients were hospitalized and 14 were included. Three patients were diagnosed for PID, all women. One 19-year-old patient had a complete IgA deficiency and suffered from a meningococcal serogroup B meningitidis. Two IgG2 deficiency (36 and 65 years of age) who suffered from severe pneumococcal pneumonia were identifed. Five SID were detected (2 HIV with pneumocytosis and CMV pneumonia, one T-cell lymphoma revealed by an hemophagocytic syndrome, 1 myelodysplastic syndrome with disseminated tuberculosis and 1 lung cancer revealed by a severe sepsis). One patient was lost of follow up and no immunodeficiency was found in 3 patients.
Conclusion :
Severe or opportunistic infections may reveal PID in ICU despite of the absence of ESID warning sign. Our strategy may prevent recurrences.
ESID-0727 UKPID Registry: 2014 Update
C. Bangs 1, J.?D.?M. Edgar2, D. Guzman3, M. Buckland4
1Immunology, Manchester Royal Infirmary, Manchester, United Kingdom
2Immunology, The Royal Hospitals, Belfast, United Kingdom
3Immunology, University College London, London, United Kingdom
4Immunology, Royal London Hospital, London, United Kingdom
Background
Since its inception in 2009, participation in the UKPID Registry has steadily increased to the current total of over 90% of UK centres. Contribution to the registry is a requirement for UK Primary Immunodeficiency Network (UKPIN) centre accreditation.
The new ESID design will be implemented in the UK, improving data integrity and reliability, however the UK will continue collecting data on HAE and secondary hypogammaglobulinemia.
Methods
Three centres transfer data from their own database, two enter their own data and the remaining centres use the facility to have data entered by the two UKPIN funded study co-ordinators. UKPIN also funds IT support.
Results
Data from over 3100 patients with over 75 different diagnoses has been entered from 31 centres. The poster shows a selection of data including, breakdown by diagnosis, the incidence of bronchiectasis and also trends in home therapy/hospital and subcutaneous/IV immunoglobulin replacement therapy.
Discussion
In addition to the on-going task of ensuring data is up to date and of high integrity, the challenge is to promote use of data, so that centres feel a return for their efforts in consenting patients and data contribution. This means ensuring centres have access to their own data in a user friendly format and also encouraging interested parties to join together to design studies to use the data to answer clinical questions. A welcome step in this direction is the recent funding by the NIHR for rare diseases into detailed phenotyping of CVID and Complement deficiencies using UKPID registry data.
ESID-0791 Primary Immunodeficiencies in Romania – Epidemiological Data
M. Serban 1, M. Cucuruz1, A. Cochino2, B. Capalna3, M. Dumitru4, M. Marc5, Z. Ellenes6, A. Rugina7, S. Iurian8, A. Apostol9, I. Gherghina10, N. Iagaru10, M. Bataneant1
1IIIrd Pediatric Clinic, University of Medicine and Pharmacy "Victor Babes", Timisoara, Romania
2Institute for Mother and Child Care, University of medicine and Pharmacy “Carol Davila”, Bucharest, Romania
3Pediatric Allergology and Immunology, County Hospital, Targu-Mures, Romania
4Allergology and Immunology, County Hospital, Targu-Mures, Romania
5IInd Pediatric Clinic, University of Medicine and Pharmacy, Cluj, Romania
6Ambulatory, Children’s Hospital “Gavril Curteanu”, Oradea, Romania
7IInd Pediatric Clinic, University of Medicine and Pharmacy, Iasi, Romania
8Ist Pediatric Clinic, University of Medicine and Pharmacy, Sibiu, Romania
9Oncohematology, Clinical Emergency County Hospital, Constanta, Romania
10Institute for Mother and Child Care, University of Medicine and Pharmacy “Carol Davila”, Bucharest, Romania
Introduction. Primary Immunodeficiencies (PIDs) are rare disorders but more and more cases are diagnosed. Thus a national registry is important for establishing the dimension of this pathology, the investigational and therapeutical needs in order to improve the diagnosis, treatment and the quality of life for PIDs patients.
Aim of the study. To establish the number of PIDs patients, the type of disease, the level of the diagnosis, the treatment modalities.
Methods. We collected data from 9 pediatric and adult centers, based on a questionnaire including the age, type of the disease, sustaining diagnosis data, genetic diagnosis, treatment and evolution.
Results. 527 patients were reported, that represent a prevalence of 2,63/100,000 individuals. 24% are adults and 76% childrens. The spectrum of the PID was: antibody deficiencies: 58,25%, combined immunodeficiencies: 2,84%, well defined immunodeficiencies: 7,96%, immune dysregulation disorders: 1,7%, phagocyte defects: 6,45%, innate immunodeficiencies: 0,75%, autoinflammatory disorders: 5,88% and complement deficiencies: 16,12%. 30% of the patients have a definitive diagnosis (52 cases genetically confirmed and 85 cases proved by enzyme determination). 98 patients receive immunoglobulin substitution, 18 patients are treated with granulocyte-colony stimulating factor, 5 patients underwent bone marrow transplantation, 1 patient gene therapy and 1 patient thymus transplantation. The mortality rate was 7,21%.
Conclusions. In Romania there were an insufficient recognition/registration of PIDs patients (only 5,5% of the patients are identified), low level of the diagnosis-possible/probable in majority of the cases, absence of skills and infrastructure for genetic diagnosis and an insufficient/inadequate treatment.
ESID-0599 A Multicentre Approach to Document Bronchial Pathology in Computed Tomography of the Chest: Data from the Chest CT in Antibody Deficiency Group
K. Schütz1, B. Grimbacher2, J. Haddock3, I. Hartmann4, G. Driessen5, I. Quinti6, G. Serra7, V. Thon8, J. Litzman8, A. Jones9, P. Aurora9, P. Kelleher10, M. Loebinger11, D. Kumaratne12, A. Condliffe13, N. Galal14, R. Stirling15, A. Plebani16, G. Spadaro17, L.I. Gonzalez-Granado18, H. Chapel19, C.M. Farber †20, S. Hackett21, N. Screaton22, B. Gathmann23, I. Meyts24, E. de Vries25, K. Warnatz26, J. Hurst27, U. Baumann 1
1Paediatric Immunology Unit Dept. of Paediatric Pulmonology Allergology and Neonatology, Hanover Medical School, Hanover, Germany
2Clinical Immunology, Royal Free Hospital, London, United Kingdom
3Radiology, Royal Free Hospital, London, United Kingdom
4Department of Radiology, Erasmus MC Sophia Children's Hospital, Rotterdam, Netherlands
5Paediatric Immunology, Erasmus MC Sophia Children's Hospital, Rotterdam, Netherlands
6Department of Molecular Medicine, Sapienza University of Rome, Rome, Italy
7Department of Radiology, Università degli Studi di Roma La Sapienza, Rome, Italy
8Dept. of Clinical Immunology and Allergy Faculty of Medicine, St Anne´s University Hospital, Brno, Czech Republic
9Paediatric Immunology, Great Ormond Street Hospital, London, United Kingdom
10Immunology, Royal Brompton Hospital, London, United Kingdom
11Pulmonology, Royal Brompton Hospital, London, United Kingdom
12Immunology, Addenbrooke's Hospital, Cambridge, United Kingdom
13University of Cambridge Department of Respiratory Medicine, Addenbrooke's Hospital, Cambridge, United Kingdom
14Department of Pediatrics, Cairo University, Cairo, Egypt
15Immunology and Respiratory Medicine Monash University, Monash University The Alfred Hospital Melbourne, Melbourne, Australia
16Department of Paediatrics Clinic Department of Clinical and Experimental Sciences, University of Brescia Spedali Civili di Brescia, Brescia, Italy
17Department of Paediatrics Clinic, University of Naples Federico II Naples, Naples, Italy
18Paediatrics, Hospital 12 Octubre, Madrid, Spain
19Immunology, John Radcliffe Hospital University of Oxford, Oxford, United Kingdom
20Immunology, Cliniques Universitaires de Bruxelles Hôpital Erasme, Brussels, Belgium
21Radiology, Heartlands Hospital, Birmingham, United Kingdom
22Radiology, Papworth Hospital, Cambridge, United Kingdom
23For the ESID Registry Working Party Center for Chronic Immunodeficiency (CCI) Clinical Research Unit, University Medical Centre and University of Freiburg, Freiburg, Germany
24Paediatric Immunology and Pulmonology, University Hospitals, Leuven, Belgium
25Immunology, Jeroen Bosch Hospital, 's-Hertogenbosch, Netherlands
26Centre for Chronic Immunodeficiency, University Medical Centre and University of Freiburg, Freiburg, Germany
27UCL Respiratory Medicine, University College London, London, United Kingdom
Background: Chest CT studies in patients with primary antibody deficiency syndromes suggest a high prevalence of bronchial pathology. However, multicentre studies to assess the variety of bronchial pathology are still missing. One of the underlying reasons is the lack of a common syllabus and platform to jointly document chest CT findings.
Objectives: We aimed to establish an international platform for assessment of bronchial pathology as assessed by chest CT and to describe the range of bronchial pathologies in patients with antibody deficiency.
Methods: The ESID Online Database was used for collecting data for this study. 20 immunodeficiency centres of 9 countries participated. Bronchial pathology was described by a predefined list of potential findings.
Results: Data of n = 282 patients with primary antibody deficiency syndrome were collected. Patients with common variable immunodeficiency (CVID) comprised the largest subgroup (n = 232). Of CVID patients, 80% had evidence of bronchial pathology, including bronchiectasis at 60%, bronchial wall thickening 44% and mucus plugging 27%. In 44% of young CVID patients (
Conclusion: This study demonstrates a high prevalence of structural lung disease in patients with CVID even in patients with normal spirometry. This suggests a possible place for routine CT scans in this population.
ESID-0511 Analysis of Early and Late Complications After Bone Marrow Transplantation In Children Affected by SCID: Role of Conditioning Regimens
S. Cavagnini 1, S. Guarisco1, I. Bosio1, R. Baffelli2, A. Beghin2, F. Bolda2, A. Caruso3, A. Lanfranchi4, F. Porta4
1Children's Hospital, Oncohematology and BMT Unit, Brescia, Italy
2Children's Hospital, Lab. Cell. Stam, Brescia, Italy
3Children's Hospital, Microbiology and Virology Unit, Brescia, Italy
4Children's Hospital, Lab. Cell. Stam., Brescia, Italy
This retrospective study shows acute and chronic complications and overall mortality in SCID patients receiving BMT and treated with different conditioning regimens.Have been enrolled 96 patients affected by classical SCID and affected by ADA negative SCID treated from 1991 till the.Mortality at December 2013 has been evacuate together with incidence of acute and chronic complications.The case have been analyzed by T Student test,Chi squared by the SPSS software;p<0,05 was considered statistical significant.Patients underwent at least 1 BMT,with no chronic GVHD,divided on the base of received conditioning (29 not conditioned,40 reduced intensity conditioning [RIC],43 mieloablative conditioning [MAC];the groups were similar.We show,not considering children in clinical critical state at transplant,at diagnosis,a statistical significant difference between mortality and type of conditioning ,both in RIC (10 vs 0,p = 0,01), and in MAC(11 vs 0,p=0,015) in comparison with non conditioned.A difference has been noted in the incidence of side acute effects during conditioning in MAC versus RIC(35 vs 20,p=0,001),nevertheless without a difference in mortality related to adverse events among late complications ,we observed a difference between thyroid disfunctions observed in MAC compared with non conditioned(7 vs 0, p=0,025),while no difference among the other groups;similarly no differences in the incidence of growth in all groups of patients.The results of our survey demonstrate an increased acute and chronic morbility in patients treated by MAC.Conditioning regimes do not affect mortality in SCID patients;we hypothised that this would depend on clinical conditions at presentation before BMT that drive the choice of a donor and the type of conditioning regimen.
ESID-0502 Chronic Granulomatous Disease
A. Beghin 1, A. Soresina2, R. Baffelli1, M. Zucchi1, E. Soncini3, I. Bosio3, F. Porta3, A. Caruso4, A. Lanfranchi5
1Children's Hospital, Lab. Cell. Stam., Brescia, Italy
2Clinical and Experimental ScienceSpedali Civili di Brescia, Pediatrics Clinic and Laboratory for Molecular Medicine "A.Nocivelli", Brescia, Italy
3Children's Hospital, Oncohematology and BMT Unit, Brescia, Italy
4Children's Hospital, Microbiology and Virology Unit, Brescia, Italy
5Children's Hospital, Lab. Cell. Stam, Brescia, ItalyChronic granulomatous disease (CGD) is a rare inherited disorder of the innate immune system caused by a defect in NADPH oxidase, leaving the granulocytes unable to kill invading microorganisms. In our center we collected 11 patients affected by CGD: 8 males and 3 females. 7 patients (all males) underwent transplantation: 4 HLA-Matched Unrelated SCT, 2 HLA-Matched Related SCT and 1 Haploidentical SCT,the mean age at HSCT was 36 months (range 7 m to 10 years),3 of whom are dead.The mean number of CD34+ infused was 9.2x10^6/Kg (the data of transplantation are in table 1),after 30 days there were a complete engraftment of donor for 4 patients and a detection of mixed chimerism for 3 patients. Mutation screening for AR-CGD was performed in 4 patients displaying impaired NADPH oxidase activity: 3 female and 1 male of whom the X linked CGD pattern of inheritance was excluded.We analyzed NCF1, NCF2 and CYBA genes. 1 patients was a compound for the Tyr26HisfsX26 and Trp193X. 1 has Tyr26HisfsX26 and a snp Arg90His in heterozigousis, while 2 patients have snp Arg90His and Gly99Ser in hez.Our study confirm only one patient with mutation in NCF1 gene. These preliminary data show a characteristic cluster of snp in AR-CGD patients. Particularly all share the same Arg90His snp in heterozigousis, that Olsonn et?al dimostrated significantly reduced reactive oxygen species production. Moreover 2 patients have no mutation but share the same snp in CYBA and NCF1 gene. These preliminary data show a characteristic cluster of snp in AR-CGD patients.
| PATIENT | D.B | AGE | SCT | HPC | CD34X10^6/Kg | Engrafment +30 days | FOLLO WUP |
| C.M | 12/09/97 | 17M | HLA-Matched Unrelated | HPC-M | 4,27 | PBL=100% PMN=100% | ALIVE |
| A.Y. | 01/08/06 | 7M | HLA-Matched Related | HPC-M | 8,30 | PMN=87,70% | ALIVE |
| AA | 20/08/05 | 22M | Haploidentical | HPC-M | 9,20 | PBL=100% PMN=100% | DEAD |
| C.A. | 12/05/97 | 36M | HLA-Matched Unrelated | HPC-M | 1,83 | PBL=100% | DEAD |
| I.I. | 09/06/08 | 36M | HLA-Matched Unrelated | HPC-M | 3,97 | PBL=91,87 PMN=97,63 | ALIVE |
| V.A.G. | 27/06/02 | 10Y | Matched Unrelated | HPC-A | 33,48 | PBL=91,2% PMN=96% | DEAD |
| L.G.L. | 17/06/12 | 19M | HLA-Matched Related | HPC-M | 11,24 | PBL=100% PMN=100% | ALIVE |
ESID-0499 TCR Alpha/Beta Depletion in Children Affected by Osteopetrosis
A. Lanfranchi 1, S. Cavagnini2, F. Bolda1, R. Baffelli1, F. Schumacher2, A. Caruso3, F. Porta2
1Children's Hospital, Lab. Cell. Stam, Brescia, Italy
2Children's Hospital, Oncohematology and BMT Unit, Brescia, Italy
3Children's Hospital, Microbiology and Virology Unit, Brescia, Italy
We presented a retrospective study of 20 children affected by osteopetrosis;the disease is diagnosed at a mean age of 4 months.We found 9 children mutated in TCIRG,2 in CLCN7,3 in OSTM1 and one in RANKL and 6 children have unknown genetic defects.14 children underwent BMT,8 of whom are alive.4 children underwent 2 BMT. 6 patient didn't receive a BMT.8 children developed forms of acute GVHD. Disease free survival was 80% for HLA matched, 33% for haploidentical HLA matched donor and 60% for MUD.To improve engraftment rate and decrease intensity and severity of GvHD that in our previous experience didn't exceeded 60%, with 4 patients who required a second SCT.We applied depletion of TCR a/?ß +/CD19+ cells (CliniMacs system depletion ) as a novel SCT approach in osteopetrosis in 3 cases.We planned to infuse a high number of both CD34+ and TCR ?/?d+ cells together with a controlled number of TCRa/?ß+ cells (not exceeding 25x10^6 cells/Kg). The data on the infused cells are reported in Table?1.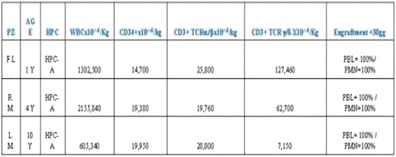
The mean number of cells was: 18x10^6 CD34+/Kg, 66x10^6TCR ?/?d+ cells /Kg and and 21.85 x10^6 TCR a/?ß /Kg obtained from the 'non target' fraction. In all 3 cases engraftment occurred in 100% donor by day 12 with no evidence of acute GvHD.This seems a promising procedure to enhance engraftment due to an high CD34+ inoculum combined with a good number of TCR ?/?d+ cells ?and a controlled number of TCR a/?ß+ cells that in our previous experience didn't provoke GVHD in the MUD setting.
ESID-0262 A 1,100 Year-Old Founder Effect Mutation in IL12B Gene is Responsible for Mendelian Susceptibility to Mycobacterial Disease in Tunisian Patients
M. Ben-Ali 1, I. Ben-Mustapha1, N. Mekki1, E. Patin2, C. Harmant2, J. Bouguila3, H. Elloumi-Zghal1, A. Harbi4, M. Bejaoui5, L. Boughammoura3, J. Chemli4, M.R. Barbouche1
1Laboratory of Transmission Control and Immunobiology of Infections, Pasteur Institute of Tunis, Tunis, Tunisia
2Unit of Human Evolutionary Genetics, Pasteur Institute, Paris, France
3Department of Pediatrics, Farhat Hached Hospital, Sousse, Tunisia
4Department of Pediatrics, Sahloul Hospital, Sousse, Tunisia
5Department of Pediatrics, Bone Marrow Transplantation Center, Tunis, Tunisia
Mendelian susceptibility to mycobacterial disease due to complete IL-12p40 deficiency is a rare syndrome predisposing to infections mainly caused by weakly virulent mycobacteria and poorly virulent Salmonella strains. IL-12p40 deficiency is an autosomal disease; all mutations are loss-of-function with an undetectable IL12p40 secretion. Interestingly, all previously reported patients originated from Middle Eastern and North African region.
In the present study, we report six Tunisian patients from 4 kindreds with complete IL12-p40 deficiency due to the same mutation in IL12B gene: 297del8. All of them originate from the same small town in Tunisia (Akouda, Central Tunisia). We have first shown, using segregation analysis, that all patients share a 5.8-MB common homozygous haplotype. We then estimated the date of the founder event responsible for this mutation to approximately 1100 years ago. Interestingly, two additional Tunisian patients living in France have also been shown to bear the same mutation.
Among the only 8 other mutant alleles of the IL12B gene which have been previously reported, three arise from a founder effect in specific populations. Founder effect has also been reported in other PIDs in Tunisia including the Major Histocompatibility Complex class II, combined immunodeficiency, Omenn syndrome as well as Leukocyte Adhesion Deficiency type I. In the Tunisian population, where endogamy is high, founder effects represent 42% of all genetic disorders with identified molecular defect. One major clinical implication of the existence of such mutations is the development of a preventive approach by genetic counseling and prenatal or preimplantation diagnosis.
ESID-0677 Data from the Slovenian National PID Registry
S. Blazina 1, G. Markelj1, M. Debeljak2, A. Jeverica Koren1, N. Toplak1, A. Ihan3, T. Avcin1
1Dpt. of Allerg. Rheum. and Clin. Immunol., University Children's Hospital, Ljubljana, Slovenia
2Dpt. of Laborat. Diagnost., University Children's Hospital, Ljubljana, Slovenia
3Institute of Microbiology and Immunology, Faculty of Medicine University of Ljubljana, Ljubljana, Slovenia
Progress in the field of primary immunodeficiencies (PIDs) is reflected in national PID registries. Data from Slovenian PID registry were analyzed. Patients’ data were collected retrospectively before 2007 and prospectively afterward. Patients were classified according to international classification and updated regularly.
Data of 202 patients with 46 different PIDs were analyzed. Interestingly, complement deficiencies are the most common, accounting for 23% of all entries. Second most common are antibody deficiencies with 19%, followed by well-defined syndromes (15%), immune dysregulation (15%), neutrophil defects (12%), combined deficiencies (8%), autoinflammatory syndromes (6%) and deficiencies of innate immunity (2%).
Prevalence of diagnosed PIDs in Slovenia has changed in the last 5 years; less complement deficiencies and more antibody deficiencies were diagnosed in comparison to previous decades. The number of new PID cases has been gradually increasing, a more prominent increase is noted in the last 10 years. The prevalence increased most for combined immunodeficiencies, CVID and autoinflammatory syndromes. The spectrum of PID entities has also widened in the last decade. Three patients with SCID were diagnosed and successfully treated in the last three years.
High prevalence of complement deficiencies reflects early implementation of good complement diagnostic facilities and awareness among infectologists. This group of patients was prospectively collected from 1987. Combined immunodeficiencies, CVID and autoinflammatory syndromes were all probably underdiagnosed before due to lack of awareness among physicians. Distribution of PID groups is more consistent with ESID registry in the last five years.
ESID-0377 Recurrent Infections due to Mycobacteria, Viruses and Pyogenic Bacteria, Plus TLR Defect: First Brazilian Case of MonoMAC and New GATA2 Mutation?
E.B. de Oliveira Junior 1, M.B. de Carvalho1, L. Pedroza2, R. A. Campos3, J. Bustamante4, J.L. Casanova5, A. Condino Neto1
1Immunology, Institute of Biomedical Sciences University of Sao Paulo, São Paulo, Brazil
2Immunogenetics Department, School of Medicine Universidad San Francisco de Quito, Quito, Ecuador
3Prof. Edgard Santos University Hopsital, Federal University of Bahia, Salvador, Brazil
4Center for the Study of Primary Immunodeficiencies, Necker-Enfants Malades Hospital, Paris, France
5St. Giles Laboratory of Human Genetics of Infectious Diseases Rockefeller Branch, The Rockefeller University, New York, USA
Introduction: The GATA2 is a transcription factor containing "zinc finger" domain and is considered a central regulator of "Hematopoietic Stem and Progenitor Cells". Recently, mutations in GATA2 gene have been associated with different clinical phenotypes, such as MonoMAC.
Methods: Whole Exome Sequencing was used to investigate a patient with the clinical features comented above. We selected a GATA2 variation as a candidate gene.
Case Report: Caucasian, female, from Brazil, 47 year, non-consanguineous family. At 21 years old, she presented cutaneous infections with abscess, and purulent secretion due to Staphylococcus aureus. Also, she developed respiratory tract infections with sinusitis. At 33 years old she presented a persistent febrile episode and diagnosis of tuberculous lymphadenitis. Two years later she presented adverse effect to yellow fever vaccine. At age 38 years she presented warts on her hands indicating viral infection. In the first episode of infection by M.?kansasii. Recently she presents several intermittent periods of anemia and lymphopenia without apparent cause; myelogram was normal.
Results: TLR function measured by TNF-alpha release after stimulation with agonists for TLR1 through TLR9 and CD62L shedding after the same stimuli was below of the limit of detection. Recently, using WES we found a heterozygous variation (p.M388T; c.1163T>C) in GATA2 gene in the Zinc Finder Domain 2. Additional experiments are being conducted.
Conclusion: We believe that affected gene leads to clinical phenotypes found. Is the first time an association between GATA2 mutation and TLRs defects.
ESID-0710 Thymic Tissue Regeneration Using Natural Collagen Scaffolds
I. Bortolomai 1, M. Sandri2, T. Di Tomaso1, M. Catucci3, A. Lombardo1, L. Sergi Sergi1, S. Panseri2, L. Naldini1, G.A. Hollander3, A. Tampieri2, A. Villa1, M. Bosticardo1
1TIGET, Ospedale San Raffaele, Milano, Italy
2ISTEC, National Research Council, Faenza, Italy
3Department of Biomedicine, University of Basel, Basel, Switzerland
Thymus transplantation has great clinical potential, but the shortage of transplant donors limits the progress of this therapy. Creation of a bioengineered thymus, where the cellular component is autologous, will overcome two major obstacles in transplantation: the lack of organs and the toxicity arising from lifelong immunosuppression. To this aim, we would need to reprogram adult TECs in order to allow the expansion of thymic epithelial progenitor-like cells (pTECs), retaining their capability to differentiate into mature TECs and potentially form a functional thymus ?in?vivo. In order to obtain bipotent TECs precursors that could differentiate into both cortical and medullary TECs, we transduced freshly isolated adult TECs with a lentiviral vector (LV) that allows for ectopic expression of Oct4. Ectopic Oct4 expression is sufficient to promote the de-differentiation of mature TECs into pTECs, inducing the expression of progenitor markers (Thy1.2, Sca1 and CD44) and decreasing the expression of maturation markers (MHCII). To improve thymic organogenesis and restore thymopoietic functions both in?vitro and ?in?vivo, we exploited 3D collagen type I scaffolds, mimicking the thymic structure. pTECs grown in 3D scaffolds displayed a high proliferation rate. To induce pTECs maturation and thymic regeneration, we are now evaluating a LV-based strategy for the inducible expression of Oct4 that allows us to switch off Oct4 expression and control pTECs differentiation. We will then perform morphological and phenotypical characterization of TECs cultured in scaffolds and evaluate their ability in restoring thymic function both in?vitro and ?in?vivo.
ESID-0153 Education Programs for Patients with Primary Immunodeficiency in France: National Survey
C. Brito de Azevedo Amaral 1, V. Millet2, A. Fischer1, N. Mahlaoui1, C. CEREDIH network1
1CEREDIH, Necker-Enfants Malades Hospital, Paris, France
2CFTDE, Necker-Enfants Malades Hospital, Paris, France
Introduction: Education of the patient, according to the WHO, aims to help the patients acquire or maintain the competencies they will need to better manage their lives with a chronic disease. In France, Health Authorities validated patients’ education programs are ascribed in Public Health Law since 2009. In this context, the French National Reference Center of PIDs (CEREDIH) has conducted a nation-wide survey aiming at establishing the state of the art in the field.
Method: A half-open questionnaire sent to 68 pediatric and adult services which are a part of the CEREDIH network in order to take a census of what is being done for PID patients.
Results: 25/32 medical units (78%) have educational activities for adult patients, children, parents and/or caregivers/family. 7/?32 (22%) do not have established educational activities but would like to develop them. For 22/25 (88%), education is offered all along the treatment (as a part of the care, or at specific moments). The main goal of these educational activities is the patients' empowerment. The group that's being engaged is multidisciplinary made primarily of doctors and nurses. In order to carry out these activities, 19/25 (76%) of the units have partnerships with the patients’ national association (IRIS) and/or with pharmaceutical sponsor.
Conclusion: For participants in the survey, education of patients is a response to their needs and promotes their compliance and autonomy. However, in order to develop this, the groups face different obstacles: lack of time, fundings and need for specialized and trained human resources.
ESID-0576 Bioinformatics Analysis of Exome Sequencing Data: Challenges and Solutions
A. Bulashevska 1, B.?A. Grüning2, R. Backofen2, B. Grimbacher1
1Center for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany
2Bioinformatics Group Department of Computer Science, University of Freiburg, Freiburg, Germany
Whole exome sequencing (WES) has become pivotal methodology for effective detection of pathogenic variations. To address bioinformatics challenges of WES data we based our workflow on the best practices quide of GATK (Genome Analysis Toolkit) which includes the following main sections:
- Data pre-processing: from raw sequence reads (FASTQ files) to analysis-ready reads (BAM files in which the reads have been aligned to reference genome)
- Variant discovery: identification of sites where the data displays variation relative to the reference genome, and calculation of genotypes for each sample at that site (generation of VCF files)
- Functional annotation (using the tool SnpEff), genotype refinement and evaluation of variants.
We have constructed our workflow using web browser-based platform Galaxy. Galaxy workflows are reusable bioinformatic protocols and can be shared between researchers.
A major challenge of WES approach is to determine the causative mutations from a substantial number of bystander variations that do not play any role in the disease etiology. Our strategy to narrow down the candidate list until the causative variation is isolated includes:
- systematic rejection of variations that are not shared between multiple cases or that do not follow the assumed mode of inheritance
- rejection of variations that have been catalogued in dbSNP or 1000 Genomes project, as these are relatively common while harmful mutations must be rare
- focusing on functional impact of the variation.
We have analysed the data of 54 sequenced exomes from 15 families and have identified four novel and one known genetic causes in five of them.
ESID-0617 Acute Viral Retinitis in a Patient with Autosomal Recessive Hyper-IGE Syndrome
J.C. Bustamante Ogando 1, M.?A. Yamazaki Nakashimada1, F. Rivas Larrauri1, M. Gómez Solana2, J.C. Juárez Echenique2
1Allergy and Immunology, Instituto Nacional de Pediatría, Mexico City, Mexico
2Ophtalmology, Instituto Nacional de Pediatría, Mexico City, Mexico
Autosomic Recessive Hyper-IgE Syndrome (DOCK-8 Deficiency) is a primary immunodeficiency characterized by elevated IgE levels, eosinophilia, eczema, recurrent sinopulmonary infections, staphylococcal skin abscesses, increased susceptibility to mucocutaneous candidiasis and severe cutaneous viral infections. It also predisposes to autoimmune disease and malignancy. Ocular involvement has been rarely described in Hyper-IgE syndromes as retinal detachment, keratoconus, staphylococcal chalazia with blepharitis, corneal ulcerations and Candida albicans endophthalmitis. We present a patient with severe unilateral viral retinitis and uveitis.
Case report. 14 years old girl with DOCK8 deficiency and history of pulmonary infections, disseminated cutaneous warts, bowel inflammatory disease, autoimmune hepatitis and pancreatitis, as well as encephalitis, treated with antibiotic prophylaxis, intravenous immunoglobulin, systemic corticosteroids and thalidomide. She presented to our clinic with conjunctival erythema in the right eye without any other symptom. Opthalmologic evaluation revealed vitreal celularity and acute retineal necrosis compatible with herpes virus infection. She received IV acyclovir for one month with resolution of retineal lesions and now she continues on oral acyclovir.
DOCK-8 deficiency predisposes to viral infections mainly involving skin, but to our knowledge there are no reports on viral eye involvement for this condition. Remarkably, this patient presented only with unilateral red eye and no pain or signs of inflammation. Clinical suspicion and prompt ophtalmologic consultation allowed us to give appropiate treatment and avoid potential complications. Severe complications can be present in this patients despite subtle clinical manifestations.
ESID-0780 The Expanding Clinical Phenotype of Adenosine Deaminase (ADA) Deficiency
R. Sokolic 1, C. Koh2, H. Komarow3, E. Garabedian1, C. Silvin1, E. Cowen4, C. Hadigan5, T. Heller2, D. Kleiner6, D. Metcalfe3, M. Hershfield7, F. Candotti1
1GMBB, NHGRI, BETHESDA, USA
2LDB, NIDDK, BETHESDA, USA
3LAD, NIAID, BETHESDA, USA
4DB, NCI, BETHESDA, USA
5CRS, NIAID, BETHESDA, USA
6LP, NCI, BETHESDA, USA
7Biochemistry, Duke University School of Medicine, Durham, USA
ADA deficiency is a metabolic disease most severely manifesting as profound immunodeficiency. Several other manifestations are becoming clinically important. We evaluated patients for signs of liver disease. Findings included fatty liver and/or hepatomegaly, increased fat content, histologic abnormalities (steatosis, non-alcoholic steatohepatitis, fibrosis, and cirrhosis), and elevated liver stiffness. Three children showed features of metabolic syndrome. Liver abnormalities correlated with ongoing enzyme replacement therapy and were less pronounced in patients successfully treated with cell therapy.
We also observed a spectrum of lung pathologies including hyperreactive and obstructive airways disease, granulomatous inflammation, fibrosis and subpleural cysts. In 7 out of 8 patients, impulse oscillometry (IOS) evaluation showed increased baseline reactance (X5) to levels 119-254% of reference. Only two patients had significant response to bronchodilators. These data suggest a pattern of small airway disease only partially reversible. Notably, changes occurred in patients with otherwise adequate red blood cell deoxynucleotide levels and independent from measures of immune function.
Also concerning is the association between ADA deficiency and dermatofibrosarcoma protuberans (DFSP), a rare skin malignancy that we have diagnosed in 9 patients. Follow-up by serial exam and photography for 2.5-7 years showed that most lesions enlarged, but none metastasized. Lesions that evolved from plaque to protuberant morphology or presented subcutaneous nodular/infiltrative pattern were resected. None recurred with a follow-up to 7 years. This conservative management approach may contain morbidity without compromising oncologic outcome.
In summary, non-immunologic manifestations of ADA deficiency are emerging that may be life-limiting and warrant comprehensive assessment in all patients.
ESID-0582 Lung Clearance Index for Early Assessment of Lung Disease in Primary Immunodeficiency
P. Pérusse1, H. El Fassy1, D. Smith1, C. Poirier1, H. Chapdelaine 1
1Medicine, Centre hospitalier universitaire de Montreal, Montreal, Canada
Introduction:
Primary immunodeficiencies (PID) are associated with various lung disease, including bronchiectasies and interstitial lung disease. Spirometric abnormalities are inconsistent and hard to interpret in the context of PID. In pediatric cystic fibrosis patients, lung clearance index (LCI) have been demonstrated as more sensitive and reliable over time than FEV1.
Objectives:
To assess if LCI is a more sensitive test for CT scan proven lung abnormality than the FVC, FEV1 and DLCO in PID patients
Methods:
Fifteen patients with proven PID were included for assessment of FEV1, LCI and DLCO. Lung CT-scan were reinterpreted by an independant radiologist. Medical files were reviewed for recurrent infections, antibiotic use and active treatment for PID.
Measurements and results:
73% of the patients included had primary humoral defects. 3 patients had biopsy-proven granulomatous and lymphocytic lung disease (GLILD), 3 patients bronchiectasis and 9 patients normal pulmonary function tests and CT scan.
The three patients with bronchiectasis had abnormal LCI. The other 12 patients, including those with GLILD, had normal LCI.
Conclusion:
Preliminary results suggest that LCI is a sensitive marker for detection of bronchiectasis in PID patients. Our data suggest that it could be a good measurement to evaluate early lung disease in PID population, avoiding unnecessary irradiation.
ESID-0524 Status of Primary Immunodeficiency Disorders in India – Single Centre Experience from North India
Y. Chopra 1, S. Nivargi1, M. Ramzan1, R. Sharma1, S. Katewa1, S. Yadav1
1Pediatric Hematology Oncology and Bone Marrow Transplant Unit, Fortis Memorial Research Institute, Gurgaon, India
Introduction: - Primary Immunodeficiency disorders (PID) are under-diagnosed and under – reported in the developing world like ours. On reviewing published Indian data on PID we came across only 4 large series reporting a total of 386 cases only with mortality as high as 51%. We are hereby presenting our own experience over last 15 months.
Methods: - We retrospectively analysed 23 children registered in our PID clinic from January 2013 to April 2014.
Results: - M: F was 17:6. Mean age was 6.02 years (3 mon -15 yrs).Three children had x –linked agammaglobunemia and are doing well on intravenous immunoglobulin.One child had pure red cell aplasia with IgM deficiency and responded well to prednisolone. One had congenital neutropenia and is being managed with G-CSF.Hyper IgE syndrome (DOCK 8 mutation) was seen in three children. ALPS was seen in three children and are doing well on mycophenolate, steroids and sirolimus (one case). Three children (2 males, 1 female) were diagnosed as case of Heme-oxygenase-1 deficiency with auto-inflammatory syndrome. Four children underwent BMT (2 matched sibling , 2 haploidentical) for Aplastic anemia with CVID , Chediak Higashi syndrome , X-linked severe combined immunodeficiency and DOCK 8 mutation respectively and two (SCID and Dock 8) died of fulminant sepsis. Hemophagocytic lymphohistiocytosis (Three acquired and One familial) was managed as per HLH 2004 protocol. Overall survival was 90.9% (20/22) with one lost to follow up.Conclusion: - This is a small series shows that improvement in survival is possible with early diagnosis and appropriate management.
ESID-0370 An Integrative Diagnostic Platform for Patients with Primary Immunodeficiencies
J. Chou 1, M.J. Massaad1, H. Jabara1, B. Cangemi1, W. Bainter1, L.D. Notarangelo1, T.A. Chatila1, R.S. Geha1
1Division of Immunology, Boston Children's Hospital, Boston, USA
Introduction. Studies using whole exome and genome sequencing (WES/WGS) as diagnostic tools identify a possible diagnosis in ~25% of patients, but lack data proving the biologic effect of identified mutations. The true accuracy and diagnostic yield of WES/WGS thus remains unknown.
Objective. To determine the diagnostic yield of WES/WGS in patients with primary immunodeficiencies (PID) by incorporating WES/WGS with functional assays.
Methods. WES/WGS was performed on 40 patients with PID. The significance of identified variants was tested using functional and genetic assays.
Results. A definitive diagnosis was achieved in 58% of patients, for whom functional assays demonstrated the pathogenic effect of the candidate mutation. An additional 10% of patients had a likely diagnosis that requires the development of novel functional assays, generating an overall discovery rate of 68%. The best predictor of diagnostic success was a history of life-threatening infections. The majority (70%) of pathogenic variants affected a boundary between non-conserved and conserved loci, compared to 52% of polymorphisms (p<0.01), suggesting that these junctions represent genomic hotspots for deleterious mutations. Variant validation using functional or genetic assays altered the outcome for 8% of patients by demonstrating either that a mutation predicted to be benign in silico had a pathogenic effect ?in?vivo, or that a mutation predicted to be pathogenic had no association with disease.
Conclusions. Our integrated diagnostic platform achieved an overall diagnostic yield of 68%. Functional assays are critical for validating WES/WGS in clinical medicine. WES/WGS is particularly successful for diagnosing patients with potentially fatal PIDs.
ESID-0060 Selective IGM Deficiency: Clinical and Laboratory Manifestation of 17 Patients
Z. Chovancova 1, P. Kralickova2, M. Vlkova1, J. Litzman1
1Department of Clinical Immunology and Allergy, Faculty of Medicine of Masaryk University and St. Anne University Hospital in Brno, Brno, Czech Republic
2Institute of Clinical Immunology and Allergology, Charles University in Prague School of Medicine and University Hospital, Hradec Kralove, Czech Republic
Selective IgM deficiency represents a rare primary immune disorder of unknown pathogenesis, characterized by marked decrease of IgM immunoglobulin with normal IgG and IgA levels. Clinical manifestation is variable including recurrent infections and/or occurrence of autoimmune, allergic or malignant diseases.
Clinical and immunological manifestation of 17 adult patients with selective IgM deficiency, defined as IgM < 0.20 g/?l, were evaluated.
The cohort consists of 17 adult patients, 9 males (aged 22 – 70 years at the time of diagnosis) and 8 females (36 – 66 years). Five patients had undetectable serum levels of IgM (<0.05 g/?l), while in 12 patients IgM level ranged from 0.07 to 0.19 g/?l. Four patients presented with susceptibility to infections, twelve had type-one hypersensitivity diseases and four had systemic autoimmune diseases. Seven of 14 examined patients had decrease of IgG subclasses: 1 patient in IgG1 (3.4 g/?l), 1 patient in IgG2 (1.02 g/?l), 4 patients in IgG4 (<0.08 g/?l), 1 patient in IgG1 (3.54 g/?l) and IgG4 (<0.08 g/?l). All examined 10 patients had protective levels of anti-tetanic and anti-pneumococcal polysaccharide antibodies. Surprisingly 3 of them had normal isohemagglutinin levels. All patients had normal numbers of B-cells, in 9 of them including presence of surface IgM+ B-cells. In one patient the progression to panhypogammaglobulinemia with thymoma was observed.
Clinical manifestation of selective IgM deficiency is variable. Besides mild clinical immunodeficiency, also other immunopathological diseases, both allergic and autoimmune, seem to be increased. The mechanism leading to this association remains to be elucidated.
ESID-0775 Mechanisms Behind the Failure to Express HLA-B In Stem Cells and Cancer Cells
D. Christoffer 1, J. Nehlin1, T. Barington1
1KIA, Odense University Hospital, Odense, Denmark
The main function of HLA class I is to bind and present intracellularily produced peptides on the surface of cells. These peptides can either originate from the cell’s own genome, or they may result from an intracellular pathogen, e.?g. a virus. Once on the cell surface, the HLA-peptide complex is monitored by T cells from the immune system that can recognize foreign peptides and kill the infected cells that present them. Cancer cells can also be identified and killed by the immune system by the unique peptides they present. Reduced HLA expression is, however, a common evasive mechanism seen in cancer cells leading to immune escape of cancer cells. The purpose of this project is to understand one of the mechanisms controlling the expression of HLA in the cell thus hoping to provide a better understanding of this evasive mechanism of cancer.
We use an experimental system in which the human cell line HEK293T is transfected by constructs encoding the HLA class I subtypes HLA-A and HLA-B and hybrids of these. The HEK293T cell line constitutively expresses HLA-A3 (highly) and HLA-B7 (weakly) which can be discriminated by our allele-specific antibodies and constitute a point of reference for our expression studies.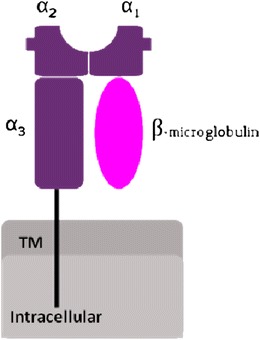
We have determined the importance of the different domains that constitute the HLA molecule in the two subtypes HLA-A2 and HLA-B8, and found that alpha 3 is especially important for the expression of HLA on the cell surface.
ESID-0733 Assessment of Mother-Child Transmission to VIH-1 in 2012 to Mali
Y. CISSE 1, D. KOITA1, A.L.O.U. SANOGO1, I. GUINDO1, M. GUINDO1, T. TIERRO1, F. MAIGA2, A.L.O.U. SYLLA3, F. BOUGOUDOGO1
1DDRB, Institute National Of Rechearch in Public Heath (INRSP), bamako, Mali
2PCR, CSLS, bamako, Mali
3CSLS, CSLS, bamako, Mali
Introduction
Mother- to child transmission of VIH constitutes a reality in the incomes countries. The goal of our study was to value mother-to-child transmission of VIH-1 during year 2012 in Mali.
Methodology
It's a study of assessment of early diagnosis at the newborns and children of less than 18 months in 2012. Their mothers were under HAART according to recommendations 2010 of WHO at exception of those no consistent ' cases out protocols '. The study variables were delay of withdrawal, type of nursing, child ARV, delay of the tests realization.
DBS achieved have been routed to the laboratory. The molecular diagnosis has been made on two platforms : HIV Amplicor 1 DNA test and qualitative m2000rt.
Results :
We achieved 1399 PCR at 900 newborns. The middle delay of withdrawal was of 4,4 months. The global middle delay of DBS routing was 19,6 days and the global median delay, of 4 days. The middle delay of realization of test was 8 days. The maternal nursing was more used with 66,4%. The children under PTME represented 63,8%.
The PCR was positive in 143 cases. The transmission was 8,3% for the cases' out protocol' and 0,7% for PTME cases, more elevated in the cases of mixed and maternal nursing with respectively 29,9% and 10,6% (p=0,000).
Conclusion :
Vertical transmission remains always a problem of public health in Mali. Aimprovement of the PTME cover would permit a early charge of cases' out Protocol '.
Key word: Early diagnosis, Newborn, PCR, PTME, VIH-1,
ESID-0740 Severe Combined Immunodeficiency in Slovakia – A 10 Year Experience
P. Ciznar 1, J. Horakova2, P. Svec2, I. Bodova2, S. Sufliarska2, L. Libai-Veghova2, M. Hricova2
11st Pediatric Department, Comenius University Faculty of Medicine, Bratislava, Slovakia
21st Pediatric Department, Children University Hospital, Bratislava, Slovakia
In the period of past 10 years total number of 10 patients has been diagnosed with SCID at Children University Hospital in Bratislava. Half of these patients have been exposed to BCG vaccine early in life (BCG+). Icidence rate of diagnosed and treated SCID in Slovakia was calculated to 1:87.000. Final diagnosis was ADA in 4 patients, IL2RG deficiency in 2 patients and complete del22q11 in 1 patient. In 3 cases genetic defect was not found. Seven out of these 10 patients underwent hematopoietic stem cell transplantation, in 5 the hematopoietic stem cell source was a matched unrelated donor. Favorite outcome was achieved in 6 of them. Patients vaccinated with BCG faced severe complications and organ damage due to generalized skin and organ abscess formation, requiring prolonged hospital care and complex antibiotic therapy with more than four types of anti-mycobacterium drugs, for more than 2 years. Average length of hospital care for BCG exposed patient was 10, 8 months vs. 2, 7 months in non-exposed group (p<0, 05). No statistical difference was found between the time of recognized first symptoms, and time of diagnosis in BCG+ and BCG- group. The clinical presentation of non-BCG vaccinated patient differs in the initial symptoms when failure to thrive and pneumonia at 4 months was the most common finding. BCG and late diagnosis prolongs time for hospital care, immune reconstitution and carries severe complications, consequently it increase the costs of health care and decrease the quality of patients’ life.
ESID-0553 Nationwide One Year Study “DIHOSP” Evaluating Emergency Hospitalizations in Children and Adults with Primary Immunodeficiency
H. Coignard-Biehler 1, N. Mahlaoui2, P. Brosselin3, N. de Vergnes3, M. Debré2, M. Malphettes4, I. Pellier5, V. Barlogis6, E. Jeziorski7, O. Lortholary1
1Infectious Disease, Necker Hospital, Paris, France
2Paediatric Immuno-Hematology and Rheumatology, Necker Hospital, Paris, France
3CEREDIH, Necker Hospital, Paris, France
4Immuno-hematology, Saint Louis Hospital, Paris, France
5Pediatrics, Angers Hospital, Paris, France
6Paediatric Haematology, Timone Enfants Hospital, Paris, France
7Pediatrics, Arnaud de Villeneuve Hospital, Paris, France
Introduction and method: This study aimed to provide the reasons of hospitalizations in emergency of children and adults presenting a PID. Other parameters were registered: details about the hospitalization, final diagnosis, precisions about type of follow-up of the immunodeficiency and associated complications. This observational multicenter prospective study has been conducted in France between October 2010 and November 2011. Results: A total of 200 hospitalizations of 137 different patients had been notified. There were 64 patients below 16 (46.7% of the whole population). Sixteen HSCT recipients have been hospitalized 30 times. Comparison of the characteristics of our series with the data of the French national Registty of PID held by the CEREDIH during the same period showed similar characteristics. Infections were the leading cause of hospitalization for 149/170 of them (85,1%). Other diagnoses were haematological, pulmonary, digestive or other, and represent 14,8% of the hospitalizations. Regarding treatments, anti infectious drugs were administered in 92 patients, and 12 patients have initiated Ig replacement therapy (4 children and 8 adults). We noticed that 28 patients were not totally cured at the issue of hospitalisation with persistent of some symptoms. Thirteen needed to be transferred in an intensive care unit. Twelve patients (8.8%, 5 children and 7 adults) died during a hospital stay at a median age of 19.2. Conclusion: This work highlights the incidence of infectious complications in patients with PID. We noticed a significant mortality (8.8%). Analyses are ongoing to determine a profile of patients at risk of severe complications.
ESID-0017 Clinical and Biological Features in Two Cases of PFAPA Syndrome
M. Cucuruz 1, E. Boeriu1
1IIIrd Paediatric Clinic, University of Medicine and Pharmacy “Victor Babes”, Timisoara, Romania
PFAPA is an acronym for the most common characteristics of the disease: periodic fever, aphtous stomatitis, pharangytis and cervical adenitis. We present two pediatric cases which correspond to the diagnostic criteria.
The cases (one boy 4 years old, one girl 3 years old) were without immunological changes, but had complete diagnostic criteria for PFAPA syndrome. The treatment was mainly supportive, with reduction of symptoms using NSADs. For the girl steroid administration was necessary because of the persistent high fever. In this case tonsillectomy leaded to cessation of the disease.
In the other case the symptoms disappeared within four years (when the child was 8 years old).
We put in discussion if tonsillectomy (with or without adenoidectomy) can be considered treatment for patients with PFAPA syndrome.
ESID-0129 Gamma Chain is Prominently Overexpressed in B-PRE Acute Lymphoblastic Leukemia Cells
R. Romano1, R. D'Assante 1, G. Bianchino2, V. Grieco2, R. Parasole3, V. Poggi3, L. Palamaro1, A. Fusco1, G. Scotto Di Marco1, C. Pignata1
1Translational Medical Sciences, "Federico II" University, Naples, Italy
2Laboratory of Clinical Research and Advanced Diagnostics, "Centro di Riferimento Oncologico della Basilicata" (CROB) IRCCS, Rionero in Vulture Potenza, Italy
3Department of Pediatric Hemato-Oncology, Santobono-Pausilipon Hospital, Naples, Italy
INTRODUCTION: The ?-chain (?c) is a transducing element shared between several cytokine receptors, that plays a prominent role in immunological functions and cell survival. It is directly involved in the regulation of self-sufficient growth and cell cycle progression in continuous human malignant hematopoietic cell lines, in a concentration dependent manner.
OBJECTIVE: To evaluate the role of ?c in the biology of different types of leukemia, through the analysis of ?c-expression profile and ?c-signaling in different leukemic cells.
METHODS: Leukemic cells were obtained from the bone marrow of 39 newly diagnosed patients with CML, AML, B-pre ALL and T-ALL and healthy controls. Total RNA was extracted and IL2RG, D-type cyclins, BCL-XL and BECLIN-1expression levels were evaluated through Real-Time PCR. The amount of BECLIN-1 protein was evaluated through western blot, using the specific primary antibody.
RESULTS: IL2RG expression was increased, as compared to controls, in ALL cells and, in particular, in B-pre ALL (2.55 fold increase vs 1.57 in T-ALL). A positive correlation between IL2RG and cyclins D2 (R = 0.82) and D3 (R = 0.76) expression levels was found. BCL-XL expression was 57% than controls, while BECLIN-1 expression was 4.73-times higher than controls, suggesting the involvement of autophagy in ?c-induced cell survival.
CONCLUSIONS: The ?c is prominently over-expressed in B-pre ALL, with a direct correlation with D-type cyclins. The up-regulation of ?c also associates with increased BECLIN-1 expression, which is a potent mediator of autophagy.
ESID-0163 Aberrant Autophagic Vesicles in the Lymphocytes from Patients Affected with Ataxia-Telangiectasia
A. Fusco1, E. Polishchuk2, R. D'Assante 1, L. Palamaro1, A. Ballabio1, C. Pignata1
1Translational Medical Sciences, "Federico II" University, Naples, Italy
2Advanced Microscopy and Imaging Core, Telethon Institute of Genetics and Medicine (TIGEM), Naples, Italy
INTRODUCTION: Ataxia-Telangiectasia (AT) is a rare disorder mostly characterized by cerebellar neurodegeneration and immunodeficiency. AT is caused by mutations in the Ataxia-Telangiectasia-Mutated (ATM) gene encoding a kinase, mostly localized in the nucleus and involved in cell-cycle control and DNA repair. ATM also displays a cytoplasmic localization, where its role is still poorly defined.
OBJECTIVE: To evaluate potential abnormalities in the autophagic vesicle formation, which could be responsible for an inappropriate cell-clearance.
METHODS: The ultrastructure of autophagic structures, such as autophagosomes (AP) and autolysosomes (AL), was analyzed by transmission electron microscopy (TEM) in lymphocytes obtained from AT patients and healthy controls and cultured at basal conditions or in a serum-starved medium. A quantitative analysis was also performed.
RESULTS: In the patients at basal conditions, we found that the number of APs was 7 times higher than in the controls (14.00 vs 2.00 average number/100 µm2), while the number of ALs was more than two times lower compared to the healthy subjects (2.00 vs 4.80 average number/100 µm2). Under basal conditions, APs/ALs ratio was much higher in the patients than in the controls (7 vs 0.5). Under starved conditions, in the patients the number of APs did not further increase, resulting in an APs/ALs ratio comparable to the controls.
CONCLUSIONS: Our data suggest an aberrant pattern of autophagic structures in lymphocytes from AT patients, characterized by an imbalance between autolysosomes and autophagosomes, suggesting an impairment in the cell clearance network.
ESID-0594 Long Term Clinical Outcomes of Patients with X-Linked Hyper IGM Syndrome, an International Collaborative Study
M. de la Morena 1, D. Leonard2
1Pediatrics and Internal Medicine, University of Texas Southwestern Medical Center, Dallas, USA
2Statistics, Children' Medical Center, Dallas, USA
The X linked form of Hyper IgM (XHIGM) syndrome is a primary immunodeficiency (PID) caused by mutations in the gene for CD40Ligand (CD40L; CD154). Treatment includes prophylactic antimicrobials, intravenous gamma-globulin and, in some centers, hematopoietic cell transplantation (HCT).
Objectives: To compare the long term clinical outcomes of patients with XHIGM treated with or without HCT.
Methods: Clinical data were surveyed online using REDCap. Post-diagnosis survival for patients treated with/? without HCT was compared using the Kaplan-Meier method with log-rank testing. In the HCT subgroup, log-rank tests were used to assess risk factors on post-transplant survival.
Results: 189 subjects were entered in REDCap from August 2012 to September 2013. 14 subjects (7%) were excluded due to invalid follow–up time or vital status information. Median age at diagnosis was 1 year, and mean follow-up time was 8.4 ± 7.0 years.
At last follow-up, 143 subjects were surviving. Estimated median survival time was 21.7 years. Of the 32 subjects who died, opportunistic infections (n=7, 22%) and malignancy (n=6, 19%) were the most common cause of death. Comparing subjects who received HCT to others, median survival time was similar although median Lansky/Karnofsky scores were higher in surviving HCT subjects (rank-sum p<0.001).
67 subjects underwent HCT. At last follow-up, 56 subjects were surviving. Neither age or transplant year, donor relationship, nor stem cell source, were found to be associated with post-transplant survival.
Conclusion: Long term survival for patients with XHIGM remains poor. Collaborative prospective studies are needed to identify best outcome variables.
ESID-0172 Epidemiology of PID: Analysis of More than 5000 Patients in the French National Registry of PID (CEREDIH)
N. Mahlaoui1, N. De Vergnes 1, V. Courteille1, L. Costes1, C. Brito de Azevado1, L. Gérard2, J. Jais1, I. Pellier3, V. Barlogis4, S. Blanche1, C. Picard1, F. Suarez1, O. Lortholary1, O. Hermine1, J. Donadieu5, E. Oksenhendler2, A. Fischer1
1CEREDIH, AP HP Hôpital Necker Enfants Malades, PARIS Cedex 15, France
2CEREDIH, AP HP Hôpital Saint Louis, PARIS Cedex 10, France
3CEREDIH, CHU Angers, Angers Cedex 09, France
4CEREDIH, Hôpital de la Timone, Marseille, France
5CEREDIH, AP HP Hôpital Trousseau, PARIS Cedex 12, France
Introduction: Registries are useful tools for better understanding of rare diseases and particularly of PID diseases. The French national Reference Center for PID (CEREDIH) runs a nationwide registry for PID and fully contributes to the ESID online database since Jan. 2006.
Objective: Better understanding of the epidemiology of PID at a nationwide level. Improvement of access to care.
Methods: National registry dedicated to PID diseases with the collaboration of the French national registry for severe chronic neutropenia and the French DEFI study group.
Results: As of May, 12th 2014, the total number of patients registered since Jan. 2006 in France is 5,425 out of 19,358 (28%) patients registered in the ESID online database. It is the largest nationwide database worlwide to date. Mean prevalence and incidence are estimated to be 5.96 per 100.000 inhabitants (French population: 65 million inhabitants) and 200 new cases each year (# live births: 830,000/year). Predominantly B cell deficiencies, T cell deficiencies Innate immunity deficiency accounted for 45.8%, 34.5% and 18.6%, respectively. Detailed epidemiological data will be presented.
Conclusion: Although the completeness of registration in good for patients followed in the national network of University Teaching Hospitals, we have performed epidemiological investigations for comprehensiveness (searching for ‘ignored’ cases). There are still some patients who are not followed in the frame of this gold standard network and thus should beneficiate of the best standard of care. Registries for rare diseases are useful indicators of measure and efficacy
ESID-0382 Complicated Mycobacterial Diseases in Patients with Primary Immunodeficiencies (PIDS) from One Argentinean Centre
A. Gómez Raccio 1, D. Di Giovanni1, A. Seminario1, M.S. Caldirola1, D. Comas1, C. Cerqueiro2, I. Squassi2, M.I. Gaillard1, L. Bezrodnik1
1Immunology, Hospital de Niños R. Gutierrez, Ciudad Autónoma de Buenos Aire, Argentina
2Phthisiology, Hospital de Niños R. Gutierrez, Ciudad Autónoma de Buenos Aire, Argentina
Patients with specific PIDs are more susceptible to severe mycobacterial infections: tuberculosis (TB), atypical mycobacterial disease (AM) and dissemination following live-attenuated M.?bovis bacille Calmette-Guérin (BCG) vaccination (BCGosis). Tuberculosis is a common infectious disease. The BCG vaccine given routinely is one of the strategies to control meningeal and disseminated tuberculosis. Objective: to report and describe complicated mycobacterial diseases in PID patients from our centre. Material and Methods: retrospective analysis of medical records. Results: 22 patients with PID and mycobacterial diseases (8 CGD, 4 CID, 2 HIGE, 2 XLA, 2 IL12RB1, 1 STAT1, 1 IKBalpha, 1 HIGM, 1 A-T). 18 males/ 4 females, median age 29 months (r: 3-228). 6/?22 (25%) < 1 year old presented BCGosis. 9 Complicated TB, 2 AM disease (CID) and 11 BCGosis patients (7 CGD). 82% BCGosis with lung and lymph compromise. The antituberculous therapy in 18/22 patients consisted of 4-5 drugs. Two patients (HIGM and IL12RB1) also received IFNgamma. In 12 patients mycobacterial infection let us arrive to PID diagnose. Conclusions: The most related PID with mycobacterium diseases is CGD. In younger PID patients these infections were due to BCG; certainly related with the routinely BCG vaccine indication at birth. The mayority of TB and AM occurred in not CGD PIDs. PID should be suspected in severe TB. Most infections responded to multiple antibacterial drugs and two patients required additional immunomodulatory therapy.
ESID-0430 Two in One: A Newborn with Chronic Inflammatory Bowel Disease (IBD) with Homozygous Mutations in IL10RB and NCF2
G. Dueckers 1, M. Hoenig2, C. Schuetz2, K. Schwarz3, J. Berrang4, M. Sirin2, F. Kropp2, T. Niehues1, A. Schulz2
1Department of Pediatrics and Adolescent Medicine, HELIOS Hospital, Krefeld, Germany
2Department of Pediatrics and Adolescent Medicine, University Medical Center, Ulm, Germany
3Institute for Transfusion Medicine University Ulm and Institute for Clinical Transfusion Medicine and Immunogenetics, German Red Cross Blood Service Baden-Württemberg – Hessen, Ulm, Germany
4Department for pediatric gastroenterology, Children's Hospital, Dortmund, Germany
A six week old male patient presented with oral and anal aphthous lesions, severe colitis, chronic diarrhoea, failure to thrive, chronic pustulous dermatitis and recurrent episodes of fever. He is the fifth child of a consanguineous family of Turkish origin. Two sisters of the patient deceased within the first years of life, one with meningitis at 38 days of life, the other with undefined immunodeficiency and severe colitis. Repeated DHR Test in the newborn showed an impaired but not absent oxidative burst. Identification of a homozygous mutation (c.74C>G, p.Ala25Gly) in NCF2 led to the diagnosis of chronic granulomatous disease. Surprisingly, the healthy HLA identical brother (11 years of age) carries the same homozygous NCF2 mutation. Revaluation of the newborn revealed a homozygous splice site mutation (IVS2 c.173+2T>G) in IL10RB, leading to the diagnosis of primary chronic IBD due to IL-10 receptor deficiency. The healthy brother carries a heterozygous splice mutation in IL10RB.
The patient was transplanted with peripheral blood stem cells (MUD, 10/10 match; conditioning regimen: targeted Busulfan (AUC 65 mg/l*h), Fludarabine (160 mg/m2), Thiotepa (10 mg/kg) and alemtuzumab (0.7 mg/kg)). The dermatitis and the diarrhoea rapidly resolved after conditioning. The patient is currently two months after transplantation with complete donor chimerism. CMV reactivation is treated with ganciclovir and acute GvHD is well controlled with steroids, CSA and MTX.
The mutation in IL10RB most likely explains the phenotype of chronic IBD. However the mutation in NCF2 might have contributed to the clinical course.
ESID-0269 The Diverse Clinical Spectrum of an XIAP (BIRC4) Nonsense Mutation in a Single Large Caucasian Family
M. Dziadzio 1, S. Ammann2, A. Hassan3, M. Elawad4, C. Cale3, B.K. Fiil5, M. Gyrd-Hansen5, U. Salzer2, C. Speckmann2, B. Grimbacher1
1Immunology, Royal Free Hospital, London, United Kingdom
2CCI-Center for Chronic Immunodeficiency, University of Freiburg, Freiburg, Germany
3Pediatric Immunology, Great Ormond Street Hospital, London, United Kingdom
4Pediatric Gastroenterology, Great Ormond Street Hospital, London, United Kingdom
5Department of Disease Biology, University of Copenhagen, Copenhagen, Denmark
An association between primary immunodeficiency diseases and inflammatory bowel disease (IBD) is not uncommon. Recently, inflammatory manifestations including IBD have been reported in individual patients with an X-linked inhibitor of apoptosis (XIAP) deficiency. The clinical phenotypes of the carriers of the XIAP mutation have not been well characterised.
We describe a large Caucasian family with XIAP deficiency due to a mutation in BIRC4.
The absence of detectable XIAP protein was confirmed by immunoblotting and flow cytometry. Genetic analysis identified a new nonsense mutation (c.dupT672/p.Pro225Serfs*2) in BIRC4. XIAP protein was present in the carriers who showed skewed XIAP inactivation towards the healthy allele in T, B, NK-cells and monocytes. A critical impairment of the NOD2 pathway was detected by an L18-MDP flow cytometric assay in our XIAP deficient patients but not in the carriers.
Clinically, affected individuals showed large phenotypical diversity and a variable clinical course. The index case presented in childhood and died despite BMT. Other affected individuals presented with symptoms ranging from mild to moderate IBD occurring in childhood or in adolescence, to an acute, probably virally-induced, catastrophic haemophagocytic lymphohistiocytosis (HLH) in early childhood. Most carriers in this family have experienced recurrent episodes of erythema nodosum and a variable degree of gastrointestinal symptoms.
In summary, IBD can be the main and/or presenting feature of XIAP deficiency. The clinical spectrum is wide and includes HLH, severe IBD and mild colitis. Carriers may be symptomatic with gastrointestinal manifestations and erythema nodosum.
ESID-0709 The German PID-Net Registry
S.M. El-Helou 1, B. Gathmann1, G. Kindle1, B. Grimbacher1
1German PID-NET Registry Working Group - Center for Chronic Immunodeficiency (CCI) Clinical Research Unit (CRU), University Medical Center Freiburg and University of Freiburg, Freiburg, Germany
Since 2009, the German Registry for Primary Immunodeficiencies (PID) exists within the PID-NET consortium - sponsored by the German Ministry of Education and Research (BMBF). Patient data are directly entered into the ESID Online Registry of the European Society for Immunodeficiencies, the German PID-NET is therefore a subset of the ESID data.
One of its aims is to determine the prevalence of PID in Germany. With 1,999 patients registered in May 2014, the prevalence is about 2:100,000 inhabitants. This number is below the expected number of at least 4:100,000, when compared to other countries like France. Therefore, it is assumed that a lot of patients are also treated at non-university hospitals and primary care practitioners. With 63%, antibody deficiencies represent the biggest group of PID patients.
Currently, 37 German documenting centers take part (May 2014). The documenting process however is slowed down due to the decentralized ethic approval in Germany. Another reason for the slow registering process is the lack of study nurses in most hospitals. Therefore, in November 2009, a medical data entry clerk was employed for PID-NET. She travels all over Germany and also helps the centers with their ethics application.
The registry also offers the possibility for collaborations between the centers. Clinical and genetic parameters identify patients for specific studies. It is essential that the German PID-NET data is also accessible in the ESID Registry. Data from German centers has already been included in several multi centre studies like DOCK8, PedPAD, CVID, NBS, and ChestCT.
ESID-0403 Griscelli Syndrome Type 2: Different Pattern of Clinical Presentation
N. Radwan 1, R. Elfeky1, S. Hassanein2, S. Reda1
1Paediatric Allergy and Clinical Immunology, Ain Shams University, Cairo, Egypt
2Paediatric Neurology, Ain Shams University, Cairo, Egypt
Griscelli type 2 (GS2) is a rare disorder induced by mutations in RAB27A gene . Herein we report two consanguineous families with variable initial clinical presentation. In both families, affected siblings had clinical stigmata of GS2 ;albinism, silver hair, large irregular melanin and normal immunological profile. A RAB27A mutation was identified; (c.337delT;p.Trp113Glyfs*2) in family A , (c 148 149delinsC; p Arg50GlnfsX35) in family B. No suitable donor was found for affected siblings.
Family A: The elder son presented with refractory HLH at the age of 10 months and died a month later. The younger daughter had recurrent gastroenteritis since her first birthday and died two years later of HLH.
Family B: The elder daughter presented at 9 months with rough plaques over her skin, skin biopsy showed lymphohistiocytic infiltrates, cured with topical steroids. Three months later, she developed bronchopneumonia, ataxia, brain MRI showed diffuse cerebellitis. In both occasions, no laboratory evidence of HLH was elicited. She had poor response to IV methylprednisolone but responded to high doses of IVIG, discharged within a month with a residual neurological deficit . She was maintained on monthly IVIG till her death at the age of 4 years of HLH. Her younger brother died of HLH with multisystem organ failure at age of four months.
GS2 is a fatal disease unless HSCT is commenced. Age at presentation and severity of disease varies within the same family members. Boys might have a severe disease than girls. IVIG might delay the development of HLH in GS2.
ESID-0321 In Search of the TACI Modifiers in Common Variable Immunodeficiency
Z. Eskandarian 1, S. Zhang2, A. Schäffer3, U. Salzer4, N. Knoll4, E. Vries5, L. Hammarström6, T. Español7, I. Quinti8, B. Grimbacher9
1Center for Chronic Immunodeficiency, Medical University Center, Freiburg, Germany
2Graduate Program in Bioinformatics, Boston University, Boston, USA
3NIH, NCBI, Bethesda, USA
4Medical University Center, Center for Chronic Immunodeficiency, Freiburg, Germany
5Department of Pediatrics, Jeroen Bosch Hospital, 's-Hertogenbosch, Netherlands
6Division of Clinical Immunology, Karolinska Institutet at the Karolinska University Hospital, Huddinge, Sweden
7Immunology Unit, Vall d’Hebron University Hospital Hospital, Barcelona, Spain
8Department of Molecular Medicine, Sapienza University, Rome, Italy
9Center for Chronic Immunodeficiency, Medical University Cente, Freiburg, Germany
TNFRSF13B (which encodes TACI) is mutated in 8-10% of patients with Common Variable Immuunodeficiency. Two distinct TACI mutations are common among CVID patients: C104R and A181E. However, these two mutations can also be seen in the healthy population, albeit with a much lesser frequency of about 1%.In this study, 126 individuals, all carrying at least one TACI mutation, from 42 families with CVID were recruited. Of these, 70 were diagnosed with hypogammaglobulinemia or CVID and 56 family members with the very same TACI mutation were considered healthy. A combination of genetic linkage and association analysis was performed to identify loci harboring potential modifier genes for TACI deficiency causing or protecting from the CVID phenotype. Possible candidate genes were sequenced on genomic DNA and cDNA and the expression was studied by qPCR and Western blotting.
Three possible linkage regions with an HLOD score above 3 were identified. The best-scoring regions did not depend on whether the TACI mutation was C104R, A181E, or something else. The first two we explored are both on chromosome 5q, between 130-145?Mb and between 60-75 Mb, both acting under recessive modes of inheritance, but with very different optimal frequencies for the modifier (see Figure). Among the interesting, genes located in these two intervals were: CAMLG (encodes CAML, a known binding partner of TACI), Interleukin-4, and GTF2H2. There was no genetic polymorphism within CAMLG which segregated with the phenotype; moreover, CAMLG RNA expression was not significantly different when CVID and healthy mutation carriers were compared.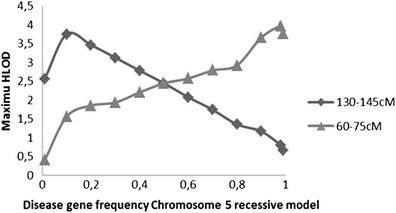
ESID-0560 A Case of Wiskott-Aldrich Syndrome with a New Mutation C121T (R41X)
S. Esteghamati 1, S. Alavi1, S. Safaei2, Z. Pourpak3
1Pediatric Congenital Hematologic Disorders Research Center, Shahid Beheshti University of Medical Sciences., Tehran, Iran
2Tehran University of Medical Sciences, Asthma and Allergy Research Institute, Tehran, Iran
3Children’s Medical Center Tehran University of Medical Sciences Tehran Iran, Immunology Asthma and Allergy Research Institute, Tehran, Iran
Introduction:
Wiskott-Aldrich syndrome (WAS) is an X-linked recessive disease characterized by thrombocytopenia, eczema, and recurrent infections due to immunodeficiency. The responsible gene is located on Xp11.23 and consists of 12 exons that encode a 502-amino acid protein called WASP. Here we report a patient with a new mutation; C121T (R41X).
Case Report:
A 15-month-old, born of consanguineous parents with a history of refractory thrombocytopenia was referred for further evaluation. He was diagnosed with idiopathic thrombocytopenic purpura (ITP) at the age of 9 months. On physical examination, mild eczematous lesions were noted on the trunk and extremities. Complete blood count was remarkable for thrombocytopenia, mean platelet volume of 6.0?f. and small platelets on the peripheral blood. IgM level was decreased at 17.5 mg/dL (normal range: 58 ± 23 mg/dL). Flowcytometry revealed reduced amounts of the glycoprotein IIb/IIIa (about 15% of normal controls). Examination of the bone marrow was inconsistent with the original diagnosis of ITP. A presumptive diagnosis of WAS was established and genetic analysis for WASP gene mutations was performed. All the 12 exons of the WASP gene were individually amplified using PCR primers. A C?T mutation in exon 1 (C121T) was identified.
Conclusion:
The patient was a case of WAS with a new mutation in exon 1 (C121T). He underwent successful allogeneic stem cell transplantation from his sister, followed by a complete resolution of thrombocytopenia. Four years after the transplantation, he has no clinical problems and his platelet count is within normal limits.
Keywords: mutation; syndrome; thrombocytopenia; Wiskott-Aldrich
ESID-0646 A Report from Iranian Primary Immunodeficiency Registry (IPIDR), New Trend in Diagnosis of PID
M.R. Fazlollahi 1, A.A. Hamidieh2, M. Movahedi3, I. Mohammadzadeh4, Z. Chavoshzadeh5, M. Nabavi6, F. Zandieh7, M.H. Bemanian8, M. Mansouri5, L. Atarod9, M. Mahloujirad1, A. Fayezi10, M. Mahdaviani5, N. Kalantari11, N. Bazargan12, F. Behmanesh13, G. Khotaei14, S. Arshi6, Z. Pourpak1, M. Moin1
1IAARI, ImunologyAsthma and Allergy Research Institiute Tehran University of Medical Sciences, Tehran, Iran
2Hematology-Oncology and Stem Cell Transplantation Research Center, Shariati HospitalTehran University of Medical Sciences, Tehran, Iran
3Department of Allergy and Clinical Immunology, Children's Medical Center Tehran University of Medical Sciences, Tehran, Iran
4Department of Immunology and Allergy, Amirkola Hospital Babol University of Medical Sciences, Babol, Iran
5Department of Allergy and Clinical Immunology, Mofid Hospital Shahid Beheshti University of Medical Sciences, Tehran, Iran
6Department of Allergy and Clinical Immunology, Iran University of Medical Sciences, Tehran, Iran
7Department of Allergy and Clinical Immunology, Bahrami Hospital Tehran University of Medical Sciences, Tehran, Iran
8Department of Allergy and Clinical Immunology, Iran University of Medical Sciences, Tehran, Iran
9Department of Pediatrics, Imam Khomeini Hospital Tehran University of Medical Sciences, Tehran, Iran
10Department of Allergy and Clinical Immunology, Ahvaz University of Medical Sciences, Ahvaz, Iran
11Department of Allergy and Clinical Immunology, Golestan University of Medical Sciences, Golestan, Iran
12Department of Allergy and Clinical Immunology, Kerman University of Medical Sciences, Kerman, Iran
13Department of Allergy and Clinical Immunology, Mashhad University of Medical Sciences, Mashhad, Iran
14Department of Pediatrics, Children's Medical Center Tehran University of Medical Sciences, Tehran, Iran
Introduction: Primary immunodeficiency disorders (PID) are rare and heterogeneous group of disease. In attempts to address and to provide an estimate of the precise prevalence of these disorders in Iran, the Iranian Primary Immunodeficiency Registry (IPIDR) was established in 1999. The Registry is currently being expanded and is a part of IAARI (Immunology, Asthma & Allergy Research Institute).
Method: During (2006-2013), 1857 patients who were referred to IAARI with possible diagnosis of PID were enrolled in this study .Primary and advanced screening tests were done and after confirmation of diagnosis all patients registered in IPIDR (Ipidr.tums.ac.ir)
Results: fifty hundred and sixty-five patients (354 M/?211F) had definite or probable diagnosis of PID; Consanguinity rate and family history of previous child with PID were 58% and 24.6%, respectively. Genetic studies were done in 50.8% of patients and in 37.3% of them specific mutation was found. The most common form of immunodeficiency was congenital defects of phagocyte number, function, or both. (43.5%), mainly chronic granulomatous disease, followed by Well-defined syndromes with immunodeficiency (32.6%), predominantly antibody disorders (16%), combined immunodeficiencies (9.7%), Complement deficiencies (8.8%), Diseases of immune dysregulation (7.2%) and defects of innate immunity (2.1%)
Conclusion: Recent development in molecular diagnosis can help in early and definite diagnosis of PID. Accurate diagnosis of these conditions are essential for evaluation of PID burden , better decision in prevention and treatment strategies and research studies.
ESID-0045 A Novel Mutation in the SERPING1 Gene in Patients with Hereditary Angioedema and a Coexistent Complement Deficiency
D. Firinu 1, C. Zizzo2, P. Colomba2, R. Meleddu1, M.P. Barca1, P.E. Manconi1, S.R. Del Giacco1, R. Messineo2, M. Filogamo2, G. Duro2
1Department of Medical Sciences "M. Aresu", University of Cagliari, Monserrato, Italy
2Institute of Biomedicine and Molecular Immunology “A. Monroy”, National Research Council, Palermo, Italy
Hereditary angioedema (HAE) due to C1 inhibitor (C1-INH) deficiency is an autosomal dominant disorder caused by mutations at C1-INH locus (SERPING1 gene). These mutations may result in a quantitative (type I, low C1-INH antigen) or functional (type II, normal C1-INH antigen) defect which affects the complement, coagulation and contact cascades, leading to overproduction of bradikinin. Diagnosis of HAE requires clinical criteria and assay of C1-INH. Serum C1q levels are usually normal in this disorder. In HAE due to C1-INH, more than 200 different mutations are known, with high genetic heterogeneity and occurrence of de novo mutations. We analyzed the exons of the SERPING1 gene and their flanking regions, using High Resolution Melting Analysis and sequencing of DNA samples, from a single type I HAE family. The analysis of SERPING1 revealed a deletion, in heterozygous state, of five nucleotides in exon 6. This mutation, that involves 5 bases from nucleotide 1020 to nucleotide 1024 in the cDNA sequence, is a novel mutation reported here for the first time and was named c.1020_1024delGAAAG. It causes a frameshift mutation from the beginning of the open reading frame up to the amino-acid 366 and a premature termination that probably leads to the degradation of the protein. Unexpectedly, C1q levels were reduced in some family members without evidence of anti-C1q, anti-C1INH autoantibodies and lymphoproliferative disorders. We describe for the first time in HAE type I the mutation c.1020_1024delGAAAG and the rare simultaneous occurrence of C1q deficiency among some of the family members.
ESID-0401 Functional Natural Killer (NK) Cell Deficiency in Patients with Signal Transducer and Activator of Transcription (STAT1) Gain of Function Mutation (GOF)
L.R. Forbes 1, A. Vargas-Hernandez1, E.M. Mace1, A.F. Freeman2, S. Rosenzweig2, S.M. Holland2, J.S. Orange1
1Pediatrics Immunology Rheumatology & Allergy, Baylor College of Medicine Center for Human Immunobiology, Houston, USA
2National Institute of Allergy and Infectious Diseases, NIH, Bethesda, USA
Introduction: NK cells are required for human host defense, particularly against viral infections. STAT1 is required for cytokine signaling and subsequent defense against viral infection. Heterozygous autosomal GOF mutations in STAT1 have been linked to chronic mucocutaneous candidiasis and impairment of IL-17 mediated immunity. We describe a functional NK cell deficiency in patients with STAT1 GOF mutation and herpesviral susceptibility.
Objective: To evaluate NK cell phenotype and function in patients with STAT1 GOF mutations.
Methods: NK cells from four patients with confirmed STAT1 GOF mutations were studied phenotypically by FACS and evaluated for NK cell activating, inhibiting, and maturation markers as well as intracellular cytokines and lytic granule content. NK cell cytotoxic function was evaluated by standard Cr51 release assay. Survival and proliferation were measured by in?vitro culture of patient NK cells and FACS analysis.
Results: Patient NK cells displayed a seemingly immature phenotype, which had lower expression of terminal maturation markers such as CD57and CD16. Perforin was also markedly decreased, suggesting both an immature phenotype and a lowered ability for target cell lysis. Functionally, both natural cytotoxicity against susceptible target cells and ADCC were severely reduced. Proliferation and overall survival in?vitro culture were also decreased compared to normal controls.
Conclusion: Our data highlight a novel and distinct NK cell phenotype and dysfunction in patients with GOF mutations in STAT1. These NK defects likely contribute to the pathophysiology and clinical manifestations of this disease.
ESID-0756 The Extended Clinical Phenotype of 36 Patients with Chronic Mucocutaneous Candidiasis Due to Gain-of-Function Mutations in STAT1
N. Frede 1, M. Depner1, J. Raabe1, R. Doffinger2, E. Gkrania-Klotsas3, D. Kumararatne2, T.P. Atkinson4, H.W. Schroeder5, T. Niehues6, G. Dückers7, J. Puck8, E.M. Eisenstein9, A. Stray-Pedersen10, U. Baumann11, R.E. Schmidt12, J.L. Franco13, J.C. Orrego13, M. Ben-Shoshan14, C. McCusker14, C.M. Jacob15, M. Carneiro-Sampaio16, L.A. Devlin17, J.?D. Edgar18, P. Henderson19, T. Dyrsø20, S.L. Seneviratne21, J. Wanders21, H. Stauss21, I. Meyts22, L. Moens23, M. Jesenak24, B. Grimbacher1
1Centre of Chronic Immunodeficiency, University Medical Centre Freiburg, Freiburg, Germany
2Department of Immunology, Addenbrooke's Hospital, Cambridge, United Kingdom
3Infectious Diseases, Addenbrooke's Hospital, Cambridge, United Kingdom
4Department of Pediatrics, University of Alabama at Birmingham, Birmingham, USA
5Division Clinical Immunology & Rheumatology, University of Alabama at Birmingham, Birmingham, USA
6Department of Pediatrics, Helios Klinikum, Krefeld, Germany
7Pediatric Immunology and Rheumatology, Helios Klinikum, Krefeld, Germany
8Department of Pediatrics, University of California San Francisco, San Francisco, USA
9Department of Pediatrics, Hadassah-Hebrew University Medical Center Mount Scopus Campus, Jerusalem, Israel
10Department of Medical Genetics, Oslo University Hospital, Oslo, Norway
11Department of Paediatric Pulmonology and Neonatalogy, Medical University of Hannover, Hannover, Germany
12Department of Immunology and Rheumatology, Medical University of Hannover, Hannover, Germany
13Group of Primary Immunodeficiencies, Universidad de Antioquia, Medellin, Colombia
14Montreal Children's Hospital, McGill University, Montreal, Canada
15Allergy and Immunology Unit, University of São Paulo, São Paulo, Brazil
16Department of Pediatrics, University of São Paulo, São Paulo, Brazil
17Immunology Day Centre, Royal Group of Hospitals, Belfast, United Kingdom
18Department of Immunology, Royal Hospitals, Belfast, United Kingdom
19Department of Child Life and Health, University of Edinburgh, Edinburgh, United Kingdom
20Department of Clinical Genetics, Vejle Hospital, Vejle, Denmark
21Royal Free Hospital, University College London, London, United Kingdom
22Department of Pediatrics, University Hospitals Leuven, Leuven, Belgium
23Experimental Laboratory Immunology, Catholic University of Leuven, Leuven, Belgium
24Department of Pediatrics, University Hospital in Martin Jessenius Faculty of Medicine of Comenius University in Bratislava, Martin, Slovakia
The identification of genetic defects in patients with CMC now offers the opportunity to confirm the diagnosis in both familial and sporadic CMC cases, to provide genetic counselling, and to enable a more precise classification of CMC due to additional information. In this study we have explored the frequency of STAT1 mutations in a large cohort of 99 individuals from 12 unrelated CMC families and sporadic cases. We analysed 57 patients and 42 healthy family members in order to identify the unknown underlying genetic defect.
Of the 57 patients with clinical suspicion of CMC in our cohort, we identified a total of 36 patients with heterozygous STAT1 mutations. Twenty-eight of these patients came from 10 unrelated families, and eight were sporadic patients. Hence, the frequency of STAT1 mutations among all the CMC patients in our cohort is 63%. Twenty-eight out of 41 familial cases with the clinical diagnosis of CMC carried STAT1 mutations (68%) and 10 out of 12 CMC families with autosomal dominant (AD) inheritance passed on STAT1 mutations (83%). Focussing on the sporadic patients with isolated CMC, we detected STAT1 mutations in eight out of 16 sporadic patients (50%).
We will provide an in depth description of the STAT1 clinical CMC phenotype in order to provide a clear clinical picture for this specific condition.
ESID-0415 Spectrum of Mutations in Czech Patients with Wiskott-Aldrich Syndrome and X-Linked Thrombocytopenia
T. Freiberger 1, B. Ravcukova1, L. Grodecka2, E. Mejstrikova3, R. Formankova3, A. Sediva4, D. Pospisilova5, P. Sedlacek3, J. Litzman6
1Molecular Genetics Laboratory, Centre for Cardiovascular Surgery and Transplantation, Brno, Czech Republic
2Molecular Immunology and Microbiology RG, Ceitec MU, Brno, Czech Republic
3Department of Pediatric Hematology and Oncology, 2nd Medical Faculty Charles University and University Hospital Motol, Brno, Czech Republic
4Department of Clinical Immunology, 2nd Medical Faculty Charles University and University Hospital Motol, Brno, Czech Republic
5Department of Pediatrics, Faculty of Medicine and Dentistry Palacky University and University Hospital, Olomouc, Czech Republic
6Department of Clinical Immunology and Allergology, Medical Faculty Masaryk University and St. Anne's University Hospital, Brno, Czech Republic
Introduction: Wiskott-Aldrich syndrome (WAS) is a severe condition characterized by eczema, low number of small-sized platelets, immune deficiency and increased susceptibility to the development of autoimmune disease or malignancy. Genetic cause resides in defective WASP gene coding for protein Wasp, important player in regulation of actin cytoskeleton. X-linked thrombocytopenia (XLT) results from mutations in the same gene as WAS.
Material and Methods: Authors collected a set of 20 patients (from 16 families) with clinical and laboratory features consistent with diagnosis of WAS (15) and XLT (5). All 12 exons of the WASP gene, including adjacent intronic regions, were analyzed using direct sequencing.
Results: Sixteen different mutations were identified, vast majority leading to premature stop codon. Small frameshift deletion, small frameshift insertion and nonsense mutation were detected each of them in 4 cases, together accounting for 75% of mutations. One missense mutation, 1 small inframe deletion and 2 splicing affecting mutations represented remaining 25% of mutations. Nine mutations were novel.
Conclusion: Molecular basis in WAS and XLT Czech patients were determined in all 20 patients and genetic counseling including prenatal diagnostics could be offered in affected families.
ESID-0409 Allergic Asthma and Common Variable Immunodeficiency (CVID): Diagnostic Challenge
R. Morariu 1, L. Marinangeli2, C. Gelardi1, S. Gambini1, M.G. Danieli1, L. Antonicelli3
1Medicina Interna, Clinica Medica, Ancona, Italy
2Allergologia ed immunologia, Immunologia Clinica, Ancona, Italy
3Allergologia ed immunologia, Allergologia e Malattie dell'apparato respiratorio, Ancona, Italy
Common variable immunodeficiency (CVID) is a condition characterised by low immunoglobulin levels and inadequate antibodies production. Patients suffering from CVID show recurrent infections, granulomatous and autoimmune diseases.
We here report the case of a 44 years old man, admitted to our department for recurrent respiratory infections, which required several hospitalizations. Pneumonia and bronchiectasis were detected. The laboratory tests showed a severe hypogammaglobulinemia and lack of specific antibody response consistent with the diagnosis of CVID. He has been started replacement therapy with intravenous immunoglobulin (IVIg) at dose 25?g every 3 weeks, with partial clinical benefit. After six months of therapy, despite normal serum levels of immunoglobulins, respiratory symptoms persisted. Spirometry showed reversible airflow obstruction. The total IgE measured were abnormal (>130 IU/ml). Once asthma treated with LABA and ICS, we decided to treat the patient with omalizumab, an anti-IgE monoclonal antibody, which has been shown to be effective in a large number of patients with severe allergic asthma. The patient started monthly omalizumab injections with improvement in respiratory symptoms and decreased exacerbations rate.
Some patients have respiratory symptoms that might represent only the infectious complications of CVID or might effectively be manifestations of respiratory allergy. Asthma can be masked by recurrent respiratory infections. However, asthma patients are more susceptible to protracted respiratory infections, which might delay a possible diagnosis of humoral immunodeficiency.
ESID-0077 Searching for a Primary Immunodeficiency After an Invasive Pneumococcal Disease
J. Gaschignard1, C. Lévy2, M. Chrabieh1, J.L. Casanova1, A. Puel1, C. Picard 1
1Institut Imagine - Laboratory of Human Genetics of Infectious Diseases, Institut National de la Santé et de la recherche Médicale, Paris, France
2Groupe de Pathologie Infectieuse Pédiatrique, Association Clinique et Thérapeutique Infantile du Val de Marne, Paris, France
Introduction: Although some primary immunodeficiencies (PIDs) are known to confer predisposition to invasive pneumococcal diseases (IPD) in children, no systematic search for these PIDs has ever been performed in children presenting with IPD, despite a 10% mortality rate.
Methods: We prospectively collected pediatric cases of IPD requiring hospitalization, between 2005 and 2011, in pediatric units throughout France. IPD was defined as a positive pneumococcal culture, PCR result and/or soluble antigen detection at a normally sterile site. The immunological assessment included abdominal ultrasound, whole-blood counts and smears, determinations of plasma Ig and complement levels, and the evaluation of pro-inflammatory cytokines.
Results: We included 163 children with IPD (M/?F sex ratio: 1.3, median age: 13 months). Seventeen children had recurrent IPD and 10 were from consanguineous families. Meningitis was the most frequent type of infection (87%). One patient with recurrent meningitis had a congenital cerebrospinal fluid fistula. The results of immunological explorations were abnormal in 26 children (16%) and a PID was identified in 17 patients (10%), including one case of MyD88 deficiency, three of complement fraction C2 or C3 deficiencies, one of isolated congenital asplenia and two of Bruton's agammaglobulinemia. Nine children had a transient deficiency in their immunoglobulin production. The proportion of PIDs was much higher in children older than two years than in younger children (26% vs 3%, p < 0.001).
Conclusions: Children with IPD should undergo immunological investigations, particularly those older than two years, as PIDs may be discovered in up to 26% of cases.
ESID-0232 Clinical Implementation of Targeted Next Generation Sequencing for Patients with Primary Immune Deficiency and VEO-IBD Expands Genotype-Phenotype Correlation and Increases Diagnostic Yield
S. Drury1, S. Bibi1, J. Kammermeier2, S. Loughlin1, L. Jenkins1, N. Shah2, C. Cale3, N. Lench1, K. Gilmour 3
1North East Thames Regional Genetics, Great Ormond Street Hospital, London, United Kingdom
2Gastroenterology, Great Ormond Street Hospital, London, United Kingdom
3Immunology, Great Ormond Street Hospital, London, United Kingdom
Primary immunodeficiencies (PID) and very early onset inflammatory bowel disease (VEO-IBD) are life-threatening, genetically heterogeneous disorders with considerable phenotypic overlap. Within the North East Thames Regional Genetics Laboratory we have developed and introduced into routine practice targeted next generation sequencing to simultaneously investigate 98 PID and VEO-IBD associated genes with a sensitivity of 97.6%. 19 PID genetic diagnoses have been made, including in genes for which there was previously no diagnostic genetic service (e.?g. CD3e, CD3d, ZAP70). 7 VEO-IBD diagnoses have been made in 6 genes. Novel, atypical presentations of PIDs and VEO-IBD have been identified including presentations of IL7R (presented with T-B-NK+ SCID), MAGT1-XMEN (presented with ALPs), and STAT1 (presented with HLH) among others. This panel is faster and more cost effective than individual gene screening (e.?g. £1500 per panel compared to an average of £500 per gene). Gene panel screening has played a critical role in determining appropriate treatment and clinical management of patients; for example, in PID and VEO-IBD, cases have been identified where haematopoietic stem cell transplant (HSCT) may be curative (e.?g. mutations in IL10 and DOCK8) and the HSCT has been avoided in a VEO-IBD patient where a mutation in EPCAM was identified.
ESID-0785 Strategies for Screening, Diagnosis and Monitoring of Primary Immunodeficiencies in Medellin Colombia, by Group of Primary Inmunodeficiencies Grip
D. Gongora 1, J. Orrego1, J. Franco1, S. Ortiz1
1University of Antioquia, Group of primary inmunodeficiencies, Medellin, Colombia
In 1994, we began a surveillance inmunodeficiencies program for abnormal recurrent infection in Aburrá Valley (Antioquia, Colombia) and since then the task of detection of primary immunodeficiencies in our region has been provided. However the best diagnostic scope has been achieved in the last 5 years and the GIDP has managed to approach the rate of diagnosis of IP described in the international literature, from 6 to 10 per 100,000 inhabitants. This has been made possible by three pillars in the group: 1 health care area: Consultation and clinical laboratory , achieving over the past 5 years, a 300% growth in consultations regarding the last years, improving the diagnostic rate by population in the metropolitan area, across 25 tests were implemented in our laboratory. 2 academic field: research and teaching, where 50 undergraduates and 105 posgraduate has been receiving education in immunology by six teachers who were assigned to the medical faculty of the University of Antioquia, authors of a number of 42 publications in national and international magazines. 3 The social support of the patients, diagnosed by the foundation for IP Diana Garcia de Olarte , fundraising in total 5,000 dollars per year, by providing laboratory tests , internal and external medical consultation, advising the patient and family through 5 events and programs to include the patient and family in the process of health and disease. All this has given us recognition as a reference center for diagnosis, monitoring and training for primary immunodeficiency in our country (Colombia)
ESID-0784 Strategies for Screening, Diagnosis and Monitoring of Primary Immunodeficiencies in Medellin Colombia, by Group of Primary Inmunodeficiencies Grip
D. Gongora 1, J. Orrego1, J. Franco1, S. Ortiz1
1University of Antioquia, Group of primary inmunodeficiencies, Medellin, Colombia
In 1994, we began a surveillance inmunodeficiencies program for abnormal recurrent infection in Aburrá Valley (Antioquia, Colombia) and since then the task of detection of primary immunodeficiencies in our region has been provided. However the best diagnostic scope has been achieved in the last 5 years and the GIDP has managed to approach the rate of diagnosis of IP described in the international literature, from 6 to 10 per 100,000 inhabitants. This has been made possible by three pillars in the group: 1 health care area: Consultation and clinical laboratory , achieving over the past 5 years, a 300% growth in consultations regarding the last years, improving the diagnostic rate by population in the metropolitan area, across 25 tests were implemented in our laboratory. 2 academic field: research and teaching, where 50 undergraduates and 105 posgraduate has been receiving education in immunology by six teachers who were assigned to the medical faculty of the University of Antioquia, authors of a number of 42 publications in national and international magazines. 3 The social support of the patients, diagnosed by the foundation for IP Diana Garcia de Olarte , fundraising in total 5,000 dollars per year, by providing laboratory tests , internal and external medical consultation, advising the patient and family through 5 events and programs to include the patient and family in the process of health and disease. All this has given us recognition as a reference center for diagnosis, monitoring and training for primary immunodeficiency in our country (Colombia)
ESID-0742 Clinical Features in was Mexican Patients
M. Gonzalez-Serrano 1, P. Rubio-Mortera1, E. Cruz-Jaramillo2, H. Gomez-Tello3, S.?C. Scheffler-Mendoza1, E.G. Lopez-Rocha4, N.H. Segura-Mendez4, N.D. Lopez-Lara5, M.?A. Yamazaki-Nakashimada6, S.E. Espinosa-Padilla1, S.O. Lugo-Reyes1
1Immunodeficiency Research Unit, Instituto Nacional de Pediatría, Mexico City, Mexico
2Medicine, Universidad Autonoma Metropolitana, Mexico City, Mexico
3Allergy and Clinical Immunology, Hospital del Niño Poblano, Mexico City, Mexico
4Immunology, CMN SXXI, Mexico City, Mexico
5Immunology, Hospital del Niño de Toluca, Mexico City, Mexico
6Immunology, Instituto Nacional de Pediatría, Mexico City, Mexico
In our clinical series we collected data from 15 male patients with WAS compatible phenotype from 15 unrelated families seen at four referal centers between 2005 to 2014.
Results are reported as median and range.
Age at clinical onset 2 (1-26), diagnosis 14 (2-57) and delay 8 (0-54) months respectively.
Earliest diagnosis achieved due to sibiling previously diagnosed. The background of male affected in the family was presented in 20 percent of the patients.
28% of patients had recurrent infections at diagnosis, eccema and thrombocytopenia was present in all the patients.
The most common infections in the patients: sepsis 75%, pneumonia 46%, gastroenteritis 46%, otitis media 33%, sinusitis 13%. Chronic diarrhea in 40%.
At least 60% and 46% of patients were hospitalized because an infectious process and bleeding before the diagnosis, respectively. Three patients deceased, because of bleeding.
Seric immunoglobuline levels (mg/dL) at diagnosis were IgG 984 (695-1560), IgM 79 (17-173), IgA 143 (40-428), IgE UI/L 170 (1-1360)
Ten of the patients had WASP determination by flow cytometry in CD3+ cells, expression was abscent in 8, 1 patient shows bimodal pattern and 1 patient shows diminished expression. Four of the patients has genetic diagnosis.
4 patients presented neutropenia associated with concurrent infections.
Splenectomy was performed on two patients, whom presented autoimmune manifestations: reactive arthritis (1/?15) and hemolitic anemia (1/?15). HSCT was performed on 7 patients, among them two were successful. 9 patiens received GGIV replacement specially those diagnosed within the last 5 years. 11 patients received antibiotic prophilaxis.
ESID-0477 Transition of Primary Immunodeficiency Patients into Adult Health Care
K. gotoh 1, M. Teramachi1, K. Ishimoto1, Y. Tanaka1, Y. Ohtsu1, N. Tsumura1, T. Matsuishi1
1pediatrics and child health, Kurume University, Fukuoka, Japan
With advances in antibiotics, IVIG and bone marrow transplantation, many primary immunodeficiency (PID) children can survive into adulthood. To provide the optimal health care for these youth and adult, subspecialty pediatricians have to do planning for transition of care to adult health care providers.
We have 5 PID patients over 18 years old. Those of three are Bruton-type agammaglobulinemia patients and two are chronic granulomatous disease patients (The age of 18 to 43 years). For every patient, we are trying to do planning for transition of care to adult health care. But two patients rejected transition because of their good and long term confidence of pediatrician. And the cases of being completed of transition, two patients came back to our hospital from adult health care within 1 year. We have experienced one case of successful transition to adult health care. From our experience, we suggest some opinions.
Referring providers should have a written policy for the transfer of PID youth to adult care, which will guide in the development of an individualized plan for each patient. Assessment of developmental milestones is important to define the readiness of the youth in assuming responsibility for their own care before initiating the transfer. Communication among all providers is essential and should include both personal contact and a written medical summary. Progress toward the transition should be tracked and, once completed, should be documented and assessed.
ESID-0743 A Successful Application of Next Generation Sequencing to the Diagnosis of Adaptive Primary Immunodeficiency
P. Gray 1, C. Walsh2, G. Elakis2, M. Buckley2, J. Ziegler1, T. Roscioli3
1Immunology and Infectious diseases, Sydney Children's Hospital, Sydney, Australia
2Genetics laboratory, SEALS, Sydney, Australia
3School of Women's and Children's Health, University of New South Wales, Sydney, Australia
Adaptive Primary immunodeficiencies (A-PIDs) include more than 100 known single gene diseases which affect lymphocyte function, and which are associated with predisposition to infection. Next generation sequencing (NGS) technologies have demonstrated increasing utility in the diagnosis of single gene disorders including A-PID. We aimed to assess the clinical utility of the haloplex target enrichment system, in the identification of disease causing mutations in genes associated with A-PID. DNA was collected from 6 patients with A-PID caused by known mutations in genes associated with X-linked and autosomal recessive A-PIDs, and from 6 patients with an A-PID where the causative gene had not been identified. Samples were analysed using a haloplex panel including 100 known primary immunodeficiency genes.
All 6 known disease-associated variants were identified. One of the unknown patients received a new diagnosis of RAG1 deficiency, associated with a previously reported homozygous 2 base pair deletion. No diagnosis was made in the other 5 patients. A number of variants were discounted as artefacts by parallel analysis of multiple patients. The haloplex system provided good coverage of adaptive Primary immunodeficiency genes with an excellent diagnostic yield. We recommend parallel analysis with multicalling of samples as a method to reduce systematic errors within NGS processes.
ESID-0366 New Insights into the Immune System Abnormalities in Nijmegen Breakage Syndrome Versus Ataxia-Telangiectasia in Aspect of Malignancy Predisposition
H. Gregorek 1, B. Wolska-Kusnierz2, B. Pietrucha2, A. Zapasnik3, D. Smolka-Afiffi3, E. Heropolitanska-Pliszka2, E. Bernatowska2, K. Chrzanowska4
1Microbiology and Clinical Immunology, The Children's Memorial Helath Institute, Warsaw, Poland
2Immunology Department, The Children's Memorial Helath Institute, Warsaw, Poland
3Microbiology and Immunology Department, The Children's Memorial Helath Institute, Warsaw, Poland
4Department of Clinical Genetics, The Children's Memorial Helath Institute, Warsaw, Poland
Introduction. Nijmegen breakage syndrome (NBS) and ataxia-telangiectasia (AT) are rare, autosomal recessive chromosomal instability disorders characterized by combined immunodeficiency, commonly termed as “profound”. Given that NBN and ATM proteins play related, but disparate, functional roles in the maturation of immunocompetent cells one might expect that the profile and severity of the immune system disturbances in those syndromes must differ significantly.
Objective. To characterize malignancies, profile and severity of the immune system deteriorations in both disorders in a long-term follow-up study.
Material. Clinical and immune status of 50 NBS and 40 AT patients were followed. Serum Ig’s, IgG subclasses, B and T cell subpopulations, lymphocyte proliferation, C3, C4, CIC, viral infections, BCR/TCR gene rearrangements, monoclonal gammopathy were systematically monitored.
Results. Although in both groups of patients similar frequency of the immune system abnormalities was observed, there were evident differences in the profile of deficient Ig’s isotypes, absolute number of B and T cell subpopulations, frequency and type of BCR/TCR clonal gene rearrangements, and frequency of lymphatic malignancies. Generally, abnormalities in patients with AT were less severe and more stable over the time. In contrast to NBS, EBV-DNA load in AT patients was low, stable or even no detectable at the end of study.
Conclusions. This study clearly showed fundamental differences in malignancy incidence and abnormalities of the immune parameters between NBS and AT patients, which confirmed different biological function of NBN and ATM proteins in the lymphocyte maturation.
This study was supported by grant no. S123/?2012 funded by CMHI, Warsaw, Poland
ESID-0054 Serum Free Light Chain Assay as an Adjunct to Immunofixation Electrophoresis for Early Detection of Monoclonal B-Cell Disorders in Patients with Nijmegen Breakage Syndrome
H. Gregorek1, B. Pietrucha 2, K.H. Chrzanowska3, A. Zapasnik1, E. Heropolitanska-Pliszka1, A. Wakulinska4, W. Kaminska1, K. Dzierzanowska-Fangrat1
1Microbiology and Clinical Immunology, The Children's Memorial Health Institute, Warsaw, Poland
2Immunology, The Children's Memorial Health Institute, Warsaw, Poland
3Clinical Genetics, The Children's Memorial Health Institute, Warsaw, Poland
4Oncology, The Children's Memorial Health Institute, Warsaw, Poland
Introduction. Nijmegen breakage syndrome (NBS) is a rare chromosomal instability disorder clinically characterized by microcephaly, immunodeficiency, and a high predisposition to lymphoid malignancies with generally fatal course. Therefore, the search for new biomarkers useful for early detection of changes preceding malignant transformation seems to be of great clinical importance.
Objective. To assess whether a sensitive serum free light chain (sFLC) immunoassay and sFLC ratio (sFLCr) may improve early detection of monoclonal gammopathy in patients with NBS.
Material and Methods. Serum samples were collected from 38 patients with defined NBS at a time when they did not need anti-cancer treatment, and afterwards at 6 months intervals during control visit. In each sample sFLC were measured by immunonephelometry, and immunofixation electrophoresis (IFE) was performed by the agarose gel method.
Results. Monoclonal components (MC) were detected in 14 (37%) of 38 patients; 78% (11/14) were sFLC-positive and 43% (6/?14) were IFE-positive. MC was detectable only by sFLC analysis in 57% (8/?14) and only by IFE in 21% (3/?14). During observation, 15 (39%) patients developed histopathologically confirmed lymphatic malignancies. All patients who developed B-NHL (20%) and two patients who died without confirmed diagnosis were positive for monoclonal serum FLC.
Conclusions. The emergence of new immunoassay specific for the detection of serum FLC may provide a more sensitive laboratory tool to detect monoclonal proteins in NBS patients at the early stage of B-cell clonality.
This study was supported by internal grants no. S123/?2012 and 219/12 funded by CMHI, Warsaw, Poland
ESID-0668 A Case of Chronic Lymphocytic Leukaemia with Expression of CD8 Antigen on Leukemic Cells, IGG2 Subclass Deficiency, and Co-Infection with Epstein-Barr Virus
E. Grywalska 1, A. Surdacka1, M. Maj1, A. Grafka1, M. Pasiarski2, A. Stelmach-Goldys2, P. Steckiewicz2, S. Gozdz3, J. Rolinski1
1Department of Clinical Immunology and Immunotherapy, Medical University of Lublin, Lublin, Poland
2Department of Hematology, Holycross Cancer Center, Kielce, Poland
3Department of Chemotherapy and Clinical Oncology, Holycross Cancer Center, Kielce, Poland
Chronic lymphocytic leukaemia (CLL) is phenotypically characterized by the co-expression of CD19, CD5, CD20, and CD23 antigens. The concomitant presence of surface markers characteristic for other leukocyte subsets is uncommon. CD8 antigen is present on the surface of cytotoxic-suppressor T cells, NK cells, thymocytes, and is undetectable on normal B cells. CLL expressing the CD8 antigen is described with the frequency lower than 0.5% of all cases. These cases are not fully characterized.
Herein we report a case of a CD8+ CLL patient with an extensive phenotypical characterization, immunoglobulin serum levels assessment, as well as virological studies.
A 55-yr-old man was diagnosed as having CLL stage 1 according to Rai classification, ZAP-70- and CD38-negative. The patient had 86.5% CD19+/CD5+/CD8+ leukemic lymphocytes. Lymphocyte doubling time was 6 months, lymphadenopathy and organomegaly appeared after 7 months and the treatment (chemoimmunotherapy – FCR –fludarabine, cyclophosphamide, rituximab) was introduced 9 months after the diagnosis. The patient finished 6 courses of FCR, however the progression was noted 12 months after the treatment with enlargement of superficial lymph nodes. Immunoglobulin assessment revealed IgG2 deficiency. Primary symptom was recurrent fever 38-38.5% of unknown aetiology but the patient had no serious bacterial infections. Virological screening revealed 244 Epstein-Barr virus (EBV) DNA copies/?ug DNA. The percentage of CD19+/CD5+/CD38+ cells amounted to 86%, and CD19+/CD5+/CD8+ - 90.7%. We show that the co-expression of the CD8 antigen is more than a coincidental phenomenon. It seems that this abnormality may be related to aggressive course of CLL, subclass deficiency and EBV reactivation.
ESID-0736 Education and PID Awareness Among Medical Students
S. Guner 1, N. Reisli1, E. Hazar Sayar1, I. Reisli1
1Pediatric Allergy and Immunology, Meram Medical School, Konya, Turkey
The objective of this study was to assess PID awereness before and after clinical immunogy education among medical students. One hundred and thirty-two questionnaires with 71 items were distributed to seventh somestre medical students and 116 (88%) completed questionnaires were evaluated before and after their education about clinical immunology courses for 6 hours.
Mean of the first QS was 37.4 ± 3.7 and second QS was 42 ± 4.3 (p<0.05). The correct responses rate less than 50% before education were 10 of 39 questions. All participants corrected their responses after education. The best improvement was detected in the responses of the clinical signs related with PID. It was remarkable that the participants have known the family history related with PID excellent before education.Chronic diarrhea due to giardiasis, neonatal tetany, petechia, dermal scarring lesions, microcephaly, short limb dwarfism and tetralogy of Fallot were not accepted as a part of PID before education.80% believed that a lymphocyte count of 2500/?mm3 was related to immunodeficiency. NBT and CH50 test were not found to be related with PID before education. The participants showed confusion about the evaluation of innate immunity because of lack of information concerning major clinical features of these together with some problems in the evaluation of laboratory tests such as NBT and CH50. It is also important to increase the awareness of PID among the the physicians during their education in medical school and more comprehensive education in PID appears to be useful for medical students
ESID-0592 Clinical Presentation of Hyper-IGE Syndrome in a Family with Impaired IL-22 Production and STAT3 Phosphorylation
F. Haerynck 1, D. Bogaert2, E. De Baere3, B. Lambrecht4, K. Vermaelen5, V. De Backer5, K. Van Schil3, F. De Baets1, P. Schelstraete1, V. Bordon6, M. Dullaers5
1Pediatric Pulmonology and Immunology, University Hospital Ghent, Ghent, Belgium
2Clinical Immunology Research lab - Pediatric Pulmonology and Immunology, University Hospital Ghent, Ghent, Belgium
3Center of Medical Genetics, University Hospital Ghent, Ghent, Belgium
4Department of Respiratory Medicine, University Hospital Ghent- VIB Inflammation Research Center, Ghent, Belgium
5Department of Respiratory Medicine- Clinical Immunology Research lab, Ghent University, Ghent, Belgium
6Pediatric Hemato-oncology and pediatric bone marrow transplant center, University Hospital Ghent, Ghent, Belgium
Recurrent bacterial and fungal infections, eczema, and increased serum IgE levels characterize patients with Hyper-IgE syndrome (HIES). Known genetic causes of HIES are mutations in signal transducer and activator of transcription 3 (STAT3) and dedicator of cytokinesis 8 (DOCK8).
We describe a non-consanguineous family with the clinical presentation of HIES. Th17 immunity was analyzed in peripheral blood mononuclear cells and STAT3 and DOCK8 gene sequencing was performed.
The father presents with recurrent skin infections and cauda equina syndrome related to a spinal cord abcedation. The 10-years old boy represents with recurrent skin and teeth abcedations, sinusitis, oral candidiasis, retained primary teeth, eczema, recurrent bone fractures after minor trauma, and slightly increased serum levels of IgE (500 kU/l) with a HIES score of 42. The younger sister represents with recurrent skin abcedation, eczema, and candidiasis. While IL-17 production was on the lower end but still within normal range, IL-22 production was impaired in all 3 family members. In addition, STAT3 phosphorylation upon IL-6 stimulation was defect in all patients monocytes but normal in lymphocytes. DOCK8 and STAT3 gene sequencing did not return any causative mutations. Currently, whole exome sequencing is pending. Although the boy has the highest HIES score, all three family members have significant impaired IL-22 production and deficient STAT3 phosphorylation.
We represent a family with the clinical presentation of hyper-IgE syndrome and impaired IL-22 production and deficient STAT3 phosphorylation in monocytes but normal in lymphocytes. No DOCK8 and STAT3 mutations were found. Further immunologic and genetic analysis is ongoing.
ESID-0586 Association of Common Variable Immunodeficiency (CVID) with a Rapidly Progressive and Widespread Central Nervous System Inflammation
S. Hanson 1, M.?A. Ibrahim1
1Department of Clinical Immunology, King's College Hospital, London, United Kingdom
Common Variable Immunodeficiency (CVID) are a heterogeneous group of primarily humoral immune deficiencies, characterised by failure to produce appropriate circulating immunoglobulin. A subset of individuals diagnosed with CVID manifest autoimmune disease predominantly autoimmune haemolytic anaemia, idiopathic thrombocytopenia purpura, rheumatoid arthritis and pernicious anaemia.
We present the case of an individual diagnosed with CVID of early onset, harbouring a heterozygous mutation in the NFKB2 gene and experiencing a rare rapidly progressive neuroinflammatory process. Extensive Inflammation within the central nervous system with severe involvement of the brain stem, basal ganglia, spinal cord and cerebellum, exhibited a constellation of neurological signs and symptoms including visual impairment, ataxia, spasticity of the lower limbs, choreiform movements of the upper and lower limbs, dysphagia and cognitive disturbance with hypomania.
Bacterial and/or viral drivers of inflammation were not identified by either culture or nucleic acid amplification test (NAAT) methodologies.
Immunosuppression in the form of corticosteroids induced a temporary remission in symptoms, further attempts at halting the inflammatory process using cyclophosphamide were complicated by neutropenia and respiratory infection. Targeted B-cell therapy using an anti-CD20 monoclonal antibody (rituximab) resulted in a Type I hypersensitivity reaction, a fully human anti-CD20 monoclonal antibody Ofatumumab was administered without incident.
The advanced nature of the CNS inflammatory process and intervention required to support the patients feeding, unfortunately resulted in the patient’s death.
This case highlights the importance of rapidly excluding potential infective causes of neurological signs and symptoms, aggressively seeking evidence of neurological inflammation and initiation of appropriate immunosuppression.
ESID-0241 Clinical, Laboratory Features and Outcomes of Patients with DOCK 8 Deficiency: Report of 6 Cases
S. Haskologlu 1, F. Dogu1, S. Keles2, C. Aytekin3, I. Reisli2, A. Ikinciogullari4, T. Chatila5, A. Ikinciogullari1
1Pediatric Immunology and Allergy, Ankara University Faculty of Medicine, Ankara, Turkey
2Pediatric Immunology and Allergy, Meram University Faculty of Medicine, Konya, Turkey
3Pediatric Immunology and Allergy, Ankara Dr. Sami Ulus Child Health and Disease Training and Research Hospital, Ankara, Turkey
4Pediatric Immunology and Allergy, Ankara Numune Training and Research Hospital, Ankara, Turkey
5Division of Immunology, Harvard Medical School, Boston, USA
Introduction: DOCK8 deficiency is a combined immunodeficiency(CID) characterized by recurrent viral infections, severe atopy, and early onset malignancy. Immune assessment showed T cell lymphopenia, hyperIgE, hypoIgM, and eosinophilia. Diagnosis can be established by demonstration of large homozygous or compound heterozygous deletions, although point mutations and splice site mutations. The only definitive treatment option is allogeneic hematopoietic stem cell transplantation (HSCT).
Material ve Methods: Medical records of six patients diagnosed with DOCK8 deficiency, between 2008 - 2014, were evaluated retrospectively.
| Clinical and Laboratory Features of Patients | ||||||
| Patients | P 1 | P 2 | P 3 | P4 | P 5 | P 6 |
| Gende r/?Age at diagnosis(year) | M/3 | F/4 | M/4 | F/1 | M/14 | F/4 |
| Consanguinity | + | + | + | + | + | + |
| Atopic dermatitis | + | + | + | + | + | + |
| RTI | + | + | + | + | + | + |
| Malignancy | - | - | Plasmocytoma | - | Plasmocytoma | - |
| High serum IgE | + | + | + | + | + | + |
| Eosinophilia | + | + | + | + | + | + |
| Decreased T cells | + | + | + | + | + | |
| Low NK cells | - | + | + | - | - | - |
| Low serum IgM | + | + | + | + | - | + |
| LPR to PHA | Low | Low | Low | Low | Low | Low |
| Site of DOCK 8 gene mutation | Exon 18 | Exon 28 | Exon 26 | Exon 18 | Exon 5 | Proceeding |
| Treatment | HSCT | HSCT | HSCT | IVIG, IFN a | IVIG, IFN a | IVIG, IFN a |
| Outcomes | *A/W | A/W | A/W | A/W | A/W | A/W |
*:Alive / Well
CONCLUSION: DOCK8 deficiency is a common form of CID in Turkey. HSCT should be performed prior to development of malignancy and fatal infections.
ESID-0344 Purine Nucleoside Phosphorylase (PNP) Deficiency: Clinical and Immunologic Features of 4 Cases and Outcome
S. Haskologlu 1, F. Dogu1, C. Aytekin1, F. Cipe1, M. Yüksek1, M. Hershfield2, A. Ikinciogullari1
1Pediatric Immunology and Allergy, Ankara University Faculty of Medicine, Ankara, Turkey
2Department of Biochemistry, Duke University Medical Center, Durham, USA
Introduction:Purine nucleoside phosphorylase (PNP) deficiency is a rare form of autosomal recessive combined primary immunodeficiency caused by an enzyme defect leading to the accumulation of purine metabolites in all cells, especially lymphocytes. PNP deficiency characterized by recurrent infections, neurological dysfunction, and autoimmunity. Treatments are available and curative for PNP deficiency, but their efficacy depends on the early approach.
| Clinical features and treatment of the patients | ||||
| Case I | Case II | Case III | Case IV | |
| Gender Age at diagnosis | M / 4 yrs | M / 13mo (brother of 1st case) | F / 13mo | F / 11mo |
| Parental Consanguinity | + | + | - | + |
| Infections | Diarrhea Pneumonia Sepsis | - | Disseminated varicella |
RTI Diarrhea |
| Autoimmunity | No | No | No | No |
| Neuro-developmental symptoms |
Truncal hypotonia Developmental delay |
Spasticity Developmental delay |
Hypotonia Spasticity |
Hypotonia Spasticity |
| Uric acid(UA) levels (mg/dl) | 0,1 | 0,1 | 0,2 | 0,5 |
| Genetic defect | c286-18G>A | c286-18G>A | c286-18G>A | c.172C>T |
| Treatment | IVIG | HSCT | HSCT | HSCT |
| Outcome | Died with sepsis and MOF |
A/W Some neurological problems At posttransplant 10 yrs |
A/W At posttransplant 3,5 yrs |
A/W At posttransplant 8 mo |
*: Alive and well
CONCLUSION: Recurrent infections, autoimmune diseases, neurological findings and low serum UA levels should be considered PNP deficiency. Early diagnosis and HSCT reverse the poor prognosis.
ESID-0644 Wiskott-Aldrich Syndrome – A Delayed Diagnosis
M. Hatzistilianou 1, M. Tzanoudaki2, M. Massaad3, A. O'Connel3, E. Kapravelou1, E. Papadopoulou1, V. Doulioglou1, M. Kanariou2, R. Geha3
12nd Department of Paediatrics, Aristotle University of Thessaloniki, Thessaloniki, Greece
2Department of Immunology - Histocompatibility Specialized Center & Referral Center for Primary Immunodeficiencies - Paediatric Immunology, "Aghia Sophia" Children's Hospital Athens Greece, Athens, Greece
3Division of Allergy/ Immunology/Rheumatology/ Dermatology Children's Hospital, Harvard Medical School Boston MA USA, Boston, USA
Introduction: Wiskott-Aldrich syndrome is an X linked primary immunodeficiency, characterized by a malfunction of both T and B lymphocytes. The mean age of diagnosis is 21 months, but delayed diagnosis is still of great concern.
Case report: We report the case of a 5-year-old boy with past medical history of thrombocytopenia since the age of 40 days of life, bearing the diagnosis of ‘idiopathic thrombocytopenic purpura”. He also was reported to have recurrent infections, and bleeding from nose, ear and gastrointestinal tract. The boy was admitted in our unit for the first time at the age of 4.5 years, because of thrombocytopenia and bleeding from his right ear. He had no family history of hereditary diseases. Examination revealed petechiae and ecchymoses at the upper and lower limbs and oral mucosa, eczema at his face with crusting from pruritus. Laboratory results: IgM 17 mg/dl, IgG 917 mg/dl, IgA 281 mg/dl, total IgE 402 IU/ml. The peripheral blood lymphocyte immune phenotype showed: CD3+ 47%, CD3+/CD4+ 22%, CD3+/CD8+ 14%, CD19+ 21% and CD3-/CD16+/CD56+ 28%. He had a borderline proliferative response to PHA and anti-CD3. WAS protein expression on the patient’s T cells was totally undetectable by flow cytometry. In the sequence analysis a homozygous deletion was identified of 8?bp in exon 7 (48545236-48545243), leading to a frameshift and premature stop of codon 9 after the deletion.
Conclusion: This case report underlines the delayed diagnosis problems of the Wiskott – Aldrich Syndrome.
ESID-0248 Reversal of the Ability to Respond to Polysaccharide Antigens as a Possible Cause of Recurrent Respiratory Tract Infections in the Elderly
M.H. Haverkamp 1, E. van de Vosse1, J.T. van Dissel1
1Infectious Diseases, Leiden University Medical Center, Leiden, Netherlands
Background
Children under age two respond poorly to capsular polysaccharides but mount adequate humoral responses to protein antigens. Whether the capacity to respond to polysaccharides, subsequently acquired, remains permanent over life or may be lost in late adulthood is unknown. Such loss may play a role in susceptibility to respiratory tract infections (RTI) in the elderly.
Methods
Patients aged over 45 with recurrent RTI (>2 documented infections in the last year, but no recurrent RTI in adolescence or early adulthood) were retrospectively selected from our outpatient database. We obtained antibody responses to 11 polysaccharides in Pneumovax-23, before and one month after vaccine administration, and anamnestic responses to protein antigens (e.?g., VZV, rubella). In addition, we determined gammaglobuline levels, complement and CD4-counts. Patients with IgG<5 g/?L were excluded.
Results
Over a period of 4 years, an estimated 22 patients fulfilled the selection criteria. Of these, ten patients (45%, median age 57 years (r.45-76)) were deficient in responses to Pneumovax-23 with antibody titers < 1 µg/ml to > 5:11 serotypes, despite normal IgG (mean 10,2 g/?L). Four out of six patients had either no or low protection to H.?influenza. All patients responded to protein antigens and had normal CD4-counts. Five patients were MBL deficient.
Conclusion
Elderly patients with ‘late onset’ RRTI may be deficient in their ability to mount humoral responses to capsular polysaccharides despite normal IgG and humoral immunity to proteins. This suggests that polysaccharide responses may be lost during adult life, and helps explain susceptibility to RRTI in some.
ESID-0367 Secondary Combine Immunodeficiency in Patients with Hypoplastic Left Heart Syndrome - 2 Case Reports
E. Hlavackova 1, M. Vlkova1, D. Humlova2, H. Jicinska2, J. Litzman1
1Department of Clinical Immunology and Allergy, St Anne University Hospital in Brno, Brno, Czech Republic
2Department of Pediatrics - Pediatric Cardiology, The University Hospital Brno, Brno, Czech Republic
Hypoplastic left heart syndrome (HLHS) is a rare congenital cardiac defect. Univentricular circulation after series of palliative surgeries leads in 12-25% patients to protein-losing enteropathy.
We present two patients suffering from HLHS complicated by secondary intestinal lymphangiectasia with clinical signs of protein-losing enteropathy. Laboratory results revealed combined immunodeficiency.
Patients were referred at the age of 9 and 17 for recurrent uncomplicated respiratory tract infections without opportunistic or sever bacterial, viral or fungal infections history.
Immunological examination revealed severe hypogamaglobulinaemia, mainly of IgG and also IgA isotypes. IgM levels decrease was present only in one patient. Severe lymphopenia was attributed mainly to depletion of CD4+ cells, these cells were almost exclusively CD45R0 positive. Both patients had very low levels of antibodies against tetanus toxoid and pneumococcal capsular polysaccharide antigens with insufficient response to vaccination against both antigens in one vaccinated patient. Progressive hypoalbuminaemia and hypoproteinaemia correlated with the protein-losing entheropathy severity. Clinically edema, malnutrition and grow retardation was present.
Introduction of antibiotic prophylaxis and subcutaneous immunoglobulin substitution led to decrease of infection courses; however one of the patients died aged 17 years of heart failure during Campylobater jejuni enteritis. The second patient is currently waiting for heart transplantation.
Protein-losing enteropathy is a life expectancy limiting complication of the patients with HLHS. Patients with HLHS need a multidisciplinary care and should be followed by immunologist. Antibiotic prophylaxis and subcutaneous immunoglobulin substitution, when indicated, improves the life quality of these patients.
ESID-0699 Safety and Efficacy of Tetanus Vaccination in Hematopoietic Cell Transplant Recipients Due to Primary Immunodeficiency Diseases
V. Horakova 1, J. Skovrankova1, A. Sediva1, P. Keslova2, R. Formankova2
1Institute of Immunology, Faculty Hospital Motol, Prague, Czech Republic
2Department of Pediatric Hematology and Oncology, Faculty Hospital Motol, Prague, Czech Republic
Vaccination is a routine part of post-transplant care.
Inactivated vaccines can be administered 6-12 months after HSCT.
We retrospectively reviewed all children with primary immunodeficiency diseases who have undergone allogeneic HSCT in the University Hospital Motol between 1997-2012 and have been vaccinated in our center. Our aim was to evaluate tetanus post vaccination response of those patients. We observed the significant rise in specific antibody levels or rise to level considered protective during follow-up.
32 patients were enrolled. Patient diagnoses were: severe combined immunodeficiency (SCID) (11), T cell immunodeficiency: Omenn´s syndrome (1) Wiskott-Aldrich syndrome (4) hyper IgM syndrome (3), X-linked lymphoproliferative disease (1), phagocytic cell disorders: familial hemophagocytic lymphohistiocytosis (5), Chediak Higashi syndrome (1), chronic granulomatous disease (5), leucocyte adhesion defect (1). Immunization was postponed if patients were treated with rituximab (a mouse-human chimeric anti CD 20 monoclonal antibody) until at least six months after the last dose of rituximab due to depletion of B cells.
Results:
Majority of our patients acquired or maintained titers in the protective range.
There were no differences between groups with respect to type of stem cells source (umbilical cord blood, peripheral blood stem cells, bone marrow) or diagnosis. Repetitive administration of rituximab due to autoimmune hemolytic anemia was often associated with vaccination failure though the patients were vaccinated at least six months after the last dose of rituximab and had a normal B cell count. Only one patient from this group developed a vaccine side reaction, a toxoallergic exanthema.
ESID-0659 Keeping Up with Progress in the Field of Immunology and Primary Immunodeficiencies. Results of a Survey Among ESID Members
P. Olbrich 1, L.I. Gonzalez-Granado2, J. Feberova3, M. Rizzi4, A. Janda5
1Department of Pediatric Infectious Diseases and Immunopathologies, Hospital Infantil Universitario Virgen del Rocío, Sevilla, Spain
2Immunodeficiencies and Pediatric Infectious Diseases Division, Hospital 12 Octubre, Madrid, Spain
3Department of Science Information, 2nd Medical Faculty of Medicine Charles University, Prague, Czech Republic
4Center for Chronic Immunodeficiency (CCI), University Medical Center Freiburg, Freiburg, Germany
5Center for Pediatric and Adolescent Medicine, University Medical Center Freiburg, Freiburg, Germany
Introduction: The field of immunology and primary immunodeficiencies (PID) is rapidly evolving and keeping up with the latest knowledge is challenging.
Objective: To collect and analyse data from professionals regarding their use of internet-based multimedia tools.
Methods: A brief online survey has been designed and distributed via email among all ESID members. Anonymous data collection was performed using a commercial internet platform.
Results: Thirty-one percent (n=136) of the current ESID members (median age 39 years, range from 24 to 77) participated in the survey. The majority were physicians (81%), and active in patient care (77%). Seventy-three percent of the respondents regularly checked electronic journals, majority via electronic table of content, only a few (8%) used more advanced techniques as RSS, printed journals were checked by 39%, most of whom were older than 50 years of age (71% versus 31%, p = 0.013); 46% used alert services, 37% followed meta-journals. Evidence-based resources were used by 44% of the respondents, most of whom were younger than 50 years of age (53% versus 15%, p=0.015). Podcast were very scarcely applied (4%), while employment of online networking service was indicated in 56%. Automatic services for following citations of own publications were used by 36% respondents.
Conclusion: Our data show that the vast possibilities of online information services are not always fully utilized. In our presentation details on the current experience of the ESID members as well as an overview of easy-to-use and relevant resources will be provided, stressing their potency for PID professionals.
ESID-0737 Poor CD4+CD25HIGH Lymphocytes Recovery Post Hematopoietic Stem Cells Transplantation Associated with Donor CMV IGG Antibodies Negativity and Patients Immunoglobulin Reconstitution and Affects Patients Outcome
E. Jaskula 1, D. Dlubek1, K. Suchnicki2, A. Tarnowska2, J. Lange2, A. Borowik2, M. Mordak-Domagala2, M. Sedzimirska2, S. Mizia2, A. Lange1
1Clinical Immunology, L. Hirszfeld Institute of Immunology and Experimental Therapy Polish Academy of Sciences/Lower Silesian Center for Cellular Transplantation with National Bone Marrow Donor Registry, Wroclaw, Poland
2Clinical Immunology, Lower Silesian Center for Cellular Transplantation with National Bone Marrow Donor Registry, Wroclaw, Poland
Among risk factors associated with aGvHD and survival post HSCT the recipient/donor IgG CMV serostatus plays a significant role. Ninety-nine patients with hematological malignancies receiving HSCT were followed to assess the risk of Herpes viruses reactivation. The presence of herpes viruses (CMV, EBV or/and HHV6) reactivation events were associated with the presence of aGvHD (16/44 vs. 11/55, p?=?0.069). The risk of both aGvHD (0.420±0.0512% vs. 0.488±0.032%, p=0.033; 1.692±0.225x10^6 cells/l vs. 3.778±0.484x10^6 cells/l, p<0.001) and Herpes viruses reactivation (2.180±0.349x10^6 vs. 3.482±0.477x10^6 cells/l, p=0.006; 0.370±0.030% vs. 0.539±0.045%, p<0.001) were associated with low values of CD4+CD25high lymphocytes in the first wave of lymphocytes reconstitution post-transplant. The levels of CD4+CD25high lymphocytes were screened for their association with the HSCT outcome. Patients having <0.4% of CD4+CD25high lymphocytes at hematologic recovery had inferior survival.
The low levels of CD4+CD25high lymphocytes in the blood at hematologic recovery were associated with the donation from CMV IgG antibodies negative individuals (22/47 vs. 11/50 p=0.018). Patients receiving IgG immunoglobulins support before 100?day post HSCT had lower number of CD4+CD25high lymphocytes (1.934±0.284x10^6 vs.3.554±0.473x10^6 cells/l, p=0.007).
CD4+CD25high lymphocytes present in the blood at hematologic recovery originate from the transplant material and may therefore reflect the level of regulatory cells in donors prior to donation. Poor CD4+CD25high lymphocytes recovery post HSCT associated with a lack of the immune responsiveness against CMV in donors affect the immunological reconstitution post HSCT seen as a higher risk of Herpes viruses reactivation, a higher demand of IgG support and poor survival.
Supported by the NN402430039 grant
ESID-0559 Common Variable Immune Deficiency Case
I. Jaunalksne1, T. Romanova 1, N. Sirotina1, M. Jersova1
1Clinical immunology center, Pauls Stradins Clinical University Hospital, Riga, Latvia
Common variable immune deficiency can present with multiple phenotypes all of which are characterized by hipogammaglobulinemia.
Our aim was to monitor patient suffering from CVID during sc Ig therapy. We use laser flow cytofluorimeter method for lymphocyte subpopulation evaluation and nephelometric method for humoral immunity parameter detection.
Our patient had traditional clinical signs- rhinitis, frequent ARVI,bronchitis, recurrent pneumonia, allergic reactions to antibiotics, anemia. Cellular immunity was decreased by CD4, CD16 absolute counts with inverted CD4/CD8 ratio. Humoral immunity was characterized by reduced CD19, IgG, IgA, IgM levels and hypogammaglobulinemia. Patient received sc immunoglobulin treatment for the last 3 years. Despite the therapy IgG levels only it increase up to 7.5 g/?l. Patient was followed by b2 microglobulin, it stayed elevated during the monitoring and patient was still leukopenic, neutropenic. Question is if there is still a ongoing autoimmune process against leukocytes, neutrophils? And is additional treatment required?
ESID-0483 Report on the Moroccan Registry of Primary Immunodeficiencies
F. Ailal 1, N. Amenzoui1, L. Jeddane1, H. Ouair2, N. El Hafidi2, I. Benhsaien1, J. Najib1, S. Benmiloud2, A. Kili2, A. Aglaguel2, Z. Aadam2, L. Ait Baba2, N. Benajiba2, N. Rada2, R. Abilkassem2, M. Hida2, J. El Bakkouri2, H. Salih Alj3, A.A. Bousfiha1
1Clinical Immunology Unit, Faculty of Medicine and Pharmacy, Casablanca, Morocco
2Registry Study Group, Moroccan Society for Primary Immunodeficiency (MSPID), Casablanca, Morocco
3Laboratory of Biology and Health URAC34—Metabolic and Immunologic pathology Research Team, Faculty of Science of Ben M'sik King Hassan II University, Casablanca, Morocco
Purpose: Primary immunodeficiencies (PID) are a large group of diseases, characterized by an increased susceptibility to infections. We report here actual data on the Moroccan Registry for Primary Immunodeficiencies. Methods: A registry was established in 2008 and gathered data on PID patients diagnosed since 1998. Results: A total of 469 patients were diagnosed between 1998 and 2013. Globally, 28.1% were classed as combined immunodeficiency with associated or syndromic features; 22.4% as predominantly antibody deficiencies; 19.4% as T- and B-cell combined Immunodeficiencies; and 17.3% as phagocyte disorders. Parental consanguinity within patients reached 44.1% and a median time to diagnosis of 2.0 years was observed. Mortality reached 28.1%, and only 14 patients received bone marrow transplantation. Conclusions: The PID prevalence observed is 0.8/100,000 inhabitants in Morocco, which show a large underdiagnosis in our country. The PID distribution is different of what we observe in Occident, especially with emergence of autosomal recessive diseases and overrepresentation of severe phenotype.
ESID-0713 Experiences with Icatibant in Adolescent Girl with Hereditary Angioedema
M. Jesenak 1, P. Banovcin1
1Centre for Hereditary Angioedema Department of Pediatrics, Comenius University in Bratislava Jessenius Faculty of Medicine in Martin, Martin, Slovakia
Hereditary angioedema (HAE) is a rare disease presented with recurrent skin or mucosal swelling. Several medicament have been developed for treatment of acute attacks, however, in some drugs data for adolescents and especially children are missing. Acute attacks could be in adolescent age treated either with plasma derived C1-inhibitor or the application of ecallantide since the age of 16. Self-administration of the drugs by patient during acute attacks is challenging. Icatibant is one of the newest drugs for the treatment of acute attacks and it is approved only for adult patients, however, the efficacy and advantages for adolescents could be expected.
We report a case of 16-years old girl with positive family history for hereditary angioedema and with the diagnosis of HAE type I. The symptoms started at the age of 14 with the facial skin sweeling. The frequency of the attacks is approximately 4 per year involving limbs swelling and abdominal symptoms. Due to poor access to the health care at home (living in the country side), we decided after the achievement of Approval from Ministry of Health of Slovak Republic, Health Insurance Company and Institutional Ethic Committee to recommend icatibant for rescue therapy. Till today, 4 application of the injections have been performed. The relief of the symptoms was within 2.5 hours on average. The therapy was well tolerated, without any significant local or systemic side effects. Icatibant could enlarge the possible therapeutic options for adolescent patients with HAE, however, controlled trials are missing and should be performed.
ESID-0156 Clinical and Molecular Features of 38 Children with Chronic Granulomatous Disease in Mainland China
L. Jiang 1, H. Xu1, W. Tian1, L. Zhang1, X. Tang2, Z. Zhang2, X. Zhao1, X. Yang1
1clinical immunology laboratory, Children's Hospital of Chongqing Medical University, Chongqing, China
2Department of Immunology and Nephrology, Children's Hospital of Chongqing Medical University, Chongqing, China
Purpose: Chronic granulomatous disease (CGD) is an inherited disorder, with phagocytes failing to produce antimicrobial superoxide due to deficient NADPH oxidase activity. Mutations in the gene encoding CYBB are responsible for the majority of the CGD cases. To date, there have been no reports on large samples of children with CGD in China. Therefore, in this study, we describe the clinical and molecular features of 38 suspected CGD patients from 36 unrelated Chinese families.
Methods: Clinical diagnosis was performed using dihydrorhodamine assays detected by flow cytometry. Molecular analysis was used to identify underlying CGD-causative genes.
Results: The mean age of onset in our 38 patients was 3.4 months, while the mean age at diagnosis was 31.7 months. Apart from recurrent pneumonia and abscesses, tuberculosis (TB) and Bacille Calmette-Guerin (BCG) infections were notable features in our cohort. Overall, 17 cases died and patient 1 did not participate in the follow-up period. In total, we identified 29 different CYBB gene mutations in 31 patients. We found NCF1 and CYBA mutations in 3 and 2 patients, respectively. In addition, we identified 30 carriers and prenatally diagnosed 4 CGD and 4 healthy fetuses.
Conclusions: The results of our study demonstrate that children with BCG infections or recurrent TB infections should have immune function screening tests performed. Moreover, newborns with family histories of primary immunodeficiency diseases should avoid of BCG vaccination. Molecular analysis is an important tool for identifying patients, carriers, and high-risk CGD fetuses.
ESID-0197 Lymphomas in Common Variable Immunodefficiency: A Single Center Experience
M. Bouzani1, I. Kakkas 2, G. Kourti1, F. Michelis1, E. Grigoriou2, A. Tsirogianni2, T. Karmiris1, N. Harhalakis1
1HAEMATOLOGY-LYMPHOMA, EVAGGELISMOS HOSPITAL, ATHENS, Greece
2IMMUNOLOGY-HISTOCOMPATIBILITY, EVAGGELISMOS HOSPITAL, ATHENS, Greece
Common Variable Immunodeficiency (CVID) patients are prone to autoimmune disorders, granulomatous diseases and malignancies. In the past thirty years, many studies established the association between CVID and lymphoma. The cumulative risk of Hodgkin's Lymphoma (HL) and Non Hodgkin Lymphoma (NHL) affecting the patients with CVID is estimated to range between 2-9%.
In the past 25 years, 75 CVID patients were diagnosed in our hospital. During the same period, 400 patients with HL and 800 patients with NHL were also diagnosed in our hospital. In this study we describe seven cases of patients with CVID, who developed Lymphoproliferative Diesease (LPD, either HL or NHL). All the patients were males aged 16 to 55 years old, and CVID had been identified several years before the diagnosis of LPD. Four HL and three NHL (Diffuse Large B Cell) were developed. In all cases the malignancy was extensive, presenting multiple extranodal infiltrations. Three patients with HL achieved complete response and remain disease free after the end of treatment. One patient with HL had refractory disease. From patients with NHL, one achieved a complete response and the disease reappeared four years later and the other two had only a partial response of the disease. Four patients, one with HL and all the three with NHL have died with progressive disease one to three years after the diagnosis of the LPD.
Conclusively, it is obvious that CVID patients necessitate a close follow up for the prompt diagnosis of a LPD.
ESID-0340 The Early Recognition Program of Primary Immunodeficiencies in the Ural Region
M. Karakina 1, I. Tuzankina1
1Laboratory of inflammation immunology, Institute of Immunology and Physiology, Yekaterinburg, Russia
The majority of Primary immunodeficiencies (PID) is rare diseases which poorly identification in routine clinical practice. Therefore we have developed and now we are implementing the early recognition program of primary immunodeficiencies in our region. It includes:
1. Analysis of PID - debut clinical manifestation;
2. Analysis of PID clinical manifestation;
3. Evaluation of the death causes in PID patients;
4. The population laboratory testing (in the framework of the annual preventive examinations) to identify any cytopenias and hypogammaglobulinemia;
5. Investigations in so-called "target" groups:
- Chromosome 22q11 deletion syndrome – the relatives of patients with already identified chromosome 22q11 deletion syndrome, the persons with schizophrenia, patients with certain phenotypic features of this syndrome (congenital heart disease, "cleft palate" dysplastic facial features, nasal speech, hypocalcemic seizures, persons who had any problems in education (pupils of speech schools and nursery schools)) and persons with low levels of TREC / KREC;
- Wiskott-Aldrich syndrome – the persons with thrombocytopenia, impaired platelet adhesion and aggregation, mikrotrombocytosis.
- Nijmegen syndrome – the persons with microcephaly;
- The relatives of children with a reduced TREC / KREC in neonatal screening survey;
- The persons with different localization infectious during the application of monoclonal antibodies to different links of immunity factors.
ESID-0095 Analysis of Adult Patients with Primary Immunodeficiency Pedigrees with Using Genealogical Markers
M. Karakina 1, V. Shershnev2, I. Tuzankina1, O. Nicolaenco3
1Laboratory of inflammation immunology, Institute of Immunology and Physiology, Yekaterinburg, Russia
2Instituion of PromEcology, Ural Branch on Russian Academy of Science, Yekaterinburg, Russia
3Therapeutic Department, Medical Center "Zdorovie 365", Yekaterinburg, Russia
The investigation of pedigrees in patients with primary immunodeficiency (PID) is an important method for determining the inheritance of the disease. This method is also used in genetic counseling of families with PID members. However, the families of PID patients may have some members with individual phenotypic features of these diseases without the development of typical clinical manifestations. Therefore, we have identified such phenotypic features as severe infectious diseases, allergic, autoimmune diseases, tumours, reproductive pathology, child deaths, congenital malformations and angioedema in the group of so-called genealogical markers to which we should pay special attention in the genealogy investigation and PID- suspicion.
The goal of this study: identification of genealogical markers in adult patients with PID.
Methods: we have analyzed the pedigrees of 92 Ural Region register PID adult patients and 218 adults without PID. We measured the percentage of relatives with some pathologies for at least four generations.
We have carried out considered quartile analysis and definition of statistical significance of differences between the groups with and without PID (Kruskal - Wallis test).
Results: the most informative markers are severe infections and allergies (p <0,001). Such markers as tumour and reproductive pathology were not detected (p>0,4).
ESID-0029 The Study of Wasp-Gene Mutations in the Relatives of One Patient Having Wiskott-Aldrich Syndrome
M. Karakina 1, S. Deriabina1, E. Vlasova2, I. Tuzankina1
1Laboratory of inflammation immunology, Institute of Immunology and Physiology, Yekaterinburg, Russia
2Depatment of clinical immunology, Region Children Hospital 1, Yekaterinburg, Russia
In our Center of Clinical Immunology we are observing one patient with Wiskott-Aldrich syndrome. This is a boy who is 10. The diagnosis - Primary immunodeficienty: Wiskott-Aldrich syndrome was made at the age of 8 months. The Genetic identification of WASP-gene mutations was performed in the Primary Immunodeficiency Center (Debrecen, Hungary), in 2009. The name of the mutation is splace site WASP mutation (IVS3+1g>a). The boy has a big family, his parents are young and healthy. He has an older brother and a younger sister and a cousin sister related to the mother and cousins ?? related to his father.
The goal of this study is to recognize WASP-gene mutations with the relatives of the patient with Wiskott-Aldrich syndrome for the prognosis of the disease they could have.
Methods: genealogical, genetic investigations. For search and identification of this mutation we sequenced exon 3 of WASP-gene in 8 relatives of the proband.
Results: we don't have any WASP-gene mutations in exon 3 in the relatives of the patient with Wiskott-Aldrich syndrome. Therefore this mutation is sporadic in our patient and his family.
ESID-0726 Insulin-Like Growth Factor -1- and Interleukin -6- as Key Cytokines in Pathogenesis of Multiple Myeloma
N. Kassem 1, H. Kassem1, M. Erian2, O. Shaker3
1Clinical pathology department, Kasr Al Ainy, cairo, Egypt
2oncology department, Kasr Al Ainy, cairo, Egypt
3Biochemistry department, Kasr Al Ainy, cairo, Egypt
Rationale: Increased angiogenesis has recently been recognized in active multiple myeloma (MM) and is associated with poor prognosis. The underlying mechanism for increased angiogenesis in MM remains unclear, with various factors implicated such as interleukin-6 (IL6) and insulin Growth Factor-1 (IGF1) which promote the proliferation and survival of myeloma cells. Purpose: The present work is intended to study the level of IL-6 and IGF-1, their role in the pathogenesis of MM and to define the effect of thalidomide on BM angiogenesis and angiogenic cytokines when used as initial therapy. Materials and method: This study includes 40 newly diagnosed MM patients referred to Kasr AL Aini Centre of Clinical Oncology and Radiation-Cairo University during the period 2010-2012. ELISA technique was used to measure IGF-1and IL-6 in subjects' sera. Results: This study showed that IGF-1 and IL-6 post treatment were lower than pretreatment levels but IGF-1 level was more significantly responding to treatment (102.8 ± 61 ng/ml before vs. 65.7 ± 51.4 ng/ml after, P value 0.028). Conclusion: Our study delineates the importance of anti-angiogenic drugs such as thalidomide against MM and further suggests the clinical utility of novel treatment paradigms targeting not only the tumor cell directly, but also cellular interactions and cytokine secretion in the BM milieu. We suggest that IL6 and IGF-I should be further studied in future clinical trials as useful monitoring biomarkers for MM.
ESID-0800 Novel Mutations in Two Korean Boys with Wiskott-Aldrich Syndrome and Normal Range of Mean Platelet Volume
J.M. Kim1, J.M. Kang1, J.H. Kim1, K.H. Yoo1, K.M. Ahn1, Y.H. Choe1, C.H. Park2, E.S. Kang2, H.J. Kim2, Y.J. Kim 1
1Pediatrics, Samsung Medical Center Sungkyunkwan University School of Medicine, Seoul, Korea
2Laboratory Medicine and Genetics, Samsung Medical Center Sungkyunkwan University School of Medicine, Seoul, Korea
Background: Wiskott Aldrich Syndrome (WAS) is defined as an X-linked hereditary disorder associated with adaptive and innate immune deficiency, microthrombocytopenia, eczema, and an increased risk of autoimmune disorders and malignancy.
Cases: Patient 1 presented with thrombocytopenia and known CMV viremia from outside hospital. He was initially diagnosed with idiopathic thrombocytopenic purpura (ITP) and was tread with steroid and intravenous immunoglobulin (IVIG). At our hospital, he developed respiratory distress for which Pneumocystis jirovicii pneumonia (PCP) was confirmed and was treated with TMP/SMX. For persistent CMV viremia, he received ganciclovir. His WAS gene test revealed a novel nonsense mutation, NM_000377.2:c.321C>G (p.Tyr107*). His current condition is stable and is due for allogeneic hematopoietic stem cell transplantation (HCT)
Patient 2 initially presented with blood stool and eczema. He was evaluated for milk allergy but eventually diagnosed with WAS with persistent thrombocytopenia. His WAS gene test revealed a novel nonsense mutation, NM_000377.2: c.308C>A (p.Ser103*). This patient’s parents were non-compliant and he presented to pediatric intensive care unit with hematocrit of 10.4% and hypovolemic shock due to massive gastrointestinal bleeding following an ear infection. He was successfully resuscitated and was empirically treated with acyclovir and broad-spectrum antibiotics for suspected HSV or pyogenic meningoencephalitis since he also developed seizure. He restarted TMP/SMX prophylaxis. His current condition is stable and is due for HCT. Interestingly, mean platelet volume (MPV) in both patients was within normal ranges (7.5 f. and 8 fL).
Conclusion: We report two novel nonsense mutations in the WAS gene which were identified in Korean boys.
ESID-0154 HIES Clinical and Immunological Presentations (Report of Single Center Experience)
I. Kondratenko 1, O. Pashchenko2, S. Vakhlyarskaya3, B. Bragvadze2, A. Bologov3
1Clinical Immunology, Russian Clinical Children's Hospital, Moscow, Russia
2Immunology, Russian National Scientific Medical University, Moscow, Russia
3Clinical Immunology, Russian Clinical Children’s Hospital, Moscow, Russia
The autosomal dominant Hyper IgE syndrome (AD-HIES) is characterized by recurrent staphylococcal soft tissues, lung and liver infections, skeletal abnormalities, markedly elevated IgE levels. We observed 12 patients with AD-HIES confirmed by clinical and laboratory criteria. All patients had total HIES HIN score >50 points. In 5 of 5 evaluated STAT3 mutations were found. IgE levels: <500 IU/ml – 2, 500-2000 IU/ml – 2, >2000 IU/ml – 8; elevated eosinophils counts- in 8. Cold soft tissue abscesses developed in all patients: skin – 9, lymph nodes – 3, liver – 3, lung – 1. Pneumonia developed in 9 with pneumatocele formation in 6, enterocolitis – 2, meningitis – 1, sepsis – 8, mucocutaneous candidiasis - 7, BCG-associated abscess in 1 patient. Kidneys abscesses developed in two patients, in one case after pyelonephritis, on other case it was found occasionally. In last case 14 years girl was observed for 9 years. She is a second child in non consanguineous family, old sister is healthy. Since 7 days of life recurrent bacterial skin, INT, lymph nodes and lungs infections, skin candidiasis. HIES was diagnosed in 5 years (total score – 65, mutation of STAT3: c.1144C>T). Treatment: prophylaxis IVIG, trimetoprim-sulphometocsazole, itraconazole, parenteral antibacterial drugs while infectious episodes. During a routine examination at the age of 14 years was found kidney abscess without clinic and laboratory inflammatory symptoms. Drainage of the abscess was performed. Patient received IVIG and parenteral antibiotics with a good effect.Our observation confirm that HIES patients should regularly undergo clinical and instrumental examination even if they do not have any complaints.
ESID-0145 Anti-Interferon-Gamma Autoantibodies in Adults with Disseminated Nontuberculous Mycobacterial Infections are Associated with HLA-DRB1*16:02 AND DQB1*05:02
C.Y. Chi1, C.H. Lin2, C.L. KU 2
1Graduate Institute of Clincal Medical Science, China Medical University, Taichung, Taiwan
2Graduate Institute of Clincal Medical Sciences, Chang Gung University, Taoyuan, Taiwan
Adult patients with disseminated nontuberculous mycobacterial (dNTM) infections usually have severe immune system defects. Recently, several reports have shown that anti-IFN- gamma autoantibodies may play an important role in the pathogenicity of dNTM infections. A considerable proportion of reported cases of anti-IFN-gamma autoantibodies show either clinical or laboratory evidence of autoimmune disease. In this study, we identified 19 formerly healthy adults who later developed dNTM infections, and 17 were further investigated immunologically. High-titer anti-IFN-gamma autoantibodies capable of inhibiting IL-12 production in vitro were found in the plasma of all of these patients. In addition to dNTM infection, 35% and 71% of our patients also suffered from salmonellosis and herpes zoster, respectively. This observation suggests that IFN-gamma may be crucial in controlling salmonella infection and reactivating latent varicella-zoster virus (VZV) infection in humans. Two HLA alleles, DRB1*16:02 DQB1*05:02 (odds ratios 8.68, 95% CI: 3.47-21.90, P=1.1×10-6, Pc=3.08×10-5 and 7.16, 95% CI: 3.02-17.05, P=1×10-7, Pc=1.4×10-6, respectively), were found in 82% (14/17) of our patients. In conclusion, our data suggest that anti-IFN-gamma autoantibodies may play a critical role in the pathogenesis of dNTM infections and reactivation of latent VZV infection and are associated with HLA-DRB1*16:02 and DQB1*05:02.
ESID-0673 The Outcome of Allogeneic Hematopoietic Stem Cell Transplantation Following Liver Transplantation in Children with Immune Deficiencies: A Single Institution Experience
Z. Kucuk 1, J.J.H. Bleesing1, M. Grimley1, S. Jodele1, M. Alonso2, G.M. Tiao2, J.J.H. Bucuvalas3, S. Kocoshis3, N. Yazigi3, L. Filipovich1
1Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children's Hospital Medical Center, Cincinnati, USA
2Pediatric General and Thoracic Surgery, Cincinnati Children's Hospital Medical Center, Cincinnati, USA
3Gastroenterology Hepatology and Nutrition, Cincinnati Children's Hospital Medical Center, Cincinnati, USA
Background: Case reports of allogeneic hematopoietic stem cell transplantation (HSCT) following liver transplantation (LT) in children with immunodeficiencies have been published in the past and have described correction of the underlying disease.
The objective of this study is to report the clinical outcomes of a series of pediatric recipients of HSCT following previous liver transplantation.
Method: Retrospective analysis of 8 patients who underwent HSCT following previous liver transplantation between 2003-2012. IRB was approved.
Results: Seven patients (4 males, 3 females) underwent orthotropic LT for fulminant or chronic liver disease of unknown etiology; 1 male patient received segmental LT for alpha-1 antitrypsin deficiency. Seven patients received unrelated donor HSCT and 1 patient had a matched sibling donor. Six of 8 patients received reduced intensity conditioning (RIC), 2/8 received myeloablative conditioning prior to HSCT. All patients were later diagnosed with hemophagocytic lymphohistiocytosis (HLH), 7/8 with negative genetic studies for known genes of HLH, 1 patient with XIAP deficiency. All patients engrafted. Three patients died before day 100, 1 patient who is currently known to be alive has been lost to follow up 19 months after HSCT, 4 patients are alive for +4 years with good life quality. Two patients still receive low dose of immune suppressive drugs for liver transplant.
Conclusion: HLH may present with fulminant or cryptogenic liver failure requiring correction with LT prior to HSCT. LT followed by unrelated donor HSCT using RIC can lead to complete immune reconstitution, discontinuation of anti-rejection medications and resolution of HLH.
ESID-0352 Optimising the Investigation of Chronic Diarrhoea in Common Variable Immune Deficiency
L. Leeman 1, C.A. Bethune1, C. Symons1, T. Trump1, J. Gilliam1, T. O'Shea1
1Clinical Immunology and Allergy Service, Derriford Hospital, Plymouth, United Kingdom
Gastrointestinal (GI) conditions are common in patients with common variable immune deficiency (CVID) who are at increased risk of GI infection as well as numerous disease related complications, e.g, villous atrophy, nodular lymphoid hyperplasia, lymphoma and GI autoimmune conditions. Patients with CVID related enteropathy have been shown to have a poorer prognosis than other CVID phenotypes. Experience from our clinic indicated that a large number of our patient cohort suffered with chronic diarrhoea of un-determined cause and that this incidence may well be under-reported due to associated stigma and the patients’ perception that diarrhoea is unrelated to immune deficiency. We also lacked a clear, effective and timely investigation plan for these complications due to unclear boundaries of care between the immunology and gastroenterology departments.
A number of service developments were therefore initiated with the following aims;
Increasing awareness among patients and staff that chronic diarrhoea is a recognised CVID complication, has significant quality of life implications and that certain aetiologies can impact upon disease prognosis.
Assessing prevalence of chronic diarrhoea and malabsorption within our cohort.
Develop a multi-stage investigation plan for chronic diarrhoea, which is tailored to CVID, cost effective, acceptable to patients, easily instigated by staff and can be used to identify patients needing specialist review.
The poster will display our current investigation plan for chronic diarrhoea in CVID and clinical outcomes for a series of patients. We identify areas for further development and reflect upon positive clinical outcomes and difficulties encountered.
ESID-0625 Evaluation of Reversal of IGAD: Relevance for the Diagnosis in Children
C. Lim 1, C. Dahle2, K. Elvin3, B. Andersson4, J. Rönnelid5, L. Truedsson6, L. Hammarström1
1Laboratory Medicine, Karolinska Institutet, Stockholm, Sweden
2Clinical and Experimental Medicine, Linköping University, Linköping, Sweden
3Medicine, Karolinska Institutet, Stockholm, Sweden
4Immunology, Sahlgrenska University Hospital, Gothenburg, Sweden
5Immunology Genetics and Pathology, Uppsala University, Uppsala, Sweden
6Laboratory Medicine, Lund University, Lund, Sweden
Immunoglobulin A deficiency (IgAD) is the most common primary immunodeficiency in the general population. It is defined as a serum IgA levels below 0.07 g/l in the presence of normal levels of other immunoglobulin isotypes in an individual older than 4 years of age. A few cases of reversion of IgAD have previously been observed, especially in pediatric patients. This study aimed to investigate the frequency of reversal in order to evaluate present definition of IgAD so as to propose an improved definition to prevent premature diagnosis and unnecessary treatment in pediatric patients. Clinical laboratory records from pediatric patients where IgA levels were routinely measured were retrieved from the Karolinska hospital in Stockholm, the Sahlgrenska hospital in Gothenburg, the University hospital in Lund, the University hospital in Linköping and the Academic hospital in Uppsala. In total, 9 out of 39 (23.1%) children who were identified as IgAD at 4 years of age increased their serum IgA level above 0.07 g/L at an average age of 9.53±2.91 years of age. In addition, 30 out of 131 (22.9%) children diagnosed as IgAD between 5 – 9.99 years of age reversed their serum IgA level as they grew older. The average age of reversal was 12.21 ± 3.43 years of age. The results indicate that the use of four years of age as a cut off for IgAD may not be suitable. The present observations may contribute to the development of more efficient diagnostic workups for childeren with suspected IgAD.
ESID-0316 Atopic Dermatitis as a Manifestation of Serious Immunodeficiency
M. Liska 1, T. Votava2, A. Sediva3, P. Sedlacek4
1Department of Immunology and Allergology, Medical Faculty Hospital Plzen Charles University Prague, Plzen, Czech Republic
2Department of Paediatrics, Medical Faculty Hospital Plzen Charles University Prague, Plzen, Czech Republic
3Institute of Immunology, Medical Faculty Hospital Motol Charles University Prague, Prague, Czech Republic
4Department of Paediatric Haematology and Oncology, Medical Faculty Hospital Motol Charles University Prague, Prague, Czech Republic
Omenn syndrome is primary immunodeficiency assigned to severe combined immunodeficiency (SCID) group. It is an autosomal recessive disease characterized by presence of oligoclonal activated T cells which infiltrate skin and gut.
2 months old boy was admitted to Department of Paediatrics of University Hospital in Pilsen for impetiginization of atopic dermatitis. Microbiological examination proved massive colonization with Staphylococcus aureus and the treatment with intravenous antibiotics was started. Immunological examination showed low levels of immunoglobulin, decreased B cell counts and decreased proliferation response of T cells to mitogen stimulation. Treatment with intravenous immunoglobulins was added. More detailed examination of lymphocytes showed increased proportion of activated T cells, low Treg counts and significantly decreased TCR excision circles (TRECs) and B cell kappa chain excision circles (KRECs). Genetic examination proved RAG2 mutation and Omenn syndrome. The boy was transferred to Department of Paediatric Haematology and Oncology of University Hospital in Prague-Motol and allogeneic cord blood stem cells transplantation was successfully performed. Unfortunately, the patient died 5 month later due to cardiac arrest.
Omenn syndrome is a rare primary immunodeficiency which rapidly leads to death without bone marrow transplantation. It is accompanied with atopic dermatitis or erythroderma, so it is important to consider possible primary immunodeficiency in children with severe and atypically manifesting atopic dermatitis.
ESID-0171 Anti-GAL IGG, IGM and IGA Antibodies in Sera of the Patients with Primary Hypogammaglobulinemia, Newborns and Adult Blood Donors
M. Hamanova1, Z. Chovancova1, V. Thon1, J. Litzman1, J. Lokaj 1
1Department of Clinical Immunology and Allergy, Faculty of Medicine Masaryk University St. Anne University Hospital in Brno, Brno, Czech Republic
Natural antibodies to a variety of different antigens are commonly detected in normal individuals. Anti-Gal (Galalpha1-3Galbeta1-4GlcNAc-R) antibodies are the most prominent in humans; they are continuously produced throughout the life as an immunological response to antigenic stimulation by the gut microbiota.
Quantitative, isotype-specific (IgM, IgG, IgA) determination of anti-Gal by a new enzyme immunoassay in 33 patients with CVID (18 females, 12 males) and 30 patients with sIgAD (18 females, 12 males), 348 healthy adult blood donors (158 females, 190 males) was performed. Anti-Gal antibodies were also determined in cord blood samples obtained from newborns during delivery (n = 41), from these infants aged 6 months (n=26), 12 months (n = 16) and 24 months (n= 23).
Anti-Gal in sera from healthy blood donors showed wide interindividual variability. IgM, but not IgG and IgA anti-Gal antibody concentrations showed significant decrease with age.
In patients with CVID we observed a significant decrease of anti-Gal IgM and IgA antibodies, the normal levels of IgG anti-Gal reflects replacement therapy of the patients. SIgAD patients had undetectable concentration of IgA, but levels of IgM and IgG were within the physiological range. Evaluating cord blood no detectable anti-Gal IgM and IgA were present. The increase towards the normal adult levels in IgM isotype was markedly quicker than in IgG and IgA.
Determination of anti-Gal antibodies may broaden the spectrum of available specific antibodies that can be used for determination of specific antibody response in physiological and pathological situations.
ESID-0177 Skin Involvement in Selective IGA Deficiency (SIGAD) and Common Variable Immunodeficiency (CVID): An Underestimated Cause of Co-Morbidity
V. Lougaris 1, G. Gualdi2, T. Lorenzini3, M. Andolini4, P. Monari2, G.P. Calzavara-Pinton2, A. Plebani3
1Department of Clinical and Experimental Sciences, Pediatrics Clinic & Ins.for Molecular Medicine A.NocivelliUniversity of Brescia, Brescia, Italy
2Department of Clinical and Experimental Sciences, Dermatology Clinic University of Brescia, Brescia, Italy
3Department of Clinical and Experimental Sciences, Pediatrics Clinic & Ins. for Molecular Medicine A.NocivelliUniversity of Brescia, Brescia, Italy
4Department of Clinical and Experimental Sciences, Pediatrics Clinic & Ins. for Molecular Medicine A.NocivellUniversity of Brescia, Brescia, Italy
Skin manifestations may represent an important part of the clinical presentation in several forms of primary immunodeficiencies such as granulocyte defects, Hyper-IgE syndrome or severe combined immunodeficiencies (SCIDs). However, data regarding detailed skin manifestations in primary humoral immunodeficiencies are scarse. We decided therefore to evaluate the type and prevalence of skin manifestations in Selective IgA deficiency (SIgAD) and Common Variable Immunodeficiency (CVID) in a longitudinal, prospective study. One hundred two patients with SIgAD (M:F 0,55: 0,45; median age at diagnosis: 8,6 years) and forty seven patients with CVID (M: F 0,57: 0,43; median age at diagnosis 23,4 years) were included in this study. Routine dermatological evaluation included clinical evaluation and skin biopsy where necessary; the Hanifin-Rajka criteria for atopic dermatitis were applied.
Regarding SIgAD, 59/102 patients (58%) presented atopic dermatitis (AD), 6/102 (6%) acne and 2/102 (2%) skin infections. Regarding CVID, 9/47 (20%) patients presented atopic dermatis (AD), 9/47 (20%) presented psoriasis, 7/47 (15%) recurrent skin infections, 6/47 (7%) acne, while another 9/47 (20%) patients presented recurrent aphthosis, alopecia areata and vitiligo. It appears evident that certain manifestations such as AD and psoriasis are particularly frequent in SIgAD and CVID respectively, with higher prevalence than previously reported, and are appropriately diagnosed only when dermatological specialist evaluation is performed during follow-up.
Routine clinical management of patients with SIgAD and CVID should take into consideration the elevated prevalence of skin involvement in these disorders.
ESID-0217 Transmembrane Activator and CAML Interactor (TACI) Mutations in Pediatric-Onset Common Variable Immunodeficiency (CVID)
V. Lougaris 1, M. Vitali1, M. Baronio1, T. Lorenzini1, M. Andolini1, G. Tampella1, A. Soresina1, R. Badolato1, A. Plebani1
1Department of Clinical and Experimental Sciences, Pediatrics Clinic & Ins.for Molecular Medicine A.NocivelliUniversity of Brescia, Brescia, Italy
Mutations in TRANSMEMBRANE ACTIVATOR AND CAML INTERACTOR (TACI) are present in 8-10% of Common Variable Immunodeficiency (CVID) patients. However, no studies have been published yet regarding TACI mutations in exclusively pediatric-onset CVID patients. We investigated 51 pediatric-onset CVID patients for mutations in TACI and correlated clinical and immunological data with genetic alterations in TACI. Ten out of 51 patients (19,6%) presented disease associated mutations in TACI. Mean follow-up ranged from 9 years for TACI mutated patients to 22.5 years for wild type ones. Mean age of onset and immunoglobulin serum levels at diagnosis between mutated and wild type patients were similar. Invasive infections were present in the wild type patients (3/41) but absent in the TACI mutated ones. The frequency of lung infections (pneumonia) was particulary high among wild type patients (19/41) (46,3%) when compared to mutated ones (1/10) (10%). Autoimmune phaenomena such as autoimmune haemolytic anemia, celiac disease, scleroderma and others were only present in the wild type CVID patients (16/41) and absent in the TACI mutated ones. Finally, splenomegaly was present in 6/10 (60%) TACI mutated patients and 16/41 (39%) wild type ones. Our data suggest that pediatric-onset CVID patients carrying disease-associated mutations in TACI present a milder clinical history with absence of known causes of co-morbidity such as autoimmune phaenomena and recurrent lung infections.
The clinical consequences of these findings are still a matter of debate.
ESID-0236 Sensorineural Hearing Loss (SNHL) in Common Variable Immunodeficiency (CVID): Immunological and Clinical Features
V. Lougaris 1, M. Berlucchi2, M. Baronio1, F. Pellegrini3, S. Calzavara1, D. Moratto1, M. Vitali1, G. Tampella1, C. Balzanelli4, A. Plebani1
1Department of Clinical and Experimental Sciences, Pediatrics Clinic & Ins.for Molecular Medicine A.NocivelliUniversity of Brescia, Brescia, Italy
2Spedali Civili di Brescia Brescia Italy, Pediatric Otorinolaringoiatry Unit Spedali Civili di Brescia Brescia Italy, Brescia, Italy
3University of Insubria, Pediatrics Clinic Varese Circle Hospital, Varese, Italy
4Spedali Civili di Brescia Italy, Audiology Unit Otorinolaringoiatric Clinic, Brescia, Italy
Sensorineural hearing loss (SNHL) was recently reported by our group in a limited number of patients affected with Primary Antibody Deficiencies (PADs). Considering the limited available data in the literature, we decided to better characterize this cause of co-morbidity in patients affected with Common Variable Immunodeficiency (CVID). Fifty five patients with CVID were included in this study with a male:female ratio of 0,53: 0,47. The mean age during evaluation was 31,6 years, while the mean age at diagnosis was 18,8 years. SNHL was diagnosed in 18/55 patients (32,7%). No correlation with SNHL in CVID and immunoglobulin serum levels at diagnosis, history of otitis media, ototoxic drugs or autoimmune phaenomena emerged from this study. However, the age at diagnosis was higher among CVID patients with SNHL (p=0,0087), and the diagnostic delay between symptom onset and diagnosis was longer for CVID patients with SNHL (p=0,0003). Follow-up over time showed that SNHL in CVID tends to be progressive: 4 out of 18 patients diagnosed with SNHL showed a deterioration of their hearing loss, while 2 patients that were intially normoacusic developed SNHL. Lymphocyte subset evaluation in these patients showed that CVID patients with SNHL are mainly characterized by expansion of the CD21lo subset with a concomitant reduction of CD4 naive T cells and Recent Thymic Emigrants (RTE). Our data suggest that Sensorineural Hearing Loss (SNHL) is an underestimated cause of co-morbidity in CVID and should be taken into consideration during patients' management.
ESID-0374 Clinical and Immunological Heterogeneity in a Family with the E1021K Activating Mutation in Phosphoinositide 3-Kinase Delta?
D. Lowe 1, N. Verma1, M. Dziadzio1, A. Symes1, S. Workman1, J. Curtis2, V. Plagnol3, A. Jones4, S. Nejentsev2, S. Burns1
1Department of Clinical Immunology, UCL Institute of Immunity and Transplantation, London, United Kingdom
2Department of Medicine, University of Cambridge, Cambridge, United Kingdom
3UCL Genetics Institute, University College London, London, United Kingdom
4Department of Immunology, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
The E1021K activating mutation in p110δ, the catalytic subunit of Phosphoinositide 3-kinase δ (PI3Kδ), was recently demonstrated to cause Activated PI3Kδ Syndrome (APDS, MIM615513), an immunodeficiency characterised by respiratory infections often with elevated serum IgM. We describe three siblings who all possess the heterozygous E1021K mutation but display diverse clinical phenotypes.
The eldest, a male now aged 26, presented with severe upper respiratory tract infections in childhood. He was found to be IgA- and borderline IgG-deficient with normal IgM. He received IVIG replacement but now no longer requires infusions, suffers only occasional chest infections and has minimal bronchiectasis. The second sibling, a 21 year-old female, has suffered since early childhood with recurrent lower respiratory infections, chronic rhino-sinusitis and colitis. Initially diagnosed with ‘Common Variable Immunodeficiency (CVID)’, she remains dependent on IVIG infusions plus regular antibiotics and has moderate bronchiectasis. The youngest female sibling, now eighteen, was also diagnosed with CVID in childhood. She experiences chronic rhinitis and occasional chest infections on IVIG replacement, but has a normal CT Chest: her clinical picture is dominated by acquired pancreatic exocrine dysfunction. Both sisters had normal serum IgM at diagnosis.
This clinical heterogeneity correlates with differences in immunological findings, for example in the proportion of CD27+ IgD- class switched memory B-cells. Our report emphasises variability of the APDS phenotype, even within one family. Importantly, APDS may present without elevated IgM and be diagnosed as CVID. Screening of CVID patients for the E1021K mutation is important given the prospects of specific treatment with p110δ inhibitors.
ESID-0583 Multivariate Logistic Regression and Linear Discriminant Analysis to Assist the Clinical Diagnosis of Primary Immunodeficiencies
C. Murata1, J. Morales2, S. Lugo Reyes 3
1Research Methodology Department, Instituto Nacional de Pediatría, Mexico City, Mexico
2Mathematics Department, Instituto Tecnológico Autónomo de México, Mexico City, Mexico
3Immunodeficiencies Research Unit, Instituto Nacional de Pediatría, Mexico City, Mexico
Features in clinical history direct the differential diagnosis through attribute selection and pattern recognition. Machine learning methods build predictive models that turn data into knowledge. We aimed to identify the features that best explain membership of pediatric PID patients to a group defect or disease.
Methods: Logistic regression (MLR) and discriminant analysis (LDA) were employed to explore a database with records of 168 patients followed from 1980-2010, and explain their diagnoses for the most prevalent 5 group defects (I through V) and 4 diseases (XLA, CVID, AT, CGD). Data included demographics, family history, infection sites and germs, associated features, complications, serum immunoglobulins and blood counts. 30 features were selected after a preliminary run (4 demographic, 10 clinical, 10 laboratory, and 6 germs) to be included in stepwise models. These were then tested for sensitiviy, specificity, accuracy of prediction, and kappa coefficient.
Results: The features incorporated in models (number of features ranged from 3 to 16), and their performance (accuracy 80-93%) are shown in Table 1. Example: Immune dysregulation was predicted by "no infection site", susceptibility to virus, and lymphopenia, with a 50% sensitivity, 93.6% specificity, 92% accuracy, 0.28 kappa. CVID by Age at dx/low IgG.
Discussion: The selected attributes have clinical plausibility, given what we know in terms of heritability, onset, germ susceptibility, and initial lab findings. The encouraging results of this exploratory study suggest feasibility of a more complete extensive approach to predict and assist the clinical diagnosis of PID through these and other machine learning methods.
| GROUP/DIS. | METHOD | FEATURES include in the model | SENS | SPEC | ACCUR | KAPPA |
| Combined | LDA | Age at dx, GI infections, lymphadenitis, “no infection site”, allergy, leukopenia, low IgM | 100% | 91% | 91% | 0.9 |
| Antibody | LDA | Age at dx, lymphadenitis, “no infect. site”. Gram negative isolate, leukopenia, low IgG and low IgA. | 78.4% | 94.6% | 88.8% | 0.75 |
| Well-defined | LDA | current age, age at dx, lymphadenitis, fungi, lymphopenia, eosinophilia, thrombocytopenia, and low IgG. | 82.9% | 78.1% | 79.5% | 0.55 |
| Immune disregulation | LDA | “no infect. site”, susceptibility to virus, lymphopenia | 50% | 93.6% | 92% | 0.28 |
| Phagocyte | MLR | Sex, age, consanguinity, lung infect., urinary tract infect., allergy, low IgA, high IgA, cellulitis/osteomyelitis, “no infect. site”. mycobacterial susceptibility, fungi susceptibility mycobacterial isolate, neutropenia, eosinophilia, thrombocytopenia. | 90% | 99.1% | 97.2% | 0.914 |
| XLA | LDA | Sex, low IgG. low IgM, protozoo isolate, Gram negative isolate. | 91.7% | 86% | 86.9% | 0.62 |
| CVID | LDA | Age at Dx, low IgG. | 84.6% | 81.2% | 81.5% | 0.37 |
| AT | LDA | “no infect. site”, autoimmunity, cancer, anemia, neutropenia, lymphopenia, low IgG, low IgM, low IgA. | 82.6% | 95% | 93.1% | 0.75 |
| CGD | MLR | Lung infection, urinary tract infect. lymphadenitis, cellulitis/osteomielitis, allergy, neutropenia, eosinophilia, thrombocytopenia, low IgG, low IgA | 90% | 90.3% | 90.3% | 0.66 |
ESID-0009 Prenatal Diagnosis of Primary Immunodeficiency (PID) Disorders by Flow Cytometry: A Rapid and Sensitive Tool
M. Madkaikar 1, M. Gupta1, A. Mishra1, S. Mhatre1, M. Desai2, K. Ghosh3
1Paediatric Immunology and Leukocyte Biology, National Institute of Immunohaematology ICMR, Mumbai, India
2Immunology, Bai Jerbai Wadia Hospital for Children, Mumbai, India
3Director, National Institute of Immunohaematology ICMR, Mumbai, India
Primary Immunodeficiency diseases (PID) are a heterogeneous group of inherited disorders of immune system. Prenatal diagnosis (PND) forms an important component of management in families affected with severe PID. PIDs are diverse and molecular diagnostic facilities for each of them are not yet widely available and may not be possible to perform in all the index cases. To help the affected families in such scenario we opted for phenotypic PND by cordocentesis where index case had immunophenotypically well characterized PID.
Normal reference ranges of lymphocyte subsets, CD 18/CD11 integrins on leukocytes, MHC class II expression and oxidative burst activity of fetal neutrophils at 18 weeks of gestation were previously established on 30 cord blood samples. Second trimester PND by cordocentesis was performed in 18 families: 5 with leukocyte adhesion deficiency (LAD-I), 8 with severe combined immunodeficiency diseases (SCID), 3 with chronic granulomatous disease (CGD) and 1 with X-linked agammaglobulinemia (XLA). Maternal contamination was ruled out by VNTR analysis.
Out of 17 fetuses, four were unaffected (one with T-B+NK-SCID, one with MHC class II deficiency, one with T-B-NK+ SCID and one with LAD-I), 12 were unaffected and in one family diagnosis could not be offered due to maternal contamination. Diagnosis was confirmed by testing the cord blood samples after delivery and further follow-up of the children. No procedure related complications were observed.
Flowcytometry offers rapid and sensitive method for prenatal diagnosis and genetic counseling for selected phenotypically well characterized PIDs when molecular diagnostic facilities are not available.
ESID-0166 Incidence, Presentation and Prognosis of Malignancies in Ataxia-Telangiectasia : Final Report from the French National Registry of Primary Immune Deficiencies (CEREDIH)
F. SUAREZ 1, N. MAHLAOUI1, D. CANIONI2, C. Andriamanga1, C. DUBOIS D ENGHIEN3, N. BROUSSE2, J. JAIS4, A. FISCHER1, O. HERMINE1, D. STOPPA LYONNET3
1CEREDIH (French National Reference Center for PID), Necker-Enfants Malades Hospital, Paris, France
2Pathology Laboratory, Necker-Enfants Malades Hospital, Paris, France
3Department of Genetics, Curie Institute, Paris, France
4Biostatistics Department, Necker-Enfants Malades Hospital, Paris, France
Purpose: Bi-allelic mutations in ATM cause Ataxia-Telangiectasia (AT), a rare inherited disease with a high incidence of cancer. Precise estimates of the risk, presentation and outcomes of cancer in AT need to be addressed in large series. Patients and Methods: In this large retrospective cohort, 69 patients with cancers were identified among 279 patients with AT (24.5%). Centralized review was performed on 60% of the lymphomas. Incidence rates were compared to the French population and risk factors were analyzed. Results: Eight patients developed acute leukemias (including 4T-cell acute lymphoblastic leukemia), 12 Hodgkin's lymphoma (HL), 38 non-hodgkin lymphoma (NHL), 3T-cell prolymphocytic leukemia (PLL) and 8 carcinoma at a median age of 8.3, 10.6, 9.7, 24.2 and 31.4 respectively (p<10-4). The majority of NHL were aggressive B-cell NHL. Epstein-Barr virus was associated with all of the HL and 50% of the NHL. Overall survival (OS) was shorter in AT patients who developed cancer (15 vs. 24 years, p<0.001). Cancer survival was improved in patients who achieved a major response to treatment (2.8 vs. 0.75 years for major and minor responses respectively, p=0.002). Immunodeficiency was associated with increased risk of cancer. ATM mutation type was associated with a difference in survival in the entire cohort but not with cancer incidence or cancer survival. Interpretation: B-cell NHL, HL and ALL occur at a very high rate and earlier age than carcinomas in AT. T-PLL are rarer than initially reported. Prognosis is poor but patients benefit from treatment with an improved survival.
ESID-0459 Epidemiology of PID: Innovative New Way to Identify Patients in the CEREDIH Registry Through a Medical Data Warehouse
N. GARCELON2, V. COURTEILLE1, A. FISCHER1, N. MAHLAOUI 1
1CEREDIH (French National Reference Center for PID), Necker-Enfants Malades Hospital, Paris, France
2Imagine Institute Paris Descartes – Sorbonne Paris Cité University, Necker Enfants malades University Hospital, Paris, France
Introduction: Identification and enrolment of patients in medical cohorts is based on (i) physicians' goodwill and memory and (ii) the extraction from the Hospital Discharge Database (HDD, using ICD10 codes - which do not provide information for outpatients. Identification of patients with PID is thus suboptimal. Moreover, physicians produce a huge quantity of information inside the Electronic Health Record (EHR) that can help find new patients otherwise unidentifiable through the previous ways.
Method: Full text search engine on Imagine Institute/Necker Hospital's medical data warehouse filled with all the available EHR in Necker hospital. In order to improve the search engine's precision and recall, we will use Natural Language Processing methods to exclude negative formulations and family history. We will also enrich each document with synonyms and subsumption semantic relation by using the Unified Medical Language System Metathesaurus developed by the NLM. Thus searching term « Bruton » will also display document containing terms « X-linked Agammaglobulinemia ». To evaluate the gain of our method we will cross-reference our findings with other databases.
Preliminary Results: 189,000 EHR available from 2010 to mid-2014. For Bruton Agammaglobulinemia, 8.5% of patients were picked-up to be registered in the French National Registry for PID. Further results will be presented.
Conclusion and Perspectives: Our method provides an innovative new way to enrol patient in our PID registry, and help fostering clinical and translational research. HDD codes will be added in the data warehouse in order to combine both methods ICD10 codes and full text information.
ESID-0442 Mycobacterial, Viral and Fungal Infections in Distinct Type of STAT1 Mutation
H. MAO 1, P.P.W. Lee2, W. Yang2, J. Yang2, K.W. Chan2, M.H.K. Ho2, T.L. Lee2, W. Tu2, Y.L. Lau2
1Department of Pediatrics, The university of Hong Kong-Shenzhen Hospital, Shenzhen, China
2Department of Pediatrics and Adolescent Medicine, LKS Faculty of Medicine The university of Hong Kong, Hong Kong, China
Signal transducer and activator of transcription 1 (STAT1) is crucial in interferon-mediated immunity against microbial infections by regulating the expressions of IFN-responsive genes. Different infections caused by mycobacterium, virus or fungus had been separately described in distinct type of STAT1 mutation. Here we reported two Chinese girls (P1 and P2), who had distinct STAT1 mutation, but presented with similar infection phenotype.
Both patients had recurrent mucocutaneous candidiasis since infancy, which was refractory to (P1) or improved with (P2) antifungal treatment. At 5 years old, they developed granulomatous lymphadenitis that was caused by Mycobacterium fortuitum in P1 and acid-fast bacilli in P2 respectively. They also had herpes zoster over right L2 (P1) and left trigeminal 3rd division (P2) dermatome. P1 further had recurrent Salmonella gastroenteritis and type 1 diabetes mellitus.
Heterozygous STAT1 mutations in linker domain (E559K) and coiled coil domain (T288I) were identified in P1 by exome sequencing and P2 by targeted sequencing respectively. In response to IFNα and IFNγ stimulation, the mutation in P1 impaired STAT1 phosphorylation, leading to decreased expressions of interferon-inducible genes. In contrast, the mutation in P2 caused increased STAT1 phosphorylation and IFN-stimulated monokine expressions. IL17 production and IL12-induced IFNγ expression in P2 were downregulated.
We identified the first mutation in the linker domain of STAT1. Our findings indicate that the infectious disease susceptibility and phenotypic spectrum caused by STAT1 mutation are wider than previously believed. The diversity of infectious and immunological phenotypes suggests the many divergent roles of STAT1 in host-pathogen interaction and immunity.
ESID-0651 Going with the Flow: Detection of PI3K Activation in Germline PIK3CD Mutants
B. Martinez1, S.M. Holland1, G. Uzel 1
1Lab of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases, Bethesda, USA
Germline gain-of-function mutations in PIK3CD, encoding the PI(3)K catalytic subunit p110, result in constitutive activation of PI3K/AKT/mTORC1 axis and lead to immune dysregulation and immunodeficiency. Seeking for an in vitro assay to study and monitor the activation of PI3K/AKT/mTORC1 axis, we have utilized flow cytometry and phospho-specific antibodies directed to Thr308 and Ser473 activation sites of AKT as well as S6, which is downstream of mTOR kinase MTORC1. We have used freshly isolated peripheral blood mononuclear cells from healthy controls and affected patients before and during in vivo treatment with rapalogues, and measured B and T cell pAKT and pS6 levels with and without anti-CD3/CD28/IL-2 or IgMFab2 stimulation. We have found consistently and reproducibly elevated B cell pS6 at baseline in patients as compared to healthy controls (mean: 3.5% vs 13.5%, n: 6, 12 consecutively; p: 0.087). We also observed further increased levels of pS6 (mean 3.5% vs 34.8%, p:0.001) in controls with BCR activation. In patients, even though we observed further activation, the increase was not statistically significant given high baseline levels (mean: 13.4% vs 31.2%). Patients receiving treatment using mTOR inhibitors displayed significantly diminished baseline activation of S6 (median: 0.33% vs 13.5%, p<0.001), proving effective ex-vivo inhibition of this axis with in-vivo treatment. In conclusion, of the phospho-specific sites and lymphocyte subsets we studied, monitoring B cell S6 phosphorylation with a flow cytometry based approach proved the most consistent and reproducible method in screening and monitoring treatment in patients with PIK3CD GOF mutations.
ESID-0769 Common Variable Immunodeficiency and Nutritional Status of Adult Patients. Cohort of 23 Patients
A. MELENDEZ 1, M. MENDOZA1, J. BARRIENTOS1, F. CAMPOS2, A. AMAYA2, N. SEGURA2
1Alergia e Inmunología Hospital de Especialidades CMN SXXI, Universidad Autónoma Metropolitana - Xochimilco, Mexico City, Mexico
2Alergia e Inmunología Hospital de Especialidades CMN SXXI, Instituto Mexicano del Seguro Social, Mexico City, Mexico
Introduction. An adequate nutritional status, allows the organism to maintain its physiological balance, to ensure a better life quality. There is little information known about their nutritional status. The aim of this assessment is to show the nutritional complications presented on adult CVI patients.
Materials ans Methods: 23 CVI adult patients were assessed, average age was 37.6 years. Were 9 men and 13 women, average of height and weight of 1.62 m and 59 kg respectively. The clinical histories were analyzed for each of the patients and they all underwent through a nutritional interview, anthropometric assessment and laboratory studies. The average BMI was 22.3 and waist circumference was 82.3. Patient population showed skeletal muscle depletion, this obtained by the results of arm circumference and tricipital skinfold on the Gurney nomogram.
Results: 44% of the CVI patients showed skeletal muscle depletion, 18% low weight, 52% normal weight, 26% overweight and 4% obesity. 46.6% have cardiovascular disease risk, this according to their waist circumference. None of the patients had any information about their nutritional status, their energy requirement or the diet personal options that matched their commorbidities.The complications nutritional status were: Vitamin D level in all of the cases, dyslipidemia in 3 cases, hypothyroidism 2 cases, celiac sprue, 2 cases of colitis, 2 cases of hypophosphatemia and 7 with damage kidney.
A patient with a healty nutritional status, lower the risks of presenting commorbidities as well as the gammaglobulin IV requirement, maintaining an adequate cost/benefit relationship.
ESID-0794 Molecular Diagnostics of Severe Congenital Neutropenia and Shwachman-Diamond Syndrome in Seven Croatian Families
A. Merkler 1, J. Kelecic2, D. Tjesic-Drinkovic2, I. Baric2, J. Vukovic2, V. Sarnavka2, E. Bilic2, L. Omerza2, M. Cuk2, D. Petkovic-Ramadza2, J. Sertic1
1Department of Laboratory Diagnostics, University Hospital Centre Zagreb, Zagreb, Croatia
2Department of Pediatrics, University Hospital Centre Zagreb, Zagreb, Croatia
Introduction: Severe congenital neutropenia (SCN) is an autosomal dominant disease characterized by chronic neutropenia which leads to severe recurrent bacterial or mycotic infections. Shwachman-Diamond syndrome (SDS) is an autosomal recessive disease that is characterized by exocrine pancreatic insufficiency, dysfunction of the bone marrow and predisposition to develop myelodysplastic syndrome or leukemia.
Objective: The objective was to confirm clinical diagnosis in several Croatian families at genetic level by analyzing their DNA samples.
Methods: 20 samples of genomic DNA were analyzed: 7 samples of patients with clinical diagnosis of SCN or SDS and 13 samples of their family members. Coding regions of ELANE and SBDS gene were amplified by PCR with specific primers. PCR fragments were bidirecty sequenced using Big Dye® Terminator v3.1 Cycle Sequencing Kit and Applied Biosystems 3130xl Genetic analyzer.
Results: In all 4 patients with SCN, mutations were found in ELANE gene. These mutations were de novo, because in their parents' samples mutations were not found. In all 3 patients with SDS, mutations were found on both alleles, which were inherited from their parents. Two new variants were found, one in ELANE gene (c.213C>G; p.Cys71Trp) and one in SBDS gene (c.41A>G; p.Asn14Ser). Both variants were analyzed with PolyPhen-2 software and were predicted as probably damaging.
Conclusion: Mutations in ELANE gene were reported in 38-80% of individuals with SCN, and in SBDS gene in more that 90% of individuals with SDS. In our case, clinical diagnoses of SCN or SDS were confirmed in all analyzed patients.
ESID-0613 Primary Immunodeficiency Disorders (PID) in a Specialized Dermatology Outpatient Unit in São Paulo, Brazil
D. Moraes Vasconcelos 1, M. Domingues-Ferreira1, N. Chuffi-Barros1, T.A. Bezerra1, D.L. Bertolini1, R. Muniz Junior1, L.E. Prestes-Carneiro1, A.J. Duarte1
1Dermatology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
Although Primary Immunodeficiency Diseases (PIDs) have been classically considered as diseases of infants and children, increasing numbers of affected adults have been identified. Here we describe the experience of a specialized dermatology ambulatory in an university hospital in São Paulo, Brazil, with both adult and pediatric PID patients. HC-FMUSP is the biggest public hospital of the country and represents the main national reference center for rare and complex diseases.
In our outpatient unit the most common diagnoses are: Symptomatic IgA deficiency-63 patients; Hereditary angioedema-68 pts; CVID-56 patients; CMC-38; Transient hypogammaglobulinemia-17; Idiopathic CD4 lymphocytopenia-17; Complement deficiencies (excluding HAE)-15; Phagocyte G6PD deficiency-18; MSMD-16; IgM deficiency-10; CGD-11 patients, among many other diseases. Of these patients 44 died, mainly due to septic episodes and neoplasias. The distributions are as follows: Males: 209; Females: 213; 204 children and adolescents; 233 adults; As a whole there are more than 1600 patients in follow-up nowadays, with 437 patients with well defined PIDD.
Our casuistry shows an extreme diversity of different cases, with several rare diagnoses. Due to the fact that we are located in the dermatology department our patients present mainly dermatological manifestations, such as HAE, CMC, XP, EVER, Netherton etc. Primary immunodeficiencies as a whole comprise an important health problem considering that their frequency is comparable to hematological cancer or cystic fibrosis, for example. Our specialized unit has a specific set of different diseases not commonly found in other primary immunodeficiencies ambulatories.
ESID-0389 Dual Response to Human Papilloma Virus Vaccine in a Whim-Like Disorder: Resolution of Foot Cutaneous Warts and Persistence of Condylomas
G. Moscato 1, A. Criscuolo2, M.L. Romiti3, S. Di Cesare4, G. Di Matteo5, R. Carsetti6, P. Di Bonito7, A. Mauriello8, M. Ciotti9, M. Andreoni10, V. Moschese11
1Experimental Medicine and Surgery, University of Rome Tor Vergata, Rome, Italy
2Surgery and Gynaecology, Policlinico Tor Vergata, Rome, Italy
3Medicine of Sistems, University Tor Vergata, Rome, Italy
4Pediatrics, Bambin Gesu' Children Hospital, Rome, Italy
5Medicine of Systems, University Tor Vergata, Rome, Italy
6Immunology Research Area, Bambin Gesu' Children Hospital, Rome, Italy
7Infectious Parasitic and Immunomediated Diseases, National Institute of Health, Rome, Italy
8Biomedicine and Prevention, Policlinico Tor Vergata, Rome, Italy
9Laboratory of Molecular Virology, Policlinico Tor Vergata, Rome, Italy
10Head of Infectious Diseases Unit, Policlinico Tor Vergata, Rome, Italy
11Head of Pediatrics Allergology and Immunology, Policlinico Tor Vergata - University Tor Vergata, Rome, Italy
A caucasian 24 year old woman with a pelvic inflammatory disease was referred to our department because of acquired pneumonia after surgery for abdominal abscess. Physical examination revealed severe warts on the feet and disseminated genital condilomas; increased liver and spleen volume. Her brother died from pneumonia in the first months of life. Since childhood she had developed recalcitrant cutaneous warts and, over the next few years, an extensive genital involvement occurred despite long term treatment with laser ablation, imiquimod and retinoic acid derivatives. Immunological investigations revealed B cell lymphopenia (2%), normal IgG and IgA values with mild IgM hypergammaglobulinemia, and a protective titer after pneumococcus vaccination. Mutations of CXCR4, GATA2, NEMO and CD40L were excluded by direct sequencing. BM evaluation showed a myeloid and eosinophil hypercellularity, unclear signs of myelokathesis, absence from pro-B to transitional cells. HPV genotyping identified 18 and 33 (with E6/E7 mRNA) subtypes in her cervix and 6 subtype in her foot skin. CIN1 rapidly evolving into CIN3 required prompt treatment . To attempt control of HPV infection a quadrivalent HPV vaccine was administered at months 0, 2 and 6 with dramatic resolution of her cutaneous warts after the second dose. Conversely, no change was observed in the cervix since genital warts persisted and CIN1 reappeared. To our knowledge, this is the first report on the exclusive efficacy of HPV vaccine on cutaneous warts. HPV humoral and cellular immunity are under investigation to highlight the unexpected dual response to HPV vaccine in our immunodeficient patient.
ESID-0720 22Q11.2 Deletion Syndrome. Association of Thymus Size Measured by Ultrasound and Clinical Response to Infection
E. NAVARRETE RODRIGUEZ 1, B. DEL RIO NAVARRO1, S. PADILLA ESPINOSA2, J.U.A.N. ESPINOSA GARDUÑO1, O. SAUCEDO RAMIREZ1, N. MOGUEL MOLINA1
1ALLERGY AND CLINICAL IMMUNOLOGY, HOSPITAL INFANTIL DE MEXICO FEDERICO GOMEZ, Mexico City, Mexico
2IMMUNODEFICIENCIES RESEARCH UNIT, INSTITUTO NACIONAL DE PEDIATRIA, Mexico City, Mexico
Background.- Del22q11.2 syndrome has an estimated incidence of 1/3,800 newborns , the TBX1 gene is the main responsible for the phenotypic characteristics and plays a crucial role in thymus development involved in the maturation of T lymphocytes.
High percentage of 22q11.2 syndrome patients have a hypoplasic thymus, although it is not clear the proportion, it has been observed that 80% of them have diminished T cells. Up to 40% of children with 22q11.2 deletion syndrome have repetitive upper respiratory infections with an increased risk of bacterial colonization.
Objective.- Relate thymus size measured by ultrasound with the clinical response to infection in children with del22q11.2 syndrome
Methods.- We included patients aged 3 to 14 years old with del22q11.2 followed in Hospital Infantil de Mexico Federico Gómez. We reviewed our records and ask them for infectious illnesses in the past year and hospitalizations; later thymus size was measured by ultrasound with transverse, longitudinal and anteroposterior axis.
Results.- We studied 24 children with del22q11.2 diagnosed by FISH, the mean age was 7.8 years. Thymus was not found in 5 children. There was a statistically significant difference in the volumes reported by USG and those expected for age in Mexican population with a mean volume difference of 1.47 cm3. The average of infections in patients with thymus hypoplasia was 5.77 while in the group of absent thymus was 6.40.
Conclusion.- Thymic hypoplasia is inherent to del22q11.2, immunological abnormalities are frequent and those children have increased susceptibility to infections illnesses.
ESID-0648 Fatal Granulibacter Bethesdensis Meningitis in XL-CGD. How Bad Is the Bug?
J. Neves 1, A. Zelazny2, S. Holland3, A. Cordeiro1, C. Gouveia1, C. Neves1, L. Ding2
1Pediatrics, Hospital Dona Estefânia, Lisbon, Portugal
2National Institute of Allergy and Infectious Diseases NIH, National Institute of Allergy and Infectious Diseases NIH, Bethesda, USA
3Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases NIH, Bethesda, USA
Introduction Granulibacter bethesdensis (Gb) is a Gram-negative bacteria that belongs to the Acetobacteraceae family. First described in 2006, it has been shown to cause infection in Chronic Granulomatous Disease (CGD) patients.
Case report In June 2012 a boy with XL-CGD was admitted for deep cervical abscess and treated with an appropriate 6 week IV antibiotic regimen, followed by oral therapy. He had complete clinical and radiological resolution. In October 2012 he was readmitted for pneumonia and treated with IV antibiotics and voriconazole with rapid resolution. No microorganism was identified in either episode, despite agressive research. In December 2012 he presented with fever and vomiting and a diagnosis of meningitis was made. He was treated with IV antibiotics (including anti-mycobacterial) and antifungals but his condition worsened and he ultimately passed away in April 2013. The CSF analysis led to the identification of Granulibacter bethesdensis by 16S rRNA sequencing, resistant to all tested antibiotics.
Discussion To our knowledge, 12 CGD patients with Gb infections have been identified. The 10 US patients had relatively mild infections and none died. The 2 European patients have both succumbed to their Gb infections. Interestingly, both European Gb are genetically similar and different from the US Gb. Mouse experiments performed in both gp91 KO and p47 KO showed higher lethality in those inoculated with the European Gb.
Previously known as causing mild infections in CGD patients, Gb should also be suspected in life-threatening infections in CGD patients, especially in Europe.
ESID-0517 Newborn Screening of Primary Immunodeficiency Using TREC/KREC Assay: An Experience from Iran
M. Nourizadeh 1, S. Borte2, M. Fazlollahi1, M. Moin1, F. Taghizade Mortezaee1, Z. Pourpak1, L. Hammarström3
1Immunology Asthma and Allergy Research Institute, Tehran University of Medical Sciences, Tehran, Iran
2Jeffrey Modell Diagnostic and Research Center for Primary Immunodeficiencies, The Municipal Hospital St. Georg, Leipzig, Germany
3Division of Clinical Immunology Department of Laboratory Medicine, Karolinska University Hospital Huddinge, Stockholm, Sweden
Introduction: Early diagnosis of PID patients can be applicable using a standard screening test. PCR-based detection of T-cell receptor excision circles (TRECs) and κ-deleting excision circles (KRECs), has proven to be a valuable tool for identifying the patients.
Methods: We investigated 100 blood samples of healthy newborns taken during the first 72 hours after birth on Guthrie cards (DNA was extracted from the paper's punches) and 31 frozen DNA samples of Severe Combined Immunodeficiency (14), X-linked agammaglobulinemia (7) and Wiskott–Aldrich syndrome (10) patients referred to Immunology, Asthma and Allergy Research Institute (IAARI). After running on Multiplex Real time PCR, copy numbers of TREC/KREC were deduced based on the standard curve of plasmid dilutions. This test was done in Leipzig/Germany.
Results: Zero TREC and KREC values were not seen among healthy newborns. But for the SCID patients, zero number of TREC/KREC in 10 out of 14 revealed SCID T-B- phenotype and zero number of TREC and higher but not normal number of KRECs in the remaining samples pointed out to T-B+ phenotype. Four of seven XLA patients had no KREC copy numbers and just one was negative for both TREC and KREC. Among ten WAS patients, very low copy numbers of TREC were seen in four cases. These findings were greatly in accordance with laboratory and clinical manifestations of patients as well as their genetic analysis.
Conclusion: This molecular method can easily be used for early newborn screening for PID especially SCID and further evaluation in a timely manner.
ESID-0614 B-Cell Compartment Disturbance in Bone Marrow and Peripheral Blood is the Most Distinct Feature of GATA-2 Deficiency
M. Novakova 1, A. Janda2, M. Wlodarski3, M. Kubricanova Zaliova1, E. Fronkova1, T. Kalina1, M. Sukova4, J. Stary4, O. Hrusak1, E. Mejstrikova1
1CLIP-Department of Paediatric Haematology and Oncology, 2nd Faculty of Medicine and University Hospital Motol, Prague, Czech Republic
2Center for Chronic Immunodeficiency (CCI), University Medical Center Freiburg, Freiburg im Breisgau, Germany
3Center for Pediatric and Adolescent Medicine, University Medical Center Freiburg, Freiburg im Breisgau, Germany
4Department of Paediatric Haematology and Oncology, 2nd Faculty of Medicine and University Hospital Motol, Prague, Czech Republic
Germline GATA-2 mutation was described in patients with sporadic and familiar forms of myelodysplastic syndrome (MDS), hereditary lymphedema and immunodeficiency. Immunophenotypic abnormalities are frequently found in peripheral blood (PB) and bone marrow (BM).
Lymphoid and myeloid subpopulations were analyzed in PB and BM in 11 children with proven GATA2 mutation and in 122 patients with either defined other primary immunodeficiency or bone marrow failure (MDS or AA). KREC and TREC levels were investigated to assess the proliferation history of T and B cells. The primary manifestation of GATA-2 deficiency was MDS in 7 patients, immunodeficiency in 2 patients, 1 patient was diagnosed with intersticial lung process and chronic active EBV infection. One healthy carrier with no clinical symptoms was analyzed as well.
The most frequent aberrancy detected within hematopoietic cells in GATA2 mutated patients was a lower count of B cells in PB and BM with no decrease of immunoglobulins in most patients. Bone marrow compartment was characterized by a lack of B cell progenitors and an increase of plasma cells, which were aberrant in some patients either by CD19 negativity or CD56 expression. Stable relative monocytopenia was found in 2 patients. KREC levels were significantly lower compared to other MDS patients.
Changes in B cell compartment are the most distinctive aberrancies in GATA-2 deficiency, which can help to establish the diagnosis. Lower copies of KRECs are probably caused by proliferation of mature B cells and low production of immature B cells.
Supported by GAUK802214, IGA NT14534-3, GACR P301/10/1877, UNCE204012
ESID-0011 Long-Term Follow-Up and Outcome of Patients with Chronic Granulomatous Disease on Jeju Island, Korea
M.S. Oh 1, G. Hwang1, K. Shin1
1Pediatrics, Jeju National University School of Medicine, Jeju, Korea
Introduction: This study investigated the long-term clinical course of patients with chronic granulomatous disease (CGD) and the effect of prophylaxis with interferon-gamma (IFN-?).
Methods: The records of Jeju National University Hospital between 2001 and 2012 were reviewed and 21 patients with a diagnosis of CGD were identified. The clinical presentations were reviewed retrospectively from the medical records of 15 patients with CGD. The efficacy of prophylaxis was evaluated by comparing the incidence of severe infections before and after starting continuous prophylaxis with IFN-?.
Results: At the time of the analysis, 14 patients were alive, with a median age of 14.3 (range 1.1–25.2) years. The diagnosis of CGD was made at a median of 2.4 (range 0.1–13.1) years. Thirteen (87%) of 15 patients with CGD had their first severe infection within the first year of life. The survival rate was 93% at 20 years of age. Pneumonia was the most common infection, and the most common infectious agent was Aspergillus species. Chronic conditions associated with CGD were hepatomegaly, splenomegaly, short stature, and anemia of chronic disease. Long-term prophylaxis with IFN-? did not significantly affect the frequency of severe infection per patient-year.
Conclusions: Considering the data on the role of p22-phox in the NOX family, patients with CGD on Jeju Island did not show obviously different clinical manifestations from X-linked CGD or other autosomal recessive forms of CGD.
ESID-0118 Screening of Neonatal Spanish Dried Blood Spots Using TREC-KREC Screening Assay: A Prospective Pilot Study
B. de Felipe1, P. Olbrich2, C. Delgado-Pecellin3, B. Sanchez-Sanchez4, J.M. Lucena5, P. Rojas6, J. Marquez-Fernandez7, L. Ancio1, S. Borte8, O. Neth 2
1Laboratory of Paediatric Infectious Diseases and Immunology, Hospital Virgen del Rocio, Sevilla, Spain
2Department of Paediatric Infectious Diseases and Immunology, Hospital Virgen del Rocio, Sevilla, Spain
3Department of Metabolic Diseases, Hospital Virgen del Rocio, Sevilla, Spain
4Department of Immunology, Hospital Virgen del Valme, Sevilla, Spain
5Department of Immunology, Hospital Virgen del Rocio, Sevilla, Spain
6Department of Neonatal Critical Care and Neonatology, Hospital Virgen del Rocio, Sevilla, Spain
7Department of Neonatology, Hospital Virgen del Valme, Sevilla, Spain
8Department of Clinical Immunology and Transfusion Medicine, University of Leipzig, Leipzig, Germany
Introduction: Early diagnosis of primary immunodeficiency such as severe combined immunodeficiency (SCID) and X-linked agammaglobulinemia (XLA) improves outcome of affected infants/children. T cell receptor excision circles (TRECS) and ka ppa-deleting recombination excision circles (KRECS) measurement can identify neonates with severe T or B-cell lymphopenia.
Objectives: To determine TRECs and KRECs levels from prospectively collected dried blood spot samples (DBS) and to correctly identify severe T and B-cell lymphopenia.
Methods: TRECs and KRECs determination by multiplex PCR from neonates, born between 02/2014 and 05/2014 in two hospitals (Seville, Spain). Parents' written informed consent and ethical committees' approval were obtained. PCR cut-off levels: TRECs<15copies/μl, KRECs<10copies/μl, ACTB (b-actin)>1000copies/μl. Internal (XLA) and external (SCID) controls were included.
Results: 1087 neonates (mean GA 39 weeks (38-40) and BW 3256 g (2950-3540) were enrolled. Mean (median, min/max) copies/μl were as following: TRECs 144 (74, 8/503), KRECs 81 (51, 7/381) and ACTB 2811 (2757, 997/7710). Samples were insufficient in 15 neonates (1,4%) and 8 neonates (0,7%) showed values below the chosen cut-off (Figure 1). Retesting in all samples confirmed normal results. Lower cut-offs (TRECs<8 and KRECs<4 copies/μl) as recently suggested by S Borte (unpublished data) resulted in normal determination of all the samples tested. No neonate needed neither re-bleeding nor clinical examination. Controls were correctly identified.
Discussion: First prospective pilot study in Spain using TREC/KREC/ACTB-assay. Initial higher cut-off levels were chosen in order not to miss any patients. The ideal cut-off for our population remains to be established. Quality of sampling, storage and preparation needs to be further optimized.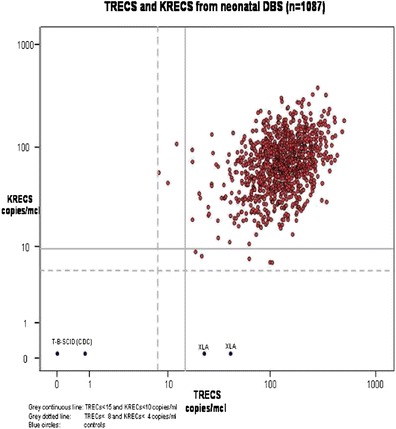
ESID-0540 Convergence of Two X-Linked Rare Diseases
A. Partida-Gaytan 1, L. Blancas-Galicia1, L. Santos-Argumedo2, M. Yamazaki-Nakashimada3, S. Espinosa-Padilla1, F. Espinosa-Rosales1
1Primary Immunodeficiency Research Unit, Instituto Nacional de Pediatría, Mexico City, Mexico
2Molecular Biomedicine, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional (CINVESTAV), Mexico City, Mexico
3Clinical Immunology, Instituto Nacional de Pediatría, Mexico City, Mexico
Introduction: The concurrence of two different X-linked diseases in one same family is not a common finding. Chronic granulomatous disease (CGD) and hypohidrotic ectodermal dysplasia (EDA) are two diseases with genetic heterogeneity, though both are most frequently caused by genes located in chromosome X.
Case presentation: We describe a non-consanguineous family conformed by a 3 years old boy diagnosed with CGD; his sister, a 7 years old girl with EDA and CGD carrier state; and their mother and grandmother, both carriers of CGD and with no EDA signs. Clinical and immunological features of both siblings are presented, a clinical and genetic approach is discussed including known disease-causing-genes and plausible mechanisms involved in the presence of two different X-linked diseases in the same family. Although nuclear factor kappa-light-chain-enhancer of activated B cells essential modulator (NEMO) is a gene that comes to mind when these two diseases are mentioned in the same line (CGD & EDA) we dispute against this explanation.
Conclusions: To our knowledge this is the first family reported to mingle X-linked CGD and EDA. When we confront an inheritable disease on an index case, extending the study to family members is an opportunity to offer genetic advice, and from time to time encounter with rare and interesting associatons
ESID-0443 Gastrointestinal Manifestations in Patients with Primary Immunodeficiencies
L. Pinho1, R. Lima2, F. Pereira2, L. Marques 1
1Pediatrics, Centro Hospitalar do Porto, Porto, Portugal
2Pediatric Gastroenterology, Centro Hospitalar do Porto, Porto, Portugal
PIDs present with a variety of symptoms, some of which involve the gastrointestinal (GI) system.
Retrospective review of 48 patients followed at a paediatric immunology unit of a tertiary care portuguese hospital during 2012, to analyse GI manifestations both at presentation and during the follow-up period (median of 70 months).
Fifteen girls and 33 boys (1:2,2) were included (median age of 9y7m). Thirty-three had a predominantly antibody deficiency (16 selective IgA deficiency, 8 transient hypogammaglobulinemia of infancy, 6 congenital agammaglobulinemia, 2 common variable immunodeficiency syndrome, 1 IgA with IgG subclass deficiency), 13 had a combined immunodeficiency (6 DiGeorge anomaly, 4 ataxia-telangiectasia (AT), 1 AT-like disease, 2 SCID) and 2 had a defect of phagocyte function (CGD). Eleven patients (8 selective IgA deficiency, 2 CGD and 1 AT) presented with GI symptoms, which in 6 of them were the first major complaints, the most frequent being diarrhea (7), followed by recurrent abdominal pain and oral aphtas (2 each), granulomatous colitis and diarrhea/constipation (1 each). During follow-up, 16 cases (6 selective IgA deficiency, 4 congenital agammaglobulinemia, 3 AT, 2 CGD and 1 SCID) were complicated with GI manifestations: diarrhea (11), abdominal pain (8) and vomiting (4). A diagnosis was established in 4 of them: intestinal parasitosis (2), leishmaniasis (1) and granulomatous colitis (1). Growth impairment was observed in 7 patients, 5 of whom had GI symptoms.
GI complications are common among PIDs patients. Early diagnosis is important to allow appropriate treatment and to improve the quality of life of these patients.
ESID-0007 Mechanism of Cellular Immune Response to Strange Objects
M. Ponizovskiy 1, M. Ponizovskiy1
1Head of Biochemical and Toxicological Laboratory, Kiev Regular P/N Hospital, Nuernberg, Germany
The interactions of cellular transport across cellular wall promote stability Internal Energy of a cell via creating extracellular and intracellular chemical potentials which induce different electrical charges on external and internal cellular membranes of cellular wall. The formed different cellular capacitors into cellular wall are functioned. The mechanism of mutual interactions between cellular capacitors promote remote reactions across distance for immune responses on strange objects. Interactions between all cells result in cellular capacitors operations via production of resonance waves. Penetration of strange high-molecular substance into an organism creates local change of chemical potential and promotes remote reactions of cellular capacitors via cellular waves on common molecular wave of strange high-molecular substance, due to the wave function of any molecule which is determined as the total wave functions of the nuclear orbitals, according to Schreodinger (wave) equation of linear combination of atomic orbitals (MO LCAO). The forming resonance waves cause attraction the immune cells to strange high-molecular substance and create the contact reaction of decomposing the high-molecular substance of the strange object.
Conclusion: Biophysical mechanism of immune cells remote reactions transit into contact biochemical immune reactions [Phagocytosis, Autophagy etc.] to ruin strange object.
Reference:
1. Ponisovskiy M.R., (2011), Driving mechanisms of passive and active transport across cellular membranes as the mechanisms of cell metabolism and development as well as the mechanisms of cellular distance reactions on hormonal expression and the immune response, Critical Reviews in Eukaryotic Gene Expression, vol.21 (3), 267-290.
ESID-0427 The Importance of Protein Studies to Confirm Novel Sequence Variants
E. Ralph 1, S. Bibi2, L. Jenkins2, N. Lench2, C. Cale1, K. Gilmour1
1Immunology Laboratory, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
2North East Thames Regional Molecular Genetics Laboratory, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
Next generation sequencing carried out at Great Ormond Street Hospital has identified novel sequence variants in a number of genes including STAT1, CD3delta and CD3epsilon. These novel variants have been demonstrated to be pathogenic by examining the protein expression from these genes. The two cases presented here highlight the importance of such work, both in terms of expanding our knowledge of disease causing mutations in rare primary immune deficiencies, and for enabling appropriate genetic counseling of affected families.
In the first case, a homozygous missense mutation in ZAP-70 that had not previously been reported was identified, and it was unclear whether the mutation was pathogenic. Unfortunately, there was no sample stored on this patient prior to their HSCT. Although the clinical background and immunophenotyping was consistent with a diagnosis of ZAP-70 SCID, the genetic results could only be reported as showing a sequence variant that was likely pathogenic; with protein work it may have been classified as pathogenic.
The second case was a compound heterozygote for two mutations in ZAP-70. Only one of these mutations had been previously reported in ZAP-70 SCID. The second mutation was, however, predicted in silico to be probably disease causing. To confirm this, ZAP-70 expression was examined by flow cytometry on PBMCs stored prior to the patient undergoing HSCT. Expression of ZAP-70 was found to be significantly reduced, thus confirming the pathogenicity of the identified mutations.
ESID-0130 Monitoring Plasma Levels of Interferon Gamma in a Child with Complete Interferon Gamma Receptor 1 Deficiency: Might it Be Helpful?
E.M. Ruga 1, D. Donà1, V. Dal Cengio1, J. Bustamante2, J.L. Casanova2
1Division of Pediatric Infectious Diseases Department for Woman and Child Health, University-Hospital of Padua, Padova, Italy
2The Laboratory of Human Genetics of Infectious Diseases Necker Branch Inserm U980, Necker Medical School Paris France, Paris, France
Introduction: Autosomal recessive complete IFN-γ R1 is a rare primary immunodeficiency associated with early-onset and recurrent disseminated mycobacterial infections. The finding of high plasma levels of IFN-γ in patients with this immunodeficiency has been described but data on its monitoring are lacking.
Objective: To describe the pattern of INF-γ levels detected in a patient with a complete INF-γ R1 deficiency through nine-year follow- up.
Methods: An eleven-year-old patient with complete IFN-γ R1 deficiency has been regularly followed at our Division of Pediatric Infectious Diseases from the age of two years. We retrospectively evaluated IFN-γ plasma levels detected during hospital admissions due to mycobacterial infections and at any follow-up evaluations.
Results: Plasma levels of IFN-γ significantly and progressively increased (highest value, range: 477 – 934 ng/L) within three months before the onset of mycobacterial disease clinical findings in all mycobacterial disseminated infections (3 diagnosed episodes) and rapidly decreased after starting antimicrobial therapy. In addition no significant increase of IFN-γ levels was demonstrated before and during adenoviral interstitial pneumonia.
Conclusions: Monitoring of IFN-γ plasma levels might be helpful in early diagnosis and treatment of disseminated mycobacterial disease in patients with complete IFN-γ R1 deficiency. Moreover a rapid decrease might confirm the efficacy of the ongoing therapy.
ESID-0786 Cutis Marmarato Telangiectatica Congenita and Associations
M. sadeghi shabestari 1
1pediatrics, children hospital, Tabriz, Iran
Cutis marmarato telangiectatica congenita or CMTC is a rare congenital vascular disorder that usually manifests in affecting the blood vessels of the skin. CMTC usually observed at birth or shortly thereafter in 94% of patients. The prognosis is favorable with an isolated cutaneous abnormality necessary unless complicated with other associated anamolies and is good with improvement of the skin lesions especially during the first two years of life.
Case presentation: An 18-month girl was admitted because of fever and respiratory distress .She was the first child of non-consanguineous marriage. On physical examination, she had skin changes in the form of hyperpigmented macules of reticular pattern as well as telangiectasia that were observed from little days after birth. These skin lesions were located on the abdomen, whole of the back, both of the gluteus, legs, and hands that were prominent in cold temperature .She had also limb asymmetry ,FTT and history of recurrent infections without neurologic and developmental abnormalities. Abdomen ultrasound and echocardiogram findings were unremarkable.
Biopsy specimens showed an increase in the number and size of the blood vessels in affected sites. Laboratory analysis was in normal range.
Conclusion: The disorder is self-limiting with a good prognosis of skin changes and treatment is not necessary unless complicated with other associated anomalies.So, comprehensive examination of all the organs, annual controls of skin changes and psychomotor development of these children warranted and Consultation with neurologists and ophthalmologists is necessary along with vascular cosmetic surgeons in some cases.
Key words: Vascular lesions, Cutis marmarato
ESID-0475 The Diagnosis of Hyper Immunoglobulin E Syndrome Based on Project Management
S. Saghafi 1, Z. Pourpak1, C. Glocker2, F. Nussbaumer2, A. Babamahmoodi3, B. Grimbacher 2, M. Moin 1
1Immunology Asthma and Allergy Research Institute (IAARI), Tehran University of Medical Sciences, Tehran, Iran
2Centre of Chronic Immunodeficiency (CCI), University Medical Center Freiburg and University of Freiburg, Freiburg, Germany
3Health Management Research Center, Baqiyatallah University of Medical Sciences, Tehran, Iran
Background: Hyperimmunoglobulin E Syndrome (HIES) is a complex primary immunodeficiency characterized by both immunologic and non-immunologic manifestations. High serum IgE levels, eosinophilia, eczema, recurrent skin and lung infections constitute the immunologic profile of HIES, whereas characteristic facial appearance, scoliosis, retained primary teeth, joint hyperextensibility, bone fractures following minimal trauma and craniosynostosis are the main non-immunologic manifestations. The diagnosis of HIES cannot be made by routine immunologic tests. As the only characteristic laboratory abnormalities of this syndrome are highly elevated serum IgE levels and eosinophilia; both features have a broad spectrum of differential diagnosis. The purpose of this essay is presenting the best way for diagnosis management of HIES.
Methods: Based on the genetic reports of patients of the Center for Chronic Immunodeficiency (CCI) as a single center experience, and applying project management (PM) in health care research projects, we sought the best way for a rapid diagnosis of HIES.
Results: The combination of project management principles with immunologic and genetic knowledge to better define the laboratory and clinical diagnosis leads to an improvement of the management of patients with HIES. These results are shown in one "Decision Tree" which is based on 342 genetic reports of the CCI during the past ten years.
Conclusion: It is necessary to facilitate the diagnostic analysis of suspected HIES patients; applying project management in health care research projects provides a better and more accurate diagnosis eventually leading to a better patients’ care.
Keywords: Diagnosis, Hyperimmunoglobulin E Syndrome, Project Management
ESID-0457 Hyper Immunogobulin E Syndrome: A Report on DOCK8 Mutations in Iranian Patients
S. Saghafi 1, C. Glocker 2, Z. Pourpak 1, F. Nussbaumer2, M. Fazlollahi1, A.A. Hamidieh3, M.H. Bemanian4, M. Nabavi4, N. Parvaneh5, B. Grimbacher2, M. Moin1
1Immunology Asthma and Allergy Research Institute (IAARI), Tehran University of Medical Sciences, Tehran, Iran
2Centre of Chronic Immunodeficiency (CCI), University Medical Center Freiburg and University of Freiburg, Freiburg, Germany
3Hematology-Oncology and Stem Cell Transplantation Research Center, Tehran University of Medical Sciences, Tehran, Iran
4Department of Allergy and Clinical Immunology Hazrat Rasoul-e-Akram Hospital, Tehran University of Medical Sciences, Tehran, Iran
5Department of Immunology and Allergy Children's Medical Center, Tehran University of Medical Sciences, Tehran, Iran
Background: Homozygous mutations in the dedicator of cytokinesis 8 (DOCK8) gene can lead to autosomal-recessive hyper-IgE syndrome (AR-HIES), a rare primary immunodeficiency, characterized by severe and persistent viral skin infections as well as systemic infections, eczema and allergy. Patients with DOCK8-deficiency present multiple abnormalities of the immune system and are usually found to have highly elevated serum IgE levels, eosinophilia, lymphopenia as well as defective T cell function.
Objectives: We aimed to identify the molecular basis of six Iranian patients suffering from AR-HIES.
Methods: Six patients referred to the Immunology, Asthma and Allergy Research Institute (IAARI) between 2006-2013 due to their elevated IgE level, along with persistent viral infections, were enrolled in this study. Homozygosity mapping at the DOCK8 locus was performed with all six patients, followed by PCR amplification and analysis of the 48 exons encoding for DOCK8. To exclude failed PCR amplifications results were confirmed by examination of cDNA (for two alive patients).
Results: The lack of exonic PCR products suggested deletions including parts of the DOCK8 gene in all patients. We identified two large deletions, one encompassing the whole DOCK8 gene (exons 1-48) and one spanning exons 1-44 in two patients respectively. The other four patients showed smaller deletions, one of exons 11-13, one of exons 11-14 and two unrelated patients both carried a deletion of exons 25 and 26.
Conclusion: The data presented here further expand the spectrum of mutations in DOCK8 leading to AR-HIES in the Iranian population.
ESID-0015 Combined NIH Score and TH17 Cell Numbers Is a Better Indicator of STAT3 Mutation in Hyper-IGE Syndrome
B. Saikia 1, S. Sharma1, S. Goel1, A. Rawat2, R. Minz1, D. Suri2, S. Singh2
1Immunopathology, Post Graduate Institute of Medical Education and Research, Chandigarh, India
2Pediatrics, Post Graduate Institute of Medical Education and Research, Chandigarh, India
Background: Hyper-IgE syndrome (HIES) is a rare primary immunodeficiency disorder characterized by skin abscesses, skin rash resembling atopic dermatitis (AD), elevated serum IgE levels, recurrent infections, and skeletal/ connective tissue abnormalities. A majority of those with autosomal dominant and sporadic forms have heterozygous mutations in STAT3 gene and resultant impaired TH17 differentiation
Objective: To assess blood TH17 cells in patients with HIES, AD and cases with helminthic infestation (HI) and correlate with clinical and laboratory parameters and NIH scores.
Methods: The clinical phenotypes with NIH scores ≥40 and <40, and STAT3 mutation status were analysed for absolute eosinophil count (AEC), serum IgE levels and TH17 cell numbers in 19 patients with suspected HIES and compared AD (n=16), HI with eosinophilia (n=7) and healthy controls (HC, n=20).
Results: Two patients with NIH score ≥40 were STAT3 mutation negative. AEC was highest in HI (1336/Cumm) whereas it was comparable in AD and all HIES groups. Serum IgE in patients with STAT3 mutation was comparable to those with NIH score ≥40 and HI whereas mutation negative patients or those with NIH score <40 had IgE levels similar to AD. TH17 cells were markedly low in HIES with STAT3 mutation compared to all other groups. TH17 cells were raised in AD compared to all other groups.
Conclusion: ow TH17 cell numbers together with NIH scores can have a better predictive value for presence of STAT3 mutations. Cases with Atopic dermatitis have raised TH17 cell numbers which help differentiating AD from HIES with dermatitic presentation.
ESID-0801 Invalid Assessment Cases in the PCR Results Quality of the National Institute of Research Public Health Virology Lab in Mali from 2009 to 2013; Challenges
A. SANOGO 1, Y. CISSE1, S. COULIBALY1, I. GUINDO1, D. KOITA1, M. GUINDO1, A. KEITA1, T. THIERO1, F. BOUGOUDOGO1, F. MAIGA2, A. SYLLA2
1Biological Diagnosis Depatment of Research, National Institute of Research in Public health, Bamako, Mali
2Health Ministry, Sectoriel Cell of Fight against AIDS, Bamako, Mali
SUMMARY
Introduction Sectoral Cell of Fight against AIDS of National Heath in collaboration with INRSP through the financial support of several partners helped organize periodic supervisory training to improve the early infant diagnosis quality. Despite efforts, we find a large invalid cases number affecting the PCR results quality. The Dried Blood Spot (DBS) should be evaluated through the results quality.
Our goal is to evaluate the invalid results rate on the PCR qualitative quality.
Methodology We conducted from January 2009 to December 2013 a cross-sectional study of 6067 PCR evaluative referred from health centers in references for 4 regions and Bamako's District. Majority of these were technical DBS from kits AMPLICOR HIV- 1 DNA test at the Bamako's national reference laboratory according the extraction protocol. Molecular diagnosis was performed with the same algorithm as kits three identical PCR. Almost all cases have been disabled by a second test in order to improve the PCR results quality.
Results Totally 6067 PCR was performed with 554 HIV-1 positive of 9.13% of positive cases and 153 invalid or 2.52% disability degree. According to the protocol used, 71.90% of disabilities were due to an amplification problem and 28.10% were due to the wells positive proximity. These disabilities can be explained by the DBS crafted quality, ELISA technique and DBS conservation.
Conclusion PCR remains the EID safe way. However, regular monitoring of monitoring sampling sites, the new use, and more efficient platforms can improve the EID results quality.
Keywords : HIV-1 -DBS - PCR – DNA
ESID-0671 Progressive Hepatic Disease in Griscelli Syndrome Type 2 an Enigmatic Association: Case Report
S. Scheffler-Mendoza 1, S. Lugo-Reyes1, M.A. Yamazaki-Nakashimada2
1Immunodeficiency Research Unit, Instituto Nacional de Pediatría, Mexico City, Mexico
2Clinical Immunology Department, Instituto Nacional de Pediatría, Mexico City, Mexico
Griscelli syndrome Type 2 (GS) is an autosomal recessive disorder characterized by partial albinism associated with immunodeficiency and disregulation of immune system.
Case report We present a case of a 3 year-old girl with GS type 2 and progresive chronic hepatic disease. She was the second child of healthy consanguineous parents. She was born at term with gray-silver hair. She began manifestations at 15 months of age with skin lesion in low extremities, consistent with chronic granulomatous necrotizing ulcerative dermatitis and granulomatous folliculitis. At 16 months she had neurological manifestations characterized by seizures, and lost of motor control. She coursed with bilateral papiledema and ophtalmic vasculitis; brain resonance imaging showed vasculitis. Treatment started with intravenous immunoglobulin, methylprednisolone and cyclosporine, with remission and improvement of neurological and ophthalmic manifestations. We did not find mutation at exons 2-6 of RAB27a gene.
At the age of 2 years, because of the severe manifestations, hematopoietic stem cell transplantation (HSCT) was performed from a matching sibling donor but it was not successful. After HSCT she had progressive elevation of hepatic enzymes; we discarded infections, adverse drug reactions and autoimmune disease. Liver biopsy revealed intracellular intracanalicular cholestasis and nonspecific portal inflammation. Nowadays the patient is waiting for a second HSCT.
Conclusion. Our patient has atypical manifestations of GS. She debuted with neurological manifestations without hemophagocytosis infiltrate in nervous system and the cholestasic chronic hepatic disease. This last condition has not been reported previously. A possible etiology could be progressive familial intrahepatic cholestasis.
ESID-0468 How to Diagnose and Treat Neuropathic Osteopetrosis
A. Schulz 1, G. Lahr1, C. Schuetz1, M. Hoenig1, H. Bode1, K.M. Debatin1, B. Winter1
1Department of Pediatrics and Adolescent Medicine, University Medical Center Ulm, Ulm, Germany
Six pts originating from 5 independent families of Arabic (n=5) and German (n=1) origin with neuropathic infantile osteopetrosis and biallelic mutations in CLCN7 (n=5) and OSTM1 (n=1) were treated in our institution between 2011 and 2013. After a mostly unremarkable postnatal period all infants developed failure to thrive due to feeding difficulties, mild anemia and thrombocytopenia during their first months of life. After this period, progressive neurological symptoms appeared starting with mild muscular hypotonia and ended up with severe spasticity and general epilepsy in the first year of life. Accordingly, there was a gradual deterioration of EEG findings with a characteristic pattern of multifocal epileptiform activity. In MRI, brain atrophy, hypoplasia of the corpus callosum and delay of myelinisation were salient findings. Remarkably, the clinical picture as well as EEG and MRI investigations were found to be normal at very early time points. EEG changes seem to be the first measurable signs of neuropathy.
One pt with CLCN7 mutation was transplanted from a MSD after myeloablative conditioning at the age of 11 weeks before any signs of neuropathy were observed. Unfortunately, during the transplantation the pt developed these typical clinical symptoms and EEG abnormalities and died despite of full donor engraftment several months after transplantation.
Progressive neurodegeneration with dismal prognosis is found in a subgroup of infantile osteopetrosis pts with OSTM1 and CLCN7 mutations. Since these pts do not benefit from stem cell transplantation, a careful workup regarding neurological abnormalities is mandatory in pts with mutations in these genes.
ESID-0472 Immunodeficiency, Osteopetrosis and Bleeding – A Diagnostic Challenge
A. Schulz 1, I. Furlan1, H. Schulze2, B. Zieger3, J. Zustin4, M. Hoenig1, C. Schuetz1, R. Crazzolara5
1Department of Pediatrics and Adolescent Medicine, University Medical Center Ulm, Ulm, Germany
2Center of Experimental Biomedicine, University Clinic of Wuerzburg, Wuerzburg, Germany
3Department of Pediatric Hematology and Oncology, University Medical Center Freiburg, Freiburg, Germany
4Institute of Pathology, University Hospital Muenster, Muenster, Germany
5Department of Pediatrics, Innsbruck University Clinic, Innsbruck, Austria
We report on a 1.5 year old girl, who adapted poorly after birth and developed neonatal sepsis. Substantial leukocytosis in association with mild anemia and thrombocytopenia was detected, which persisted after successful treatment of the infection. The patient experienced recurrent episodes of bacterial infections and mucosal bleeding episodes in her first weeks of life. Because of increased bone density in the initial X-rays infantile osteopetrosis was suspected.
Bone marrow histology confirmed significant focal bone marrow fibrosis compatible with osteopetrosis.In addition, giant and dysfunctional osteoclasts were described. However, no mutations were detected in genes usually involved in infantile osteopetrosis such as TCIRG1, CLCN7, SNX10, RANK and RANKL. Since the patient showed severe mucosal hemorrhage despite of only mildly decreased platelet counts, a thrombocytopathic disorder was supposed and verified by in vitro assays. This combination of frequent bacterial infections, platelet dysfunction and osteopetrosis was consistent with the phenotype described for Leukocyte Adhesion Deficiency Type III, a very rare disorder with autosomal recessive inheritance. Sequencing of FERMT3 encoding for Kindlin-3 revealed a homozygous nonsense-mutation. Accordingly, no Kindlin-3 expression was found in blood cells of the patient.
The patient was treated by allogeneic stem cell transplantation from a matched unrelated donor after myeloablative conditioning. One year after transplantation she is alive and well without signs of the underlying disease.
ESID-0519 Abnormal Natural Killer Cell Functions and Clinical Heterogeneity of Chronic Mucocutaneus Candidiasis Disease in Patients with Heterozygous Mutations in the STAT1 Gene
O. Scomodon 1, D. Vairo1, L. Tassone1, S. Parolini2, G. Tabellini2, O. Patrizi2, M. Giacomelli1, G. Maggiore1, A. Soresina1, V. Lougaris1, A. Plebani1, S. Giliani1, R. Badolato1
1Pediatrics Clinic and Laboratory for Molecular Medicine "Angelo Nocivelli" Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy
2Department of Molecular and Traslational Medicine, University of Brescia, Brescia, Italy
We have analyzed six patients with Chronic Mucocutaneus Candidiasis Disease (CMCD) affected by persistent and recurrent infections of nails, skin, oral or genital mucosae caused by Candida albicans. One patient also showed cryptococcal and Leishmania infections. We identified a missense heterozygous mutation in STAT1 coiled-coil domain, L283M and three different mutations in DNA-Binding domain, T385M, L351F and L400V.
In order to investigate how these mutations could affect the ability of the immune system to response against Candida albicans, we characterized STAT1 phosphorylation in response to interferon-alpha and interferon-gamma, the capacity of CD4+ T-lymphocytes to polarize to Th17 and the ability to determinate luciferase activity of a reporter gene under control of the GAS promoter after different stimulations. Our results confirm the Gain of function trait of all mutations.
Analysis of Natural Killer (NK) cells in four patients revealed abnormal functions when compared with healthy donors. Cytoflorimetric analysis of peripheral blood lymphocytes revealed low percentage of NK cells in one patient only. Regarding expression of activating receptors on different subsets of NK cells CD56dim and CD56bright we did not observe altered expression. In three of six patients, CD56dim CD16+ NK cell showed lower expression of KIR molecules and of maturation marker CD57. Moreover, on CD56dim NK cells, we noted higher expression of activating marker CD69 when compared with normal donors NK cells. Finally, interferon-gamma production was also defective in STAT1-mutated patients when compared with healthy donors. These results suggest that abnormal NK functions might account for clinical heterogeneity of CMCD patients.
ESID-0327 Disseminated Mycobacterial Infection in One Patient with CD 40 Ligand Deficiency
A. SEMINARIO 1, A. Bravo Kleiman1, M. Esnaola Azcoiti1, D. Comas1, A. Gomez Raccio1, D. Di Giovanni1, M. Gaillard1, L. Bezrodnik1
1Immunology, R. Gutierrez Children Hospital, CABA, Argentina
Introduction: Disorders pathway interleukin 12 (IL12)/ gamma interferon (IFNγ) has greater susceptibility to mycobacterial infections. The involvement of CD40/CD40L in NFKB signaling pathway is important for the production of IL12 for protection against mycobacterial infections. Objectives: Report a case of disseminated mycobacterial infection in patient with CD40L deficiency (CD40LD). Material and Methods: Retrospective analysis of medical history of a six years old boy diagnosed with CD40LD.Results: At 9 months of life was diagnosed of CD40LD because of pseudomona pneumonia and chronic neutropenia. Began prophylaxis treatment with trimethoprim sulfametoxasole, intravenous gammaglobulin and granulocytes stimulating factors.His evolution was stable and persists with neutropenia. After 5 years he presented cervical adenomegaly with positive tuberculosis complex PCR and granulomas were presented in the pathological anatomy so began treatment antituberculostatics drugs. Despite of a correct treatment after 15 days he presented typhlitis and abundant caseous has been observed in the bowel mucous membrane. He needed surgeries and parenteral nutrition because of his bowel compromise. As his evolution was going bad we added IFNγ for 6 months with clinical improvement. Nowadays he is waiting for stem cell transplantation. No donors are available up today. Conclusion: Here we report a complication in the bowel that has not been reported in these patients before.CD40/CD40L interaction is thought to be important in the handling of mycobacterial infections. Usually this germ is relatively uncommon in this deficiency
ESID-0378 Effects of Rat Mesenchymal Stem Cells Supernatant Derived from Bone Marrow and Adipose Tissue on Peripheral Blood Mononuclear Cell Viability
H. Sevim 1, E. Akbay1, D. Perver1, O.A. Gurpinar1, M.A. Onur1
1Biology, Hacettepe University, Ankara, Turkey
Introduction Mesenchymal stem cells (MSCs) are a small population of multipotent progenitor cells are isolated from various tissues like bone marrow and adipose tissue. MSCs are important source for regenerative therapy. They also secrete mediators for reducing inflammation and accelerating tissue regeneration, growth factors for differentiating the cells. Peripheral blood mono nuclear cells (PBMC) are the most common tolls for immunological research but their lifespan in tissue culture is very limited. Although all MSCs express the same factors or have a similar secretion profile is still controversial, our aim is to study the effect of secreted factors from MSCs from two different tissue on PBMC proliferation.
Methods Density gradient centrifugation was used to isolate bone marrow mesenchymal stem cell (BM-MSCs), primary explants culture technique is used to isolate adipose tissue derived mesenchymal stem cells (AD-MSCs) . Supernatant of cells collected 24, 48 and 72 hours of incubation and filtered. PBMC were treated with supernatant for 5 days normal media used as a control. Cell viability were assessed with PrestoBlue assay at 1st, 3rd and 5th days of incubation.
Results and Conclusion Cell proliferation was higher BM-MSCs supernatant from 48 hours than control and AD-MSCs group. Also PBMC showed standart cell proliferation profile. MSCs from different tissues have different secretion profiles according to their source. These abilities can be further used for the research areas of immunology.
Figure 1. PrestoBlue assay results showed tha cell has higher viabilty BM-MSCs 48 hours supernatant. Values are expressed as average ±ss of three replicates.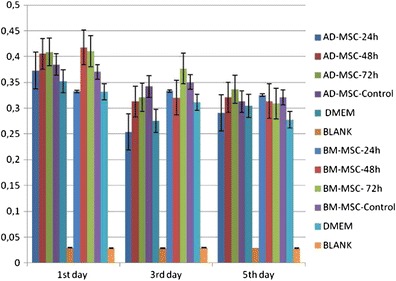
ESID-0636 Enigmas Left in Diagnosis: Chronic Epstein Barr Virus Lymphoproliferation or Lymphoma
R. Sherkat 1, N. Reisi1, M. Nazem1
1Aquired Immunodeficiency Research Center, Isfahan University of Medical Sciences, Isfahafan, Iran
In primary B cells, the expression of the catalytically active form of activation-induced cytidine deaminase has been shown to lead to MYC/IGH translocations, similar to those which occur in Burkitt’s lymphoma (BL) within a matter of hours. These translocations are normally prevented by the tumour suppressor genes like ATM consistent with the ability of these genes to inhibit progression through the cell cycle and to initiate DNA repair or apoptosis in the presence of DNA damage.As a result in the absence of ATM most people with Ataxia Telangiectasia (A-T) have broken pieces of DNA in chromosomes involved in the above mentioned rearrangements and a tendency to recombine with other genes (translocation), making the cells prone to the development of cancer (lymphoma and leukemia).
EBV-containing cells must reach the memory cell compartment in order to survive throughout the life of the individual. Normally, cells that do not produce high affinity antibodies do not survive this passage, and are induced to undergo apoptosis. EBV, however, prevents this, and in doing so may also enhance the likelihood of survival of rare translocation-containing cells.
We present a girl with A.T has got acute EBV infection at the age of four . Burkeit Lymphoma , positive in-situ hybridization for EBV-encoded RNA with immunostaining against CD20 , after few months of EBV infection . Tumour has been subsided without proper chemotherapy but larg cell lymphoma developed after 3 years with diffuse lymphadenopathy and consequently the lymph nodes and spleen has been regressed again after few months later.
ESID-0637 New Notions on Platelet Biology in Wiskott-Aldrich Syndrome (WAS)
R. Sokolic 1, K. Nghiem2, N. Oden3, E. Garabedian1, J. Lozier2, F. Candotti1
1Disorders of Immunity Section, National Institutes of Health, Bethesda, USA
2Department of Laboratory Medicine, National Institutes of Health, Bethesda, USA
3Department of Statistics, EMMES corporation, Rockville, USA
Microthrombocytopenia is a clinical feature of WAS. However, mechanisms leading to low platelet counts and bleeding complications remain incompletely defined. Increased peripheral platelets destruction in WAS is widely accepted, while less information is available about abnormalities of platelet production. We collected data on immature platelet function (IPF), a marker of platelet production, in 18 WAS patients and 30 patients with immune thrombocytopenic purpura (ITP). Consistent with reduced production, we found a reduction of IPF values in WAS patients compared to ITP patients (2% vs. 6%, p= 0.0063). Because children with WAS are often misdiagnosed as having ITP, we took advantage of this observation to develop a diagnostic rule by which WAS should be considered if 75*IPF+PLT
We performed thromboelastography in 16 WAS patients. Most parameters were normal in citrated whole blood, although the time to initiate a clot (R) was decreased. When Kaolin was added to the blood, R normalized, but parameters of clot strength (K, α, MA and G) decreased. This mechanical weakness was attenuated but not completely reversed, in patients with >100,000 platelets/mcL after splenectomy. Similarly, clotting parameters in platelet rich plasma from two eusplenic patients improved but didn't normalize, suggesting that factors beyond thrombocytopenia contribute to clot weakness in WAS.
ESID-0758 Tracking the Development of the Humoral and Cellular Fetal Immunity
E. Rechavi1, A. Lev1, N. Amariglio1, G. Rechavi1, A.J. Simon1, Y.L. Lee2, L.D. Notarangelo2, B. Weisz1, R. Somech 1
1Pediatrics Immunology, Sheba Medical Center, Ramat Gan, Israel
2Children's Hospital Harvard Medical School, Division of Immunology and Manton Center for Orphan Disease Research, Boston Mass, USA
Extending our knowledge on the development of the human fetal immune system is critical for understanding of primary immune deficiencies, intrauterine infections and maternal-fetal interactions. This field remains largely uncharted territory. In this study, we document adaptive immunity developmental processes in human fetuses as young as 12 weeks gestational age (GA) and describe their progression throughout gestation. Blood samples from fetuses 12-26 weeks GA were collected and analyzed for lymphocyte subset counts, TCR rearrangement excision circle (TREC) and Kappa-deleting recombination excision circle (KREC) copy numbers, TCR-Vb families spectratyping, and finally, TCR and BCR repertoire analysis by PCR and next generation sequence (NGS) techniques. Both TRECs and KRECs could be detected in small numbers as early as 12 weeks GA and rose in copy numbers along with GA. Cell repertoires showed an olygoclonal pattern in early weeks and gradually evolve to form a clear polyclonal pattern in later weeks. These findings were confirmed when NGS was used to sequence thousands of TCRs and BCRs. NGS results could also shed more lights on other diversity mechanisms including DNA recombination preferences and somatic hyper mutation. In terms of both number of excision circles and clonal diversity, B-cell development precedes that of T-cells. These findings suggest partial fetal immunocompetence as early as mid-second trimester. This may enable earlier recognition of primary immune deficiencies and could promote our understanding of the role the fetal immune system plays in abortions versus maternal-fetal immune tolerance.
ESID-0041 Psychological Thearpy for Adults with Immunodeficiency: An Update
M. Campbell 1, A. Clarke2, A. Symes1, S. Workman1, S. Burns1, R. Chee1, D. Lowe1, S. Seneviratne1, H. Stauss3, D. Webster3
1Clinical Immunology, Royal Free Hampstead NHS Foundation Trust, London, United Kingdom
2Plastic & Reconstructive Surgery, Royal Free Hampstead NHS Foundation Trust, London, United Kingdom
3Institute of Immunity & Transplantation, University College London, London, United Kingdom
For some individuals, living with an immunodeficiency can have a significant impact on their quality of life (Quinti et al., 2012; Booker et al., 2007; Edwards et al., 2003). For three years, a Specialist Immunology Service in the UK has been screening and offering short-term psychological treatment to patients to lessen this impact and improve overall wellbeing. A case series analysis examining the effectiveness of the treatment in improving mental wellbeing, its impact on medical services usage, and acceptability with the model of treatment will be presented. The implications of these findings and suggestions for future research and service development will be discussed.
ESID-0332 Primary Immunodeficiency Diseases with Mucocutaneous Candidiasis: “Experiments of Nature”
A. TAHIAT 1, R. DJIDJIK1, S. CHERIK1, R. BELLOUNI1, A. BENSENOUCI2, M.E. KHIARI3, M. GHAFFOR1
1Medical Biology Laboratory, CHU béni-Messous, algiers, Algeria
2Department of Pediatrics "B", CHU béni-Messous, algiers, Algeria
3Department of Pediatrics "A", CHU béni-Messous, algiers, Algeria
Introduction. Mucocutaneous candidiasis occurs either in isolation or alongside other symptoms in patients with various primary immunodeficiency diseases (PID). The primary purpose of this study is to identify PIDs with susceptibility to candidiasis and a secondary goal is to determine the critical pathways in human immunity against Candida species.
Patient and methods. This retrospective study was conducted on 140 patients.
Results. Among the 140 PIDs, oral and mucocutaneous candidiasis were observed in 19 (13.6%) patients who can be divided in two groups. The first group (n=13) include patients with T-cell defects: SCID (n=6), DiGeorge syndrome (n=1), MHC—Class II deficiency (n=5), and DOCK8 deficiency (n=1). The second group (n=6) is very heterogeneous with regard to the nature of the immunodeficiency but has a common feature that is neutrophils defect: congenital neutropenias (n=1), chronic granulomatous disease (n=1), hyper-IgM syndrome (n=1), X-linked agammaglobulinemia (n=2) and Chediak—Higashi syndrome (n=1). The present work highlights the role of T-cell, in particular TH17 cells and neutrophils in host defense against Candida species. TH17 cells secrete IL-17 and IL-22, which promote activation and recruitment of neutrophils. Neutrophils mediate microbial killing through phagocytosis, degranulation, and neutrophil extracellular traps.
Conclusions. The delineation of the critical pathways in human host defense against against Candida species will not only lead to an improved risk stratification in affected patients but will also lead to improved novel therapeutic management strategies by strengthening the IL-17/IL-22 axis in patients at risk for or already having overt disease.
ESID-0107 Wiskott-Aldrich Syndrome in a Girl Caused by Heterozygous Wasp Mutation and Extremely Skewed X-Chromosome Inactivation: An Association of Non-Random X-Chromosome Inactivation and Uniparental Isodisomy 6
H. Takada 1, T. Takimoto2, M. Ishimura2, M. Urata2, T. Morio3, T. Hara2
1Department of Perinatology and Pediatric Medicine, Graduate School of Medical Sciences Kyushu University, Fukuoka, Japan
2Department of Pediatrics, Graduate School of Medical Sciences Kyushu University, Fukuoka, Japan
3Department of Pediatrics and Developmental Biology, Graduate School of Medicine Tokyo Medical and Dental University, Tokyo, Japan
Wiskott-Aldrich syndrome (WAS) is an X-linked disease characterized by microthrombocytopenia, eczema, and various degrees of immune deficiency, caused primarily by mutations in WASP (Wiskott-Aldrich syndrome protein) gene. Female carriers of the disorder have no clinical signs because of the preferential selection of the normal, nonmutated X-chromosome in their hematopoietic cells. Here we report a girl with WAS, who manifested the signs of thrombocytopenia and inflammatory bowel disease since early infancy. The DNA sequencing analysis of the WASP gene revealed a heterozygous nonsense mutation in exon 10. On the other hand, peripheral blood cells of the patient lacked the expressions of WASP and normal WASP mRNA. X-chromosome inactivation pattern of the patient was investigated by using methylation-sensitive restriction enzyme HpaII on the human androgen receptor gene located in X-chromosome. We found non-random inactivation of paternally-derived X-chromosome on which normal WASP gene was located. Surprisingly, maternal uniparental isodisomy of chromosome 6 (UPD6) was confirmed in this patient by single nucleotide polymorphism microarray analysis and by analyzing polymorphic variable number of tandem repeat regions (VNTR) from chromosome 6 of the patient and her parents. Our results indicate the importance of WASP evaluation in females with neonatal thrombocytopenia. In addition, further analysis on the pathophysiology of this rare association may provide a clue to clarify the mechanism of X-chromosome inactivation and pathogenesis of the development of X-linked disorders in females.
ESID-0326 Agammaglobulinaemia in a Male Patient Due to Mutations in BTK and WAS Genes
S. Tantou 1, A.C. Asplund2, N. Wang3, M. Tzanoudaki1, C.I.E. Smith2, L. Hammarström3, M. Kanariou1
1Specialized Center & Referral Center for Primary Immunodeficiencies – Paediatric Immunology, "Aghia Sophia" Children's Hospital, Athens, Greece
2Department of Laboratory Medicine Clinical Research Center, Karolinska Institute, Stockholm, Sweden
3Div. of Clinical Immunology Dept. of Laboratory Medicine, Karolinska Institute at KUS Huddinge SE-14186, Stockholm, Sweden
Introduction: X-linked agammaglobulinaemia and Wiskott - Aldrich syndrome are two Primary Immunodeficiency Diseases caused by individual genes located on the X -chromosome.
Case Report: We report a 30 year-old male patient with a combination of BTK and WAS mutations. During his infancy he was hospitalized because of respiratory infections and an episode of septicaemia. At late childhood, undetectable levels of IgG and IgA, normal IgM and slightly decreased B cells were found. The diagnosis was Hyper-IgM Syndrome and immunoglobulin replacement therapy was started. At his middle twenties he was referred to our center; B cells in peripheral blood were almost absent, and IgG, IgA low with declining IgM levels. CD40L was expressed normally. Further investigation revealed extremely low BTK expression on monocytes by FACS as compared to his mother and a healthy control. WAS mRNA expression was higher than the control sample. At 28 years of age, he was diagnosed to have a novel intronic mutation in BTK (a novel intronic mutation in BTK (78 bp downstream of exon 15) and an uncommon mutation in WAS gene (E131K / SNP rs146220228). The intronic BTK mutation is of unknown significance, while analysis of RNA revealed the absence of exons 2 and 3. The patient remains in a good clinical condition.
Conclusion: This is the first reported patient with a combination of BTK and WAS mutations. The mechanism and the clinical importance of this finding remains unclear. This combination might potentially be involved in the pathogenesis of the patient's unique phenotype.
ESID-0147 Clinical and Cost-Effectiveness Prospective Study of Neonatal Screening for Severe Combined Immunodeficiency Using the T-Cell Receptor Excision Circles Assay in a French Multicentre Study
C. Thomas 1, S. Mirallie2, I. durand-zaleski3, V. Sebille4, N. Mahlaoui5, A. Fischer6, M. Audrain7
1paediatric immunology and haematology, Hôpital mère-enfant CHU Nantes, Nantes, France
2neonatal screening lab, Hôpital mère-enfant CHU Nantes, Nantes, France
3URC-eco, APHP Paris, paris, France
4Biometry Platforme, University, nantes, France
5CEREDIH, Necker Hospital, Paris, France
6CEREDIH Institut Imagine, Necker Hospital, Paris, France
7Immunology lab, CHU, Nantes, France
Objective: To evaluate the clinical utility and the cost-effectiveness of the TREC quantification assay for neonatal screening of SCID in France.
Methods: One group of 200 000 newborns who will benefit from the screening for SCID, all over France during 2 years, representing 1birth out of 8. The method used is the Enlite™ TREC kit.
A control group comprising all newborns diagnosed with a SCID in the same period who have not benefited from screening.
The screening will be organized using the architecture already used in France for other screening. Briefly, a national organization is responsible of screening which is performed in 23 regional centres.
A supplemental Guthrie card will be prepared at the same time that the original card, after information of parents and signature of consent. TREC assay will be performed in 2 laboratories.
We propose to study the clinical utility and cost effectiveness ratio , and SCID screening to Demonstrate That Could result in a broad benefit to Individuals detected, making screening Relatively cost-effective in Spite of the low incidence of the disease.
This study is supported by a grand from the French Ministry of Health (PRME 2013 13-0265)
ESID-0781 Immunological and Clinical Features in Two Cartilage-Hair Hypoplasia Patients with Compound Heterozygous RMRP Mutations
J.M. TORRES CANIZALES 1, P. MORALES2, B. Sagastizabal3, R. Rodriguez1, A. Ferreira1, J.T. Ramos3, L.M. Allende2, E. Lopez-Granados1
1Clinical Immunology, Hospital Universitario La Paz-IdiPAZ, MADRID, Spain
2Immunology, Hospital 12 de Octubre, MADRID, Spain
3Deparment of Pediatrics, Hospital de Getafe, MADRID, Spain
Background: Cartilage-hair hypoplasia (CHH) is a rare autosomal recessive disease caused by mutations in the RMRP gene. Clinical variability is remarkable, due to different degrees of combined immunodeficiency, autoimmunity, and malignancy. Deciding that a patient could benefit from Hematopoietic stem cell transplantation (HSCT) is challenging, but can improve prognosis by restoring immunity. We present two unrelated patients with distinct manifestations.
Case 1. A 11-years-old girl, presented at 2 years of age with recurrent respiratory infections and diarrheas. At 5 years of age she developed immune thrombocytopenic purpura (ITP). She had short stature, fine and sparse hair, and metaphyseal chondrodysplasia. Immunological evaluation showed CD3+ lymphopenia, mostly of naive T cells, with normal B and NK cells. Lymphoproliferative responses were normal. Immunoglobulins were normal, but specific antibodies were low. Heterozygous RMRP mutations: g.215G/A; g.251A/C were found. ITP became refractory to treatment and HSCT with HLA-matched related donor was undertaken.
Case 2. A 3-years-old girl was born with metaphyseal chondrodysplasia, and just presented delayed growth her first 2 years of life. Then she commenced with recurrent diarrhea, steatorrhea, rectal prolapse, and sclerosing cholangitis. She developed Coombs+ hemolytic anemia. Immunological findings included CD3+ lymphopenia, low naive T and memory B cells. Lymphoproliferative responses were low. IgA was undetectable but IgG and IgM were normal. Heterozygous RMRP mutations: g.5C/T; g.32C/T were identified. She is on antibacterial prophylaxis, immunoglobulin replacement and awaiting HSCT.
Conclusions: Patients with CHH should have continuous immunological assessment, and HSCT considered in some to prevent significant organ damage or malignancy.
ESID-0440 Primary Hemophagocytic Lymphohistiocytosis in Patient with Isolated Neurological Findings
S. Unal 1, M. Cetin1, Y. Bryceson2, F. Gumruk3
1Division of Pediatric Hematology, Hacettepe University Medical School, ANKARA, Turkey
2Department of Medicine, Karolinska University Hospital Huddinge, Stockholm, Sweden
3Division of Pediatric Hematology, Hacettepe University, Ankara, Turkey
Hemophagocytic lymphohistiocytosis (HLH) is the clinical syndrome of an overstimulated but ineffective immune response associated with hypercytokinemia and may be either primary or secondary. A 2.5 year-old girl presented with afebrile convulsions, somnolence to another center 2 months ago and was thought to have encephalitis. Cerebrospinal fluid (CSF) examination was unrevealing at that time and MRI revealed cerebral edema. Cefotaxime, vancomycin and aciclovir was initiated. During the course she had dysphagia and gastrostomy was performed. Spasticity developed, she was unable to contol her neck, was unable to sit or talk. She was referred to our center after replacement of a ventriculoperitoneal shunt. At admission to our center, MRI revealed cerebellar edema, bilateral hyperintense lesions in the cerebellar hemispheres and vermis at the T2 weighted images, micronodular, irregular, periventricular, cortical and subcortical lesions with restricted diffusion in the cerebral hemispheres, in addition to diffuse hemorrhagic lesions in the white and grey matter of cerebrum and cerebellum. CSF protein 440 g/dL with lymphomononuclear cells. Tests for viral or mycobacterial etiologies were negative. She had no fever, splenomegaly or cytopenias. Ferritin: 167.9 ng/mL, fibrinogen: 227 mg/dL, triglyceride 172 mg/dL. Cerebellar biopsy revealed T cell infiltration, bone marrow revealed no hemophagocytosis. Molecular testing exhibited compound heterozygous mutation in PRF1 gene. HLH 2004 protocol was initiated, HSCT was planned. The diagnostic criteria for HLH has been well-established. Herein, we describe a patient who presented with neurological findings solely and was diagnosed to have primary HLH, without fulfillment of the diagnostic criteria.
ESID-0455 A Standardized and Validated Screening Tube for PID Diagnostics: The Euroflow Approach
M. van der Burg 1, T. Kalina2, M. Perez-Andres3, E. Lopez-Granados4, A. Kienzler5, M. Vlkova6, J. Torres-Canizales4, J. Stuchly2, M. Wentink1, E. Mejstríková2, M.C. van Zelm1, A. Orfao3, J.J.M. Van Dongen on behalf of the EuroFlow consortium - PID Work Package1
1Dept. of Immunology, Erasmus MC, Rotterdam, Netherlands
2Dept. of Pediatric Hematology and Oncology, Charles University, Prague, Czech Republic
3Dept. of Medicine-Serv. Cytometry, Univ. of Salamanca, Salamanca, Spain
4Dept. of Immunology, Hospital Universitario La Paz, Madrid, Spain
5Nuffield Department of Medicine Experimental Medicine Division, University of Oxford, Oxford, United Kingdom
6Institute of Clinical Immunology and Allergology, Masaryk University and St Anne`s University Hospital Brno, Brno, Czech Republic
Flowcytometric immunophenotyping of peripheral blood has become central in diagnostics and research on primary immunodeficiencies (PID). Many centers have developed multi-color flowcytometric protocols, but differences in antibody panels, sample handling, instrument setup and data analysis hamper exchange of data between centers. With the EuroFlow PID consortium consisting of six laboratories, we developed an 8 color – 12 parameter screening tube using a standardized protocol for sample handling, instrument setup, antibody titers and uniform analysis. With this screening tube all B, T and NK cell subpopulations and their maturation stages can be analyzed. We have evaluated 52 genetically defined PID of 9 disease groups (18 SCID, 14 DiGeorge Syndrome, 4 CD40L, 4 ALPS, 6 BTK, 2 DOCK8, 1 GATA2, 1 WAS, 2 XIAP) with the EuroFlow PID screening tube (PST).
We used the C4.5 classification tree algorithm to find whether we can achieve full classification of the disease groups for cases evaluated with the PST at distinct centers. Full classification was possible into 10 groups, where classifiers for all disease categories were found, only DOCK8 patients were not classified into a common group. Ten of the 12 markers appeared useful for current classification, the other two being useful for identification of cell subsets. In addition, we could visualize the (missing) lymphocyte subsets in multidimensional space using Infinicyt software (e.g. APS view).
In conclusion, the standardized EuroFlow PID screening tube facilitates fast and standardized immunophenotypic diagnosis of PID and therapeutic monitoring and most importantly, allows full exchange of data between different centers.
ESID-0445 Implementation of Targeted NGS Based PID Screening for Routine Diagnostic Use
J. van Montfrans 1, L. van de Corput2, P. van Zon3, M.G. Elferink3, W. Harts3, J. Frenkel1, H. Leavis4, P. Ellerbroek4, A. van Royen- Kerkhof5, W. Janssen5, M. Boes5, J. Ploos van Amstel3, M.E. Gijn3
1Pediatric Immunology and Infectious Diseases, University Medical Center Utrecht, Utrecht, Netherlands
2Medical Immunology, University Medical Center Utrecht, Utrecht, Netherlands
3Medical Genetics, University Medical Center Utrecht, Utrecht, Netherlands
4Infectious Diseases Rheumatology and Immunology, University Medical Center Utrecht, Utrecht, Netherlands
5Pediatric Immunology and Infectious diseases UMC Utrecht the Netherlands, University Medical Center Utrecht, Utrecht, Netherlands
Primary immunodeficiency (PID) disorders are a genetically heterogeneous group monogenetic immune defects. To date, mutations in over 210 different genes causing PID have been described. The genotype-phenotype correlation can be highly variable which makes a genetic diagnosis in PID patients complex and laborious.
We previously developed a diagnostic method to facilitate genetic evaluation of 184 known PID related genes using Next Generation Sequencing (NGS). We applied targeted in-solution enrichment, a SOLID sequencing platform and an in-house developed bioinformatic pipeline to analyze changes in the selected genes. Detected variants were confirmed by Sanger sequencing.
Since May 2013, 89 patients were tested, identifying a genetic diagnosis in 17 cases. In 9 additional patients the genetic results need follow-up by functional testing. In 28 patients, variants of unknown significance (VUS) or only single heterozygous pathogenic mutations were detected. No mutations were detected in 35 patients. Approximately 50% of the patients with a confirmed genetic diagnosis had an atypical presentation of previously described PIDs (including mutations in STAT3, STAT1, NLRP12, and NLRP3). Interestingly, we detected with NGS mosaicism for a pathogenic NLRP3 mutation which was missed with Sanger sequencing. This further supports the application of NGS based testing.
To conclude, our results indicate that this NGS-based approach is an efficient strategy for rapid mutation detection in PID. Moreover, the increased yield will give more insight in the genotype phenotype relationship for the different PID disorders enabling earlier diagnosis and better treatment.
ESID-0544 Progressive Multifocal Leucoencephalopthy in a CVID Patient
N. Verma 1, D. Kidd2, M. Dziadio1, S.L. Seneviratne1, S.O. Burns1, R. Chee1
1Immunology, Royal Free Hospital, London, United Kingdom
2Neurology, Royal Free Hospital, London, United Kingdom
Introduction Progressive Multifocal Leucoencephalopathy (PML) is a rare and fatal disease of the CNS leading to virally driven inflammatory white matter changes within the brain. PML occurs secondary to JC virus reactivation in immunocompromised individuals. The literature is scarce on the occurrence of PML in Common Variable Immunodeficient (CVID) patients.
Method We report on a CVID patient who developed PML.
Results The 65-year-old female newly diagnosed with CVID (pre-IVIG IgG 0.5 g/l) and awaiting to be commenced on intravenous immunoglobulin (IVIG) replacement, presented with aphasia, progressive right sided weakness and cognitive impairment. CT and MRI head evidenced subcortical and periventricular white matter lesions, mainly within the frontal lobe. She was HIV negative by PCR. Lumbar puncture confirmed the presence of the JC virus (170,000 JCV DNA copies/ml) and she was treated with 5 day regime of ultra-high dose IVIG (5 g/kg). Despite the initial clinical neurological improvement, she subsequently developed PCP pneumonia and her PML deteriorated on MRI with new lesions in the cerebellum, parietal lobes, corpus callosum and midbrain. She passed away after a prolonged hospital admission.
Conclusion PML is uncommon in CVID patients established on immunoglobulin replacement despite the underlying immunodeficiency. There is currently no effective treatment for JC virus infection, although one report suggested CNS viral clearance with high dose IVIG. IVIG should be commenced urgently if the pre-IVIG IgG is very low to avoid re-activation of JC virus in CVID patients.
ESID-0109 Different Leaky Phenotype in Two Siblings with X-Linked Severe Combined Immunodeficiency
T. Wada 1, T. Toma1, M. Yasui2, M. Inoue2, K. Kawa2, K. Imai3, T. Morio3, A. Yachie1
1Pediatrics, Kanazawa University Graduate School of Medical Sciences, Kanazawa, Japan
2Hematology/Oncology, Osaka Medical Center and Research Institute for Maternal and Child Health, Osaka, Japan
3Pediatrics, Tokyo Medical and Dental University, Tokyo, Japan
X-linked severe combined immunodeficiency (XSCID) is caused by mutations of the common gamma chain (γc) and usually characterized by the absence of T and NK cells (T-NK-B+). However, atypical cases of XSCID presenting with autologous T and/or NK cells have been described. Here we report two siblings with atypical XSCID who showed different clinical and immunological features. The elder brother had autologous T and NK cells (T+NK+B+) and Omenn syndrome-like manifestation including erythroderma, lymphadenopathy, hepatosplenomegaly, eosinophilia, low serum IgG, and elevated serum IgE. The younger brother was diagnosed with XSCID at birth due to the positive family history. He had autologous NK cells but not T cells (T-NK+B+), and showed no manifestations of Omenn syndrome. These patients carried the same splice-site mutation (IVS1+5G>A) that caused most of the mRNA to be incorrectly spliced but produced normally spliced transcript in lesser amount. Similar levels of residual γc expression were observed in their lymphocytes. The leaky splice-site mutation likely permitted NK-cell development to occur in our patients; however, the reason underlying the appearance of autologous T cells only in the elder brother is presently unclear. His T cells showed activated phenotype, a moderate restricted T-cell receptor repertoire, and no detectable levels of T-cell receptor excision, suggesting that his T cells were generated from few T-cell progenitors. These findings suggest that leaky γc mutations may lead to diverse spectrum of clinical and immunological phenotype.
ESID-0484 Lymphoma in Adult Patient with Nijmegen Breakage Syndrom (NBS) – The Diagnostic Challenges
E. Wiesik-Szewczyk 1, A. Kucharczyk1, K. Jahnz-Rozyk1, E. Bernatowska2
1Clinical Immunology and Allergology, Military Institute of Medicine, Warsaw, Poland
2Clinical Immunology, Children's Memorial Health Institute, Warsaw, Poland
Background: Nijmegen breakage syndrome (NBS) is a rare autosomal recessive condition of chromosomal instability that is a strong predisposition to lymphoid malignancy. NBS is caused by mutations in the NBN/NBS1 gene involved in end-processing of physiological and mutagenic DNA double-strand breaks and maintenance of the chromosomal stability.
Objectives: To show insidious course of lymphoma in NBS patient.
Case: On March 2013, 20-years old NBS female patient on continuous IVIG therapy, presented with cervical lymphadenopathy, moderate weight loss, and persistent ulcerative skin lesions (Fig 1.). Laboratory tests revealed anemia, thrombocytopenia, ESR/CRP elevation. Subject`s brother, also diagnosed with NBS, was successfully treated for lymphoma at the age of nine. Diagnostic tests excluded infection (including EBV) or lymphoma (cytometry of lymph node). The PET-CT scan revealed active metabolic pattern with general lymph-node and some organ and focal skin involvement. Other tests: histo-pathology of the whole lymph-node, bone marrow trepan biopsy, and skin biopsy suggested non-malignant lymphocyte B proliferation and non-specific granulomatous inflammation. On further follow-up, abdominal and chest MRI was performed and revealed small tumor in muscle, which was tested on histopathology. Eventually on March 2014 aggressive B-cell lymphoma was proven. The suitable hematooncology treatment was introduced.
Conclusion: The lymphoma should be actively search in NBS patients, despite repetitive negative results. This is the matter of debate if specific treatment, rituximab or even bone marrow transplantation should be performed electively, earlier on the stage of increasing lymphocyte B proliferation, with gene rearrangements even not fulfilling criteria for the clonal neoplasia.
ESID-0489 Primary Immunodeficiency Care for Adults – First Experiences from a New Center
E. Wiesik-Szewczyk 1, A. Kucharczyk1, K. Jahnz-Rozyk1, E. Bernatowska2
1Clinical Immunology and Allergology, Military Institute of Medicine, Warsaw, Poland
2Clinical Immunology, Children's Memorial Health Institute, Warsaw, Poland
Background: More than 200 primary immune deficiencies have been described. In adults their identification can be difficult due to lack of specific centers of expertise, delayed referral or overlooking of PID in differential diagnosis by physicians. Moreover achievements in care of PID by pediatricians lead to survival of patients beyond the age of 18. There is a need to better understand the medical problems in this age group.
Objectives: To analyze the clinical spectrum and problems to overcome of adult patients with PID.
Methods: We conducted retrospective analysis of adult patients referred to our center established in Aug 2012.
Results: From Aug 2012 to Apr 2014, twenty seven patients with relevant PID (exluding IgA deficiency) were diagnosed as immune deficient. Among them 19 had a prior diagnosis and were referred for follow-up care from pediatric center. Eight patients were newly diagnosed mainly with CVID. The patients spectrum involved: CVID/hipogammaglobulinemia – 14 cases, NBS- 8 cases, Bloom syndrome-2 cases, hiper IgE syndrome – 1 case, CGD -2 cases. During follow-up 6 cases of lymphoma were identified: 4 in NBS patients, 1 in hiper IgE syndrome, 1 in CVID patient.
Conclusion: In our early experience there is still a gap between estimated and newly diagnose number of adult patient with CVID, what underscore the lack of knowledge of PID in adults among physicians. On follow-up care of PID patients the most common complication was occurrence of neoplasia, what highlight the need for their systematic control.
ESID-0463 Targeted Next Generation Re-Sequencing for Hyper-IGE Syndrome and Chronic Mucocutaneous Candidiasis
N. Frede1, F. Nussbaumer1, M. Buchta1, M. Depner1, C. Glocker1, L.L. Yang 1, B. Grimbacher1
1CCI-Center of Chronic Immunodeficiency, University Medical Centre Freiburg, Freiburg, Germany
The hyper-IgEsyndromes (HIES) are a collection of primaryimmunodeficiencies predominantly leading to elevated serum IgE levels, recurrent staphylococcal skin abscesses, eczema and pulmonary infections. However, the clinical presentation of HIES is variable and presents overlapping, non-specific features shared by other immunodeficiencies. Throughout recent years, a growing number of mutations underlying this clinical phenotype have been identified.
For the molecular diagnosis of HIES and related immunodeficiencies we developed a multigene, next generation sequencing panel for targeted re-sequencing. The first panel comprised 15 genes associated with chronic mucocutaneous candidiasis (CMC) or HIES, such as STAT3, DOCK8, PGM3 and TYK2, while additional genes were added in subsequent designs. For target enrichment and sequencing, Agilent’s HaloPlex and Illumina’sMiSeq technologies were used.The average sequencing depth amounted to 1037 reads. 98% of the target regions were covered at least 20 times, whereas 91% had a 100-fold coverage.
In our first run, 23 patients with the clinical diagnosis of HIES, and 8 with CMC were analyzed. Among these patients, we identified five patients carrying known heterozygous mutations in STAT3, one patient had a homozygous mutation in DOCK8, and two patients had heterozygous mutations in STAT1. Furthermore, we detected several homo- and heterozygous sequence variations in TYK2, SPINK5, IL17RA and IL17F. However, the effect of these variations is unclear and requires further functional investigation. All detected mutations were confirmed by Sanger sequencing.
We conclude that targeted re-sequencing provides a reliable, time- and cost-efficient (250€ per sample) method for the establishment of a genetic diagnosis.
ESID-0297 Assessment of Infection Rates and Health Resource Utilization Among Patients with Primary Immunodeficiency (PID) Prior to Diagnosis
C. Rabbat1, D. Ito2, Y. Xiong2, X. Ye 3, J. Li-McLeod2, P. Faivre4
1Medical Affairs, Baxter HealthCare, Westlake Village, USA
2Medical Outcomes Research and Economics, Baxter HealthCare, Westlake Village, USA
3Medical Outcomes Research and Economics, Baxter HealthCare, Deerfield, USA
4Health Economics and Market Access, Baxter HealthCare, Zurich, Switzerland
Background: Undiagnosed PID may burden the patient with repeated infections and the health system with substantial resource utilization. This analysis assessed infection rates and healthcare utilization among patients in the years leading up to a PID diagnosis.
Methods: This retrospective analysis utilized the Truven Marketscan Database, 2002-2012. Patients who had at least 1 inpatient or ER diagnosis or at least 2 outpatient diagnoses of PID (279.xx) and at least one of the following PID diagnoses: 279.04, 279.01, 279.05, or 279.06 and 5 years of continuous health plan enrollment were identified. The first PID diagnosis (279.xx) was identified as the index date and data preceding the index date were examined. Incidence rates of pneumonia, sinusitis, bronchitis, otitis, as well as hospitalizations, outpatient visits and outpatient drug utilization were analyzed.
Results: A total of 1388 patients were undiagnosed with PID for at least 5 years, of these, 84 were undiagnosed for at least 10 years, suggesting that many are undiagnosed for years. The average percent increase in rates of pneumonia, sinusitis, bronchitis and otitis per year over the 10 years prior to diagnosis were 39%, 20.4%, 20.2%, 14.2%, respectively. Hospitalizations, outpatient visits and outpatient drug utilization increased on average 29.1%, 10.5% and 5.3% per year, respectively during the same time frame. This considerable increase in infections and hospitalizations suggests the condition may be increasing in severity over time prior to diagnosis.
Conclusion: The results suggest a more timely PID diagnosis may ease the burden for both patients and the healthcare system.
ESID-0294 Patient Preferences for Recombinant Human Hyaluronidase (RHUPH20)-Facilitated Subcutaneous (SC) Infusion of Immunoglobulin G (IGHY) in Adults with Primary Immunodeficiencies (PI): Phase 3 Study Results
D. Ito1, X. Ye 2, Y. Xiong1, J. Li-McLeod1, R. Schiff1, P. Faivre3
1Medical Outcomes Research and Economics, Baxter HealthCare, Westlake Village, USA
2Medical Outcomes Research and Economics, Baxter HealthCare, Deerfield, USA
3Health Economics and Market Access, Baxter HealthCare, Westlake Village, Switzerland
Background: Compared to intravenous (IV) or SC infusion of immunoglobulin (IG) for PI patients, a new method of facilitated SC infusion of IgG using rHuPH20 (IGHy) offers the option to self-administer at home on a 3-4 week basis. This analysis assessed treatment preference of patients who switched to IGHy after previously receiving IVIG or SCIG.
Methods: Treatment preference was assessed within a prospective, non-controlled study of PI patients aged 18 or older who were first treated with IVIG for 3 months or previously received SCIG, then IGHy for a 1 to 3-4 week ramp-up followed by a 12 month follow-up. At the end of study, patients completed a 5-point preference scale (from 'dislike very much' to 'like very much') to assess a range of treatment attributes, such as convenience, infusion time and frequency of IGHy.
Results: A total of 61 PI patients aged ≥ 18 years were enrolled, with 49 completing the questionnaire. Of these, 38 (78%) patients indicated they would choose to continue receiving IGHy over IV or SC. The majority liked/liked very much most aspects of IGHy, especially the ability to fit treatment into their schedules (78%), overall convenience (76%), administration frequency (71%) of IGHy and total time spent on treatment per month (67%). The lowest rated item was change in physical appearance (17%), however 58% had no preference.
Conclusion: Most patients preferred IGHy over IV or SC. Future research might focus on how this innovative administration method may facilitate treatment adherence.
ESID-0299 Differences in Infection Rates Between Outpatient Hospital, Clinic and Home Infusion Settings for Patients with Primary Immunodeficiency Disorder (PID)
D. Ito1, Y. Xiong1, X. Ye 2, J. Li-McLeod1, P. Faivre3
1Medical Outcomes Research and Economics, Baxter HealthCare, Westlake Village, USA
2Medical Outcomes Research and Economics, Baxter HealthCare, Deerfield, USA
3Health Economics and Market Access, Baxter HealthCare, Zurich, Switzerland
Background: PID patients may benefit from home-based infusion of intravenous immunoglobulin (IVIG) therapy compared to outpatient hospital (OH) or clinic setting due to the potentially reduced risk of exposure to infections. This analysis compared infection rates among PIDD patients who infused in the home, clinic or OH.
Methods: Patients who had at least 1 inpatient or ER claim or at least 2 outpatient claims with PID ICD-9 code 279.xx and had at least 6 months of continuous IVIG claims from the same site of care (home, clinic or OH) were identified from the US Truven Marketscan Database, years 2002 -2012. Incidence rates of pneumonia and bronchitis were calculated for each site of care. Regression analyses were conducted to compare age and gender-adjusted infection rates between sites.
Results: A total of 2758 patients infused in the clinic, 2006 in the home, and 1919 in the OH setting. Numbers of pneumonia per person per year were 0.67, 0.55 and 1.04 and chronic bronchitis were 0.38, 0.24 and 0.59 for the clinic, home and OH settings, respectively. The age and sex-adjusted rate of pneumonia was found to be significantly higher in the OH setting than the home (p=0.008). The difference in the rate of pneumonia between home and clinic was not significant. Rates of chronic bronchitis were significantly higher in both clinic (p=0.0144) and OH (p<0.0001) compared to home.
Conclusion: Home infusion is associated with significantly lower rates of pneumonia and bronchitis compared to the OH among PID patients in the US.
ESID-0073 Using Exome Sequencing for Clinical Diagnoses of Syndromic and Non-Syndromic Immunodeficiency Disorders in Pediatric Patients
K. Zhang 1, A. Husami1, D. Kissell1, A. Valencia1, F. Zou1, D. Neilson1, A. Lindsley1, R. Marsh1, M. Jordan1, A. Filipovich1
1Pediatrics, Cincinnati Children's Hospital Medical Center, Cincinnati, USA
The Next-generation sequencing (NGS), an ultra-high-throughput sequencing technologies, have facilitated the discoveries of many novel genetic causes in recent years. As part of the clinical validation and testing, we tested twelve patients with unknown immunodeficiency disorders by whole exome sequencing. All of these patients have been through extensive clinical work up and resulted in no definitely genetic diagnoses. We used the NimbleGen V3 whole exome targets capture and the Illumina HiSeq 2500 sequencing system to achieve 100 basepair (bp) paired-end reads at a minimum coverage of 10X of 95% of the target regions. We also developed a bioinformatics analysis suite that includes duel analyzing pipelines of of NextGene V2.3.3 and GATK/Golden Helix V7.7.4 software, as well as laboratory developed tools. Clinical relevant variants were confirmed by Sanger sequencing. In addition, a series of quality measurements were implemented to meet the CLIA and CAP requirement for establishing a clinical test.
From this study, we confirmed the clinical diagnosis in five of twelve patients with the following disorders: 1) Combined immune deficiency (CID) with alopecia universalis and impaired T cell antigen response; 2) Lymphocytic Interstitial Lung Disease; 3) Otofaciocervical syndrome II with T-, B+, NK+ combined immunodeficiency (CID); 4) Primary B-cell immunodeficiency with autoimmune hepatitis; 5) Novel Combined immunodeficiency with B- ,T+ and CD57iNKT-.
In conclusion, with a robust analyses pipeline and affordable sequencing cost, direct exome sequencing provides a clinically useful tool for clinical genetic diagnoses in families with rare syndromic and non-syndromic immunodeficiency.
ESID-0302 Rapid Diagnosis of Dedicator of Cytokinesis 8 Deficiency Using Targeted Deep Sequencing and Flow Cytometry
T. Qin 1, Y. An1, J. Wu1, R. Dai1, D. Liu1, X. Li1, L. Jiang1, D. Wu1, X. Tang1, W. Song2, T. Wang3, X. Zhao1
1Ministry of Education Key Laboratory of Child Development and Disorders, Children's Hospital of Chongqing Medical University, Chongqing, China
2Department of Cell Biology & Molecular Genetics, University of Maryland, Maryland, USA
3Immunology, Fujian Provincial Hospital, Fuzhou, China
Background—Dedicator of cytokinesis 8 (DOCK8) deficiency is a primary combined immunodeficiency characterized by recurrent infections, elevated serum IgE levels, eosinophilia, and a high incidence of food allergies. Mutations of the gene responsible, DOCK8, are usually complex, including copy-number variations, large deletions, insertions, and point mutations; thus genetic diagnosis of DOCK8 deficiency is difficult and time-consuming.
Objective—We sought to find the rapid and reliable diagnosis method in Hyper IgE Syndrome patients caused by mutations in DOCK8 and to describe clinical characteristics of this syndrome in Chinese population.
Methods—Patients with clinical features of HIES and absence of STAT3 mutation were subjected to deep sequencing targeting DOCK8 and flow cytometry to detect expression of DOCK8 protein. Clinical features and laboratory findings were evaluated in the patients with a confirmed diagnosis of DIDS in details.
Results—All 5 patients had most of the typical features of DOCK8 deficiency, but 2 patients had retained primary teeth and 1 patient had metacarpophalangeal joint hyperextension. Targeted deep sequencing revealed large deletions and point mutations precisely and efficiently. DOCK8 was absent from the peripheral mononuclear cells of all 5 patients by flow cytometric analysis, whereas control cells from peripheral blood and lymphoid organs of donors constitutively expressed DOCK8 protein.
Conclusion—The clinical features of DOCK8 deficiency can vary among ethnic populations. Targeted deep sequencing and flow cytometry is a reliable combination strategy to rapidly diagnose DOCK8 deficiency.
ESID-0557 AK-2 Prevents From Oxidative Stress During HSC Development and Hematopoietic Differentiation
A. Rissone 1, K. Weinacht2, G. la Marca3, K. Bishop4, E. Giocaliere5, J. Jagadeesh1, K. Felgentreff6, K. Dobbs6, W. Al-Herz7, M. Jones8, S. Chandrasekharappa8, Y. Itan9, A. Schambach10, R. Sood4, L.D. Notarangelo6, F. Candotti1
1GMBB, NHGRI, BETHESDA, USA
2Division Hematology Oncology, Boston Children's Hospital, BOSTON, USA
3Department of Neurosciences Psychology Pharmacology and Child Health, University of Florence, FLORENCE, Italy
4Zebrafish Core, NHGRI, BETHESDA, USA
5University Hospital, Meyer Children's University Hospital, FLORENCE, Italy
6Division Immunology, Boston Children's Hospital, BOSTON, USA
7Pediatric Department Faculty of Medicine, Kuwait University, Kuwait City, Kuwait
8Genomics Core, NHGRI, BETHESDA, USA
9St. Giles Laboratory of Human Genetics of Infectious Diseases, The Rockefeller University, NEW YORK CITY, USA
10Institute of Experimental Hematology, Hannover Medical School, Hannover, Germany
Adenylate kinase-2 (AK2) is a mitochondrial phosphotransferase enzyme that regulates the cellular adenine nucleotide composition and has a critical role in cell energy homeostasis. In humans, mutations of the AK2 gene are responsible for reticular dysgenesis (RD), one of the most profound forms of severe combined immunodeficiency (SCID). To gain insights into the pathophysiology of RD, we studied the effects of AK2 deficiency using the zebrafish model and induced pluripotent stem cells (iPSCs) derived from fibroblasts of a patient affected with RD. In zebrafish, AK2 deficiency resulted in severe impairment of hematopoietic stem cells (HSC) development associated with increased levels of reactive oxygen species (ROS), oxidative stress and apoptosis. AK2-deficient iPSCs showed abnormalities in adenine nucleotide levels and recapitulated the characteristic granulocytic maturation arrest at the promyelocyte stage observed in the bone marrow of RD patients. Importantly, antioxidant treatment rescued the hematopoietic phenotypes in vivo in ak2-mutant zebrafish and restored differentiation of AK2-deficient iPSCs into mature granulocytes. Overall, our results support a mechanistic hypothesis involving abnormal redox state in the HSCs and multilineage abnormalities observed in AK2 deficiency and point to the potential of antioxidants as supportive therapeutic modality for patients affected by RD.
ESID-0730 Microbiota/Host Interactions in Human IGA Deficiency
J. Fadlallah 1, C. JUSTE2, L. Galicier3, E. Oksenhendler3, Z. Amoura4, S.D. Ehrlich5, S.P. Kennedy5, C. Fieschi3, G. Gorochov1, M. Larsen1
1Cellular and molecular immunology of chronic inflammatory diseases, Hôpital Pitié Salpêtrière- INSERM 1135 CIMI Paris, Paris, France
2MICALIS, INRA, Jouy en Josas, France
3Immunologie clinique, Hôpital Saint Louis, Paris, France
4Médecine Interne, Hôpital Pitié Salpêtrière, Paris, France
5Metagenopolis, INRA, Jouy en Josas, France
IgA deficiency (IgAd), either isolated in selective IgA deficiency (sIgAd), or combined with other antibody deficiencies, in common variable immunodeficiency (CVID), is defined by undetectable seric IgA levels. Autoimmunity and recurrent mucosal infections are associated with this condition. Host/microbiota homeostasis is partially controlled by secretory IgA (sIgA), as IgA knock-out mice suffer from gut dysbiosis and auto-immunity. However, the impact of IgAd on digestive immunity and host/microbiota commensalism in humans has never been studied. Viable microbiota from healthy controls (HC, n=35) and IgA-deficient adults (sIgAd: n=14, CVID: n=12) was gradient purified from stool. Ig-opsonized bacteria were quantified using flow cytometry. Gut microbiota composition was determined using deep sequencing and metagenomic analysis. We observed that : (i) seric IgAd is indeed associated with digestive IgAd, (ii) secretory IgM (sIgM) levels are significantly elevated in sIgAd compared to HC (180 μg [0-872] vs 3.74 μg [0-77] /g feces, p=0,01), (iii) sIgA targets a restricted subset of gut bacteria in HC (8.179% [0-18.8]), while a sIgM-opsonized subset is detected in sIgAd (7,99%[3,24-20,3]). Finally, metagenomic analysis revealed that IgAd patients have an aberrant microbiota composition with a tendency towards an aberrant distribution of proteobacteria, and also a highly different enterotype distribution compared to HC. In summary, while sIgA appears to survey only a restricted fraction of gut microbiota in HC, its absence is associated with dysbiosis, even when compensatory sIgM are present. Further identification of bacteria inducing local, but also systemic immunity might lead to a new understanding of disease pathogenesis and to novel therapeutic approaches.
ESID-0319 Identification of Two Novel Genetic Alterations as Possible Causes of Combined Immunodeficiency
M. Fuehrer 1, U. Pannicke1, M. Hönig2, B. Baumann3, A. Enders4, A. Enders4, K. Holzmann5, C. Schütz2, W. Friedrich2, A. Schulz2, K. Schwarz6
1Institute for Transfusion Medicine, University of Ulm, Ulm, Germany
2Department of Pediatrics, University Medical Center Ulm, Ulm, Germany
3Institute for Physiological Chemistry, University of Ulm, Ulm, Germany
4Department of Immunology John Curtin School of Medical Research, The Australian National University, Canberra, Australia
5Genomics Core Facility, University of Ulm, Ulm, Germany
6Institute for Clinical Transfusion Medicine and Immunogenetics, German Red Cross Blood Service Baden-Württemberg - Hessen, Ulm, Germany
Introduction: The female patient reported here is the only offspring of a consanguineous marriage and presented with eczema, hepato-splenomegaly, pneumonia, chronic diarrhoea, and a failure to thrive at the age of 4 months. According to the lymphocyte subsets the patient was classified as a TlowBlowNKlow CID. At the age of three years the patient after conditioning received a transplant from her HLA-identical mother. Although a complete chimerism was achieved the patient continued to suffer from recurrent infections.
Objective and Methods: Homozygosity mapping and whole exome sequencing approaches were used to narrow down the genetic causes of the disease. The molecular analysis of candidate mutations was followed by RT-PCR, Western Blotting, EMSA and apoptosis assays.
Results: Two alterations, a missense mutation in SEC16A and a nonsense mutation in RIPK1 were detected. While SEC16A was expressed in patient fibroblasts, RIPK1 was expressed only weakly in a truncated form lacking its RIP homotypic interaction motif (RHIM) and death domain. RIPK1 is involved in NF-κB activation and apoptosis or necroptosis induction after tumor necrosis factor (TNF)α stimulation. Whereas RIPK1 was shown to be dispensable in Jurkat-cells to TNFα-induced apoptosis, human fibroblasts were not sensitive to TNFα-induced cell death. Patient´s fibroblasts and Jurkat-cells lacking RIPK1 showed reduced NF-κB activation after TNFα stimulation.
Conclusion: We were able to detect two plausible genetic causes potentially causative for the phenotype, which were not connected to CID up to now. Functional testing is ongoing and will reveal the role of the genetic variants for the disease.
ESID-0811 Altered Usage of Proximal and Distal Immunoglobulin Variable Genes in Patients with the Cohesinopathy Cornelia De Lange Syndrome
A. Björkman1, D. Dunn Walters2, D. Kipling3, Q. Pan-Hammarström 1
1Department of Laboratory Medicine, Karolinska Institutet at Karolinska University Hospital Huddinge, SE-14186, Sweden
2Department of Immunobiology, King's College London School of Medicine London, UK
3Institute of Cancer and Genetics, School of Medicine, Cardiff University, Cardiff, UK
Cornelia de Lange syndrome (CdLS) is a genetic disease that affects several systems and organs during development. A major cause of morbidity and mortality in the patients are infections, however, whether these are associated with immunodeficiency have not been extensively studied. CdLS is caused by mutations in genes encoding members of the cohesin complex/pathway that is involved in sister chromatid cohesion, gene regulation and homologous recombination. We have previously shown that the cohesin loader NIPBL is involved in the non-homologous end-joining process during immunoglobulin (Ig) class switch recombination, a deletion/recombination process that result in a change of Ig isotype. Here we have studied whether NIPBL and the associated cohesin pathway is also functional in the two Ig diversification processes V(D)J recombination and somatic hypermutation (SHM), which result in Ig variable region assembly and antibody affinity maturation, respectively.
The IGHV regions and corresponding V(D)J recombination junctions were amplified by PCR from NIPBL- and SMC1-deficient CdLS patient cells and analyzed by the 454 or Illumina Hiseq2000 high throughput sequencing platform. Whereas the SHM pattern was largely normal, the V gene usage for V(D)J recombination was skewed in the patients. Notably, V genes located in proximity to the D and J genes were preferentially used at the expense of V genes located in the 5’ distal end of the IGH locus. Similar results were also obtained in analyzing the V(D)J recombination pattern at the TCRbeta locus. This implies an involvement of cohesin in processes affecting long range interactions during V(D)J recombination in human B- and T- cells.
ESID-0068 Active Downregulation of AK1 Protein Expression in Lymphocytes Might Be an Explanation for the Phenotype of Reticular Dysgenesis in AK2-Deficient Patients
R. Waldmann 1, U. Pannicke1, J. Högel2, K. Schwarz1
1Institute for Transfusion Medicine, Ulm University, Ulm, Germany
2Institute for Human Genetics, University Hospital of Ulm, Ulm, Germany
Introduction: Reticular dysgenesis, caused by defects in the adenylate kinase 2 gene (AK2), is the most severe form of inborn SCID. It is characterized by leukopenia and bilateral sensorineural deafness.
A previous study indicates that leukocytes may be susceptible to defects caused by the lack of AK2, as they do not express adenylate kinase 1 (AK1) in sufficient amounts to compensate for the AK2 functional deficits (Pannicke et al., Nature Genetics 2009).
Objective: The aim of this study was to investigate the expression and regulation of AK1 in hematopoietic cell lines.
Methods: Western blot and qRT-PCR analysis of AK1 expression in hematopoietic cell lines and Western blot analysis after reconstitution experiments on the basis of RNA and miRNA inhibitor transfection.
Results: Human T lymphocytes, postfollicular B lymphocytes and progenitor cells with myeloid surface markers have only marginal AK1 RNA levels and do not express AK1 protein. The AK1 expression can only be reconstituted if AK1 RNA without the genuine 5`untranslated region (5`UTR) is transfected. The 5`UTR of AK1 contains two potential miRNA binding sites and the results of site-directed mutagenesis experiments of these sites, will be presented.
Conclusion: The AK1 protein expression in T lymphocytes, postfollicular B lymphocytes and progenitor cells with myeloid surface markers is actively suppressed in a 5`UTR dependent matter and not due to promoter regulation.
Topic: B Cell
ESID-0470 Clinical Features and Mutation Analysis of Iranian Patients with Congenital Agammaglobulinemia
H. Abolhassani1, B. Mirminachi2, M. Vitali3, V. Lougaris3, T. Cheraghi4, H. Khazaei5, A. Plebani3, N. Rezaei2, N. Parvaneh2, A. Aghamohammadi 2
1Division of Clinical Immunology Department of Laboratory Medicine, Karolinska Institutet at the Karolinska University Hospital Huddinge, Stockholm, Sweden
2Research Center for Immunodeficiencies, Pediatrics Center of Excellence Children's Medical Center Tehran University of Medical Sciences, Tehran, Iran
3Department of Clinical and Experimental Sciences, Pediatrics Clinic and Institute for Molecular Medicine A. Nocivelli, Brescia, Italy
4Department of Pediatrics, 17th Shahrivar Children's Hospital Guilan University of Medical Sciences, Rasht, Iran
5Department of Immunology and Hematology, Zahedan University of Medical Sciences, Zahedan, Iran
Background: Congenital agammaglobulinemia is a primary immunodeficiency disorder characterized by early onset of recurrent bacterial infections, extremely low levels of peripheral B lymphocytes and reduced levels of all immunoglobulin isotypes. Mutation in Bruton's tyrosine kinase (BTK) gene are responsible in majority of patients with congenital agammaglobulinemia.
Methods: Eighty-five Iranian patients with congenital agammaglobulinemia with an age range of 1-43 years were included in this study. The correlation between responsible mutation and several factors including clinical severity and immunologic phenotypes were evaluated in available patients. Clinical severity was indicated as age of disease onset and development of severe complications.
Results: From 85 patients, molecular investigation were performed in 43 cases and 33 different mutations were identified in 36 unrelated families including 32 BTK deficiency (1 novel mutation), 6 μ heavy chain defect (2 novel mutation), and 1 patient with Igα deficiency. Muations were classified into severe and mild based on structural and functional consequence by bioinformatics analysis. Age at diagnosis and diagnostic delay was lower significantly in non-BTK deficient patients and the survival rate of BTK deficiency with premature stop codons mutation was lowest.
Conclusion: There is no comprehensive correlation between type of responsible mutation for agammaglobulinemia and severity of clinical phenotype.
ESID-0731 Selective IGA Deficiency: New Insights in Pathogenesis and Classification
A. Aghamohammadi 1, H. Abolhassani2, L. Hammarström2
1Research Center for Immunodeficiencies, Pediatrics Center of Excellence Children's Medical Center Tehran University of Medical Science, Tehran, Iran
2Division of Clinical Immunology Department of Laboratory Medicine, Karolinska Institutet at the Karolinska University Hospital Huddinge, Stockholm, Sweden
Selective IgA deficiency (SIgAD) is the most common primary immunodeficiency, ranging from 1:155 to 1:18550 in different ethnic groups. SIgAD is defined as serum IgA levels equal or less than 0.07 g/l and normal serum IgG and IgM levels in a patient older than 4 years. The defect is presumed to result from impaired switching to IgA or a maturational failure of IgA-producing lymphocytes, but the nature of basic defect is unknown.
Most affected individuals with SIgAD are clinically asymptomatic; however, selected patients suffer from recurrent infections, allergies and autoimmune disorders. Moreover, different immunological abnormalities have been described in patients with SIgAD, including IgG subclass deficiency , specific antibody deficiency , low switched memory B cells and diminished number of regulatory T cells. In this review, we suggest a new classification based on clinical and immunological characteristics of SIgAD patients.
ESID-0481 IGG Anti-IGA Antibodies in Patients with Primary Antibody Deficiency Receiving Intravenous Immunoglobulin
B. Mirminachi1, B. Torabi Sagvand1, H. Abolhassani2, T. Shokouhfar1, T. Keihanian1, E. Hedayat 1, A. Amirzargar3, A. Mahdaviani4, A. Aghamohammadi1
1Research Center for Immunodeficiencies, Pediatrics Center of Excellence Children's Medical Center Tehran University of Medical Science, Tehran, Iran
2Division of Clinical Immunology Department of Laboratory Medicine, Karolinska Institutet at the Karolinska University Hospital Huddinge, Tehran, Iran
3Department of Immunology and Biology, Tehran University of Medical Science, Tehran, Iran
4Pediatric Respiratory Diseases Research Center, National Research Institute of Tuberculosis and Lung Diseases Shahid Beheshti University of Medical Science, Tehran, Iran
Background: Immunoglobulin replacement therapy is the mainstay treatment of patients with primary antibody deficiency (PAD). Traces level of IgA (ranged from 0.4 to 2500 mg/ml), which exists in all immunoglobulin products, could lead to an increased proneness to adverse reactions in PAD patients. Furthermore, the exact mechanism which stimulates the anti-IgA antibody production in PAD is still unknown. This study was conducted to evaluate IgG anti-IgA antibodies in PAD patients receiving intravenous immunoglobulin (IVIg) and its predisposing factors.
Methods: A total of sixty seven PAD patients, who underwent regular IVIg replacement therapy in our center were included in the study. Control group includes 24 healthy individuals as the negative control and 8 symptomatic patients with IgA deficiency as the positive control. IgG anti-IgA antibodies level was measured by the enzyme-linked immunosorbent assay (ELISA) method.
Results: A significant difference was observed between Anti-IgA level of common variable immunodeficiency (CVID) and other PAD groups (p=0.02). Moreover, 6 CVID patients were seropositive for the IgG anti-IgA antibody, with higher susceptibility to the adverse reactions (p<0.001). IgG anti-IgA level has a negative relationship with serum IgA level (r=-0.06) and IVIg treatment duration (r=-0.006).
Conclusion: There was a significant correlation between anti-IgA antibody presence and the adverse reactions, especially in CVID patients with higher susceptibility to produce this constitutional antibody.
Keywords: Anti IgA, Adverse reaction, Common variable immunodeficiency, Intravenous immunoglobulin
ESID-0388 The Clinical Effectiveness of Immunoglobulin Infusion Therapy in Specific Antibody Deficiency (SAD)
H. Alachkar 1, M. Arslan Anwar2
1Immunology, Salford Royal Hospital, Manchester, United Kingdom
2Medical School, Manchester University, Manchester, United Kingdom
SAD is the most frequently diagnosed PID, characterized by an impaired response to polysaccharide antigens/vaccines with normal immunoglobulin levels.
Patients are susceptible to sinopulmonary infections with polysaccharide encapsulated bacteria. The recurrent infections may lead to bronchiectasis and chronic sinus disease. Immunoglobulin infusion is one of treatment options for SAD.
The aim of this study was to assess the clinical effectiveness of immunoglobulin infusion therapy in SAD patients.
Methods- Retrospective data collection. We looked at age of diagnosis, delay in diagnosis, occurrence of pneumonia and respiratory comorbidities (COPD, asthma, and bronchiectasis). The outcome measures were: number of chest infections and hospital admissions per year after immunoglobulin therapy started and patient’s perception of well-being.
Results- we identified 45 patients (70% females), who fulfilled the criteria of “isolated” SAD diagnosis.
Two groups were recognized according to the number of infections after starting treatment; Group 1 (G1=25 patients) continued to have ≥3 respiratory infection per year (mean of 4.4), and Group 2 (G2=20) had <3 infections (mean of 1.6). Mean number of admissions to hospital was 1.6 /patient/year for G1 and 0.8 for G2. 48% of G1 patients felt better after treatment, while 85% of G2 did so. In the G1, 68% had bronchiectasis, 40% had COPD and 45% had asthma, comparing with 50%, 20% and 50%, respectively, of G2 to have these comorbidities.
Conclusion- Immunoglobulin therapy helped reducing respiratory infections in almost half of the treated SAD patients. Immunoglobulin therapy was less effective in SAD patients who had bronchiectasis and COPD.
ESID-0097 Two Novel Mutations in DNA Binding Domain in Patients with Hyper IGE Syndrome
J. Alcantara-Montiel 1, T. Staines Boone2, G. Lopez Herrera3, S. Espinosa Padilla3, F. Espinosa Rosales3, L. Santos-Argumedo1
1Molecular Biomedicine, Centro de Investigacion y de Estudios Avanzados del IPN, Mexico City, Mexico
2Department of Clinical Immunology, Hospital of High Specialties of IMSS Number 25, Monterrey Nuevo Leon, Mexico
3Department of Clinical Immunology, National Institute of Pediatrics, Mexico City, Mexico
Introduction: The hyper-IgE syndrome (HIES) is a rare primary immunodeficiency characterized by recurrent skin abscesses, pneumonia, and highly elevated levels of serum IgE. HIES includes non-immunologic manifestations like: characteristic face, pathologic dentition, scoliosis, bone alterations and hyper-extensible joints. HIES can be transmitted as an autosomal dominant trait with variable expressivity. Objective: In-silico and functional characterization of new mutations. Methods: Two patients with established diagnosis of autosomal dominant HIES and more than 40 points in the NIH scoring system were included. This protocol has been approved by the ethics committee of the hospital. The patients read and signed informed consent. Patients were previously analyzed to discard known genetic causes of autosomal recessive HIES (DOCK8, TYK2). To evaluate the pathways of IL-6, IL-21, PBMCs were stimulated with rhIL-6 and rhIL-21 for 15 minutes and phosphorylation of STAT-3 was evaluated by flow cytometry. The electrophoretic mobility shift assay (EMSA) was done to evaluate if STAT-3 was able to bind to the DNA sequence. Immunoprecipitation (IP) was performed to evaluate heterodimers with STAT-1. Results: The prediction in-silico suggested disruption of the conformation on the DNA binding domain loops. The patients showed low phosphorylation of STAT-3 after 15 minutes of stimulation with rhIL-6 and rhIL-21. EMSA showed low complex formation and IP showed low formation of heterodimers with STAT-1. Conclusion: After three functional assays, we characterized two non-reported mutations on the DNA binding domain that impact directly on STAT-3 function.
ESID-0416 Isohemagglutinis as Early Diagnostic Criteria for Primary Humoral Immundeficiency Compared with Vaccine Responses in Children with Recurrent Infections
M. Olaya1, L. Alsina 1, M.A. Martín Mateos1, M. Piquer1, M.T. Giner1, A.M. Plaza1
1Allergy and Clinical Immunology, Hospital Sant Joan de Déu, BARCELONA, Spain
Introduction: Diagnosis of humoral deficiency is performed through the quantitation of immunoglobulin isotypes (IgG and subclasses/IgA/IgM) and their functional evaluation (isohemagglutinins and/or presence of antibodies against past infections or vaccination). Our aim was to evaluate the performance of isohemagglutinins in the diagnosis of humoral PID.
Methods: This is a retrospective and prospective study between 2011 and 2013. Patients 1 to 18 years-old evaluated for recurrent infections and followed for at least one year in the Clinical Immunology Clinic, with determinations of immunoglobulins, isohemagglutinis and vaccine responses (diphtheria, tetanus, pneumococcus), were eligible. After the follow up and the tests, patients were given the following diagnosis: group 1: no PID, group 2: humoral PID. Finally, group 2 was divided into mild humoral PID and severe-moderate humoral PID (hypoIgG+/-IgA, IgM, absent isohemagglutinins and/or poor response to vaccines).
Results: 87 patients fulfilled diagnostic criteria. 4 had AB blood, two in each group, so 83 were analysed: 63% were males, median age was 9.6yo. 43/83 (51.8%) of isohemagglutinins were positive. Of the non-PID group, 24/28 isohemagglutinins (85.7%) were positive; 30/30 patients had also positive vaccine responses. Of the PID group, 30/55 (54.5%) had negative isohemagglutinins, 18/39 and 12/15 in mild and severe-moderate humoral PID, respectively. Vaccine responses were negative in 4/40 and 9/13 of mild and severe-moderate humoral PID, respectively.
Conclusions: Isohemagglutinins are useful in the identification of humoral PID in patients with recurrent infections. Correlation with vaccine responses is high. It yields a sensitivity of 80.0% (56.4-100.0) and a specificity of 85.7% (70.9-100.0).
ESID-0421 B Regulatory Cells: A New Player in Fetal Immune Tolerance?
A. Esteve-Sole 1, M. Antón-Iborra2, I. Teixidó3, M. Piquer4, A.M. Plaza4, M. Juan2, L. Alsina4
1Allergy and Clinical Immunology, Fundació Sant Joan de Déu, BARCELONA, Spain
2Immunology Department-CDB, Hospital Clínic, BARCELONA, Spain
3Gynecology Department, Casa Maternitat. Hospital Clínic, BARCELONA, Spain
4Allergy and Clinical Immunology, Hospital Sant Joan de Déu, BARCELONA, Spain
Introduction: Pregnancy is characterized by a tolerogenic immune state of the mother to her haploidentical fetus. Maternal-fetal tolerance is achieved through different mechanisms including B regulatory cells (Breg), which have been shown to be increased during pregnancy. Bregs are a cell subtype involved in the regulation of the immune system, by secreting IL10 and inhibiting T cell proliferation and cytokine secretion. These cells are defined as CD19+CD38hiCD24hi cells. No studies have been done on B regs in umbilical cord blood (UCB).
Objectives: To study Breg cell population number and function in UCB of healthy neonates.
Materials and methods: Breg cell number was determined by flow cytometry of CD19+CD24hiCD38hi in 8 UCB from heallhy newborns and compared to 5 adult peripheral blood (aPB). Functional assays were performed by studying B cell capacity to produce IL10 and assessing T cell proliferation and cytokine production inhibition by sorted Breg cells.
Results: We observed significant increases (p<0.0001) in Breg cells in UCB of healthy neonates (71.33% + - 9.42 of B cells; mean+-SD) when compared to healthy aPB (11.07%+- 4.455 of B cells; mean+-SD). We also observed reduced proliferation in response to T cell-oriented polyclonal stimuli in UCB compared to adult PBMCs. We set up the experimental conditions to assess Breg cell inhibitory capacity, which are are ongoing.
Discussion: Given these results, we hypothesize Bregs isolated from UCB will inhibit T cells proliferation and pro-inflammatory cytokine secretion. In this context, we propose Bregs as a new player in the tolerogenic equilibrium of the neonatal immune system.
ESID-0624 The New Zealand CVID/ Hypogammaglobulinemia Study
R. Ameratunga 1, M. Brewerton2, A. Jordan3, R. Barker4, P. Storey3, S.T. Woon4, W. Koopmans4, B. McGettigan5, K. Lindsay3, A. Baker3, C. Slade6, R. Steele7
1Immunology, Auckland hospital, Auckland, New Zealand
2Immunology, Melobourne Hospital, Melbourne, Australia
3Immunology, Auckland, Auckland, New Zealand
4VIM, Auckland, Auckland, New Zealand
5Immunology, Christchurch, Auckland, New Zealand
6Immunology, Melbourne Hospital, Melbourne, Australia
7Immunology, Wellington Hospital, Wellington, New Zealand
Background Patients with hypogammaglobulinemia are often encountered in clinical practice. In 2006 we established a long term study to determine the natural history of hypogammaglobulinemia in patients who did not qualify or accept treatment with IVIG.
Methods Adult patients (>16 years) with hypogammaglobulinemia with IgG levels < 6.5 g/l were eligible to enroll in this study. After written consent a detailed history was obtained with an interview assisted questionnaire. Computerised clinical notes and laboratory data were also reviewed.
Results There are now 120 patients in this long-term study. There was wide variation in IgG levels ranging from 1.7 g/l to 6.5 g/l. In several patients with IgG levels between 5-6 g/l there was spontaneous normalization without obvious explanation. In others a secondary cause was subsequently identified. It is also clear that some patients with IgG levels < 2 g/l are in excellent health, as several have refused IVIG/SCIG treatment and remain well several years after diagnosis. There is also a subgroup of adults who were given a diagnosis of “transient hypogammaglobulinemia of infancy” because their reduced immunoglobulin levels date back to infancy.
Conclusions This long term study will be very useful in identifying biomarkers for progression to CVID. The study will also be used to compare various diagnostic criteria for CVID.
ESID-0633 Comparison of Diagnostic Criteria for Common Variable Immunodeficiency Disorder
R. Ameratunga 1, M. Brewerton2, C. Slade2, A. Jordan3, D. Gillis4, R. Steele5, W. Koopmans6, S.T. Woon6
1Immunology, Auckland hospital, Auckland, New Zealand
2Immunology, Melbourne Hospital, Melbourne, Australia
3Immunology, Auckland Hospital, Auckland, New Zealand
4Immunology, Royal Brisbane Hospital, Brisbane, Australia
5Immunology, Wellington Hospital, Wellington, New Zealand
6VIM, Auckland Hospital, Auckland, New Zealand
Common Variable Immunodeficiency disorders (CVID) are the most frequent symptomatic primary immune deficiency in adults. The genetic basis for the condition is not known and no single clinical feature or laboratory test can establish the diagnosis: it has been a diagnosis of exclusion. In areas of uncertainty, diagnostic criteria can provide valuable clinical information. Here we compare the revised European Society of Immune Deficiencies (ESID) registry (2014) criteria with the diagnostic criteria of Ameratunga et al (2013) and the original ESID/ Pan American Group for Immune Deficiency (ESID/PAGID 1999) criteria. The ESID/PAGID (1999) criteria either require absent isohemmagglutinins or impaired vaccine responses to establish the diagnosis in patients with primary hypogammaglobulinemia. Although commonly encountered, infective and autoimmune sequelae of CVID were not part of the original ESID/PAGID (1999) criteria. Also excluded were a series of characteristic laboratory and histological abnormalities, which are useful when making the diagnosis. The diagnostic criteria of Ameratunga et al (2013) for CVID are based on these markers. The revised ESID registry (2014) criteria for CVID require the presence of symptoms as well as laboratory abnormalities to make the diagnosis. Once validated, criteria for CVID will improve diagnostic accuracy and will result in more equitable and judicious use of Intravenous (IVIG) or subcutaneous (SCIG) immunoglobulin therapy.
ESID-0745 Quantification of Specific Antibodies Against Pneumococcal Polysaccharides: Experience at the National Institute of Pediatrics in Mexico
S. Arablin 1, E.A. Medina-Torres1, S.E. Espinosa-Padilla1, M.A. Yamazaki-Nakashiada2, F.E. Espinosa-Rosales1
1Immunodeficiency Research Unit, National Institute of Pediatrics, Mexico City, Mexico
2Immunology Department, National Institute of Pediatrics, Mexico City, Mexico
Some patients have normal levels of lmmuoglobulins and all forms of IGG, but do not produce sufficient specific IGG antibodies to protect them from some viruses and bacteria. This condition is known as Specific Antibody Deficiency (SAD). Patients with SAD against Streptococcus Pneumoniae suffer from recurrent sinopulmonary infections, such as otitis media, bronchitis, and pneumonia.
In Mexico, the identification of patients with primary immunodeficiency has spread in the last few years, so the number of cases of SAD as well.
In this study, we analize the epidemiological and clinical characteristics of patients referred to the Jeffrey Modell Immunodeficiency Research Unit, at the National Institute of Pediatrics in Mexico, since May 2012 to May 2014. We determinate the quantity of antibodies against 14 serotypes of Streptococcus pneumoniae (1, 3, 4, 5, 6A, 6B, 8, 9V, 11A, 14, 18C, 19F, 19A and 23) before and after the application of 23 valent polysaccharide pneumococcal vaccine. Those patients who did not showed a post-immunization antibody concentration ≥1.3 mcg/mL in more than 50 or 70% percent of serotypes evaluated (depending on the age), or those who did not showed an 4 fold increase of the basal antibody concentration, were diagnosed with SAD. We have evaluated 150 referred patients. We discuss which are the main clinical and epidemiological clues to identify patients with suspected SAD.
ESID-0040 FCG Receptor Polymorphisms in Patients with Transient Hypogammaglobulinemia of Infancy Presenting Mild and Severe Infections
E. Azarsiz 1, N. Kutukculer1, N.E. Karaca1, G. Aksu1, A. Berdeli2
1Pediatric Immunology, Ege University Faculty of Medicine, Izmir, Turkey
2Molecular Genetics, Ege University Faculty of Medicine, Izmir, Turkey
A total of 20% of patients with transient hypogammaglobulinemia of infancy (THI) have more severe and recurrent infections and may need hospitalization. In order to define the pathogenetic mechanisms in these patients, it has been suggested that there might be additional immunologic or environmental factors and intravenous immunoglobulin (IVIg) replacement therapy is prefered to stop the vicious circle of infection- immunodeficiency condition. IVIg may influence the effect of Fc receptor-mediated activity of immune cells by interacting with Fc regions, acts to block activating receptors and induce inhibitory factors. We aimed to search out the Fcγ receptor polymorphisms in severely infected and hospitalized THI patients (treated with IVIg, n: 18) and in patients who were asymptomatic or had mild infections (treated with antibiotics/ no medication, n: 25) in order to evalute the association between these heterogeneous clinical pictures and polymorphisms.
FcγR polymorphisms (H/H-131, H/R-131 and R/R-131 for FcγIIa; V/V-158, V/F-158 and F/F-158 for FcγRIIIa; NA1/NA1, NA1/NA2 and NA2/NA2 for FcγRIIIb) were analyzed in both groups.
Genotypic distributions between two groups did not deviate significantly from the Hardy- Weinberg equilibrium expectations (p>0.05) and odds ratios for “disease risk estimate” did not show any dominance for any genotype. Nor allele frequencies showed any significant difference (p>0.05) neither the number of infections per year for each FcγR genotype was significantly different between two groups.
In conclusion, there is no association between heterogenous clinical picture of THI patients and FcγR polymorphisms making why some THI patients are severely symptomatic and need IVIg.
ESID-0554 Molecular Studies of Different Types of HIGM Patients
S.E. Banijamali 1, Z. Alizadeh1, M.R. Fazlollahi1, L. Atarod2, Z. Pourpak1
1Imunology Asthma &Allergy Research Institute, Tehran University of Medical Sciences, Tehran, Iran
2Vali-Asr Hospital, Tehran University of Medical Sciences, Tehran, Iran
Background: Hyper IgM syndrome (HIgM) is a rare primary immunodeficiency disorder; so far five types of HIgM have been charachterized. The aim of this study was to determine genetic types (type I and II) of HIgM in patients with recurrent infections and related phenotypes. Mutations in CD40L and AID are responsible for HIgM type I and II respectively.
Methods: Patients with clinical and laboratory diagnosis of HIgM who referred to IARRI between 2006 to 20013 were enrolled in our study. Whole blood samples in EDTA were collected from patients and genomic DNA was extracted. Mutations in AID and CD40L were investigated by PCR followed by direct sequencing and analyzed for finding mutation.
Results: Five patients were included in this study. We found two different mutations in CD40L which included a new mutation in exon 1(c.93delT) leading to a stop codon and a reported mutation in exon 5(G198R) and also, a repoted mutation was found in AID(E122X) respectively.
Conclusion: Although HIgM is a rare genetic disorder, evaluating the types and prevalence of principle mutations could be useful for deciding better treatments in early stage and HSCT. Also, diagnosis of specific gene defects is important in embryonic phase and prenatal diagnosis.
Keywords Hyper IgM Syndrom, CD40L, AID
ESID-0238 Investigation of the Molecular Basis of Agammaglobulinemia in Patients from Highly Consanguineous Tunisian Population
M. Ben-Ali 1, K.W. Chan2, I. Ben-Mustapha1, J. Gamara1, B. Larguèche1, M. Bejaoui3, F. Mellouli3, Y.L. Lau2, M.R. Barbouche1
1Laboratory of Transmission Control and Immunobiology of Infections, Pasteur Institute of Tunis, Tunis, Tunisia
2Department of Paediatrics and Adolescent Medicine, University of Hong Kong, Hong Kong, China
3Department of Pediatrics, Bone Marrow Transplantation Center, Tunis, Tunisia
Agammaglobulinemia is a rare primary immunodeficiency characterized by absent peripheral B cells and severe hypogammaglobulinemia. About 80-85% of patients have mutations in BTK, the gene responsible for the X-linked agammaglobulinemia (XLA). Approximately, half of the remaining patients have mutations in the genes required for the pre-BCR assembly or for its signaling cascade including genes encoding for the m heavy chain, Igα, Igβ, λ5, BLNK, p85α and E47.
The objective of our study is to characterize the molecular basis of agammaglobulinemia in Tunisian patients. Sixteen male and two female patients presenting with an early onset of bacterial infections, less than 0.5% circulating B lymphocytes and low immunoglobulin levels were investigated and analysed by direct sequencing of candidate genes.
Sequencing analysis of BTK gene revealed the presence of 9 different mutations including 5 previously unreported in 12 male patients. Four of them are premature stop codons, three are missense mutations and two are splice site mutations. The remaining six patients assigned to autosomal-recessive agammaglobulinemia were investigated for IGHM, CD79A, CD79B, IGLL1, Vpre-B and BLNK mutations. Only one female patient was found to be homozygous for a complex 2-pb insertion and 5-bp deletion in IGHM gene leading to a frameshift and a premature stop codon.
X-linked agammaglobulinemia is present in ~66% of patients. The autosomal recessive forms are more frequent in Tunisia compared to European series, probably due to high prevalence of parental consanguinity. Molecular basis remains unknown in a majority of the latter, for whom total exome sequencing is underway.
ESID-0428 Spectrum of AICDA Gene Mutation Observed in Tunisian Patients with Class Switch Recombination Deficiencies
I. Ben-Mustapha 1, H. Ouadhani1, L. Ben-Khemis1, B. Larguèche1, H. Masmoudi2, M. Ben-Ali1, M.R. Barbouche1
1Immunology, Pasteur Institute of Tunis, Tunis, Tunisia
2Immunology, Habib Bourguiba, Sfax, Tunisia
Introduction: Immunoglobulin class switch recombination deficiencies (CSR-D), also named hyper-IgM syndromes are a group of primary immunodeficiency disorders, characterized by normal or elevated serum levels of IgM and low levels of other types of immunoglobulins. CSR-D is a heterogeneous syndrome and several molecular causes have been defined. Among the autosomal recessive forms, HIGM2 caused by AICDA gene mutation, is the most frequent.
Objective: To investigate molecular genetic defects in patients with clinical and immunological phenotype of AR-CSR-D.
Methods: Nine patients including 6 females and 3 males were screened for AICDA gene mutations by PCR and DNA direct sequencing.
Results: We identified 3 mutations which sit at different regions of AICDA in 8/9 patients.
One patient carried a missense mutation in exon 2 (c.91T>C) leading to the replacement of Tyrosine by Histidine (p.Tyr31His) within the dimerization domain.
Two patients carried the same missense mutation in exon 3 (c.389A>C) inducing an Histidine to Proline substitution (p. His130Pro) in the APOBEC-like domain.
Interestingly, the third mutation (c.156_c166ins72) located in the catalytic domain and inducing the generation of a stop codon was identified in 5 patients belonging to 3 unrelated families.
Conclusion: The absence of AICDA genetic defects in one patient should prompt the investigation of other autosomal recessive genes. The recurrence of the same mutation in Tunisian patients strongly suggests the existence of a potential founder effect. Such results have been observed in our highly consanguineous population for several primary immunodeficiencies, thus facilitating routine molecular screening of patients with PIDs in our settings.
ESID-0296 Association of Clinical Manifestations and B Cell Subsets with Freiburg and Paris Classifications in Patients with Common Variable Immunodeficiency (CVID)
L. BERRÓN-RUIZ 1, A. Vargas-Hernández2, G. López-Herrera1, M.A. Yamazaki-Nakashimada3, D. Mogica-Martínez4, N.H. Segura-Méndez5, N. Gómez-Hernández6, F.J. Espinosa-Rosales1, L. Santos-Argumedo2
1Unidad de Investigación en Inmunodeficiencias, Instituto Nacional de Pediatría, Mexico City, Mexico
2Departamento de Biomedicina Molecular, Centro de Investigación y Estudios Superiores IPN, Mexico City, Mexico
3Servicio de Inmunología y Alergia, Instituto Nacional de Pediatría, Mexico City, Mexico
4Servicio de Alergía e Inmunología Clínica, Centro Médico Nacional "La Raza" IMSS., Mexico City, Mexico
5Servicio de Alergía e Inmunología Clínica, Hospital de Especialidades CMN Siglo XXI IMSS, Mexico City, Mexico
6Servicio de Alergía e Inmunología Clínica, IMSS Centro Médico de Occidente, Guadalajara, Mexico
Introduction: CVID comprises a heterogeneous group characterized by hypogammaglobulinemia leading to recurrent infections; other manifestations are autoimmune disease and malignancies. The Classification schemes have been proposed based on the relative frequency of various memory B cell subsets and their association with different clinical manifestations. Objective: To determine which classification (Freiburg or Paris) reflect a better correlation between clinical manifestations with B cell subsets in a cohort of CVID patients. Methods: This study comprised 48 patients and 40 healthy controls. Relative and absolute numbers of B cells, transitional, naive, non class-switched memory, class-switched memory, double negative, CD21 low B cells and plasmacytoid cells were determined by flow cytometry, based on differential expression of CD19, IgM, IgD, CD24, CD27, CD21 and CD38. Patients were grouped according the Freiburg classification which explores memory class-switched B cells and CD21 expression and the Paris classification which is based on the assessment of memory class-switched B cells and total CD27+ B cells. Results: We found a higher incidence of autoimmune disease in Paris MBO (p=0.0120) and Freiburg Ia (p=0.05). In Infective pneumonia we found incidence in Paris MBO (p=0.0214) and Freiburg Ib (0.0157). We report abnormalities into the distribution of peripheral blood lymphocytes in these classifications. Conclusions: Classification of CVID patients by determination of switched memory B cells could be useful to predict clinical prognosis of these patients. Acknowledgments: CONACYT # 161089; # 154472; FUNEMI.
ESID-0738 Clinical and Laboratory Characteristics of Slovene Patients with CVID
S. Blazina 1, M. Kemperle1, G. Markelj1, A. Pecnik Pikelj2, P. Kopac3, M. Debeljak4, A. Ihan5, T. Avcin1
1Dpt. of Allerg. Rheum. and Clin. Immunol., University Children's Hospital University Medical Centre, Ljubljana, Slovenia
2Dpt. of Infectious Diseases and Febrile Illnesses, University Medical Centre, Ljubljana, Slovenia
3University Clinic of Pulmonary and Allergic Diseases Golnik, University Clinic of Pulmonary and Allergic Diseases Golnik, Golnik, Slovenia
4Dpt. of Laborat. Diagnost., University Children's Hospital University Medical Centre, Ljubljana, Slovenia
5Institute of Microbiology and Immunology, Faculty of Medicine University of Ljubljana, Ljubljana, Slovenia
Common variable immunodeficiency is a heterogenous group of diseases. The aim of study was to determine clinical and laboratory characteristics of Slovene patients with CVID. Ten out of twelve patients from Slovene national registry were assessed.
Clinical assessment with a prepared questionnaire and physical examination was performed. Some laboratory investigations were performed in all patients, with additional according to the clinical assessment. Diagnosis of CVID was re-evaluated according to the new ESID diagnostic criteria for CVID. Hypogammaglobulinemia before IgG supplementation and block in differentiation of B cells were present in all patients.
Mean age was 26,1 years (8 to 50 years), gender distribution was equal. All have had recurrent bacterial infections before diagnosis, but none has suffered from severe bacterial infection in the last year.
Before inclusion in the study five patients have had lung disease (3 bronchiectasis, 4 abnormal lung function tests, 1 lymphocytic lung disease). Three patients have been diagnosed with autoimmune disease before inclusion (1 AI thyroiditis, 1 rheumatoid arthritis, 1 severe AI haemolytic anaemia). None has been diagnosed with malignancy.
Previously undiscovered lung disease was not found found in any patient. However, autoimmune disease was newly diagnosed in 4 patients (3 AI thyroiditis, 1 rheumatoid arthritis) during the study. Six patients had signs of lymphoproliferative disorder (4 splenomegaly). Eight patients were receiving immunoglobulins, of these 2 subcutaneously.
Slovene patients with CVID have had recurrent bacterial infections before immunoglobulin supplementation. Besides lung disease, concealed autoimmune disease and lymphoproliferative disorders are common and should be actively sought.
ESID-0175 Recurrent Invasive Pneumococcal Infections with Anti-Pneumococcal Antibody Deficiency Due to Dedicator of Cytokinesis 8 (DOCK8) Mutation
A. Broides 1, R. Somech2, J. Levy1, A.B.. Mandola1, A.J. Simon2, G. Shubinsky3
1ambulatory care unit, Soroka University Medical Center and Ben-Gurion University of the Negev Beer-Sheva, Beer-Sheva, Israel
2Pediatric Immunology, Safra Children's Hospital Sheba Medical Center Tel Hashomer Sackler School of Medicine Tel Aviv University Israel, Tel-Aviv, Israel
3flow cytometry unnit Hematology laboratory, Soroka University Medical Center and Ben-Gurion University of the Negev Beer-Sheva, Beer-Sheva, Israel
Background: Recurrent invasive pneumococcal infections as the main manifestation of immunodeficiency are common in patients with TLR/IRAK4/MYD88/NEMO defects. Patients with mutations in DOCK8 suffer from combined immunodeficiency with eczema and elevated IgE.
Objective: To describe an anti-pneumococcal antibody deficiency patient who suffered solely from recurrent pneumococcal infections and diagnosed with a mutation in DOCK8.
Case report: A 17 year old Bedouin male, from a consanguineous family, presented with recurrent infections since the age of 4 months, he suffered from 7 separate invasive pneumococcal diseases (2 pneumonias, 2 bacteremia/sepsis, septic arthritis, meningitis and osteomyelitis). He was also diagnosed as having asthma and celiac disease. He does not have eczema although he has disseminated verruca plana. His immunological evaluation revealed normal/elevated IgG, elevated IgA and IgE, and lack of appropriate anti-pneumococcal antibody production after pneumococcal vaccines. TLR/IRAK4/MYD88/NEMO defects were ruled out by a functional assay. Immunophenotyping revealed severe CD4+ and moderate CD8+ T-lymphopenia. While a total B-cell count remained normal, only few IgM+/CD27+ cells were found. Switched IgM-/CD27+ B-memory cells were significantly reduced. CD5 was found in many CD27-negative B-cells and absent in CD27+ B-cells. Pneumococcal infections did not occur after the institution of intravenous immunoglobulin therapy. Eventually a mutation in the DOCK8 gene was identified (p.S1711X,c.C5134A).
Conclusions: DOCK8 patients may present with invasive recurrent pneumococcal disease with anti-pneumococcal antibody deficiency as the main manifestation of immunodeficiency, and this clinical phenotype may reflect a lack of B-lymphocytes with a non-switched memory/splenic marginal zone phenotype associated with increase of CD5+ transitional B cells.
ESID-0634 Humoral Alteration in 22Q11.2 Deletion Syndrome Patients
C. Cancrini 1, S. Di Cesare1, P. Puliafito1, D. Montin2, R. Rondelli3, A. Soresina4, C. Pignata5, M. Pietrogrande6, R. Carsetti7, P. Rossi 1 for IPINET group8
1University Department of Pediatrics, Pediatric Hospital Bambino Gesu' and Tor Vergata University, Roma, Italy
2Department of Pediatrics, University of Turin, Turin, Italy
3Department of Pediatrics, University of Bologna, Bologna, Italy
4Pediatrics Clinic and Institute of Molecular Medicine "A. Nocivelli", University and Spedali Civili, Brescia, Italy
5Department of Pediatrics, "Federico II" University of Naples, Naples, Italy
6Department of Pediatrics, Ca' Granda IRCCS Foundation University of Milan, Milan, Italy
7Immunology Area Research Center, Bambino Gesù Children's Hospital, Rome, Italy
8Italian Network for, Primary Immunodeficiencies, ., Italy
Objective: To investigate humoral compartment at diagnosis and during follow-up in patients with 22q11.2 Deletion Syndrome (22q11DS).
Methods: A retrospective and prospective multicenter study was conducted with 165 patients in the context of the Italian Network for Primary Immunodeficiencies. Patients were investigated for serum immunoglobulin, testing specific antibody titers to recall antigens and proliferative responses to phytohemagglutinin (PHA) and OKT3. Lymphocyte subsets were assessed by flow cytometric analysis.
Results: The cohort consisted of 96 males and 69 females; median age at diagnosis was 4 months (range 0 to 35 years). Eighty out of 165 patients had a history of recurrent and/or severe infections and 41% patients received at least one prophylactic treatment. During follow-up the frequency of autoimmune manifestations increased, severe infections and antibiotic prophylaxis decreased. We reported hypogammaglobulinemia in 34% of patients at diagnosis, confirmed during follow-up. Total lymphocytes and T-cells subsets were decreased in most of 22q11DS patients, although these immunological abnormalities did not correlate to risk of infections. Lymphocyte proliferation in response to mitogens and antigens was normal/ in the lower levels of normality, in most of patients. Our preliminary data showed a decrease of B cell memory.
Conclusions: Not only T-cell but also B- cell compartment should be routinely investigated in 22q11DS patients with an history of recurrent infections and autoimmune manifestations. These data could help to better define, in these patients, follow-up and clinical management with a specific antimicrobial treatment and/or intravenous immunoglobulin therapy.
ESID-0227 The Origin and Function of Innate Memory B Cells
R. Carsetti 1, M. Rosado1, A. Aramburu1, A. Disabatino2, G.R. Corazza2, I. Quinti3
1Immunology and Pharmacotherapy, Bambino Gesù Children Hospital, Rome, Italy
2Internal Medicine, Univ.of Pavia S.Matteo Hospital, Rome, Italy
3Internal Medicine, Univ. La Sapienza Rome, Rome, Italy
Secretory IgA regulates the microbiota and prevents pathogen invasion at mucosal sites.
Here we show that IgM memory B cells are indispensable for the production of IgA plasma cells in response to innate mechanisms in vitro and in vivo.
Innate signals generated by TLRs and the receptor TACI on B lymphocytes induce the differentiation of IgM memory into IgA plasma cells. SIgA is absent in patients lacking IgM memory B cells. The mechanism leading to SIgA production is triggered in the neonate by bacterial colonization, inducing APRIL production by epithelial cells and recruitment of IgM memory B cells, followed by production of SIgA. This function of IgM memory B cells is unique and cannot be replaced by other B-cell types.
ESID-0765 Challenges of Raising Awareness of Bruton’s Disease in Bosnia and Herzegovina-Earlier Diagnosis Leads to Reduce Morbidity and Better Outcomes
A. Cengic 1, A. Omercahic Dizdarevic1, V. Selmanovic1
1Allergology rheumatology and clinical immunology, Children's hospital Clinical Center University in Sarajevo, Sarajevo, Bosnia and Herzegovina
X linked agammaglobulinemia (XLA), also known as Bruton’s disease, is a humoral immunodeficiency disease characterized by recurrent bacterial infections due to low levels or absence of serum immunoglobulins. It is caused by mutations in the gene coding for Bruton tyrosine kinase (BTK) that is critical to the maturation of pre–B cells to differentiating mature B cells. The incidence of XLA is reported to be around 1/250,000 male births. In Jeffrey Modell centre in Sarajevo, we have only one registered child with XLA. Statistically, according to the size of our population, we should have approximately 10 diagnosed patients. That means that the majority of children and adults are not recognized as a result of inadequate knowledge of primary immunodeficiency. Our patient is now 11 years old boy who developed recurrent pneumonia at age of 3, after enrollment in kindergarten. Immunological studies revealed decreased serum immunoglobulins, (IgG 0.05 g/l; IgA 0.01 g/l; IgM 0.19 g/l), nearly total absence of circulating B cells (CD 19 0.01 %), increased CD3 (87 %) and CD4 (60 %). In march 2006 genetic testing confirmed C763 T mutation (effect R255X). Following diagnosis, substitution therapy with intravenous immunoglobulins was started with uneventful follow up. Curiosity is that our patient has well developed tonsils and that he had been immunized with live polio vaccine at 2, 4, 6 and 18 months of age without any complication. Conclusion: Increasing the knowledge and awareness of XLA will lead to earlier diagnosis, which will increase the quality of life and reduce morbidity and mortality.
ESID-0603 Symptomatic and Asymptomatic HPER IGM Syndrome in a Family (New Mutation)
S. armin 1, Z. chavoshzadeh natanzy2, K. ramezani3, N. Rezaei4, B.S. shamsian5
1infectios, Shahid beheshti university of medical sciences Pediatric Infections Research Ce, Tehran, Iran
2allergy, Shahid beheshti university of medical sciences Pediatric Infections Research Ce, Tehran, Iran
3Allergy, Shahid beheshti university of medical sciences, Tehran, Iran
4immunodeficiency, Tehran university of medical sciences, Tehran, Iran
5hematology, Shahid Beheshti University of Medical Sciences, Tehran, Iran
The hyper-IgM syndrome genetically heterogeneous and characterized by normal or elevated serum levels of IgM and low levels of other immunoglobulin classes.
We report two cases of Hyper- IgM, two sons from identical twin sisters who are married to two brothers (their cousins).one of them is a 1 year-old boy, from related parents (first cousin). He admitted with nausea, vomiting, tachypnea, respiratory distress and bilateral alveolar infiltration in chest X ray. Because of chronic diarrhea and eczema that have been started from four months age he was treated as a case of food allergy, avoided from milk and egg protein , although it was not effective and his growth chart showed failure to thrive from 7 months old.
He was treated as a case of presumptive Pneumocystis carinii infection and symptoms recovered fully with prompt treatment.
Immune system evaluation compatible with Hyper IgM syndrome.
second case was his cousin , a 2 year-old boy with neutropenia in a CBC which was done for checkup, he had no sign or symptom ,because of neutropenia and family history of immunodeficiency, immunologic system was evaluated that was compatible with hyper IgM syndrome ,complementary evaluation showed new mutation in these two relative cases
ESID-0491 Increased IL-21-Induced STAT3 Phosphorylation on CD27+ B-Cells from Common Variable Immunodeficiency Disease (CVID) Patients
A. Clemente 1, J. Pons1, N. Lanio1, V. Cunill1, N. Matamoros1, J.M. Ferrer1
1Immunology Department, Son Espases Hospital, Palma de Mallorca, Spain
Introduction CVID is a humoral immunodeficiency characterized by hypogammaglobulineamia and defective response to vaccination. Patients suffer from recurrent infections, autoimmune and/or haematological disorders. Several alterations on B-cells functionality have been associated with CVID, but the underlying molecular defect that impairs B-cells differentiation remains to be elucidated.
STAT3 activation plays a critical role on B-cells development and its defective expression or phosphorylation leads to immunodeficiency. IL-21 costimulation through STAT3 induces different outcomes on B-cells, depending upon their maturation status and the stimulus used to activate them.
Objective To evaluate STAT3 activation (expression and phosphorylation) on naïve and memory B-cells in response to BCR, TLR-9 or CD40 engagement and IL-21 costimulation, in a subgroup of CVID patients with low memory B-cells.
Material and methods Purified B-cells from CVID patients and controls were stimulated with anti-IgM (5 mg/ml), ODN (0.6 μg/ml) or anti-CD40 (1 μg/ml), with or without IL-21 (100 ng/ml). Intracellular STAT3 and phospho-STAT3 levels were evaluated by flow cytometry on CD27- and CD27+ B-cells.
Results IL-21 costimulation upmodulates STAT3 expression and phosphorylation on CD27- and CD27+ B-cells, both in patients and controls. CD27- B-cells from patients showed normal STAT3 and phospho-STAT3 levels, independently of the stimulus used. In contrast, anti-IgM- or anti-CD40-stimulated CD27+ B-cells from patients showed significant higher levels of phospho-STAT3 than those from controls in response to IL-21 costimulation.
Conclusion CD27+ B-cells from CVID patients with defective memory B-cells differentiation show higher levels of STAT3 phosphorylation. This could contribute to hypogammaglobulineamia as well as autoimmune and/or haematological processes characteristics of these patients.
ESID-0453 Serum Free Light Chain in Primary and Secondary Hypogammaglobulinemia: A New Diagnostic Tool in Common Variable Immunodeficiency?
N. Compagno 1, F. Cinetto1, G. Malipiero1, C. Agostini1
1Department of Medicine, University of Padua, Padova, Italy
Introduction: Serum free light chains (sFLC) are widely used as a prognostic marker in monoclonal gammapathy of undetermined significance, multiple myeloma, solitary plasmocytoma, light chain amyloidosis, chronic lymphocytic leukemia; in all these conditions a raise of either kappa or lambda chain is a marker of clonality. Furthermore, polyclonal increase in serum FLC have been identified in autoimmune disease, as a marker of immune stimulation. In most cases common variable immunodeficiency (CVID) diagnosis is an exclusion diagnosis. In particular, in adult patients differential diagnosis between CVID and lymphoproliferative disorders or non secreting multiple myeloma may represent a challenge.
Methods: in this study sFLC levels were determined in 56 patients with primary immune deficiency (43 CVID, 2 X-linked agammaglobulinemia, 1 IgA deficiency, 10 IgG subclass deficiency) and in 41 patients with secondary hypogammaglobulinemia (24 chronic lymphocytic leukemia, 15 non secreting multiple myeloma, 1 nephrotic syndrome, 1 protein losing enteropathy).
Results: in 39 out of 43 patients with CVID and 2 patients with XLA a reduction of either kappa or lambda light chains was observed. By contrast, sFLC were normal in IgA deficiency and IgG subclass deficiency. More importantly, in all patients with secondary hypogammaglobulinemia sFLC were normal, or, alternatively, a raise of either kappa or lambda chains were detected.
Conclusion: sFLC can be a powerful tool in the differential diagnosis of hypogammaglobulinemia, allowing to discriminate with high sensitivity and specificity patients with CVID.
ESID-0686 Molecular Characterization and Clinical Aspects of Bruton's Tirosine Kinase Deficient Patients in a Single Center in Brazil
L.A.O. Cunha 1, J.B. Oliveira2, M.L. Silva3, J.A. Pinto1
1Pediatrics, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
2Immunology, IMIP, Recife, Brazil
3Immunology, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
Clinical, molecular and laboratorial data of seventeen boys diagnosed as Brutons Tirosine Kinase deficient patients, ten from four family clusters and seven sporadic cases, followed at a single center in a Brazilian State (Minas Gerais) were analyzed. The estimated prevalence of X-linked agammaglobulinemia in Minas Gerais according to the investigated population is 1 in 1.211374 inhabitants. The most frequent infections are respiratory tract infections, followed by gastrointestinal manifestations. Before the diagnosis, 94% of patients had needed hospitalization and after the diagnosis and instauration of treatment only 35% needed hospitalization. The treatment with immunoglobulin replacement was well tolerated, with only one patient presenting reaction requiring medication before infusion. . Different types of genes abnormalities were detected in patients, premature stop codons, initiation codon variant, deletions, insertions, frameshift deletion, intronic variant, missense variants. Five new mutations were found. One patient presented BTK expression at flow cytometry and mutation described as pathogenic.
The prevalence of X-linked agammaglobulinemia in Minas Gerais, despite not differing from the USA or Europe should be underestimated because it is based on patients who manage to be referred to a tertiary service. The genetic testing is important to genetic counselling especially in patients who present BTK at flow cytometry, but the history and clinical presentation are suggestive of X-linked agammaglobulinemia. The diagnosis and correct treatment contribute to a decrease in the need of hospitalization.
ESID-0083 Recurrent Bacterial Prostatitis as Clinical Presentation in a Patient with Common Variable Immunodeficiency (CVID)
V.A.S.H. Dalm 1, W.J. Kirkels2, M. van der Burg1, M.W. van der Ent3, P.M. van Hagen1
1Immunology, Erasmus MC, Rotterdam, Netherlands
2Urology, Erasmus MC, Rotterdam, Netherlands
3Internal Medicine, Erasmus MC, Rotterdam, Netherlands
Common variable immunodeficiency (CVID) is a primary immunodeficiency defined by markedly reduced serum levels of IgG, low levels of IgA and/or IgM, and poor or absent response to immunizations. Patients typically present with recurrent infections affecting the upper and lower respiratory tracts and gastrointestinal tract.
Here we report a 37 year old patient with a medical history of non-specific colitis, who was referred by the department of urology because of recurrent bacterial prostatitis with repetitive urinary cultures demonstrating E. Coli. He suffered from episodes of dysuria, hematuria, pain with ejaculation, perianal discomfort and lower abdominal pain, accompanied by fever. There were no significant recurrent infections at other sites. Rectal examination was very painful and prostatic tenderness was reported. No trauma, stones, functional or anatomical anomalies were found.
Immunological evaluation revealed low IgG (3.9 g/l), IgA (0.43 g/l) and IgM (0.26 g/l), with low pre-vaccination titers against Str. Pneumonia and H. Influenza and no significant increase in antibody titers after vaccination. Flowcytometry analysis on peripheral blood demonstrated normal B-, T- and NK-cell numbers.
As no other specified immunodeficiency state could be defined, diagnosis was compatible with CVID. Monthly intravenous immunoglobulin (IvIg) suppletion therapy (0.4 g/kg body weight) was initiated and after 6 months serum IgG was increased to 10.5 g/l. Until now, 12 months on IvIg, no episodes of bacterial prostatitis occurred.
This uncommon case shows that CVID may present with atypical infections and stresses the importance of immunological evaluation in patients with recurrent bacterial prostatitis with no urological explanation.
ESID-0596 Vaccination Responses in a Cohort of 'Mildly' Antibody Deficient Adults Suggests Unconjugated Pneumococcal Response Analysis is Most Relevant Clinically
R.M.J. Schamp 1, S. Christiaanse1, T.H. Macken2, J.F.M. Pruijt3, E. de Vries1
1Pediatrics, Jeroen Bosch Hospital, 's-Hertogenbosch, Netherlands
2Pulmonology, Jeroen Bosch Hospital, 's-Hertogenbosch, Netherlands
3Internal Medicine, Jeroen Bosch Hospital, 's-Hertogenbosch, Netherlands
Background: In the Jeroen Bosch hospital (JBH), immunologist EdV cooperates with pulmonologists and internists in a Care Pathway to improve detection of primary immunodeficiency (PID).
Objective: What is the contribution of vaccination responses in the analysis of primary antibody deficiency (PAD) in adults.
Methods: Diphtheria (D), tetanus (T) and unconjugated pneumococcal (uP) responses were determined.
Results: In 75 referred adults (≥20 yrs; 54 women; Jan'12-April'14) PAD was found in 42 (29 women; 21 new ESID working definition 'unclassified immunodeficiency', 9 CVID, 9 'unclassified hypogammaglobulinaemia', 3 selective IgA-deficiency). T and D responses were >0.1 IU/ml in all(T) and 87%(D), >1.0 IU/ml in 97%(T) and 49%(D), uninfluenced by ESID working definition or treatment chosen by the physician (none, antibiotic prophylaxis, immunoglobulin substitution). uP responses to 11 serotypes showed none to be specifically useful for differentiation; the physician's choice to treat seems to increase with decreasing number of uP-serotype responses >1.0 IU/ml (Pearson's Chi-square:p=0.053), which suggests these were clinically more severe. The degree of hypogammaglobulinaemia in this non-university cohort is 'milder' (see above), however, there is serious clinical impact (8/15 bronchiectasis on HRCTscan).
Discussion: Especially uP responses seem to be impaired in these milder antibody deficient adults. Further studies are important to analyse this further. This would have implications for the usefulness of the new ESID working diagnoses: all patients with abnormal uP-responses fall under 'unclassified immunodeficiency'. It might be more useful for further studies to group all non-CVID, non-agammaglobulinaemia, non-class-switch-recombination-problem patients together as 'unclassified antibody deficiency'.
ESID-0048 Hepatitis B Vaccination Response is Less Well Sustained in Down Syndrome Children
M.I. Hollegien 1, L.A. Bok2, M. Wojciechowski3, T. Boiy3, M. Hilbink4, A.C.A.P. Leenders5, E. de Vries1
1Pediatrics, Jeroen Bosch Hospital, 's-Hertogenbosch, Netherlands
2Pediatrics, Máxima Medical Centre, Veldhoven, Netherlands
3Pediatrics, University Hospital, Antwerp, Belgium
4Jeroen Bosch Academy, Jeroen Bosch Hospital, 's-Hertogenbosch, Netherlands
5Laboratory for Medical Microbiology, Jeroen Bosch Hospital, 's-Hertogenbosch, Netherlands
Background Several studies describe lower vaccination responses in Down syndrome (DS). Post-vaccination titers after routine hepatitis B (HBV-)vaccination are adequate but lower in DS; long-term follow-up data on anti-HBs-titer are lacking.
Objective To compare long-term anti-HBs-titers and prevalence of long-term post-vaccination HBV-protection (anti-HBs-titer ≥ 10 IU/l) in DS versus non-DS children.
Method Retrospective study in the Jeroen Bosch Hospital (JBH) ´s-Hertogenbosch and Máxima Medical Centre Veldhoven, the Netherlands, and University Hospital Antwerp, Belgium. Parents (n=398) were approached by mail; 120 gave informed consent. Stored serum of 100 DS-children was available; 81 of these were HBV-vaccinated (parental information). The anti-HBs-titer was determined by chemiluminescence-immunoassay (UniCel-DxI-600; JBH). Results were compared to data of non-DS children from the literature [Zanetti et al. 2005].
Results All 0 to 2-year-old vaccinated DS-children had an anti-HBs-titer of ≥ 10 IU/l (geometric mean (GM) 254.5 IU/l; 95%-CI: 117.1-552.8). Only 1 in 3 DS-children ≥ 7 years of age maintained adequate protection (anti-HBs-titer ≥ 10 IU/l) (GM 5.9 IU/l; 95 %-CI: 2.9-11.9). In non-DS children 2 in 3 have adequate protection for > 10 years (GM 32.1 IU/l; 95%-CI: 28.6-36.0).
Conclusion Only 1 in 3 children with DS≥7 years of age had maintained a protective anti-HBs-titer long after vaccination. Follow-up studies are needed to determine the rise in titer after a booster vaccination and to decide whether the unprotective levels we found are clinically relevant enough to advise routine follow-up titer(s) with booster vaccination(s), if needed, in DS.
ESID-0034 Clinical Findings in Patients with Common Variable Immunodeficiency in Two Hospitals in Mexico City
R. Dorbeker-Azcona 1, M.D. Mojica-Martinez2, M. Becerril-Angeles2, M. Guevara-Cruz3, S.E. Espinosa-Padilla4, M.A. Yamazaki-Nakashimada5, L. Blancas-Galicia4
1Pediatria, Hospital Central Sur de Alta Especialidad Petroleos Mexicanos, Mexico City, Mexico
2Alergia e Inmunología, Instituto Mexicano del Seguro Social Hospital de Especialidades CMN La Raza, Mexico City, Mexico
3Fisiologia de la Nutricion, Instituto Nacional de Ciencias Medicas y Nutricion Salvador Subiran, Mexico City, Mexico
4Unidad de Investigación en Inmunodeficiencias, Instituto Nacional de Pediatria, Mexico City, Mexico
5Inmunologia, Instituto Nacional de Pediatria, Mexico City, Mexico
Background: Common variable immunodeficiency is one of the most common antibody deficiencies, is characterized by low serum immunoglobulins, impaired antibody response and increased susceptibility to chronic and recurrent infections.
Objectives: present the clinical characteristics of patients with CVID in two hospitals in Mexico City
Methods: Retrospective study in patients with diagnostic criteria for CVID. The data collected was: demographic, age at onset, age at diagnosis, family history, infection, autoimmunity, lymphoproliferative disease, allergy, malignancy, immunoglobulin levels at diagnosis, route of administration, dosage and frequency of IVIG of each patient. Data were analyzed with descriptive statistics.
Results: We report findings in 26 patients who met CVID criteria, 14 men and 12 women. The mean diagnosis delay was 48 months (22-128), serum immunoglobulins at diagnosis in mg / dl were IgG 216 (114-316), IgM 21 (12-121), IgA 21 (6-26) and IgE 4.6 (1.8) IU / ml. 81% of patients had pneumonia. There was a decrease in the number of pneumonias before and after treatment with gammaglobulin (p ?? = 0.028). 27% of the patients had autoimmune diseases, 35% allergies, 35% chronic diarrhea, 62% bronchiectasis, 73% chronic cough, 50% lymphadenopathy. One patient had lymphoproliferative disease and none developed malignancy.
Conclusions: We found that the delay in the diagnosis and initiation of gammaglobulin replacement affects the occurrence of complications such as bronchiectasis.
ESID-0265 Diagnostic Agreement Between Pneumococcal 23-Valent IGG ELISA and 13-Valent Serotype-Specific Multiplex Assay in Patients with Suspected Antibody Deficiency
M. Dziadzio 1, R. Doffinger2, J. Harvey1, R. Smith3, S. Burns1, F. Tahami1, L. Ceron –Gutierrez2, G. Barcenas-Morales4, S.L. Seneviratne1, D. Kumararatne2, R. Chee1, H. Baxendale2
1Immunology, Royal Free Hospital, London, United Kingdom
2Immunology, Addenbrooke's and Papworth Hospitals, Cambridge, United Kingdom
3Medical Electronics, Royal Free Hospital, London, United Kingdom
4Immunology, UNAM, FES-Cuautitlan, Mexico
Background: Vaccination responses to polysaccharides guide diagnosis and therapeutic interventions in patients with suspected immunodeficiency. The robustness of Pneumococcal serology in diagnosing clinically relevant antibody deficiency is controversial.
Objective: To compare Pneumococcal 23vELISA and 13-valent serotype-specific multiplex assay in evaluating responses to Pneumovax® in new patients referred with suspected immunodeficiency.
Methods: Pneumococcal IgG levels from 64 patients were analysed before and after test-vaccination with Pneumovax® using both assays. Adequate ELISA response was defined as a ≥4-fold increase in total Pneumococcal IgG levels from baseline. Adequate multiplex response was defined as serotype-specific IgG threshold of >0.35 mg/l for ≥70% (≥9/13) of serotypes tested. Serotype-specific IgG thresholds of >0.5 mg/l and ≥1.3 mg/l were also analysed.
Results: Abnormal vaccine response was found in 43.7% of patients by ELISA, 32.8% by multiplex at >0.35 mg/, 45.3% for >0.5 mg/l threshold and 76.5% for ≥1.3 mg/l. Assay agreement was good for thresholds >0.35 mg/l (79.6%, Cohen's κ=0.57) and >0.5 mg/l (81.2%, κ=0.58) but poor for ≥1.3 mg/l threshold due to significant positive bias on multiplex (p<0.01). No patients with undetectable Pneumococcal IgG by ELISA at baseline had normal vaccine response by either method.
Conclusions: In our hands, 23vELISA and multiplex show approximately 80% diagnostic agreement using a serotype-specific IgG threshold of >0.35 mg/l and >0.5 mg/l for ≥9/13 serotypes and total Pneumococcal IgG threshold of 4-fold rise. When a threshold of ≥1.3 mg/l is used, agreement between the assays is poor. Undetectable baseline Pneumococcal IgG predicted lack of Pneumovax® responses as determined by both assays.
ESID-0273 Salmonella Typhim VI® Vaccine (Sanofi Pasteur MSD) for the Investigation of Antibody Deficiency to Polysaccharide: Preliminary Results of UK Study
C.S. Evans 1, A.C. Cullinane1, E.A.L. Bateman1, J. Miller2, F. Dhalla2, D. Arnold2, R. Jain2, S.Y. Patel2, S.A. Misbah2, B.L. Ferry1
1Clinical Immunology Laboratory Churchill Hospital, Oxford University Hospitals NHS Trust, Oxford, United Kingdom
2Clinical Immunology Department John Radcliffe Hospital, Oxford University Hospitals NHS Trust, Oxford, United Kingdom
Evaluation of antigen-specific antibody responses is important in diagnosing suspected antibody deficiency. It has been suggested that the licensed polysaccharide vaccine Salmonella Typhim Vi® may be an appropriate substitute for the current Pneumovax test vaccine. Limited data defining vaccine response ranges to Salmonella Typhim Vi® exist. Here, we compared responses to Salmonella Typhim Vi® vaccine in healthy controls and antibody deficient patients with the aim of determining whether it would be possible to replace the Pneumovax test vaccine with Salmonella Typhim Vi®. Ethical Permission (10/H0604/75) to recruit healthy controls from Oxford University Travel Clinic, and antibody deficient patients from the Clinical Immunology Department was acquired. Serum samples were obtained prior to vaccination with Salmonella Typhim Vi® vaccine and 3-4 weeks post-vaccination. Antibody levels were quantified using the VaccZyme Anti-S typhi Vi IgG ELISA Kit (The Binding Site Group Ltd). Antibody deficient patients (n=10) showed minimal responsiveness to Salmonella Typhim Vi® (mean fold increase of 1.3) compared to a significantly higher response in healthy controls (mean fold increase of 11.8). Low responsiveness to Salmonella Typhim Vi® in antibody deficient patients correlated with a similar unresponsiveness to Pneumovax (mean fold increase of 1.8) indicating that it may be possible to replace Pneumovax test vaccine with Salmonella Typhim Vi®. This study is ongoing, with patients and controls being recruited continuously and our data constantly being updated. The potential implications of these findings (using updated numbers of patients and controls) will be discussed with reference to the diagnosis of suspected antibody deficient patients.
ESID-0626 X-Linked Agammaglobulinemia: A Portuguese National Survey
A. Fernandes 1, C. Teixeira1, S. Lopes da Silva2, S. Pereira da Silva2, I. Esteves2, J. Gonçalo Marques2, C. Neves3, J. Farela Neves3, E. Faria4, M. Guedes1, S. Lemos4, L. Marques1
1Pediatria, Centro Hospitalar do Porto, Oporto, Portugal
2Pediatria, Centro Hospitalar Lisboa Ocidental, Lisbon, Portugal
3Pediatria, Centro Hospitalar Lisboa Central, Lisbon, Portugal
4Pediatria, Centro Hospitalar Universitario de Coimbra, Coimbra, Portugal
Introduction: X-Linked agammaglobulinemia (XLA) is a primary immunodeficiency that causes recurrent bacterial infections in affected males. Appropriate lifelong immunoglobulin replacement therapy (IRT) is indicated to prevent infections and their complications. The aim of this study is to characterize the Portuguese experience on XLA.
Materials and Methods: A national survey of children and adolescents diagnosed with XLA was accomplished referred to the 1st May 2014. Data was collected retrospectively by review of clinical files.
Results: Thirty-three children with XLA were diagnosed and followed in 4 centres. Two children (6%) had a positive family history. Infection led to diagnosis in 31 (94%) at an average age of 36 months (range:7-108 months). The average age of onset was 15 months. Infections included acute otitis media (23/33), pneumonia (19/33) (recurrent in 11), septic arthritis (11/33), giardiasis (8/33) and sepsis (5/33). Haemophilus influenza was identified in nine (27%) and Pseudomonas aeruginosa in 4 (12%). All fulfilled diagnostic criteria for XLA with a Btk mutation identified in 29 (88%). Six (18%) presented neutropenia at diagnosis. All were on IRT: the majority on subcutaneous IgG and 36% on intravenous IgG. The average time of IRT is 13.5 years (cumulative time: 324 years). One death occurred not associated to XLA. During follow-up 25 (76%) cases presented infectious complications: sinusitis (17/33), giardiasis (8/33), conjunctivitis (7/33) and pneumonia (5/33). Seven (21%) patients developed bronchiectasis.
Discussion: The overall prognosis of XLA is good as long as patients are diagnosed and treated early. However, even with adequate IgG replacement therapy, complications can occur.
ESID-0184 A New Gene Involved in an Autosomal Dominant Form of Common Variable Immunodeficiency
V. Di Pierro1, R. Zuntini1, E. Bacchelli2, A. Schaffer3, B. Grimbacher4, I. Quinti5, S. Ferrari 1
1Medical Genetics Lab, University Hospital S. Orsola-Malpighi, Bologna, Italy
2FaBiT, University of Bologna, Bologna, Italy
3National Center for Biotechnology Information, National Institutes of Health, Bethesda, USA
4Center for Chronic Immunodeficiency, UniversitatsKlinikum, Freiburg, Germany
5Center for Primary Immunodeficiency, University of Roma La Sapienza, Roma, Italy
Common variable immunodeficiency (CVID, MIM#607594) is the most common symptomatic primary antibody deficiency in adults. It includes a heterogeneous group of disorders characterized by defects in the terminal stage of B lymphocyte differentiation, whose underlying genetic defects remain unknown in the majority of cases.
We studied a five-generation Italian family with an autosomal dominant form of CVID through whole exome sequencing, giving priority to the variants within a 9.2 Mb candidate genomic interval on chromosome 3q27.2-q29 previously identified by genome-wide linkage analysis. Since no causative mutation was identified by whole exome sequencing, we performed a whole genome high resolution SNP array analysis, which allowed to detect a ~880 kb tandem duplication in 3q27.3, located inside the candidate linkage region and involving 8 genes: ST6GAL1, RPL39L, RTP1, MASP1, RTP4, SST, RTP2 and BCL6.
The expression pattern analysis in control peripheral blood lymphocytes (PBL) of all genes included in the duplicated region revealed that only ST6GAL1, RPL39L, RTP4 and BCL6 are expressed in mature circulating lymphocytes, allowing us to exclude the remaining four genes from further investigations. Preliminary qRT-PCR analyses conducted on affected vs unaffected subjects showed a significant upregulation of RTP4 in affected members (t-test, p < 0.0001). This finding suggests RTP4 overexpression as a possible pathogenetic mechanism underlying CVID in this family. RTP4 is a Golgi chaperone and we hypothesize that it might be involved in the regulation of the Unfolding Protein Response (UPR), a key process of plasma cell differentiation.
ESID-0322 Next Generation Sequencing Identifies a New Astrovirus Variant in a 14-Year Old Boy with X-Linked Agammaglobulinemia and Encephalitis
M.L. Frémond 1, G. Cros1, M. Eloit2, M. Lecuit3, N. Mahlaoui1, D. Seilhean4, I. Desguerre5, C. Picard6, B. Neven1, S. Blanche1
1Unité d’immunologie hématologie et rhumatologie pédiatriques, Hôpital Necker, Paris, France
2Laboratoire découverte de pathogènes, Institut Pasteur, Paris, France
3Service des maladies infectieuses et tropicales, Hôpital Necker, Paris, France
4Service de neuropathologie, Hôpital La Pitié-Salpêtrière, Paris, France
5Unité de neuropédiatrie, Hôpital Necker, Paris, France
6Centre d'étude des déficits immunitaires, Hôpital Necker, Paris, France
Background Viral encephalitis is a major cause of morbidity and mortality in hereditary agammaglobulinemia patients. Conventional diagnosis methods frequently fail to identify their aetiology.
Case report A 14-year boy with X-linked agammaglobulinemia (XLA) and epilepsy was admitted with progressive cognitive decline and seizures recurrence. Examination found global motor decline and cerebellar syndrome. MRI showed diffuse cortico-subcortical brain atrophy and periventricular hypersignals. CSF examination, real-time PCR and RT-PCR were negative, especially for enterovirus and human astrovirus. Histologic findings on brain biopsy were consistent with acute pan-encephalitis, all standard PCR for neurotropic viruses were negative. Total RNAs were extracted from frozen brain tissue and analyzed by next generation sequencing (NGS). Large fragments of an astrovirus genome were identified, the strain come from a clade distant to astroviruses screened by standard RT-PCR. A specific RT-PCR was designed against this strain of astrovirus and was positive on the biopsy sample but not on previous and concomitant CSF samples. Patient received high doses of intravenous immunoglobulin (IVIG), 3 bolus of methylprednisolone and is currently under daily ribavirin and weekly pegylated interferon (PEG-IFN) alpha-2b. Follow-up is too short to evaluate the efficacy of these therapeutic intervention.
Conclusion Astrovirus should be considered as a possible cause of encephalitis in XLA even if conventional RT-PCR is negative, specific RT-PCR with primers aimed at amplifying this new variant should be performed. Ribavirin plus PEG-IFN could be a possible therapy associated to high-dose of IVIG in Astrovirus-associated encephalitis in XLA.
ESID-0447 Clinical and Phenotypic Characterization of a Cohort of Argentinean Common Variable Immune Deficiency (CVID) Patients
M.I. Gaillard 1, M.S. CALDIROLA1, D. Di Giovanni1, A. Gomez Raccio1, M. Esnaola Azcoiti1, I. Moreira1, A. Lucas1, G. Seminario1, L. Regairaz2, L. Bezrodnik3
1Clinical Immunology Group, R. Gutierrez Children Hospital, CABA, Argentina
2Clinical Immunology Group, Immunology Group, CABA, Argentina
3Clinical Immunology Group, R. Gutierrez Children Hospital-Immunology Group, CABA, Argentina
CVID is characterized by hypogammaglobulinemia, defective antibody responses and recurrent infections. It is associated with an increased susceptibility to autoimmune disorders, lymphoproliferation, granulomas and malignancies. It´s usually diagnosed in second/third decade of life, but a proportion of children with hypogammaglobulinemia develop CVID. Objetives: Clinical and phenotypic characterization of a cohort of CVID patients. Materials and Methods: 50 patients with CVID (ESID criteria), male:28/female:22.Clinical and laboratory data were collected retrospectively from medical records. According to the age at diagnosis, we divide in 2 groups: G1 (4-11 years) n=22, G2 (>11 years) n=28. We analized B compartment (Bc): transitional, naïve, IgM+memory, class switched memory, and plasma cells. TCD4 compartment: naïve and memory. Results: Median age at onset of disease was 3.04 years (0.16-38.56) and 29/50 (58%) had their first symptom before 5 years. Median age at diagnosis was 14.84 years (3.89-57.83). Infections were reported in 98% of patients, autoimmunity in 28%, lymphoproliferation in 18%, Granulomas in 8% and malignancies in 8%. G2 have more autoimmunity than G1: (39% vs 14%), malignancies (14% vs 0%) and granulomas (11 vs 5%). Regarding laboratory: no differences in memoryTCD4 (CD45RO) G1:51.9±18.2/G2:61.4±17.0. Bc (EUROclass criteria) altered in 73% G1 and 48% G2. 27% of G1 associated complications vs 55% of G2. Conclusion: Infections have been the most frequent early onset of CVID before 5 years. 44% of our cohort had been diagnosed during the first decade of life. Even though the proportion of B compartment alterations in youngers was higher, complications were more frequent in the older group.
ESID-0587 LRBA Deficiency: Clinical, Immunologic and Genetic Characterization
L. Gamez-Diaz 1, D. August1, M. Kanariou2, S. Seneviratne3, C. Speckmann1, M. Seidel4, M. Noriko5, T. Morio5, M. Jordan6, P. Stepensky7, B. Grimbacher1
1Center for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany
2Department of Immunology, Aghia Sophia Children's Hospital, Athens, Greece
3UCL Centre for Immunodeficiency, Royal Free Hospital Foundation Trust, London, United Kingdom
4Department of Pediatrics and Adolescent Medicine, Medical University Graz, Graz, Austria
5Department of Pediatrics and Developmental Biology Graduate School, Tokyo Medical and Dental University, Tokyo, Japan
6Cincinnati Children's Hospital Medical Center, University of Cincinnati Medical School, Cincinnati, USA
7Pediatric Hematology-Oncology and Bone Marrow Transplantation, Hadassah Hebrew University Hospital, Jerusalem, Israel
LRBA deficiency is caused by loss-of-protein-expression of LRBA, which can be due to either homozygous or compound heterozygous mutations in LRBA gene. The clinical spectrum of LRBA deficiency has become very variable. Therefore, we have started to collect and characterize a world-wide LRBA deficiency cohort since 2012. Based on our inclusion criteria, we have recruited 107 patients who were tested for LRBA protein expression by either immunoblotting or flow cytometry. Twenty-five patients lacked LRBA protein. Out of the 25, twenty-two had been sequenced by exome sequencing, next generation sequencing or Sanger sequencing. Eight patients had either homozygous or compound heterozygous mutations in the LRBA gene whereas four had mutations in a different gene (unpublished). The remaining 11 LRBA-deficient patients had only common variants within the coding region of LRBA. The clinical manifestations of our 21 novel diagnosed LRBA deficiency patients and the 11 previously published cases, we observed 53% female and 56% consanguinity. The most frequent age of onset was before the age of 2 years (45%). The main clinical manifestations were hypogammaglobulinemia (85%), autoimmunity (73%), recurrent infections (60%) and inflammatory bowel disease (45%). Laboratory findings showed normal counts of NK and total T cells in 93% of LRBA-deficient patients. In contrast, B cell counts were reduced in 60% of patients. In conclusion, here we present a summary of the clinical phenotype, laboratory and genetic findings of 32 LRBA- deficient patients in order to facilitate the early diagnosis of LRBA deficiency.
ESID-0593 Rapid Diagnosis Test for LRBA Deficiency
L. Gamez-Diaz 1, E. Sigmund1, B. Grimbacher1
1Center for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany
LRBA deficiency is a primary immunodeficiency (PID) caused by the loss-of-protein-expression of LRBA. Diagnosis of LRBA deficiency has relied on immunoblotting of stimulated PBMCs and/or genome sequencing approaches that are expensive and time-consuming. LRBA deficiency is a quite severe PID with early manifestations, bad quality of life and a very much reduced life expecting. Therefore the development of an accurate, rapid, sensitive and cost-effective diagnostic test is needed. Here, we present a flow cytometry assay as an alternative tool for the diagnosis of LRBA deficiency. In addition, we show the best timing and stimulus for LRBA expression in the different lymphocytes subsets. Finally, we compared the expression of LRBA before and after HSCT. Following stimulation, permeabilization and fixation of PBMCs, intracellular expression of LRBA was measured using flow cytometry. A time- course of LRBA expression upon several stimuli revealed that T lymphocytes showed the highest expression upon stimulation with PMA + Ionomycin for 24 hours whereas IgM+CpG for 2 days was the best time and stimulus for B lymphocytes. Next, we compared flow cytometry against immunoblotting in EBV cells and PBMCs from a healthy control and LRBA-deficient patient. LRBA expression was undetectable in cells from patient contrasting to the control. In conclusion, here we provide an alternative test for the screening of LRBA deficiency in patients with unknown genetic defect and child-onset hypogammaglobulinemia, autoimmune manifestations, inflammatory bowel disease, lymphoproliferative syndrome and recurrent infections.
ESID-0284 Novel BTK Mutations in Patients with X-Linked Agammaglobulinemia (XLA)
E. García García 1, A. Aponte López2, L. Berrón Ruíz3, T. Staines Boone4, M. Yamazaki Nakashimada3, F. Espinosa Rosales3, G. López Herrera3
1UNIDAD DE INVESTIGACIÓN EN INMUNODEFICIENCIAS, INSTITUTO NACIONAL DE PEDIATRÍA/UNIVERSIDAD AUTÓNOMA METROPOLITANA, MÉXICO, Mexico
2UNIDAD DE INVESTIGACIÓN EN INMUNODEFICIENCIAS, UNIVERSIDAD AUTÓNOMA METROPOLITANA, MÉXICO, Mexico
3UNIDAD DE INVESTIGACIÓN EN INMUNODEFICIENCIAS, INSTITUTO NACIONAL DE PEDIATRÍA, MÉXICO, Mexico
4UNIDAD DE INVESTIGACIÓN EN INMUNODEFICIENCIAS, MÉDICA DE ALTA ESPECIALIDAD NÚMERO 25 DEL INSTITUTO MEXICANO DEL SEGURO SOCIAL (UMAE-IMSS). MONTERREY NL., MÉXICO, Mexico
Introduction XLA is a primary immunodeficiency characterized by a defect in early B cell maturation, with absent peripheral B cells and lack of immunoglobulins in sera. XLA is due to mutations in BTK (Bruton´s Tyrosine Kinase), which is involved in the signaling through the B cell receptor.
Methods This study includes patients with clinical diagnosis of XLA, peripheral mononuclear cells (PBMCs) were obtained from total blood and Btk expression by Western-Blot were performed. Additionally, RNA was purified and cDNA was obtained from all patients. BTK transcript was amplified by PCR using four pairs of primers, the PCR products were cloned and automated sequencing was performed. Sequences obtained were compared with a reference sequence ENST00000308731 (www.ensembl.org/).
Results Protein expression was absent in all patients, mutation analysis showed that two patients had a change c.G1056A, affecting the SH2 domain of Btk, causing an amino acid change R288Q. In a third and fourth patient, c.C1776G and c.C1983T, affecting the kinase domain, protein changes are R525G and P597L, respectively. In two other patients an frameshift deletion of exons 10-11 was found, introducing a premature stop codon. Finally, c.T857C cause an amino acid change L222P in SH3 domain, this domain has been reported to be refractory to missense mutations being important to evaluate the possible implications of this change in protein functionality as L222 residue is located next to an important phosphorylation site (Y223).
Conclusions Mutations described are novel, affect protein expression, indicating the importance of these mutations in protein stability.
ESID-0420 Clinical Picture and Treatment of 2212 Patients with Common Variable Immunodeficiency (CVID)
B. Gathmann1, N. Mahlaoui2, K. Warnatz1, T.W. Kuijpers3, D. Guzman4, P. Soler-Palacín5, T. Witte6, J. Litzman7, S. Borte8, D. Kumararatne9, C. Feighery10, H. Longhurst11, M. Helbert12, A. Szaflarska13, A. Sediva14, B.H. Belohradsky15, A. Jones16, U. Baumann17, I. Meyts18, N. Kutukculer19, P. Wågström20, N.M. Galal21, J. Roesler22, E. Farmaki23, N. Zinovieva24, P. Ciznar25, E. Papadopoulou-Alataki26, K. Bienemann27, S. Velbri28, B. Grimbacher 1
1Center for Chronic Immunodeficiency (CCI), University Medical Centre Freiburg and University of Freiburg, Freiburg, Germany
2Centre de Référence Déficits Immunitaires Héréditaires (CEREDIH), Hôpital Universitaire Necker-Enfants Malades AP-HP, Paris, France
3Dutch Working Party for Immunodeficiencies (WID), WID, Amsterdam, Netherlands
4UCL Medical School Royal Free Campus and Royal Free Hospital, NHS Foundation Trust, London, United Kingdom
5Pediatric Infectious Diseases and Immunodeficiencies Unit., Hospital Universitari Vall d'Hebron. Universitat Autònoma de Barcelona, Barcelona, Spain
6Clinic for Immunology and Rheumatology, Medical University Hanover, Hannover, Germany
7Dept. Clin. Immunol. Allergol Faculty of Medicine, Masaryk University and St. Anne's University Hospital, Brno, Czech Republic
8Children's Hospital, Municipal Hospital 'St Georg' Academic Teaching Hospital of the University of Leipzig, Leipzig, Germany
9Department of Clinical Biochemistry and Immunology, Cambridge University Hospitals NHS Foundation Trust, Cambridge, United Kingdom
10Department of Immunology, St. James's Hospital, Dublin, Ireland
11Department of Immunology, Barts Health NHS Trust, London, United Kingdom
12Central Manchester and Manchester Children's University Hospitals NHS Trust, Manchester, Manchester, United Kingdom
13Department of Clinical Immunology and Transplantology, Jagiellonian University Medical College Children University Hospital, Cracow, Poland
14Department of Immunology, Charles University and University Hospital Motol, Prague, Czech Republic
15Dr v. Haunersches Kinderspital, Ludwig Maximilians University, Munich, Germany
16Institute of Child Health, Great Ormond Street Hospital, London, United Kingdom
17Immunology Unit Paediatric Pulmonology Allergy and Neonatology, Hanover Medical School, Hannover, Germany
18Department of Pediatrics, University Hospital Gasthuisberg, Leuven, Belgium
19Department of Pediatric Immunology, Ege University Faculty of Medicine, Izmir, Turkey
20Department of Infectious Diseases, County Hospital Ryhov, Jönköping, Sweden
21Primary Immunodeficiency Clinic Department of Pediatrics, Cairo University, Cairo, Egypt
22Dept. of Pediatrics, University Hospital Dresden, Dresden, Germany
23Pediatric Immunology and Rheumatology Referral Centre First Dept of Pediatrics, Ippokration Hospital Aristotle University of Thessaloniki, Thessaloniki, Greece
24Research and Clinical Centre for Pediatric Hematology Oncology Immunology, Moscow, Moscow, Russia
251st Pediatric Department Faculty of Medicine, Comenius University and Children University Hospital, Bratislava, Slovakia
26Fourth Department of Pediatrics Papageorgiou Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
27Pediatric Oncology Hematology and Clinical Immunology, Medical Faculty Heinrich Heine University, Düsseldorf, Germany
28Tallinn Children's Hospital, Tallinn, Tallinn, Estonia
Background: Common variable immunodeficiency (CVID) is an antibody deficiency with an equal gender distribution and a high variability in its clinical presentation. Main features include respiratory tract infections and its associated complications, enteropathy, autoimmunity, and lymphoproliferative disorders.
Objective: This study analyzes the clinical presentation, the association between clinical features, and differences and effects of immunoglobulin (Ig) treatment in Europe.
Methods: Data on 2212 CVID patients from 28 medical centers contributing to the European ESID Database were analyzed retrospectively.
Results: Early disease onset (
Conclusion: CVID patients are being differently managed throughout Europe, affecting various outcome measures. Clinically, CVID is a truly variable antibody deficiency syndrome.
ESID-0270 RAG Mutations in Adults with Predominant Deficiency in T-Independent Antibody Production
C.B. Geier 1, A. Piller1, A. Linder1, M.M. Eibl1, H.M. Wolf1
1Immunologische Tagesklinik, Immunologische Tagesklinik, Vienna, Austria
We describe two unrelated adult patients, with predominantly antibody deficiency. Patient A had moderate hypogammaglobulinemia, patient B had normal IgG an IgG1 serum levels but decreased IgA, IgM, IgG 2, 3, and 4. Both patients presented intact production of T cell dependent IgG antibody but defective T-independent IgG response to bacterial polysaccharide antigens (eg. Pn23). The patients experienced recurrent pneumonia, sinusitis, otitis media and one patient recurrent cutaneous vasculitis. Following intravenous immunoglobulin substitution therapy the patients’ susceptibility to infection improved.
CD3+ T-cell count of both patients was within the normal range, as well as CD4+, CD8+ T-cells and CD56+ NK-cells. However naïve CD4+-CD45RA+ T cells were significantly reduced in both patients. We employed TCR Vb spectratyping to assess the clonality of the TCR repertoire in patient A and observed no restriction of the TCR repertoire.
Resequencing of a multitude of known primary immunodeficiencies genes resulted in the detection of compound heterozygous mutation in RAG 1 in patient A and RAG 2 in patient B. We identified 2 novel hypomorphic mutations, resulting in aberrant gene expression of RAG1 and altered gene function of RAG 2, as known form the mouse model.
In the present study we describe selective antibody deficiency as a novel immunological phenotype of leaky RAG1 and 2 deficiencies. Thus our findings contribute to the known complexity of phenotypic manifestations of hypomorphic RAG mutations.
ESID-0276 TLR9 Signaling in Patients with Ectodermal Dysplasia and Immunodeficiency Associated with Nuclear Factor Essential Modulator (NEMO) Mutations
G. Giardino 1, R. Naddei1, E. Cirillo1, V. Gallo1, T. Esposito1, F. Fusco2, I. Quinti3, M.V. Ursini2, R. Carsetti4, C. Pignata5
1Department of Translational Medical Science, Università "Federico II", Naples, Italy
2International Institute of Genetics and Biophysics, CNR, Naples, Italy
3Department of Molecular Medicine, La Sapienza University, Rome, Italy
4Research Center, Ospedale Pediatrico Bambino Gesù (IRCCS), Rome, Italy
5Department of Translational Medical Sciences, Università "Federico II", Naples, Italy
Background: Hypohidrotic ectodermal dysplasia (HED) is a group of disorders of ectodermal tissues, resulting from mutations in the ectodysplasin-A (EDA) pathway. Hypomorphic mutations in the NF-κB essential modulator (NEMO) result in HED with immunodeficiency (HED-ID, OMIM 300291), characterized by susceptibility to encapsulated bacteria, mycobacteria, and herpes virus infections.
Objective: To analyze B-cell compartment and TLR9 signaling in HED-ID patients.
Methods: Two HED-ID patients and a patient with HED caused by EDA gene mutation (XLHED) to confirm the implication of NFkB in this pathway were studied.
Results: In HED-ID, differently from XLHED, only few B cells acquired the phenotype of IgM memory and differentiated into plasma cells upon TLR9 stimulation. Memory B cells did not produce IgG and IgA, and only small amounts of IgM in vitro.
Conclusion: In HED-ID patients, TLR9 signaling is abnormal, in keeping with the lack of IgM memory B cells and natural antibodies. In individuals at a high risk of developing pneumococcal diseases, increased susceptibility to Streptococcus pneumonia infections and poor response to polysaccharide antigens have been associated with the lack of IgM memory B cells, required for the T-independent response toward encapsulated bacteria, whose differentiation from transitional B cells is under TLR9 control. This finding helps explain the susceptibility to infections by encapsulated bacteria.
ESID-0556 Influence of WAS Gene Alterations on the Maturation of the B Cell Compartment
G. Lanzi 1, D. Moratto1, L. Gazzurelli1, C. Mazza1, L.D. Notarangelo2, L.D. Notarangelo3, S. Giliani1
1c/o Spedali Civili, A.Nocivelli Institute for Molecular Medicine, Brescia, Italy
2c/o Spedali Civili, Pediatric Oncoematology, Brescia, Italy
3Childrens' Hospital, Harvard medical School Division of Immunology, Boston, USA
The Wiskott-Aldrich syndrome protein (WASp) is a multifunctional domain protein expressed in hematopoietic cell lineages. Mutations in WASp coding sequence are causative of X-linked disorders, among which Wiskott-Aldrich syndrome (WAS) and X-linked thrombocytopenia (XLT) are the most frequent. Both these diseases are characterized by microthrombocytopenia, eczema and immunological defects, affecting also humoral immunity.
Our analysis on 40 XLT/WAS patients showed a reduction of unswitched memory cells (IgD+CD27-), while generation of switched cells (IgD-CD27+) correlate with the severity of WASp mutations. Moreover, the presence of this impairment was confirmed in two different in vivo systems in which wt and mutated cells coexist. In fact, we observed a progressive increase of the ratio between WASp+ and WASp- cells during B-cell differentiation both in female carriers of WAS mutations and in WAS patients who display a mixed chimerism in the B cell compartment after hematopoietic cell transplantation.
Despite the inability to form an adequate memory B-cell compartment, we could demonstrate that the percentage of effector B-cells (CD20-CD38bright) in our cohort of WAS/XLT patients was in the normal range when not increased. The hypothesis of a normal capacity to generate Ig secreting cells is supported also by the generation of an adequate amount of WASp- plasma cells (CD138+) in female carriers of WAS mutations in an in vitro differentiation experiment.
We demonstrate that WAS gene alterations are detrimental for the differentiation into memory B cells, while they do not seem to influence differentiation into effector cells, likely generated through a WASp independent pathway.
ESID-0504 Study of Interstitial Lung Disease in Patients with Primary Antibody Deficiency (STILPAD)
S. Goldacker 1, M. Malphettes2, S. Seneviratne3, H. Longhurst4, J.F. Viallard5, E. Scharbatke6, C. Neumann7, H.P. Tony6, B. Grimbacher1, E. Ocksenhendler2, C. Winterhalter1, A. Uhlmann1, V. Reiser8, W. Vach8, A. Prasse9, G. Kayser10, H. Tiddens11, K. Warnatz1
1Centre for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany
2Département d'Immunologie, Hôpital Saint-Louis, Paris, France
3Department of Clinical Immunology, Royal Free Hospital, London, United Kingdom
4Department of Immunology and Department of Haemato-Oncology, Barts Health NHS Trust The Royal London Hospital, London, United Kingdom
5Department of Internal Medicine, Centre Hospitalier Universitaire of Bordeaux, Bordeaux, France
6Rheumatology/Clinical Immunology, University Hospital of Würzburg, Würzburg, Germany
7Division of Rheumatology and Clinical Immunology, University of Munich, München, Germany
8Institute of Medical Biometry and Medical Informatics, University Medical Center Freiburg, Freiburg, Germany
9Department of Pneumology, University Medical Center Freiburg, Freiburg, Germany
10Center for Pathology, University Medical Center Freiburg, Freiburg, Germany
11Department of Pediatric Respiratory Medicine and Department of Radiology, Erasmus Medical Center-Sophia Children's Hospital, Rotterdam, Netherlands
Objective: Interstitial lung disease (ILD) can severely impact morbidity and mortality in patients with common variable immunodeficiency (CVID). Little is known about the immunopathology, natural course, potential biomarkers and optimized therapeutic approaches. To address these questions we initiated a non-interventional, prospective, 5-year international multicenter study (NIS) in adult patients with CVID and ILD. Methods: STILPAD is a prospective 5-year NIS. Here we present the analysis of retrospective data. CVID Patients diagnosed according to ESID criteria and ILD defined by chest CT were evaluated at baseline visit for clinical history, previous lung function, serum sIL2R levels, chest CT scans, bronchoalveolar lavage values, pulmonary histology, IgG substitution therapy and use of immunosuppressants. Chest CTs and histologies are scored by blinded experts.
Results: 149 patients were recruited from 14 centers in Germany, France and the UK. This retrospective analysis includes data from 89 patients. In 71 patients longitudinal lung function parameters are available. 53 patients have sIL2R values. 277 chest CTs are analysed. 55 patients underwent histological workup. 29 patients had no immunosuppression, 40 patients received exclusively steroid treatment and 28 patients were on DMARDs or biologicals.
Conclusion: STILPAD recruited 149 CVID patients with ILD. The retrospective analysis will provide important information on the heterogeneity of the highly relevant complication. The distribution of patients with and without therapy will allow a first comparison between the different groups. A better understanding of clinical and diagnostic presentation and a detailed acquisition of the impact on management are mandatory for future interventional trials.
ESID-0532 Clinical Experience in X-Linked Agammaglobulinemia from Mexican Patients
M. Gonzalez-Serrano 1, M.A. Gonzalez-Cruz2, P. Rubio-Mortera1, D. Mogica-Martinez3, M.A. Yamazaki-Nakashimada4, L. Berron-Ruiz1, G. Lopez-Herrera1, S.E. Espinosa-Padilla1, F.J. Espinosa-Rosales1
1Immunodeficiency Research Unit, Instituto Nacional de Pediatría, Mexico City, Mexico
2Pediatrics, Instituto Nacional de Pediatría, Mexico City, Mexico
3Allergy, Medical Center “La Raza” IMSS, Mexico City, Mexico
4Immunology, Instituto Nacional de Pediatría, Mexico City, Mexico
We included 40 patients with XLA dianosis (ESID criteria) from 38 unrelated families.
Results are reported as median and range.
Age at the first symptom, diagnosis and delay in diagnosis were 12 (1-69), 66.5 (1-69) and 49.5 (0-182) months respectively. The earlier diagnosis was at birth related with a sibiling previously diagnosed. Male affected in the family was presented in 65%.
Infections were: pneumonia 85%, sinusitis 70%, otitis media 57.5%, meningitis 27.5%, septic arthritis 25%, celulitis and abscess 12.5%, conjunctivitis 12.5%, sepsis 7.5% and UTI 7.5%. Other infections were osteomyelitis, giardiasis, varicela, measles and hepatitis. Gastrointestinal manifestations were presented in 22.5%, 7.5% with malabsortion syndrome.
Tonsils and lymph nodes were absent in 70 and 65% respectively.
Complications were: sinopulmonary syndrome 60%, bronchiectasis 50%, chronic sinusitis 30%, chronic otitis media 20%, mastoiditis 7.5% and failure to thrive 22.5%.
Autoimmune manifestations were: dermatomyositis like syndrome (1/40), reactive arthritis (1/40) and hemolitic anemia (1/40).
Seric immunoglobuline levels (mg/dL) at diagnosis were IgG 40 (0-282), IgM 16.8 (0-60), IgA 8 (0-27). Porcentaje of B cells on peripheral blood was 0.11 (0-1.9). 52% of the patients (21/40) had Btk determination by flow cytometry, Btk MFI on monocytes was of 15.86 (0-99).
Neutropenia was presented in 17%, thrombocytopenia was presented in 10%, both associated with infections. Anemia was presented in 30%, one case hemolitic.
Agents isolated were: Streptococcus pneumoniae 22.5%, HiB 15%, Giardia 10%, Pseudomona aeruginosa 10%, Aspergillus fumigatus 10%, Serratia marcescens 7.5%. Other agents were Moraxella catarrhalis, Neisseria spp, rotavirus, Ascaris lumbricoides and Bordetella pertussis.
ESID-0735 A Patient with Bruton Agammaglobulinemia with High IGE Level
I. Reisli 1, S. Guner1, E. Sayar Hazar1, B. Gokturk1, M. Kirac1, M. van der Burg2
1Pediatric Allergy and Immunology, Meram Medical School, Konya, Turkey
2Immunology, Erasmus University, Rotterdam, Netherlands
Bruton agammaglobulinemia (XLA), is the prototypic humoral immunodeficiency. Function-loss mutations in BTK lead to a block in B-cell maturation, a near total absence of B cells in the periphery, and pan-hypogammaglobulinemia. Although patients begin to suffer recurrent infections by age 1 year, with antibiotics and good hygiene it is not uncommon to delay suspicion of the diagnosis until well into mid-childhood. Seven years old boy admitted to hospital with bronchiolitis and pericardial effusion. He had history of recurrent pneumonia and otitis media since he was two years old. There was no consanguinity between his parents. It was determined the high IgE level while IgG, IgA, IgM levels, isohemagglutinin titer were very low. In the flow cytometric analysis CD19 + B lymphocyte was absent. In his genetic analysis, it was identified a mutation in the BTK gene. After IVIG therapy, IgE levels were decreased above to the 100 IU/ml. With this patient, we would like to discuss how and where these IgE molecules were produced.
ESID-0279 Clinical Case of Agammaglobulinemia in Identical Twins
M. Guseva 1, A. Semenov2, N. Kalinina3, A. Totolian2
1consulting center, Saint-Petersburg Pediatrical Medical Academy, St-Petersburg, Russia
2consulting center, Saint-Petersburg Pasteur Institute, St-Petersburg, Russia
3consulting center, Russian Center of Emergency and Radiation Medicine, St-Petersburg, Russia
Clinical case of agammaglobulinemia in identical twins
Interesting clinical case was observed in St-Petersburg PID center. Identical siblings S.A. and S.B. (born at 2008), diagnosed in 07. 2013. Clinical symptoms are discordant and more severe in S.B. patient (recurrent otitis, bronchitis, gonasynovitis, coxarthritis, osteomyelitis with 3 surgeries, two times pneumonia per year) than in S.A. twin (recurrent otitis and arthritis). Laboratory examination shows identical levels of IgA, IgM and IgG (0.01; 0.02; and <0.004 g/l respectively) as well as multicolor flow cytometry data (see Figure 1) that demonstrate absence of B-cells (CD 45+/19+).
Post-vaccine IgG are 0,003 IU/ml for anti-diphtheria antitoxin IgG (susceptible to diphtheria) and 1:10 titer of tetanus antitoxic IgG in both twins.
All 19 exons of btk-gene were amplified by PCR and analyzed with SSCR and HRM techniques. The mutation was founded in exon 17 position 1877 C-> T. It represents missense mutation in codon replacement CGG-> TGG (Arg-> Trp). Genetic analysis proved agammaglobulinemia diagnosis.
After the examination and diagnosis of patients was initiated regular intravenous immunoglobulin replacement therapy in the mode of the loading dose and then in a supporting mode.
Adequate therapy significantly improved the clinical condition of patients. More episodes of inflammatory diseases not observed. Twins began to attend public school. The quality of life for these patients was improved. Observation of the dynamics will continue.
Results: Even genetically and immunologically identical patients may show pretty different clinical symptoms.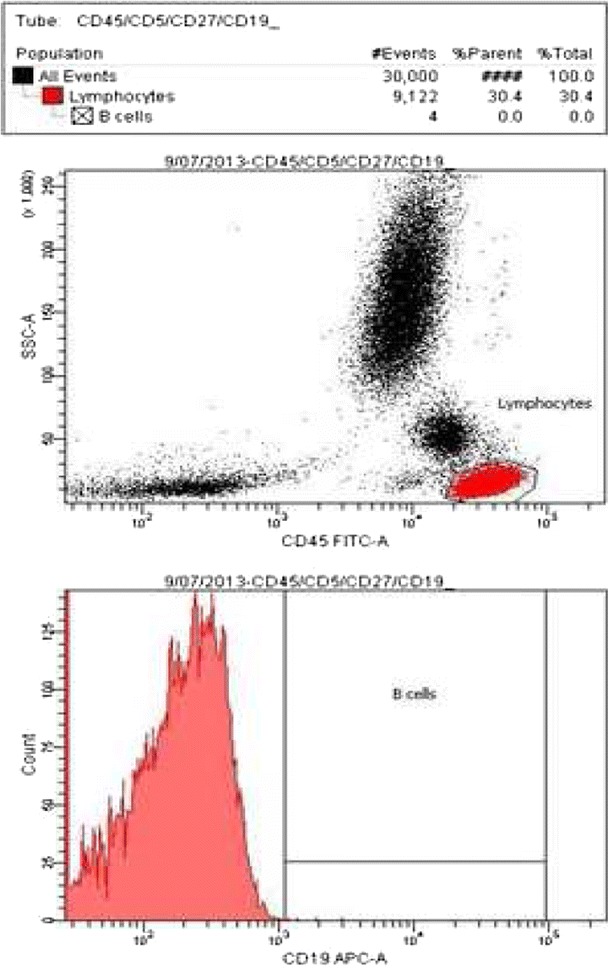
ESID-0707 Pneumococcal IGG-, IGA- and IGM-Responses Allow Further Distinction of Patients with Hypogammaglobulinemia
L. Hanitsch 1, J.F. Mieves1, N. Unterwalder2, C. Meisel2, K. Wittke1, U. Kölsch2, P. Grabowski1, H.D. Volk1, C. Scheibenbogen1
1Charité University Hospital, Institute for Medical Immunology, Berlin, Germany
2Immunology Department, Labor Berlin, Berlin, Germany
In 28 patients with combined hypogammaglobulinemia (reduced IgG, IgA and/or IgM) we assessed the specific pneumococcal (PcP) IgG-, IgA- and IgM-responses 4-6 weeks after vaccination with Pneumovax23 using a commercially available ELISA (Binding Site) and correlated clinical and immunophenotyping data. 7/28 patients (group 1) had no specific IgG-, IgA- or IgM-response, fulfilling CVID criteria. 8/28 patients (group 2) had a >4-fold increase in IgG- as well as in IgA- und IgM response. 13/28 patients showed an intermediate IgG response with a 2–4–fold increase. Of those 13 patients, 6 patients (group 3) showed an impaired specific IgM response (<2-fold) and in 4/6 cases an impaired specific IgA-response. The remaining 7 patients (group 4) had a specific IgA- and IgM increase of >2-fold. Autoimmunity is found at similar rates in groups 1,2 and 3 (1/2/3/4: 43%/25%/33%/0%), splenomegaly is predominant in group 1 (non-responder) and group 3 (IgM-nonresponder) (1/2/3/4: 29%/0%/67%/14%). Infectious rates were higher in group 1 and 3 (recurrent sinusitis groups 1/2/3/4: 43%/12%/83%/14%; recurrent pneumonia 14%/0%/17%/0%). Regarding B cell phenotypes group 1 (non-responder) express the lowest percentage of switched memory and MZ-like B cells and the highest percentage of transitional B cells. Both non-responder and IgM-non-responder have a higher percentage of activated B cells. Thus patients with intermediate IgG but impaired IgM response have a similar clinical course as patients with no PcP-IgG response. Based on these data we suggest to assess specific PcP-IgM/IgA response in patients with intermediate PcP-IgG-response, which may help to define subgroups with distinct clinical course.
ESID-0286 Phenotype Variability in Patients with Selective IGA Deficiency
S.I. Iurian 1, S. Iurian2, D. Fintina3
1Pediatric Clinic, "Lucian Blaga" University Pediatric Hospital, Sibiu, Romania
2Pediatric Hospital Pediatric Clinic, Clinical Laboratory, Sibiu, Romania
3Pediatric Clinic, Pediatric Hospital, Sibiu, Romania
Background. The most frequent primary antibody deficiency is selective IgA deficiency (SIgAD). Aims. Authors emphasize the phenotype diversity among SIgAD patients included in our casuistry. Methods. Authors analyzed clinical and biochemical peculiarities of patients diagnosed with SIgAD. They’ve been evaluated concerning previous respiratory and gastro-intestinal diseases severity (necessity for intravenous antibiotics and/or intensive care measures). Serology investigations performed: complete blood count, evaluations for immunoglobulin isotypes, IgG subclasses, total IgE, specific IgE for cow’s milk precipitins. Autoimmune status estimation included antibodies against: red blood cells, basement membrane, smooth muscle, thyroglobulin, thyroid microsomal antigens, transglutaminase(IgG) / endomysium and nuclear proteins. Authors accomplished microbiological tests for patients with sinopulmonary infections and analyzed stools in order to exclude Giardia/ Cryptococcus infections. Brochiectasis susceptibility justified chest X-ray accomplishing. Study was approved by Institutional Review Board that waived the need for informed consent. Results. Among 9 patients followed in our clinic, 3 patients were considered as asymptomatic. The others were diagnosed with: recurrent knee arthritis in context of Epstein-Barr virus reactivation confirmed by quantitative PCR for DNA-EBV in serum (1 patient), juvenile recurrent parotitis (1 patient), celiac disease (2 patients), giardiasis (2 patients), asthma and allergic rhinitis (1 patient), cow milk allergy (1 patient). Severe sinopulmonary infections/ purulent otitis were identified for all symptomatic cases. One patient complained about tonsil phlegmon. Conclusions. 1.In spite of small SIgAD patients group, authors revealed a quite large range of clinical manifestations: from asymptomatic status to severe infections; 2.Four symptomatic patients have presented more than 1 clinical feature.
ESID-0010 Diagnosis Difficulties in a Case with Hyper-IGM Syndrome
S. Iurian 1, C. Picard2, S. Kracker3, S. Iurian4, D. Fintina5
1Pediatric Clinic, "Lucian Blaga" University Pediatric Hospital, Sibiu, Romania
2Centre d'étude des Déficits Immunitaires, Hôpital Necker-Enfants Malades, Paris, France
3Laboratoire de Lympho-Hématopoïèse Humaine, Institut Imagine, Paris, France
4Clinical Laboratory, Pediatric Hospital, Sibiu, Romania
5Pediatric Clinic, Pediatric Hospital, Sibiu, Romania
Background. Hyper-IgM syndrome (HIGM) is characterized by diminished serum levels of immunoglobulins G and A and normal or elevated levels of immunoglobulin M. HIGM syndrome genetic background includes variable genetic defects (X-linked type is the most common).
Aims. Authors emphasize diagnosis peculiarities in an infant with hyper-IgM phenotype.
Methods. Authors present a 9 month-old male infant admitted for airway infection symptoms.
Family history: no consanguinity.
Clinical exam: fever, impaired nutritional status (weight below 3rd percentile), coughing, lung crackles, hepato-splenomegaly, oral candidiasis, purulent secretions at external ear canals, severe hypotonia.
Results. Hematological evaluation revealed: anemia, B lymphopenia, CD8 lymphopenia, NK high levels. Serum immunological profile: low values for IgA/IgG (below 3rd percentile) and high IgM value. Microbiological tests identified Staphylococcus aureus in ear secretions. Quantitative polymerase-chain-reaction didn’t detect DNA-Epstein-Barr virus in serum.
Genetic background assessment included: CD40/CD40L deficiencies (normal expression of CD40-L on T cells and CD40 on B cells), NEMO deficiency (no ectodermal dysplasia clinical features), ataxia-teleangiectasia (serum α feto-protein normal level). Patient wasn’t evaluated for UNG/AID deficiencies, SH2D1A mutation. Suspicion for PIK3CD mutation justified gene testing that excluded E1021K hot-spot mutation involved in gene gain function.
Evolution: in spite of immunoglobulin replacement therapy (IRT), patient continued suffering severe respiratory infections; IgM serum level have progressively increased to 20 times higher than normal; in addition, period of time between IRT also increased.
Conclusions.
1.Authors described a patient with HIGM phenotype and unknown molecular defect;
2.Even though the patient rarely necessitated IRT, IgM serum values have gradually increased.
ESID-0663 Autoimmune Lymphoproliferative Syndrome Reveals a Critical Role of FAS in Selection of Human Memory B Cell
A. Janda 1, K. Schwarz2, M. van der Burg3, H. Ijspeert3, M.R. Lorenz2, W. Vach4, M. Elgizouli1, K. Pieper1, P. Fisch5, J. Hagel1, R. Lorenzetti1, M. Seidl5, J. Rösler6, F. Hauck7, C. Speckmann8, A. Rensing-Ehl1, S. Ehl1, H. Eibel1, M. Rizzi1
1Center for Chronic Immunodeficiency (CCI), University Medical Center Freiburg, Freiburg, Germany
2Institute for Transfusion Medicine, University of Ulm, Ulm, Germany
3Department of Immunology, Erasmus MC University Medical Center Rotterdam, Rotterdam, Netherlands
4Institute of Medical Biometry and Medical Statistics, University Medical Center Freiburg, Freiburg, Germany
5Department of Pathology and Molecular Pathology, University Medical Center Freiburg, Freiburg, Germany
6Department of Pediatrics, University Clinic Carl Gustav Carus, Dresden, Germany
7Department of Pediatric Hematology and Oncology, Dr von Hauner Children's Hospital Ludwig-Maximilians-University, Munich, Germany
8Centre for Pediatrics and Adolescent Medicine, University Medical Center Freiburg, Freiburg, Germany
Autoimmune lymphoproliferative syndrome (ALPS) is characterized by lymphoproliferative disease, autoimmune cytopenias and increased susceptibility to lymphoid malignancies. Most patients harbor heterozygous germline or somatic mutations in FAS or a combination of both. While impaired FAS mediated apoptosis is associated with accumulation of atypical “double-negative” T cells, the impact of defective FAS signaling on B cells and the pathogenesis of autoimmunity remain unclear. While FAS has been shown to be essential for deletion of autoreactive B cells in the germinal center in murine models, evidence from humans is missing. We analyzed patients with somatic FAS mutation or germline FAS mutation plus somatic loss-of-heterozygosity (LOH) allowing comparison of B cells with impaired versus normal FAS signaling and determination of their distribution among naive and memory B cells within the same individual. A high prevalence of FAS mutated B cells within the memory compartment indicated defective FAS dependent deletion. Moreover, we found increased amount of sequences of the variable region of the immunoglobulin receptor associated with autoimmunity and higher antibody polyreactivity in the memory B-cell pool of the patients indicating impaired autoreactivity checkpoint in the germinal center. Our data confirm an important role of FAS in negative selection and provide insights into the pathogenesis of autoimmunity in ALPS patients.
Dr. Janda is a recipient of an unrestricted fellowship grant from ESID provided by Baxter.
ESID-0568 Measurement of Pneumococcal Polysaccharide Vaccine Responses for Immunodeficiency Diagnostics: Combined IGG Responses Compared to Serotype Specific IGG Responses
W.J.M. Janssen 1, A.C. Bloem2, P. Vellekoop3, G.J. Driessen4, M. Boes3, J.M. van Montfrans3
1LTI, Wilhelmina Children's Hospital University Medical Center Utrecht, Utrecht, Netherlands
2Medical Immunology, University Medical Center Utrecht, Utrecht, Netherlands
3Pediatric Immunology, Wilhelmina Children's Hospital University Medical Center Utrecht, Utrecht, Netherlands
4Department of Pediatrics subdivision of Pediatric Infectious Disease and Immunology, Erasmus MC, Rotterdam, Netherlands
Purpose: Two distinct methods for the quantification of PnPS vaccination responses are now in use for immunodeficiency diagnostics, those that involve IgG measurement of separate serotypes by a luminex method, and total IgG measurement of the Pn-PS vaccine specific serotypes by combined ELISA. We here compare the usefulness of the combined PnPS IgG response measurement with specific serotype IgG response measurement in a defined cohort of immunodeficiency patients and control patients.
Methods: Patients analysed were CVID or specific antibody deficient (SAD) and as controls we used children with recurrent infections without underlying immunodeficiency. The combined IgG responses and specific serotype IgG responses after PnPS vaccination were measured respectively by ELISA and Luminex.
Conclusions: Our data show that combined PnPS IgG response measurement can mask the presence of immunodeficiency in up to 42% of patients. The specific serotype method is superior in detection of Pn-PS vaccination responses and thereby for immunodeficiency diagnostics.
ESID-0549 Antigen-Specific IGA Titers After 23-Valent Pneumococcal Vaccine Differentiate Transient from Persistent Antibody Deficiency Disease
W.J.M. Janssen 1, S. Nierkens2, M. Boes3, J.M. van Montfrans3
1LTI, Wilhelmina Children's Hospital University Medical Center Utrecht, Utrecht, Netherlands
2Department of Medical Immunology, University Medical Center Utrecht, Utrecht, Netherlands
3Pediatric Immunology, Wilhelmina Children's Hospital University Medical Center Utrecht, Utrecht, Netherlands
In young children diagnosed with antibody deficiency, it can be difficult to prognosticate persistent immunodeficiency versus delayed maturation of humoral immunity. We hypothesized that vaccination-induced IgA and IgM anti-pneumococcal polysaccharide (PnPS) responses differentiate transient from persistent disease course.
We performed a retrospective cohort study on 67 serum samples of children (3-18 yrs) vaccinated with Pneumovax 23® during immunodeficiency screening. Fifty-one of 67 patients had antibody deficiency disease, and in 16/67 subjects with recurrent infections no diagnosis of immunodeficiency was made (control group). Of the 51 antibody deficient patients at initial screening, 15 subjects displayed transient and 36 persistent antibody deficiency. We measured pre- and post vaccination anti-PnPS IgM, IgA and IgG titers by luminex for 11 PnPS serotypes.
Controls had significantly higher IgM and IgA anti-PnPS responses than antibody deficiency patients. Delayed maturation of humoral immunity (transient antibody deficiency) was observed up to 6 years of age, however several of the 6-14 year old patients also displayed further maturation of humoral immunity. Across all ages, transient patients displayed significantly higher anti-PnPS IgA responses on individual serotypes than persistent antibody deficiency patients. IgM anti-PnPS titers were significantly higher in the transient disease group, but only in children < 6 years of age. The IgG anti-PnPS response of transient patients showed high variability on serotypes and could thus not be used to predict the further disease course.
Thus, anti-PnPS IgA responses can stratify transient versus persistent antibody deficiency disease in children. These data may be of use for immunodeficiency disease diagnostics.
ESID-0567 Silent Progression of Pulmonary Disease in Paediatric CVID Patients at Risk for Pulmonary Complications
W.J.M. Janssen 1, F.A.A. Mohamed Hoesein2, P.A. de Jong2, J.M. van Montfrans3
1LTI, Wilhelmina Children's Hospital University Medical Center Utrecht, Utrecht, Netherlands
2Radiology, University Medical Center Utrecht, Utrecht, Netherlands
3Pediatric Immunology, Wilhelmina Children's Hospital University Medical Center Utrecht, Utrecht, Netherlands
Pulmonary disease is frequently observed in common variable immunodeficiency (CVID) patients. We sought to determine the optimal follow-up interval for high resolution CT (HRCT) scanning for monitoring of pulmonary complications in CVID.
We retrospectively evaluated our institutional protocol for HRCT-scanning in CVID patients. In this protocol patients are scanned 3 or 5 years after initial diagnosis, depending on their risk for development of pulmonary complications. Pulmonary complications were scored using a previously published CVID specific scoring system (Ven et al, Chest 2010).
The current study describes the 3 year follow up scans in 13 'high risk' patients, with airway disease and/or interstitial lung disease (ILD), reduced memory B-cell percentages, active auto-immunity, and/or recurrent pneumonias despite prophylaxis.
Twelve out of 13 patients (92%) showed worsening of the pulmonary CT-scan score for at least 2 out of 6 lung segments. Conventional CT scan reports (i.e., without using the predefined scoring system) described deterioration of the pulmonary condition in only 6 out of 13 patients. The mean ILD score of the 13 patients was 7.5 at initial HRCT-scanning and 12.5 on the 3 years follow up scanning (p=0.0005). Two out of 13 CVID patients had active autoimmunity and 2 out of 13 patients progressed into combined immunodeficiency disease (CID) for which HSCT was necessary.
We conclude that in paediatric CVID patients with high risk for pulmonary disease, silent progression of pulmonary disease occurs frequently. Further follow up is needed, as well as intervention studies to prevent pulmonary damage in CVID patients.
ESID-0623 Agammaglobulinemia in Morocco
Z. Aadam1, S. El Atiqi2, L. Jeddane 2, I. Benhsaien2, J. Najib2, F. Ailal2, J. El Bakkouri3, A.A. Bousfiha2, H. Salih Alj1
1Laboratory of Biology and Health URAC34—Metabolic and Immunologic pathology Research Team, Faculty of Science of Ben M'sik King Hassan II University, Casablanca, Morocco
2Clinical Immunology Unit, Faculty of Medicine and Pharmacy, Casablanca, Morocco
3Laboratory of Immunology, Averroes University Hospital, Casablanca, Morocco
Agammaglobulinemia represents a major primary immunodeficiency. They are of X-linked transmission in 90% of cases ( Bruton 's disease), and autosomal recessive in 10% of cases. They are caused by a blockade in B cells maturation, leading to an inability to produce immunoglobulins.
The aim of our study was to describe the characteristics of these diseases and their treatment in Morocco.
Through the period from January 1997 to October 2013, our study collected 102 patients with antibody deficiencies, including 28 cases of agammaglobulinemia.
The distribution of these patients was 13 cases of genetically confirmed Bruton's disease and 15 cases of autosomal recessive agammaglobulinemia. The mean age at diagnosis was 4.5 years in Bruton 's disease , and 6.8 years in other agammaglobulinemia. Clinical manifestations were dominated by recurrent infections, mainly of respiratory tract, ENT, bone and joint and gastrointestinal tract.
Treatment consisted in the administration of intravenous immunoglobulin and antibiotic prophylaxis. Mortality was 25%, mainly by chronic respiratory failure complicating bronchiectasis for patients diagnosed late.
Our series is characterized by a delay in diagnosis, hence the importance of awareness among physicians, especially pediatricians, for early treatment and thus a better prognosis.
ESID-0604 Clinical and Immunological Characterization of Patients with Selective IgA Deficiency (SIgAD) in Colombia
J. Jessica L. Rojas 1, M. Marcela A. Moncada1, J. Julio César Orrego1, J. José Luis Franco1
1Antioquia, Group of Primary Immunodeficiencies-Universidad de Antioquia, Medellin, Colombia
Introduction: SIgAD is the most common immune abnormality of unknown etiology. Most individuals are asymptomatic however 10-20% exhibit heterogeneous clinical manifestations mainly of the respiratory and gastrointestinal tracts as well as atopy and autoimmunity. We describe the initial immunological characterization of 7 patients with SIgAD.
Immunologic workup: Data were collected from clinical and laboratory records of patients admitted until April of 2014, who met ESID criteria for SIgAD. We evaluated sIg and performed flow cytometry to phenotype lymphocytes in peripheral blood (PB) and to evaluate lymphoproliferation to PHA, CD3+CD28, CpG, SAC and IL-21 plus CD40L.
Results: All patients born from non-consanguineous parents had a median age of onset of symptoms was at 6 (0.6 –18) and of diagnosis at 10 (2.5 – 52) years of age, respectively. Clinical findings included recurrent RT infections as the most frequent, followed by GI infections and atopy; no autoimmune abnormalities were observed. Normal IgG and IgM with undetectable IgA (<7 mg/dl) were seen in all. Phenotyping of lymphocyte populations revealed normal percentage and numbers of T, B and NK cells as well as normal numbers T and B cells subpopulations, yet IgA+ B cells were absent in all. T and B cell lymphoproliferation were normal in all patients.
Conclusion: Our preliminary findings show that our SIgAD patients consistently exhibit absent IgA+ B cells without any other detectable abnormalities.
Financial support: COLCIENCIAS grant # 111556934592.
ESID-0570 Exome Sequencing Identifies a Heterozygous Gain-of-Function Mutation in PIK3CD in a Colombian Patient with Early-Onset Combined Immunodeficiency
J.L. Rojas 1, M.A. Moncada2, C. Prando3, S.Y. Zhang2, J.C. Orrego1, M.A. Velásquez4, A. Wilches5, A. Cepeda6, J.L. Franco1
1Microbiology and Parasitology, Universidad de Antioquia, Medellín, Colombia
2St. Giles Laboratory of Human Genetics, The Rockefeller University, New York, USA
3., Instituto de Pesquisa Pelé Pequeno Príncipe, Curitiba, Brazil
4Centro de investigaciones dermatológicas (CIDERM), Universidad de Antioquia, Medellín, Colombia
5Pediatrics, San Vicente Fundación, Medellín, Colombia
6., Universidad del Norte y Universidad Metropolitana de Barranquilla, Barranquilla, Colombia
Introduction: Activating mutations in the p110 subunit of PI3K-delta are associated with Activated PI3K syndrome (APDS). We describe a 12 year-old girl with combined immunodeficiency who was found to carry a mutation in PIK3CD.
Methods: We analyzed PB lymphocytes, lymphoproliferation, cytokine production and CD69/CD40L expression by flow cytometry, and performed exome sequencing and Sanger sequencing on gDNA to find potential pathogenic variants.
Results: This girl developed since16 months GI and sinopulmonary infections, failure to thrive, chronic malabsorption, oral HPV+ warts, hepatosplenomegaly and multiple lymphadenopathies. At 7 years she was referred to us due to EBV infection, hypertrophic osteoarthropathy and CLD, and later on developed recurrent parotiditis. Serum IgG, IgM and IgE were normal with absent IgA, IgG2 and IgG4. Anti-TT, anti-pertussis and anti-Pn IgG were all negative. Total lymphocytes were normal but T cells were > 90% with inverted CD4:CD8 ratio with only 3% B cells and normal NK cells. T cell subpopulations showed low naive and central memory with high effector memory and effector T Cells. B cells subpopulations showed expansion of CD21low cells with increased transitional, normal IgD- and IgD+CD27+ and absent IgA+ B cells. Lymphoproliferation to mitogens was severely impaired in CD8+ T cells. CD69 and CD40L expression in PMA-stimulated T cells were low and IC TNF-alpha production was increased in both subpopulations. Lastly, we found a previously reported pathogenic variant in exon 24 of the PI3KCD (c.3061G>A; p.E1021K).
Conclusion: Our patient is affected with APDS. Financial support: Colciencias grant #111556934592.
ESID-0429 Anaphylactic Reactions to Ceftriaxone in a Patient with BTK-Deficiency
M.T. Giner1, L. Alsina1, M. Pascal2, M. Piquer1, M. Juan-Otero 2, A.M. Plaza1
1Allergy and Clinical Immunology Department / Functional Unit of Immunology, Hospital Sant Joan de Déu / Barcelona University, Barcelona, Spain
2Servei d’Immunologia – Centre Diagnòstic Biomèdic / Functional Unit of Immunology, Hospital Clínic – IDIBAPS, Barcelona, Spain
Introduction: Drugs are a common cause of anaphylaxis, which can be driven through an IgE, IgG and/or complement activation of basophils/mast cells. Patients with BTK-deficiency suffer from agammaglobulinemia of all isotypes. BTK seems to be required for IgE-mediated activation of human basophils.
Case report: 10 year-old male, with mutated BTK (K18fsX23 causing loss of function), presented 10 minutes after ceftriaxone infusion, mild-moderate anaphylaxis (angioedema, generalized erythroderma, pruritus, anxiety and dysphonia), on 2 occasions, 2 months apart. Allergy testing: PRICK and intradermal test for PPL, MDM, amoxicillin, ceftriaxone and cefotaxime were negative, also sIgE for penicillin and amoxicillin. Basophil activation test was performed on buffy coat cells using the Flow2CAST-kit (Bühlmann, Switzerland) to evaluate the contribution on basophil activation of complement/serum in relation to triggering and non-triggering drugs: activation was evaluated by measuring CD63 expression in patient and healthy donor basophils stimulated with patient's serum (complement inactivated or intact) alone and in combination with ceftriaxone, cefotaxime or meropenem.
Results: Patient and donor cells were reactive to positive stimulators (FMLP and anti-FcRIE receptor). None of the drugs tested caused cell-stimulation. Patient's intact serum caused activation of patient and donor cells, whereas inactivated complement didn’t induce activation, regardless of the presence of any of the 3 drugs.
Conclusions: 1- Unexpectedly, cell activation is present in BTK-deficiency. 2- Cell activation, for both patient and healthy donor, is only detected in the presence of patient’s intact complement/serum, and not with drugs-stimulation alone. More experiments are ongoing to elucidate the mechanism underlying this clinical observation.
ESID-0751 Chronic Kidney Disease in Two Patients with Common Variable Immunodeficiency
J.M. Kang1, J.M. Kim1, H.Y. Cho1, J.H. Kim1, K.H. Yoo1, K.M. Ahn1, H.J. Kim2, E.S. Kang2, Y.J. Kim 1
1Pediatrics, Samsung Medical Center Sungkyunkwan University School of Medicine, Seoul, Korea
2Laboratory Medicine and Genetics, Samsung Medical Center Sungkyunkwan University School of Medicine, Seoul, Korea
Certain patients with Common variable Immunodeficiency (CVID) have several co-morbidities. We present two female CVID patients with chronic kidney disease (CKD).
Patient 1 was diagnosed with CVID at age 10 years old when she presented with seizure, pancytopenia, renal failure, massive splenomegaly, and cytomegalovirus infection. She was diagnosed as CVID based on low immunoglobulin levels. She has a brother who has hypereosinophilc syndrome and no other immunological diseases in the family. She had urinary tract infection due to Ureaplasma urelyticum and developed granulomatous lymphocytic interstitial lung disease that dramatically responded steroid therapy. Her kidney function (KF) gradually worsened and she started continuous ambulatory peritoneal dialysis (CAPD). Her condition has been stabilized on CAPD and IVIG therapy. She also received growth hormone therapy and her height showed improvement from 133 cm to 141 cm during 20 months.
Patient 2 was diagnosed with histiocytosis X at age 3 months at hospital A. She was diagnosed with ulcerative colitis at age 14 years at hospital B. At age 24 years, she developed persistent hepatosplenomegaly, anemia, thrombocytopenia, and recurrent parotitis for which she received splenectomy at hospital B. She was diagnosed as CVID at our hospital and started IVIG therapy. She has a sister who was also diagnosed as CVID but does not have other significant co-morbidities. Her KF shows CKD features with estimated GFR at 52 mL/min/1.73 m2.
Although not common, certain CVID patients may develop CKD and KF monitoring and patient education is important during the follow-up visits.
ESID-0290 Childhood Onset Common Variable Immunodeficiency: A Single Center Experience
A. KIYKIM 1, E. KARAKOÇ-AYDINER1, S. BARIS1, A.O. ÖZEN1, I.B. BARLAN1
1Pediatric Allergy and Immunology, Marmara University Pendik Training and Research Hospital, Istanbul, Turkey
Aim: In this study we aimed to investigate the clinical and immunological features of childhood onset-CVID patients.
Methods: Medical records of 27 CVID patients (M: 18, F: 9) were reviewed, retrospectively. Data on disease onset, infectious manifestations, autoimmunity and laboratory examinations were analyzed.
Results: The mean age of patients was 18.1±10.8 yrs (min:4.7 yrs; max:57.5 yrs) and the age at diagnosis 10.6±12.1 yrs. Parental consanguinity rate was 68%. The mean lag period from disease onset until diagnosis was 7.01±6.5yrs. All patients had a history of recurrent sinopulmonary infections, whereas, 45% bronchiectasis, 37.5% failure to thrive, 29.6% dermatitis, 26.1% fungal infections and 14.8% inflammatory bowel disease (IBD). In 17 of them an autoimmune manifestation was observed; thrombocytopenia:11.1%, coeliac disease:7.4%, thyroiditis:7.4%, arthritis:3.7% and diabetes:3.7%. Two patients experienced malignancy (one with stomach adenocarcinoma, the other cholangiocarcinoma). Mean serum levels of IgG, IgM and IgA were 396±375 mg/dL, 69.2±132 mg/dl and 29.3±19 mg/dl, respectively. Ten patients had lymphopenia (37%); 4 patients CD4 lymphopenia (14.8%) and 12 low B cell counts (44%). Spirometric evaluation revealed a moderate impairment in FEV1 (72±24%) and FVC (64±20%) values. The most important determinants of quality of life were recurrent respiratory infections and IBD-like colitis.
Conclusion: In this cohort of patients with childhood-onset CVID, the diagnostic delay was longer than previously reported. The rate of autoimmunity appears higher compared to the adult onset CVID patients reported in the literature. Significantly high consanguinity rates may suggest underlying genetic defects that follow autosomal recessive mode of inheritance.
ESID-0155 Granulomatous-Lympocytic Interstitial Lung Disease (GLILD) in Common Variable Immunodeficiency
I. Kondratenko 1, O. Pashchenko2, S. Vakhlyarskaya3, A. Bologov3
1Clinical Immunology, Russian Clinical Children's Hospital, Moscow, Russia
2Immunology, Russian National Scientific Medical University, Moscow, Russia
3Clinical Immunology, Russian Clinical Children’s Hospital, Moscow, Russia
CVID is often associated with various inflammatory and autoimmune manifestations as a results of abnormal cellular immunity. Lungs are especially affected by a granulomatous lymphocytic interstitial lung disease (GLILD). We observed 14 years old boy with severe GLILD, first signs of the disease appeared at the age of 7 years: hemolytic anemia, enlarge of the lymph nodes and spleen. CVID was diagnosed at the age of 8 years after immunological evaluation: IgA <6.67 mg/dl, IgM 24 mg/dl, IgG 190 mg/dl, normal counts of T and B-cells, absence of postvaccinal antibodies. Patient recived regular replacement IVIG therapy and trimetoprim-sulphometocsazole prophylaxis since that time. At 8,7 years developed hypersplenism with anemia, leucopenia, thrombocytopenia; splenectomy was performed with good effect. At 10 years developed autoimmune hepatitis and thrombocytopenia, treated with azathioprine 2 mg/kg. Till 14 years the boy did not have infectious complications. At 14 years X-ray and SCT examination revealed multiple interstitial small foci in lungs, enlarged thoracic and abdomen lymph nodes. Lung biopsy showed massive parabronchial lymphoid infiltrates with the lymphoid follicles, follicular bronchiolitis, lymphoid hyperplasia; lymph node – follicular and parafollicular hyperplasia. Patient received abatacept 10 mg/kg once per month. After 10 month of therapy the X-ray examination showed marked reduction of number and intensivity of interstitial foci. Treatment was continued. GLILD is a severe inflammatory complication of CVID with high mortality. 10 month therapy with abatacept had a good effect in our patient. We need longer follow-up to assess the stable result of this boy.
ESID-0744 Peripheral B Cell Subpopulations and IGG Anti-Pneumococcal Polysaccharide Antibody Levels in a Cohort of Healthy Children
L.E. Leiva 1, S. Lefevre1, H. Monjure1, R.U. Sorensen1
1Pediatrics, Jeffrey Modell Center for Immunodeficiencies LSU Health Sciences Center, New Orleans, USA
Introduction: Several B cell subpopulations have been characterized and some of them are found to be impaired in Common Variable Immunodeficiency (CVID) and Specific Antibody Deficiency (SAD). Patients with CVID or SAD also fail to produce IgG anti-pneumococcal polysaccharide (PnPs) antibodies. Recent reports show that B cell subpopulations normally develop in an age-dependent manner. Therefore, age-dependent reference values for B cell subpopulations are essential for determining if patients have an impaired peripheral B cell development. Objective: In this study we determined serotype-specific IgG anti-PnPs antibody levels and percentages of peripheral B cell subpopulations in healthy children, to establish age-dependent reference values. Methods: We enrolled 100 healthy children, 2-18 years of age of both genders after obtaining consent from the subject’s legal guardian. Patients with known immunodeficiencies or immunoglobulin abnormalities were excluded. Evaluation included a detailed history of pneumococcal immunizations, determination of IgG, IgM, IgA and IgE concentrations and IgG anti-PnPs antibody levels against 14 serotypes by ELISA. Expression of CD27, IgD, IgM and CD21was analyzed on peripheral CD19+ B cells to determine percentages of total memory, naïve, class-switched memory and CD21low B cells by flow cytometry. Results: Children were divided into four age-groups: 2-5, 6-10, 11-16 and >16 years of age. Results of B cell subpopulations and IgG anti-PnPs antibody levels reveal significant variability in both parameters in each age group. Conclusions: results from this study will be used to establish age-dependent reference values, which will be useful for the evaluation of patients with suspected primary immunodeficiency affecting B cells.
ESID-0024 Serum Cytokine Signature in Common Variable Immunodeficiency
J. Litzman 1, R.P.H. Huijbregts2, J. Nechvatalova1, M. Vlkova1, J. Xu2, Z. Hel2
1Dept. Clin. Immunol. Allergol., St Anne´s University Hospital, Brno, Czech Republic
2Dept. Pathol. and Dept. Microbiol., University of Alabama at Birmingham, Birmingham, USA
Although common variable immunodeficiency (CVID) is the most frequent symptomatic primary hypogammaglobulinaemia, the pathogenetic mechanisms leading to immunodeficiency are not fully resolved. A plethora of abnormalities in lymphocyte subpopulations was described in CVID patients, including decreased numbers of circulating CD4+ T-cells and increased expression of T-cell activation markers. Also, an increased concentration of soluble CD14 (sCD14) and other factors indicating limited microbial translocation was described.
To address the mechanisms of chronic immune activation in CVID, we performed a detailed analysis of cytokine serum levels in 35 CVID patients, 53 patients with selective IgA deficiency (IgAD), and 63 healthy volunteers. We show that CVID is associated with elevated serum levels of TNF-α, IL-10, IL-12 p40, G-CSF, IL-1R antagonist, CXCL-10/IP-10, CCL-2/MCP-1, and CCL-11/eotaxin. The detected cytokine profile is consistent with an activation of cells of myeloid lineage. The expression of cytokines that are primarily produced by activated cells of myeloid lineage including TNF-α, G-CSF, CXCL-10/IP-10, Flt-3L, and IL-10 directly correlated with the expression of lymphocyte activation markers (expression of HLA-DR and CD45RO on CD4+ and CD8+ T cells). In contrast, the levels of cytokines typically produced by Th1 (IFN-γ, IL-2), Th2 (IL-9, IL-13), and Th17 (IL-17) subsets were suppressed in CVID patients compared to healthy donors.
In individuals with IgAD, serum levels of CXCL-10/IP-10, IL-10, and G-CSF were significantly higher compared to healthy donors.
Presented data suggest that CVID is associated with chronic activation of cells of myeloid lineage, which may contribute to secondary immunodeficiency in these patients.
ESID-0682 LRBA Deficiency in Mexican Patients with Common Variable Immunodeficiency
G. LOPEZ HERRERA 1, L. BERRON RUIZ1, D. MOGICA MARTINEZ2, M.A. YAMAZAKI NAKASHIMADA3, N.H. SEGURA-MENDEZ4, L. SANTOS ARGUMEDO5, F.J. ESPINOSA ROSALES1
1IMMUNODEFICIENCY RESEARCH UNIT, NATIONAL INSTITUTE OF PEDIATRICS, Mexico City, Mexico
2ALLERGY AND CLINICAL IMMUNOLOGY, NATIONAL MEDICAL CENTER LA RAZA, Mexico City, Mexico
3ALLERGY AND CLINICAL IMMUNOLOGY, NATIONAL INSTITUTE OF PEDIATRICS, Mexico City, Mexico
4ALLERGY AND CLINICAL IMMUNOLOGY, NATIONAL MEDICAL CENTER SIGLO XXI, Mexico City, Mexico
5BIOMEDICINA MOLECULAR, CENTRO DE INVESTIGACION Y DE ESTUDIOS AVANZADOS, Mexico City, Mexico
Introduction: LRBA deficiency is an autosomal recessive defect associated with immunodeficiency and autoimmunity, B and T cells from LRBA deficiency show activation defects, peripheral B cell compartment is altered. Up to date, new cases of LRBA deficiency have been described. Objective: To screen out LRBA deficiency in patients with CVID diagnosis. Methods: Peripheral mononuclear cells (PBMCs) from 45 individuals with CVID diagnosis were stimulated with phytohaemagglutinin (PHA) for 72 hours. After activation, protein extracts were used to analyze LRBA expression by Western-blot. As T cells from LRBA-deficiency show activation defects, EBV cell lines were generated from individuals in which negative expression in PHA-stimulated PBMCs to confirm the protein deficiency. Mutation detection was performed by sequencing LRBA cDNA from PHA-stimulated PBMCs. Analysis of peripheral B cell subpopulations were performed by flow cytometry. Results: Consanguinity was unknown in most patients, , all patients had hypogammaglobulinemia with variable number of B cells. LRBA expression was negative in 6 of patients, 3 of them presented autoimmunity and 5 suffer from not-infectious chronic diarrhea. Mutation analysis up to now, showed that two of these patients carry homozygous detelerious mutations in LRBA, screening for mutations is currently being performed, autoimmunity and chronic diarrhea was present in half and all showed lack memory B cells. Conclusions: LRBA protein expression is absent in 15%of CVID individuals analyzed in this work, suggesting that LRBA-deficiency might have a high incidence compared with other genetic deffects already described for CVID. Acknowledgements: This project is supported by CONACY #161089 and 154472.
ESID-0354 Multidimensional Analysis of B Cell Subpopulations in CVID Patients Based on EuroFlow PID 8 Color Flow Cytometry Panels
E. Lopez-Granados 1, J.M. Torres-Canizales1, E. Blanco2, M. Perez-Andres2, T. Kalina3, A.K. Kienzler4, M. van der Burg5, M. Vlkova6, M. Wentink5, J.J.M. van Dongen5, A. Orfao2
1Clinical Immunology, La Paz University Hospital-IdiPAZ, Madrid, Spain
2Dep. Medicine-Serv. Cytometry, Cancer Research Center (IBMCC-CSIC/USAL) and Univ. of Salamanca, Salamanca, Spain
3Pediatric Hematology and Oncology, Charles University in Prague School of Medicine and University Hospital Hradec Kralove, Prague, Czech Republic
4Nuffield Department of Medicine, Experimental Medicine Division University of Oxford, Oxford, United Kingdom
5Dept. of Immunology, Erasmus MC, Rotterdam, Netherlands
6Institute of Clinical Immunology and Allergology, Masaryk University and St Anne`s University Hospital, Brno, Czech Republic
Common variable immunodeficiency disorders (CVIDs) are the most frequent symptomatic antibody deficiencies. Several attempts for subclassification have been based on immunophenotyping, but pathogenic causes and a correlation between clinical and immunological phenotypes are yet unknown. EuroFlow B-cell-specific panels and flow cytometry approaches were used for multicentric unsupervised multidimensional analysis of peripheral blood B cell subsets from CVID patients vs healthy controls.
Absence of plasma cells (2 cells/uL), decreased naïve B-cells (
We explored the utility of multi-dimensional immunophenotyping of B cells using the EuroFlow strategy and PID panels to address differences in B-cell composition in CVID patients. Overall, three CVID B-cell profiles were identified, indicating that this EuroFlow-PID approach could be instrumental for obtaining further pathogenic insight and association with clinical phenotypes and prognosis.
ESID-0012 X-Linked Hyper IGM Syndrome: Clinical, Immunological and Molecular Features in Patients from India
M. Madkaikar 1, M. Gupta1, A. Dalvi1, K. Italia1, M. Desai2, N. Radhakrishnan3, K. Ghosh4
1Paediatric Immunology and Leukocyte Biology, National Institute of Immunohaematology ICMR, Mumbai, India
2Immunology, Bai Jerbai Wadia Hospital for Children, Mumbai, India
3Pediatric Hematology Oncology & BMT Unit, Sir Ganga Ram Hospital, New Delhi, India
4Director, National Institute of Immunohaematology ICMR, Mumbai, India
Background: Hyper-Immunoglobulin M (HIGM) syndrome is a heterogeneous group of primary immunodeficiency disorders, characterized by recurrent infections associated with decreased serum levels of IgG, IgA and IgE and normal to increased levels of IgM. Mutations in five genes have so far been associated to the disease; of these X-linked HIGM caused due to CD40 ligand (CD40LG) mutations is the most common. The aim of this study was to analyze the nature of mutations in Indian patients diagnosed with X-linked HIGM and correlate with their clinical and immunological features.
Method: The entire coding region and intronic splice sites of the CD40LG were sequenced from the genomic DNA of seven patients diagnosed with X-linked HIGM.
Results: The median age at diagnosis and age of onset was found to be 2.6 years and 6 months respectively suggesting significant delay in specific diagnosis. Recurrent sino-pulmonary infections and diarrhea were the commonest clinical manifestations. All patients had low levels of serum IgA and IgG, five patients had elevated serum IgM levels and two patients showed normal level of IgM. Six patients had significant neutropenia. We identified seven mutations; three nonsense (c.172delA, c.A229T, c.C478T), one missense (c.A506G) and three splice site [c.346+2(T>C), c.289-1(G>C), c.346+1(G>T)] out of which five were novel.
Conclusion: This study not only shows high heterogeneity in mutational profile of the patient but also provides important information for prenatal diagnosis in affected families.
ESID-0355 Atypical and Agressive Presentation of Gastric Cancer in a Patient with Common Variable Disease
L. mendonca 1, C. Tavares de Alcântara1, C. Kokron1, M. Barros1, F. Mascarenhas1, N. Siqueira de Castro1, K. Latella e Boufleur1, G. Garcia1, L.A. Marcondes Fonseca1
1clinical immunology and allergy, HCFMUSP, Sao Paulo, Brazil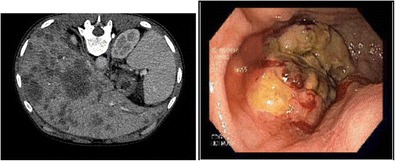
In patients with common variable immunodeficiency (CVID) gastrointestinal disorders and malignancies occur in higher than expected in the general population rate. Kalha and Kellin, 2004, published that in these patients, there is a risk of 50 times higher than the population for the development of gastric cancer, what do reinforces the importance of screening for premature diagnosis and treatment. The objective of this paper is to present a case of a patient with CVID with an atypical and very aggressive presentation of gastric cancer. M.S.S., 32 female patient, followed with CVID since she was 21 years old, was hospitalized with 30 days of cought, fever unresponsive to oral antibiotics. Despite intravenous broad-spectrum of antibiotics there was no clinical improvement. The patient developed ascites and a computed tomography of the thorax was made showing multiple liver abscesses. In the investigation of the origin of these abscesses two gastric ulcers and one rectal ulcer was found. The pathologic analysis of the gastric and rectum byopsis and the cytologic analysis of the paracentesis showed adenocarcinoma of the stomach with peritoneal carcinomatosis. The patient died on the 24th day of hospitalization.
ESID-0331 Descriptive Review of Clinical Data from 186 Records of Outpatients with IgA Deficiency Accompanied at a Quaternary Hospital in Brazil
L. mendonca 1, F. Mascarenhas1, A. Cohon1, C. Kokron1, O. Grecco1, J. Kalil1, M. Barros1
1clinical immunology and allergy, HCFMUSP, Sao Paulo, Brazil
IgA Deficiency (IgAD) is the most common immunoglobulin deficiency, with an approximate incidence of 1 in 400 to 3000 individuals in the general population. Selective IgA deficiency is defined as serum IgA levels less than 7 mg/dL with normal serum IgM and IgG levels. A subgroup of individuals, with more than 7 mg/dL but less than 30 mg/dL of serum IgA, are defined as partial IgA deficient patients (pIgAD). Many are clinically asymptomatic, however some patients have increased incidence of infections, as well as presence of atopy, autoimmunity and cancer. Our objective was to report clinical data observed in a cohort of patients followed between 1994 and 2014. The most common associated condition found was atopic disease in about 60% of the patients, specially rhinitis (in t IgAD and pI gAD). The recurrent infection prevalence was 36%: 20% of sinusitis, otitis and amigdalitis, 10% of recurrent pneumonia and 18% of recurrent diarrhea. Autoimmunity condition was observed in 18% of the patients, like thyroiditis ( the most prevalent, 13%), autoimmune hemolytic anemia, idiopathic thrombocytopenic purpura, rheumatoid arthritis, celiac-like disease, systemic lupus erythematosus, Sjögren’s disease, juvenile rheumatoid arthritis, autoimmune hepatitis and vitiligo; 9% of them had history of medication reaction and 8% had contact dermatitis. About 9% of the IgAD patients had malignancies or pre neoplasic conditions like MGUS. The exact value of deaths among these patients is difficult to calculate because many patients lose up for unknown reasons, but so far only one death was reported, and was attributed to sepsis.
ESID-0094 Predominant Generation of IgA+ Plasma and Memory B Cells in Response to Pneumovax23
A. Meyer-Bahlburg 1, A. Roth1, S. Gläsener1, K. Schütz1
1Pediatric Pneumology and Neonatology, Hannover Medical School, Hannover, Germany
CVID patients are in particular prone for infections with encapsulated bacteria including S. pneumoniae. The polysaccharide capsular of these bacteria induces a T cell independent (TI) immune response, thereby directly stimulating marginal zone (MZ) and B-1 B cells. In mice, both subsets have been characterized in great detail. In humans, IgM+ memory B cells in peripheral blood have been suggested to represent equivalents of splenic MZ B cells, whereas human B-1 B cells have been recently described as CD20+CD43+CD27+CD5- B cells. However, both views remain controversial. We therefore aimed to further characterize B cell subsets in a TI immune response with the pneumococcal polysaccharide (PnPS) vaccine Pneumovax23.
A group of healthy adults (n=30) was immunized with Pneumovax23. Blood samples were obtained pre-vaccination and on several time-points post-vaccination. Phenotypes of B cell subsets were assessed by FACS and PnPS-specific antibody secreting cells and production by EliSpot and ELISA.
On day 7 post-vaccination all healthy subjects showed a significant increase in plasma cells and proposed CD20+CD43+CD27+ B-1 B cells. However, no significant change of IgM+ cthroughout the whole study. Instead, we found a significant increase, both in relative and absolute numbers, in IgA+ memory B cells. In addition, PnPS-specific immunoglobulin production was mainly of the IgA isotype. Because B-1 B cells, in particular B-1b B cells, have been describe to produce large amounts of IgA we are now investigating if IgA+ memory B cells generated in response to Pneumovax23 resemble more characteristics of B-1 B cells.
ESID-0239 Novel STAT3 Amino Acid Substitution in a Patient with Low IgE Serum Levels
L. Moens 1, G. Frans1, H. Schaballie1, G. Wuyts1, C. De Boeck2, X. Bossuyt1, I. Meyts2
1Microbiology and Immunology, Catholic University Leuven, Leuven, Belgium
2Pediatrics, University Hospital Leuven, Leuven, Belgium
Hyper-IgE syndromes (HIES) are primary immunodeficiency disorders characterized by Staphylococcus aureus abscesses, recurrent pneumonia, eczema, increased number of eosinophils and increased serum IgE levels. Autosomal dominant (AD)-HIES is caused by heterozygous signal transducer and activator of transcription 3 (STAT3) mutations. The National Institutes of Health (NIH) HIES clinical phenotype score (>30) in patients with elevated IgE serum levels facilitate early diagnosis of AD-HIES to initiate appropriate therapy [Woellner et al. JACI. 2010. 125(2)].
An 8 year old Caucasian boy with a history of severe eczema from birth and extensive diaper dermatitis is presented. Other past medical concerns include gingivostomatitis, perlèche, frequent upper and lower respiratory tract infections and 6 episodes of febrile seizures. Extraction of some of his primary teeth was necessary for normal eruption of his permanent teeth. His wrist was fractured twice after minor trauma. Recently, he was hospitalized with a S. aureus sepsis and pleuropneumonia which led to the suspicion of STAT3 deficiency. However, laboratory data showed an eosinophil count variation between 0,1-0,6x109 cells/L and the highest IgE serum level measured was 180 kU/L. Because of the strong clinical suspicion, the NIH HIES score was calculated (=36.64) and subsequently STAT3 gene was sequenced. A heterozygous cytosine to adenine replacement resulting in a new histidine to proline substitution at position 332 in the DNA-binding domain of STAT3 was identified.
We can conclude that in cases with overwhelming clinical evidence for AD-HIES, sequencing of STAT3 gene has to be considered regardless of low IgE serum levels.
ESID-0397 Phenotypic and Genotypic Changes in CVID Patients
D. Moraes Vasconcelos 1, T.A. Bezerra2, A.C. Collanieri2, T.P. Oliveira2, C.C. Barreto2, A.J. Duarte2
1d, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
2Dermatology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
In recent years, the discovery of genes related to Common variable immunodeficiency (CVID) has broadened, including TACI, ICOS, CD19, CD20, CD81 and BAFF-R.
We selected 20 patients previously diagnosed as CVID and 20 healthy individuals. Cell activation was performed to evaluate the expression of CD154 molecule in T cells, phenotyping of surface markers in B cells (CD19, CD20, CD21, CD40, BAFF-R, CD5, CD27, IgM and IgD), PBMC culture using PHA and OKT3 for stimulation, and sequencing of the TACI, BAFF and BAFF-R genes.
All patients had reduced immunoglobulin levels, recurrent infections and in 30% of these patients, we found autoimmune manifestations. CVID patient may present higher T CD8+ counts, as well as a reduction in the expression of CD27+ in B cells. The genotyping of TACI showed that 5/17 patients presented allelic variants, cited below: a non-pathogenic allelic variant in exon 2 (G>A in g19525, c94), a previously described mutation in exon 3 (g23216T>C;c310 T>C;p C104R), one intronic SNP in g23376 A>C, a heterozygous SNP at position g31695 A>T, possibly pathogenic and not previously described in exon 4, and a heterozygous SNP g32491 T>C. The genotyping of BAFF showed one previously unpublished heterozygous allelic variant (g336A>G, c69A>G) predicted to be a non-pathogenic polymorphism. The evaluation of BAFF-R genotype showed an intronic homozygous genetic variant in g1027 T>A, between exons 2 and 3, not previously described and predicted as pathogenic, affecting the splice-site of exon 3.
ESID-0396 CD27 Expression in T Cells as a Surrogate Marker for Autoimmunity Among Common Variable Immunodeficiency Patients
D. Moraes Vasconcelos 1, A.C. Collanieri1, M.C.S. Pereira1, T.P. Oliveira1, A. Santos Dias1, A.J. Duarte1
1Dermatology, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
Common variable immunodeficiency (CVID) is the most common symptomatic primary immunodeficiency. The absence of adequate levels of antibodies in CVID patients result in recurrent bacterial infections mainly in the respiratory and digestive tract, leading to sinusal and lung sequels. Over the past 10 years the discovery of genes related to CVID began, such as the genes of TACI, BAFF-R, CD19 and ICOS. Among the immunological changes, there is impairment of memory B cells (CD19+/IgM-IgD-CD27+), leading to disturbance of isotypic switching and reduced secretion of immunoglobulins. Currently this feature has been used to classify CVID. During the present study we observed that patients with CVID present changes in the expression of CD27 not only in B cells, but also in T cells, and reduced lymphoproliferative response to PHA. CD27 molecule is a member of the TNF family present constitutively in T cells, and after activation in B cells. Its importance in the immune response is related to the proliferation and coactivation of specific T cells that act in T-B interaction, in the T cell dependent B cells response. Thus disturbances in the CD27 pathway can result in defects in isotypic switch and differentiation of germinal center B cells, as well as decrease of memory cells. These characteristics can be observed in murine models of CD27/CD70 deficiency. Our findings allow a new approach for the study of CVID. The evaluation of defects in CD27 expression in T cells showed an interesting relation between low levels of this molecule and autoimmune manifestations.
ESID-0552 Agammaglobulinemia: 30 Years Follow Up in One Center in Argentina
I. Moreira 1, A. Seminario1, M. Esquivel1, A. Bravo Kleiman1, A. Gómez Raccio1, D. Di Giovanni1, L. Bezrodnik1
1Immunology Group, Hospital de Niños R Gutierrez, Buenos Aires, Argentina
Introduction: Agammaglobulinemia is a primary immunodeficiency characterized by early onset of recurrent bacterial infections, very low immunoglobulins levels and fewer than 2% circulating B cells.
Methods: Retrospective analyze of medical records of patients with agammaglobulinemia (ESID criteria) from one center in Argentina, followed since 1994.
Results: 31 patients with agammaglobulinemia were studied, all of them male. 26 patients present BTK mutations, 1 presents a μ heavy chain mutation, 2 are currently being study and in other 2 there was not found any mutation.
Mean age of the first symptoms: 9,6 months. Mean age at diagnosis: 31,9 months (Excluding one patient diagnosed at 80 years old). 25 patients were diagnosed because of recurrent infections and 6 because of positive family history. Patients with family history were diagnosed earlier than those without it. (Mean: 20 and 34,9 months, respectively).
Pulmonary infections were the most frequent infections before diagnosis. Bacterial pneumonias, otitis, sinusitis, meningitis, arthritis and sepsis were reduced after an adequate immunoglobulin (IG) treatment, also hospitalizations.
10 patients have bronquiectasis, 7 patients another sequelae.
At this time 4 patients were died and 8 are over 18 years old. 23 are under Intravenous IG (IVIG) treatment and 4 patients are using Subcutaneous IG (SCIG). Mean doses: 599 mg/k/month with IVIG and 793 mg/k/month with SCIG. Mean serum IgG level: 784 mg/dl with IVIG and 1060 mg/dl with SCIG.
Conclusion: During this time the patients have presented an acceptable survive. Nowadays some of them are reaching adulthood. An adequate treatment signified a decrease in severe infections and hospitalizations.
ESID-0729 Antibody Response to Pneumococcal Vaccination in Children with Transient Hypogammaglobulinemia of Infancy (THI)
F.M. Cavaliere1, S. Graziani2, F. Pulvirenti1, G. Granata1, M.L. Romiti2, L. Chini2, V. Moschese 2, I. Quinti1
1Clinical Immunology, Sapienza University of Rome, Rome, Italy
2Pediatric Allergology and Immunology Unit, University of Tor Vergata, Rome, Italy
Transient hypogammaglobulinemia of infancy (THI) is a primary immunodeficiencies of the first few years of life characterized by a delay in the natural antibody production. The diagnosis is usually made a posteriori when the normalization of IgG values occurs. THI is generally characterized by a benign clinical course and few relevant immunologic defects have been reported. Particularly, rare THI children have a transient reduction in the vaccine response, which may resolve at an older age. The present study focuses on the specific antibody response to pneumococcus vaccine in THI patients to better define B cell function in this condition.
Biological samples from 20 THI patients (aged 12–36 months) and 11 age-matched healthy controls (HC) have been analyzed for specific IgG, IgA and IgM response to pneumococcal conjugate vaccine (PCV). No differences in pre-vaccine and after the first dose of Anti-Pr IgG, IgM and IgA titers were found. The mean IgG, IgA and IgM specific antibody response (U/ml) after the second PCV dose was significantly lower in THI than in HC (23 vs 259, p<0.001; 8 vs 232, p<0.001 ; 72 vs 438, p<0.002; respectively).
Our preliminary observation that, after the second dose of PCV, the intensity of specific antibody response is reduced in THI children raises the question whether a B-cell memory defect might alter their antibody efficiency. Further dissection of B cell response in THI children might contribute to devising B cell development as well as optimal vaccine schedule/dose in specific settings.
ESID-0362 Concentration of IGG Antibodies Specific to Salmonella Typhi Vi in Adult Blood Donors
A. Parker 1, O. Hemmings2, S. Allen2, S. Harding3
1Head of Scientific Affairs, The Binding Site Group Ltd, Birmingham, United Kingdom
2Clinical Chemistry, The Binding Site Group Ltd, Birmingham, United Kingdom
3Director of Research and Development, The Binding Site Group Ltd, Birmingham, United Kingdom
Background Salmonella Typhi Vi IgG ELISA has been developed.Here we measure the serum concentrations of Typhi Vi antibodies in adult blood donors.
Methods Serum samples were obtained from blood donors (n=215; 123 males & 92 females; median age 40 years, range 18-90 years) and Typhi Vi concentrations measured using Typhi Vi IgG ELISA (The Binding Site, Birmingham, UK). Patients with serum CRP concentrations (>10 mg/L) were excluded.
Results 153/215 (71%) donors had Typhi Vi antibody concentrations <7.4 U/mL, corresponding to the bottom of the assay measuring range. 62/215 donors (29%) had antibody concentrations >7.4 U/ml (median 20.5 U/mL, range 7.4-226 U/mL; p<0.0001). There were no significant differences between median ages of the two populations <7.4 U/ml (31 years, range 18-90 years) v >7.4 U/ml (46 years, range 18-87 years; p=0.07).
The population with antibody concentrations >7.4 U/ml showed an age related bias to the distribution of the antibodies. 18-21 years (n=8; median 21 U/ml), 22-31 years (n=9; median 14 U/ml), 32-41 years (n=6; median 43 U/mL), 42-51 years (n=16; median 29 U/mL), 52-61 years (n=5; median 26 U/mL), 62-71 years (n=9; median 13 U/mL), 72-81 years (n=6; median concentration 27 U/mL), and 82-90 years (n=3; median 14 U/mL). There was a higher median antibody titre in the 32-41 years group (p=0.036) compared to those aged 22-31 years.
Conclusion The data suggests that Salmonella typhoid immunity is only present in 28% of random blood donors and the highest concentrations are reached at between 32-41 years and 72-81 years of age.
ESID-0405 Flow Cytometric BTK Screening in B-Cell Deficient Patients for Identification of Patients with X-Linked Agammaglobulinemia
B. Piatosa 1, M. van der Burg2, M. Pac3, K. Siewiera4, B. Pietrucha3, E. Heropolitanska-Pliszka3, E. Bernatowska3, J.J.M. van Dongen2
1Histocompatibility Laboratory, The Children's Memorial Health Institute, Warsaw, Poland
2Immunology, Erasmus MC, Rotterdamm, Netherlands
3Clinical Immunology, The Childrens' Memorial Health Institute, Warsaw, Poland
4Histocompatibility Laboratory, The Childrens' Memorial Health Institute, Warsaw, Poland
Introduction X-linked agammaglobulinemia (XLA) is a congenital deficiency caused by mutations in the BTK gene. Patients lacking Bruton's tyrosine kinase (BTK) demonstrate B-cell deficiency, hypogammaglobulinemia and susceptibility to recurrent infections. Definitive diagnosis requires demonstration of mutation or protein deficiency in monocytes or platelets, but diagnostic procedures are time-consuming and costly. The study was performed to verify the usefulness of a new monoclonal antibody for fast diagnosis of XLA patients.
Material and methods Twenty XLA patients with a defined genetic defect in BTK gene, 10 mothers (including one proven carrier), and 31 healthy male controls were screened for presence of Btk in B lymphocytes and monocytes. Btk presence was evaluated by four color flow cytometry in permeabilized B lymphocytes and monocytes. Mouse IgG2a anti-human Btk, clone 53/BTK, obtained by immunization with human N-terminal Btk 2-172 aminoacid protein (i.e. PH and Btk-kinase domains) was used to identify the presence of Btk in B lymphocytes and monocytes.
Results Median 2.5% of monocytes and 2.0% of B lymphocytes from XLA patients demonstrated Btk expression. Among healthy individuals median 90.5% (84.3-95.5%, 25-75 percentile) B lymphocytes and 80.5% (72.4-90%) monocytes expressed Btk. Single genetically confirmed carrier, as well as 7 other mothers, demonstrated significantly reduced, while 2 mothers demonstrated normal Btk expression.
Conclusion Flow cytometric detection of Btk expression with clone Btk/53 monoclonal antibody is an effective tool for discrimination of XLA from non-XLA patients and carriers.
The study was sponsored by grant no N N407 146338 from the Ministry of Science and Education (Poland).
ESID-0609 New LRBA-Mutation in a Patient with Severe Reduction in IgG, IgM and IgA with Normal Number of B-Cells at Diagnosis and Previously Classified as CVID
M. Piquer 1, G. de Valles2, E. González3, A. Esteve1, M.A. Martín-Mateos1, J.I. Aróstegui3, L. Alsina1, M. Juan-Otero3, F. Casals4
1Allergy and Clinical Immunology Pediatric Department / Functional Unit of Immunology SJD-Clinic, Hospital Sant Joan de Déu / Barcelona University / Functional Unit of Immunology, Barcelona, Spain
2IBE-Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra., Barcelona, Spain
3Servei d'Immunologia – Centre Diagnòstic Biomèdic / Functional Unit of Immunology SJD-Clínic, Hospital Clínic - IDIBAPS, Barcelona, Spain
4IBE-Institute of Evolutionary Biology (UPF-CSIC) / Genomics Core Facility, Universitat Pompeu Fabra., Barcelona, Spain
Introduction: The recent description of mutated genes as a cause of some hypogammaglobulinemias with normal levels of B-cells, is leading to review old cases previously labelled as common variable immunodeficiencies (CVID) to identify whether these genes define the patient's phenotype. LRBA (Lipopolysaccharide Responsive Beige-like Anchorprotein) is one of these newly involved genes (Lopez-Herrera G et al 2012).
Case report: North African female patient, from consanguineous parents, suffered of chronic diarrhoea since early infancy. At 8-yo (2005), she moved to Spain and, after detection of hypogammaglobulinemia and anaemia, she was derived to our paediatric hospital where lymphadenopathy/bilateral-rales/splenomegaly and an immune work-up led us to label the patient as CVID (decrease IgG/IgM/IgA, and negative isohemagglutinins next to 11% B-cells); treatment was initiated with IGIV. Interestingly intestinal biopsy revealed minimum duodenal lesions with normal villi and lymphocytic infiltration. Lung biopsy confirmed a low-grade marginal-zone lymphoma with a positive PCR to EBV, and Rituximab treatment was performed. At 14-yo, she had a severe flare of CMV-positive colitis with poor response to different treatments, requiring total colectomy (pathologist reported indeterminate inflammatory disease). Flow-cytometry analyses reveal no switched-memory B-cells (0.0%) as the most relevant data, but clear levels of (non-malignant) marginal (12.3%) and transitional B-cells (23,1%) are observed.
Results: By next generation sequencing and genotyping, we detected a new homozygous mutation of LRBA in the exon 4 coding for BEACH domain (C6607T->R2203STOP). Patient's clinical phenotype only partially coincides with reported cases.
Conclusions: Additional analyses are ongoing to elucidate mechanisms underlying this new LRBA-mutation.
ESID-0649 The Quantification of T-Cell Receptor Recombination Excision Circles (TREC) and IG Kappa –Deleting Recombination Excision Circles (KREC) in Children with B-Cell Immunodeficiencies
I. Korsunskiy1, M. Gordukova1, N. Davydova1, A. Smirnova1, O. Barabanova1, T. Kosacheva1, Y. Konopliannikova1, S. Zimin1, N. Zinoveva1, M. Filipenko2, A. Prodeus 1
1Moscow City Center for Pediatric Immunology and Allergy, Speranskiy Children's Hospital #9, Moscow, Russia
2Laboratory of Pharmacogenomics, Institute of Chemical Biology and Fundamental Medicine, Novosibirsk, Russia
Introduction: We have recently developed real-time PCR based quantification of TRECs and KRECs and applied this method to test patients with antibody deficiency syndromes: X-linked agammaglobulinemia (XLA), common variable immunodeficiency (CVID) and hyper IgM syndrome with CD40L deficiency.
Patients and methods: Five X-linked agammaglobulinemia patients, ten CVID and three hyper IgM were included in this study. A quantitative triplex qPCR protocol for simultaneous detection of TRECs, KRECs and IL17RA gene as a housekeeping gene in DNA from peripheral blood was used.
Results: All five patients with XLA were KREC negative and TREC positive. The CVID patients varied in levels of TRECs and KRECs. They were divided into four groups: two patients had both low TRECs and KRECs, two patients had normal KRECs and low TRECs, four patients conversely had normal TRECs and low KRECs and two patients had normal values of both. The patients with hyper IgM syndrome did not differ from normal.
Conclusions: The combined measurement of TRECs and KRECs improves the diagnostic of X-linked agammaglobulinemia. These markers can be used for screening of XLA patients. The different levels TRECs and KRECs with CVID patients reflect a heterogeneity of this group of syndromes and can be used for the understanding of the clinical severity, prognosis and pathogenesis.
ESID-0467 Adaptive Immunity in GATA2 Deficiency with a Focus on B Cells
C. Wehr1, A. Janda1, M. Wlodarski2, E. Mejstrikova3, R. Dräger1, C. Niemeyer2, K. Warnatz1, M. Rizzi1, U. Salzer 1
1Center for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany
2Division of Pediatric Hematology-Oncology Department of Pediatric and Adolescent Medicine, University of Freiburg, Freiburg, Germany
3Department of Pediatric Hematology and Oncology 2nd Faculty of Medicine, Charles University in Prague and University Hospital Motol, Prague, Czech Republic
Heterozygous germline mutations in the transcription factor GATA2 result a complex variable clinical phenotype: the most prevalent findings are myelodysplastic syndrome, syndromal aspects and immunodeficiency with susceptibility to infections. The defect results in a reduction of monocytes, T, B, NK and dendritic cells in the peripheral blood.
We aimed to analyse T and B-cell subpopulation and vaccination responses in patients with GATA2 deficiency.
Nine patients with proven mutations in the GATA2 gene underwent detailed immunological analyses of the lymphocyte compartment in the peripheral blood. Immunoglobulin levels and vaccination titers were documented.
A reduction in NK, B and CD4+ T cells was present in the majority of patients. T-cell phenotyping revealed mostly normal findings, but some patients showed expansions of gamma/delta T-cells and CD8+ effector cells. B cell phenotyping showed a consistent profound reduction of transitional B cells and an expansion of CD21low B cells. We found low IgG levels in two and skewed IgG subclass distribution in 4 patients. Antibody levels to peptide antigens (Tetanus) were normal in all tested patients but vaccination response to polysaccharides was absent in one patient.
Although CD4+ T cells are reduced in GATA-2 deficiency, T-cell subsets appear normal in most patients. In addition to marked B cell cytopenia, B cell subsets show more consistent abnormalities but immunoglobulin levels are largely maintained in GATA2 deficient patients, although minor deficiencies do occur occasionally.
ESID-0439 Long-Term B Cell Reconstitution After Hematopoietic Stem Cell Transplantation in a Group of Patients with Primary Immunodeficiency
A. Scarselli 1, S. Di Cesare1, C. Capponi2, S. Cascioli3, M.L. Romiti1, A. Finocchi1, M. Caniglia4, R. Carsetti3, A. Aiuti5, C. Cancrini1
1Department of Pediatrics (DPUO), Pediatric Hospital Bambino Gesu', Roma, Italy
2Department of Laboratories, Pediatric Hospital Bambino Gesu', Roma, Italy
3Immunology Area, Pediatric Hospital Bambino Gesu', Roma, Italy
4 Department of Paediatric Haematology-OncologyCurrent affiliation:Pediatric Haematology and Oncology S. Maria della Misericordia Perugia Italy, Pediatric Hospital Bambino Gesu', Roma, Italy
5Pediatric Immunohematology and Bone Marrow Transplant Unit and San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET), Scientific Institute HS Raffaele, Milan, Italy
Introduction and aim: Functional recovery of B-lymphocytes after hematopoietic cell transplantation (HCT) is often slow and suboptimal. Noteworthy, a deficiency in B-cell compartment, mostly in circulating B-cell memory, may be associated to an increased risk of late bacterial infections. Our aim was to identify and characterize the variables affecting kinetic of post-HCT B-cell recovery in different Primary Immunodeficiencies (PID).
Methods: B-cell development and function were studied in a cohort of 17 patients affected by PID transplanted since 1997 to 2010. The follow-up ranged from 0.7 to 15 years. Multiple approaches were used, and naïve and antigen-experienced B-cell subsets at different times were studied and compared with B-cell subsets of 74 healthy children collected in Bambino Gesù Children’s Hospital.
Results and discussion: Total memory B-cells count remained below normal levels for most of the follow up, mostly due to IgM memory that persists under the normal range for age in most of patients. However, in those with good in vivo humoral function, a progressive increase of memory B-cells, mainly switched B-cells, related to the recovery of CpG-response, was observed. We found that recovery kinetic of naïve and antigen experienced B-cells differed between patients, and a good and complete B-cell reconstitution was usually associated with donor B-cells chimerism, supported by myeloablative conditioning. The monitoring of phenotypic and functional changes on B-cells may prove clinically relevant, and gives suggestions to tailor patients’ care.
ESID-0460 B Cell Characterization in ADA2 Deficiency Patients
F. SCHENA 1, S. Volpi2, R. Caorsi3, A. Omenetti2, C. Pastorino3, P. Picco3, I. Aksentijevich4, A. Martini3, E. Traggiai5, M. Gattorno3
1Pediatric Division 2, G.Gaslini Institute, Genoa, Italy
2DINOGMI, University of Genoa, Genoa, Italy
3Pediatric Division 2, G. Gaslini Institute, Genoa, Italy
4National Human Genome Research Institute, National Institute of Health, Bethesda, USA
5B cell and antibody discovery Lab, Novartis Institute for Research in Biomedicine, Basel, Switzerland
Background and Objectives ADA2 deficiency, a recently described disease, is characterized by systemic vasculopathy and episodes of strokes. The defect is due to a loss of function mutation of CECR1 gene, codifying for Adenosine Deaminase 2 protein. This protein regulates the catabolism of extracellular adenosine, which we have recently shown is an important regulator of Class Switch Recombination in B lymphocytes. Accordingly DADA2 patients can present hypogammaglobulinemia. Therefore we decided to characterize peripheral B-cell compartment of two patients to directly address if ADA2 mutation affects B-cell function.
Patients and Methods Two brothers (15 and 7 years old) carrying mutations in CECR1 were followed up from the age of two. They showed similar clinical history with livedo reticularis, fever, vasculitis and neurological symptoms caused by haemorragic strokes. Both presented early hypogammaglobulinemia requiring intravenous immunoglobulin replacement therapy. Remarkably after etanercept treatment serum Igs slowly increased to a normal level. We analyzed peripheral B-cell phenotype by flow cytometry, in vitro B-cell proliferation and differentiation to plasmacells in response to CpG, BCR and T cell help.
Results and Conclusions Flow cytometer analysis showed a significative reduction of total B cells compared with age matched controls. Intriguingly a defect in the memory B-cell compartment (CD19+CD27+) was observed. We found that the rate of proliferation and differentiation to Igs secreting cells is lower as compared to normal controls and it is not supported by autologous T cells. Our findings suggest that ADA2 defect could lead to a reduced formation of T cell dependent memory B cells.
ESID-0204 Clinical and Immunological Features of 31 Pediatric Patients with Common Variable Immunodeficiency
N. serdar 1, H. çokugras1, N. Akcakaya1, Y. Camcioglu1
1pediatric Allergy and Immunology, Istanbul UNiversity Cerrahpasa Medical Faculty, Istanbul, Turkey
Common variable immunodeficiency is one of the most common primary immunodeficiences which characterized by hypogammaglobulinemia, poor response to vaccines or absent isohemagglutinins and onset of immunodeficiency at greater than 2 years of age.
We ought to analyze clinical and immunological features and comorbid diseases of 31 CVID (20 males, 11 females ) patients retrospectively who were followed at Cerrahpasa Medical School, Pediatric Immunology Department between 1992-2011. Patients were followed up from 3 to 18 years (8.9±3.1 years, Median:9).
The median age at diagnosis was 96 months, onset of symptoms was 12 months with a diagnostic delay of approximately 45 months. Mean immunoglobulins levels of patients were IgG:341,5±249,4 ( median:276), Ig A:19.5±28,4 (median:13), IgM:33,7±22,9 (median:25).The patients were mostly referred to our clinic with lower (61%) and upper respiratory tract infections (12.9%) and acute gastroenteritis 12.9%. 25 patients had comorbid disorders such as bronchiectasis(n:18), growth retardation(4), asthma(4), IBD(3), hypothyroidism(2), epilepsy(2), cataract(1), diabetes mellitus type 1(1) . Patients had been hospitalized 65 times, due to respiratory infection 71%, acute gastroenteritis 6%, meningitis 3%, sepsis 1.5%, congestive heart failure 1.5%, hemolytic anemia 1.5%, tuberculous pleurisy 1.5%, hepatic ensefalopathy 1.5%, cerebellitis 1.5%, lymphoma 1.5% and febrile neutropenia 1.5%. Median hospitalization per patient per year was 3(3.5±1.6) and low IgM level was correlated with increased hospitalization(p:0,009). Diagnostic delay was significant in patients with bronchiectasis(p:0.001). 11(28%) patients who had low serum IgA levels had adverse reactions to IVIG at least once , severe reactions(anaphlaxis, hypotension ,bronchospasm) were observed in 5 CVID patients.
ESID-0770 Presentation of Infections and Immune Dysregulations in Common Variable Immunodeficiency (CVID) Patients and its Relation to Their Clinical and Immunological Characteristics in Isfahan
R. Sherkat 1, M. Hasanzadeh1, M. Ganjalikhani-Hakemi2, N. Eskandari2
1Aquired Immunodeficiency Research Center, Isfahan University of Medical Sciences, Isfahafan, Iran
2Cellular & Molecular Immunology Research Center, Isfahan University of Medical Sciences, Isfahafan, Iran
Background: CVID is a heterogeneous collection of disorders and mostly characterized by hypogammaglobulinaemia and recurrent bacterial infections , however immundysregulations like autoimmunity , granulomatouse disease , malignancy and allergy could occur as a part of the syndrome.
Methods: Based on a retrospective study 25 patients diagnosed as CVID according to the diagnostic criteria in Immunology clinic of Isfahan Alzahra University hospital , between 1997 and 2013 were evaluated.
Results: Median age of disease onset was 5.22 years and diagnosis was 12.5 years .The median percentage of CD19+ was 8.35%, CD20+ 8% and CD3+ 80.88, CD4+ 28.72, CD8+ 38.88, CD16+CD56 11.08. Recurrent infections like Sinusitis in 79% , pneumonia in 85% and acute otitis media in 40% of patients and were the most common manifestations. one case with disseminated Tuberculosis, one Progressive multifocal leucoencephalopathy and one with sever herpes encephalitis , also two case with nodular lymphoid hyperplasia have been detected. Bronchiectasis was present in 25% of the patients. Autoimmunity like Autoimmune thrombocytopenia , reumatoid like disease , ulcerative colitis and autoimmune hepatitis was present in 33% of the patients , allergy in more than 85% of patients and allergic rhinitis as the most frequent types of allergic symptoms .One patients developed malignancy with two of her siblings have died of CVID and lymphoma as well.
Conclusion: The defects in different components of humoral and cellular immunity could be the etiology of this heterogeneity. The insights to exact genetic disturbance or polymorphism of CVID could be explaining the enigmas of the phenotype variety and polymorphism.
ESID-0112 Should First-Degree Relatives of Patients with Selective IgA Deficiency Be Screened?
P. Soler-Palacin 1, E. Cobos-Carrascosa1, A. Martin-Nalda1, F. Caracseghi1, M. Hernandez2, C. Figueras2
1Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d'Hebron Institut de Recerca Vall d'Hebron Universitat Autonoma de Barcelona, Barcelona, Spain
2Immunology Division, Hospital Universitari Vall d'Hebron Institut de Recerca Vall d'Hebron Universitat Autonoma de Barcelona, Barcelona, Spain
Background: Selective IgA deficiency (SIgAD) is the most common primary immunodeficiency being commonly asymptomatic. High family aggregation has been described but neither a causative genetic defect nor its inheritance mechanisms have been demonstrated.
Objectives: To determine the usefulness of screening first-degree relatives (FDR) of SIgAD patients to assess whether familial cases have more severe clinical and immunological phenotype and whether FDR with SIgAD have significant morbidity.
Patients and methods: Descriptive cross-sectional study (October 2010-September 2011) of all pediatric patients with SIgAD according to the ESID criteria, with review of demographic, clinical and laboratory data. Family case (FC) was defined as anyone with at least one FDR with SIgAD. FDR were defined as affected (A-FDR) when they fulfilled SIgAD ESID criteria. Otherwise they were considered as unaffected (U-FDR).
Results: One-hundred thirty participants were included: 42 patients with SIgAD and 88 FDR. Thirteen FC (31%) and 14 A-FDR (16%) were diagnosed. One out of each 6 tested FDR was affected. There were no clinical differences between FC and spontaneous cases. There was a higher proportion of gut disorders (p=0.001, OR=9.57 95% CI 2.59 to 35.3), hospital admission (p=0.045, OR=4.01, 95% CI 1.10 to 14.67] and the need for chronic treatment (p=0.006, OR=5.5, 95% CI 1.57 to 19.54) in the A-FDR group with regard to U-FDR.
Conclusions: Despite the absence of a more severe phenotype in FC, the high prevalence of A-FDR with significant morbidity could justify the systematic introduction of this screening program in clinical practice.
ESID-0508 Dissecting B Cell Memory and Serum Anti-Cytokines Antibody Repertoire in APECED Patients
E. Traggiai 1, A. Magnani2, M. Marchant1, M. Scharenberg1, A. Meloni3
1Novartis Biologics, Novartis Institute for Research in Biomedicine, Basel, Switzerland
2Immunogenetics of pediatric autoimmune diseases Frédéric Rieux-Laucat Immunogenetics of pediatric autoimmune diseases, Imagine Institute Des Maladies Genetiques Paris France, Paris, France
3Clinica Pediatria II, Clinica Pediatrica II Ospedale Microcitemico, Cagliari, Italy
Background and Objectives Anticytokine autoantibodies are becoming recognized as important regulators of disease pathogenesis of several life-threatening diseases. One of these conditions is autoimmune polyendocrinopathy, candidiasis, ectodermal dystrophy syndrome (APECED), caused by mutations in the autoimmune regulator (AIRE) gene. Indeed it has been proposed that chronic mucocutaneous candidiasis (CMC) in APECED is due to the production of anti-cytokines autoantibodies. In the present work we analyze by Gyros assay the presence of serum autoantibodies IgG against IL-17A/F, A/A, F/F and IL-22 in APECED patients compared with age matched control. We then dissect the memory B cell repertoire specific for cytokines in one selected patients.
Patients and Methods A cohort of 17 Sardinian patients with APECED syndrome followed at the Pediatric Clinic II, Ospedale Microcitemico and Department of Biomedical and Biotechnological Science, University of Cagliari, Sardinia (Italy) was studied. Serum IgG auto-antibodies against IL-17A/A, F/F, A/F and IL-22 was analysed by Gyros technology. Analysis of the B cell memory repertoire was done with EBV immortalization of primary human memory B cells of different isotypes.
Results and Conclusions Serum IgG autoantibodies against IL-17A/F are present at significant higher frequency in APECED patients respect to age matched normal donors.
IgG auto-antibodies against IL-17A are not significantly different respect to age matched control, indicating that IL-17F or IL-22 could play a more crucial role in protection from candida
Frequency of IgG memory B cells specific for IL-17A, F, A/F and 22 are high in APCED patients compared to healthy controls
ESID-0277 X-Linked Agammaglobulinemia
L. Tricas Aizpun 1, M. Iglesias1, D. Gonzalez2, A.C. Asplund3, C.I.E. Smith3
1Inmunology, Hospital Universitario Central de Asturias, Oviedo, Spain
2Paediatrics, Hospital Carmen y Severo Ochoa, Cangas del Narcea, Spain
3Karolinska Institutet, Clinical Research Center at Novum, Stockholm, Sweden
The patient is an 2-year-old boy without family consanguinity and no similarly affected relatives.
Patient History: At the age of 12 months acute gastroenteritis, at 18 months severe respiratory infection.
Immunological findings: IgG: < 0.33 g/L (normal range (nr): 3.5-8.6 g/L), IgA<0.07 g/L (nr: 0.17-0.96 g/L), IgM: < 0.04 g/L (nr: 0.30-1.83 g/L), IgD: 36 U/ml (nr: 0-70 U/ml) and IgE: 9.0 KU/L (nr: 0-100 KU/L). The functional activity of complement was normal. In peripheral blood we found absence of B lymphocytes (CD19: 0%,). T lymphocytes: 91%, TCD4+ lymphocytes: 69%, TCD8+ lymphocytes: 21% and NK lymphocytes: 5%. Btk expression in monocytes was negative.
Genetic findings: Direct sequencing of the BTK gene revealed the mutation Arginine 255 Stop ( c.C763T). The mother is a carrier of her son´s mutation, but the grandmother is not a carrier of her grandson’s mutation.
Treatment: The patient is receiving gammaglobulin IV replacement and he is doing well.
ESID-0438 Pure Red Cell Aplasia Progressed to Hypoplastic MDS in a Patient with CD 19 Deficiency
M. Cetin 1, S. Unal1
1Division of Pediatric Hematology, Hacettepe University Medical School, ANKARA, Turkey
Herein, we report a currently 16 year-old girl, who presented initially at one month-old with anemia. There was no consanguinity between parents. Bone marrow aspiration was compatible with Diamond-Blackfan anemia (DBA)with erythroid hypoplasia. Parvovirus PCR was negative. CD19 was found to be 0% in lymphocyte subset evaluation. She was transfused and corticosteroid treatment was initiated. She was responsive and was free of transfusions during the course. At follow-up visit of 6 years of age she was found to have decrease in WBC and platelets, in addition to anemia: Hgb 7.1 g/dl, MCV 94 fl, WBC 2500/mm3, platelets 99000/mm3, ANC 900/mm3 and ALS 1400/mm3. Bone marrow aspiration and biopsy was hypocellular. Hepatoplenomegaly was absent. Cytogenetics was normal. Molecular testing for DBA revealed no mutation. Throughout the course CD19 on lymphocytes was 0-1%, CD3: 95%,CD4: 36%, CD8: 57%, CD16+56: 2%. Serum Ig A <6.67 mg/dl, IgG 666 mg/dl, Ig M 48 mg/dl. Corticosteroid was re-initiated but she was unresponsive. Cyclosporine was added, but cytopenias gradually worsened. Hematopoetic stem cell transplantation was performed form unrelated donor and she is currently free of symptoms at +4th year of transplantation. Transient low T cells, reduced T cell repsonses, low B cell number serum immunoglobulins have previously been reported. Our patient is interestin with having low to absent B cell numbers since neonatal period. Ribosomopahies such as DBA have many mysteries and the only impact may not be on eryhtroid lienage.
ESID-0807 DNA Repair Defects Give New Insights in Immune Receptor Repertoire Formation
H. IJspeert1, E.A. Faqeih2, D. van Zessen1, A.P. Stubbs3, K. Schwarz4, A. Jawad1, G. Stewart5, J. Murray6, A.P. Jackson6, M. van der Burg 1
1Dept. of Immunology, Erasmus MC, Rotterdam, Netherlands
2Dept. of Pediatrics, King Saud bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia
3Dept. of Bioinformatics, Erasmus MC, Rotterdam, Netherlands
4Institute for Transfusion Medicine, Ulm University, Ulm, Germany
5Dept. of Cancer Genetics, University of Birmingham, Birmingham, United Kingdom
6MRC Human Genetics Unit Institute of Genetics & Molecular Medicine, University of Edinburgh, Edingburgh, United Kingdom
The antigen receptor repertoires of B and T cells form the basis of the adaptive immune system. They are generated through V(D)J recombination, which is initiated by introduction of DNA double strand breaks by RAG1/RAG2 and repair via the non-homologous end joining (NHEJ) pathway, which consists of DNA end processing (i.e. hairpin opening) and ligation. Patients with inherited defects in NHEJ (Artemis, DNA-PKcs, LIG4, XLF or XRCC4) suffer from immunodeficiency and various neurological abnormalities, although there is considerable clinical heterogeneity.
The aim of our study was to elucidate how genetic defects in NHEJ affect formation of the antigen receptor repertoire, especially the diversity of the complementarity determining region (CDR3) involved in antigen binding.
We performed IGH repertoire analysis using 454 sequencing in 16 patients with genetic defects in NHEJ genes. The data were analyzed using our antigen receptor analysis application (http://galaxyproject.org/).
Artemis and DNA-PKcs deficiencies result in increased numbers of palindromic nucleotides as a result of asymmetric hairpin opening as previously described. Mutations in LIG4, XLF and XRCC4, affecting ligation, all affected the CDR3 length, although in a different manner: large stretches of nucleotide deletions were found (LIG4) or the number of non-templated nucleotides inserted by TdT was significantly reduced (XLF/XRCC4). We are currently analyzing the potential impact on antigen binding and linking the repertoire data to immunophenotype and clinical presentation via integrative analysis.
In conclusion, this study contributes to better understanding of the formation of antigen receptor repertoire diversity, which is the basis of the adaptive immune system.
ESID-0513 Human IgE+ B Cells Are Derived from T Cell-Dependent and –Independent Pathways
M.A. Berkowska1, J.J. Heeringa2, E. Hajdarbegovic3, M. van der Burg1, H.B. Thio3, P.M. van Hagen4, L. Boon5, A. Orfao6, J.J.M. van Dongen1, M.C. van Zelm 1
1Immunology, Erasmus MC, Rotterdam, Netherlands
2Pediatrics, Erasmus MC, Rotterdam, Netherlands
3Dermatology, Erasmus MC, Rotterdam, Netherlands
4Internal Medicine, Erasmus MC, Rotterdam, Netherlands
5Bioceros, BV, Utrecht, Netherlands
6Centro de Investigacion del Cancer and Service of Cytometry Department of Medicine, University of Salamanca, Salamanca, Spain
Background: The prevalence of IgE-mediated diseases increases worldwide. Still, the IgE-expressing B cells are poorly characterized, mainly due to their scarcity and low membrane IgE levels.
Objective: To study the immunobiology of human IgE-expressing B cells in health and allergic disease.
Methods: Stepwise approach for flow cytometric detection and purification of human IgE-expressing B cells in controls, CD40L-deficient patients and patients with atopic dermatitis. Molecular analysis of replication histories, somatic hypermutations (SHM) and Ig class switching.
Results: Using multi-color flow cytometry, we reliably detected IgE-expressing plasma cells and two IgE-expressing memory B-cell subsets. These IgE-expressing cells showed molecular and phenotypic signs of antigen responses. The replication history and SHM levels of IgE+ plasma cells and CD27+IgE+ memory B cells fitted with a germinal center (GC-)dependent pathway, often via an IgG intermediate, as evidenced from Sγ remnants in Sμ-Sε switch regions. CD27-IgE+ cells showed limited proliferation and SHM, and were present in CD40L-deficient patients, indicating a GC-independent origin. Patients with atopic dermatitis had normal numbers of blood IgE+ plasma cells and CD27+IgE+ memory B cell, but increased CD27-IgE+ memory B cells with high SHM loads as compared to healthy controls and patients with psoriasis.
Conclusions: We delineated GC-dependent and GC-independent IgE+ B-cell responses in health, and indicated involvement of the GC-independent pathway in a human IgE-mediated disease. These findings provide new insights into the pathogenesis of IgE-mediated diseases, and may contribute to accurate monitoring of IgE+ cells in patients with severe disease undergoing anti-IgE treatment.
ESID-0196 Primary Antibody Deficiencies in Children with Juvenile Arthritis
S. Velbri 1, K. Uibo2
1immunology, Tallinn Children's Hospital, Tallinn, Estonia
2Rheumatology, Tallinn Children's Hospital, Tallinn, Estonia
IgA and/or IgG subclass deficiency can cause recurrent infections but is also a risk factor for autoimmune and allergic diseases. In our former study we have found autoimmune diseases in 19,5% of children with IgA and/or IgG subclass deficiency.
Aim. To analyse the frequency of juvenile idiopathic arthritis (JIA) in children with antibody deficiencies and the frequency of antibody deficiencies in children with JIA.
Material and methods. 88 children with antibody deficiencies were analysed retrospectively and 80 children (52 girls, 28 boys, 2-20 years of age) with JIA were studied visiting in 2014. JIA children were on steady treatment during 0,5-18 years. Children with JIA were studied clinically and IgG, IgA, IgM, IgG subclasses were determined.
Results. In children with antibody deficiency there was found JIA in 6 cases (6,8%): in one with IgA deficiency, in 3 with IgA/IgG subclass deficiency and in 2 cases with IgG subclass deficiency. In children with JIA there was found antibody deficiency in 25 cases (31%): IgA deficiency in 4 children (5%) besides in 3 cases IgA/ IgG subclass deficiency. Low level of IgG subclasses was ascertained in 21 children (26%) besides in 2 cases was also low IgG and IgA level. More pronounced cases of antibody deficiencies were ascertained in boys.
Conclusions. JIA was diagnosed in 6,8% of cases in children with antibody deficiencies.
Antibody deficiency was found in 31% of JIA children.
Antibody deficiency can be one of the risk factors for development of JIA or sometimes caused by treatment.
ESID-0755 Monocyte XBP1 Hyperexpression and Differential BTK Gene Expression in XLA Patients
V.D. Ramallho1, M.A. Teocchi 1, B.M. Abramczuk1, T.N. Mazzola1, M.M. Vilela2
1Center for Investigation in Pediatrics CIPED UNICAMP, University of Campinas, Campinas SP, Brazil
2Pediatrics Immunology Division and Center for Investigation in Pediatrics CIPED UNICAMP, University of Campinas, Campinas SP, Brazil
Key-Words: immunodeficiency; B cells; X-linked agammaglobulinemia; BTK; unfolded protein response.
Introduction: X-linked agammaglobulinemia (XLA) is characterized by a B lymphocyte differentiation block in the bone marrow, leading to hypogammaglobulinemia with few, or the absence of, peripheral B lymphocytes. Mutations in the BTK gene are responsible for XLA and in most cases lead to low protein expression. Misfolded proteins can trigger stress pathways in the endoplasmic reticulum (ER).
Methods: We evaluated eight male Brazilian patients whose diagnosis was based on recurrent infections, markedly reduced levels of IgM, IgG and IgA, and circulating B cell numbers <2%. BTK mutations were identified by sequencing and the mRNA expression of BTK and ER stress markers was assessed with real-time quantitative PCR (RT-qPCR) technology.
Results: We detected four missense mutations, one nonsense mutation, two frameshifts and one splice site defect. Quantitative real-time detection PCR measurements showed a reduced expression of BTK mRNA in patients with mutations that result in a stop codon. However, we found that missense mutations do not affect BTK mRNA expression. XLA patients showed an increased level of XBP1 mRNA, which could be a mechanism to revert cellular ER stress.
Conclusions: This is the first study relating XBP1 with BTK mutations and XLA. Our data suggest that defective BTK might affect XBP1 expression in monocytes. As a multifunctional transcriptional factor, our finding on XBP1 upregulation in XLA patients opens several possible avenues of research that will help us to understand the complex pathophysiology in XLA.
FINANCIAL SUPPORT: FAPESP and CAPES.Brazil.
ESID-0371 Intravenous Immunoglobulin Replacement Therapy Primes B Cells of Common Variable Immunodeficiency Patients to an Apoptotic Programme
M. Visentini 1, M. Mitrevski1, R. Marrapodi1, L. Todi1, I. Quinti2, M. Fiorilli1
1clinical medicine, Sapienza University of Rome, Rome, Italy
2molecular medicine, Sapienza University of Rome, Rome, Italy
Background: Intravenous immunoglobulin (IVIg) is used as replacement therapy as well as for the treatment of several immune-mediated disorders, although its immunomodulatory effects in vivo are relatively poorly understood. In vitro studies suggest that IVIg modulates a broad range of B-cell responses including signaling, proliferation and apoptosis.
Objective: To investigate whether IVIg administration at replacement dosage modulates BCR/ERK signaling, influences B cell phenotype and promotes B-cell apoptosis in vivo.
Methods: B-cell count and phenotype were analyzed from blood samples obtained immediately before and after IVIg infusion from 16 patients with common variable immunodeficiency (CVID) undergoing IVIg replacement therapy. BCR-induced ERK phosphorylation was measured by PhosFlow. After over-night culture in the absence of stimuli, B-cell apoptosis (measured by flow-cytometry by Annexin-V and 7AAD staining) and expression of CD21 were analyzed.
Results: Immediately after IVIg treatment, circulating B cell counts, unlike those of T cells and NK cells, decreased significantly (p=0,0146), while the percentage of CD21low B cells (p=0.0019) and the level of constitutive ERK signaling increased significantly. After overnight culture, the percentage of CD21low B cells further increased (p<0.0001); the rate of B-cell apoptosis correlated with that of newly generated CD21low B-cells (p=0,0118, r2=0,375).
Conclusion: IVIg induces in vivo anergy-like changes and apoptosis of CVID B cells.
ESID-0542 B-Regulatory Cells in CVID Patients
M. Vlkova 1, O. Ticha1, J. Nechvatalova1, J. Litzman1
1Institute of Clinical Immunology and Alergology, St Anne University Hospital in Brno, Brno, Czech Republic
B-regulatory cells (Breg cells) were described in human as B-cells with the phenotype CD19+CD24++CD38++IL10+. This phenotype that has been previously associated with immature transitional B cells, it comprises the highest fraction of IL-10-producing B-cells upon CD40 stimulation in human peripheral blood in healthy individuals. Breg cells suppress production of TNFα+ and IFNγ+ by CD4+ T-cell. CD40 signalling and the engagement of CD80 and CD86 are pivotal for the generation and function of Breg cells.
Defect of CD40 ligand expression on T-cells and low levels of CD80 on B-cells was described in CVID patients. For these reasons we have examined the number and function of Breg cells in CVID patients. We have investigated the production of IL10+ in B-cells and intracellular TNFα+ and IFNγ+CD4+ T-cells in 28 CVID patients and 24 healthy controls.
We did not find differences between IL10 positive B-cells in CVID patients and control group. When comparing CVID patients with healthy controls the frequency of CD19+CD38++CD24++IL10+ Breg cells were decreased in patients with CVID (p
Our experiment showed that the population of IL10+ B-cells but not CD19+CD38++CD24++IL10+ B-cells inhibit proinflammatory cytokine production by CD4+ T-cells in CVID patients. This inhibition is proportional to the increase of number of IL10+ B-cells in CVID patients.
ESID-0660 Expanding Clinical Spectrum of RAG Deficiencies - Diagnostic Challenge Not Only for Immunologist
B. Wolska-Kusnierz 1, M. van der Burg2, N. Irga3, N. Dabrowska-Leonik4, K. Bernat-Sitarz4, B. Piatosa5, E. Bernatowska4
1Immunology, The Children's Memorial Health Institute, Warsaw, Poland
2Department of Immunology, Erasmus MC University Medical Center Rotterdam, Rotterdam, Netherlands
3Department of Pediatrics Hematology Oncology and Endocrinology, Medical University of Gdansk, Gdansk, Poland
4Immunology, Children's Memorial Health Institute, Warsaw, Poland
5Histocompatibility Laboratory, Children's Memorial Health Institute, Warsaw, Poland
Introduction Hypomorphic mutations in RAG genes with residual V(D)J recombination activity result in expanding spectrum of immunologic and clinical phenotypes including classical and incomplete Omenn syndrome, RAG deficiency ( RAGD) with gammadelta T-cell expansion, RAGD with granulomas. Autoimmune manifestation, most often autoimmune cytopenias, are observed in these 'leaky' SCID or CID phenotypes.
Objective. To present obstacles in establishing early, correct diagnosis of RAGD patients with dominance of autoimmune presentation.
Material. The case histories of two patients aged 3,5 and 4 years were presented. Most attention was paid to analyse prolonged diagnostic process before making suspicion of primary immunodeficiency. Obstacles in in proper assesement interpretation of immune parameter in patients on long-term immunosupression treatment were shown. The low B cell numbers as a consequence of therapy with anti-CD20 in one of the patient was particularly difficult to interprete.
Results. In above cases implementation of long-term immunosuppressive therapy postponed current diagnosis of RAGD. Infectious complications observed in children were explained by secondary immune deficiency. As a consequence, implementation of correct treatment of primary immunodeficiency, including stem cell transplantation, was delayed.
Conclusions Increasing knowledge about clinical spectrum of RAGD becomes a real diagnostic challenge. Awareness of these important issues should be awaken among other specialists. We postulate that every child with autoimmune complications should be routinely screened towards PIDs before any immunosuppressive treatment is implemented.
ESID-0350 Impaired Long-Lived Plasma Cell Generation Results in Hypogammaglobulinaemia in Common Variable Immunodeficiency (CVID).
G. Wong 1, L. Morton1, S. Maisuria2, J. Jones3, M. Goodall4, A. Huissoon3, S. Savic2, R. Tooze2, M. Cobbold1
1School of Immunity and Infection, University of Birmingham, Birmingham, United Kingdom
2Section of Experimental Haematology, University of Leeds, Leeds, United Kingdom
3West Midlands primary immunodeficiency centre, Birmingham Heartlands Hospital, Birmingham, United Kingdom
4Clinical Immunology, University of Birmingham, Birmingham, United Kingdom
Introduction: CVID is considered to be a disorder of terminal B-cell differentiation and many studies have suggested various stages of blockage using immunophenotyping and immunohistochemistry. Recent advances in plasma cell culture provide us with an excellent opportunity to examine the terminal B-cell differentiation of CVID under direct observation.
Methods: Ten healthy donors, fourteen CVID patients and four disease controls were recruited into the study. Plasmablasts (PB: CD20-CD27+CD38++CD138-) and long-lived plasma cells (LLPC: CD20-CD27+CD38++CD138+) were generated from both naïve and memory B-cells using a transwell-system supported by bone marrow fibroblasts. Immunoglobulin output was measured using an in-house sandwich ELISA.
Results: The majority of the differences were noted from the memory B-cell cultures. Compare to healthy controls, memory B-cells from CVID patients were less able to generate PB and LLPCs with time (p=0.0093 and p=0.0022 respectively). In vitro IgG production was significantly lower in CVIDs when compared to healthy controls (p<0.0001), however IgM production was well preserved. A subgroup of CVID patients (n=5) had normal LLPC generation but with impaired IgG production suggesting a more terminal defect. Failure of LLPC generation was more closely associated with clinical inflammatory complications (p=0.021).
Conclusion: By observing the terminal differentiation of B-cells over time in vitro, we identified the failure of LLPC generation from circulating B-cells in the majority of CVID patients. These data suggest that this cellular defect may be present in vivo resulting in attenuated humoral responses. Further work is required to pinpoint the molecular mechanisms leading to this observation.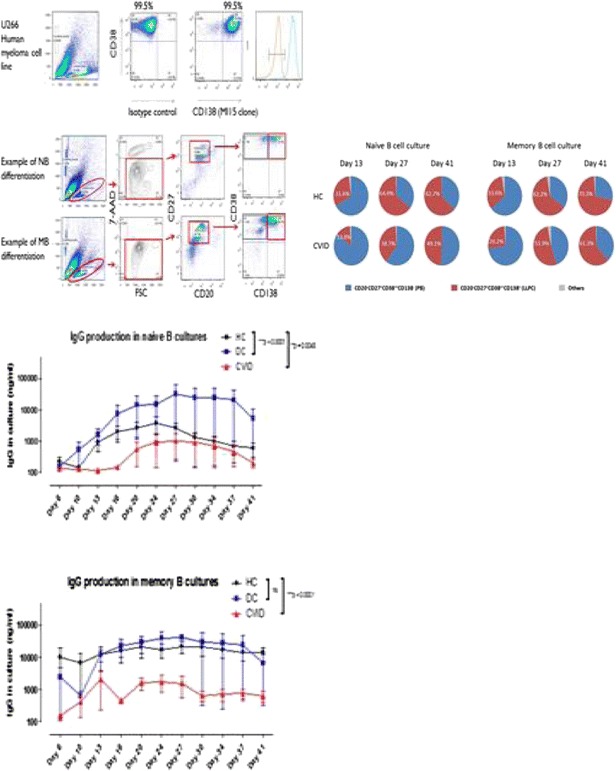
ESID-0761 Comparison of B Cell Precursors Between Bone Marrow Samples From "Infections Only" CVID Patients and Healthy Controls
C. Anzilotti 1, H. Chapel1, A.K. Kienzler1, E. Lopez-Granados1, S. Gooding1, B. Davies2, A. Price2, H. Pandit2, M. Lucas1, T. Littlewood1, M. van der Burg3, S. Patel1
1Nuffield Dept of Medicine, University of Oxford, Oxford, United Kingdom
2Nuffield Dept of Orthopaedics Rheumatology and Musculoskeletal Sciences, University of Oxford, Oxford, United Kingdom
3Dept. of Immunology Erasmus Medical Centre, University of Rotterdam, Rotterdam, Netherlands
Patients with Common Variable Immunodeficiency Disorders (CVID) fail to produce protective amounts of antigen-specific antibodies. This may be caused by defects anywhere along the B cell maturation pathway in bone marrow, blood or secondary lymphoid tissues. In the only previous study of B cell precursors in CVID bone marrow samples, 9 of 25 CVID patients showed a partial arrest in early B cell development but only patients with severe disease were studied as part of clinical management.
We report, for the first time, early bone marrow-dependent B cell development in 10 volunteer CVID patients presenting without any disease-related complications and two with cytopenias. These results are compared with those from samples from individuals without immune disease undergoing orthopaedic surgery or investigated for haematological malignancy. The samples from otherwise healthy donors showed remarkable B cell precursor homogeneity. The phenotypically homogenous 'infections only' CVID patient group showed highly heterogeneous B cell precursor compartments, highlighting the pathophysiological heterogeneity of the commonest/simplest phenotype in CVID. These data add to the view that several different immune mechanisms may be involved in this polygenic condition(1).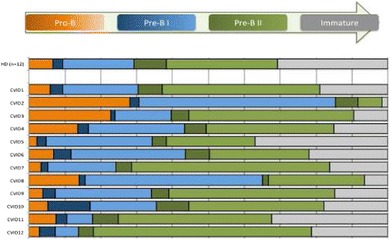
Figure: Composition of the precursor B-cell compartment in CVID patients compared with healthy donors. The precursor B-cell compartment was set at 100% after exclusion of mature B cells. CVID patients show great variability in the size of populations, suggesting partial block in the maturation process at different stages.
1. Driessen G, van Zelm, M., van Hagen, PM., Hartwig, NG., Trip, M., Warris, A., de Vries, E., Barendregt, BH., Pico, I., Hop, W., van Dongen, JJ., van der Burg, M. B-cell replication history and somatic hypermutation status identify distinct pathophysiologic backgrounds in common variable immunodeficiency. Blood 2011;118:6814-23.
ESID-0505 A Primitive Extra-Follicular Pathway of IgM Memory B-Cell Development in Severe Combined Immune Deficiency
A. Aranburu 1, E. Piano Mortari1, F. Capolunghi1, E. Giorda1, V. Marcellini1, S. Cascioli1, F.R. Monzo1, E. Danielli2, C. Capponi2, S. Ceccarelli2, L. Putignani3, F. Del Chierico3, L. Nicolosi4, A. Lanfranchi5, F. Porta5, C. Cancrini6, A. Scarselli6, G. Di Matteo6, A. Finocchi6, P. Rossi6, A. Bertaina7, F. Locatelli7, R. Carsetti8
1B cell development, Children's Hospital Bambino Gesu IRCCS, Rome, Italy
2Diagnostic Immunology Unit Department of Laboratories, Children's Hospital Bambino Gesu IRCCS, Rome, Italy
3Parasitology Unit Department of Laboratories, Children's Hospital Bambino Gesu IRCCS, Rome, Italy
4Department of Pediatrics, Children's Hospital Bambino Gesu IRCCS, Rome, Italy
5Department of Paediatrics, University of Brescia, Brescia, Italy
6DPUO University Department of Pediatrics, Children's Hospital Bambino Gesu IRCCS and University of Tor Vergata School of Medicine, Rome, Italy
7Department of Oncohaematology, Children's Hospital Bambino Gesu IRCCS, Rome, Italy
8B cell development and Diagnostic Immunology Unit Department of Laboratories, Children's Hospital Bambino Gesu IRCCS, Rome, Italy
Mutations of the ILR2G gene cause X-linked SCID (Severe combined immunodeficiency), characterized by lack of NK and T cells with normal B cell numbers. Haematopoietic cell transplantation is the elective treatment that replaces T and NK cells. Insufficient conditioning leads to chimerism with persistence of host B cells that are unable respond to IL-21 and cannot build germinal centers. We have studied the molecular features and function of IgM memory B cells of SCID patients before and after transplantation, developing respectively without or with normal T cells. We measured the function of IgM memory B cells in vitro (response to CpG) and in vivo (antibody production). We extracted RNA from sorted populations and studied VH family usage and frequency of SHM. As comparison we used age-matched controls and transplanted JAK3-SCID patients with complete or partial host engraftment. We report that IgM memory B cells, developed in the absence of T cells, are functional, secrete IgM in vitro and in vivo and carry somatically mutated VH genes. In transplanted X-SCID patients, analysed at 10 years of age, the frequency of SHM increases, but at the molecular level IgM memory B cells remain similar to those of control infants. In 10-year-old controls and in transplanted patients with complete host engraftment, IgM memory B cells have more and complex mutations and show molecular signs of antigen selection suggesting that, after a first wave of differentiation that is independent of T-cells and of IL-21, IgM memory B cells refine their repertoire in the GC.
ESID-0423 Altered B Cell Tolerance and Bone Marrow Egress in ADA-Deficient Mice
N. Carriglio 1, A.V. Sauer1, R. Jofra Hernandez1, B. Di Lorenzo1, V. Di Marzo2, A. Aiuti1
1HSR-TIGET, Ospedale San Raffaele, Milano, Italy
2Endocannabinoid Research Group, Institute of Biomolecular Chemistry - CNR, Pozzuoli, Italy
Genetic defects in the adenosine deaminase (ADA) gene are among the most common causes for severe combined immunodeficiency (SCID). ADA-SCID patients suffer from lymphopenia, absent cellular and humoral immunity, recurrent infections and autoimmune manifestations in milder forms. Currently available therapeutic options for this otherwise fatal disorder include bone marrow transplantation, enzyme replacement therapy or hematopoietic stem cell gene therapy.
B-cell tolerance defects and autoantibody production, characteristic of many autoimmune diseases, have also been associated with autoimmune manifestations in ADA-SCID patients. However bone marrow (BM) B-cell development is less characterized in ADA-deficient mice. We therefore studied their B-cell phenotype focusing on the fate of developing B-cells and in particular the role of the Cannabinoid receptor 2 (CB2) in BM egress. CB2 is a receptor of the endocannabinoid system involved in B-cell differentiation and migration. Newly emigrating B-cells were analysed using an in-vivo pulse labelling technique that allows to distinguish immature and recirculant B-cells present in synovial vessels from B cells present in the parenchyma of the bone. The results showed a significant increase of B-cell egress in ADA-/- compared to wildtype mice. ADA-/- mice also showed an increase of 2-AG levels, the endogenous ligand of CB2 in BM plasma. In-vitro, 2-AG induced a significant increase of ADA-deficient B-cell migration. Under these conditions in vivo, 2-AG stimulation may allow B cells to overcome the central tolerance checkpoint prematurely. Autoreactive B-cells may thereby escape from the central B-cell tolerance selection process in the BM and contribute to the onset of autoimmunity.
ESID-0433 Expression of IgD on Mature Lymphocytes Is Dependent on Zinc-Finger Protein ZFP318
A. Enders 1, A. Short1, L.A. Miosge1, H. Bergmann1, S.J. Lambe1, Y. Sontani1, M. Yabas1, E.M. Bertram2, B. Whittle2, B. Balakishnan2, K. Yoshida3, M.A. Field1, T.D. Andrews1, H. Hagiwara3, C.C. Goodnow1
1Department of Immunology, John Curtin School of Medical Research Australian National University, Canberra, Australia
2Australian Phenomics Facility, John Curtin School of Medical Research Australian National University, Canberra, Australia
3Biomedical Engineering Center, Toin University of Yokohama, Yokohama, Japan
IgM and IgD, the two forms of the B cell receptor on naïve mature B cells, are generated by alternative splicing of long primary RNA transcripts from the immunoglobulin heavy chain (Igh) locus. While immature B cells only express IgM, mature follicular or marginal zone B cells co-express IgM and IgD and self-reactive anergic B cells further down-regulate IgM and mainly express IgD. Despite great efforts, the factor regulating this alternative splicing has not been identified. Through a flow cytometry screen in mice after ENU mutagenesis we identified zinc-finger protein ZFP318 as a critical regulator of IgD expression. Similar to the expression of IgD, expression of Zfp318 mRNA is developmentally regulated with low expression in pro-B cells in the bone marrow, intermediate levels in immature B cells and high expression in mature follicular B cells. Mice with a targeted Zfp318 null allele had a largely normal B cell development into the major mature B cell subsets despite a complete absence of IgD on mature B cells. These findings identify ZFP318 as a crucial factor regulating the expression of the two major antibody isotypes on the surface of most mature B cells.
ESID-0064 Novel NFKB2 Mutation in Early-Onset CVID
Y. Liu 1, S. Hanson1, P. Gurugama1, A. Jones2, B. Clark1, M. Ibrahim1
1Department of Immunological Medicine, King's College Hospital, London, United Kingdom
2Immunology Department, Great Ormond Street Hospital for Children, London, United Kingdom
Common variable immunodeficiency (CVID) is heterogeneous, clinically, immunologically and genetically. The majority of genetic mechanisms leading to CVID remain elusive.
We studied a Greek Cypriot family of non-consanguineous parents and three children. Two children were diagnosed with CVID at an early age. The mother and the eldest sibling are unaffected. Whole exome sequencing of the two affected siblings, unaffected sibling and mother were performed, and revealed 8 bp deletion in the C-terminal part of NFKB2 gene, which leads to a frameshift (p.Asp865Valfs*17) altering 17 C-terminal amino acids from residue 865, and creating a premature stop-codon resulting in a truncated protein, 19 amino acids shorter than wild type (p100Δ19). We validated the results via Dye-termination sequencing and Western blot, and confirmed that the conserved residue at 866 is mutated from serine to arginine in p100Δ19, so the protein expressed from the mutant allele cannot be phosphorylated at this position. As a result, there was less p52 (the mature form of NFκB2/p100) production and nuclear translocation. Using flow cytometry, we further demonstrated that there was a reduction in B cells (CD19+), switched memory B cells (CD27+IgD-) and T follicular helper (Tfh) cells (both CD4+CXCR5+ and CD4+CXCR5Hi) in CVID patient, compared to healthy controls.
These data support the notion that the non-canonical NFκB pathway plays an important role in B cell differentiation, and demonstrate the requirement of an intact non-canonical NFκB pathway for the development of Tfh cells. This paves the way for better understanding of the pathology of CVID.
ESID-0514 Altered B Cell Responses Are Key to the Hyper IgE Phenotype in Job’s Syndrome
F. Rucci1, S. Volpi1, K. Moffitt2, K. Ching3, S. Blasi3, M. Divij1, Y. Fujiwara4, R. Malley2, L. Notarangelo1, J. Manis 5
1Division of Immunology and Manton Center for Orphan Disease Research, Boston Children's Hospital, Boston, USA
2Division of Infectious Diseases, Boston Children's Hospital, Boston, USA
3Division of Laboratory Medicine, Boston Children's Hospital, Boston, USA
4Division of Hematology and Oncology, Boston Children's Hospital, Boston, USA
5Division of Laboratory Medicine/Department of Pathology, Boston Children's Hospital/Harvard Medical School, Boston, USA
Autosomal Dominant Hyper-IgE syndrome (AD-HIES) is a multisystem disorder with immunodeficiency characterized by abnormal susceptibility to skin and pulmonary infections, markedly elevated serum IgE and abnormalities of soft tissues, bones, and teeth, and usually caused by a dominant negative mutation of STAT3. Defects in the Th17 pathway and humoral response are a hallmark of immunological defects observed in Job’s syndrome patients who suffer from recurrent infections commonly due to Staphylococcus, Pneumococcus and/or Candida. The mechanism underlying the humoral immunodeficiency and the characteristic feature of elevated serum IgE in AD-HIES patients remains unsolved. We generated a tissue specific mouse model of AD-HIES and found defective IgG responses to a Th17-dependent S. Pneumoniae vaccine. Serum IgE was massively increased only in mutant mice beginning 7 days after vaccination, but not in response to other T-dependent or T-Independent antigens. Impaired IgG responses after immunization were seen when mutated STAT3 was restricted to either T or B cells, but surprisingly, the increased IgE response was dependent solely on mutant B cells. These findings uncover a previously unrecognized intrinsic B cell defect in HIES causal for elevated serum IgE.
ESID-0323 Mutations in Bruton's Tyrosine Kinase Impair IgA Responses
N. Mitsuiki 1, X. Yang2, S. Bartol3, Y. Kosaka4, H. Takada5, K. Imai1, H. Kanegane2, S. Mizutani1, M. van der Burg3, M. van Zelm3, O. Ohara6, T. Morio1
1Department of Pediatrics and Developmental Biology, Tokyo Medical and Dental University Graduate School of Medical and Dental Sciences, Tokyo, Japan
2Department of Pediatrics, Graduate School of Medicine and Pharmaceutical Sciences Toyama University, Toyama, Japan
3Department of Immunology, Erasmus MC University Medical Center, Rotterdam, Netherlands
4Department of Hematology/Oncology, Hyogo Prefectural Children's Hospital, Hyogo, Japan
5Department of Pediatrics, Graduate School of Medical Sciences Kyushu University, Fukuoka, Japan
6Laboratory for Immunogenomics, RIKEN Research Center for Allergy and Immunology, Kanagawa, Japan
X-linked agammaglobulinemia (XLA) is a primary immunodeficiency caused by mutations in Bruton's tyrosine kinase gene (BTK) and is characterized by decreased peripheral blood B cells and an absence of immunoglobulins. We performed whole exome sequencing in an IgA-deficient-boy. Genetic analysis revealed a BTK mutation (Thr316Ala). The mutation resulted in B cells with reduced BTK expression and high IgM expression. Equal proportions of CD19low and CD19normal were observed; and both included naïve and memory B cells. Calcium influx and phospholipase Cγ2 phosphorylation upon IgM stimulation were marginally impaired in CD19low but not in CD19normal cells. IgA transcripts of B cells with the mutation showed low somatic hypermutation levels, which were also observed in other XLA patients. We concluded the BTK mutation (Thr316Ala) likely underlay the disease in the IgA-deficient-boy, and suggest hypomorphic BTK mutations can result in normal circulating B cell numbers but specifically impaired IgA responses.
ESID-0725 Three Sisters with Agammaglobulinemia Revealing CD79? Deficiency
B. neven 1, P. Skendros2, V. Bertrand2, I. Theodorou2, C. picard3
1pediatric immunology, AP HP Hôpital Necker Enfants Malades, PARIS Cedex 15, France
2immunology, Pitié Salpétrière hospital, PARIS Cedex 15, France
3Study Center of Primary Immunodeficiency APHP, Necker Hospital, PARIS Cedex 15, France
We report here the sixth kindreds of autosomal recessive agammaglobulinemia (ARA) due to a novel mutation in CD79A gene. Pre-B cell receptor assembly is an essential step for B cell differentiation in the bone marrow. Mutations in components of the pre-BCR or in the downstream signaling cascade have been reported to result in the rare forms of ARA (IGHM, CD79A, CD79B, BLNK, IGLL1 and PIK3R1 genes). These primary immunodeficiencies are characterized by absent B cells due to a developmental arrest at the pro-B to pre-B stage. Five patients with AR CD79a deficiency have been previously reported. We report here three sisters from consanguineous Algerian parents. One of them died at the age of 6 months due to respiratory infection, allowing diagnosis of agammaglobulinemia. Due to positive familial history, agammaglobulinemia was diagnosed in the 2 asymptomatic sisters. These both patients have normal T and NK cells counts, but complete absence of the B cells subset. They are well on IV immunoglobulins. The immunological phenotype, the female sex and the parental consanguinity of these patients prompted us to investigate the known genes responsible for ARA. Genetic analysis of the patients revealed a novel nonsense homozygous mutation in CD79A gene, c.197G>A which leads to a premature stop codon, p.T66X, located in the extracellular domain. The healthy parents are both heterozygous for the mutation. In conclusion, this report highlights the role of CD79A in B cell differentiation. Even though ARAs are rare conditions, precise molecular diagnosis is important to allow genetic counseling.
ESID-0441 Expression and Function of SOX5 During Human Late B Cell Development
M. Rakhmanov 1, H. Sic2, M. Rizzi2, M. Seidl3, S. Unger4, H.H. Peter5, R.E. Voll5, U. Salzer5, H. Eibel5, K. Warnatz5
1Advanced Diagnostics Unit, Centre for Chronic Immunodeficiency, Freiburg, Germany
2Eibel Lab, Centre for Chronic Immunodeficiency, Freiburg, Germany
3Department of Pathology, Centre for Chronic Immunodeficiency, Freiburg, Germany
4Warnatz Lab, Centre for Chronic Immunodeficiency, Freiburg, Germany
5Department of Rheumatology, Centre for Chronic Immunodeficiency, Freiburg, Germany
The transcription factor SOX5 plays essential roles in chondrogenesis, neurogenesis and melanogenesis. In humans, it is encoded by at least five different transcript variants. Previous studies showed that SOX5 gene is highly expressed in human tonsillar FCRL4+ memory B-cell subpopulation and innate-like CD21low B-cell subpopulation of patients with common variable immunodeficiency and patients with hepatitis C virus-associated mixed cryoglobulinemia. However, the function of SOX5 in B cells still remains elusive. Therefore, we aimed in this study to investigate the expression and function of SOX5 in human B-cell biology. We identified two new splice variants of SOX5 in human B cells, encoding the known L-SOX5B isoform and a new shorter isoform L-SOX5F. The SOX5 transcripts are highly expressed during late stages of B-cell differentiation, including atypical memory B cells, activated CD21low B cells and germinal center B cells of tonsils. In tonsillar sections SOX5 expression was predominantly polarized to centrocytes within the light zone. After in vitro stimulation, SOX5 expression was down-regulated during proliferation while high expression levels were permissible for plasmablast differentiation. Overexpression of L-SOX5F in human primary B lymphocytes resulted in reduced proliferation, less survival of CD138neg B cells, but comparable numbers of CD138+CD38hi plasmablasts compared to control cells. Thus, our findings describe for the first time a functional role of SOX5 during late B cell development reducing the proliferative capacity and thus potentially affecting the differentiation of B cells during the germinal center response.
ESID-0778 Clinical and Immunological Features of Patients with Gain-Of-Function PIK3CD Mutations in Japan
T. Takashima 1, Y. Tsujita2, T.W. Yeh1, N. Mitsuiki1, H. Kanegane3, S. Kracker4, A. Durandy4, S. Nonoyama2, T. Morio1, K. Imai1
1Pediatrics, Tokyo Medical and Dental University, Tokyo, Japan
2Pediatrics, National Defense Medical College, Saitama, Japan
3Pediatrics, University of Toyama, Toyama, Japan
4Unitè 768, Institut National de la Santé et de la Recherche Médicale (INSERM) Université Paris V René-Descartes, Paris, France
Introduction: PIK3CD is the gene encoding the p110δ subunit of phosphatidylinositol-3-kinase, which is critical for lymphocyte proliferation and survival. Autosomal dominant gain-of-function mutations in PIK3CD gene have been recently reported in patients with hyper IgM syndrome (HIGM) and common variable immunodeficiency (CVID).
Objective: To clarify the clinical and immunological phenotype of patients with gain-of-function PIK3CD mutations in Japan.
Methods: Immunological status was evaluated by 10-color flow cytometry and quantitative analysis of T cell receptor excision circles (TRECs) and signal joint Ig kappa chain recombination excision circles (KRECs).
Results: We identified eleven patients who had a gain-of-function PIK3CD mutation (E1021K) among 175 hypogammaglobulinemic patients (6.3%). Recurrent respiratory tract infections were the most frequent clinical manifestation, followed by lymphoid organ hyperplasia including gastrointestinal tracts. Some patients also had bronchiectasis, opportunistic infections or mild mental retardation. Flow cytometric analysis on peripheral blood revealed elevated proportions of regulatory T cells, follicular helper T cells, transitional B cells, and plasmablasts. CD4+ T cells, naïve T cells, and memory B cells were decreased. Most of patients had low TRECs levels and normal KRECs levels.
Conclusion: Combining these clinical and immunological features, it may be possible to identify gain-of-function PIK3CD mutations from HIGM and CVID patients.
Topic: T Cell
ESID-0032 A Consanguineous Egyptian Family with Hyper Immunoglobulin E Syndrome
D. Abd Elaziz 1, R. Alkady1, S. Meshaal2, R. El Hawary2, N. Galal1, J. Azmy1, A. Elmarsafy1, T. Freiberger3, J. Litzman4, B. Grimbacher5
1Pediatric, Cairo University Hospital, Cairo, Egypt
2Clinical Pathology, Cairo University Hospital, Cairo, Egypt
3Molecular biology and immunology, Central Europe Technology Institute, Brno, Czech Republic
4Clinical immunology and Allerogology, Faculty of Medicine, Brno, Czech Republic
5Rheumatology and clincal immunology, CCI, Freiburg, Germany
Hyper Immunoglobulin E Syndrome (HIE) is a rare primary immunodeficiency disorder characterize by elevated IgE level, recurrent staphylococcal skin abscesses, eczema and pulmonary infections. Autosomal dominant and autosomal recessive forms of this disorder had been described. Most autosomal dominant HIES (AD-HIES) have been found to be due to mutations in STAT3 (Signal transducer and activator of transcription 3); whereas DOCK8 (Dedicator of cytokinesis 8) mutations have been described in patients with autosomal recessive HIES (AR-HIES).
We describe here a consanguineous Egyptian family with three affected siblings ( two males 18 & 16 years old and one female 6 years old ); their clinical picture is that of classical HIGE syndrome; eczema, recurrent staphylococcal skin abscesses that required surgical drainage, lymphadenitis , recurrent chest infections and coarse features. All of them had high eosinophilia > 2 SD for age, CD4 lymphopenia , their IgE levels were 6000 IU/ml, 20,000 IU/ml, and 500 IU/ml respectively. Whereas their HIES scores were (42, 52 and 32 respectively)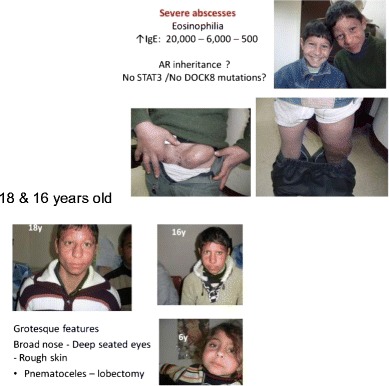
The family was tested for STAT3 and DOC8 mutations and both were negative.
Several cases of HIES with unknown genetic basis are encountered with the challenge of pinning the underlying defect highlighting the need for whole exome sequencing In selective cases.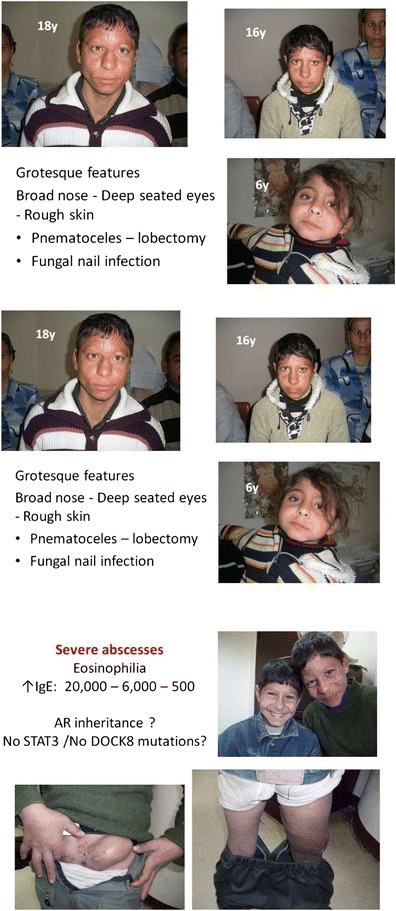
ESID-0071 Severe Combined Immunodeficiency: A Cohort Study of 2 Years Duration from a Single Center (Cairo University Children Hospital)
D. Abd Elaziz 1, S. Meshaal2, R. El Hawary2, R. Alkady1, N. Galal1, J. Azmy1, A. Elmarsafy1
1Pediatric, Cairo University Hospital, Cairo, Egypt
2Clinical and Chemical Pathology, Cairo University Hospital, Cairo, Egypt
Severe combined immunodeficiency (SCID) is a medical emergency, which consists of a heterogeneous group of genetic disorders of humoral and cell mediated immunity, it is characterized by an arrest in T-lymphocyte development. Overall incidence is estimated to be 1 in 75,000 births.
SCID patients are classified according to their peripheral B-lymphocyte population as T-B+and T+B+.
We present 42(26 male 62%, 16 female 38%) patients from 41 unrelated families diagnosed in our center during the past 2 years. The consanguinity rate was 76% among our patients.
SCID patients with T-B- were 55% while with T-B+ were 45%. 23 patients (55%) had history of sibling deaths. The mean age of diagnosis was 8.9 months while the mean age of presenting symptoms was 2.7 months. 3 patients with T-B- SCID presented with Omenn Syndrome like features. Few patients were molecularly diagnosed and the results will be shown. Pneumonia and diarrhea were present in 100% of the patients, 3 cases were complicated with disseminated BCGosis as a complication of BCG vaccine.
The outcome was 100% mortality (4 patients from TB infection as a complication from the BCG vaccine post bone marrow transplantation , the rest of the patients as a complication from the severe infections and disseminated BCGosis) pointing to the need for increased awareness among physicians for early diagnosis as well as early access to transplantation services.
ESID-0503 Clinical Features and Outcomes of 86 Severe Combined Immunodeficiency Patients
M. Afarideh1, A. Ghajar1, B. Mirminachi1, H. Abolhassani2, A. Havaei1, B. Torabi Sagvand1, T. Shokouhfar1, M. Yeganeh1, M.H. Asgardoon 1, M. Gharagozlou3, M. Movahedi3, A.A. Hamidieh4, N. Parvaneh1, N. Rezaei1, A. Aghamohammadi1
1Research Center for Immunodeficiencies, Pediatrics Center of Excellence Children's Medical Center Tehran University of Medical Sciences, Tehran, Iran
2Division of Clinical Immunology Department of Laboratory Medicine, Karolinska Institutet at the Karolinska University Hospital Huddinge, Tehran, Iran
3Department of Allergy and Clinical Immunology, Pediatrics Center of Excellence Children's Medical Center Tehran University of Medical Sciences, Tehran, Iran
4Hematology Oncology and Stem Cell Transplantation Research Centre, Tehran University of Medical Sciences, Tehran, Iran
Background: Severe combined immunodeficiency (SCID) is considered as a heterogeneous group of diseases with different forms of inheritance. It is critical to improve timely diagnosis and effective treatment of SCID patients especially in the oriental countries without screening system and developed public cord blood bank.
Methods: 86 patients diagnosed with SCID were included in the study. A questionnaire was designed to contain all the patients' demographic, clinical and immunological information. The eligible criteria were compromised from the patients with well-established immunodeficiency and the clinical manifestations corresponding to their diagnosis.
Results:The median age at onset was 2 months (range 0 to 48 months) .The median age at diagnosis was 5 months (range 0 to 48 months).The most common presenting manifestations were persistent recurrent oral candidiasis (n=33, 38%), pneumonia (n=32, 37%), failure to thrive (n=29, 34%) and diarrhea. The immunophenotype of 74 patients was recorded in IPIDR registry; 44 were B-cell positive and 30 were B-cell negative.
Conclusion: TCR disarrangement represents the most common form of SCID in Iran.
Keywords: Severe combined immunodeficiency, Mortality, Morbidity
ESID-0764 Type 1 Hyper IgM Syndrome with Novel Mutation from India
R. merchant 1, J. ahmed1, C. Picard2
1pediatric, nanavati hospital, Mumbai, India
2immunology, Necker Hospital, paris, France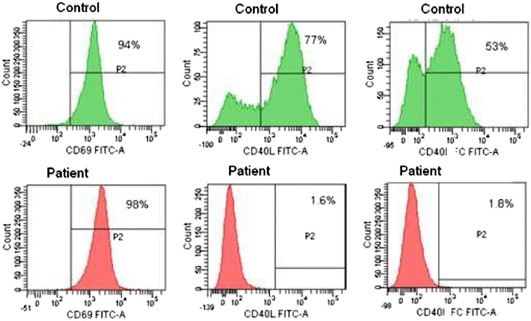
2.5 year child presented for evaluation of recurrent infections and neutropenia. Born to non consanguineous marriage developed pneumonia at age of 1 year with septicemia requiring prolonged ICU stay and mechanical ventilation. His total count was 5400 with 36% neutrophil that time. Child responded to antibiotics but neutropenia persisted. Child used to get recurrent febrile nutropenic ulcers and skin infections (7 episode till 2.5 years).
At the age of 2.5 years immunoglobulin panel was suggestive of Hyper IgM syndrome. Investigation was send for CD40L expression. Child lymphocyte does not express CD40L at all after stimulation. Gene sequencing showed a novel nonsens mutation (at nucleotide level A250T and at protein level R77X) which is never describe till now . This is first case report of mutation study from India in case of Hyper IgM syndrome.
CD40LG has five coding exons and four introns that span over 13 kb. Different allelic variants in intron does not leads to any pathology but 165 pathological CD40L mutation have been described in all the 5 exons. but are particularly common in the TNF-homology domain(exon 5):
? missense mutations (26%)
? nonsense mutations (20%)
? deletions/insertions, splicing mutations, and partial- or whole-gene deletions or large insertions.
Child was started on regular immunoglobulin replacement therapy preparing for transplant and does not have any further infection or neutropenic episodes (9 month of observation). His growth parameter also normalized. He underwent successful 6/6 matched genoidentical sibling transplant and is off transfusion or infection for past 2.5 years with 80% chimerism
ESID-0717 Mechanism of Down Regulation of Treg Cells Induced by Hyeproxia Among Neonates
M. Ahmed 1, H. Patel1, A. Leon Hernandez1, M.A.R.K. Pezzano2
1Pediatrics/Neonatology, Cohen Chidlren hospitals at New york, New York, USA
2Biology Department, The City College of NY, New York, USA
Oxygen is the most common therapy used in NICU. The potential effect of hyperoxic exposure has become a focus of concern. In our previous studies, we found decreased levels of CD4+ cells and Tregs (FOXP3+) in the thymus of hyperoxia exposed neonatal mice. Chemokines such as CCL19 and CCL21 and their receptors CCR7, CCR9 and CxCR4 are crucial for development of Treg cell in the thymus. In this study, we try to find out the mechanism by which hyperoxia decreases Treg cells in neonatal mice thymus.
Neonate mice were housed either at room air (FiO2 21%), or hyperoxia (FiO2 95%) for five days. After exposure, thymus immunostainined for FOXP3, CD4, CCL19 and K14. Chemokines CCL19 & 21 expression was assayed using RT-qPCR. CCR7, CCR9, CxCR4 and FOXp3 were assayed using for flow cytometery.
Immunostaining showed arrest of FOXP3 positive cells at the cortico-medullary junction and increased expression of CCL19 in cortical area of thymus of hyperoxia group. CCL19 and CCL21 mRNA levels were higher in hyperoxia group. CCR7, CCR9 and CxCR4 chemokine receptors were significant decrease on double negative (CD4-CD8-) cells. CCR7 expression was also significantly decreased on FOXP3 expressing double positive cells.
Suppression effect of hyperoxia on Treg development and maturation is possibly due to down regulation of receptor (CCR7, CCR9 and CXCR4) expression in the early developmental stage. Treg cells arrest in the cortico-medullary junction, even with higher expression of chemkines, is possibly related to low CCR7 expression on CD4+ CD8+ double positive cells.
ESID-0790 Screening Di George Syndrome with “A Single Tube of Blood” in Cases with Congenital Cardiopathies
G. Aksu 1, N. Edeer Karaca1, O. Altun Koroglu2, E. Karaca3, E. Toprak Kanik4, E. Levent5, F. Ozkinay3
1Pediatric Immunology, Ege University Faculty of Medicine, Izmir, Turkey
2Neonatology, Ege University Faculty of Medicine, Izmir, Turkey
3Medical Genetics, Ege University Faculty of Medicine, Izmir, Turkey
4Pediatrics, Celal Bayar University Faculty of Medicine, Manisa, Turkey
5Pediatric Cardiology, Ege University Faculty of Medicine, Izmir, Turkey
Di George syndrome (DGS) is caused by a defect in chromosome 22 (22q11.2 deletion) and include congenital heart defects, hypoparathyroidism and thymic hypoplasia or aplasia leading to T-cell immunodeficiency. We aimed to screen and determine the incidence of Di George syndrome with "only one tube of blood sample" in children with congenital heart anomalies in our population.
Children who were found to have a cardiac defect during routine visits in pediatric cardiology and neonatology departments were included . Cases with known genetic syndromes and newborns younger than 32 gestational weeks of age and small for gestational age (birth weight <2500 gr) were excluded. A total of 136 patients were included (F/M: 42/58%). Ages ranged between 0-80 months (10.2±15.9 months). Parental consanguinity was 17% (n=23) in the study group. The majority of patients were diagnosed after a murmur was heard during the routine physical examination (n=62, 46%). Five patients (3%) were diagnosed antenatally. Remaining clinical signs on admission were; respiratory distress (n=33, 24%), tachycardia (n=20, 15%) and central cyanosis (n=16, 12%).
22q11.2 deletion was ascertained in 7 (5.1%) patients; these patients were diagnosed to have Tetralogy of Fallot (28.6%), truncus arteriosus (14.6%), Double outlet right ventricle (14.3%), VSD (14.3%) and VSD-ASD (14.3%).
Our data showed that the frequency of 22q11.2 deletion was 5% in patients with known cardiac defects. A single tube of blood is enough for flow cytometric and genetic analyses, sudies with higher number of subjects will give the exact incidence of DGS in children with cardiopathies.
ESID-0509 Upregulation of DUSP6 in Naïve CD4 T-Cells of Adults Submitted to Total Thymectomy Early in Life
A.S. Albuquerque 1, S.L. Silva1, P. Matoso1, B. Charmeteau2, R. Anjos3, M. Abecasis3, R. Cheynier2, R.M. Victorino1, A.E. Sousa1
1Centro de Imunodeficiências Primárias, Instituto de Medicina Molecular Faculdade de Medicina Universidade de Lisboa Portugal, Lisbon, Portugal
2Département d'Immunologie-Hematologie, Institut Cochin INSERM U1016-CNRS UMR8104-Université Paris Descartes Paris France, Paris, France
3Departamento de Cirurgia Cardíaca Carnaxide Portugal, Hospital de Santa Cruz, Lisbon, Portugal
Thymic output contributes for the expansion of peripheral T-cell pool during childhood and its life-long maintenance. Nevertheless, individuals thymectomized early in infancy feature relatively preserved naïve T-cells during adulthood.
We investigated possible underlying mechanisms by studying adults with total thymectomy (TT) based on surgical report and sjTREC levels below the lower limit of age-matched healthy controls (HC). Patients submitted to partial thymectomy (PT) and HC were evaluated in parallel. Despite the significant contraction of the naïve compartment as compared to both PT and HC, TT patients remain without clinical manifestations of immunodeficiency.
Naïve CD4 T-cells were purified in order to quantify the expression of molecules relevant for cell quiescence, survival and cycling, namely: KLF-2, Foxp1, p21, DUSP4, DUSP6, Bim, Bcl-2 and Ki-67.
The frequency of cycling cells within naïve CD4 was significantly higher in TT as compared to both HC and PT, without differences in Bim transcriptional levels, suggesting no increased susceptibility to apoptosis.
The most significant difference in TT was elevated DUSP6 transcriptional levels, a marker of immunesenescence in aged individuals, which were similar in PT and HC. By targeting ERK, this phosphatase raises the threshold for T-cell activation, preventing cells to be activated.
In conclusion, we showed that the lack of thymus was associated with increased levels of DUSP6 in naïve CD4 T-cells, which on one hand may reduce the purge of naïve cells into memory pool, but on the other hand may limit the ability of these young adults to mount responses to pathogens or vaccines.
ESID-0471 Digeorge Syndrome-Like Phenotype with Severe Transient Combined Immunodeficiency: A Case Report
A. ALI 1, A.A. BILKIS1, J. ROHANA1, A.L. HASNIAH1, A.H. ABDUL LATIFF2, L.M. NOH1
1Department of Pediatric, Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia
2Pantai Hospital Kuala Lumpur, Bangsar, Kuala Lumpur, Malaysia
Introduction: We report the case of transient combined immunodeficiency in DiGeorge syndrome (DGS)-like phenotype girl with abnormal skin condition.
Case Presentation: A 2-year-old Malay girl of non-consanguineous parents presented with recurrent infection and ventricular septal defect since birth. She was dysmorphic with abnormal skin appearance. She had lymphopenia in early neonatal period with T-cell defect (T-B+NK+) and absent thymus, complicated with post-infective bronchiolitis obliterans. Her serum immunoglobulin was unremarkable at birth but at 6 months of age with discontinuation of IVIg for 3 months, the IgG level was reduced (262 mg/dL). A diagnosis of severe combined immunodeficiency (SCID) was entertained. By 12 months of age, her lymphocyte immunophenotype had normalized. It was argued that she could also have a transient hypogammaglabolinemia as well. At age 22 months Isohemeaglutinin A titres were at 1:16 dilution . At this point a cessation of IVIg was attempted. She remained well with no reduction of her serum IgG below the normal range for more than 3 months. We were convinced that her T-cell defect and hypogammaglobulinemia was transient. Genetic study for DGS showed no visible deletion of loci N25 and TUPLE 1 at region of Chromosome 22. SCID mutation analysis for JAK3, RAG1, RAG2 and Artmis were also negative.
Conclusion: We are possibly describing a variant mutation of DGS with transient immunodeficiency and abnormal skin appearance. Further genetic analysis for DGS-like phenotypes associated with mutations in genes not included in del22q11 need to be explored.
ESID-0392 Mucosal-Associated Invariant T Cells Participate in the Development of Autoimmune and Inflammatory Complications in Common Variable Immunodeficiency
S. Arduini 1, D.G. Doherty1, C. Feighery2
1Department of Immunology, Trinity College Dublin, Dublin, Ireland
2Department of Immunology, St. James's Hospital, Dublin, Ireland
Common Variable ImmunoDeficiency (CVID) is a primary immunodeficiency characterized by low immunoglobulin titres, recurrent infections and frequent idiopathic complications. We investigated the role of innate T cells in CVID, as their contribution to the disease has hitherto been largely overlooked.
After obtaining written informed consent, blood was collected from 27 healthy controls and 23 CVID patients attending St. James's Hospital, Dublin. The frequencies of B and T cell populations, including Vδ1, Vδ2 and Vδ3 γδ T cells, invariant Natural Killer T (iNKT) cells and Mucosal-Associated Invariant T (MAIT) cells, were determined by flow cytometry. MAIT cell functionality was assessed stimulating whole blood with anti-CD3 and -CD28 antibodies or PMA and ionomycin. Clinical data were collected from medical records.
γδ T cells were found at normal frequencies in patient blood, however iNKT and MAIT cells were depleted (p<0.0001 and p<0.01, respectively). The frequencies of MAIT cells, but not iNKT cells, were especially low in patients with complicated CVID (p<0.05). Regardless of complication status, MAIT cells from patients appeared more activated in unstimulated blood and produced IL-17a, IL-22 and TNFα significantly more frequently than healthy controls following stimulation with CD3 and CD28.
MAIT cells possess recently discovered anti-bacterial functions, while IL-17a and IL-22 are key both in infection resolution and in the development of inflammatory and autoimmune conditions. We propose MAIT cells are drawn to the peripheral tissues of CVID patients due to the high availability of bacterial antigen. There, their strong cytokine secretion would contribute to the onset of complications like psoriasis and enteropathy.
ESID-0402 Severe Combined Immune Deficiency Due To Heterozygous Compound Mutation in Rag1 Gene
H. Artac 1, H. Ozdemir1, A. Ceylan1, V. Uygun2, A. Yesilipek2, S. Guner3, I. Reisli3, M. van der Burg4
1Pediatric Immunology and Allergy, Selcuk University Medical Faculty, Konya, Turkey
2Pediatric Hematology and Oncology, Antalya Medical Park Hospital, Antalya, Turkey
3Pediatric Immunology and Allergy, Necmettin Erbakan University Meram Medical Faculty, Konya, Turkey
4Molecular Immunology, Erasmus Medical Center, Rotterdam, Netherlands
Introduction: Genotype-phenotype correlation in patients with RAG deficiency reported in the literature and compound heterozygous mutation generally lead to classical Omenn syndrome. We report a male with severe combined immunodeficiency (SCID) caused by heterozygous compound mutations in Rag1 gene.
Case: The 4-month-old patient presented with bronchopneumonia complicated by respiratory failure. He had no erythrodermic desquamation and organomegaly. He is the first child of consanguineous parents. He had no lymphopenia at time of diagnosis (5.2x109/l). High levels of peripheral blood CMV DNA (36500 copy/ml) copy was detected. Immunological evaluation showed agammaglobulinemia and absence of CD19+ B cell. Flow-cytometric analysis revealed a reduced absolute number of T cell of 0.3x109/l (normal range 2.4-8.1x109/l). He has diagnosed as severe combined immunodeficiency and started intravenous replacement therapy. His genetic analysis demonstrated the mutations c.433delG and c.2326C>T in Rag1 gene. Bone marrow transplantation was done from his mother and he is still on follow up.
Discussion: This case illustrates that patients with compound heterozygous mutations in RAG1 gene could be presented with classical SCID phenotype.
ESID-0194 Protective Vaccination Responses in an Infant with Severe LAD1 Deficiency
K. Assing 1, C. Nielsen1, M.A. Jakobsen1, U. Hartling2, N. Fisker2
1Department of Clinical Immunology, Odense University Hospital, Odense, Denmark
2H.C.Andersen Children's Hospital, Odense University Hospital, Odense, Denmark
Lack of anti-protein vaccination responses has been reported in complete LFA-1 deficiency. We report a first born boy, to unrelated healthy Danish parents, who, following a normal pregnancy, presented with leuko-/granulocytosis (48.6/30.6 x 109/L), B (4.2 x 109/L) and T (9.6 x 109/L) lymphocytosis. Omphalitis was ongoing for two months and the umbilical cord did not separate until age five weeks. At 11 months, the boy presented a truncal hypergranulating and partly necrotic ulceration complicated by perforation and bleeding, but no pus formation. Biopsy showed inflammation with necrosis but lack of granulocytes. A lymphocyte marker study, at 12 months, revealed normal percentages of CD19+CD27+IgD- switched memory B cells (1%) and CD4+CD45RO+ memory T cells (8%). Peripheral T follicular helper cell (CD4+ CD45RA- CCR7+ CXCR5+) levels (22% of CD4+) slightly exceeded those of an adult control (17% of CD4+). Vaccination status, at 12 months, showed protective repsonses to Haemophilus influenzae type B, Clostridium tetani, tridecavalent Strep. pneumonia and VZV. Flow cytometry revealed the total absence (0%) of CD18 (integrin β2 chain) expression on neutrophil granulocytes. Father and boy carried a c.817G>A mutation leading to a Gly273Arg amino acid shift not supportive of CD11a/CD18 heterodimerization. No mutations were found in the ITGB2 gene (chromosome 21q22.3) derived from the mother. Genetic investigations will determine whether the mother carries a deletion or whether the boy is a carrier of uniparental isodisomi on chromosome 21. To our knowledge, this is the first case of a severe LAD1 deficiency mounting protective anti-protein vaccination responses.
ESID-0363 Quantitative Analysis of Antigen-Specific T-Cell Responses to Common Pathogens in CVID Patients
E. Bateman 1, R. Sadler1, S. Patel2, J. Miller2, S. Misbah2, B.L. Ferry1
1Department of Immunology, Oxford University Hospitals NHS Trust, Oxford, United Kingdom
2Clinical Immunology, Oxford University Hospitals NHS Trust, Oxford, United Kingdom
T-cell defects have been identified in CVID patients, as well as the traditionally recognised B-cell defects. These defects include reduced naive CD4 and CD8 T-cells, and reduced proliferative and cytokine responses to mitogens and recall antigens. There has been poor overall assessment in the literature of quantitative antigen-specific CD4+ T-cell responses to multiple common pathogens in CVID patients. To further investigate this, antigen-specific CD4 T-cell responses were measured in a whole blood flow cytometric assay utilising dual expression of OX40 and CD25. Antigen-specific responses to CMV, EBV, Candida, JCV, Streptococcus pneumonia and PHA as a mitogen were assessed in 43 CVID patients and 43 age and gender matched healthy controls. Cytokine production was assayed on supernatants by multiplex technology. T-cell memory population analysis was performed by flow cytometry.
CMV, S.pneumoniae, JCV and Candida responses were all significantly lower in the CVID group compared to the control group, with CMV responses showing the most significant decrease (p<0.001). There was a significant reduction in IFN-γ production in the CVID group compared to controls. There were significant correlations of CD4+ effector (r=0.68 p<0.002) and CD8+ naive memory (r=-0.84 p<0.0001) with CMV responses in healthy controls not replicated in CVID patients (r=0.15 p>0.05 and r=0.33 p>0.05 respectively).
This cohort of CVID patients shows significant changes to their antigen-specific CD4+ T-cell repertoire to a number of pathogens. CMV responses were the most significantly different compared with controls. This may indicate a lack of regulation and development of the T-cell memory compartment to chronic pathogens.
ESID-0348 Underlying Molecular Genetic Defects of Severe Combined Immunodeficiency in Tunisia
I. Ben-Mustapha 1, A. Tezeghdenti1, L. Al Baik2, S. Al-Hissi2, B. Larguèche1, A. Sammoud3, N. Ben-Jaballah3, F. Mellouli4, M. Béjaoui4, H. Al-Mousa5, A. Hawwari2, M.R. Barbouche1
1Immunology, Pasteur Institute of Tunis, Tunis, Tunisia
2Section of ImmunoGenetics Department of Genetics, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
3Pediatrics, Children Hospital, Tunis, Tunisia
4Pediatrics, National Bone Marrow Transplantation Center, Tunis, Tunisia
5Allergy and Immunology Department of Pediatrics, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
Introduction: Severe combined immunodeficiency (SCID) is the most severe form of PID, characterized by complete absence of T cell-mediated immunity, resulting in a broad-spectrum susceptibility to multiple pathogens. There is considerable genetic heterogeneity, as 14 different conditions all resulting in a SCID have been fully characterized.
Objective: To identify the molecular genetic defects of SCID patients in Tunisia.
Methods: Among forty-four SCID patients diagnosed, 16 patients for whom genetic material was available have been studied. Based on the immunological phenotype, patients were analyzed for gene mutations using specific primers followed by direct sequencing of the coding regions of the different candidate genes (RAG1, RAG2, ARTEMIS, ADA, IL2RG, JAK3 and IL7RA).
Results: T-B+ was the predominant phenotype found in 13/16 patients (81%). Three different IL2RG mutations have been found in eight patients belonging to three families. They include two previously reported mutations (p.R289X p.Trp237X) and a novel one (p.Trp74X). IL7Ra defect was identified in three patients from a multiplex family. The same p.C446W RAG2 mutation was found in three T-B-patients belonging to two unrelated families.
Conclusion: Despite the high rate of consanguinity in Tunisia favoring autosomal recessive forms, X-linked inheritance remains well represented among Tunisian SCID patients. No genetic defects were identified in 2 patients screened for the 7 candidate genes for whom total exome sequencing should be attempted.
ESID-0250 During HIV Infection Immune Reconstitution Restores the Quality of Vgamma9Vdelta2 T Cell Response
R. Casetti 1, G. De Simone1, A. Sacchi1, A. Rinaldi1, D. Viola1, C. Agrati1, V. Bordoni1, E. Cimini1, N. Tumino1, F. Martini1
1Laboratory of Cellular Immunology, National Institute for Infectious Diseases "L.Spallanzani", Roma, Italy
Scientific background. Alteration of gammadelta T-cell in peripheral blood is among the earliest defects in HIV infection. We asked whether polyfunctionality is also affected according to treatment failure. We performed a cross-sectional study analyzing HAART-treated HIV+ individuals.
Methods. To evaluate functional- Vgamma9Vdelta2 T-cell of HAART-treated HIV+ persons and healthy donors (HD), cytokine/chemokine secretion (IFNγ, IL-2, TNFα/MIP1-β) and cytotoxicity (CD170A) were assessed by flow-cytometry.
Results. We found positive correlation between Vgamma9Vdelta2 T-cells and functional- Vgamma9Vdelta2 T-cell. Functional- Vgamma9Vdelta2T-cell resulted correlated to CD4 T-cells. Moreover, functional- Vgamma9Vdelta2T-cell was lower in HIV patients than HD. Three-, bi- and mono-functional Vgamma9Vdelta2 T-cell subsets were lower in HIV+ patients as well as CD107A- and IFNγ-producing Vgamma9Vdelta2 T-cell subsets. In particular 3+(CD107A+IFNγ+MIP1-β+), 2+(CD107A+IFNγ+), 2+(CD107A+MIP1-β+), 1+(CD107A+)Vgamma9Vdelta2 T-cell subsets resulted lower. Grouping the HIV+ patients according to CD4 cell-count, H(igh)-CD4 and L(ow)-CD4, functional Vgamma9Vdelta2 T-cell was lower in L-CD4 patients if compared to H-CD4 patients and HD. L-CD4 patients expressed a lower absolute number of bi-functional (IFNγ+MIP1-β+)Vgamma9Vdelta2 T-cell subset respect to H-CD4 patients and HD. This population is also lower in progressor patients and in immunological non-responder individuals if compared to HD and virological non-responder patients.
Conclusion. Our results have shown that functional Vgamma9Vdelta2 T-cells were lower in patients with low CD4 T-cell count demonstrating a correlation between functional- Vgamma9Vdelta2 T-cell and immunological reconstitution. These findings support the view that intact Vgamma9Vdelta2 T-cell response capability is important in controlling HIV disease. Recovering Vgamma9Vdelta2-T cell integrity could be a goal for new therapeutical strategy to fight HIV infection.
ESID-0590 A Case Series of Severe Combined Immunodeficiency (10 Cases) from Mofid Children's Hospital in Iran
Z. chavoshzadeh natanzy 1, M. mansouri2, D. babaei2, M. mesdaghi2, M. amirmoeini2, A. karimi3, S. armin3, A. fahimzad3, B. bashardoust2
1allergy, Shahid beheshti university of medical sciences Pediatric Infections Research Ce, Tehran, Iran
2allergy, shahid beheshti university of medical sciences, Tehran, Iran
3infectious, shahid beheshti university of medical sciences, Tehran, Iran
Severe combined immunodeficiency is a group of disorders that leads to early childhood death as a result of severe infections . The different forms of SCID are classified to the presence or absence of T,B, and NK cells. Survival ultimately depends on the reconstitution of immune system with BMT.
We report 10 cases of SCID with some special clinical and laboratory presentations.
Methods: 10 patients with SCID diagnosis was selected and clinical and laboratory results of them studied.
Results: Patients were 6 male and 4 female and between 2 -12 months age. Age of their presentations was between 1-7 months. 60% of patients had cousin parents . 4 cases were diagnosed due to history SCID in other siblings. Clinical manifestations include :FTT(70%), severe lung infections(60%), necrotic lesion in mouth in one patient ,disseminated BCG infections in 3 patients( one of them had granulomatosis skin lesions with AFB in culture ,pesos abscess in the another one).
All of the patients had lymphopenia. 90% had low level of IgG,IgM,IgA . Flucytometery showed (T ¯ B+K ¯ in 3 patients, T ¯ B ¯ K+ in 4 cases, T ¯ B ¯ K ¯ in one cases .T ¯ B+K+ in 2 patients).
10 patients were referred to BMT ,but 6 patients were died due to severe infections before BMT.
Conclusion: Report these numbers of cases from one center guided us to importance of early diagnosis and treatment of these patients with BMT and need to use screening tests for early diagnosis before infection complications.
ESID-0368 A Novel Mutation in ICOS Presenting with Hypogammaglobulinemia, Enteropathy, and Susceptibility to Opportunistic Pathogens
J. Chou 1, M.J. Massaad1, B. Cangemi1, W. Bainter1, W. Al-Herz2, R.S. Geha1
1Division of Immunology, Boston Children's Hospital, Boston, USA
2Department of Pediatrics Faculty of Medicine, Kuwait University, Kuwait City, Kuwait
Introduction. Combined immunodeficiencies (CID) are characterized by impaired lymphocyte function and susceptibility to opportunistic infections.
Objective. To determine the molecular cause of CID in a patient with hypogammaglobulinemia, reduced memory B cells, and normal lymphocyte proliferation, who presented with persistent enteropathy and Pneumocystis jiroveci pneumonia.
Methods. Whole exome sequencing (WES) was performed on the patient. Flow cytometry was used to evaluate protein expression and immunophenotyping.
Results. WES revealed a homozygous 1 base-pair deletion in ICOS causing a frameshift and premature truncation in exon 2, which was confirmed by Sanger sequencing in the patient and his sister, who has hypogammaglobulinemia, chronic diarrhea, low memory B cells, and low CD4+CXCR5+CD45RO+ T follicular helper (Tfh) cells. The mutation abolishes ICOS expression in both patients. ICOS is needed for the development of germinal center (GC)-associated CXCR5+ Tfh cells and GC B cells. The patient’s enteropathy resolved after HSCT, a novel finding indicating that his colitis was driven by hematopoietic cells. In contrast, his sister, who has remained untransplanted, has persistent diarrhea.
Conclusion. Although this mutation caused classical features of ICOS deficiency, including hypogammaglobulinemia, low memory B cells, and low CXCR5+ Tfh cells, chronic enteropathy and the proband’s opportunistic infection suggest that ICOS deficiency is not solely a disease of defective class-switching. The ICOS-ICOSL interaction may be important for macrophage clearance of P. jiroveci and a loss-of-function ICOSL polymorphism is associated with Crohn’s ileitis. It is important to consider antibiotic prophylaxis for opportunistic infections in ICOS-deficient patients and HSCT for those with severe enteropathy.
ESID-0200 Digeorge-Like Syndrome in a Child with a 3p12.3 Deletion Involving MiRNA-4273 Born to a Diabetic Mother
E. Cirillo 1, V. Gallo1, G. Giardino1, G. Galasso1, R. Romano1, R. D'Assante1, R. Genesio2, A. Baldini2, L. Nitsch2, C. Pignata1
1Translational Medical Sciences, Università "Federico II", Naples, Italy
2Molecular Medicine and Medical Biotechnology, Università "Federico II", Naples, Italy
Introduction: Chromosome 22q11.2 deletion is the most commonly chromosomal alteration associated with DiGeorge syndrome (DGS). However, 22q11.2 deletion is not the only underlying cause of DGS. Recently it has been reported that miRNA may modulate the expression of critical T-box transcriptional regulators during midface development and the Bmp-signalling.
Objective: To describe the immunological and molecular phenotype of a child with DGS-like syndrome born to a diabetic mother.
Methods: Cytogenetic and molecular analysis includes fluorescent in situ hybridization (FISH), Array-CGH and TBX1 sequencing. Lymphocyte subpopulations were studied by flow-cytometry and PBMC proliferation by standard method.
Results: a 6-year-old Caucasian male was admitted for thymic aplasia, recurrent airway infections, hypoparathyroidism, renal agenesis, patent oval foramen, septum pellucidum cyst, language delay and dysmorphisms. The immunological evaluation revealed normal serum Ig levels. The immunophenotype revealed a reduction of CD3+ (373cell/mm3), CD4+ (163cells/mm3), CD8+ (388cells/mm3), CD3+CD4+CD45RA (47cells/mm3), CD3+CD4+CD45RO (116cells/mm3) cells. B-lymphocytes were increased (1352cells/mm3), while CD56+ were normal. The proliferative response to PHA and Pockweed was decreased, corresponding to the 45% and 39% of the control, respectively. FISH analysis and TBX1 sequencing were negative. Array CGH revealed a 371Kb-interstitial deletion at 3p12.3 involving the ZNF717, MiRNA-1243 and 4273 genes. Among the MiRNA-4273 predicted target-genes, we found Bone Morphogenetic protein-3 (BMP-3), involved in several steps of embryogenesis, as in kidney and lung organogenesis.
Conclusions: We report on a novel association between a DG-like phenotype and a 3p12.3 chromosomal deletion in an infant born to a diabetic mother, although the causal relationship remains to be proved.
ESID-0683 A Founder Splicing Mutation in CD3D in a Tab-Tgd+B+NK+SCID Pedigree of Ecuadorian Descent
L. Martinez-Martinez 1, R. Cedeño2, A.V. Marin3, M.V. Rubiales1, A. Jiménez-Reinoso3, M.J. Recio3, P. Cabrera4, I. Badell5, J.R. Regueiro3, O. de la Calle-Martín6
1Immunology, Hospital de Sant Pau, Barcelona, Spain
2Pediatrics, Hospital Oncológico Dr. Julio Villacreces Colmont, Portoviejo, Ecuador
3Immunology, School of Medicine Complutense University, Madrid, Spain
4Cytometry, Cytolab, Cuenca, Ecuador
5Pediatrics, Hospital de Sant Pau - Universitat Autonoma de Barcelona, Barcelona, Spain
6Immunology, Hospital de Sant Pau - Universitat Autonoma de Barcelona, Barcelona, Spain
Introduction: Severe Combined Immunodeficiency (SCID) is a group of rare and fatal-inherited disorders characterized by early onset of infections and diminished T cell numbers and/or strongly impaired T cell function.
Aims: To identify the genetic and molecular alteration in an Ecuadorian patient with T±B+NK+SCID.
Methods: The patient, a 6-month-old girl of nonconsanguineous parents, presented with fever, eczema, and respiratory distress. CMV pneumonia was diagnosed, and active CMV infection also caused chorioretinitis. The patient also presented regional lymphadenitis following BCG vaccination (BCGitis). Lymphopenia, and severe hypogammaglobulinemia were observed. Lymphocyte subpopulations were determined by flow cytometry. CD3 gamma and delta genes were analyzed in DNA from patient and parents's peripheral blood leukocytes.
Results: The patient showed a severe selective reduction in peripheral blood αβ T lymphocyte numbers (both CD4+ and CD8+). In contrast, γδ T cells as well as B and NK lymphocytes were detected in normal numbers (Tαβ-Tγδ+B+NK+ phenotype). TCR and CD3 expression levels were strongly affected. Genomic DNA sequencing detected a homozygous G-to-A mutation at position +5 in the 5' splice donor site of intron 2 (IVS2+5G>A). The patients' parents were carriers of the same CD3D mutation. This mutation has been previously described in two non-related nonconsanguineous families of Ecuadorian descent. The 3 families reported to date are unrelated, but had a similar geographic origin (Manabí region), indicating that they likely shared a frequent founder mutant allele.
Conclusions: We report a T±B+NK+SCID patient belonging to a complex pedigree with a homozygous splicing mutation in the CD3 delta gene (c.274+5G>A).
ESID-0414 The Role of STAT3 in the Differentiation and Function of Unconventional T Cells
E.K. Deenick 1, M.L. Ives1, R.P. Wilson1, G. Uzel2, S.G. Tangye1
1Immunology, Garvan Institute of Medical Research, Darlinghurst, Australia
2Laboratory of Clinical Infectious Diseases, NIAID NIH, Bethesda, USA
Introduction: Unconventional or innate-like T cells include γδ T, natural killer T (NKT) and mucosal-associated invariant T (MAIT) cells. MAIT and γδ T cells are a major component of the human immune system, comprising 1-15% of peripheral blood lymphocytes and have been implicated in responses to many pathogens. However, our knowledge of their precise role in protective immunity and the molecular mechanisms regulating their development, activation and function is limited. Human primary immunodeficiencies caused by loss-of-function mutations in single genes provide a unique opportunity to assess the requirement for particular molecules as regulators of human unconventional T-cell activation and differentiation.
Objective: To determine the role of STAT3 in regulating the differentiation and function of unconventional T cells.
Methods: Autosomal dominant-hyper IgE syndrome (AD-HIES) is caused by mutations in STAT3. We have used lymphocytes from these patients to assess the roles of STAT3 in NKT, MAIT and γδ T cell differentiation in vivo and in vitro.
Results: Patients with STAT3 mutations displayed normal numbers of γδ T cells but reduced numbers of MAIT and NKT cells ex vivo. Further, γδ T cells and MAIT cells from these patients also displayed altered responses to in vitro stimulation.
Conclusion: STAT3 plays an important role in regulating unconventional T cells. Defects in these cells may contribute to the clinical phenotype observed in AD-HIES.
ESID-0529 Opportunistic Infections in a Patient with X-Men Disease
F. Dhalla 1, S. Murray1, R. Sadler1, J. Miller1, E. Soilleux2, M. Bhole3, B. Ferry1, H.M. Chapel1, S.Y. Patel1
1Department of Immunology, Oxford University Hospitals NHS Trust, Oxford, United Kingdom
2Department of Histopathology, Oxford University Hospitals NHS Trust, Oxford, United Kingdom
3Department of Immunology, Russells Hall Hospital, Dudley, United Kingdom
X-MEN disease (X-linked immunodeficiency with Magnesium defect, Epstein-Barr virus infection and Neoplasia) is a novel primary immune deficiency caused by loss of function mutations in MAGT1, which encodes a selective magnesium transporter found to be important for T-cell receptor signalling and for NKG2D expression on NK- and CD8 T-cells. To date only 7 patients, ranging from 3-45 years of age, have been described in the literature, identifying chronic infection with Epstein-Barr virus (EBV), EBV-driven lymphoma, CD4 T-cell lymphopaenia, and dysgammaglobulinaemia as clinical features. Here we present the case of a 58 year-old man with MAGT1 deficiency and hope to add to the clinical phenotype of this newly described primary immune deficiency by naming Pneumocystis pneumonia and progressive multifocal leucoencephalopathy (PML) as possible complications, particularly in the context of treatment with chemotherapeutic agents and Rituximab.
ESID-0771 Chronic Mucocutaneous Candidiasis: Lessons Learnt from the Study of One Pedigree
F. Dhalla 1, R. Sadler1, B. Ferry1, C. Anziolotti1, E. Davenport2, J. Knight2, H.M. Chapel1, S.Y. Patel1
1Department of Immunology, Oxford University Hospitals NHS Trust, Oxford, United Kingdom
2Genetics, Wellcome Trust Centre for Human Genetics, Oxford, United Kingdom
Chronic mucocutaneous candidiasis is characterised by recurrent and persistent superficial infections with Candida albicans, affecting the mucous membranes, skin and nails. It can be caused by acquired and primary immune deficiencies, in particular those that impair IL-17 and IL-22 immunity. Mutations in the coiled-coil (CC-) and DNA-binding domains of STAT1 have been found to cause autosomal dominant CMC due to a gain of STAT1 function, which hinders the development and function of IL-17 producing T-cells.
Here we present a family in which chronic mucocutaneous candidiasis segregates with oropharyngeal cancer as an autosomal dominant trait. Whole exome sequencing using DNA from seven members of the family (6 affected and 1 healthy member) identified a variant in STAT1 (R274W). Interestingly the variant was not identified in 2 of the 6 ‘affecteds’; it was noted that they had a much milder phenotype and their diagnoses have subsequently been reviewed. Most patients were started on imidazole prophylaxis as adults, however the 3 youngest patients started prophylaxis in childhood, and all developed varying degrees of resistance to this class of antifungals.
We also present laboratory data showing evidence of impaired Th17 responses using an ex vivo technique measuring cell surface markers (CCR6+/CCR4+/CD161+/CXCR3-). We show that this assay gives results which correlate with the measurement of IL-17A producing T-cells via intracellular cytokine staining, making it attractive as a routine testing technique for the detection of Th17 dysregulation.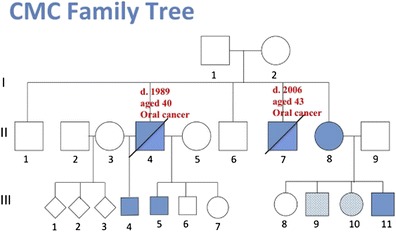
ESID-0424 Defects in Thymic Central Tolerance and T Cell Development Contribute to Autoimmunity in Adenosine Deaminase (ADA)-Deficiency
B. Di Lorenzo 1, A.V. Sauer1, M. Bosticardo1, I. Brigida1, N. Carriglio1, R.J. Hernandez1, S. Giannelli1, P.L. Poliani2, G.A. Holländer3, A. Aiuti1
1HSR-TIGET, Hospital San Raffaele, Milan, Italy
2Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy
3Department of Biomedicine, University of Basel and Basel University Children’s Hospital, Basel, Switzerland
Adenosine deaminase (ADA) is an enzyme of the purine salvage pathway. ADA deficiency in humans and mice is characterized by immunodeficiency, recurrent infection, impaired lymphocyte development and function. Particularly ADA-deficient patients with late-onset forms and after treatment may manifest immune dysregulation.
In order to improve treatment outcome, we aim at providing new insights into the pathogenesis and underlying mechanisms causing autoimmunity. Proper establishment of thymic cortical or medullary compartments and clonal deletion of autoreactive T cells require interaction between thymic epithelial cells (TECs) and developing thymocytes. We therefore studied alterations in thymic function and central tolerance in untreated ADA-/- mice, which closely resemble the human disease phenotype. Their thymus is severely compromised by the absence of ADA and substrate accumulation, affecting both thymocyte and stromal components.
Medullary TECs, which mediate negative selection via the expression of tissue specific antigens (TSAs), were significantly reduced in number and their TSA expression was 2-fold decreased. Only few thymocytes overcome a block at the double-negative stage of thymocyte development. They receive insufficient survival signals and emigrate as immature T cells due to defects during negative selection. Peripheral mature T cells showed alterations in their Vβ-TCR repertoire and in response to multiple stimuli.
We therefore hypothesize that the thymic microarchitecture in ADA-/- mice is altered due to a block of thymocyte differentiation, thereby interfering with the establishment of central tolerance mechanisms. Hence, selection defects and subsequent egress of autoreactive T cells may contribute to autoimmune manifestations in this disease.
ESID-0464 Overview of 15 Years Severe Combined Immunodeficiency in The Netherlands: Towards Neonatal Blood Spot Screening
P.J. Pagter 1, R.G. Bredius2, J. Tramper3, T.W. Kuijpers4, M. van der Burg5, J.M. van Montfrans6, G.J. Driessen1
1Infectious Disease and Immunology, Erasmus MC Sophia Children's Hospital, Rotterdam, Netherlands
2Pediatric HSCT Unit, Leiden University Medical Center, Leiden, Netherlands
3Infectious Disease and immunology, Erasmus MC Sophia Children's Hospital, Rotterdam, Netherlands
4Department of Pediatric Hematology Immunology and Infectious Disease, Emma Children's Hospital Academic Medical Center, Amsterdam, Netherlands
5Immunology, Erasmus MC, Rotterdam, Netherlands
6Department of Pediatric Immunology and Infectious Diseases, University Medical Center Utrecht/Wilhelmina Children's Hospital, Utrecht, Netherlands
Severe combined immunodeficiency (SCID) is a fatal primary immunodeficiency presenting with (opportunistic) infections and failure to thrive. Hematopoietic stem cell transplantation (HSCT) or gene therapy are curative treatment options.
The objective of the study was to assess the morbidity, mortality, diagnostic and therapeutic delay in children with SCID.
In the period 1998-2013, 43 SCID patients were diagnosed in the Netherlands, 11 of whom were atypical SCID (presentation more than 365 days after birth). The median interval between first symptom and diagnosis was 60 days (range 0-3228) and the median interval between diagnosis and treatment or death was 73 days (range 0-4742). During the interval between first symptoms and curative treatment or mortality, 22 patients suffered from severe systemic viral/bacterial infections and 25/43 patients suffered from opportunistic infections. Of these 25 patients, 17 were successfully treated and 8 deceased at a median of 12 (range 0-88) days after diagnosis of the opportunistic infection(s), despite adequate antimicrobial treatment. The total mortality rate was 41,8%, irrespective of delay between first symptom and treatment. All patients who were not eligible for HSCT because of a poor general condition deceased, due to severe infectious complications (n=9). In total, 32 patients were treated with HSCT of whom 8 deceased.
Because of the substantial mortality in this cohort of SCID patients, neonatal screening based on quantification of TRECs in neonatal blood spots and pre-emptive HSCT (<3 months of life) is likely to improve survival of children with SCID in the Netherlands.
ESID-0168 A Novel Stat1 Mutation Leading To Hyperphosphorylation in a Patient with Pneumocystis Jjiroveci and Chronic Mucocutaneous Candidiasis
M. Dullaers 1, J. Staal2, S. Gerlo3, E. Vandermarliere4, V. Debacker1, V. Bordon5, K. De Waele6, I. Meyts7, L. Moens8, X. Bossuyt8, F. De Baets9, B.N. Lambrecht10, K.Y. Vermaelen11, F. Haerynck9
1Clinical Immunology Research lab, University Hospital Ghent, Ghent, Belgium
2Molecular Signal Transduction in Inflammation VIB, University Hospital Ghent, Ghent, Belgium
3Cytokine Receptor lab Department Medical Protein Research VIB, Ghent University, Ghent, Belgium
4Computational Omics and Systems Biology Group Department Medical Protein Research VIB, Ghent University, Ghent, Belgium
5Pediatric Hematology and Oncology, University Hospital Ghent, Ghent, Belgium
6Pediatric Endocrinology, University Hospital Ghent, Ghent, Belgium
7Pediatric Immunology, University Hospital Leuven, Leuven, Belgium
8Experimental Laboratory Immunology, University Hospital Leuven, Leuven, Belgium
9Pediatric Immunology and Pulmonology, University Hospital Ghent, Ghent, Belgium
10Lab for Immunoregulation and Mucosal Immunology VIB, Ghent University, Ghent, Belgium
11Pulmonary Medicine Clinical Immunology Research Lab, University Hospital Ghent, Ghent, Belgium
We report on a girl who presented at the age of 4 months with SCID-like symptoms. She had pneumonia from which Pneumocystis jiroveci, cytomegalovirus and Candida albicans were isolated. However T and B lymphocyte number and lymphocyte proliferation upon mitogens and candida were normal. Two years later she developed chronic mucocutaneous candidiasis (CMC) and autoimmune hypothyroidism pointing towards an autoimmune polyendocrine (APECED)-like syndrome. This was ruled out because no deleterious mutations were found in the AIRE gene.
CMC is characterized by recurrent/persistent Candida albicans infections and has been linked to various defects in anti-fungal Th17 immunity. CMC can also be caused by defects in dectin-1, involved in fungal recognition and Stat1, counteracting Th17-inducing Stat3. Dectin-1 expression by peripheral blood mononuclear cells (PBMC) was normal, as was the cytokine response to dectin-1 stimulation. After super-antigen stimulation of PBMC, Th17 cytokines were strongly impaired, suggesting a defect downstream of fungus recognition.
Sequencing of the STAT1 gene revealed a deletion (p.Leu301del) in the coiled-coil region. Several gain-of-function mutations in this domain have been described to cause CMC by a mechanism of defective dephosphorylation of STAT1.We confirmed that this mutation increased STAT1 reporter activation. Patient PBMC displayed profound hyperphosphorylation of STAT1 upon IFN stimulation, which was not due to a dephosphorylation defect.
We conclude that our patient has a dominant gain-of-function mutation in STAT1 resulting in an impaired Th17 response as the basis of CMC and pneumocystis pneumonia. We are currently investigating the molecular mechanisms underlying the immunological abnormalities.
ESID-0173T Cell Proliferation Rather than T Cell Effector Function Is the Limiting Factor of Antiviral Immunity in Mice with a Partial Defect in TCR Signaling
K. Hillen1, R. Gather1, A. Enders2, H. Pircher3, P. Aichele3, P. Fisch4, B. Blumenthal5, W. Schamel5, T. Straub3, C. Goodnow2, S. Ehl 1
1University Medical Center and University of Freiburg, Center for Chronic Immunodeficiency, Freiburg, Germany
2John Curtin School of Medical Research The Australian National University, Department of Immunology, Canberra, Australia
3University of Freiburg, Institute of Medical Microbiology and Hygiene, Freiburg, Germany
4University Medical Center Freiburg, Institute of Pathology, Freiburg, Germany
5University of Freiburg, BIOSS Centre for Biological Signaling Studies, Freiburg, Germany
Defining the minimal thresholds for effective antiviral T cell immunity is important for clinical decisions in immunodeficient patients. TCR signaling is critical for T cell development, activation and effector functions. Here, we analyzed which of these TCR-mediated processes is limiting for antiviral immunity in a mouse strain with reduced expression of SLP-76 (twp mice). Despite severe T cell activation defects in vitro, twp mice generated a normal proportion of antiviral effector T cells following infection with Lymphocytic Choriomeningitis Virus (LCMV). Moreover, TCR dependent antiviral effector functions including cytokine production and cytotoxicity were largely normal. The main limiting factor in the antiviral response of twp mice was impaired T cell proliferation leading to a 5-10-fold reduction of antiviral T cells at the peak of the immune response. This was still sufficient to control infection with LCMV-ARM, but the more rapidly replicating LCMV-WE induced T cell exhaustion and viral persistence. Thus, under conditions of impaired TCR signaling, reduced T cell proliferation was the limiting factor in antiviral immunity. Conventional in vitro tests of T cell immunity were poorly predictive for the extent of immunodeficiency. These findings have implications for understanding and evaluating antiviral immunity in patients with T cell deficiencies.
ESID-0138 RAG1 and RAG2 Mutations in Egyptian SCID Patients
A. Elmarsafy 1, S. Meshaal2, R. El-hawary2, M. El-sharkawy2, R. Khaled2, R. Jan2, D. Abdel Aziz2, R. Alkady1, N. Galal1, J. Boutros1
1Pediatric Department, Cairo University - Faculty of Medicine, Cairo, Egypt
2Clinical and Chemical Pathology Department, Cairo University - Faculty of Medicine, Cairo, Egypt
Mutations in RAG1 and RAG2 genes comprise approximately 10% of all SCID cases and 50% of AR-SCID. In Egypt, RAG mutations would probably be responsible for higher percentages of SCID patients as a result of high rate of consanguinity.
In this study RAG1/2 mutations were analyzed in 12 SCID/OS infants by Sanger sequencing. We detected 4 different RAG mutations in 4 patients and 2 previously reported polymorphisms in 5 patients. One novel homozygous nonsense mutation in RAG1 gene was detected in a patient diagnosed as Omenn Syndrome (Q812X), one previously reported homozygous missense mutation in RAG1 gene in another Omenn Syndrome patient (R975Q), a hypomorphic mutation in RAG2 (R229Q) in a homozygous form and lastly one novel missense mutation in RAG2 gene (S260N) which cannot alone explain the disease status being heterozygous in the patient. Three silent mutations were detected in 2 patients.
Mutations in RAG1/2 are not uncommon in Egyptian SCID patients. Early diagnosis could dramatically affect the outcome of the disease by establishing bone marrow transplantation in a timely fashion. Genetic counseling, preimplantation and prenatal diagnosis can alter the fate of the affected Egyptian PID families.
ESID-0766 Characterization of Invariant iNKT Cells in Pheripheral Blood of Patients with Common Variable Immunodeficiencie
L.V. Erazo Borras 1, C.A. Perez Romero1, J.C. Orrego Arango1, J.L. Franco Restrepo1, C.M. Trujillo-Vargas1
1Grupo de Inmunodeficiencias Primarias, Universidad de Antioquia, Medellín, Colombia
Introduction Common Variable Immunodeficiency (CVID) is a heterogeneous primary antibody disorder with alterations in B cell subpopulations. However, a decrease in invariant NKT cells (iNKT) in peripheral blood (PB) has been also reported in these patients. Therefore, we analyzed the phenotype of iNKT cells in PB of CVID patients.
Methods Surface markers were detected by flow cytometry. iNKT cell function was evaluated by Phorbol Myristate Acetate (PMA)/Ionomycine stimulation, subsequently analyzing the expression of co-stimulatory molecules and intracellular cytokines. All these parameters were evaluated in twenty-three CVID patients classified according to EUROclass as compared to their age- and sex- matched healthy controls.
Results Reduced total and double negative NKTi cells were observed in PB of CVID patients, predominantly in the smB+ group. A tendency towards reduced Follicular helper iNKT cells as well a significant reduction in the CCR5+/CXCR3+- with a concomitant increase in the CD62L+/CCR7+ iNKT cells percentages was also observed in this patients. In addition, decreased CD4+ iNKT cells were observed in all CVID groups. After stimulation, iNKT cells from patients showed normal expression of CD69 and CD40L however, decreased percentages of intracellular IFNgamma and TNFalpha was detected again in the smB+ group.
Conclusions Phenotype defects and impaired expression of migration markers in PB iNKT cells is a common finding, specifically, in the smB+ group. Also exhibited defects in the production of intracellular IFNgamma and TNFalpha. Further experiments are necessary to if impairment in the iNKT:B cell help are also present in CVID.
ESID-0399 Atypical Combined Immunodeficiency with Immune Dysregulation
M. Esnaola Azcoiti 1, M.S. Caldirola1, A. Bravo Kleiman1, I. Moreira1, M. Esquivel1, D. Di Giovanni1, A. Gomez Raccio1, M.I. Gaillard1, L. Bezrodnik1
1Immunology, R. Gutierrez Children Hospital, CABA, Argentina
Introduction: combined immunodeficiency comprises a heterogeneous group of genetic disorders affecting the humoral and cell-mediated immunity. Hypomorphic mutations that lead to 'leaky' severe combined immunodeficiency presentation with partial protein function are increasingly being identified.
Objective: Case report of an undiagnosed atypical combined immunodeficiency (ACI).
Results: Full term baby boy without relevant perinatal history. Third child of non-consanguineous parents. Brother dead at 17 years of H1N1 with unstudied hypogammaglobulinemia, elevated IgM, mycobacterial infection, brain abscesses and chronic diarrhea and hepatosplenomegaly since birth. Healthy sister and parents. Personal History: chronic diarrhea since birth with dysentery. Hypothyroidism. Recurrent thrush. Herpes virus infection. Prolonged fever, hepatosplenomegaly and abdominal pain with anechoic images on the spleen. Perianal psoriasiform dermatitis. Failure to thrive with decreased bone age. Dysmorphic facies. Hipogammaglobulinemia and impaired response to proteins and polysaccharides. 4 yo treatment with IVIG. Negative HIV, EBV and CMV. Several parasite/fungus stool isolations. T lymphopenia, increased CD4+CD45RO+. Impaired proliferative response to mitogens. Low memory B cells, high transitional B subpopulation. Normal NK functional assays, γ-chain, DHR, CD25 and CD40L. Low Treg cells. Low IL-10 and IL-8 after stimulation. Positive ATPO. Treatment: IVIG, antibiotics, antifungal, steroids and rapamicin. Molecular studies for NEMO, CD40L and IKBα with normal results. For ACI, RAG1, RAG2 and ARTEMIS where evaluated, also with normal results.
Discussion: despite not having molecular diagnose, he is under bone marrow transplantation plan. Due to family history we suspect an X-linked PID. A panel of 200 PID related genes are being studied.
ESID-0601 Clinical Findings and Molecular Study of 55 Patients with Severe Combined Immunodeficiency, A Report from Iran
M.R. Fazlollahi 1, A.A. Hamidieh2, M. Movahedi3, Z. Chavoshzadeh4, M. Mahloujirad1, S. Arshi5, A. Khayatzadeh3, S. Dabaghzadeh3, L. Atarod6, F. Zandieh7, M. Sadeghi Shabestary8, M. Mesdaghi4, I. Mohammadzadeh9, A. Mahdaviani10, D. Babaii4, M.H. Eslamain3, M. Badalzadeh1, M. Houshmand1, Z. Pourpak1, M. Moin1
1IAARI, ImmunologyAsthma and Allergy Research InstitiuteTehran University of Medical Sciences, Tehran, Iran
2Hematology-Oncology and Stem Cell Transplantation Research Center, Shariati HospitalTehran University of Medical Sciences, Tehran, Iran
3Department of Allergy and Clinical Immunology, Children's Medical Center Tehran University of Medical Sciences, Tehran, Iran
4Department of Allergy and Clinical Immunology, Mofid Hospital Shahid Beheshti University of Medical Sciences, Tehran, Iran
5Department of Allergy and Clinical Immunology, Iran University of Medical Sciences, Tehran, Iran
6Department of Pediatrics, Imam Khomeini Hospital Tehran University of Medical Sciences, Tehran, Iran
7Department of Allergy and Clinical Immunology, Bahrami Hospital Tehran University of Medical Sciences, Tehran, Iran
8Department of Allergy and Clinical Immunology, Tabriz University of Medical Sciences, Tabriz, Iran
9Department of Immunology and Allergy, Amirkola Hospital Babol University of Medical Sciences, Babol, Iran
10Department of Immunology and Allergy, Masih Daneshvari Hospital Shahid Beheshti University of Medical Sciences, Tehran, Iran
Introduction: Severe combined immunodeficiency (SCID), a life-threatening disease, is the prototype of the primary immunodeficiency diseases .This study attempts to assess the clinical and laboratory characteristics of this rare disease in Iranian patients.
Methods: In this study, fifty five SCID patients (female: 36/male: 19) who were referred to the Immunology, Asthma and Allergy Research Institute (IAARI), a referral center for PID, from 2006 to 2013 were enrolled. Molecular, immunological and clinical data were evaluated.
Results: According to immunologic phenotypes, twenty one patients were classified as T-B-NK+ SCID; ten patients as T-B-NK-, ten patients as T – B+NK +, ten patients as T-B+NK- and four patients were classified as atypical SCID.
The mean age diagnosis was 3.6±3.02 and mean diagnostic delay was 2.2 month. Consanguinity rate and family history of SCID were 85.5% and 56.4%, respectively. Vaccination rate of BCG was 72.7% and disseminated BCG infection occurred in 20% of patients. Mutations in RAG1&2 and IL7R genes were the most common genetic cause of SCID. Until now 27.3% of our patients are alive. Three of seven patients survived after hematopoietic stem cell transplantation.
Conclusion: Based on these results, because of the high rate of consanguinity, autosomal recessive SCID is the most inherited type in Iranian patients. Delay diagnosis and lack of suitable donor are the main cause of high mortality rate.
ESID-0235 A Novel STAT1-Mutation in a Patient with Chronic Mucocutaneous Candidiasis
J. Fischer 1, S. Okada2, A. Puel3, J.L. Casanova2, M. Laaß1, J. Roesler1
1Klinik für Kinder- und Jugendmedizin, Medizinische Fakultät Carl Gustav Carus an der TU Dresden, Dresden, Germany
2St. Giles Laboratory of Human Genetics of Infectious Diseases Rockefeller Branch, The Rockefeller University, New York, USA
3Laboratory of Human Genetics of Infectious Diseases, Necker Medical School and University Paris Descartes, Paris, France
The 7-year-old boy suffered from recurrent impetigo, and chronic and recurrent fungal infections. Mucosal candidiasis led to an oesophageal stenosis. In the past, immunodeficiency had repeatedly been suspected, but conventional diagnostics did not show any crucial abnormalities. However, no antibody formation against Candida was found. The clinical course was further characterized by recurrent infections, bronchiectasis, failure to thrive, moderate mental retardation, and pancreatic insufficiency. The STAT1 gene was sequenced because many of the symptoms were compatible with chronic mucocutaneous candidiasis disease (CMCD). A new mutation (P293L) in the coiled-coil domaine of STAT-1 was found. Recently, hypermorphic mutations in this domaine have been described as a cause of autosomal-dominant chronic mucocutaneous candidiasis. This form of CMCD can present with severe infections in addition to candida and with autoimmunity such as thyreoiditis. The mutations found in CMCD impair the nuclear dephosphorylation of activated STAT-1. This gain-of-function leads to increased cellular responses to IFN-α/β, IFN-γ, and IL-27 and thereby to an impairment of the development of IL-17- producing T-cell. In addition to its anti-viral activity, interferon-alpha is known to play a role in autoimmunity. Under prophylactic treatment with itraconazole and cotrimoxazole the infections could almost be abolished and the general condition could be essentially improved.
ESID-0324 The Results of Combined TREC/KREC Monitoring in Patients with Digeorge Syndrome
E. Fronkova 1, M. Svaton1, A. Klocperk2, M. Kotrová1, P. Keslova1, F. Votava3, H. Vinohradská4, T. Freiberger5, E. Mejstrikova1, J. Trka1, A. Sediva2
1CLIP Department of Pediatric Hematology/Oncology, 2nd Medical School Charles University Prague and University Hospital Motol, Prague 5, Czech Republic
2Department of Immunology, 2nd Medical School Charles University Prague and University Hospital Motol, Prague 5, Czech Republic
3Department of Pediatrics, 3rd Medical School Charles University Prague and University Hospital Kralovske Vinohrady, Prague 5, Czech Republic
4Department of Clinical Biochemistry, Children Hospital Faculty of Medicine Masaryk University, Brno, Czech Republic
5Department of Clinical Immunology and Allergology Medical Faculty and Central European Institute of Technology Masaryk University Brno, Molecular Genetics Lab Centre for Cardiovascular Surgery and Transplantation Brno, Brno, Czech Republic
DiGeorge syndrome presents with a wide spectrum of thymic pathologies. Neonatal screening using TREC repeatedly identifies patients with severe DiGeorge syndrome. We tested what proportion of DiGeorge syndrome patients could be identifed at birth by combined TREC and KREC screening. Furthermore, we followed TREC/KREC levels in peripheral blood (PB) to monitor postnatal changes in lymphocyte production.
Methods: TREC/KREC copies were assessed by qPCR and were related to the albumin control gene in dry blood spots (DBS) from control (n=56), severe immunodeficiency syndrome (SCID, n=10) and DiGeorge syndrome (n=13) newborns. PB was evaluated in children with DiGeorge syndrome (n=32), in diagnostic samples from infants with SCID (n=5) and in 35 controls.
Results: Only one patient (with complete DiGeorge syndrome) had TREC levels outside the normal range, albeit quantitative TREC values were significantly lower in DiGeorge syndrome cohort. One patient had slightly reduced KRECs at birth. Postnatal DiGeorge syndrome samples revealed reduced TREC numbers in 5 of 32 (16%) patients, together with normal KREC copy numbers. The patient with positive neonatal screening received repeated DLI´s in infancy; the patients with reduced TREC levels had repeated infections in infancy and developed allergy and/or autoimmunity, but not strikingly different from other DiGeorge patients. In 12 patients with paired DBS and blood samples, the TREC/KREC levels were mostly stable or increased.
Conclusions: The combined TREC/KREC approach with correction via control gene identified 8% of DiGeorge syndrome patients at birth. The majority of patients had normal TREC/KREC levels, which remained stable or increased postnatally.
Support: P302/12/G101.
ESID-0093 Differentially Impaired Cytokine Signaling Causes Severe Combined Immunodeficiency in T+/Low NK+IL2 Receptor Gamma Chain Deficiency
S. Fuchs 1, A. Rensing-Ehl1, M. Erlacher1, T. Vraetz2, L. Hartjes1, A. Janda1, M. Rizzi1, K. Gilmour3, G. de Saint-Basile4, C. Roifman5, S. Cheuk6, A. Gennery7, A. Thrasher3, I. Fuchs1, C. Speckmann1, S. Ehl1
1University Medical Center and University of Freiburg, Center for Chronic Immunodeficiency, Freiburg, Germany
2University Medical Center and University of Freiburg, Center of Pediatric and Adolescent Medicine, Freiburg, Germany
3University College London (UCL), Molecular Immunology Unit Institute of Child Health (ICH), London, United Kingdom
4INSEM UMR 1163, Laboratory of Normal and pathological homeostasis of the immune system, Paris, France
5Hospital for Sick Children and the University of Toronto, The Canadian Centre for Primary Immunodeficiency, Toronto, Canada
6n/d, n/d, Calgary, Canada
7Newcastle University, Institute of Cellular Medicine, Newcastle, United Kingdom
X-linked severe combined immunodeficiency (X-SCID) leads to a T-NK-B+ immunophenotype and is caused by mutations in the gene encoding the IL-2 receptor γ-chain (IL2RG). IL2RGR222C leads to atypical SCID with a severe early onset phenotype despite largely normal NK and T-cell numbers. To address this discrepancy, we performed a detailed analysis of T, B and NK-cells, including quantitative STAT phosphorylation and functional responses to the cytokines IL-2, IL-4, IL-15 and IL-21 in a patient with the IL2RGR222C mutation. Moreover, we identified 9 additional unpublished patients with the same mutations, all with a full SCID phenotype and confirmed selected immunological observations. T-cell development was variably affected, but led to borderline T-cell receptor excision circles (TREC) levels and a normal repertoire. T-cells showed moderately reduced proliferation, failing enhancement by IL-2. While NK-cell development was normal, IL-2 enhancement of NK-cell degranulation and IL-15 induced cytokine production were absent. IL-2 or IL-21 failed to enhance B-cell proliferation and plasmablast differentiation. These functional alterations were reflected by a differential impact of IL2RGR222C on cytokine signal transduction with a gradient IL-4<IL-2/IL-15<IL-21. Thus, IL2RGR222C causes a consistently severe clinical phenotype that is not predicted by the variable and moderate impairment of T-cell immunity or TREC analysis.
ESID-0383 Chemosensitive Combined Immunodeficiency with EBV Lymphoproliferation – A Problem of Homologous Recombination?
S. Ghosh 1, A. Hönscheid2, G. Dückers3, H. Gohlke4, M. Kuhlen2, H.J. Laws2, R. Linka2, R. Meisel2, T. Niehues3, D. Schindler5, D. Schneider6, F.R. Schuster2, K. Schwarz7, C. Speckmann8, A. Borkhardt2
1Department of Pediatric Oncology Hematology and Clinical Immunology / Molecular and Cellular Immunology Section, Heinrich-Heine-University / UCL Institute of Child Health, Düsseldorf / London, Germany
2Department of Pediatric Oncology Hematology and Clinical Immunology, Heinrich Heine University Medical Faculty, Düsseldorf, Germany
3Department of Pediatrics, Helios Hospital Krefeld, Krefeld, Germany
4Institute for Pharmaceutical and Medicinal Chemistry, Heinrich Heine University, Düsseldorf, Germany
5Institute for Human Genetics, Institute for Human Genetics, Würzburg, Germany
6Clinic of Pediatrics, Municipal Hospital Dortmund, Dortmund, Germany
7Institute for Transfusion Medicine / Institute for Clinical Transfusion Medicine and Immunogenetics Ulm, University Hospital Ulm / German Red Cross Blood Service Baden-Württemberg – Hessen, Ulm, Germany
8Centre of Pediatrics, Centre for Chronic Immunodeficiency (CCI) and University of Freiburg, Freiburg, Germany
Reports on hypomorphic mutations in classical SCID genes in adolescents and adults with susceptibility to severe infections and immune dysregulation are continuously emerging; however a growing number of patients with combined immunodeficiency are without a genetic diagnosis.
We report on a 19-year-old male from non-consanguineous German descent. Since birth he had been suffering from splenomegaly, recurrent respiratory tract infections and bacterial as well as varicella meningitis. A unifying diagnosis was never established due to repeated loss to follow-up in different hospitals. At the age of 17 years we first saw him with severe dermal necrotizing varicella infection, vasculitis, EBV associated B cell lymphoproliferation and leukopenia with few lymphocytes only consisting mainly of γδ (gammadelta) T cells. Interestingly, fibroblasts showed increased mitomycin C (MMC) sensitivity, but normal radiosensitivity. Due to the rapid deteriorating clinical course, the patient had to undergo haploidentical hemopoetic stem cell transplantation (HSCT). Currently, more than 500 days post-HSCT the patient is in a very good clinical condition and fully immune-reconstituted. Whole exome sequencing results of the patient and his family (including the mother, who died from breast cancer at age 42), MMC sensitivity and in-silico prediction hint towards a possible new primary immunodeficiency due to a failure of proper homologous recombination in DNA repair.
ESID-0108 Regulation of Adaptive Mucosal Response in the Small Intestine by Microbiota-Derived Adenosine Triphosphate
F. Grassi 1
1T cell development, Institute for Research in BIomedicine, Bellinzona, Switzerland
Adenosine triphosphate (ATP) is a ubiquitous extracellular messenger, which activates purinergic receptors in the plasma membranes of eukaryotic cells termed P2 receptors. P2X 1-7 receptors are ATP-gated cation channels, whereas P2Y1, 2, 4, 6, 11-14 are guanine nucleotide-binding protein (G protein)-coupled receptors (GPCRs), which bind also ADP, UDP, UTP or UDP-glucose. ATP released during tissue damage acts as danger associated molecular pattern (DAMP) for cells of the innate immune system through stimulation of P2 receptors. P2rx7, which encodes the ATP-gated P2X7 receptor, is a signature gene of effector T cell subsets. We have previously shown that P2X7 activity regulates the immunosuppressive function of regulatory T cells (Tregs), and persistent P2X7 stimulation by ATP results in pyroptosis-like cell death of Tregs. In fact, sustained P2X7 activation leads to the formation of a pore permeable to molecules up to 900 Da. We found that p2rx7 is selectively and highly expressed in T follicular helper (TFH) cells of the Peyer’s patches (PPs) in the small intestine. Microbiota-derived ATP provokes TFH cell death via P2X7, thus limiting T-dependent IgA responses. Lack of P2X7 enhanced germinal centre reaction and secretory IgA, which affected microbiota abundance and composition. Evidence is provided that ATP is a metabolite released by commensals, which shapes adaptive mucosal immunity in the gut and systemic immune homeostasis.
ESID-0670 Large Granular Lymphocytes as Predictors of Morbidity and Early Mortality in GATA2 Deficiency
J. Grossman 1, M. Spinner2, L. Sanchez2, A. Hsu2, K. Olivier2, C. Zerbe2, K. Calvo3, S. Holland2, S. Rosensweig3
1Department of Medicine Section of Hematology, Alberta Health Services, Calgary, Canada
2National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, USA
3Department of Laboratory Medicine, National Institutes of Health, Bethesda, USA
Mutations in GATA2 are known to cause a syndrome featuring an increased susceptibility to infections and predisposition to myelodysplastic syndrome (MDS). Clonal CD3+, CD8+, CD16+ and CD57+ large granular lymphocytes (LGLs) have been noted in some patients with GATA2 deficiency and the significance of this finding has yet to be determined.
Data on a cohort of 34 patients diagnosed with GATA2 deficiency and tested for the presence of LGLs at the NIH was retrospectively analyzed.
Of the 34 patients tested, 8 (24%) were positive for LGLs (LGL+). These patients had a higher incidence of MDS (8/8 versus 21/26), but cytopenias were less common in the LGL+ group. Only 2/8 LGL+ patients were neutropenic versus 17/26 LGL- patients. The LGL+ group was more susceptible to invasive fungal infections (4/8 versus 3/26, p=0.03). Autoimmune disorders were more frequent in the LGL+ group and 6 out of 8 tested patients in each group were positive for lupus anticoagulant. All 6 patients in the LGL+ group developed a clot but only 1/6 in the LGL- group did (p=0.01). Surprisingly, the presence of LGLs was also associated with early mortality (6/8 vs 4/26; p=0.003).
Our preliminary data suggest a relationship between the presence of T-cell LGLs and increased morbidity as well as early mortality in patients with GATA2 deficiency. Further retrospective and prospective analysis is necessary to determine the role of this finding in predicting disease progression and severity.
ESID-0258 Intractable Molluscum Contagiosum Treated Successfully with Pegylated-Interferon Alpha
N.P. Gurugama 1, S. Hanson1, A. Grammatikos1, D. Grosse-Kreul1, R. Morris-Jones2, M.A.A. Ibrahim1
1Department of Immunological Medicine, King's College Hospital, London, United Kingdom
2Dermatology, King's College Hospital, London, United Kingdom
Molluscum contagiosum (MC) is usually a self-limiting paediatric skin disease caused by a pox virus, but patients with primary and/or secondary immunodeficiencies may present with disseminated and difficult to treat infections. We report a patient with reduced TCR repertoire and T cell proliferative defects with extensive MC treated successfully with pegylated-Interferon alpha (IFNα).
A 33-year old Slovakian woman, presented with a 3-year history of widespread giant MC with no past history of recurrent infections, lymphadenopathy or organomegaly. She was HIV negative. FBC, Immunoglobulins, IgG sub-classes, C3/C4, MBL, NBT, auto antibodies were all normal. Lymphocyte subsets were normal except for reduced NK cells. Lymphocyte proliferative function was low with PHA, PWM, anti-CD3, PPD and Candida. Red cell lysate enzyme assays and red cell nucleotide profile were normal. TCRVB spectratyping showed reduced T cell receptor repertoire and skewed distribution in VB1, VB4, VB5, VB6a/b, VB7, VB8, VB9, VB13a, VB15, VB16 and VB22. Adequate antibody levels to tetanus, pneumococci, haemophilus, rubella, mumps and varicella were detectable, whereas CMV and EBV viral DNA was undetectable.
Her skin lesions didn't respond at all to conventional medications including topical Crystacide, Retin-A gel, Cidofovir 0.3% cream, electrocautery, Imiquimod 5%, Molludab and oral Isotretinoin over a period of 3 years. Subcutaneous pegylated-IFNα three millions units, three times-a-week for 9 months successfully resolved many of the lesions leaving some post-inflammatory hyperpigmentation.
This case report and other cases in the literature suggest that IFNα should be considered in the treatment of chronic viral infections, especially in patients with proven/suspected immunodeficiency.
ESID-0346 SYK Expression Endows ZAP-70 Deficient Cytotoxic T Cells with Residual TCR Signalling
F. Hauck 1, B. Blumenthal2, S. Fuchs2, M. Audrain3, C. Speckmann2, K. Schwarz4, W.W.A. Schamel2, S. Ehl2, C. Picard5, A. Rensing-Ehl2, S. Latour6
1Hematology and Oncology, Dr. von Hauner Children's Hospital, Munich, Germany
2Centre for Chronic Immunodeficiency, University Medical Center, Freiburg, Germany
3Laboratoire d’Immunologie, CHU de Nantes, Nantes, France
4Institute for Transfusion Medicine, University Hospital Ulm, Ulm, Germany
5Centre d’Étude des Déficits Immunitaires, Hôpital Necker-Enfants Malades, Paris, France
6Laboratory of Lymphocyte Activation and Susceptibility to EBV infection, Institut IMAGINE, Paris, France
Background: Autosomal recessive human ZAP-70 deficiency is a rare cause of combined immune deficiency (CID) characterized by functionally impaired CD4+ helper T cells (TH) and profound CD8+ cytotoxic T cell (TCyt) lymphopenia. Most T cell antigen receptor (TCR) signalling studies stem from zap70 knockout mice that are differing from its human counterpart as they display a combined TH and TCyt lymphopenia.
Objective: To analyze molecular genetics, T cell phenotypes, TCR signalling and T cell function in two patients with ZAP-70 deficiency.
Methods: Characterization of genotypes, clinical and immunological phenotypes, and biochemical signalling properties of ZAP-70 deficient T cells by molecular genetics, flow cytometry, and biochemistry.
Results: Patients present with infant or toddler CID with severe systemic VZV infection or vaccination strain infections and are cured by allogeneic hematopoietic stem cell transplantation (alloHSCT) or decease thereafter, respectively. Retrospective TCR excision circle (TREC) newborn screening (NBS) is normal in both patients. We report one new homozygous ZAP70 splice site (p.A495fsX75) and one known homozygous mutation (p.A507V). Especially, oligoclonal TCyt show SYK overexpression and residual TCR signalling such as protein tyrosine/threonine phosphorylation and Ca2+-flux.
Conclusion: ZAP-70 deficiency cannot be diagnosed by TREC NBS. Polyclonal TH and oligoclonal TCyt development with residual TCR signalling only in the latter might be driven by SYK. Like in murine T cell development, consecutive ZAP70 upregulation and increasing TCR signalling strenght might overcome the signalling threshold necessary for human TCyt development. However, due to more delayed SYK downregulation kinetics, human ZAP-70 deficiency might have oligoclonal TCyt development.
ESID-0075 Significant Augmentation of Regulatory T Cells in the Early Neonatal Period
S. hayakawa 1, K. maeno2, N. ohno1, S. okada1, Y. nishimura2, M. hayashidani2, M. kobayashi1
1department of pediatrics, Hiroshima University Graduate School of Biomedical & Health Sciences, Hiroshima, Japan
2department of neonatology, Hiroshima City Hospital, Hiroshima, Japan
Introduction: Newborns are exposed to ubiquitous antigens immediately after birth. Regulatory T cells (Tregs) have suppressive functions and control immune responses. There are currently few studies investigating Tregs in the neonatal period.
Objective: To elucidate the developmental fluctuation of Tregs and Treg subpopulations in neonates.
Methods: Mononuclear leukocytes in cord and peripheral blood were analyzed in 28 premature newborns admitted to Hiroshima University Hospital (12 boys and 16 girls, gestational age 35.4±1.9 weeks, birth weight 2,168±362 g). This study was approved by the Ethics/International Review Board of Hiroshima University. Tregs were identified by flow cytometry based on the expression of CD4, CD45RA and FOXP3. Cord blood cells were cultured in vitro with CD3 mAb for 5 days to study changes in Treg populations.
Results: CD4+ T cells were classified into resting-Tregs (CD45RA+ FOXP3low), activated-Tregs (CD45RA- FOXP3high), and non-Tregs (CD45RA- FOXP3low) by flow cytometry. Compared with cord blood, the total number of Tregs, and all subpopulations, transiently and significantly increased in the early neonatal period. The proportion of activated-Tregs most markedly increased among Treg subpopulations. Moreover, CD4+ CD25high Tregs from early neonates expressed high CCR4 and low CCR7, suggesting activation of Tregs in early neonates. An increased activated-Treg subpopulation was also observed in in vitro cultures.
Conclusions: We observed transient and striking increases in activated-Tregs in early neonates. Considering that neonates are exposed to ubiquitous antigens immediately after birth, increased activated-Tregs in early neonates may reflect immunological regulation to suppress excessive T cell activation.
ESID-0580 A Patient with Combined Immunodeficiency and Hemolytic Anemia
E. Hazar Sayar 1, S.N. Guner1, M. Emiroglu2, G. De Saint-Basile3, I. Reisli1
1Pediatric Allergy and Immunolgy, Necmettin Erbakan University, Konya, Turkey
2Pediatric Infection, Selcuk University, Konya, Turkey
3Centre d'études des déficits immunitaires, Hospital Necker-Enfants Malades, Paris, France
Mutations in the CD3D, CD3E, CD3G, and CD3Z genes have been described in humans. Homozygous mutations leading to complete deficiencies of either CD3δ, CD3ε, or CD3ζ protein produce a form of severe combined immunodeficiency (SCID) characterized by severe T-cell lymphopenia, but the presence of phenotypically normal B cells and NK cells (T-B+NK+SCID). Interestingly, mutations in CD3G leading to deficiency of CD3γ produces considerable clinical heterogeneity ranging from severe immunodeficiency in infants to mild forms of autoimmunity in adulthood. A six month old girl presented with lymphadenopathy in her left axilla. In her past medical history, she had a skin infection on the scalp at two months of age, a bronchiolitis at three months of age and an otitis at six months of age. Vaccines were applied according to her age. BCG vaccine was applied four months ago. Her parents were consanguineous. There was a solid, mobile, 2x2 cm lymphadenopathy in the left axilla. She also had lymphopenia, her CD3+ T lymphocyte, CD19+ B lymphocyte and TCR A/B ratio were detected as low, and TCR G/D ratio and sedimentation were detected as high. Antituberculosis treatment was started for mediastinal lymphadenopathy. In the CD3 Gamma gene analysis, two polymorphisims were detected and no mutation was identified in any of CD3D, CD3E and CD3Z. In the follow up, autoimmune hemolytic anemia was detected when she was sixteen months old. We would like to discuss if the two polymorphisims detected in CD3G gene of this patient could be related with her clinical condition.
ESID-0149 Rare STAT1 GOF Mutation in Dna Binding Domain Causing Mild CMC
P. Hsu 1, N. Wong2, D.T. Avery2, J. Torpy2, P.E. Gray3, D.A. Fulcher4, G. Uzel5, A. Williams1, M. Wong1, D.E. Campbell1, S.G. Tangye2
1Allergy and Immunology, The Children's Hospital at Westmead, Sydney, Australia
2Immunology Research Program, Garvan Institute of Medical Research, Sydney, Australia
3Immunology and Infectious diseases, Sydney Children's Hospital, Sydney, Australia
4Department of Immunology and Immunopathology, Institute for Clinical Pathology and Medical Research, Sydney, Australia
5Laboratory of Clinical and Infectious Diseases, National Institute of Health, Bethesda, USA
Introduction Gain of function (GOF) mutations in the coiled-coil domains (CCD) and DNA-binding domains (DBD) of STAT1 have been shown to cause chronic mucocutaneous candidiasis (CMC). Several DBD mutations have been reported (p.T385M, p.M390T, p.P329L), and patients with the p.T385M and p.M390T mutations have a severe CMC phenotype with associated recurrent sinopulmonary infections and bronchiectasis.
Case details We report two male children and their father with CMC and p.P329L mutation in the DBD of STAT1. P1 (3 yrs) presented with bilateral cervical necrotising lymphadenitis and recurrent oral candidiasis and ulceration, P2 (9 yrs) with recurrent oral and nail candidiasis and their father (P3, 37 yrs) with recurrent oral candidiasis, previous cutaneous furunculosis and pneumonia with bronchiectasis.
Methods and Results Sanger sequencing identified the mutation (c.986C>T, p.P329L) in the DBD of STAT1 in all 3 affected individuals, but not in other family members. Functional testing revealed immune defects similar to other STAT1 GOF patients, including impaired IL-17/IL-22 expression and production ex vivo and Th17 differentiation in vitro, and defective T follicular helper cell development associated with reduced memory B cell differentiation.
Conclusion On the basis of the functional studies, we confirm that the mutation p.P329L results in a clinical CMC phenotype consistent with STAT1-GOF. Unlike other patients with STAT1-GOF DBD mutations, our patients appear to have a milder phenotype comparable to the two other reported patients with the same mutation. The variable clinical phenotypes of patients with DBD mutations suggest potential genotype-phenotype correlation or additional disease-modifying factors.
ESID-0157 Immune Phenotype of Patients with Charge Syndrome
P. Hsu 1, A. Ma2, M. Wilson2, C. Munns3, M. Wong1, C.M. van Ravenswaaij-Arts4, G. Williams5, J. Curotta6, S. Mehr1
1Allergy and Immunology, The Children's Hospital at Westmead, Sydney, Australia
2Clinical Genetics, The Children's Hospital at Westmead, Sydney, Australia
3Endocrinology, The Children's Hospital at Westmead, Sydney, Australia
4Department of human genetics, Radboud University Nijmegen Medical Centre, Nijmegen, Netherlands
5Paediatrics, St George and Hurstville Private Hospitals, Sydney, Australia
6Ear Nose and Throat, The Children's Hospital at Westmead, Sydney, Australia
Introduction Dominant negative mutations in the CHD7 gene cause CHARGE syndrome, which shares many clinical features with 22q11.2 deletion syndrome, including the rare occurrence of severe combined immunodeficiency (SCID). The purpose of this study was to examine the immune phenotype of patients with CHARGE syndrome.
Methods We established a multi-disciplinary CHARGE clinic involving general paediatrician, endocrinologist, geneticist, ENT specialist and immunologist. Patients with CHARGE syndrome at our hospital (n=22) were invited to attend. Lymphocyte subsets, antibody levels and vaccine responses were measured.
Results The median age of the subjects was 9 years. 68% had recurrent acute otitis media (AOM) and upper respiratory tract infections (URTI) in early childhood. 59% had atopic disease. None had autoimmune disease. 21/22 patients had mutations in the CHD7 gene. All but one (IgA deficiency) patient had normal antibody levels, as well as protein and polysaccharide vaccine responses. Whilst the lymphocyte counts were within reference ranges, CHARGE patients had significantly lower CD8 and NK cell count, but normal CD4 and B cell count compared to healthy aged matched controls.
Conclusion Although SCID can rarely occur in both CHARGE and 22q11.2 deletion syndrome, in contrast to patients with 22q11.2 deletion syndrome, CD4 lymphopenia, autoimmune association and antibody defects (including specific antibody deficiency) is uncommon in CHARGE syndrome. However, a relative CD8 and NK cell lymphopenia appears to be a common finding though it does not apparently increase infection susceptibility. The cause and significance of this finding require further investigations.
ESID-0208 Clinical Presentation and Allogeneic Haematopoietic Stem Cell Transplantation in Cartilage Hair Hypoplasia
W. Ip 1, A. Pitts2, H.B. Gaspar1, E.G. Davies1, P. Veys3, M.A. Slatter2, A.R. Gennery2, W. Qasim1
1Molecular and Cellular Immunology, Institute of Child Health, London, United Kingdom
2Institute of Cellular Medicine, Newcastle University, Newcastle, United Kingdom
3Bone Marrow Transplantation Department, Great Ormond Street Hospital, London, United Kingdom
Introduction Cartilage-Hair Hypoplasia (CHH) is a rare autosomal recessive disorder with a highly variable phenotype, including metaphyseal dysplasia, short stature, bone marrow failure, immunodeficiency, and increased predisposition to malignancies. It is caused by mutations in the non-coding RMRP (RNA component of ribonuclease mitochondrial RNA processing) gene. Allogeneic haematopoietic stem cell transplantation (HSCT) can treat the immunodeficiency, which can present in early infancy as severe combined immunodeficiency (SCID).
Objective To describe clinical and immunologic features and outcome in children with CHH/RMRP mutations treated with HSCT.
Methods Thirteen children with CHH/RMRP mutations received HSCT at two tertiary UK centres between 1999-2013. Patients were monitored for infectious complications, immune reconstitution, chimerism, growth and development. Transplant procedures for each patient were examined.
Results Four patients presented in infancy with features of severe combined immunodeficiency, two of whom had no characteristic skeletal features at presentation. Seventeen allogeneic procedures were performed on 13 patients including 2 second transplants. Six patients were transplanted in early childhood before the onset of radiological changes of CHH and were found to have RMRP mutations as part of genetic screening for molecular characterisation of SCID. Eleven patients are alive at the last follow-up from transplantation (median 50 months; range 14-142 months) with full donor chimerism achieved in 10 patients.
Conclusions Cartilage Hair Hypoplasia caused by RMRP mutations has a highly variable phenotype. Diagnosis should be considered even in the absence of skeletal changes in infants. Allogeneic stem cell transplantation is a highly effective corrective therapy for the associated immunodeficiency.
ESID-0485 Ataxia-Telangiectasia in Moroccan Patients
L. JEDDANE 1, F. Ailal1, C. Dubois-d'Enghien2, O. Abidi3, I. Benhsaien4, A. Kili5, S. Chaouki6, Y. Kriouile5, N. El Hafidi5, R. Abilkassem7, N. Rada8, J. Najib4, A. Barakat3, D. Stoppa-Lyonnet2, A.A. Bousfiha4
1Clinical Immunology Unit, Faculty of Medicine and Pharmacy, Casablanca, Morocco
2Department of Genetics, Curie Institute, Paris, France
3Human Molecular Genetic Laboratory, Pasteur Insitute, Casablanca, Morocco
4Clinical Immunology Unit, Faculty of Medicine and Pharmacy , Casablanca, Morocco
5Children Hopsital, Avicennes University Hospital, Rabat, Morocco
6Department of Pediatrics, Hassan II University Hospital, Fez, Morocco
7Department of Pediatrics, Mohamed V Military Hospital, Rabat, Morocco
8Department of Pediatrics, Mohamed VI University Hospital, Marrakech, Morocco
Background Ataxia-telangiectasia (AT) is a rare autosomal recessive disease, affecting neurologic and immune system. Numerous mutations are described in the ATM gene in several populations. However, in Morocco, few data were available concerning this condition. Objective Our main goal is to determine clinical, immunological and molecular presentation of Moroccan patients with AT. Methods We analysed 42 patients for clinical presentation. We screened 27 patients, out of 22 unrelated families, for ATM gene mutations. Results All our patients showed ataxia, ocular telangiectasia and immunodeficiency, as well as elevated serum alphafoetoprotein levels. Mean age at diagnosis were 6.88 years and consanguinity rate was 63.3%. Mean age at onset was 2.6 years. Mortality reached 66.6%, with a mean age at death of 11.5 years. We found 14 different mutations in 19 unrelated families, of which 7 were not reported. Conclusion Our results showed that c.5644C>T mutation was the most common in our series. However, further studies are required to demonstrate a founder effects on ATM gene in Moroccan patients, who showed mutational heterogeneity otherwise. Our data indicate that direct sequencing of coding exons is sufficient for a high detection rate in ATM in Moroccan population.
ESID-0494 ADA Deficiency: About 3 Cases
I. Benhsaien1, F. Ailal 1, I. Brigida2, J. El Bakkouri3, L. Jeddane1, J. Najib1, A. Aiuti2, A.A. Bousfiha1
1Clinical Immunology Unit, Faculty of Medicine and Pharmacy, Casablanca, Morocco
2San Raffaele Telethon Institute for Gene Therapy (TIGET), San Raffaele Scientific Institute, Milan, Italy
3Laboratory of Immunology, Averroes University Hospital, Casablanca, Morocco
Adenosine Deaminase (ADA) deficiency lead to a severe combined immunodeficiency (SCID), of autosomal recessive transmission. Generally, this disorder is expressed in the first months of life by severe infections (bacterial, viral or fungal).
Here, we report the observations of three infants, aged respectively 6, 9 and 2 months. Parental consanguinity was reported in 2 patients. The 2 first cases consulted for long-winded respiratory infections, associated to a BCGitis in the second case. The 3rd patient was admitted for diffuse oral and esophageal candidiasis. Only one patient showed anomaly in skeleton. CBC showed profound lymphopenia in all patients and lymphocyte numeration suggested T-B-NK- SCID. Diagnosis confirmation was done by erythrocyte ADA activity assay in all patients, and genetic analysis in one patient. Outcome was fatal for the first patient. One of the last two was treated by bone marrow transplantation from his sister and the last one is on PEG-ADA, waiting for gene therapy.
In conclusion, ADA deficiency is a rare form of SCID, who impairs lymphocyte development and function. This work show the severity and diversity of presentation for this disease, hence the importance of early diagnostis confirmation in order to benefit from the treatment, namely bone marrow transplantation, gene therapy or the administration of polyethylene glycol-ADA complex.
ESID-0495 Severe Combined Immunodeficiency Complicated By Hodgkin Disease
F. Ailal 1, J. El Bakkouri2, N. Khadre1, C. Mansour1, L. Jeddane1, J. Najib1, A.A. Bousfiha1
1Clinical Immunology Unit, Faculty of Medicine and Pharmacy, Casablanca, Morocco
2Laboratory of Immunology, Averroes University Hospital, Casablanca, Morocco
Introduction : Severe Combined Immunodeficiency (SCID) is a severe defect in differenciation and function of T cells, associated or not with B or NK cells impairment. Here, we report a case of SCID associated with Hodgkin disease in mandibular location, and complicated by hemophagocytose lymphohistiocytosis (HLH).
Case report: This is about a 11 months-old boy, born from 1st-degree cousins, followed since 6 months-old for SCID, associated with a mandibular tumor. T-B+ SCID diagnosis was retained based on clinical features: several episodes of febrile dysentery, recurrent respiratory tract infections and oral candidiasis since 1 month-old; and laboratory analysis showing agammaglobulinemia, severe lymphopenia (300/mm3) and only B-cells on lymphocyte numeration. Maxillofacial CT was in favor of a malignant bone tumor of the ramus of the mandible. This aspect required several biopsies, the 1st one being inconcluding, and the second one in favor of a diffuse lymphoma proliferation or myeloid sarcome. Clinical evolution was marked at 10 months-old by emergence of an HLH confirmed by osteomedullar biopsy. The 3rd biopsy confirmed an Hodgkin type lymphoproliferation, and EBV PCR was negative. The infant was treated by HLH-2004 protocol with immunoglobulin perfusion every 2 weeks. Initially, the patient showed clinical and biological amelioration. However, at D-10 of treatment, he developped a diffuse pneumopathy, then later died from severe respiratory failure.
Conclusion: SCID is considered as the most severe primary immunodeficiency and requires bone marrow transpantation. The association with HLH is exceptional and this is the first case reporting Hodgkin disease as a complication of SCID.
ESID-0313 Dedicator of Cytokinesis 8 (DOCK8) Deficiency and Sclerosing Cholangitis: A New Association
T. Shah1, A. Jones 2, N. Hadzic3, C. Cale2
1Immunology and Infectious Diseases, Great Ormond Street Hospital, London, United Kingdom
2Immunology, Great Ormond Street Hospital, London, United Kingdom
3Paediatric Centre for Hepatology Gastroenterology and Nutrition, King’s College Hospital, London, United Kingdom
Introduction Dedicator of cytokinesis 8 deficiency is an autosomal recessive combined immunodeficiency, caused by a heterogeneous group of defects in the DOCK8 gene. Two children with DOCK8 deficiency and sclerosing cholangitis were reported in 2012 [1]. We describe two further cases providing evidence to support this association.
Patient Characteristics The first child had bronchiectasis, skin infections and atopy. He had CD4 lymphopenia, low IgM, normal IgA and IgG, eosinophilia and elevated IgE. Aged 13, he presented with weight loss, reduced appetite, abdominal pain, mildly elevated inflammatory markers and liver enzymes. Endoscopic retrograde cholangio-pancreatography (ERCP) and liver biopsy confirmed sclerosing cholangitis and Crypotosporidium was isolated from bile fluid. Symptoms resolved with conservative management and he awaits hematopoietic stem cell transplant (HSCT).
The second child had bronchiectasis, septic arthritis, septicaemia, molluscum contagiosum, chronic diarrhoea secondary to Cryptosporidium and atopy. He had CD4 lymphopenia, low IgM, raised IgA, normal IgG, and elevated IgE. Aged 8, although asymptomatic and with normal liver function, ERCP and liver biopsy were performed prior to HSCT. Mild intrahepatic cholangiopathy was demonstrated that worsened until he underwent HSCT and thereafter improved.
Discussion The 4 children with DOCK8 and sclerosing cholangitis had no common mutation, represent 3 ethnicities and all had Cryptosporidium infection. Sclerosing cholangitis should be borne in mind as a potential complication of DOCK8 deficiency, even in the absence of overt cryptosporidiosis.
1. Sanal O et al: Additional diverse findings expand the clinical presentation of DOCK8 deficiency. Journal of clinical immunology 2012, 32(4):698-708.
ESID-0724 New ADA-Mutation Defining an Alternative Splicing in a Patient with SCID T-B-NK+
E.A. González1, M. Piquer2, E. Ruiz1, E. González1, E. Giocaliere3, G. La Marca3, J. Garcia-Villoria4, A.M. Plaza2, J. Yagüe1, I. Badell5, A. Ribes4, J.I. Arostegui1, L. Alsina2, M. Juan-Otero 1
1Servei d’Immunologia – Centre Diagnòstic Biomèdic / Functional Unit of Immunology SJD-Clínic, Hospital Clínic - IDIBAPS, Barcelona, Spain
2Allergy and Clinical Immunology Pediatric Department / Functional Unit of Immunology SJD-Clinic, Hospital Sant Joan de Déu / Barcelona University, Barcelona, Spain
3Newborn Screening Biochemistry and Pharmacology Laboratories., Clinic of Pediatric Neurology Meyer University Children’s Hospital, Florence, Italy
4Secció d’Errors Congènits del Metabolisme Servei de Bioquímica i Genètica Molecular-CDB, Hospital Clínic – IDIBAPS - CIBERER, Barcelona, Spain
5Pediatric BMT Unit, Hospital de Sant Pau, Barcelona, Spain
Introduction: About 20% of human SCIDs (severe combined immunodeficiency) are the result of homozygous mutation in adenosine deaminase (ADA) gene, leading usually to a severe lymphopenia with T-B-NK-phenotype.
Case report: At 17 days of birth, a North-African girl (with consanguineous parents and 2 healthy siblings) developed an interstitial pneumonia without an evidenced infectious origin, associated to a severe lymphopenia (300 cells/μl). Preauricular and laterocervical fistulae were also present. Flow-cytometry analysis revealed only 3.2% of T-cells (CD4/CD8 ratio 1.2 , 47.9% were DN; 98.3% CD45RO+), 63% NK (absolute 189/mm3) but no B-cells (0.9%) and absent PHA proliferation, thus stating SCID T-B-NK+ diagnosis. One month later, a SCT from his matched-sibling donor was performed with success. In order to assess this SCID, tandem mass-spectrometry of adenosine metabolites and genomic DNA sequencing were performed.
Results: Adenosine and 2-deoxiadenosine levels were markedly elevated in newborn dried spot blood from the patient. By sequencing ADA gene, a previously non-described 4 bp homozygous deletion in the first four nucleotides of intron 4 (c.362+1_+4del4bp), responsible for the binding of splicing machinery, was detected. To state functional implication of this mutation, RNA was extracted from frozen blood, and cDNA was sequenced (exon 2-12), confirming a deletion of exons 4-6 and surprisingly also of exon 9, confirming a defect in splicing process.
Conclusions: Genetic defects affecting the splicing process in ADA gene can define an analytical T-B-NK+ phenotype. This report reinforces the relevance of assessing splicing-sites (and in general, non-coding parts) of primary immunodeficiencies involved genes.
ESID-0181 In Vitro and In Vivo Functional Evaluation of T-Cells Generated from WT1-TCR Transduced Human Hematopoietic Stem Cells Using the OP9-DL1 Coculture System
S. Bonte1, S. Snauwaert1, G. Goetgeluk2, H. Stauss3, R. Stripecke4, M. Heemskerk5, Y. Van Caeneghem2, G. Verstichel2, S. Vanhee2, B. Vandekerckhove2, T. Kerre 6
1Immunology - Hematology, Ghent University Hospital, Ghent, Belgium
2Immunology, Ghent University Hospital, Ghent, Belgium
3Immunology, University College London, London, United Kingdom
4Regenerative Immune Therapies Applied, Hannover Medical School, Hannover, Germany
5Laboratory of Experimental Hematology, Leids Universitair Medisch Centrum, Leiden, Netherlands
6Immunology - Hematology, Ghent University Hospital, Gent, Belgium
Chemotherapy leads to cure of AML in less than half of the patients. Stem cell transplantation carries a high risk of toxicity and mortality, and not all patients have a suitable donor. We have developed a novel immunotherapeutic treatment directed against WT1, a tumor antigen that is overexpressed on 70% of the AMLs.
UCB CD34+ cells were cultured on OP9-DL1 in the presence of IL-7, Flt3-L and SCF for 2 w, transduced with a WT1-TCR, and again cocultured until CD4+CD8+ cells were abundantly present. Then, the agonist peptide WT1126-134 was added, together with IL-7, IL-2, IL-15, IL-21 or combinations. Five days later, cells were harvested and expanded polyclonally. T-cells were evaluated in vitro using a 51chromium release assay, and, upon activation, for production of IFN? (ELISA). IL-15 combined with IL-7 gave the highest number of phenotypically mature CD1a- T-cells, and resulted in a similar amount of CD4+ and CD8+ SP T cells. We could show specificity of the T-cells.
For the in vivo evaluation, NSG mice were irradiated (200 cGy), and 24 hours later injected IV with 10E6 K562-A2-WT1-fLuc cells, together with 5x106 or 107 WT1+ T-cells or CMV+ T-cells (negative control). Mice were evaluated using the IVIS bioluminescence assay. From day 16 after injection on, a clear bioluminescent signal could be observed. Iintravenously injected K562 cells preferably engraft in mice ovaria, central nervous system (CNS) and liver. Mice receiving WT1-specific T-cells generally live longer compared to mice receiving control T-cells or no T-cells at all (Figure 1).
ESID-0328 Neonatal T-Cell Receptor Excision Circles and Phenotypic Features in 22q11.2 Deletion Syndrome
G. Kiran 1, T. Øverland2, L. Osnes3, L. Baumbusch3, R. Pettersen4, K. Lima5, T. Abrahamsen6
1Pediatric research Institute, Oslo University Hospital, Oslo, Norway
2Pediatric Medicine, Oslo University Hospital, Oslo, Norway
3Immunologi, Oslo University Hospital, Oslo, Norway
4Norwegian national unit of newborn screening, Oslo University Hospital, Oslo, Norway
5Endocrinology, Akershus University Hospital, Oslo, Norway
6Pediatric Medicine University oslo, Oslo University Hospital, Oslo, Norway
Background: Newborns with 22q11.2 deletion syndrome often have T-cell lymphopenia and low number of TRECs (T-cell receptor excision circles). So far, only few studies have investigated the correlation between TRECs and phenotypes in 22q11.2 deletion syndrome.
Methods: In this national survey neonatal TREC levels of 46 patients have been measured and correlated with development of different phenotypes in 22q11.2 deletion syndrome. TREC have been analyzed using dried blood from newborn screening filter cards.
Results: Two of the 46 patients in the survey had thymus aplasia. The other patients were divided in quartiles of TREC levels. A significant correlation (p=0.010) was found between lowest TREC quartile (<71 TREC/ul) and severe cardiac defects. Eight of eleven patients in the lowest quartile had an urgent need for cardiac intervention or operation within 2 weeks of life or they died of the heart defect. Furthermore, a correlation was found between low TREC quartiles and abnormal thymus feature (no thymus or remnant thymus, p=0.022). The proportion of patients with CD3+CD4+ T-cells below the 5th percentile (p=0.027) also correlated with low TREC quartile.
Conclusion: Our study demonstrates a correlation between severe cardiac defects and low neonatal TREC levels. There is no clear explanation for this, but embryological factors may play a role. Our results further indicate that low TREC levels correlate with low CD3+CD4+ T-cells and abnormal thymus features.
ESID-0480 Helios Expression in T-Regulatory Cells in Patients with diGeorge Syndrome
A. Klocperk 1, J. Kayserová1, J. Grecová1, K. ?i?mová1, E. Fronková1, A. ?edivá1
1Ústav imunologie 2.LF UK a FN Motol, FN Motol, Praha 6, Czech Republic
Purpose Syndrome diGeorge is associated amongst other clinical signs with various degrees of thymic dysplasia, related immunodeficiency and autoimmune disorders. Helios, a transcription factor from Ikaros family, has been proposed as a marker for thymus derived Tregs. We therefore examined Helios+ Tregs in a cohort of patients with genetically proven diGeorge syndrome with typical T cell lymphopenia due to thymic pathology.
Methods T cells, FoxP3+ Tregs and Helios+ FoxP3+ Tregs were examined in 52 samples from 37 patients. One patient with diGeorge/CHARGE syndrome with total thymic aplasia was also included. Statistical analysis was performed using a linear regression comparison.
Results Total Tregs were significantly lower in diGeorge patients as compared to controls in all age groups (0-20 years) (p=0.0016). The difference was more expressed during first four years of age. Relative Treg count expressed as the percentage of Tregs in CD4+ T-cells, however, was not different in patients and controls in all age groups (p=0.661), neither could we find any significant difference in the percentage of Helios+ Tregs between patients and controls (p=0.238). Helios+ Tregs were still present in a patient with diGeorge/CHARGE syndrome with complete athymia 7 years after partially matched unrelated repeated T lymphocytes infusions.
Conclusion Our findings show that while there was a significant decrease in absolute numbers of Tregs in patients with diGeorge syndrome, the relative percentage of this population did not differ between patients and controls. Low absolute Tregs thus reflected typical T cells lymphopenia in patients. Helios expression was not affected in diGeorge syndrome.
ESID-0795 Mucocutanous Candidiasis and Combined Variable Immunodeficiency in a Malnourished Boy with Gain of Function Mutation in the STAT1 Gene
R. Kobbe 1, F. Brinkert1, D. Nolkemper1, H. Schäfer2, B. Grimbacher3
1Department of Paediatrics, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
2Institute for Pathology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
3Centre of Chronic Immunodeficiency, University Medical Centre Freiburg, Freiburg, Germany
Introduction Dominant gain-of-function mutations in signal transducer and activator of transcription 1 (STAT1) have been associated with chronic mucocutaneous candidiasis (CMC). We present a 10-year old boy with CMC, combined variable immunodeficiency (CVID) and failure to thrive.
Case report Initially, the boy was admitted with typical symptoms of celiac disease and elevated anti-gliadin IgG antibodies. However, endoscopy showed no typical histology of celiac disease, but severe candida esophagitis. Subsequently, FACS analysis of CD4+ cells showed low IL17 expression, but normal IFN-gamma signals after stimulation. V-beta receptor repertoir was normal. Interestingly, the patient also showed diminished responses to vaccines, B cell lymphopenia and low serum levels of IgG, IgA and IgM, fulfilling criteria for CVID. The B cell compartment showed expansion of naive (IgM+IgGD+/CD27-), CD21low- and transitional B cells, while marginal zone (IgD+CD27+IgM) and switched memory- B- and plasma cells (IgD-CD27+) were reduced. Sequencing of STAT1 identi?ed a heterozygous mutation in the coiled-coil domain (c.514T>C, p.Phe172Leu), previously only described in an adult with a history of recurrent histoplasmosis in childhood and late onset CMC.
Treatment with fluconazol, trimethoprim/sulfmethoxazole and immuneglobulines resulted in partial response, but oral candidiasis frequently reoccured. Although, anti-gliadin antibodies declined, because of failure to thrive and non-typical histological duodenitis with patchy villous atrophy in combination with the boys`genetic background (HLA-DQ2) celiac disease was reconsidered and gluten-free diet initiated. Clinical and histological response is pending and will be reported.Conclusion
Mutations in the STAT1 gene can result in heterogeneous clinical phenotypes and pitfalls in diagnosis and treatment.
ESID-0281 The Three Copies of Interferon Receptors Underlie a T Cell Immunodeficiency in Patients with Down Syndrome
X.F. Kong 1, A. Puel2, M. Goulet3, Y. Itan1, J. Bustamante4, S. Boisson-Dupuis1, L. Abel2, C. Picard4, S. Lyonnet5, J.L. Casanova1
1St. Giles Laboratory of infectious disease, The Rockefeller University, New York, USA
2Laboratoire de Génétique Humaine des Maladies Infectieuses INSERM U1163, Institut Imagine, Paris, France
3Département de Génétique, Institut Imagine, Paris, France
4Centre d'Etudes des Déficits Immunitaires (CEDI), Institut Imagine, Paris, France
5Département de Génétique et Unité INSERM U-781, Institut Imagine, Paris, France
Patients with Down Syndrome (DS) are prone to various infectious diseases, the pathogenesis of which is poorly understood. The four interferon receptor (IFN-Rs) genes, IFNAR1, IFNAR2, IFNGR2 and IL-10RB are clustered together on chromosome 21. Patients heterozygous for gain-of-function (GOF) STAT1 mutations suffer from overlapping infectious diseases. Accordingly, we found that EBV-B cells and monocytes from DS patients show increased IFN-Rs expression and enhanced cellular responses to IFN-γ and IFN-α/β. Ex vivo study of monocytes revealed higher levels of basal STAT1 expression and STAT1 Tyrosine-701 phosphorylation. In contrast, CD8+ and CD4+ naïve T cell showed an impaired IFN-α response. Ex-vivo whole transcriptome analysis of the monocytes, CD4+ and CD8+ T cells revealed an interferon signature and immune dysregulation. DS patients also have diminished CD4+ naïve T cells with selectively decreased IFNAR1 and IFNAR2 mRNA, which suggest that in vivo selection may favor the survival of IFN-α hypo-responsive T cells. We confirmed the selective pressure exerted by IFN-α, as CD8+ T cells from GOF STAT1 patients display the same phenotype. Overall, the two genetic models of DS and GOF STAT1 show that enhanced IFN responses are detrimental to certain T cells but not to monocytes. These findings provide a molecular and cellular basis for the infections see in both DS and GOF STAT1 patients.
ESID-0023 CD4+CD25+Foxp3+ T Regulatory Cells, Th1 (CCR5, IL-2, IFN-G) and Th2 (CCR4, IL-4, IL-13) Type Chemokine Receptors and Intracellular Cytokines in Children with Common Variable Immunodeficiency
N. Kutukculer 1, N.E. Karaca1, E. Azarsiz1
1Department of Pediatrics, Ege University Facultyof Medicine, Izmir, Turkey
Abnormal lymphocyte trafficking, dysregulated cellular responses to chemokines and uncontrolled T cell polarization may be involved in the pathogenesis of CVID. We evaluated T helper subsets (Chemokine receptors-CCR4, CCR5 and CCR7-expressions on T lymphocytes, intracellular cytokines - IL-2, IL-4, IL-13, IFN- γ-on CD4+ T cells, expression of CD4+CD25+ Foxp3+ regulatory T cells of 20 CVID patients and 26 healthy controls. Autoimmune clinical findings and other complications were also determined.
Percentages, and absolute numbers of CD4+CD25+ Foxp3+ cells did not show any significant difference between CVID cases and controls and also between severe and moderate patients. The only significant difference regarding Th1 and Th2 intracellular cytokines is the decreased numbers of CD3+CD4+IL4+ cells in CVID cases. There are some findings about T helper type dominance in CVID patients such as positive correlation between hepatomegaly and high IL-2 and IFN-γ in CD3+CD4+ cells and very high expression of CCR5 (Th1) on CD3+CD4+ cells in patients with granuloma. Th1 (CCR5) and Th2 (CCR4) type chemokine receptors did not show any dominance in CVID cases. However, frequencies of CCR7 expressing CD3+ T cells, CD3+CD4+ T helper cells and CD3+CD8+ T cytotoxic cells were significantly lower in severe CVID patients. In addition, presence of autoimmune clinical findings was negatively correlated with CCR7+ cells.
As CCR7 is a key mediator balancing immunity and tolerance in the immune system, the abnormality of this mediator might contribute to the profound immune dysregulation seen in CVID. In addition, Th1 cells seem to be more involved in disease pathogenesis than Th2 cells.
ESID-0228 Moesin Mutation in a New Form of X-Linked Primary Immunodeficiency (PID)
C. Lagresle-Peyrou 1, S. Luce2, F. Ouchani2, H. Sadek2, C. Picard3, V. Gandemer4, D. Monnier5, C. Hejmans6, N. Malhaoui7, J.L. Stephan8, A. Fischer9, M. Cavazzana2, I. André-Schmutz2
1CIC BT502, Imagine - INSERM, Paris, France
2Human Lympho-Hematopoiesis Lab, Imagine - INSERM U1163, Paris, France
3CEDI, AP-HP, Paris, France
4Unité d'hémato-oncologie et greffes de moelle, CHU Rennes, Rennes, France
5ITeCH, CHU Rennes, Rennes, France
6Oncologie-Hématologie, Hopital Universitaire des enfants Reine Fabiola, Bruxelles, Belgium
7CEREDIH, Hôpital Necker-Enfants Malades, Paris, France
8Unité d'Immuno-hématologie et Oncologie pédiatrique, CHU de Saint Etienne, Saint Etienne, France
9Unité d'Immuno-hématologie pédiatrique, Hôpital Necker-Enfants Malades, Paris, France
Five pediatric (1.3- 14 years) and 1 adult (39 years) patients from 4 different families presented similar clinical and biological features. During childhood, most of them developed severe varicella, pneumopathies and recurrent pulmonary infections. In the peripheral blood, we observed a profound leucopenia and neutropenia. All the patients have a hypogammaglobulinaemia and a poor response to vaccinal Ag. The naïve T cell compartment was particularly low while CD57+ senescent T cells were overrepresented as compared to healthy controls or others PID. Despite the severe leucopenia, Igs and prophylactic treatment appeared sufficient to prevent severe infections. Using exome and sequencing analysis, we have identified in all patients the same missense mutation in the moesin gene (Xq11.1) that introduces an amino acid change into the highly conserved FERM domain of the protein (p.R171W). Moesin is a member of the ERM protein families, which link plasma membrane proteins with actin–based cytoskeletons and are implicated in various cellular functions such as survival, adhesion, migration and activation. The p.R171W mutation was associated with a heterogeneous expression in lymphocytes. Upon in vitro activation, proliferation and survival capacity of patient's T cells was impaired and they harbored a decreased expression of some chemokine receptors and increased expression adhesion molecules. Complementation assays on patients' T cells confirmed the main role of MSN in T cell activation/survival. This report highlights for the first time the association of a defect in an ERM protein and a primary immune deficiency affecting both innate and adaptive immunity.
ESID-0341 Human Naive-Derived T Memory Stem Cells Preferentially Reconstitute the T Cell Immunodeficient Host
E. Lugli 1, A. Roberto1, V. Zanon1, R. Crocchiolo2, J. McLaren3, B. Sarina2, S. Bramanti2, S. Gandolfi2, K. Ladell3, D.A. Price3, M. Roederer4, L. Castagna2, D. Mavilio1
1Clinical and Experimental Immunology, Humanitas Clinical and Research Center, Rozzano, Italy
2Humanitas Cancer Center, Humanitas Clinical and Research Center, Rozzano, Italy
3Infection and Immunity, Cardiff University, Cardiff, United Kingdom
4Vaccine Research Center, National Institutes of Health, Bethesda, USA
Current models of peripheral T cell differentiation indicate early differentiated T cells, and above all the human T memory stem cells (TSCM), as endowed with the highest capacity to reconstitute immunodeficient hosts, but a direct demonstration is lacking. By combining 18-color flow cytometry, cellular technologies and antigen-specific assays, we define a role for TSCM cells in immune recovery in lymphoma patients (n=26) undergoing haploidentical bone marrow transplantation (BMT) and post-transplant cyclophosphamide (Cy). All patients reached 100% peripheral donor chimerism, indicating a pivotal role for donor T cells in reconstitution. Following transfer in the lymphopenic host, donor memory T cells undergo rapid proliferation and are thus depleted by Cy treatment. Conversely, due to their delayed activation kinetics, naïve T cells survive Cy and subsequently differentiate into TSCM cells. Naïve–derived TSCM dominate the T cell compartment early after transplantation (~70% of all donor T cells) while central memory and effector memory T cells appear later, suggesting the differentiation of TSCM into more differentiated subsets. Importantly, antigen-specific memory T cells that are transferred with the graft cannot be found in the recipients, indicating their poor contribution to T cell recovery. Conversely, naïve T cells specific for the self antigens MelanA/Mart1 and Wilms’ Tumor 1 (WT1) are readily detectable in the host up to 90 days post transplantation and differentiate into effector cells. Our data indicate the pivotal role of naive-derived TSCM cells in mediating T cell recovery and suggest that induction of TSCM cell differentiation should be favoured to overcome immunodeficiency.
ESID-0076 Determining the Molecular Requirements for Generating Human T Follicular Helper (Tfh) Cells
C.S. Ma 1, N. Wong1, D.T. Avery1, J. Torpy1, J.L. Stoddard2, M. Kobayashi3, E.K. Deenick1, J.L. Casanova4, G. Uzel5, S.G. Tangye1
1Immunology, Garvan Institute of Medical Research, Darlinghurst, Australia
2Clinical Center, NIAID NIH, Bethesda, USA
3Department of Pediatrics, Hiroshima University Graduate School of Biomedical ad Health Sciences, Hiroshima, Japan
4Laboratory of Human Genetics of Infectious Diseases Rockefeller Branch, The Rockefeller University, New York, USA
5Laboratory of Clinical Infectious Diseases, NIAID NIH, Bethesda, USA
The generation of protective antibodies by B cells following natural infection or vaccination requires 'help' from CD4+ T cells. T follicular helper (Tfh) cells are the specialized CD4+ T cell subset that has evolved appropriate mechanisms to induce the differentiation of B cells into memory and plasma cells. As such, it is important to determine the mechanisms that regulate Tfh cell generation and function, as excessive Tfh cell activity has been implicated in the development of autoimmune diseases, while under-activation is associated with immunodeficiency. Improved understanding of the regulation of these cells may also be invaluable to facilitate improved vaccine development. Characteristics of human Tfh cells include expression of CXCR5, IL-21, CD40L, ICOS, and PD-1. We have previously shown that IL-12 is capable of inducing some of these characteristics in vitro. We have now extended this work to determine the molecular requirements for generating human Tfh cells by investigating patients with specific defects in key signaling pathways. Circulating Tfh cells were reduced in patients with loss-of function mutations in STAT3, CD40LG, ICOS, NEMO and BTK. Furthermore, the ability to generate Tfh-like cells in vitro was compromised by loss-of function mutations in STAT3, IL21R/IL21,CD40LG, NEMO, BTK and IL12R, as well as gain-of function mutations in STAT1. These findings reveal critical roles for signaling through IL-12R and IL-21R, as well as cellular interactions between naïve CD4+ T cells and Ag-presenting cells in generating human Tfh cells, thereby providing new insights into human Tfh cell biology.
ESID-0287 Experience at the Immunodeficiencies Research Unit of the National Institute of Pediatrics in the Quantification of TRECs Obtained from Neonatal Screening Samples in a Mexican Population
E. Medina_Torres 1, E. González-Serrano1, A. Garay-González2, F. Espinosa-Rosales1, S. Espinosa-Padilla1
1Immunodeficiencies Research Unit, Instituto Nacional de Pediatría, Mexico City, Mexico
2Methodology Department, Instituto Nacional de Pediatría, Mexico City, Mexico
Introduction In 2010, severe combined immunodeficiency (SCID) disorder became the first new condition to be added to the US HHS Recomended Uniform Screening Panel. The assay used to detect SCID is a quantitative PCR that measures the amount of-Tcell receptor excision circles (TREC) from a punch from a newborn dried blood spot. We have implemented this technique in our laboratory and so far we have analyzed 600 newborns samples. The objective of this paper is to present the results obtained until now.
Methods DNA was extracted with the DNA extraction kit PrepFiler Express from blood samples on Guthrie card, complete lysis automated equipment was used to extract DNA Express.
Healthy newborns samples were obtained through an agreement with the health authorities of the state of Tabasco, Mexico.
To validate the method, we used samples from patients clinically diagnosed as SCID, healthy adults and patients with CID in single-blind trials.
Results The quantization values of 1161 healthy new borns ??ranged from 36-8010 TRECs / mL with median 285 TRECs/mL (percentile 25-75 96-552 TRECs/mL).
The single-blinded trials where we analyzed samples from patients with clinical diagnosis of SCID along with samples from healthy newborns resulted in identification of all SCID patients. Moreover, we confirmed the diagnosis of SCID in samples from patients with CID, except for one case in which the diagnosis was confirmed to CID.
Conclusions The DNA extraction is a key process in this assay and may affect the efficiency, reproducibility and costs of TRECs quantification.
ESID-0561 Prevalence of Immunosenescence Profiles in Patients with CVID
E. Naumova 1, S.M. Mihailova1, N. Gesheva1, M.I. Ivanova1, S. Lesichkova1, A.P. Mihaylova1
1Department of Clinical Immunology, University Hospital Alexandrovska, Sofia, Bulgaria
Common variable immunodeficiency (CVID) is a heterogeneous antibody deficiency syndrome with alterations in T-cell regulation and function. Our previous observations revealed shared immune profiles between CVID patients and elderly healthy. Thus, the objective of this study was to evaluate the immune risk markers leading to accelerated immunosenescence in CVID. Ten patients with CVID were characterized according to their cell phenotype and cytokine gene expression profiles. Our data demonstrated a number of immunological abnormalities, including low CD4/CD8 rati?, shift of phenotype from naïve and early differentiated toward terminally differentiated CD8+ T cells, higher levels of T-cell activation (CD8+HLA-DR+,CD8+CD38+) and senescence (CD8+CD28- and CD8+CD57+), decreased switched memory (IgM-IgD-CD27+), circulating mature (CD21+CD24+) B cells and NK cells. In elderly we have previously described similar results: decreased B cells and naive T cells, accumulation of late-differentiated (CD28-57+) CD8 subsets. However, unlike of CVID, the number of NK cells increased and the expression of cell activation markers decreased with aging. Extended cytokine profiles, strongly related with cytokine secretion, showed low IFN-gamma and high TNF-a expression level among patients which benefit pro-inflammatory induced immunosenesence. In conclusion, our data suggested an association of the CVID with accelerated immunological ageing due to a prevalence of immune risk phenotype. Since this phenotype is linked to poor clinical outcomes in the elderly it may also contribute to the clinical consequences in CVID patients. Furthermore the immunosenescent phenomena seem to be already present early in the disease process and are partially modulated by IVIG treatment.
ESID-0728 Anuric Renal Failure Revealing Materno-Foetal Graft Versus Host Disease in a Child with ILR2G Related Severe Combined Immunodeficiency
M. chomton1, S. pierrepon2, G. mortamet3, L. de saint Blancat3, A. lim4, C. picard5, R. Salomon6, A. Fischer7, B. neven 1
1pediatric immunology, AP HP Hôpital Necker Enfants Malades, PARIS Cedex 15, France
2pediatric nephrology, AP HP Hôpital Necker Enfants Malades, PARIS Cedex 15, France
3pediatric intensive care unit, AP HP Hôpital Necker Enfants Malades, PARIS Cedex 15, France
4immunology, Institut Pasteur, PARIS Cedex 15, France
5centr d'étude des déficits immunitaires, Necker Hospital, PARIS Cedex 15, France
6pediatric nephrology, Necker Hospital, PARIS Cedex 15, France
7pediatric immunology, Necker Hospital, PARIS Cedex 15, France
We report on a male patient born from non-consanguineous parents who presented a rhinovirus bronchiolitis at the age of 6 months complicated by anuric renal failure requiring peritoneal dialysis within few days. Kidney biopsy showed massive lymphohistiocytic infiltrates destroying normal tissue architecture. Due to mild. lymphopenia (1700 to 2200 lymphocytes/μl) and agammaglobulinemia, immune deficiency was suspected and confirmed by the immunophenotype. The following displayed T cells lymphopenia (900 CD3/ μl) with 93% of γδ T cells, low B cells (70/ μl) and NK cells (10/ μl)) counts compatible with a diagnosis of severe combined immune deficiency (SCID). Surprisingly, γδ T cells were from maternal origin. Immunoscope showed oligoclonal profile of T cells with a dominant Vd1/Vg3 clone. Molecular diagnostic study showed hemizygous mutation in IL2RG gene (c.G344A, p.C115Y). Hematopoietic stem cell transplantation was performed from a matched sibling donor without conditioning regimen, allowing a fast immune reconstitution and a rapid clearance of circulating maternal γδ T cells (within 2 weeks). Three months post HSCT, anuric renal failure persists and a kidney transplant is pending. We hypothesize that renal failure was related to inflammation driven by infiltrating maternal T cells. Materno-foetal GVHD driven by γδ T cells and targeting kidneys are both very unusual and worthwhile to be reported.
ESID-0212 Immunodeficiency Caused by a Non-Sense Mutation of IKBKB
C. Nielsen 1, M.A. Jakobsen1, M.J. Larsen2, A.C. Müller1, S. Hansen3, S.T. Lillevang1, N. Fisker4, T. Barington1
1Clinical Immunology, Odense University Hospital, Odense, Denmark
2Clinical Genetics, Odense University Hospital, Odense, Denmark
3Institute of Molecular Medicine, University of Southern Denmark, Odense, Denmark
4Hans Christian Andersen Children’s Hospital, Odense University Hospital, Odense, Denmark
We report an infant of consanguineous parents of Turkish decent with a novel immunodeficiency caused by homozygosity for a non-sense mutation of the gene encoding Inhibitor of nuclear factor kappa-B (NF-κB) kinase subunit beta (IKKβ). At five months, she presented with respiratory insufficiency and Pneumocystis jirovecii pneumonia which was successfully treated. At nine months, iatrogenic systemic infection with Mycobacterium bovis was found and eventually led to her death at age 14 months. Laboratory findings were reminiscent of hyper-IgM syndrome, but genetic testing gave no explanation before whole exome sequencing revealed a novel mutation abrogating signaling through the canonical NF-κB pathway.
ESID-0406 Immune Defect in Schimke Syndrome is Partially Due to Defective T Cell Production in Thymus
B. Piatosa 1, E. Heropolitanska-Pliszka2, B. Pietrucha2, K. Siewiera1, A. Rekawek1, K. Tkaczyk1, W. Jarmuzek3, J. Rubik3, E. Bernatowska2, R. Grenda3
1Histocompatibility Laboratory, The Children's Memorial Health Institute, Warsaw, Poland
2Clinical Immunology, The Children's Memorial Health Institute, Warsaw, Poland
3Nephrology and Kidney Transplantation, The Children's Memorial Health Institute, Warsaw, Poland
Introduction: Schimke immunoosseus dysplasia is an autosomal recessive multisystem disorder caused by mutation in SMARCAL1 gene. HARP, the product of defective gene, is responsible for chromatin remodelling therefore affecting replication, recombination, methylation, as well as DNA repair and transcription. The majority of affected individuals present with T cell deficiency and risk of opportunistic infections.
The purpose of the study was to evaluate the effectiveness of T and B cell maturation process in order to evaluate the cause and scope of immune deficiency in patients with Schimke syndrome.
Material and methods: Three patients aged 7-10 y.o. with Schimke syndrome on stable, reduced to two drugs, immunosuppression, (6 months, 2 years, 3 years after kidney transplantation respectively), were submitted to immunological investigations due to atypically frequent serious recurrent infectious phenomena. Flow cytometric evaluation of T and B cell maturation was performed by four color flow cytometry and results were compared to normal controls.
Results: Irrespective from suppressive regimen applied after transplantation, significantly reduced proportion of recent thymic emigrants and CD4:CD8 ratio, as well as elevated proportion of antigen-experienced cells (RO+) were found. Despite participation in DNA repair defective HARP did not affect peripheral B cell maturation process.
Conclusion: Immune deficiency in patients with SMARCAL1 mutations results at least in some part from ineffective thymic lymphocyte production.
ESID-0408 First Case of Bare Lymphocyte Syndrome in Poland
B. Piatosa 1, B. Wolska-Kusnierz2, K. Bernat-Sitarz2, M. Ambroziak-Tyminska3, K. Siewiera1, K. Tkaczyk1, A. Rekawek1, M. Pac2, H. Wieteska-Klimczak3, E. Bernatowska2
1Histocompatibility Laboratory, The Children's Memorial Health Institute, Warsaw, Poland
2Clinical Immunology, The Children's Memorial Health Institute, Warsaw, Poland
3Pediatrics Nutrition and Metabolic Diseases, The Children's Memorial Health Institute, Warsaw, Poland
MHC class II deficiency is a combined immunodeficiency with absent cellular and humoral immune, resulting in extreme susceptibility to viral, bacterial and fungal infections. The disease is usually fatal in early childhood. Diagnosis is established based on defective expression MHC class II antigens on B-cells or monocytes and/or molecular identification of mutation in any of CIITA/RFX-B/RFX-5 or RFX-AP genes. Most patients originate from the Mediterranean region. We report the first case diagnosed in Poland.
Male child aged 5 months, with irrelevant family history, was referred to our institution due to protracted fever, hepatitis of unknown origin, sepsis, and history of urinary tract infection at age of 2 weeks. Severe complications developing during following weeks, skin lesions developing into exfoliating erythrodermia with low serum IgE levels, agammaglobulinemia, and poor response to mitogens, suggested severe combined immune deficiency (SCID).
Four color flow cytometry was used to evaluate lymphocyte subsets, peripheral T cell maturation, expression of TCR, distribution of TCRVβ chains, and expression of MHC class I and II antigens.
Lymphopenia with low T-lymphocytes affecting mostly T-helper cells, low normal recent thymic emigrants with increased proportion of memory T cells, and polyclonal distribution of TCRVβ chains suggested T+SCID. Although most nucleated cells expressed MHC class I antigens, fluorescence intensity was significantly lower than on control cells. MHC class II expression was found on 0.1% of B cells and 0.4% of monocytes and did not rise after stimulation. The patient was diagnosed as bare lymphocyte syndrome with negative expression of MHC class II.
ESID-0522 Recurrent Respiratory Infections Revealing CD8Alpha Deficiency
E. Dumontet1, J. Osman1, N. Guillemont-Lambert2, G. Cros3, D. Moshous 3, C. Picard1
1Study center of immunodeficiency, Necker Hospital, Paris, France
2Pneumology pediatric unit, Trousseau Hospital, Paris, France
3Immunology hematology unit, Necker Hospital, Paris, France
We have characterized the third kindred with CD8α deficiency in a 14 years old Portuguese patient caused by the same homozygous c.331G>A missense mutation as previously reported. Patient’s medical history was characterized by recurrent ENT and lung infections since first months of life. He developed progressive chronic obstructive pulmonary disease and bilateral bronchiectasis. Laboratory tests revealed normal levels of immunoglobulins (Ig) and specific antibodies. Absolute lymphocyte counts were normal. Immunophenotyping showed normal TCD4+, B and NK cells counts, but complete absence of the TCD8+ subset. The patient has an increased of double negative T cells, but normal repartition of TCRγδ and αβ. The Vβ repertory was normal. T cell proliferations were normal upon stimulation with mitogens and antigens. The diagnosis of CD8a deficiency may be a challenge for the clinician as the basic immunological investigations can be completely normal as in the patient reported here, who has normal absolute lymphocyte counts and normal humoral responses. This underlines the importance to perform an immunophenotyping in patients with clinical suspicion of primary immunodeficiency in order to confirm or to discard the absence of CD8+ T lymphocytes. Antibioprophylaxis is recommended. IgIV substitution should be considered despite apparently normal humoral immunity as CD8α deficiency may have an impact on the quality of the immune response and as the prognosis seems to be variable ranging from mild and even asymptomatic forms to fatal pulmonary insufficiency justifying optimal treatment. Further studies are warranted to determine the eventual benefit of hematopoietic stem cell transplantation in CD8α-deficient patient.
ESID-0125 Digeorge Syndrome: Phenotypic Spectrum of a Paediatric Cohort Enrolled in IPINET (Italian Primary Immunodeficiency Network) Database
C. Caccia Dominioni 1, L.A. Baselli1, R.M. Dellepiane1, L.V. Beilis1, E. Clerici1, M. Raimondi1, M.C. Pietrogrande1
1Department of Paediatrics University of Milan, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milano, Italy
DiGeorge syndrome occurs in 1:4000-1:6000 births. It can be inherited in autosomal dominant fashion, however it is mostly due to a de novo mutation.
The phenotypic spectrum is variable including, in patients diagnosed before two years of age, congenital heart defects, neonatal hypocalcaemia due to hypoparathyroidism, dysmorphic features. After two years of age atypical symptoms such as recurrent infections, otorinolaryngoiatric and gastrointestinal manifestations, autoimmune disease led to diagnosis. Developmental delay is reported in 82-100% cases. Cardiovascular complications resulted to be the main cause of mortality. Thymic aplasia/hypoplasia were reported in 75% cases, characterised by different immunological alteration, ranging from mild decrease of T cell with tendency to normalisation to total absence of T cell. Variable defects in humoral compartment have been also reported.
Fifteen patients (6M, 9F) were enrolled detected by FISH, median age at diagnosis 11 months. Precocious diagnosis was associated to major cardiac defect and/or neonatal hypocalcaemia. Prenatal diagnosis was made in two cases. Two patients diagnosed after 12 months had only dysmorphic features. Eight presented with otorinolaryngoiatric manifestations, mainly nasal reflux. Four revealed thymic aplasia, CD4+ T cells were decreased in 12 patients of whom 5 also had PHA response to mitogen decreased. Four received antibiotic prophylaxis until CD4+ normalisation. Eleven patients had cardiac defects, 4 major, the other ASD/VSD, respectively. Neuropsychiatric manifestations frequently required therapy over time.
In conclusion, this clinical picture suggests that a multidisciplinary management is necessary for a proper diagnosis and treatment, while preventing a delay of diagnosis.
ESID-0176 STAT3 Mutational Spectrum of Newly Diagnosed Patients with HIES from Eastern and Central European Countries
Z. Pistár 1, I. Kondratenko2, Z. Maszárovics3, L. Chernyshova4, A. Bondarenko4, M. Guseva5, L. Kostyuchenko6, Y. Romanyshyn6, B. Soltész1, A.K. Sarkadi1, L. Maródi1, B. Tóth1
1Department of Infectious and Pediatric Immunology, University of Debrecen, Debrecen, Hungary
2Department of Clinical Immunology, Russian Clinical Children’s Hospital, Moscow, Russia
3Department of Infectology, Markhot Ferenc Hospital, Eger, Hungary
4Department of Pediatric Infectious Diseases and Immunology, Medical Academy for Postgraduate Education, Kiev, Ukraine
5Consulting Centre, State Pediatric Medical Academy, St. Petersburg, Russia
6Western-Ukrainian Specialized Children’s, Medical Center, Lviv, Ukraine
The autosomal dominant hyper-IgE syndrome (AD-HIES) is a primary immunodeficiency disorder characterized by eczema, skin abscesses, recurrent staphylococcal infections of the lung and skin and elevated serum levels of IgE. Mutations in signal transducer and activator of transcription 3 (STAT3) have been implicated in the pathogenesis of AD-HIES. The mutational spectrum of patients in Eastern and Central European countries with AD-HIES was first published in 2008 (Mol. Immunol. 46: 202-206). Since then 12 more patients were genetically analysed in our PID Center. Sanger sequencing was used to detect sequences of coding exons by using genomic DNA. We have examined the pathogenicity of mutation by cDNA analyses and flow cytometry. We found 9 different mutations, two of which (c.1145G>C, c.1110-3C>G) are reported for the first time. The c.1110-3C
ESID-0630 The Quantification of T-Cell Receptor Recombination Excision Circles (TREC) and Ig Kappa –Deleting Recombination Excision Circles (KREC) in Children with Predominant T-Cell Immunodeficiency in Russia
M. Gordukova1, I. Korsunskiy1, N. Davydova1, A. Smirnova1, O. Barabanova1, O. Shvech1, S. Zimin1, N. Zinoveva1, M. Filipenko2, A. Prodeus 1
1Moscow City Center for Pediatric Immunology and Allergy, Speranskiy Children's Hospital #9, Moscow, Russia
2Laboratory of Pharmacogenomics, Institute of Chemical Biology and Fundamental Medicine, Novosibirsk, Russia
Introduction: We have recently developed real-time PCR based quantification of TREC and KREC and decided to apply this method to test patients with predominant T-cell deficiency
Patients and methods: Six severe combined immunonodeficiency (SCID), thirteen chromosome 22q11 deletion patients and four patients with chromosome instability syndrome - ataxia-telangiectasiya (AT) were included in this study. Multiplex quantitative PCR of frozen peripheral blood, immunophenotyping and clinical information were analyzed.
Results: Three SCID patients were T-B+NK- and three had phenotype T-B+NK+. All SCID patients had very low or negative TRECs. Only one patient was KRECs negative and two had low KRECs. In ten chromosome 22q11 deletion patients TRECs were undetectable. In two patients TRECs were significantly low. In four patients KRECs were undetectable and four of them had low KRECs. Only one patient had normal TRECs and KRECs. Among four patients with AT syndrome two were TRECs and KRECs negative. One was TRECs normal and KRECs negative and one had normal values of both analytes.
Conclusions: The combined measurement of TRECs and TRECs is a sensitive and specific test for diagnosis of T-cell disorders. These markers can be used for the understanding of the clinical severity, prognosis and pathogenesis together with other methods. Also this test can be applied for the early diagnosis of such patients.
ESID-0298 A Female with Late and Atypical Presentation of SCID
N. Radwan 1, S. Youssef2, S. Reda1, L. Notarangelo3, J. Chou3, M. Massad3, Z. Awad1, E. Hossny1, S. Saad1, D. Helmy1, R. Hassan1, Z. Ibrahim1
1Pediatric Allergy and Immunology, Ain Shames University, Cairo, Egypt
2Nutrition, Ain Shames University, Cairo, Egypt
3Boston Children's Hospital, Harvard Medical School, Boston, USA
Background: Severe combined immunodeficiency (SCID) is a life-threatening condition characterized by recurrent, severe and persistent infections. It is caused by several molecular defects affecting the number and function of T-cells, B-cells, and occasionally natural killer cells. Most patients present before three months of age.
Case Presentation: We report a three-years old girl, 6th in order of birth and of remote consanguineous parents who started repeated chest infections and frequent hospital admissions at 8-months of age. Her elder three brothers died before the age of 6-months due to pneumonia and gastroenteritis, the other siblings (three elder sisters and a younger brother) were healthy. The patient received all scheduled infant’s immunization including BCG without complications. On presentation, she had failure to thrive, no dysmorphic features and no organomegaly. Chest X-Ray showed absent thymus. The laboratory tests revealed anemia(Hb was 9.8 gm/dl), WBC count was 4.6x109/L with lymphopenia(2.5x109/L),platelets:250x109/L.All serum immunoglobulin levels were low (IgG:250 mg/dl,IgM:20 mg/dl,IgA:17 mg/dl,IgE:10 IU/ml).Lymphocyte subsets were: CD3: 75.8%,CD4:7.24% , CD8: 88.1%, CD19:6.65% and CD56:2.51%. Most of T-cells were memory CD45RO. Maternal engraftment was ruled out. There was absent recent thymic emigrants and CD8 T cells were effector memory. There was absent proliferation to PHA, anti-CD3, and PMA/Io. The diagnosis of SCID was put forward and she started regular IVIG treatment. Unfortunately, she died of pneumonia at the age of 3 years. Whole exome sequencing revealed a homozygous mutation in the JAK3 gene, causing A58T amino acid change (novel mutation), but a previously demonstrated different missense mutation at the same codon (A58P) was found.
ESID-0799 Omenn-Like Syndrome Associated with Mild Defects in V(D)J Recombination and TCR Signaling
M. Recio 1, R. Ramírez-Muñoz1, J. Torres-Canizales2, B. Garcillán1, M. Muñoz-Ruiz1, A.C. Briones1, E. López-Granados2, J.R. Regueiro1
1Immunology, Universidad Complutense de Madrid School of Medicine, Madrid, Spain
2Clinical Immunology, Hospital Universitario La Paz-IdiPaz, Madrid, Spain
Introduction Omenn syndrome (OS) is characterized by severe combined immunodeficiency associated with autoimmune features leading to erythrodermia, alopecia, lymphoadenopathy, hepatosplenomegaly, and intractable diarrhea. OS is genetically heterogeneous with clasical forms due to hypomorphic defects in genes involved in V(D)J recombination such as RAG or Artemis and OS-like forms associated with mutations in genes involved in other lymphocyte maturation steps. OS with both types of defects have not been reported.
Case Report A 3-month-old male admitted with failure to thrive, dermatitis, and severe respiratory infection was diagnosed as OS and received a successful HLA-matched unrelated cord blood transplantation
Results The immunological work-up showed low T and B cells numbers, eosinophilia and hypogammaglobulinemia. T cells showed impaired TCR-induced responses (CD25 and CD69 induction, Ca2+ mobilization, TCR down-modulation and proliferation using anti-CD3). Fibroblasts showed radiosensitivity and low Double-Strand Breaks (DSB) repair capacity both early and up to 72 h after irradiation. Pathogenic mutations in RAG and Artemis were excluded, but a polymorphism was found in the metallo beta-lactamase domain of Artemis (p.T.89A) that could impact on its function and explain the observed radiosensitivity and DSB repair defect.
Conclusions We report a patient with clinical features of OS closely resembling those of Artemis hypomorphic mutations patients and mild defects in both V(D)J recombination and TCR signaling.
ESID-0748 Complete Deficiency of CD247, the Zeta Chain of the T-Cell Antigen Receptor and the NK Fc Gamma Receptor III
M. Recio 1, C. Aydogmus2, A.V.M. Marin1, S. Haskologlu3, A. Jimenez-Reinoso1, F. Cipe2, A.C. Briones1, F. Dogu3, G. Esteso4, B. Garcillán1, J. Gil5, M. Muñoz-Ruiz1, M. Valés-Gómez4, H.T. Reyburn4, A. Ikinciogullari3, J.R. Regueiro1
1Immunology, Universidad Complutense de Madrid School of Medicine, Madrid, Spain
2Pediatric Immunology, Istanbul Kanuni Sultan Süleyman Hospital, Istambul, Turkey
3Pediatric Immunology-Allergy, Ankara University School of Medicine, Ankara, Turkey
4Immunology and Oncology, National Centre for Biotechnology CSIC, Madrid, Spain
5Immunology, Hospital Gregorio Marañón, Madrid, Spain
Introduction The CD247 dimer is required for assembly and signalling by the TCR and several NK receptors. Two CD247-deficient Caribbean cases reported previously showed some disparate T and NK immunological features, thus new cases can help to define the role of CD247 and the clinical and immunological range of this rare disorder.
Case report A 20 month-old Turkish girl born to consanguineous parents was admitted at 2 months of age with fever and pancytopenia after routine vaccination. Immunodeficiency was suspected because CMV-DNA did not decrease with treatment. TCR deficiency was initially suspected by extracellular flow cytometry.
Results CD247 deficiency was diagnosed by intracellular flow cytometry (no iCD247, but normal iCD3g, d or e) and ascertained by RT-PCR and sequencing as due to a c.2T>C (p.M1T) mutation. The patient had normal total T, B and NK lymphocyte numbers, with high CD8+ but very low CD4+ T cells and recent thymus emigrants, despite a normal-size thymus. T cells showed a severe surface TCR expression defect, with Mendelian partial defects in parents and several family members. Both CD4+ and CD8+ T cells showed impaired responses to TCR-dependent stimuli. A few T cells were iCD247+ due to somatic mutations and, in contrast to prior cases, showed carrier-level surface TCR and phosphorylation after anti-CD3 stimulation.
Conclusions Intracellular flow cytometry is useful for the rapid diagnosis of CD247 and other TCR immunodeficiencies. CD4+, but not CD8+, T cell development was strongly impaired in this case, although both were non-functional except when reverted.
ESID-0452 A Case of Combined Immunodeficiency Associated to Immune Dysregulation and Thrombocytopenia
S. Ricci 1, C. Canessa1, F. Lippi1, L. Bianchi1, F. Romano1, C. Azzari1
1Pediatric Immunology, Anna Meyer Children's Hospital, Florence, Italy
We report the case of a 4 years-old child, affected by an unknown primary immunodeficiency associated to immune dysregulation. Since he was 3 months old, he presents extended simil-vasculitic lesions. He is prone to persistent viral infections, as Rotavirus gastroenteritis and respiratory infections by Adenovirus and Syncitial Respiratory Virus. His immunological assessment shows a B-T+NK- picture. However, his exhibits hypergammaglobulinemia, with very high levels of IgA.Cytokines pattern is characterized by high values of IL-6 and depletion of IFN-γ. Autoimmunity exams are negative. He developed slight microcitic anemia and trombocytopenia progressively, and fluctuation of transaminases with normal liver function. In the last year, platelet count rapidly decreased. Myeloaspirate showed the absence of megacariocytes. All genetic tests, including array CGH, analysis of RAG1, RAG2, ZAP70, IL-2α receptor, GATA-2, WASP genes resulted negative. We suppose that the child presents a new gene defect, involved in inflammation pathway, immunity and platelets development.
ESID-0448 Adenosine Deaminase Deficiency: Genotype-Phenotype Correlation Based on Toxic Metabolite Levels Measured by Tandem Mass Spectrometry
C. Canessa1, F. Lippi1, S. Ricci1, F. Romano 1, E. Giocaliere2, G. la Marca2, C. Azzari1
1Pediatric Immunology, Anna Meyer Children's Hospital, Florence, Italy
2Newborn Screening Biochemistry and Pharmacology Laboratory Paediatric Neurology Unit and Laboratories Neuroscience Department Meyer Children's Hospital Firenze Department of Neurosciences Psychology Pharmacology and Child Health, Anna Meyer Children's Hospital, Florence, Italy
Adenosine deaminase (ADA)-severe combined immunodeficiency (SCID) is caused by genetic variants that disrupt the function of ADA and cause accumulation of toxic metabolites adenosine and 2'-deoxyadenosine. Quantification of T-cell receptor excision circles (TRECs) or tandem mass spectrometry analysis on dried blood spots (DBSs) collected at birth can identify newborns with early-onset ADA-SCID and are used in screening programs. Recently we demonstrated that only tandem-MS and not TRECs can be used to identify newborns with delayed- and late-onset ADA deficiencies. In a single blind study, we measured adenosine and 2'-deoxyadenosine levels using tandem mass spectrometry on 20 DBSs from affected patients, carriers and healthy subjects. Levels of metabolites were compared with correspondent genotypes and enzymatic activity. We observed that adenosine and 2'-deoxyadenosine do not increase during life, then we suppose that thymic damage is caused by chronic toxicity. Metabolites levels better than clinical phenotype, correlate with genotype. Our data suggest that analysis of toxic metabolites by tandem mass spectrometry could be used to assign patient to the correct phenotype, early or late-onset ADA-SCID.
ESID-0164 Partial vs. Complete DOCK8 Deficiency
R. Ruiz-García 1, R. Rodríguez-Pena2, L. Martínez-Lostao3, S. Mora-Díaz1, E. Paz-Artal1, J. Ruiz-Contreras4, A. Anel3, E. López-Granados2, L.I. González-Granado4, L.M. Allende1
1Inmunología, Hospital Universitario 12 de Octubre, Madrid, Spain
2Inmunología, Hospital Universitario La Paz, Madrid, Spain
3Bioquímica Biología Molecular y Celular., Universidad de Zaragoza, Zaragoza, Spain
4Pediatria Inmunodeficiencias, Hospital Universitario 12 de Octubre, Madrid, Spain
Introduction: DOCK8-deficiency is defined as profound combined immunodeficiency produced by the classical loss-of-function/expression of DOCK8 that present an exhausted T-cell phenotype with increased susceptibility to viral infections and a compromised immunosurveillance. Abnormalities in the T and NK-cell compartment have been described in complete and also here in partial DOCK8-deficiency.
Objectives: We compare the clinical, immunophenotypical and functional spectrum of two patients with partial and complete DOCK8-deficiency (patient1 and 2, respectively).
Methods: Patients suspected of having DOCK8-deficiency were screened for DOCK8 protein expression by western blot and DNA sequencing. Phenotype, degranulation and cytotoxicity of CD8 and NK-cells were assessed by flow-cytometry.
Results: Patient1 showed defective thymopoiesis, decreased naïve CD4+ T-cells but T-cell proliferation was unaffected. This could be related to the non-exhausted phenotype showed by the patient1, with normal levels of TEMRA CD8+ T-cells, in contrast to classical loss-of-function/expression of DOCK8 in patient2 presenting an exhausted phenotype with high levels of TEMRA CD8+ T-cells impairing partially lymphocyte proliferation. Phenotypically, both patient´s CD8+ T cells showed dysregulated expression of some markers implicated during differentiation from naïve to effector cells: CD27, CD127, CD57, CD95 and perforin. Functionally, patient1’s CD8+ T-cells were less effective at killing R69 target cells contrasting with the intact cytotoxicity in complete DOCK8 patient2. Additionally, NK degranulation and cytotoxicity were lower in both patients than in controls.
Conclusions: Partial DOCK8 deficiencies could lead to milder forms of autosomal recessive hyper IgE syndrome and help to refine DOCK8-dependent features and extend the range of immunodeficiency cases that should be screened for DOCK8 mutations
ESID-0703 Establishment of a Healthy Human Range for the Whole Blood “OX40” Assay for the Detection of Antigen Specific CD4+ T Cells by Flow Cytometry
R. Sadler 1, E. Bateman1, V. Heath1, S. Patel1, P. Schwingshackl1, A. Cullinane1, L. Ayers1, B. Ferry1
1Immunology, Oxford University Hospitals NHS Trust, Oxford, United Kingdom
Clinical investigation of antigen-specific T cells in suspected immunodeficient patients is an important and often challenging aspect of patient diagnostic work up. Methods for detection of microbial exposure to the T-cell compartment exist but are laborious and time consuming. Recently, a whole blood technique involving flow cytometry and detection of CD25 and OX40 (CD134) expression on the surface of activated CD4 T cells was shown to be accurate and concordant when compared with more traditional methods of antigen-specific T-cell detection.
Methods: Whole heparinized blood was collected from 63 healthy donors and set up using the “OX40”
assay to detect antigen-specific CD4 T-cell responses to Varicella Zoster Virus, Epstein-Barr Virus
(EBV), Cytomegalovirus, Candida albicans, and Streptococcus pneumoniae.
Results: The “OX40” assay technique was clinically validated for routine use in an NHS clinical immunology laboratory by analysis of incubation length (40–50 h), sample transport time (up to 24 h at room temperature). Concordance with all serology testing and CFSE proliferation was >94%. Correlation of OX40 responses and interferon-gamma production was significant with EBV and CMV stimulation. In addition, 63 healthy controls (age range 21–78) were tested for responses to generate a healthy control reference range, this was shown to have clinical utility by comparison of EBV specific responses in 2 XIAP defective patients to the generated normal range.
Conclusions: The OX40 assay, as presented in this report, represents an economical, rapid, robust
whole blood technique to detect antigen-specific T cells, which is suitable for clinical immunology diagnostic laboratory use.
ESID-0295 Human Neonatal CD8 T Cells are Upregulated in Tolerance-Associated Genes
M.A. SANTANA 1, A.O. GALINDO-ALBARRAN1, A. HERNANDEZ-MENDOZA1, S. SPICUGLIA2
1FACULTAD DE CIENCIAS, UNIVERSIDAD AUTONOMA DEL ESTADO DE MORELOS, Cuernavaca Morelos, Mexico
2INSERM U1090, Technological Advances for Genomics and Clinics, Marseille, France
Newborn infants are very susceptible to intracellular pathogens and respond with an inadequate memory to several vaccines. Immune response is often tolerant or skewed towards the Th2 phenotype. CD8 T cells are responsible for eliminating infected cells in an antigen-dependent manner. To gain an insight on the particularities of this cell population in neonates, we evaluated the transcriptome of human neonate CD8 T cells as compared to the corresponding adult naïve cells. A very specific gene profile was found in the neonatal cells, including a lower expression of effector functions associated genes. Recently, Schietinger et. al. (Science 2012; 335:723) reported a tolerance-specific gene profile in a mouse model of tolerance. They found 8 gene clusters associated with functional T cells and 2 tolerance-specific clusters.
We compared the data of neonate-enriched transcribed genes from our CD8 T cells trancriptome with the enriched genes of the clusters reported by Schietinger et. al. We found that neonatal genes were enriched in the expression of the two tolerance-cluster associated genes and down regulated in functional genes. Our results may explain the low functionality of neonatal CD8 T cells and point to a tolerance-specific genetic program that is at least partially conserved between species.
ESID-0008T Cell Receptor Ligation Causes Wiskott-Aldrich Syndrome Protein Degradation and F-Actin Assembly Downregulation in T Cells
Y. Sasahara 1 , Y. Watanabe 1 , S. Kure 1 , M.J. Massaad 2 , N. Ramesh 2 , R.S. Geha 2
1 Pediatrics, Tohoku University Graduate School of Medicine, Sendai, Japan
2 Immunology, Children's Hospital Boston, Boston, USA
Background: Wiskott-Aldrich Syndrome Protein (WASP) links T cell receptor (TCR) signaling to the actin polymerization. WASP is normally protected from degradation by its interaction with WASP interacting protein, WIP.
Objective: We investigated the mechanism of WASP protein degradation following TCR ligation and whether its degradation downregulates F-actin assembly caused by TCR ligation.
1Method: Primary T cells, Jurkat T cells and transfected 293T cells were used in experiments. Intracellular F-actin content was measured in splenic T cells from wild type (WT), WASP deficient and c-Casitas B-lineage Lymphoma-b (Cbl-b) deficient mice by flow cytometry. Calpeptin and MG132 were used to inhibit calpain and the proteasome, respectively.
Results: A fraction of WASP in T cells was degraded by calpain and by the ubiquitin-proteasome pathway following TCR ligation both in vitro and in vivo. Cbl-b and c-Cbl E3 ubiquitin ligases associated with WASP in time dependent manner following TCR ligation and caused WASP ubiquitination. Inhibition of calpain and lack of Cbl-b resulted in a significantly more sustained increase in F-actin content following TCR ligation in WT T cells, but not in WASP deficient T cells.
Conclusion: TCR ligation causes WASP to be degraded by calpain and to be ubiquinated by Cbl family E3 ligases, which targets it for degradation by the proteasome. Inhibition of WASP degradation results in sustained F-actin content in T cells. WASP degradation may provide a mechanism for regulating WASP-dependent TCR-driven assembly of F-actin.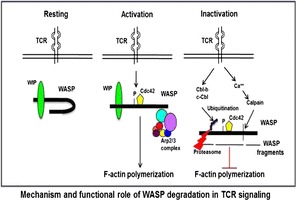
ESID-0033 Skin Sensitization Model Based on Only Animal Data by Qualitative Structure-Toxicity Relationships (QSTR) Approach
K. Sato 1, K. Yuta2, Y. Kusaka1
1Department of Environmental Health, University of Fukui School of Medicine, Fukui, Japan
2In Silico Data, In Silico Data Co Ltd, Narashino, Japan
[Objectives]T cell proliferation is one of the key events of adverse outcome pathway (AOP) for skin sensitization. Contact dermatitis is by far the most common form of occupational skin illness. In silico assessment of skin sensitization is increasingly needed owing to the problems concerning animal welfare, as well as excessive time consumed and cost involved in the development and testing of new chemicals.
[Materials and methods]We previously made skin sensitization model from human and animal data and reported. Its accuracy was 61.2% (sensitivity 60.7%, specificity 62.8%) by external validation. This time we made skin sensitization QSTR model from only animal data (LLNA, 471 chemicals,) by using K-step Yard sampling (KY) methods (U.S. Patent No. 7725413, 2010) and 1 model KY methds (US Patent Application).
[Results]A total of 320 compounds (212 positive sensitizers and 108 negative sensitizers) were used in this study. 288 compounds were used to make a QSTR model and external validation study were performed by 32 compounds. The concordance of QSTR prediction for LLNA data were 71.9% (sensitivity 54.5%, specificity 81%) and better than previous report.
[Dicscussion]The concordance was better than previous time and indicate that the data of human and animal study were gualitatively different from each other.
[Acknowledgment]This work was supported by JSPS KAKENHI Grant Number 25293148.
ESID-0588 Defective T-Cell Lymphoproliferative Response and Muscular Hypotonia: Identification of ORAI1 Mutation
L. Schejbel 1, L.P. Ryder1, K. Müller2, H.V. Marquart1, M. Ifversen2, C. Heilmann2
1Dept. of Clinical Immunology, Rigshospitalet Copenhagen University Hospital, Copenhagen, Denmark
2Dept. of Paediatrics, Rigshospitalet Copenhagen University Hospital, Copenhagen, Denmark
Introduction: We present a patient with normal T cell counts, functional T cell immunodeficiency and muscular hypotonia caused by a previously undescribed ORAI1 mutation.
Case presentation: The patient was admitted to the hospital at the age of 5 weeks on suspicion of Staphylococcal Scalded Skin Syndrome (SSSS). From the age of two months he developed a cough and lymphocytosis, he was tested negative for Pertussis (By PCR). Three months old, he was admitted on suspicion of sepsis and received antibiotic treatment, no microbial agent was identified. At this time point, muscular hypotonia was noted and a biopsy was taken. At the age of 4½ months Pneumocystis Jirovecii was detected in BAL and immunological evaluation was performed.
Results: The patient had normal concentrations of T, B, and NK cells. Stimulation of CD4 T cells with PWM or with CD3/CD28/CD2 beads, caused most of the cells to die, while unstimulated cells survived in culture. The proliferative response was restored by addition of exogenous IL-2.
The combination of abolished lymphoproliferative response in the absence of IL-2 and muscular hypotonia lead to the suspicion of ORAI1 deficiency. This was confirmed by genetic analyses that revealed homozygousity for a 2 bp insertion in the ORAI1 gene causing a frameshift and premature stopcodon (g.19618-19619insAG, NG_007500, p.Ser152ArgfsX37).
Conclusion: In patients with a clinical phenotype of SCID in spite of normal T cell numbers, T cell death upon stimulation can be an important sign of functional T cell deficiency. The patient is now allocated for Hematopoietic stem cell transplantation.
ESID-0607 Association of a Heterozygous STAT4 Mutation with Susceptibility to Pacaroccidioidomycosis
L. Schimke 1, O. Cabral Marques1, T.A. Khan1, R. de Souza Cavalvanti2, E. Borges Junior1, C. Feriotti1, T. Costa1, J. Bustamante3, J.L. Casanova4, R. Poncio Mendes2, V.L. Garcia Calich1, H.D. Ochs5, T. Torgerson5, A. Condino Neto1
1Immunology, Biomedical Science, Sao Paulo, Brazil
2Clinical Infectiology and Immunology, Clinical Hospital University Medical SchoolUNESP, Botucatu, Brazil
3Necker Hospital, Study Center of Primary Immunodeficiencies, Paris, France
4The Rockefeller University, St. Giles Laboratory of Human Genetics of Infectious Diseases, New York, USA
5Department of Pediatrics at University of Washington, Seattle Children's Research Institute, Seattle, USA
We investigated a Brazilian female patient from a non consanguineous family who developed at 20 years of age (currently 30 years old) acute Paracoccidioidomycosis manifesting with generalized lymphadenopathy. Granulomatous lesions were identified in gastrointestinal lymphnodes. The patient was over three years on Sulfamethoxazole treatment to improve the clinical manifestations. Whole-exome sequencing identified the heterozygous mutation p. Glu651Val in the signal transducer and activator of transcription 4 (STAT4) gene. This mutation affects a conserved amino acid located in the Src Holology 2 (SH2) domain near residues critical for binding to the phosphotyrosine residue on the receptor. Bioinformatic computational analysis by Mutstion Taster predicted that this mutation is disease causing. The mutation was confirmed by sanger sequencing and was not found to be a single-nucleotide polymorphism (SNP) in dbSNP. Further functional tests are underinvestigation. Our preliminary data suggests that STAT4 deficiency is a new primary immunodeficiency disease leading to susceptibility to Paracoccidioidomycosis.
ESID-0380 Symptomatic Immunodeficiency in a CD40L Mutation Carrier Due to Skewed X-Inactivation
S.L. Seneviratne 1, M. Dziadzio1, N. Verma1, D. Webster1, S. Loughlin2, S.O. Burns1, R. Chee1, K. Gilmour3
1UCL Centre for Immunodeficiency, Royal Free Hospital, London, United Kingdom
2Molecular Genetics, Great Ormond Street Hospital for Children, London, United Kingdom
3Molecular Immunology, Great Ormond Street Hospital for Children, London, United Kingdom
Background: XHIM is a rare immunodeficiency disorder due to a mutation in the CD40L gene. We report a female patient with recurrent infections and complete IgA and IgG1 subclass deficiency due to a CD40L mutation and skewed X-inactivation.
Case report: A 47 year old lady was found to have complete IgA deficiency following family screening when her brother was diagnosed with CD40L deficiency. Since 2003, she had persistent fatigue, recurrent respiratory tract infections and shingles. In 2011, her infection burden increased and IgG1 subclass deficiency, low serotype-specific pneumococcal antibodies and a poor response to tetanus immunisation were found. She was started on immunoglobulin replacement and is clinically well. She was initially screened for expression of CD40L on activated CD4+ T cells and expression was found to be very abnormal. As her brother has been diagnosed with XHIM, genetic analysis was performed which showed her to be a carrier of CD40L gene mutation. X-inactivation assays were undertaken in her whole blood, T and B cells. Analysis of the androgen receptor locus in all cell types showed extreme X inactivation. Further analysis showed that the active X was the same one her brother inherited and carried the CD40L mutation.
Conclusion: We report an XHIM carrier with clinically significant immunodeficiency due to extreme non-random inactivation of the normal X chromosomes. Female relatives of XHIM patients with recurrent infections should be investigated for humoral or combined immune defects and have their X-inactivation assessed to determine if they are manifesting carriers.
ESID-0528 Successful Hematopoietic Stem Cell Transplantation (HSCT) in Patient with Wiskott-Aldrich Syndrome (WAS), Complicated by Castleman Disease
N. Kuzmenko1, A. Kozlova1, D. Abramov2, D. Balashov3, N. Myakova4, Y. Skvortcova3, S. Zimin5, A. Maschan6, A. Shcherbina 1
1immunology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
2pathology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
3transplantology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
4oncology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
5immunology, Children`s Clinical Hospital #9, Moscow, Russia
6hematology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
Wiskott-Aldrich syndrome (WAS) is a combined immunodeficiency, characterized by thrombocytopenia, eczema, infections and propensity to develop autoimmunity and tumors. We describe a case of WAS, complicated Castleman disease (CD) - rare lymphoproliferative disorder with massive growth of lymphoid tissue, follicular hyperplasia, vascular proliferation and plasmocytosis. Patients with multicentric CD (MCD) have multiple lesions, affecting lymph nodes, liver, spleen, with prolonged fever and hematologic abnormalities.
Our patient has been diagnosed with WAS at the age of 2 based on typical clinical symptoms of severe WAS and confirmed by identifying c.982insCC mutation in WASP gene. The family declined HSCT at the time. At the age of 4 years progressive cervical lymphadenopathy was observed, followed by hepatomegaly, frequent episodes of fever and neuropathy. Lymph node biopsy demonstrated MCD, hyaline vascular type. Later this condition was complicated by POEMS syndrome.
The patient was treated experimentally with tocilizumab (2 infusions) without effect: hydrothorax, ascites, vasculitis, anemia, thrombocytopenia, hypocoagulation progressed. The patient was then started on combination of cyclophosphamide, vincristine, prednisone and rituximab with partial effect. 6 months later HSCT from fully matched unrelated donor was performed, with good engraftment, complete resolution of symptoms of both diseases and minor complications post-transplant (current follow-up is 18 months).
To our knowledge, it is the first report of multicentric Castleman disease in a patient with WAS. HSCT might be considered in severe cases of multicentric Castleman disease in other patients.
ESID-0531 Thrombopoietin (TPO) Receptor Agonist Romiplostim Is Effective in Treatment of Thrombocytopenia in Wiskott-Aldrich Syndrome (WAS)
D.E. Pershin1, E.S. Mamzerova1, E.A. Viktorova1, E.Y. Osipova2, S.A. Kashpor2, S.A. Plyasynova2, O.I. Illarionova2, T.A. Varlamova3, V.O. Bobrynina3, A.A. Maschan4, A. Shcherbina 1
1Immunology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
2laboratory immunology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
3molecular biology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
4Hematology, Federal Research Center of Pediatric Hematology Oncology Immunology, Moscow, Russia
Wiskott -Aldrich syndrome caused by mutations of WASP gene, and characterized by thrombocytopenia and combined immunodeficiency. Some patients, usually with missense mutations in exon 1 and 2, have a milder disease, mostly manifested by thrombocytopenia (termed by some X-linked thrombocytopenia – XLT).
Most WAS patients require stem cell transplantation (HSCT), yet XLT patients are often followed conservatively. Bleeding is the major cause of morbidity\ mortality in XLT patients, as well as in classic WAS patients, awaiting HSCT.
Romiplostim, TPO receptor agonist, has demonstrated its efficacy in patients with various thrombocytopenias, including immune thrombocytopenia(ITP).
We demonstrate that TPO level in 17 WAS patients (mean 119,2 pg/ml), as well as in 5 ITP patients (mean 110 pg/ml), was comparable to that of normal controls (120.5 pg/ml), which, considering expected higher levels of TPO in thrombocytopenia, can be classified as relative TPO deficiency.
We studied romiplostim efficacy at a dose of 9 ug/kg of body weight weekly in 17 WAS patients. In all patients significant increase in platelet numbers was noted (from mean 19.5\ul before to mean 86.3 \ul by the second week of treatment). Upon prolonged treatment the effect was less dramatic (mean 52.2\ul after 12 weeks of treatment), yet all patients showed clinical improvement and drastic reduction of hemorrhagic phenomena.
In conclusion, TPO receptor agonists can be beneficial in reducing bleeding complications in WAS patients preparing for HSCT and for XLT patients during the exacerbations of bleeding. Long-term efficacy and side effects of such therapy are yet to be established.
ESID-0541 DNA Ligase 1 Deficiency - A Novel Cause of Severe Combined Immunodeficiency with Multicystic Dysplastic Kidneys and Refractory Anaemia
J. Silva 1, A. Jones2, W. Qasim2, H. Gaspar2, R. Chiesa1, O. Nikolajeva1, S. Bibi3, P. Amrolia1, P. Veys1, A. Worth2
1Bone marrow transplant, Great Ormond Street Hospital, London, United Kingdom
2Immunology, Great Ormond Street Hospital, London, United Kingdom
3Genetics, Great Ormond Street Hospital, London, United Kingdom
DNA ligase1 is critical for DNA single strand break repair. One patient with mutations in the DNA ligase1 gene (LIG1) has been previously described with combined immunodeficiency, in vitro hypersensitivity to DNA-damaging agents and impaired Okazaki fragment joining. Here we report two brothers with homozygous Arg771Trp mutations in the catalytic domain of LIG1, described in the previously reported patient. Additionally they have an undescribed homozygous Pro529Leu LIG1 variant. Patient 1 presented at 2 months with eczema, recurrent severe respiratory infections, and failure to thrive. Investigations showed anaemia, lymphopenia, multicystic dysplastic right kidney and multiple enteropathic viral infections. Immunophenotyping revealed low T and B cells, with normal NK cell numbers (CD3+ 0.73x109/L, CD3+CD4+ 0.41x109/L, CD3+CD8+ of 0.05x109/L, CD19+ 0.12x109/L, CD16+CD56+ 0.61x109/L), raised γδ T cells (18%) and impaired PHA proliferative responses. The proportion of CD4+CD45RA+CD27+ T cells (20%) and TRECs (1015 /106 T cells) were low. Cultured fibroblasts showed normal radiosensitvity. He was successfully stem cell transplanted using a match family donor and minimal intensity conditioning (anti-CD45 monoclonal antibody based). Patient 2 was diagnosed at birth following prospective screening. He demonstrated a more severe SCID immunophenotype (CD3+ 0.55x109/L, CD3+CD4+ 0.09x109/L, CD+3CD8+ 0.02x109/L, CD19+ 0.03x109/L) with highly raised γδ T cells (93%). He also has a multicystic dysplastic kidney and severe anaemia requiring blood transfusion support. He is well on supportive treatment awaiting stem cell transplant. DNA ligase 1 deficiency is a novel cause of NK+ SCID, and should be particularly considered in the presence of multicystic dysplastic kidneys and refractory anaemia.
ESID-0628 SJ/Beta TREC Ratio in Adults Submitted to Total or Partial Thymectomy in Early Infancy
S.L. Silva 1, A.S. Albuquerque1, B. Charmeteau2, R. Anjos3, M. Abecasis3, R.M.M. Victorino1, R. Cheynier2, A.E. Sousa1
1Centro de Imunodeficiências Primárias, Instituto de Medicina Molecular. Faculdade de Medicina / Universidade de Lisboa, Lisboa, Portugal
2Département d'Immunologie-Hematologie, Institut Cochin INSERM U1016-CNRS UMR8104-Université Paris Descartes, Paris, France
3Departamento de Cirurgia Cardíaca, Hospital de Santa Cruz, Carnaxide, Portugal
Long-term immunological impact of total and partial thymectomy performed during corrective cardiac surgery in early infancy remains debatable. We investigated possible factors contributing to the degree of maintenance of the naive T-cell compartment during adulthood.
β and single-joint (sj) T-cell receptor (TCR) excision circles (TREC), sequential by-products of TCR rearrangement during T-cell development in the thymus, were quantified in circulating T-cells of total and partial thymectomized, as well as age-matched healthy adults.
The sj/βTREC ratio is thought to reflect intra-thymic cell division which correlates with thymic output, and to be not subsequently altered by peripheral T-cell division.
Therefore, in individuals submitted to total thymectomy (based on surgical reports and severely reduced levels of sjTREC/μl) this ratio would represent an estimation of the thymic activity at the time of thymus removal. We found no correlation between the sj/βTREC ratio and the size of both naive CD4 and CD8 T-cell compartments, suggesting that peripheral mechanisms may compensate the differences existing at age of thymectomy. Notably, the frequency of cycling cells was significantly increased, although naive T-cell depletion was still observed.
Conversely, in partial thymectomized individuals the sj/βTREC ratio would provide an estimation of current thymic activity. We found that this ratio was comparable to age-matched healthy individuals supporting thymus recovery after partial thymectomy. This is in agreement with the lack of significant reduction in both numbers and frequency of naive CD4 T-cells in these individuals.
Our data provide evidence to implement the adoption of surgical strategies that allow partial thymus preservation.
ESID-0650 Human Naïve Regulatory T-Cells are Maintained by IL-7
S.L. Silva 1, A.S. Albuquerque1, A. Serra-Caetano1, R.B. Russell2, A.R. Pires2, S.F. Fernandes2, J. Ferreira2, R.M.M. Victorino1, I. Caramalho2, J.T. Barata2, A.E. Sousa1
1Centro de Imunodeficiências Primárias, Instituto de Medicina Molecular. Faculdade de Medicina. Universidade de Lisboa, Lisboa, Portugal
2Instituto de Medicina Molecular, Faculdade de Medicina. Universidade de Lisboa, Lisboa, Portugal
Naïve FoxP3-expressing regulatory T-cells (Treg) play an essential role in preventing autoimmunity via continuous replenishment of the activated Treg pool with thymus-committed Tregs. Despite their clinical relevance, the mechanisms underlying human naïve-Treg maintenance throughout life, in parallel with the age-associated thymic involution, remain unclear.
We found that circulating naïve-Tregs featured higher levels of the pro-survival molecule Bcl-2 and increased proportions of cycling-cells as compared to conventional naïve CD4 T-cells. Their significant turnover in human tonsils was supported by ex-vivo 5-ethynyl-2'-deoxy-uridine (EdU) incorporation.
Naïve-Tregs featured the highest ex-vivo levels of STAT5 phosphorylation (pSTAT5) amongst the studied CD4 T-cell subsets, indicative of γc-cytokine stimulation, and recovered IL-7Rα expression upon IL-7 deprivation, consistent with ongoing in-vivo responsiveness to IL-7.
Finally, in-vitro exposure of naïve-Tregs to IL-7 led to up-regulation of pSTAT5 and Bcl-2, and induced the proliferation of both naïve-Tregs and FoxP3+ CD4 single-positive thymocytes, in a phosphatidylinositol 3-kinase (PI3K)-dependent manner, whilst preserving their naïve phenotype.
Overall, our data revealed a role for IL-7 in human naïve-Treg homeostasis, highlighting a need for reappraisal of strategies for immune reconstitution targeting IL-7.
Silva S.L. and Albuquerque A.S. are co-first authors
ESID-0759 Correlation Between Thymic Function-Related Markers Within the Thymus and Peripheral Blood
A. Lev1, A.J. Simon1, D. Machnes-Maayan1, R. Somech 1
1Pediatrics Immunology, Sheba Medical Center, Ramat Gan, Israel
The thymus is a highly specialized organ for T-cell receptor rearrangements and selection mechanisms that ensures the formation of functional and self-tolerant cells. Little is known of how peripheral blood assessment of thymic function is indeed a reflective picture of the inside of thymus activity. The aim of this study was to compare thymic function-related markers within the thymus and peripheral blood. We have concomitantly studied thymic as well as peripheral blood samples from immunocompetent infants who underwent cardiac surgery that involved thymectomy. The studied thymic markers included T cell receptor (TCR) excision circles (TRECs), 4 different TCR gene rearrangements, TCR repertoire, regulatory T cells (defined as CD3+CD4+CD25+FOXP3+ cells) and mRNA expression of AIRE and FOXP3. Twenty patients were enrolled in this study. The mean age at the time of the surgery was 3 months and 5 days (±3 months and 18 days). A significant correlation was found between thymic and peripheral blood levels of TREC, all 4 TCR gene rearrangements and the amount of regulatory T cells (Tregs). The level of these parameters was significantly higher than that detected at the peripheral blood. The TCR repertoire distribution in thymic samples and peripheral blood was similar. FOXP3 but not AIRE mRNA levels in the thymus and peripheral blood correlated well. A correlation between peripheral blood and intrathymic activity parameters during infancy exists. Assessment of these parameters at the peripheral blood can be accurately used to estimate different intrathymic capacities in order to assess T cell function in health and disease.
ESID-0105 Analysis of the Relationship Between Peripheral Blood Follicular Helper T Cells and Switched Memory B Cells in Common Variable Immunodeficiency
R. Steven 1, P. Gurugama1, S. Hanson1, A. Grammatikos1, K. Bramhall2, M. Burgess2, A. Bennett2, T. El-shanawany2, P. Williams2, S. Jolles2, E. Davies1, M.A.A. Ibrahim1
1Department of Immunological Medicine, King's College London King's Health Partners Kings College Hospital NHS Foundation Trust School of Medicine Division of Asthma Allergy and Lung Biology Denmark Hill London SE5 9RS UK, London, United Kingdom
2Department of Medical Biochemistry and Immunology, Immunodeficiency Centre for Wales University Hospital of Wales Heath Park Cardiff CF14 4XW UK, Cardiff, United Kingdom
The interaction between T and B cells is essential for long lived humoral immune responses. T cells provide help with the process of somatic hypermutation and class switch recombination of immunoglobulins, resulting in the generation of high affinity antibodies. Analysis of B cell subsets has proven useful in the prognostic classification of common variable immunodeficiency (CVID). Although altered B cell subset populations are well recognised as a feature of CVID (decreased switched memory B cells (SWM) and increased transitional B cells), less is known of the T cell biology in this condition. Recent studies have demonstrated the importance of T follicular helper cells (Tfh) in the generation of SWM B cells and high affinity antibodies. Therefore, this study aimed to assess the relationship of circulating Tfh cells in CVID and SWM B. A flow cytometric method for the quantification of Tfh in peripheral blood was developed and measurements from CVID patients were compared with those from healthy controls and secondary immunodeficiency patients due to infection with the human immunodeficiency virus (HIV). Results revealed that circulating Tfh cells were significantly reduced in CVID compared to healthy controls and HIV patients. Furthermore, the relationship between Tfh and SWM in CVID differed when compared with healthy controls and HIV patients. Our results suggest that both the Tfh and SWM compartments may be dysregulated in CVID. Further investigation of this area of immunological interaction may advance the understanding of pathogenesis of this disease.
ESID-0734 DOCK8 Regulates Lymphocyte Shape Integrity for Skin Antiviral Immunity
H. Su 1, Q. Zhang1, C. Dove1, J. Hor2, H. Murdock1, D. Strauss-Albee1, J. Garcia1, J. Mandl3, R. Grodick1, H. Jing1, D. Chandler-Brown1, E. Kim1, T. Lenardo1, G. Crawford4, H. Matthews5, A. Freeman6, R. Cornall4, R. Germain3, S. Mueller2
1Laboratory of Host Defenses, NIAID NIH, Bethesda, USA
2Microbiology and Immunology, University of Melbourne, Parkville Victoria, Australia
3Laboratory of Systems Biology, NIAID NIH, Bethesda, USA
4Nuffield Department of Medicine, Oxford University, Oxford, United Kingdom
5Laboratory of Immunology, NIAID NIH, Bethesda, USA
6Laboratory of Clinical Infectious Diseases, NIAID NIH, Bethesda, USA
Introduction: DOCK8 mutations result in an inherited combined immunodeficiency characterized by increased susceptibility to skin infections. Patients often suffer from disseminated and persistent viral skin infections caused by herpes simplex virus, human papillomavirus, molluscum contagiosum, and varicella-zoster virus.
Objective: To determine whether defective lymphocyte migration could contribute to the increased susceptibility of DOCK8-deficient patients to viral skin infections.
Methods: Lymphocytes were genetically deficient in DOCK8, transfected with siRNA or fluorescently-tagged constructs, or treated with pharmacologic inhibitors. Cell migration was evaluated in vitro within foreskin tissue or collagen gel matrices, and in vivo within skin using a mouse model of herpes simplex virus infection. Live cell video-microscopy, confocal imaging, intravital two-photon microscopy, flow cytometry, biochemistry, and quantitation of virus replication were performed.
Results: When DOCK8-deficient T and NK cells migrated through confined spaces, they developed cell shape and organelle abnormalities that contributed to a distinct form of catastrophic cell death we term cytothripsis. Such defects were associated with spatial and temporal dyscoordination of microtubule function during lymphocyte migration in collagen-dense tissues, and arose from DOCK8 regulation of microtubule dynamics through a CDC42-dependent pathway. Cytothripsis of DOCK8-deficient cells led to decreased generation of long-lived skin-resident memory CD8 T cells, which in turn impaired control of herpesvirus skin infections.
Conclusions: DOCK8-regulated shape integrity of lymphocytes prevents cytothripsis and promotes antiviral immunity in the skin. These findings are likely to have general relevance for skin immunity to other pathogens and tumor surveillance in the skin.
ESID-0335 Immunophenotyping of Severe Combined Immunodeficiency in Algeria
R. DJIDJIK1, A. TAHIAT 1, R. BELLOUNI1, S. CHERIK1, N. CHERIF2, M. CHAOU3, M.E. KHIARI3, A. BENSENOUCI2, M. GHAFFOR1
1Medical Biology Laboratory, CHU béni-Messous, algiers, Algeria
2Department of Pediatrics "B", CHU béni-Messous, algiers, Algeria
3Department of Pediatrics "A", CHU béni-Messous, algiers, Algeria
Objective. Severe combined immunodeficiency (SCID) is a group of disorders that leads to early childhood death as a result of severe infections. High consanguineous rate in Algeria, allow us to suggest a high prevalence of autosomal recessive SCIDs.
Patients and Methods. We analysed a series of 9 SCID, recruited on a 5 years period [2009—2014]. Analysis included a blood count, Immunoglobulins A, G and M dosage by nephelometry (BNProSpec/Siemens, Germany) and lymphocyte subpopulations count by flow cytometry (FACSCanto/BD Biosciences, U.S.A.).
Results. The frequency of SCID among primary immunodeficiencies (n=140) diagnosed in our laboratory is 6.4%. On the 9 SCID, 6 (66.7%) were T-B-NK+ SCID, 2 were T-B+NK- SCID and one was T-B-NK- SCID. Parental consanguinity was observed in 7 cases (77.8%) and most of the patients were females (7/9).
Conclusion. Our results show a predominance of autosomal recessive SCID such as T-B-NK+ SCID which probably due to RAG1/2 deficiency. Molecular analysis is able to confirm this hypothesis or possibly to identify new molecular abnormalities.
ESID-0642 Invasive Adenocarcinoma of Large Intestine and Gallbladder in a Patient with X-Linked Hyper-IgM Syndrome
S. Tantou 1, G. Stylianidis2, T. Argyrakos3, M. Tzanoudaki4, T. Makatsoris5, M. Liatsis4, M. Kanariou4
1Specialized Center & Referral Center for Primary Immunodeficiencies – Paediatric Immunology, "Aghia Sophia" Children's Hospital, Athens, Greece
22nd Surgical Department, “Evaggelismos” Hospital, Athens, Greece
3Department of Pathology, “Evaggelismos” Hospital, Athens, Greece
4Specialized Center & Referral Center for Primary Immunodeficiencies – Paediatric Immunology, "Aghia Sophia" Children's Hospital, Athens, Greece
5Division of Oncology Department of Internal Medicine, University of Patra, Patra, Greece
Introduction: X-linked Hyper IgM Syndrome is a rare Primary Immunodeficiency with increased susceptibility to infections, autoimmunity, but to lymphoid hyperplasia / carcinomas, as well. One of the prevalent causes of death is the adenocarcinomas mainly in liver, bile duct and pancreas.
Case Report: We present a patient who was diagnosed with X-linked Hyper-IgM at the age of 2. Since then, he was under immunoglobulin replacement therapy with great compliance. At early infancy he presented with episodes of cytopenias and Cryptococcus meningitis with good response to relative management and remained asymptomatic until the age of 29 years, when he showed rectal bleeding. Digital Rectal examination showed a polyp. Histological examination revealed rectal invasive adenocarcinoma arising from an underlying ulcerated tubular adenoma with severe epithelial dysplasia. Staging radiological examination revealed also a gallbladder polyp. The patient underwent neoadjuvant chemoradiation for the rectal cancer, which was followed by ultra low anterior resection and cholecystectomy. From the histological examination gallbladder adenocarcinoma and sclerosing cholangitis were diagnosed. Three months later excision of part V of the liver and extended lymphadenectomy of the hepatodeodenum ligament was performed. The procedure was followed by adjuvant chemotherapy with satisfied response. Six months later, the patient deceased due to cardiac arrest.
Conclusion: In our knowledge, this is the first reported patient with Hyper IgM syndrome, who had been diagnosed and treated for adenocarcinoma of rectum. The mechanisms for this malignancy are unclear.
ESID-0515 Epidemiological, Clinical and Biological Features in Major Histocompatibility Complex Class II Deficiency
F. Mellouli1, S. THRAYA 1, M. Ben Khaled1, M. Ouederni1, A. Haoua1, R. Hassouna1, M. Barbouche2, M. Bejaoui3
1Department of Pediatric Immuno-Hematology, National Bone Marrow Transplant Center, Tunis, Tunisia
2Laboratory of Immunology, Pasteur Institute of Tunis, Tunis, Tunisia
3Department of Pediatric Immuno-Hematology, National Bone Marrow transplant Center, Tunis, Tunisia
I-Introduction: Major histocompatibility complex class II(MHC class II) deficiency is a rare autosomal recessive primary combined immunodeficiency. The prevalence of this deficiency is the highest in Mediterranean areas, especially North Africa. We describe in this work the epidemiological, clinical and biological characteristics of this rare condition in a Tunisian Pediatric Immuno-Hematology unit.
II-Methods:Retrospective study over 16 years (March 1998- March 2013), including patients with a MHC class II deficiency, followed within the National Center of Bone-Marrow Transplant of Tunis. The expression of MHC class II antigens was studied on the residual peripheral mononuclear cells and PHA stimulated cells.
III-Results: Forty-two patients were included. Their average age was 20 months(2 – 72 months). The mean age at onset of disease was eight months (2 days-48 months). Clinical signs were dominated by infectious(n=40), growth retardation(n=32), allergy(n=6), lymphoproliferation(n=8), BCGitis(n=3). Infectious sites were: bronco-pulmonary(n=33), gastrointestinal (n=33), oral candidiasis(n=25) and cutaneous infections(n=11). Hypogammaglobulinemia was found in 30 cases and CD4 lymphopenia in all cases. MHC class II molecules expression was absent in all cases. Nineteen patients died during follow-up. Three of the 10 patients who underwent hematopoietic stem cell transplantation (HSCT) were cured, with the recovery of almost normal immune functions. All patients were transplanted beyond 2 years-old had died (n=6).
IV-Conclusion:MHC II antigens deficiency is a relatively common primary immunodeficiency in Tunisia. The diagnosis should be suspected in recurrent infections, hypogammaglobulinemia and CD4 lymphopenia. Prognosis is poor. More precociously HSCT is performed, better are the results.
ESID-0521 A Hypomorphic Coronin-1A Mutation that Abolishes Oligomerization and Cytoskeletal Association Is Associated with Impaired CD4+ T Cell Survival
C. Yee 1, O. Sanal2, A.N. Akarsu3, C. Ayetkin4, D.C. Ayvaz2, R.S. Geha1, J.S. Chou1
1Division of Immunology, Boston Children's Hospital, Boston, USA
2Immunology Division, Hacettepe University and Ihsan Dogramaci Children's Hospital, Ankara, Turkey
3Department of Medical Genetics, Hacettepe University and Ihsan Dogramaci Children's Hospital, Ankara, Turkey
4Department of Pediatric Immunology, Sami Ulus Children's Hospital, Ankara, Turkey
Coronin-1A (CORO1A) is an actin regulator important for T cell homeostasis. Coronin-1A deficiency has been observed in patients with T-B+NK+ SCID and impaired NK cell function. We present two siblings with disseminated varicella, cutaneous warts, and CD4+ T cell lymphopenia. T cell function in vitro was relatively preserved, but the percentage of annexin V+ apoptotic CD4+ peripheral T cells was increased at baseline and upon stimulation with PHA or anti-CD3 compared to controls, indicating increased susceptibility to cell death. Whole genome sequencing of the patients and their mother identified a single nucleotide insertion in CORO1A (1191_1192insC). Sanger sequencing of genomic DNA confirmed that both patients were homozygous for the mutation, while the mother was heterozygous. Sequencing of cDNA by 3' RACE confirmed that the mutation would result in the replacement of the last 61 C-terminal a.a. of the protein by a novel 91 a.a. sequence. The mutant protein was expressed in the patients' cells, but failed to self-associate or to co-immunoprecipitate with wild-type Coronin-1A in transiently transfected 293T cells. The mutant protein showed decreased association with cytoskeletal components in insoluble cell fractions. Lentiviral reconstitution of CORO1A -/- T cells with mutant CORO1A cDNA failed to rescue CD4+ T cells from increased cell death. We describe a novel mutation in CORO1A in two siblings with immunodeficiency which preserves protein expression but abolishes oligomerization and impairs cytoskeletal association. These functions of Coronin-1A are linked with decreased CD4+ T cell survival and impaired resistance to viral pathogens.
ESID-0301 TCR Diversity is Selectively Skewed in T-Cell Populations of Wiskott-Aldrich Syndrome Patients
J. Wu 1, D. Liu1, W. Tu2, W. Song3, X. Zhao1
1Ministry of Education Key Laboratory of Child Development and Disorders, Children's Hospital of Chongqing Medical University, Chongqing, China
2Department of Paediatrics & Adolescent Medicine, LKS Faculty of Medicine the University of Hong Kong, Hongkong, China
3Department of Cell Biology & Molecular Genetics, University of Maryland, Maryland, USA
Background -— Wiskott-Aldrich syndrome (WAS) is a severe disorder characterized by thrombocytopenia, eczema, immunodeficiency, and increased risk of autoimmune disease and lymphoid malignancies. The immunodeficiency caused by lack of WAS protein (WASP) expression has been mainly attributed to defective T-cell functions. Whether WAS mutations differentially influence the T cell receptor (TCR) diversity of different T-cell subsets is unknown.
Objective- — We aimed to identify the degree and the pattern of skewing in the variable region of the β-chain (Vβ) TCR repertoire in different T-cell subsets from patients with WAS.
Methods —The TCR repertoire diversity in total peripheral T cells, sorted CD4+ and CD8+ T cells, CD45RA+(CD45RA+CD45RO-) and CD45RO+(CD45RA-CD45RO+) CD4+ and CD8+ T cells from patients with WAS and age-matched healthy controls were analyzed and compared, using spectratyping, of complementarity-determining region 3 (CDR3); and the CDR3 region of TCRβ transcripts in CD45RA+CD4+ and CD8+ T cells, CD45RO+CD4+ T cells, CD8+ terminally differentiated effctor memory T cells (Temra) and naïve CD8+T (CD8+CD45RO-CCR7+) cells from patients and controls were analyzed and compared by high-throughput sequencing (HTS).
Results — The TCR repertoire diversity in CD45RO+CD4+ T cells and CD8+ Temra of WAS patients was significantly skewed, compared to that in age-matched controls.
Conclusion — Our results indicate that WAS gene mutations selectively influence the repertoire development or expansion of CD45RO+ (memory) CD4+ T cells.
ESID-0193 Omenn Syndrome Associated with a Functional Reversion Due to a Somatic Second-Site Mutation in CARD11 Deficiency
S. Fuchs 1, A. Rensing-Ehl1, U. Pannicke2, M. Lorenz2, Y. Jeelall3, J. Rohr1, C. Speckmann1, T. Vraetz4, S. Farmand5, A. Schmitt-Graeff6, P. Fisch6, B. Strahm4, P. Henneke1, A. Enders3, K. Horikawa3, C. Goodnow3, K. Schwarz2, S. Ehl1
1University Medical Center and University of Freiburg, Centre for Chronic Immunodeficiency, Freiburg, Germany
2University of Ulm, Institute for Transfusion Medicine, Ulm, Germany
3John Curtin School of Medical Research The Australian National University, Department of Immunology, Canberra, Australia
4University Medical Center and University of Freiburg, Center of Pediatric and Adolescent Medicine, Freiburg, Germany
5Karolinska Institutet, Department of Microbiology Tumor and Cell Biology, Stockholm, Sweden
6University Medical Center Freiburg, Institute of Pathology, Freiburg, Germany
Background: Omenn syndrome (OS) is a severe immunodeficiency associated with erythroderma, lymphoproliferation, eosinophilia and elevated IgE caused by paradoxically hyperactive oligoclonal T-cells. Two siblings from a consanguineous family presented with severe CMV and pneumocystis jirovecii infections and multiple episodes of sepsis. One of them developed OS, but no mutations were found in the genes so far associated with OS.
Objective: To define the molecular defect and to determine factors leading to OS in only one of the two siblings.
Methods: T-cell proliferation, activation, cytokine expression and NFκB signalling were analyzed by flow cytometry. Genetic analysis was performed by targeted Sanger sequencing in sorted cell populations. Patient T-cells and Jurkat cells were reconstituted by retroviral transduction. Functional properties of mutant alleles in B-cells were investigated in the IgHEL mouse model.
Results: Both patients carried a homozygous germline loss of function CARD11 mutation (C150X). IL-2 production and NFκB signaling were impaired and could be restored by retroviral reconstitution with wild-type CARD11. In the oligoclonally expanded T cells of the patient with OS, we detected a second site mutation in the affected codon leading to a missense mutation (C150L) and restoration of protein expression. Expression of C150L in CARD11 deficient Jurkat cells partly reconstituted NFκB signaling. No gain-of-function effect was demonstrated after introduction of the mutation into murine IgHel B-cells. We describe a novel genetic cause of OS, associated with a somatic second-site mutation in CARD11, presumably providing a proliferative advantage to revertant T cells in a CARD11 deficient background.
Topic: Late Breaking Abstracts
ESID-0811 Altered Usage of Proximal and Distal Immunoglobulin Variable Genes in Patients with the Cohesinopathy Cornelia de Lange Syndrome
A. Björkman1, D. Dunn Walters2, D. Kipling3, Q. Pan-Hammarström 1
1Department of Laboratory Medicine, Karolinska Institutet at Karolinska University Hospital Huddinge, SE-14186, Sweden
2Department of Immunobiology, King's College London School of Medicine London, UK
3Institute of Cancer and Genetics, School of Medicine, Cardiff University, Cardiff, UK
Cornelia de Lange syndrome (CdLS) is a genetic disease that affects several systems and organs during development. A major cause of morbidity and mortality in the patients are infections, however, whether these are associated with immunodeficiency have not been extensively studied. CdLS is caused by mutations in genes encoding members of the cohesin complex/pathway that is involved in sister chromatid cohesion, gene regulation and homologous recombination. We have previously shown that the cohesin loader NIPBL is involved in the non-homologous end-joining process during immunoglobulin (Ig) class switch recombination, a deletion/recombination process that result in a change of Ig isotype. Here we have studied whether NIPBL and the associated cohesin pathway is also functional in the two Ig diversification processes V(D)J recombination and somatic hypermutation (SHM), which result in Ig variable region assembly and antibody affinity maturation, respectively.
The IGHV regions and corresponding V(D)J recombination junctions were amplified by PCR from NIPBL- and SMC1-deficient CdLS patient cells and analyzed by the 454 or Illumina Hiseq2000 high throughput sequencing platform. Whereas the SHM pattern was largely normal, the V gene usage for V(D)J recombination was skewed in the patients. Notably, V genes located in proximity to the D and J genes were preferentially used at the expense of V genes located in the 5’ distal end of the IGH locus. Similar results were also obtained in analyzing the V(D)J recombination pattern at the TCRbeta locus. This implies an involvement of cohesin in processes affecting long range interactions during V(D)J recombination in human B- and T-cells.
ESID-0817 Inherited BCL10 Deficiency in a Child with Innate and Adaptive Defects
R. Perez deDiego 1, R. Martinez-Barricarte2, J. Torres3, S. García-Gómez1, S. Mazariegos4, Y. Itan2, R. Álvarez3, E. López-Granados3, R. Rodríguez3, A. Ferreira3, J.L. Unzueta-Roch5, E. López-Collazo6, S. Sánchez-Ramón7, J.R. Regueiro4, J.L. Casanova2
1Laboratory of Immunogenetics of Diseases, UNIVERSITY HOSPITAL LA PAZ, Madrid, Spain
2St. Giles Laboratory of Human Genetics of Infectious Diseases, The Rockefeller University, New York, USA
3Immunology Unit, UNIVERSITY HOSPITAL LA PAZ, Madrid, Spain
4Immunology Department, Faculty of Medicine Complutense University, Madrid, Spain
5Intensive Care Unit, Niño Jesus Hospital, Madrid, Spain
6Innate Immunity Group, UNIVERSITY HOSPITAL LA PAZ, Madrid, Spain
7Clinical Immunology Department, San Carlos Clinical Hospital, Madrid, Spain
BCL10, MALT1, and CARD family adaptors form heterotrimers, which are involved in NF-κB activation, and both the innate and adaptive arms of mouse immunity. Inherited defects of human MALT1, CARD9, and CARD11 however display different immunological and clinical phenotypes. Here, we report autosomal recessive, complete BCL10 deficiency in a child with a broad immunodeficiency including innate and adaptive defects. The patient died at 3 years of age and an older sister had died in similar conditions at 6 months years. The patient is homozygous for a loss-of-expression, loss-of-function BCL10 mutation. The impact of BCL10 depends on the signalling pathways, and, for some pathways, on the cell types. Despite similarities with Bcl10-deficient mice, including a deficient adaptive immune response, human BCL10 deficiency displays specific features, such as normal response in myeloid cells and a newly defined impact in fibroblasts. Inherited BCL10 deficiency should be considered in patients with a broad immunodeficiency involving hematopoietic and non-hematopoietic immunity.
ESID-0819 IGM and IGA Anti-Pneumococcal Capsular Polysaccharide Antibodies Assessed by a New Elisa Assay: A Potential Biomarker of Clinical Outcome in CVID
J. Carbone 1, M. Jaramillo1, L. Calahorra1, A. Gallego1, J. Navarro1, E. Sarmiento1
1Immunology, Gregorio Marañon Hospital, Madrid, Spain
We aimed to evaluate anti-polysaccharide responses as a potential biomarker in CVID. Serum samples of 35 primary antibody deficiency (PAD) patients (18 CVID, 5 agammaglobulinemia, 2 hyper-IgM, 10 other PAD) were tested. We used a new anti-PS23 IgM and IgA ELISA assay, which evaluates response to 23 pneumococcal polysaccharides. Clinical follow-up to correlate with clinical outcomes was performed up to July 2014 for CVID patients. Serum samples were obtained before development of clinical complications.
Anti-PS23 IgM and IgA antibody concentrations were similar in CVID and agammaglobulinemic patients. Both groups disclosed significantly lower concentration of anti-PS23 IgM and IgA antibody concentrations than other PAD. Patients with hyper-IgM disclosed similar concentrations of IgA anti-PS23 but higher of IgM anti-PS23 as compared to other groups. CVID patients with splenomegaly (n=8) or bronchiectasis (n=9) disclosed lower concentrations of IgA and IgM anti-PS23 (0.18±0.031 vs 0.31±0.12 IU/mL, p=0.06, 0.19±0.02 vs 0.37±0.09 IU/mL, p=0.09 and 0.18±0.02 vs 0.38±0.13 IU/mL, p=0.050, 0.2±0.02 vs 0.39±0.1 IU/mL, p=0.04, respectively). Antibody titers were not correlated to development of recurrent infections, intestinal compromise or death during follow-up in CVID patients. Anti-PS23 antibody titers did not correlate with the percentage of circulating CD19+CD27+IgM-IgD- B-cells in CVID patients.
IgA and IgM anti-PS23 assessed in CVID patients on IgG replacement therapy might be useful as a surrogate biomarker of clinical outcome. Low IgM antibody response to pneumococcal antigens has been demonstrated in older adults with a low capacity to opsonize bacteria. Low IgA anti-polysaccharide responses might have a role in the pathogenesis of bronchiectasis.
ESID-0820 Linking PID Therapies with Solid Organ Transplantation: A New Strategy to Prevent Severe Infection in Heart Transplantation
J. Carbone 1, P. Diez2, M. Arraya1, M. Jaramillo1, L. Calahorra1, J. Fernandez-Yanez2, J. Palomo2, A. Villa2, I. Souza2, P. Muñoz3, J. Hortal4, A. Gonzalez Pinto5, J. Navarro1, J. Rodriguez Molina1, E. Sarmiento1
1Immunology, Gregorio Marañon Hospital, Madrid, Spain
2Cardiology, Gregorio Marañon Hospital, Madrid, Spain
3Microbiology, Gregorio Marañon Hospital, Madrid, Spain
4Anestesiology, Gregorio Marañon Hospital, Madrid, Spain
5Heart Surgery, Gregorio Marañon Hospital, Madrid, Spain
Objective. Post heart transplant IgG hypogammaglobulinemia (HGG) is a risk factor for severe infections that can be modulated. In a clinical trial we evaluated the efficacy and safety of intravenous immunoglobulin (IVIG) for prevention of severe infection in heart recipients with post-transplant HGG.
Methods: 12 adult heart recipients who developed HGG (IgG
Results: Severe infection was detected in 3 of 12 IVIG-treated recipients and in 9 of 12 controls (2-sided Fisher´s exact test, p=0.039). Cytomegalovirus infection that required antiviral treatment developed in 1 recipient with IVIG vs in 7 non-IVIG recipients (p =0.027). No severe IVIG-related side effects occurred.
Conclusion. The data of this pilot study demonstrate that prophylactic use of IVIG replacement therapy is safe, and decreases the incidence of severe infections in heart transplant recipients with secondary HGG.
ESID-0821 Leukocyte Adhesion Defect Presenting with Diffuse Necrotic Skin LessionS
S. hancerli torun 1, H. akturk1, M. sutcu1, U. aydin2, S. orhan3, A. somer4, N. salman4
1pediatric infectious disease and clinical immunology, Istanbul University Istanbul Medical Faculty, Istanbul, Turkey
2Plastic Surgery Department, Istanbul University Istanbul Medical Faculty, Istanbul, Turkey
3Ear Nose Throat and Head&Neck Surgery Department, Istanbul University Istanbul Medical Faculty, Istanbul, Turkey
4pediatric infectious disease and clinicial immunology, Istanbul University Istanbul Medical Faculty, Istanbul, Turkey
Purpose: Leukocyte adhesion deficiency is a rare autosomal recessive disorder of leukocyte function and is characterized by recurrent bacterial and fungal infections and depressed inflammatory responses despite striking blood neutrophilia. Skin infection may progress to large choronic ulcers that often require plastic surgical grafting.
We hereby present a case of leukocyte adhesion deficiency with widespread necrosis of the tissues, which have seen promising results after plastic surgical grafting.
Case: 6 year old old female patient diagnosed with leukocyte adhesion deficiency was admitted to our clinic for widespread necrotic skin lessions and sepsis. She had malodorous ulcereted lesion on each side of thorasic wall, left shoulder, left thigh, right gluteal region on physical examination. Broad spectrum antibiotic therapy (teicoplanin, meropenem and flucanosole) was started. She had undergone surgical debridement after basic life support strategies for septic shock were held. Her wound swab cultures were positive for E.coli and P.aeruginosa. Graft surgery was performed 2 weeks after the debridement procedure. Wound care was done with reguler dressing. She had complete recovery 1,5 months after admission.
Conclusions: Leukocyte adhesion deficiency is characterized by local progressive soft tissue infection. Untreated patients can result in septic shock. Supportive treatment, proper antibiotics early debridement and graft surgery can be life-saving.
ESID-0822 Fatal Association of Chronic Granulomatous Disease and Acquired Hemophagocytic Lymphohistiocytosis
S. hancerli torun 1, S. sapmaz1, A. somer1, G. sik2, S. karaman3, N. salman4, A.G.O.P. citak2, C. aleattin5, O.M.E.R. devecioglu3
1pediatric infectious disease and clinical immunology, Istanbul University Istanbul Medical Faculty, Istanbul, Turkey
2Pediatric Intensive Care Unit, Istanbul University Istanbul Medical Faculty, Istanbul, Turkey
3Pediatric Haemotology and Oncology, Istanbul University Istanbul Medical Faculty, Istanbul, Turkey
4pediatric infectious disease and clinicial immunology, Istanbul University Istanbul Medical Faculty, Istanbul, Turkey
5Pediatric Surgery, Istanbul University Istanbul Medical Faculty, Istanbul, Turkey
Background: Chronic granulomatous disease (CGD) is a rare phagocyte disorder. Here we report a 9-year-old girl with CGD hospitalized for a liver abscess due to Staphylococcus aureus developed hemophagocytic lymphohistiocytosis.
Case: 9-year-old girl with CGD was admitted to our clinic for fever, wounds on legs and fatigue. She had been diagnosed as CGD by presence of skin abscess and the result of 0% of the nitroblue-tetrazolium (NBT) test at age of one year old.
Basic laboratory studies revealed leukocytosis (18,900/mm3), elevated levels of C-reactive protein (291 mg/dL). Granulocyte suspension was given five times in consecutive days. CT showed that atelectasis on the right lower lobe, subdiaphragmatic fluid and heterogeneous, multilocular lesion in the liver. Surgical drainage was performed by laparotomy and 500 cc subdiaphragmatic abscesses were drained. Methicillin resistant Staphylococcus aureus was isolated.
On the 28th day, the patient had hyponatremic convulsion and became hypoxemic. Splenomegaly, pancytopenia, hypofibrinogenemia, elevated ferritin level and hemophagocytosis on bone marrow aspiration were developed Intravenous immunglobulin and steroid treatment were initiated. She had focal convulsions. Cranial CT showed excessive subarachnoid hemorrhage. On the 34th day of admission, she died from multiple organ failure.
Conclusion: Chronic granulomatous disease can be associated with hemophagocytic lymphohistiocytosis. Physicians should therefore pay particular attention to this mortal complication.
ESID-0824 The Ameliorative Effect of Bone Marrow-Derived Stem Cells on Injured Liver of Mice Infected with Schistosoma Mansoni
W. Mansour 1, M. El-Mahdi2, O. Hammam3, T. Hasan4
1Immunology, Theodor Bilharz Research Institute (TBRI), Giza, Egypt
2Paracitology, Faculty of Science Cairo University, Giza, Egypt
3Pathology, Theodor Bilharz Research Institute (TBRI), Giza, Egypt
4Immunology, Faculty of Science Cairo University, Giza, Egypt
Abstract
Objectives: Schistosomiasis leads to liver fibrosis, cirrhosis and finally malignancy. The technique of hepatocytes transplantation has recently improved in order to bridge the time between whole-organ liver transplantation. Methods: In the study, mice stem cells were isolated from bone marrow cells harvested from the tibia and femoral marrow compartments of male mice, then cultured in DMEM with and without hepatocyte growth factor, then transplanted into Schistosoma mansoni-infected female mice on their 12th week post-infection via tail injections. Mice were sacrificed monthly until the third month of bone marrow transplantation, serum was collected, albumin concentration, ALT, AST and alkaline phosphatase (ALP) were assayed. Immunohistopathological and immunohistochemical changes of granuloma size and number, collagen content and OV-6 cells were detected for identification of liver fibrosis. BMSCs were shown to differentiate into hepatocyte-like tissue after HGF treatment. Results: Serum ALT, AST and ALP were markedly reduced in the group of mice treated with BMSCs than in the BM-untreated control group. Also, granuloma showed a marked decrease in size and number as compared to the BMSCs untreated Schistosoma infected group. Collagen content showed marked decrease after the third month of treatment with BMSCs. The expression of OV-6 cells increased detecting the presence of newly formed hepatocytes after BMSCs treatment. In conclusion, BMSCs transplantation into Schistosoma mansoni infected mice found to restore liver function and reduce liver fibrosis. The curative action of stem cells would be considered as a potential strategy for future liver treatment, including malignant liver.
ESID-0825 Analysis of the Expression and Interrelationship Between the CD74 and CD44 Receptors Expressed on Human Breast Cancer Derived Cells
H. Al Ssadh 1, N. Fernandez1, W. alabdulmenaim1
1School of Biological Sciences, University of Essex, Colchester, United Kingdom
CD74 is a transmembrane protein that functions as a chaperone of MHC class II and is also thought to be involved in signaling via MIF[FN1] and CD44. CD44 is a transmembrane glycoprotein that acts as thereceptor for hyaluronan and it is considered as a member of of cell adhesion molecules . The relationship between these two proteins is not well understood; one hypothesis postulates that the expression of CD74 and CD44 is associated with inflammatory disorders including cancer. We have examined the interaction of CD74 and CD44 by flow cytometry and western blot in two breast tumor cell lines, CAMA-1 and MDA-MB-231. The cell surface membrane expression of CD44 is higher than CD74 in both cell lines MDA-MB-231 and CAMA-1 studied. On the other hand, the level of expression of CD74 in CAMA-1 cell lines was higher than in MDA-MB-231. To evaluate the physical association of these two proteins, co-localization experiments using bioimaging were carried out. It was observed that CD74 and CD44 are highly co-localized suggesting a possible mode of function in facilitating signalling.
ESID-0827 Spectrum and Management of Complement Immunodeficiencies (Excluding HAE) Across Europe
A. Fryer1, B. Gathmann2, C. Bangs3, M. Bradbury4, S. Seneviratne5, E. Azarsiz6, H. Alachkar7, S. Hambleton8, H. Ritterbusch9, P. Kralickova10, L. Marodi11, M. Seidel12, G. Dueckers13, J. Roesler14, A. Huissoon15, H. Baxendale16, J. Litzman17, P. Arkwright 18
1Paediatric Immunology, University of Manchester, Manchester, United Kingdom
2for the ESID Working Party Center for Chronic Immunodeficiency, Universitatsklinkkum, Freiburg, Germany
3Immunology, Royal Manchester Infirmary, Manchester, United Kingdom
4Paediatric Nephrology, Royal Manchester Children's Hospital, Manchester, United Kingdom
5Immunology, Royal Free Hospital, London, United Kingdom
6Paediatric Immunology, Ege University, Izmir, Turkey
7Immunology, Salford Royal Foundation Trust, Manchester, United Kingdom
8Paediatric Immunology, University of Newcastle, Newcastle, United Kingdom
9Center for Chronic Immunodeficiency, Universitatsklinkkum, Freiburg, Germany
10Allergology & Clinical Immunology, Charles University, Prague, Czech Republic
11Medical & Health Science Center, University of Debrecen, Debrecen, Hungary
12Pediatric Hematology-Oncology, Medical University, Graz, Austria
13Helios Clinic, Children's Hospital, Krefeld, Germany
14Kinder und Jugendmedizin, Universitatsklinkkum Carl Gustav Carus, Dresden, Germany
15Immunology, Birmingham Heartlands Hospital, Birmingham, United Kingdom
16Immunology, Addenbrookes Hospital, Cambridge, United Kingdom
17Clinical Immunology & Allergology, St. Anne´s University Hospital, Brno, Czech Republic
18Paediatric Immunology, Royal Manchester Children's Hospital, Manchester, United Kingdom
Background: Complement immunodeficiencies (excluding HAE) are rare. Published literature consists largely of case reports and small series. We collated data on complement PIDs from the ESID registry to provide an overview of disease impact and management across Europe.
Methods: 62 patients aged 2 to 66 years (median 22) from 14 centres in the UK and the Continent were recruited through the ESID registry. Clinical and laboratory information was collected onto standardised forms and analyzed statistically using SPSS software. The study had multi-centre ethics approval.
Results: 44% of patients had defects in the classical (C1, C2, C4), 32% in the alternative (C3, factors D, H, I) and 24% in the terminal (C5 to C9) complement pathways. Prevalence of classical and terminal pathways were evenly spread between regions, while 18 of 20 patients with alternative pathway defects (59% with renal disease) were referred by Nephrologists in Manchester, UK. Two patients had died. 31% were Asian, the rest Caucasian. Patients with a history of infections pre-diagnosis were more likely to receive prophylactic antibiotics (80% versus 48%, P <0.02), but not more likely to receive quadrivalent meningoccal vaccination (P =0.1). There was no significant difference in post-diagnosis infection rates in relation to either prophylactic antibiotic use or Menveo vaccination.
Conclusions: Complement PIDs are rare but associated with a high rate of infection. The role of prophylactic antibiotics and meningococcal vaccine in preventing infections is not confirmed by this study and larger cohorts are required to allow for definitive evidence based recommendations.
ESID-0828 Gene Expression Profiling in Peripheral Blood Mononuclear Cells of Patients with Common Variable Immunodeficiency: Modulation of Immune Response Following Intravenous Immunoglobulin Therapy.
G. Patuzzo 1, M. Dolcino2, A. Barbieri1, G. Argentino1, R. Beri1, M. Rizzi1, A. Ottria3, C. Lunardi1, A. Puccetti2
1INTERNAL MEDICINE, University of Verona, VERONA, Italy
2immunology, G.Gaslini Institute, Genoa, Italy
3MEDICINE, University of GENOVA, GEnoa, Italy
Intravenous immunoglobulin (IVIG) treatment is used to replace antibody deficiency in primary immunodeficiency diseases; however the therapeutic effect seems to be related not only to antibody replacement but also to an active role in the modulation of the immune response. Common variable immunodeficiency (CVID) is the most frequent primary immunodeficiency seen in clinical practice. We have studied the effect of intravenous immunoglobulin replacement in patients with CVID by evaluating the gene-expression profiles from Affimetrix HG-U133A. The gene array results were validated by real time RT-PCR and by the measurement of circulating cytokines and chemokines by ELISA. Moreover FACS analysis of blood mononuclear cells from the patients enrolled in the study was performed. A series of genes involved in immune responses were markedly up- or down-modulated before therapy. Such genes included CD14, CD36, LEPR, IRF-5, RGS-1, CD38, TNFRSF25, IL-4, CXCR4, CCR3, IL-8. Most of these modulated genes showed an expression similar to that of normal controls after immunoglobulin replacement. Real time RT-PCR of selected genes and serum levels of IL-4, CXCR4 before and after therapy changed accordingly to gene array results. FACS analysis showed a marked decrease of CD8+T cells and an increase of CD4+T cells following treatment. Moreover we observed a marked increase of CD23-CD27-IgM-IgG- B cells (centrocytes). Our results are in accordance with previous reports and provide support to the hypothesis that the benefits of IVIG are not only related to antibody replacement but also to its ability to modulate the immune response in common variable immunodeficiency.
ESID-0829 Diagnostic Value of Co-Screening for IGA Deficiency with Anti-Tissue Transglutaminase Assays for the Diagnosis of Coeliac Disease
C. Philbey 1, I. Thopte2
1General Medicine, Harrogate District Hospital, Leeds West Yorkshire, United Kingdom
2Paediatric Gastroenterology, York District Hospital, York, United Kingdom
Background: Many physicians, especially paediatricians, repeat the anti-tissue transglutaminase assay for diagnosis of coeliac disease if the first test is negative. This is because the gold standard of jejunal biopsy is very invasive.
Yet, the assay is reliant on IgA based immunity and IgA deficiency has an overall prevalence of 0.4% in the general population rising to 2.5% in those with Coeliac Disease.
This project evaluates the diagnostic value of 5 years of co-screening for IgA deficiency (via IgG to tTG, IgA total levels and IgG to EMA) versus simply repeating the IgA anti-tTG assay on an initially seronegative patient.
Method: Case-series of 1409 paediatric patients from a large district hospital and regional centre that were under investigation for coeliac disease over a 5 year period. Selected for initial and repeated diagnostic screening and progress to definitive diagnosis via the gold standard of jejunal biopsy. Approved for retrospective analysis compared to known standard by York District Hospital.
The sensitivity, specificity and likelihood ratios for each method were then calculated and accuracy to jejunal biopsy was compared via pearson's chi-square test.
Results: We found that co-screening raised the sensitivity of a single test to 99.97%, the specificity to 99.99% and also found that the population examined had an IgA deficiency prevalence of 15% amongst its Coeliac Disease patients. Co-screening reduced biopsy requirement by 88%, p<0.05
Conclusions: 1. Implement co-screening of IgA deficiency with anti-tTG assays as the first step of diagnosis
2. Next diagnostic step: Obtain a biopsy for histological diagnosis if still uncertain.
ESID-0830 Salmonella Enterica in CVID as a Probe Host-Bacteria Interaction in Inflammatory Bowel Disease.
P. Zdziarski 1, G. Dworacki2, E. Jaskula3
1Dept Infectious Dis and Clinical Immunology Hospital Infection Control Team, Lower Silesian center for Cellular Transplantation L. Hirszfeld Institute of Immunology and Experimental Therapy, Wroclaw-46, Poland
2Deopt Clin Immunol, Poznan University of Medical Sciences, Poznan, Poland
3Deopt Clin Immunol, L. Hirszfeld Institute of Immunology and Experimental Therapy, Wroclaw, Poland
Inflammatory bowel disease is caused by complex interaction of genetic and environmental factors. Important pathways in IBD development are linked to the polymorphism in innate or adaptive regulations gene. Although known NOD2/CARD15 polymorphism is crucial in Crohn Disease pathogenesis, the microbial flora and environmental factors, exactly defined bacterial infection are elusive.
54-year old woman with chronic, progressive transmural inflammatory process with mucosal damage was diagnosed as a CD and treated with anti-inflammatory agents. Immunosuppressive therapy accumulation was ineffective: chronic diarrhea, abdominal pain, weight loss, fatigue and fever was still observed.
Histopathology picture revealed in mucosa features of chronic lymphocytic inflammation with a pattern of prominent lymphoid aggregates mainly as a lymphoid hyperplasia, increased rate of apoptosis, but lack of final B cell maturation i.e. presence of lymphoplasmocytic differentiation including presence of plasma cells. Besides an increased rate of intraepithelial lymphocytes, presence of intraepithelial neutrophils and noncaseating granulomas formation were found.
Among the SNP polimophism in NOD2 gene SNP8 [2104C> T Arg702Trp] was observed. Further analysis show lack of isoagglutinins, IgM, IgA below detection limit and IgG-147mg/dl, recurrent Salmonella spp. infection.
Conclusion: This case report show that initial IBD development may be linked to aberrancies of adaptive immune response to Enterobacteriaceae. CD is a consequence of immune deficiency i.e. abnormal ability of lymphoid tissue (MALT) to respond to salmonella carbohydrates and an extreme manifestation of the immune dysregulation that are source of asymmetric lymphocyte development. TNF/TACI abnormalities observed in CVID may be immunogenetic background of CD and source of ineffective anti-TNF therapy.
ESID-0831 CD46 Links Complement and Metabolic Reprogramming in Human TH1 Responses
M. Kolev1, S. Dimeloe 2, G. Le Friec1, G. Arbore1, G. Povoleri1, M. Fischer2, L. Razik2, J. Watson3, L. Couzi4, B. Afzali1, P. Lavender3, C. Hess2, C. Kemper1
1Division of Transplant Immunology and Mucosal Biology MRC Centre for Transplantation, King's College London, London, United Kingdom
2Immunobiology Department of Biomedicine, University Hospital Basel, Basel, Switzerland
3MRC and Asthma UK Centre in Allergic Mechanisms of Asthma, King's College London, London, United Kingdom
4Nephrology Transplantation CHU Bordeaux Hospital Pellegrin, CNRS UMR 1564, Bordeaux, France
The complement regulator CD46 provides an important costimulatory signal for T cells. CD4+ T cells from CD46-deficient patients are characterized by defective Th1 induction. Such differentiation of CD4+ T cells requires significant metabolic reprogramming. Activated T cells increase rates of aerobic glycolysis and mitochondrial oxidative phosphorylation (OXPHOS), meeting increased demands for ATP and biosynthetic intermediates and additionally facilitating cytokine production through generation of mitochondrial reactive oxygen species and glucose-dependent post-transcriptional and epigenetic pathways. However, in vivo signals instructing this metabolic adaptation remain poorly defined in humans.
Here we found that CD46, and specifically its intracellular domain CYT-1, is critical for these metabolic changes. CD46 co-ligation upon T cell receptor (TCR) activation triggered significant upregulation of the glucose transporter GLUT1, and the amino acid (AA) transporter LAT1 and increased uptake of glucose, leucine and phenylalanine, compared to cells activated via the TCR alone or with CD28 co-ligation. Simultaneously, CD46 signaling specifically drove expression of LAMTOR5, which mediates assembly of the AA-sensing Ragulator-Rag/mTORC1 complex at the lysosome. Lysosomal tethering activated mTORC1, resulting in increased glycolysis and OXPHOS rates. Inhibition of glucose or AA uptake, mTORC1 activity or LAMTOR5 expression prevented CD4+ T cell activation and IFN-γ secretion. Importantly, T cells from CD46-deficient patients failed to upregulate the molecular components of this metabolic program as well as glycolysis and OXPHOS, and IFN-γ production could be reinstated by retrovirus-mediated CD46 CYT-1 expression. These data establish CD46 as a key player in linking immunometabolic adaption with T cell effector function.
ESID-0832 Functional Dichotomy of Human ISG15 – Mycobacterial Disease and Interferonopathy
S. Speer1, X. Zhang2, B. Payelle-Brogard3, V. Francois-Newton3, O. Sanal4, D. Mansouri5, G. Rice6, L. Abel7, L. Notarangelo8, Y. Crow9, S. Boisson-Dupuis10, J.L. Casanova10, S. Pellegrini3, D. Bogunovic 1
1Microbiology, Icahn School of Medicine at Mount Sinai, New York City, USA
2Key Laboratory of Molecular Biophysics, College of Life Science and Technology Huazhong University of Science and Technology, Wuhan, China
3Cytokine Signaling Unit CNRS URA 1961, Institut Pasteur, Paris, France
4Pediatric Chest Disease Department, Hacettepe University Children’s Hospital, Ankara, Turkey
5Division of Infectious Diseases, Shahid Beheshti University of Medical Sciences, Teheran, Iran
6Genetic Medicine, Manchester Academic Health Science Centre University of Manchester, Manchester, United Kingdom
7Laboratory of Human Genetics of Infectious Diseases, Institut Imagine- Necker Hospital, Paris, France
8Division of Immunology, Children's Hospital Boston, Boston, USA
9Laboratory of Neurogenetics and Neuroinflammation, Institut Imagine, Paris, France
10Laboratory of Human Genetics of Infectious Diseases, The Rockefeller University - Howard Hughes Medical Institute, New York City, USA
ISG15 is primarily known as an IFN-α/β-inducible intracellular ubiquitin-like anti-viral molecule involved in ISGylation. However, it exists in three different states: free extracellular, free intracellular, and conjugated via a covalent bond to target protein lysine residues (ISGylation). We previously reported humans with inherited ISG15 deficiency but without unusually severe viral diseases. These patients were prone to mycobacterial disease due to lack of free extracellular human ISG15, responsible for IFN-γ-induction in lymphocytes. Interestingly, ISG15-deficient patients also display unanticipated cellular, immunological, and clinical signs of enhanced IFN-α/β immunity, reminiscent of the Mendelian auto-inflammatory interferonopathies Aicardi-Goutières syndrome and spondyloenchondrodysplasia. Absence of free intracellular ISG15 in the patients’ cells prevents the accumulation of USP18, a potent negative regulator of IFN-α/βR signaling, resulting in the enhancement of IFN-α/β responses. Human ISG15 is, therefore, important for induction of IFN-γ as an extracellular molecule, and as a key negative regulator of IFN-α/β immunity as an intracellular molecule.
ESID-0833 Rapid MALDI-TOF Profiling of Unfractionated Serum and Plasma for Albumin/IgG Ratio.
A. Cepica 1
1Pathology and Microbiology, University of Prince Edward Island, Charlottetown, Canada
Hyper-gammaglobulinemia and ensuing decrease of albumin (A) – IgG ratio are hallmarks of autoimmune disorders, persistent and chronic viral, bacterial, protozoal infections, and of myelomas. In addition to multiple diagnostic applications, a rapid, quantitative, inexpensive, and high throughput test for A/ IgG would be valuable for monitoring of progression of the mentioned disorders and therapies. We developed and validated such test of large m.w. (40-190 kDa) MALDI-TOF profiling using unfractionated mink serum and plasma. Similarly to human B19 infection, Aleutian disease (AD) parvovirus leads to hypergammaglobulinemia and immune complex disease, even in immunologically competent individuals. In principle, the relative signal intensities within the mass spectrum of a complex protein sample are quantitatively reproducible. We sought the method of MALDI-TOF monitoring of hypergammaglobulinemia through A/ IgG, because such ratios are not only quantitative, but they are more biologically significant than mere IgG w/v quantities. The developed test was reproducible, and it was validated by protein electrophoresis. The A/ IgG ratios were found to be stable over wide range of specimen dilutions (undiluted to 1:500), indicating that in vivo variation in plasma volume do not affect this parameter. In the preliminary experiments on human sera and plasma, including a multiple myeloma patient, no differences in the albumin and IgG mass spectra, compared to mink serum and plasma, were discernible. The method is expected to be useful for diagnosis of primary hypoalbuminemia in the absence of hyper γ-globulinemia, and for monitoring of other proteins of interests in the m.w. range 40-190 kDa, e.g. CRP.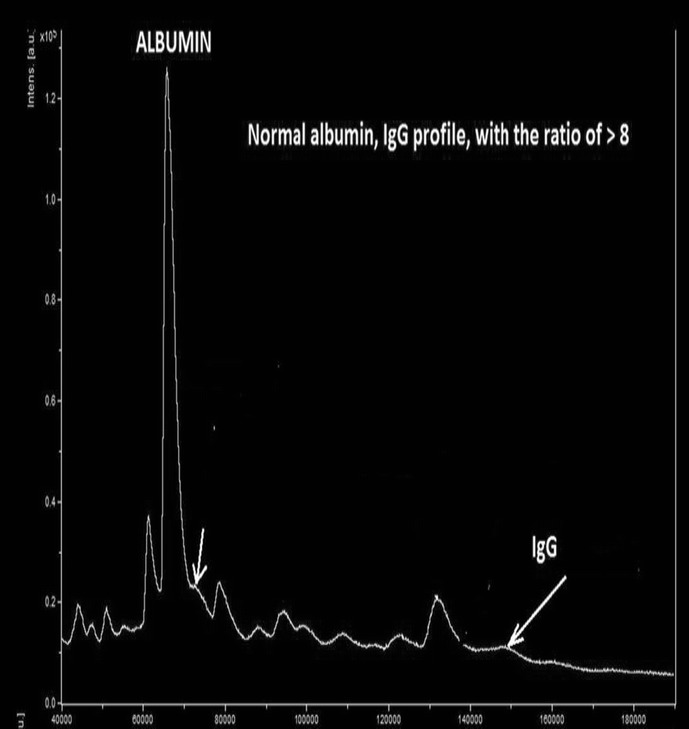
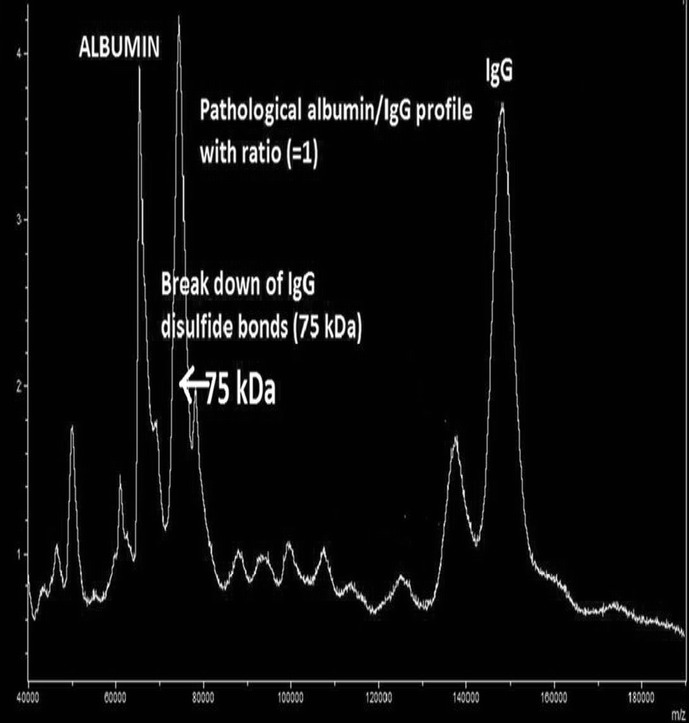
ESID-0834 Morbidity and Mortality in CVID: Correlation with Laboratory Parameters
J. Brent1, D. Guzman2, C. Bangs3, B. Grimbacher4, M. Buckland5, A. Huissoon6, C. Bethune7, H. Alachkar8, S. Patel9, M. Thomas10, D. Kumararatne11, H. Baxendale12, D. Edgar13, M. Helbert14, S. Hambleton15, P. Arkwright 1
1Paediatric Immunology, University of Manchester, Manchester, United Kingdom
2Immunology, UK-PIN, London, United Kingdom
3Immunology, UK-PIN, Manchester, United Kingdom
4Immunology, Royal Free Hospital, London, United Kingdom
5Immunology, St Batholomew's Hospital, London, United Kingdom
6Immunology, Birmingham Heartland Hospital, Birmingham, United Kingdom
7Immunology, Derriford Hospital, Plymouth, United Kingdom
8Immunology, Salford Royal Foundation Trust, Manchester, United Kingdom
9Immunology, John Radcliffe Hospital, Oxford, United Kingdom
10Immunology, NHS Greater Glasgow & Clyde, Glasgow, United Kingdom
11Immunology, Addenbrooke's Hospital, Cambridge, United Kingdom
12Immunology, Papworth Hospital, Cambridge, United Kingdom
13Immunology, Royal Hospitals, Belfast, United Kingdom
14Immunology, Manchester Royal Infirmary, Manchester, United Kingdom
15Paediatric Immunology, University of Newcastle, Newcastle, United Kingdom
Background: Although CVID is classified as an antibody immunodeficiency, a number of the common complications do not relate to pure B-cell dysfunction. We studied the association between cellular and humoral laboratory parameters and both morbidity and mortality in CVID.
Methods: 734 patients aged 5 to 96 years (median 52) from nine UK cities were recruited through the UK-PIN registry. Clinical (demographics and clinical complications) and laboratory information (full blood count and differential, lymphocyte subsets and initial and latest serum immunoglobulin levels) were collected onto standardised forms and analyzed using Kaplan-Meier statistics. The study had multi-centre ethics approval.
Results: 41% of patients in the cohort had bronchiectiasis, 14% splenomegaly, 8% autoimmune disease, 8% cancers of which 4% were lymphoproliferative and 5% granulomatous disease. 7% had died. [IgG] of p <0.001). [IgM] of p <0.001). Low [IgG] on treatment was not associated with bronchiectasis. Patients with cancer (non-lymphoid) were more likely to have a CD3
Conclusions: Although low trough [IgG] predicts overall survival in CVID, other humoral and cellular parameters correlate with common complications. Considering CVID as not just a B-cell disorder may help in our unravelling of its complex pathogenesis.
ESID-0835 Homozygous CCBE1 Mutation in Two Siblings with a Novel Phenotype: Severe Intestinal Lymphangiectasia and Chylous Ascites in the Absence of Cholestasis
C. Jackson 1,2, L. Lorenzo3, J.L. Casanova2,3,4,5,6, J. Wacker7, T. Harrer7
1Department of Pediatrics, The Memorial Sloan Kettering Cancer Center, New York, USA
2St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY, USA
3Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Hospital for Sick Children, INSERM, Paris, France, EU
4Howard Hughes Medical Institute, USA
5Paris Descartes University, Imagine Institute, Paris, France, EU
6Pediatric Hematology-Immunology Unit, Necker Hospital for Sick Children, Paris, France, EU
7Department for Internal Medicine III, University Hospital Erlangen, FriedrichAlexander-University, Erlangen, Germany
Aagenaes (lymphedema-cholestasis) and Hennekam (lymphedema-lymphangiectasia) syndromes are characterized by widespread congenital lymphatic dysplasia in humans. Additional clinical features of Hennekam syndrome include, 1) hypogammaglobulinemia and lymphocytopenia secondary to intestinal lymphangiectasia, 2) facial dysmorphy and 3) intellectual disability. These two syndromes are both genetically heterogeneous and allelic, as bi-allelic mutations in CCBE1 account for a minority of both cases. We report two siblings (a 21-year-old sister and a 20-year-old brother) born to consanguineous parents of Turkish ancestry, with a distinctive clinical phenotype marked by relapsing chylous ascites and severe intestinal lymphangiectasia requiring intravenous IgG therapy. The two siblings had a history of mild cognitive delay and limb lymphedema in early childhood. However, they do not have cholestasis or facial dysmorphy. Whole exome and Sanger sequencing identified the homozygous CCBE1 p.C174Y mutation in both siblings. The CCBE1 gene encodes a putative extracellular matrix protein crucial for lymphatic development. This mutation affects a cysteine residue, highly conserved across species, located in the calcium-binding EGF domain. Homozygous CCBE1 p.C174Y had been previously reported in a 2-year-old girl with lymphedema, lymphangiectasia, intellectual disability, and facial dysmorphy without chylous ascites. Our results extend the phenotype associated with CCBE1 mutations to include intestinal lymphangiectasia, limb lymphedema, chylous ascites, and intellectual disability, in the absence of cholestasis or facial dysmorphy. Our findings suggest that genetic modifiers may be responsible for the phenotypic heterogeneity of mutant CCBE1-caused lymphatic dysplasias.
ESID-0836 A Homozygous CARD9 Mutation (R101S) in a Brazilian Patient with Deep Dermatophytosis
A. Grumach1, F. Queiroz Telles Filho2, M. Migaud3, F. Lanternier3, N. Rosario Filho4, S. Palma5, R.N. Constantino-Silva6, J.L. Casanova7, A. Puel 8
1Center of Research & Outpatient group of Recurrent Infections, Faculty of Medicine ABC, Santo Andre, Brazil
2Department of Health Community, Federal University of Parana, Curitiba, Brazil
3Laboratory of Human Genetics of Infectious Diseases Necker Branch Institut National de la Santée de la Recherche Médicale, Institut Imagine Université Paris Descartes, Paris, France
4Department of Pediatrics, Federal University of Parana, Curitiba, Brazil
5Department of Pediatrics, Faculty of Medicine ABC, Sao Paulo, Brazil
6Center of Research, Faculty of Medicine ABC, Santo Andre, Brazil
7Laboratory of Human Genetics of Infectious Diseases Necker Branch & St Giles Laboratory of Human Genetics of Infectious Diseases, Universite Paris Descartes & The Rockefeller University, Paris New York, USA
8Laboratory of Human Genetics of Infectious Diseases Necker Branch Institut National de la Santée de la Recherche Médicale, Universite Paris Descartes & The Rockefeller University, Paris, France
Deep dermatophytosis has been described in HIV and immunosupressed patients. Recently, CARD9 deficiency was reported in individuals with deep dermatophytosis previously classified as “immunocompetent”. We report a 24-year-old Brazilian male with deep dermatophytosis born to an apparently non-consanguineous family. The symptoms started with oral candidiasis when he was 3 years old, persistent although treated. At 11 years old well delimitated, descamative and pruriginosus skin lesions appeared; ketoconazol and itraconazole were maintained for 5 years. At 12 years old, the lesions affected initially the face, turning out ulcerative, secretive and painful, spreading to several regions of the body (15cm of diameter); terbinafine was introduced without improvement. Trichophyton mentagrophytes was isolated from the skin lesions. Neutrophil evaluation showed specific impaired fungal killing. A homozygous mutation in CARD9 exon 3 (R101S) was identified in the patient. Both parents, one brother (symptomatic) and one sister were heterozygous for this mutation, while another brother was found to be homozygous for the CARD9 wildtype allele. His symptomatic brother presents persistent superficial dermatophytosis. This is the first report of CARD9 deficiency (R101S mutation) in a Brazilian family.
ESID-0837 Coexisting Unrelated Cerebellar Disorders in a Child with Ataxia-Telangiectasia
A. Molinaro1, A. Soresina 2, R. Micheli3, S. Costa3, A. Milanesi2, M. Maffeis2, L. Pinelli4, A. Plebani2
1School in Reproductive and Developmental Science, University of Trieste and University of Brescia, Brescia, Italy
2Pediatrics Clinic Department of Clinical and Experimental Sciences, Spedali Civili and University of Brescia, Brescia, Italy
3Unit of Child Neurology and Psychiatry, Spedali Civili and University of Brescia, Brescia, Italy
4Department of Neuroradiology, Spedali Civili Brescia, Brescia, Italy
Ataxia–telangiectasia (AT) is a genetic disorder caused by mutations in the ATM gene and characterized by progressive neurodegeneration, immunodeficiency, and susceptibility to cancer. Here we report a 2-year-old female patient diagnosed as having AT by clinical, laboratory and genetic testing. At diagnosis, brain magnetic resonance imaging (MRI) showed a signal intensity alteration in the left paravermian cerebellar cortex, consistent with low-flow vascular malformation. Longitudinal neurological, immunological and neuroradiological evaluations were performed with no signs of disease progression (except for AT) up to the age of 6 years, when a lesion enlargement on brain MRI was suspected as low-grade glioma, histologically confirmed as pilocytic astrocytoma after a complete surgical resection. After 5 months acute dysarthria and a worsening in ataxia were noted, spontaneously recovered in 3 days. Brain MRI performed 1 month later revealed a right intraparenchymal malacic area, contralateral to the glioma resection, consistent with ischemic stroke (IS). Common risk factor for IS, both genetic and acquired, were excluded. In summary, we report a child with AT, an extremely rare condition, who developed, in addition, a left cerebellar astrocytoma and a right cerebellar infarction, considered as two independent events. Children with AT have an increased risk of developing cancer, but, only few cases of glioma are reported and, to our knowledge, no other cases of unrelated cerebellar glioma and cerebellar infarction in children with AT have been described. Further analysis is required to better define the role of the ATM gene mutation in the pathogenesis of these coexisting conditions.
ESID-0838 Dual Impairment of IL-17 and IFN-G Immunity Cause Infections by Candida and Mycobacterium in Humans with RORC Deficiency
J. Markle 1, S. Okada1, E. Deenick2, F. Mele3, N. Wong2, C.S. Ma2, C. Soudais4, D. Averbuch5, M. Alzahrani6, M. Lagos7, M. Gonzalez7, R. Martinez-Baricarte1, C. Okada1, M. Migaud8, Y. Itan1, C. Picard8,9,10, O. Lantz4, L. Abel1,8, D. Engelhard5, S. Al-Muhsen6, F. Sallusto3, S. Boisson-Dupuis1,8, A.N.N.E. Puel8, J. Bustamante8, S. Tange2, J.L. Casanova1,8,9,10,11
1St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY, USA
2Garvan Institute of Medical Research, 384 Victoria St, Darlinghurst, NSW, Australia
3Institute for Research in Biomedicine, Università della Svizzzera Italiana, Via Vincenzo Vela 6, Bellinzona, Switzerland
4Institut Curie, Inserm U932, Paris, France
5Department of Pediatrics, Hadassah University Hospital, 91120 Jerusalem, Israel
6Department of Pediatrics, College of Medicine, King Saud University, and King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
7Departamento de Preclínicas, Escuela de Medicina, Universidad de Valparaíso, Chile
8Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Hospital for Sick Children, INSERM, Paris, France, EU
9Paris Descartes University, Imagine Institute, Paris, France, EU
10Pediatric Hematology-Immunology Unit, Necker Hospital for Sick Children, Paris, France, EU
11Howard Hughes Medical Institute, New York, USA
Inborn errors of human IL-17 immunity underlie mucocutaneous candidiasis, whereas inborn errors of IFN-γ immunity underlie mycobacterial disease. We report the discovery of autosomal recessive complete RORC deficiency in six patients, from three kindreds of diverse ethnic origins, with both candidiasis and mycobacteriosis. As predicted by the mouse model, and in agreement with our previous findings in IL17RA deficient patients, the lack of functional RORγt protein prevented the development of IL-17-producing T cells, accounting for the patients’ chronic candidiasis. RORC deficient patients had altered T cell repertoires including a complete absence of mucosal associated invariant T (MAIT) cells and invariant natural killer T cells (iNKT), which normally produce IFN-γ. Leukocytes from RORC deficient patients fail to produce IFN-γ in response to BCG challenge, and this defect is attributable to functional defects in RORγt+ dual IFN-γ– and IL-17-producing CD4+ T cells, previously shown to specifically respond to mycobacterial antigens, and γδT cells, as well as the absence of other cellular sources of IFN-γ (MAIT and iNKT). Heterozygous carriers respond normally to BCG challenge, therefore only autosomal recessive RORC deficiency is disease-causing. This experiment of Nature suggests that human RORγt is not only essential for the development of IL-17-producing T cells and protective mucocutaneous immunity to Candida, but also for the development of BCG-responsive, IFN-γ-producing conventional and innate T cells and systemic immunity against mycobacteria. RORγt seems to be otherwise largely redundant in host defense against other pathogens in humans.
ESID-0839 All Parties and Training Programs for PID Care in a New Post-Graduate Specialization Era
J. Carbone 1
1Immunology, Gregorio Marañon Hospital, Madrid, Spain
On July 25, 2014, a new law (Real Decreto de Troncalidad de Formacion Sanitaria) was approved in Spain that will reorganize training programs in all specialties. This will affect specific training programs of distinct medical and multidisciplinary specialties for management of immune based diseases including primary immunodeficiencies (PID). Important changes will have to be introduced in the following years to maintain and improve training programs and strategies for PID care with the aim of improving awareness, diagnosis and integral management of PID. We discuss opportunities to partner with all parties involved in PID management in designing collaborative solutions. Primary care level. Active participation of PID patient organizations, specific training programs for nurses involved in PID care and the development of training programs for patients in self-care are important components which should be fully developed. Post-graduate specialization level. The maintenance of specific clinical immunology contents within Immunology training programs, the inclusion of PID care learning objectives for non medical immunology specialists, the design of a new Clinical Immunology Specific Training Program with participation of the distinct specialists involved in PID care, are important issues in the new organization and innovation of training programs for all medical and non medical specialists. The potential role for industry-sponsored education and the use of new learning technologies and telemedicine services, are other opportunities to take into consideration.
ESID-0840 Efficacy and Safety of Subcutaneous Inmonuglobulin in Adult and Pediatric Patients: Novel Experience in Colombia
M. Ortega-Lopez 1, M. Leon-Pinilla2, J. Garay-Fernandez3, S. Uribe-Granados4
1Departamento de Pediatría Alergia e Inmunología, Hospital Universitario Infantil de San José Bogotá - Colombia / Fundación Universitaria de Ciencias de la Salud, Bogotá, Colombia
2Departamento de Enfermería HUISJ, Hospital Universitario Infantil de San José Bogotá - Colombia, Bogotá, Colombia
3Epidemiólogo Clínico, Pontificia Universidad Javeriana Bogotá - Colombia, Bogotá, Colombia
4Departamento de Pediatría Alergia e Inmunología Residente de II año Pediatría, Hospital Universitario Infantil de San José Bogotá - Colombia / Fundación Universitaria de Ciencias de la Salud, Bogotá, Colombia
Introduction: Subcutaneous immunoglobulin (SCIg) replacement therapy for primary immune deficiency disease (PIDD) is a convenient alternative for pediatric and adult patients with PPID and autoimmune diseases (AID). Since 2013, Bogotá - Colombia began managing pediatric patients with subcutaneous immunoglobulin (SCIg) at Hospital Universitario Infantil de San José.
Objective: We aimed to describe effectiveness, safety and tolerability of SCIg (Gammanorm®) therapy by infusion pump in pediatric and adult patients.
Material and methods: This is a descriptive and retrospective study of patients whom received Gammanorm®. “Personalized Program of Clinical Immunology of Hospital Universitario Infantil de San José”. Database, bacterial infections, adverse reactions and safely information, were obtained from medical records.
Results: We described 43 adult and pediatric patients (31 male, 12 female; mean age 21.3 y/o; range: 3-81 y; SD=21.8 y) that received SCIg since 2013. In the PIDD group, specific antibody deficiency was the most common (11/33, 33%). The second one was common variable immunodeficiency (8/33 or 24%). In the AID group, chronic inflammatory demyelinating polyneuropathy was the most common (4/10, 10%). Mean SCIg dose was 6.9 g/month (range, 1.98-33 g/month; SD=7 g/month). Serious bacterial infections were observed in 7 (21%) patients, of which bacterial pneumonia (3, 9%) was the most common. There were no serious adverse events (AEs) related to any treatment. Product-related adverse reactions were reported in 2/10 (20%) patients.
Conclusion: Our interim analysis of ongoing study on Gammanorm® in children indicate that self-administered SCIg has provided effective protection and favorable tolerability in PIDD.
ESID-0841 EV5 Deficiency: The Key to Understand Molecular Pathogenesis of Epidermodysplasia Verruciformis?
S. de Jong 1, A. Crequer1,2, L. Lorenzo3,4, A. D' Amico1, A. Arias7, N. Portilla Maya5, X. Rueda Cadena6, J.L. Franco7, B. Burger8, G. Orth9, E. Jouanguy1,3,4, J.L. Casanova3,4,10,11
1St. Giles Laboratory of Human Genetics of Infectious Diseases, The Rockefeller University, New York, NY, USA
2Department of Pathology, NYU School of Medicine, New York, USA
3Paris Descartes University, Imagine Institute, Paris, France, EU
4INSERM UMR 1163, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, Necker Hospital for Sick Children, INSERM, Paris, France, EU
5University El Bosque, Bogota, Colombia
6Dermatology/Oncology - Skin Cancer Unit, National Cancer Institute, Bogota, Colombia
7Grupo de Inmunodeficiencias Primarias, Universidad de Antioquia, Medellin, Colombia
8Department of Biomedicine, University Hospital Basel, Basel, Switzerland
9Department of Virology, Pasteur Institute, Paris, France, EU
10Pediatric Hematology-Immunology Unit, Necker Hospital for Sick Children, Paris, France, EU
11Howard Hughes Medical Institute, USA
Mendelian predisposition to persistent human beta-papillomavirus (EV-HPV) infections in otherwise healthy patients is known as epidermodysplasia verruciformis (EV). EV is a rare genodermatosis characterized by pityriasis-versicolor-like skin warts and a high risk of non-melanoma skin cancer caused by bi-allelic loss-of-function mutations in EVER1/TMC6 and EVER2/TMC8. Although EVER1/2-defects are believed to impact keratinocyte-intrinsic immunity by modulating intracellular zinc fluxes, the cellular and molecular pathogenesis of EV remains largely elusive. Moreover, about 25% of EV patients lack a genetic etiology. Combining genome-wide-linkage analysis and whole-exome sequencing in five EV patients from two related Colombian families, we identified an unreported frameshift variation in the gene EV5. The mutant allele segregates with EV as an autosomal recessive trait and shows complete clinical penetrance. By targeted Sanger sequencing two additional unreported frameshift variations were identified in a French and two Swiss EV patients. EV5 is a pleiotropic regulator of cellular signal transduction but has not been related to antiviral immunity or keratinocyte function. The three mutations lead to a complete loss of endogenous EV5 protein expression in patient-derived B-cells and keratinocytes. Interestingly, EV5 protein expression is also strongly reduced EVER1- and EVER2-deficient patient-derived cell lines, potentially linking EV5 and EVER1/2. The identification of a EV5:EVER1:EVER2 complex via co-immunoprecipitation in HEK293T cells supports this notion. We therefore propose that this complex controls keratinocyte-intrinsic immunity to EV-HPV infection. Further characterization of EV5- and EVER1/2-deficiency - in particular with regard to antiviral zinc signaling - shall enable a better understanding of immunity against EV-HPV infection and EV pathogenesis.
ESID-0842 Generation of CMV-Specific Cytotoxic T-Lymphocytes for Adoptive Immunotherapy from Virus-Naïve Donors
M. Keller 1, P. Hanley2, C. Bollard2
1Allergy/Immunology, Children's National Medical Center, Washington DC, USA
2Center for Cancer and Immunology Research, Children's National Medical Center, Washington DC, USA
Background: Adoptive immunotherapy using virus-specific cytotoxic T-lymphocyte (VSTs) products has been successful in restoring antiviral immunity after hematopoietic stem cell transplantation (HSCT), and has been used to treat several dozen patients with primary immunodeficiency. New protocols allow CTL generation from virus-naïve donors through naïve T-cell selection, and will extend the donor pool for CTL therapy.
Objective: To compare the cellular phenotype and anti-viral function of VSTs derived from CMV+ versus CMV-naïve donors.
Design/Methods: VSTs derived from CMV+ donors are expanded using antigen presenting cells (APC) pulsed with overlapping peptides spanning the CMV antigens pp65 and IE1. CMV-specific T-cells derived from CMV-naive donors utilizes a similar approach after first selecting CD45RA+ T-cells for stimulation.
Results: Central memory T-cells were detected in CMV-naïve and CMV+ donor-derived VST products (39% versus 48% respectively) without evidence of exhaustion (PD1low/CD45RA-/2B4-). Interferon-g production in response to CMV pp65 was comparable between the groups (405 ±200 SPC versus 159±72 respectively, p=0.24). Cytolytic activity of CTL against pp65/IE1-pulsed PHA blasts in vitro ranged from 2-25% at an E:T ratio of 20:1 with normal perforin/granzyme expression in all VST products. A higher percentage of Natural Killer (NK) cells were found in CMV naïve donor-derived CTL (11-56% versus <1%), though they lack CD16 and NKG2C expression.
Conclusions: VSTs derived from the two donor groups are CMV-specific and non-alloreactive, but VSTs derived from CMV-naïve donors also contain immature NK cells. The safety and clinical efficacy of these VSTs are being evaluated in high-risk patients after HSCT.
ESID-0843 Inherited IL-12Rβ2 Complete Deficiency Predisposes to BCG Disease and Tuberculosis
R. Martinez-Barricarte 1 , C. Aytekin 2 , J. Markle 1 , X.F. Knog 1 , B. Fleckenstein 3 , S. Tangye 4 , E. van de Vosse 5 , S. Boisson-Dupuis 1 , J. Bustamante 6 , J.L. Casanova 1
1St. Giles Laboratory of Human Genetics of Infectious Diseases, The Rockefeller University, NYC, USA
2Department of Pediatric Immunology, Dr. Sami Ulus Maternity and Children's Health and Diseases Training and Research Hospital, Ankara, Turkey
3Institute for Clinical and Molecular Virology, University Erlangen-Nuremberg, Erlangen-Nuremberg, Germany
4Immunology Program, Garvan Institute of Medical Research, Sydney, Australia
5Medical Center, Leiden University, Leiden, Netherlands
6Laboratory of Human Genetics of Infectious Diseases, Necker Hospital for Sick Children, Paris, France
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare syndrome predisposing individuals to severe infection by non-pathogenic mycobacteria like BCG-vaccines and environmental mycobacteria in otherwise healthy individuals. These individuals are also susceptible to the more virulent mycobacteria M. tuberculosis. To date, nine morbid genes (IFNGR1, IFNGR2, STAT1, IRF8, NEMO, CYBB, ISG15, IL12B and IL12RB1), all involved in the IFN-γ-IL-12/IL-23 loop, have been identified. The most prevalent genetic etiology of MSMD is complete IL-12Rβ1 deficiency resulting in abolished cellular responses to IL-12 and IL-23. We have identified, in a consanguineous Turkish family by whole exome sequencing, a homozygous nonsense mutation at position Q138 in IL12RB2. Three homozygous carriers showed a complete lack of expression of IL-12Rβ2 at the T cell surface. One homozygous mutant individual suffered from localized BCG disease, another from bona fide tuberculosis, and the third one remains asymptomatic. All heterozygous and WT family members were healthy. We compared the capacity of leukocytes from healthy donors, IL-12Rβ1 deficient, and IL-12Rβ2 Q138*/Q138* patients to respond to IL-12 and IL-23. Like IL-12Rβ1 deficient patients, IL-12Rβ2 patient-derived cells did not respond to BCG and IL-12 by inducing IFN-γ. Contrary to IL-12Rβ1 deficient patients, their response to IL-23 was intact. These data suggest that autosomal recessive IL-12Rβ2 deficiency is a novel genetic etiology of MSMD and tuberculosis, due to impaired IL-12-dependent induction of IFN-γ.
ESID-0845 Haploinsufficiency of the NFkB1 Subunit P50 Causes CVID
M. Fliegauf1, N. Frede 1, S. Winzer1, A.A. Schäffer2, J.W.M. van der Meer3, B. Grimbacher1
1Centre of Chronic Immunodeficiency, University Medical Centre Freiburg, Freiburg, Germany
2Department of Health and Human Services, NCBI NIH, Bethesda, USA
3Department of Medicine, Radboud University Medical Centre, Nijmegen, Netherlands
Common variable immunodeficiency (CVID) is characterized by recurrent infections due to hypogammaglobulinemia, with deficiency in IgG and at least one of IgA or IgM. 20–25% of cases are familial, presenting predominantly with an autosomal-dominant inheritance pattern. The milder selective IgA deficiency (sIgAD) may also occur in CVID families, indicating a genetic association.
In a five-generation family (AD-inheritance) with ten CVID and four sIgAD cases, we identified a novel heterozygous splice donor site mutation in NFkB1 (g.103500200A>G) within the 18.7Mb linkage interval on 4q by whole exome sequencing.
NFkB1 encodes p105, which is processed to p50 upon activation. The heteromeric p50/p65 translocates to the nucleus and activates its target genes (canonical pathway). The novel mutation c.730+4A>G causes in-frame skipping of exon 8 and thus, a deletion of 53aa (p.Asp191_Lys244delinsGlu).
The truncated p105DEx8 is expressed at reduced levels in EBV B-cells from patients and is not processed to p50DEx8 but rapidly degraded. Residual p105/p50, expressed from the non-mutated allele, is normal. Ectopic expression of GFP-p105 and GFP-p105DEx8 fusion proteins in human HEK293T cells or murine NIH3T3 fibroblasts confirmed that p105DEx8 is not tolerated. Since mutant p50DEx8 is not produced and thus, cannot mediate signaling, we conclude that CVID is caused by functional haploinsufficiency of NFkB1.
In five additional CVID families, targeted Next Generation Sequencing identified two novel splice-site, two frame-shift and one missense mutation in NFkB1, respectively, all predicted to cause functional impairment. Hence, these heterozygous NFkB1 mutations could account for the CVID phenotype in these families.
ESID-0848 The Therapeutic Effects of Lactococcus sp. KR-050L on Inflammatory Bowel Diseases
C. Lee 1, C.S. Park1, M.C. Rho1
1Eco-friendly Materials Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Jeongeup-si, Korea
Inflammatory bowel diseases (IBD), namely crohn’s diseases and ulcerative colitis, are a recurrent inflammatory disease occurred by a complex interaction of immune-regulatory factors. Up to date, extensive scientific and clinical studies of IBD have been carried out, however, the pathogenic mechanisms of IBD still remain unclear. Also, despite effectual remedy of IBD, it has been involved critical problems such as selectivity of the patients, numerous side effects, and the cost. Alternatively, recent studies have been indicated that administration of beneficial probiotics and inhibitors of IL-6/STAT3 activation can be suggested to be a promising agent for the treatment of human immune and inflammatory diseases, including IBD. In our continuing search for the remedy of IBD from probiotics, we found that the EtOAc extract of lactococcus sp. KR-050L possessed potent inhibitory effect on IL-6 induced STAT3 promoter activity in Hep3B cells. It also showed potent therapeutic effect of 3% DSS-induced IBD mouse model. Herein, we described the identification of KR-050L as well as its therapeutic effect on animal experiment.
ESID-0849 The Effect of KR-600, Salvia Plebeia Extracts, on Osteoporosis in Ovariectomized Mice Model
K. Jung 1, M.H. Kim1, M.C. Rho1
1Eco-friendly Materials Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Jeongeup-si, Korea
Osteoporosis is a disease of low bone mass most often caused by an increased bone resorption on estrogen deficiency and inflammatory disease. Salvia plebeia, which is an annual or biennial plant, naturally grows on a wide region of Asia including Korea. Recently, It is known to have an anti-inflammation and anti-allergic activity. However therapeutic effect of Salvia plebeia extracts on osteoporosis is unknown. In this study, we show that KR-600, Salvia plebeia extracts, has anti-osteoporotic bone resorption through the suppression of osteoclast formation. In vitro experiments, KR-600 treatment markedly inhibited the differentiation of mouse bone marrow derived-osteoclast by RANKL stimulation. Moreover, we ovariectomized 7 weeks age C57BL/6 mice and then treated with KR-600 for 5 weeks. The ovariectomized mice showed a decrease in total bone calcium content and bone mass density of femur bone in comparison with the sham group. Systemic treatment of KR-600 successfully prevented bone loss in ovariectomized mice model. These results suggest that KR-600 is a potent inhibitor of osteoclast differentiation and will be a therapeutic agent for postmenopausal osteoporosis.
ESID-0850 Identifying Genes Involved in Common Variable Immune Deficiency Disorders: An Integrated Whole Genome Sequencing and RNA Sequencing Approach.
P.A. van Schouwenburg 1, E.E. Davenport2, A.K. Kienzler1, I. Marwah1, B. Wright2, M. Lucas3, H. Martin2, . WGS500 Consortium2, H. Chapel1, J.C. Knight2, S.Y. Patel1
1Nuffield Department of Clinical Medicine and Oxford NIHR Biomedical Research Centre, University of Oxford, Oxford, United Kingdom
2Wellcome Trust Centre for Human Genetics and Nuffield Department of Clinical Medicine, University of Oxford, Oxford, United Kingdom
3Nuffield Department of Clinical Medicine, University of Oxford, Oxford, United Kingdom
Common Variable Immune Deficiency disorders (CVIDs) are the most clinically significant antibody failures. The genetic basis remains unresolved in ~95% of the patients, the majority of cases being heterogeneous, non-familial and polygenic. We present the first whole genome sequencing (WGS) analysis of sporadic CVID in the context of RNA-seq profiling of B cells.
Illumina WGS was performed for 31 sporadic CVID patients. Read mapping (Stampy) (average coverage 27-40X) and variant calling (Platypus) identified 14,457,884 single nucleotide variants and small in-dels across the genomes. Functional filtering (Ingenuity Variant Analysis) resolved 4,422 rare, likely pathogenic variants. This includes variants in genomic regions associated with CVID through GWAS (n=42 variants), genes associated with Primary Immunodeficiency Disorders (n=52), variants present in five or more patients (n=12) and genes linked to described CVID genes in the literature (n=106) (summarized in figure 1). The diversity between patients highlights the polygenic nature of sporadic CVID.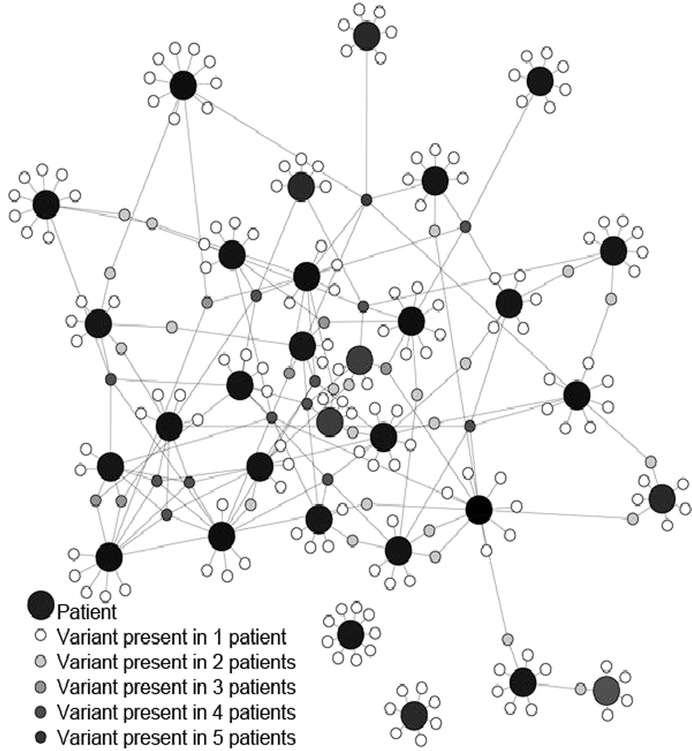
[Fig 1]
Analysis revealed heterozygous variants in LRBA, PRRC2A, IKBKB, MRE11, BLK, PLCG1, ARID3A and IL21R. We also identified heterozygous variants in RAG2, DCLRE1C, and PRKDC which are previously associated with SCID and VDJ recombination.
RNA-seq of B-cells (three of the CVID patients and three healthy controls) resolved 262 differentially expressed genes. Integrated analysis identified potentially disease causing regulatory variants and 24 pathways enriched for both WGS variants and differential gene expression including CD28 and iCOS-iCOSL signalling.
This work illustrates a combined approach using WGS and RNA-seq to identify novel potentially functional variants and pathways involved in the pathogenesis of CVID.
ESID-0851 High Fatigue Levels in XL-CGD Carriers Associated with Raised Serum IL-8
A. Battersby 1, A. Martin1, J. Tarn1, C. Cale2, D. Goldblatt3, A. Gennery1
1Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
2Clinical Immunology, Great Ormond Street Hospital, London, United Kingdom
3Institute of Child Health, University College, London, United Kingdom
Background: Chronic Granulomatous Disease (CGD) is a rare primary immunodeficiency, of which 70% of cases are X-linked (XL). We have recently found that XL-CGD suffer from inflammatory complications of CGD (ESID-0185)
Methods: During a study of health of XL-CGD carriers we investigated symptoms of excessive fatigue using the Multidimensional Fatigue Symptom Inventory Short Form (MFSI-SF). Serum levels of pro-inflammatory cytokines (IL-1α, IL-5, IL-8, IL-10, IL-17, TGFβ1, IFNα and IFN-γ) were measured on stored serum from 54 subjects, collected during the original research study, by Cytometric Bead Array (CBA) immunoassay using a BD Biosciences LSRFortessa™ cell analyser. Results were compared with inflammatory disease control groups of 10 high and 10 low fatigue Sjogren's disease patients and 15 healthy controls. XL-CGD carriers were divided into two groups; those who reported fatigue and those that did not.
Results: 50% of XL-CGD carriers reported suffering excessive fatigue, associated with significantly higher scores in the MFSI-SF (p<0.01). XL-CGD carriers had significantly higher serum IL-8 levels than normal controls (p=0.015), and patients with Sjogren's disease (p=0.031). XL-CGD carriers reporting high levels of fatigue had significantly higher IL-8 levels, than those not reporting fatigue (p=0.017). No other cytokines reached statistically significant different levels, between groups.
Discussion: XL-CGD carriers reported higher fatigue levels than expected, associated with raised IL8 suggesting a biological explanation for the symptoms. IL1β levels were not significantly raised. Previous studies have associated pro-inflammatory states with excessive fatigue. Further research is required to evaluate this finding in XL-CGD carriers.
ESID-0852 KR-301, Triterpene Acetate from Vigna Angularis, Exerts Anti-Inflammatory Effects Through the Inhibition of TLR3 and TLR7/8 Inflammatory Signaling
H. Oh 1, M. Rho1
1Eco-friendly Biomaterial Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Jeongeup-si, Korea
Previously we have reported the inhibitory effect of Vigna angularis extract on the Toll-like receptor (TLR) activation and TLR-mediated inflammatory response. In this study, we isolated KR-301 from V. angularis and investigated the signaling pathways involved in the anti-inflammatory effects of KR-301 in human monocyte THP-1 cells. The effect of KR-301 on TLR activity was evaluated by measuring NF-κB and AP-1 inducible secreted embryonic alkaline phosphatase (SEAP) activity after the treatment of TLR agonist. KR-301 significantly inhibited SEAP activity induced by TLR3 and TLR7/8 activation in a dose-dependent manner. Furthermore, KR-301 effectively inhibited mRNA expressions of pro-inflammatory cytokines IL-1β, IL-8, IL-6, MCP-1 and inflammatory adhesive molecules ICAM-1 and VACM-1 mediated by TLR3 and TLR7/8 activation. Examination of the effects of the KR-301 on NF-κB signaling showed that KR-301 inhibits the phosphorylation of IKKα/β and IκBα, preventing the nuclear translocation of the NF-κB p65 subunit. However, KR-301 did not inhibited phosphorylation of transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1), an upstream signaling molecule required for IKKα/β activation. Thus, our results suggest that KR-301 has inhibitory effect on TLR3 and TLR7/8 signaling and their inflammatory response and it can be further developed as therapeutic agent for endosomal TLR-related inflammatory diseases.
ESID-0853 Defects in CD8 Cytolysis in Immunodeficiencies with Impaired EBV Clearance
P. Schwartzberg 1, J. Cannons1, P. Zhao1, J. Reilley1, S. Kapnick1, S. Tangye2, G. Uzel3
1Genetic DIsease Research Branch, NHGRI, BETHESDA, USA
2Immunology, Garvan Institute, Sydney, Australia
3NIAID, NIH, Bethesda, USA
CD8+ cytotoxic T-lymphocytes (CTLs) help eliminate virally infected cells: defective CTL leads to lymphoproliferative diseases with hemophagocytic syndromes. We are examining cells from patients and mouse models affecting CTL function, including mutations of SH2D1a (encoding SAP), ITK and PI3KCD. These mutations lead to immunodeficiencies associated with impaired responses to Epstein-Barr Virus. We have previously shown that CTLs from SAP-deficient mice show impaired cytolysis of B cell targets, despite normal cytolysis on other cells. This defect results from recruitment of the SHP-1 phosphatase to SLAM family receptors in the absence of SAP, leading to abnormal interactions between T and B cells and immune synapse formation. Inhibition of SHP-1 or blocking SLAM family members prevented the cytolytic defects, providing mechanistic insight and potential approaches to rescue these defects. In contrast, cells from Itk-deficient mice show abnormal killing of multiple cellular targets. Surprisingly, despite defects in early TCR signaling, evaluation of distinct stages of CTL function revealed that Itk-deficient murine cells show normal T cell adherence to targets, actin organization and centrosome polarization toward the target contact. However Itk-deficient cells showed defective release of secretory granules, revealing unexpected defects in late stages of cytolysis. Finally, we are examining cells from patients expressing activating mutations of PI3KCD that are associated with immunodeficiency and elevated EBV levels. Together, these studies are aimed towards deciphering defects for potential therapeutic insight.
ESID-0855 A Complete Immunoreconstitution After Hematopoietic Stem Cell Transplantation Can Be Affected by an Abnormal Microenvironment in Anhidrotic Ectodermal Dysplasia with Immunodeficiency
A. Scarselli 1, S. Cascioli2, M.L. Romiti1, A. Aiuti3, R. Carsetti4, C. Cancrini1
1Department of Pediatrics (DPUO), Bambino Gesù Children’s Hospital University of Rome Tor Vergata, Rome, Italy
2Immunology and Pharmacotherapy Area, Bambino Gesù Children’s Hospital, Rome, Italy
3San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET), Scientific Institute HS Raffaele, Milan, Italy
4Immunology and Pharmacotherapy Area, Bambino Gesù Children's Hospital, Rome, Italy
We describe a 16-years-old-boy with Anhidrotic ectodermal dysplasia with immunodeficiency (IkBα mutations), who underwent haploidentical hematopoietic stem cell transplantation (HSCT) at 1 year of age. Despite a full donor engraftment and normal T cell recovery, humoral function remained defective. Indeed the patient still requires Ig replacement therapy (IgRT) because of impaired specific antibody production to Pneumococcus and severe bacterial infections when IgRT was temporarily discontinued. We detected an abnormal B phenotype despite the B cells were completely maternal-derived. To clarify the humoral dysfunction, we tested in vitro B lymphocyte from the patient and the mother. FACS analysis showed normal maternal B phenotype and good response to CpG proliferation whereas B-lymphocytes from patient revealed moderate proliferation, manly IgM-memory differentiation and only IgM production after CpG. The B cell alterations were not due to an intrinsic alteration of B cells that in the mother appeared perfectly normal. Because of the NF-KB pathway is involved in lymphoid organogenesis and maintenance of lymphoid structures and lymphonodes and palatine tonsils were not detectable in the patient, we speculated that structural abnormalities of secondary lymphoid organs could have influenced B cell function. Furthermore the patient presented severe planar warts after HSCT. NF-kB is an important regulator of keratinocyte proliferation then we assumed altered local anti-HPV immunity due to a defective host keratinocytes. In conclusion, our results indicate that the most severe immunologic EDA-ID abnormalities can be resolved by HSCT, but impaired NF-kB signaling in non-hematopoietic cells limits a complete immunological restoration (humoral and skin innate immunity).
ESID-0856 Trigger Infections Associated with Pediatric Kawasaki Disease
G. Jugulete 1, M. Luminos1, S. Schiopu1, M. Merisescu1
1Pediatric, Institute of Infectious Diseases "Prof. dr. Matei Bals", Bucharest, Romania
Introduction: Kawasaki Disease (KD) is a febrile multisystem syndrome of early childhood that is characterized by systemic inflammation and thrombosis. Even thought autoimmune reactions, genetic predisposition and envinmental factors are considered to be the etiologic cause of the KD, frequently most of the epidemiologic and immunologic evidence indicates that the causative agent is infectious.
Objective: In the present paper we aimed to identify the pediatric cases of KD triggered by an infectious agent.
Material and method We conducted a clinic-based retrospective surveillance over the pediatric KD cases submitted at 'The National Institute for Infectious Diseases 'Prof. Matei Bals' of Bucharest – Pediatric Department, during the period of 2010 – 2014.
Results: During the studied period we identified 27 cases of pediatric KD. We observed that the disease had a male predominance and we identified the most affected group to be the 1 - 4 years group. We were able to identify an infectious trigger for the disease in 48.1 % of the cases. From the infectious disease being associated with KD we mention: bacterial induced (Group A Beta-hemolytic Streptococcus, Haemophilus influenzae, Yersinia), viral induced (Rotavirus, Parainfluenzae, ECHO, Herpes simplex).
We did not register any deaths. All the monitored cases evolved favorably under intravenous treatment with human immunoglobulins. A percent of 11.1 of the cases were severe forms of the disease, with cardiac echocardiography proof of the coronaries affection.
Conclusions The infectious trigger is one of the most important factors that induce the autoimmune changes incriminated in the KD at the pediatric population.
ESID-0857 Regulatory T-Cells in Systemic Juvenile Idiopathic Arthritis
D. Mairiang 1, S. Vilaiyuk1, S. Soponkanaporn1, N. Apiwattanakul2
1Pediatrics Allergy/immunogy and rheumatology, Faculty of Medicine Ramathibodi Hospital Mahidol University, Bangkok, Thailand
2Pediatrics infectious disease, Faculty of Medicine Ramathibodi Hospital Mahidol University, Bangkok, Thailand
Background: The impairment of regulatory T-cell (Treg) function leads to the development of juvenile idiopathic arthritis (JIA). Currently, role of Tregs in SJIA is still unclear.
Objective: To determine Treg populations in SJIA patients.
Methods: This is a cross-sectional study. All SJIA patients who came in rheumatology clinic, Ramathibodi Hospital in August 2014 were enrolled. Patients were classified into three groups according to their disease course; polycyclic (recurrent episodes of active disease), monocyclic (active disease <12 months), and persistent (active disease >12 months). Active disease was patient who had fever and/or arthritis and/or elevation of acute phase reactants, whereas inactive disease was patient who had no symptom and normal laboratory results. Blood was drawn to measure CD4+, CD25+, FOXP3+ T cells (Treg) and CD4+ T cells by flow cytometry method.
Results: There were 10 SJIA patients with mean age of 8.9 ± 3.8 years. Patients in persistent group and polycyclic group had active disease, whereas patients in monocyclic group had inactive disease. The mean Treg populations (% of CD4+ T-lymphocytes) in polycyclic group (n=5), monocyclic group (n=3), and persistent group (n=2) were 0.22 ± 0.12, 0.06 ± 0.01, and 0.01 ± 0.001 respectively with statistical difference (p= 0.022).
Conclusions: The increment of Treg populations was found in patients with active disease. However, patients who had persistent active course had the lowest Treg populations when compared to polycyclic and monocyclic group. Thus T-reg and immune dysregulation may play the important roles in SJIA.
ESID-0858 Portulaca oleracea L. Extract and Isolated Compounds Inhibit IL-6-Induced Cellular Signaling and Ameliorate DSS-Induced Colitis in a Mouse Model
M. Rho 1, H. Oh1
1Eco-friendly Biomaterial Research Center, Korea Research Institute of Bioscience and Biotechnology, Jeongeup-si, Korea
IL-6 plays an inflammatory and regenerative role in inflammatory bowel disease (IBD). The levels of IL-6 in sera positively correlate with disease severity in IBD. It has reported that IL-6 receptor (IL-6R) monoclonal antibodies prevented the development of colitis in an acquired immunity-dependent T cell transfer colitis mouse model. In the present study, we investigated the effects of Portulaca oleracea L. extract and isolated compounds on IL-6-induced cellular signaling and experimental murine colitis. Hexane and ethyl acetate (EtOAc) extracts of Portulaca oleracea L. showed potent inhibitory effect on pSTAT3-inducible luciferase activity and STAT3 tyrosine phosphorylation induced by IL-6. Although various compounds were isolated from EtOAc extract, Kaempferol and apigenin exhibited the most strong inhibitory activity on IL-6 signaling from Janus kinases to the STAT3 tyrosine phosphorylation as well as STAT3-inducible luciferase activity. In agreement with the inhibitory activity on IL-6 signaling, EtOAc extract, kaempferol, and apigenin indeed showed protective effects against DSS-induced colitis including colon shortening, weight loss and histological damage, and decreased the production of the effector cytokines such as IFN-gamma, IL-4, and IL-17. Our data suggest that the inhibitor of IL-6/STAT3 activation can be an effective drug for IBD.
ESID-0859 Auto-Immune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED) Without Polyendocrinopathy and Candidiasis
L. Chernyshova 1, A. Bondarenko1, A. Gilfanova1, S. Sharapova2, I. Guryanova2
1Dept. of Pediatric Infectious Diseases and Clinical Immunology, National Medical Academy for Post-graduate Education, Kiev, Ukraine
2Research department, Belarusian Research Center for Pediatric Oncology Hematology and Immunology, Minsk, Belarus
We present clinical case of APECED associated with mutation in gene of autoimmune regulation (AIRE), which was diagnosed in absence of any of major criteria.
Hemorrhagic syndrome manifested in early neonatal period as nasal bleeding, at age 1 month cerebral hemorrhage (left hemisphere) with reversible hemiparesis. During first year depigmentation alternated with hyperpigmentation, total alopecia, xerosis, hyperkeratosis of palms and soles, anhidrotic ectodermal dysplasia was diagnosed. Anemia, dakriocystitis, bilateral madarosis, recurrent conjunctivitis, total caries, gingival submucosal abscess developed since one year of age, later accompanied by recurrent purulent sinusitis, otitis media and chronic active hepatitis. At 3.5 years microcephaly was revealed. Steatorrhea, 5-fold reduction in fecal elastase, positive antinuclear antibodies (1:80), periodic decrease in prothrombin index (70%), prolongation of activated partial thromboplastin (4-fold) and prothrombin (12-fold), irregularly hypocalcaemia and hyperphosphatemia were found in child. Such diagnoses as hemophilia, cystic fibrosis, glycogenosis, galactosemia, tyrosinemia, Bloch-Sulzberger syndrome, Rothman-Thomson syndrome, Sjogren's syndrome, Bayler disease, primary sclerosing cholangitis, hemochromatosis were excluded by appropriate laboratory and genetic studies. Routine immunological examination did not reveal any significant abnormalities. During year boy received immunoglobulin replacement therapy that did not affect infectious syndrome. Despite absence of major typical manifestations AIRE gene sequencing was performed with identification of homozygous misens-mutation ?. 834?®G. The child had never signs of mucocutaneous candidiasis, adrenal hormones were within normal ranges. After obtaining genetic data minimal decrease in parathormone and intermittent violation in calcium phosphorus metabolism were found.
Molecular analysis of AIRE is critical for diagnosis, especially in cases of atypical manifestations of APECED.
ESID-0860 Association of Common Nucleotide-Binding Oligomerization Domain 2 (NOD2) Polymorphisms with the Clinical Phenotype of Antibody Deficiencies
A. Michopoulos1, A. Papadopoulou-Alataki2, E. Farmaki3, A.E. Germenis1, M. Speletas 1
1Department of Immunology & Histocompatibility, University Of Thessaly School of Health Sciences Faculty of Medicine, LARISSA, Greece
2D' Pediatric Clinic, Aristotle University of Thessaloniki Papageorgiou Hospital, Thessaloniki, Greece
3B' Pediatric Clinic, Aristotle University of Thessaloniki Ippokrateion Hospital, Thessaloniki, Greece
Introduction: The intracellular pattern recognition receptor NOD2 plays an important role in innate immunity as sensor of bacterial peptidoglycans. NOD2 polymorphisms have been associated with several chronic inflammatory and granulomatous conditions, especially Crohn's disease. Recently, a correlation of NOD2 polymorphisms with autoimmunity and enteropathy in CVID patients has been reported. Our aim was to explore the contribution of NOD2 polymorphisms in the clinical phenotype of antibody deficiencies, including CVID.
Methods: Nighty-four patients (34 with CVID, 38 with IgA deficiency [IgAD], 18 with selective IgG subclass deficiencies, and 4 with transient hypogammaglobulinemia of infancy) were enrolled in the study. Data regarding their clinical manifestations (location/frequency of infections, autoimmune manifestations, granulomas development or enteropathy) were recorded. The NOD2-Arg702Trp, Gly908Arg, Leu1007fs polymorphisms were detected by PCR-RFLP and sequencing.
Results: Frequencies of NOD2 polymorphisms were similar with those previously reported in a healthy Greek population, but were less frequent than other European populations. Particularly, 4 patients (2 with CVID and 2 twins with IgAD) were heterozygotes for the Gly908Arg polymorphism, an asymptomatic IgG4D patient was heterozygous for the Arg702Trp and no patient carried the Leu1007fs (allele frequencies 0.021, 0.005 and 0.000, respectively). Both CVID patients carrying the Gly908Arg polymorphism displayed lymphadenopathy (p=0.048) and enteropathy (p=0.009), while one of them (carrying additionally the TACI-C104R mutation) displayed also autoimmune cytopenias. No patient with any NOD2 polymorphism displayed granulomatous disease.
Conclusions: NOD2 polymorphisms may affect the clinical phenotype of patients with antibody deficiencies, especially in CVID, since they are associated with an increased risk of enteropathy.
ESID-0861 Association of a Sting Gain of Function Mutation with a Familial Systemic Inflammatory and Autoimmune Syndrome.
N. Jeremiah1, B. Neven2, M. Gentili3, I. Callebaut4, Y. Crow1, N. Manel3, A. Fischer2, B. Bader-Meunier2, F. Rieux-Laucat 1
1INSERM U1163, Institut Imagine, PARIS, France
2INSERM U1163, Institut Imagine and UIHR Necker, PARIS, France
3INSERM U932 Paris France, Institut Curie, PARIS, France
4UMR CNRS 7590 UPMC Sorbonne Paris Cité ., IMPMC Sorbonne University, PARIS, France
We describe a dominant gain of function mutation in TMEM173, encoding STING (Stimulator of type-I Interferon Gene) - a key signaling molecule in cytosolic DNA sensing pathways, segregating as a dominant trait in a non-consanguineous Caucasian family. The four affected individuals demonstrate a systemic inflammatory and autoimmune condition with variable clinical expression. STING activation normally requires dimerization induced by 2'3'-cyclic GMP-AMP (cGAMP) produced by the cyclic GMP-AMP synthase (cGAS) following binding to cytosolic DNA. Structural modeling supports constitutive activation of the mutant protein based on stabilized dimerization. In agreement with this, we found that the STING mutant spontaneously localizes in the Golgi of patient fibroblasts, and is constitutively active in the absence of exogenous 2'3'-cGAMP in vitro. Accordingly, we observed elevated serum interferon activity and a type-I interferon signature in peripheral blood. These findings highlight the key role of STING in regulating inflammation and autoimmunity in humans.
ESID-0862 The Therapeutic Role of Bone Marrow-Derived Stem Cells on Some Immunological Parameters of Murine Schistosoma Mansoni
W. Mansour 1, M. El-Mahdi2, O. Hammam3, T. Hussein4
1Immunology, Theodor Bilharz Research Institute (TBRI), Giza, Egypt
2Parasitology, Faculty of Science Cairo University, Giza, Egypt
3Pathology, Theodor Bilharz Research Institute (TBRI), Giza, Egypt
4Immunology, Faculty of Science Cairo University, Giza, Egypt
The present study was designed to evaluate the effect of bone marrow-derived stem cells on improvement of the Schistosoma mansoni-induced liver fibrosis in murine model. Animals were divided into four groups: uninfected negative control, S. mansoni infected, S. mansoni infected mice subjected to BMSCs infusion and S. mansoni infected mice subjected to BMSCs infusion in combination with HGF. Animals were sacrified after the 1st, 2nd and 3rd month post treatment. Liver histopathology and immuno-histochemistry investigations were performed. Moreover, OV-6 cells were detected for identification of newly formed hepatocytes. Cytokines levels (IL-2, IL-10, TNF-α and IFN-γ) that correlates positively with tissue fibrosis as well as nitric oxide level were assessed. Granuloma showed a marked decrease in size and number in BMSCs treated mice compared to the untreated Schistosoma infected group. Treatment of S. mansoni infected mice with BMSCs and BMSCs-HGF induced significant decreases in the level of IL-2 only at the 3rd month post treatment (PT) compared to its level in the infected control group, whereas the level of IL-10 in the treated groups showed significant increase at the 1st and 2nd month PT compared to that of the infected control group. Reduction in level of IFN-γ as well as TNF-α in comparison to the infected controls was detected in both BMSCs and BMSCs-HGF treated groups. A significant decrease in NO level was detected.
In conclusion, BMSCs and BMSCs-HGF infusion ameliorates Schistosoma mansoni induced liver fibrosis through the decrease in granuloma size and number and improvement in NO & cytokines levels.
ESID-0863 A Need for Revised Clinical Warning Signs for PID?
P. Wågström 1, M. Bengnér1, C. Dahle2, A. Nilsdotter-Augustinsson3, T. Neumark4, L. Brudin5, J. Björkander6
1Department of Infectious Diseases, Ryhov County Hospital Ryhov, Jönköping, Sweden
2Department of Clinical and Experimental Medicine, Division of Clinical Immunology, Linköping, Sweden
3Department of Clinical and Experimental Medicine, Division of Infectious Diseases, Linköping, Sweden
4County of Kalmar, Primary Health Centre in Lindsdal, Kalmar, Sweden
5Kalmar County Hospital, Department of Clinical Physiology, Kalmar, Sweden
6Ryhov County Hospital Ryhov, Department of Internal Medicine, Jönköping, Sweden
Background: Clinical warning signs with high accuracy could be of help to find individuals with primary immune deficiency (PID). When early diagnosed and treated, long-term complications due to repeated infections can be reduced. We have evaluated a commonly used warning sign for PID, “four or more antibiotic treated respiratory tract infections (RTIs) annually for three or more consecutive years”, in a primary health care setting where RTI:s are the most common indication for consulting and obtaining an antibiotic prescription.
Methods: After ethical approval of the study and written consent, 53 cases with “four or more antibiotic-treated RTIs annually for three or more consecutive years” from a Swedish primary health care registry were compared with 66 age and sex-matched control subjects who had a maximum of one antibiotic-treated RTI during the study period. Levels of IgG, IgA, IgM, IgG-subclasses, IgG antibodies against Haemophilus influenzae and Streptococcus pneumoniae were determined.
Results: IgG subclass deficiencies (IgGsd) were found in 5/53 (9.4%) cases and in 7/66 (10.6%) controls (p=0.99). The most frequent deficiency was IgG3sd with a lower mean level in the control group (p=0.02). The RTI-related warning sign had a specificity of 42% and a sensitivity of 55% to detect IgGsd. The positive predictive value was 9% and the negative predictive value was 89%.
Conclusion: This study showed that repeated RTIs did not predict an increased risk for PID in primary health care and that physicians should not rely solely on the frequency of antibiotic-treated RTIs as a predictor for humoral immune deficiencies.
ESID-0865 Chromosomal Aberrations and Immunodeficiency: First Results of the PID-CS Survey Study
E. de Vries 1, M.J. Browning2, J.F. Neves3, L. Alsina4, P. Soler-Palacin5, D.C. Vinh6, A.S. Grumach7, M.G. Seidel8, T.G. Boyce9, F. Celmeli10, E. Goudouris11, G.R. Hayman12, R.M.J. Herriot13
1Pediatrics & Tranzo, Jeroen Bosch Hospital & Tilburg University, 's-Hertogenbosch & Tilburg, Netherlands
2Pediatrics, University Hospitals of Leicester NHS Trust, Leicester, United Kingdom
3Pediatrics, Hospital Dona Estefania Centro Hospitalar de Lisboa Central CEDOC, Lisbon, Portugal
4Allergy and Clinical Immunology, Hospital Sant Joan de Deu, Barcelona, Spain
5Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d'Hebron, Barcelona, Spain
6Division of Infectious Diseases Infectious Disease Susceptibility Program, McGill University Health Centre, Montreal, Canada
7Pediatrics & Dermatology, Faculty of Medicine ABC, São Paulo, Brazil
8Pediatric Hematology-Oncology, Medical University Graz, Graz, Austria
9Mayo Clinic, Mayo Clinic, Rochester, USA
10Pediatric Immunology and Allergy, Antalya Education and Research Hospital, Antalya, Turkey
11Department of Pediatrics Medicine School, IPPMG/Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, Brazil
12Immunology, Epsom & St Helier University Hospitals NHS Trust, Epsom & Carshalton, United Kingdom
13NHS Grampian, Aberdeen Royal Infirmary, Aberdeen, United Kingdom
Introduction: Patients with chromosomal aberrations often experience respiratory infections, generally ascribed to aspiration. However, chromosomal aberrations can be associated with immunodeficiency; QoL can be improved by appropriate treatment.
Methods: All ESID members were invited to share their cases in the (ongoing) PID-CS survey study.
Results: Until 1-Sep-2014, 28 cases (17male; 1-69[median9]yrs) were reported by 13 centres (1-5cases/centre). Reports concerned 15 different chromosomes (X 6x; chr18 5x; chr16 4x; chr19&11 3x, chr6 2x, chr2,7,8,9,10,12,13,14,22 1x). Seventeen predominantly presented with recurrent ENT/airway infections, 4 with failure-to-thrive, 4 with unusual/unusually severe infections, 3 with auto-immunity; only 3 did not at all suffer from recurrent ENT/airway infections. All but 4 showed developmental delay, all but 5 dysmorphic features. None had granulocytopenia, 4 lymphocytopenia. IgG-A-M were decreased in 3, IgG-A in 6, IgG-M in 3, IgA-M in 1, IgG-only in 3, IgA-only in 6; 6 had normal Ig-isotypes. IgG-subclasses were determined in 14, and abnormal in 9 (7 with normal IgG). Absolute counts of NK-cells, B- and T-lymphocytes were below age-related normal values in 4, 7 and 5 (out of 20), respectively, with decreased CD3/CD4+ in 2 and CD3/CD8+ in 5. Granulocyte and lymphocyte function tests were slightly decreased in 2/10 and 0/9 cases, respectively. Pneumococcal and tetanus vaccination responses were decreased in 7/14 and 2/11 cases.
Discussion&conclusion The PID-CS survey study shows a wide variety of chromosomal aberrations can be associated with immunodeficiency. Grouping these rare cases together (as in the ESID platform) will bring more information on their diagnosis and management.
ESID-0866 Excellent Outcome for Adults and Older Adolsecents Following Reduced Intensity Allogeneic Haematopoietic Stem Cell Transplantation for Inherited Primary Immune Deficiencies (PID).
E. Morris 1, R. Hough2, S. Grace3, A. Worth4, S. Workman5, K. Thomson6, K. Peggs6, R. Chee5, V. Grandage2, K. Roberts6, M. Collin7, A. Fielding3, S. Mackinnon3, C. Murray8, A. Thrasher4, H. Gaspar4, S. Denovan3, M. Campbell5, A. Symes5, R. Chakraverty1, S. Burns1
1Immunology, UCL Institute of Immunity and Transplantation, London, United Kingdom
2Haematology (TYA), University College London Hospital, London, United Kingdom
3Haematology, Royal Free London Hospital, London, United Kingdom
4Immunology, Great Ormond Street Hospital For sick Children, London, United Kingdom
5Immunology, Royal Free London Hospital, London, United Kingdom
6Haematology, University College London Hospital, London, United Kingdom
7Haematology, Royal Victoria Hospital, Newcastle, United Kingdom
8Gastroenterology, Royal Free London Hospital, London, United Kingdom
Introduction: Until recently Allo HSCT in adults with PID has been avoided due to unacceptably high transplant related mortalities. This study reports the outcome of 26 consecutive patients treated with (RI) Allo HSCT for PID at a single centre.
Methods: A total of 26 patients (21 males, 5 females), with a median age of 22 years (range 14-34) were transplanted. Nineteen were ≥18 years at transplant. Underlying diagnoses were X-linked CGD (n=12), CVID (n=3), JIA (n=2), Autoimmune LPD (one with Fas mutation) (n=2), variant CGD with early onset severe Crohn's disease (n=1), DCML Deficiency (one with Gata2 mutation) (n=2), Common gamma chain SCID (n=1), Undefined SCID (n=1), NK deficiency with IgA subclass deficiency and seronegative arthritis (n=1), and IL12 receptor beta deficiency with recurrent salmonella sepsis (n=1). Most patients received stems cells from matched or mismatched unrelated donors (MUD n=11, 1Ag MMUD n=5), 9 from matched sibling donors (Sib n=9) and one a 10/10 paternal donor. Stem cell source was BM (n=8) or mobilized PBSC (n=18). RI conditioning regimens were Fludarabine, Melphalan, Alemtuzumab (n=16), Fludarabine, Bulsulphan, Alemtuzumab (n=6) or Fludarabine, Busulphan, ATG (n=4).
Results: 22 patients are alive with a median follow up of 28 months (range: 7d to 9yrs 6m). The estimated overall survival is 79% (95% CI: 57-99%). Cause of death was sepsis with multiorgan failure (n=2), pulmonary toxoplasmosis (n=1) and severe refractory chronic extensive GVHD (n=1). Lineage specific chimerism and immune reconstitution studies have been analysed on all patients.
ESID-0867 Highly Efficient Lentivirus-Based Vector for Gene Delivery in Immunodeficiency Purposes
A. Karimi 1, N. Masoud1, B. Pourghaysari1, H. Shirzad1
1Medicinal Plant research centere, Shahrekord Medical University of Science, Shahrekord, Iran
Background and aims: Viral vectors are valuable tools to deliver genetic materials into cells. Vectors derived from human immunodeficiency virus type 1 are being widely used for gene delivery mainly because they are able to transduce both dividing and non-dividing cells which leads to stable and long term gene expression. Therefore, this work was aimed to produce lentivirus-based vector for using in gene delivery purposes related to immunodeficiency.
Materials and methods: To produce this system, the eGFP marker and the required plasmids include eGFP marker and the vector plasmids were co-transfected into packaging cell line (293T). The produced vector was harvested, purified, titrated, and transduced into the HEK-293T, CHO, HepG2, MCF-7, MEFs and Jurkat cell lines.
Results: Lentivirus vector at titers of up to 2 9 108 transducing units/ml (TU/ml) was successfully generated and its transduction efficacy was improved by seven to over 20-fold in various cell types. We demonstrated the applicability of this vector for the efficient transduction of dividing and nondividing cells including HEK-293T, CHO, HepG2, MCF-7, MEFs and Jurkat cell line. Transduction efficiency yielded titers of (6.3 ± 1.2) 105 TU/ml. Furthermore, lentivirus transferred transgene was expressed at high level in the target cells and expression was followed until 90 days after transduction.
Conclusion: Vector generated in this work, might be able to deliver the transgene into a wide range of mammalian cells.
ESID-0868 Granulocyte-Specific Inherited Human NRAMP1 Deficiency: Impaired Respiratory Burst and Pyogenic Infectious Diseases
J. Bustamante 1, M. Hubeau2, G. Vogt2, C. Deswarte2, L. Abel2, M. Cellier3, C. Picard4, J. Casanova5
1CEDI, AP HP Hôpital Necker Enfants Malades, PARIS Cedex 15, France
2INSERM 1163, . Laboratory of Human Genetics of Infectious Diseases, PARIS Cedex 15, France
3INRS, Institut Armand-Frappier, Laval, Canada
4CEDI, AP-HP Hôpital Necker-Enfants Malades, PARIS Cedex 15, France
5St. Gilles Laboratory of Human Genetics of Infectious Diseases, The Rochefeller University, New York, USA
The human genetic determinism of pyogenic infections of childhood remains largely unknown. We report the study of a patient with recurrent upper respiratory tract infections and lymphadenitis due to pyogenic bacteria. As detected by genome-wide linkage and whole exome sequencing, the patient carries a bi-allelic mutation in NRAMP1, V484M, that segregates with disease as an autosomal recessive trait. This missense mutation however impairs NRAMP1 protein expression in granulocytes but not in monocytes and monocyte-derived macrophages. The lack of NRAMP1 in granulocytes impairs both the NADPH oxidase activity and the in vitro killing of Staphylococcus aureus. No functional anomalies were detected in monocytes and monocyte-derived macrophages. Overall, we identified a patient with a granulocyte-specific form of inherited NRAMP1 deficiency, which impairs NADPH oxidase activity and results in pyogenic bacterial infections. These findings establish an essential role for human NRAMP1 in granulocyte NADPH oxidase activity and protective immunity against pyogenic bacteria. They further suggest that staphylococcal disease in patients with chronic granulomatous diseases involves a respiratory defect in granulocytes.
ESID-0869 Mucosal-Associated Invariant T Cells in Primary and Secondary Immunodeficiency Patients
Y. Gao 1, K.A. Ramakrishnan1, G. Barcenas-Morales2, R. Döffinger3, R. Pengelly4, S. Ennis4, S.N. Faust5, C. Ottensmeier6, A. Williams7
1Cancer Science Unit, University of Southampton, Southampton, United Kingdom
2Department of Clinical Immunology, Addenbrookes Hospital, Cambridge, United Kingdom
3Department of Clinical Immunology, Addenbrookes Hospital, cambridge, United Kingdom
4Human Genetics and Genomic Medicine, university of Southampton, Southampton, United Kingdom
5Academic Unit of Clinical and Experimental Sciences, university of Southampton, Southampton, United Kingdom
6Cancer Science Unit, university of Southampton, Southampton, United Kingdom
7Department of Clinical Immunology, university of Southampton, Southampton, United Kingdom
Primary immunodeficiency diseases (PID) represent over 150 different disorders. An understanding of such conditions has provided important insights into the non-redundant immune pathways that are necessary for control of divergent pathogens and immune interventions. Mucosal- associated invariant T cells (MAITs) are an example of innate T cells that act as ‘interface immunity’ T cell compartment, possessing an invariant T cell receptor and broad effector response. We developed a flow cytometry panel to phenotype MAIT cells using the invariant V alpha 7.2 receptor antibody (3C10), CD8 beta, CD8 alpha and CD161. Their functional properties were evaluated by intracellular cytokine staining (IL-17 and interferon-gamma) following mitogen activation. MAIT cells were found to be absent in several primary and secondary immune deficiency disorders, typically with frequencies of less than 0.3% of T cells compared to >1% in healthy controls. Functional analysis of MAIT cytokine production demonstrated absent IL-17 production in PID that were typically associated with a susceptibility to fungal infection (IL-17 producing MAIT <0.1% compare to 3% in healthy individual). An interesting group of Thymoma patients with cytokine autoantibodies were studied. MAIT cells were found to be absent in all patients with IL-12/23 autoantibodies (<0.1% compared to 2% in healthy controls). The analysis of MAIT cells in PID suggests that these are an important cell type for the prevention of mucosal infection. The recent identification of their specific ligand provides further opportunity to evaluate and target this arm of immunity in future vaccination strategies to prevent fungal and bacterial disease.
ESID-0870 First Human Combined Immunodeficiency Associated with Dysfunctional CD28 Costimulation in T Lymphocytes.
Y. Wang1, Y. Ling1, L. Lorenzo1, A. Belkadi1, S. Jacquot2, A.A. Bousfiha3, Y. Camcioglu4, F. Ailal3, A. Puel1, C. Picard5, S. Boisson-Dupuis6, L. Abel1, J.L. Casanova6, V. Béziat 1, E. Jouanguy1
1INSERM U1163, Imagine, PARIS, France
2Service d'immunopathologie, CHU CH Nicolle, Rouen, France
3Clinical Immunology unit Casablanca children's hospital Ibn Rochd Medical School, King Hassan II University, Casablanca, Morocco
4Cerrahpasa Medical School, Istanbul University, Istanbul, Turkey
5Study Center for Primary Immunodeficiencies, Necker-Enfants malades Hospital, Paris, France
6St. Giles Laboratory of Human Genetics of Infectious Diseases, The Rockefeller University, New York, USA
T cell immunodeficiency results in SCID or CID phenotypes depending on the severity of the impairment of T cell number and/or function. Combining Genome Wide Linkage and Whole Exome Sequencing strategies, we report the discovery of autosomal recessive mutations of a gene in 4 young adults from 3 independent families originating from Morocco, Tunisia and Turkey. The genetic variants consisted in two distinct homozygous missense mutations in two boys and one premature homozygous stop codon in two sisters. The four patients presented with strikingly similar inflammatory skin phenotype and a wide spectrum of severe infections including tuberculosis (n=2), skin abscess (staphylococcus aureus; n=2) and a suspected viral encephalitis (n=1). Other noticeable clinical phenotypes included recurrent otitis (n=2) and bronchiectasis (n=3). Variants expression in 293T cells revealed that the alleles carrying the two missense mutations were normally expressed while the nonsense mutation resulted in a truncated protein. It was previously demonstrated in mice that the affected gene plays a crucial role in the CD28 costimulation pathway. The main immunological feature of these patients was a drastic reduction of CD4+ memory T cell number while the CD8+ T cell compartment was seemingly normal. Interestingly, we showed that patient’s CD4+ T cells responded normally to CD3 stimulation but did not respond to costimulation in presence of anti-CD28 antibodies, suggesting that all mutations were deleterious. In conclusion, to our knowledge, it is the first report of a human T cell primary immunodeficiency resulting of defects in the CD28 costimulation pathway.
ESID-0871 Validation of the QuantiFERON-CMV Assay for Monitoring of Cytomegalovirus-Specific CD8+ T-Cell Reconstitution in Pediatric Patients After Allogeneic Hematopoietic Stem Cell Transplantation
A. Soldatou 1, B. Paouri1, A. Paisiou2, G. Avgerinou2, K. Zisaki2, S. Graphakos2, E. Goussetis2
1Second Department of Pediatrics University of Athens Medical School, P. & A. Kyriakou Children?s Hospital, ATHENS, Greece
2Stem Cell Transplant Unit, Aghia Sophia Children’s Hospital, ATHENS, Greece
Background: Reactivation of cytomegalovirus (CMV) is a major cause of morbidity and mortality after allogeneic hematopoietic stem cell transplantation (HSCT). Monitoring CMV-specific CD8+ T-cell immunity is useful for predicting CMV infection and directing targeted antiviral therapy. However, conventional flow cytometry based assays are relatively complex to implement in a routine clinical setting. Objective: To evaluate the clinical utility of a new whole blood interferon-γ assay, known as QuantiFERON-CMV. Population and Methods: CMV-specific CD8+ T-cell reconstitution in a cohort of 37 pediatric allogeneic HSCT recipients at 30, 90, 180, 270, and 360 days after allogeneic hematopoietic stem cell transplantation (HSCT) was analyzed prospectively. HSCT recipients were classified as high risk (Donor (-) / Recipient (+)) (n =12), intermediate risk (D(+)/R(+) and D(+)/R(-)) (n =18), and low risk (D(-)/R(-)) (n =7). Results: The cumulative incidence of CMV reactivation was 57% for the high and intermediate risk patients with evidence of CMV-disease in 2 patients only. Fifteen patients showed stable CMV-specific immune reconstitution at average time of 82 days. The cumulative incidence of CMV infection in patients after they developed CMV-specific immunity was lower than those who did not (15% versus 53.3%; p=0.023). Conclusion: These results demonstrate that the QuantiFERON-CMV provides an accurate evaluation of CMV specific CD8+ T-cell immunity and may be useful in identifying HSCT recipients at risk of CMV-infection and determining more specific CMV monitoring by PCR and timing of therapy.
ESID-0872 Study of Anti-HCMV T Cell Responses and CXCL9 Production of Patients After Transplantation of Hematopoetic Stem Cells
S. Nemeckova 1, K. Babiarova1, P. Hainz1, J. Krystofova1, J. Mackova1, V. Sroller1, M. Stastna2
1Department of Experimental Virology, Institute of Hematology and Blood Transfusion, Prague 2, Czech Republic
2Transplantation Ward, Institute of Hematology and Blood Transfusion, Prague 2, Czech Republic
AIMS: Human cytomegalovirus (HCMV) reactivation in immunosuppressed patients is associated with significant morbidity and mortality. The testing HCMV specific T cell response can help to find the patients which are in a high risk of HCMV disease. Such patients would profit from adoptive transfer of HCMV specific T cells. Cellular immune response against HCMV of 26 patients after allogeneic SCT was monitored for one year by ELISPOT-interferon gamma against HCMV antigens with the aim to find patients with insufficient control of HCMV reactivation. The recipients of SCT were classified according to the anti-HCMV T cell response, the presence of HCMV DNA in the blood, GVHD incidence and CXCL9 (MIG) concentration in plasma.
METHODS: Cell mediated immunity was determined by ELISPOT-interferon gamma using the 15mer peptide pools of pp65, IE-1, US3, UL36, US29, UL55 antigens or lysate of fibroblasts infected with the CMV AD169 virus. CXCL9 concentration in plasma was measured by ELISA.
RESULTS: (i) The antigens US3, gB (UL55) and UL36 can be favourably included in test of T cell response against HCMV. (ii) Monitoring of dynamics of the response can improve prediction of HCMV reactivation. (iii) The T cell response against CMV lysate is of major importance for protection, however it is missing in 70% recipients. (iv) Patients with GVHD who had obtained the graft from the seronegative donor have an increased risk of HCMV reactivation. (v) Patients with GVHD have increased levels of CXCL9.
Supported by NT/13898/2012 grant from IGA MZ CR.
ESID-0873 Extrapulmonary Non-Tuberculous Mycobacterial Infection in a Child with Autosomal Dominant Hyper IgE Syndrome
P. Gray 1, C. Walsh2, Y. Zhu2, G. Elakis2, G. Mullan2, W. Lo2, E. Lee2, M. Cowley3, M. Dinger3, M. Buckley2, J. Ziegler1, S. Tangye4, T. Roscioli5
1Immunology and Infectious diseases, Sydney Children's Hospital, Sydney, Australia
2SEALS Genetics Laboratory, Prince of Wales Hospital, Sydney, Australia
3The Kinghorn Centre for Clinical Genomics, Garvan Institute of Medical Research, Sydney, Australia
4Immunology Program, Garvan Institute of Medical Research, Sydney, Australia
5School of Women's and Children's Health, University of New South Wales, Sydney, Australia
Background: With the emergence of mass genetic sequencing technologies, the phenotypes of many Primary immunodeficiency diseases are being extended. Heterozygous mutations in STAT3 underlie Autosomal Dominant Hyper IgE (AD-HIGE) syndrome, and more recently have been associated with early onset autoimmunity. However, while non-tuberculous mycobacteria have been found in the lungs of AD-HIGE patients with bronchiectasis, an immune predisposition to extrapulmonary mycobacterial infection is not reported.
Case report: We report an 11 year old girl who presented in early life with a pustular scalp rash and went on to have episodes of pneumonia and a localised skin rash on her arm. At age 3 she developed an eyelid mass which was refractory to conventional antibiotics and which extended to involve the extra-conal intra-orbital structures. Surgical biopsies identified granulomatous inflammation and Mycobacterium Fortuitum Complex was identified by culture of tissues. Excision and treatment with macrolide antibiotics resulted in full resolution of the episode. Based on the mycobacterial infection and functional testing she was treated for a possible interferon gamma pathway defect. By age 11 when she attended our clinic she had a high IgE and delayed secondary dentition. Whole exome sequencing demonstrated a previously reported 1909G>A, Val637Met mutation in STAT3, which was confirmed as a de Novo variant.
Conclusion: This is the first report of extra-pulmonary non-tuberculous mycobacterial infection occurring in AD STAT3 deficiency. It demonstrates the importance of mass sequencing technologies for extending the phenotypes of well-described genetic diseases.
ESID-0874 First Treatment with Rituximab for Granulomatous-Lymphocytic Interstitial Lung Disease (GLILD) in X-Linked Lymphoproliferative Syndrome (XLP) Type 2.
M.P. Dore 1, C. Steele1, N. Conlon1, L. Devlin1, H. Shendi1, B. Gasper2, D. Downey3, C. Speckmann4, S.O. Burns5, J.D.M. Edgar1
1Regional Immunology Dept, The Royal Victoria Hospitals Belfast, Belfast, United Kingdom
2Paediatric Immunology, Great Ormond Street Hospital, London, United Kingdom
3Regional Respiratoy Centre, Belfast City Hospital, Belfast, United Kingdom
4Centrum fuer Chronische Immundefizienz, Universitaet Freiburg, Freiburg, Germany
5Department of Immunology Royal Free London NHS Foundation, University College London Institute of Immunity and Transplantation, London, United Kingdom
Introduction: This case illustrates diagnostic and therapeutic challenges in a 22 year old male. Since aged 4 he has presented with recurrent non-infective febrile episodes associated with lymphoproliferation and splenomegaly. Investigations reveal an associated inflammatory response, cytopenias, a pan-hypogammaglobinemia and a novel XIAP (X-linked inhibitor of apoptosis protein) mutation (c.497G-A variant in Exon 2). Florid granulomatous disease with T and B cell infiltration was identified on lung and liver biopsy.
Case description: No infectious agents were identified with the exceptions at the age of 4 (Epstein-Barr virus (EBV)) and at 17 (Parvovirus B19). Investigations at 17 years old revealed a pan-cytopenia, ferritin 16,750ug/L, Triglycerides 3.01mmmol/L, LDH 2000U/L, and CRP 90.4mg/L. This and all subsequent episodes have responded well to high dose oral steroids (60mg initially) improving over 2-4 weeks. Immunoglobulin replacement therapy was commenced in 2011. His novel XIAP mutation is shared by two siblings who are phenotypically well (one was shown to be EBV IgG positive). Functional studies suggest reduced XIAP expression levels as well as reduced function. Prednisolone (60mg) and subsequently Sirolimus failed to improve his splenomegaly. Rituximab and Azathioprine were commenced in early 2014, with good effect, following the current treatment guidelines for GLILD in common variable immune deficiency (CVID).
Conclusion: This case highlights diagnostic and therapeutic uncertainty and probably outlines XLP2 phenotypic overlap with CVID with GLILD. It also suggests considerable genotype phenotype variability within the family. Despite the apparent response to Rituximab and Azathioprine, matched sibling bone marrow transplant is being considered as definitive treatment.
ESID-0875 Mutations in XRCC4 Associated with Microcephaly, Severe DNA Double-Strand Breakage Repair Defect, Normal VDJ Recombination, Increased Microhomology During Class Switch Recombination, But No Clinical Immunodeficiency
A. Gennery 1, A. Björkman2, T. Ogi3, A. Lehmann4, P.A. Jeggo4, Q. Pan-Hammarström2
1Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
2Clinical Immunology, Karolinska University Hospital, Stockholm, Sweden
3Research Centre for Genomic Instability and Carcinogenesis, Nagasaki University, Nagasaki, Japan
4Genome Damage and Stability Centre, University of Sussex, Brighton, United Kingdom
NHEJ defects, impairing DNA-DSB repair, associate with microcephaly, developmental delay and immunodeficiency due to NHEJ requirement in VDJ and class switch (CS) recombination. Defects are described in LIG4, NHEJ1, DCLRE1C, PRKDC. We describe a young adult with defective XRCC4.
The patient had heterozygous missense XRCC4 mutations (p.R225X - an inactivating mutational change causing NMD; p.D254fs68 - resulting in protein with the terminal 68 amino acids being distinct to those of WT) causing protein instability. Radio-sensitivity and DSB repair kinetics were examined using standard methods. VDJ recombination was examined using an in vitro NHEJ plasmid assay. CSR was examined by sequencing amplified Sμ–Sα and Sμ–Sγ junctions from genomic PBMC DNA.
The patient had microcephaly, diabetes mellitus, hypothyroidism, progeria-like features and developmental delay, but no infection history. Immunoglobulin levels, vaccine antigen antibody responses, lymphocyte sub-type numbers were normal, and autoantibodies negative. The mutations reduced XRCC4 (~1%) and DNA ligase IV (~20%) expression. Fibroblasts displayed increased frequency of direct joining at V(D)J recombination coding joints, and diminished microhomology-based end joining as well as deletions at junctions. A CSR assay revealed increased use of microhomology at the recombination junctions.
This XRCC4 defect clinically resembled Ligase 4/cernunnos-XLF defects, with microcephaly but no clinical immunodeficiency. Surprisingly, the cells show a marked DSB repair defect but a subtle impact on VDJ and CS recombination. The patient displayed marked neuronal abnormalities but normal immunological parameters suggesting that these XRCC4 mutations confer a separation of function impact, more marked on radiation- than RAG-induced DSB repair.
ESID-0876 Robust Calcium Flux and Unimpaired Effector Functions of Human Neutrophils with Store-Operated Calcium Entry (SOCE) Deficiency
R. Elling1, B. Keller1, C. Weidinger2, I. Zee2, C. Speckmann1, S. Ehl1, K. Schwarz3, S. Feske2, P. Henneke 1
1Center for Chronic Immunodeficiency, University Freiburg Medical Center, Freiburg, Germany
2Department of Pathology, New York University School of Medicine, New York, USA
3Institute for Transfusion Medicine and Immunogenetics German Red Cross Blood Service Baden-Wuerttemberg – Hessen, University Ulm, Ulm, Germany
Cytosolic calcium is a central second messenger molecule for many cell types and known to critically regulate the adaptive immune response. Mutations of the intracellular calcium sensor STIM1 or the plasma membrane calcium channel protein ORAI1 ablate store-operated calcium entry (SOCE) and result in a complex syndrome consisting of severe immunodeficiency, immune dysregulation and various non-hematological symptoms. Data in mice and human cell lines indicate that store-operated calcium entry (SOCE) is also important for the functions of human polymorphonuclear neutrophils (PMN). Here, we analyzed calcium flux patterns and effector functions of PMN derived from a STIM1-deficient and an ORAI1-deficient patient. Unexpectedly, we did not observe defects in calcium signaling in PMN after agonist-induced stimulation with TLR ligands and only partial defects in SOCE after Ionomycin and Thapsigargin stimulation. Moreover, analysis of effector functions did not reveal defects in SOCE-deficient primary human neutrophils. These findings strongly suggest that non-SOCE mechanisms are the chief mediators of calcium signals in human PMN. As a consequence, our data do not support SOCE proteins as promising drug targets to control neutrophil-mediated inflammation in humans.
ESID-0877 Homozygous Mucosa-Associated Lymphoid Tissue 1 (MALT1) Mutation in Two Siblings with Immunodeficiency and Chronic Enteropathy
A. Koren Jeverica1, F. Charbit-Henrion2, B. Begue2, P. Nitschke3, C. Bole4, G. Markelj 1, M. Homan5, F. Reummele6, N. Cerf-Bensussan2, T. Avcin1, A. Ihan1
1Dpt. for Allergy Rheumatology and Clinical Immunology, University Children's Hospital Ljubljana, Ljubljana, Slovenia
2Laboratory of Intestinal Immunity, Universite Paris-Descartes-Paris Sorbonne Centre and Institut Imagine, Paris, France
3Bioinformatics platform, Universite Paris-Descartes-Paris Sorbonne Centre and Institut Imagine, Paris, France
4Genomic platform Institut Imagine, Universite Paris-Descartes-Paris Sorbonne Centre and Institut Imagine, Paris, France
5Dpt. for Gastroenterology Hepatology and Nutricionistics, University Children's Hospital Ljubljana, Ljubljana, Slovenia
6Dpt. of Pediatric Gastroenterology, Hopital Necker, Paris, France
Background and objectives Whole exome sequencing was applied to identify the molecular defect causing combined immunodeficiency and severe chronic enteropathy in two siblings from consaguineous parents.
Observation and results The girl (6 year-old) and her brother (4 year-old) displayed severe dermatitis since birth and progressively developed facial dysmorphy, a wide spectrum of viral, fungal and bacterial infections and severe chronic inflammatory enteropathy. Infections were most severe in the girl with notably repeated HSV-1 keratitis, EBV gut dissemination, and life-threatening pulmonary infections with adenovirus, Pneumocystis jirovecii and EBV. Immunological parameters were high IgE, low IgM, normal B cell counts but variable antibody titers to vaccination, elevated counts of activated/memory T lymphocytes, but reduced frequencies of Th1, Th2, Th17 and Treg. Whole exome sequencing identified a single autosomal recessive nucleotide variation (c.550G>T) in exon 4 of MALT1 predicting a deleterious p.Asp184Tyr amino acid change in the first immunoglobulin domain of MALT-1. MRNA expression was not affected but MALT-1 protein was undetectable in PHA-blasts from both children. In agreement with the indispensable role of MALT-1 in the NFkB cascade downstream the T cell receptor, peripheral blood lymphocytes failed to proliferate to anti-CD3/28 stimulation. Degradation of IkB and production of IL-2 were abolished in PHA-blasts activated by PMA and ionomycin.
Conclusion We describe the third case of loss-of-function mutation in the gene encoding MALT1, a component of the CARMA1-BCL10-MALT1 signalosome complex as a cause of combined immunodeficiency and chronic enteropathy. We report for the first time EBV and Pneumocystis jirovecii infections in this immunodeficiency.
ESID-0878 Frequency of Cytokine Producing CD8+ Tcells in Major Beta Thalassemia Patients in Shahr-E-Kord
A. Karimi 1, B. pourghasari1
1medicine, Shahrekord Medical University of Science, Shahrekord, Iran
Background: Infection, which may reflect the immune system incompetency, is the second cause of mortality in the major beta talasemia patients. There are some reports indicating immune suppression and earlier aging of immune system in these patients. Therefore, this study was aimed to evaluate earlier aging and suppression of immune system with measurement of intracellular cytokines.
Materials and methods: In this analytical-descriptive study, a total of 26 patients with major major thalasemia were studied and compared with the same number of healthy individuals with age and sex matched, as the control. In the both groups, the percentage of IL2 and INFγ producing TCD8+ cells were measured intracellulary using flowcytometry.
Results: Statistical analysis of the results showed that the percentage of IL2 producing TCD8+ cells in the patients were significantly lower than those of the healthy individuals (P≤0.05). Also, the proportion of IL2 to INF produced in the T cells, was significantly lower in the patients compared with the control (P=0.063).
Conclusion: Significantly reducing proportion of IL2 producing T cells and also reducing the proportion of the cytockines may indicate earlier aging of TCD8 cells in these patients
ESID-0879 Dissecting the Role of IgD in Naive B-Cell Homeostasis and Antibody Responses in Individuals with Heterozygous IgD Deficiency
J. Nechvatalova 1, S.J.W. Bartol2, Z. Chovancova1, M. Vlkova1, J. Litzman1, M.C. van Zelm2
1Department of Clinical Immunology and Allergology, St Anne’s Faculty Hospital and Faculty of Medicine Masaryk University, Brno, Czech Republic
2Department of Immunology, Erasmus MC University Medical Center Rotterdam, Rotterdam, Netherlands
Surface immunoglobulin D (IgD) is co-expressed with IgM on naïve mature B cells. Although the role of IgM has been extensively studied, the role of surface IgD remains enigmatic. Recently, we identified four members of one family who carried a heterozygous germline mutation in the IGHD gene. Because these individuals contained both IgD+ and IgD- B cells, we judged this a unique setting to study the role of surface IgD in naïve B-cell homeostasis and antibody responses.
Flow cytometry was employed to study the blood B-cell compartments. Furthermore, IgD+ and IgD- transitional and naïve mature B cells were FACS-purified for replication history analysis using KRECs. IgG and IgA transcripts were sequenced to study molecular signs of antibody maturation.
All individuals had normal numbers of B cells, but about half of the naïve and the IgM+CD27+ memory B cells lacked IgD. These individuals carried heterozygous nonsense mutations in their germline DNA encoding exon 1 of IGHD. IgD+ and IgD- naïve mature B cells showed similar immunophenotypes. The replication histories of IgD- transitional and naïve mature B cells were similar to their IgD+ counterparts and those of controls. All individuals carried normal IgM, IgA and IgG levels, IgA and IgG transcripts were normally mutated.
Surface IgD seems to be redundant for naïve B-cell homeostasis and heterozygous loss of IgD does not impair humoral immunity. Currently, single cell molecular analyses are ongoing to study the role of IgD in central and peripheral tolerance, as well as in induction and selection of somatic hypermutations.
ESID-0880 Immunodeficiency and Severe Susceptibility to Bacteria Associated with a Loss-of-Function Homozygous Mutation of MKL1
J. Record1, D. Malinova1, H.L. Zenner2, V. Plagnol3, K. Nowak1, D. Moulding1, G. Bouma1, J. Curtis2, K. Gilmour4, S. Hackett5, S. Nejentsev2, A.J. Thrasher1, S.O. Burns 6
1Institute of Child Health, University College London, London, United Kingdom
2Department of Medicine, University of Cambridge, Cambridge, United Kingdom
3Genetics Institute, University College London, London, United Kingdom
4Department of Immunology, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
5Department of Paediatrics, Birmingham Heartland Hospital, Birmingham, United Kingdom
6Institute of Immunity and Transplantation, University College London, London, United Kingdom
Megakaroyblastic Leukemia 1 (MKL1) protein, also known as MAL or MRTF-A, is a transcriptional coactivator of the serum response factor (SRF). Through binding to monomeric G-actin, MKL1 monitors cytoskeletal dynamics and regulates transcription of actin and the actin cytoskeleton-related genes. Here we report a four year old girl with susceptibility to severe bacterial infection caused by a homozygous nonsense mutation in the MKL1 gene resulting in the loss of MKL1 protein expression. Her clinical presentation was in the first year of life with Pseudomonas septic shock associated with meningitis, otitis media, and florid cutaneous and subcutaneous abscesses. We show that the absence of MKL1 in the patient led to a dramatic decrease in F-actin content in lymphoid and myeloid lineage immune cells. Patient neutrophils displayed reduced phagocytosis and almost complete abrogation of migration. Silencing MKL1 in neutrophil-like HL-60 cell lines confirmed that the cellular phenotype resulted from the loss of MKL1 expression. MKL1 depletion in HL60-derived neutrophils was associated with an uropod retraction defect and down-regulation of MYL9 expression, suggesting that MKL1 regulates neutrophil cytoskeletal dynamics through the myosin II complex. Additionally, the F-actin cytoskeleton was disrupted in dendritic cells derived from patient monocytes and MKL1-silenced THP1 cell lines, evidenced by impaired spreading and loss of the F-actin rich podosomes. Our results show that MKL1 is a non-redundant regulator of cytoskeleton-associated functions in myeloid immune cells and that its depletion underlies a novel primary immunodeficiency.
ESID-0881 The Registry of Rare Diseases in Piedmont and Valle D'aosta, an Epidemiological and Healtcare Tool for Primitive Immunodeficiencies (PIDs)
S. Baldovino 1, S. Martino2, D. Montin2, U. Ramenghi3, G.B. Ferrero4, A.L. Spicuglia1, M.S. Galati1, F. Marletto1, K. Giancaspero1, V. Tomaselli5, P.P. Sainaghi6, A. Anania7, F. Avanzi8, M. Maspoli8, D. Roccatello9
1Department of Clinical and Biological Sciences - Turin University, Center of Research of Immunopathology and Rare Diseases, TORINO, Italy
2Division of Pediatric Immunology and Rheumatology, Department of Public Health and Pediatric - Turin University, TORINO, Italy
3Division of Pediatric Hematology, Department of Public Health and Pediatric - Turin University, TORINO, Italy
4Division of Human Genetic, Department of Public Health and Pediatric - Turin University, TORINO, Italy
5Division of Internal Medicine, Cardinal Massaia Hospital, ASTI, Italy
6Division of Internal Medicine, Maggiore della Carità Hospital, TORINO, Italy
7Division of ortho-rheumatology, Department of Medical Science - Turin University, TORINO, Italy
8Piedmont Region, Health Directorate, TORINO, Italy
9Center of Research of Immunopathology and Rare Diseases, Department of Public Health and Pediatric - Turin University, TORINO, Italy
Introduction: In Italy the attention to rare diseases (RDs) stems from the Ministerial Decree 279/2001. It established a national network for RDs, cost exemptions for related health service provisions for many RDs, and a National Registry of patients affected by many RDs, including PIDs. In Piedmont and Valle d’Aosta, two regions situated in northwest Italy, with a population of about 4,774,000, a network and a registry for RDs were set from 2004 [1].
Methods: The registry collects reported cases from all hospitals in the two regions. We analyzed data collected from 2005 to now. Our analysis covers PIDs, including chronic granulomatous disease and autoinflammatory syndromes, but excludes congenital C1 esterase deficiency. We have also estimated pharmaceutical costs collecting data from health accounting flows during a sample years (2012).
Results: During 9 years of activity we collected 258 cases of PID (249 of which are residents). We can estimate an overall prevalence of 5.22 per 100,000. Health care costs not related to hospitalization in 2012, amounted to 178,000 € (average cost / patient per year: 715 €). More than 50% of costs were due to different formulations of immunoglobulins.
Conclusions A registry that covers the whole population of a region, and that collects data from several healthcare centers is useful (1) to enable wide range epidemiological studies, (2) to promote proper healthcare planning, and (3) to assess the costs of PIDs.
[1] Baldovino S et al, Blood Transfus. 2014 Apr;12 Suppl 3:s617-20
ESID-0882 Development of a Shared Clinical Pathway for Taking Charge of Patients with Hereditary and Acquired C1 Estherase Inhibitor Deficiency Angioedema
G. Zanierato1, S. Baldovino 2, G. Rolla3, S. Martino4, P. Borrelli5, G. Guida6, D. Roccatello2
1Ospedale degli Infermi, Division of Allergology, BIELLA, Italy
2Department of Clinical and Biological Sciences - Turin University, Center of Research of Immunopathology and Rare Diseases, TORINO, Italy
3Department of Medical Sciences, Division of Allergology and Immunology - Mauriziano Hospital, TORINO, Italy
4Department of Public Health and Pediatric, Division of Pediatric Immunology and Rheumatology, TORINO, Italy
5Division of Internal Medicine, Parini Hospital, Aosta, Italy
6Division of Internal Medicine II, Birago di Vische Hospital, TORINO, Italy
Introduction: In Italy the attention to rare diseases (RDs) stems from the Ministerial Decree 279/2001. From 2004 Piedmont and Valle d’Aosta, two regions situated in northwest Italy, established a widespread network for rare diseases characterized by the involvement of the majority of health professionals.[1]. One of the peculiarity of the network of RDs in Piedmont and Aosta Valley is the presence of multidisciplinary working groups, called Consortia, dedicated to specific RDs or to RDs with similar therapeutic or diagnostic problems. Here we describe the consortium that is developing a clinical pathway for patients with angioedema from C1 esterase inhibitor deficiency.
Premises From 2005 to now the Piedmont and Valle d’Aosta registry of RDs collected 74 cases of congenital C1 esterase inhibitor deficiency and 16 cases of acquired C1 esterase inhibitor deficiency. The estimated prevalence of congenital form is 1.55 per 100,000 (only 2/3 of prevalence reported in literature)[2].
Aim: The aim of the consortium is to develop a clinical pathway that allows to (1) increase the diagnosis, (2) to improve the management of patients, especially in emergency rooms, by the ENT, the dental surgeons, the allergists, the dermatologists, the gastroenterologist etc. and (3) to optimize the use of therapies, to include plasma derivatives, biotechnological inhibitors, and anti bradykinin.
[1] Baldovino S et al, Blood Transfus. 2014 Apr;12 Suppl 3:s617-20
[2] Gompels MM et al, Clin Exp Immunol. 2005 March; 139(3): 379–394
INGID 2014 Oral Presentations
ESID-0237 Patients with Complex Medical Conditions: Is Home Therapy an Option?
F. Ashworth 1, D. hughes1, A.N.N.A. shrimpton1, M. York1
1clinical Immunology & Allergy Unit, Sheffield Teaching Hospitals, Sheffield, United Kingdom
This 59 year old female with secondary antibody deficiency and other multiple serious health issues was referred to the Clinical Immunology & Allergy unit to start immunoglobulin replacement therapy with a view to going on Home Therapy.
This case study will explore Mary’s (not her real name) journey to achieve her ultimate goal of self-infusing at home. She has a long list of medical conditions including:
Cerebral Vascular Accident leaving her with left hemiparesis.
Ischemic heart Disease Recurrent venous thrombo-embolic disease – on long term Enoxaparin Untreatable Hypertension
Rheumatoid arthritis on Rituximab
Immunodeficiency requiring immunoglobulin replacement therapy This lady has also been is under the care of a psychiatrist for many years. An incident from her past has coloured her perception of hospitals and resulted in a dislike of attending almost daily hospital appointments for all her other follow up assessments. Mary also has extremely poor venous access. In August 2012 Mary and her husband who is also her main carer attended the unit to start replacement treatment. Initially she commenced intravenous infusions but due to her poor venous access it was agreed she would swap to subcutaneous infusions but with a view to transferring to home therapy. Was it possible to under take Home therapy training and give Mary her wish to reduce hospital visits and increase her independence? The answer is yes but with many barriers to overcome.
This case study demonstrates the hurdles encountered during her preparation and training for Home Therapy but also the eventual benefits in using this strategy.
ESID-0067 Importance of the Transition Process to Children and Adults with PID
C. Benson 1, B. Boardman2, C. Simon3
1Immunology, Alder Hey Children's NHS Foundation Trust, Liverpool, United Kingdom
2Immunology, Royal Manchester Children's Hospital, Manchester, United Kingdom
3Immunology, Royal Liverpool & Broadgreen University Hospitals NHS Trust, Liverpool, United Kingdom
Background: A formal transition process is thought to be important for patients, particularly those with complex medical conditions such as PID. Most guidelines have been developed by health care professionals. This study investigated the views of PID patient’s regarding transition at two major tertiary immunology centres.
Methods: Children and adults who had or were about to go through the transition process were asked to fill out standardised questionnaires.
Results: 26 PID patients (11 children and 15 adults) completed the questionnaires. Median age was 17 years and 65% were male. Only 19% felt that just a transfer letter was adequate. Patients wanted the transition process to start at 14 and by this age wanted to be seen without their parents. They wanted to be transitioned at 17 years old. Over 75% of patients thought it very important/essential that they (i) a named contact in the adult service, and (ii) the paediatrician to attend their first appointment with the adult physician. Only 45% wanted their parents to be present at their first appointment with the physician. There were no significant differences between the desires of children and adults.
Conclusions: 80% of patients felt that a formal transition process was very useful or essential. Patients wanted transition to take place around the time of leaving school. A key requirement was a named contact in the adult service. They felt that it was more important that the paediatrician attended the first appointment at the adult centre, rather than the adult physician attending paediatric appointments.
ESID-0496 A Single Centre Audit of Home Visit Outcomes in Pad Patients Self-Infusing Immunoglobulin
E. Carne 1, C. Kingdon1, T. El-Shanawany1, S. Jolles1
1Immunodeficiency Centre for Wales, University Hospital of Wales, Cardiff, United Kingdom
Background Home visits by Immunology clinical specialist nurses (CNS) are a key component of the support and care for patients who self-infuse immunoglobulin at home. At the Immunodeficiency Centre for Wales sixty nine percent (97) of patients self infuse at home and nurse home visits are carried out annually for the first two years then biannually. These visits assess all aspects of the home infusions in ‘real life’ rather than the more focussed competencies gained during the in hospital training sessions. While the benefits of home visits may appear self evident it remains important, with increasing pressures on nursing time, to establish whether these visits add significant value to patient care.
Methods In order to assess the impact and outcomes of home visits a single centre retrospective audit of all home visits undertaken in 2011 and repeated in 2013. The notes from the home visits were analysed and data regarding any changes or modifications made to the overall infusion process were collated.
Results Ninety patients were included in the audit, 86% infused subcutaneous immunoglobulin (SCIg), 3% hyaluronidase facilitated SCIg and 11% intravenous immunoglobulin (IVIg). Home visits identified that 25% of patients experienced problems requiring change in management of their infusions, altered infusion technique and/or provision of further information or training.
Conclusion Home visits were shown to add value with important changes made in many cases both solving problems and preventing others. The visits were valued by patients reducing the need to contact the centre and enhancing independence.
ESID-0224 Melding Two Databases to Guide Research and Outcomes Measurments in Patients with Primary Immunodeficiency
C. Duff 1, M. Boyle2, E. Murphy3, E. Hovermale4
1Pediatrics, University of South Florida, St. Petersburg, USA
2President, Immune Deficiency Foundation, Towson, USA
3MSL, CSL Behring Inc, King of Prussia, USA
4Public Policy, Immune Deficiency Foundation, Towson, USA
Background Rare disease patient registries, like United Stated Immunodeficiency Network (USIDNET), allow data to be collected and aggregated to trend epidemiology and disease, assess needs, and improve patient care. Electronic personal health record (ePHR) data provide benefit for accessing clinical data in a real life setting.
The Immune Deficiency Foundation (IDF) received an award from the Patient Centered Outcomes Research Institute to develop a health data network for primary immunodeficiency disease (PI), titled PI CONNECT, to be part of PCORnet: the National Patient-Centered Clinical Research Network.
Methods: In 2011, IDF launched an online ePHR for the PI community to track their clinical experiences. Consideration during development included disease specific parameters. Since launch, 900 accounts with 48 different PI types have been established. Patients using the ePHR vary in age (27% 17 years or younger, 30% 18-44 years, 35% 45-64 years) with a majority of female users (63%).
With PI CONNECT, the IDF ePHR will allow individuals to update information in the USIDNET registry, creating a database of combined patient-generated and clinical data. This enhanced approach will contribute to large rare disease population with consistent data to facilitate research across the entire spectrum of disorders. PI CONNECT will also create a venue for all clinicians and researchers and patients to discuss research involving the network data.
Conclusion PI CONNECT will meld the USIDNET registry with the IDF ePHR. This combined dataset will provide valuable information to help prioritize research efforts and illuminate patient input on treatment, quality of life and healthcare resource utilization.
ESID-0652 University PID/SID Education for Nurses
A. Gardulf 1, B. Steger2
1Dept of Laboratory Medicine Karolinska Institutet, Division of Clinical Immunology, Stockholm, Sweden
2Falu Hospital, Division of Clinical Allergology and Immunology, Falun, Sweden
Background Nurses are vital in the care and treatment of children/adults with primary immunodeficiencies (PID) and secondary immunodeficiencies (SID). For patients on long-lasting/life-long treatments, such as e g IgG replacement therapy, the nurses are often the ones who support self-care of the patients/families, take the practical decisions about the IgG therapy and the necessary equipment, and take responsibility for therapy follow-ups.
The PID/SID University Education Project The education will be at advanced level, evidence-based and comprise 7.5 credit points (nursing science 4.5, medical science 3.0 points). A web based teaching and learning platform will be used, thus making it possible for nurses living/working far away from the University to participate. The medical education will cover e g pathophysiology including genetics, symptoms and diagnostic procedures, microbiology and treatment of infections; nursing education will cover e g research-based knowledge about immunodeficiency treatments, motivational dialogue techniques, patient-/family-education including compliance, and quality of life. The education will be held for the first time during spring semester 2015.
Future education and graduation This is a unique University education for nurses in Swedish/Scandinavian speaking countries. Still, the education should be considered a first stepping-stone in creating a formal graduation as an Immunology/PID Nurse. The opportunity for further courses deepening the knowledge in different areas will hopefully be possible within the European Union Higher Educational System, and possibly globally, involving INGID as the coordinating organisation.
ESID-0030 Give Choice to the Patient
M. Levasseur 1, E. HADDAD1, C. Samaan1
1Immunologie-Rhumatologie, CHu Sainte-Justine, Montreal, Canada
Purpose: Criteria governing the choice between intravenous (IV) and subcutaneous (SC) routes for immunoglobulin (Ig) substitution are not well defined. We assessed the consequences of giving the choice to the patient.
Methods: We retrospectively analyzed 143 patients with primary immunodeficiency, followed in a single center, which were offered the choice of IVIg or SCIg. We analyzed the route more frequently chosen, and the consequences on compliance. In a first cohort (n=51, average follow up 52 months), patients already on IVIg were offered the choice to stay on IVIg or to switch to SCIg (switch cohort). In a second cohort (n=92, average follow up 11 months), newly diagnosed patients were offered the choice between IVIg and SCIg before the first injection (new cohort).
RESULTS: In the switch cohort, 50/51 chose to switch to SCIg. Of these, 90% remained on SCIg. In the new cohort, 44/92 chose SCIg, of which 95% remained on SCIg. Among the 48 patients who chose IVIg, 73% switched to SCIg. Compliance issues were observed in only 10 patients.
CONCLUSION: Home-based SCIg is much more frequently chosen than hospital-based IVIg. Giving patients the choice in treatment modality is a safe strategy in terms of compliance. We believe that given the equal efficacy and safety between hospital-based IVIg and home-based SCIg, all patients should be given the choice regardless of physician belief of “idealness” of the candidate.
ESID-0339 Subcutaneous Administration of Gammanorm in Immune-Deficient Patients by Rapid Push: Report of the Belgian Experience
A. Reniers 1, C. Heijmans2, N. Nols3, B. Rombaut4, R. Peché5
1Home Nursing, Home Nursing Reniers An, Galmaarden, Belgium
2Pediatric Hemato-Oncology, Queen Fabiola Children University Hospital, Brussels, Belgium
3Hematology, Centre Hospitalier de Mouscron, Mouscron, Belgium
4Pneumology, Algemeen Stedelijk Ziekenhuis, Aalst, Belgium
5Pneumology, University Hospital Charleroi - André Vésale, Charleroi, Belgium
Substitutive treatment of immune-deficient (ID) patients with subcutaneous immunoglobulin (SCIg) is a safe, effective and convenient alternative to treatment with intravenous immunoglobulin (IVIg). Usually administered by compact portable infusion pumps, SCIg can also be administered by rapid push technique. This method has been reported to be convenient and cost-effective.
Gammanorm was introduced to the BE market in April 2011. On the 22 patients currently on Gammanorm who were offered the choice between pump and rapid push, 15 (12 adults and 3 children) are using rapid push and 7 (4 adults and 3 children) pump(s).
Main characteristics of this cohort (previous treatment, dose regimen, site(s) and duration of administration, local tolerance, duration of treatment, compliance…) will be presented and the importance of the nurse for treatment initiation and follow-up, compliance and information of the treating physicians will be discussed.
Patients may greatly profit from the availability of several modes of administration of immunoglobulin. Choosing the most appropriate route of administration for a patient has to be based on an analysis of the actual patients’ needs. Nurses have a pivotal sentinel role during initiation and follow-up of the treatment allowing to confirm the correctness of the choice of the technique of administration or to suggest moving to another one when needed for patients’ comfort and compliance. Our experience indicates that rapid push has a place in the treatment of ID patients and should be offered to these patients in the same way as other techniques of administration.
ESID-0115 Immunology Nursing in the Czech Republic Today
I. Tepla 1, D. Ruckova2, P. Kralickova3, A. Sediva2
1Institute of Allergology and Clinical Immunology, University Hospital Hradec Králové and Medical Faculty Charles University in Hra, Hradec Králové, Czech Republic
2Institute of Immunology, University Hospital Motol, Prague, Czech Republic
3Institute of Allergology and Clinical Immunology, University Hospital Hradec Králové and Medical Faculty Charles University in Hradec Králové, Hradec Kralove, Czech Republic
Immunoglobulin supplementation therapy represents a corner stone in the treatment of patients with disturbance of specific antibody production. Czech PID Registry was founded in 2011. There were enrolled 594 patients by the end of year 2013. The highest number of patients (413 patients) are patients with humoral immunodeficiency (70.8%). Intravenous immunoglobulis are used in 214 patients and subcutaneous in 137 patients. PID patients are predominantly concentrated in the immunologic centres. The intravenous as well as subcutaneous immunoglobulins are available in the Czech Republic. The way of administration, dosage, frequency and the route of administration are individual. It is the result of discussion between doctor, patient and nurses. Nowadays the subcutaneous route of administration is frequently preferred. Patients benefit from more independence, comfort and less frequent adverse events. The pump administration and recently the direct rapid push method are used. Both methods have comparable efficacy and the frequency of adverse events. However, there are still specific groups of patients which consider intravenous route of administration more convenient. Nurses play the key role in education and practical demonstrations. The correctly guided treatment leads to the decrease of severe infections frequency and significantly improve the quality of life.
ESID-0203 Immunity & Children: The Developmental Changes in the Immune System from Birth to Adulthood
P. Vickers 1
1Nursing & Midwifery, University of Hertfordshire, Hatfield, United Kingdom
Major changes occur in the immune system during childhood; the most striking probably being the development of acquired immunity as the child comes into contact with the many infectious microorganisms which surround us. This builds upon, and adds to, the immune protection built up within the fetus. Thus, during infancy and early childhood, there are many changes within the immune system as the immune system learns to cope with the many immunological/infectious challenges it meets. After adolescence, the immune system starts to go into a gradual decline. The numbers of T- and B-cell lymphocytes, along with lymphocyte proliferation potential are all maximised early in infancy, and reach levels 1.5 – 2.5 times higher than they will be during adolescence; then gradually decline during adulthood (McDade 2003). Throughout infancy, childhood and adolescence, immunoglobulin levels rise quite rapidly and reach adult levels by the age of 12 years. During these stages, the thymus also grows and reaches its maximum size by the age of 10 years, after which – and certainly after puberty – the thymus starts to atrophy and the thymus cortex and medulla start to gradually be replaced by fat. These are just some of the many changes that take place within the immune system during childhood, and this presentation will explore these, and explain the consequences for the child and adult of these changes.
Reference: McDade TW (2003) Life history theory and the immune system: Steps towards a human ecological immunology. Yearbook of Physical Anthropology 46: 100-125
INGID 2014 Poster Presentations
ESID-0676 The Nurse´s Role in Caring for PID Patients in Slovenia
M. CAMERNIK 1, M. HREN1
1Dpt. of alergology rheumatology and clinical immunology, University Children's Hospital University Medical Centre Ljubljana, Ljubljana, Slovenia
University children´s hospital´s Department of alergology, rheumatology and clinical immunology is the only center for treating PID patients in Slovenia and is a JMF diagnostic and research center for PID in Slovenia.Here are treated mostly children and teenagers up to 19 years of age. But transition process and care for adult PID patients is lacking.
In last few years there was a big step forward in treating immune deficiencies in Slovenia, so nurses had to follow the progress. Now our patients are on subcutaneous immunoglobulin treatment at home, and nurses had to learn everything about treatment so that we can teach our patients and their parents to do this themselves.
We also have presentations of what we do in meetings organized by Slovenian nurses chamber.
Our patients, even though Slovenia is a relatively small,live in different parts of the country. That is why we established a Society for patients with immune diseases for patients to get in touch with each other and with medical professionals out of the hospital. The Society organizes two major social event per year, where patients, their families, doctors and nurses prepare social and scientific program.We take an active role in organizing, advertise and presenting lectures.
ESID-0722 The Nursing Management of WAS Children Treated With Gene Therapy
M. Casiraghi 1, R. Tresoldi1, A. Ciceri1, M. Milanovic1, C. Moser1, M.P. Cicalese1, F. Ferrua1, M.G. Roncarolo1, A. Aiuti1, C. Soliman1
1Pediatric Immuno Hematology, San Raffaele Telethon Institute for Gene Therapy, Milano, Italy
Background From 2010 till 2013 a dedicated medical-nurse team at the Pediatric Immuno-Hematology Unit treated with Gene Therapy 6 patients affected by Wiskott-Aldrich Syndrome (WAS). Patients were treated with an infusion of autologous CD34+ bone marrow cells transduced with a lentiviral vector encoding WASp, after nonmyeloablative conditioning. One patient received also transduced mobilised peripheral blood CD34+ cells (Aiuti et al, Science 2013).
Aim To describe the nursing management during the clinics, hospitalization and follow up. To underline the nursing activities for planning, achievement and evaluation of adequate intervention to prevent infections and bleeding with involvement of families.
Method The nursing assistance was carried out through the logic of total care and the centrality of the kid and family. The activity was realized through the Evidence Based Practice and the JACIE standards. The instruments include: nursing documentation and standards operating procedures, health education, family training and educational brochures.
Results The experience with patients and families combined to the nurses training allowed to elaborate a dedicated nursing plan. The process started from the identification of problems, moved to the definition of the assistance priorities and the result indicators, allowing to perform the required interventions supported by scientific motivations.
Conclusion Planning of the nursing care guarantees the cures with respect to the needs of personalization. Following this method helps to reduce the unevenness of the nursing practice. The complete involvement allows to consider clinical and emotional problems, socio-cultural differences and to obtain the best grade of quality and safety during the care.
ESID-0498 Patient Experience of Different Immunoglobulin Replacement Flexible Dosing Regimens
C. Kingdon 1, S. Jolles1, T. El-Shanawany1, E. Carne1
1Immunodeficiency Centre for Wales, University Hospital of Wales, Cardiff, United Kingdom
Background Replacement immunoglobulin therapy can be administered via intravenous, subcutaneous, facilitated subcutaneous, rapid push and historically intramuscularly routes. Infusions or injections can be given daily, weekly, three weekly, monthly or to an individually tailored regime. Infusions can be self-administered or clinician-led and given in a variety of settings including home. With an increasing focus on individualisation of treatment options and flexible dosing, we sought to explore the factors influencing patient choice and subsequent outcomes including regime adherence.
Methods Four informative case studies are presented considering the personal reasons behind the choices made by four patients on the following regimes: SCIg infusion every fortnight; three weekly IVIg; facilitated subcutaneous infusion (fSCIg) and daily rapid push injections.
Results The case studies highlight the individual reasons for choosing a particular method of administration and how that choice was affected by a variety of factors such as the patients’ personal lives and outside commitments, clinical condition and prior healthcare experience. The outcome of that choice on patient satisfaction is discussed. The results are contextualised by an analysis of the treatment choices of the Welsh national cohort of PAD patients.
Conclusion The results confirm that the choice for PAD patients is no longer a binary decision of SCIg versus IVIg and that different and varied factors influence individual treatment decisions with a clear impact on patient experience and outcome.
ESID-0169 Sanquin Homeservice; Providing IVIg and SCIg Home Therapy
J.C. Zwiers 1, M.A. Muijs1, M.I.P. Dros2, C.M. Kramer1
1Division of plasma products, Sanquin Blood Supply, Amsterdam, Netherlands
2Pharmacy, Mediq Pharma Services B.V., Nieuwegein, Netherlands
Immunoglobulin is used for antibody replacement or immune modulation therapy in patients with immune deficiencies, but also in autoimmune and chronic inflammatory diseases. Some of these diseases need prophylactic and life long therapy. To some extent this can be inconvenient for patients: they have to visit the hospital regularly. Sanquin developed a service to provide home therapy with IVIg and SCIg.
The service of the Sanquin HomeService, initiated in 2005, consists of several modules. The modules are development and implementation of a transfer and treatment protocol, reimbursement of the home treatment by the insurance company, intake of patients during last visit at hospital, distribution of product and medical devices to the patient, the administration of immunoglobulin by a dedicated nurse and training the patient or partner how to administer immunoglobulin (mostly in case of SCIg) and, if applicable, assessment of IgG trough level.
At present 530 patients receive their immunoglobulin treatment through Sanquin HomeService of which 77% receive IVIg and the remaining 23% SCIg. The distribution in gender is equal and patient age ranges from 11 months to 86 years. Conditions vary from primary and secondary immune deficiencies, neurology, gynaecology and autoimmune disorders. The experience built up over the past years resulted in creative and practical solutions to ensure that also more complex clinical cases can benefit from home therapy.
Patients can be treated at home safely. However, this requires an established network of specialized nurses, good communication with the treating physicians and an overall solid infrastructure.
ESID-0591 A Case of Once per Week SCIg in a Total of 36 Minutes
B. Sealfon 1, C. Soderman2
1Patient Programs, RMS Medical Products, Chester, USA
2Clinical Programs, RMS Medical Products, Uppsala, Sweden
Introduction We present the case of a patient transitioning successfully from 7 hour IVIg with rate shock, poor venous access, and infiltration; to 20 minute weekly self-administered SCIg without site reactions.
Objective To describe the steps in one case toward an optimal personalized SCIg delivery.
Methods Patient provided infusion records from 2007 to present, augmented by subjective history.
Results Patient on a mechanical pump and rate-control tubing aborted Week 1 SCIg after 5 hours with local reactions. Longer needles and faster flow tubing were selected. By week 3 the patient recorded infusing 8gm, 50 mL (16%), using an F275 flow tubing set, in 3 hour infusion. Severe site reactions were resolved through dry needle insertion and non-rotated infusion sites. With serum IgG rising, dose was gradually reduced to 6.8gm 40 mL by week 12. Rate was increased gradually (F600, F1200, F2400). The patient adopted a four site 26 g needle set and reported reduced infusion time with more comfortable insertion and removal. Patient now consistently achieves 13 to 20 minutes with 6gm, 30 mL (20%) with no reactions. Patient experiences a setup/cleanup time of 16 minutes. Total monthly commitment to therapy is 2 hours 24 minutes.
Conclusion Many patients miss an important opportunity to pursue personalized SCIg delivery. An SCIg decision support algorithm is in development to support clinical providers in administration troubleshooting.Further study might both inform a standard of practice to optimize therapy to individual patient needs, and quantify the impact of this process on patient success and perception of therapy.
ESID-0074 Pflegenetzwerk Immunologie Immunology Nursing Network Group
G. Strotmann 1
1Immundeficiency Unit, University Children's Hospital at Dr. v. Haunersches Kinderspital Ludwig Maximi, München, Germany
Gaby Strotmann, Mary-Louise Daly, Nadine Fender, Franziska Isenring, Marion Klima, Nathalie Piraud, Susanne Ringger, Henrike Ritterbusch, Isabelle Zenklusen, Steffi Schlieben
Background: Caring for patients with a primary immune deficiency is a very specialized area of care. Often nurses working in this field are the only nurses in the specialized clinics. In 2013 nurses from Germany and the German speaking part of Switzerland joint together to form a support network for nurses looking after patients with a primary immune defect.
Aim:
Our aims and plans are:
- Networking
- Professional Development
- Care Pathways
- Care Guidelines
- Public Relations
- Regular Interchange
- Patient Care Improvement
- Project Report
Results: We have 1 yearly meeting, as well as linking up when we attend international or national conferences. Otherwise the majority of the communication is via email.
We have developed a patient ID card which patients can choose to use if they want.
We have developed a flyer to provide a point of contact for other nurses who are interested.
Did we awaken you interest? Than we will be very happy if you get in contact with us
mary-louise.daly@usb.ch or
gaby.strotmann@med.uni-muenchen.de
Because together we can make a difference
ESID-0121 Discussion and Treatment of Chronic Norovirus II Infestation in Patients with Primary and Secondary Antibody Deficiency
A. Symes 1, N. Verma1, S. Workman1, S. Burns1
1Clinical immunology, Royal Free Hospital, London, United Kingdom
We discuss the case history of several antibody deficient patients, both primary and secondary, currently treated by a specialist immunology centre (Royal Free London NHS Foundation Trust), presenting with chronic Norovirus II infection. Norovirus is usually eradicated without treatment by most people after several days of symptoms, typically diarrhoea and vomiting, followed by a short period of recuperation. The affected patients, however, repeatedly return positive stool cultures (despite not always being symptomatic) and it is this prolonged infective period, despite varied attempts at symptom managment, which lead to weight loss, poor quality of life and possible poor prognosis. These treatments will be discussed along with their implications for future management.
ESID-0038 Patient Experience of Immunoglobulin Treatment Within a Specialist Immunology Service
A. Symes 1, S. Workman1, S. Burns1, M. Campbell1
1Clinical immunology, Royal Free Hospital, London, United Kingdom
This poster will present the abridged results of a patient experience questionnaire conducted by a Specialist Immunology service within the UK. Patients on immunoglobulin replacement therapy, administered by subcutaneous (SC) or intravenous (IV) route, at home, local hospital or specialist centre, provided information on the impact of the illness on daily life (including time off work and number of infections), their experience of treatment (including satisfaction with route of administration and feelings of control over treatment), the type of information they would find helpful in future and engagement with the team. The information gathered and implications for service development and improved patient care will be discussed.
ESID-0345 Introducing the International Hereditary Angioedema Nurses Organisation (IHNO)
C. Symons 1, K. Andritschke2, J. Dempster3, I. Leibovich4, M. Steinicke5
1Immunology, Derriford Hospital, Plymouth, United Kingdom
2Haemophilia, HZRM, Mörfelden-Walldorf, Germany
3Immunology, Barts, London, United Kingdom
4Immunology, Sheba Medical Center, Tel-Hashomer, Israel
5Immunology, Charite, Berlin, Germany
The International Hereditary Angioedema (HAE) Nurses Organisation (IHNO) was established in May 2013, during the 8th C1-Inhibitor Deficiency Workshop, in Budapest, Hungary.
All participating nurses have an extensive experience with patients with Hereditary Angioedema (C1 inhibitor deficiency) in our countries. The main purpose of this initiative is to increase the impact of expert nurses on patient management and implementation of new treatment protocols. Professor Henrietta Farkas from Hungary kindly agreed to become the Honorary President of IHNO.
The main issues discussed were:
Is there a need for a nurse-specialist in HAE? What would be her role?
Does self-treatment introduce a real revolution in patients' life?
Was a decrease in hospitalization obtained in countries where self-treatment is implemented?
Is it the role of professional teams to interact with HAE support groups?
Goals: To help as many HAE patients as possible to lead independent and productive lives
To develop training programs directed at self treatment with the available intravenous and subcutaneous medications.
To help HAE patients get easy access to all available therapies
To raise awareness of this rare genetic disease
To work hand-in-hand with national and international HAE and Immune Deficiency experts, and with patient associations (i.e. HAEi)
We realise that nurses who work with HAE patients comes from different disciplines such as Dermatology, Haemophilia, and Allergy etc. We would like to encourage other nurses, working across these settings to join us in our new initiative.
Please approach us during this meeting.
ESID-0304 Immunoglobulin Replacement Therapy in Primary Immunodeficiencies and Quality of Life-A Case Report
E. Szynkiewicz 1
11. Chair and Clinic of Allergology Clinical Immunology and Internal Diseases 2. Department of Nursing in Internal Diseases Collegium Medicum Nicolaus Copernicus University, University Hospital No.2., Bydgoszcz, Poland
Introduction
Chronic diseases affect various aspects of human life. Therefore, quality of life research, which are used to evaluate them, are becoming increasingly important in medical sciences. Treatment with subcutaneously administered immunoglobulin has become more and more popular in recent years.
Aim
Comparison of quality of life and measurement of the general degree of self-assessment in the patient with primary immunodeficiency during treatment with intravenous immunoglobulin substitution and after the transition to subcutaneous administration (after 9 months of use).
Material and methods
Patient, aged 30, with a diagnosis of congenital agammaglobulinemia, COPD, hepatitis C, left-sided lobectomy, chronic sinusitis, cholelithiasis. BMI 17.3.
For 18 years treated with the immunoglobulin replacement therapy administered intravenously. Nine months ago the route of administration was changed into subcutaneous one.
The study was conducted using the questionnaire WHOQOL-Bref and M. Rosenberg SES scale.
Results
Rosenberg Self-Esteem Scale SES, scores 20 vs. 24 (maximum score 40)
WHOQOL-Bref
In the studies the answers are presented on a five- point Likert scale where higher values correspond to higher quality of life
• Individual overall perception of quality of life 4 vs. 4
• Individual overall perception of one’s own health 2 vs. 4
• Physical domain 3 vs 3.8
• Psychological domain 2.5 vs. 3.6
• Social relationships 2.6 vs. 3.6
• Environment 3.3 vs. 4.2
Conclusions
Quality of life and patient’s self-assessment after the change of the form of substitution therapy from intravenous to subcutaneous have significantly improved.
Keywords: immune deficiencies, quality of life, self-assessment.
ESID-0116 Immunoglobulin Supplementation Therapy – One Centre Experience
I. Tepla 1, S. Cerna1, I. Krcmova1, P. Kralickova1
1Institute of Allergology and Clinical Immunology, University Hospital Hradec Králové and Medical Faculty Charles University in Hra, Hradec Králové, Czech Republic
The immunologic centre in Faculty Hospital Hradec Kralove, Czech Republic was founded in 1992. Here we take care of adults patients mainly with humoral immunodeficiency. Forty-seven patients are treated with regularly immunoglobulin supplementation therapy in 2014. We provide intravenous immunoglobulin (IVIG) administration at outpatient department in 27/47 patients. Other twenty patients undergo the home subcutaneous therapy (9/20 with pump, 11/20 by rapid push).
We followed the adverse events within the last five years. We noticed 23 adverse reactions in 13 patients during 2005 intravenous administration (frequency 1.15%). No severe anaphylactoid reaction was reported, only four adverse reactions were moderate (shivers, dizziness, hypotension), nineteen reactions were mild (chills, shivers). The causes were: 13/23 the change of product, 6/23 infection, 2/23 initiation of supplementation and 2/23 unknown origin.
The subcutaneous administration was connected with very low frequency of adverse reactions. We noticed only one case of intolerance (flu-like syndrome, fatigue). Other side effects were local and infrequent, mainly during the first weeks after initiation of therapy. One patient was switched back to the intravenous because of lower compliance.
Conclusion: The frequency of adverse reactions in subcutaneous therapy is very low in comparison with intravenous. Higher risk of adverse reactions in IVIG was especially connected with change of product or presence of infection.
IPOPI 2014 Poster Presentations
ESID-0141 Malaysia WPIW Campaign 2014: The Media Frenzy Affair
A. ALI 1, A.H. ABDUL LATIFF2, L.M. NOH1, I.H. ISMAIL3, S. MARTIN4, K. KOH4, N. AWANG4, M. ZULKIFLI4, B. LIM4
1Department of Pediatric, Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia
2Pantai Hospital Kuala Lumpur, Bangsar, Kuala Lumpur, Malaysia
3Department of Pediatric, Universiti Putra Malaysia, Serdang, Malaysia
4Malaysia Patients' Organization for Primary Immunodeficiencies (MyPOPI), Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia
Background: World Primary Immunodeficiencies Week (WPIW) is a global annual event. Malaysia participated in this event for the last few years through the Malaysia Society of AIlergy & Immunology (MSAI) online campaign. This year, Malaysia moved a step forward with our newly established Malaysia Patients' Organization for Primary Immunodeficiencies (MyPOPI).
Objective: To heighten awareness of the public and the policy makers through the voice of MyPOPI. Through the joint effort of MSAI and MyPOPI, Malaysia's 2014 WPIW celebration was aimed towards awareness and better understanding of PID nationwide with the cooperation of the local mass media.
Results: The Malaysia WPIW Campaign 2014 kick-started with a live national radio interview, whereby both the MyPOPI's President and Medical Advisor focused on creating awareness on PID and the role of MyPOPI. A press conference involving the MyPOPI's members from patients, patients' families and doctors which followed two days later, received warm welcoming response from the mass media. It did not end there as MyPOPI had been invited and participated in a number of television and radio interviews. The Medical Advisors had been approached for press interviews to highlight on the burden of PID diseases not just in Malaysia but in the South East Asia and internationally.
Conclusion: The Malaysia WPIW Campaign 2014, had definitely been an amazing 'tsunami' of media affair and was an excellent start for MyPOPI in creating awareness and providing support for PID patients and their families in Malaysia and South East Asia region.
ESID-0140 The Birth of MyPOPI: Beginning of a Hopeful Journey
A. ALI 1, A.H. ABDUL LATIFF2, L.M. NOH1, I.H. ISMAIL3, I.J. ABDUL HAMID4, A.W. ASRUL5, M.J. FAIZAH6, K. KOH7, S. MARTIN7, N. AWANG7, M. ZULKIFLI7, B. LIM7
1Department of Pediatric, Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia
2Pantai Hospital Kuala Lumpur, Bangsar, Kuala Lumpur, Malaysia
3Department of Pediatric, Universiti Putra Malaysia, Serdang, Malaysia
4Advanced Medical & Dental Institute, Universiti Sains Malaysia, Penang, Malaysia
5Department of Microbiology, Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia
6Department of Pediatric, Hospital Serdang, Serdang, Malaysia
7Malaysia Patients' Organization for Primary Immunodeficiencies (MyPOPI), Universiti Kebangsaan Malaysia Medical Center, Kuala Lumpur, Malaysia
Background: Patients with primary Immunodeficiencies (PID) were detected as early as 35 years ago in Malaysia. Despite the increasing numbers of Clinical Immunologists (4 in 2014), the scenario is still gloomy with delays in diagnosis and poor outcome.
Objective: Believing in the need of a better environment for PID, the patients' support group was established.
Results: In October 2013, the first Malaysia PID Patients' Meeting & Workshop was held in collaboration with Malaysia Society of Allergy Immunology (MSAI) and IPOPI during the 6th NACLIS (National Clinical Immunology Symposium) International Conference. In January 2014, the MyPOPI interim committee was formed and by February it became an associate member of IPOPI. MyPOPI is a patient-driven, physician-supported organization, committed towards increasing awareness of PID, while improving conditions and providing support for patients, in Malaysia and regionally. Since its inception, MyPOPI has embarked on its objective within the community through road shows, meetings with medical policy makers and even the Health Minister. MyPOPI's online website and Facebook page were established, and recruitment drive for members initiated. MyPOPI had bridged out to its neighboring countries, helping with their efforts towards similar objectives. MyPOPI and MSAI had also jointly celebrated the World PID Week with the mission to create awareness of PID through involvements of the mass media.
Conclusion: The birth of MyPOPI has definitely yielded a hopeful light to the future of PID with its advocating and supporting roles in providing a better environment for PID patients, both in Malaysia and South East Asia.
ESID-0449 High Dose Immunoglobulin Treatment- a Patient’s Experience
A. Green1, M. Bhole 1
1Immunology Department, Russells Hall Hospital, Dudley West Midlands, United Kingdom
Immunoglobulins are increasingly being used in high immunomodulatory doses in the management of various neurological and autoimmune conditions. Here we describe the personal experience of a young lady with a rare neurological condition currently managed with high dose immunoglobulin therapy.
This lady developed symptoms of acute onset visual loss in one eye at the age of 14 years. This was initially diagnosed and managed as optic neuritis. Over the next few years she developed further symptoms including visual loss in the other eye and other neurological signs. In 2009 she was diagnosed as Neuromyelitis Optica (NMO) with typical features of long-segmental demyelination on spinal MRI. Antibody to aquaporin-4 was negative but she was later found to have antibodies to myelin oligodendrocyte glycoprotein (anti-MOG antibodies).
In the period between initial symptoms and 2012 relapses were treated and managed with various immunosuppressant agents including high dose steroids, steroid sparing agents like methotrexate and azathioprine and a trial of anti-CD20 monoclonal antibody (Rituximab). Despite these she continued to have frequent relapses and was registered visually impaired.
In 2012, whilst under the care of the neurology team at the Specialist NMO centre, she was started on high dose intravenous immunoglobulin for immunomodulation. The treatment proved beneficial and she has remained relapse-free to date. However, she has had treatment related issues such as mild side effects and access problems necessitating further changes and modifications to the treatment regimen. Her personal journey and experience with high-dose immunoglobulin treatment will be discussed.
ESID-0391 Monitoring Procalcitonin in Primary Immunodeficiency: What Is its Utility for the Diagnosis of Acute Respiratory Infection?
H.L. ELFASSY 1, H. Chapdelaine2
1internal medecin, Centre Hospitalier de l'Université de Montréal, Montréal, Canada
2allergy-immunology, Centre Hospitalier de l'Université de Montréal - Hôpital Notre-Dame, Montréal, Canada
2Department of Paediatrics and Developmental Disorders in Childrens and Adolescens, Medical University, Warsaw, Poland
3Immunoprotect, Immunoprotect, Lodz, Poland
INTRODUCTION:
Infectious manifestations may be minimal and non-specific in patients with primary immunodeficiency (PID), making diagnosis and treatment of respiratory infections a challenge.
Procalcitonin is an inflammatory marker whose clinical relevance has been studied and proven in the context of severe acute bacterial infections in immunocompetent adults and HIV patients.
In this study, we try to demonstrate the relevance of this marker in the context of suspected acute respiratory infection in adults with PID.
METHODS:
It is a retrospective case-control study of adults with PID presenting signs or symptoms of acute upper/lower respiratory infection. Co-morbidities, antibiotics or corticosteroids use, leukocytosis, physical exam and imaging were analyzed.
RESULTS:
Preliminary results shows that patients with high level of procalcitonin are three times more likely to receive antibiotics than those with normal ranges (
IntroductionPolish Working Group on Primary Immunodeficiencies (PGR PNO) - a network of cooperating national centers for diagnosis and treatment of PID was established in 2005. As a result of joint efforts of the Group the number of centers actively working in the diagnosis and treatment of primary immunodeficiencies increased. Immunoprotect, patient's organization, work on expanding awareness of immunodeficiencies also.
Material and methods
PGR PNO includes 15 pediatric centers, and 11 centers for adults with total number of 3 368 PID patients. The retrospective analysis of replacement therapy in primary antibody deficient (PAD) patients is presented.
Results
According to the 2013 annual report of PGR PNO 1684 patients have primary antibody deficiency (PAD). Five hundreds ninety three patients require immunoglobulin replacement therapy. Children constitute 58% (344) patients of that group. Three fourth of patients (445) receive intravenous gammaglobulin preparations (IVIG), while the rest (148) - subcutaneous immunoglobulin (SCIG) . In children subcutaneous immunoglobulins are more often used than in adults (105 vs 43). The adverse events on SCIG are mild and self-limiting. Severe systemic adverse events (ie anaphylaxis, aseptic meningitis, trombotic complications, hemolytic anemia and other) on both IVIG and SCIG are the subject of the research analysis and will be presented during the meeting.
Conclusion
Cooperation of all centers as well as development and dissemination of new diagnostic and therapeutic standards contributed significantly to the increase of PID's detection and implementation of treatment, including gamma globulin replacement therapy. An important role in all these efforts plays Immunoprotect.
ESID-0087 Risk of Anxiety/Depression and Risk of Death in Patients with Primary Immunodeficiencies Evaluated with SF-36 and GHQ-12 Questionnaires, A Longitudinal Study
I. Quinti1, C. Milito 1, P. Giannantoni2, S. Tabolli2
1Molecular Medicine, Sapienza University of Rome, Rome, Italy
2Health Services Research, IDI IRCCS, Rome, Italy
CVID patients despite immunoglobulins replacement reported a low quality of life. The study aim was to identify if variations in quality of life are predictive of the risk to be anxious/depressed and of mortality. 96 patients were enrolled and followed for a six year period. A court of 66 patients were followed for three assessments at one and six years from the baseline. Generic health status tolls were used : 36-Items Short-Form Health Survey (SF-36) and General health Questionnaire 12 items (GHQ-12) questionnaires. Evaluation of disease severity were reported by physicians and directly by patients. 11 over 66 patients reported a worsening GHQ-12 status and negative variation in SF-36 (physical functioning, vitality, social functioning and mental) scales. A decrement of 1-point in each of these scales increase the risk of developing anxiety/depression from 3 to 5%. 18 patients died. Physical (PF) and social functioning (SF) as well as role emotional at the baseline assessment of these patients were remarkably lower than survivors. The results were confirmed when adjusted by age. The RR of death for PF and SF were 0.98 and 0.97 respectively. Each point of increase in the SF-36 scales, independently by age reduce the risk of death of 2-3%. In survival analysis PF score <50 (cut-off) were associated with a RR of 4.4 (CI 1.7-11.3, p<0.03) and SF score < 37.5 (cut-off) had a RR of 10 (CI: 2.6-37.9, p<0.001). General health status questionnaires such as SF-36 and GHQ-12 documented the outcomes of clinical management, therapeutic intervention.
ESID-0622 Patient-Driven SCIg Administration - Potential for Better Health Outcomes Through Weekly Delivery
B. Sealfon 1, A. Sealfon2, C. Soderman3
1Patient Programs, RMS Medical Products, Chester, USA
2CEO, RMS Medical Products, Chester, USA
3Clinical Programs, RMS Medical Products, Uppsala, Sweden
Introduction Weekly SCIg offers a powerful set of tools to the clinician interested in patient-centered outcomes. In a 2012 European Commission study on Patient Involvement, the need for choice was a key finding. Despite patient preference for fewer or no needles, currently patients must use needles to deliver Ig products. The similar efficacy of IVIg and SCIg suggests a natural choice, yet studies show quality of life is often better with SCIg. With the ability to adjust delivery to suit patient lifestyle, SCIg provides an opportunity to empower the patient with decisions regarding their therapy. This delivers improved quality of life for the patient and economic benefits for the payer.
Objective To outline several common examples of patient choice in optimizing an SCIg therapy regimen, when switching from 3 times per week SCIg to 1 time per week.
Methods A theoretical 60 kg 185 cm patient on 10 g 20% Ig 3X/week 4 g, 2 g, 4 g into two needle sites over 1 h, was chosen as a baseline reference. Five scenarios for infusion were then modeled using a commercially-available excel-based software.
Conclusion The five challenges and identified solutions were integrated into a reference table. Patient choice and empowerment are key to satisfaction and compliance. By using a short checklist and excel-based calculator, these five examples provide a ‘toolkit’ of adjustments a clinician can make to increase patient compliance. These options work to engage the patient’s needs and desires to adjust the treatment plan,, resulting in higher patient satisfaction.
ESID-0661 PFAPA and Other Autoinflammatory Syndomes – How Can We Help Each Other?
B. Wolska-Kusnierz 1, E. Bernatowska1
1Immunology, The Children's Memorial Health Institute, Warsaw, Poland
Introduction.
Autoinflamatory disesase ( AIDs) become important challenge in clinical practice of both immunologist and rheumatologist. PFAPA (periodic fever syndrome, aphthous stomatitis, pharyngitis, adenitis) syndrome still remains a puzzle disease of unknown pathogenesis and genetic background., but is the most often diagnosed entity in this group. Educational activities are needed to improve the recognition of the disease and the quality of care for patients.
Objective. To increase knowledge about PFAPA syndrome among patients, parents and first contact physicians.
Material.
We analyzed the most frequent questions asked by patients and their parents with suspected PFAPA syndrome. On the basis, the booklet giving reasonable answers to these questions has been written.
Results.
A booklet about the PFAPA syndrome is practical and helpful tool in facilitating everyday cooperation between the patients, parents and physicians. On the initiative of the parents an online platform also has been started, where parents can discuss and exchange experiences and practical advices on PFAPA and other AIDs. On initiative of the forum annual integration meetings of 'FEBRILE' has been organised.
Conclusions.
The initiative to increase awareness and improve care on patients with autoinflammatory diseases in Poland has been started. The first results are the dissemination of educational materials on PFAPA syndrome and set up of supportive group for patients with AIDs and their families.