

Abstract
The 8ab protein of SARS-CoV is a group-specific accessory protein, which is lost when the virus was transmitted from animals to humans due to a 29-nucleotide deletion in the ORF8ab region. Here we found that 8ab protein is associated with ER membrane at luminal surface. 8ab protein was found to up-regulate the synthesis of endogenous ER-resident chaperons involved in protein folding through the activation of the transcription factor ATF6, while it showed no effect on the CHOP induction and XBP1 splicing associated with the unfolded protein response (UPR). When ectopically expressed in mammalian cells, 8ab induced the proteolysis of ATF6 and the translocation of its cleaved DNA-binding and transcription-activation domains from the ER to the nucleus. Finally, we showed that 8ab binds to the luminal domain of ATF6. These findings suggest that 8ab could modulate the UPR by activating ATF6 to facilitate protein folding and processing. Thus, the loss of 8ab in SARS-CoV through viral evolution in animals may play a role in its pathogenicity.
Keywords: SARS-CoV, 8ab protein, ER-resident chaperones, UPR, ATF6
Introduction
Severe acute respiratory syndrome (SARS) caused profound impact in over 30 countries worldwide in 2003 (Peiris et al., 2003a). A new coronavirus SARS-CoV was identified as its causative agent (Peiris et al., 2003a, Peiris et al., 2003b). It is an enveloped, positive-strand RNA virus with a genome of approximately 30 kb. Its genome contains five major open reading frames (ORFs), which encode the replicase polyproteins, the spike protein (S), envelope protein (E), membrane (M) and nucleocapsid (N) proteins (Marra et al., 2003, Rota et al., 2003). These proteins are homologous to those of other coronaviruses and are essential for viral replication and maturation. Besides these major proteins, SARS-CoV genome also encodes five to eight putative accessory proteins which show little homology with other coronaviruses and are unique to SARS-CoV (Stadler et al., 2003, Tan et al., 2006). These accessory proteins are dispensable for virus replication (de Haan et al., 2002, Yount et al., 2005). However, emerging evidence indicated that the accessory proteins could modulate cellular processes and interfere with the virus–host interaction, thereby contributing to viral virulence and pathogenesis (Sarma et al., 2002, Sola et al., 2003).
Among the accessory proteins of SARS-CoV, proteins encoded by ORF8 have drawn particular attention. The genetic and epidemiological studies suggested a potential role of ORF8ab in zoonotic transmission of SARS-CoV (Guan et al., 2003). The genomes of SARS-CoV isolated from civet cats and some human isolates of the early-stage epidemic contain a single ORF8ab, while viruses isolated from the middle-late phase of the epidemic contain a 29-nucleotide deletion that results in two separated ORFs, ORF8a and ORF8b (Fig. 1A). The single ORF8ab encodes a protein of 122 amino acids, while ORF8a and ORF8b encode two small proteins of 39 and 84 amino acids, respectively (Fig. 1B) (Chinese, 2004, Guan et al., 2003). These observations suggested that proteins 8a, 8b and 8ab might have different biochemical properties or functions and thereby contributed differently to zoonotic transmission and pathogenesis of SARS-CoV. Recently it was shown that the 8ab protein is a functional viral protein that is lost when SARS-CoV was transmitted to the human population. In contrast, proteins 8a and 8b are unstable and probably non-functional (Oostra et al., 2007). The 8ab protein has been shown to be translocated to the ER lumen, become N-glycosylated and remain stably in the ER (Oostra et al., 2007). However, the biological function of 8ab remains unknown.
Fig. 1.

Schematic representation of ORF8 region in the genome of SARS-CoV isolated from civet cats and humans. (A) A schematic diagram shows the different genome organization in ORF8 region of SARS-CoV isolated from civet cats and humans infected during the middle/late phase of the SARS epidemic. The ORF8 region of the animal SARS-CoV contains the intact ORF8 and encodes a single protein 8ab, whereas that of the human isolates splits into two ORFs and encodes two proteins 8a and 8b. (B) Alignment of the deduced amino acid sequences of 8a, 8b and 8ab proteins. The signal sequences (a.a. 1–16) are indicated in bold and the predicted transmembrane domain (a.a. 2–24) is underlined by a black line. The predicted N-glycosylation site is indicated by a black star.
The endoplasmic reticulum is the place for folding and assembly of newly synthesized secretory or transmembrane proteins. Accumulation of unfolded or misfolded proteins activates multiple cell-signaling pathways, collectively known as the unfolded protein response (UPR), to decrease the protein load in the ER lumen (Schroder and Kaufman, 2005). The UPR alleviates the stress by attenuation of translation in general and up-regulation of transcription of the genes encoding proteins that facilitate protein folding and degradation (Rutkowski and Kaufman, 2004, Schroder and Kaufman, 2005). Three ER-resident transmembrane proteins, activating transcription factor 6 (ATF6), PKR-like ER kinase (PERK) and inositol-requiring enzyme 1 (IRE 1) have been identified as the transducers of the UPR (Harding et al., 2000, Shen et al., 2002, Wang et al., 1998). The inhibition of protein synthesis is mediated by PERK, which is an eIF2α protein kinase and is activated very rapidly following ER stress (Harding et al., 2000). PERK also promotes the selective translation of transcription factor ATF4, which, in turn, stimulates the transcription of cyclic AMP (cAMP) response element binding transcription factor homologous protein (CHOP; also known as growth arrest and DNA damage-inducible gene 153 (GADD153)), which is implicated in cell death induced by ER stress (Oyadomari and Mori, 2004, Zinszner et al., 1998). IRE1 is a type I transmembrane protein, and its C-terminal cytosolic fragment contains an endoribonuclease domain that directs the splicing of XBP1 mRNA (Lee et al., 2002, Wang et al., 1998, Yoshida et al., 2001). The spliced XBP1(S) yields an active transcription factor, which up-regulates the transcription of the selective genes involved in ER-associated protein degradation (ERAD) to degrade misfolded proteins (Lee et al., 2002, Yoshida et al., 2003). The third mediator for transcriptional induction by ER stress is the b-ZIP transcription factor ATF6. ATF6 is a type II transmembrane protein with its N-terminal DNA-binding domain facing cytosol and its C-terminus located to the ER lumen (Haze et al., 1999). Upon ER stress, ATF6 translocates to the Golgi, where it is processed to its active form through sequential cleavages by S1P and S2P proteases at the luminal and the transmembrane domains (Chen et al., 2002, Shen et al., 2002, Ye et al., 2000). The N-terminal cytosolic fragment of ATF6 is thus released, and then moves to the nucleus, along with the constitutively expressed transcription factor NF-Y, transactivating the target genes containing the ER stress response element (ERSE) (Li et al., 2000, Yoshida et al., 1998, Yoshida et al., 2000). The ERSE sequence is a bipartite sequence of CCAAT-N9-CCACG, which can be found in the promoters of ER chaperons and other proteins that are required for protein folding (Yoshida et al., 1998). Consequently, the activation of ATF6 in the UPR facilitates protein folding. During the replication of SARS-CoV, a large amount of viral proteins are processed in the ER (Stadler et al., 2003); the accumulation of nascent and unfolded SARS-CoV proteins in the ER lumen may cause ER stress (Chan et al., 2006). Since 8ab is retained in the ER (Oostra et al., 2007), we set out to investigate the possibility that 8ab may modulate the UPR.
In this study, we first confirmed that the 8ab is an ER-resident protein; however, different from the published report (Oostra et al., 2007), we found that 8abHA is strongly associated with ER membrane and behaves like an integral membrane protein. We next demonstrated that the 8ab protein could up-regulate the synthesis of ER chaperons, including GRP78, GRP94 and CRT, which are known for facilitating protein folding (Rutkowski and Kaufman, 2004). Furthermore, we presented evidence that 8ab induced the proteolysis of ATF6 and nuclear translocation of the processed N-terminus of ATF6. However, 8ab showed no effect on activation of the other two UPR transducers, PERK and IRE1. Taken these results together, our findings suggested that 8ab could modulate the UPR by specifically activating ATF6 and implicated the biological significance of the 29-nucleotide deletion in ORF8ab of SARS-CoV.
Results
8ab is an ER-luminal-oriented membrane-associated protein
The 8ab protein has been reported to localize to the ER and retained in the ER lumen as a soluble protein (Oostra et al., 2007). However, the function of 8ab remained unknown. So far, the attempts to raise antibodies using full-length recombinant 8ab or peptide antigens have failed. Thus, to investigate the biochemical properties and function of 8ab, HA epitope was tagged at the C-terminus of 8ab. We first performed confocal laser scanning microscopy to determine the subcellular localization of 8abHA. As shown in Fig. 2A, 8abHA protein was co-localized with an ER marker calnexin, suggesting its localization to the ER. Treatment with peptide-N-glycosidase F (PNGase F) or Endo H reduced the molecular mass of 8abHA by around 3 kD (Fig. 2B), indicating that 8abHA is a glycoprotein and is retained in the ER but not transported to the Golgi complex, in agreement with the published results (Oostra et al., 2007). Therefore, the HA tag did not affect the behavior of 8ab protein.
Fig. 2.

8ab is an ER-resident membrane-associated protein. (A) 8abHA is localized to the ER. VeroE6 cells were transiently transfected with pCI8abHA encoding full-length 8ab tagged with HA at its C-terminus. Cells were harvested at 36 h after transfection and then labeled with antibodies against HA epitope and antibodies against calnexin. After mounting, cells were visualized by confocal microscopy. (B). Endoglycanase susceptibility of 8abHA. Cell lysates of HeLa transfected with pCI-8abHA for 30 h were subjected to N-glycosidase F (PNGase F) or Endoglycosidase H (Endo H) digestion. The digested samples were subjected to immunoblot analysis using anti-HA antibodies. (C). Detergent solubilization of 8abHA from microsomal extracts. Microsomes purified from 8abHA-transfected HeLa cells were incubated with PBS containing no detergent, 0.1% digitonin, 0.2% digitonin or 1% deoxycholate (DOC), respectively. After incubation at 4 °C for 30 min, samples were centrifuged, and both pellets (P) and supernatant (S) were subjected to SDS-PAGE and analyzed by Western blot with anti-HA, anti-calnexin and anti-calreticulin antibodies. (D) 8ab is resistant to carbonate extraction. Microsomal extracts were re-suspended in PBS, 1 M NaCl or 100 mM Na2CO3, and incubated for 30 min at 4 °C. Samples were centrifuged, and supernatant (S) and pellet (P) were analyzed with anti-HA and anti-calnexin antibodies.
8ab was characterized as a protein soluble in the ER lumen, with its signal sequence (a.a. 1–16) being cleaved in the reported study, based on the analysis of the in vitro translated protein (Oostra et al., 2007). However, the N-terminus (a.a. 2–24) of ORF8ab is predicted to be a transmembrane domain based on several topology prediction programs, including TopPred, TMpred and SOSUI (data not shown). To investigate the possible membrane association of 8ab protein, microsomes prepared from HeLa cells transfected with pCI8abHA were treated with various detergents (Shen and Hendershot, 2005). An ER-resident membrane protein calnexin (CRN) and a soluble ER luminal protein calreticulin (CRT) were used as controls. As an integral membrane protein, calnexin remained partially associated with ER vesicles in both 0.1 and 0.2% digitonin, whereas CRT, a soluble ER luminal protein, was completely released into soluble fraction by the same treatments. Similar to the pattern observed for calnexin, the 8abHA remained partially associated with microsomes in 0.1% and 0.2% digitonin and was solubilized in the stronger detergent 1% DOC (Fig. 2C). These results suggested that 8abHA was associated with ER membranes. Microsomes prepared from 8abHA-expressing cells were also subjected to treatment with 1 M NaCl and 100 mM Na2CO3, pH 11.5. High-salt extraction will weaken the interaction of peripheral proteins with membrane, and treatment with 100 mM sodium carbonate will release the peripheral proteins by transforming microsomes into membrane sheets. As shown in Fig. 2D, 8abHA remained predominantly associated with microsomes when treated with 1 M NaCl. Although a small amount of 8abHA was extracted by sodium carbonate treatments, the majority of 8abHA remained in pellets fractions (Fig. 2D). Taken together, these observations indicated that 8abHA behaved like an integral membrane protein rather than a soluble protein.
We next determined the orientation of 8abHA in the ER by examining selective accessibility of 8ab in various detergents. We tagged an HA epitope at C-terminus and 3XFLAG epitope at N-terminus of 8ab (Fig. 3A). Saponin causes total permeabilization, whereas low concentrations of digitonin selectively permeabilize the plasma membrane while leaving the ER membrane intact. Calreticulin (CRT), a chaperone found in the ER lumen, was used as a control. As shown in Fig. 3B, after treatment with 0.05% saponin, both epitopes HA and FLAG were revealed by immunofluorescence. CRT was also recognized by anti-CRT antibody after this treatment. In contrast, when treated with low concentrations of digitonin, only FLAG-tagged 8ab, but not C-terminally HA-tagged 8ab, was detected, indicating that the N-terminus of 8ab was cytosolically oriented, whereas its C-terminus is buried within the ER. Under this condition, CRT could not be recognized either. These results indicated that the epitope tagged to the C-terminus of 8ab is oriented to the ER lumen whereas that of the N-terminus is exposed to the cytosolic side.
Fig. 3.

8ab protein is ER-luminal-oriented. (A). Diagram of constructs used for determining the orientation of 8ab within ER. A 3XFLAG epitope was tagged at the N-terminus of pCI8ab, and an HA epitope was tagged at the C-terminus. (B) 8ab is ER-luminal-oriented. HeLa cells were transiently transfected with pCI, pCI-3XFLAG8ab or pCI-8abHA as indicated. After 30 h, cells were fixed and subjected to total permeabilization with 0.05% saponin or selective permeabilization with 0.000125% digitonin, and then labeled with anti-Calreticulin, anti-HA and anti-FLAG antibodies. After mounting, cells were visualized by confocal microscopy.
Up-regulation of ER-resident chaperons by 8abHA
Having identified 8ab protein as an ER luminal-oriented membrane-associated protein, we speculated that 8ab protein might be involved in the regulation of UPR to overcome the cellular responses during SARS-CoV infection. Up-regulation of the genes encoding ER chaperones that participate in protein folding and assembly to prevent the accumulation and aggregation of unfolded proteins in the ER is an indicative event of UPR (Rutkowski and Kaufman, 2004). One of the best characterized ER chaperon proteins is glucose regulated protein 78 (GRP78). Therefore, we first measured the GRP78 promoter (nt− 304–+ 7) (Yoshida et al., 1998) activity in the presence and absence of 8abHA. As seen in Fig. 4A, 8abHA activated the expression of luciferase from the GRP78 promoter by 3.8-fold when compared with the control plasmid. Thapsigargin (TG), which blocks the calcium ATPase pump, was used as the positive control. Treatment of cells with TG led to a 4.4-fold increase of luciferase activity over that of untreated cells. These results indicated that protein 8abHA could induce the expression of the genes with the GRP78 promoter. We further investigated the induction of endogenous GRP78 by 8abHA using indirect immunofluorescence study. As shown in Fig. 4B, Vero E6 cells that express 8abHA showed stronger staining of endogenous GRP78. These data indicated that 8abHA increases the expression level of endogenous GRP78. Immunoblotting analysis also showed that the expression of 8abHA was accompanied by a higher level of endogenous GRP78 (Fig. 4C. top panel). In addition, we found that two other ER-resident chaperons, GRP94 and CRT, were also up-regulated in 8ab-expressing cells (Fig. 4C. second and third panels). These results suggested that protein 8ab is capable of up-regulating the synthesis of ER-resident chaperons.
Fig. 4.

8ab up-regulates the synthesis of ER-resident chaperons. (A) 8ab protein activates GRP78 promoter. HeLa cells were transiently co-transfected with pCI8abHA and a reporter plasmid containing firefly luciferase gene under the control of GRP78 promoter for 40 h (gray bar). Control cells were co-transfected with reporter plasmids and empty pCI vector for 24 h, and then either treated or untreated with TG 500 nM for an additional 16 h (black or white bar). In each case, pRL-TK encoding Renilla luciferase was co-transfected as an internal control. The cell lysates were harvested and assayed for firefly and Renilla luciferase activities. The results were normalized with Renilla luciferase activities and were averaged with S.E. of three experiments. (B). Indirect immunofluorescence analysis of GRP78 in 8ab-expressing cells. Vero E6 cells were transiently transfected with pCI8abHA for 30 h, and labeled with anti-HA and anti-GRP78 antibodies. Bound primary antibodies were revealed with Alexa Fluor 568- and Alexa Fluor 488-conjugated secondary antibodies, respectively. (C) Immunoblotting analysis of ER-resident chaperons in transfected cells. HeLa cells were transiently transfected with pCI-8abHA for 30 h, or pCI for 24 h, then treated or untreated with TG 500 nM for an additional 6 h. Cell lysates were used for Western blot analysis using anti-KDEL (for GRP78 and GRP94) and anti-calreticulin antibodies. Expression level of actin was used as loading control.
Activation of ATF6 by 8ab
The promoters of ER chaperones contain the ER stress response element (ERSE); the binding of ATF6 to ERSE is required for the transcriptional activation of these chaperons (Li et al., 2000, Yoshida et al., 1998). Since 8ab was demonstrated to up-regulate the synthesis of ER-resident chaperons (Fig. 4A), we further determined whether 8ab could induce the ATF6-dependent transcription using a luciferase reporter, p5XATF6-GL3. The luciferase gene in the p5XATF6-GL3 is under the control of the c-fos minimal promoter and five tandem copies of the ATF6 consensus binding site TGACGTGG, which is similar to the opposite strand of the 3′ half of ERSE (Wang et al., 2000). In Hela cells transiently transfected with 8abHA, the luciferase activity of p5XATF6-GL3 reporter increased about five-fold as compared to cells without 8ab (Fig. 5A). As a control, the transcription of p5XATF6-GL3 reporter was stimulated by treatment with TG by about fourteen-fold (Fig. 5A). Although the effect of 8abHA on p5XATF6-GL3 reporter is relatively modest, this finding, nevertheless, indicates that 8ab can induce the ATF6-dependent transcription and suggests that 8ab can activate ATF6.
Fig. 5.

Activation of ATF6 by 8ab. (A) 8ab protein induces the ATF6-dependent transcriptional activity. HeLa cells were transiently co-transfected with pCI8abHA and the reporter plasmid p5XATF6-GL3 for 40 h (gray bar). Control cells were co-transfected with reporter plasmids and empty pCI vector for 24 h, and then induced with or without TG 500 nM for an additional 16 h (black or white bar). In each case, pRL-TK encoding Renilla luciferase was co-transfected as an internal control. The cell lysates were harvested for dual luciferase assay as described. (B) Immunoblotting analysis of the proteolysis of ATF6. HeLa cells were co-transfected with 0.5 μg of 3XFLAG-ATF6 and different amounts of pCI-8abHA as indicated (lanes 3–5). After 30 h, cell lysates were harvested for Western blot analysis with anti-FLAG and anti-HA antibodies. Control cells were co-transfected with 0.5 μg of 3XFLAG-ATF6 and empty vector for 24 h, and then treated or untreated with TG 500 nM for an additional 6 h (lane 2 and lane1). The full-length 3XFLAG-ATF6 (90kD) and the processed N-terminal domain of 3XFLAG-ATF6 (50 kD) are indicated. (C) Indirect immunofluorescence analysis of the nuclear translocation of 3XFLAG-ATF6. HeLa cells were transiently co-transfected with 3XFLAG-ATF6 and pCI8abHA for 30 h (panels i–l), or transfected with 3XFLAG-ATF6 and empty vector for 20 h, and then treated (panels e–h) or untreated with 5 μg/ml of tunicamycin for an additional 4 h (panels a–d). After being fixed and permeabilized, cells were labeled with anti-HA and anti-FLAG antibodies. Cells were co-stained with DAPI to reveal the nucleus. In panels e and i, white arrows point to the cells exhibiting nuclear translocation of processed 3XFLAG-ATF6, and gray arrows point to cells in which ATF6 exhibits the ER distribution pattern. At higher magnification (panel m), partial co-localization of ATF6 and 8ab in the ER is revealed.
The activation of ATF6 depends on the proteolysis of ATF6 and nuclear movement of its cleaved N-terminal fragment, which contains the b-ZIP DNA-binding and transcriptional activation domains. In response to ER stress, ATF6 translocates from the ER to the Golgi, where it is cleaved by S1P and S2P to its active form (Chen et al., 2002, Shen et al., 2002). The cleaved N-terminal domain of ATF6 is then translocated to the nucleus, where it activates the transcription of selective genes containing ERSE (Chen et al., 2002, Wang et al., 2000). We therefore investigated the possible proteolysis of ATF6 by 8abHA. We first tried to determine whether 8ab could induce proteolytic cleavage of the endogenous ATF6, but could not get unequivocal results because of the lack of an antibody that could specifically recognize the N-terminus of ATF6. Therefore, ATF6 tagged with three tandem copies of the FLAG epitope at its N-terminus was used (Chen et al., 2002, Shen et al., 2002, Shen and Prywes, 2005). In unstressed conditions, the 3XFLAG-ATF6 was synthesized as a 90 kD protein; when treated with TG, it was cleaved to generate a cytosolic fragment (50 kD) detectable with anti-FLAG antibody (Fig. 5B, lane 1 and lane 2). In the presence of 8ab, the 50 kD fragment was also detected; its amount correlated with the expression level of 8abHA (Fig. 5B, lanes 3, 4 and 5). These results suggested that 8abHA is capable of inducing cleavage of ATF6 to release its N-terminal cytosolic fragment.
The cleaved N-terminal fragment of ATF6 has been shown to move to the nucleus and activate the expression of ER stress target genes by binding to the ERSE (Chen et al., 2002, Shen et al., 2002, Ye et al., 2000). We therefore examined the possible nuclear translocation of the N-terminal cytosolic fragment of ATF6. Immunofluorescence studies showed that the 3XFLAG-ATF6 was present in the perinuclear and cytoplasmic regions in a manner consistent with its ER localization (Fig. 5C, panels a–d). Tunicamycin treatment for 4 h caused the movement of the N-terminal domain of ATF6 to the nucleus as expected (Fig. 5C, panels e–h, white arrows). When co-expressed with 8abHA, without the tunicamycin treatment, nuclear translocation of N-terminal domain of ATF6 was also observed in the cells expressing 8abHA (Fig. 5C, panels i–l, white arrows). In contrast, in those cells without 8ab, ATF6 was localized mainly in the cytoplasm (Fig. 5C, panels i–l, gray arrows). These data indicated that 8abHA can induce the proteolytic cleavage of ATF6 and also the movement of the cleaved N-terminal domain to the nucleus.
Association of 8abHA and ATF6
The immunofluorescence study revealed a substantial co-localization of ATF6 and 8ab at the ER (Fig. 5C, m). We therefore investigated the possibility of association of ATF6 and 8ab by co-immunoprecipitation. For this purpose, we used a full-length ATF6 bearing point mutations at its S1P cleavage site, ATF6S1P−, to prevent the possible loss of binding with 8abHA due to S1P cleavage when co-immunoprecipitation was performed (Chen et al., 2002, Shen et al., 2002, Ye et al., 2000). In immunofluorescence study, the FLAG-tagged ATF6S1P− was localized to the perinuclear and cytoplasmic region in a manner consistent with the ER distribution pattern under normal condition, similar to the wild-type ATF6 (Fig. 6A, a–d). When treated with TM for 4 h, 3XFLAG-ATF6S1P− did not move to the nucleus; instead, it showed a Golgi-like distribution (Fig. 6A, panels e–h, white arrows). This observation indicated that the cleavage of ATF6, and consequently the release of its N-terminal fragment, is impaired by the mutation at the S1P cleavage site. When co-transfected with 8abHA, 3XFLAG-ATF6S1P− also moved to the Golgi and was retained there in 8ab-expressing cells (Fig. 6A, panels i–l, white arrows). Additionally, immunofluorescence study also showed co-localization of 8ab and ATF6S1P− at the site adjacent to the Golgi apparatus, probably the ER exiting site (Fig. 6A. panel m, gray arrowheads). This observation further suggested the possibility of the interaction between 8ab and ATF6. To investigate the association of 8ab and ATF6 further, we co-expressed 3XFLAG-ATF6S1P− and 8abHA in HeLa cells and then immunoblotted the anti-FLAG immunoprecipitates with monoclonal anti-HA antibodies. To confirm the specificity of the interaction between 8ab and ATF6, a hepatitis C virus (HCV) protein NS4B, which is an integral ER membrane protein (Lundin et al., 2003), was used for comparison. HCV NS4B was tagged with HA at its C-terminus and co-transfected with 3XFLAG-ATF6S1P− into cells. As shown in Fig. 6B, 8abHA was coprecipitated with ATF6S1P−; in contrast, HCV NS4B was not. These results indicated that 8ab protein is specifically associated with ATF6.
Fig. 6.

Association of 8ab with ATF6. (A) Indirect immunofluorescence analysis of 3XFLAG-ATF6S1P−. HeLa cells were transiently co-transfected with 3XFLAG-ATF6S1P− and pCI8abHA for 30 h (panels i–l), or transfected with 3XFLAG-ATF6S1P− and empty vector for 20 h, and then treated with (panels e–h) or without 5 μg/ml of tunicamycin (panels a–d) for an additional 4 h. Indirect immunofluorescence assay was performed as described. In panels e and i, white arrows point to the cells in which ATF6 exhibits Golgi localization, and gray arrows point to the cells in which ATF6 exhibits ER localization. In panel m, white arrowheads point to the Golgi, and gray arrowheads point to the site adjacent to the Golgi where 8ab co-localized with ATF6. (B) Co-immunoprecipitation of ATF6 and 8ab. Left panel, HeLa cells were transiently co-transfected with 8abHA and 3XFLAG-ATF6S1P− or empty vector. After 24 h, cell lysates were harvested and immunoprecipitated with anti-FLAG antibodies. 8abHA and ATF6 in the immunoprecipitates were detected using anti-HA or anti-FLAG antibodies as indicated. Right panel, HeLa cells were transiently co-transfected with HCV NS4B-HA and 3XFLAG-ATF6S1P− or empty vector. NS4B-HA or ATF6 contents in the immunoprecipitates were analyzed as left panel.
Since 8ab is localized to the ER lumen, we suspected that 8ab interacts with ATF6 through the luminal domain of ATF6. To test this possibility, we constructed 3XFLAG-ATF6 (418) (lacking the majority of the C-terminal luminal domain but retaining the transmembrane domain) and 3XFLAG-ATF6 (373) (containing only the N-terminal cytoplasmic domain) (Fig. 7A) and determined whether these two ATF6 mutants could bind to 8abHA. As shown in Fig. 7B, the full-length ATF6S1P− was found to be associated with 8ab (Fig. 7B, lane 6), while ATF6 (418) and ATF6 (373) (Fig. 7B, lane 7, lane 8) were not. This result indicated that neither the cytoplasmic domain nor transmembrane domain is required for its association with 8ab. Therefore, the luminal domain of ATF6 is the likely binding domain.
Fig. 7.

The luminal domain of ATF6 is required for the association with 8ab. (A) Diagram of constructs encoding ATF6S1P−, ATF6ATF6 (475), ATF6 (430), ATF6 (418), and ATF6 (373). 3XFLAG was tagged at N-terminus of each protein. The transcriptional activation (TAD), basic leucine zipper (b-ZIP) and transmembrane domain (TM) are indicated. (B) ATF6 (418) or ATF6 (317) are not co-immunoprecipitated with 8ab. HeLa cells transiently transfected with the indicated plasmids were immunoprecipitated with anti-FLAG antibodies and immunoblotted with either anti-FLAG (top) or anti-HA antibodies (bottom). (C) Co-immunoprecipitation of ATF6 (475) and 8ab. HeLa cells transiently transfected with the indicated plasmids were immunoprecipitated with anti-FLAG antibodies and immunoblotted with either anti-FLAG (top) or anti-HA antibodies (bottom).
To further map 8ab-binding region in the luminal domain of ATF6, we constructed two other ATF6 mutants, ATF6 (430) (lacking a.a. 431–670), and ATF6 (475) (lacking a.a. 476–670), and then determined their interaction with 8abHA. We found that ATF6 (475) and the full-length ATF6, could bind with 8abHA (Fig. 7C, lane 7 and lane 8), whereas the shorter proteins ATF6 (430) and ATF6 (418) had significantly reduced binding activities (Fig. 7C, lanes 9 and lane 10). This result indicated that the region a.a. 430–475 of the luminal domain of ATF6 is necessary and sufficient for its interaction with 8abHA. Combining these results together, we concluded that 8abHA could bind to the region a.a. 431–475 of the luminal domain of ATF6, causing the movement of the latter into the Golgi and inducing the proteolytic cleavage, thereby releasing its N-terminal fragment to the nucleus.
No evidence for CHOP induction or XBP-1 splicing by 8ab
To determine if 8ab modulates UPR by activation of ATF6 specifically, we assessed the effects of 8ab on the other two regulators of UPR, PERK and XBP-1. Activation of PERK will lead to phosphorylation of eukaryotic initiation factor 2α (eIF2α), resulting in the attenuation of general protein translation. However, ATF4 can bypass the translation inhibition and induce the expression of CHOP, which promotes apoptotic cell death (Oyadomari and Mori, 2004, Zinszner et al., 1998). To determine whether the expression of CHOP is induced by 8ab, we analyzed the expression levels of CHOP protein by indirect immunofluorescence study and Western blotting. In indirect immunofluorescence staining, we found that CHOP was barely detectable in 8ab-expressing cells (Fig. 8A, panels d–f). In contrast, treatment with TG leads to induction of CHOP and its accumulation on the nucleus (Fig. 8A, panels d–f). This result was confirmed by Western blotting. No CHOP induction could be observed (Fig. 8B, the second panel), although the synthesis of GRP78 was up-regulated (Fig. 8B, the top panel), in 8ab-expressing cell lysates. That over-expressing of 8ab could not induce the synthesis of CHOP suggested that 8ab did not activate the PERK-ATF4-CHOP signaling pathway.
Fig. 8.

No CHOP induction in 8ab-expressing cells. (A) Indirect immunofluorescence analysis of CHOP. HeLa cells were transiently transfected with pCI8abHA for 30 h (panels g–i), or transfected with pCI for 24 h, and then treated (panels d–f) or untreated with 5 μg/ml of tunicamycin for an additional 4 h (panels a–c). After being fixed and permeabilized, cells were labeled with anti-HA and anti-CHOP antibodies. Cells were co-stained with DAPI to reveal the nucleus. (B) Immunoblotting analysis of CHOP and GRP78 in 8ab-expressing cells. HeLa cells were transiently transfected with pCI8abHA for 30 h, or with pCI for 24 h, then treated or untreated with TG 500 nM for an additional 6 h. Cell lysates were used for Western blot analysis using anti-GRP78, anti-CHOP and anti-HA antibodies. Expression level of actin was used as loading control.
Next, we asked whether 8ab causes the activation of the endoribonuclease IRE1. Activation of IRE1 leads to unconventional mRNA splicing of the transcription factor XBP-1 by removing a 26-bp intron from the xbp-1 transcript (Lee et al., 2002). The spliced XBP1 mRNA results in the expression of the active form of XBP1 protein, which is involved in the transcriptional induction of the genes involved in ERAD pathway (Rutkowski and Kaufman, 2004). To determine whether 8ab activates IRE1, leading to alternative splicing of XBP1, we performed reverse transcription-PCR to detect possible presence of the spliced XBP-1 mRNA. As shown in Fig. 9 , while the treatment with TG resulted in the induction of an alternatively spliced form of XBP1, the expression of 8ab did not (Fig. 9, top panel, lanes 2 and 3). This result indicates that 8ab did not have an effect on the activation of IRE1 and XBP1 splicing. Taking these results together, we proposed that 8ab could modulate UPR by specifically inducing the proteolysis of ATF6, but had no effects on CHOP induction and XBP1 splicing.
Fig. 9.
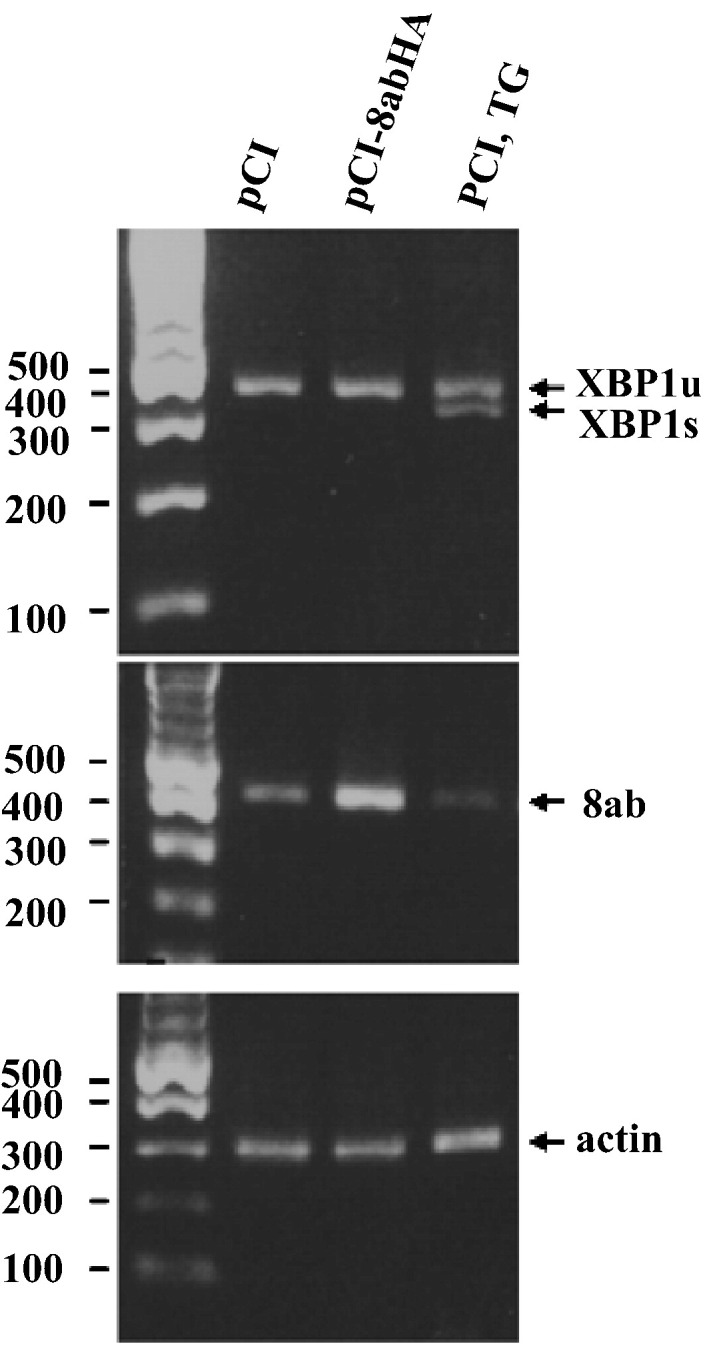
No XBP1-splicing in 8ab-expressing cells. Total RNA was prepared from HeLa cells transfected with pCI8abHA, or pCI or treated with 5 μg/ml of tunicamycin for 4 h, and subjected to reverse transcription-PCR to amplify cDNA of XBP1. The PCR products of XBP1 were separated by 2% agarose gel electrophoresis. Unspliced and spliced forms of XBP1 transcript were indicated.
Discussion
The data presented in this study indicated that 8ab protein of SARS-CoV binds to the ER-localized transcription factor ATF6 and activates its proteolysis and nuclear translocation, thereby increasing the expression of endogenous cellular ER-resident chaperones. We also showed that over-expression of 8ab had no effects on CHOP induction and splicing of XBP1 mRNA. Our results suggested that 8ab protein could differentially modulate UPR by inducing the proteolysis of ATF6 to facilitate viral protein folding during virus replication, and are important for investigating the biological significance of the loss of protein 8ab in SARS-CoV upon transmission from animals to humans.
The 8ab protein can be stably expressed in cell culture system and is thus likely a functional glycoprotein (Oostra et al., 2007). In contrast, two small proteins 8a and 8b found in the human SARS-CoV isolates during the later phase of SARS epidemic are probably non-functional, since their expression is hardly detected in mammalian cells (Oostra et al., 2007). Our experiments also showed that proteins 8a and 8b were very unstable in mammalian cells, but could be stabilized by the addition of proteasome inhibitor, MG132 (data not shown). These results are consistent with the notion that the 29-nt deletion disrupts the functional expression of ORF8 (Oostra et al., 2007). Therefore, we focused our experiments on investigating the potential function of protein 8ab. In previously published data, ER localization of 8ab was determined by using 8ab-EGFP fusion protein (Oostra et al., 2007). In our study, HA-tagged 8ab was used to determine its cellular localization (Fig. 2A). Both studies indicated that the recombinant 8ab is localized to the ER. Moreover, both studies found that 8ab was glycosylated and sensitive to Endo H digestion (Fig. 2B). These observations demonstrated that 8ab bearing an HA tag or fused to EGFP is targeted to and retained in the ER. The previous study suggested that the signal sequence of 8ab is cleaved off from the in vitro transcribed/translated 8ab and concluded that the protein was soluble in the ER lumen (Oostra et al., 2007). However, the ER retention signals which are usually found in the sequences of soluble proteins in the ER lumen are absent from the sequences of protein 8ab. In contrast to the published conclusions, we found in this study that 8abHA was extracted from microsome only by the strong detergent 1% DOC, suggesting a strong association of 8ab with the ER membrane, in a manner very similar to that of calnexin (Fig. 2C). Furthermore, treatment with sodium carbonate did not significantly release 8ab from membranes (Fig. 2D). These results suggested that 8abHA is associated with ER membrane and behaves like an integral membrane protein. The N-terminal sequence (2–24 a.a.) of 8ab protein appears to be highly hydrophobic and predicted to be a transmembrane domain based on several topology prediction programs. It is likely that the N-terminal hydrophobic domain causes 8ab to anchor to the ER membrane. Alternatively, 8ab may be associated with the membrane through interaction with other integral membrane proteins. We further established the orientation of 8ab by the selective permeability technique, and confirmed that the C-terminus of 8ab is facing toward the ER lumen (Fig. 3B). Therefore, 8ab protein is probably attached to the luminal surface of the ER membrane, and thus is well-suited to interact with ER sensor GRP78 or luminal domain of UPR transducers to modulate the UPR.
As a factory for protein folding and post-translation modification, the ER is an essential organelle for viral replication and maturation. During productive infection, large amounts of viral proteins are synthesized in the ER, where unfolded or misfolded viral proteins cause stress and induce the UPR. It has been observed that virus infection causes ER stress, and that viruses modulate the UPR by various strategies which could lead to either cell survival or cell death (He, 2006, Tardif et al., 2005). Therefore, differential regulation of the UPR affects viral pathogenesis and/or replication. As an example, hepatitis C virus (HCV) replication is associated with the ER, where it causes stress, and HCV also manipulates UPR to promote HCV persistence and pathogenesis (Tardif et al., 2005). A previous report indicated that, when ectopically expressed in mammalian cells, the E2 protein from HCV activates the promoter of GRP78 and is stably bound to GRP78 (Liberman et al., 1999). It has also been reported that the E2 protein binds to PERK and inhibits PERK phosphorylation and kinase activity, thereby enhancing protein translation (Pavio et al., 2003). HCV also modulates ATF6 and IRE-XBP-1 pathways to promote the viral protein translation (Tardif et al., 2004, Tardif et al., 2002). During the replication of SARS-CoV, the accumulation of nascent and unfolded SARS-CoV proteins in the ER lumen causes ER stress (Chan et al., 2006). Recent work by Chan et al. (2006), showed that the spike (S) protein of SARS-CoV induces transcriptional activation of ER chaperones GRP78/94 by differentially regulating the UPR. It is indicated that S protein specifically stimulates PKR-like ER kinase but shows no effect on ATF6 and XBP1, and it is proposed that SARS may use this strategy to enhance the folding and processing of SARS-CoV proteins during viral replication. In this study, we demonstrated that 8ab protein of SARS-CoV stimulates the proteolysis of ATF6 (Fig. 5B) and the transition of the cleaved N-terminal domain from the ER to the nucleus (Fig. 5C). Consistent with this result, we also observed the transcriptional activation of GRP78 and an elevated level of ER-resident chaperons which is regulated by ATF6 in 8ab-expressing cell (Fig. 4C). This result suggested that 8ab might represent another strategy of facilitating viral protein folding to combat unfavorable cellular response during SARS-CoV replication.
We found that 8ab could induce the proteolysis of wild-type ATF6 (Fig. 5B) and the Golgi retention of ATF6S1P− (Fig. 6A). These observations indicated that 8ab can trigger ATF6 exit from the ER to the Golgi, where it is cleaved by proteases S1P and S2P. It has been demonstrated that the luminal domain of ATF6 is necessary and sufficient to cause the translocation of ATF6 from the ER to the Golgi (Chen et al., 2002, Shen et al., 2005). GRP78, which binds to three distinct regions (a.a. 430–475, a.a. 476–550, and a.a. 551–670) of the luminal domain of ATF6, is the regulator of ATF6 activation (Shen et al., 2002). Binding of GRP78 masks two independent Golgi localization signals (GLSs) mapped to the a.a. 430–475 and a.a. 476–500 on the C-terminal luminal domain of ATF6, and thus blocks its translocation in unstressed cells (Shen et al., 2002). Upon ER stress, dissociation of GRP78 from ATF6 exposes GLSs in the ATF6 and allows its translocation to the Golgi (Shen et al., 2002). Our results indicated that the 8ab-binding region on the ATF6 overlaps with one of the GRP78-binding regions; the interaction between 8ab and ATF6 probably affects the inhibitory interaction of GRP78 and ATF6, so that GLSs become accessible to the ER export machinery. Alternatively, since we found that 8ab co-localized with ATF6 in the ER exiting site and bound to one of the GLS, 8ab may be an adaptor linking ATF6 with ER exporting machinery and concentrates the exporting-complex at the ER exiting site, thus facilitating its trafficking to the Golgi. However, further experiments are needed to elucidate the precise mechanism by which 8ab induces ATF6 trafficking from the ER to the Golgi.
In this study, we found that 8ab could up-regulate the synthesis of ER chaperones including GRP78, GRP94 and calreticulin via inducing the proteolysis of ATF6. However, the other two branches of UPR mediated by PERK and IRE1 are not activated by over-expression of 8ab. ER chaperons, CHOP, IRE1 and XBP1 play different, and sometimes opposing, roles in UPR on regulation of cell death and cell survival. The capability of 8ab to differentially modulate UPR would be beneficial to the replication of SARS-CoV in the infected cells. Up-regulating the synthesis of ER chaperones can facilitate viral protein processing, while avoiding induction of PERK and IRE1 can prevent the inhibition of viral protein translation and degradation of viral proteins. In addition, activation of CHOP and IRE1 could be cytopathic due to the activation of proapoptotic signaling pathway. CHOP is a proapoptotic transcription factor, which triggers cell apoptosis by repressing the expression of Bcl-2 (McCullough et al., 2001), and activation of IRE1 recruits the tumor necrosis factor receptor-associated factor 2 (TRAF2) and c-Jun N-terminal kinase (c-JNK), thus transmitting the signal via apoptotic signaling kinase c-JNK (Urano et al., 2000). Thus, the lack of CHOP induction and IRE1 activation by 8ab protein may avoid the potentially deleterious effects of ER stress-induced apoptosis during virus infection. Therefore, we suggest that protein 8ab plays a role for alleviating ER stress by specifically activating ATF6 to facilitate protein folding and processing but exerting no effect on PERK and IRE1. However, further studies are required to investigate how 8ab affects cell fate by differentially modulating UPR and to understand whether the loss of 8ab protein during the evolution of SARS-CoV from civet cats to humans contributes to its enhanced pathogenicity in humans.
Materials and methods
Cell culture and transfection
HeLa and Vero E6 cells were grown in Dulbecco's modified Eagle medium containing 10% fetal bovine serum, 100 U/ml penicillin G and 100 mg/ml streptomycin. All the transfections were carried out using Lipofectamine-2000 (Invitrogen) according to the manufacturer's instructions. Due to the high sensitivity of 3XFLAG system, the amount of 3XFLAG-ATF6 plasmid used to transfect HeLa cells was only 1/5–1/10 amount of other mammalian expression plasmids.
Materials and plasmid construction
Thapsigargin, Tunicamycin and anti-FLAG M2 antibody were obtained from Sigma. Protein G Sepharose, protease inhibitor cocktail for mammalian cell, rat monoclonal anti-HA antibody were from Roche. Horseradish peroxidase-conjugated goat anti-mouse, anti-rabbit and anti-rat IgG sera and ECL plus reagents were from Pierce. Rabbit polyclonal anti-GRP78 antibody, rabbit polyclonal anti-calreticulin antibody and rabbit polyclonal anti-CHOP antibody were from Santa Cruz. Mouse monoclonal anti-KDEL antibody was from Stressgen, and mouse monoclonal anti-calnexin antibody was from Chemicon. Alexa Fluor 594-conjugated anti-rabbit, Alexa Fluor 568-conjugated anti-rat, Alexa Fluor 488-conjugated anti-mouse or anti-rabbit antibodies were from Molecular Probes. Endoglycosidase H (EndoH) and peptide-N-glycosidase F (PNase F) were from New England Biolab.
RNA extracted from a SARS patient throat swap specimen was kindly provided by Taiwan Centers for Disease Control. The cDNA fragment containing ORF8 was produced by RT-PCR using primer 27147F (GGATTGCTATTTGGAATCTT) and primer 28142R (GTAATGCGGGGGGCATACGTTG). The PCR products were cloned into TOPO TA cloning vector (Invitrogen), and the sequences were confirmed by sequencing. This cloned ORF8 contained the 29-nucleotide deletion in ORF8 region. To express proteins 8a and 8b, the cDNA of ORF8a and ORF8b were amplified by PCR and cloned into the mammalian expression vector pCI-HA modified from pCI. For construction of the plasmid expressing 8ab protein, primers were designed based on an early-phase human SARS-CoV isolate, GZ02 (Genbank accession number AY390556), which contains the 29-nucleotide insertion in ORF8 (Keng et al., 2006). Jumping and sequential PCR were used to insert the 29-nt to construct pCI-8abHA. For the construction of 3XFLAG-8ab, cDNA of 8ab was amplified then cloned into p3XFLAG-CMV vector (Sigma). The 3XFLAG-ATF6 containing three tandem copies of FLAG tagged at N-terminus of ATF6 was kindly provided by Dr. Ron Prywes (Columbia university New York, New York) (Shen et al., 2002). Mutants of 3XFLAG-ATF6S1P−, 3XFLAG-ATF6 (418) and 3XFLAG-ATF6 (373) were made using a QuickChange site-directed mutagenesis kit (Strategene). The GRP78-luciferase reporter driven by base pairs (bp) − 304 to + 7 of the Grp78 promoter was kindly provided by Dr. Kazutoshi Mori (Kyoto Research Park, Kyoto, Japan) (Yoshida et al., 1998). The 5XATF6-GL3 in which the luciferase gene is under the control of the c-fos minimal promoter and five tandem copies of the ATF6 consensus binding sites was kindly provided by Dr. Ron Prywes (Columbia university New York, New York) (Wang et al., 2000).
RNA preparation and RT/PCR
Total RNA from culture cells were prepared with TRIZOL Reagent (Invitrogen), and cDNA was reverse transcribed from 1 μg of total RNA with random hexamers using Superscript II (Invitrogen). To amplify xbp1 mRNA, PCR was performed for 30 cycles (94 °C for 30 s, 50 °C for 30 s, and 72 °C for 1 min), using pair of primers corresponding to nucleotides 412–431 (CCTTGTAGTTGAGAACCAGG) and 834–853 (GGGGCTTGGTATATATGTT) of XBP1 cDNA. Primers 5′-TCACCCACACTGTGCCCATCTACG-3′ and 5′-CAGCGGAACCGCTCATTGCCAATGG-3′ were used for amplification of β-actin mRNA. Primers 5′-AACTTCTCATTGTTTTGACTT-3′, and 5′-ACAGCGTAATCTGGAACATC-3′were used for amplification of 8ab mRNA.
Immunoprecipitation, western blotting and glycosidase digestion
Cells were lysed in HEPES-Triton-X 100 buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 1% Triton-X 100, 1 mM EDTA and 10% glycerol) supplemented with 2 mM phenylmethylsulfonyl fluorides and Complete protease inhibitor cocktail (Roche). After rotating at 4 °C for 1 h, cell lysates were harvested by centrifugation at 13,000 ×g for 10 min at 4 °C. 3XFLAG-ATF6 or 8abHA was immunoprecipitated by anti-FLAG M2 antibodies plus 30 μl of protein A Sepharose in IP buffer. After being washed three times with IP buffer, the immunoprecipitates were resolved in SDS-PAGE and then transferred to polyvinylidene difluoride (PVDF) membrane (GE Healthcare). The proteins were then probed with appropriate antibodies and followed by horseradish peroxidase-conjugated secondary antibodies. The blots were visualized by ECL plus reagent. For glycosidase digestion, 20 μg of cell lysates were directly subjected to PNGase F (New England Biolabs) and Endo H digestion (New England Biolabs) in the buffer provided by the manufacturer at 37 °C for 2 h. Digested samples were mixed with an equal volume of 2 × Laemmli sample buffer and analyzed by Western blot.
Luciferase reporter assay
HeLa cells in 24-well plates were transfected with 600 ng of a GRP78-luciferase reporter or 5XATF6-GL3 reporter plasmid with 300 ng of pCI-8abHA or empty vector and 20 ng of pRL-TK carrying the Renilla luciferase gene as an internal control for transfection efficiency. The cells were incubated for 24 h, and were either left untreated or treated with 500 nM thapsigargin for 16 h. Cells were then lysed and assayed for firefly Renilla luciferase activities using Dual-Glo™ Luciferase Assays System (Promega). Relative activity was defined as the ratio of firefly luciferase activity and Renilla luciferase activity. All experiments were performed with triplicate plates, and data are shown as the average of three independent experiments.
Confocal immunofluorescence microscopy
Vero E6 or HeLa cells transfected in multiwell chambers were fixed with 4% paraformaldehyde in PBS and then permeabilized with cold methanol for 5 min at room temperature. Cells were blocked with PBS containing 3% BSA at room temperature for 1 h, and then incubated with appropriate primary antibodies for 1 h at room temperature. After washing with PBS containing 0.1% Tween 20, cells were incubated with Alexa Fluor 568, 488-conjugated secondary antibodies (Molecular Probes) for 1 h at room temperature. The slides were then mounted with Prolong Gold anti-fade reagent (Molecular Probes) and visualized by confocal laser scanning microscopy (Zeiss, LSM 510). For epitope orientation study, cells were fixed with 2% paraformaldehyde for 40 min at 20 °C, and then subjected to total permeabilization or selective permeabilization as described (Ivashkina et al., 2002). For total permeabilization, cells were permeabilized with 0.05% saponin (Sigma) for 10 min at 20 °C, whereas selective permeabilization was performed for 15 min at 4 °C with 0.000125% digitonin (Calbiochem) in a buffer containing 10 mM HEPES (pH 6.8), 1 mM EDTA, 0.3 M sucrose, 0.1 M KCl, and 2.5 mM MgCl2. Indirect immunofluorescence microscopy was then performed as described previously. Anti-HA, anti-calreticulin, and anti-FLAG antibodies were used as primary antibodies, and Alexa Fluor 488-conjugated antibodies (Molecular Probes) were used as secondary antibodies.
Microsomes preparation and solubilization of 8ab proteins
HeLa cells (6 × 107) transiently transfected with pCI-8abHA were homogenized in hypotonic buffer with Dounce homogenizer, and microsomes were prepared as described (Shen and Hendershot, 2005). Briefly, the crude homogenates were centrifuged at 500 ×g for 10 min to pellet cell debris and nuclei. The supernatant consisting of ER microsomes and cytosol was divided into four aliquots and centrifuged at 16,000 ×g to pellet the microsomes. The ER microsomes were subsequently re-suspended in 100 μl of phosphate-buffered saline (PBS) buffer alone or PBS containing either 0.1% or 0.2% digitonin, or 0.1% deoxycholic acid. After incubation at 4 °C for 1 h, samples were centrifuged at 16,000 ×g for 5 min to sediment residual membrane. The supernatant and pellets were separately loaded into SDS-PAGE and analyzed by Western blot. For 1 M NaCl and sodium bicarbonate treatments, microsomes were re-suspended gently in PBS, 1 M NaCl or 100 mM sodium carbonate, pH 11.5 and incubated at 4 °C for 1 h. After centrifuging at 16,000 ×g for 5 min to sediment residual membrane, supernatant and pellets were analyzed as previously described (Shen and Hendershot, 2005).
References
- Chan C.P., Siu K.L., Chin K.T., Yuen K.Y., Zheng B., Jin D.Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006;80(18):9279–9287. doi: 10.1128/JVI.00659-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Shen J., Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 2002;277(15):13045–13052. doi: 10.1074/jbc.M110636200. [DOI] [PubMed] [Google Scholar]
- Chinese S.M.E.C. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303(5664):1666–1669. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- de Haan C.A., Masters P.S., Shen X., Weiss S., Rottier P.J. The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology. 2002;296(1):177–189. doi: 10.1006/viro.2002.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L., Chan K.W., Lim W., Shortridge K.F., Yuen K.Y., Peiris J.S., Poon L.L. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science. 2003;302(5643):276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Harding H.P., Zhang Y., Bertolotti A., Zeng H., Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell. 2000;5(5):897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Haze K., Yoshida H., Yanagi H., Yura T., Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 1999;10(11):3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006;13(3):393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- Ivashkina N., Wolk B., Lohmann V., Bartenschlager R., Blum H.E., Penin F., Moradpour D. The hepatitis C virus RNA-dependent RNA polymerase membrane insertion sequence is a transmembrane segment. J. Virol. 2002;76(24):13088–13093. doi: 10.1128/JVI.76.24.13088-13093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keng C.T., Choi Y.W., Welkers M.R., Chan D.Z., Shen S., Gee Lim S., Hong W., Tan Y.J. The human severe acute respiratory syndrome coronavirus (SARS-CoV) 8b protein is distinct from its counterpart in animal SARS-CoV and down-regulates the expression of the envelope protein in infected cells. Virology. 2006;354(1):132–142. doi: 10.1016/j.virol.2006.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K., Tirasophon W., Shen X., Michalak M., Prywes R., Okada T., Yoshida H., Mori K., Kaufman R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16(4):452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Baumeister P., Roy B., Phan T., Foti D., Luo S., Lee A.S. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell. Biol. 2000;20(14):5096–5106. doi: 10.1128/mcb.20.14.5096-5106.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman E., Fong Y.L., Selby M.J., Choo Q.L., Cousens L., Houghton M., Yen T.S. Activation of the grp78 and grp94 promoters by hepatitis C virus E2 envelope protein. J. Virol. 1999;73(5):3718–3722. doi: 10.1128/jvi.73.5.3718-3722.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin M., Monne M., Widell A., Von Heijne G., Persson M.A. Topology of the membrane-associated hepatitis C virus protein NS4B. J. Virol. 2003;77(9):5428–5438. doi: 10.1128/JVI.77.9.5428-5438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The genome sequence of the SARS-associated coronavirus. Science. 2003;300(5624):1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- McCullough K.D., Martindale J.L., Klotz L.O., Aw T.Y., Holbrook N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001;21(4):1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostra M., de Haan C.A., Rottier P.J. The 29-nucleotide deletion present in human but not in animal severe acute respiratory syndrome coronaviruses disrupts the functional expression of open reading frame 8. J. Virol. 2007;81(24):13876–13888. doi: 10.1128/JVI.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S., Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Pavio N., Romano P.R., Graczyk T.M., Feinstone S.M., Taylor D.R. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J. Virol. 2003;77(6):3578–3585. doi: 10.1128/JVI.77.6.3578-3585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Chu C.M., Cheng V.C., Chan K.S., Hung I.F., Poon L.L., Law K.I., Tang B.S., Hon T.Y., Chan C.S., Chan K.H., Ng J.S., Zheng B.J., Ng W.L., Lai R.W., Guan Y., Yuen K.Y. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361(9371):1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361(9366):1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Rutkowski D.T., Kaufman R.J. A trip to the ER: coping with stress. Trends Cell. Biol. 2004;14(1):20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Sarma J.D., Scheen E., Seo S.H., Koval M., Weiss S.R. Enhanced green fluorescent protein expression may be used to monitor murine coronavirus spread in vitro and in the mouse central nervous system. J. Neurovirol. 2002;8(5):381–391. doi: 10.1080/13550280260422686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M., Kaufman R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Shen J., Chen X., Hendershot L., Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell. 2002;3(1):99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- Shen J., Prywes R. ER stress signaling by regulated proteolysis of ATF6. Methods. 2005;35(4):382–389. doi: 10.1016/j.ymeth.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Shen J., Snapp E.L., Lippincott-Schwartz J., Prywes R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell. Biol. 2005;25(3):921–932. doi: 10.1128/MCB.25.3.921-932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Hendershot L.M. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP's interactions with unfolded substrates. Mol. Biol. Cell. 2005;16(1):40–50. doi: 10.1091/mbc.E04-05-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola I., Alonso S., Zuniga S., Balasch M., Plana-Duran J., Enjuanes L. Engineering the transmissible gastroenteritis virus genome as an expression vector inducing lactogenic immunity. J. Virol. 2003;77(7):4357–4369. doi: 10.1128/JVI.77.7.4357-4369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H.D., Rappuoli R. SARS—beginning to understand a new virus. Nat. Rev. Microbiol. 2003;1(3):209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y.J., Lim S.G., Hong W. Understanding the accessory viral proteins unique to the severe acute respiratory syndrome (SARS) coronavirus. Antiviral. Res. 2006;72(2):78–88. doi: 10.1016/j.antiviral.2006.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardif K.D., Mori K., Kaufman R.J., Siddiqui A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004;279(17):17158–17164. doi: 10.1074/jbc.M312144200. [DOI] [PubMed] [Google Scholar]
- Tardif K.D., Mori K., Siddiqui A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 2002;76(15):7453–7459. doi: 10.1128/JVI.76.15.7453-7459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardif K.D., Waris G., Siddiqui A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005;13(4):159–163. doi: 10.1016/j.tim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H.P., Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Wang X.Z., Harding H.P., Zhang Y., Jolicoeur E.M., Kuroda M., Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. Embo J. 1998;17(19):5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Shen J., Arenzana N., Tirasophon W., Kaufman R.J., Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 2000;275(35):27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- Ye J., Rawson R.B., Komuro R., Chen X., Dave U.P., Prywes R., Brown M.S., Goldstein J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell. 2000;6(6):1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Haze K., Yanagi H., Yura T., Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998;273(50):33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Hosokawa N., Kaufman R.J., Nagata K., Mori K. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell. 2003;4(2):265–271. doi: 10.1016/s1534-5807(03)00022-4. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M., Mori K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000;20(18):6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount B., Roberts R.S., Sims A.C., Deming D., Frieman M.B., Sparks J., Denison M.R., Davis N., Baric R.S. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J. Virol. 2005;79(23):14909–14922. doi: 10.1128/JVI.79.23.14909-14922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R.T., Remotti H., Stevens J.L., Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12(7):982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]